User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
From paranoid fear to completed homicide
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
Delusions, hypersexuality, and a steep cognitive decline
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
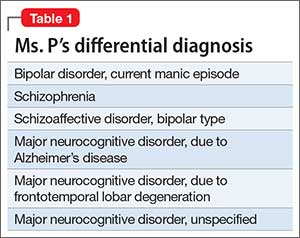
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2

Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
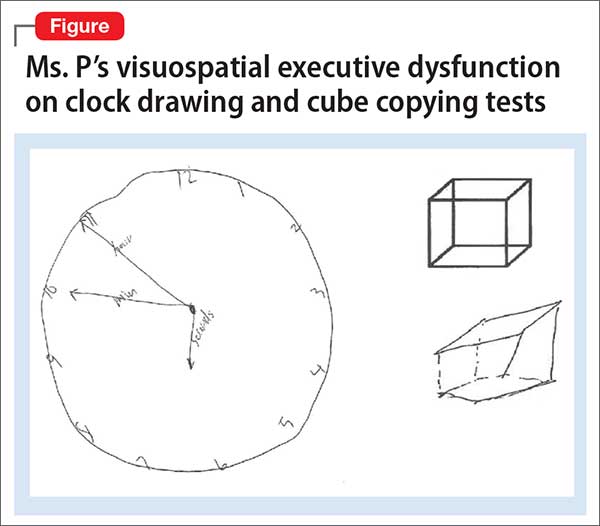
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.

TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2
Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.
TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
CASE Inconsistent stories
Ms. P, age 56, is an Asian American woman who was brought in by police after being found standing by her car in the middle of a busy road displaying bizarre behavior. She provides an inconsistent story about why she was brought to the hospital, saying that the police did so because she wasn’t driving fast enough and because her English is weak. At another point, she says that she had stopped her car to pick up a penny from the road and the police brought her to the hospital “to experience life, to rest, to meet people.”
Upon further questioning, Ms. P reveals that she is experiencing racing thoughts, feels full of energy, has pressured speech, and does not need much sleep. She also is sexually preoccupied, talks about having extra-marital affairs, and expresses her infatuation with TV news anchors. She says she is sexually active but is unable to offer any further details, and—while giggling—asks the treatment team not to reveal this information to her husband. Ms. P also reports hearing angels singing from the sky.
Chart review reveals that Ms. P had been admitted to same hospital 5 years earlier, at which time she was given diagnoses of late-onset schizophrenia (LOS) and mild cognitive impairment. Ms. P also had 3 psychiatric inpatient admissions in the past 2 years at a different hospital, but her records are inaccessible because she refuses to allow her chart to be released.
Ms. P has not taken the psychiatric medications prescribed for her for several months; she says, “I don’t need medication. I am self-healing.” She denies using illicit substances, including marijuana, smoking, and current alcohol use, but reports occasional social drinking in the past. Her urine drug screen is negative.
The most striking revelation in Ms. P’s social history is her high premorbid functional status. She has 2 master’s degrees and had been working as a senior accountant at a major hospital system until 7 years ago. In contrast, when interviewed at the hospital, Ms. P reports that she is working at a child care center.
On mental status exam, Ms. P is half-draped in a hospital gown, casual, overly friendly, smiling, and twirling her hair. Her mood is elevated with inappropriate affect. Her thought process is bizarre and illogical. She is alert, fully oriented, and her sensorium is clear. She has persistent ambivalence and contradictory thoughts regarding suicidal ideation. Recent and remote memory are largely intact. She does not express homicidal ideation.
What could be causing Ms. P’s psychosis and functional decline?
a) major neurocognitive disorder
b) schizophrenia
c) schizoaffective disorder
d) bipolar disorder, current manic episode
HISTORY Fired from her job
According to Ms. P’s chart from her admission 5 years earlier, police brought her to the hospital because she was causing a disturbance at a restaurant. When interviewed, Ms. P reported a false story that she fought with her husband, kicked him, and spat on his face. She said that her husband then punched her in the face, she ran out of the house, and a bystander called the police. At the time, her husband was contacted and denied the incident. He said that Ms. P had gone to the store and not returned, and he did not know what happened to her.
Her husband reported a steady and progressive decline in function and behavior dating back to 8 years ago with no known prior behavioral disturbances. In the chart from 5 years ago, her husband reported that Ms. P had been a high-functioning senior executive accountant at a major hospital system 7 years before the current admission, at which time she was fired from her job. He said that, just before being fired, Ms. P had been reading the mystery novel The Da Vinci Code and believed that events in the book specifically applied to her. Ms. P would stay up all night making clothes; when she would go to work, she was caught sleeping on the job and performing poorly, including submitting reports with incorrect information. She yelled at co-workers and was unable to take direction from her supervisors.
Ms. P’s husband also reported that she believed people were trying to “look like her,” by having plastic surgery. He reported unusual behavior at home, including eating food off the countertop that had been out for hours and was not fit for consumption.
Ms. P’s husband could not be contacted during this admission because he was out of country and they were separated. Collateral information is obtained from Ms. P’s mother, who lives apart from her but in the same city and speaks no English. She confirms Ms. P’s high premorbid functioning, and reports that her daughter’s change in behavior went back as far as 10 years. She reports that Ms. P had problems controlling anger and had frequent altercations with her husband and mother, including threatening her with a knife. Self-care and hygiene then declined strikingly. She began to have odd religious beliefs (eg, she was the daughter of Jesus Christ) and insisted on dressing in peculiar ways.
No family history of psychiatric disorders, such as schizophrenia, bipolar disorder, or dementia, was reported (Table 1).
The authors’ observations
The existence of LOS as a distinct subtype of schizophrenia has been the subject of discussion and controversy as far back as Manfred Bleuler in 1943 who coined the term “late-onset schizophrenia.”1 In 2000, a consensus statement by the International Late-Onset Schizophrenia Group standardized the nomenclature, defining LOS as onset between age 40 and 60, and very-late-onset schizophrenia-like psychosis (VLOS) as onset after age 60.2 Although there is no diagnostic subcategory for LOS in DSM, DSM-5 notes that (1) women are overrepresented in late-onset cases and (2) the course generally is characterized by a predominance of psychotic symptoms with preservation of affect and social functioning.3 DSM authors comment that it is not yet clear whether LOS is the same condition as schizophrenia diagnosed earlier in life. Approximately 23% of schizophrenia cases have onset after age 40.4
Cognitive symptoms in LOS
The presence of cognitive deficits in schizophrenia is common and well-recognized. The intellectual impairment is generalized and global, and there also is specific impairment in a range of cognitive functions, such as executive functions, memory, psychomotor speed, attention, and social cognition.5 Typically these cognitive impairments are present before onset of psychotic symptoms. Although cognitive symptoms are not part of the formal diagnostic criteria, DSM-5 acknowledges their presence.3 In a systematic review on nature and course of cognitive function in LOS, Rajji and Mulsant6 report that global deficits and specific deficits in executive functions, visuospatial constructional abilities, verbal fluency, and psychomotor speech have been found consistently in studies of LOS, although the presence of deficits in memory, attention, and working memory has been less consistent.
