User login
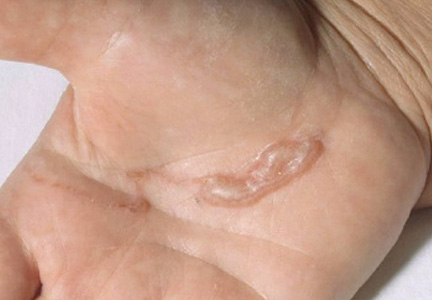
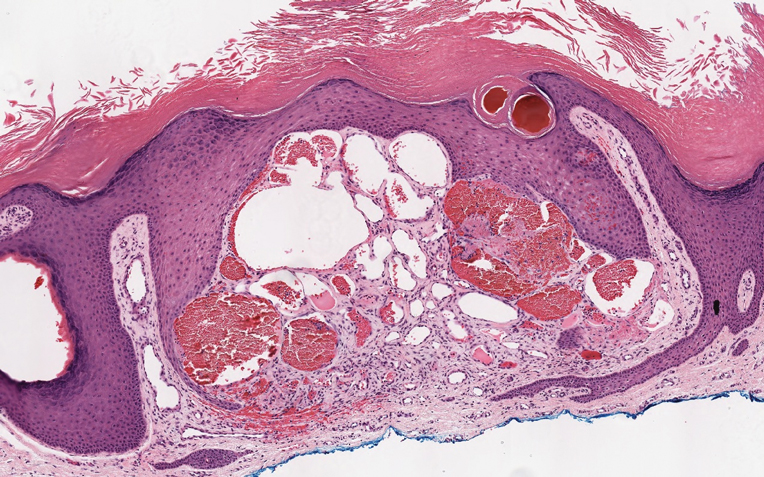
Soft Nodule on the Forearm
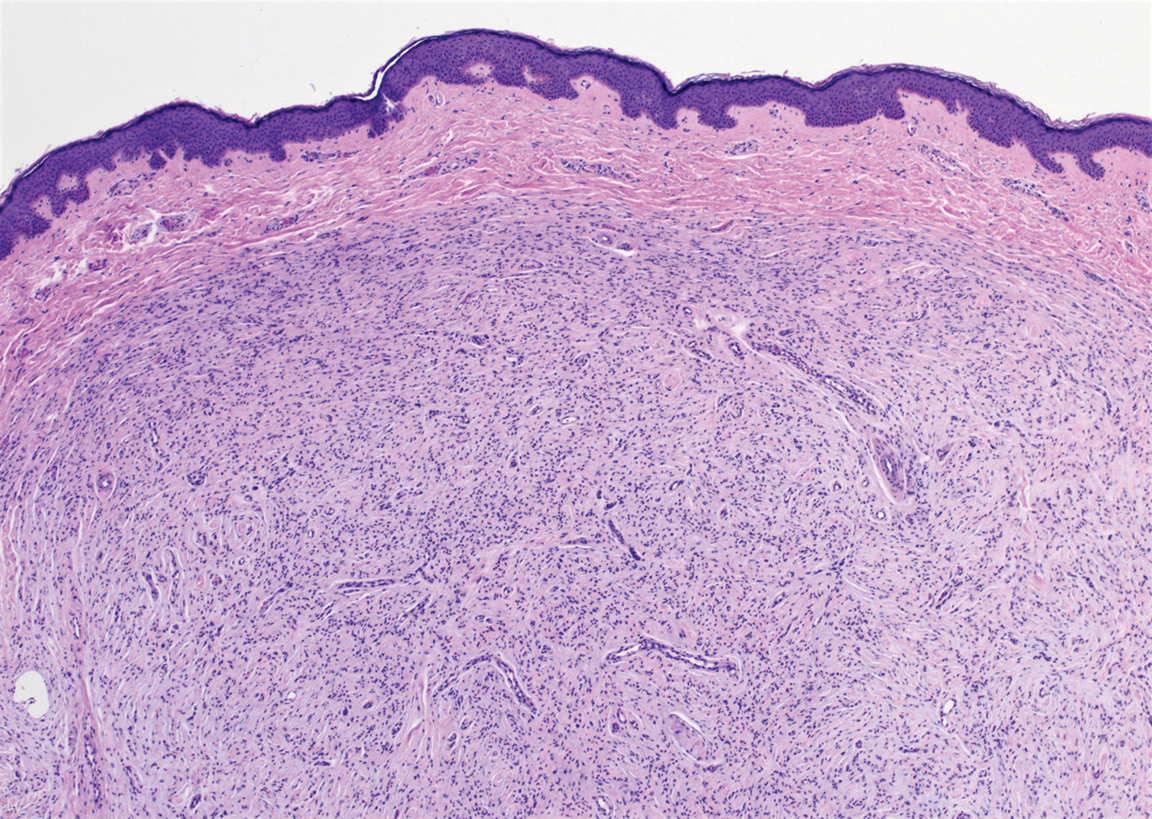
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
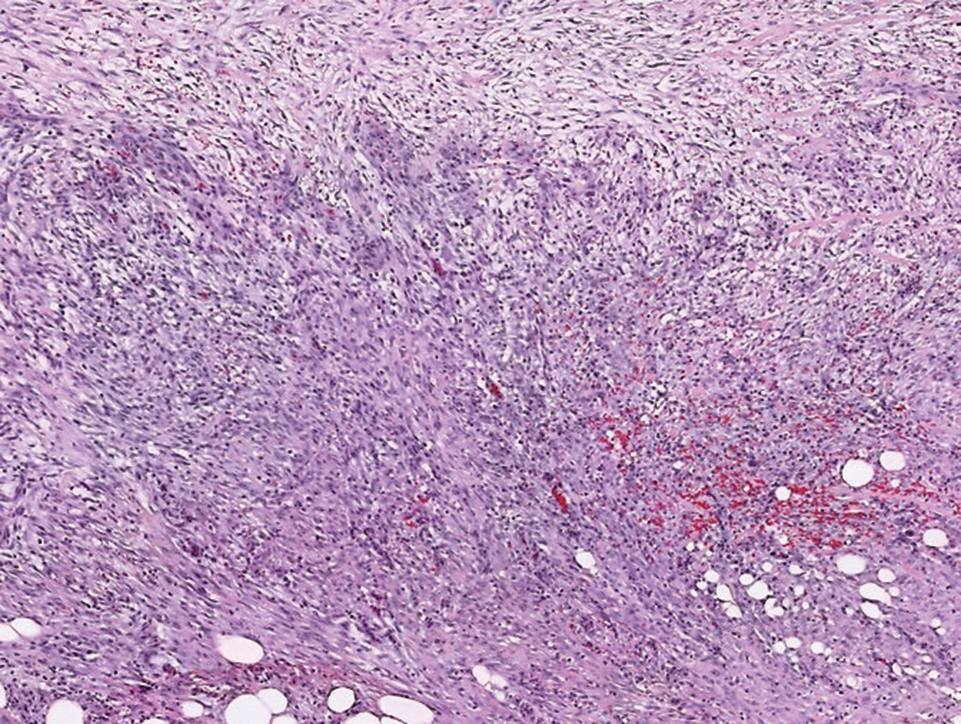
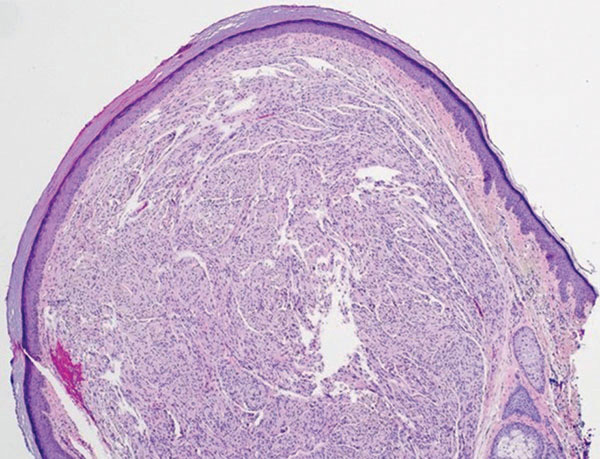
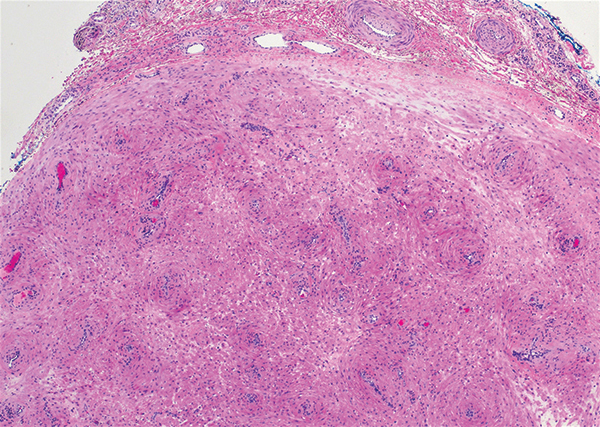
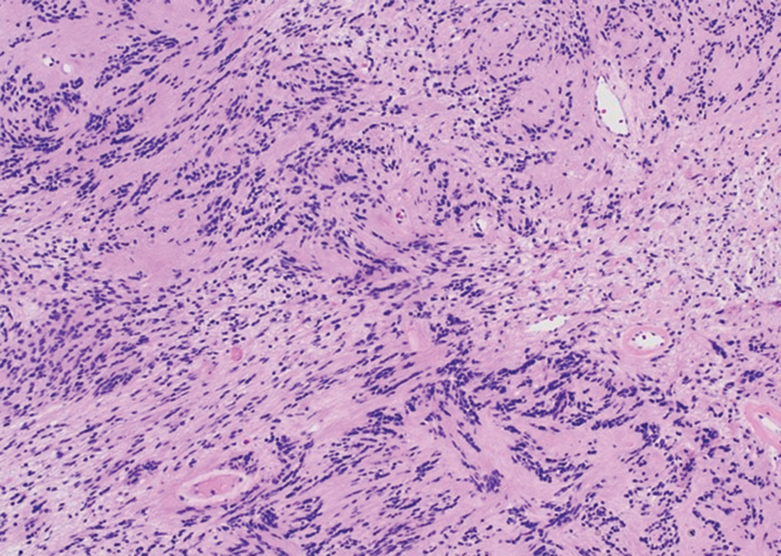
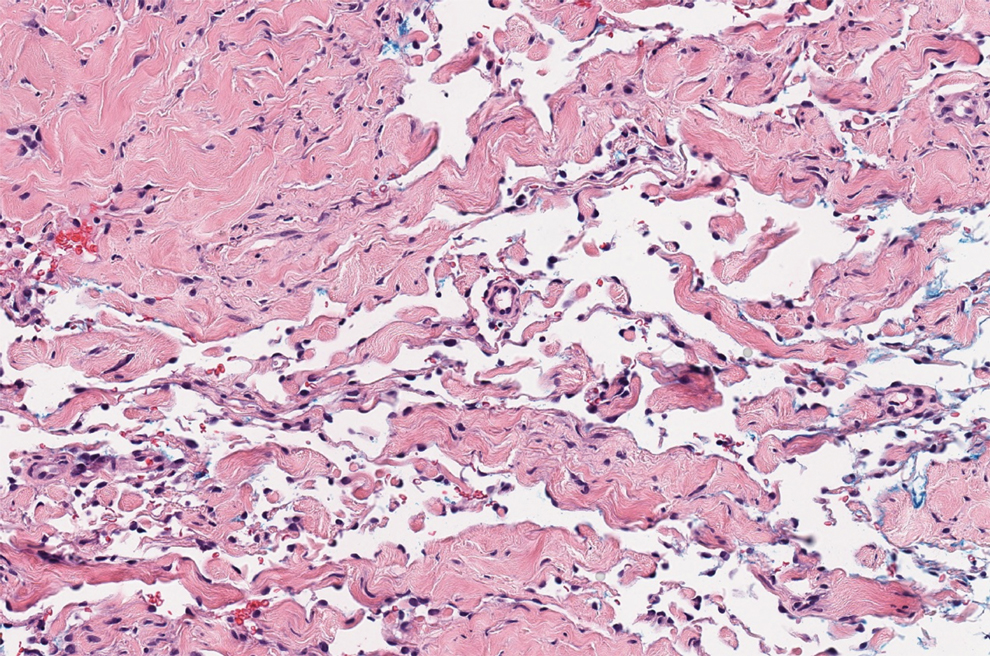
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
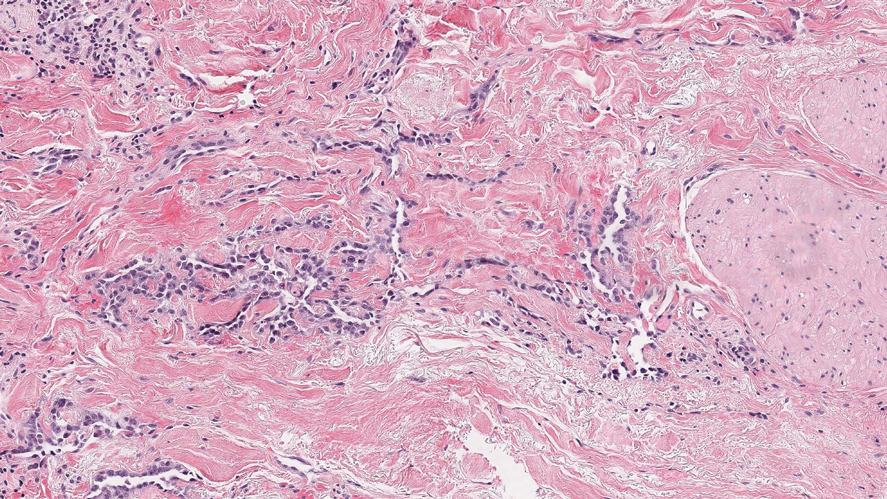
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
The Diagnosis: Schwannoma
Schwannoma, also known as neurilemmoma, is a benign encapsulated neoplasm of the peripheral nerve sheath that presents as a subcutaneous nodule.1 It also may present in the retroperitoneum, mediastinum, and viscera (eg, gastrointestinal tract, bone, upper respiratory tract, lymph nodes). It may occur as multiple lesions when associated with certain syndromes. It usually is an asymptomatic indolent tumor with neurologic symptoms, such as pain and tenderness, in the lesions that are deeper, larger, or closer in proximity to nearby structures.2,3
Histologically, a schwannoma is encapsulated by the perineurium of the nerve bundle from which it originates (quiz image [top]). The tumor consists of hypercellular (Antoni type A) and hypocellular (Antoni type B) areas. Antoni type A areas consist of tightly packed, spindleshaped cells with elongated wavy nuclei and indistinct cytoplasmic borders. These nuclei tend to align into parallel rows with intervening anuclear zones forming Verocay bodies (quiz image [bottom]).4 Verocay bodies are not seen in all schwannomas, and similar formations may be seen in other tumors as well. Solitary circumscribed neuromas also have Verocay bodies, whereas dermatofibromas and leiomyomas have Verocay-like bodies. Antoni type B areas have scattered spindled or ovoid cells in an edematous or myxoid matrix interspersed with inflammatory cells such as lymphocytes and histiocytes. Vessels with thick hyalinized walls are a helpful feature in diagnosis.2 Schwann cells of a schwannoma stain diffusely positive with S-100 protein. The capsule stains positively with epithelial membrane antigen due to the presence of perineurial cells.2
The morphologic variants of this entity include conventional (common, solitary), cellular, plexiform, ancient, melanotic, epithelioid, pseudoglandular, neuroblastomalike, and microcystic/reticular schwannomas. There are additional variants that are associated with genetic syndromes, such as multiple cutaneous plexiform schwannomas linked with neurofibromatosis type 2, psammomatous melanotic schwannoma presenting in Carney complex, schwannomatosis, and segmental schwannomatosis (a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb). Either presentation may have alteration or deletion of the neurofibromatosis type 2 gene, NF2, on chromosome 22.2,5
Nodular fasciitis is a benign tumor of fibroblasts and myofibroblasts that usually arises in the subcutaneous tissues. It most commonly occurs in the upper extremities, trunk, head, and neck. It presents as a single, often painful, rapidly growing, subcutaneous nodule. Histologically, lesions mostly are well circumscribed yet unencapsulated, in contrast to schwannomas. They may be hypocellular or hypercellular and are composed of uniform spindle cells with a feathery or fascicular (tissue culture–like) appearance in a loose, myxoid to collagenous stroma. There may be foci of hemorrhage and conspicuous mitoses but not atypical figures (Figure 1). Immunohistochemically, the cells stain positively for smooth muscle actin and negatively for S-100 protein, which sets it apart from a schwannoma. Most cases contain fusion genes, with myosin heavy chain 9 ubiquitin-specific peptidase 6, MYH9-USP6, being the most common fusion product.6
