User login
A minimally invasive treatment for early GI cancers
The treatment of early esophageal, gastric, and colorectal cancer is changing.1 For many years, surgery was the mainstay of treatment for early-stage gastrointestinal cancer. Unfortunately, surgery leads to significant loss of function of the organ, resulting in increased morbidity and decreased quality of life.2
Endoscopic techniques, particularly endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), have been developed and are widely used in Japan, where gastrointestinal cancer is more common than in the West. This article reviews the indications, complications, and outcomes of ESD for early gastrointestinal neoplasms, so that readers will recognize the subset of patients who would benefit from ESD in a Western setting.
ENDOSCOPIC MUCOSAL RESECTION AND SUBMUCOSAL DISSECTION
Since the first therapeutic polypectomy was performed in Japan in 1974, several endoscopic techniques for tumor resection have been developed.3
EMR, one of the most successful and widely used techniques, involves elevating the lesion either with submucosal injection of a solution or with cap suction, and then removing it with a snare.4 Most lesions smaller than 20 mm can be removed in one piece (en bloc).5 Larger lesions are removed in multiple pieces (ie, piecemeal). Unfortunately, some fibrotic lesions, which are usually difficult to lift, cannot be completely removed by EMR.
ESD was first performed in the late 1990s with the aim of overcoming the limitations of EMR in resecting large or fibrotic tumors en bloc.6,7 Since then, ESD technique has been standardized and training centers have been created, especially in Asia, where it is widely used for treatment of early gastric cancer.3,8–10 Since 2012 it has been covered by the Japanese National Health Insurance for treatment of early gastric cancer, and since 2014 for treatment of colorectal malignant tumors measuring 2 to 5 cm.11
Adoption of ESD has been slow in Western countries, where many patients are still referred for surgery or undergo EMR for removal of superficial neoplasms. Reasons for this slow adoption are that gastric cancer is much less common in Western countries, and also that ESD demands a high level of technical skill, is difficult to learn, and is expensive.3,12,13 However, small groups of Western endoscopists have become interested and are advocating it, first studying it on their own and then training in a Japanese center and learning from experts performing the procedure.
Therefore, in a Western setting, ESD should be performed in specialized endoscopy centers and offered to selected patients.1
CANDIDATES SHOULD HAVE EARLY-STAGE, SUPERFICIAL TUMORS
Ideal candidates for endoscopic resection are patients who have early cancer with a negligible risk of lymph node metastasis, such as cancer limited to the mucosa (stage T1a).7 Therefore, to determine the best treatment for a patient with a newly diagnosed gastrointestinal neoplasm, it is mandatory to estimate the depth of invasion.
The depth of invasion is directly correlated with lymph node involvement, which is ultimately the main predictive factor for long-term adverse outcomes of gastrointestinal tumors.4,14–17 Accurate multidisciplinary preprocedure estimations are mandatory, as incorrect evaluations may result in inappropriate therapy and residual cancer.18
Other factors that have been used to predict lymph node involvement include tumor size, macroscopic appearance, histologic differentiation, and lymphatic and vascular involvement.19 Some of these factors can be assessed by special endoscopic techniques (chromoendoscopy and narrow-band imaging with magnifying endoscopy) that allow accurate real-time estimation of the depth of invasion of the lesion.5,17,20–27 Evaluation of microsurface and microvascular arrangements is especially useful for determining the feasibility of ESD in gastric tumors, evaluation of intracapillary loops is useful in esophageal lesions, and assessment of mucosal pit patterns is useful for colorectal lesions.21–29
Endoscopic ultrasonography is another tool that has been used to estimate the depth of the tumor. Although it can differentiate between definite intramucosal and definite submucosal invasive cancers, its ability to confirm minute submucosal invasion is limited. Its use as the sole tumor staging modality is not encouraged, and it should always be used in conjunction with endoscopic evaluation.18
Though the aforementioned factors help stratify patients, pathologic staging is the best predictor of lymph node metastasis. ESD provides adequate specimens for accurate pathologic evaluation, as it removes lesions en bloc.30
All patients found to have risk factors for lymph node metastasis on endoscopic, ultrasonographic, or pathologic analysis should be referred for surgical evaluation.9,19,31,32
ENDOSCOPIC SUBMUCOSAL DISSECTION
Before the procedure, the patient’s physicians need to do the following:
Determine the best type of intervention (EMR, ESD, ablation, surgery) for the specific lesion.3 A multidisciplinary approach is encouraged, with involvement of the internist, gastroenterologist, and surgeon.
Plan for anesthesia, additional consultations, pre- and postprocedural hospital admission, and need for special equipment.33
During the procedure

Define the lateral extent of the lesion using magnification chromoendoscopy or narrow-band imaging. In the stomach, a biopsy sample should be taken from the worst-looking segment and from normal-looking mucosa. Multiple biopsies should be avoided to prevent subsequent fibrosis.33 In the colon, biopsy should be avoided.34
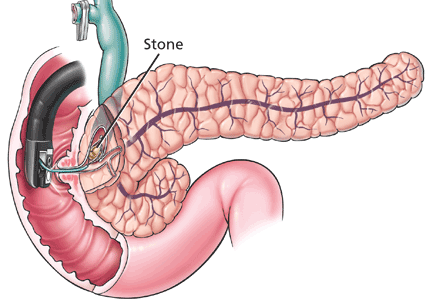
Identify and circumferentially mark the target lesion. Cautery or argon plasma coagulation can be used for making markings at a distance of 5 to 10 mm from the edges.33 This is done to recognize the borders of the lesion, because they can become distorted after submucosal injection.14 This step is unnecessary in colorectal cases, as tumor margins can be adequately visualized after chromoendoscopy.16,35
Lift the lesion by injecting saline, 0.5% hyaluronate, or glycerin to create a submucosal fluid cushion.19,33
Perform a circumferential incision lateral to the mucosal margins to allow for a normal tissue margin.33 Partial incision is performed for esophageal and colorectal ESD to avoid fluid leakage from the submucosal layer, achieving a sustained submucosal lift and safer dissection.16
Submucosal dissection. The submucosal layer is dissected with an electrocautery knife until the lesion is completely removed. Dissection should be done carefully to keep the submucosal plane.33 Hemoclips or hemostat forceps can be used to control visible bleeding. The resected specimen is then stretched and fixed to a board using small pins for further histopathologic evaluation.35
Postprocedural monitoring. All patients should be admitted for overnight observation. Those who undergo gastric ESD should receive high-dose acid suppression, and the next day they can be started on a liquid diet.19
STOMACH CANCER
Indications for ESD for stomach cancer in the East
The incidence of gastric cancer is higher in Japan and Korea, where widespread screening programs have led to early identification and early treatment of this disease.36
Pathology studies37 of samples from patients with gastric cancer identified the following as risk factors for lymph node metastasis, which would make ESD unsuitable:
- Undifferentiated type
- Tumors larger than 2 cm
- Lymphatic or venous involvement
- Submucosal invasion
- Ulcerative change.

Based on these findings, the situations in which there was no risk of lymph node involvement (ie, when none of the above factors are present) were accepted as absolute indications for endoscopic resection of early gastric cancer.38 Further histologic studies identified a subset of patients with lesions with very low risk of lymph node metastasis, which outweighed the risk of surgery. Based on these findings, expanded criteria for gastric ESD were proposed,39,40 and the Japanese gastric cancer treatment guidelines now include these expanded preoperative indications9,17 (Table 1).

The Japanese Gastric Cancer Association has proposed a treatment algorithm based on the histopathologic evaluation after resection (Figure 2).9
Outcomes
In the largest series of patients who underwent curative ESD for early gastric cancer, the 5-year survival rate was 92.6%, the 5-year disease-specific survival rate was 99.9%, and the 5-year relative survival rate was 105%.41
Similarly, in a Japanese population-based survival analysis, the relative 5-year survival rate for localized gastric cancer was 94.4%.42 Rates of en bloc resection and complete resection with ESD are higher than those with EMR, resulting in a lower risk of local recurrence in selected patients who undergo ESD.8,43,44
Although rare, local recurrence after curative gastric ESD has been reported.45 The annual incidence of local recurrence has been estimated to be 0.84%.46

ESD entails a shorter hospital stay and requires fewer resources than surgery, resulting in lower medical costs (Table 2).44 Additionally, as endoscopic resection is associated with less morbidity, fewer procedure-related adverse events, and fewer complications, ESD could be used as the standard treatment for early gastric cancer.47,48
The Western perspective on endoscopic submucosal dissection for gastric cancer
Since the prevalence of gastric cancer in Western countries is significantly lower than in Japan and Korea, local data and experience are scarce. However, experts performing ESD in the West have adopted the indications of the Japan Gastroenterological Endoscopy Society. The European Society of Gastrointestinal Endoscopy recommends ESD for excision of most superficial gastric neoplasms, with EMR being preferred only in lesions smaller than 15 mm, Paris classification 0 or IIA.5,32
Patients with gastric lesions measuring 15 mm or larger should undergo high-quality endoscopy, preferably chromoendoscopy, to evaluate the mucosal patterns and determine the depth of invasion. If superficial involvement is confirmed, other imaging techniques are not routinely recommended.5 A surgery consult is also recommended.
ESOPHAGEAL CANCER
Indications for ESD for esophageal cancer in the East
Due to the success of ESD for early gastric cancer, this technique is now also used for superficial esophageal neoplasms.19,49 It should be done in a specialized center, as it is more technically difficult than gastric ESD: the esophageal lumen is narrow, the wall is thin, and the esophagus moves with respiration and heartbeat.50 A multidisciplinary approach including an endoscopist, a surgeon, and a pathologist is highly recommended for evaluation and treatment.

EMR is preferred for removal of mucosal cancer, in view of its safety profile and success rates. ESD can be considered in cases of lesions larger than 15 mm, poorly lifting tumors, and those with the possibility of submucosal invasion (Table 3).5,45,49,51
Circumference involvement is critical when determining eligible candidates, as a defect involving more than three-fourths of the esophageal circumference can lead to esophageal strictures.52 Controlled prospective studies have shown promising results from giving intralesional and oral steroids to prevent stricture after ESD, which could potentially overcome this size limitation.53,54
Outcomes for esophageal cancer
ESD has been shown to be safe and effective, achieving en bloc resection in 85% to 100% of patients.19,51 Its advantages over EMR include en bloc resection, complete resection, and high curative rates, resulting in higher recurrence-free survival.2,55,56 Although the incidence of complications such as bleeding, perforation, and stricture formation are higher with ESD, patients usually recover uneventfully.2,19,20
ESD in the esophagus: The Western perspective
As data on the efficacy of EMR vs ESD for the treatment of Barrett esophagus with adenocarcinoma are limited, EMR is the gold standard endoscopic technique for removal of visible esophageal dysplastic lesions.5,51,57 ESD can be considered for tumors larger than 15 mm, for poorly lifting lesions, and if there is suspicion of submucosal invasion.5
Patients should be evaluated by an experienced endoscopist, using an advanced imaging technique such as narrow-band imaging or chromoendoscopy. If suspicious features are found, endoscopic ultrasonography should be considered to confirm submucosal invasion or lymph node involvement.5
COLORECTAL CANCER
Indications for ESD for colorectal cancer in the East
Colon cancer is one of the leading causes of cancer-related deaths worldwide.58 Since ESD has been found to be effective and safe in treating gastric cancer, it has also been used to remove large colorectal tumors.59 However, ESD is not universally accepted in the treatment of colorectal neoplasms due to its greater technical difficulty, longer procedural time, and higher risk of perforating the thinner colonic wall compared with EMR.21,60