The presence of cognitive symptoms in LOS is less well-studied and understood (Table 2). The International Consensus Statement reported that no difference in type of cognitive deficit has been found in early–onset cases (onset before age 40) compared with late-onset cases, although LOS is associated with relatively milder cognitive deficits. Additionally, premorbid educational, occupational, and psychosocial functioning are less impaired in LOS than they are in early-onset schizophrenia.2
Rajji et al7 performed a meta-analysis comparison of patients with youth-onset schizophrenia, adults with first-episode schizophrenia, and those with LOS on their cognitive profiles. They reported that patients with youth-onset schizophrenia have globally severe cognitive deficits, whereas those with LOS demonstrate minimal deficits on arithmetic, digit symbol coding, and vocabulary but larger deficits on attention, fluency, global cognition, IQ, and visuospatial construction.7
There are conflicting views in the literature with regards to the course of cognitive deficits in schizophrenia. One group of researchers believes that there is progressive deterioration in cognitive functioning over time, while another maintains that cognitive impairment in schizophrenia is largely “a static encephalopathy” with no significant progression of symptoms.8 A number of studies referenced by Rajji and Mulsant6 in their systematic review report that cognitive deficits seen in patients with LOS largely are stable on follow-up with an average duration of up to 3 years. However, 2 studies with longer follow-up report evidence of cognitive decline.9,10
Relevant findings from the literature. Brodaty et al9 followed 27 patients with LOS without dementia and 34 otherwise healthy participants at baseline, 1 year, and 5 years. They reported that 9 patients with LOS and none of the control group were found to have dementia (5 Alzheimer type, 1 vascular, and 3 dementia of unknown type) at 5-year follow-up. Some patients had no clinical signs of dementia at baseline or at 1-year follow-up, but were found to have dementia at 5-year follow-up. The authors speculated that LOS might be a prodrome of Alzheimer-type dementia.
Kørner et al10 studied 12,600 patients with LOS and 7,700 with VLOS, selected from the Danish nationwide registry; follow-up was 3 to 4.58 years. They concluded that patients with LOS and VLOS were at 2 to 3 times greater risk of developing dementia than patients with osteoarthritis or the general population. The most common diagnosis among patients with schizophrenia was unspecified dementia, with Alzheimer’s dementia (AD) being the most common diagnosis in control groups. The findings suggest that dementia in LOS and VLOS has a different basis than AD.
Zakzanis et al11 investigated which neuropsychological tests best differentiate patients with LOS and those with AD or frontotemporal dementia. They reported that Wechsler Adult Intelligence Scale-Revised (WAIS-R) Similarities subtest and the California Verbal Learning Test (both short- and long-delay free recall) can differentiate LOS from AD, and a test battery comprising the WAIS-R Vocabulary, Information, Digit Span, and Comprehension subtests, and the Hooper Visual Organization test can differentiate LOS and frontotemporal dementia.12
EVALUATION Significant impairment
CT head and MRI brain scans without contrast suggest mild generalized atrophy that is more prominent in frontal and parietal areas, but the scans are otherwise unremarkable overall. A PET scan is significant for hypoactivity in the temporal and parietal lobes but, again, the images are interpreted as unremarkable overall.
Ms. P scores 21 on the Montreal Cognitive Assessment (MoCA), indicative of significant cognitive impairment (normal score, ≥26). This is a 3-point decline on a MoCA performed during her admission 5 years earlier.
Ms. P scores 8 on the Middlesex Elderly Assessment of Mental State, the lowest score in the borderline range of cognitive function for geriatric patients. She scores 13 on the Kohlman Evaluation of Living Skills, indicating that she needs maximal supervision, structure, and support to live in the community. Particularly notable is that Ms. P failed 5 out of 6 subtests in money management—a marked decline for someone who had worked as a senior accountant.
Given Ms. P’s significant cognitive decline from premorbid functioning, verified by collateral information, and current cognitive deficits established on standardized tests, we determine that, in addition to a diagnosis of schizoaffective disorder, she might meet DSM-5 criteria for unspecified major neurocognitive disorder if her functioning does not improve with treatment.
The authors’ observations
There is scant literature on late-onset schizoaffective disorder. Webster and Grossberg13 conducted a retrospective chart review of 1,730 patients age >65 who were admitted to a geriatric psychiatry unit from 1988 to 1995. Of these patients, 166 (approximately 10%) were found to have late life-onset psychosis. The psychosis was attributed to various causes, such as dementia, depression, bipolar disorder, medical causes, delirium, medication toxicity. Two patients were diagnosed with schizophrenia and 2 were diagnosed with schizoaffective disorder (the authors did not provide additional information about the patients with schizoaffective disorder). Brenner et al14 reports a case of late-onset schizoaffective disorder in a 70-year-old female patient. Evans et al15 compared outpatients age 45 to 77 with a diagnosis of schizoaffective disorder (n = 29), schizophrenia (n = 154), or nonpsychotic mood disorder (n = 27) and concluded that late-onset schizoaffective disorder might represent a variant of LOS in clinical symptom profiles and cognitive impairment but with additional mood symptoms.16
How would you begin treating Ms. P?
a) start a mood stabilizer
b) start an atypical antipsychotic
c) obtain more history and collateral information
d) recommend outpatient treatment
The authors’ observations
Given Ms. P’s manic symptoms, thought disorder, and history of psychotic symptoms with diagnosis of LOS, we assigned her a presumptive diagnosis of schizoaffective disorder, bipolar type. From the patient report, collateral information from her mother, earlier documented collateral from her husband, and chart review, it was apparent to us that Ms. P’s psychiatric history went back only 10 years—therefore meeting temporal criteria for LOS.
Clinical assessment (Figure) and standardized tests revealed the presence of neurocognitive deficits sufficient to meet criteria for major neurocognitive disorder (Table 33). The pattern of neurocognitive deficits is consistent with an AD-like amnestic picture, although no clear-cut diagnosis was present, and the neurocognitive disorder was better classified as unspecified rather than of a particular type. It remains uncertain whether cognitive deficits of severity that meet criteria for major neurocognitive disorder are sufficiently accounted for by the diagnosis of LOS alone. Unless diagnostic criteria for schizophrenia are expanded to include cognitive deficits, a separate diagnosis of major neurocognitive disorder is warranted at present.
TREATMENT Pharmacotherapy
On the unit, Ms. P is observed by nursing staff wandering, with some pressured speech but no behavioral agitation. Her clothing had been bizarre, with multiple layers, and, at one point, she walks with her gown open and without undergarments. She also reports to the nurses that she has a lot of sexual thoughts. When the interview team enters her room, they find her masturbating.
Ms. P is started on aripiprazole, 10 mg/d, titrated to 20 mg/d, and divalproex sodium, 500 mg/d. The decision to initiate a cognitive enhancer, such as an acetylcholinesterase inhibitor or memantine, is deferred to outpatient care to allow for the possibility that her cognitive features will improve after the psychosis is treated.
By the end of first week, Ms. P’s manic features are no longer prominent but her thought process continues to be bizarre, with poor insight and judgment. She demonstrates severe ambivalence in all matters, consistently gives inconsistent accounts of the past, and makes dramatic false statements.
For example, when asked about her children, Ms. P tells us that she has 6 children—the youngest 3 months old, at home by himself and “probably dead by now.” In reality, she has only a 20-year-old son who is studying abroad. Talking about her marriage, Ms. P says she and her husband are not divorced on paper but that, because they haven’t had sex for 8 years, the law has provided them with an automatic divorce.
OUTCOME Significant improvement
Ms. P shows significant response to aripiprazole and divalproex, which are well tolerated without significant adverse effects. Her limitations in executive functioning and rational thought process lead the treatment team to consider nursing home placement under guardianship. Days before discharge, however, reexamination of her neuropsychiatric state suggests significant improvement in thought process, with improvement in cognitive features. Ms. P also becomes cooperative with treatment planning.
The treatment team has meetings with Ms. P’s mother to discuss monitoring and plans for discharge. Ms. P is discharged with follow-up arranged at community mental health services.
Bottom Line
Global as well as specific cognitive deficits are associated with late-onset schizophrenia. Studies have reported increased risk of dementia in these patients over the course of 3 to 5 years, usually unspecified or Alzheimer’s type. It is imperative to assess patients with schizophrenia, especially those age ≥40, for presence of neurocognitive disorder by means of neurocognitive testing.