Solitary circumscribed neuroma (palisaded encapsulated neuroma) is a benign, usually solitary dermal lesion. It most commonly occurs in middle-aged to elderly adults as a small (<1 cm), firm, flesh-colored to pink papule on the face (ie, cheeks, nose, nasolabial folds) and less commonly in the oral and acral regions and on the eyelids and penis. The lesion usually is unilobular; however, other growth patterns such as plexiform, multilobular, and fungating variants have been identified. Histologically, it is a well-circumscribed nodule with a thin capsule of perineurium that is composed of interlacing bundles of Schwann cells with a characteristic clefting artifact (Figure 2). Cells have wavy dark nuclei with scant cytoplasm that occasionally form palisades or Verocay bodies causing these lesions to be confused with schwannomas. Immunohistochemically, the Schwann cells stain positively with S-100 protein, and the perineurium stains positively with epithelial membrane antigen, Claudin-1, and Glut-1. Neurofilament protein stains axons throughout neuromas, whereas in schwannoma, the expression often is limited to entrapped axons at the periphery of the tumor.7
Angioleiomyoma is an uncommon, benign, smooth muscle neoplasm of the skin and subcutaneous tissue that originates from vascular smooth muscle. It most commonly presents in adult females aged 30 to 60 years, with a predilection for the lower limbs. These tumors typically are solitary, slow growing, and less than 2 cm in diameter and may be painful upon compression. Similar to schwannoma, angioleiomyoma is an encapsulated lesion composed of interlacing, uniform, smooth muscle bundles distributed around vessels (Figure 3). Smooth muscle cells have oval- or cigar-shaped nuclei with a small perinuclear vacuole of glycogen. Immunohistochemically, there is strong diffuse staining for smooth muscle actin and h-caldesmon. Recurrence after excision is rare.2,8
Neurofibroma is a common, mostly sporadic, benign tumor of nerve sheath origin. The solitary type may be localized (well circumscribed, unencapsulated) or diffuse. The presence of multiple, deep, and plexiform lesions is associated with neurofibromatosis type 1 (von Recklinghausen disease) that is caused by germline mutations in the NF1 gene. Histologically, the tumor is composed of Schwann cells, fibroblasts, perineurial cells, and nerve axons within an extracellular myxoid to collagenous matrix (Figure 4). The diffuse type is an ill-defined proliferation that entraps adnexal structures. The plexiform type is defined by multinodular serpentine fascicles. Immunohistochemically, the Schwann cells stain positive for S-100 protein and SOX10 (SRY-Box Transcription Factor 10). Epithelial membrane antigen stains admixed perineurial cells. Neurofilament protein highlights intratumoral axons, which generally are not found throughout schwannomas. Transformation to a malignant peripheral nerve sheath tumor occurs in up to 10% of patients with neurofibromatosis type 1, usually in plexiform neurofibromas, and is characterized by increased cellularity, atypia, mitotic activity, and necrosis.9
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.
- Ritter SE, Elston DM. Cutaneous schwannoma of the foot. Cutis. 2001;67:127-129.
- Calonje E, Damaskou V, Lazar AJ. Connective tissue tumors. In: Calonje E, Brenn T, Lazar AJ, et al, eds. McKee’s Pathology of the Skin. 5th ed. Vol 2. Elsevier Saunders; 2020:1698-1894.
- Knight DM, Birch R, Pringle J. Benign solitary schwannomas: a review of 234 cases. J Bone Joint Surg Br. 2007;89:382-387.
- Lespi PJ, Smit R. Verocay body—prominent cutaneous leiomyoma. Am J Dermatopathol. 1999;21:110-111.
- Kurtkaya-Yapicier O, Scheithauer B, Woodruff JM. The pathobiologic spectrum of schwannomas. Histol Histopathol. 2003;18:925-934.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514.
- Yeung CM, Moore L, Lans J, et al. Angioleiomyoma of the hand: a case series and review of the literature. Arch Bone Jt Surg. 2020; 8:373-377.
- Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin North Am. 2004;15:157-166.

A 54-year-old woman presented with an enlarging mass on the right volar forearm. Physical examination revealed a 1-cm, soft, mobile, subcutaneous nodule. Excision revealed tan-pink, indurated, fibrous, nodular tissue.
Dermatopathology Etiquette 101
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
Violaceous Papule With an Erythematous Rim
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
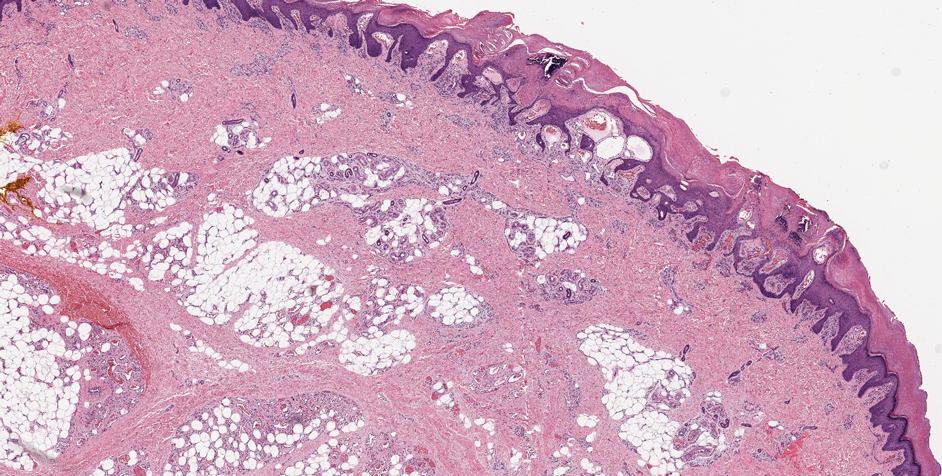
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.