Outcomes for colorectal cancer

Tumor size of 50 mm or larger is a risk factor for complications, while a high procedure volume at the center is a protective factor.60
Endoscopic treatment of colorectal cancer: The Western perspective
EMR is the gold standard for removal of superficial colorectal lesions. However, ESD can be considered if there is suspicion of superficial submucosal invasion, especially for lesions larger than 20 mm that cannot be resected en bloc by EMR.32 ESD can also be used for fibrotic lesions not amenable to complete EMR removal, or as a salvage procedure after recurrence after EMR.67 Proper selection of cases is critical.1
Patients who have a superficial colonic lesion should be evaluated by means of high-definition endoscopy and chromoendoscopy to assess the mucosal pattern and establish feasibility of endoscopic resection. If submucosal invasion is suspected, staging with endoscopic ultrasonography or magnetic resonance imaging should be considered.5
FOLLOW-UP AFTER ESD
Endoscopic surveillance after the procedure is recommended, given the persistent risk of metachronous cancer after curative ESD due to its organ-sparing quality.68 Surveillance endoscopy aims to achieve early detection and subsequent endoscopic resection of metachronous lesions.
Histopathologic evaluation assessing the presence of malignant cells in the margins of a resected sample is mandatory for determining the next step in treatment. If margins are negative, follow-up endoscopy can be done every 6 to 12 months. If margins are positive, the approach includes surgery, reattempting ESD or endoscopic surveillance in 3 or 6 months.3,32 Although the surveillance strategy varies according to individual risk of metachronous cancer, it should be continued indefinitely.68
COMPLICATIONS OF ESD
The most common procedure-related complications of ESD are bleeding, perforation, and stricture. Most intraprocedural adverse events can be managed endoscopically.69
Bleeding
Most bleeding occurs during the procedure or early after it and can be controlled with electrocautery.49,69 No episodes of massive bleeding, defined as causing clinical symptoms and requiring transfusion or surgery, have been reported.20,43,55
In gastric ESD, delayed bleeding rates have ranged from 0 to 15.6%.69 Bleeding may be prevented with endoscopic coagulation of visible vessels after dissection has been completed and by proton pump inhibitor therapy.70,71 Excessive coagulation should be avoided to lower the risk of perforation.33
In colorectal ESD the bleeding rate has been reported to be 2.2%; applying coagulation to an area where a blood vessel is suspected before cutting (precoagulation) may prevent subsequent bleeding.21
Perforation
For gastric ESD, perforation rates range from 1.2% to 5.2%.69 Esophageal perforation rates can be up to 4%.49 In colorectal ESD, perforation rates have been reported to be 1.6% to 6.6%.60,72
Although most of the cases were successfully managed with conservative treatment, some required emergency surgery.60,73
Strictures
In a case series of 532 patients undergoing gastric ESD, stricture was reported in 5 patients, all of whom presented with obstructive symptoms.74 Risk factors for post-ESD gastric stenosis are a mucosal defect with a circumferential extent of more than three-fourths or a longitudinal extent of more than 5 cm.75
Strictures are common after esophageal ESD, with rates ranging from 2% to 26%. The risk is higher when longer segments are removed or circumferential resection is performed. As previously mentioned, this complication may be reduced with ingestion or injection of steroids after the procedure.53,54
Surprisingly, ESD of large colorectal lesions involving more than three-fourths of the circumference of the rectum is rarely complicated by stenosis.76
LIMITATIONS OF ESD
ESD requires a high level of technical skill, is time-consuming, and has a higher rate of complications than conventional endoscopic resection. A standardized ESD training system is needed, as the procedure is more difficult than EMR. Training in porcine models has been shown to confer competency in ESD in a Western setting.13,16,33
Colorectal ESD is an even more challenging procedure, given the potential for complications related to its anatomy. Training centers in Japan usually have their trainees first master gastric ESD, then assist in more than 20 colorectal ESDs conducted by experienced endoscopists, and accomplish 30 cases before performing the procedure safely and independently.
As the incidence of gastric cancer is low in Western countries, trainees may also begin with lower rectal lesions, which are easier to remove.77 Incorporation of ESD in the West would require a clear treatment algorithm. It is a complex procedure, with higher rates of complications, a prolonged learning curve, and prolonged procedure time. Therefore, it should be performed in specialized centers and under the special situations discussed here to ensure that the benefits for the patients outweigh the risks.
VALUE OF ENDOSCOPIC SUBMUCOSAL DISSECTION
The optimal method for resecting gastrointestinal neoplasms should be safe, cost-effective, and quick and should also completely remove the lesion. The best treatment strategy takes into account the characteristics of the lesion and the comorbidities and wishes of the patient. Internists should be aware of the multiple options available to achieve the best outcome for the patient.1
Endoscopic resection of superficial gastrointestinal neoplasms, including EMR and ESD, has been a subject of increasing interest due to its minimally invasive and potentially curative character. However, cancer can recur after endoscopic resection because the procedure is organ-sparing.
ESD allows resection of early gastrointestinal tumors with a minimally invasive technique. It can achieve higher curative resection rates and lower recurrence rates compared with EMR. Compared with surgery, ESD leads to less morbidity, fewer procedure-related complications, and lower medical costs. Indications should be rigorously followed to achieve successful treatments in selected patients.
Multiple variables have to be taken into account when deciding which treatment is best, such as tumor characteristics, the patient’s baseline condition, physician expertise, and hospital resources.48 Less-invasive treatments may improve the prognosis of patients. No matter the approach, patients should be treated in specialized treatment centers.
Internal medicine physicians should be aware of the advances in treatments for early gastrointestinal cancer so appropriate options can be considered.
- Burgess NG, Bourke MJ. Endoscopic resection of colorectal lesions: the narrowing divide between East and West. Dig Endosc 2016; 28:296–305.
- Kim DH, Jung HY, Gong EJ, et al. Endoscopic and oncologic outcomes of endoscopic resection for superficial esophageal neoplasm. Gut Liver 2015; 9:470–477.
- Draganov PV, Gotoda T, Chavalitdhamrong D, Wallace MB. Techniques of endoscopic submucosal dissection: application for the Western endoscopist? Gastrointest Endosc 2013; 78:677–688.
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011; 14:101–112.
- Pimentel-Nunes P, Dinis-Ribeiro M, Ponchon T, et al. Endoscopic submucosal dissection: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy 2015; 47:829–854.
- Farhat S, Chaussade S, Ponchon T, et al; SFED ESD Study Group. Endoscopic submucosal dissection in a European setting. A multi-institutional report of a technique in development. Endoscopy 2011; 43:664–670.
- Gotoda T, Jung H. Endoscopic resection (endoscopic mucosal resection/endoscopic submucosal dissection) for early gastric cancer. Dig Endosc 2013; 25(suppl 1):55–63.
- Chung IK, Lee JH, Lee SH, et al. Therapeutic outcomes in 1000 cases of endoscopic submucosal dissection for early gastric neoplasms: Korean ESD Study Group multicenter study. Gastrointest Endosc 2009; 69:1228–1235.
- Japanese Gastric Cancer Association. Japanese gastric cancer treatment guidelines 2010 (ver. 3). Gastric Cancer 2011; 14:113–123.
- Ono H. Endoscopic submucosal dissection for early gastric cancer. Chin J Dig Dis 2005; 6:119–121.
- Watanabe T, Itabashi M, Shimada Y, et al; Japanese Society for Cancer of the Colon and Rectum. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2014 for treatment of colorectal cancer. Int J Clin Oncol 2015; 20:207–239.
- Oyama T, Yahagi N, Ponchon T, Kiesslich T, Berr F. How to establish endoscopic submucosal dissection in Western countries. World J Gastroenterol 2015; 21:11209–11220.
- Bhatt A, Abe S, Kumaravel A, et al. SU1575 Western skill training in endoscopic submucosal dissection (ESD)—an international remote video based study—the WEST ESD Study. Gastrointest Endosc 2015; 81(suppl):AB335–AB336.
- Sano T, Sasako M, Kinoshita T, Maruyama K. Recurrence of early gastric cancer follow-up of 1475 patients and review of the Japanese literature. Cancer 1993; 72:3174–3178.
- Japan Esophageal Society. Japanese classification of esophageal cancer, tenth edition: part I. Esophagus 2009; 6:1–25.
- Bhatt A, Abe S, Kumaravel A, Vargo J, Saito Y. Indications and techniques for endoscopic submucosal dissection. Am J Gastroenterol 2015; 110:784–791.
- Eleftheriadis N, Inoue H, Ikeda H, et al. Definition and staging of early esophageal, gastric and colorectal cancer. J Tumor 2014; 2:161–178.
- Yoshinaga S, Oda I, Nonaka S, Kushima R, Saito Y. Endoscopic ultrasound using ultrasound probes for the diagnosis of early esophageal and gastric cancers. World J Gastrointest Endosc 2012; 4:218–226.
- Stahl M, Mariette C, Haustermans K, Cervantes A, Arnold D; ESMO Guidelines Working Group. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi51–vi56.
- Higuchi K, Tanabe S, Azuma M, et al. A phase II study of endoscopic submucosal dissection for superficial esophageal neoplasms (KDOG 0901). Gastrointest Endosc 2013; 78:704–710.
- Sakamoto T, Mori G, Yamada M, et al. Endoscopic submucosal dissection for colorectal neoplasms: a review. World J Gastroenterol 2014; 20:16153–16158.
- Ohta A, Tominaga K, Sakai Y. Efficacy of magnifying colonoscopy for the diagnosis of colorectal neoplasia: comparison with histopathological findings. Dig Endosc 2004; 16:308–314.
- Katagiri A, Fu K, Sano Y, et al. Narrow band imaging with magnifying colonoscopy as diagnostic tool for predicting histology of early colorectal neoplasia. Aliment Pharmacol Ther 2008; 27:1269–1274.
- Fu KI, Kato S, Sano Y, et al. Staging of early colorectal cancers: magnifying colonoscopy versus endoscopic ultrasonography for estimation of depth of invasion. Dig Dis Sci 2008; 53:1886–1892.
- Uraoka T, Saito Y, Ikematsu H, Yamamoto K, Sano Y. Sano’s capillary pattern classification for narrow-band imaging of early colorectal lesions. Dig Endosc 2011; 23(suppl 1):112–115.
- Ikematsu H, Matsuda T, Emura F, et al. Efficacy of capillary pattern type IIIA/IIIB by magnifying narrow band imaging for estimating depth of invasion of early colorectal neoplasms. BMC Gastroenterol 2010;10:33.
- Matsuda T, Fujii T, Saito Y, et al. Efficacy of the invasive/non-invasive pattern by magnifying chromoendoscopy to estimate the depth of invasion of early colorectal neoplasms. Am J Gastroenterol 2008; 103:2700–2706.
- Sato H, Inoue H, Ikeda H, et al. Utility of intrapapillary capillary loops seen on magnifying narrow-band imaging in estimating invasive depth of esophageal squamous cell carcinoma. Endoscopy 2015; 8:122–128.
- Muto M, Yao K, Kaise M, et al. Magnifying endoscopy simple diagnostic algorithm for early gastric cancer (MESDA-G). Dig Endosc 2016; 28:379–393.
- Waddell T, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D; European Society for Medical Oncology (ESMO); European Society of Surgical Oncology (ESSO); European Society of Radiotherapy and Oncology (ESTRO). Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi57–vi63.
- Kuwano H, Nishimura Y, Ohtsu A, et al. Guidelines for diagnosis and treatment of carcinoma of the esophagus. April 2007 edition: part I - Edited by the Japan Esophageal Society. Esophagus 2008; 5:61–73.
- Tanaka S, Kashida H, Saito Y, et al. JGES guidelines for colorectal endoscopic submucosal dissection/endoscopic mucosal resection. Dig Endosc 2015; 27:417–434.
- Gotoda T, Ho KY, Soetikno R, Kaltenbach T, Draganov P. Gastric ESD: current status and future directions of devices and training. Gastrointest Endosc Clin North Am 2014; 24:213–233.
- Saito Y, Sakamoto T, Nakajima T, Matsuda T. Colorectal ESD: current indications and latest technical advances. Gastrointest Endosc Clin N Am 2014; 24:245–255.
- Saito Y, Otake Y, Sakamoto T, et al. Indications for and technical aspects of colorectal endoscopic submucosal dissection. Gut Liver 2013; 7:263–269.
- Saragoni L. Upgrading the definition of early gastric cancer: better staging means more appropriate treatment. Cancer Biol Med 2015; 12:355–361.
- Tsujitani S, Oka S, Saito H, et al. Less invasive surgery for early gastric cancer based on the low probability of lymph node metastasis. Surgery 1999; 125:148–154.
- Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003; 57:567–579.
- Hirasawa T, Gotoda T, Miyata S, et al. Incidence of lymph node metastasis and the feasibility of endoscopic resection for undifferentiated-type early gastric cancer. Gastric Cancer 2009; 12:148–152.
- Gotoda T, Yanagisawa A, Sasako M, et al. Incidence of lymph node metastasis from early gastric cancer: estimation with a large number of cases at two large centers. Gastric Cancer 2000; 3:219–225.
- Suzuki H, Oda I, Abe S, et al. High rate of 5-year survival among patients with early gastric cancer undergoing curative endoscopic submucosal dissection. Gastric Cancer 2016; 19:198–205.
- Matsuda T, Ajiki W, Marugame T, Ioka A, Tsukuma H, Sobue T; Research Group of Population-Based Cancer Registries of Japan. Population-based survival of cancer patients diagnosed between 1993 and 1999 in Japan: a chronological and international comparative study. Jpn J Clin Oncol 2011; 41:40–51.
- Ahn JY, Jung HY, Choi KD, et al. Endoscopic and oncologic outcomes after endoscopic resection for early gastric cancer: 1370 cases of absolute and extended indications. Gastrointest Endosc 2011; 74:485–493.
- Kim Y, Kim YW, Choi IJ, et al. Cost comparison between surgical treatments and endoscopic submucosal dissection in patients with early gastric cancer in Korea. Gut Liver 2015; 9:174–180.
- Abe S, Oda I, Nakajima T, et al. A case of local recurrence and distant metastasis following curative endoscopic submucosal dissection of early gastric cancer. Gastric Cancer 2015; 18:188–192.
- Hahn KY, Park JC, Kim EH, et al. Incidence and impact of scheduled endoscopic surveillance on recurrence after curative endoscopic resection for early gastric cancer. Gastrointest Endosc 2016; 84:628–638.e1.
- Wang S, Zhang Z, Liu M, Li S, Jiang C. Endoscopic resection compared with gastrectomy to treat early gastric cancer: a systematic review and meta-analysis. PLoS One 2015; 10:e0144774.
- Kondo A, de Moura EG, Bernardo WM, et al. Endoscopy vs surgery in the treatment of early gastric cancer: systematic review. World J Gastroenterol 2015; 21:13177–13187.
- Kothari S, Kaul V. Endoscopic mucosal resection and endoscopic submucosal dissection for endoscopic therapy of Barrett’s esophagus-related neoplasia. Gastroenterol Clin North Am 2015; 44:317–335.
- Yamashita T, Zeniya A, Ishii H, et al. Endoscopic mucosal resection using a cap-fitted panendoscope and endoscopic submucosal dissection as optimal endoscopic procedures for superficial esophageal carcinoma. Surg Endosc 2011; 25:2541–2546.
- Kagemoto K, Oka S, Tanaka S, et al. Clinical outcomes of endoscopic submucosal dissection for superficial Barrett’s adenocarcinoma. Gastrointest Endosc 2014; 80:239–245.
- Katada C, Muto M, Manabe T, Boku N, Ohtsu A, Yoshida S. Esophageal stenosis after endoscopic mucosal resection of superficial esophageal lesions. Gastrointest Endosc 2003; 57:165–169.
- Hanaoka N, Ishihara R, Takeuchi Y, et al. 1139: A single session of intralesional steroid injection to prevent esophageal stricture after endoscopic submucosal dissection for esophageal squamous cell carcinoma. Gastrointest Endosc 2012; 75(suppl):AB175.
- Yamaguchi N, Isomoto H, Nakayama T, et al. Usefulness of oral prednisolone in the treatment of esophageal stricture after endoscopic submucosal dissection for superficial esophageal squamous cell carcinoma. Gastrointest Endosc 2011; 73:1115–1121.
- Ono S, Fujishiro M, Niimi K, et al. Long-term outcomes of endoscopic submucosal dissection for superficial esophageal squamous cell neoplasms. Gastrointest Endosc 2009; 70:860–866.
- Katada C, Muto M, Manabe T, Ohtsu A, Yoshida S. Local recurrence of squamous-cell carcinoma of the esophagus after EMR. Gastrointest Endosc 2005; 61:219–225.
- Hirasawa K, Kokawa A, Oka H, et al. Superficial adenocarcinoma of the esophagogastric junction: long-term results of endoscopic submucosal dissection. Gastrointest Endosc 2010; 72:960–966.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
- Nakajima T, Saito Y, Tanaka S, et al. Current status of endoscopic resection strategy for large, early colorectal neoplasia in Japan. Surg Endosc 2013; 27:3262–3770.
- Saito Y, Uraoka T, Yamaguchi Y, et al. A prospective, multicenter study of 1111 colorectal endoscopic submucosal dissections (with video). Gastrointest Endosc 2010; 72:1217–1225.
- Tanaka S, Saitoh Y, Matsuda T, et al; Japanese Society of Gastroenterology. Evidence-based clinical practice guidelines for management of colorectal polyps. J Gastroenterol 2015; 50:252–260.
- Oka S, Tanaka S, Saito Y, et al; Colorectal Endoscopic Resection Standardization Implementation Working Group of the Japanese Society for Cancer of the Colon and Rectum, Tokyo, Japan. Local recurrence after endoscopic resection for large colorectal neoplasia: a multicenter prospective study in Japan. Am J Gastroenterol 2015; 110:697–707.
- Saito Y, Fukuzawa M, Matsuda T, et al. Clinical outcome of endoscopic submucosal dissection versus endoscopic mucosal resection of large colorectal tumors as determined by curative resection. Surg Endosc 2010; 24:343–352.
- Makazu M, Sakamoto T, So E, et al. Relationship between indeterminate or positive lateral margin and local recurrence after endoscopic resection of colorectal polyps. Endosc Int Open 2015; 3:E252–E257.
- Belderbos TD, Leenders M, Moons LM, Siersema PD. Local recurrence after endoscopic mucosal resection of nonpedunculated colorectal lesions: systematic review and meta-analysis. Endoscopy 2014; 46:388–402.
- Fujiya M, Tanaka K, Dokoshi T, et al. Efficacy and adverse events of EMR and endoscopic submucosal dissection for the treatment of colon neoplasms: a meta-analysis of studies comparing EMR and endoscopic submucosal dissection. Gastrointest Endosc 2015; 81:583–595.
- Rahmi G, Tanaka S, Ohara Y, et al. Efficacy of endoscopic submucosal dissection for residual or recurrent superficial colorectal tumors after endoscopic mucosal resection. J Dig Dis 2015; 16:14–21.
- Abe S, Oda I, Suzuki H, et al. Long-term surveillance and treatment outcomes of metachronous gastric cancer occurring after curative endoscopic submucosal dissection. Endoscopy 2015; 47:1113–1118.
- Oda I, Suzuki H, Nonaka S, Yoshinaga S. Complications of gastric endoscopic submucosal dissection. Dig Endosc 2013; 25(suppl 1):71–78.
- Takizawa K, Oda I, Gotoda T, et al. Routine coagulation of visible vessels may prevent delayed bleeding after endoscopic submucosal dissection—an analysis of risk factors. Endoscopy 2008; 40:179–183.
- Uedo N, Takeuchi Y, Yamada T, et al. Effect of a proton pump inhibitor or an H2-receptor antagonist on prevention of bleeding from ulcer after endoscopic submucosal dissection of early gastric cancer: a prospective randomized controlled trial. Am J Gastroenterol 2007; 102:1610–1616.
- Hayashi N, Tanaka S, Nishiyama S, et al. Predictors of incomplete resection and perforation associated with endoscopic submucosal dissection for colorectal tumors. Gastrointest Endosc 2014; 79:427–435.
- Suzuki H, Oda I, Sekiguchi M, et al. Management and associated factors of delayed perforation after gastric endoscopic submucosal dissection. World J Gastroenterol 2015; 21:12635–12643.
- Tsunada S, Ogata S, Mannen K, et al. Case series of endoscopic balloon dilation to treat a stricture caused by circumferential resection of the gastric antrum by endoscopic submucosal dissection. Gastrointest Endosc 2008; 67:979–983.
- Coda S, Oda I, Gotoda T, Yokoi C, Kikuchi T, Ono H. Risk factors for cardiac and pyloric stenosis after endoscopic submucosal dissection, and efficacy of endoscopic balloon dilation treatment. Endoscopy 2009; 41:421–426.
- Abe S, Sakamoto T, Takamaru H, et al. Stenosis rates after endoscopic submucosal dissection of large rectal tumors involving greater than three quarters of the luminal circumference. Surg Endosc 2016; 30:5459–5464.
- Sakamoto T, Saito Y, Fukunaga S, Nakajima T, Matsuda T. Learning curve associated with colorectal endoscopic submucosal dissection for endoscopists experienced in gastric endoscopic submucosal dissection. Dis Colon Rectum 2011; 54:1307–1312.
The treatment of early esophageal, gastric, and colorectal cancer is changing.1 For many years, surgery was the mainstay of treatment for early-stage gastrointestinal cancer. Unfortunately, surgery leads to significant loss of function of the organ, resulting in increased morbidity and decreased quality of life.2
Endoscopic techniques, particularly endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), have been developed and are widely used in Japan, where gastrointestinal cancer is more common than in the West. This article reviews the indications, complications, and outcomes of ESD for early gastrointestinal neoplasms, so that readers will recognize the subset of patients who would benefit from ESD in a Western setting.
ENDOSCOPIC MUCOSAL RESECTION AND SUBMUCOSAL DISSECTION
Since the first therapeutic polypectomy was performed in Japan in 1974, several endoscopic techniques for tumor resection have been developed.3
EMR, one of the most successful and widely used techniques, involves elevating the lesion either with submucosal injection of a solution or with cap suction, and then removing it with a snare.4 Most lesions smaller than 20 mm can be removed in one piece (en bloc).5 Larger lesions are removed in multiple pieces (ie, piecemeal). Unfortunately, some fibrotic lesions, which are usually difficult to lift, cannot be completely removed by EMR.
ESD was first performed in the late 1990s with the aim of overcoming the limitations of EMR in resecting large or fibrotic tumors en bloc.6,7 Since then, ESD technique has been standardized and training centers have been created, especially in Asia, where it is widely used for treatment of early gastric cancer.3,8–10 Since 2012 it has been covered by the Japanese National Health Insurance for treatment of early gastric cancer, and since 2014 for treatment of colorectal malignant tumors measuring 2 to 5 cm.11
Adoption of ESD has been slow in Western countries, where many patients are still referred for surgery or undergo EMR for removal of superficial neoplasms. Reasons for this slow adoption are that gastric cancer is much less common in Western countries, and also that ESD demands a high level of technical skill, is difficult to learn, and is expensive.3,12,13 However, small groups of Western endoscopists have become interested and are advocating it, first studying it on their own and then training in a Japanese center and learning from experts performing the procedure.
Therefore, in a Western setting, ESD should be performed in specialized endoscopy centers and offered to selected patients.1
CANDIDATES SHOULD HAVE EARLY-STAGE, SUPERFICIAL TUMORS
Ideal candidates for endoscopic resection are patients who have early cancer with a negligible risk of lymph node metastasis, such as cancer limited to the mucosa (stage T1a).7 Therefore, to determine the best treatment for a patient with a newly diagnosed gastrointestinal neoplasm, it is mandatory to estimate the depth of invasion.
The depth of invasion is directly correlated with lymph node involvement, which is ultimately the main predictive factor for long-term adverse outcomes of gastrointestinal tumors.4,14–17 Accurate multidisciplinary preprocedure estimations are mandatory, as incorrect evaluations may result in inappropriate therapy and residual cancer.18
Other factors that have been used to predict lymph node involvement include tumor size, macroscopic appearance, histologic differentiation, and lymphatic and vascular involvement.19 Some of these factors can be assessed by special endoscopic techniques (chromoendoscopy and narrow-band imaging with magnifying endoscopy) that allow accurate real-time estimation of the depth of invasion of the lesion.5,17,20–27 Evaluation of microsurface and microvascular arrangements is especially useful for determining the feasibility of ESD in gastric tumors, evaluation of intracapillary loops is useful in esophageal lesions, and assessment of mucosal pit patterns is useful for colorectal lesions.21–29
Endoscopic ultrasonography is another tool that has been used to estimate the depth of the tumor. Although it can differentiate between definite intramucosal and definite submucosal invasive cancers, its ability to confirm minute submucosal invasion is limited. Its use as the sole tumor staging modality is not encouraged, and it should always be used in conjunction with endoscopic evaluation.18
Though the aforementioned factors help stratify patients, pathologic staging is the best predictor of lymph node metastasis. ESD provides adequate specimens for accurate pathologic evaluation, as it removes lesions en bloc.30
All patients found to have risk factors for lymph node metastasis on endoscopic, ultrasonographic, or pathologic analysis should be referred for surgical evaluation.9,19,31,32
ENDOSCOPIC SUBMUCOSAL DISSECTION
Before the procedure, the patient’s physicians need to do the following:
Determine the best type of intervention (EMR, ESD, ablation, surgery) for the specific lesion.3 A multidisciplinary approach is encouraged, with involvement of the internist, gastroenterologist, and surgeon.
Plan for anesthesia, additional consultations, pre- and postprocedural hospital admission, and need for special equipment.33
During the procedure
Define the lateral extent of the lesion using magnification chromoendoscopy or narrow-band imaging. In the stomach, a biopsy sample should be taken from the worst-looking segment and from normal-looking mucosa. Multiple biopsies should be avoided to prevent subsequent fibrosis.33 In the colon, biopsy should be avoided.34
Identify and circumferentially mark the target lesion. Cautery or argon plasma coagulation can be used for making markings at a distance of 5 to 10 mm from the edges.33 This is done to recognize the borders of the lesion, because they can become distorted after submucosal injection.14 This step is unnecessary in colorectal cases, as tumor margins can be adequately visualized after chromoendoscopy.16,35
Lift the lesion by injecting saline, 0.5% hyaluronate, or glycerin to create a submucosal fluid cushion.19,33
Perform a circumferential incision lateral to the mucosal margins to allow for a normal tissue margin.33 Partial incision is performed for esophageal and colorectal ESD to avoid fluid leakage from the submucosal layer, achieving a sustained submucosal lift and safer dissection.16
Submucosal dissection. The submucosal layer is dissected with an electrocautery knife until the lesion is completely removed. Dissection should be done carefully to keep the submucosal plane.33 Hemoclips or hemostat forceps can be used to control visible bleeding. The resected specimen is then stretched and fixed to a board using small pins for further histopathologic evaluation.35
Postprocedural monitoring. All patients should be admitted for overnight observation. Those who undergo gastric ESD should receive high-dose acid suppression, and the next day they can be started on a liquid diet.19
STOMACH CANCER
Indications for ESD for stomach cancer in the East
The incidence of gastric cancer is higher in Japan and Korea, where widespread screening programs have led to early identification and early treatment of this disease.36
Pathology studies37 of samples from patients with gastric cancer identified the following as risk factors for lymph node metastasis, which would make ESD unsuitable:
- Undifferentiated type
- Tumors larger than 2 cm
- Lymphatic or venous involvement
- Submucosal invasion
- Ulcerative change.
Based on these findings, the situations in which there was no risk of lymph node involvement (ie, when none of the above factors are present) were accepted as absolute indications for endoscopic resection of early gastric cancer.38 Further histologic studies identified a subset of patients with lesions with very low risk of lymph node metastasis, which outweighed the risk of surgery. Based on these findings, expanded criteria for gastric ESD were proposed,39,40 and the Japanese gastric cancer treatment guidelines now include these expanded preoperative indications9,17 (Table 1).
The Japanese Gastric Cancer Association has proposed a treatment algorithm based on the histopathologic evaluation after resection (Figure 2).9
Outcomes
In the largest series of patients who underwent curative ESD for early gastric cancer, the 5-year survival rate was 92.6%, the 5-year disease-specific survival rate was 99.9%, and the 5-year relative survival rate was 105%.41
Similarly, in a Japanese population-based survival analysis, the relative 5-year survival rate for localized gastric cancer was 94.4%.42 Rates of en bloc resection and complete resection with ESD are higher than those with EMR, resulting in a lower risk of local recurrence in selected patients who undergo ESD.8,43,44
Although rare, local recurrence after curative gastric ESD has been reported.45 The annual incidence of local recurrence has been estimated to be 0.84%.46
ESD entails a shorter hospital stay and requires fewer resources than surgery, resulting in lower medical costs (Table 2).44 Additionally, as endoscopic resection is associated with less morbidity, fewer procedure-related adverse events, and fewer complications, ESD could be used as the standard treatment for early gastric cancer.47,48
The Western perspective on endoscopic submucosal dissection for gastric cancer
Since the prevalence of gastric cancer in Western countries is significantly lower than in Japan and Korea, local data and experience are scarce. However, experts performing ESD in the West have adopted the indications of the Japan Gastroenterological Endoscopy Society. The European Society of Gastrointestinal Endoscopy recommends ESD for excision of most superficial gastric neoplasms, with EMR being preferred only in lesions smaller than 15 mm, Paris classification 0 or IIA.5,32
Patients with gastric lesions measuring 15 mm or larger should undergo high-quality endoscopy, preferably chromoendoscopy, to evaluate the mucosal patterns and determine the depth of invasion. If superficial involvement is confirmed, other imaging techniques are not routinely recommended.5 A surgery consult is also recommended.
ESOPHAGEAL CANCER
Indications for ESD for esophageal cancer in the East
Due to the success of ESD for early gastric cancer, this technique is now also used for superficial esophageal neoplasms.19,49 It should be done in a specialized center, as it is more technically difficult than gastric ESD: the esophageal lumen is narrow, the wall is thin, and the esophagus moves with respiration and heartbeat.50 A multidisciplinary approach including an endoscopist, a surgeon, and a pathologist is highly recommended for evaluation and treatment.
EMR is preferred for removal of mucosal cancer, in view of its safety profile and success rates. ESD can be considered in cases of lesions larger than 15 mm, poorly lifting tumors, and those with the possibility of submucosal invasion (Table 3).5,45,49,51
Circumference involvement is critical when determining eligible candidates, as a defect involving more than three-fourths of the esophageal circumference can lead to esophageal strictures.52 Controlled prospective studies have shown promising results from giving intralesional and oral steroids to prevent stricture after ESD, which could potentially overcome this size limitation.53,54
Outcomes for esophageal cancer
ESD has been shown to be safe and effective, achieving en bloc resection in 85% to 100% of patients.19,51 Its advantages over EMR include en bloc resection, complete resection, and high curative rates, resulting in higher recurrence-free survival.2,55,56 Although the incidence of complications such as bleeding, perforation, and stricture formation are higher with ESD, patients usually recover uneventfully.2,19,20
ESD in the esophagus: The Western perspective
As data on the efficacy of EMR vs ESD for the treatment of Barrett esophagus with adenocarcinoma are limited, EMR is the gold standard endoscopic technique for removal of visible esophageal dysplastic lesions.5,51,57 ESD can be considered for tumors larger than 15 mm, for poorly lifting lesions, and if there is suspicion of submucosal invasion.5
Patients should be evaluated by an experienced endoscopist, using an advanced imaging technique such as narrow-band imaging or chromoendoscopy. If suspicious features are found, endoscopic ultrasonography should be considered to confirm submucosal invasion or lymph node involvement.5
COLORECTAL CANCER
Indications for ESD for colorectal cancer in the East
Colon cancer is one of the leading causes of cancer-related deaths worldwide.58 Since ESD has been found to be effective and safe in treating gastric cancer, it has also been used to remove large colorectal tumors.59 However, ESD is not universally accepted in the treatment of colorectal neoplasms due to its greater technical difficulty, longer procedural time, and higher risk of perforating the thinner colonic wall compared with EMR.21,60
Outcomes for colorectal cancer
Tumor size of 50 mm or larger is a risk factor for complications, while a high procedure volume at the center is a protective factor.60
Endoscopic treatment of colorectal cancer: The Western perspective
EMR is the gold standard for removal of superficial colorectal lesions. However, ESD can be considered if there is suspicion of superficial submucosal invasion, especially for lesions larger than 20 mm that cannot be resected en bloc by EMR.32 ESD can also be used for fibrotic lesions not amenable to complete EMR removal, or as a salvage procedure after recurrence after EMR.67 Proper selection of cases is critical.1
Patients who have a superficial colonic lesion should be evaluated by means of high-definition endoscopy and chromoendoscopy to assess the mucosal pattern and establish feasibility of endoscopic resection. If submucosal invasion is suspected, staging with endoscopic ultrasonography or magnetic resonance imaging should be considered.5
FOLLOW-UP AFTER ESD
Endoscopic surveillance after the procedure is recommended, given the persistent risk of metachronous cancer after curative ESD due to its organ-sparing quality.68 Surveillance endoscopy aims to achieve early detection and subsequent endoscopic resection of metachronous lesions.
Histopathologic evaluation assessing the presence of malignant cells in the margins of a resected sample is mandatory for determining the next step in treatment. If margins are negative, follow-up endoscopy can be done every 6 to 12 months. If margins are positive, the approach includes surgery, reattempting ESD or endoscopic surveillance in 3 or 6 months.3,32 Although the surveillance strategy varies according to individual risk of metachronous cancer, it should be continued indefinitely.68
COMPLICATIONS OF ESD
The most common procedure-related complications of ESD are bleeding, perforation, and stricture. Most intraprocedural adverse events can be managed endoscopically.69
Bleeding
Most bleeding occurs during the procedure or early after it and can be controlled with electrocautery.49,69 No episodes of massive bleeding, defined as causing clinical symptoms and requiring transfusion or surgery, have been reported.20,43,55
In gastric ESD, delayed bleeding rates have ranged from 0 to 15.6%.69 Bleeding may be prevented with endoscopic coagulation of visible vessels after dissection has been completed and by proton pump inhibitor therapy.70,71 Excessive coagulation should be avoided to lower the risk of perforation.33
In colorectal ESD the bleeding rate has been reported to be 2.2%; applying coagulation to an area where a blood vessel is suspected before cutting (precoagulation) may prevent subsequent bleeding.21
Perforation
For gastric ESD, perforation rates range from 1.2% to 5.2%.69 Esophageal perforation rates can be up to 4%.49 In colorectal ESD, perforation rates have been reported to be 1.6% to 6.6%.60,72
Although most of the cases were successfully managed with conservative treatment, some required emergency surgery.60,73
Strictures
In a case series of 532 patients undergoing gastric ESD, stricture was reported in 5 patients, all of whom presented with obstructive symptoms.74 Risk factors for post-ESD gastric stenosis are a mucosal defect with a circumferential extent of more than three-fourths or a longitudinal extent of more than 5 cm.75
Strictures are common after esophageal ESD, with rates ranging from 2% to 26%. The risk is higher when longer segments are removed or circumferential resection is performed. As previously mentioned, this complication may be reduced with ingestion or injection of steroids after the procedure.53,54
Surprisingly, ESD of large colorectal lesions involving more than three-fourths of the circumference of the rectum is rarely complicated by stenosis.76
LIMITATIONS OF ESD
ESD requires a high level of technical skill, is time-consuming, and has a higher rate of complications than conventional endoscopic resection. A standardized ESD training system is needed, as the procedure is more difficult than EMR. Training in porcine models has been shown to confer competency in ESD in a Western setting.13,16,33
Colorectal ESD is an even more challenging procedure, given the potential for complications related to its anatomy. Training centers in Japan usually have their trainees first master gastric ESD, then assist in more than 20 colorectal ESDs conducted by experienced endoscopists, and accomplish 30 cases before performing the procedure safely and independently.
As the incidence of gastric cancer is low in Western countries, trainees may also begin with lower rectal lesions, which are easier to remove.77 Incorporation of ESD in the West would require a clear treatment algorithm. It is a complex procedure, with higher rates of complications, a prolonged learning curve, and prolonged procedure time. Therefore, it should be performed in specialized centers and under the special situations discussed here to ensure that the benefits for the patients outweigh the risks.
VALUE OF ENDOSCOPIC SUBMUCOSAL DISSECTION
The optimal method for resecting gastrointestinal neoplasms should be safe, cost-effective, and quick and should also completely remove the lesion. The best treatment strategy takes into account the characteristics of the lesion and the comorbidities and wishes of the patient. Internists should be aware of the multiple options available to achieve the best outcome for the patient.1
Endoscopic resection of superficial gastrointestinal neoplasms, including EMR and ESD, has been a subject of increasing interest due to its minimally invasive and potentially curative character. However, cancer can recur after endoscopic resection because the procedure is organ-sparing.
ESD allows resection of early gastrointestinal tumors with a minimally invasive technique. It can achieve higher curative resection rates and lower recurrence rates compared with EMR. Compared with surgery, ESD leads to less morbidity, fewer procedure-related complications, and lower medical costs. Indications should be rigorously followed to achieve successful treatments in selected patients.
Multiple variables have to be taken into account when deciding which treatment is best, such as tumor characteristics, the patient’s baseline condition, physician expertise, and hospital resources.48 Less-invasive treatments may improve the prognosis of patients. No matter the approach, patients should be treated in specialized treatment centers.
Internal medicine physicians should be aware of the advances in treatments for early gastrointestinal cancer so appropriate options can be considered.
The treatment of early esophageal, gastric, and colorectal cancer is changing.1 For many years, surgery was the mainstay of treatment for early-stage gastrointestinal cancer. Unfortunately, surgery leads to significant loss of function of the organ, resulting in increased morbidity and decreased quality of life.2
Endoscopic techniques, particularly endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), have been developed and are widely used in Japan, where gastrointestinal cancer is more common than in the West. This article reviews the indications, complications, and outcomes of ESD for early gastrointestinal neoplasms, so that readers will recognize the subset of patients who would benefit from ESD in a Western setting.
ENDOSCOPIC MUCOSAL RESECTION AND SUBMUCOSAL DISSECTION
Since the first therapeutic polypectomy was performed in Japan in 1974, several endoscopic techniques for tumor resection have been developed.3
EMR, one of the most successful and widely used techniques, involves elevating the lesion either with submucosal injection of a solution or with cap suction, and then removing it with a snare.4 Most lesions smaller than 20 mm can be removed in one piece (en bloc).5 Larger lesions are removed in multiple pieces (ie, piecemeal). Unfortunately, some fibrotic lesions, which are usually difficult to lift, cannot be completely removed by EMR.
ESD was first performed in the late 1990s with the aim of overcoming the limitations of EMR in resecting large or fibrotic tumors en bloc.6,7 Since then, ESD technique has been standardized and training centers have been created, especially in Asia, where it is widely used for treatment of early gastric cancer.3,8–10 Since 2012 it has been covered by the Japanese National Health Insurance for treatment of early gastric cancer, and since 2014 for treatment of colorectal malignant tumors measuring 2 to 5 cm.11
Adoption of ESD has been slow in Western countries, where many patients are still referred for surgery or undergo EMR for removal of superficial neoplasms. Reasons for this slow adoption are that gastric cancer is much less common in Western countries, and also that ESD demands a high level of technical skill, is difficult to learn, and is expensive.3,12,13 However, small groups of Western endoscopists have become interested and are advocating it, first studying it on their own and then training in a Japanese center and learning from experts performing the procedure.
Therefore, in a Western setting, ESD should be performed in specialized endoscopy centers and offered to selected patients.1
CANDIDATES SHOULD HAVE EARLY-STAGE, SUPERFICIAL TUMORS
Ideal candidates for endoscopic resection are patients who have early cancer with a negligible risk of lymph node metastasis, such as cancer limited to the mucosa (stage T1a).7 Therefore, to determine the best treatment for a patient with a newly diagnosed gastrointestinal neoplasm, it is mandatory to estimate the depth of invasion.
The depth of invasion is directly correlated with lymph node involvement, which is ultimately the main predictive factor for long-term adverse outcomes of gastrointestinal tumors.4,14–17 Accurate multidisciplinary preprocedure estimations are mandatory, as incorrect evaluations may result in inappropriate therapy and residual cancer.18
Other factors that have been used to predict lymph node involvement include tumor size, macroscopic appearance, histologic differentiation, and lymphatic and vascular involvement.19 Some of these factors can be assessed by special endoscopic techniques (chromoendoscopy and narrow-band imaging with magnifying endoscopy) that allow accurate real-time estimation of the depth of invasion of the lesion.5,17,20–27 Evaluation of microsurface and microvascular arrangements is especially useful for determining the feasibility of ESD in gastric tumors, evaluation of intracapillary loops is useful in esophageal lesions, and assessment of mucosal pit patterns is useful for colorectal lesions.21–29
Endoscopic ultrasonography is another tool that has been used to estimate the depth of the tumor. Although it can differentiate between definite intramucosal and definite submucosal invasive cancers, its ability to confirm minute submucosal invasion is limited. Its use as the sole tumor staging modality is not encouraged, and it should always be used in conjunction with endoscopic evaluation.18
Though the aforementioned factors help stratify patients, pathologic staging is the best predictor of lymph node metastasis. ESD provides adequate specimens for accurate pathologic evaluation, as it removes lesions en bloc.30
All patients found to have risk factors for lymph node metastasis on endoscopic, ultrasonographic, or pathologic analysis should be referred for surgical evaluation.9,19,31,32
ENDOSCOPIC SUBMUCOSAL DISSECTION
Before the procedure, the patient’s physicians need to do the following:
Determine the best type of intervention (EMR, ESD, ablation, surgery) for the specific lesion.3 A multidisciplinary approach is encouraged, with involvement of the internist, gastroenterologist, and surgeon.
Plan for anesthesia, additional consultations, pre- and postprocedural hospital admission, and need for special equipment.33
During the procedure
Define the lateral extent of the lesion using magnification chromoendoscopy or narrow-band imaging. In the stomach, a biopsy sample should be taken from the worst-looking segment and from normal-looking mucosa. Multiple biopsies should be avoided to prevent subsequent fibrosis.33 In the colon, biopsy should be avoided.34
Identify and circumferentially mark the target lesion. Cautery or argon plasma coagulation can be used for making markings at a distance of 5 to 10 mm from the edges.33 This is done to recognize the borders of the lesion, because they can become distorted after submucosal injection.14 This step is unnecessary in colorectal cases, as tumor margins can be adequately visualized after chromoendoscopy.16,35
Lift the lesion by injecting saline, 0.5% hyaluronate, or glycerin to create a submucosal fluid cushion.19,33
Perform a circumferential incision lateral to the mucosal margins to allow for a normal tissue margin.33 Partial incision is performed for esophageal and colorectal ESD to avoid fluid leakage from the submucosal layer, achieving a sustained submucosal lift and safer dissection.16
Submucosal dissection. The submucosal layer is dissected with an electrocautery knife until the lesion is completely removed. Dissection should be done carefully to keep the submucosal plane.33 Hemoclips or hemostat forceps can be used to control visible bleeding. The resected specimen is then stretched and fixed to a board using small pins for further histopathologic evaluation.35
Postprocedural monitoring. All patients should be admitted for overnight observation. Those who undergo gastric ESD should receive high-dose acid suppression, and the next day they can be started on a liquid diet.19
STOMACH CANCER
Indications for ESD for stomach cancer in the East
The incidence of gastric cancer is higher in Japan and Korea, where widespread screening programs have led to early identification and early treatment of this disease.36
Pathology studies37 of samples from patients with gastric cancer identified the following as risk factors for lymph node metastasis, which would make ESD unsuitable:
- Undifferentiated type
- Tumors larger than 2 cm
- Lymphatic or venous involvement
- Submucosal invasion
- Ulcerative change.
Based on these findings, the situations in which there was no risk of lymph node involvement (ie, when none of the above factors are present) were accepted as absolute indications for endoscopic resection of early gastric cancer.38 Further histologic studies identified a subset of patients with lesions with very low risk of lymph node metastasis, which outweighed the risk of surgery. Based on these findings, expanded criteria for gastric ESD were proposed,39,40 and the Japanese gastric cancer treatment guidelines now include these expanded preoperative indications9,17 (Table 1).
The Japanese Gastric Cancer Association has proposed a treatment algorithm based on the histopathologic evaluation after resection (Figure 2).9
Outcomes
In the largest series of patients who underwent curative ESD for early gastric cancer, the 5-year survival rate was 92.6%, the 5-year disease-specific survival rate was 99.9%, and the 5-year relative survival rate was 105%.41
Similarly, in a Japanese population-based survival analysis, the relative 5-year survival rate for localized gastric cancer was 94.4%.42 Rates of en bloc resection and complete resection with ESD are higher than those with EMR, resulting in a lower risk of local recurrence in selected patients who undergo ESD.8,43,44
Although rare, local recurrence after curative gastric ESD has been reported.45 The annual incidence of local recurrence has been estimated to be 0.84%.46
ESD entails a shorter hospital stay and requires fewer resources than surgery, resulting in lower medical costs (Table 2).44 Additionally, as endoscopic resection is associated with less morbidity, fewer procedure-related adverse events, and fewer complications, ESD could be used as the standard treatment for early gastric cancer.47,48
The Western perspective on endoscopic submucosal dissection for gastric cancer
Since the prevalence of gastric cancer in Western countries is significantly lower than in Japan and Korea, local data and experience are scarce. However, experts performing ESD in the West have adopted the indications of the Japan Gastroenterological Endoscopy Society. The European Society of Gastrointestinal Endoscopy recommends ESD for excision of most superficial gastric neoplasms, with EMR being preferred only in lesions smaller than 15 mm, Paris classification 0 or IIA.5,32
Patients with gastric lesions measuring 15 mm or larger should undergo high-quality endoscopy, preferably chromoendoscopy, to evaluate the mucosal patterns and determine the depth of invasion. If superficial involvement is confirmed, other imaging techniques are not routinely recommended.5 A surgery consult is also recommended.
ESOPHAGEAL CANCER
Indications for ESD for esophageal cancer in the East
Due to the success of ESD for early gastric cancer, this technique is now also used for superficial esophageal neoplasms.19,49 It should be done in a specialized center, as it is more technically difficult than gastric ESD: the esophageal lumen is narrow, the wall is thin, and the esophagus moves with respiration and heartbeat.50 A multidisciplinary approach including an endoscopist, a surgeon, and a pathologist is highly recommended for evaluation and treatment.
EMR is preferred for removal of mucosal cancer, in view of its safety profile and success rates. ESD can be considered in cases of lesions larger than 15 mm, poorly lifting tumors, and those with the possibility of submucosal invasion (Table 3).5,45,49,51
Circumference involvement is critical when determining eligible candidates, as a defect involving more than three-fourths of the esophageal circumference can lead to esophageal strictures.52 Controlled prospective studies have shown promising results from giving intralesional and oral steroids to prevent stricture after ESD, which could potentially overcome this size limitation.53,54
Outcomes for esophageal cancer
ESD has been shown to be safe and effective, achieving en bloc resection in 85% to 100% of patients.19,51 Its advantages over EMR include en bloc resection, complete resection, and high curative rates, resulting in higher recurrence-free survival.2,55,56 Although the incidence of complications such as bleeding, perforation, and stricture formation are higher with ESD, patients usually recover uneventfully.2,19,20
ESD in the esophagus: The Western perspective
As data on the efficacy of EMR vs ESD for the treatment of Barrett esophagus with adenocarcinoma are limited, EMR is the gold standard endoscopic technique for removal of visible esophageal dysplastic lesions.5,51,57 ESD can be considered for tumors larger than 15 mm, for poorly lifting lesions, and if there is suspicion of submucosal invasion.5
Patients should be evaluated by an experienced endoscopist, using an advanced imaging technique such as narrow-band imaging or chromoendoscopy. If suspicious features are found, endoscopic ultrasonography should be considered to confirm submucosal invasion or lymph node involvement.5
COLORECTAL CANCER
Indications for ESD for colorectal cancer in the East
Colon cancer is one of the leading causes of cancer-related deaths worldwide.58 Since ESD has been found to be effective and safe in treating gastric cancer, it has also been used to remove large colorectal tumors.59 However, ESD is not universally accepted in the treatment of colorectal neoplasms due to its greater technical difficulty, longer procedural time, and higher risk of perforating the thinner colonic wall compared with EMR.21,60
Outcomes for colorectal cancer
Tumor size of 50 mm or larger is a risk factor for complications, while a high procedure volume at the center is a protective factor.60
Endoscopic treatment of colorectal cancer: The Western perspective
EMR is the gold standard for removal of superficial colorectal lesions. However, ESD can be considered if there is suspicion of superficial submucosal invasion, especially for lesions larger than 20 mm that cannot be resected en bloc by EMR.32 ESD can also be used for fibrotic lesions not amenable to complete EMR removal, or as a salvage procedure after recurrence after EMR.67 Proper selection of cases is critical.1
Patients who have a superficial colonic lesion should be evaluated by means of high-definition endoscopy and chromoendoscopy to assess the mucosal pattern and establish feasibility of endoscopic resection. If submucosal invasion is suspected, staging with endoscopic ultrasonography or magnetic resonance imaging should be considered.5
FOLLOW-UP AFTER ESD
Endoscopic surveillance after the procedure is recommended, given the persistent risk of metachronous cancer after curative ESD due to its organ-sparing quality.68 Surveillance endoscopy aims to achieve early detection and subsequent endoscopic resection of metachronous lesions.
Histopathologic evaluation assessing the presence of malignant cells in the margins of a resected sample is mandatory for determining the next step in treatment. If margins are negative, follow-up endoscopy can be done every 6 to 12 months. If margins are positive, the approach includes surgery, reattempting ESD or endoscopic surveillance in 3 or 6 months.3,32 Although the surveillance strategy varies according to individual risk of metachronous cancer, it should be continued indefinitely.68
COMPLICATIONS OF ESD
The most common procedure-related complications of ESD are bleeding, perforation, and stricture. Most intraprocedural adverse events can be managed endoscopically.69
Bleeding
Most bleeding occurs during the procedure or early after it and can be controlled with electrocautery.49,69 No episodes of massive bleeding, defined as causing clinical symptoms and requiring transfusion or surgery, have been reported.20,43,55
In gastric ESD, delayed bleeding rates have ranged from 0 to 15.6%.69 Bleeding may be prevented with endoscopic coagulation of visible vessels after dissection has been completed and by proton pump inhibitor therapy.70,71 Excessive coagulation should be avoided to lower the risk of perforation.33
In colorectal ESD the bleeding rate has been reported to be 2.2%; applying coagulation to an area where a blood vessel is suspected before cutting (precoagulation) may prevent subsequent bleeding.21
Perforation
For gastric ESD, perforation rates range from 1.2% to 5.2%.69 Esophageal perforation rates can be up to 4%.49 In colorectal ESD, perforation rates have been reported to be 1.6% to 6.6%.60,72
Although most of the cases were successfully managed with conservative treatment, some required emergency surgery.60,73
Strictures
In a case series of 532 patients undergoing gastric ESD, stricture was reported in 5 patients, all of whom presented with obstructive symptoms.74 Risk factors for post-ESD gastric stenosis are a mucosal defect with a circumferential extent of more than three-fourths or a longitudinal extent of more than 5 cm.75
Strictures are common after esophageal ESD, with rates ranging from 2% to 26%. The risk is higher when longer segments are removed or circumferential resection is performed. As previously mentioned, this complication may be reduced with ingestion or injection of steroids after the procedure.53,54
Surprisingly, ESD of large colorectal lesions involving more than three-fourths of the circumference of the rectum is rarely complicated by stenosis.76
LIMITATIONS OF ESD
ESD requires a high level of technical skill, is time-consuming, and has a higher rate of complications than conventional endoscopic resection. A standardized ESD training system is needed, as the procedure is more difficult than EMR. Training in porcine models has been shown to confer competency in ESD in a Western setting.13,16,33
Colorectal ESD is an even more challenging procedure, given the potential for complications related to its anatomy. Training centers in Japan usually have their trainees first master gastric ESD, then assist in more than 20 colorectal ESDs conducted by experienced endoscopists, and accomplish 30 cases before performing the procedure safely and independently.
As the incidence of gastric cancer is low in Western countries, trainees may also begin with lower rectal lesions, which are easier to remove.77 Incorporation of ESD in the West would require a clear treatment algorithm. It is a complex procedure, with higher rates of complications, a prolonged learning curve, and prolonged procedure time. Therefore, it should be performed in specialized centers and under the special situations discussed here to ensure that the benefits for the patients outweigh the risks.
VALUE OF ENDOSCOPIC SUBMUCOSAL DISSECTION
The optimal method for resecting gastrointestinal neoplasms should be safe, cost-effective, and quick and should also completely remove the lesion. The best treatment strategy takes into account the characteristics of the lesion and the comorbidities and wishes of the patient. Internists should be aware of the multiple options available to achieve the best outcome for the patient.1
Endoscopic resection of superficial gastrointestinal neoplasms, including EMR and ESD, has been a subject of increasing interest due to its minimally invasive and potentially curative character. However, cancer can recur after endoscopic resection because the procedure is organ-sparing.
ESD allows resection of early gastrointestinal tumors with a minimally invasive technique. It can achieve higher curative resection rates and lower recurrence rates compared with EMR. Compared with surgery, ESD leads to less morbidity, fewer procedure-related complications, and lower medical costs. Indications should be rigorously followed to achieve successful treatments in selected patients.
Multiple variables have to be taken into account when deciding which treatment is best, such as tumor characteristics, the patient’s baseline condition, physician expertise, and hospital resources.48 Less-invasive treatments may improve the prognosis of patients. No matter the approach, patients should be treated in specialized treatment centers.
Internal medicine physicians should be aware of the advances in treatments for early gastrointestinal cancer so appropriate options can be considered.
- Burgess NG, Bourke MJ. Endoscopic resection of colorectal lesions: the narrowing divide between East and West. Dig Endosc 2016; 28:296–305.
- Kim DH, Jung HY, Gong EJ, et al. Endoscopic and oncologic outcomes of endoscopic resection for superficial esophageal neoplasm. Gut Liver 2015; 9:470–477.
- Draganov PV, Gotoda T, Chavalitdhamrong D, Wallace MB. Techniques of endoscopic submucosal dissection: application for the Western endoscopist? Gastrointest Endosc 2013; 78:677–688.
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011; 14:101–112.
- Pimentel-Nunes P, Dinis-Ribeiro M, Ponchon T, et al. Endoscopic submucosal dissection: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy 2015; 47:829–854.
- Farhat S, Chaussade S, Ponchon T, et al; SFED ESD Study Group. Endoscopic submucosal dissection in a European setting. A multi-institutional report of a technique in development. Endoscopy 2011; 43:664–670.
- Gotoda T, Jung H. Endoscopic resection (endoscopic mucosal resection/endoscopic submucosal dissection) for early gastric cancer. Dig Endosc 2013; 25(suppl 1):55–63.
- Chung IK, Lee JH, Lee SH, et al. Therapeutic outcomes in 1000 cases of endoscopic submucosal dissection for early gastric neoplasms: Korean ESD Study Group multicenter study. Gastrointest Endosc 2009; 69:1228–1235.
- Japanese Gastric Cancer Association. Japanese gastric cancer treatment guidelines 2010 (ver. 3). Gastric Cancer 2011; 14:113–123.
- Ono H. Endoscopic submucosal dissection for early gastric cancer. Chin J Dig Dis 2005; 6:119–121.
- Watanabe T, Itabashi M, Shimada Y, et al; Japanese Society for Cancer of the Colon and Rectum. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2014 for treatment of colorectal cancer. Int J Clin Oncol 2015; 20:207–239.
- Oyama T, Yahagi N, Ponchon T, Kiesslich T, Berr F. How to establish endoscopic submucosal dissection in Western countries. World J Gastroenterol 2015; 21:11209–11220.
- Bhatt A, Abe S, Kumaravel A, et al. SU1575 Western skill training in endoscopic submucosal dissection (ESD)—an international remote video based study—the WEST ESD Study. Gastrointest Endosc 2015; 81(suppl):AB335–AB336.
- Sano T, Sasako M, Kinoshita T, Maruyama K. Recurrence of early gastric cancer follow-up of 1475 patients and review of the Japanese literature. Cancer 1993; 72:3174–3178.
- Japan Esophageal Society. Japanese classification of esophageal cancer, tenth edition: part I. Esophagus 2009; 6:1–25.
- Bhatt A, Abe S, Kumaravel A, Vargo J, Saito Y. Indications and techniques for endoscopic submucosal dissection. Am J Gastroenterol 2015; 110:784–791.
- Eleftheriadis N, Inoue H, Ikeda H, et al. Definition and staging of early esophageal, gastric and colorectal cancer. J Tumor 2014; 2:161–178.
- Yoshinaga S, Oda I, Nonaka S, Kushima R, Saito Y. Endoscopic ultrasound using ultrasound probes for the diagnosis of early esophageal and gastric cancers. World J Gastrointest Endosc 2012; 4:218–226.
- Stahl M, Mariette C, Haustermans K, Cervantes A, Arnold D; ESMO Guidelines Working Group. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi51–vi56.
- Higuchi K, Tanabe S, Azuma M, et al. A phase II study of endoscopic submucosal dissection for superficial esophageal neoplasms (KDOG 0901). Gastrointest Endosc 2013; 78:704–710.
- Sakamoto T, Mori G, Yamada M, et al. Endoscopic submucosal dissection for colorectal neoplasms: a review. World J Gastroenterol 2014; 20:16153–16158.
- Ohta A, Tominaga K, Sakai Y. Efficacy of magnifying colonoscopy for the diagnosis of colorectal neoplasia: comparison with histopathological findings. Dig Endosc 2004; 16:308–314.
- Katagiri A, Fu K, Sano Y, et al. Narrow band imaging with magnifying colonoscopy as diagnostic tool for predicting histology of early colorectal neoplasia. Aliment Pharmacol Ther 2008; 27:1269–1274.
- Fu KI, Kato S, Sano Y, et al. Staging of early colorectal cancers: magnifying colonoscopy versus endoscopic ultrasonography for estimation of depth of invasion. Dig Dis Sci 2008; 53:1886–1892.
- Uraoka T, Saito Y, Ikematsu H, Yamamoto K, Sano Y. Sano’s capillary pattern classification for narrow-band imaging of early colorectal lesions. Dig Endosc 2011; 23(suppl 1):112–115.
- Ikematsu H, Matsuda T, Emura F, et al. Efficacy of capillary pattern type IIIA/IIIB by magnifying narrow band imaging for estimating depth of invasion of early colorectal neoplasms. BMC Gastroenterol 2010;10:33.
- Matsuda T, Fujii T, Saito Y, et al. Efficacy of the invasive/non-invasive pattern by magnifying chromoendoscopy to estimate the depth of invasion of early colorectal neoplasms. Am J Gastroenterol 2008; 103:2700–2706.
- Sato H, Inoue H, Ikeda H, et al. Utility of intrapapillary capillary loops seen on magnifying narrow-band imaging in estimating invasive depth of esophageal squamous cell carcinoma. Endoscopy 2015; 8:122–128.
- Muto M, Yao K, Kaise M, et al. Magnifying endoscopy simple diagnostic algorithm for early gastric cancer (MESDA-G). Dig Endosc 2016; 28:379–393.
- Waddell T, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D; European Society for Medical Oncology (ESMO); European Society of Surgical Oncology (ESSO); European Society of Radiotherapy and Oncology (ESTRO). Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi57–vi63.
- Kuwano H, Nishimura Y, Ohtsu A, et al. Guidelines for diagnosis and treatment of carcinoma of the esophagus. April 2007 edition: part I - Edited by the Japan Esophageal Society. Esophagus 2008; 5:61–73.
- Tanaka S, Kashida H, Saito Y, et al. JGES guidelines for colorectal endoscopic submucosal dissection/endoscopic mucosal resection. Dig Endosc 2015; 27:417–434.
- Gotoda T, Ho KY, Soetikno R, Kaltenbach T, Draganov P. Gastric ESD: current status and future directions of devices and training. Gastrointest Endosc Clin North Am 2014; 24:213–233.
- Saito Y, Sakamoto T, Nakajima T, Matsuda T. Colorectal ESD: current indications and latest technical advances. Gastrointest Endosc Clin N Am 2014; 24:245–255.
- Saito Y, Otake Y, Sakamoto T, et al. Indications for and technical aspects of colorectal endoscopic submucosal dissection. Gut Liver 2013; 7:263–269.
- Saragoni L. Upgrading the definition of early gastric cancer: better staging means more appropriate treatment. Cancer Biol Med 2015; 12:355–361.
- Tsujitani S, Oka S, Saito H, et al. Less invasive surgery for early gastric cancer based on the low probability of lymph node metastasis. Surgery 1999; 125:148–154.
- Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003; 57:567–579.
- Hirasawa T, Gotoda T, Miyata S, et al. Incidence of lymph node metastasis and the feasibility of endoscopic resection for undifferentiated-type early gastric cancer. Gastric Cancer 2009; 12:148–152.
- Gotoda T, Yanagisawa A, Sasako M, et al. Incidence of lymph node metastasis from early gastric cancer: estimation with a large number of cases at two large centers. Gastric Cancer 2000; 3:219–225.
- Suzuki H, Oda I, Abe S, et al. High rate of 5-year survival among patients with early gastric cancer undergoing curative endoscopic submucosal dissection. Gastric Cancer 2016; 19:198–205.
- Matsuda T, Ajiki W, Marugame T, Ioka A, Tsukuma H, Sobue T; Research Group of Population-Based Cancer Registries of Japan. Population-based survival of cancer patients diagnosed between 1993 and 1999 in Japan: a chronological and international comparative study. Jpn J Clin Oncol 2011; 41:40–51.
- Ahn JY, Jung HY, Choi KD, et al. Endoscopic and oncologic outcomes after endoscopic resection for early gastric cancer: 1370 cases of absolute and extended indications. Gastrointest Endosc 2011; 74:485–493.
- Kim Y, Kim YW, Choi IJ, et al. Cost comparison between surgical treatments and endoscopic submucosal dissection in patients with early gastric cancer in Korea. Gut Liver 2015; 9:174–180.
- Abe S, Oda I, Nakajima T, et al. A case of local recurrence and distant metastasis following curative endoscopic submucosal dissection of early gastric cancer. Gastric Cancer 2015; 18:188–192.
- Hahn KY, Park JC, Kim EH, et al. Incidence and impact of scheduled endoscopic surveillance on recurrence after curative endoscopic resection for early gastric cancer. Gastrointest Endosc 2016; 84:628–638.e1.
- Wang S, Zhang Z, Liu M, Li S, Jiang C. Endoscopic resection compared with gastrectomy to treat early gastric cancer: a systematic review and meta-analysis. PLoS One 2015; 10:e0144774.
- Kondo A, de Moura EG, Bernardo WM, et al. Endoscopy vs surgery in the treatment of early gastric cancer: systematic review. World J Gastroenterol 2015; 21:13177–13187.
- Kothari S, Kaul V. Endoscopic mucosal resection and endoscopic submucosal dissection for endoscopic therapy of Barrett’s esophagus-related neoplasia. Gastroenterol Clin North Am 2015; 44:317–335.
- Yamashita T, Zeniya A, Ishii H, et al. Endoscopic mucosal resection using a cap-fitted panendoscope and endoscopic submucosal dissection as optimal endoscopic procedures for superficial esophageal carcinoma. Surg Endosc 2011; 25:2541–2546.
- Kagemoto K, Oka S, Tanaka S, et al. Clinical outcomes of endoscopic submucosal dissection for superficial Barrett’s adenocarcinoma. Gastrointest Endosc 2014; 80:239–245.
- Katada C, Muto M, Manabe T, Boku N, Ohtsu A, Yoshida S. Esophageal stenosis after endoscopic mucosal resection of superficial esophageal lesions. Gastrointest Endosc 2003; 57:165–169.
- Hanaoka N, Ishihara R, Takeuchi Y, et al. 1139: A single session of intralesional steroid injection to prevent esophageal stricture after endoscopic submucosal dissection for esophageal squamous cell carcinoma. Gastrointest Endosc 2012; 75(suppl):AB175.
- Yamaguchi N, Isomoto H, Nakayama T, et al. Usefulness of oral prednisolone in the treatment of esophageal stricture after endoscopic submucosal dissection for superficial esophageal squamous cell carcinoma. Gastrointest Endosc 2011; 73:1115–1121.
- Ono S, Fujishiro M, Niimi K, et al. Long-term outcomes of endoscopic submucosal dissection for superficial esophageal squamous cell neoplasms. Gastrointest Endosc 2009; 70:860–866.
- Katada C, Muto M, Manabe T, Ohtsu A, Yoshida S. Local recurrence of squamous-cell carcinoma of the esophagus after EMR. Gastrointest Endosc 2005; 61:219–225.
- Hirasawa K, Kokawa A, Oka H, et al. Superficial adenocarcinoma of the esophagogastric junction: long-term results of endoscopic submucosal dissection. Gastrointest Endosc 2010; 72:960–966.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
- Nakajima T, Saito Y, Tanaka S, et al. Current status of endoscopic resection strategy for large, early colorectal neoplasia in Japan. Surg Endosc 2013; 27:3262–3770.
- Saito Y, Uraoka T, Yamaguchi Y, et al. A prospective, multicenter study of 1111 colorectal endoscopic submucosal dissections (with video). Gastrointest Endosc 2010; 72:1217–1225.
- Tanaka S, Saitoh Y, Matsuda T, et al; Japanese Society of Gastroenterology. Evidence-based clinical practice guidelines for management of colorectal polyps. J Gastroenterol 2015; 50:252–260.
- Oka S, Tanaka S, Saito Y, et al; Colorectal Endoscopic Resection Standardization Implementation Working Group of the Japanese Society for Cancer of the Colon and Rectum, Tokyo, Japan. Local recurrence after endoscopic resection for large colorectal neoplasia: a multicenter prospective study in Japan. Am J Gastroenterol 2015; 110:697–707.
- Saito Y, Fukuzawa M, Matsuda T, et al. Clinical outcome of endoscopic submucosal dissection versus endoscopic mucosal resection of large colorectal tumors as determined by curative resection. Surg Endosc 2010; 24:343–352.
- Makazu M, Sakamoto T, So E, et al. Relationship between indeterminate or positive lateral margin and local recurrence after endoscopic resection of colorectal polyps. Endosc Int Open 2015; 3:E252–E257.
- Belderbos TD, Leenders M, Moons LM, Siersema PD. Local recurrence after endoscopic mucosal resection of nonpedunculated colorectal lesions: systematic review and meta-analysis. Endoscopy 2014; 46:388–402.
- Fujiya M, Tanaka K, Dokoshi T, et al. Efficacy and adverse events of EMR and endoscopic submucosal dissection for the treatment of colon neoplasms: a meta-analysis of studies comparing EMR and endoscopic submucosal dissection. Gastrointest Endosc 2015; 81:583–595.
- Rahmi G, Tanaka S, Ohara Y, et al. Efficacy of endoscopic submucosal dissection for residual or recurrent superficial colorectal tumors after endoscopic mucosal resection. J Dig Dis 2015; 16:14–21.
- Abe S, Oda I, Suzuki H, et al. Long-term surveillance and treatment outcomes of metachronous gastric cancer occurring after curative endoscopic submucosal dissection. Endoscopy 2015; 47:1113–1118.
- Oda I, Suzuki H, Nonaka S, Yoshinaga S. Complications of gastric endoscopic submucosal dissection. Dig Endosc 2013; 25(suppl 1):71–78.
- Takizawa K, Oda I, Gotoda T, et al. Routine coagulation of visible vessels may prevent delayed bleeding after endoscopic submucosal dissection—an analysis of risk factors. Endoscopy 2008; 40:179–183.
- Uedo N, Takeuchi Y, Yamada T, et al. Effect of a proton pump inhibitor or an H2-receptor antagonist on prevention of bleeding from ulcer after endoscopic submucosal dissection of early gastric cancer: a prospective randomized controlled trial. Am J Gastroenterol 2007; 102:1610–1616.
- Hayashi N, Tanaka S, Nishiyama S, et al. Predictors of incomplete resection and perforation associated with endoscopic submucosal dissection for colorectal tumors. Gastrointest Endosc 2014; 79:427–435.
- Suzuki H, Oda I, Sekiguchi M, et al. Management and associated factors of delayed perforation after gastric endoscopic submucosal dissection. World J Gastroenterol 2015; 21:12635–12643.
- Tsunada S, Ogata S, Mannen K, et al. Case series of endoscopic balloon dilation to treat a stricture caused by circumferential resection of the gastric antrum by endoscopic submucosal dissection. Gastrointest Endosc 2008; 67:979–983.
- Coda S, Oda I, Gotoda T, Yokoi C, Kikuchi T, Ono H. Risk factors for cardiac and pyloric stenosis after endoscopic submucosal dissection, and efficacy of endoscopic balloon dilation treatment. Endoscopy 2009; 41:421–426.
- Abe S, Sakamoto T, Takamaru H, et al. Stenosis rates after endoscopic submucosal dissection of large rectal tumors involving greater than three quarters of the luminal circumference. Surg Endosc 2016; 30:5459–5464.
- Sakamoto T, Saito Y, Fukunaga S, Nakajima T, Matsuda T. Learning curve associated with colorectal endoscopic submucosal dissection for endoscopists experienced in gastric endoscopic submucosal dissection. Dis Colon Rectum 2011; 54:1307–1312.
- Burgess NG, Bourke MJ. Endoscopic resection of colorectal lesions: the narrowing divide between East and West. Dig Endosc 2016; 28:296–305.
- Kim DH, Jung HY, Gong EJ, et al. Endoscopic and oncologic outcomes of endoscopic resection for superficial esophageal neoplasm. Gut Liver 2015; 9:470–477.
- Draganov PV, Gotoda T, Chavalitdhamrong D, Wallace MB. Techniques of endoscopic submucosal dissection: application for the Western endoscopist? Gastrointest Endosc 2013; 78:677–688.
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011; 14:101–112.
- Pimentel-Nunes P, Dinis-Ribeiro M, Ponchon T, et al. Endoscopic submucosal dissection: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy 2015; 47:829–854.
- Farhat S, Chaussade S, Ponchon T, et al; SFED ESD Study Group. Endoscopic submucosal dissection in a European setting. A multi-institutional report of a technique in development. Endoscopy 2011; 43:664–670.
- Gotoda T, Jung H. Endoscopic resection (endoscopic mucosal resection/endoscopic submucosal dissection) for early gastric cancer. Dig Endosc 2013; 25(suppl 1):55–63.
- Chung IK, Lee JH, Lee SH, et al. Therapeutic outcomes in 1000 cases of endoscopic submucosal dissection for early gastric neoplasms: Korean ESD Study Group multicenter study. Gastrointest Endosc 2009; 69:1228–1235.
- Japanese Gastric Cancer Association. Japanese gastric cancer treatment guidelines 2010 (ver. 3). Gastric Cancer 2011; 14:113–123.
- Ono H. Endoscopic submucosal dissection for early gastric cancer. Chin J Dig Dis 2005; 6:119–121.
- Watanabe T, Itabashi M, Shimada Y, et al; Japanese Society for Cancer of the Colon and Rectum. Japanese Society for Cancer of the Colon and Rectum (JSCCR) guidelines 2014 for treatment of colorectal cancer. Int J Clin Oncol 2015; 20:207–239.
- Oyama T, Yahagi N, Ponchon T, Kiesslich T, Berr F. How to establish endoscopic submucosal dissection in Western countries. World J Gastroenterol 2015; 21:11209–11220.
- Bhatt A, Abe S, Kumaravel A, et al. SU1575 Western skill training in endoscopic submucosal dissection (ESD)—an international remote video based study—the WEST ESD Study. Gastrointest Endosc 2015; 81(suppl):AB335–AB336.
- Sano T, Sasako M, Kinoshita T, Maruyama K. Recurrence of early gastric cancer follow-up of 1475 patients and review of the Japanese literature. Cancer 1993; 72:3174–3178.
- Japan Esophageal Society. Japanese classification of esophageal cancer, tenth edition: part I. Esophagus 2009; 6:1–25.
- Bhatt A, Abe S, Kumaravel A, Vargo J, Saito Y. Indications and techniques for endoscopic submucosal dissection. Am J Gastroenterol 2015; 110:784–791.
- Eleftheriadis N, Inoue H, Ikeda H, et al. Definition and staging of early esophageal, gastric and colorectal cancer. J Tumor 2014; 2:161–178.
- Yoshinaga S, Oda I, Nonaka S, Kushima R, Saito Y. Endoscopic ultrasound using ultrasound probes for the diagnosis of early esophageal and gastric cancers. World J Gastrointest Endosc 2012; 4:218–226.
- Stahl M, Mariette C, Haustermans K, Cervantes A, Arnold D; ESMO Guidelines Working Group. Oesophageal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi51–vi56.
- Higuchi K, Tanabe S, Azuma M, et al. A phase II study of endoscopic submucosal dissection for superficial esophageal neoplasms (KDOG 0901). Gastrointest Endosc 2013; 78:704–710.
- Sakamoto T, Mori G, Yamada M, et al. Endoscopic submucosal dissection for colorectal neoplasms: a review. World J Gastroenterol 2014; 20:16153–16158.
- Ohta A, Tominaga K, Sakai Y. Efficacy of magnifying colonoscopy for the diagnosis of colorectal neoplasia: comparison with histopathological findings. Dig Endosc 2004; 16:308–314.
- Katagiri A, Fu K, Sano Y, et al. Narrow band imaging with magnifying colonoscopy as diagnostic tool for predicting histology of early colorectal neoplasia. Aliment Pharmacol Ther 2008; 27:1269–1274.
- Fu KI, Kato S, Sano Y, et al. Staging of early colorectal cancers: magnifying colonoscopy versus endoscopic ultrasonography for estimation of depth of invasion. Dig Dis Sci 2008; 53:1886–1892.
- Uraoka T, Saito Y, Ikematsu H, Yamamoto K, Sano Y. Sano’s capillary pattern classification for narrow-band imaging of early colorectal lesions. Dig Endosc 2011; 23(suppl 1):112–115.
- Ikematsu H, Matsuda T, Emura F, et al. Efficacy of capillary pattern type IIIA/IIIB by magnifying narrow band imaging for estimating depth of invasion of early colorectal neoplasms. BMC Gastroenterol 2010;10:33.
- Matsuda T, Fujii T, Saito Y, et al. Efficacy of the invasive/non-invasive pattern by magnifying chromoendoscopy to estimate the depth of invasion of early colorectal neoplasms. Am J Gastroenterol 2008; 103:2700–2706.
- Sato H, Inoue H, Ikeda H, et al. Utility of intrapapillary capillary loops seen on magnifying narrow-band imaging in estimating invasive depth of esophageal squamous cell carcinoma. Endoscopy 2015; 8:122–128.
- Muto M, Yao K, Kaise M, et al. Magnifying endoscopy simple diagnostic algorithm for early gastric cancer (MESDA-G). Dig Endosc 2016; 28:379–393.
- Waddell T, Verheij M, Allum W, Cunningham D, Cervantes A, Arnold D; European Society for Medical Oncology (ESMO); European Society of Surgical Oncology (ESSO); European Society of Radiotherapy and Oncology (ESTRO). Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013; 24(suppl 6):vi57–vi63.
- Kuwano H, Nishimura Y, Ohtsu A, et al. Guidelines for diagnosis and treatment of carcinoma of the esophagus. April 2007 edition: part I - Edited by the Japan Esophageal Society. Esophagus 2008; 5:61–73.
- Tanaka S, Kashida H, Saito Y, et al. JGES guidelines for colorectal endoscopic submucosal dissection/endoscopic mucosal resection. Dig Endosc 2015; 27:417–434.
- Gotoda T, Ho KY, Soetikno R, Kaltenbach T, Draganov P. Gastric ESD: current status and future directions of devices and training. Gastrointest Endosc Clin North Am 2014; 24:213–233.
- Saito Y, Sakamoto T, Nakajima T, Matsuda T. Colorectal ESD: current indications and latest technical advances. Gastrointest Endosc Clin N Am 2014; 24:245–255.
- Saito Y, Otake Y, Sakamoto T, et al. Indications for and technical aspects of colorectal endoscopic submucosal dissection. Gut Liver 2013; 7:263–269.
- Saragoni L. Upgrading the definition of early gastric cancer: better staging means more appropriate treatment. Cancer Biol Med 2015; 12:355–361.
- Tsujitani S, Oka S, Saito H, et al. Less invasive surgery for early gastric cancer based on the low probability of lymph node metastasis. Surgery 1999; 125:148–154.
- Soetikno RM, Gotoda T, Nakanishi Y, Soehendra N. Endoscopic mucosal resection. Gastrointest Endosc 2003; 57:567–579.
- Hirasawa T, Gotoda T, Miyata S, et al. Incidence of lymph node metastasis and the feasibility of endoscopic resection for undifferentiated-type early gastric cancer. Gastric Cancer 2009; 12:148–152.
- Gotoda T, Yanagisawa A, Sasako M, et al. Incidence of lymph node metastasis from early gastric cancer: estimation with a large number of cases at two large centers. Gastric Cancer 2000; 3:219–225.
- Suzuki H, Oda I, Abe S, et al. High rate of 5-year survival among patients with early gastric cancer undergoing curative endoscopic submucosal dissection. Gastric Cancer 2016; 19:198–205.
- Matsuda T, Ajiki W, Marugame T, Ioka A, Tsukuma H, Sobue T; Research Group of Population-Based Cancer Registries of Japan. Population-based survival of cancer patients diagnosed between 1993 and 1999 in Japan: a chronological and international comparative study. Jpn J Clin Oncol 2011; 41:40–51.
- Ahn JY, Jung HY, Choi KD, et al. Endoscopic and oncologic outcomes after endoscopic resection for early gastric cancer: 1370 cases of absolute and extended indications. Gastrointest Endosc 2011; 74:485–493.
- Kim Y, Kim YW, Choi IJ, et al. Cost comparison between surgical treatments and endoscopic submucosal dissection in patients with early gastric cancer in Korea. Gut Liver 2015; 9:174–180.
- Abe S, Oda I, Nakajima T, et al. A case of local recurrence and distant metastasis following curative endoscopic submucosal dissection of early gastric cancer. Gastric Cancer 2015; 18:188–192.
- Hahn KY, Park JC, Kim EH, et al. Incidence and impact of scheduled endoscopic surveillance on recurrence after curative endoscopic resection for early gastric cancer. Gastrointest Endosc 2016; 84:628–638.e1.
- Wang S, Zhang Z, Liu M, Li S, Jiang C. Endoscopic resection compared with gastrectomy to treat early gastric cancer: a systematic review and meta-analysis. PLoS One 2015; 10:e0144774.
- Kondo A, de Moura EG, Bernardo WM, et al. Endoscopy vs surgery in the treatment of early gastric cancer: systematic review. World J Gastroenterol 2015; 21:13177–13187.
- Kothari S, Kaul V. Endoscopic mucosal resection and endoscopic submucosal dissection for endoscopic therapy of Barrett’s esophagus-related neoplasia. Gastroenterol Clin North Am 2015; 44:317–335.
- Yamashita T, Zeniya A, Ishii H, et al. Endoscopic mucosal resection using a cap-fitted panendoscope and endoscopic submucosal dissection as optimal endoscopic procedures for superficial esophageal carcinoma. Surg Endosc 2011; 25:2541–2546.
- Kagemoto K, Oka S, Tanaka S, et al. Clinical outcomes of endoscopic submucosal dissection for superficial Barrett’s adenocarcinoma. Gastrointest Endosc 2014; 80:239–245.
- Katada C, Muto M, Manabe T, Boku N, Ohtsu A, Yoshida S. Esophageal stenosis after endoscopic mucosal resection of superficial esophageal lesions. Gastrointest Endosc 2003; 57:165–169.
- Hanaoka N, Ishihara R, Takeuchi Y, et al. 1139: A single session of intralesional steroid injection to prevent esophageal stricture after endoscopic submucosal dissection for esophageal squamous cell carcinoma. Gastrointest Endosc 2012; 75(suppl):AB175.
- Yamaguchi N, Isomoto H, Nakayama T, et al. Usefulness of oral prednisolone in the treatment of esophageal stricture after endoscopic submucosal dissection for superficial esophageal squamous cell carcinoma. Gastrointest Endosc 2011; 73:1115–1121.
- Ono S, Fujishiro M, Niimi K, et al. Long-term outcomes of endoscopic submucosal dissection for superficial esophageal squamous cell neoplasms. Gastrointest Endosc 2009; 70:860–866.
- Katada C, Muto M, Manabe T, Ohtsu A, Yoshida S. Local recurrence of squamous-cell carcinoma of the esophagus after EMR. Gastrointest Endosc 2005; 61:219–225.
- Hirasawa K, Kokawa A, Oka H, et al. Superficial adenocarcinoma of the esophagogastric junction: long-term results of endoscopic submucosal dissection. Gastrointest Endosc 2010; 72:960–966.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
- Nakajima T, Saito Y, Tanaka S, et al. Current status of endoscopic resection strategy for large, early colorectal neoplasia in Japan. Surg Endosc 2013; 27:3262–3770.
- Saito Y, Uraoka T, Yamaguchi Y, et al. A prospective, multicenter study of 1111 colorectal endoscopic submucosal dissections (with video). Gastrointest Endosc 2010; 72:1217–1225.
- Tanaka S, Saitoh Y, Matsuda T, et al; Japanese Society of Gastroenterology. Evidence-based clinical practice guidelines for management of colorectal polyps. J Gastroenterol 2015; 50:252–260.
- Oka S, Tanaka S, Saito Y, et al; Colorectal Endoscopic Resection Standardization Implementation Working Group of the Japanese Society for Cancer of the Colon and Rectum, Tokyo, Japan. Local recurrence after endoscopic resection for large colorectal neoplasia: a multicenter prospective study in Japan. Am J Gastroenterol 2015; 110:697–707.
- Saito Y, Fukuzawa M, Matsuda T, et al. Clinical outcome of endoscopic submucosal dissection versus endoscopic mucosal resection of large colorectal tumors as determined by curative resection. Surg Endosc 2010; 24:343–352.
- Makazu M, Sakamoto T, So E, et al. Relationship between indeterminate or positive lateral margin and local recurrence after endoscopic resection of colorectal polyps. Endosc Int Open 2015; 3:E252–E257.
- Belderbos TD, Leenders M, Moons LM, Siersema PD. Local recurrence after endoscopic mucosal resection of nonpedunculated colorectal lesions: systematic review and meta-analysis. Endoscopy 2014; 46:388–402.
- Fujiya M, Tanaka K, Dokoshi T, et al. Efficacy and adverse events of EMR and endoscopic submucosal dissection for the treatment of colon neoplasms: a meta-analysis of studies comparing EMR and endoscopic submucosal dissection. Gastrointest Endosc 2015; 81:583–595.
- Rahmi G, Tanaka S, Ohara Y, et al. Efficacy of endoscopic submucosal dissection for residual or recurrent superficial colorectal tumors after endoscopic mucosal resection. J Dig Dis 2015; 16:14–21.
- Abe S, Oda I, Suzuki H, et al. Long-term surveillance and treatment outcomes of metachronous gastric cancer occurring after curative endoscopic submucosal dissection. Endoscopy 2015; 47:1113–1118.
- Oda I, Suzuki H, Nonaka S, Yoshinaga S. Complications of gastric endoscopic submucosal dissection. Dig Endosc 2013; 25(suppl 1):71–78.
- Takizawa K, Oda I, Gotoda T, et al. Routine coagulation of visible vessels may prevent delayed bleeding after endoscopic submucosal dissection—an analysis of risk factors. Endoscopy 2008; 40:179–183.
- Uedo N, Takeuchi Y, Yamada T, et al. Effect of a proton pump inhibitor or an H2-receptor antagonist on prevention of bleeding from ulcer after endoscopic submucosal dissection of early gastric cancer: a prospective randomized controlled trial. Am J Gastroenterol 2007; 102:1610–1616.
- Hayashi N, Tanaka S, Nishiyama S, et al. Predictors of incomplete resection and perforation associated with endoscopic submucosal dissection for colorectal tumors. Gastrointest Endosc 2014; 79:427–435.
- Suzuki H, Oda I, Sekiguchi M, et al. Management and associated factors of delayed perforation after gastric endoscopic submucosal dissection. World J Gastroenterol 2015; 21:12635–12643.
- Tsunada S, Ogata S, Mannen K, et al. Case series of endoscopic balloon dilation to treat a stricture caused by circumferential resection of the gastric antrum by endoscopic submucosal dissection. Gastrointest Endosc 2008; 67:979–983.
- Coda S, Oda I, Gotoda T, Yokoi C, Kikuchi T, Ono H. Risk factors for cardiac and pyloric stenosis after endoscopic submucosal dissection, and efficacy of endoscopic balloon dilation treatment. Endoscopy 2009; 41:421–426.
- Abe S, Sakamoto T, Takamaru H, et al. Stenosis rates after endoscopic submucosal dissection of large rectal tumors involving greater than three quarters of the luminal circumference. Surg Endosc 2016; 30:5459–5464.
- Sakamoto T, Saito Y, Fukunaga S, Nakajima T, Matsuda T. Learning curve associated with colorectal endoscopic submucosal dissection for endoscopists experienced in gastric endoscopic submucosal dissection. Dis Colon Rectum 2011; 54:1307–1312.
KEY POINTS
- ESD is a minimally invasive endoscopic technique with curative potential for patients with superficial GI neoplasia.
- ESD preserves the integrity of the organ while achieving curative resection of large neoplasms.
- ESD is indicated rather than surgery in patients with early GI lesions with a negligible risk of lymph node metastasis.
- Complications of the procedure include bleeding, perforation, and stenosis. Most of these respond to endoscopic treatment.
- Successful ESD requires supportive teamwork among internists, gastroenterologists, pathologists, and surgeons.
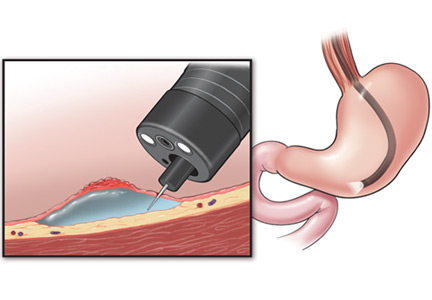
Necrotizing pancreatitis: Diagnose, treat, consult
Acute pancreatitis accounted for more than 300,000 admissions and $2.6 billion in associated healthcare costs in the United States in 2012.1 First-line management is early aggressive fluid resuscitation and analgesics for pain control. Guidelines recommend estimating the clinical severity of each attack using a validated scoring system such as the Bedside Index of Severity in Acute Pancreatitis.2 Clinically severe pancreatitis is associated with necrosis.
Acute pancreatitis results from inappropriate activation of zymogens and subsequent autodigestion of the pancreas by its own enzymes. Though necrotizing pancreatitis is thought to be an ischemic complication, its pathogenesis is not completely understood. Necrosis increases the morbidity and mortality risk of acute pancreatitis because of its association with organ failure and infectious complications. As such, patients with necrotizing pancreatitis may need admission to the intensive care unit, nutritional support, antibiotics, and radiologic, endoscopic, or surgical interventions.
Here, we review current evidence regarding the diagnosis and management of necrotizing pancreatitis.
PROPER TERMINOLOGY HELPS COLLABORATION
Managing necrotizing pancreatitis requires the combined efforts of internists, gastroenterologists, radiologists, and surgeons. This collaboration is aided by proper terminology.
A classification system was devised in Atlanta, GA, in 1992 to facilitate communication and interdisciplinary collaboration.3 Severe pancreatitis was differentiated from mild by the presence of organ failure or the complications of pseudocyst, necrosis, or abscess.
The original Atlanta classification had several limitations. First, the terminology for fluid collections was ambiguous and frequently misused. Second, the assessment of clinical severity required either the Ranson score or the Acute Physiology and Chronic Health Evaluation II score, both of which are complex and have other limitations. Finally, advances in imaging and treatment have rendered the original Atlanta nomenclature obsolete.
In 2012, the Acute Pancreatitis Classification Working Group issued a revised Atlanta classification that modernized the terminology pertaining to natural history, severity, imaging features, and complications. It divides the natural course of acute pancreatitis into early and late phases.4
Early vs late phase

In the early phase, findings on computed tomography (CT) neither correlate with clinical severity nor alter clinical management.6 Thus, early imaging is not indicated unless there is diagnostic uncertainty, lack of response to appropriate treatment, or sudden deterioration.
Moderate pancreatitis describes patients with pancreatic necrosis with or without transient organ failure (organ dysfunction for ≤ 48 hours).
Severe pancreatitis is defined by pancreatic necrosis and persistent organ dysfunction.4 It may be accompanied by pancreatic and peripancreatic fluid collections; bacteremia and sepsis can occur in association with infection of necrotic collections.
Interstitial edematous pancreatitis vs necrotizing pancreatitis
The revised Atlanta classification maintains the original classification of acute pancreatitis into 2 main categories: interstitial edematous pancreatitis and necrotizing pancreatitis.