Related Resources
- Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245-253.
- Radhakrishnan R, Butler R, Head L. Dementia in schizophrenia. Adv Psychiatr Treat. 2012;18(2):144-153.
Drug Brand Names
Aripiprazole • Abilify
Divalproex sodium • Depakote
Mematine • Namenda
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturer of competing products.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
1. Bleuler M. Die spätschizophrenen Krankheitsbilder. Fortschr Neurol Psychiatr. 1943;15:259-290.
2. Howard R, Rabins PV, Seeman MV, et al. Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am J Psychiatry. 2000; 157(2):172-178.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Harris MJ, Jeste DV. Late-onset schizophrenia: an overview. Schizophr Bull. 1988;14(1):39-55.
5. Tandon R, Keshavan MS, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 part 1: overview. Schizophr Res. 2008;100(1):4-19.
6. Rajji TK, Mulsant BH. Nature and course of cognitive function in late-life schizophrenia: a systematic review. Schizophr Res. 2008;102(1-3):122-140.
7. Rajji TK, Ismail Z, Mulsant BH. Age at onset and cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2009;195(4):286-293.
8. Goldberg TE, Hyde TM, Kleinman JE, et al. Course of schizophrenia: neuropsychological evidence for a static encephalopathy. Schizophr Bull. 1993;19(4):797-804.
9. Brodaty H, Sachdev P, Koschera A, et al. Long-term outcome of late-onset schizophrenia: 5-year follow-up study. Br J Psychiatry. 2003;183(3):213-219.
10. Kørner A, Lopez AG, Lauritzen L, et al. Late and very-late first‐contact schizophrenia and the risk of dementia—a nationwide register based study. Int J Geriatr Psychiatry. 2009;24(1):61-67.
11. Zakzanis KK, Andrikopoulos J, Young DA, et al. Neuropsychological differentiation of late-onset schizophrenia and dementia of the Alzheimer’s type. Appl Neuropsychol. 2003;10(2):105-114.
12. Zakzanis KK, Kielar A, Young DA, et al. Neuropsychological differentiation of late onset schizophrenia and frontotemporal dementia. Cognitive Neuropsychiatry. 2001;6(1):63-77.
13. Webster J, Grossberg GT. Late-life onset of psychotic symptoms. Am J Geriatr Psychiatry. 1998;6(3):196-202.
14. Brenner R, Campbell K, Konakondla K, et al. Late onset schizoaffective disorder. Consultant. 2014;53(6):487-488.
15. Evans JD, Heaton RK, Paulsen JS, et al. Schizoaffective disorder: a form of schizophrenia or affective disorder? J Clin Psychiatry. 1999;60(12):874-882.
16. Jeste DV, Blazer DG, First M. Aging-related diagnostic variations: need for diagnostic criteria appropriate for elderly psychiatric patients. Biol Psychiatry. 2005;58(4):265-271.
Chronic pain and psychiatric illness: Managing comorbid conditions
Pain is one of the most common symptoms for which patients seek medical care, with an associated estimated annual cost of $600 billion.1 Using a multimodal approach to care—thorough evaluation, cognitive-behavioral and psychophysiological therapy, physical therapy, medications, and other interventions—can help patients effectively manage their condition and achieve healthier outcomes.
Evaluating a patient with pain
When developing a safe, comprehensive, and effective treatment plan for patients with chronic pain, first perform a thorough history and physical exam using the following elements:
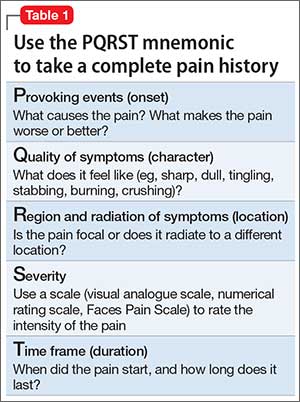
Pain history. The PQRST mnemonic (Table 1) can help you obtain critical information and assist in determining the appropriate diagnosis and cause of the patient’s pain complaints.
Psychiatric history. Document the mental health history of the patient and first-degree relatives.
Medical history. Knowing the medical history could reveal comorbidities contributing to a patient’s pain complaint.
Treatment history. Listing past and current treatments for pain, including effectiveness, helps the clinician understand if an existing treatment plan should be modified.
Functional status. Document current level of daily activity, how life activities are affected by pain; strategies used to help cope with pain; level of physical and emotional support provided in home, work, and school environments; and active stressors (eg, financial, interpersonal).
Psychosocial history. Document historical information related to coping skills, trauma history, family of origin, abuse, interpersonal relationships, social support, and academic and vocational functioning.
Substance use or abuse. Assess for use of controlled substances (ie, early refills; lost medications; obtaining medications from multiple prescribers, friends, families, or strangers; use of prescribed and non-prescribed medications for non-medical and medical purposes), nicotine, alcohol, illicit substances, and caffeine. A thorough inventory can help to identify substances a patient is using that could affect daily functioning and pain level.
Behavioral observations. Assessing mental status (eg, insight, pain behavior, cooperation) can be useful. Paying attention to pain behaviors, such as complaints of pain, decreased activity, increased medication intake, or altered facial expressions or body posture, can help the clinician gain insight to the extent that pain affects the patient’s quality of life.
The information gathered in the patient evaluation can be used to design a multimodal treatment plan to achieve maximum effectiveness.
Assessing psychiatric illness
Current approaches to pain evaluation and treatment recommend use of a biopsychosocial orientation because psychological, behavioral, and social factors can influence the experience and impact of pain, regardless of the primary cause.2 A comprehensive psychiatric evaluation, diagnosis, and treatment plan should consider the broader context in which a patient’s pain occurs.
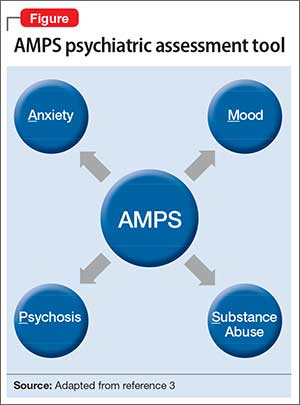
Regarding psychiatric illness, past and current symptoms, treatment history, and risk assessment should all be included. Using the “AMPS approach” (Figure)3—assessing Anxiety, Mood (depression and mania), Psychotic symptoms (paranoid ideation and hallucinations), and Substance use—helps screen for comorbid psychiatric conditions in patients with chronic pain.
Sleep assessment
Chronic pain patients often experience significant sleep disturbance that could be caused by physiological aspects of the pain condition, environmental factors (eg, uncomfortable bedding), a comorbid sleep disorder (eg, sleep apnea), a psychiatric disorder, or a combination of the above.
Obstructive and central sleep apnea are characterized by nighttime hypoxia, which leads to frequent disruption of the sleep-wake cycle and often manifests as daytime fatigue, irritability, depression, drowsiness, headaches, and increased pain sensitivity. Changes in sleep arousal can lead to neuropsychological changes during the day, such as decreased attention, memory problems, impaired executive functioning, and reduced impulse control.
Screen patients for central and obstructive sleep apnea before prescribing opioids or benzodiazepines for pain because these medications can cause or exacerbate underlying sleep apnea. Although many screening tools, such as the Epworth Sleepiness Scale, assess daytime somnolence,4 the STOP-BANG questionnaire is a quick, validated, and efficient screening tool that often is used to assess sleep apnea risk5,6 (Table 2). The presence of ≥3 risk factors identifies patients at increased risk and warrants consideration for further workup by a sleep specialist.7,8

Pharmacotherapy for chronic pain
Non-opioid medications. Pain can be broadly categorized as neuropathic or nociceptive. Neuropathic pain can be described by patients as numbness, burning, electric-like, and tingling, and is associated with nerve damage. Nociceptive pain commonly is described as similar to a toothache with descriptors such as stabbing, sharp, or a dull aching sensation; it is often, but not always, associated with acute injury or ongoing trauma to tissue. Drug treatment is most successful when the appropriate class of medication is matched to the specific type of pain.