A 35-year-old man presented with a reddish brown papule on the left upper chest of 1 year’s duration that had changed color to reddish purple. Physical examination revealed a 6-mm violaceous papule with an erythematous rim.
Distinct Violaceous Plaques in Conjunction With Blisters
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
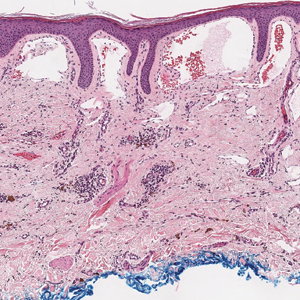
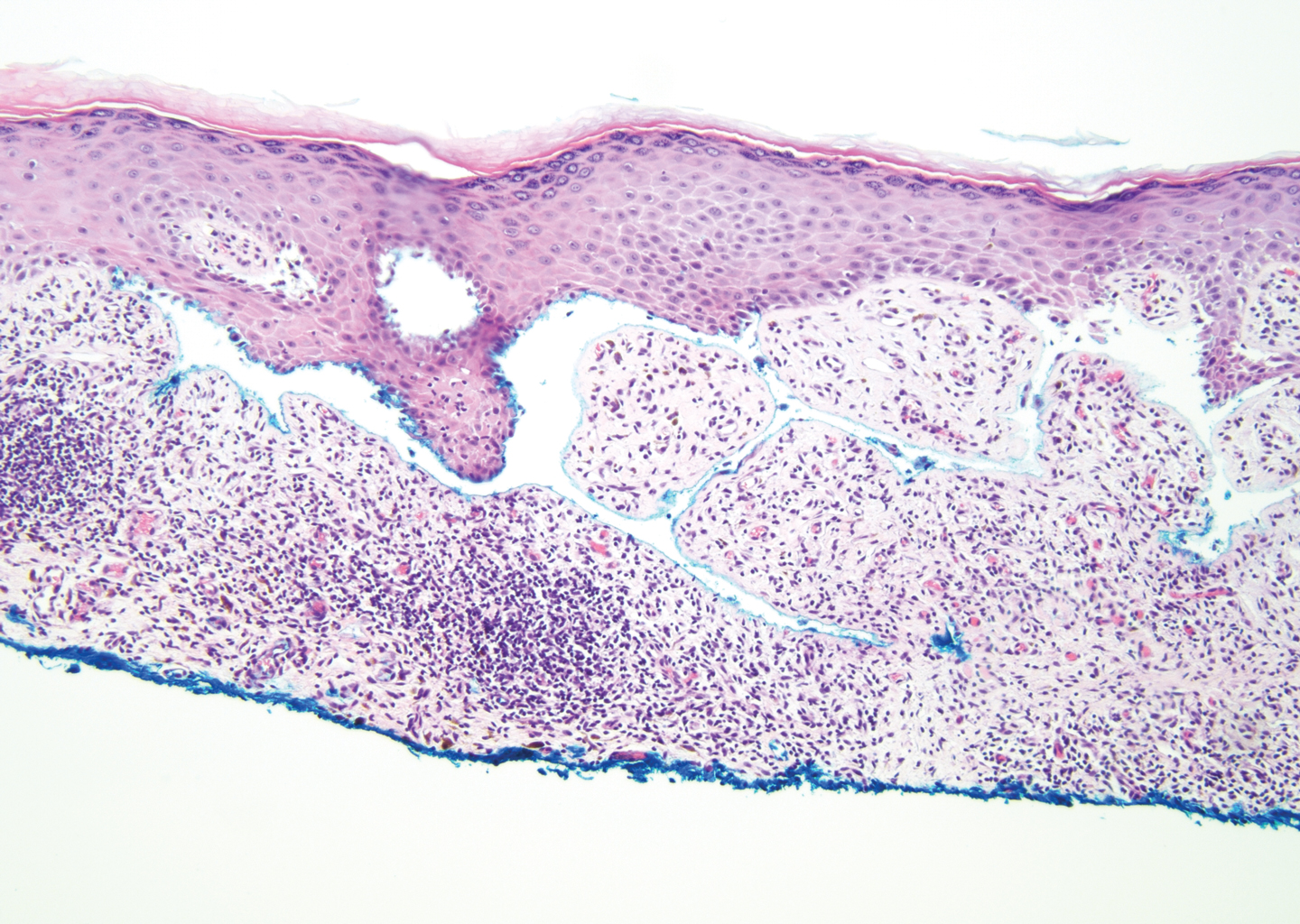
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
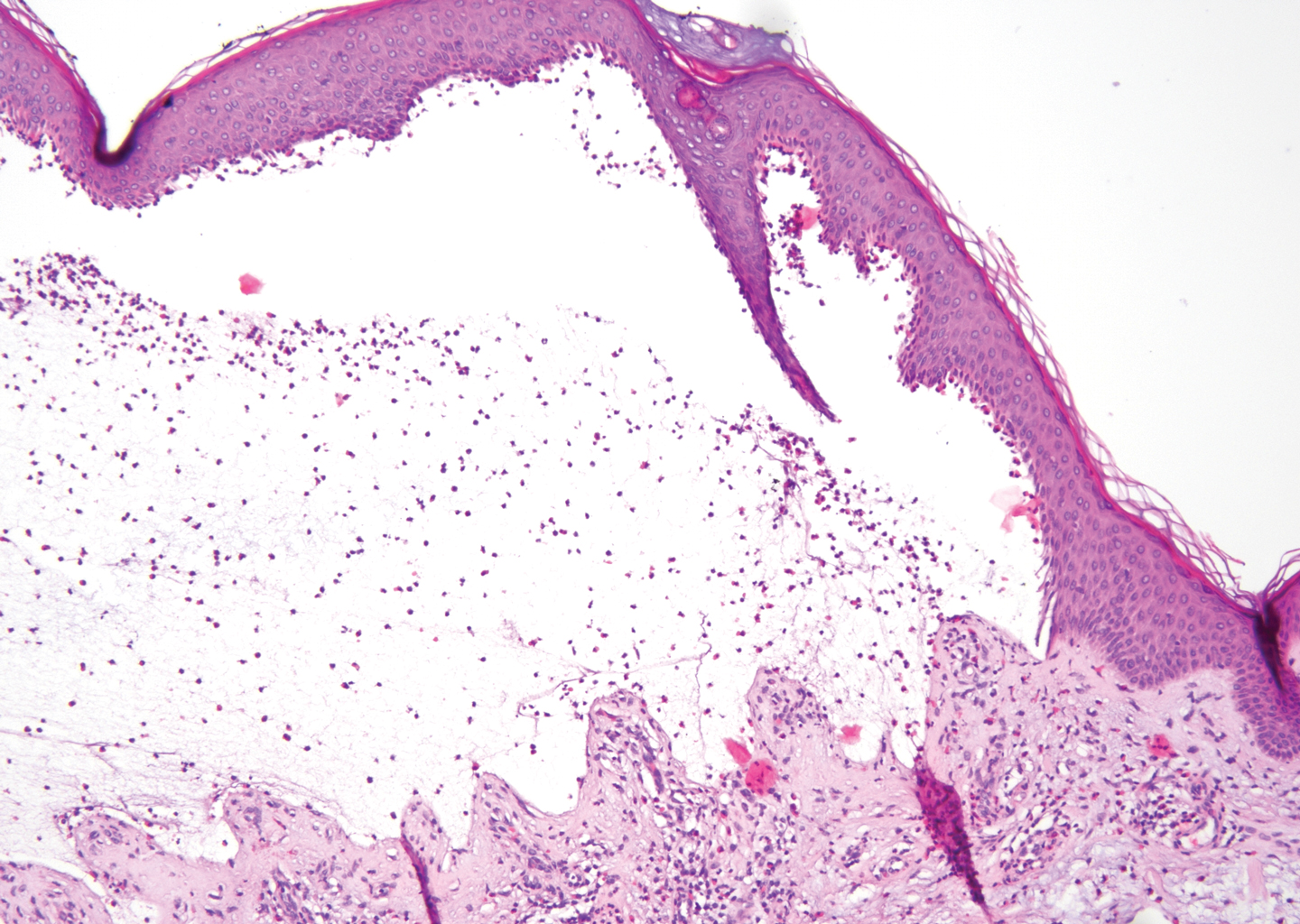
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11

Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12

Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
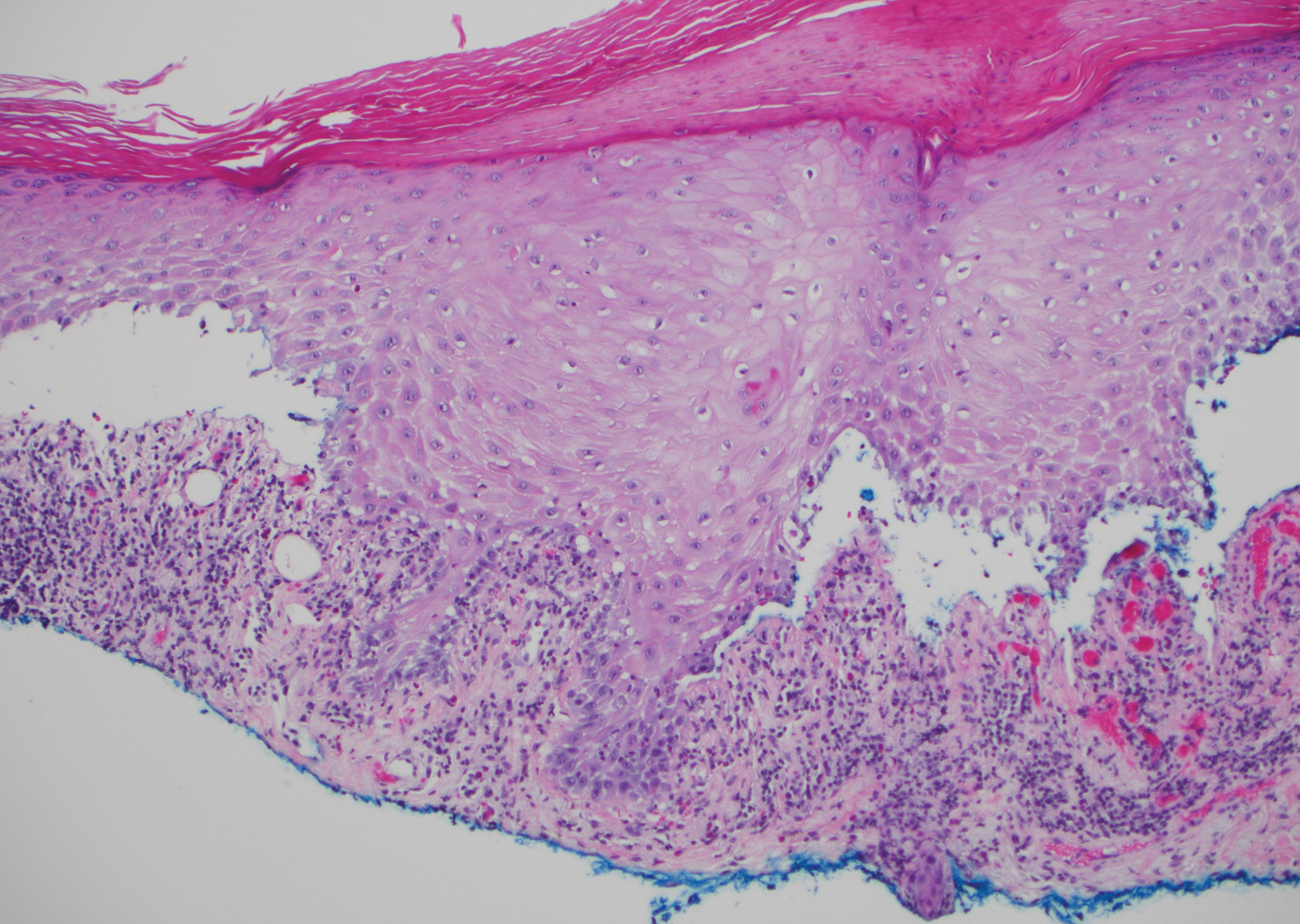
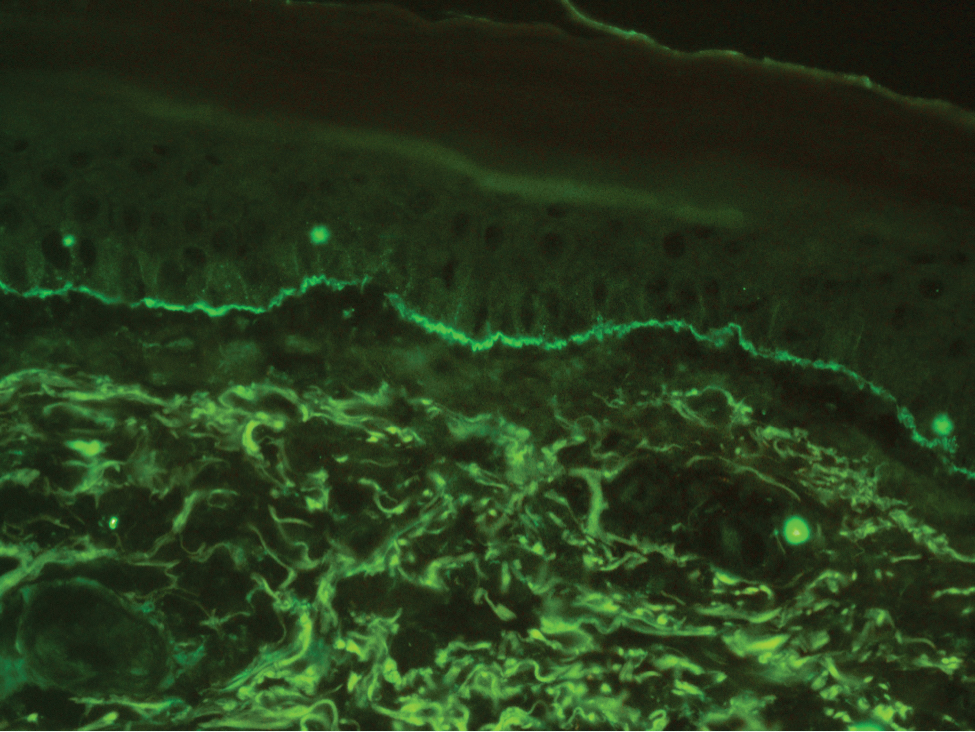