Necrotizing pancreatitis is further divided into 3 subtypes based on extent and location of necrosis:
- Parenchymal necrosis alone (5% of cases)
- Necrosis of peripancreatic fat alone (20%)
- Necrosis of both parenchyma and peripancreatic fat (75%).
Peripancreatic involvement is commonly found in the mesentery, peripancreatic and distant retroperitoneum, and lesser sac.
Of the three subtypes, peripancreatic necrosis has the best prognosis. However, all of the subtypes of necrotizing pancreatitis are associated with poorer outcomes than interstitial edematous pancreatitis.
Fluid collections

Acute pancreatic fluid collections contain exclusively nonsolid components without an inflammatory wall and are typically found in the peripancreatic fat. These collections often resolve without intervention as the patient recovers. If they persist beyond 4 weeks and develop a nonepithelialized, fibrous wall, they become pseudocysts. Intervention is generally not recommended for pseudocysts unless they are symptomatic.

ROLE OF IMAGING
Radiographic imaging is not usually necessary to diagnose acute pancreatitis. However, it can be a valuable tool to clarify an ambiguous presentation, determine severity, and identify complications.
The timing and appropriate type of imaging are integral to obtaining useful data. Any imaging obtained in acute pancreatitis to evaluate necrosis should be performed at least 3 to 5 days from the initial symptom onset; if imaging is obtained before 72 hours, necrosis cannot be confidently excluded.8
COMPUTED TOMOGRAPHY
CT is the imaging test of choice when evaluating acute pancreatitis. In addition, almost all percutaneous interventions are performed with CT guidance. The Balthazar score is the most well-known CT severity index. It is calculated based on the degree of inflammation, acute fluid collections, and parenchymal necrosis.9 However, a modified severity index incorporates extrapancreatic complications such as ascites and vascular compromise and was found to more strongly correlate with outcomes than the standard Balthazar score.10
Contrast-enhanced CT is performed in 2 phases:
The pancreatic parenchymal phase
The pancreatic parenchymal or late arterial phase is obtained approximately 40 to 45 seconds after the start of the contrast bolus. It is used to detect necrosis in the early phase of acute pancreatitis and to assess the peripancreatic arteries for pseudoaneurysms in the late phase of acute pancreatitis.11
Pancreatic necrosis appears as an area of decreased parenchymal enhancement, either well-defined or heterogeneous. The normal pancreatic parenchyma has a postcontrast enhancement pattern similar to that of the spleen. Parenchyma that does not enhance to the same degree is considered necrotic. The severity of necrosis is graded based on the percentage of the pancreas involved (< 30%, 30%–50%, or > 50%), and a higher percentage correlates with a worse outcome.12,13
Peripancreatic necrosis is harder to detect, as there is no method to assess fat enhancement as there is with pancreatic parenchymal enhancement. In general, radiologists assume that heterogeneous peripancreatic changes, including areas of fat, fluid, and soft tissue attenuation, are consistent with peripancreatic necrosis. After 7 to 10 days, if these changes become more homogeneous and confluent with a more mass-like process, peripancreatic necrosis can be more confidently identified.12,13
The portal venous phase
The later, portal venous phase of the scan is obtained approximately 70 seconds after the start of the contrast bolus. It is used to detect and characterize fluid collections and venous complications of the disease.
Drawbacks of CT
A drawback of CT is the need for iodinated intravenous contrast media, which in severely ill patients may precipitate or worsen pre-existing acute kidney injury.
Further, several studies have shown that findings on CT rarely alter the management of patients in the early phase of acute pancreatitis and in fact may be an overuse of medical resources.14 Unless there are confounding clinical signs or symptoms, CT should be delayed for at least 72 hours.9,10,14,15
MAGNETIC RESONANCE IMAGING
Magnetic resonance imaging (MRI) is not a first-line imaging test in this disease because it is not as available as CT and takes longer to perform—20 to 30 minutes. The patient must be evaluated for candidacy, as it is difficult for acutely ill patients to tolerate an examination that takes this long and requires them to hold their breath multiple times.
MRI is an appropriate alternative in patients who are pregnant or who have severe iodinated-contrast allergy. While contrast is necessary to detect pancreatic necrosis with CT, MRI can detect necrosis without the need for contrast in patients with acute kidney injury or severe chronic kidney disease. Also, MRI may be better in complicated cases requiring repeated imaging because it does not expose the patient to radiation.
On MRI, pancreatic necrosis appears as a heterogeneous area, owing to its liquid and solid components. Liquid components appear hyperintense, and solid components hypointense, on T2 fluid-weighted imaging. This ability to differentiate the components of a walled-off pancreatic necrosis can be useful in determining whether a collection requires drainage or debridement. MRI is also more sensitive for hemorrhagic complications, best seen on T1 fat-weighted images.12,16
Magnetic resonance cholangiopancreatography is an excellent method for ductal evaluation through heavily T2-weighted imaging. It is more sensitive than CT for detecting common bile duct stones and can also detect pancreatic duct strictures or extravasation into fluid collections.16
SUPPORTIVE MANAGEMENT OF EARLY NECROTIZING PANCREATITIS
In the early phase of necrotizing pancreatitis, management is supportive with the primary aim of preventing intravascular volume depletion. Aggressive fluid resuscitation in the first 48 to 72 hours, pain control, and bowel rest are the mainstays of supportive therapy. Intensive care may be necessary if organ failure and hemodynamic instability accompany necrotizing pancreatitis.
Prophylactic antibiotic and antifungal therapy to prevent infected necrosis has been controversial. Recent studies of its utility have not yielded supportive results, and the American College of Gastroenterology and the Infectious Diseases Society of America no longer recommend it.9,17 These medications should not be given unless concomitant cholangitis or extrapancreatic infection is clinically suspected.
Early enteral nutrition is recommended in patients in whom pancreatitis is predicted to be severe and in those not expected to resume oral intake within 5 to 7 days. Enteral nutrition most commonly involves bedside or endoscopic placement of a nasojejunal feeding tube and collaboration with a nutritionist to determine protein-caloric requirements.
Compared with enteral nutrition, total parenteral nutrition is associated with higher rates of infection, multiorgan dysfunction and failure, and death.18
MANAGING COMPLICATIONS OF PANCREATIC NECROSIS
Necrotizing pancreatitis is a defining complication of acute pancreatitis, and its presence alone indicates greater severity. However, superimposed complications may further worsen outcomes.
Infected pancreatic necrosis
Infection occurs in approximately 20% of patients with necrotizing pancreatitis and confers a mortality rate of 20% to 50%.19 Infected pancreatic necrosis occurs when gut organisms translocate into the nearby necrotic pancreatic and peripancreatic tissue. The most commonly identified organisms include Escherichia coli and Enterococcus species.20
This complication usually manifests 2 to 4 weeks after symptom onset; earlier onset is uncommon to rare. It should be considered when the systemic inflammatory response syndrome persists or recurs after 10 days to 2 weeks. Systemic inflammatory response syndrome is also common in sterile necrotizing pancreatitis and sometimes in interstitial pancreatitis, particularly during the first week. However, its sudden appearance or resurgence, high spiking fevers, or worsening organ failure in the later phase (2–4 weeks) of pancreatitis should heighten suspicion of infected pancreatic necrosis.
Imaging may also help diagnose infection, and the presence of gas within a collection or region of necrosis is highly specific. However, the presence of gas is not completely sensitive for infection, as it is seen in only 12% to 22% of infected cases.
Before minimally invasive techniques became available, the diagnosis of infected pancreatic necrosis was confirmed by percutaneous CT-guided aspiration of the necrotic mass or collection for Gram stain and culture.
Antibiotic therapy is indicated in confirmed or suspected cases of infected pancreatic necrosis. Antibiotics with gram-negative coverage and appropriate penetration such as carbapenems, metronidazole, fluoroquinolones, and selected cephalosporins are most commonly used. Meropenem is the antibiotic of choice at our institution.
CT-guided fine-needle aspiration is often done if suspected infected pancreatic necrosis fails to respond to empiric antibiotic therapy.
Debridement or drainage. Generally, the diagnosis or suspicion of infected pancreatic necrosis (suggestive signs are high fever, elevated white blood cell count, and sepsis) warrants an intervention to debride or drain infected pancreatic tissue and control sepsis.21
While source control is integral to the successful treatment of infected pancreatic necrosis, antibiotic therapy may provide a bridge to intervention for critically ill patients by suppressing bacteremia and subsequent sepsis. A 2013 meta-analysis found that 324 of 409 patients with suspected infected pancreatic necrosis were successfully stabilized with antibiotic treatment.21,22 The trend toward conservative management and promising outcomes with antibiotic therapy alone or with minimally invasive techniques has lessened the need for diagnostic CT-guided fine-needle aspiration.
Hemorrhage
Spontaneous hemorrhage into pancreatic necrosis is a rare but life-threatening complication. Because CT is almost always performed with contrast enhancement, this complication is rarely identified with imaging. The diagnosis is made by noting a drop in hemoglobin and hematocrit.
Hemorrhage into the retroperitoneum or the peritoneal cavity, or both, can occur when an inflammatory process erodes into a nearby artery. Luminal gastrointestinal bleeding can occur from gastric varices arising from splenic vein thrombosis and resulting left-sided portal hypertension, or from pseudoaneurysms. These can also bleed into the pancreatic duct (hemosuccus pancreaticus). Pseudoaneurysm is a later complication that occurs when an arterial wall (most commonly the splenic or gastroduodenal artery) is weakened by pancreatic enzymes.23
Prompt recognition of hemorrhagic events and consultation with an interventional radiologist or surgeon are required to prevent death.
Inflammation and abdominal compartment syndrome
Inflammation from necrotizing pancreatitis can cause further complications by blocking nearby structures. Reported complications include jaundice from biliary compression, hydronephrosis from ureteral compression, bowel obstruction, and gastric outlet obstruction.
Abdominal compartment syndrome is an increasingly recognized complication of acute pancreatitis. Abdominal pressure can rise due to a number of factors, including fluid collections, ascites, ileus, and overly aggressive fluid resuscitation.24 Elevated abdominal pressure is associated with complications such as decreased respiratory compliance, increased peak airway pressure, decreased cardiac preload, hypotension, mesenteric and intestinal ischemia, feeding intolerance, and lower-extremity ischemia and thrombosis.
Patients with necrotizing pancreatitis who have abdominal compartment syndrome have a mortality rate 5 times higher than patients without abdominal compartment syndrome.25
Abdominal pressures should be monitored using a bladder pressure sensor in critically ill or ventilated patients with acute pancreatitis. If the abdominal pressure rises above 20 mm Hg, medical and surgical interventions should be offered in a stepwise fashion to decrease it. Interventions include decompression by nasogastric and rectal tube, sedation or paralysis to relax abdominal wall tension, minimization of intravenous fluids, percutaneous drainage of ascites, and (rarely) surgical midline or subcostal laparotomy.
ROLE OF INTERVENTION
The treatment of necrotizing pancreatitis has changed rapidly, thanks to a growing experience with minimally invasive techniques.
Indications for intervention
Infected pancreatic necrosis is the primary indication for surgical, percutaneous, or endoscopic intervention.
In sterile necrosis, the threshold for intervention is less clear, and intervention is often reserved for patients who fail to clinically improve or who have intractable abdominal pain, gastric outlet obstruction, or fistulating disease.26
In asymptomatic cases, intervention is almost never indicated regardless of the location or size of the necrotic area.
In walled-off pancreatic necrosis, less-invasive and less-morbid interventions such as endoscopic or percutaneous drainage or video-assisted retroperitoneal debridement can be done.
Timing of intervention
In the past, delaying intervention was thought to increase the risk of death. However, multiple studies have found that outcomes are often worse if intervention is done early, likely due to the lack of a fully formed fibrous wall or demarcation of the necrotic area.27
If the patient remains clinically stable, it is best to delay intervention until at least 4 weeks after the index event to achieve optimal outcomes. Delay can often be achieved by antibiotic treatment to suppress bacteremia and endoscopic or percutaneous drainage of infected collections to control sepsis.
Open surgery
The gold-standard intervention for infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis is open necrosectomy. This involves exploratory laparotomy with blunt debridement of all visible necrotic pancreatic tissue.
Methods to facilitate later evacuation of residual infected fluid and debris vary widely. Multiple large-caliber drains can be placed to facilitate irrigation and drainage before closure of the abdominal fascia. As infected pancreatic necrosis carries the risk of contaminating the peritoneal cavity, the skin is often left open to heal by secondary intention. An interventional radiologist is frequently enlisted to place, exchange, or downsize drainage catheters.
Infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis often requires more than one operation to achieve satisfactory debridement.
The goals of open necrosectomy are to remove nonviable tissue and infection, preserve viable pancreatic tissue, eliminate fistulous connections, and minimize damage to local organs and vasculature.
Minimally invasive techniques


Video-assisted retroperitoneal debridement has been described as a hybrid between endoscopic and open retroperitoneal debridement.28 This technique requires first placing a percutaneous catheter into the necrotic area through the left flank to create a retroperitoneal tract. A 5-cm incision is made and the necrotic space is entered using the drain for guidance. Necrotic tissue is carefully debrided under direct vision using a combination of forceps, irrigation, and suction. A laparoscopic port can also be introduced into the incision when the procedure can no longer be continued under direct vision.29,30
Although not all patients are candidates for minimal-access surgery, it remains an evolving surgical option.
Endoscopic transmural debridement is another option for infected pancreatic necrosis and symptomatic walled-off pancreatic necrosis. Depending on the location of the necrotic area, an echoendoscope is passed to either the stomach or duodenum. Guided by endoscopic ultrasonography, a needle is passed into the collection, allowing subsequent fistula creation and stenting for internal drainage or debridement. In the past, this process required several steps, multiple devices, fluoroscopic guidance, and considerable time. But newer endoscopic lumen-apposing metal stents have been developed that can be placed in a single step without fluoroscopy. A slimmer endoscope can then be introduced into the necrotic cavity via the stent, and the necrotic debris can be debrided with endoscopic baskets, snares, forceps, and irrigation.9,31
Similar to surgical necrosectomy, satisfactory debridement is not often obtained with a single procedure; 2 to 5 endoscopic procedures may be needed to achieve resolution. However, the luminal approach in endoscopic necrosectomy avoids the significant morbidity of major abdominal surgery and the potential for pancreaticocutaneous fistulae that may occur with drains.
In a randomized trial comparing endoscopic necrosectomy vs surgical necrosectomy (video-assisted retroperitoneal debridement and exploratory laparotomy),32 endoscopic necrosectomy showed less inflammatory response than surgical necrosectomy and had a lower risk of new-onset organ failure, bleeding, fistula formation, and death.32
Selecting the best intervention for the individual patient
Given the multiple available techniques, selecting the best intervention for individual patients can be challenging. A team approach with input from a gastroenterologist, surgeon, and interventional radiologist is best when determining which technique would best suit each patient.
Surgical necrosectomy is still the treatment of choice for unstable patients with infected pancreatic necrosis or multiple, inaccessible collections, but current evidence suggests a different approach in stable infected pancreatic necrosis and symptomatic sterile walled-off pancreatic necrosis.
The Dutch Pancreatitis Group28 randomized 88 patients with infected pancreatic necrosis or symptomatic walled-off pancreatic necrosis to open necrosectomy or a minimally invasive “step-up” approach consisting of up to 2 percutaneous drainage or endoscopic debridement procedures before escalation to video-assisted retroperitoneal debridement. The step-up approach resulted in lower rates of morbidity and death than surgical necrosectomy as first-line treatment. Furthermore, some patients in the step-up group avoided the need for surgery entirely.30
SUMMING UP
Necrosis significantly increases rates of morbidity and mortality in acute pancreatitis. Hospitalists, general internists, and general surgeons are all on the front lines in identifying severe cases and consulting the appropriate specialists for optimal multidisciplinary care. Selective and appropriate timing of radiologic imaging is key, and a vital tool in the management of necrotizing pancreatitis.
While the primary indication for intervention is infected pancreatic necrosis, additional indications are symptomatic walled-off pancreatic necrosis secondary to intractable abdominal pain, bowel obstruction, and failure to thrive. As a result of improving technology and inpatient care, these patients may present with intractable symptoms in the outpatient setting rather than the inpatient setting. The onus is on the primary care physician to maintain a high level of suspicion and refer these patients to subspecialists as appropriate.
Open surgical necrosectomy remains an important approach for care of infected pancreatic necrosis or patients with intractable symptoms. A step-up approach starting with a minimally invasive procedure and escalating if the initial intervention is unsuccessful is gradually becoming the standard of care.
- Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic disease in the United States. Gastroenterology 2015; 149:1731–1741e3.
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108:1400–1416.
- Bradley EL 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993; 128:586–590.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 1995; 23:1638–1652.
- Kadiyala V, Suleiman SL, McNabb-Baltar J, Wu BU, Banks PA, Singh VK. The Atlanta classification, revised Atlanta classification, and determinant-based classification of acute pancreatitis: which is best at stratifying outcomes? Pancreas 2016; 45:510–515.
- Singh VK, Bollen TL, Wu BU, et al. An assessment of the severity of interstitial pancreatitis. Clin Gastroenterol Hepatol 2011; 9:1098–1103.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Mortele KJ, Wiesner W, Intriere L, et al. A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. AJR Am J Roentgenol 2004; 183:1261–1265.
- Verde F, Fishman EK, Johnson PT. Arterial pseudoaneurysms complicating pancreatitis: literature review. J Comput Assist Tomogr 2015; 39:7–12.
- Shyu JY, Sainani NI, Sahni VA, et al. Necrotizing pancreatitis: diagnosis, imaging, and intervention. Radiographics 2014; 34:1218–1239.
- Thoeni RF. The revised Atlanta classification of acute pancreatitis: its importance for the radiologist and its effect on treatment. Radiology 2012; 262:751–764.
- Morgan DE, Ragheb CM, Lockhart ME, Cary B, Fineberg NS, Berland LL. Acute pancreatitis: computed tomography utilization and radiation exposure are related to severity but not patient age. Clin Gastroenterol Hepatol 2010; 8:303–308.
- Vitellas KM, Paulson EK, Enns RA, Keogan MT, Pappas TN. Pancreatitis complicated by gland necrosis: evolution of findings on contrast-enhanced CT. J Comput Assist Tomogr 1999; 23:898–905.
- Stimac D, Miletic D, Radic M, et al. The role of nonenhanced magnetic resonance imaging in the early assessment of acute pancreatitis. Am J Gastroenterol 2007; 102:997–1004.
- Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect (Larchmt) 2010; 11:79–109.
- Petrov MS, Kukosh MV, Emelyanov NV. A randomized controlled trial of enteral versus parenteral feeding in patients with predicted severe acute pancreatitis shows a significant reduction in mortality and in infected pancreatic complications with total enteral nutrition. Dig Surg 2006; 23:336–345.
- Petrov MS, Shanbhag S, Chakraborty M, Phillips AR, Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 2010; 139:813–820.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006; 4:CD002941.
- Baril NB, Ralls PW, Wren SM, et al. Does an infected peripancreatic fluid collection or abscess mandate operation? Ann Surg 2000; 231:361–367.
- Mouli VP, Sreenivas V, Garg PK. Efficacy of conservative treatment, without necrosectomy, for infected pancreatic necrosis: a systematic review and meta-analysis. Gastroenterology 2013; 144:333–340.e2.
- Kirby JM, Vora P, Midia M, Rawlinson J. Vascular complications of pancreatitis: imaging and intervention. Cardiovasc Intervent Radiol 2008; 31:957–970.
- De Waele JJ, Hoste E, Blot SI, Decruyenaere J, Colardyn F. Intra-abdominal hypertension in patients with severe acute pancreatitis. Crit Care 2005; 9:R452–R457.
- van Brunschot S, Schut AJ, Bouwense SA, et al; Dutch Pancreatitis Study Group. Abdominal compartment syndrome in acute pancreatitis: a systematic review. Pancreas 2014; 43:665–674.
- Bugiantella W, Rondelli F, Boni M, et al. Necrotizing pancreatitis: a review of the interventions. Int J Surg 2016; 28(suppl 1):S163–S171.
- Besselink MG, Verwer TJ, Schoenmaeckers EJ, et al. Timing of surgical intervention in necrotizing pancreatitis. Arch Surg 2007; 142:1194–1201.
- van Santvoort HC, Besselink MG, Horvath KD, et al; Dutch Acute Pancreatis Study Group. Videoscopic assisted retroperitoneal debridement in infected necrotizing pancreatitis. HPB (Oxford) 2007; 9:156–159.
- van Santvoort HC, Besselink MG, Bollen TL, Buskens E, van Ramshorst B, Gooszen HG; Dutch Acute Pancreatitis Study Group. Case-matched comparison of the retroperitoneal approach with laparotomy for necrotizing pancreatitis. World J Surg 2007; 31:1635–1642.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Thompson CC, Kumar N, Slattery J, et al. A standardized method for endoscopic necrosectomy improves complication and mortality rates. Pancreatology 2016; 16:66–72.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
Acute pancreatitis accounted for more than 300,000 admissions and $2.6 billion in associated healthcare costs in the United States in 2012.1 First-line management is early aggressive fluid resuscitation and analgesics for pain control. Guidelines recommend estimating the clinical severity of each attack using a validated scoring system such as the Bedside Index of Severity in Acute Pancreatitis.2 Clinically severe pancreatitis is associated with necrosis.
Acute pancreatitis results from inappropriate activation of zymogens and subsequent autodigestion of the pancreas by its own enzymes. Though necrotizing pancreatitis is thought to be an ischemic complication, its pathogenesis is not completely understood. Necrosis increases the morbidity and mortality risk of acute pancreatitis because of its association with organ failure and infectious complications. As such, patients with necrotizing pancreatitis may need admission to the intensive care unit, nutritional support, antibiotics, and radiologic, endoscopic, or surgical interventions.
Here, we review current evidence regarding the diagnosis and management of necrotizing pancreatitis.
PROPER TERMINOLOGY HELPS COLLABORATION
Managing necrotizing pancreatitis requires the combined efforts of internists, gastroenterologists, radiologists, and surgeons. This collaboration is aided by proper terminology.
A classification system was devised in Atlanta, GA, in 1992 to facilitate communication and interdisciplinary collaboration.3 Severe pancreatitis was differentiated from mild by the presence of organ failure or the complications of pseudocyst, necrosis, or abscess.
The original Atlanta classification had several limitations. First, the terminology for fluid collections was ambiguous and frequently misused. Second, the assessment of clinical severity required either the Ranson score or the Acute Physiology and Chronic Health Evaluation II score, both of which are complex and have other limitations. Finally, advances in imaging and treatment have rendered the original Atlanta nomenclature obsolete.
In 2012, the Acute Pancreatitis Classification Working Group issued a revised Atlanta classification that modernized the terminology pertaining to natural history, severity, imaging features, and complications. It divides the natural course of acute pancreatitis into early and late phases.4
Early vs late phase
In the early phase, findings on computed tomography (CT) neither correlate with clinical severity nor alter clinical management.6 Thus, early imaging is not indicated unless there is diagnostic uncertainty, lack of response to appropriate treatment, or sudden deterioration.
Moderate pancreatitis describes patients with pancreatic necrosis with or without transient organ failure (organ dysfunction for ≤ 48 hours).
Severe pancreatitis is defined by pancreatic necrosis and persistent organ dysfunction.4 It may be accompanied by pancreatic and peripancreatic fluid collections; bacteremia and sepsis can occur in association with infection of necrotic collections.
Interstitial edematous pancreatitis vs necrotizing pancreatitis
The revised Atlanta classification maintains the original classification of acute pancreatitis into 2 main categories: interstitial edematous pancreatitis and necrotizing pancreatitis.
Necrotizing pancreatitis is further divided into 3 subtypes based on extent and location of necrosis:
- Parenchymal necrosis alone (5% of cases)
- Necrosis of peripancreatic fat alone (20%)
- Necrosis of both parenchyma and peripancreatic fat (75%).
Peripancreatic involvement is commonly found in the mesentery, peripancreatic and distant retroperitoneum, and lesser sac.
Of the three subtypes, peripancreatic necrosis has the best prognosis. However, all of the subtypes of necrotizing pancreatitis are associated with poorer outcomes than interstitial edematous pancreatitis.
Fluid collections
Acute pancreatic fluid collections contain exclusively nonsolid components without an inflammatory wall and are typically found in the peripancreatic fat. These collections often resolve without intervention as the patient recovers. If they persist beyond 4 weeks and develop a nonepithelialized, fibrous wall, they become pseudocysts. Intervention is generally not recommended for pseudocysts unless they are symptomatic.
ROLE OF IMAGING
Radiographic imaging is not usually necessary to diagnose acute pancreatitis. However, it can be a valuable tool to clarify an ambiguous presentation, determine severity, and identify complications.
The timing and appropriate type of imaging are integral to obtaining useful data. Any imaging obtained in acute pancreatitis to evaluate necrosis should be performed at least 3 to 5 days from the initial symptom onset; if imaging is obtained before 72 hours, necrosis cannot be confidently excluded.8
COMPUTED TOMOGRAPHY
CT is the imaging test of choice when evaluating acute pancreatitis. In addition, almost all percutaneous interventions are performed with CT guidance. The Balthazar score is the most well-known CT severity index. It is calculated based on the degree of inflammation, acute fluid collections, and parenchymal necrosis.9 However, a modified severity index incorporates extrapancreatic complications such as ascites and vascular compromise and was found to more strongly correlate with outcomes than the standard Balthazar score.10
Contrast-enhanced CT is performed in 2 phases:
The pancreatic parenchymal phase
The pancreatic parenchymal or late arterial phase is obtained approximately 40 to 45 seconds after the start of the contrast bolus. It is used to detect necrosis in the early phase of acute pancreatitis and to assess the peripancreatic arteries for pseudoaneurysms in the late phase of acute pancreatitis.11
Pancreatic necrosis appears as an area of decreased parenchymal enhancement, either well-defined or heterogeneous. The normal pancreatic parenchyma has a postcontrast enhancement pattern similar to that of the spleen. Parenchyma that does not enhance to the same degree is considered necrotic. The severity of necrosis is graded based on the percentage of the pancreas involved (< 30%, 30%–50%, or > 50%), and a higher percentage correlates with a worse outcome.12,13
Peripancreatic necrosis is harder to detect, as there is no method to assess fat enhancement as there is with pancreatic parenchymal enhancement. In general, radiologists assume that heterogeneous peripancreatic changes, including areas of fat, fluid, and soft tissue attenuation, are consistent with peripancreatic necrosis. After 7 to 10 days, if these changes become more homogeneous and confluent with a more mass-like process, peripancreatic necrosis can be more confidently identified.12,13
The portal venous phase
The later, portal venous phase of the scan is obtained approximately 70 seconds after the start of the contrast bolus. It is used to detect and characterize fluid collections and venous complications of the disease.
Drawbacks of CT
A drawback of CT is the need for iodinated intravenous contrast media, which in severely ill patients may precipitate or worsen pre-existing acute kidney injury.
Further, several studies have shown that findings on CT rarely alter the management of patients in the early phase of acute pancreatitis and in fact may be an overuse of medical resources.14 Unless there are confounding clinical signs or symptoms, CT should be delayed for at least 72 hours.9,10,14,15
MAGNETIC RESONANCE IMAGING
Magnetic resonance imaging (MRI) is not a first-line imaging test in this disease because it is not as available as CT and takes longer to perform—20 to 30 minutes. The patient must be evaluated for candidacy, as it is difficult for acutely ill patients to tolerate an examination that takes this long and requires them to hold their breath multiple times.
MRI is an appropriate alternative in patients who are pregnant or who have severe iodinated-contrast allergy. While contrast is necessary to detect pancreatic necrosis with CT, MRI can detect necrosis without the need for contrast in patients with acute kidney injury or severe chronic kidney disease. Also, MRI may be better in complicated cases requiring repeated imaging because it does not expose the patient to radiation.
On MRI, pancreatic necrosis appears as a heterogeneous area, owing to its liquid and solid components. Liquid components appear hyperintense, and solid components hypointense, on T2 fluid-weighted imaging. This ability to differentiate the components of a walled-off pancreatic necrosis can be useful in determining whether a collection requires drainage or debridement. MRI is also more sensitive for hemorrhagic complications, best seen on T1 fat-weighted images.12,16
Magnetic resonance cholangiopancreatography is an excellent method for ductal evaluation through heavily T2-weighted imaging. It is more sensitive than CT for detecting common bile duct stones and can also detect pancreatic duct strictures or extravasation into fluid collections.16
SUPPORTIVE MANAGEMENT OF EARLY NECROTIZING PANCREATITIS
In the early phase of necrotizing pancreatitis, management is supportive with the primary aim of preventing intravascular volume depletion. Aggressive fluid resuscitation in the first 48 to 72 hours, pain control, and bowel rest are the mainstays of supportive therapy. Intensive care may be necessary if organ failure and hemodynamic instability accompany necrotizing pancreatitis.
Prophylactic antibiotic and antifungal therapy to prevent infected necrosis has been controversial. Recent studies of its utility have not yielded supportive results, and the American College of Gastroenterology and the Infectious Diseases Society of America no longer recommend it.9,17 These medications should not be given unless concomitant cholangitis or extrapancreatic infection is clinically suspected.
Early enteral nutrition is recommended in patients in whom pancreatitis is predicted to be severe and in those not expected to resume oral intake within 5 to 7 days. Enteral nutrition most commonly involves bedside or endoscopic placement of a nasojejunal feeding tube and collaboration with a nutritionist to determine protein-caloric requirements.
Compared with enteral nutrition, total parenteral nutrition is associated with higher rates of infection, multiorgan dysfunction and failure, and death.18
MANAGING COMPLICATIONS OF PANCREATIC NECROSIS
Necrotizing pancreatitis is a defining complication of acute pancreatitis, and its presence alone indicates greater severity. However, superimposed complications may further worsen outcomes.
Infected pancreatic necrosis
Infection occurs in approximately 20% of patients with necrotizing pancreatitis and confers a mortality rate of 20% to 50%.19 Infected pancreatic necrosis occurs when gut organisms translocate into the nearby necrotic pancreatic and peripancreatic tissue. The most commonly identified organisms include Escherichia coli and Enterococcus species.20
This complication usually manifests 2 to 4 weeks after symptom onset; earlier onset is uncommon to rare. It should be considered when the systemic inflammatory response syndrome persists or recurs after 10 days to 2 weeks. Systemic inflammatory response syndrome is also common in sterile necrotizing pancreatitis and sometimes in interstitial pancreatitis, particularly during the first week. However, its sudden appearance or resurgence, high spiking fevers, or worsening organ failure in the later phase (2–4 weeks) of pancreatitis should heighten suspicion of infected pancreatic necrosis.
Imaging may also help diagnose infection, and the presence of gas within a collection or region of necrosis is highly specific. However, the presence of gas is not completely sensitive for infection, as it is seen in only 12% to 22% of infected cases.
Before minimally invasive techniques became available, the diagnosis of infected pancreatic necrosis was confirmed by percutaneous CT-guided aspiration of the necrotic mass or collection for Gram stain and culture.
Antibiotic therapy is indicated in confirmed or suspected cases of infected pancreatic necrosis. Antibiotics with gram-negative coverage and appropriate penetration such as carbapenems, metronidazole, fluoroquinolones, and selected cephalosporins are most commonly used. Meropenem is the antibiotic of choice at our institution.
CT-guided fine-needle aspiration is often done if suspected infected pancreatic necrosis fails to respond to empiric antibiotic therapy.
Debridement or drainage. Generally, the diagnosis or suspicion of infected pancreatic necrosis (suggestive signs are high fever, elevated white blood cell count, and sepsis) warrants an intervention to debride or drain infected pancreatic tissue and control sepsis.21
While source control is integral to the successful treatment of infected pancreatic necrosis, antibiotic therapy may provide a bridge to intervention for critically ill patients by suppressing bacteremia and subsequent sepsis. A 2013 meta-analysis found that 324 of 409 patients with suspected infected pancreatic necrosis were successfully stabilized with antibiotic treatment.21,22 The trend toward conservative management and promising outcomes with antibiotic therapy alone or with minimally invasive techniques has lessened the need for diagnostic CT-guided fine-needle aspiration.
Hemorrhage
Spontaneous hemorrhage into pancreatic necrosis is a rare but life-threatening complication. Because CT is almost always performed with contrast enhancement, this complication is rarely identified with imaging. The diagnosis is made by noting a drop in hemoglobin and hematocrit.
Hemorrhage into the retroperitoneum or the peritoneal cavity, or both, can occur when an inflammatory process erodes into a nearby artery. Luminal gastrointestinal bleeding can occur from gastric varices arising from splenic vein thrombosis and resulting left-sided portal hypertension, or from pseudoaneurysms. These can also bleed into the pancreatic duct (hemosuccus pancreaticus). Pseudoaneurysm is a later complication that occurs when an arterial wall (most commonly the splenic or gastroduodenal artery) is weakened by pancreatic enzymes.23
Prompt recognition of hemorrhagic events and consultation with an interventional radiologist or surgeon are required to prevent death.
Inflammation and abdominal compartment syndrome
Inflammation from necrotizing pancreatitis can cause further complications by blocking nearby structures. Reported complications include jaundice from biliary compression, hydronephrosis from ureteral compression, bowel obstruction, and gastric outlet obstruction.
Abdominal compartment syndrome is an increasingly recognized complication of acute pancreatitis. Abdominal pressure can rise due to a number of factors, including fluid collections, ascites, ileus, and overly aggressive fluid resuscitation.24 Elevated abdominal pressure is associated with complications such as decreased respiratory compliance, increased peak airway pressure, decreased cardiac preload, hypotension, mesenteric and intestinal ischemia, feeding intolerance, and lower-extremity ischemia and thrombosis.
Patients with necrotizing pancreatitis who have abdominal compartment syndrome have a mortality rate 5 times higher than patients without abdominal compartment syndrome.25
Abdominal pressures should be monitored using a bladder pressure sensor in critically ill or ventilated patients with acute pancreatitis. If the abdominal pressure rises above 20 mm Hg, medical and surgical interventions should be offered in a stepwise fashion to decrease it. Interventions include decompression by nasogastric and rectal tube, sedation or paralysis to relax abdominal wall tension, minimization of intravenous fluids, percutaneous drainage of ascites, and (rarely) surgical midline or subcostal laparotomy.
ROLE OF INTERVENTION
The treatment of necrotizing pancreatitis has changed rapidly, thanks to a growing experience with minimally invasive techniques.
Indications for intervention
Infected pancreatic necrosis is the primary indication for surgical, percutaneous, or endoscopic intervention.
In sterile necrosis, the threshold for intervention is less clear, and intervention is often reserved for patients who fail to clinically improve or who have intractable abdominal pain, gastric outlet obstruction, or fistulating disease.26
In asymptomatic cases, intervention is almost never indicated regardless of the location or size of the necrotic area.
In walled-off pancreatic necrosis, less-invasive and less-morbid interventions such as endoscopic or percutaneous drainage or video-assisted retroperitoneal debridement can be done.
Timing of intervention
In the past, delaying intervention was thought to increase the risk of death. However, multiple studies have found that outcomes are often worse if intervention is done early, likely due to the lack of a fully formed fibrous wall or demarcation of the necrotic area.27
If the patient remains clinically stable, it is best to delay intervention until at least 4 weeks after the index event to achieve optimal outcomes. Delay can often be achieved by antibiotic treatment to suppress bacteremia and endoscopic or percutaneous drainage of infected collections to control sepsis.
Open surgery
The gold-standard intervention for infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis is open necrosectomy. This involves exploratory laparotomy with blunt debridement of all visible necrotic pancreatic tissue.
Methods to facilitate later evacuation of residual infected fluid and debris vary widely. Multiple large-caliber drains can be placed to facilitate irrigation and drainage before closure of the abdominal fascia. As infected pancreatic necrosis carries the risk of contaminating the peritoneal cavity, the skin is often left open to heal by secondary intention. An interventional radiologist is frequently enlisted to place, exchange, or downsize drainage catheters.
Infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis often requires more than one operation to achieve satisfactory debridement.
The goals of open necrosectomy are to remove nonviable tissue and infection, preserve viable pancreatic tissue, eliminate fistulous connections, and minimize damage to local organs and vasculature.
Minimally invasive techniques
Video-assisted retroperitoneal debridement has been described as a hybrid between endoscopic and open retroperitoneal debridement.28 This technique requires first placing a percutaneous catheter into the necrotic area through the left flank to create a retroperitoneal tract. A 5-cm incision is made and the necrotic space is entered using the drain for guidance. Necrotic tissue is carefully debrided under direct vision using a combination of forceps, irrigation, and suction. A laparoscopic port can also be introduced into the incision when the procedure can no longer be continued under direct vision.29,30
Although not all patients are candidates for minimal-access surgery, it remains an evolving surgical option.
Endoscopic transmural debridement is another option for infected pancreatic necrosis and symptomatic walled-off pancreatic necrosis. Depending on the location of the necrotic area, an echoendoscope is passed to either the stomach or duodenum. Guided by endoscopic ultrasonography, a needle is passed into the collection, allowing subsequent fistula creation and stenting for internal drainage or debridement. In the past, this process required several steps, multiple devices, fluoroscopic guidance, and considerable time. But newer endoscopic lumen-apposing metal stents have been developed that can be placed in a single step without fluoroscopy. A slimmer endoscope can then be introduced into the necrotic cavity via the stent, and the necrotic debris can be debrided with endoscopic baskets, snares, forceps, and irrigation.9,31
Similar to surgical necrosectomy, satisfactory debridement is not often obtained with a single procedure; 2 to 5 endoscopic procedures may be needed to achieve resolution. However, the luminal approach in endoscopic necrosectomy avoids the significant morbidity of major abdominal surgery and the potential for pancreaticocutaneous fistulae that may occur with drains.
In a randomized trial comparing endoscopic necrosectomy vs surgical necrosectomy (video-assisted retroperitoneal debridement and exploratory laparotomy),32 endoscopic necrosectomy showed less inflammatory response than surgical necrosectomy and had a lower risk of new-onset organ failure, bleeding, fistula formation, and death.32
Selecting the best intervention for the individual patient
Given the multiple available techniques, selecting the best intervention for individual patients can be challenging. A team approach with input from a gastroenterologist, surgeon, and interventional radiologist is best when determining which technique would best suit each patient.
Surgical necrosectomy is still the treatment of choice for unstable patients with infected pancreatic necrosis or multiple, inaccessible collections, but current evidence suggests a different approach in stable infected pancreatic necrosis and symptomatic sterile walled-off pancreatic necrosis.
The Dutch Pancreatitis Group28 randomized 88 patients with infected pancreatic necrosis or symptomatic walled-off pancreatic necrosis to open necrosectomy or a minimally invasive “step-up” approach consisting of up to 2 percutaneous drainage or endoscopic debridement procedures before escalation to video-assisted retroperitoneal debridement. The step-up approach resulted in lower rates of morbidity and death than surgical necrosectomy as first-line treatment. Furthermore, some patients in the step-up group avoided the need for surgery entirely.30
SUMMING UP
Necrosis significantly increases rates of morbidity and mortality in acute pancreatitis. Hospitalists, general internists, and general surgeons are all on the front lines in identifying severe cases and consulting the appropriate specialists for optimal multidisciplinary care. Selective and appropriate timing of radiologic imaging is key, and a vital tool in the management of necrotizing pancreatitis.
While the primary indication for intervention is infected pancreatic necrosis, additional indications are symptomatic walled-off pancreatic necrosis secondary to intractable abdominal pain, bowel obstruction, and failure to thrive. As a result of improving technology and inpatient care, these patients may present with intractable symptoms in the outpatient setting rather than the inpatient setting. The onus is on the primary care physician to maintain a high level of suspicion and refer these patients to subspecialists as appropriate.
Open surgical necrosectomy remains an important approach for care of infected pancreatic necrosis or patients with intractable symptoms. A step-up approach starting with a minimally invasive procedure and escalating if the initial intervention is unsuccessful is gradually becoming the standard of care.
Acute pancreatitis accounted for more than 300,000 admissions and $2.6 billion in associated healthcare costs in the United States in 2012.1 First-line management is early aggressive fluid resuscitation and analgesics for pain control. Guidelines recommend estimating the clinical severity of each attack using a validated scoring system such as the Bedside Index of Severity in Acute Pancreatitis.2 Clinically severe pancreatitis is associated with necrosis.
Acute pancreatitis results from inappropriate activation of zymogens and subsequent autodigestion of the pancreas by its own enzymes. Though necrotizing pancreatitis is thought to be an ischemic complication, its pathogenesis is not completely understood. Necrosis increases the morbidity and mortality risk of acute pancreatitis because of its association with organ failure and infectious complications. As such, patients with necrotizing pancreatitis may need admission to the intensive care unit, nutritional support, antibiotics, and radiologic, endoscopic, or surgical interventions.
Here, we review current evidence regarding the diagnosis and management of necrotizing pancreatitis.
PROPER TERMINOLOGY HELPS COLLABORATION
Managing necrotizing pancreatitis requires the combined efforts of internists, gastroenterologists, radiologists, and surgeons. This collaboration is aided by proper terminology.
A classification system was devised in Atlanta, GA, in 1992 to facilitate communication and interdisciplinary collaboration.3 Severe pancreatitis was differentiated from mild by the presence of organ failure or the complications of pseudocyst, necrosis, or abscess.
The original Atlanta classification had several limitations. First, the terminology for fluid collections was ambiguous and frequently misused. Second, the assessment of clinical severity required either the Ranson score or the Acute Physiology and Chronic Health Evaluation II score, both of which are complex and have other limitations. Finally, advances in imaging and treatment have rendered the original Atlanta nomenclature obsolete.
In 2012, the Acute Pancreatitis Classification Working Group issued a revised Atlanta classification that modernized the terminology pertaining to natural history, severity, imaging features, and complications. It divides the natural course of acute pancreatitis into early and late phases.4
Early vs late phase
In the early phase, findings on computed tomography (CT) neither correlate with clinical severity nor alter clinical management.6 Thus, early imaging is not indicated unless there is diagnostic uncertainty, lack of response to appropriate treatment, or sudden deterioration.
Moderate pancreatitis describes patients with pancreatic necrosis with or without transient organ failure (organ dysfunction for ≤ 48 hours).
Severe pancreatitis is defined by pancreatic necrosis and persistent organ dysfunction.4 It may be accompanied by pancreatic and peripancreatic fluid collections; bacteremia and sepsis can occur in association with infection of necrotic collections.
Interstitial edematous pancreatitis vs necrotizing pancreatitis
The revised Atlanta classification maintains the original classification of acute pancreatitis into 2 main categories: interstitial edematous pancreatitis and necrotizing pancreatitis.
Necrotizing pancreatitis is further divided into 3 subtypes based on extent and location of necrosis:
- Parenchymal necrosis alone (5% of cases)
- Necrosis of peripancreatic fat alone (20%)
- Necrosis of both parenchyma and peripancreatic fat (75%).
Peripancreatic involvement is commonly found in the mesentery, peripancreatic and distant retroperitoneum, and lesser sac.
Of the three subtypes, peripancreatic necrosis has the best prognosis. However, all of the subtypes of necrotizing pancreatitis are associated with poorer outcomes than interstitial edematous pancreatitis.
Fluid collections
Acute pancreatic fluid collections contain exclusively nonsolid components without an inflammatory wall and are typically found in the peripancreatic fat. These collections often resolve without intervention as the patient recovers. If they persist beyond 4 weeks and develop a nonepithelialized, fibrous wall, they become pseudocysts. Intervention is generally not recommended for pseudocysts unless they are symptomatic.
ROLE OF IMAGING
Radiographic imaging is not usually necessary to diagnose acute pancreatitis. However, it can be a valuable tool to clarify an ambiguous presentation, determine severity, and identify complications.
The timing and appropriate type of imaging are integral to obtaining useful data. Any imaging obtained in acute pancreatitis to evaluate necrosis should be performed at least 3 to 5 days from the initial symptom onset; if imaging is obtained before 72 hours, necrosis cannot be confidently excluded.8
COMPUTED TOMOGRAPHY
CT is the imaging test of choice when evaluating acute pancreatitis. In addition, almost all percutaneous interventions are performed with CT guidance. The Balthazar score is the most well-known CT severity index. It is calculated based on the degree of inflammation, acute fluid collections, and parenchymal necrosis.9 However, a modified severity index incorporates extrapancreatic complications such as ascites and vascular compromise and was found to more strongly correlate with outcomes than the standard Balthazar score.10
Contrast-enhanced CT is performed in 2 phases:
The pancreatic parenchymal phase
The pancreatic parenchymal or late arterial phase is obtained approximately 40 to 45 seconds after the start of the contrast bolus. It is used to detect necrosis in the early phase of acute pancreatitis and to assess the peripancreatic arteries for pseudoaneurysms in the late phase of acute pancreatitis.11
Pancreatic necrosis appears as an area of decreased parenchymal enhancement, either well-defined or heterogeneous. The normal pancreatic parenchyma has a postcontrast enhancement pattern similar to that of the spleen. Parenchyma that does not enhance to the same degree is considered necrotic. The severity of necrosis is graded based on the percentage of the pancreas involved (< 30%, 30%–50%, or > 50%), and a higher percentage correlates with a worse outcome.12,13
Peripancreatic necrosis is harder to detect, as there is no method to assess fat enhancement as there is with pancreatic parenchymal enhancement. In general, radiologists assume that heterogeneous peripancreatic changes, including areas of fat, fluid, and soft tissue attenuation, are consistent with peripancreatic necrosis. After 7 to 10 days, if these changes become more homogeneous and confluent with a more mass-like process, peripancreatic necrosis can be more confidently identified.12,13
The portal venous phase
The later, portal venous phase of the scan is obtained approximately 70 seconds after the start of the contrast bolus. It is used to detect and characterize fluid collections and venous complications of the disease.
Drawbacks of CT
A drawback of CT is the need for iodinated intravenous contrast media, which in severely ill patients may precipitate or worsen pre-existing acute kidney injury.
Further, several studies have shown that findings on CT rarely alter the management of patients in the early phase of acute pancreatitis and in fact may be an overuse of medical resources.14 Unless there are confounding clinical signs or symptoms, CT should be delayed for at least 72 hours.9,10,14,15
MAGNETIC RESONANCE IMAGING
Magnetic resonance imaging (MRI) is not a first-line imaging test in this disease because it is not as available as CT and takes longer to perform—20 to 30 minutes. The patient must be evaluated for candidacy, as it is difficult for acutely ill patients to tolerate an examination that takes this long and requires them to hold their breath multiple times.
MRI is an appropriate alternative in patients who are pregnant or who have severe iodinated-contrast allergy. While contrast is necessary to detect pancreatic necrosis with CT, MRI can detect necrosis without the need for contrast in patients with acute kidney injury or severe chronic kidney disease. Also, MRI may be better in complicated cases requiring repeated imaging because it does not expose the patient to radiation.
On MRI, pancreatic necrosis appears as a heterogeneous area, owing to its liquid and solid components. Liquid components appear hyperintense, and solid components hypointense, on T2 fluid-weighted imaging. This ability to differentiate the components of a walled-off pancreatic necrosis can be useful in determining whether a collection requires drainage or debridement. MRI is also more sensitive for hemorrhagic complications, best seen on T1 fat-weighted images.12,16
Magnetic resonance cholangiopancreatography is an excellent method for ductal evaluation through heavily T2-weighted imaging. It is more sensitive than CT for detecting common bile duct stones and can also detect pancreatic duct strictures or extravasation into fluid collections.16
SUPPORTIVE MANAGEMENT OF EARLY NECROTIZING PANCREATITIS
In the early phase of necrotizing pancreatitis, management is supportive with the primary aim of preventing intravascular volume depletion. Aggressive fluid resuscitation in the first 48 to 72 hours, pain control, and bowel rest are the mainstays of supportive therapy. Intensive care may be necessary if organ failure and hemodynamic instability accompany necrotizing pancreatitis.
Prophylactic antibiotic and antifungal therapy to prevent infected necrosis has been controversial. Recent studies of its utility have not yielded supportive results, and the American College of Gastroenterology and the Infectious Diseases Society of America no longer recommend it.9,17 These medications should not be given unless concomitant cholangitis or extrapancreatic infection is clinically suspected.
Early enteral nutrition is recommended in patients in whom pancreatitis is predicted to be severe and in those not expected to resume oral intake within 5 to 7 days. Enteral nutrition most commonly involves bedside or endoscopic placement of a nasojejunal feeding tube and collaboration with a nutritionist to determine protein-caloric requirements.
Compared with enteral nutrition, total parenteral nutrition is associated with higher rates of infection, multiorgan dysfunction and failure, and death.18
MANAGING COMPLICATIONS OF PANCREATIC NECROSIS
Necrotizing pancreatitis is a defining complication of acute pancreatitis, and its presence alone indicates greater severity. However, superimposed complications may further worsen outcomes.
Infected pancreatic necrosis
Infection occurs in approximately 20% of patients with necrotizing pancreatitis and confers a mortality rate of 20% to 50%.19 Infected pancreatic necrosis occurs when gut organisms translocate into the nearby necrotic pancreatic and peripancreatic tissue. The most commonly identified organisms include Escherichia coli and Enterococcus species.20
This complication usually manifests 2 to 4 weeks after symptom onset; earlier onset is uncommon to rare. It should be considered when the systemic inflammatory response syndrome persists or recurs after 10 days to 2 weeks. Systemic inflammatory response syndrome is also common in sterile necrotizing pancreatitis and sometimes in interstitial pancreatitis, particularly during the first week. However, its sudden appearance or resurgence, high spiking fevers, or worsening organ failure in the later phase (2–4 weeks) of pancreatitis should heighten suspicion of infected pancreatic necrosis.
Imaging may also help diagnose infection, and the presence of gas within a collection or region of necrosis is highly specific. However, the presence of gas is not completely sensitive for infection, as it is seen in only 12% to 22% of infected cases.
Before minimally invasive techniques became available, the diagnosis of infected pancreatic necrosis was confirmed by percutaneous CT-guided aspiration of the necrotic mass or collection for Gram stain and culture.
Antibiotic therapy is indicated in confirmed or suspected cases of infected pancreatic necrosis. Antibiotics with gram-negative coverage and appropriate penetration such as carbapenems, metronidazole, fluoroquinolones, and selected cephalosporins are most commonly used. Meropenem is the antibiotic of choice at our institution.
CT-guided fine-needle aspiration is often done if suspected infected pancreatic necrosis fails to respond to empiric antibiotic therapy.
Debridement or drainage. Generally, the diagnosis or suspicion of infected pancreatic necrosis (suggestive signs are high fever, elevated white blood cell count, and sepsis) warrants an intervention to debride or drain infected pancreatic tissue and control sepsis.21
While source control is integral to the successful treatment of infected pancreatic necrosis, antibiotic therapy may provide a bridge to intervention for critically ill patients by suppressing bacteremia and subsequent sepsis. A 2013 meta-analysis found that 324 of 409 patients with suspected infected pancreatic necrosis were successfully stabilized with antibiotic treatment.21,22 The trend toward conservative management and promising outcomes with antibiotic therapy alone or with minimally invasive techniques has lessened the need for diagnostic CT-guided fine-needle aspiration.
Hemorrhage
Spontaneous hemorrhage into pancreatic necrosis is a rare but life-threatening complication. Because CT is almost always performed with contrast enhancement, this complication is rarely identified with imaging. The diagnosis is made by noting a drop in hemoglobin and hematocrit.
Hemorrhage into the retroperitoneum or the peritoneal cavity, or both, can occur when an inflammatory process erodes into a nearby artery. Luminal gastrointestinal bleeding can occur from gastric varices arising from splenic vein thrombosis and resulting left-sided portal hypertension, or from pseudoaneurysms. These can also bleed into the pancreatic duct (hemosuccus pancreaticus). Pseudoaneurysm is a later complication that occurs when an arterial wall (most commonly the splenic or gastroduodenal artery) is weakened by pancreatic enzymes.23
Prompt recognition of hemorrhagic events and consultation with an interventional radiologist or surgeon are required to prevent death.
Inflammation and abdominal compartment syndrome
Inflammation from necrotizing pancreatitis can cause further complications by blocking nearby structures. Reported complications include jaundice from biliary compression, hydronephrosis from ureteral compression, bowel obstruction, and gastric outlet obstruction.
Abdominal compartment syndrome is an increasingly recognized complication of acute pancreatitis. Abdominal pressure can rise due to a number of factors, including fluid collections, ascites, ileus, and overly aggressive fluid resuscitation.24 Elevated abdominal pressure is associated with complications such as decreased respiratory compliance, increased peak airway pressure, decreased cardiac preload, hypotension, mesenteric and intestinal ischemia, feeding intolerance, and lower-extremity ischemia and thrombosis.
Patients with necrotizing pancreatitis who have abdominal compartment syndrome have a mortality rate 5 times higher than patients without abdominal compartment syndrome.25
Abdominal pressures should be monitored using a bladder pressure sensor in critically ill or ventilated patients with acute pancreatitis. If the abdominal pressure rises above 20 mm Hg, medical and surgical interventions should be offered in a stepwise fashion to decrease it. Interventions include decompression by nasogastric and rectal tube, sedation or paralysis to relax abdominal wall tension, minimization of intravenous fluids, percutaneous drainage of ascites, and (rarely) surgical midline or subcostal laparotomy.
ROLE OF INTERVENTION
The treatment of necrotizing pancreatitis has changed rapidly, thanks to a growing experience with minimally invasive techniques.
Indications for intervention
Infected pancreatic necrosis is the primary indication for surgical, percutaneous, or endoscopic intervention.
In sterile necrosis, the threshold for intervention is less clear, and intervention is often reserved for patients who fail to clinically improve or who have intractable abdominal pain, gastric outlet obstruction, or fistulating disease.26
In asymptomatic cases, intervention is almost never indicated regardless of the location or size of the necrotic area.
In walled-off pancreatic necrosis, less-invasive and less-morbid interventions such as endoscopic or percutaneous drainage or video-assisted retroperitoneal debridement can be done.
Timing of intervention
In the past, delaying intervention was thought to increase the risk of death. However, multiple studies have found that outcomes are often worse if intervention is done early, likely due to the lack of a fully formed fibrous wall or demarcation of the necrotic area.27
If the patient remains clinically stable, it is best to delay intervention until at least 4 weeks after the index event to achieve optimal outcomes. Delay can often be achieved by antibiotic treatment to suppress bacteremia and endoscopic or percutaneous drainage of infected collections to control sepsis.
Open surgery
The gold-standard intervention for infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis is open necrosectomy. This involves exploratory laparotomy with blunt debridement of all visible necrotic pancreatic tissue.
Methods to facilitate later evacuation of residual infected fluid and debris vary widely. Multiple large-caliber drains can be placed to facilitate irrigation and drainage before closure of the abdominal fascia. As infected pancreatic necrosis carries the risk of contaminating the peritoneal cavity, the skin is often left open to heal by secondary intention. An interventional radiologist is frequently enlisted to place, exchange, or downsize drainage catheters.
Infected pancreatic necrosis or symptomatic sterile walled-off pancreatic necrosis often requires more than one operation to achieve satisfactory debridement.
The goals of open necrosectomy are to remove nonviable tissue and infection, preserve viable pancreatic tissue, eliminate fistulous connections, and minimize damage to local organs and vasculature.
Minimally invasive techniques
Video-assisted retroperitoneal debridement has been described as a hybrid between endoscopic and open retroperitoneal debridement.28 This technique requires first placing a percutaneous catheter into the necrotic area through the left flank to create a retroperitoneal tract. A 5-cm incision is made and the necrotic space is entered using the drain for guidance. Necrotic tissue is carefully debrided under direct vision using a combination of forceps, irrigation, and suction. A laparoscopic port can also be introduced into the incision when the procedure can no longer be continued under direct vision.29,30
Although not all patients are candidates for minimal-access surgery, it remains an evolving surgical option.
Endoscopic transmural debridement is another option for infected pancreatic necrosis and symptomatic walled-off pancreatic necrosis. Depending on the location of the necrotic area, an echoendoscope is passed to either the stomach or duodenum. Guided by endoscopic ultrasonography, a needle is passed into the collection, allowing subsequent fistula creation and stenting for internal drainage or debridement. In the past, this process required several steps, multiple devices, fluoroscopic guidance, and considerable time. But newer endoscopic lumen-apposing metal stents have been developed that can be placed in a single step without fluoroscopy. A slimmer endoscope can then be introduced into the necrotic cavity via the stent, and the necrotic debris can be debrided with endoscopic baskets, snares, forceps, and irrigation.9,31
Similar to surgical necrosectomy, satisfactory debridement is not often obtained with a single procedure; 2 to 5 endoscopic procedures may be needed to achieve resolution. However, the luminal approach in endoscopic necrosectomy avoids the significant morbidity of major abdominal surgery and the potential for pancreaticocutaneous fistulae that may occur with drains.
In a randomized trial comparing endoscopic necrosectomy vs surgical necrosectomy (video-assisted retroperitoneal debridement and exploratory laparotomy),32 endoscopic necrosectomy showed less inflammatory response than surgical necrosectomy and had a lower risk of new-onset organ failure, bleeding, fistula formation, and death.32
Selecting the best intervention for the individual patient
Given the multiple available techniques, selecting the best intervention for individual patients can be challenging. A team approach with input from a gastroenterologist, surgeon, and interventional radiologist is best when determining which technique would best suit each patient.
Surgical necrosectomy is still the treatment of choice for unstable patients with infected pancreatic necrosis or multiple, inaccessible collections, but current evidence suggests a different approach in stable infected pancreatic necrosis and symptomatic sterile walled-off pancreatic necrosis.
The Dutch Pancreatitis Group28 randomized 88 patients with infected pancreatic necrosis or symptomatic walled-off pancreatic necrosis to open necrosectomy or a minimally invasive “step-up” approach consisting of up to 2 percutaneous drainage or endoscopic debridement procedures before escalation to video-assisted retroperitoneal debridement. The step-up approach resulted in lower rates of morbidity and death than surgical necrosectomy as first-line treatment. Furthermore, some patients in the step-up group avoided the need for surgery entirely.30
SUMMING UP
Necrosis significantly increases rates of morbidity and mortality in acute pancreatitis. Hospitalists, general internists, and general surgeons are all on the front lines in identifying severe cases and consulting the appropriate specialists for optimal multidisciplinary care. Selective and appropriate timing of radiologic imaging is key, and a vital tool in the management of necrotizing pancreatitis.
While the primary indication for intervention is infected pancreatic necrosis, additional indications are symptomatic walled-off pancreatic necrosis secondary to intractable abdominal pain, bowel obstruction, and failure to thrive. As a result of improving technology and inpatient care, these patients may present with intractable symptoms in the outpatient setting rather than the inpatient setting. The onus is on the primary care physician to maintain a high level of suspicion and refer these patients to subspecialists as appropriate.
Open surgical necrosectomy remains an important approach for care of infected pancreatic necrosis or patients with intractable symptoms. A step-up approach starting with a minimally invasive procedure and escalating if the initial intervention is unsuccessful is gradually becoming the standard of care.
- Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic disease in the United States. Gastroenterology 2015; 149:1731–1741e3.
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108:1400–1416.
- Bradley EL 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993; 128:586–590.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 1995; 23:1638–1652.
- Kadiyala V, Suleiman SL, McNabb-Baltar J, Wu BU, Banks PA, Singh VK. The Atlanta classification, revised Atlanta classification, and determinant-based classification of acute pancreatitis: which is best at stratifying outcomes? Pancreas 2016; 45:510–515.
- Singh VK, Bollen TL, Wu BU, et al. An assessment of the severity of interstitial pancreatitis. Clin Gastroenterol Hepatol 2011; 9:1098–1103.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Mortele KJ, Wiesner W, Intriere L, et al. A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. AJR Am J Roentgenol 2004; 183:1261–1265.
- Verde F, Fishman EK, Johnson PT. Arterial pseudoaneurysms complicating pancreatitis: literature review. J Comput Assist Tomogr 2015; 39:7–12.
- Shyu JY, Sainani NI, Sahni VA, et al. Necrotizing pancreatitis: diagnosis, imaging, and intervention. Radiographics 2014; 34:1218–1239.
- Thoeni RF. The revised Atlanta classification of acute pancreatitis: its importance for the radiologist and its effect on treatment. Radiology 2012; 262:751–764.
- Morgan DE, Ragheb CM, Lockhart ME, Cary B, Fineberg NS, Berland LL. Acute pancreatitis: computed tomography utilization and radiation exposure are related to severity but not patient age. Clin Gastroenterol Hepatol 2010; 8:303–308.
- Vitellas KM, Paulson EK, Enns RA, Keogan MT, Pappas TN. Pancreatitis complicated by gland necrosis: evolution of findings on contrast-enhanced CT. J Comput Assist Tomogr 1999; 23:898–905.
- Stimac D, Miletic D, Radic M, et al. The role of nonenhanced magnetic resonance imaging in the early assessment of acute pancreatitis. Am J Gastroenterol 2007; 102:997–1004.
- Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect (Larchmt) 2010; 11:79–109.
- Petrov MS, Kukosh MV, Emelyanov NV. A randomized controlled trial of enteral versus parenteral feeding in patients with predicted severe acute pancreatitis shows a significant reduction in mortality and in infected pancreatic complications with total enteral nutrition. Dig Surg 2006; 23:336–345.
- Petrov MS, Shanbhag S, Chakraborty M, Phillips AR, Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 2010; 139:813–820.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006; 4:CD002941.
- Baril NB, Ralls PW, Wren SM, et al. Does an infected peripancreatic fluid collection or abscess mandate operation? Ann Surg 2000; 231:361–367.
- Mouli VP, Sreenivas V, Garg PK. Efficacy of conservative treatment, without necrosectomy, for infected pancreatic necrosis: a systematic review and meta-analysis. Gastroenterology 2013; 144:333–340.e2.
- Kirby JM, Vora P, Midia M, Rawlinson J. Vascular complications of pancreatitis: imaging and intervention. Cardiovasc Intervent Radiol 2008; 31:957–970.
- De Waele JJ, Hoste E, Blot SI, Decruyenaere J, Colardyn F. Intra-abdominal hypertension in patients with severe acute pancreatitis. Crit Care 2005; 9:R452–R457.
- van Brunschot S, Schut AJ, Bouwense SA, et al; Dutch Pancreatitis Study Group. Abdominal compartment syndrome in acute pancreatitis: a systematic review. Pancreas 2014; 43:665–674.
- Bugiantella W, Rondelli F, Boni M, et al. Necrotizing pancreatitis: a review of the interventions. Int J Surg 2016; 28(suppl 1):S163–S171.
- Besselink MG, Verwer TJ, Schoenmaeckers EJ, et al. Timing of surgical intervention in necrotizing pancreatitis. Arch Surg 2007; 142:1194–1201.
- van Santvoort HC, Besselink MG, Horvath KD, et al; Dutch Acute Pancreatis Study Group. Videoscopic assisted retroperitoneal debridement in infected necrotizing pancreatitis. HPB (Oxford) 2007; 9:156–159.
- van Santvoort HC, Besselink MG, Bollen TL, Buskens E, van Ramshorst B, Gooszen HG; Dutch Acute Pancreatitis Study Group. Case-matched comparison of the retroperitoneal approach with laparotomy for necrotizing pancreatitis. World J Surg 2007; 31:1635–1642.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Thompson CC, Kumar N, Slattery J, et al. A standardized method for endoscopic necrosectomy improves complication and mortality rates. Pancreatology 2016; 16:66–72.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
- Peery AF, Crockett SD, Barritt AS, et al. Burden of gastrointestinal, liver, and pancreatic disease in the United States. Gastroenterology 2015; 149:1731–1741e3.
- Tenner S, Baillie J, DeWitt J, Vege SS; American College of Gastroenterology. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol 2013; 108:1400–1416.
- Bradley EL 3rd. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993; 128:586–590.
- Banks PA, Bollen TL, Dervenis C, et al; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62:102–111.
- Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 1995; 23:1638–1652.
- Kadiyala V, Suleiman SL, McNabb-Baltar J, Wu BU, Banks PA, Singh VK. The Atlanta classification, revised Atlanta classification, and determinant-based classification of acute pancreatitis: which is best at stratifying outcomes? Pancreas 2016; 45:510–515.
- Singh VK, Bollen TL, Wu BU, et al. An assessment of the severity of interstitial pancreatitis. Clin Gastroenterol Hepatol 2011; 9:1098–1103.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Mortele KJ, Wiesner W, Intriere L, et al. A modified CT severity index for evaluating acute pancreatitis: improved correlation with patient outcome. AJR Am J Roentgenol 2004; 183:1261–1265.
- Verde F, Fishman EK, Johnson PT. Arterial pseudoaneurysms complicating pancreatitis: literature review. J Comput Assist Tomogr 2015; 39:7–12.
- Shyu JY, Sainani NI, Sahni VA, et al. Necrotizing pancreatitis: diagnosis, imaging, and intervention. Radiographics 2014; 34:1218–1239.
- Thoeni RF. The revised Atlanta classification of acute pancreatitis: its importance for the radiologist and its effect on treatment. Radiology 2012; 262:751–764.
- Morgan DE, Ragheb CM, Lockhart ME, Cary B, Fineberg NS, Berland LL. Acute pancreatitis: computed tomography utilization and radiation exposure are related to severity but not patient age. Clin Gastroenterol Hepatol 2010; 8:303–308.
- Vitellas KM, Paulson EK, Enns RA, Keogan MT, Pappas TN. Pancreatitis complicated by gland necrosis: evolution of findings on contrast-enhanced CT. J Comput Assist Tomogr 1999; 23:898–905.
- Stimac D, Miletic D, Radic M, et al. The role of nonenhanced magnetic resonance imaging in the early assessment of acute pancreatitis. Am J Gastroenterol 2007; 102:997–1004.
- Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect (Larchmt) 2010; 11:79–109.
- Petrov MS, Kukosh MV, Emelyanov NV. A randomized controlled trial of enteral versus parenteral feeding in patients with predicted severe acute pancreatitis shows a significant reduction in mortality and in infected pancreatic complications with total enteral nutrition. Dig Surg 2006; 23:336–345.
- Petrov MS, Shanbhag S, Chakraborty M, Phillips AR, Windsor JA. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 2010; 139:813–820.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006; 4:CD002941.
- Baril NB, Ralls PW, Wren SM, et al. Does an infected peripancreatic fluid collection or abscess mandate operation? Ann Surg 2000; 231:361–367.
- Mouli VP, Sreenivas V, Garg PK. Efficacy of conservative treatment, without necrosectomy, for infected pancreatic necrosis: a systematic review and meta-analysis. Gastroenterology 2013; 144:333–340.e2.
- Kirby JM, Vora P, Midia M, Rawlinson J. Vascular complications of pancreatitis: imaging and intervention. Cardiovasc Intervent Radiol 2008; 31:957–970.
- De Waele JJ, Hoste E, Blot SI, Decruyenaere J, Colardyn F. Intra-abdominal hypertension in patients with severe acute pancreatitis. Crit Care 2005; 9:R452–R457.
- van Brunschot S, Schut AJ, Bouwense SA, et al; Dutch Pancreatitis Study Group. Abdominal compartment syndrome in acute pancreatitis: a systematic review. Pancreas 2014; 43:665–674.
- Bugiantella W, Rondelli F, Boni M, et al. Necrotizing pancreatitis: a review of the interventions. Int J Surg 2016; 28(suppl 1):S163–S171.
- Besselink MG, Verwer TJ, Schoenmaeckers EJ, et al. Timing of surgical intervention in necrotizing pancreatitis. Arch Surg 2007; 142:1194–1201.
- van Santvoort HC, Besselink MG, Horvath KD, et al; Dutch Acute Pancreatis Study Group. Videoscopic assisted retroperitoneal debridement in infected necrotizing pancreatitis. HPB (Oxford) 2007; 9:156–159.
- van Santvoort HC, Besselink MG, Bollen TL, Buskens E, van Ramshorst B, Gooszen HG; Dutch Acute Pancreatitis Study Group. Case-matched comparison of the retroperitoneal approach with laparotomy for necrotizing pancreatitis. World J Surg 2007; 31:1635–1642.
- van Santvoort HC, Besselink MG, Bakker OJ, et al; Dutch Pancreatitis Study Group. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med 2010; 362:1491–1502.
- Thompson CC, Kumar N, Slattery J, et al. A standardized method for endoscopic necrosectomy improves complication and mortality rates. Pancreatology 2016; 16:66–72.
- Bakker OJ, van Santvoort HC, van Brunschot S, et al; Dutch Pancreatitis Study Group. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: a randomized trial. JAMA 2012; 307:1053–1061.
KEY POINTS
- Selective and appropriate timing of radiologic imaging is vital in managing necrotizing pancreatitis. Protocols are valuable tools.
- While the primary indication for debridement and drainage in necrotizing pancreatitis is infection, other indications are symptomatic walled-off pancreatic necrosis, intractable abdominal pain, bowel obstruction, and failure to thrive.
- Open surgical necrosectomy remains an important treatment for infected pancreatic necrosis or intractable symptoms.
- A “step-up” approach starting with a minimally invasive procedure and escalating if the initial intervention is unsuccessful is gradually becoming the standard of care.
Total pancreatectomy and islet cell autotransplantation: Definitive treatment for chronic pancreatitis
For some patients with chronic pancreatitis, the best option is to remove the entire pancreas. This does not necessarily doom the patient to diabetes mellitus, because we can harvest the islet cells and reinsert them so that, lodged in the liver, they can continue making insulin. However, this approach is underemphasized in the general medical literature and is likely underutilized in the United States.
Here, we discuss chronic pancreatitis, the indications for and contraindications to this procedure, its outcomes, and the management of patients who undergo it.
CHRONIC PANCREATITIS IS PROGRESSIVE AND PAINFUL
Chronic pancreatitis is a progressive condition characterized by chronic inflammation, irreversible fibrosis, and scarring, resulting in loss of both exocrine and endocrine tissue.
According to a National Institutes of Health database, pancreatitis is the seventh most common digestive disease diagnosis on hospitalization, with annual healthcare costs exceeding $3 billion.1 Although data are scarce, by some estimates the incidence of chronic pancreatitis ranges from 4 to 14 per 100,000 person-years, and the prevalence ranges from 26.4 to 52 per 100,000.2–4 Moreover, a meta-analysis5 found that acute pancreatitis progresses to chronic pancreatitis in 10% of patients who have a first episode of acute pancreatitis and in 36% who have recurrent episodes.
Historically, alcoholism was and still is the most common cause of chronic pancreatitis, contributing to 60% to 90% of cases in Western countries.6,7 However, cases due to nonalcoholic causes have been increasing, and in more than one-fourth of patients, no identifiable cause is found.6,8 Smoking is an independent risk factor.6,8,9 Some cases can be linked to genetic abnormalities, particularly in children.10
The clinical manifestations of chronic pancreatitis include exocrine pancreatic insufficiency (leading to malnutrition and steatorrhea), endocrine insufficiency (causing diabetes mellitus), and intractable pain.11 Pain is the predominant clinical symptom early in the disease and is often debilitating and difficult to manage. Uncontrolled pain has a devastating impact on quality of life and may become complicated by narcotic dependence.
The pain of chronic pancreatitis is often multifactorial, with mechanisms that include increased intraductal pressure from obstruction of the pancreatic duct, pancreatic ischemia, neuronal injury, and neuroimmune interactions between neuronal processes and chronic inflammation.12
Treatment: Medical and surgical
In chronic pancreatitis, the aim of treatment is to alleviate deficiencies of exocrine and endocrine function and mitigate the pain. Patients who smoke or drink alcohol should be strongly encouraged to quit.
Loss of exocrine function is mainly managed with oral pancreatic enzyme supplements, and diabetes control is often attained with insulin therapy.13 Besides helping digestion, pancreatic enzyme therapy in the form of nonenteric tablets may also reduce pain and pancreatitis attacks.14 The mechanism may be by degrading cholecystokinin-releasing factor in the duodenum, lowering cholecystokinin levels and thereby reducing pain.12
Nonnarcotic analgesics are often the first line of therapy for pain management, but many patients need narcotic analgesics. Along with narcotics, adjunctive agents such as tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors, and gabapentinoids have been used to treat chronic pancreatitis pain, but with limited success.15
In patients for whom medical pain management has failed, one can consider another option, such as nerve block, neurolysis, or endoscopic or surgical therapy. Neuromodulators are often prescribed by pain clinics.15 Percutaneous and endoscopic celiac ganglion blocks can be an option but rarely achieve substantial or permanent pain relief, and the induced transient responses (on average 2 to 4 months) often cannot be repeated.14–17
Surgical options to relieve pain try to preserve pancreatic function and vary depending on the degree of severity and nature of pancreatic damage. In broad terms, the surgical procedures can be divided into two types:
- Drainage procedures (eg, pseudocyst drainage; minimally invasive endoscopic duct drainage via sphincterotomy or stent placement, or both; pancreaticojejunostomy)
- Resectional procedures (eg, distal pancreatectomy, isolated head resection, pancreaticoduodenectomy, Whipple procedure, total pancreatectomy).
In carefully selected patients, total pancreatectomy can be offered to remove the cause of the pain.18 This procedure is most often performed in patients who have small-duct disease or a genetic cause or for whom other surgical procedures have failed.11
HISTORY OF THE PROCEDURE
Islet cell transplantation grew out of visionary work by Paul Lacy and David Scharp at the University of Washington at Seattle, whose research focused on isolating and transplanting islet cells in rodent models. The topic has been reviewed by Jahansouz et al.19 In the 1970s, experiments in pancreatectomized dogs showed that transplanting unpurified pancreatic islet tissue that was dispersed by collagenase digestion into the spleen or portal vein could prevent diabetes.20,21 In 1974, the first human trials of transplanting islet cells were conducted, using isolated islets from cadaveric donors to treat diabetes.19
In the past, pancreatectomy was performed to treat painful chronic pancreatitis, but it was viewed as undesirable because removing the gland would inevitably cause insulin-dependent diabetes.22 That changed in 1977 at the University of Minnesota, with the first reported islet cell autotransplant after pancreatectomy. The patient remained pain-free and insulin-independent long-term.23 This seminal case showed that endocrine function could be preserved by autotransplant of islets prepared from the excised pancreas.24
In 1992, Pyzdrowski et al25 reported that intrahepatic transplant of as few as 265,000 islets was enough to prevent the need for insulin therapy. Since this technique was first described, there have been many advances, and now more than 30 centers worldwide do it.
PRIMARY INDICATION: INTRACTABLE PAIN
Interest has been growing in using total pancreatectomy and islet autotransplant to treat recurrent acute pancreatitis, chronic pancreatitis, and hereditary pancreatitis. The rationale is that removing the offending tissue eliminates pancreatitis, pain, and cancer risk, while preserving and replacing the islet cells prevents the development of brittle diabetes with loss of insulin and glucagon.26
Proposed criteria for total pancreatectomy and islet autotransplant
Bellin et al14 proposed five criteria for patient selection for this procedure based on imaging studies, pancreatic function tests, and histopathology to detect pancreatic fibrosis. Patients must fulfill all five of the following criteria:
Criterion 1. Diagnosis of chronic pancreatitis, based on chronic abdominal pain lasting more than 6 months with either at least one of the following:
- Pancreatic calcifications on computed tomography
- At least two of the following: four or more of nine criteria on endoscopic ultrasonography described by Catalano et al,27 a compatible ductal or parenchymal abnormality on secretin magnetic resonance cholangiopancreatography; abnormal endoscopic pancreatic function test (peak HCO2 ≤ 80 mmol/L)
- Histopathologically confirmed diagnosis of chronic pancreatitis
- Compatible clinical history and documented hereditary pancreatitis (PRSS1 gene mutation)
OR
- History of recurrent acute pancreatitis (more than one episode of characteristic pain associated with imaging diagnostic of acute pancreatitis or elevated serum amylase or lipase > 3 times the upper limit of normal).
Criterion 2. At least one of the following:
- Daily narcotic dependence
- Pain resulting in impaired quality of life, which may include inability to attend school, recurrent hospitalizations, or inability to participate in usual age-appropriate activities.
Criterion 3. Complete evaluation with no reversible cause of pancreatitis present or untreated.
Criterion 4. Failure to respond to maximal medical and endoscopic therapy.
Criterion 5. Adequate islet cell function (nondiabetic or C-peptide-positive). Patients with C-peptide-negative diabetes meeting criteria 1 to 4 are candidates for total pancreatectomy alone.
The primary goal is to treat intractable pain and improve quality of life in selected patients with chronic pancreatitis or recurrent acute pancreatitis when endoscopic and prior surgical therapies have failed, and whose impairment due to pain is substantial enough to accept the risk of postoperative insulin-dependent diabetes and lifelong commitment to pancreatic enzyme replacement therapy.15,26 Patients with a known genetic cause of chronic pancreatitis should be offered special consideration for the procedure, as their disease is unlikely to remit.
CONTRAINDICATIONS
Total pancreatectomy and islet autotransplant should not be performed in patients with active alcoholism, illicit drug use, or untreated or poorly controlled psychiatric illnesses that could impair the patient’s ability to adhere to a complicated postoperative medical regimen.
A poor support network may be a relative contraindication in view of the cost and complexity of diabetic and pancreatic enzyme replacement therapy.18,26