Nociceptive pain often is successfully treated with non-steroidal anti-inflammatory drugs and acetaminophen. Non-selective COX inhibitors (eg, ibuprofen, indomethacin, ketorolac) and COX-2 selective inhibitors (eg, celecoxib) have been associated with cardiovascular, gastrointestinal, and renal disease; acetaminophen is associated with liver dysfunction.9-11 However, the absolute risk for complications in healthy patients is low.12 To minimize risk, use these agents for the shortest duration and at the lowest effective dosage possible.
Neuropathic pain can be addressed with certain antidepressants13—specifically, those that increase serotonin and norepinephrine (eg, tricyclic antidepressants [TCAs] and serotonin-norepinephrine reuptake inhibitors [SNRIs]), or medications that block ion channels (eg, anticonvulsants). TCAs (eg, desipramine, nortriptyline, amitriptyline) are among the best studied and most cost effective medications for treating neuropathic pain,14,15 but they can have sedating and anticholinergic effects, as well as cardiac adverse effects (ie, prolongation of the QTc interval). SNRIs (eg, venlafaxine, desvenlafaxine, duloxetine, and milnacipran) can be effective and often are better tolerated than TCAs.14
Some newer anticonvulsants (eg, gabapentin and pregabalin) have been found to be more effective than placebo for a variety of neuropathic pain conditions.16,17 Although they have few drug-drug interactions, anticonvulsants can cause dizziness, forgetfulness, and sedation. These adverse effects can be minimized by starting at a low dosage and titrating carefully. Because hepatic or renal impairment can affect metabolism or excretion of these drugs, review the prescribing information to determine safe dosing.
Targeted injection of medications to major pain generators (eg, an epidural steroid for radicular neck and back pain; facet injections for facet-related neck and back pain; trigger point injections for myofascial pain; occipital nerve blocks for occipital neuralgia; and botulinum toxin A injections for chronic migraine headache) can be effective in reducing discomfort and increasing function in patients with chronic pain. A detailed discussion of such therapies is beyond the scope of this article, but have been reviewed extensively elsewhere.18,19
Opioids. Although there is little evidence of long-term efficacy with chronic opioid therapy for most patients, a trial of opioids might be warranted for select patients who do not respond to other medications. Because the risk–benefit ratio for chronic opioid therapy is high,20-22 a decision to initiate a trial of a low-dosage opioid should be made only after careful consideration of those risks. It is generally agreed that treatment of chronic pain with low-dosage opioid therapy is more likely to be successful when it is used as an adjuvant to non-opioid modalities (eg, physical reconditioning, injection therapies, spinal cord stimulation, neurobehavioral interventions, non-opioid medications).
The Federation of State Medical Boards has stated that excessive reliance on opioid medications for treating chronic pain is a deviation from best practices.23 To maximize benefit and minimize risk, clinicians should carefully select appropriate patients, establish functional goals, and regularly monitor for efficacy and compliance. Thoroughly document these steps in the patient’s record for later reference.23
After establishing a clinical diagnosis for the cause of the pain, you should determine the risk of opioid abuse or misuse by using any one of the available risk assessment tools (Box). Understand, however, that no single tool has been shown to be more effective than others.

Although patients and some clinicians tend to overvalue the benefits of chronic opioid therapy, many do not fully appreciate the risks (eg, respiratory depression and death), which can be exacerbated if the patient is using other substance that suppress respiration (eg, benzodiazepines, alcohol, and illicit substances). Written informed consent and treatment agreement is highly recommended; components of such a document are listed in Table 3.23

Develop a treatment plan that emphasizes functional goals based on the patient’s physical limitations and that incorporates some type of daily, atraumatic physical activity. This plan should be documented and reviewed regularly to help monitor treatment effectiveness.
After an initial trial of a few weeks, the patient and clinician should meet to review the 5 “A”s (Table 4)24 to determine the success of the opioid regimen. Consulting your state’s prescription drug monitoring program (if one is available), obtaining a random urine drug test, and doing a pill count can provide useful, objective data. If the patient has not made progress but has experienced no adverse effects, then a small dosage increase might be warranted. If any of the 5 “A”s indicates lack of improvement or increased risk, consider stopping opioid therapy and exploring non-opioid options to manage chronic pain.
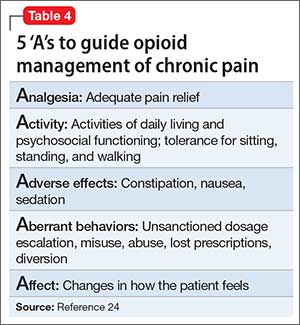
Referrals to a pain specialist or an addiction specialist, or both, might be needed, depending on the patient’s condition at any given follow-up visit. Such referral decisions, as well as all treatment plans, should be documented clearly in the medical record to prevent any misunderstanding, false accusations, or medicolegal repercussions regarding the rationale for continuing or terminating opioid-based treatment.
Non-pharmaceutical therapy for treating pain
The pain management field has successfully integrated the biopsychosocial model into regular practice. This model advocates the use of multimodal non-drug interventions in conjunction with opioid and non-opioid medications. Such interventions address behavioral, cognitive, sociocultural (psychosocial), lifestyle, and physiological dimensions of pain. A partial list of non-drug interventions is provided in Table 5.
Integration of these interventions within a biopsychosocial framework can assist you in making a comprehensive treatment plan. For example, patients with focal myofascial shoulder and back pain might derive only transient benefit from trigger point injection. However, concurrent referral to a pain psychologist and physical therapist could substantially improve functional outcomes by addressing factors that directly and indirectly influence myofascial pain. Inclusion of cognitive-behavioral therapy (addressing psychosocial and lifestyle dimensions), surface electromyography, psychophysiological interventions/biofeedback (addressing psychosocial, lifestyle, and physiological dimensions), and physical therapy (addressing lifestyle and physiological dimensions) allows the patient to learn coping skills, decrease physiological arousal that can lead to unnecessary tensing of muscles, and strengthen core muscle groups.
1. Institute of Medicine. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://www.iom.edu/~/media/Files/Report%20 Files/2011/Relieving-Pain-in-America-A-Blueprint-for- Transforming-Prevention-Care-Education-Research/ Pain%20Research%202011%20Report%20Brief.pdf. Published June 2011. Accessed April 15, 2015.
2. Jensen MP, Moore MR, Bockow TB, et al. Psychosocial factors and adjustment to chronic pain in persons with physical disabilities: a systematic review. Arch Phys Med Rehabil. 2011;92(1):146-160.
3. McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams & Wilkins; 2009.
4. Abrishami A, Khajehdehi A, Chung F. A systematic review of screening questionnaires for obstructive sleep apnea. Can J Anaesth. 2010;57(5):423-438.
5. Boynton G, Vahabzadeh A, Hammoud S, et al. Validation of the STOP-BANG questionnaire among patients referred for suspected obstructive sleep apnea. J Sleep Disord Treat Care. 2013;2(4). doi: 10.4172/2325-9639.1000121.
6. Vana KD, Silva GE, Goldberg R. Predictive abilities of the STOP-Bang and Epworth Sleepiness Scale in identifying sleep clinic patients at high risk for obstructive sleep apnea. Res Nurs Health. 2013;36(1):84-94.
7. Chung F, Elsaid H. Screening for obstructive sleep apnea before surgery: why is it important? Curr Opin Anaesthesiol. 2009;22(3):405-411.
8. Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology. 2008;108(5):812-821.
9. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167(4):394-399.