A 72-year-old woman presented to our dermatology clinic with a rash of several months' duration that began as itchy bumps on the wrists and spread to involve the legs. Approximately 2 months prior to presentation, she noted blisters on the feet and legs. She initially went to her primary care physician, who prescribed levofloxacin, cephalexin, and a 5-day course of prednisone. The prednisone initially helped; however the rash worsened on discontinuation. In our clinic, the patient had scattered tense bullae and numerous erosions with crust on the dorsum of the feet and legs, some of which were in conjunction with violaceous papules and plaques. There also was hypertrophic scale on the soles of the feet. A potassium hydroxide preparation of skin scrapings from the feet was negative for fungal elements. Two shave biopsies of a violaceous plaque and bulla as well as a perilesional punch biopsy from the leg were obtained.
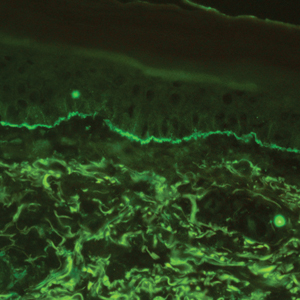
Multiple Pink Papules on the Chest and Upper Abdomen
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6


Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
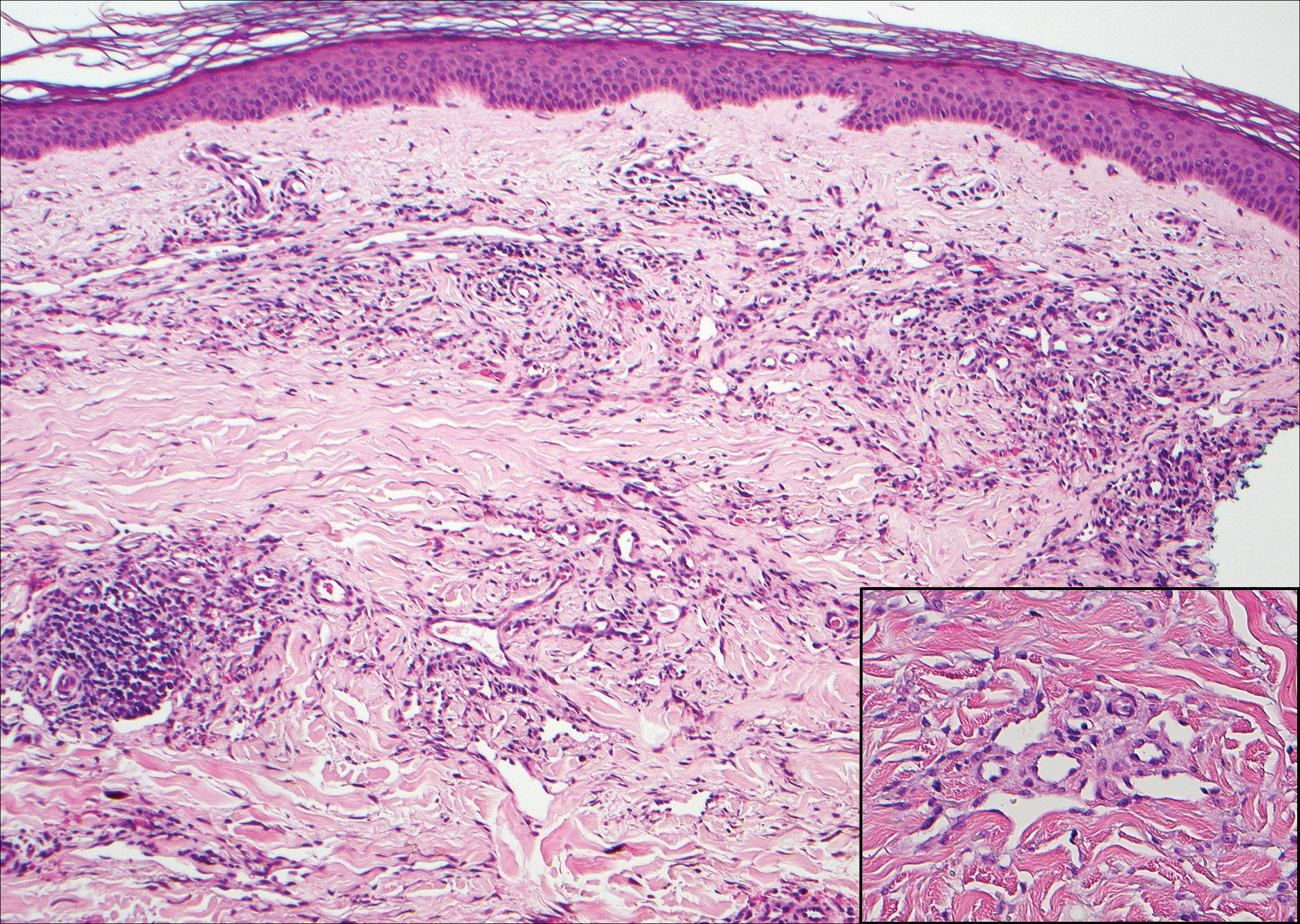
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
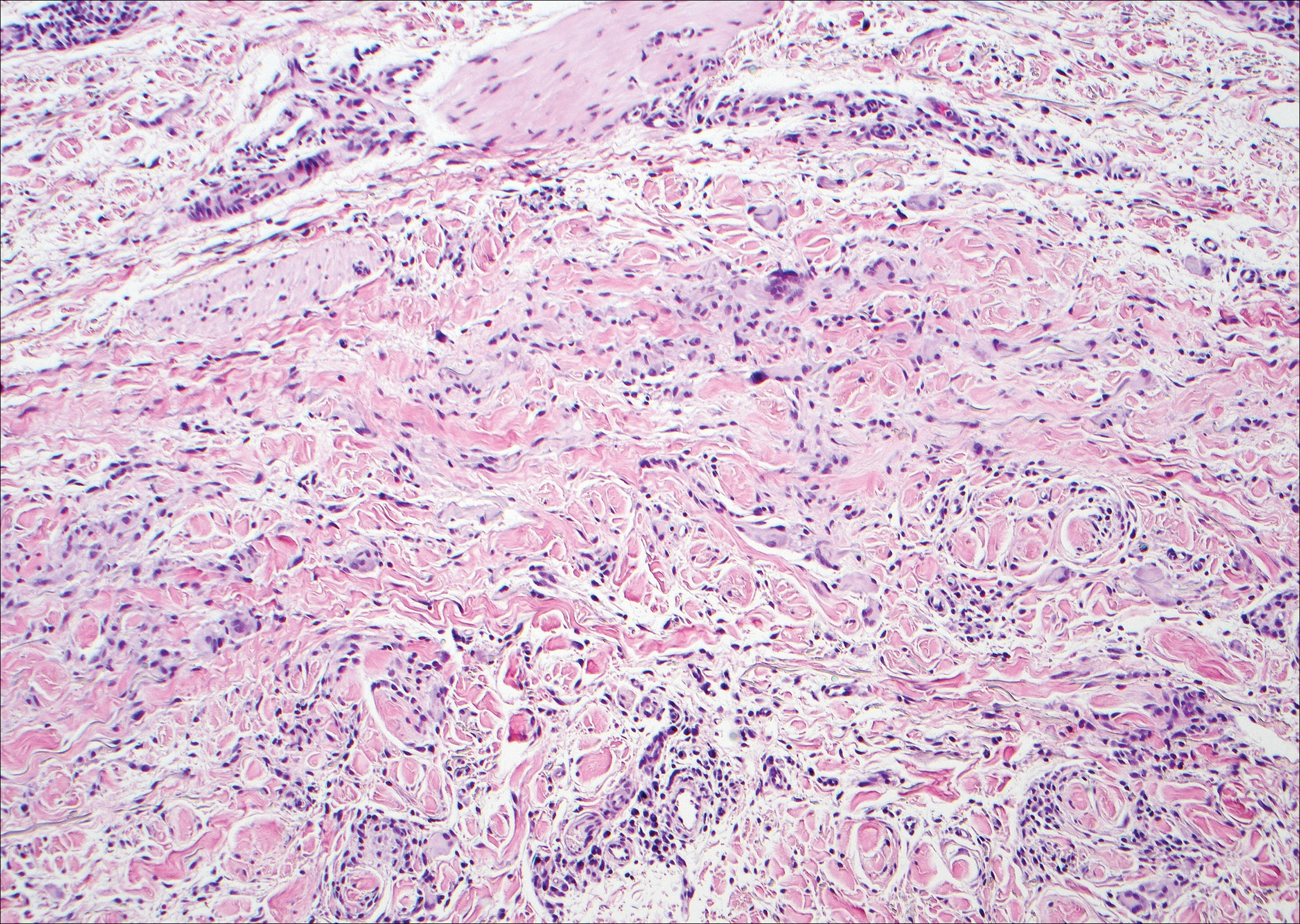
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
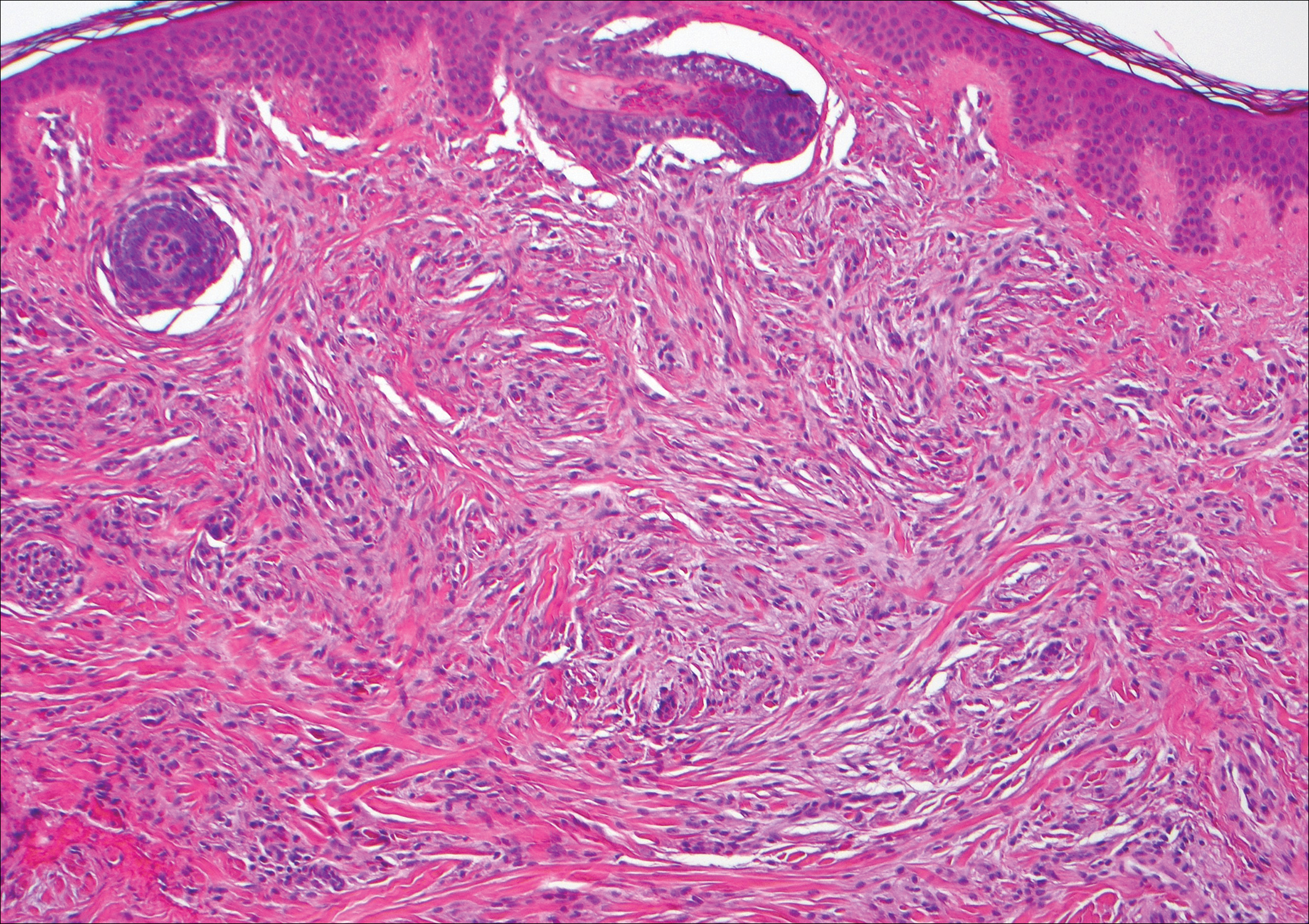
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
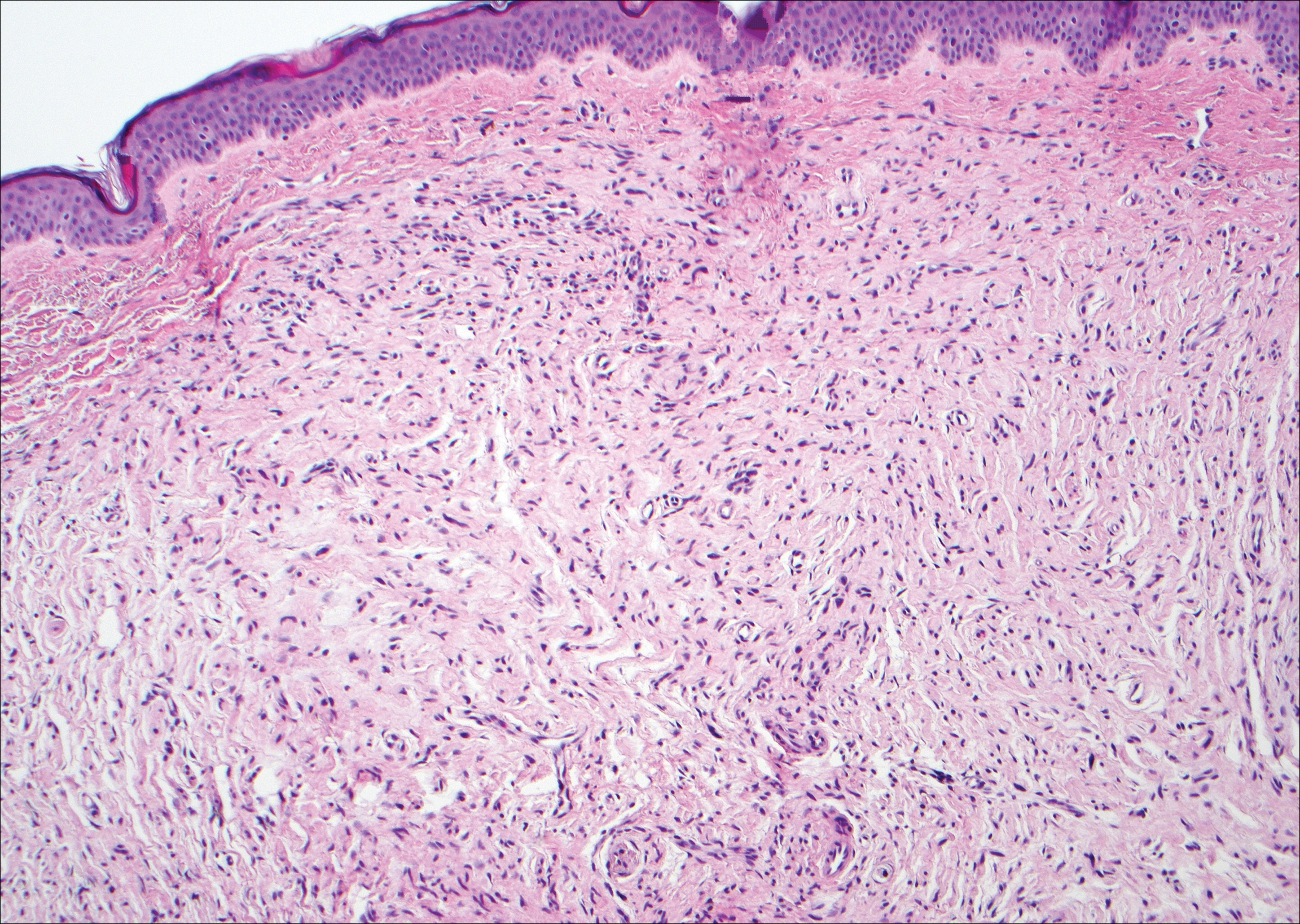
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6
Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6
Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
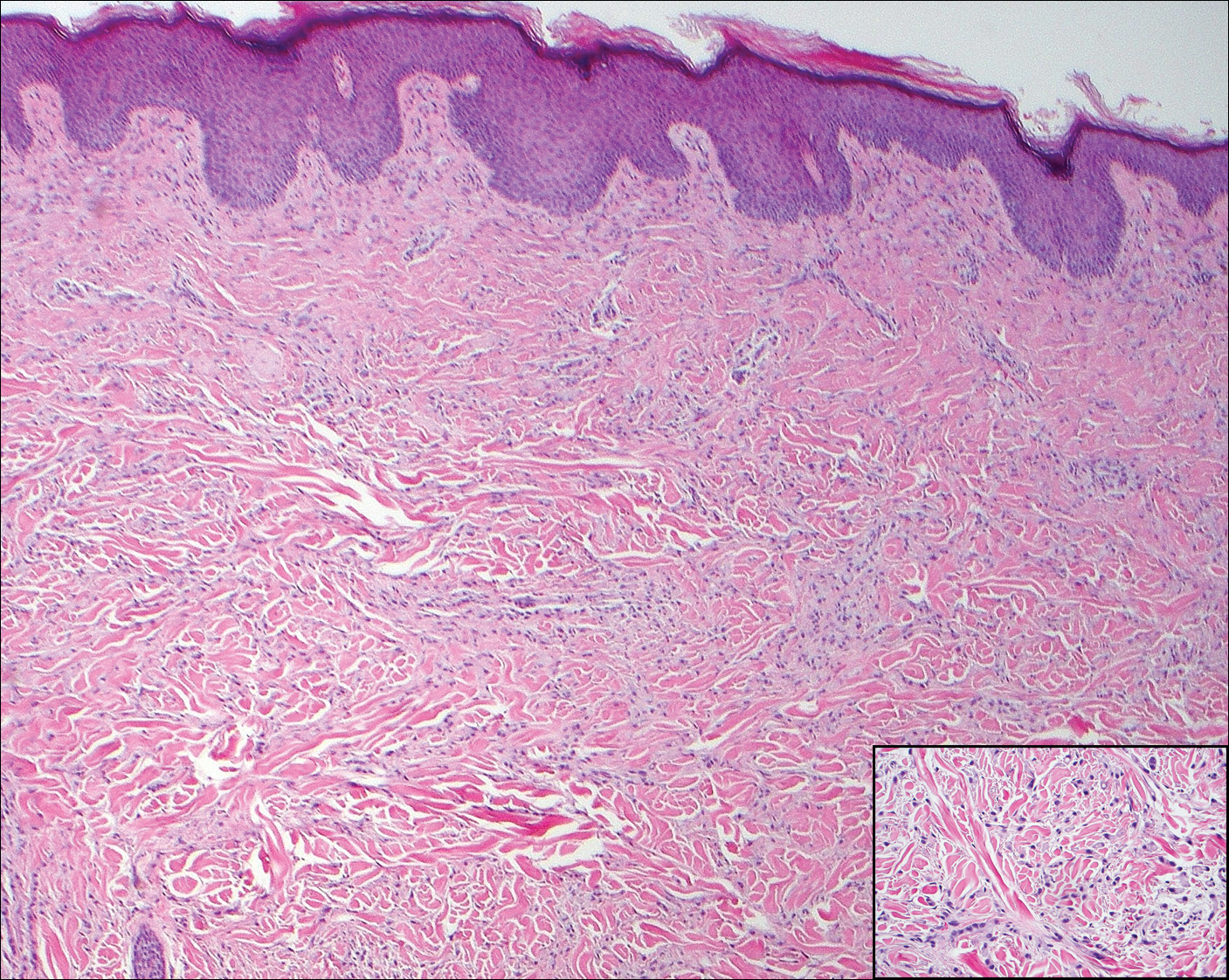
A 56-year-old woman presented with multiple asymptomatic lesions of 2 months' duration. On physical examination firm pink papules were noted dispersed across the upper abdomen, chest, and back. A 5-mm punch biopsy was obtained.