Islet cell autotransplant is contraindicated in patients with conditions such as C-peptide-negative or type 1 diabetes or a history of portal vein thrombosis, portal hypertension, significant liver disease, high-risk cardiopulmonary disease, or pancreatic cancer (Table 1).26
WHEN TO CONSIDER REFERRAL FOR THIS PROCEDURE
The choice of total pancreatectomy and islet autotransplant vs conventional surgery must be individualized on the basis of each patient’s anatomy, comorbidities, symptom burden, presence or degree of diabetes, and rate of disease progression. The most important factors to consider in determining the need for and timing of this procedure are the patient’s pain, narcotic requirements, and impaired ability to function.26
Sooner rather than later?
An argument can be made for performing this procedure sooner in the course of the disease rather than later when all else has failed. First, prolonged pain can result in central sensitization, in which the threshold for perceiving pain is lowered by damage to the nociceptive neurons from repeated stimulation and inflammation.28
Further, prolonged opioid therapy can lead to opioid-induced hyperalgesia, which may also render patients more sensitive to pain and aggravate their preexisting pain.26,28
In addition, although operative drainage procedures and partial resections are often considered the gold standard for chronic pancreatitis management, patients who undergo partial pancreatectomy or lateral pancreaticojejunostomy (Puestow procedure) have fewer islet cells left to harvest (about 50% fewer) if they subsequently undergo total pancreatectomy and islet cell autotransplant.22,26
Therefore, performing this procedure earlier may help the patient avoid chronic pain syndromes and complications of chronic opioid use, including hyperalgesia, and give the best chance of harvesting enough islet cells to prevent or minimize diabetes afterward.11
REMOVING THE PANCREAS, RETURNING THE ISLET CELLS
During this procedure, the blood supply to the pancreas must be preserved until just before its removal to minimize warm ischemia of the islet cells.18,29 Although there are several surgical variations, a pylorus-preserving total pancreatectomy with duodenectomy is typically performed, usually with splenectomy to preserve perfusion to the body and tail.30
The resected pancreas is taken to the islet isolation laboratory. There, the pancreatic duct is cannulated to fill the organ with a cold collagenase solution, followed by gentle mechanical dispersion using the semiautomated Ricordi method,31 which separates the islet cells from the exocrine tissue.32
The number of islet cells is quantified as islet equivalents; 1 islet equivalent is equal to the volume of an islet with a diameter of 150 µm. Islet equivalents per kilogram of body weight is the unit commonly used to report the graft amount transplanted.33
After digestion, the islet cells can be purified or partially purified by a gradient separation method using a Cobe 2991 cell processor (Terumo Corporation, Tokyo, Japan),34 or can be transplanted as an unpurified preparation. In islet cell autotransplant for chronic pancreatitis, purification is not always necessary due to the small tissue volume extracted from the often atrophic and fibrotic pancreas.32 The decision to purify depends on the postdigest tissue volume; usually, a tissue volume greater than 0.25 mL/kg body weight is an indication to at least partially purify.18,35
The final preparation is returned to the operating room, and after heparin is given, the islets are infused into the portal system using a stump of the splenic vein, or alternatively through direct puncture of the portal vein or cannulation of the umbilical vein.32,36 If the portal vein pressure reaches 25 cm H2O, the infusion is stopped and the remaining islets can be placed in the peritoneal cavity or elsewhere.18 Transplant of the islets into the liver or peritoneum allows the islets to secrete insulin into the hepatic portal circulation, which is the route used by the native pancreas.22
CONTROLLING GLUCOSE DURING AND AFTER THE PROCEDURE
Animal studies have shown that hyperglycemia impairs islet revascularization,37 and glucose toxicity may cause dysfunction and structural lesions of the transplanted islets.11,38
Therefore, during and after the procedure, most centers maintain euglycemia by an intravenous insulin infusion and subsequently move to subcutaneous insulin when the patient starts eating again. Some centers continue insulin at discharge and gradually taper it over months, even in patients who can possibly achieve euglycemia without it.
OUTCOMES
Many institutions have reported their clinical outcomes in terms of pain relief, islet function, glycemic control, and improvement of quality of life. The largest series have been from the University of Minnesota, Leicester General Hospital, the University of Cincinnati, and the Medical University of South Carolina.
Insulin independence is common but wanes with time
The ability to achieve insulin independence after islet autotransplant appears to be related to the number of islets transplanted, with the best results when more than 2,000 or 3,000 islet equivalents/kg are transplanted.39,40
Sutherland et al18 reported that of 409 patients who underwent islet cell autotransplant at the University of Minnesota (the largest series reported to date), 30% were insulin-independent at 3 years, 33% had partial graft function (defined by positive C-peptide), and 82% achieved a mean hemoglobin A1c of less than 7%. However, in the subset who received more than 5,000 islet equivalents/kg, nearly three-fourths of patients were insulin-independent at 3 years.
The Leicester General Hospital group presented long-term data on 46 patients who underwent total pancreatectomy and islet cell autotransplant. Twelve of the 46 had shown periods of insulin independence for a median of 16.5 months, and 5 remained insulin-free at the time of the publication.41 Over the 10 years of follow-up, insulin requirements and hemoglobin A1c increased notably. However, all of the patients tested C-peptide-positive, suggesting long-lasting graft function.
Most recently, the University of Cincinnati group reported long-term data on 116 patients. The insulin independence rate was 38% at 1 year, decreasing to 27% at 5 years. The number of patients with partial graft function was 38% at 1 year and 35% at 5 years.42
Thus, all three institutions confirmed that the autotransplanted islets continue to secrete insulin long-term, but that function decreases over time.
Pancreatectomy reduces pain
Multiple studies have shown that total pancreatectomy reduces pain in patients with chronic pancreatitis. Ahmad et al43 reported a marked reduction in narcotic use (mean morphine equivalents 206 mg/day before surgery, compared with 90 mg after), and a 58% reduction in pain as demonstrated by narcotic independence.
In the University of Minnesota series, 85% of the 409 patients had less pain at 2 years, and 59% were able to stop taking narcotics.18
The University of Cincinnati group reported a narcotic independence rate of 55% at 1 year, which continued to improve to 73% at 5 years.42
Although the source of pain is removed, pain persists or recurs in 10% to 20% of patients after total pancreatectomy and islet cell autotransplant, showing that the pathogenesis of pain is complex, and some uncertainty exists about it.26
Quality of life
Reports evaluating health-related quality of life after total pancreatectomy and islet autotransplant are limited.
The University of Cincinnati group reported the long-term outcomes of quality of life as measured by the Short Form 36 Health Survey.42 Ninety-two percent of patients reported overall improvement in their health at 1 year, and 85% continued to report improved health more than 5 years after the surgery.
In a series of 20 patients, 79% to 90% reported improvements in the seven various domains of the Pain Disability Index. In addition, 60% showed improvement in depression and 70% showed improvement in anxiety. The greatest improvements were in those who had not undergone prior pancreatic surgery, who were younger, and in those with higher levels of preoperative pain.30
Similarly, in a series of 74 patients, the Medical University of South Carolina group reported significant improvement in physical and mental health components of the Short Form 12 Health Survey and an associated decrease in daily narcotic requirements. Moreover, the need to start or increase the dose of insulin after the surgery was not associated with a lower quality of life.44
OFF-SITE ISLET CELL ISOLATION
Despite the positive outcomes in terms of pain relief and insulin independence in many patients after total pancreatectomy and islet cell autotransplant, few medical centers have an on-site islet-processing facility. Since the mid-1990s, a few centers have been able to circumvent this limitation by working with off-site islet cell isolation laboratories.45,46
The University of California, Los Angeles, first reported on a series of nine patients who received autologous islet cells after near-total or total pancreatectomy using a remote islet cell isolation facility, with results comparable to those of other large institutions.45
Similarly, the procedure has been performed at Cleveland Clinic since 2007 with the collaboration of an off-site islet cell isolation laboratory at the University of Pittsburgh. A cohort study from this collaboration published in 2015 showed that in 36 patients (mean follow-up 28 months, range 3–26 months), 33% were insulin-independent, with a C-peptide-positive rate of 70%. This is the largest cohort to date from a center utilizing an off-site islet isolation facility.47
In view of the positive outcomes at these centers, lack of a local islet-processing facility should no longer be a barrier to total pancreatectomy and islet cell autotransplant.
PATIENT CARE AFTER THE PROCEDURE
A multidisciplinary team is an essential component of the postoperative management of patients who undergo total pancreatectomy and islet cell autotransplant.
For patients who had been receiving narcotics for a long time before surgery or who were receiving frequent doses, an experienced pain management physician should be involved in the patient’s postoperative care.
Because islet function can wane over time, testing for diabetes should be done at least annually for the rest of the patient’s life and should include fasting plasma glucose, hemoglobin A1c, and C-peptide, along with self-monitored blood glucose.26
All patients who have surgically induced exocrine insufficiency are at risk of malabsorption and fat-soluble vitamin deficiencies.48 Hence, lifelong pancreatic enzyme replacement therapy is mandatory. Nutritional monitoring should include assessment of steatorrhea, body composition, and fat-soluble vitamin levels (vitamins A, D, and E) at least every year.26 Patients with chronic pancreatitis are at increased risk for low bone density from malabsorption of vitamin D and calcium; therefore, it is recommended that a dual-energy x-ray absorptiometry bone density scan be obtained.26,49
Patients who undergo splenectomy as part of their procedure will require appropriate precautions and ongoing vaccinations as recommended by the US Centers for Disease Control and Prevention.26,50,51
WHAT TO EXPECT FOR THE FUTURE
The National Institute of Diabetes and Digestive and Kidney Diseases has reviewed the potential future research directions for total pancreatectomy and islet autotransplant.15
Patient selection remains challenging despite the availability of criteria15 and guidelines.26 More research is needed to better assess preoperative beta-cell function and to predict postoperative outcomes. Mixed meal-tolerance testing is adopted by most clinical centers to predict posttransplant beta-cell function. The use of arginine instead of glucagon in a stimulation test for insulin and C-peptide response has been validated and may allow more accurate assessment.52,53
Another targeted area of research is the advancement of safety and metabolic outcomes. Techniques to minimize warm ischemic time and complications are being evaluated. Islet isolation methods that yield more islets, reduce beta-cell apoptosis, and can isolate islets from glands with malignancy should be further investigated.54 Further, enhanced islet infusion methods that achieve lower portal venous pressures and minimize portal vein thrombosis are needed.
Unfortunately, the function of transplanted islet grafts declines over time. This phenomenon is at least partially attributed to the immediate blood-mediated inflammatory response,55,56 along with islet hypoxia,57 leading to islet apoptosis. Research on different strategies is expanding our knowledge in islet engraftment and posttransplant beta-cell apoptosis, with the expectation that the transplanted islet lifespan will increase. Alternative transplant sites with low inflammatory reaction, such as the omental pouch,58 muscle,59 and bone marrow,60 have shown encouraging data. Other approaches, such as adjuvant anti-inflammatory agents and heparinization, have been proposed.15
Research into complications is also of clinical importance. There is growing attention to hypoglycemia unrelated to exogenous insulin use in posttransplant patients. One hypothesis is that glucagon secretion, a counterregulatory response to hypoglycemia, is defective if the islet cells are transplanted into the liver, and that implanting them into another site may avoid this effect.61
- Everhart JE. Pancreatitis. In: Everhart JE, editor. The Burden of Digestive Diseases in the United States. US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of
- Diabetes and Digestive and Kidney Diseases. Washington, DC: US Government Printing Office; 2008. www.niddk.nih.gov/about-niddk/strategic-plans-reports/Pages/burden-digestive-diseases-in-united-states-report.aspx. Accessed May 10, 2016.
- Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol 2011; 106:2192–2199.
- Lévy P, Barthet M, Mollard BR, Amouretti M, Marion-Audibert AM, Dyard F. Estimation of the prevalence and incidence of chronic pancreatitis and its complications. Gastroenterol Clin Biol 2006; 30:838–844.
- Hirota M, Shimosegawa T, Masamune A, et al; Research Committee of Intractable Pancreatic Diseases. The seventh nationwide epidemiological survey for chronic pancreatitis in Japan: clinical significance of smoking habit in Japanese patients. Pancreatology 2014; 14:490–496.
- Sankaran SJ, Xiao AY, Wu LM, Windsor JA, Forsmark CE, Petrov MS. Frequency of progression from acute to chronic pancreatitis and risk factors: a meta-analysis. Gastroenterology 2015; 149:1490–1500.e1.
- Coté GA, Yadav D, Slivka A, et al; North American Pancreatitis Study Group. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:266–273.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon 2014; 60:530–550.
- Frulloni L, Gabbrielli A, Pezzilli R, et al; PanCroInfAISP Study Group. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig Liver Dis 2009; 41:311–317.
- Talamini G, Bassi C, Falconi M, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci 1999; 44:1303–1311.
- Schwarzenberg SJ, Bellin M, Husain SZ, et al. Pediatric chronic pancreatitis is associated with genetic risk factors and substantial disease burden. J Pediatr 2015; 166:890–896.e1.
- Blondet JJ, Carlson AM, Kobayashi T, et al. The role of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Surg Clin North Am 2007; 87:1477–1501.
- Lieb JG 2nd, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther 2009; 29:706–719.
- Lin YK, Johnston PC, Arce K, Hatipoglu BA. Chronic pancreatitis and diabetes mellitus. Curr Treat Options Gastroenterol 2015; 13:319–331.
- Bellin MD, Gelrud A, Arreaza-Rubin G, et al. Total pancreatectomy with islet autotransplantation: summary of a National Institute of Diabetes and Digestive and Kidney diseases workshop. Pancreas 2014; 43:1163–1171.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part II: diagnosis, complications, and management. Dis Mon 2015; 61:5–37.
- Warshaw AL, Banks PA, Fernández-Del Castillo C. AGA technical review: treatment of pain in chronic pancreatitis. Gastroenterology 1998; 115:765–776.
- Chauhan S, Forsmark CE. Pain management in chronic pancreatitis: a treatment algorithm. Best Pract Res Clin Gastroenterol 2010; 24:323–335.
- Sutherland DE, Radosevich DM, Bellin MD, et al. Total pancreatectomy and islet autotransplantation for chronic pancreatitis. J Am Coll Surg 2012; 214:409–426.
- Jahansouz C, Jahansouz C, Kumer SC, Brayman KL. Evolution of beta-cell replacement therapy in diabetes mellitus: islet cell transplantation. J Transplant 2011; 2011:247959.
- Kretschmer GJ, Sutherland DE, Matas AJ, Cain TL, Najarian JS. Autotransplantation of pancreatic islets without separation of exocrine and endocrine tissue in totally pancreatectomized dogs. Surgery 1977; 82:74–81.
- Kretschmer GJ, Sutherland DR, Matas AJ, Payne WD, Najarian JS. Autotransplantation of pancreatic fragments to the portal vein and spleen of totally pancreatectomized dogs: a comparative evaluation. Ann Surg 1978; 187:79–86.
- Bellin MD, Sutherland DE, Robertson RP. Pancreatectomy and autologous islet transplantation for painful chronic pancreatitis: indications and outcomes. Hosp Pract (1995) 2012; 40:80–87.
- Najarian JS, Sutherland DE, Baumgartner D, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis. Ann Surg 1980; 192:526–542.
- Sutherland DE, Matas AJ, Najarian JS. Pancreatic islet cell transplantation. Surg Clin North Am 1978; 58:365–382.
- Pyzdrowski KL, Kendall DM, Halter JB, Nakhleh RE, Sutherland DE, Robertson RP. Preserved insulin secretion and insulin independence in recipients of islet autografts. N Engl J Med 1992; 327:220–226.
- Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet autotransplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology 2014; 14:27–35.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 2006; 104:570–587.
- Bramis K, Gordon-Weeks AN, Friend PJ, et al. Systematic review of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Br J Surg 2012; 99:761–766.
- Walsh RM, Saavedra JR, Lentz G, et al. Improved quality of life following total pancreatectomy and auto-islet transplantation for chronic pancreatitis. J Gastrointest Surg 2012; 16:1469–1477.
- Ricordi C, Lacy PE, Scharp DW. Automated islet isolation from human pancreas. Diabetes 1989; 38(suppl 1):140–142.
- Witkowski P, Savari O, Matthews JB. Islet autotransplantation and total pancreatectomy. Adv Surg 2014; 48:223–233.
- Bellin MD, Beilman GJ, Dunn TB, et al. Islet autotransplantation to preserve beta cell mass in selected patients with chronic pancreatitis and diabetes mellitus undergoing total pancreatectomy. Pancreas 2013; 42:317–321.
- Anazawa T, Matsumoto S, Yonekawa Y, et al. Prediction of pancreatic tissue densities by an analytical test gradient system before purification maximizes human islet recovery for islet autotransplantation/allotransplantation. Transplantation 2011; 91:508–514.
- Lake SP, Bassett PD, Larkins A, et al. Large-scale purification of human islets utilizing discontinuous albumin gradient on IBM 2991 cell separator. Diabetes 1989; 38(suppl 1):143–145.
- Bellin MD, Freeman ML, Schwarzenberg SJ, et al. Quality of life improves for pediatric patients after total pancreatectomy and islet autotransplant for chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:793–799.
- Andersson A, Korsgren O, Jansson L. Intraportally transplanted pancreatic islets revascularized from hepatic arterial system. Diabetes 1989; 38(suppl 1):192–195.
- Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 1992; 15:442–455.
- Bellin MD, Carlson AM, Kobayashi T, et al. Outcome after pancreatectomy and islet autotransplantation in a pediatric population. J Pediatr Gastroenterol Nutr 2008; 47:37–44.
- White SA, Davies JE, Pollard C, et al. Pancreas resection and islet autotransplantation for end-stage chronic pancreatitis. Ann Surg 2001; 233:423–431.
- Webb MA, Illouz SC, Pollard CA, et al. Islet auto transplantation following total pancreatectomy: a long-term assessment of graft function. Pancreas 2008; 37:282–287.
- Wilson GC, Sutton JM, Abbott DE, et al. Long-term outcomes after total pancreatectomy and islet cell autotransplantation: is it a durable operation? Ann Surg 2014; 260:659–667.
- Ahmad SA, Lowy AM, Wray CJ, et al. Factors associated with insulin and narcotic independence after islet autotransplantation in patients with severe chronic pancreatitis. J Am Coll Surg 2005; 201:680–687.
- Dorlon M, Owczarski S, Wang H, Adams D, Morgan K. Increase in postoperative insulin requirements does not lead to decreased quality of life after total pancreatectomy with islet cell autotransplantation for chronic pancreatitis. Am Surg 2013; 79:676–680.
- Tai DS, Shen N, Szot GL, et al. Autologous islet transplantation with remote islet isolation after pancreas resection for chronic pancreatitis. JAMA Surg 2015; 150:118–124.
- Rabkin JM, Olyaei AJ, Orloff SL, et al. Distant processing of pancreas islets for autotransplantation following total pancreatectomy. Am J Surg 1999; 177:423–427.
- Johnston PC, Lin YK, Walsh RM, et al. Factors associated with islet yield and insulin independence after total pancreatectomy and islet cell autotransplantation in patients with chronic pancreatitis utilizing off-site islet isolation: Cleveland Clinic experience. J Clin Endocrinol Metab 2015; 100:1765–1770.
- Dresler CM, Fortner JG, McDermott K, Bajorunas DR. Metabolic consequences of (regional) total pancreatectomy. Ann Surg 1991; 214:131–140.
- Duggan SN, O’Sullivan M, Hamilton S, Feehan SM, Ridgway PF, Conlon KC. Patients with chronic pancreatitis are at increased risk for osteoporosis. Pancreas 2012; 41:1119–1124.
- Rubin LG, Levin MJ, Ljungman P, et al; Infectious Diseases Society of America. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:e44–e100.
- Di Sabatino A, Carsetti R, Corazza GR. Post-splenectomy and hyposplenic states. Lancet 2011; 378:86–97.
- Robertson RP, Raymond RH, Lee DS, et al; Beta Cell Project Team of the Foundation for the NIH Biomarkers Consortium. Arginine is preferred to glucagon for stimulation testing of beta-cell function. Am J Physiol Endocrinol Metab 2014; 307:E720–E727.
- Robertson RP, Bogachus LD, Oseid E, et al. Assessment of beta-cell mass and alpha- and beta-cell survival and function by arginine stimulation in human autologous islet recipients. Diabetes 2015; 64:565–572.
- Balzano G, Piemonti L. Autologous islet transplantation in patients requiring pancreatectomy for neoplasm. Curr Diab Rep 2014; 14:512.
- Naziruddin B, Iwahashi S, Kanak MA, Takita M, Itoh T, Levy MF. Evidence for instant blood-mediated inflammatory reaction in clinical autologous islet transplantation. Am J Transplant 2014; 14:428–437.
- Abdelli S, Ansite J, Roduit R, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 2004; 53:2815–2823.
- Olsson R, Olerud J, Pettersson U, Carlsson PO. Increased numbers of low-oxygenated pancreatic islets after intraportal islet transplantation. Diabetes 2011; 60:2350–2353.
- Berman DM, O’Neil JJ, Coffey LC, et al. Long-term survival of nonhuman primate islets implanted in an omental pouch on a biodegradable scaffold. Am J Transplant 2009; 9:91–104.
- Sterkers A, Hubert T, Gmyr V, et al. Islet survival and function following intramuscular autotransplantation in the minipig. Am J Transplant 2013; 13:891–898.
- Maffi P, Balzano G, Ponzoni M, et al. Autologous pancreatic islet transplantation in human bone marrow. Diabetes 2013; 62:3523–3531.
- Bellin MD, Parazzoli S, Oseid E, et al. Defective glucagon secretion during hypoglycemia after intrahepatic but not nonhepatic islet autotransplantation. Am J Transplant 2014; 14:1880–1886.
For some patients with chronic pancreatitis, the best option is to remove the entire pancreas. This does not necessarily doom the patient to diabetes mellitus, because we can harvest the islet cells and reinsert them so that, lodged in the liver, they can continue making insulin. However, this approach is underemphasized in the general medical literature and is likely underutilized in the United States.
Here, we discuss chronic pancreatitis, the indications for and contraindications to this procedure, its outcomes, and the management of patients who undergo it.
CHRONIC PANCREATITIS IS PROGRESSIVE AND PAINFUL
Chronic pancreatitis is a progressive condition characterized by chronic inflammation, irreversible fibrosis, and scarring, resulting in loss of both exocrine and endocrine tissue.
According to a National Institutes of Health database, pancreatitis is the seventh most common digestive disease diagnosis on hospitalization, with annual healthcare costs exceeding $3 billion.1 Although data are scarce, by some estimates the incidence of chronic pancreatitis ranges from 4 to 14 per 100,000 person-years, and the prevalence ranges from 26.4 to 52 per 100,000.2–4 Moreover, a meta-analysis5 found that acute pancreatitis progresses to chronic pancreatitis in 10% of patients who have a first episode of acute pancreatitis and in 36% who have recurrent episodes.
Historically, alcoholism was and still is the most common cause of chronic pancreatitis, contributing to 60% to 90% of cases in Western countries.6,7 However, cases due to nonalcoholic causes have been increasing, and in more than one-fourth of patients, no identifiable cause is found.6,8 Smoking is an independent risk factor.6,8,9 Some cases can be linked to genetic abnormalities, particularly in children.10
The clinical manifestations of chronic pancreatitis include exocrine pancreatic insufficiency (leading to malnutrition and steatorrhea), endocrine insufficiency (causing diabetes mellitus), and intractable pain.11 Pain is the predominant clinical symptom early in the disease and is often debilitating and difficult to manage. Uncontrolled pain has a devastating impact on quality of life and may become complicated by narcotic dependence.
The pain of chronic pancreatitis is often multifactorial, with mechanisms that include increased intraductal pressure from obstruction of the pancreatic duct, pancreatic ischemia, neuronal injury, and neuroimmune interactions between neuronal processes and chronic inflammation.12
Treatment: Medical and surgical
In chronic pancreatitis, the aim of treatment is to alleviate deficiencies of exocrine and endocrine function and mitigate the pain. Patients who smoke or drink alcohol should be strongly encouraged to quit.
Loss of exocrine function is mainly managed with oral pancreatic enzyme supplements, and diabetes control is often attained with insulin therapy.13 Besides helping digestion, pancreatic enzyme therapy in the form of nonenteric tablets may also reduce pain and pancreatitis attacks.14 The mechanism may be by degrading cholecystokinin-releasing factor in the duodenum, lowering cholecystokinin levels and thereby reducing pain.12
Nonnarcotic analgesics are often the first line of therapy for pain management, but many patients need narcotic analgesics. Along with narcotics, adjunctive agents such as tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors, and gabapentinoids have been used to treat chronic pancreatitis pain, but with limited success.15
In patients for whom medical pain management has failed, one can consider another option, such as nerve block, neurolysis, or endoscopic or surgical therapy. Neuromodulators are often prescribed by pain clinics.15 Percutaneous and endoscopic celiac ganglion blocks can be an option but rarely achieve substantial or permanent pain relief, and the induced transient responses (on average 2 to 4 months) often cannot be repeated.14–17
Surgical options to relieve pain try to preserve pancreatic function and vary depending on the degree of severity and nature of pancreatic damage. In broad terms, the surgical procedures can be divided into two types:
- Drainage procedures (eg, pseudocyst drainage; minimally invasive endoscopic duct drainage via sphincterotomy or stent placement, or both; pancreaticojejunostomy)
- Resectional procedures (eg, distal pancreatectomy, isolated head resection, pancreaticoduodenectomy, Whipple procedure, total pancreatectomy).
In carefully selected patients, total pancreatectomy can be offered to remove the cause of the pain.18 This procedure is most often performed in patients who have small-duct disease or a genetic cause or for whom other surgical procedures have failed.11
HISTORY OF THE PROCEDURE
Islet cell transplantation grew out of visionary work by Paul Lacy and David Scharp at the University of Washington at Seattle, whose research focused on isolating and transplanting islet cells in rodent models. The topic has been reviewed by Jahansouz et al.19 In the 1970s, experiments in pancreatectomized dogs showed that transplanting unpurified pancreatic islet tissue that was dispersed by collagenase digestion into the spleen or portal vein could prevent diabetes.20,21 In 1974, the first human trials of transplanting islet cells were conducted, using isolated islets from cadaveric donors to treat diabetes.19
In the past, pancreatectomy was performed to treat painful chronic pancreatitis, but it was viewed as undesirable because removing the gland would inevitably cause insulin-dependent diabetes.22 That changed in 1977 at the University of Minnesota, with the first reported islet cell autotransplant after pancreatectomy. The patient remained pain-free and insulin-independent long-term.23 This seminal case showed that endocrine function could be preserved by autotransplant of islets prepared from the excised pancreas.24
In 1992, Pyzdrowski et al25 reported that intrahepatic transplant of as few as 265,000 islets was enough to prevent the need for insulin therapy. Since this technique was first described, there have been many advances, and now more than 30 centers worldwide do it.
PRIMARY INDICATION: INTRACTABLE PAIN
Interest has been growing in using total pancreatectomy and islet autotransplant to treat recurrent acute pancreatitis, chronic pancreatitis, and hereditary pancreatitis. The rationale is that removing the offending tissue eliminates pancreatitis, pain, and cancer risk, while preserving and replacing the islet cells prevents the development of brittle diabetes with loss of insulin and glucagon.26
Proposed criteria for total pancreatectomy and islet autotransplant
Bellin et al14 proposed five criteria for patient selection for this procedure based on imaging studies, pancreatic function tests, and histopathology to detect pancreatic fibrosis. Patients must fulfill all five of the following criteria:
Criterion 1. Diagnosis of chronic pancreatitis, based on chronic abdominal pain lasting more than 6 months with either at least one of the following:
- Pancreatic calcifications on computed tomography
- At least two of the following: four or more of nine criteria on endoscopic ultrasonography described by Catalano et al,27 a compatible ductal or parenchymal abnormality on secretin magnetic resonance cholangiopancreatography; abnormal endoscopic pancreatic function test (peak HCO2 ≤ 80 mmol/L)
- Histopathologically confirmed diagnosis of chronic pancreatitis
- Compatible clinical history and documented hereditary pancreatitis (PRSS1 gene mutation)
OR
- History of recurrent acute pancreatitis (more than one episode of characteristic pain associated with imaging diagnostic of acute pancreatitis or elevated serum amylase or lipase > 3 times the upper limit of normal).
Criterion 2. At least one of the following:
- Daily narcotic dependence
- Pain resulting in impaired quality of life, which may include inability to attend school, recurrent hospitalizations, or inability to participate in usual age-appropriate activities.
Criterion 3. Complete evaluation with no reversible cause of pancreatitis present or untreated.
Criterion 4. Failure to respond to maximal medical and endoscopic therapy.
Criterion 5. Adequate islet cell function (nondiabetic or C-peptide-positive). Patients with C-peptide-negative diabetes meeting criteria 1 to 4 are candidates for total pancreatectomy alone.
The primary goal is to treat intractable pain and improve quality of life in selected patients with chronic pancreatitis or recurrent acute pancreatitis when endoscopic and prior surgical therapies have failed, and whose impairment due to pain is substantial enough to accept the risk of postoperative insulin-dependent diabetes and lifelong commitment to pancreatic enzyme replacement therapy.15,26 Patients with a known genetic cause of chronic pancreatitis should be offered special consideration for the procedure, as their disease is unlikely to remit.
CONTRAINDICATIONS
Total pancreatectomy and islet autotransplant should not be performed in patients with active alcoholism, illicit drug use, or untreated or poorly controlled psychiatric illnesses that could impair the patient’s ability to adhere to a complicated postoperative medical regimen.
A poor support network may be a relative contraindication in view of the cost and complexity of diabetic and pancreatic enzyme replacement therapy.18,26
Islet cell autotransplant is contraindicated in patients with conditions such as C-peptide-negative or type 1 diabetes or a history of portal vein thrombosis, portal hypertension, significant liver disease, high-risk cardiopulmonary disease, or pancreatic cancer (Table 1).26
WHEN TO CONSIDER REFERRAL FOR THIS PROCEDURE
The choice of total pancreatectomy and islet autotransplant vs conventional surgery must be individualized on the basis of each patient’s anatomy, comorbidities, symptom burden, presence or degree of diabetes, and rate of disease progression. The most important factors to consider in determining the need for and timing of this procedure are the patient’s pain, narcotic requirements, and impaired ability to function.26
Sooner rather than later?
An argument can be made for performing this procedure sooner in the course of the disease rather than later when all else has failed. First, prolonged pain can result in central sensitization, in which the threshold for perceiving pain is lowered by damage to the nociceptive neurons from repeated stimulation and inflammation.28
Further, prolonged opioid therapy can lead to opioid-induced hyperalgesia, which may also render patients more sensitive to pain and aggravate their preexisting pain.26,28
In addition, although operative drainage procedures and partial resections are often considered the gold standard for chronic pancreatitis management, patients who undergo partial pancreatectomy or lateral pancreaticojejunostomy (Puestow procedure) have fewer islet cells left to harvest (about 50% fewer) if they subsequently undergo total pancreatectomy and islet cell autotransplant.22,26
Therefore, performing this procedure earlier may help the patient avoid chronic pain syndromes and complications of chronic opioid use, including hyperalgesia, and give the best chance of harvesting enough islet cells to prevent or minimize diabetes afterward.11
REMOVING THE PANCREAS, RETURNING THE ISLET CELLS
During this procedure, the blood supply to the pancreas must be preserved until just before its removal to minimize warm ischemia of the islet cells.18,29 Although there are several surgical variations, a pylorus-preserving total pancreatectomy with duodenectomy is typically performed, usually with splenectomy to preserve perfusion to the body and tail.30
The resected pancreas is taken to the islet isolation laboratory. There, the pancreatic duct is cannulated to fill the organ with a cold collagenase solution, followed by gentle mechanical dispersion using the semiautomated Ricordi method,31 which separates the islet cells from the exocrine tissue.32
The number of islet cells is quantified as islet equivalents; 1 islet equivalent is equal to the volume of an islet with a diameter of 150 µm. Islet equivalents per kilogram of body weight is the unit commonly used to report the graft amount transplanted.33
After digestion, the islet cells can be purified or partially purified by a gradient separation method using a Cobe 2991 cell processor (Terumo Corporation, Tokyo, Japan),34 or can be transplanted as an unpurified preparation. In islet cell autotransplant for chronic pancreatitis, purification is not always necessary due to the small tissue volume extracted from the often atrophic and fibrotic pancreas.32 The decision to purify depends on the postdigest tissue volume; usually, a tissue volume greater than 0.25 mL/kg body weight is an indication to at least partially purify.18,35
The final preparation is returned to the operating room, and after heparin is given, the islets are infused into the portal system using a stump of the splenic vein, or alternatively through direct puncture of the portal vein or cannulation of the umbilical vein.32,36 If the portal vein pressure reaches 25 cm H2O, the infusion is stopped and the remaining islets can be placed in the peritoneal cavity or elsewhere.18 Transplant of the islets into the liver or peritoneum allows the islets to secrete insulin into the hepatic portal circulation, which is the route used by the native pancreas.22
CONTROLLING GLUCOSE DURING AND AFTER THE PROCEDURE
Animal studies have shown that hyperglycemia impairs islet revascularization,37 and glucose toxicity may cause dysfunction and structural lesions of the transplanted islets.11,38
Therefore, during and after the procedure, most centers maintain euglycemia by an intravenous insulin infusion and subsequently move to subcutaneous insulin when the patient starts eating again. Some centers continue insulin at discharge and gradually taper it over months, even in patients who can possibly achieve euglycemia without it.
OUTCOMES
Many institutions have reported their clinical outcomes in terms of pain relief, islet function, glycemic control, and improvement of quality of life. The largest series have been from the University of Minnesota, Leicester General Hospital, the University of Cincinnati, and the Medical University of South Carolina.
Insulin independence is common but wanes with time
The ability to achieve insulin independence after islet autotransplant appears to be related to the number of islets transplanted, with the best results when more than 2,000 or 3,000 islet equivalents/kg are transplanted.39,40
Sutherland et al18 reported that of 409 patients who underwent islet cell autotransplant at the University of Minnesota (the largest series reported to date), 30% were insulin-independent at 3 years, 33% had partial graft function (defined by positive C-peptide), and 82% achieved a mean hemoglobin A1c of less than 7%. However, in the subset who received more than 5,000 islet equivalents/kg, nearly three-fourths of patients were insulin-independent at 3 years.
The Leicester General Hospital group presented long-term data on 46 patients who underwent total pancreatectomy and islet cell autotransplant. Twelve of the 46 had shown periods of insulin independence for a median of 16.5 months, and 5 remained insulin-free at the time of the publication.41 Over the 10 years of follow-up, insulin requirements and hemoglobin A1c increased notably. However, all of the patients tested C-peptide-positive, suggesting long-lasting graft function.
Most recently, the University of Cincinnati group reported long-term data on 116 patients. The insulin independence rate was 38% at 1 year, decreasing to 27% at 5 years. The number of patients with partial graft function was 38% at 1 year and 35% at 5 years.42
Thus, all three institutions confirmed that the autotransplanted islets continue to secrete insulin long-term, but that function decreases over time.
Pancreatectomy reduces pain
Multiple studies have shown that total pancreatectomy reduces pain in patients with chronic pancreatitis. Ahmad et al43 reported a marked reduction in narcotic use (mean morphine equivalents 206 mg/day before surgery, compared with 90 mg after), and a 58% reduction in pain as demonstrated by narcotic independence.
In the University of Minnesota series, 85% of the 409 patients had less pain at 2 years, and 59% were able to stop taking narcotics.18
The University of Cincinnati group reported a narcotic independence rate of 55% at 1 year, which continued to improve to 73% at 5 years.42
Although the source of pain is removed, pain persists or recurs in 10% to 20% of patients after total pancreatectomy and islet cell autotransplant, showing that the pathogenesis of pain is complex, and some uncertainty exists about it.26
Quality of life
Reports evaluating health-related quality of life after total pancreatectomy and islet autotransplant are limited.
The University of Cincinnati group reported the long-term outcomes of quality of life as measured by the Short Form 36 Health Survey.42 Ninety-two percent of patients reported overall improvement in their health at 1 year, and 85% continued to report improved health more than 5 years after the surgery.
In a series of 20 patients, 79% to 90% reported improvements in the seven various domains of the Pain Disability Index. In addition, 60% showed improvement in depression and 70% showed improvement in anxiety. The greatest improvements were in those who had not undergone prior pancreatic surgery, who were younger, and in those with higher levels of preoperative pain.30
Similarly, in a series of 74 patients, the Medical University of South Carolina group reported significant improvement in physical and mental health components of the Short Form 12 Health Survey and an associated decrease in daily narcotic requirements. Moreover, the need to start or increase the dose of insulin after the surgery was not associated with a lower quality of life.44
OFF-SITE ISLET CELL ISOLATION
Despite the positive outcomes in terms of pain relief and insulin independence in many patients after total pancreatectomy and islet cell autotransplant, few medical centers have an on-site islet-processing facility. Since the mid-1990s, a few centers have been able to circumvent this limitation by working with off-site islet cell isolation laboratories.45,46
The University of California, Los Angeles, first reported on a series of nine patients who received autologous islet cells after near-total or total pancreatectomy using a remote islet cell isolation facility, with results comparable to those of other large institutions.45
Similarly, the procedure has been performed at Cleveland Clinic since 2007 with the collaboration of an off-site islet cell isolation laboratory at the University of Pittsburgh. A cohort study from this collaboration published in 2015 showed that in 36 patients (mean follow-up 28 months, range 3–26 months), 33% were insulin-independent, with a C-peptide-positive rate of 70%. This is the largest cohort to date from a center utilizing an off-site islet isolation facility.47
In view of the positive outcomes at these centers, lack of a local islet-processing facility should no longer be a barrier to total pancreatectomy and islet cell autotransplant.
PATIENT CARE AFTER THE PROCEDURE
A multidisciplinary team is an essential component of the postoperative management of patients who undergo total pancreatectomy and islet cell autotransplant.
For patients who had been receiving narcotics for a long time before surgery or who were receiving frequent doses, an experienced pain management physician should be involved in the patient’s postoperative care.
Because islet function can wane over time, testing for diabetes should be done at least annually for the rest of the patient’s life and should include fasting plasma glucose, hemoglobin A1c, and C-peptide, along with self-monitored blood glucose.26
All patients who have surgically induced exocrine insufficiency are at risk of malabsorption and fat-soluble vitamin deficiencies.48 Hence, lifelong pancreatic enzyme replacement therapy is mandatory. Nutritional monitoring should include assessment of steatorrhea, body composition, and fat-soluble vitamin levels (vitamins A, D, and E) at least every year.26 Patients with chronic pancreatitis are at increased risk for low bone density from malabsorption of vitamin D and calcium; therefore, it is recommended that a dual-energy x-ray absorptiometry bone density scan be obtained.26,49
Patients who undergo splenectomy as part of their procedure will require appropriate precautions and ongoing vaccinations as recommended by the US Centers for Disease Control and Prevention.26,50,51
WHAT TO EXPECT FOR THE FUTURE
The National Institute of Diabetes and Digestive and Kidney Diseases has reviewed the potential future research directions for total pancreatectomy and islet autotransplant.15
Patient selection remains challenging despite the availability of criteria15 and guidelines.26 More research is needed to better assess preoperative beta-cell function and to predict postoperative outcomes. Mixed meal-tolerance testing is adopted by most clinical centers to predict posttransplant beta-cell function. The use of arginine instead of glucagon in a stimulation test for insulin and C-peptide response has been validated and may allow more accurate assessment.52,53
Another targeted area of research is the advancement of safety and metabolic outcomes. Techniques to minimize warm ischemic time and complications are being evaluated. Islet isolation methods that yield more islets, reduce beta-cell apoptosis, and can isolate islets from glands with malignancy should be further investigated.54 Further, enhanced islet infusion methods that achieve lower portal venous pressures and minimize portal vein thrombosis are needed.
Unfortunately, the function of transplanted islet grafts declines over time. This phenomenon is at least partially attributed to the immediate blood-mediated inflammatory response,55,56 along with islet hypoxia,57 leading to islet apoptosis. Research on different strategies is expanding our knowledge in islet engraftment and posttransplant beta-cell apoptosis, with the expectation that the transplanted islet lifespan will increase. Alternative transplant sites with low inflammatory reaction, such as the omental pouch,58 muscle,59 and bone marrow,60 have shown encouraging data. Other approaches, such as adjuvant anti-inflammatory agents and heparinization, have been proposed.15
Research into complications is also of clinical importance. There is growing attention to hypoglycemia unrelated to exogenous insulin use in posttransplant patients. One hypothesis is that glucagon secretion, a counterregulatory response to hypoglycemia, is defective if the islet cells are transplanted into the liver, and that implanting them into another site may avoid this effect.61
For some patients with chronic pancreatitis, the best option is to remove the entire pancreas. This does not necessarily doom the patient to diabetes mellitus, because we can harvest the islet cells and reinsert them so that, lodged in the liver, they can continue making insulin. However, this approach is underemphasized in the general medical literature and is likely underutilized in the United States.
Here, we discuss chronic pancreatitis, the indications for and contraindications to this procedure, its outcomes, and the management of patients who undergo it.
CHRONIC PANCREATITIS IS PROGRESSIVE AND PAINFUL
Chronic pancreatitis is a progressive condition characterized by chronic inflammation, irreversible fibrosis, and scarring, resulting in loss of both exocrine and endocrine tissue.
According to a National Institutes of Health database, pancreatitis is the seventh most common digestive disease diagnosis on hospitalization, with annual healthcare costs exceeding $3 billion.1 Although data are scarce, by some estimates the incidence of chronic pancreatitis ranges from 4 to 14 per 100,000 person-years, and the prevalence ranges from 26.4 to 52 per 100,000.2–4 Moreover, a meta-analysis5 found that acute pancreatitis progresses to chronic pancreatitis in 10% of patients who have a first episode of acute pancreatitis and in 36% who have recurrent episodes.
Historically, alcoholism was and still is the most common cause of chronic pancreatitis, contributing to 60% to 90% of cases in Western countries.6,7 However, cases due to nonalcoholic causes have been increasing, and in more than one-fourth of patients, no identifiable cause is found.6,8 Smoking is an independent risk factor.6,8,9 Some cases can be linked to genetic abnormalities, particularly in children.10
The clinical manifestations of chronic pancreatitis include exocrine pancreatic insufficiency (leading to malnutrition and steatorrhea), endocrine insufficiency (causing diabetes mellitus), and intractable pain.11 Pain is the predominant clinical symptom early in the disease and is often debilitating and difficult to manage. Uncontrolled pain has a devastating impact on quality of life and may become complicated by narcotic dependence.
The pain of chronic pancreatitis is often multifactorial, with mechanisms that include increased intraductal pressure from obstruction of the pancreatic duct, pancreatic ischemia, neuronal injury, and neuroimmune interactions between neuronal processes and chronic inflammation.12
Treatment: Medical and surgical
In chronic pancreatitis, the aim of treatment is to alleviate deficiencies of exocrine and endocrine function and mitigate the pain. Patients who smoke or drink alcohol should be strongly encouraged to quit.
Loss of exocrine function is mainly managed with oral pancreatic enzyme supplements, and diabetes control is often attained with insulin therapy.13 Besides helping digestion, pancreatic enzyme therapy in the form of nonenteric tablets may also reduce pain and pancreatitis attacks.14 The mechanism may be by degrading cholecystokinin-releasing factor in the duodenum, lowering cholecystokinin levels and thereby reducing pain.12
Nonnarcotic analgesics are often the first line of therapy for pain management, but many patients need narcotic analgesics. Along with narcotics, adjunctive agents such as tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors, and gabapentinoids have been used to treat chronic pancreatitis pain, but with limited success.15
In patients for whom medical pain management has failed, one can consider another option, such as nerve block, neurolysis, or endoscopic or surgical therapy. Neuromodulators are often prescribed by pain clinics.15 Percutaneous and endoscopic celiac ganglion blocks can be an option but rarely achieve substantial or permanent pain relief, and the induced transient responses (on average 2 to 4 months) often cannot be repeated.14–17
Surgical options to relieve pain try to preserve pancreatic function and vary depending on the degree of severity and nature of pancreatic damage. In broad terms, the surgical procedures can be divided into two types:
- Drainage procedures (eg, pseudocyst drainage; minimally invasive endoscopic duct drainage via sphincterotomy or stent placement, or both; pancreaticojejunostomy)
- Resectional procedures (eg, distal pancreatectomy, isolated head resection, pancreaticoduodenectomy, Whipple procedure, total pancreatectomy).
In carefully selected patients, total pancreatectomy can be offered to remove the cause of the pain.18 This procedure is most often performed in patients who have small-duct disease or a genetic cause or for whom other surgical procedures have failed.11
HISTORY OF THE PROCEDURE
Islet cell transplantation grew out of visionary work by Paul Lacy and David Scharp at the University of Washington at Seattle, whose research focused on isolating and transplanting islet cells in rodent models. The topic has been reviewed by Jahansouz et al.19 In the 1970s, experiments in pancreatectomized dogs showed that transplanting unpurified pancreatic islet tissue that was dispersed by collagenase digestion into the spleen or portal vein could prevent diabetes.20,21 In 1974, the first human trials of transplanting islet cells were conducted, using isolated islets from cadaveric donors to treat diabetes.19
In the past, pancreatectomy was performed to treat painful chronic pancreatitis, but it was viewed as undesirable because removing the gland would inevitably cause insulin-dependent diabetes.22 That changed in 1977 at the University of Minnesota, with the first reported islet cell autotransplant after pancreatectomy. The patient remained pain-free and insulin-independent long-term.23 This seminal case showed that endocrine function could be preserved by autotransplant of islets prepared from the excised pancreas.24
In 1992, Pyzdrowski et al25 reported that intrahepatic transplant of as few as 265,000 islets was enough to prevent the need for insulin therapy. Since this technique was first described, there have been many advances, and now more than 30 centers worldwide do it.
PRIMARY INDICATION: INTRACTABLE PAIN
Interest has been growing in using total pancreatectomy and islet autotransplant to treat recurrent acute pancreatitis, chronic pancreatitis, and hereditary pancreatitis. The rationale is that removing the offending tissue eliminates pancreatitis, pain, and cancer risk, while preserving and replacing the islet cells prevents the development of brittle diabetes with loss of insulin and glucagon.26
Proposed criteria for total pancreatectomy and islet autotransplant
Bellin et al14 proposed five criteria for patient selection for this procedure based on imaging studies, pancreatic function tests, and histopathology to detect pancreatic fibrosis. Patients must fulfill all five of the following criteria:
Criterion 1. Diagnosis of chronic pancreatitis, based on chronic abdominal pain lasting more than 6 months with either at least one of the following:
- Pancreatic calcifications on computed tomography
- At least two of the following: four or more of nine criteria on endoscopic ultrasonography described by Catalano et al,27 a compatible ductal or parenchymal abnormality on secretin magnetic resonance cholangiopancreatography; abnormal endoscopic pancreatic function test (peak HCO2 ≤ 80 mmol/L)
- Histopathologically confirmed diagnosis of chronic pancreatitis
- Compatible clinical history and documented hereditary pancreatitis (PRSS1 gene mutation)
OR
- History of recurrent acute pancreatitis (more than one episode of characteristic pain associated with imaging diagnostic of acute pancreatitis or elevated serum amylase or lipase > 3 times the upper limit of normal).
Criterion 2. At least one of the following:
- Daily narcotic dependence
- Pain resulting in impaired quality of life, which may include inability to attend school, recurrent hospitalizations, or inability to participate in usual age-appropriate activities.
Criterion 3. Complete evaluation with no reversible cause of pancreatitis present or untreated.
Criterion 4. Failure to respond to maximal medical and endoscopic therapy.
Criterion 5. Adequate islet cell function (nondiabetic or C-peptide-positive). Patients with C-peptide-negative diabetes meeting criteria 1 to 4 are candidates for total pancreatectomy alone.
The primary goal is to treat intractable pain and improve quality of life in selected patients with chronic pancreatitis or recurrent acute pancreatitis when endoscopic and prior surgical therapies have failed, and whose impairment due to pain is substantial enough to accept the risk of postoperative insulin-dependent diabetes and lifelong commitment to pancreatic enzyme replacement therapy.15,26 Patients with a known genetic cause of chronic pancreatitis should be offered special consideration for the procedure, as their disease is unlikely to remit.
CONTRAINDICATIONS
Total pancreatectomy and islet autotransplant should not be performed in patients with active alcoholism, illicit drug use, or untreated or poorly controlled psychiatric illnesses that could impair the patient’s ability to adhere to a complicated postoperative medical regimen.
A poor support network may be a relative contraindication in view of the cost and complexity of diabetic and pancreatic enzyme replacement therapy.18,26
Islet cell autotransplant is contraindicated in patients with conditions such as C-peptide-negative or type 1 diabetes or a history of portal vein thrombosis, portal hypertension, significant liver disease, high-risk cardiopulmonary disease, or pancreatic cancer (Table 1).26
WHEN TO CONSIDER REFERRAL FOR THIS PROCEDURE
The choice of total pancreatectomy and islet autotransplant vs conventional surgery must be individualized on the basis of each patient’s anatomy, comorbidities, symptom burden, presence or degree of diabetes, and rate of disease progression. The most important factors to consider in determining the need for and timing of this procedure are the patient’s pain, narcotic requirements, and impaired ability to function.26
Sooner rather than later?
An argument can be made for performing this procedure sooner in the course of the disease rather than later when all else has failed. First, prolonged pain can result in central sensitization, in which the threshold for perceiving pain is lowered by damage to the nociceptive neurons from repeated stimulation and inflammation.28
Further, prolonged opioid therapy can lead to opioid-induced hyperalgesia, which may also render patients more sensitive to pain and aggravate their preexisting pain.26,28
In addition, although operative drainage procedures and partial resections are often considered the gold standard for chronic pancreatitis management, patients who undergo partial pancreatectomy or lateral pancreaticojejunostomy (Puestow procedure) have fewer islet cells left to harvest (about 50% fewer) if they subsequently undergo total pancreatectomy and islet cell autotransplant.22,26
Therefore, performing this procedure earlier may help the patient avoid chronic pain syndromes and complications of chronic opioid use, including hyperalgesia, and give the best chance of harvesting enough islet cells to prevent or minimize diabetes afterward.11
REMOVING THE PANCREAS, RETURNING THE ISLET CELLS
During this procedure, the blood supply to the pancreas must be preserved until just before its removal to minimize warm ischemia of the islet cells.18,29 Although there are several surgical variations, a pylorus-preserving total pancreatectomy with duodenectomy is typically performed, usually with splenectomy to preserve perfusion to the body and tail.30
The resected pancreas is taken to the islet isolation laboratory. There, the pancreatic duct is cannulated to fill the organ with a cold collagenase solution, followed by gentle mechanical dispersion using the semiautomated Ricordi method,31 which separates the islet cells from the exocrine tissue.32
The number of islet cells is quantified as islet equivalents; 1 islet equivalent is equal to the volume of an islet with a diameter of 150 µm. Islet equivalents per kilogram of body weight is the unit commonly used to report the graft amount transplanted.33
After digestion, the islet cells can be purified or partially purified by a gradient separation method using a Cobe 2991 cell processor (Terumo Corporation, Tokyo, Japan),34 or can be transplanted as an unpurified preparation. In islet cell autotransplant for chronic pancreatitis, purification is not always necessary due to the small tissue volume extracted from the often atrophic and fibrotic pancreas.32 The decision to purify depends on the postdigest tissue volume; usually, a tissue volume greater than 0.25 mL/kg body weight is an indication to at least partially purify.18,35
The final preparation is returned to the operating room, and after heparin is given, the islets are infused into the portal system using a stump of the splenic vein, or alternatively through direct puncture of the portal vein or cannulation of the umbilical vein.32,36 If the portal vein pressure reaches 25 cm H2O, the infusion is stopped and the remaining islets can be placed in the peritoneal cavity or elsewhere.18 Transplant of the islets into the liver or peritoneum allows the islets to secrete insulin into the hepatic portal circulation, which is the route used by the native pancreas.22
CONTROLLING GLUCOSE DURING AND AFTER THE PROCEDURE
Animal studies have shown that hyperglycemia impairs islet revascularization,37 and glucose toxicity may cause dysfunction and structural lesions of the transplanted islets.11,38
Therefore, during and after the procedure, most centers maintain euglycemia by an intravenous insulin infusion and subsequently move to subcutaneous insulin when the patient starts eating again. Some centers continue insulin at discharge and gradually taper it over months, even in patients who can possibly achieve euglycemia without it.
OUTCOMES
Many institutions have reported their clinical outcomes in terms of pain relief, islet function, glycemic control, and improvement of quality of life. The largest series have been from the University of Minnesota, Leicester General Hospital, the University of Cincinnati, and the Medical University of South Carolina.
Insulin independence is common but wanes with time
The ability to achieve insulin independence after islet autotransplant appears to be related to the number of islets transplanted, with the best results when more than 2,000 or 3,000 islet equivalents/kg are transplanted.39,40
Sutherland et al18 reported that of 409 patients who underwent islet cell autotransplant at the University of Minnesota (the largest series reported to date), 30% were insulin-independent at 3 years, 33% had partial graft function (defined by positive C-peptide), and 82% achieved a mean hemoglobin A1c of less than 7%. However, in the subset who received more than 5,000 islet equivalents/kg, nearly three-fourths of patients were insulin-independent at 3 years.
The Leicester General Hospital group presented long-term data on 46 patients who underwent total pancreatectomy and islet cell autotransplant. Twelve of the 46 had shown periods of insulin independence for a median of 16.5 months, and 5 remained insulin-free at the time of the publication.41 Over the 10 years of follow-up, insulin requirements and hemoglobin A1c increased notably. However, all of the patients tested C-peptide-positive, suggesting long-lasting graft function.
Most recently, the University of Cincinnati group reported long-term data on 116 patients. The insulin independence rate was 38% at 1 year, decreasing to 27% at 5 years. The number of patients with partial graft function was 38% at 1 year and 35% at 5 years.42
Thus, all three institutions confirmed that the autotransplanted islets continue to secrete insulin long-term, but that function decreases over time.
Pancreatectomy reduces pain
Multiple studies have shown that total pancreatectomy reduces pain in patients with chronic pancreatitis. Ahmad et al43 reported a marked reduction in narcotic use (mean morphine equivalents 206 mg/day before surgery, compared with 90 mg after), and a 58% reduction in pain as demonstrated by narcotic independence.
In the University of Minnesota series, 85% of the 409 patients had less pain at 2 years, and 59% were able to stop taking narcotics.18
The University of Cincinnati group reported a narcotic independence rate of 55% at 1 year, which continued to improve to 73% at 5 years.42
Although the source of pain is removed, pain persists or recurs in 10% to 20% of patients after total pancreatectomy and islet cell autotransplant, showing that the pathogenesis of pain is complex, and some uncertainty exists about it.26
Quality of life
Reports evaluating health-related quality of life after total pancreatectomy and islet autotransplant are limited.
The University of Cincinnati group reported the long-term outcomes of quality of life as measured by the Short Form 36 Health Survey.42 Ninety-two percent of patients reported overall improvement in their health at 1 year, and 85% continued to report improved health more than 5 years after the surgery.
In a series of 20 patients, 79% to 90% reported improvements in the seven various domains of the Pain Disability Index. In addition, 60% showed improvement in depression and 70% showed improvement in anxiety. The greatest improvements were in those who had not undergone prior pancreatic surgery, who were younger, and in those with higher levels of preoperative pain.30
Similarly, in a series of 74 patients, the Medical University of South Carolina group reported significant improvement in physical and mental health components of the Short Form 12 Health Survey and an associated decrease in daily narcotic requirements. Moreover, the need to start or increase the dose of insulin after the surgery was not associated with a lower quality of life.44
OFF-SITE ISLET CELL ISOLATION
Despite the positive outcomes in terms of pain relief and insulin independence in many patients after total pancreatectomy and islet cell autotransplant, few medical centers have an on-site islet-processing facility. Since the mid-1990s, a few centers have been able to circumvent this limitation by working with off-site islet cell isolation laboratories.45,46
The University of California, Los Angeles, first reported on a series of nine patients who received autologous islet cells after near-total or total pancreatectomy using a remote islet cell isolation facility, with results comparable to those of other large institutions.45
Similarly, the procedure has been performed at Cleveland Clinic since 2007 with the collaboration of an off-site islet cell isolation laboratory at the University of Pittsburgh. A cohort study from this collaboration published in 2015 showed that in 36 patients (mean follow-up 28 months, range 3–26 months), 33% were insulin-independent, with a C-peptide-positive rate of 70%. This is the largest cohort to date from a center utilizing an off-site islet isolation facility.47
In view of the positive outcomes at these centers, lack of a local islet-processing facility should no longer be a barrier to total pancreatectomy and islet cell autotransplant.
PATIENT CARE AFTER THE PROCEDURE
A multidisciplinary team is an essential component of the postoperative management of patients who undergo total pancreatectomy and islet cell autotransplant.
For patients who had been receiving narcotics for a long time before surgery or who were receiving frequent doses, an experienced pain management physician should be involved in the patient’s postoperative care.
Because islet function can wane over time, testing for diabetes should be done at least annually for the rest of the patient’s life and should include fasting plasma glucose, hemoglobin A1c, and C-peptide, along with self-monitored blood glucose.26
All patients who have surgically induced exocrine insufficiency are at risk of malabsorption and fat-soluble vitamin deficiencies.48 Hence, lifelong pancreatic enzyme replacement therapy is mandatory. Nutritional monitoring should include assessment of steatorrhea, body composition, and fat-soluble vitamin levels (vitamins A, D, and E) at least every year.26 Patients with chronic pancreatitis are at increased risk for low bone density from malabsorption of vitamin D and calcium; therefore, it is recommended that a dual-energy x-ray absorptiometry bone density scan be obtained.26,49
Patients who undergo splenectomy as part of their procedure will require appropriate precautions and ongoing vaccinations as recommended by the US Centers for Disease Control and Prevention.26,50,51
WHAT TO EXPECT FOR THE FUTURE
The National Institute of Diabetes and Digestive and Kidney Diseases has reviewed the potential future research directions for total pancreatectomy and islet autotransplant.15
Patient selection remains challenging despite the availability of criteria15 and guidelines.26 More research is needed to better assess preoperative beta-cell function and to predict postoperative outcomes. Mixed meal-tolerance testing is adopted by most clinical centers to predict posttransplant beta-cell function. The use of arginine instead of glucagon in a stimulation test for insulin and C-peptide response has been validated and may allow more accurate assessment.52,53
Another targeted area of research is the advancement of safety and metabolic outcomes. Techniques to minimize warm ischemic time and complications are being evaluated. Islet isolation methods that yield more islets, reduce beta-cell apoptosis, and can isolate islets from glands with malignancy should be further investigated.54 Further, enhanced islet infusion methods that achieve lower portal venous pressures and minimize portal vein thrombosis are needed.
Unfortunately, the function of transplanted islet grafts declines over time. This phenomenon is at least partially attributed to the immediate blood-mediated inflammatory response,55,56 along with islet hypoxia,57 leading to islet apoptosis. Research on different strategies is expanding our knowledge in islet engraftment and posttransplant beta-cell apoptosis, with the expectation that the transplanted islet lifespan will increase. Alternative transplant sites with low inflammatory reaction, such as the omental pouch,58 muscle,59 and bone marrow,60 have shown encouraging data. Other approaches, such as adjuvant anti-inflammatory agents and heparinization, have been proposed.15
Research into complications is also of clinical importance. There is growing attention to hypoglycemia unrelated to exogenous insulin use in posttransplant patients. One hypothesis is that glucagon secretion, a counterregulatory response to hypoglycemia, is defective if the islet cells are transplanted into the liver, and that implanting them into another site may avoid this effect.61
- Everhart JE. Pancreatitis. In: Everhart JE, editor. The Burden of Digestive Diseases in the United States. US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of
- Diabetes and Digestive and Kidney Diseases. Washington, DC: US Government Printing Office; 2008. www.niddk.nih.gov/about-niddk/strategic-plans-reports/Pages/burden-digestive-diseases-in-united-states-report.aspx. Accessed May 10, 2016.
- Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol 2011; 106:2192–2199.
- Lévy P, Barthet M, Mollard BR, Amouretti M, Marion-Audibert AM, Dyard F. Estimation of the prevalence and incidence of chronic pancreatitis and its complications. Gastroenterol Clin Biol 2006; 30:838–844.
- Hirota M, Shimosegawa T, Masamune A, et al; Research Committee of Intractable Pancreatic Diseases. The seventh nationwide epidemiological survey for chronic pancreatitis in Japan: clinical significance of smoking habit in Japanese patients. Pancreatology 2014; 14:490–496.
- Sankaran SJ, Xiao AY, Wu LM, Windsor JA, Forsmark CE, Petrov MS. Frequency of progression from acute to chronic pancreatitis and risk factors: a meta-analysis. Gastroenterology 2015; 149:1490–1500.e1.
- Coté GA, Yadav D, Slivka A, et al; North American Pancreatitis Study Group. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:266–273.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon 2014; 60:530–550.
- Frulloni L, Gabbrielli A, Pezzilli R, et al; PanCroInfAISP Study Group. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig Liver Dis 2009; 41:311–317.
- Talamini G, Bassi C, Falconi M, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci 1999; 44:1303–1311.
- Schwarzenberg SJ, Bellin M, Husain SZ, et al. Pediatric chronic pancreatitis is associated with genetic risk factors and substantial disease burden. J Pediatr 2015; 166:890–896.e1.
- Blondet JJ, Carlson AM, Kobayashi T, et al. The role of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Surg Clin North Am 2007; 87:1477–1501.
- Lieb JG 2nd, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther 2009; 29:706–719.
- Lin YK, Johnston PC, Arce K, Hatipoglu BA. Chronic pancreatitis and diabetes mellitus. Curr Treat Options Gastroenterol 2015; 13:319–331.