10. Sudano I, Flammer AJ, Périat D, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789-1796.
11. U.S. Food and Drug Administration. Questions and answers about oral prescription acetaminophen products to be limited to 325 mg per dosage unit. http://www.fda.gov/ drugs/drugsafety/informationbydrugclass/ucm239871. htm. Updated December 11, 2014. Accessed February 23, 2015.
12. Bhala N, Emberson J, Merhi A, et al; Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769-779.
13. Sullivan MD, Robinson JP. Antidepressant and anticonvulsant medication for chronic pain. Phys Med Rehabil Clin N Am. 2006;17(2):381-400, vi-vii.
14. Sindrup SH, Otto M, Finnerup NB, et al. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96(6):399-409.
15. Pilowsky I, Hallett EC, Bassett DL, et al. A controlled study of amitriptyline in the treatment of chronic pain. Pain. 1982;14(2):169-179.
16. Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573-581.
17. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
18. Manchikanti L, Abdi S, Atluri S, et al. An update of comprehensive evidence-based guidelines for interventional techniques in chronic spinal pain. Part II: guidance and recommendations. Pain Physician. 2013;16(suppl 2):S49-S283.
19. Singh V, Trescot A, Nishio I. Injections for chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):249-261.
20. Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers— United States, 1999–2008. MMWR Morb Mortal Wkly Rep. 2011;60(43):1487-1492.
21. Jones CM, Mack KA, Paulozzi LJ. Pharmaceutical overdose deaths, United States, 2010. JAMA. 2013;309(7):657-659.
22. Chen L, Vo T, Seefeld L, et al. Lack of correlation between opioid dose adjustment and pain score change in a group of chronic pain patients. J Pain. 2013;14(4):384-392.
23. Federation of State Medical Boards. Model policy for the use of opioid analgesics in the treatment of chronic pain. http:// www.fsmb.org/Media/Default/PDF/FSMB/Advocacy/ pain_policy_july2013.pdf. Published July 2013. Accessed December 18, 2015.
24. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
Pain is one of the most common symptoms for which patients seek medical care, with an associated estimated annual cost of $600 billion.1 Using a multimodal approach to care—thorough evaluation, cognitive-behavioral and psychophysiological therapy, physical therapy, medications, and other interventions—can help patients effectively manage their condition and achieve healthier outcomes.
Evaluating a patient with pain
When developing a safe, comprehensive, and effective treatment plan for patients with chronic pain, first perform a thorough history and physical exam using the following elements:
Pain history. The PQRST mnemonic (Table 1) can help you obtain critical information and assist in determining the appropriate diagnosis and cause of the patient’s pain complaints.
Psychiatric history. Document the mental health history of the patient and first-degree relatives.
Medical history. Knowing the medical history could reveal comorbidities contributing to a patient’s pain complaint.
Treatment history. Listing past and current treatments for pain, including effectiveness, helps the clinician understand if an existing treatment plan should be modified.
Functional status. Document current level of daily activity, how life activities are affected by pain; strategies used to help cope with pain; level of physical and emotional support provided in home, work, and school environments; and active stressors (eg, financial, interpersonal).
Psychosocial history. Document historical information related to coping skills, trauma history, family of origin, abuse, interpersonal relationships, social support, and academic and vocational functioning.
Substance use or abuse. Assess for use of controlled substances (ie, early refills; lost medications; obtaining medications from multiple prescribers, friends, families, or strangers; use of prescribed and non-prescribed medications for non-medical and medical purposes), nicotine, alcohol, illicit substances, and caffeine. A thorough inventory can help to identify substances a patient is using that could affect daily functioning and pain level.
Behavioral observations. Assessing mental status (eg, insight, pain behavior, cooperation) can be useful. Paying attention to pain behaviors, such as complaints of pain, decreased activity, increased medication intake, or altered facial expressions or body posture, can help the clinician gain insight to the extent that pain affects the patient’s quality of life.
The information gathered in the patient evaluation can be used to design a multimodal treatment plan to achieve maximum effectiveness.
Assessing psychiatric illness
Current approaches to pain evaluation and treatment recommend use of a biopsychosocial orientation because psychological, behavioral, and social factors can influence the experience and impact of pain, regardless of the primary cause.2 A comprehensive psychiatric evaluation, diagnosis, and treatment plan should consider the broader context in which a patient’s pain occurs.
Regarding psychiatric illness, past and current symptoms, treatment history, and risk assessment should all be included. Using the “AMPS approach” (Figure)3—assessing Anxiety, Mood (depression and mania), Psychotic symptoms (paranoid ideation and hallucinations), and Substance use—helps screen for comorbid psychiatric conditions in patients with chronic pain.
Sleep assessment
Chronic pain patients often experience significant sleep disturbance that could be caused by physiological aspects of the pain condition, environmental factors (eg, uncomfortable bedding), a comorbid sleep disorder (eg, sleep apnea), a psychiatric disorder, or a combination of the above.
Obstructive and central sleep apnea are characterized by nighttime hypoxia, which leads to frequent disruption of the sleep-wake cycle and often manifests as daytime fatigue, irritability, depression, drowsiness, headaches, and increased pain sensitivity. Changes in sleep arousal can lead to neuropsychological changes during the day, such as decreased attention, memory problems, impaired executive functioning, and reduced impulse control.
Screen patients for central and obstructive sleep apnea before prescribing opioids or benzodiazepines for pain because these medications can cause or exacerbate underlying sleep apnea. Although many screening tools, such as the Epworth Sleepiness Scale, assess daytime somnolence,4 the STOP-BANG questionnaire is a quick, validated, and efficient screening tool that often is used to assess sleep apnea risk5,6 (Table 2). The presence of ≥3 risk factors identifies patients at increased risk and warrants consideration for further workup by a sleep specialist.7,8
Pharmacotherapy for chronic pain
Non-opioid medications. Pain can be broadly categorized as neuropathic or nociceptive. Neuropathic pain can be described by patients as numbness, burning, electric-like, and tingling, and is associated with nerve damage. Nociceptive pain commonly is described as similar to a toothache with descriptors such as stabbing, sharp, or a dull aching sensation; it is often, but not always, associated with acute injury or ongoing trauma to tissue. Drug treatment is most successful when the appropriate class of medication is matched to the specific type of pain.
Nociceptive pain often is successfully treated with non-steroidal anti-inflammatory drugs and acetaminophen. Non-selective COX inhibitors (eg, ibuprofen, indomethacin, ketorolac) and COX-2 selective inhibitors (eg, celecoxib) have been associated with cardiovascular, gastrointestinal, and renal disease; acetaminophen is associated with liver dysfunction.9-11 However, the absolute risk for complications in healthy patients is low.12 To minimize risk, use these agents for the shortest duration and at the lowest effective dosage possible.
Neuropathic pain can be addressed with certain antidepressants13—specifically, those that increase serotonin and norepinephrine (eg, tricyclic antidepressants [TCAs] and serotonin-norepinephrine reuptake inhibitors [SNRIs]), or medications that block ion channels (eg, anticonvulsants). TCAs (eg, desipramine, nortriptyline, amitriptyline) are among the best studied and most cost effective medications for treating neuropathic pain,14,15 but they can have sedating and anticholinergic effects, as well as cardiac adverse effects (ie, prolongation of the QTc interval). SNRIs (eg, venlafaxine, desvenlafaxine, duloxetine, and milnacipran) can be effective and often are better tolerated than TCAs.14
Some newer anticonvulsants (eg, gabapentin and pregabalin) have been found to be more effective than placebo for a variety of neuropathic pain conditions.16,17 Although they have few drug-drug interactions, anticonvulsants can cause dizziness, forgetfulness, and sedation. These adverse effects can be minimized by starting at a low dosage and titrating carefully. Because hepatic or renal impairment can affect metabolism or excretion of these drugs, review the prescribing information to determine safe dosing.