Red-Brown Patches in the Groin
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
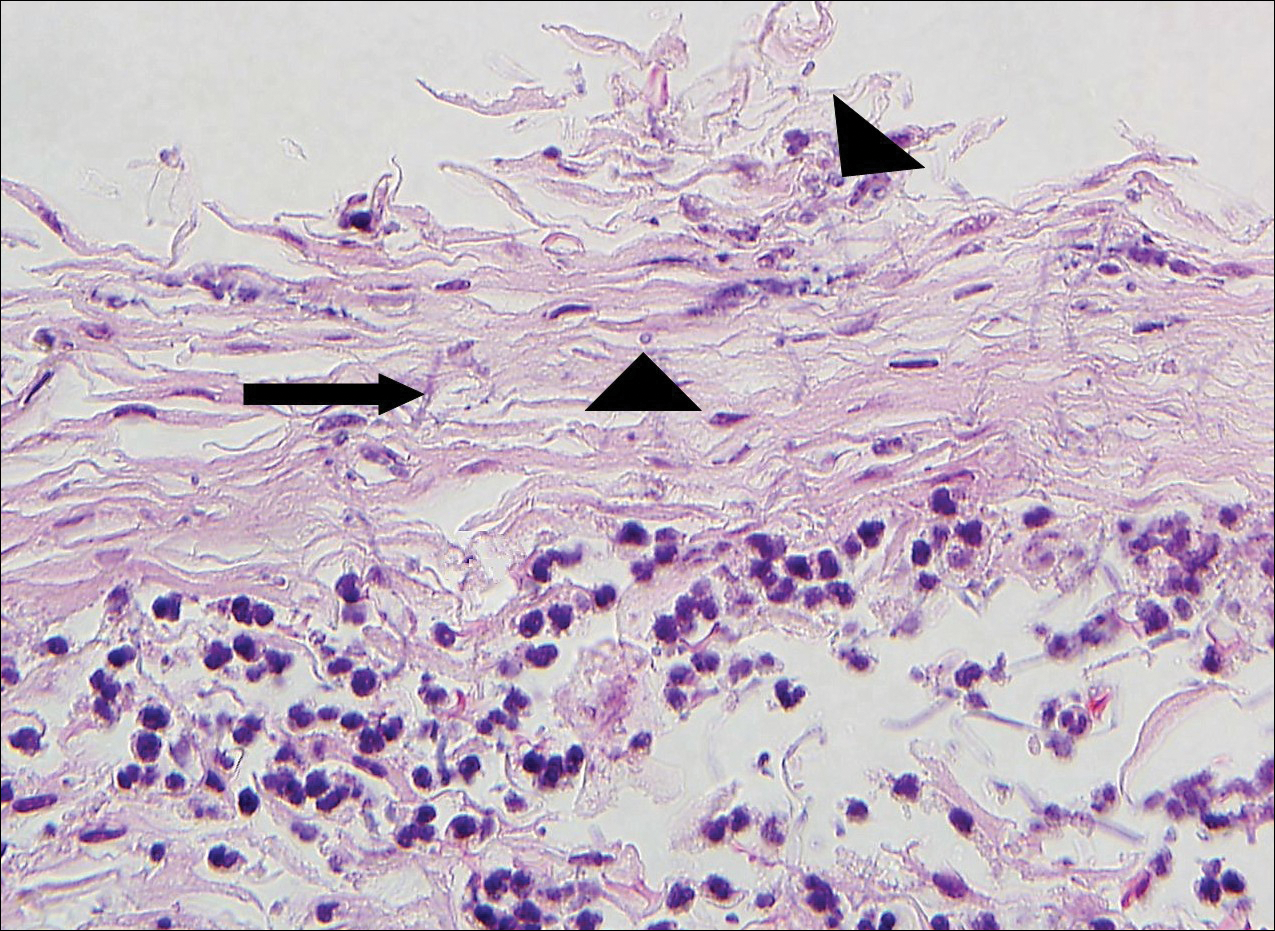
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
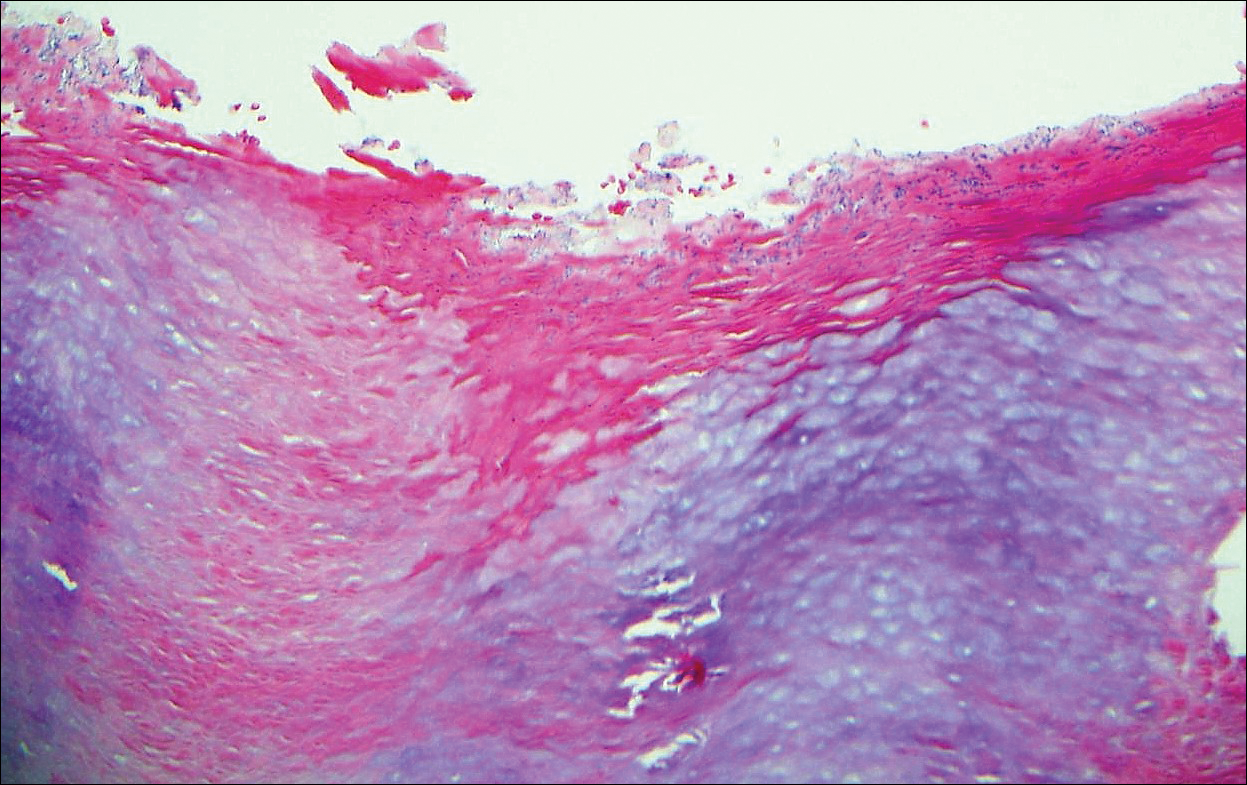
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
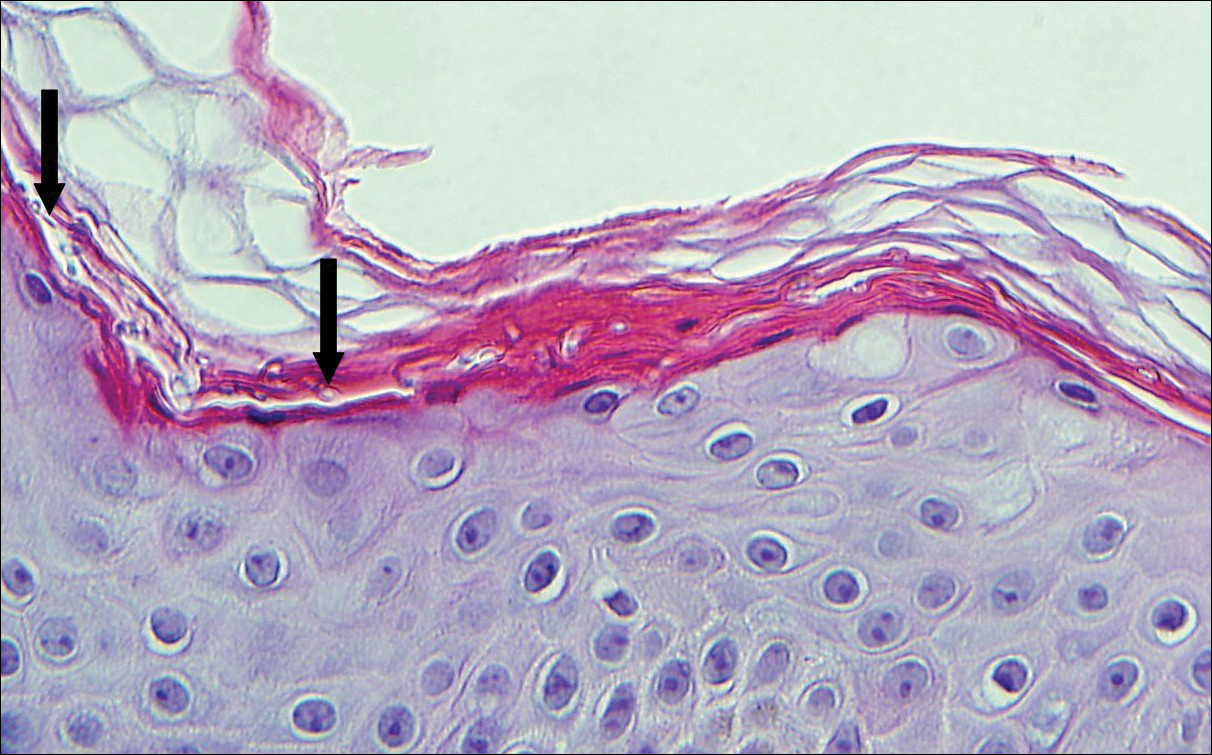
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
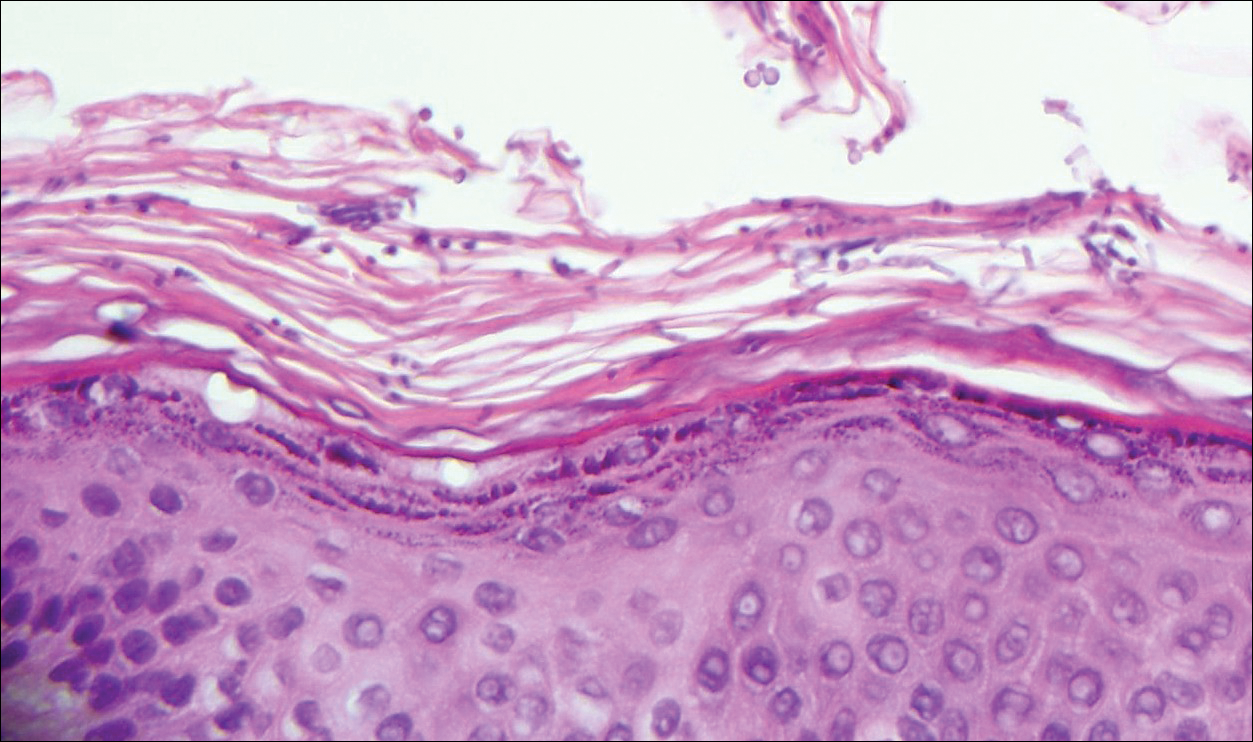
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.

A 66-year-old man presented with reddish arciform patches in the inguinal area.
Red-Blue Nodule on the Scalp
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
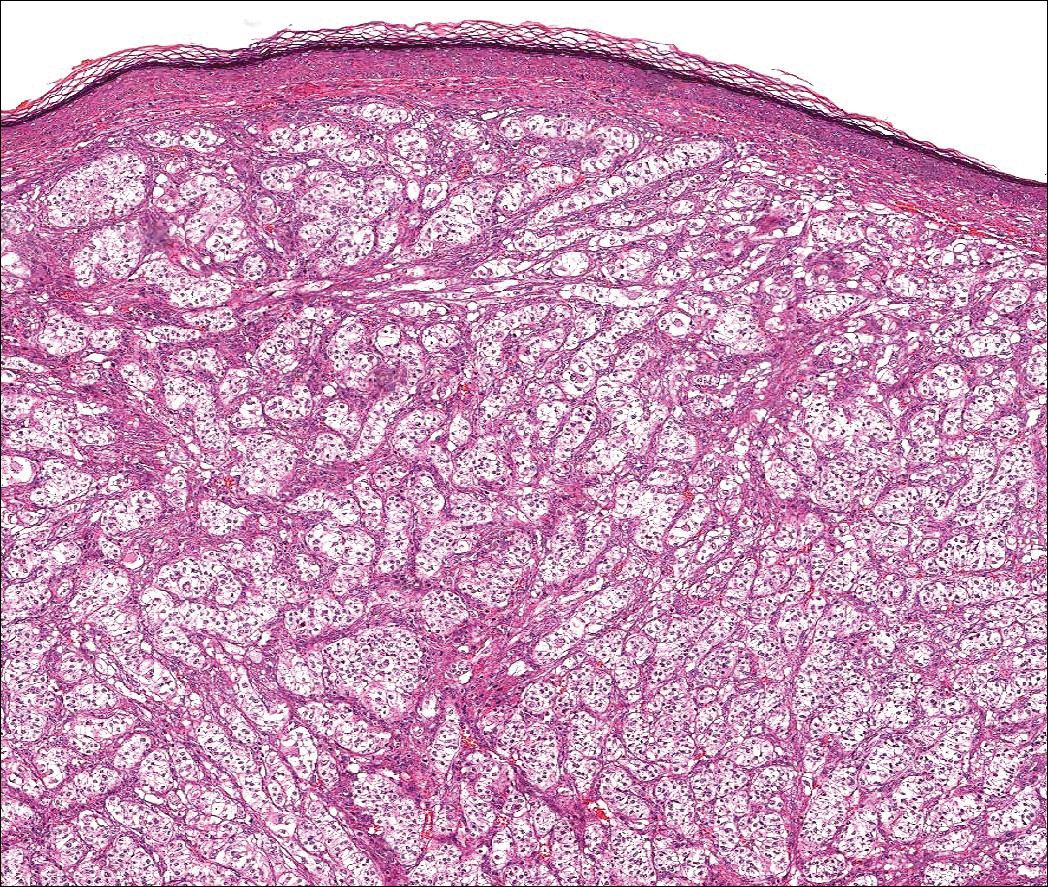
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