- Bellin MD, Gelrud A, Arreaza-Rubin G, et al. Total pancreatectomy with islet autotransplantation: summary of a National Institute of Diabetes and Digestive and Kidney diseases workshop. Pancreas 2014; 43:1163–1171.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part II: diagnosis, complications, and management. Dis Mon 2015; 61:5–37.
- Warshaw AL, Banks PA, Fernández-Del Castillo C. AGA technical review: treatment of pain in chronic pancreatitis. Gastroenterology 1998; 115:765–776.
- Chauhan S, Forsmark CE. Pain management in chronic pancreatitis: a treatment algorithm. Best Pract Res Clin Gastroenterol 2010; 24:323–335.
- Sutherland DE, Radosevich DM, Bellin MD, et al. Total pancreatectomy and islet autotransplantation for chronic pancreatitis. J Am Coll Surg 2012; 214:409–426.
- Jahansouz C, Jahansouz C, Kumer SC, Brayman KL. Evolution of beta-cell replacement therapy in diabetes mellitus: islet cell transplantation. J Transplant 2011; 2011:247959.
- Kretschmer GJ, Sutherland DE, Matas AJ, Cain TL, Najarian JS. Autotransplantation of pancreatic islets without separation of exocrine and endocrine tissue in totally pancreatectomized dogs. Surgery 1977; 82:74–81.
- Kretschmer GJ, Sutherland DR, Matas AJ, Payne WD, Najarian JS. Autotransplantation of pancreatic fragments to the portal vein and spleen of totally pancreatectomized dogs: a comparative evaluation. Ann Surg 1978; 187:79–86.
- Bellin MD, Sutherland DE, Robertson RP. Pancreatectomy and autologous islet transplantation for painful chronic pancreatitis: indications and outcomes. Hosp Pract (1995) 2012; 40:80–87.
- Najarian JS, Sutherland DE, Baumgartner D, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis. Ann Surg 1980; 192:526–542.
- Sutherland DE, Matas AJ, Najarian JS. Pancreatic islet cell transplantation. Surg Clin North Am 1978; 58:365–382.
- Pyzdrowski KL, Kendall DM, Halter JB, Nakhleh RE, Sutherland DE, Robertson RP. Preserved insulin secretion and insulin independence in recipients of islet autografts. N Engl J Med 1992; 327:220–226.
- Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet autotransplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology 2014; 14:27–35.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 2006; 104:570–587.
- Bramis K, Gordon-Weeks AN, Friend PJ, et al. Systematic review of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Br J Surg 2012; 99:761–766.
- Walsh RM, Saavedra JR, Lentz G, et al. Improved quality of life following total pancreatectomy and auto-islet transplantation for chronic pancreatitis. J Gastrointest Surg 2012; 16:1469–1477.
- Ricordi C, Lacy PE, Scharp DW. Automated islet isolation from human pancreas. Diabetes 1989; 38(suppl 1):140–142.
- Witkowski P, Savari O, Matthews JB. Islet autotransplantation and total pancreatectomy. Adv Surg 2014; 48:223–233.
- Bellin MD, Beilman GJ, Dunn TB, et al. Islet autotransplantation to preserve beta cell mass in selected patients with chronic pancreatitis and diabetes mellitus undergoing total pancreatectomy. Pancreas 2013; 42:317–321.
- Anazawa T, Matsumoto S, Yonekawa Y, et al. Prediction of pancreatic tissue densities by an analytical test gradient system before purification maximizes human islet recovery for islet autotransplantation/allotransplantation. Transplantation 2011; 91:508–514.
- Lake SP, Bassett PD, Larkins A, et al. Large-scale purification of human islets utilizing discontinuous albumin gradient on IBM 2991 cell separator. Diabetes 1989; 38(suppl 1):143–145.
- Bellin MD, Freeman ML, Schwarzenberg SJ, et al. Quality of life improves for pediatric patients after total pancreatectomy and islet autotransplant for chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:793–799.
- Andersson A, Korsgren O, Jansson L. Intraportally transplanted pancreatic islets revascularized from hepatic arterial system. Diabetes 1989; 38(suppl 1):192–195.
- Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 1992; 15:442–455.
- Bellin MD, Carlson AM, Kobayashi T, et al. Outcome after pancreatectomy and islet autotransplantation in a pediatric population. J Pediatr Gastroenterol Nutr 2008; 47:37–44.
- White SA, Davies JE, Pollard C, et al. Pancreas resection and islet autotransplantation for end-stage chronic pancreatitis. Ann Surg 2001; 233:423–431.
- Webb MA, Illouz SC, Pollard CA, et al. Islet auto transplantation following total pancreatectomy: a long-term assessment of graft function. Pancreas 2008; 37:282–287.
- Wilson GC, Sutton JM, Abbott DE, et al. Long-term outcomes after total pancreatectomy and islet cell autotransplantation: is it a durable operation? Ann Surg 2014; 260:659–667.
- Ahmad SA, Lowy AM, Wray CJ, et al. Factors associated with insulin and narcotic independence after islet autotransplantation in patients with severe chronic pancreatitis. J Am Coll Surg 2005; 201:680–687.
- Dorlon M, Owczarski S, Wang H, Adams D, Morgan K. Increase in postoperative insulin requirements does not lead to decreased quality of life after total pancreatectomy with islet cell autotransplantation for chronic pancreatitis. Am Surg 2013; 79:676–680.
- Tai DS, Shen N, Szot GL, et al. Autologous islet transplantation with remote islet isolation after pancreas resection for chronic pancreatitis. JAMA Surg 2015; 150:118–124.
- Rabkin JM, Olyaei AJ, Orloff SL, et al. Distant processing of pancreas islets for autotransplantation following total pancreatectomy. Am J Surg 1999; 177:423–427.
- Johnston PC, Lin YK, Walsh RM, et al. Factors associated with islet yield and insulin independence after total pancreatectomy and islet cell autotransplantation in patients with chronic pancreatitis utilizing off-site islet isolation: Cleveland Clinic experience. J Clin Endocrinol Metab 2015; 100:1765–1770.
- Dresler CM, Fortner JG, McDermott K, Bajorunas DR. Metabolic consequences of (regional) total pancreatectomy. Ann Surg 1991; 214:131–140.
- Duggan SN, O’Sullivan M, Hamilton S, Feehan SM, Ridgway PF, Conlon KC. Patients with chronic pancreatitis are at increased risk for osteoporosis. Pancreas 2012; 41:1119–1124.
- Rubin LG, Levin MJ, Ljungman P, et al; Infectious Diseases Society of America. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:e44–e100.
- Di Sabatino A, Carsetti R, Corazza GR. Post-splenectomy and hyposplenic states. Lancet 2011; 378:86–97.
- Robertson RP, Raymond RH, Lee DS, et al; Beta Cell Project Team of the Foundation for the NIH Biomarkers Consortium. Arginine is preferred to glucagon for stimulation testing of beta-cell function. Am J Physiol Endocrinol Metab 2014; 307:E720–E727.
- Robertson RP, Bogachus LD, Oseid E, et al. Assessment of beta-cell mass and alpha- and beta-cell survival and function by arginine stimulation in human autologous islet recipients. Diabetes 2015; 64:565–572.
- Balzano G, Piemonti L. Autologous islet transplantation in patients requiring pancreatectomy for neoplasm. Curr Diab Rep 2014; 14:512.
- Naziruddin B, Iwahashi S, Kanak MA, Takita M, Itoh T, Levy MF. Evidence for instant blood-mediated inflammatory reaction in clinical autologous islet transplantation. Am J Transplant 2014; 14:428–437.
- Abdelli S, Ansite J, Roduit R, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 2004; 53:2815–2823.
- Olsson R, Olerud J, Pettersson U, Carlsson PO. Increased numbers of low-oxygenated pancreatic islets after intraportal islet transplantation. Diabetes 2011; 60:2350–2353.
- Berman DM, O’Neil JJ, Coffey LC, et al. Long-term survival of nonhuman primate islets implanted in an omental pouch on a biodegradable scaffold. Am J Transplant 2009; 9:91–104.
- Sterkers A, Hubert T, Gmyr V, et al. Islet survival and function following intramuscular autotransplantation in the minipig. Am J Transplant 2013; 13:891–898.
- Maffi P, Balzano G, Ponzoni M, et al. Autologous pancreatic islet transplantation in human bone marrow. Diabetes 2013; 62:3523–3531.
- Bellin MD, Parazzoli S, Oseid E, et al. Defective glucagon secretion during hypoglycemia after intrahepatic but not nonhepatic islet autotransplantation. Am J Transplant 2014; 14:1880–1886.
- Everhart JE. Pancreatitis. In: Everhart JE, editor. The Burden of Digestive Diseases in the United States. US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of
- Diabetes and Digestive and Kidney Diseases. Washington, DC: US Government Printing Office; 2008. www.niddk.nih.gov/about-niddk/strategic-plans-reports/Pages/burden-digestive-diseases-in-united-states-report.aspx. Accessed May 10, 2016.
- Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol 2011; 106:2192–2199.
- Lévy P, Barthet M, Mollard BR, Amouretti M, Marion-Audibert AM, Dyard F. Estimation of the prevalence and incidence of chronic pancreatitis and its complications. Gastroenterol Clin Biol 2006; 30:838–844.
- Hirota M, Shimosegawa T, Masamune A, et al; Research Committee of Intractable Pancreatic Diseases. The seventh nationwide epidemiological survey for chronic pancreatitis in Japan: clinical significance of smoking habit in Japanese patients. Pancreatology 2014; 14:490–496.
- Sankaran SJ, Xiao AY, Wu LM, Windsor JA, Forsmark CE, Petrov MS. Frequency of progression from acute to chronic pancreatitis and risk factors: a meta-analysis. Gastroenterology 2015; 149:1490–1500.e1.
- Coté GA, Yadav D, Slivka A, et al; North American Pancreatitis Study Group. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:266–273.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon 2014; 60:530–550.
- Frulloni L, Gabbrielli A, Pezzilli R, et al; PanCroInfAISP Study Group. Chronic pancreatitis: report from a multicenter Italian survey (PanCroInfAISP) on 893 patients. Dig Liver Dis 2009; 41:311–317.
- Talamini G, Bassi C, Falconi M, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci 1999; 44:1303–1311.
- Schwarzenberg SJ, Bellin M, Husain SZ, et al. Pediatric chronic pancreatitis is associated with genetic risk factors and substantial disease burden. J Pediatr 2015; 166:890–896.e1.
- Blondet JJ, Carlson AM, Kobayashi T, et al. The role of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Surg Clin North Am 2007; 87:1477–1501.
- Lieb JG 2nd, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther 2009; 29:706–719.
- Lin YK, Johnston PC, Arce K, Hatipoglu BA. Chronic pancreatitis and diabetes mellitus. Curr Treat Options Gastroenterol 2015; 13:319–331.
- Bellin MD, Gelrud A, Arreaza-Rubin G, et al. Total pancreatectomy with islet autotransplantation: summary of a National Institute of Diabetes and Digestive and Kidney diseases workshop. Pancreas 2014; 43:1163–1171.
- Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part II: diagnosis, complications, and management. Dis Mon 2015; 61:5–37.
- Warshaw AL, Banks PA, Fernández-Del Castillo C. AGA technical review: treatment of pain in chronic pancreatitis. Gastroenterology 1998; 115:765–776.
- Chauhan S, Forsmark CE. Pain management in chronic pancreatitis: a treatment algorithm. Best Pract Res Clin Gastroenterol 2010; 24:323–335.
- Sutherland DE, Radosevich DM, Bellin MD, et al. Total pancreatectomy and islet autotransplantation for chronic pancreatitis. J Am Coll Surg 2012; 214:409–426.
- Jahansouz C, Jahansouz C, Kumer SC, Brayman KL. Evolution of beta-cell replacement therapy in diabetes mellitus: islet cell transplantation. J Transplant 2011; 2011:247959.
- Kretschmer GJ, Sutherland DE, Matas AJ, Cain TL, Najarian JS. Autotransplantation of pancreatic islets without separation of exocrine and endocrine tissue in totally pancreatectomized dogs. Surgery 1977; 82:74–81.
- Kretschmer GJ, Sutherland DR, Matas AJ, Payne WD, Najarian JS. Autotransplantation of pancreatic fragments to the portal vein and spleen of totally pancreatectomized dogs: a comparative evaluation. Ann Surg 1978; 187:79–86.
- Bellin MD, Sutherland DE, Robertson RP. Pancreatectomy and autologous islet transplantation for painful chronic pancreatitis: indications and outcomes. Hosp Pract (1995) 2012; 40:80–87.
- Najarian JS, Sutherland DE, Baumgartner D, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis. Ann Surg 1980; 192:526–542.
- Sutherland DE, Matas AJ, Najarian JS. Pancreatic islet cell transplantation. Surg Clin North Am 1978; 58:365–382.
- Pyzdrowski KL, Kendall DM, Halter JB, Nakhleh RE, Sutherland DE, Robertson RP. Preserved insulin secretion and insulin independence in recipients of islet autografts. N Engl J Med 1992; 327:220–226.
- Bellin MD, Freeman ML, Gelrud A, et al. Total pancreatectomy and islet autotransplantation in chronic pancreatitis: recommendations from PancreasFest. Pancreatology 2014; 14:27–35.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology 2006; 104:570–587.
- Bramis K, Gordon-Weeks AN, Friend PJ, et al. Systematic review of total pancreatectomy and islet autotransplantation for chronic pancreatitis. Br J Surg 2012; 99:761–766.
- Walsh RM, Saavedra JR, Lentz G, et al. Improved quality of life following total pancreatectomy and auto-islet transplantation for chronic pancreatitis. J Gastrointest Surg 2012; 16:1469–1477.
- Ricordi C, Lacy PE, Scharp DW. Automated islet isolation from human pancreas. Diabetes 1989; 38(suppl 1):140–142.
- Witkowski P, Savari O, Matthews JB. Islet autotransplantation and total pancreatectomy. Adv Surg 2014; 48:223–233.
- Bellin MD, Beilman GJ, Dunn TB, et al. Islet autotransplantation to preserve beta cell mass in selected patients with chronic pancreatitis and diabetes mellitus undergoing total pancreatectomy. Pancreas 2013; 42:317–321.
- Anazawa T, Matsumoto S, Yonekawa Y, et al. Prediction of pancreatic tissue densities by an analytical test gradient system before purification maximizes human islet recovery for islet autotransplantation/allotransplantation. Transplantation 2011; 91:508–514.
- Lake SP, Bassett PD, Larkins A, et al. Large-scale purification of human islets utilizing discontinuous albumin gradient on IBM 2991 cell separator. Diabetes 1989; 38(suppl 1):143–145.
- Bellin MD, Freeman ML, Schwarzenberg SJ, et al. Quality of life improves for pediatric patients after total pancreatectomy and islet autotransplant for chronic pancreatitis. Clin Gastroenterol Hepatol 2011; 9:793–799.
- Andersson A, Korsgren O, Jansson L. Intraportally transplanted pancreatic islets revascularized from hepatic arterial system. Diabetes 1989; 38(suppl 1):192–195.
- Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 1992; 15:442–455.
- Bellin MD, Carlson AM, Kobayashi T, et al. Outcome after pancreatectomy and islet autotransplantation in a pediatric population. J Pediatr Gastroenterol Nutr 2008; 47:37–44.
- White SA, Davies JE, Pollard C, et al. Pancreas resection and islet autotransplantation for end-stage chronic pancreatitis. Ann Surg 2001; 233:423–431.
- Webb MA, Illouz SC, Pollard CA, et al. Islet auto transplantation following total pancreatectomy: a long-term assessment of graft function. Pancreas 2008; 37:282–287.
- Wilson GC, Sutton JM, Abbott DE, et al. Long-term outcomes after total pancreatectomy and islet cell autotransplantation: is it a durable operation? Ann Surg 2014; 260:659–667.
- Ahmad SA, Lowy AM, Wray CJ, et al. Factors associated with insulin and narcotic independence after islet autotransplantation in patients with severe chronic pancreatitis. J Am Coll Surg 2005; 201:680–687.
- Dorlon M, Owczarski S, Wang H, Adams D, Morgan K. Increase in postoperative insulin requirements does not lead to decreased quality of life after total pancreatectomy with islet cell autotransplantation for chronic pancreatitis. Am Surg 2013; 79:676–680.
- Tai DS, Shen N, Szot GL, et al. Autologous islet transplantation with remote islet isolation after pancreas resection for chronic pancreatitis. JAMA Surg 2015; 150:118–124.
- Rabkin JM, Olyaei AJ, Orloff SL, et al. Distant processing of pancreas islets for autotransplantation following total pancreatectomy. Am J Surg 1999; 177:423–427.
- Johnston PC, Lin YK, Walsh RM, et al. Factors associated with islet yield and insulin independence after total pancreatectomy and islet cell autotransplantation in patients with chronic pancreatitis utilizing off-site islet isolation: Cleveland Clinic experience. J Clin Endocrinol Metab 2015; 100:1765–1770.
- Dresler CM, Fortner JG, McDermott K, Bajorunas DR. Metabolic consequences of (regional) total pancreatectomy. Ann Surg 1991; 214:131–140.
- Duggan SN, O’Sullivan M, Hamilton S, Feehan SM, Ridgway PF, Conlon KC. Patients with chronic pancreatitis are at increased risk for osteoporosis. Pancreas 2012; 41:1119–1124.
- Rubin LG, Levin MJ, Ljungman P, et al; Infectious Diseases Society of America. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis 2014; 58:e44–e100.
- Di Sabatino A, Carsetti R, Corazza GR. Post-splenectomy and hyposplenic states. Lancet 2011; 378:86–97.
- Robertson RP, Raymond RH, Lee DS, et al; Beta Cell Project Team of the Foundation for the NIH Biomarkers Consortium. Arginine is preferred to glucagon for stimulation testing of beta-cell function. Am J Physiol Endocrinol Metab 2014; 307:E720–E727.
- Robertson RP, Bogachus LD, Oseid E, et al. Assessment of beta-cell mass and alpha- and beta-cell survival and function by arginine stimulation in human autologous islet recipients. Diabetes 2015; 64:565–572.
- Balzano G, Piemonti L. Autologous islet transplantation in patients requiring pancreatectomy for neoplasm. Curr Diab Rep 2014; 14:512.
- Naziruddin B, Iwahashi S, Kanak MA, Takita M, Itoh T, Levy MF. Evidence for instant blood-mediated inflammatory reaction in clinical autologous islet transplantation. Am J Transplant 2014; 14:428–437.
- Abdelli S, Ansite J, Roduit R, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 2004; 53:2815–2823.
- Olsson R, Olerud J, Pettersson U, Carlsson PO. Increased numbers of low-oxygenated pancreatic islets after intraportal islet transplantation. Diabetes 2011; 60:2350–2353.
- Berman DM, O’Neil JJ, Coffey LC, et al. Long-term survival of nonhuman primate islets implanted in an omental pouch on a biodegradable scaffold. Am J Transplant 2009; 9:91–104.
- Sterkers A, Hubert T, Gmyr V, et al. Islet survival and function following intramuscular autotransplantation in the minipig. Am J Transplant 2013; 13:891–898.
- Maffi P, Balzano G, Ponzoni M, et al. Autologous pancreatic islet transplantation in human bone marrow. Diabetes 2013; 62:3523–3531.
- Bellin MD, Parazzoli S, Oseid E, et al. Defective glucagon secretion during hypoglycemia after intrahepatic but not nonhepatic islet autotransplantation. Am J Transplant 2014; 14:1880–1886.
KEY POINTS
- Chronic pancreatitis is caused by inflammation and results in progressive, irreversible loss of both exocrine and endocrine function.
- Total pancreatectomy with islet cell autotransplant is a definitive treatment for chronic pancreatitis, with most patients reporting less pain and better quality of life.
- Patients who have undergone this procedure need lifelong pancreatic enzyme replacement therapy along with surveillance for and treatment of diabetes.
- Research in this field is expanding our knowledge, from altered physiology to patient selection to improvement in islet yield and survival.
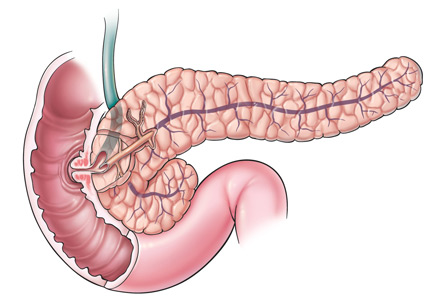
Endoscopic ultrasonography to evaluate pancreatitis
Endoscopic ultrasonography (EUS) is a minimally invasive test that provides high-resolution imaging of the pancreas.1,2 As such, it is proving useful.
Accurate diagnosis and timely intervention are essential in managing acute and chronic pancreatitis, which are often encountered in the clinic and the hospital. However, the cause of acute pancreatitis is not always easy to determine. Furthermore, recurrent bouts can progress to chronic pancreatitis if the cause is not identified and eliminated. EUS has been studied extensively in the evaluation of both acute and chronic pancreatitis, as it can identify obstructive and biliary causes of acute pancreatitis and early structural features of chronic pancreatitis.
This article will review the indications and evidence for EUS in the evaluation of acute and chronic pancreatitis.
SPECIALIZED TRAINING REQUIRED
EUS involves passage of a specialized endoscope through the esophagus and stomach and into the duodenum. The scope has a very small ultrasound probe at the tip, allowing detailed imaging of the upper gastrointestinal tract and surrounding organs.
There are two types of EUS endoscope: radial and linear. A radial scope provides a 360° range of view perpendicular to the long axis of the scope. A linear scope provides a 150° view parallel to the long axis of the scope. Many endosonographers favor linear EUS for imaging the pancreas because it permits fine-needle aspiration biopsy of masses, cysts, and lymph nodes.
Specialized training beyond the gastroenterology fellowship is usually required to become proficient in performing EUS, in recognizing the anatomy it reveals, and in performing fine-needle aspiration biopsy.
ENDOSCOPIC ULTRASONOGRAPHY IN ACUTE PANCREATITIS
Finding the cause of acute pancreatitis can be challenging in patients who do not have typical risk factors, eg, those who do not drink substantial amounts of alcohol and in whom transabdominal ultrasonography fails to reveal gallstones.
Several studies have evaluated the role of EUS in recurrent “idiopathic” pancreatitis.3–5 Causes of acute pancreatitis detectable with EUS included gallbladder and bile duct microlithiasis (stones smaller than 3 mm), cysts, intraductal papillary mucinous neoplasms, ampullary neoplasms, pancreas divisum, and pancreatic masses.
Stones, sludge. Transabdominal ultrasonography is often performed in the workup of acute pancreatitis to rule out gallbladder stones and biliary dilation. Unfortunately, it does a poor job of imaging the distal common bile duct, where culprit stones may reside.
EUS provides a high-quality view of the bile duct from the ampulla of Vater to the region of the hepatic hilum and is safer than endoscopic retrograde cholangiopancreatography (ERCP). The available evidence supports the use of EUS as a diagnostic test for bile duct stones.3–7 In fact, using ERCP as the reference standard, EUS has been found to be more sensitive than transabdominal ultrasonography for bile duct stones.4
The yield of EUS for finding biliary sludge and stones may be high in patients with unexplained pancreatitis. EUS detected sludge, microlithiasis, or both in 33 of 35 patients with idiopathic acute pancreatitis who underwent transabdominal ultrasonography with negative results.8 Furthermore, most were symptom-free at an average of 10 months after cholecystectomy, suggesting that microlithiasis was the cause of the “idiopathic” pancreatitis.
EUS can also decrease the number of unnecessary ERCP procedures in patients with suspected biliary pancreatitis. In these patients, EUS can be performed as an initial diagnostic test to exclude retained biliary stones. If a stone is present, the endoscopist can proceed to ERCP for sphincterotomy and stone removal during the same endoscopic session. If EUS is negative, the endoscopy can be concluded without cannulating the bile duct and putting the patient at risk of acute pancreatitis. In one report, this approach eliminated the need for ERCP in five of six patients with suspected biliary pancreatitis.6
Tumors and other causes of bile duct obstruction can also cause recurrent acute pancreatitis and may be difficult to detect with cross-sectional imaging. EUS, on the other hand, can detect small pancreatic masses (< 2 cm), which may be missed by conventional computed tomography. Also, a linear EUS scope, with its forward oblique view, can image the duodenum and ampulla, where obstructing inflammation, tumors, and polyps may be found. One should strongly suspect occult malignancy in elderly patients with unexplained acute pancreatitis. In those patients, repeat imaging with high-resolution dual-phase computed tomography or with EUS should be considered after a few weeks once the acute inflammation resolves.
Pancreas divisum is a relatively common congenital abnormality in which the dorsal and ventral pancreatic ducts do not properly fuse during embryonic development. To rule out pancreas divisum, the endosonographer must carefully trace the pancreatic duct from the dorsal pancreas into the ventral pancreas, where it connects with the bile duct at the duodenal wall.
In summary, EUS appears to be safe and accurate for diagnosing bile duct stones and other structural causes of idiopathic acute pancreatitis.
ENDOSCOPIC ULTRASONOGRAPHY IN CHRONIC PANCREATITIS
Chronic pancreatitis, a relatively common and sometimes debilitating cause of chronic upper abdominal pain, may be difficult to diagnose using noninvasive imaging tests. Minimal-change chronic pancreatitis is defined as a syndrome of pancreatic abdominal pain with no or slight structural changes detected on imaging but with histologic inflammation and fibrosis diagnostic of chronic pancreatitis.9
A clinical rationale for trying to detect chronic pancreatitis early in its course is that interventions can be started earlier. These include abstinence from alcohol, giving exogenous pancreatic enzymes, and advanced interventions such as celiac plexus blocks for pain control. Some patients may even benefit from resection of the pancreas if pain is severe and resistant to conservative measures.
EUS can detect both parenchymal and ductal changes that correlate with histologic fibrosis.10 Parenchymal changes include hyperechoic foci, hyperechoic strands, lobularity, cysts, and shadowing calcifications. Ductal changes include dilation of the main pancreatic duct, irregularity, hyperechoic duct margins, and visible side branches.
Several studies have evaluated the ability of EUS to diagnose early chronic pancreatitis.9,11–15 Reference standards used to determine the accuracy of EUS have included histology,10,16–18 pancreatic function testing,19–22 and ERCP.11,15,23,24
The best diagnostic test may be pancreatic histology. However, biopsy of the pancreas is impractical and exposes patients to high risk. In addition, the patchy and focal distribution of histologic changes may decrease its reliability. Fortunately, the histologic findings of fibrosis have been shown to correlate with EUS criteria in patients undergoing EUS before surgical resection in three recent studies.16–18 A threshold of four or more criteria out of a possible nine was found to provide the optimal sensitivity and specificity for histologic pancreatic fibrosis.16,17 The criteria used were four parenchymal features (hyperechoic foci, strands, hypoechoic lobules, cysts) and five ductal features (irregularity of the main pancreatic duct, dilation, hyperechoic duct walls, visible side branches, and calcifications or stones).
EUS is sensitive for chronic pancreatitis, but ‘true’ accuracy is impossible to know
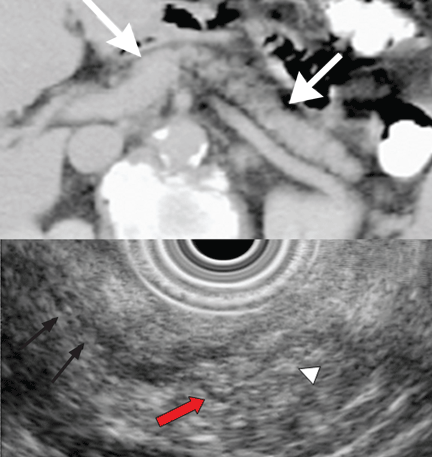
Unfortunately, greater sensitivity may come at the expense of worse specificity. Certain demographic variables may alter the EUS appearance of the pancreas. A multivariate analysis25 found several variables that predicted abnormalities on EUS even in the absence of clinically evident pancreatitis; the strongest were heavy ethanol use (odds ratio [OR] 5.1, 95% confidence interval [CI] 3.1–8.5), male sex (OR 1.8, 95% CI 1.3–2.55), clinical suspicion of pancreatic disease (OR 1.7, 95% CI 1.2–2.3), and heavy smoking (OR 1.7, 95% CI 1.2–2.4). More prospective studies are needed to further differentiate true disease from false-positive findings of chronic pancreatitis.
Also, traditional EUS scoring symptoms have counted features in an unweighted fashion and assigned an arbitrary cut point (eg, four or more features) for diagnosis. This approach fails to account for the greater importance of some features (eg, calcifications) compared with others.
Interobserver variability is another important limitation of EUS in diagnosing chronic pancreatitis.26,27 In one multicenter study of EUS interpretation, the overall kappa (agreement beyond chance) was only 0.45 for overall chronic pancreatitis diagnosis and worse for many individual criteria for chronic pancreatitis. The endosonographers disagreed most about hyperechoic strands and foci, main pancreatic duct irregularity, and visible side branches (kappa < 0.4).
The Rosemont classification
These limitations led a group of experts to meet in Chicago, IL, to develop a consensus-based and weighted EUS scoring system for the diagnosis of chronic pancreatitis, termed the Rosemont classification.
In this system, the previous parenchymal and ductal features are assigned stricter definitions and reclassified as major and minor criteria. Based on the presence of major and minor features, EUS results are stratified as “normal,” “indeterminate for chronic pancreatitis,” “suggestive of chronic pancreatitis,” or “most consistent with chronic pancreatitis.”15,28
Further validation of this scoring system is needed before it can be used widely.
ENDOSCOPIC ULTRASONOGRAPHY PLUS PANCREATIC FUNCTION TESTING
The best way to diagnose minimal-change chronic pancreatitis may be a combination of sensitive structural and functional testing. Although clinically apparent steatorrhea typically occurs late in the course of chronic pancreatitis, mild exocrine insufficiency may occur early and is detectable with hormone-stimulated pancreatic function testing. Therefore, pancreatic function tests are considered sensitive for diagnosing chronic pancreatitis.20,21,29
Endoscopic pancreatic function testing involves injecting secretin intravenously and then collecting duodenal aspirates through the endoscope. The duodenal fluid is analyzed for bicarbonate concentration as a measure of exocrine function.29
We have studied combined EUS and endoscopic pancreatic function testing in the diagnosis of chronic pancreatitis.16 The combination gives a simultaneous structural and functional assessment of the pancreas and may optimize sensitivity for detecting minimal-change chronic pancreatitis. In a small study, we found the combination had 100% sensitivity for noncalcific chronic pancreatitis compared with a histologic reference standard.16
- Sivak MV, Kaufman A. Endoscopic ultrasonography in the differential diagnosis of pancreatic disease. A preliminary report. Scand J Gastroenterol Suppl 1986; 123:130–134.
- Hisanaga K, Hisanaga A, Nagata K, Ichie Y. High speed rotating scanner for transgastric sonography. AJR Am J Roentgenol 1980; 135:627–629.
- Frossard JL, Sosa-Valencia L, Amouyal G, Marty O, Hadengue A, Amouyal P. Usefulness of endoscopic ultrasonography in patients with “idiopathic” acute pancreatitis. Am J Med 2000; 109:196–200.
- Sugiyama M, Wada N, Atomi Y, Kuroda A, Muto T. Diagnosis of acute pancreatitis: value of endoscopic sonography. AJR Am J Roentgenol 1995; 165:867–872.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Liu CL, Lo CM, Chan JK, et al. Detection of choledocholithiasis by EUS in acute pancreatitis: a prospective evaluation in 100 consecutive patients. Gastrointest Endosc 2001; 54:325–330.
- Mirbagheri SA, Mohamadnejad M, Nasiri J, Vahid AA, Ghadimi R, Malekzadeh R. Prospective evaluation of endoscopic ultrasonography in the diagnosis of biliary microlithiasis in patients with normal transabdominal ultrasonography. J Gastrointest Surg 2005; 9:961–964.
- Walsh TN, Rode J, Theis BA, Russell RC. Minimal change chronic pancreatitis. Gut 1992; 33:1566–1571.
- Bhutani MJ, Arantes VN, Verma D, et al. Histopathologic correlation of endoscopic ultrasound findings of chronic pancreatitis in human autopsies. Pancreas 2009; 38:820–824.
- Wiersema MJ, Hawes RH, Lehman GA, Kochman ML, Sherman S, Kopecky KK. Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy 1993; 25:555–564.
- Kahl S, Glasbrenner B, Leodolter A, Pross M, Schulz HU, Malfertheiner P. EUS in the diagnosis of early chronic pancreatitis: a prospective follow-up study. Gastrointest Endosc 2002; 55:507–511.
- Jones SN, Lees WR, Frost RA. Diagnosis and grading of chronic pancreatitis by morphological criteria derived by ultrasound and pancreatography. Clin Radiol 1988; 39:43–48.
- Lees WR. Endoscopic ultrasonography of chronic pancreatitis and pancreatic pseudocysts. Scand J Gastroenterol Suppl 1986; 123:123–129.
- Sahai AV, Zimmerman M, Aabakken L, et al. Prospective assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pancreatitis found by endoscopic retrograde cholangiopancreatography. Gastrointest Endosc 1998; 48:18–25.
- Albashir S, Bronner MP, Parsi MA, Walsh RM, Stevens T. Endoscopic ultrasound, secretin endoscopic pancreatic function test, and histology: correlation in chronic pancreatitis. Am J Gastroenterol 2010; 105:2498–2503.
- Varadarajulu S, Eltoum I, Tamhane A, Eloubeidi MA. Histopathologic correlates of noncalcific chronic pancreatitis by EUS: a prospective tissue characterization study. Gastrointest Endosc 2007; 66:501–509.
- Chong AK, Hawes RH, Hoffman BJ, Adams DB, Lewin DN, Romagnuolo J. Diagnostic performance of EUS for chronic pancreatitis: a comparison with histopathology. Gastrointest Endosc 2007; 65:808–814.
- Chowdhury R, Bhutani MS, Mishra G, Toskes PP, Forsmark CE. Comparative analysis of direct pancreatic function testing versus morphological assessment by endoscopic ultrasonography for the evaluation of chronic unexplained abdominal pain of presumed pancreatic origin. Pancreas 2005; 31:63–68.
- Conwell DL, Zuccaro G, Purich E, et al. Comparison of endoscopic ultrasound chronic pancreatitis criteria to the endoscopic secretinstimulated pancreatic function test. Dig Dis Sci 2007; 52:1206–1210.
- Stevens T, Conwell DL, Zuccaro G, Vargo JJ, Dumot JA, Lopez R. Comparison of endoscopic ultrasound and endoscopic retrograde pancreatography for the prediction of pancreatic exocrine insufficiency. Dig Dis Sci 2008; 53:1146–1151.
- Stevens T, Dumot JA, Parsi MA, Zuccaro G, Vargo JJ. Combined endoscopic ultrasound and secretin endoscopic pancreatic function test in patients evaluated for chronic pancreatitis. Dig Dis Sci 2010; 55:2681–2687.
- Catalano MF, Lahoti S, Geenen JE, Hogan WJ. Prospective evaluation of endoscopic ultrasonography, endoscopic retrograde pancreatography, and secretin test in the diagnosis of chronic pancreatitis. Gastrointest Endosc 1998; 48:11–17.
- Irisawa A, Katakura K, Ohira H, et al. Usefulness of endoscopic ultrasound to diagnose the severity of chronic pancreatitis. J Gastroenterol 2007; 42(suppl 17):90–94.
- Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol 2004; 2:405–409.
- Stevens T, Lopez R, Adler DG, et al. Multicenter comparison of the interobserver agreement of standard EUS scoring and Rosemont classification scoring for diagnosis of chronic pancreatitis. Gastrointest Endosc 2010; 71:519–526.
- Wallace MB, Hawes RH, Durkalski V, et al. The reliability of EUS for the diagnosis of chronic pancreatitis: interobserver agreement among experienced endosonographers. Gastrointest Endosc 2001; 53:294–299.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Stevens T, Conwell DL, Zuccaro G, et al. A prospective crossover study comparing secretin-stimulated endoscopic and Dreiling tube pancreatic function testing in patients evaluated for chronic pancreatitis. Gastrointest Endosc 2008; 67:458–466.
Endoscopic ultrasonography (EUS) is a minimally invasive test that provides high-resolution imaging of the pancreas.1,2 As such, it is proving useful.
Accurate diagnosis and timely intervention are essential in managing acute and chronic pancreatitis, which are often encountered in the clinic and the hospital. However, the cause of acute pancreatitis is not always easy to determine. Furthermore, recurrent bouts can progress to chronic pancreatitis if the cause is not identified and eliminated. EUS has been studied extensively in the evaluation of both acute and chronic pancreatitis, as it can identify obstructive and biliary causes of acute pancreatitis and early structural features of chronic pancreatitis.
This article will review the indications and evidence for EUS in the evaluation of acute and chronic pancreatitis.
SPECIALIZED TRAINING REQUIRED
EUS involves passage of a specialized endoscope through the esophagus and stomach and into the duodenum. The scope has a very small ultrasound probe at the tip, allowing detailed imaging of the upper gastrointestinal tract and surrounding organs.
There are two types of EUS endoscope: radial and linear. A radial scope provides a 360° range of view perpendicular to the long axis of the scope. A linear scope provides a 150° view parallel to the long axis of the scope. Many endosonographers favor linear EUS for imaging the pancreas because it permits fine-needle aspiration biopsy of masses, cysts, and lymph nodes.
Specialized training beyond the gastroenterology fellowship is usually required to become proficient in performing EUS, in recognizing the anatomy it reveals, and in performing fine-needle aspiration biopsy.
ENDOSCOPIC ULTRASONOGRAPHY IN ACUTE PANCREATITIS
Finding the cause of acute pancreatitis can be challenging in patients who do not have typical risk factors, eg, those who do not drink substantial amounts of alcohol and in whom transabdominal ultrasonography fails to reveal gallstones.
Several studies have evaluated the role of EUS in recurrent “idiopathic” pancreatitis.3–5 Causes of acute pancreatitis detectable with EUS included gallbladder and bile duct microlithiasis (stones smaller than 3 mm), cysts, intraductal papillary mucinous neoplasms, ampullary neoplasms, pancreas divisum, and pancreatic masses.
Stones, sludge. Transabdominal ultrasonography is often performed in the workup of acute pancreatitis to rule out gallbladder stones and biliary dilation. Unfortunately, it does a poor job of imaging the distal common bile duct, where culprit stones may reside.
EUS provides a high-quality view of the bile duct from the ampulla of Vater to the region of the hepatic hilum and is safer than endoscopic retrograde cholangiopancreatography (ERCP). The available evidence supports the use of EUS as a diagnostic test for bile duct stones.3–7 In fact, using ERCP as the reference standard, EUS has been found to be more sensitive than transabdominal ultrasonography for bile duct stones.4
The yield of EUS for finding biliary sludge and stones may be high in patients with unexplained pancreatitis. EUS detected sludge, microlithiasis, or both in 33 of 35 patients with idiopathic acute pancreatitis who underwent transabdominal ultrasonography with negative results.8 Furthermore, most were symptom-free at an average of 10 months after cholecystectomy, suggesting that microlithiasis was the cause of the “idiopathic” pancreatitis.
EUS can also decrease the number of unnecessary ERCP procedures in patients with suspected biliary pancreatitis. In these patients, EUS can be performed as an initial diagnostic test to exclude retained biliary stones. If a stone is present, the endoscopist can proceed to ERCP for sphincterotomy and stone removal during the same endoscopic session. If EUS is negative, the endoscopy can be concluded without cannulating the bile duct and putting the patient at risk of acute pancreatitis. In one report, this approach eliminated the need for ERCP in five of six patients with suspected biliary pancreatitis.6
Tumors and other causes of bile duct obstruction can also cause recurrent acute pancreatitis and may be difficult to detect with cross-sectional imaging. EUS, on the other hand, can detect small pancreatic masses (< 2 cm), which may be missed by conventional computed tomography. Also, a linear EUS scope, with its forward oblique view, can image the duodenum and ampulla, where obstructing inflammation, tumors, and polyps may be found. One should strongly suspect occult malignancy in elderly patients with unexplained acute pancreatitis. In those patients, repeat imaging with high-resolution dual-phase computed tomography or with EUS should be considered after a few weeks once the acute inflammation resolves.
Pancreas divisum is a relatively common congenital abnormality in which the dorsal and ventral pancreatic ducts do not properly fuse during embryonic development. To rule out pancreas divisum, the endosonographer must carefully trace the pancreatic duct from the dorsal pancreas into the ventral pancreas, where it connects with the bile duct at the duodenal wall.
In summary, EUS appears to be safe and accurate for diagnosing bile duct stones and other structural causes of idiopathic acute pancreatitis.
ENDOSCOPIC ULTRASONOGRAPHY IN CHRONIC PANCREATITIS
Chronic pancreatitis, a relatively common and sometimes debilitating cause of chronic upper abdominal pain, may be difficult to diagnose using noninvasive imaging tests. Minimal-change chronic pancreatitis is defined as a syndrome of pancreatic abdominal pain with no or slight structural changes detected on imaging but with histologic inflammation and fibrosis diagnostic of chronic pancreatitis.9
A clinical rationale for trying to detect chronic pancreatitis early in its course is that interventions can be started earlier. These include abstinence from alcohol, giving exogenous pancreatic enzymes, and advanced interventions such as celiac plexus blocks for pain control. Some patients may even benefit from resection of the pancreas if pain is severe and resistant to conservative measures.
EUS can detect both parenchymal and ductal changes that correlate with histologic fibrosis.10 Parenchymal changes include hyperechoic foci, hyperechoic strands, lobularity, cysts, and shadowing calcifications. Ductal changes include dilation of the main pancreatic duct, irregularity, hyperechoic duct margins, and visible side branches.
Several studies have evaluated the ability of EUS to diagnose early chronic pancreatitis.9,11–15 Reference standards used to determine the accuracy of EUS have included histology,10,16–18 pancreatic function testing,19–22 and ERCP.11,15,23,24
The best diagnostic test may be pancreatic histology. However, biopsy of the pancreas is impractical and exposes patients to high risk. In addition, the patchy and focal distribution of histologic changes may decrease its reliability. Fortunately, the histologic findings of fibrosis have been shown to correlate with EUS criteria in patients undergoing EUS before surgical resection in three recent studies.16–18 A threshold of four or more criteria out of a possible nine was found to provide the optimal sensitivity and specificity for histologic pancreatic fibrosis.16,17 The criteria used were four parenchymal features (hyperechoic foci, strands, hypoechoic lobules, cysts) and five ductal features (irregularity of the main pancreatic duct, dilation, hyperechoic duct walls, visible side branches, and calcifications or stones).
EUS is sensitive for chronic pancreatitis, but ‘true’ accuracy is impossible to know
Unfortunately, greater sensitivity may come at the expense of worse specificity. Certain demographic variables may alter the EUS appearance of the pancreas. A multivariate analysis25 found several variables that predicted abnormalities on EUS even in the absence of clinically evident pancreatitis; the strongest were heavy ethanol use (odds ratio [OR] 5.1, 95% confidence interval [CI] 3.1–8.5), male sex (OR 1.8, 95% CI 1.3–2.55), clinical suspicion of pancreatic disease (OR 1.7, 95% CI 1.2–2.3), and heavy smoking (OR 1.7, 95% CI 1.2–2.4). More prospective studies are needed to further differentiate true disease from false-positive findings of chronic pancreatitis.
Also, traditional EUS scoring symptoms have counted features in an unweighted fashion and assigned an arbitrary cut point (eg, four or more features) for diagnosis. This approach fails to account for the greater importance of some features (eg, calcifications) compared with others.
Interobserver variability is another important limitation of EUS in diagnosing chronic pancreatitis.26,27 In one multicenter study of EUS interpretation, the overall kappa (agreement beyond chance) was only 0.45 for overall chronic pancreatitis diagnosis and worse for many individual criteria for chronic pancreatitis. The endosonographers disagreed most about hyperechoic strands and foci, main pancreatic duct irregularity, and visible side branches (kappa < 0.4).
The Rosemont classification
These limitations led a group of experts to meet in Chicago, IL, to develop a consensus-based and weighted EUS scoring system for the diagnosis of chronic pancreatitis, termed the Rosemont classification.
In this system, the previous parenchymal and ductal features are assigned stricter definitions and reclassified as major and minor criteria. Based on the presence of major and minor features, EUS results are stratified as “normal,” “indeterminate for chronic pancreatitis,” “suggestive of chronic pancreatitis,” or “most consistent with chronic pancreatitis.”15,28
Further validation of this scoring system is needed before it can be used widely.
ENDOSCOPIC ULTRASONOGRAPHY PLUS PANCREATIC FUNCTION TESTING
The best way to diagnose minimal-change chronic pancreatitis may be a combination of sensitive structural and functional testing. Although clinically apparent steatorrhea typically occurs late in the course of chronic pancreatitis, mild exocrine insufficiency may occur early and is detectable with hormone-stimulated pancreatic function testing. Therefore, pancreatic function tests are considered sensitive for diagnosing chronic pancreatitis.20,21,29
Endoscopic pancreatic function testing involves injecting secretin intravenously and then collecting duodenal aspirates through the endoscope. The duodenal fluid is analyzed for bicarbonate concentration as a measure of exocrine function.29
We have studied combined EUS and endoscopic pancreatic function testing in the diagnosis of chronic pancreatitis.16 The combination gives a simultaneous structural and functional assessment of the pancreas and may optimize sensitivity for detecting minimal-change chronic pancreatitis. In a small study, we found the combination had 100% sensitivity for noncalcific chronic pancreatitis compared with a histologic reference standard.16
Endoscopic ultrasonography (EUS) is a minimally invasive test that provides high-resolution imaging of the pancreas.1,2 As such, it is proving useful.
Accurate diagnosis and timely intervention are essential in managing acute and chronic pancreatitis, which are often encountered in the clinic and the hospital. However, the cause of acute pancreatitis is not always easy to determine. Furthermore, recurrent bouts can progress to chronic pancreatitis if the cause is not identified and eliminated. EUS has been studied extensively in the evaluation of both acute and chronic pancreatitis, as it can identify obstructive and biliary causes of acute pancreatitis and early structural features of chronic pancreatitis.
This article will review the indications and evidence for EUS in the evaluation of acute and chronic pancreatitis.
SPECIALIZED TRAINING REQUIRED
EUS involves passage of a specialized endoscope through the esophagus and stomach and into the duodenum. The scope has a very small ultrasound probe at the tip, allowing detailed imaging of the upper gastrointestinal tract and surrounding organs.
There are two types of EUS endoscope: radial and linear. A radial scope provides a 360° range of view perpendicular to the long axis of the scope. A linear scope provides a 150° view parallel to the long axis of the scope. Many endosonographers favor linear EUS for imaging the pancreas because it permits fine-needle aspiration biopsy of masses, cysts, and lymph nodes.
Specialized training beyond the gastroenterology fellowship is usually required to become proficient in performing EUS, in recognizing the anatomy it reveals, and in performing fine-needle aspiration biopsy.
ENDOSCOPIC ULTRASONOGRAPHY IN ACUTE PANCREATITIS
Finding the cause of acute pancreatitis can be challenging in patients who do not have typical risk factors, eg, those who do not drink substantial amounts of alcohol and in whom transabdominal ultrasonography fails to reveal gallstones.
Several studies have evaluated the role of EUS in recurrent “idiopathic” pancreatitis.3–5 Causes of acute pancreatitis detectable with EUS included gallbladder and bile duct microlithiasis (stones smaller than 3 mm), cysts, intraductal papillary mucinous neoplasms, ampullary neoplasms, pancreas divisum, and pancreatic masses.
Stones, sludge. Transabdominal ultrasonography is often performed in the workup of acute pancreatitis to rule out gallbladder stones and biliary dilation. Unfortunately, it does a poor job of imaging the distal common bile duct, where culprit stones may reside.
EUS provides a high-quality view of the bile duct from the ampulla of Vater to the region of the hepatic hilum and is safer than endoscopic retrograde cholangiopancreatography (ERCP). The available evidence supports the use of EUS as a diagnostic test for bile duct stones.3–7 In fact, using ERCP as the reference standard, EUS has been found to be more sensitive than transabdominal ultrasonography for bile duct stones.4
The yield of EUS for finding biliary sludge and stones may be high in patients with unexplained pancreatitis. EUS detected sludge, microlithiasis, or both in 33 of 35 patients with idiopathic acute pancreatitis who underwent transabdominal ultrasonography with negative results.8 Furthermore, most were symptom-free at an average of 10 months after cholecystectomy, suggesting that microlithiasis was the cause of the “idiopathic” pancreatitis.
EUS can also decrease the number of unnecessary ERCP procedures in patients with suspected biliary pancreatitis. In these patients, EUS can be performed as an initial diagnostic test to exclude retained biliary stones. If a stone is present, the endoscopist can proceed to ERCP for sphincterotomy and stone removal during the same endoscopic session. If EUS is negative, the endoscopy can be concluded without cannulating the bile duct and putting the patient at risk of acute pancreatitis. In one report, this approach eliminated the need for ERCP in five of six patients with suspected biliary pancreatitis.6
Tumors and other causes of bile duct obstruction can also cause recurrent acute pancreatitis and may be difficult to detect with cross-sectional imaging. EUS, on the other hand, can detect small pancreatic masses (< 2 cm), which may be missed by conventional computed tomography. Also, a linear EUS scope, with its forward oblique view, can image the duodenum and ampulla, where obstructing inflammation, tumors, and polyps may be found. One should strongly suspect occult malignancy in elderly patients with unexplained acute pancreatitis. In those patients, repeat imaging with high-resolution dual-phase computed tomography or with EUS should be considered after a few weeks once the acute inflammation resolves.
Pancreas divisum is a relatively common congenital abnormality in which the dorsal and ventral pancreatic ducts do not properly fuse during embryonic development. To rule out pancreas divisum, the endosonographer must carefully trace the pancreatic duct from the dorsal pancreas into the ventral pancreas, where it connects with the bile duct at the duodenal wall.
In summary, EUS appears to be safe and accurate for diagnosing bile duct stones and other structural causes of idiopathic acute pancreatitis.
ENDOSCOPIC ULTRASONOGRAPHY IN CHRONIC PANCREATITIS
Chronic pancreatitis, a relatively common and sometimes debilitating cause of chronic upper abdominal pain, may be difficult to diagnose using noninvasive imaging tests. Minimal-change chronic pancreatitis is defined as a syndrome of pancreatic abdominal pain with no or slight structural changes detected on imaging but with histologic inflammation and fibrosis diagnostic of chronic pancreatitis.9
A clinical rationale for trying to detect chronic pancreatitis early in its course is that interventions can be started earlier. These include abstinence from alcohol, giving exogenous pancreatic enzymes, and advanced interventions such as celiac plexus blocks for pain control. Some patients may even benefit from resection of the pancreas if pain is severe and resistant to conservative measures.
EUS can detect both parenchymal and ductal changes that correlate with histologic fibrosis.10 Parenchymal changes include hyperechoic foci, hyperechoic strands, lobularity, cysts, and shadowing calcifications. Ductal changes include dilation of the main pancreatic duct, irregularity, hyperechoic duct margins, and visible side branches.
Several studies have evaluated the ability of EUS to diagnose early chronic pancreatitis.9,11–15 Reference standards used to determine the accuracy of EUS have included histology,10,16–18 pancreatic function testing,19–22 and ERCP.11,15,23,24
The best diagnostic test may be pancreatic histology. However, biopsy of the pancreas is impractical and exposes patients to high risk. In addition, the patchy and focal distribution of histologic changes may decrease its reliability. Fortunately, the histologic findings of fibrosis have been shown to correlate with EUS criteria in patients undergoing EUS before surgical resection in three recent studies.16–18 A threshold of four or more criteria out of a possible nine was found to provide the optimal sensitivity and specificity for histologic pancreatic fibrosis.16,17 The criteria used were four parenchymal features (hyperechoic foci, strands, hypoechoic lobules, cysts) and five ductal features (irregularity of the main pancreatic duct, dilation, hyperechoic duct walls, visible side branches, and calcifications or stones).
EUS is sensitive for chronic pancreatitis, but ‘true’ accuracy is impossible to know
Unfortunately, greater sensitivity may come at the expense of worse specificity. Certain demographic variables may alter the EUS appearance of the pancreas. A multivariate analysis25 found several variables that predicted abnormalities on EUS even in the absence of clinically evident pancreatitis; the strongest were heavy ethanol use (odds ratio [OR] 5.1, 95% confidence interval [CI] 3.1–8.5), male sex (OR 1.8, 95% CI 1.3–2.55), clinical suspicion of pancreatic disease (OR 1.7, 95% CI 1.2–2.3), and heavy smoking (OR 1.7, 95% CI 1.2–2.4). More prospective studies are needed to further differentiate true disease from false-positive findings of chronic pancreatitis.
Also, traditional EUS scoring symptoms have counted features in an unweighted fashion and assigned an arbitrary cut point (eg, four or more features) for diagnosis. This approach fails to account for the greater importance of some features (eg, calcifications) compared with others.
Interobserver variability is another important limitation of EUS in diagnosing chronic pancreatitis.26,27 In one multicenter study of EUS interpretation, the overall kappa (agreement beyond chance) was only 0.45 for overall chronic pancreatitis diagnosis and worse for many individual criteria for chronic pancreatitis. The endosonographers disagreed most about hyperechoic strands and foci, main pancreatic duct irregularity, and visible side branches (kappa < 0.4).
The Rosemont classification
These limitations led a group of experts to meet in Chicago, IL, to develop a consensus-based and weighted EUS scoring system for the diagnosis of chronic pancreatitis, termed the Rosemont classification.
In this system, the previous parenchymal and ductal features are assigned stricter definitions and reclassified as major and minor criteria. Based on the presence of major and minor features, EUS results are stratified as “normal,” “indeterminate for chronic pancreatitis,” “suggestive of chronic pancreatitis,” or “most consistent with chronic pancreatitis.”15,28
Further validation of this scoring system is needed before it can be used widely.
ENDOSCOPIC ULTRASONOGRAPHY PLUS PANCREATIC FUNCTION TESTING
The best way to diagnose minimal-change chronic pancreatitis may be a combination of sensitive structural and functional testing. Although clinically apparent steatorrhea typically occurs late in the course of chronic pancreatitis, mild exocrine insufficiency may occur early and is detectable with hormone-stimulated pancreatic function testing. Therefore, pancreatic function tests are considered sensitive for diagnosing chronic pancreatitis.20,21,29
Endoscopic pancreatic function testing involves injecting secretin intravenously and then collecting duodenal aspirates through the endoscope. The duodenal fluid is analyzed for bicarbonate concentration as a measure of exocrine function.29
We have studied combined EUS and endoscopic pancreatic function testing in the diagnosis of chronic pancreatitis.16 The combination gives a simultaneous structural and functional assessment of the pancreas and may optimize sensitivity for detecting minimal-change chronic pancreatitis. In a small study, we found the combination had 100% sensitivity for noncalcific chronic pancreatitis compared with a histologic reference standard.16
- Sivak MV, Kaufman A. Endoscopic ultrasonography in the differential diagnosis of pancreatic disease. A preliminary report. Scand J Gastroenterol Suppl 1986; 123:130–134.
- Hisanaga K, Hisanaga A, Nagata K, Ichie Y. High speed rotating scanner for transgastric sonography. AJR Am J Roentgenol 1980; 135:627–629.
- Frossard JL, Sosa-Valencia L, Amouyal G, Marty O, Hadengue A, Amouyal P. Usefulness of endoscopic ultrasonography in patients with “idiopathic” acute pancreatitis. Am J Med 2000; 109:196–200.
- Sugiyama M, Wada N, Atomi Y, Kuroda A, Muto T. Diagnosis of acute pancreatitis: value of endoscopic sonography. AJR Am J Roentgenol 1995; 165:867–872.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Liu CL, Lo CM, Chan JK, et al. Detection of choledocholithiasis by EUS in acute pancreatitis: a prospective evaluation in 100 consecutive patients. Gastrointest Endosc 2001; 54:325–330.
- Mirbagheri SA, Mohamadnejad M, Nasiri J, Vahid AA, Ghadimi R, Malekzadeh R. Prospective evaluation of endoscopic ultrasonography in the diagnosis of biliary microlithiasis in patients with normal transabdominal ultrasonography. J Gastrointest Surg 2005; 9:961–964.
- Walsh TN, Rode J, Theis BA, Russell RC. Minimal change chronic pancreatitis. Gut 1992; 33:1566–1571.
- Bhutani MJ, Arantes VN, Verma D, et al. Histopathologic correlation of endoscopic ultrasound findings of chronic pancreatitis in human autopsies. Pancreas 2009; 38:820–824.
- Wiersema MJ, Hawes RH, Lehman GA, Kochman ML, Sherman S, Kopecky KK. Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy 1993; 25:555–564.
- Kahl S, Glasbrenner B, Leodolter A, Pross M, Schulz HU, Malfertheiner P. EUS in the diagnosis of early chronic pancreatitis: a prospective follow-up study. Gastrointest Endosc 2002; 55:507–511.
- Jones SN, Lees WR, Frost RA. Diagnosis and grading of chronic pancreatitis by morphological criteria derived by ultrasound and pancreatography. Clin Radiol 1988; 39:43–48.
- Lees WR. Endoscopic ultrasonography of chronic pancreatitis and pancreatic pseudocysts. Scand J Gastroenterol Suppl 1986; 123:123–129.
- Sahai AV, Zimmerman M, Aabakken L, et al. Prospective assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pancreatitis found by endoscopic retrograde cholangiopancreatography. Gastrointest Endosc 1998; 48:18–25.
- Albashir S, Bronner MP, Parsi MA, Walsh RM, Stevens T. Endoscopic ultrasound, secretin endoscopic pancreatic function test, and histology: correlation in chronic pancreatitis. Am J Gastroenterol 2010; 105:2498–2503.
- Varadarajulu S, Eltoum I, Tamhane A, Eloubeidi MA. Histopathologic correlates of noncalcific chronic pancreatitis by EUS: a prospective tissue characterization study. Gastrointest Endosc 2007; 66:501–509.
- Chong AK, Hawes RH, Hoffman BJ, Adams DB, Lewin DN, Romagnuolo J. Diagnostic performance of EUS for chronic pancreatitis: a comparison with histopathology. Gastrointest Endosc 2007; 65:808–814.
- Chowdhury R, Bhutani MS, Mishra G, Toskes PP, Forsmark CE. Comparative analysis of direct pancreatic function testing versus morphological assessment by endoscopic ultrasonography for the evaluation of chronic unexplained abdominal pain of presumed pancreatic origin. Pancreas 2005; 31:63–68.
- Conwell DL, Zuccaro G, Purich E, et al. Comparison of endoscopic ultrasound chronic pancreatitis criteria to the endoscopic secretinstimulated pancreatic function test. Dig Dis Sci 2007; 52:1206–1210.
- Stevens T, Conwell DL, Zuccaro G, Vargo JJ, Dumot JA, Lopez R. Comparison of endoscopic ultrasound and endoscopic retrograde pancreatography for the prediction of pancreatic exocrine insufficiency. Dig Dis Sci 2008; 53:1146–1151.
- Stevens T, Dumot JA, Parsi MA, Zuccaro G, Vargo JJ. Combined endoscopic ultrasound and secretin endoscopic pancreatic function test in patients evaluated for chronic pancreatitis. Dig Dis Sci 2010; 55:2681–2687.
- Catalano MF, Lahoti S, Geenen JE, Hogan WJ. Prospective evaluation of endoscopic ultrasonography, endoscopic retrograde pancreatography, and secretin test in the diagnosis of chronic pancreatitis. Gastrointest Endosc 1998; 48:11–17.
- Irisawa A, Katakura K, Ohira H, et al. Usefulness of endoscopic ultrasound to diagnose the severity of chronic pancreatitis. J Gastroenterol 2007; 42(suppl 17):90–94.
- Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol 2004; 2:405–409.
- Stevens T, Lopez R, Adler DG, et al. Multicenter comparison of the interobserver agreement of standard EUS scoring and Rosemont classification scoring for diagnosis of chronic pancreatitis. Gastrointest Endosc 2010; 71:519–526.
- Wallace MB, Hawes RH, Durkalski V, et al. The reliability of EUS for the diagnosis of chronic pancreatitis: interobserver agreement among experienced endosonographers. Gastrointest Endosc 2001; 53:294–299.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Stevens T, Conwell DL, Zuccaro G, et al. A prospective crossover study comparing secretin-stimulated endoscopic and Dreiling tube pancreatic function testing in patients evaluated for chronic pancreatitis. Gastrointest Endosc 2008; 67:458–466.
- Sivak MV, Kaufman A. Endoscopic ultrasonography in the differential diagnosis of pancreatic disease. A preliminary report. Scand J Gastroenterol Suppl 1986; 123:130–134.
- Hisanaga K, Hisanaga A, Nagata K, Ichie Y. High speed rotating scanner for transgastric sonography. AJR Am J Roentgenol 1980; 135:627–629.
- Frossard JL, Sosa-Valencia L, Amouyal G, Marty O, Hadengue A, Amouyal P. Usefulness of endoscopic ultrasonography in patients with “idiopathic” acute pancreatitis. Am J Med 2000; 109:196–200.
- Sugiyama M, Wada N, Atomi Y, Kuroda A, Muto T. Diagnosis of acute pancreatitis: value of endoscopic sonography. AJR Am J Roentgenol 1995; 165:867–872.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Kotwal V, Talukdar R, Levy M, Vege SS. Role of endoscopic ultrasound during hospitalization for acute pancreatitis. World J Gastroenterol 2010; 16:4888–4891.
- Liu CL, Lo CM, Chan JK, et al. Detection of choledocholithiasis by EUS in acute pancreatitis: a prospective evaluation in 100 consecutive patients. Gastrointest Endosc 2001; 54:325–330.
- Mirbagheri SA, Mohamadnejad M, Nasiri J, Vahid AA, Ghadimi R, Malekzadeh R. Prospective evaluation of endoscopic ultrasonography in the diagnosis of biliary microlithiasis in patients with normal transabdominal ultrasonography. J Gastrointest Surg 2005; 9:961–964.
- Walsh TN, Rode J, Theis BA, Russell RC. Minimal change chronic pancreatitis. Gut 1992; 33:1566–1571.
- Bhutani MJ, Arantes VN, Verma D, et al. Histopathologic correlation of endoscopic ultrasound findings of chronic pancreatitis in human autopsies. Pancreas 2009; 38:820–824.
- Wiersema MJ, Hawes RH, Lehman GA, Kochman ML, Sherman S, Kopecky KK. Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy 1993; 25:555–564.
- Kahl S, Glasbrenner B, Leodolter A, Pross M, Schulz HU, Malfertheiner P. EUS in the diagnosis of early chronic pancreatitis: a prospective follow-up study. Gastrointest Endosc 2002; 55:507–511.
- Jones SN, Lees WR, Frost RA. Diagnosis and grading of chronic pancreatitis by morphological criteria derived by ultrasound and pancreatography. Clin Radiol 1988; 39:43–48.
- Lees WR. Endoscopic ultrasonography of chronic pancreatitis and pancreatic pseudocysts. Scand J Gastroenterol Suppl 1986; 123:123–129.
- Sahai AV, Zimmerman M, Aabakken L, et al. Prospective assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pancreatitis found by endoscopic retrograde cholangiopancreatography. Gastrointest Endosc 1998; 48:18–25.
- Albashir S, Bronner MP, Parsi MA, Walsh RM, Stevens T. Endoscopic ultrasound, secretin endoscopic pancreatic function test, and histology: correlation in chronic pancreatitis. Am J Gastroenterol 2010; 105:2498–2503.
- Varadarajulu S, Eltoum I, Tamhane A, Eloubeidi MA. Histopathologic correlates of noncalcific chronic pancreatitis by EUS: a prospective tissue characterization study. Gastrointest Endosc 2007; 66:501–509.
- Chong AK, Hawes RH, Hoffman BJ, Adams DB, Lewin DN, Romagnuolo J. Diagnostic performance of EUS for chronic pancreatitis: a comparison with histopathology. Gastrointest Endosc 2007; 65:808–814.
- Chowdhury R, Bhutani MS, Mishra G, Toskes PP, Forsmark CE. Comparative analysis of direct pancreatic function testing versus morphological assessment by endoscopic ultrasonography for the evaluation of chronic unexplained abdominal pain of presumed pancreatic origin. Pancreas 2005; 31:63–68.
- Conwell DL, Zuccaro G, Purich E, et al. Comparison of endoscopic ultrasound chronic pancreatitis criteria to the endoscopic secretinstimulated pancreatic function test. Dig Dis Sci 2007; 52:1206–1210.
- Stevens T, Conwell DL, Zuccaro G, Vargo JJ, Dumot JA, Lopez R. Comparison of endoscopic ultrasound and endoscopic retrograde pancreatography for the prediction of pancreatic exocrine insufficiency. Dig Dis Sci 2008; 53:1146–1151.
- Stevens T, Dumot JA, Parsi MA, Zuccaro G, Vargo JJ. Combined endoscopic ultrasound and secretin endoscopic pancreatic function test in patients evaluated for chronic pancreatitis. Dig Dis Sci 2010; 55:2681–2687.
- Catalano MF, Lahoti S, Geenen JE, Hogan WJ. Prospective evaluation of endoscopic ultrasonography, endoscopic retrograde pancreatography, and secretin test in the diagnosis of chronic pancreatitis. Gastrointest Endosc 1998; 48:11–17.
- Irisawa A, Katakura K, Ohira H, et al. Usefulness of endoscopic ultrasound to diagnose the severity of chronic pancreatitis. J Gastroenterol 2007; 42(suppl 17):90–94.
- Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol 2004; 2:405–409.
- Stevens T, Lopez R, Adler DG, et al. Multicenter comparison of the interobserver agreement of standard EUS scoring and Rosemont classification scoring for diagnosis of chronic pancreatitis. Gastrointest Endosc 2010; 71:519–526.
- Wallace MB, Hawes RH, Durkalski V, et al. The reliability of EUS for the diagnosis of chronic pancreatitis: interobserver agreement among experienced endosonographers. Gastrointest Endosc 2001; 53:294–299.
- Catalano MF, Sahai A, Levy M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009; 69:1251–1261.
- Stevens T, Conwell DL, Zuccaro G, et al. A prospective crossover study comparing secretin-stimulated endoscopic and Dreiling tube pancreatic function testing in patients evaluated for chronic pancreatitis. Gastrointest Endosc 2008; 67:458–466.
KEY POINTS
- EUS can identify the cause of acute pancreatitis when other imaging tests (computed tomography, transabdominal ultrasonography) are unrevealing.
- EUS can safely and accurately detect bile duct stones and other causes of recurrent acute pancreatitis. It can also detect mild and severe structural features of chronic pancreatitis.
- An endoscopic pancreatic function test may be a useful adjunct to EUS to detect mild exocrine insufficiency in early chronic pancreatitis.
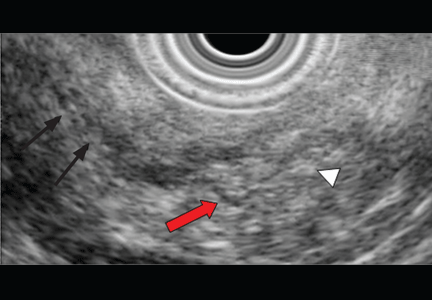
Acute pancreatitis: Problems in adherence to guidelines
Several major gastroenterological and surgical societies have issued guidelines on how to manage acute pancreatitis, based on evidence from high-quality randomized trials and nonrandomized studies as well as on expert opinion.1–3 Information is limited on how well physicians in the United States comply with these guidelines, but compliance is suboptimal in other developed countries, according to several studies,4–8 and we suspect that many US physicians are not following the guidelines either.
Acute pancreatitis is a frequent inpatient diagnosis that internists, gastroenterologists, and surgeons all confront. The most common causes are gallstones and heavy alcohol intake. Its management is typically straightforward: intravenous fluids, analgesia, and nothing by mouth. However, treatment of severe cases can be quite complex, particularly if multiple organ systems are involved or if there are local complications.
The primary aim of this article is to raise awareness of recognized deviations from established recommendations that may lead to adverse patient outcomes.
MEASURING ENZYME LEVELS DAILY ADDS COST BUT LITTLE BENEFIT
Problem: Serum amylase and lipase levels are often needlessly measured every day.
Measuring the serum amylase and lipase levels is useful in diagnosing acute pancreatitis, which requires two of the following three features1:
- Characteristic abdominal pain
- Levels of serum amylase or serum lipase, or both, that are three or more times the upper limit of normal
- Findings of acute pancreatitis on computed tomography (CT).
However, the magnitude or duration of the serum enzyme elevation does not correlate with the severity of the attack. Further, we have noticed that physicians at our hospital often order daily serum amylase and lipase levels in patients admitted with acute pancreatitis.
The American College of Gastroenterology (ACG) guidelines1 state that daily monitoring of amylase and lipase has limited value in managing acute pancreatitis. Rechecking these concentrations may be reasonable if pain fails to resolve or worsens during a prolonged hospitalization, as this may suggest a recurrent attack of acute pancreatitis or a developing pseudocyst. But in most cases of acute pancreatitis, daily serum enzyme measurements add cost but little benefit.
REGULAR ASSESSMENT IS IMPORTANT
Problem: Often, severity assessments are not performed regularly or acted on.
Most cases of acute pancreatitis are mild, with rapid recovery and excellent prognosis. However, 15% to 20% are severe and may result in a prolonged hospitalization, systemic inflammatory response syndrome (SIRS), multiorgan system failure, and death.
In severe acute pancreatitis, as pancreatic enzymes and inflammatory cytokines damage the blood vessels, a vast amount of fluid leaks out into the interstitial (“third”) space. This fluid extravasation leads to decreased effective circulating volume, local pancreatic necrosis, hemodynamic instability, and end-organ failure.
It is important to recognize severe acute pancreatitis early because the patient needs to be transferred to a step-down unit or intensive care unit to receive optimal fluid resuscitation and supportive care for organ dysfunction. After 48 to 72 hours, a prediction of severe acute pancreatitis should also prompt the physician to order CT to detect pancreatic necrosis, and also to initiate nutritional support.
Assessment of severity begins in the emergency room or on admission to the hospital. Older age, obesity, organ failure, and pulmonary infiltrates or pleural effusions are initial indicators of poor prognosis. Signs of SIRS (high or low core body temperature, tachycardia, tachypnea, low or high peripheral white blood cell count) or organ failure (eg, elevated serum creatinine) are present on admission in 21% of patients with acute pancreatitis.9
Hemoconcentration is a marker of decreased effective circulating volume in severe acute pancreatitis. A hematocrit higher than 44% at admission or that rises in the first 24 to 48 hours of admission predicts necrosis.10,11 However, a more robust marker of organ failure may be the blood urea nitrogen level.12
Clinical scoring systems
Several clinical scoring systems have been studied for assessing severity.