Targeted injection of medications to major pain generators (eg, an epidural steroid for radicular neck and back pain; facet injections for facet-related neck and back pain; trigger point injections for myofascial pain; occipital nerve blocks for occipital neuralgia; and botulinum toxin A injections for chronic migraine headache) can be effective in reducing discomfort and increasing function in patients with chronic pain. A detailed discussion of such therapies is beyond the scope of this article, but have been reviewed extensively elsewhere.18,19
Opioids. Although there is little evidence of long-term efficacy with chronic opioid therapy for most patients, a trial of opioids might be warranted for select patients who do not respond to other medications. Because the risk–benefit ratio for chronic opioid therapy is high,20-22 a decision to initiate a trial of a low-dosage opioid should be made only after careful consideration of those risks. It is generally agreed that treatment of chronic pain with low-dosage opioid therapy is more likely to be successful when it is used as an adjuvant to non-opioid modalities (eg, physical reconditioning, injection therapies, spinal cord stimulation, neurobehavioral interventions, non-opioid medications).
The Federation of State Medical Boards has stated that excessive reliance on opioid medications for treating chronic pain is a deviation from best practices.23 To maximize benefit and minimize risk, clinicians should carefully select appropriate patients, establish functional goals, and regularly monitor for efficacy and compliance. Thoroughly document these steps in the patient’s record for later reference.23
After establishing a clinical diagnosis for the cause of the pain, you should determine the risk of opioid abuse or misuse by using any one of the available risk assessment tools (Box). Understand, however, that no single tool has been shown to be more effective than others.
Although patients and some clinicians tend to overvalue the benefits of chronic opioid therapy, many do not fully appreciate the risks (eg, respiratory depression and death), which can be exacerbated if the patient is using other substance that suppress respiration (eg, benzodiazepines, alcohol, and illicit substances). Written informed consent and treatment agreement is highly recommended; components of such a document are listed in Table 3.23
Develop a treatment plan that emphasizes functional goals based on the patient’s physical limitations and that incorporates some type of daily, atraumatic physical activity. This plan should be documented and reviewed regularly to help monitor treatment effectiveness.
After an initial trial of a few weeks, the patient and clinician should meet to review the 5 “A”s (Table 4)24 to determine the success of the opioid regimen. Consulting your state’s prescription drug monitoring program (if one is available), obtaining a random urine drug test, and doing a pill count can provide useful, objective data. If the patient has not made progress but has experienced no adverse effects, then a small dosage increase might be warranted. If any of the 5 “A”s indicates lack of improvement or increased risk, consider stopping opioid therapy and exploring non-opioid options to manage chronic pain.
Referrals to a pain specialist or an addiction specialist, or both, might be needed, depending on the patient’s condition at any given follow-up visit. Such referral decisions, as well as all treatment plans, should be documented clearly in the medical record to prevent any misunderstanding, false accusations, or medicolegal repercussions regarding the rationale for continuing or terminating opioid-based treatment.
Non-pharmaceutical therapy for treating pain
The pain management field has successfully integrated the biopsychosocial model into regular practice. This model advocates the use of multimodal non-drug interventions in conjunction with opioid and non-opioid medications. Such interventions address behavioral, cognitive, sociocultural (psychosocial), lifestyle, and physiological dimensions of pain. A partial list of non-drug interventions is provided in Table 5.
Integration of these interventions within a biopsychosocial framework can assist you in making a comprehensive treatment plan. For example, patients with focal myofascial shoulder and back pain might derive only transient benefit from trigger point injection. However, concurrent referral to a pain psychologist and physical therapist could substantially improve functional outcomes by addressing factors that directly and indirectly influence myofascial pain. Inclusion of cognitive-behavioral therapy (addressing psychosocial and lifestyle dimensions), surface electromyography, psychophysiological interventions/biofeedback (addressing psychosocial, lifestyle, and physiological dimensions), and physical therapy (addressing lifestyle and physiological dimensions) allows the patient to learn coping skills, decrease physiological arousal that can lead to unnecessary tensing of muscles, and strengthen core muscle groups.
Pain is one of the most common symptoms for which patients seek medical care, with an associated estimated annual cost of $600 billion.1 Using a multimodal approach to care—thorough evaluation, cognitive-behavioral and psychophysiological therapy, physical therapy, medications, and other interventions—can help patients effectively manage their condition and achieve healthier outcomes.
Evaluating a patient with pain
When developing a safe, comprehensive, and effective treatment plan for patients with chronic pain, first perform a thorough history and physical exam using the following elements:
Pain history. The PQRST mnemonic (Table 1) can help you obtain critical information and assist in determining the appropriate diagnosis and cause of the patient’s pain complaints.
Psychiatric history. Document the mental health history of the patient and first-degree relatives.
Medical history. Knowing the medical history could reveal comorbidities contributing to a patient’s pain complaint.
Treatment history. Listing past and current treatments for pain, including effectiveness, helps the clinician understand if an existing treatment plan should be modified.
Functional status. Document current level of daily activity, how life activities are affected by pain; strategies used to help cope with pain; level of physical and emotional support provided in home, work, and school environments; and active stressors (eg, financial, interpersonal).
Psychosocial history. Document historical information related to coping skills, trauma history, family of origin, abuse, interpersonal relationships, social support, and academic and vocational functioning.
Substance use or abuse. Assess for use of controlled substances (ie, early refills; lost medications; obtaining medications from multiple prescribers, friends, families, or strangers; use of prescribed and non-prescribed medications for non-medical and medical purposes), nicotine, alcohol, illicit substances, and caffeine. A thorough inventory can help to identify substances a patient is using that could affect daily functioning and pain level.
Behavioral observations. Assessing mental status (eg, insight, pain behavior, cooperation) can be useful. Paying attention to pain behaviors, such as complaints of pain, decreased activity, increased medication intake, or altered facial expressions or body posture, can help the clinician gain insight to the extent that pain affects the patient’s quality of life.
The information gathered in the patient evaluation can be used to design a multimodal treatment plan to achieve maximum effectiveness.
Assessing psychiatric illness
Current approaches to pain evaluation and treatment recommend use of a biopsychosocial orientation because psychological, behavioral, and social factors can influence the experience and impact of pain, regardless of the primary cause.2 A comprehensive psychiatric evaluation, diagnosis, and treatment plan should consider the broader context in which a patient’s pain occurs.
Regarding psychiatric illness, past and current symptoms, treatment history, and risk assessment should all be included. Using the “AMPS approach” (Figure)3—assessing Anxiety, Mood (depression and mania), Psychotic symptoms (paranoid ideation and hallucinations), and Substance use—helps screen for comorbid psychiatric conditions in patients with chronic pain.
Sleep assessment
Chronic pain patients often experience significant sleep disturbance that could be caused by physiological aspects of the pain condition, environmental factors (eg, uncomfortable bedding), a comorbid sleep disorder (eg, sleep apnea), a psychiatric disorder, or a combination of the above.
Obstructive and central sleep apnea are characterized by nighttime hypoxia, which leads to frequent disruption of the sleep-wake cycle and often manifests as daytime fatigue, irritability, depression, drowsiness, headaches, and increased pain sensitivity. Changes in sleep arousal can lead to neuropsychological changes during the day, such as decreased attention, memory problems, impaired executive functioning, and reduced impulse control.