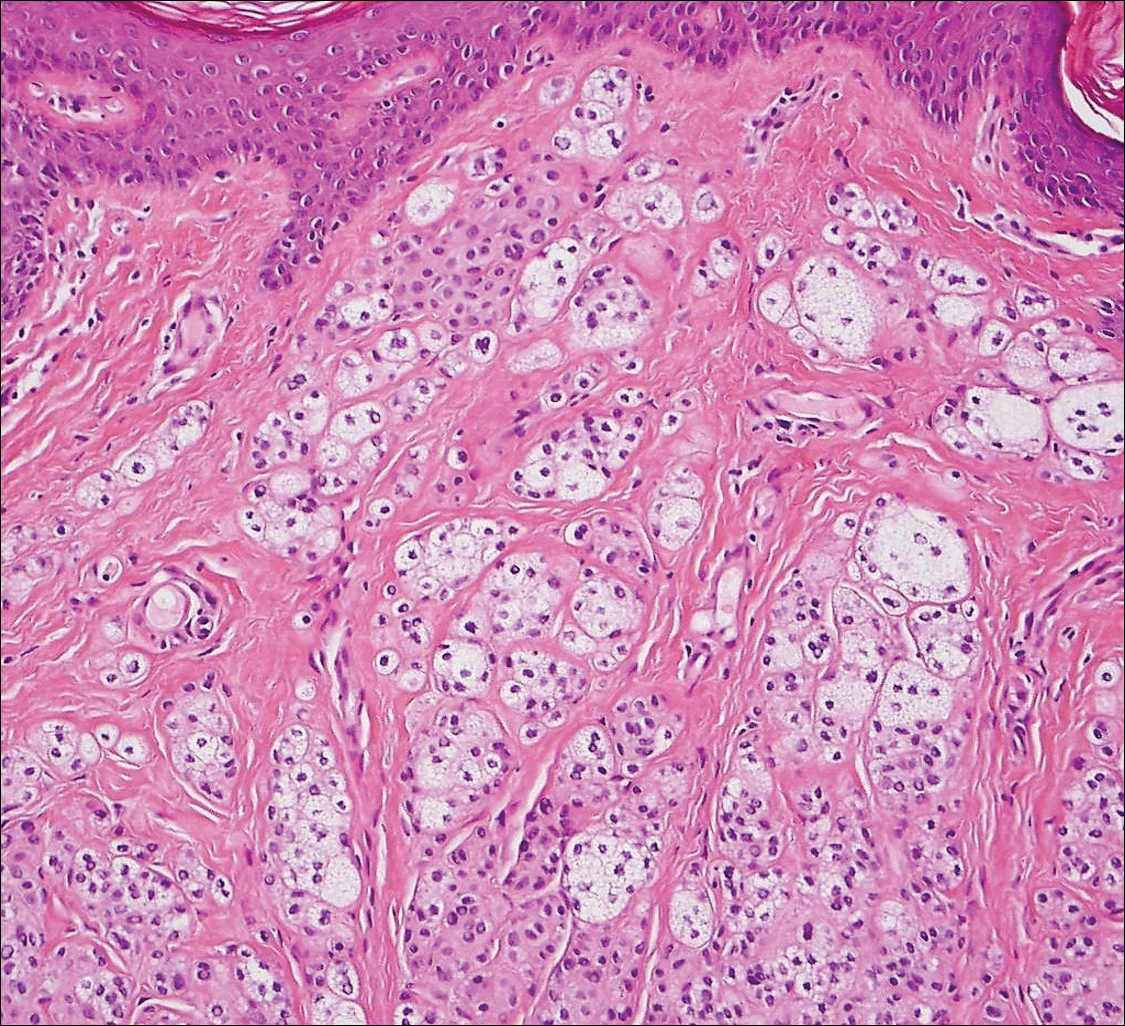
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6

Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
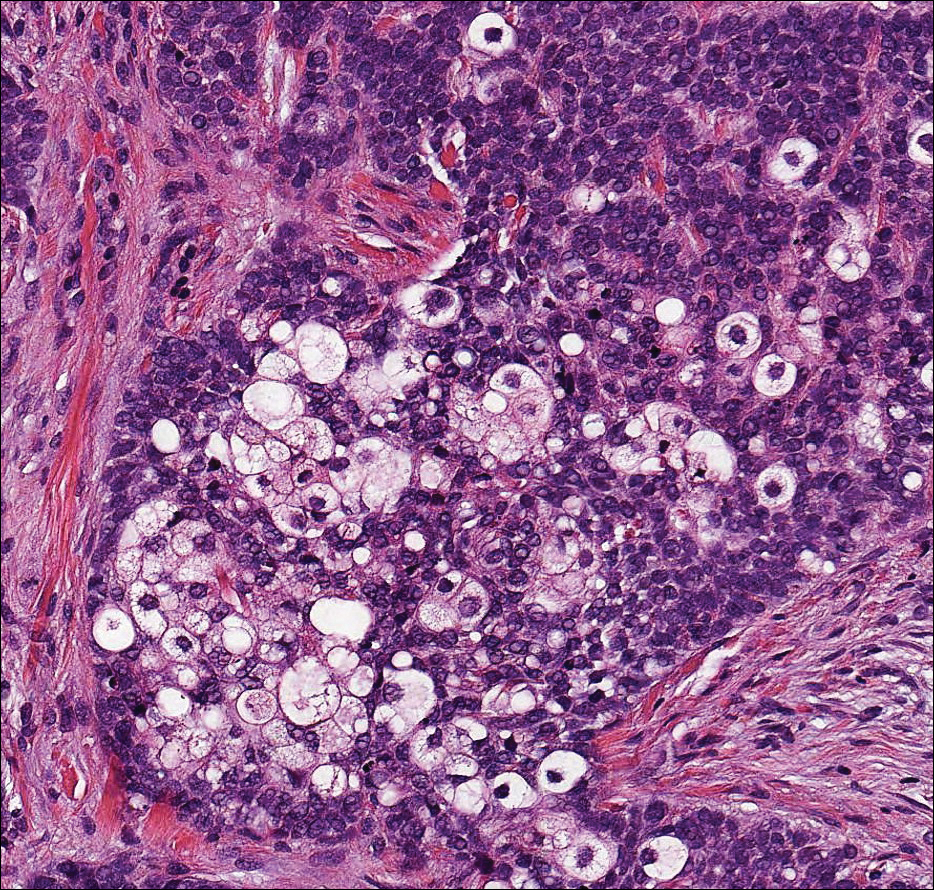
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
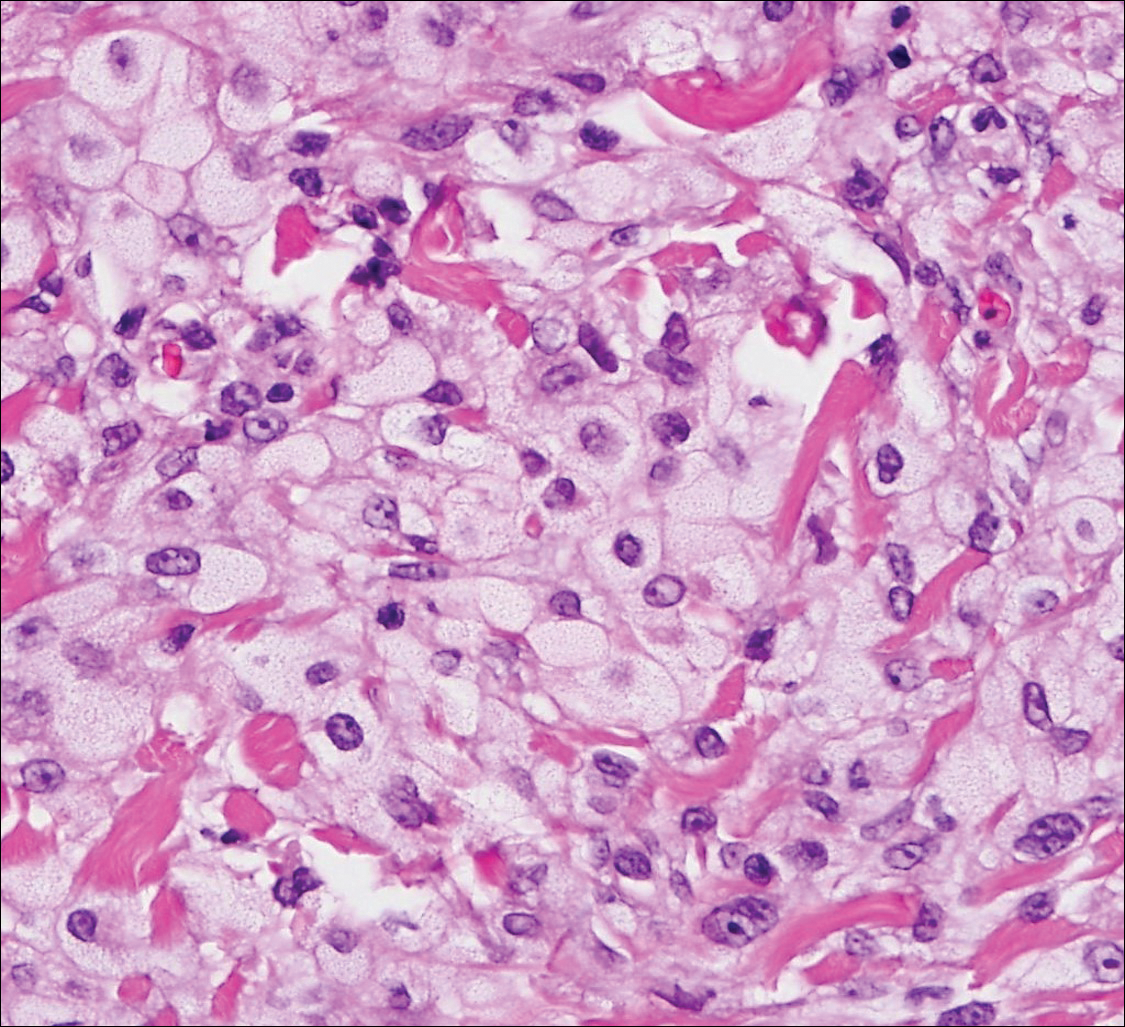
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
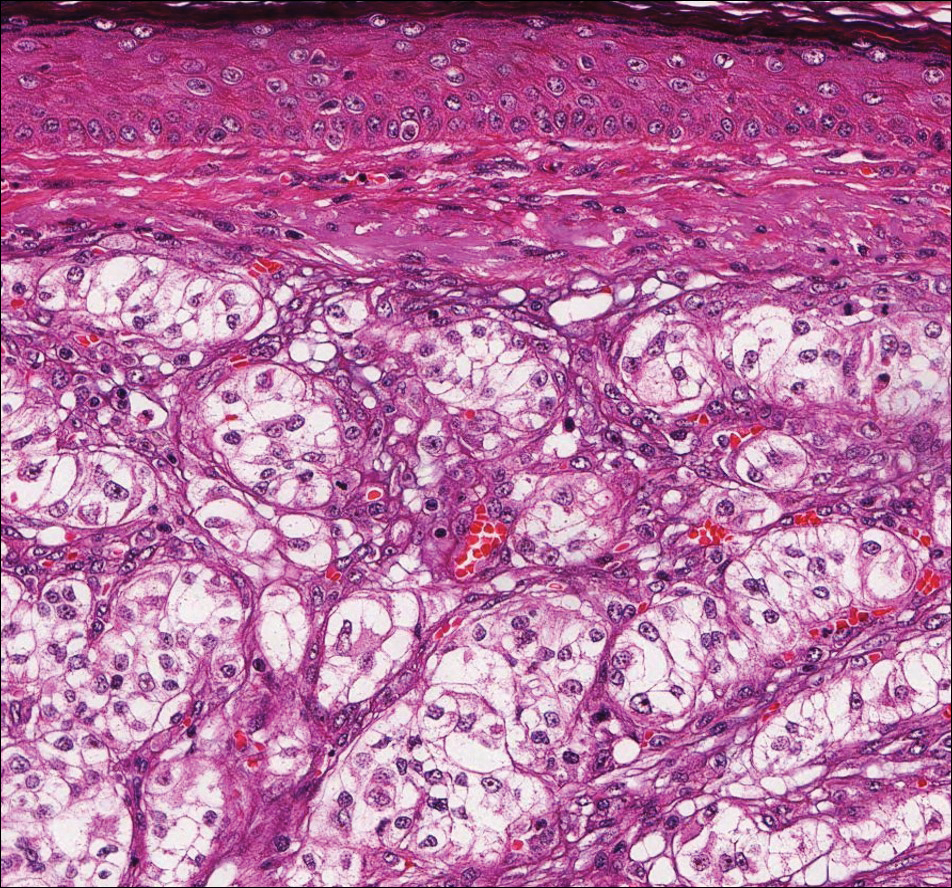
A 59-year-old man presented with a 1.5×1.0-cm asymptomatic, smooth, red-blue nodule on the left parietal scalp. The nodule had been rapidly enlarging over the last 3 weeks. After resection, the cut surface was golden yellow and focally hemorrhagic.
What’s Eating You? Cutaneous Larva Migrans
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Practice Points
- Classic cutaneous larva migrans (CLM) presents with a unilateral, serpiginous, pruritic eruption on the hands, feet, or buttocks following direct contact with sand or soil that is contaminated with Ancylostoma braziliense or Ancylostoma caninum.
- Atypical presentations of CLM include bilateral distribution; folliculitis and urticarial plaques; prolonged cases lasting up to 1 year; and Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia.
- Cutaneous larva migrans is self-limited, but treatment often is necessary due to intense pruritus. Treatment options include a single oral dose of albendazole or ivermectin, topical thiabendazole, and prolonged courses of oral albendazole in cases complicated by Löffler syndrome.