Limitations of the Ranson score are that it can only be completed after 48 hours, all the data points are not always obtained, and it cannot be repeated on a daily basis. Owing to these limitations and its less-than-optimal predictive value, the Ranson score has fallen into disuse.
The APACHE II (Acute Physiology and Chronic Health Evaluation II) score is more versatile. It is based on multiple clinical and laboratory values, and it correlates very well with the risk of death in acute pancreatitis. Death rates are less than 4% when the APACHE II score is less than 8, and 11% to 18% when it is 8 or higher.1 The trajectory of the APACHE II score in the first 48 hours is also an accurate prognostic indicator.
Previous limitations of the APACHE II score were that it was complicated and timeconsuming to calculate and required arterial blood gas measurements. Easy-to-use online calculators are now available (eg, www.globalrph.com/apacheii.htm), and the venous bicarbonate level and the oxygen saturation can be substituted for the arterial pH and oxygen partial pressure.
BISAP, a new five-point scoring system,15 was recently prospectively validated.12 “BISAP” is an acronym for the five markers it is based on, each of which has been shown to predict severe illness in acute pancreatitis:
- Blood urea nitrogen level > 25 mg/dL
- Impaired mental status
- SIRS
- Age > 60 years
- Pleural effusion.
The presence of three or more of these factors correlates with higher risk of death, organ failure, and pancreatic necrosis.12
Compared with APACHE II, BISAP has similar accuracy and is easier to calculate. Also, BISAP was specifically developed for acute pancreatitis, whereas APACHE II is a generic score for all critically ill patients.
The Atlanta criteria16 define severe acute pancreatitis as one or more of the following:
- A Ranson score of 3 or higher during the first 48 hours
- An APACHE II score of 8 or higher at any time
- Failure of one or more organs
- One or more local complications (eg, necrosis, pseudocysts, abscesses).
Recommendation: Assess severity at least daily
A severity assessment should be performed at admission and at least every day thereafter. Clinical guidelines recognize the importance of severity assessment but vary in their specific recommendations.
The ACG advises calculating the APACHE II score within 3 days of admission and measuring the hematocrit at admission, at 12 hours, and at 24 hours. The level of evidence is III, ie, “from published well-designed trials without randomization, single group prepost, cohort, time series, or matched case controlled studies”.1
The American Gastroenterological Association (AGA) provides a more generalized recommendation, that “clinical judgment” should take into account the presence of risk factors (eg, age, obesity), presence or absence of SIRS, routine laboratory values (eg, hematocrit, serum creatinine), and APACHE II score when assessing severity and making decisions.2
In a German survey, only 32% of gastroenterologists used the APACHE II score for assessing risk in acute pancreatitis, in spite of national guidelines emphasizing its importance.7 Also, not all patients with severe acute pancreatitis are transferred to a step-down unit or intensive care unit as recommended. In a British study,4 only 8 (17%) of 46 patients with predicted severe acute pancreatitis were transferred, and 8 of the 38 patients who were not transferred died.
FLUID MUST BE AGGRESSIVELY REPLACED AND MONITORED
Problem: Often, not enough fluid is replaced, or fluid status is not adequately monitored.
Fluid must be aggressively replaced to balance the massive third-space fluid losses that occur in the early inflammatory phase of acute pancreatitis. Intravascular volume depletion can develop rapidly and result in tachycardia, hypotension, and renal failure. It may also impair the blood flow to the pancreas and worsen necrosis.
Animal studies show that aggressive fluid replacement supports the pancreatic microcirculation and prevents necrosis.17 It may also support the intestinal microcirculation and gut barrier, preventing bacterial translocation.
In humans, no controlled trials have been done to test the efficacy of aggressive fluid resuscitation in acute pancreatitis. However, the notion that intravascular fluid loss contributes to poor outcomes is inferred from human studies showing more necrosis and deaths in patients with hemoconcentration. In one study, patients who received inadequate fluid replacement (evidenced by a rise in hematocrit at 24 hours) were more likely to develop necrotizing pancreatitis.18
Recommendation: Early, aggressive fluid replacement
Experts have suggested initially infusing 500 to 1,000 mL of fluid per hour in those who are volume-depleted, initially infusing 250 to 350 mL per hour in those who are not volumedepleted, and adjusting the fluid rate every 1 to 4 hours on the basis of clinical variables.19 The sufficiency of fluid replacement should be carefully monitored by vital signs, urine output, and serum hematocrit.
On the other hand, overly aggressive fluid resuscitation can be detrimental in patients at risk of volume overload or pulmonary edema. Fluid replacement should be tempered in elderly patients and those with cardiac or renal comorbidities, and may require monitoring of central venous pressure.
The ACG and AGA guidelines both recognize the need for early aggressive volume replacement in acute pancreatitis (level of evidence III), but they do not specify the exact amounts and rates. Young and healthy patients should receive a rapid bolus of isotonic saline or Ringer’s lactate solution followed by an infusion at a high initial maintenance rate.
Few studies have been done to assess physicians’ compliance with recommendations for aggressive volume replacement. In an Italian multicenter study, patients with mild or severe acute pancreatitis received an average of only 2.5 L of fluid per day (about 100 mL/hour).20 Gardner et al21 recently summarized the available evidence for fluid support in acute pancreatitis.
NUTRITIONAL SUPPORT
Problem: In many severe cases, enteral or parenteral feeding is not started soon enough.
Nutritional support entails enteral or parenteral feeding when an oral diet is contraindicated. Enteral feeding is usually via a nasojejunal tube, which may need to be placed under endoscopic or radiographic guidance. Neither parenteral nor nasojejunal feeding stimulates pancreatic secretion, and both are safe in acute pancreatitis.
Severe acute pancreatitis is an intensely catabolic state characterized by increased energy expenditure, protein breakdown, and substrate utilization. Patients may not be able to resume an oral diet for weeks or even months, particularly if local complications develop. Early nutritional support has been shown to improve outcomes in severe acute pancreatitis.22 Therefore, nutritional support should be started as soon as possible in severe acute pancreatitis based on initial clinical and radiographic indicators of severity, optimally within the first 2 or 3 days.
Enteral nutrition is preferred to parenteral nutrition in pancreatitis: it is less expensive and does not pose a risk of catheter-related infection or thrombosis or hepatic complications. Also, there is experimental evidence that enteral nutrition may preserve the gut barrier, decreasing mucosal permeability and bacterial translocation.
A number of small randomized trials compared enteral and parenteral nutrition in acute pancreatitis, but they yielded mixed results. A meta-analysis of six trials showed a lower rate of infectious complications with enteral than with parenteral nutrition. 23 However, no significant difference was found in the rates of death or noninfectious complications.
Recommendation: Enteral feeding, when possible
Nutritional support is unnecessary in most cases of mild acute pancreatitis. Pancreatic inflammation typically resolves within a few days, allowing patients to resume eating. Occasionally, patients in whom pain resolves slowly and who fast for more than 5 to 7 days need nutritional support to prevent proteincalorie malnutrition.
The ACG guidelines1 and most others suggest that, whenever possible, enteral rather than parenteral feeding should be given to those who require nutritional support. The level of evidence is II (“strong evidence from at least one published properly designed randomized controlled trial of appropriate size and in an appropriate clinical setting”).
However, not all physicians recognize the benefit of enteral feeding. In a cohort of German gastroenterologists, only 73% favored enteral over parenteral feeding in acute pancreatitis.7
COMPUTED TOMOGRAPHY
Problem: CT is not done in many patients with severe acute pancreatitis, or it is done too soon during the admission.
Dual-phase, contrast-enhanced, pancreatic-protocol CT provides a sensitive structural evaluation of the pancreas and is useful to diagnose necrotizing pancreatitis. Pancreatic necrosis is correlated with a severe clinical course, the development of single or multiorgan dysfunction, and death.