Screen patients for central and obstructive sleep apnea before prescribing opioids or benzodiazepines for pain because these medications can cause or exacerbate underlying sleep apnea. Although many screening tools, such as the Epworth Sleepiness Scale, assess daytime somnolence,4 the STOP-BANG questionnaire is a quick, validated, and efficient screening tool that often is used to assess sleep apnea risk5,6 (Table 2). The presence of ≥3 risk factors identifies patients at increased risk and warrants consideration for further workup by a sleep specialist.7,8
Pharmacotherapy for chronic pain
Non-opioid medications. Pain can be broadly categorized as neuropathic or nociceptive. Neuropathic pain can be described by patients as numbness, burning, electric-like, and tingling, and is associated with nerve damage. Nociceptive pain commonly is described as similar to a toothache with descriptors such as stabbing, sharp, or a dull aching sensation; it is often, but not always, associated with acute injury or ongoing trauma to tissue. Drug treatment is most successful when the appropriate class of medication is matched to the specific type of pain.
Nociceptive pain often is successfully treated with non-steroidal anti-inflammatory drugs and acetaminophen. Non-selective COX inhibitors (eg, ibuprofen, indomethacin, ketorolac) and COX-2 selective inhibitors (eg, celecoxib) have been associated with cardiovascular, gastrointestinal, and renal disease; acetaminophen is associated with liver dysfunction.9-11 However, the absolute risk for complications in healthy patients is low.12 To minimize risk, use these agents for the shortest duration and at the lowest effective dosage possible.
Neuropathic pain can be addressed with certain antidepressants13—specifically, those that increase serotonin and norepinephrine (eg, tricyclic antidepressants [TCAs] and serotonin-norepinephrine reuptake inhibitors [SNRIs]), or medications that block ion channels (eg, anticonvulsants). TCAs (eg, desipramine, nortriptyline, amitriptyline) are among the best studied and most cost effective medications for treating neuropathic pain,14,15 but they can have sedating and anticholinergic effects, as well as cardiac adverse effects (ie, prolongation of the QTc interval). SNRIs (eg, venlafaxine, desvenlafaxine, duloxetine, and milnacipran) can be effective and often are better tolerated than TCAs.14
Some newer anticonvulsants (eg, gabapentin and pregabalin) have been found to be more effective than placebo for a variety of neuropathic pain conditions.16,17 Although they have few drug-drug interactions, anticonvulsants can cause dizziness, forgetfulness, and sedation. These adverse effects can be minimized by starting at a low dosage and titrating carefully. Because hepatic or renal impairment can affect metabolism or excretion of these drugs, review the prescribing information to determine safe dosing.
Targeted injection of medications to major pain generators (eg, an epidural steroid for radicular neck and back pain; facet injections for facet-related neck and back pain; trigger point injections for myofascial pain; occipital nerve blocks for occipital neuralgia; and botulinum toxin A injections for chronic migraine headache) can be effective in reducing discomfort and increasing function in patients with chronic pain. A detailed discussion of such therapies is beyond the scope of this article, but have been reviewed extensively elsewhere.18,19
Opioids. Although there is little evidence of long-term efficacy with chronic opioid therapy for most patients, a trial of opioids might be warranted for select patients who do not respond to other medications. Because the risk–benefit ratio for chronic opioid therapy is high,20-22 a decision to initiate a trial of a low-dosage opioid should be made only after careful consideration of those risks. It is generally agreed that treatment of chronic pain with low-dosage opioid therapy is more likely to be successful when it is used as an adjuvant to non-opioid modalities (eg, physical reconditioning, injection therapies, spinal cord stimulation, neurobehavioral interventions, non-opioid medications).
The Federation of State Medical Boards has stated that excessive reliance on opioid medications for treating chronic pain is a deviation from best practices.23 To maximize benefit and minimize risk, clinicians should carefully select appropriate patients, establish functional goals, and regularly monitor for efficacy and compliance. Thoroughly document these steps in the patient’s record for later reference.23
After establishing a clinical diagnosis for the cause of the pain, you should determine the risk of opioid abuse or misuse by using any one of the available risk assessment tools (Box). Understand, however, that no single tool has been shown to be more effective than others.
Although patients and some clinicians tend to overvalue the benefits of chronic opioid therapy, many do not fully appreciate the risks (eg, respiratory depression and death), which can be exacerbated if the patient is using other substance that suppress respiration (eg, benzodiazepines, alcohol, and illicit substances). Written informed consent and treatment agreement is highly recommended; components of such a document are listed in Table 3.23
Develop a treatment plan that emphasizes functional goals based on the patient’s physical limitations and that incorporates some type of daily, atraumatic physical activity. This plan should be documented and reviewed regularly to help monitor treatment effectiveness.
After an initial trial of a few weeks, the patient and clinician should meet to review the 5 “A”s (Table 4)24 to determine the success of the opioid regimen. Consulting your state’s prescription drug monitoring program (if one is available), obtaining a random urine drug test, and doing a pill count can provide useful, objective data. If the patient has not made progress but has experienced no adverse effects, then a small dosage increase might be warranted. If any of the 5 “A”s indicates lack of improvement or increased risk, consider stopping opioid therapy and exploring non-opioid options to manage chronic pain.
Referrals to a pain specialist or an addiction specialist, or both, might be needed, depending on the patient’s condition at any given follow-up visit. Such referral decisions, as well as all treatment plans, should be documented clearly in the medical record to prevent any misunderstanding, false accusations, or medicolegal repercussions regarding the rationale for continuing or terminating opioid-based treatment.
Non-pharmaceutical therapy for treating pain
The pain management field has successfully integrated the biopsychosocial model into regular practice. This model advocates the use of multimodal non-drug interventions in conjunction with opioid and non-opioid medications. Such interventions address behavioral, cognitive, sociocultural (psychosocial), lifestyle, and physiological dimensions of pain. A partial list of non-drug interventions is provided in Table 5.
Integration of these interventions within a biopsychosocial framework can assist you in making a comprehensive treatment plan. For example, patients with focal myofascial shoulder and back pain might derive only transient benefit from trigger point injection. However, concurrent referral to a pain psychologist and physical therapist could substantially improve functional outcomes by addressing factors that directly and indirectly influence myofascial pain. Inclusion of cognitive-behavioral therapy (addressing psychosocial and lifestyle dimensions), surface electromyography, psychophysiological interventions/biofeedback (addressing psychosocial, lifestyle, and physiological dimensions), and physical therapy (addressing lifestyle and physiological dimensions) allows the patient to learn coping skills, decrease physiological arousal that can lead to unnecessary tensing of muscles, and strengthen core muscle groups.
1. Institute of Medicine. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://www.iom.edu/~/media/Files/Report%20 Files/2011/Relieving-Pain-in-America-A-Blueprint-for- Transforming-Prevention-Care-Education-Research/ Pain%20Research%202011%20Report%20Brief.pdf. Published June 2011. Accessed April 15, 2015.
2. Jensen MP, Moore MR, Bockow TB, et al. Psychosocial factors and adjustment to chronic pain in persons with physical disabilities: a systematic review. Arch Phys Med Rehabil. 2011;92(1):146-160.
3. McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams & Wilkins; 2009.
4. Abrishami A, Khajehdehi A, Chung F. A systematic review of screening questionnaires for obstructive sleep apnea. Can J Anaesth. 2010;57(5):423-438.
5. Boynton G, Vahabzadeh A, Hammoud S, et al. Validation of the STOP-BANG questionnaire among patients referred for suspected obstructive sleep apnea. J Sleep Disord Treat Care. 2013;2(4). doi: 10.4172/2325-9639.1000121.
6. Vana KD, Silva GE, Goldberg R. Predictive abilities of the STOP-Bang and Epworth Sleepiness Scale in identifying sleep clinic patients at high risk for obstructive sleep apnea. Res Nurs Health. 2013;36(1):84-94.
7. Chung F, Elsaid H. Screening for obstructive sleep apnea before surgery: why is it important? Curr Opin Anaesthesiol. 2009;22(3):405-411.
8. Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology. 2008;108(5):812-821.
9. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167(4):394-399.