Recommendation: CT in severe cases
Not every patient with acute pancreatitis needs to undergo CT. Most mild cases do not require routine CT, since necrosis and other local complications are infrequent in this group.
Also, CT is often ordered too soon during the hospitalization. Indicators of severity on CT are not usually evident until 2 to 3 days after admission.25 CT should be considered about 3 days after the onset of symptoms rather than immediately upon admission.
On the other hand, CT at the time of admission may be warranted to rule out other life-threatening causes of abdominal pain and hyperamylasemia (eg, bowel obstruction, viscus perforation). CT may also be useful in the late phase of acute pancreatitis (weeks after admission) to diagnose or monitor complications (eg, pseudocysts, abscesses, splenic vein thrombosis, splenic artery pseudoaneurysms). Magnetic resonance imaging with gadolinium contrast is a reasonable alternative to CT for detecting pancreatic necrosis and other local complications.
In patients who have severe acute pancreatitis and compromised renal function (serum creatinine > 1.5 mg/dL), CT can be performed without contrast to assess severity based on a limited Balthazar score (ie, without a necrosis score). Studies in rats suggest that iodinated contrast may decrease pancreatic microcirculation and worsen or precipitate necrosis,26 although published human studies do not support this contention.27,28
Guidelines uniformly recommend CT for patients with severe acute pancreatitis (the ACG guideline gives it a level of evidence of III), but this recommendation is not always followed. A study from Australia showed that CT was done in only 27% to 67% of patients with severe acute pancreatitis.5 In a British study, only 8 of 46 patients with clinically predicted severe pancreatitis underwent CT within the first 10 days of admission.4
SUSPECTED INFECTED NECROSIS
Problem: Fine-needle aspiration is not done in many cases of suspected infected necrosis.
Approximately one-third of patients with necrotizing pancreatitis develop infected necrosis. The death rate for patients with infected pancreatic necrosis is high—30%, compared with 12% in those with sterile necrosis.1 Differentiating sterile and infected necrosis is therefore essential.
Clinical signs such as fever are poor predictors of infection. Signs of SIRS can be present in both sterile and infected necrotizing pancreatitis.
Recommendation: Fine-needle aspiration of necrosis
For the reasons given above, the findings of necrosis on CT and persistent SIRS should prompt consideration of fine-needle aspiration with Gram stain and culture to differentiate sterile and infected necrosis (ACG guideline, level of evidence III).1 If infection is confirmed, surgical debridement should be strongly considered. Other less-invasive approaches such as endoscopic debridement can be used in selected cases.
Fine-needle aspiration of necrosis is too often neglected. In a cohort of German surgeons, only 55% complied with International Association of Pancreatology recommendations to perform biopsy to differentiate sterile from infected necrosis in patients with signs of sepsis.29
BROAD-SPECTRUM ANTIBIOTICS
Problem: Broad-spectrum antibiotics are often used inappropriately in patients with mild acute pancreatitis and in patients with sterile necrotizing pancreatitis who are clinically stable and have no signs of sepsis.
Antibiotics are not indicated in mild acute pancreatitis. A limited course of antibiotics is typically indicated in severe cases with suspected or proven infected necrosis (in conjunction with surgical necrosectomy). However, the use of antibiotics in sterile necrosis has been very controversial.
At least six small, nonblinded, randomized trials have evaluated the benefit of giving antibiotics prophylactically for presumed sterile necrosis. A recent Cochrane analysis of five of these trials (294 patients) suggested that patients who got antibiotics had a lower risk of death (odds ratio 0.37, 95% confidence interval [CI] 0.17–0.83) but no difference in the rates of pancreatic infection or surgery.30 These paradoxical results suggest that antibiotics may prevent death by preventing nonpancreatic infections (eg, pneumonia, line infections) rather than by preventing infection of necrotic pancreatic tissue. The five trials in the meta-analysis are limited by significant methodologic heterogeneity and by lack of double-blinding.
In spite of the overall lower death rate observed in the meta-analysis, the prophylactic use of antibiotics in sterile necrosis remains controversial. One concern is that patients given long prophylactic courses of antibiotics may develop resistant bacterial or fungal infections. However, the Cochrane and other meta-analyses have not shown a higher rate of fungal infections in those given antibiotics.31
Recommendation: No routine antibiotics for mild cases
The AGA guidelines recommend against routinely giving antibiotics in mild acute pancreatitis and do not provide strict recommendations for prophylactic antibiotic use in necrotizing acute pancreatitis.2 The guidelines state that antibiotics can be used “on demand” based on clinical signs of infection (eg, high fevers, rising leukocytosis, hypotension) or worsening organ failure.
If a purely prophylactic strategy is used, only patients at high risk of developing infection (eg, those with necrosis in more than 30% of the pancreas) should receive antibiotics. Antibiotics with high tissue-penetration should be used, such as imipenem-cilastin (Primaxin IV) or ciprofloxacin (Cipro) plus metronidazole (Flagyl).
Adherence to these guidelines is not optimal. For example, in an Italian multicenter study, 9% of patients with mild acute pancreatitis were treated with antibiotics.19 Moreover, many patients with proven infected necrosis received antibiotics that do not penetrate the pancreatic tissue very well.
ERCP IN SEVERE BILIARY ACUTE PANCREATITIS
Problem: Endoscopic retrograde cholangiopancreatography (ERCP) often is performed inappropriately in mild biliary acute pancreatitis or is not performed urgently in severe cases.
In most cases of mild biliary pancreatitis, the stones pass spontaneously, as verified by cholangiography done during laparoscopic cholecystectomy. Ongoing ampullary obstruction by impacted biliary stones can perpetuate pancreatic inflammation and delay recovery.
Two early randomized trials showed a benefit from early ERCP (within 72 hours) with sphincterotomy and stone extraction, primarily in those with severe biliary acute pancreatitis or ascending cholangitis,32,33 but a third trial failed to reveal a benefit.34 A Cochrane metaanalysis of these three trials failed to show a lower death rate with ERCP in mild or severe biliary pancreatitis.35 However, early ERCP did prevent complications in severe biliary pancreatitis (odds ratio 0.27, 95% CI 0.14–0.53).
Later, a fourth randomized trial was restricted to patients with suspected biliary pancreatitis, evidence of biliary obstruction, and no signs of cholangitis36: 103 patients were randomized to undergo either ERCP within 72 hours or conservative management. No difference was observed in rates of death or organ failure or in the CT severity index.
Recommendation: ER CP for suspected retained stones
ERCP has a limited role in patients with biliary pancreatitis, being used to clear retained bile duct stones or to relieve ongoing biliary obstruction.
The decision to perform ERCP before surgery should be based on how strongly one suspects retained stones. ERCP is most appropriate if the suspicion of retained stones and the likelihood of therapeutic intervention are high (eg, if the serum bilirubin and alkaline phosphatase levels are rising and ultrasonography shows a dilated bile duct). If there is moderate suspicion, a safer and less-invasive imaging study such as magnetic resonance cholangiopancreatography (MRCP) or endoscopic ultrasonography can be done to screen for bile duct stones before proceeding to ERCP.
The ACG guidelines suggest urgent ERCP (preferably within 24 hours) for those with severe biliary pancreatitis complicated by organ failure or those with suspicion of cholangitis. The level of evidence is I, ie, “strong evidence from at least one published systematic review of multiple well-designed randomized controlled trials.”1
Elective ERCP is recommended for those who are poor surgical candidates. ERCP is also recommended for those with rising liver enzyme values or imaging findings suggesting a retained common bile duct stone (including intraoperative cholangiography). Endoscopic ultrasonography or MRCP is recommended for those with slow clinical resolution, who are pregnant, or in whom uncertainty exists regarding the biliary etiology of pancreatitis.
Compliance rates with these and similar guidelines are not adequate. In an audit of adherence to the British Society of Gastroenterology guidelines, early ERCP was performed in only 25% of patients with severe biliary acute pancreatitis.6
LAPAROSCOPIC CHOLECYSTECTOMY FOR MILD BILIARY PANCREATITIS
Problem: Laparoscopic cholecystectomy is not done at admission or within 2 weeks in many patients with mild biliary pancreatitis.
If the gallbladder is not removed, biliary pancreatitis may recur in up to 61% of patients within 6 weeks of hospital discharge.37 This is the basis for guideline recommendations for surgery (or a confirmation of a surgery date) prior to hospital discharge.
The International Association of Pancreatology recommends early cholecystectomy (preferably during the same hospitalization) for patients with mild gallstone-associated acute pancreatitis.38 In severe gallstone-associated acute pancreatitis, cholecystectomy should be delayed until there is sufficient resolution of the inflammatory response and clinical recovery. The AGA guidelines advocate cholecystectomy as soon as possible and in no case later than 4 weeks after discharge to prevent relapse. ERCP with biliary sphinc-terotomy may also protect against relapse in those who are not fit to undergo surgery.
Recommendations for definitive management of gallstones (laparoscopic cholecystectomy or ERCP, or both) are not always followed. For example, a British study showed 70% compliance with this recommendation.4 A similar compliance audit in Germany revealed that cholecystectomy was performed during the initial hospital stay in only 23% of cases.7 In a New Zealand study, a regular compliance audit with feedback to surgeons resulted in an increase in the early cholecystectomy rate from 54% to 80%.8
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- United Kingdom guidelines for the management of acute pancreatitis. British Society of Gastroenterology. Gut 1998; 42(suppl 2):S1–S13.
- Norton SA, Cheruvu CV, Collins J, Dix FP, Eyre-Brook IA. An assessment of clinical guidelines for the management of acute pancreatitis. Ann R Coll Surg Engl 2001; 83:399–405.
- Chiang DT, Anozie A, Fleming WR, Kiroff GK. Comparative study on acute pancreatitis management. ANZ J Surg 2004; 74:218–221.
- Barnard J, Siriwardena AK. Variations in implementation of current national guidelines for the treatment of acute pancreatitis: implications for acute surgical service provision. Ann R Coll Surg Engl 2002; 84:79–81.
- Lankisch PG, Weber-Dany B, Lerch MM. Clinical perspectives in pancreatology: compliance with acute pancreatitis in Germany [letter]. Pancreatology 2005; 5:591–593.
- Connor SJ, Lienert AR, Brown LA, Bagshaw PF. Closing the audit loop is necessary to achieve compliance with evidence-based guidelines in the management of acute pancreatitis. N Z Med J 2008; 121:19–25.
- Mofidi R, Duff MD, Wigmore SJ, Madhavan KK, Garden OJ, Parks RW. Association between early systemic inflammatory response, severity of multiorgan dysfunction, and death in acute pancreatitis. Br J Surg 2006; 93:738–744.
- Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas 2000; 20:367–372.
- Lankisch PG, Mahlke R, Blum T, et al. Hemoconcentration: an early marker of severe and/or necrotizing pancreatitis? A critical appraisal. Am J Gastroenterol 2001; 96:2081–2085.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Larvin M. Assessment of clinical severity and prognosis. In:Beger HG, Warshaw AL, Buchler MW, et al, editors. The Pancreas. Blackwell Science: New York, 1998:489–502.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993, 128:586–590.
- Forgacs B, Eible G, Faulhaber J, Kahrau S, Buhr H, Foitzik T. Effect of fluid resuscitation with and without endothelin A receptor blockade on hemoconcentration and organ function in experimental pancreatitis. Eur Surg Res 2000; 32:162–168.
- Brown A, Baillargeon JD, Hughes MD, Banks PA. Can fluid resuscitation prevent pancreatic necrosis in severe acute pancreatitis? Pancreatology 2002; 2:104–107.
- Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 2007; 132:1127–1151.
- Pezzilli R, Uomo G, Gabbrielli A, et al; ProInf-AISP Study Group. A prospective multicenter survey on the treatment of acute pancreatitis in Italy. Dig Liver Dis 2007; 39:838–846.
- Gardner TB, Vege SS, Pearson RK, Chari ST. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol 2008; 6:1070–1076.
- Petrov MS, Pylypchuk RD, Emelyanov NV. Systematic review: nutritional support in acute pancreatitis. Aliment Pharmacol Ther 2008; 28:704–712.
- Marik PE, Zaloga GP. Meta-analysis of parenteral nutrition versus enteral nutrition in patients with acute pancreatitis. BMJ 2004; 328:1407.
- Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology 1990; 174:331–336.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Foitzik T, Bassi DG, Schmidt J, et al. Intravenous contrast medium accentuates the severity of acute necrotizing pancreatitis in the rat. Gastroenterology 1994; 106:207–214.
- Carmona-Sanchez R, Uscanga L, Bezaury-Rivas P, Robles-Díaz G, Suazo-Barahona J, Vargas-Vorácková F. Potential harmful effect of iodinated intravenous contrast medium on the clinical course of mild acute pancreatitis. Arch Surg 2000; 135:1280–1284.
- Uhl W, Roggo A, Kirschstein T, et al. Influence of contrast-enhanced computed tomography on couse and outcome in patients with acute pancreatitis. Pancreas 2002; 24:191–197.
- Foitzik T, Klar E. Non-compliance with guidelines for the management of severe acute pancreatitis among German surgeons. Pancreatology 2007; 7:80–85.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006;CD002941.
- Heinrich S, Schafer M, Rousson V, Clavien PA. Evidence-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg 2006; 243:154–168.
- Neoptolemos JP, Carr-Locke DL, London NJ, Bailey IA, James D, Fossard DP. Controlled trial of urgent endoscopic retrograde cholangiopancreatography and endoscopic sphincterotomy versus conservative treatment for acute pancreatitis due to gallstones. Lancet 1988; 2:979–983.
- Fan ST, Lai EC, Mok FP, Lo CM, Zheng SS, Wong J. Early treatment of acute biliary pancreatitis by endoscopic papillotomy. N Engl J Med 1993; 328:228–232.
- Folsch UR, Nitsche R, Ludtke R, Hilgers RA, Creutzfeldt W. Early ERCP and papillotomy compared with conservative treatment for acute biliary pancreatitis. The German Study Group on Acute Biliary Pancreatitis. N Engl J Med 1997; 336:237–242.
- Ayub K, Imada R, Slavin J. Endoscopic retrograde cholangiopancreatography in gallstone associated pancreatitis. Cochrane Database Syst Rev 2004;CD003630
- Oria A, Cimmino D, Ocampo C, et al. Early endoscopic intervention versus early conservative management in patients with acute gallstone pancreatitis and biliopancreatic obstruction. A randomized clinical trial. Ann Surg 2007; 245:10–17.
- Frei GJ, Frei VT, Thirlby RC, McClelland RN. Biliary pancreatitis: clinical presentation and surgical management. Am J Surg 1986; 151:170–175.
- Uhl W, Warshaw A, Imrie C, et al; International Association of Pancreatology. IAP guidelines on the surgical management of acute pancreatitis. Pancreatology 2002; 2:565–573.
Several major gastroenterological and surgical societies have issued guidelines on how to manage acute pancreatitis, based on evidence from high-quality randomized trials and nonrandomized studies as well as on expert opinion.1–3 Information is limited on how well physicians in the United States comply with these guidelines, but compliance is suboptimal in other developed countries, according to several studies,4–8 and we suspect that many US physicians are not following the guidelines either.
Acute pancreatitis is a frequent inpatient diagnosis that internists, gastroenterologists, and surgeons all confront. The most common causes are gallstones and heavy alcohol intake. Its management is typically straightforward: intravenous fluids, analgesia, and nothing by mouth. However, treatment of severe cases can be quite complex, particularly if multiple organ systems are involved or if there are local complications.
The primary aim of this article is to raise awareness of recognized deviations from established recommendations that may lead to adverse patient outcomes.
MEASURING ENZYME LEVELS DAILY ADDS COST BUT LITTLE BENEFIT
Problem: Serum amylase and lipase levels are often needlessly measured every day.
Measuring the serum amylase and lipase levels is useful in diagnosing acute pancreatitis, which requires two of the following three features1:
- Characteristic abdominal pain
- Levels of serum amylase or serum lipase, or both, that are three or more times the upper limit of normal
- Findings of acute pancreatitis on computed tomography (CT).
However, the magnitude or duration of the serum enzyme elevation does not correlate with the severity of the attack. Further, we have noticed that physicians at our hospital often order daily serum amylase and lipase levels in patients admitted with acute pancreatitis.
The American College of Gastroenterology (ACG) guidelines1 state that daily monitoring of amylase and lipase has limited value in managing acute pancreatitis. Rechecking these concentrations may be reasonable if pain fails to resolve or worsens during a prolonged hospitalization, as this may suggest a recurrent attack of acute pancreatitis or a developing pseudocyst. But in most cases of acute pancreatitis, daily serum enzyme measurements add cost but little benefit.
REGULAR ASSESSMENT IS IMPORTANT
Problem: Often, severity assessments are not performed regularly or acted on.
Most cases of acute pancreatitis are mild, with rapid recovery and excellent prognosis. However, 15% to 20% are severe and may result in a prolonged hospitalization, systemic inflammatory response syndrome (SIRS), multiorgan system failure, and death.
In severe acute pancreatitis, as pancreatic enzymes and inflammatory cytokines damage the blood vessels, a vast amount of fluid leaks out into the interstitial (“third”) space. This fluid extravasation leads to decreased effective circulating volume, local pancreatic necrosis, hemodynamic instability, and end-organ failure.
It is important to recognize severe acute pancreatitis early because the patient needs to be transferred to a step-down unit or intensive care unit to receive optimal fluid resuscitation and supportive care for organ dysfunction. After 48 to 72 hours, a prediction of severe acute pancreatitis should also prompt the physician to order CT to detect pancreatic necrosis, and also to initiate nutritional support.
Assessment of severity begins in the emergency room or on admission to the hospital. Older age, obesity, organ failure, and pulmonary infiltrates or pleural effusions are initial indicators of poor prognosis. Signs of SIRS (high or low core body temperature, tachycardia, tachypnea, low or high peripheral white blood cell count) or organ failure (eg, elevated serum creatinine) are present on admission in 21% of patients with acute pancreatitis.9
Hemoconcentration is a marker of decreased effective circulating volume in severe acute pancreatitis. A hematocrit higher than 44% at admission or that rises in the first 24 to 48 hours of admission predicts necrosis.10,11 However, a more robust marker of organ failure may be the blood urea nitrogen level.12
Clinical scoring systems
Several clinical scoring systems have been studied for assessing severity.
Limitations of the Ranson score are that it can only be completed after 48 hours, all the data points are not always obtained, and it cannot be repeated on a daily basis. Owing to these limitations and its less-than-optimal predictive value, the Ranson score has fallen into disuse.
The APACHE II (Acute Physiology and Chronic Health Evaluation II) score is more versatile. It is based on multiple clinical and laboratory values, and it correlates very well with the risk of death in acute pancreatitis. Death rates are less than 4% when the APACHE II score is less than 8, and 11% to 18% when it is 8 or higher.1 The trajectory of the APACHE II score in the first 48 hours is also an accurate prognostic indicator.
Previous limitations of the APACHE II score were that it was complicated and timeconsuming to calculate and required arterial blood gas measurements. Easy-to-use online calculators are now available (eg, www.globalrph.com/apacheii.htm), and the venous bicarbonate level and the oxygen saturation can be substituted for the arterial pH and oxygen partial pressure.
BISAP, a new five-point scoring system,15 was recently prospectively validated.12 “BISAP” is an acronym for the five markers it is based on, each of which has been shown to predict severe illness in acute pancreatitis:
- Blood urea nitrogen level > 25 mg/dL
- Impaired mental status
- SIRS
- Age > 60 years
- Pleural effusion.
The presence of three or more of these factors correlates with higher risk of death, organ failure, and pancreatic necrosis.12
Compared with APACHE II, BISAP has similar accuracy and is easier to calculate. Also, BISAP was specifically developed for acute pancreatitis, whereas APACHE II is a generic score for all critically ill patients.
The Atlanta criteria16 define severe acute pancreatitis as one or more of the following:
- A Ranson score of 3 or higher during the first 48 hours
- An APACHE II score of 8 or higher at any time
- Failure of one or more organs
- One or more local complications (eg, necrosis, pseudocysts, abscesses).
Recommendation: Assess severity at least daily
A severity assessment should be performed at admission and at least every day thereafter. Clinical guidelines recognize the importance of severity assessment but vary in their specific recommendations.
The ACG advises calculating the APACHE II score within 3 days of admission and measuring the hematocrit at admission, at 12 hours, and at 24 hours. The level of evidence is III, ie, “from published well-designed trials without randomization, single group prepost, cohort, time series, or matched case controlled studies”.1
The American Gastroenterological Association (AGA) provides a more generalized recommendation, that “clinical judgment” should take into account the presence of risk factors (eg, age, obesity), presence or absence of SIRS, routine laboratory values (eg, hematocrit, serum creatinine), and APACHE II score when assessing severity and making decisions.2
In a German survey, only 32% of gastroenterologists used the APACHE II score for assessing risk in acute pancreatitis, in spite of national guidelines emphasizing its importance.7 Also, not all patients with severe acute pancreatitis are transferred to a step-down unit or intensive care unit as recommended. In a British study,4 only 8 (17%) of 46 patients with predicted severe acute pancreatitis were transferred, and 8 of the 38 patients who were not transferred died.
FLUID MUST BE AGGRESSIVELY REPLACED AND MONITORED
Problem: Often, not enough fluid is replaced, or fluid status is not adequately monitored.
Fluid must be aggressively replaced to balance the massive third-space fluid losses that occur in the early inflammatory phase of acute pancreatitis. Intravascular volume depletion can develop rapidly and result in tachycardia, hypotension, and renal failure. It may also impair the blood flow to the pancreas and worsen necrosis.
Animal studies show that aggressive fluid replacement supports the pancreatic microcirculation and prevents necrosis.17 It may also support the intestinal microcirculation and gut barrier, preventing bacterial translocation.
In humans, no controlled trials have been done to test the efficacy of aggressive fluid resuscitation in acute pancreatitis. However, the notion that intravascular fluid loss contributes to poor outcomes is inferred from human studies showing more necrosis and deaths in patients with hemoconcentration. In one study, patients who received inadequate fluid replacement (evidenced by a rise in hematocrit at 24 hours) were more likely to develop necrotizing pancreatitis.18
Recommendation: Early, aggressive fluid replacement
Experts have suggested initially infusing 500 to 1,000 mL of fluid per hour in those who are volume-depleted, initially infusing 250 to 350 mL per hour in those who are not volumedepleted, and adjusting the fluid rate every 1 to 4 hours on the basis of clinical variables.19 The sufficiency of fluid replacement should be carefully monitored by vital signs, urine output, and serum hematocrit.
On the other hand, overly aggressive fluid resuscitation can be detrimental in patients at risk of volume overload or pulmonary edema. Fluid replacement should be tempered in elderly patients and those with cardiac or renal comorbidities, and may require monitoring of central venous pressure.
The ACG and AGA guidelines both recognize the need for early aggressive volume replacement in acute pancreatitis (level of evidence III), but they do not specify the exact amounts and rates. Young and healthy patients should receive a rapid bolus of isotonic saline or Ringer’s lactate solution followed by an infusion at a high initial maintenance rate.
Few studies have been done to assess physicians’ compliance with recommendations for aggressive volume replacement. In an Italian multicenter study, patients with mild or severe acute pancreatitis received an average of only 2.5 L of fluid per day (about 100 mL/hour).20 Gardner et al21 recently summarized the available evidence for fluid support in acute pancreatitis.
NUTRITIONAL SUPPORT
Problem: In many severe cases, enteral or parenteral feeding is not started soon enough.
Nutritional support entails enteral or parenteral feeding when an oral diet is contraindicated. Enteral feeding is usually via a nasojejunal tube, which may need to be placed under endoscopic or radiographic guidance. Neither parenteral nor nasojejunal feeding stimulates pancreatic secretion, and both are safe in acute pancreatitis.
Severe acute pancreatitis is an intensely catabolic state characterized by increased energy expenditure, protein breakdown, and substrate utilization. Patients may not be able to resume an oral diet for weeks or even months, particularly if local complications develop. Early nutritional support has been shown to improve outcomes in severe acute pancreatitis.22 Therefore, nutritional support should be started as soon as possible in severe acute pancreatitis based on initial clinical and radiographic indicators of severity, optimally within the first 2 or 3 days.
Enteral nutrition is preferred to parenteral nutrition in pancreatitis: it is less expensive and does not pose a risk of catheter-related infection or thrombosis or hepatic complications. Also, there is experimental evidence that enteral nutrition may preserve the gut barrier, decreasing mucosal permeability and bacterial translocation.
A number of small randomized trials compared enteral and parenteral nutrition in acute pancreatitis, but they yielded mixed results. A meta-analysis of six trials showed a lower rate of infectious complications with enteral than with parenteral nutrition. 23 However, no significant difference was found in the rates of death or noninfectious complications.
Recommendation: Enteral feeding, when possible
Nutritional support is unnecessary in most cases of mild acute pancreatitis. Pancreatic inflammation typically resolves within a few days, allowing patients to resume eating. Occasionally, patients in whom pain resolves slowly and who fast for more than 5 to 7 days need nutritional support to prevent proteincalorie malnutrition.
The ACG guidelines1 and most others suggest that, whenever possible, enteral rather than parenteral feeding should be given to those who require nutritional support. The level of evidence is II (“strong evidence from at least one published properly designed randomized controlled trial of appropriate size and in an appropriate clinical setting”).
However, not all physicians recognize the benefit of enteral feeding. In a cohort of German gastroenterologists, only 73% favored enteral over parenteral feeding in acute pancreatitis.7
COMPUTED TOMOGRAPHY
Problem: CT is not done in many patients with severe acute pancreatitis, or it is done too soon during the admission.
Dual-phase, contrast-enhanced, pancreatic-protocol CT provides a sensitive structural evaluation of the pancreas and is useful to diagnose necrotizing pancreatitis. Pancreatic necrosis is correlated with a severe clinical course, the development of single or multiorgan dysfunction, and death.
Recommendation: CT in severe cases
Not every patient with acute pancreatitis needs to undergo CT. Most mild cases do not require routine CT, since necrosis and other local complications are infrequent in this group.
Also, CT is often ordered too soon during the hospitalization. Indicators of severity on CT are not usually evident until 2 to 3 days after admission.25 CT should be considered about 3 days after the onset of symptoms rather than immediately upon admission.
On the other hand, CT at the time of admission may be warranted to rule out other life-threatening causes of abdominal pain and hyperamylasemia (eg, bowel obstruction, viscus perforation). CT may also be useful in the late phase of acute pancreatitis (weeks after admission) to diagnose or monitor complications (eg, pseudocysts, abscesses, splenic vein thrombosis, splenic artery pseudoaneurysms). Magnetic resonance imaging with gadolinium contrast is a reasonable alternative to CT for detecting pancreatic necrosis and other local complications.
In patients who have severe acute pancreatitis and compromised renal function (serum creatinine > 1.5 mg/dL), CT can be performed without contrast to assess severity based on a limited Balthazar score (ie, without a necrosis score). Studies in rats suggest that iodinated contrast may decrease pancreatic microcirculation and worsen or precipitate necrosis,26 although published human studies do not support this contention.27,28
Guidelines uniformly recommend CT for patients with severe acute pancreatitis (the ACG guideline gives it a level of evidence of III), but this recommendation is not always followed. A study from Australia showed that CT was done in only 27% to 67% of patients with severe acute pancreatitis.5 In a British study, only 8 of 46 patients with clinically predicted severe pancreatitis underwent CT within the first 10 days of admission.4
SUSPECTED INFECTED NECROSIS
Problem: Fine-needle aspiration is not done in many cases of suspected infected necrosis.
Approximately one-third of patients with necrotizing pancreatitis develop infected necrosis. The death rate for patients with infected pancreatic necrosis is high—30%, compared with 12% in those with sterile necrosis.1 Differentiating sterile and infected necrosis is therefore essential.
Clinical signs such as fever are poor predictors of infection. Signs of SIRS can be present in both sterile and infected necrotizing pancreatitis.
Recommendation: Fine-needle aspiration of necrosis
For the reasons given above, the findings of necrosis on CT and persistent SIRS should prompt consideration of fine-needle aspiration with Gram stain and culture to differentiate sterile and infected necrosis (ACG guideline, level of evidence III).1 If infection is confirmed, surgical debridement should be strongly considered. Other less-invasive approaches such as endoscopic debridement can be used in selected cases.
Fine-needle aspiration of necrosis is too often neglected. In a cohort of German surgeons, only 55% complied with International Association of Pancreatology recommendations to perform biopsy to differentiate sterile from infected necrosis in patients with signs of sepsis.29
BROAD-SPECTRUM ANTIBIOTICS
Problem: Broad-spectrum antibiotics are often used inappropriately in patients with mild acute pancreatitis and in patients with sterile necrotizing pancreatitis who are clinically stable and have no signs of sepsis.
Antibiotics are not indicated in mild acute pancreatitis. A limited course of antibiotics is typically indicated in severe cases with suspected or proven infected necrosis (in conjunction with surgical necrosectomy). However, the use of antibiotics in sterile necrosis has been very controversial.
At least six small, nonblinded, randomized trials have evaluated the benefit of giving antibiotics prophylactically for presumed sterile necrosis. A recent Cochrane analysis of five of these trials (294 patients) suggested that patients who got antibiotics had a lower risk of death (odds ratio 0.37, 95% confidence interval [CI] 0.17–0.83) but no difference in the rates of pancreatic infection or surgery.30 These paradoxical results suggest that antibiotics may prevent death by preventing nonpancreatic infections (eg, pneumonia, line infections) rather than by preventing infection of necrotic pancreatic tissue. The five trials in the meta-analysis are limited by significant methodologic heterogeneity and by lack of double-blinding.
In spite of the overall lower death rate observed in the meta-analysis, the prophylactic use of antibiotics in sterile necrosis remains controversial. One concern is that patients given long prophylactic courses of antibiotics may develop resistant bacterial or fungal infections. However, the Cochrane and other meta-analyses have not shown a higher rate of fungal infections in those given antibiotics.31
Recommendation: No routine antibiotics for mild cases
The AGA guidelines recommend against routinely giving antibiotics in mild acute pancreatitis and do not provide strict recommendations for prophylactic antibiotic use in necrotizing acute pancreatitis.2 The guidelines state that antibiotics can be used “on demand” based on clinical signs of infection (eg, high fevers, rising leukocytosis, hypotension) or worsening organ failure.
If a purely prophylactic strategy is used, only patients at high risk of developing infection (eg, those with necrosis in more than 30% of the pancreas) should receive antibiotics. Antibiotics with high tissue-penetration should be used, such as imipenem-cilastin (Primaxin IV) or ciprofloxacin (Cipro) plus metronidazole (Flagyl).
Adherence to these guidelines is not optimal. For example, in an Italian multicenter study, 9% of patients with mild acute pancreatitis were treated with antibiotics.19 Moreover, many patients with proven infected necrosis received antibiotics that do not penetrate the pancreatic tissue very well.
ERCP IN SEVERE BILIARY ACUTE PANCREATITIS
Problem: Endoscopic retrograde cholangiopancreatography (ERCP) often is performed inappropriately in mild biliary acute pancreatitis or is not performed urgently in severe cases.
In most cases of mild biliary pancreatitis, the stones pass spontaneously, as verified by cholangiography done during laparoscopic cholecystectomy. Ongoing ampullary obstruction by impacted biliary stones can perpetuate pancreatic inflammation and delay recovery.
Two early randomized trials showed a benefit from early ERCP (within 72 hours) with sphincterotomy and stone extraction, primarily in those with severe biliary acute pancreatitis or ascending cholangitis,32,33 but a third trial failed to reveal a benefit.34 A Cochrane metaanalysis of these three trials failed to show a lower death rate with ERCP in mild or severe biliary pancreatitis.35 However, early ERCP did prevent complications in severe biliary pancreatitis (odds ratio 0.27, 95% CI 0.14–0.53).
Later, a fourth randomized trial was restricted to patients with suspected biliary pancreatitis, evidence of biliary obstruction, and no signs of cholangitis36: 103 patients were randomized to undergo either ERCP within 72 hours or conservative management. No difference was observed in rates of death or organ failure or in the CT severity index.
Recommendation: ER CP for suspected retained stones
ERCP has a limited role in patients with biliary pancreatitis, being used to clear retained bile duct stones or to relieve ongoing biliary obstruction.
The decision to perform ERCP before surgery should be based on how strongly one suspects retained stones. ERCP is most appropriate if the suspicion of retained stones and the likelihood of therapeutic intervention are high (eg, if the serum bilirubin and alkaline phosphatase levels are rising and ultrasonography shows a dilated bile duct). If there is moderate suspicion, a safer and less-invasive imaging study such as magnetic resonance cholangiopancreatography (MRCP) or endoscopic ultrasonography can be done to screen for bile duct stones before proceeding to ERCP.
The ACG guidelines suggest urgent ERCP (preferably within 24 hours) for those with severe biliary pancreatitis complicated by organ failure or those with suspicion of cholangitis. The level of evidence is I, ie, “strong evidence from at least one published systematic review of multiple well-designed randomized controlled trials.”1
Elective ERCP is recommended for those who are poor surgical candidates. ERCP is also recommended for those with rising liver enzyme values or imaging findings suggesting a retained common bile duct stone (including intraoperative cholangiography). Endoscopic ultrasonography or MRCP is recommended for those with slow clinical resolution, who are pregnant, or in whom uncertainty exists regarding the biliary etiology of pancreatitis.
Compliance rates with these and similar guidelines are not adequate. In an audit of adherence to the British Society of Gastroenterology guidelines, early ERCP was performed in only 25% of patients with severe biliary acute pancreatitis.6
LAPAROSCOPIC CHOLECYSTECTOMY FOR MILD BILIARY PANCREATITIS
Problem: Laparoscopic cholecystectomy is not done at admission or within 2 weeks in many patients with mild biliary pancreatitis.
If the gallbladder is not removed, biliary pancreatitis may recur in up to 61% of patients within 6 weeks of hospital discharge.37 This is the basis for guideline recommendations for surgery (or a confirmation of a surgery date) prior to hospital discharge.
The International Association of Pancreatology recommends early cholecystectomy (preferably during the same hospitalization) for patients with mild gallstone-associated acute pancreatitis.38 In severe gallstone-associated acute pancreatitis, cholecystectomy should be delayed until there is sufficient resolution of the inflammatory response and clinical recovery. The AGA guidelines advocate cholecystectomy as soon as possible and in no case later than 4 weeks after discharge to prevent relapse. ERCP with biliary sphinc-terotomy may also protect against relapse in those who are not fit to undergo surgery.
Recommendations for definitive management of gallstones (laparoscopic cholecystectomy or ERCP, or both) are not always followed. For example, a British study showed 70% compliance with this recommendation.4 A similar compliance audit in Germany revealed that cholecystectomy was performed during the initial hospital stay in only 23% of cases.7 In a New Zealand study, a regular compliance audit with feedback to surgeons resulted in an increase in the early cholecystectomy rate from 54% to 80%.8
Several major gastroenterological and surgical societies have issued guidelines on how to manage acute pancreatitis, based on evidence from high-quality randomized trials and nonrandomized studies as well as on expert opinion.1–3 Information is limited on how well physicians in the United States comply with these guidelines, but compliance is suboptimal in other developed countries, according to several studies,4–8 and we suspect that many US physicians are not following the guidelines either.
Acute pancreatitis is a frequent inpatient diagnosis that internists, gastroenterologists, and surgeons all confront. The most common causes are gallstones and heavy alcohol intake. Its management is typically straightforward: intravenous fluids, analgesia, and nothing by mouth. However, treatment of severe cases can be quite complex, particularly if multiple organ systems are involved or if there are local complications.
The primary aim of this article is to raise awareness of recognized deviations from established recommendations that may lead to adverse patient outcomes.
MEASURING ENZYME LEVELS DAILY ADDS COST BUT LITTLE BENEFIT
Problem: Serum amylase and lipase levels are often needlessly measured every day.
Measuring the serum amylase and lipase levels is useful in diagnosing acute pancreatitis, which requires two of the following three features1:
- Characteristic abdominal pain
- Levels of serum amylase or serum lipase, or both, that are three or more times the upper limit of normal
- Findings of acute pancreatitis on computed tomography (CT).
However, the magnitude or duration of the serum enzyme elevation does not correlate with the severity of the attack. Further, we have noticed that physicians at our hospital often order daily serum amylase and lipase levels in patients admitted with acute pancreatitis.
The American College of Gastroenterology (ACG) guidelines1 state that daily monitoring of amylase and lipase has limited value in managing acute pancreatitis. Rechecking these concentrations may be reasonable if pain fails to resolve or worsens during a prolonged hospitalization, as this may suggest a recurrent attack of acute pancreatitis or a developing pseudocyst. But in most cases of acute pancreatitis, daily serum enzyme measurements add cost but little benefit.
REGULAR ASSESSMENT IS IMPORTANT
Problem: Often, severity assessments are not performed regularly or acted on.
Most cases of acute pancreatitis are mild, with rapid recovery and excellent prognosis. However, 15% to 20% are severe and may result in a prolonged hospitalization, systemic inflammatory response syndrome (SIRS), multiorgan system failure, and death.
In severe acute pancreatitis, as pancreatic enzymes and inflammatory cytokines damage the blood vessels, a vast amount of fluid leaks out into the interstitial (“third”) space. This fluid extravasation leads to decreased effective circulating volume, local pancreatic necrosis, hemodynamic instability, and end-organ failure.
It is important to recognize severe acute pancreatitis early because the patient needs to be transferred to a step-down unit or intensive care unit to receive optimal fluid resuscitation and supportive care for organ dysfunction. After 48 to 72 hours, a prediction of severe acute pancreatitis should also prompt the physician to order CT to detect pancreatic necrosis, and also to initiate nutritional support.
Assessment of severity begins in the emergency room or on admission to the hospital. Older age, obesity, organ failure, and pulmonary infiltrates or pleural effusions are initial indicators of poor prognosis. Signs of SIRS (high or low core body temperature, tachycardia, tachypnea, low or high peripheral white blood cell count) or organ failure (eg, elevated serum creatinine) are present on admission in 21% of patients with acute pancreatitis.9
Hemoconcentration is a marker of decreased effective circulating volume in severe acute pancreatitis. A hematocrit higher than 44% at admission or that rises in the first 24 to 48 hours of admission predicts necrosis.10,11 However, a more robust marker of organ failure may be the blood urea nitrogen level.12
Clinical scoring systems
Several clinical scoring systems have been studied for assessing severity.
Limitations of the Ranson score are that it can only be completed after 48 hours, all the data points are not always obtained, and it cannot be repeated on a daily basis. Owing to these limitations and its less-than-optimal predictive value, the Ranson score has fallen into disuse.
The APACHE II (Acute Physiology and Chronic Health Evaluation II) score is more versatile. It is based on multiple clinical and laboratory values, and it correlates very well with the risk of death in acute pancreatitis. Death rates are less than 4% when the APACHE II score is less than 8, and 11% to 18% when it is 8 or higher.1 The trajectory of the APACHE II score in the first 48 hours is also an accurate prognostic indicator.
Previous limitations of the APACHE II score were that it was complicated and timeconsuming to calculate and required arterial blood gas measurements. Easy-to-use online calculators are now available (eg, www.globalrph.com/apacheii.htm), and the venous bicarbonate level and the oxygen saturation can be substituted for the arterial pH and oxygen partial pressure.
BISAP, a new five-point scoring system,15 was recently prospectively validated.12 “BISAP” is an acronym for the five markers it is based on, each of which has been shown to predict severe illness in acute pancreatitis:
- Blood urea nitrogen level > 25 mg/dL
- Impaired mental status
- SIRS
- Age > 60 years
- Pleural effusion.
The presence of three or more of these factors correlates with higher risk of death, organ failure, and pancreatic necrosis.12
Compared with APACHE II, BISAP has similar accuracy and is easier to calculate. Also, BISAP was specifically developed for acute pancreatitis, whereas APACHE II is a generic score for all critically ill patients.
The Atlanta criteria16 define severe acute pancreatitis as one or more of the following:
- A Ranson score of 3 or higher during the first 48 hours
- An APACHE II score of 8 or higher at any time
- Failure of one or more organs
- One or more local complications (eg, necrosis, pseudocysts, abscesses).
Recommendation: Assess severity at least daily
A severity assessment should be performed at admission and at least every day thereafter. Clinical guidelines recognize the importance of severity assessment but vary in their specific recommendations.
The ACG advises calculating the APACHE II score within 3 days of admission and measuring the hematocrit at admission, at 12 hours, and at 24 hours. The level of evidence is III, ie, “from published well-designed trials without randomization, single group prepost, cohort, time series, or matched case controlled studies”.1
The American Gastroenterological Association (AGA) provides a more generalized recommendation, that “clinical judgment” should take into account the presence of risk factors (eg, age, obesity), presence or absence of SIRS, routine laboratory values (eg, hematocrit, serum creatinine), and APACHE II score when assessing severity and making decisions.2
In a German survey, only 32% of gastroenterologists used the APACHE II score for assessing risk in acute pancreatitis, in spite of national guidelines emphasizing its importance.7 Also, not all patients with severe acute pancreatitis are transferred to a step-down unit or intensive care unit as recommended. In a British study,4 only 8 (17%) of 46 patients with predicted severe acute pancreatitis were transferred, and 8 of the 38 patients who were not transferred died.
FLUID MUST BE AGGRESSIVELY REPLACED AND MONITORED
Problem: Often, not enough fluid is replaced, or fluid status is not adequately monitored.
Fluid must be aggressively replaced to balance the massive third-space fluid losses that occur in the early inflammatory phase of acute pancreatitis. Intravascular volume depletion can develop rapidly and result in tachycardia, hypotension, and renal failure. It may also impair the blood flow to the pancreas and worsen necrosis.
Animal studies show that aggressive fluid replacement supports the pancreatic microcirculation and prevents necrosis.17 It may also support the intestinal microcirculation and gut barrier, preventing bacterial translocation.
In humans, no controlled trials have been done to test the efficacy of aggressive fluid resuscitation in acute pancreatitis. However, the notion that intravascular fluid loss contributes to poor outcomes is inferred from human studies showing more necrosis and deaths in patients with hemoconcentration. In one study, patients who received inadequate fluid replacement (evidenced by a rise in hematocrit at 24 hours) were more likely to develop necrotizing pancreatitis.18
Recommendation: Early, aggressive fluid replacement
Experts have suggested initially infusing 500 to 1,000 mL of fluid per hour in those who are volume-depleted, initially infusing 250 to 350 mL per hour in those who are not volumedepleted, and adjusting the fluid rate every 1 to 4 hours on the basis of clinical variables.19 The sufficiency of fluid replacement should be carefully monitored by vital signs, urine output, and serum hematocrit.
On the other hand, overly aggressive fluid resuscitation can be detrimental in patients at risk of volume overload or pulmonary edema. Fluid replacement should be tempered in elderly patients and those with cardiac or renal comorbidities, and may require monitoring of central venous pressure.
The ACG and AGA guidelines both recognize the need for early aggressive volume replacement in acute pancreatitis (level of evidence III), but they do not specify the exact amounts and rates. Young and healthy patients should receive a rapid bolus of isotonic saline or Ringer’s lactate solution followed by an infusion at a high initial maintenance rate.
Few studies have been done to assess physicians’ compliance with recommendations for aggressive volume replacement. In an Italian multicenter study, patients with mild or severe acute pancreatitis received an average of only 2.5 L of fluid per day (about 100 mL/hour).20 Gardner et al21 recently summarized the available evidence for fluid support in acute pancreatitis.
NUTRITIONAL SUPPORT
Problem: In many severe cases, enteral or parenteral feeding is not started soon enough.
Nutritional support entails enteral or parenteral feeding when an oral diet is contraindicated. Enteral feeding is usually via a nasojejunal tube, which may need to be placed under endoscopic or radiographic guidance. Neither parenteral nor nasojejunal feeding stimulates pancreatic secretion, and both are safe in acute pancreatitis.
Severe acute pancreatitis is an intensely catabolic state characterized by increased energy expenditure, protein breakdown, and substrate utilization. Patients may not be able to resume an oral diet for weeks or even months, particularly if local complications develop. Early nutritional support has been shown to improve outcomes in severe acute pancreatitis.22 Therefore, nutritional support should be started as soon as possible in severe acute pancreatitis based on initial clinical and radiographic indicators of severity, optimally within the first 2 or 3 days.
Enteral nutrition is preferred to parenteral nutrition in pancreatitis: it is less expensive and does not pose a risk of catheter-related infection or thrombosis or hepatic complications. Also, there is experimental evidence that enteral nutrition may preserve the gut barrier, decreasing mucosal permeability and bacterial translocation.
A number of small randomized trials compared enteral and parenteral nutrition in acute pancreatitis, but they yielded mixed results. A meta-analysis of six trials showed a lower rate of infectious complications with enteral than with parenteral nutrition. 23 However, no significant difference was found in the rates of death or noninfectious complications.
Recommendation: Enteral feeding, when possible
Nutritional support is unnecessary in most cases of mild acute pancreatitis. Pancreatic inflammation typically resolves within a few days, allowing patients to resume eating. Occasionally, patients in whom pain resolves slowly and who fast for more than 5 to 7 days need nutritional support to prevent proteincalorie malnutrition.
The ACG guidelines1 and most others suggest that, whenever possible, enteral rather than parenteral feeding should be given to those who require nutritional support. The level of evidence is II (“strong evidence from at least one published properly designed randomized controlled trial of appropriate size and in an appropriate clinical setting”).
However, not all physicians recognize the benefit of enteral feeding. In a cohort of German gastroenterologists, only 73% favored enteral over parenteral feeding in acute pancreatitis.7
COMPUTED TOMOGRAPHY
Problem: CT is not done in many patients with severe acute pancreatitis, or it is done too soon during the admission.
Dual-phase, contrast-enhanced, pancreatic-protocol CT provides a sensitive structural evaluation of the pancreas and is useful to diagnose necrotizing pancreatitis. Pancreatic necrosis is correlated with a severe clinical course, the development of single or multiorgan dysfunction, and death.
Recommendation: CT in severe cases
Not every patient with acute pancreatitis needs to undergo CT. Most mild cases do not require routine CT, since necrosis and other local complications are infrequent in this group.
Also, CT is often ordered too soon during the hospitalization. Indicators of severity on CT are not usually evident until 2 to 3 days after admission.25 CT should be considered about 3 days after the onset of symptoms rather than immediately upon admission.
On the other hand, CT at the time of admission may be warranted to rule out other life-threatening causes of abdominal pain and hyperamylasemia (eg, bowel obstruction, viscus perforation). CT may also be useful in the late phase of acute pancreatitis (weeks after admission) to diagnose or monitor complications (eg, pseudocysts, abscesses, splenic vein thrombosis, splenic artery pseudoaneurysms). Magnetic resonance imaging with gadolinium contrast is a reasonable alternative to CT for detecting pancreatic necrosis and other local complications.
In patients who have severe acute pancreatitis and compromised renal function (serum creatinine > 1.5 mg/dL), CT can be performed without contrast to assess severity based on a limited Balthazar score (ie, without a necrosis score). Studies in rats suggest that iodinated contrast may decrease pancreatic microcirculation and worsen or precipitate necrosis,26 although published human studies do not support this contention.27,28
Guidelines uniformly recommend CT for patients with severe acute pancreatitis (the ACG guideline gives it a level of evidence of III), but this recommendation is not always followed. A study from Australia showed that CT was done in only 27% to 67% of patients with severe acute pancreatitis.5 In a British study, only 8 of 46 patients with clinically predicted severe pancreatitis underwent CT within the first 10 days of admission.4
SUSPECTED INFECTED NECROSIS
Problem: Fine-needle aspiration is not done in many cases of suspected infected necrosis.
Approximately one-third of patients with necrotizing pancreatitis develop infected necrosis. The death rate for patients with infected pancreatic necrosis is high—30%, compared with 12% in those with sterile necrosis.1 Differentiating sterile and infected necrosis is therefore essential.
Clinical signs such as fever are poor predictors of infection. Signs of SIRS can be present in both sterile and infected necrotizing pancreatitis.
Recommendation: Fine-needle aspiration of necrosis
For the reasons given above, the findings of necrosis on CT and persistent SIRS should prompt consideration of fine-needle aspiration with Gram stain and culture to differentiate sterile and infected necrosis (ACG guideline, level of evidence III).1 If infection is confirmed, surgical debridement should be strongly considered. Other less-invasive approaches such as endoscopic debridement can be used in selected cases.
Fine-needle aspiration of necrosis is too often neglected. In a cohort of German surgeons, only 55% complied with International Association of Pancreatology recommendations to perform biopsy to differentiate sterile from infected necrosis in patients with signs of sepsis.29
BROAD-SPECTRUM ANTIBIOTICS
Problem: Broad-spectrum antibiotics are often used inappropriately in patients with mild acute pancreatitis and in patients with sterile necrotizing pancreatitis who are clinically stable and have no signs of sepsis.
Antibiotics are not indicated in mild acute pancreatitis. A limited course of antibiotics is typically indicated in severe cases with suspected or proven infected necrosis (in conjunction with surgical necrosectomy). However, the use of antibiotics in sterile necrosis has been very controversial.
At least six small, nonblinded, randomized trials have evaluated the benefit of giving antibiotics prophylactically for presumed sterile necrosis. A recent Cochrane analysis of five of these trials (294 patients) suggested that patients who got antibiotics had a lower risk of death (odds ratio 0.37, 95% confidence interval [CI] 0.17–0.83) but no difference in the rates of pancreatic infection or surgery.30 These paradoxical results suggest that antibiotics may prevent death by preventing nonpancreatic infections (eg, pneumonia, line infections) rather than by preventing infection of necrotic pancreatic tissue. The five trials in the meta-analysis are limited by significant methodologic heterogeneity and by lack of double-blinding.
In spite of the overall lower death rate observed in the meta-analysis, the prophylactic use of antibiotics in sterile necrosis remains controversial. One concern is that patients given long prophylactic courses of antibiotics may develop resistant bacterial or fungal infections. However, the Cochrane and other meta-analyses have not shown a higher rate of fungal infections in those given antibiotics.31
Recommendation: No routine antibiotics for mild cases
The AGA guidelines recommend against routinely giving antibiotics in mild acute pancreatitis and do not provide strict recommendations for prophylactic antibiotic use in necrotizing acute pancreatitis.2 The guidelines state that antibiotics can be used “on demand” based on clinical signs of infection (eg, high fevers, rising leukocytosis, hypotension) or worsening organ failure.
If a purely prophylactic strategy is used, only patients at high risk of developing infection (eg, those with necrosis in more than 30% of the pancreas) should receive antibiotics. Antibiotics with high tissue-penetration should be used, such as imipenem-cilastin (Primaxin IV) or ciprofloxacin (Cipro) plus metronidazole (Flagyl).
Adherence to these guidelines is not optimal. For example, in an Italian multicenter study, 9% of patients with mild acute pancreatitis were treated with antibiotics.19 Moreover, many patients with proven infected necrosis received antibiotics that do not penetrate the pancreatic tissue very well.
ERCP IN SEVERE BILIARY ACUTE PANCREATITIS
Problem: Endoscopic retrograde cholangiopancreatography (ERCP) often is performed inappropriately in mild biliary acute pancreatitis or is not performed urgently in severe cases.
In most cases of mild biliary pancreatitis, the stones pass spontaneously, as verified by cholangiography done during laparoscopic cholecystectomy. Ongoing ampullary obstruction by impacted biliary stones can perpetuate pancreatic inflammation and delay recovery.
Two early randomized trials showed a benefit from early ERCP (within 72 hours) with sphincterotomy and stone extraction, primarily in those with severe biliary acute pancreatitis or ascending cholangitis,32,33 but a third trial failed to reveal a benefit.34 A Cochrane metaanalysis of these three trials failed to show a lower death rate with ERCP in mild or severe biliary pancreatitis.35 However, early ERCP did prevent complications in severe biliary pancreatitis (odds ratio 0.27, 95% CI 0.14–0.53).
Later, a fourth randomized trial was restricted to patients with suspected biliary pancreatitis, evidence of biliary obstruction, and no signs of cholangitis36: 103 patients were randomized to undergo either ERCP within 72 hours or conservative management. No difference was observed in rates of death or organ failure or in the CT severity index.
Recommendation: ER CP for suspected retained stones
ERCP has a limited role in patients with biliary pancreatitis, being used to clear retained bile duct stones or to relieve ongoing biliary obstruction.
The decision to perform ERCP before surgery should be based on how strongly one suspects retained stones. ERCP is most appropriate if the suspicion of retained stones and the likelihood of therapeutic intervention are high (eg, if the serum bilirubin and alkaline phosphatase levels are rising and ultrasonography shows a dilated bile duct). If there is moderate suspicion, a safer and less-invasive imaging study such as magnetic resonance cholangiopancreatography (MRCP) or endoscopic ultrasonography can be done to screen for bile duct stones before proceeding to ERCP.
The ACG guidelines suggest urgent ERCP (preferably within 24 hours) for those with severe biliary pancreatitis complicated by organ failure or those with suspicion of cholangitis. The level of evidence is I, ie, “strong evidence from at least one published systematic review of multiple well-designed randomized controlled trials.”1
Elective ERCP is recommended for those who are poor surgical candidates. ERCP is also recommended for those with rising liver enzyme values or imaging findings suggesting a retained common bile duct stone (including intraoperative cholangiography). Endoscopic ultrasonography or MRCP is recommended for those with slow clinical resolution, who are pregnant, or in whom uncertainty exists regarding the biliary etiology of pancreatitis.
Compliance rates with these and similar guidelines are not adequate. In an audit of adherence to the British Society of Gastroenterology guidelines, early ERCP was performed in only 25% of patients with severe biliary acute pancreatitis.6
LAPAROSCOPIC CHOLECYSTECTOMY FOR MILD BILIARY PANCREATITIS
Problem: Laparoscopic cholecystectomy is not done at admission or within 2 weeks in many patients with mild biliary pancreatitis.
If the gallbladder is not removed, biliary pancreatitis may recur in up to 61% of patients within 6 weeks of hospital discharge.37 This is the basis for guideline recommendations for surgery (or a confirmation of a surgery date) prior to hospital discharge.
The International Association of Pancreatology recommends early cholecystectomy (preferably during the same hospitalization) for patients with mild gallstone-associated acute pancreatitis.38 In severe gallstone-associated acute pancreatitis, cholecystectomy should be delayed until there is sufficient resolution of the inflammatory response and clinical recovery. The AGA guidelines advocate cholecystectomy as soon as possible and in no case later than 4 weeks after discharge to prevent relapse. ERCP with biliary sphinc-terotomy may also protect against relapse in those who are not fit to undergo surgery.
Recommendations for definitive management of gallstones (laparoscopic cholecystectomy or ERCP, or both) are not always followed. For example, a British study showed 70% compliance with this recommendation.4 A similar compliance audit in Germany revealed that cholecystectomy was performed during the initial hospital stay in only 23% of cases.7 In a New Zealand study, a regular compliance audit with feedback to surgeons resulted in an increase in the early cholecystectomy rate from 54% to 80%.8
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- United Kingdom guidelines for the management of acute pancreatitis. British Society of Gastroenterology. Gut 1998; 42(suppl 2):S1–S13.
- Norton SA, Cheruvu CV, Collins J, Dix FP, Eyre-Brook IA. An assessment of clinical guidelines for the management of acute pancreatitis. Ann R Coll Surg Engl 2001; 83:399–405.
- Chiang DT, Anozie A, Fleming WR, Kiroff GK. Comparative study on acute pancreatitis management. ANZ J Surg 2004; 74:218–221.
- Barnard J, Siriwardena AK. Variations in implementation of current national guidelines for the treatment of acute pancreatitis: implications for acute surgical service provision. Ann R Coll Surg Engl 2002; 84:79–81.
- Lankisch PG, Weber-Dany B, Lerch MM. Clinical perspectives in pancreatology: compliance with acute pancreatitis in Germany [letter]. Pancreatology 2005; 5:591–593.
- Connor SJ, Lienert AR, Brown LA, Bagshaw PF. Closing the audit loop is necessary to achieve compliance with evidence-based guidelines in the management of acute pancreatitis. N Z Med J 2008; 121:19–25.
- Mofidi R, Duff MD, Wigmore SJ, Madhavan KK, Garden OJ, Parks RW. Association between early systemic inflammatory response, severity of multiorgan dysfunction, and death in acute pancreatitis. Br J Surg 2006; 93:738–744.
- Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas 2000; 20:367–372.
- Lankisch PG, Mahlke R, Blum T, et al. Hemoconcentration: an early marker of severe and/or necrotizing pancreatitis? A critical appraisal. Am J Gastroenterol 2001; 96:2081–2085.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Larvin M. Assessment of clinical severity and prognosis. In:Beger HG, Warshaw AL, Buchler MW, et al, editors. The Pancreas. Blackwell Science: New York, 1998:489–502.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993, 128:586–590.
- Forgacs B, Eible G, Faulhaber J, Kahrau S, Buhr H, Foitzik T. Effect of fluid resuscitation with and without endothelin A receptor blockade on hemoconcentration and organ function in experimental pancreatitis. Eur Surg Res 2000; 32:162–168.
- Brown A, Baillargeon JD, Hughes MD, Banks PA. Can fluid resuscitation prevent pancreatic necrosis in severe acute pancreatitis? Pancreatology 2002; 2:104–107.
- Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 2007; 132:1127–1151.
- Pezzilli R, Uomo G, Gabbrielli A, et al; ProInf-AISP Study Group. A prospective multicenter survey on the treatment of acute pancreatitis in Italy. Dig Liver Dis 2007; 39:838–846.
- Gardner TB, Vege SS, Pearson RK, Chari ST. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol 2008; 6:1070–1076.
- Petrov MS, Pylypchuk RD, Emelyanov NV. Systematic review: nutritional support in acute pancreatitis. Aliment Pharmacol Ther 2008; 28:704–712.
- Marik PE, Zaloga GP. Meta-analysis of parenteral nutrition versus enteral nutrition in patients with acute pancreatitis. BMJ 2004; 328:1407.
- Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology 1990; 174:331–336.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Foitzik T, Bassi DG, Schmidt J, et al. Intravenous contrast medium accentuates the severity of acute necrotizing pancreatitis in the rat. Gastroenterology 1994; 106:207–214.
- Carmona-Sanchez R, Uscanga L, Bezaury-Rivas P, Robles-Díaz G, Suazo-Barahona J, Vargas-Vorácková F. Potential harmful effect of iodinated intravenous contrast medium on the clinical course of mild acute pancreatitis. Arch Surg 2000; 135:1280–1284.
- Uhl W, Roggo A, Kirschstein T, et al. Influence of contrast-enhanced computed tomography on couse and outcome in patients with acute pancreatitis. Pancreas 2002; 24:191–197.
- Foitzik T, Klar E. Non-compliance with guidelines for the management of severe acute pancreatitis among German surgeons. Pancreatology 2007; 7:80–85.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006;CD002941.
- Heinrich S, Schafer M, Rousson V, Clavien PA. Evidence-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg 2006; 243:154–168.
- Neoptolemos JP, Carr-Locke DL, London NJ, Bailey IA, James D, Fossard DP. Controlled trial of urgent endoscopic retrograde cholangiopancreatography and endoscopic sphincterotomy versus conservative treatment for acute pancreatitis due to gallstones. Lancet 1988; 2:979–983.
- Fan ST, Lai EC, Mok FP, Lo CM, Zheng SS, Wong J. Early treatment of acute biliary pancreatitis by endoscopic papillotomy. N Engl J Med 1993; 328:228–232.
- Folsch UR, Nitsche R, Ludtke R, Hilgers RA, Creutzfeldt W. Early ERCP and papillotomy compared with conservative treatment for acute biliary pancreatitis. The German Study Group on Acute Biliary Pancreatitis. N Engl J Med 1997; 336:237–242.
- Ayub K, Imada R, Slavin J. Endoscopic retrograde cholangiopancreatography in gallstone associated pancreatitis. Cochrane Database Syst Rev 2004;CD003630
- Oria A, Cimmino D, Ocampo C, et al. Early endoscopic intervention versus early conservative management in patients with acute gallstone pancreatitis and biliopancreatic obstruction. A randomized clinical trial. Ann Surg 2007; 245:10–17.
- Frei GJ, Frei VT, Thirlby RC, McClelland RN. Biliary pancreatitis: clinical presentation and surgical management. Am J Surg 1986; 151:170–175.
- Uhl W, Warshaw A, Imrie C, et al; International Association of Pancreatology. IAP guidelines on the surgical management of acute pancreatitis. Pancreatology 2002; 2:565–573.
- Banks PA, Freeman ML; Practice Parameters Committee of the American College of Gastroenterology. Practice guidelines in acute pancreatitis. Am J Gastroenterol 2006; 101:2379–2400.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- United Kingdom guidelines for the management of acute pancreatitis. British Society of Gastroenterology. Gut 1998; 42(suppl 2):S1–S13.
- Norton SA, Cheruvu CV, Collins J, Dix FP, Eyre-Brook IA. An assessment of clinical guidelines for the management of acute pancreatitis. Ann R Coll Surg Engl 2001; 83:399–405.
- Chiang DT, Anozie A, Fleming WR, Kiroff GK. Comparative study on acute pancreatitis management. ANZ J Surg 2004; 74:218–221.
- Barnard J, Siriwardena AK. Variations in implementation of current national guidelines for the treatment of acute pancreatitis: implications for acute surgical service provision. Ann R Coll Surg Engl 2002; 84:79–81.
- Lankisch PG, Weber-Dany B, Lerch MM. Clinical perspectives in pancreatology: compliance with acute pancreatitis in Germany [letter]. Pancreatology 2005; 5:591–593.
- Connor SJ, Lienert AR, Brown LA, Bagshaw PF. Closing the audit loop is necessary to achieve compliance with evidence-based guidelines in the management of acute pancreatitis. N Z Med J 2008; 121:19–25.
- Mofidi R, Duff MD, Wigmore SJ, Madhavan KK, Garden OJ, Parks RW. Association between early systemic inflammatory response, severity of multiorgan dysfunction, and death in acute pancreatitis. Br J Surg 2006; 93:738–744.
- Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas 2000; 20:367–372.
- Lankisch PG, Mahlke R, Blum T, et al. Hemoconcentration: an early marker of severe and/or necrotizing pancreatitis? A critical appraisal. Am J Gastroenterol 2001; 96:2081–2085.
- Singh VK, Wu BU, Bollen TL, et al. A prospective evaluation of the bedside index for severity in acute pancreatitis score in assessing mortality and intermediate markers of severity in acute pancreatitis. Am J Gastroenterol 2009; 104:966–971.
- Ranson JH, Rifkind KM, Roses DF, Fink SD, Eng K, Spencer FC. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974; 139:69–81.
- Larvin M. Assessment of clinical severity and prognosis. In:Beger HG, Warshaw AL, Buchler MW, et al, editors. The Pancreas. Blackwell Science: New York, 1998:489–502.
- Wu BU, Johannes RS, Sun X, Tabak Y, Conwell DL, Banks PA. The early prediction of mortality in acute pancreatitis: a large population-based study. Gut 2008; 57:1698–1703.
- Bradley EL. A clinically based classification system for acute pancreatitis. Summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg 1993, 128:586–590.
- Forgacs B, Eible G, Faulhaber J, Kahrau S, Buhr H, Foitzik T. Effect of fluid resuscitation with and without endothelin A receptor blockade on hemoconcentration and organ function in experimental pancreatitis. Eur Surg Res 2000; 32:162–168.
- Brown A, Baillargeon JD, Hughes MD, Banks PA. Can fluid resuscitation prevent pancreatic necrosis in severe acute pancreatitis? Pancreatology 2002; 2:104–107.
- Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology 2007; 132:1127–1151.
- Pezzilli R, Uomo G, Gabbrielli A, et al; ProInf-AISP Study Group. A prospective multicenter survey on the treatment of acute pancreatitis in Italy. Dig Liver Dis 2007; 39:838–846.
- Gardner TB, Vege SS, Pearson RK, Chari ST. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol 2008; 6:1070–1076.
- Petrov MS, Pylypchuk RD, Emelyanov NV. Systematic review: nutritional support in acute pancreatitis. Aliment Pharmacol Ther 2008; 28:704–712.
- Marik PE, Zaloga GP. Meta-analysis of parenteral nutrition versus enteral nutrition in patients with acute pancreatitis. BMJ 2004; 328:1407.
- Balthazar EJ, Robinson DL, Megibow AJ, Ranson JH. Acute pancreatitis: value of CT in establishing prognosis. Radiology 1990; 174:331–336.
- Balthazar EJ. Acute pancreatitis: assessment of severity with clinical and CT evaluation. Radiology 2002; 223:603–613.
- Foitzik T, Bassi DG, Schmidt J, et al. Intravenous contrast medium accentuates the severity of acute necrotizing pancreatitis in the rat. Gastroenterology 1994; 106:207–214.
- Carmona-Sanchez R, Uscanga L, Bezaury-Rivas P, Robles-Díaz G, Suazo-Barahona J, Vargas-Vorácková F. Potential harmful effect of iodinated intravenous contrast medium on the clinical course of mild acute pancreatitis. Arch Surg 2000; 135:1280–1284.
- Uhl W, Roggo A, Kirschstein T, et al. Influence of contrast-enhanced computed tomography on couse and outcome in patients with acute pancreatitis. Pancreas 2002; 24:191–197.
- Foitzik T, Klar E. Non-compliance with guidelines for the management of severe acute pancreatitis among German surgeons. Pancreatology 2007; 7:80–85.
- Villatoro E, Bassi C, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev 2006;CD002941.
- Heinrich S, Schafer M, Rousson V, Clavien PA. Evidence-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg 2006; 243:154–168.
- Neoptolemos JP, Carr-Locke DL, London NJ, Bailey IA, James D, Fossard DP. Controlled trial of urgent endoscopic retrograde cholangiopancreatography and endoscopic sphincterotomy versus conservative treatment for acute pancreatitis due to gallstones. Lancet 1988; 2:979–983.
- Fan ST, Lai EC, Mok FP, Lo CM, Zheng SS, Wong J. Early treatment of acute biliary pancreatitis by endoscopic papillotomy. N Engl J Med 1993; 328:228–232.
- Folsch UR, Nitsche R, Ludtke R, Hilgers RA, Creutzfeldt W. Early ERCP and papillotomy compared with conservative treatment for acute biliary pancreatitis. The German Study Group on Acute Biliary Pancreatitis. N Engl J Med 1997; 336:237–242.
- Ayub K, Imada R, Slavin J. Endoscopic retrograde cholangiopancreatography in gallstone associated pancreatitis. Cochrane Database Syst Rev 2004;CD003630
- Oria A, Cimmino D, Ocampo C, et al. Early endoscopic intervention versus early conservative management in patients with acute gallstone pancreatitis and biliopancreatic obstruction. A randomized clinical trial. Ann Surg 2007; 245:10–17.
- Frei GJ, Frei VT, Thirlby RC, McClelland RN. Biliary pancreatitis: clinical presentation and surgical management. Am J Surg 1986; 151:170–175.
- Uhl W, Warshaw A, Imrie C, et al; International Association of Pancreatology. IAP guidelines on the surgical management of acute pancreatitis. Pancreatology 2002; 2:565–573.
KEY POINTS
- Serum amylase and lipase levels are often needlessly measured every day.
- Often, severity assessments are not performed regularly or acted on.
- Often, not enough fluid is replaced, or fluid status is not adequately monitored.
- In many severe cases, enteral or parenteral feeding is not started soon enough.
- Computed tomography is not done in many patients with severe acute pancreatitis, or it is performed too soon.
- In many cases of suspected infected necrosis, fine-needle aspiration is not done.
- Broad-spectrum antibiotics are often used inappropriately in patients with mild acute pancreatitis and in patients with sterile necrotizing pancreatitis who are clinically stable and have no signs of sepsis.
Autoimmune pancreatitis: A mimic of pancreatic cancer
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
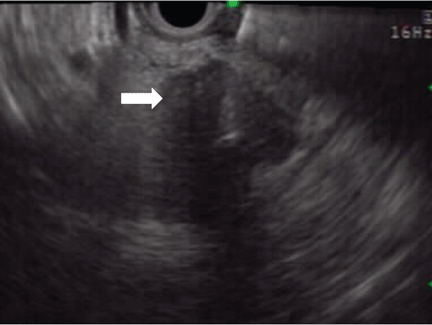
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS

The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
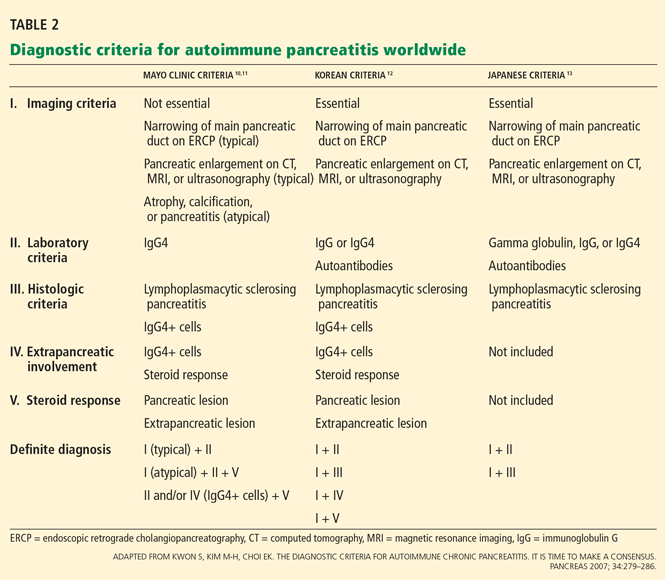
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings

Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS
The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings
Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS
The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings
Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
KEY POINTS
- Hallmark features of autoimmune pancreatitis include an elevated serum immunoglobulin G4 level, focal or diffuse pancreatic enlargement on imaging, and dense lymphoplasmacytic infiltrates on histologic study.
- The disease can be associated with extrapancreatic manifestations, including sclerosing cholangitis, sialadenitis and retroperitoneal fibrosis.
- Autoimmune pancreatitis responds dramatically to corticosteroid treatment.
Endoscopic therapy of recurrent acute pancreatitis
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
KEY POINTS
- Recurrent attacks of acute pancreatitis can be prevented only by determining and treating the underlying cause.
- Endoscopic procedures can cause anxiety and carry a risk of bleeding, perforation, and pancreatitis. The risks, benefits, and other treatment options should be discussed with the patient.
- Endoscopic therapy is now the preferred treatment of sphincter of Oddi dysfunction at centers that have experience with this technique.
- In patients with pancreas divisum and recurrent acute pancreatitis, surgical and endoscopic minor sphincterotomy are equally effective.