10. Sudano I, Flammer AJ, Périat D, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789-1796.
11. U.S. Food and Drug Administration. Questions and answers about oral prescription acetaminophen products to be limited to 325 mg per dosage unit. http://www.fda.gov/ drugs/drugsafety/informationbydrugclass/ucm239871. htm. Updated December 11, 2014. Accessed February 23, 2015.
12. Bhala N, Emberson J, Merhi A, et al; Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769-779.
13. Sullivan MD, Robinson JP. Antidepressant and anticonvulsant medication for chronic pain. Phys Med Rehabil Clin N Am. 2006;17(2):381-400, vi-vii.
14. Sindrup SH, Otto M, Finnerup NB, et al. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96(6):399-409.
15. Pilowsky I, Hallett EC, Bassett DL, et al. A controlled study of amitriptyline in the treatment of chronic pain. Pain. 1982;14(2):169-179.
16. Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573-581.
17. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
18. Manchikanti L, Abdi S, Atluri S, et al. An update of comprehensive evidence-based guidelines for interventional techniques in chronic spinal pain. Part II: guidance and recommendations. Pain Physician. 2013;16(suppl 2):S49-S283.
19. Singh V, Trescot A, Nishio I. Injections for chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):249-261.
20. Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers— United States, 1999–2008. MMWR Morb Mortal Wkly Rep. 2011;60(43):1487-1492.
21. Jones CM, Mack KA, Paulozzi LJ. Pharmaceutical overdose deaths, United States, 2010. JAMA. 2013;309(7):657-659.
22. Chen L, Vo T, Seefeld L, et al. Lack of correlation between opioid dose adjustment and pain score change in a group of chronic pain patients. J Pain. 2013;14(4):384-392.
23. Federation of State Medical Boards. Model policy for the use of opioid analgesics in the treatment of chronic pain. http:// www.fsmb.org/Media/Default/PDF/FSMB/Advocacy/ pain_policy_july2013.pdf. Published July 2013. Accessed December 18, 2015.
24. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
1. Institute of Medicine. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. http://www.iom.edu/~/media/Files/Report%20 Files/2011/Relieving-Pain-in-America-A-Blueprint-for- Transforming-Prevention-Care-Education-Research/ Pain%20Research%202011%20Report%20Brief.pdf. Published June 2011. Accessed April 15, 2015.
2. Jensen MP, Moore MR, Bockow TB, et al. Psychosocial factors and adjustment to chronic pain in persons with physical disabilities: a systematic review. Arch Phys Med Rehabil. 2011;92(1):146-160.
3. McCarron R, Xiong G, Bourgeois J. Lippincott’s primary care psychiatry. Philadelphia, PA: Lippincott Williams & Wilkins; 2009.
4. Abrishami A, Khajehdehi A, Chung F. A systematic review of screening questionnaires for obstructive sleep apnea. Can J Anaesth. 2010;57(5):423-438.
5. Boynton G, Vahabzadeh A, Hammoud S, et al. Validation of the STOP-BANG questionnaire among patients referred for suspected obstructive sleep apnea. J Sleep Disord Treat Care. 2013;2(4). doi: 10.4172/2325-9639.1000121.
6. Vana KD, Silva GE, Goldberg R. Predictive abilities of the STOP-Bang and Epworth Sleepiness Scale in identifying sleep clinic patients at high risk for obstructive sleep apnea. Res Nurs Health. 2013;36(1):84-94.
7. Chung F, Elsaid H. Screening for obstructive sleep apnea before surgery: why is it important? Curr Opin Anaesthesiol. 2009;22(3):405-411.
8. Chung F, Yegneswaran B, Liao P, et al. STOP questionnaire: a tool to screen patients for obstructive sleep apnea. Anesthesiology. 2008;108(5):812-821.
9. Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167(4):394-399.
10. Sudano I, Flammer AJ, Périat D, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789-1796.
11. U.S. Food and Drug Administration. Questions and answers about oral prescription acetaminophen products to be limited to 325 mg per dosage unit. http://www.fda.gov/ drugs/drugsafety/informationbydrugclass/ucm239871. htm. Updated December 11, 2014. Accessed February 23, 2015.
12. Bhala N, Emberson J, Merhi A, et al; Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769-779.
13. Sullivan MD, Robinson JP. Antidepressant and anticonvulsant medication for chronic pain. Phys Med Rehabil Clin N Am. 2006;17(2):381-400, vi-vii.
14. Sindrup SH, Otto M, Finnerup NB, et al. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96(6):399-409.
15. Pilowsky I, Hallett EC, Bassett DL, et al. A controlled study of amitriptyline in the treatment of chronic pain. Pain. 1982;14(2):169-179.
16. Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573-581.
17. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
18. Manchikanti L, Abdi S, Atluri S, et al. An update of comprehensive evidence-based guidelines for interventional techniques in chronic spinal pain. Part II: guidance and recommendations. Pain Physician. 2013;16(suppl 2):S49-S283.
19. Singh V, Trescot A, Nishio I. Injections for chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):249-261.
20. Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers— United States, 1999–2008. MMWR Morb Mortal Wkly Rep. 2011;60(43):1487-1492.
21. Jones CM, Mack KA, Paulozzi LJ. Pharmaceutical overdose deaths, United States, 2010. JAMA. 2013;309(7):657-659.
22. Chen L, Vo T, Seefeld L, et al. Lack of correlation between opioid dose adjustment and pain score change in a group of chronic pain patients. J Pain. 2013;14(4):384-392.
23. Federation of State Medical Boards. Model policy for the use of opioid analgesics in the treatment of chronic pain. http:// www.fsmb.org/Media/Default/PDF/FSMB/Advocacy/ pain_policy_july2013.pdf. Published July 2013. Accessed December 18, 2015.
24. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.

Stop blaming ‘demons’ for bizarre delusions or behavior!
That expression is a residue of the absurd belief during the Middle Ages that mental illness is caused by evil spirits—that justified burning the afflicted person at the stake. (Remember Joan of Arc?)
This ignorant, even maliciously unscientific, portrayal of psychiatric symptoms is an appalling disservice to all our patients who struggle with a potentially disabling neuropsychiatric disorder. Regrettably, some religious entities still propagate the fallacy of possession by an evil spirit and call for exorcism of bizarre behaviors sometimes associated with psychosis.1 What is really needed is an exorcism of unscientific and harmful misconceptions that mental illness is the nefarious work of Beelzebub or Lucifer.
Strange manifestations beget weird explanations
I can understand how ignorance about the neurologic basis of unusual delusions and behavior can trigger absurd religious explanations for their cause. Sometimes, brain pathology can have strange clinical manifestations that are beyond the ken of the average layperson, which invites metaphysical, religious, or philosophical explanation. Here are examples of neuropsychiatric symptoms in the category of “very unusual” that might summon a demonic malfeasance.
Delusion of possession or alien control. Some people complain of being possessed2; delusional people who have a strong religious background might believe they are possessed by Satan himself. Some of my patients with psychotic depression believe this, expressing great guilt and anguish about being doomed to go to Hell.
Alternately, patients with schizophrenia often think they are under the control of an “alien force” that shapes their behavior, feelings, and thoughts (a Schneiderian first-rank symptom). In a 2012 editorial, I proposed that this “alien intruder” is the unintegrated right hemispheric consciousness,3 and that disintegration of the 200 million inter-hemispheric white matter fibers of the corpus callosum might be the cause of the loss of integration of the right hemisphere into the dominant left hemisphere.
Some people attribute external control on their lives to a government agency, a foreign country, or a spiteful neighbor; others believe it is the work of evil spirits. Whereas the foundation of the delusion is brain pathology, the content of the delusion is colored by the affected person’s cultural and religious background.
Apotemnophilia. A neurologic disorder that manifests in a bizarre clinical symptom that invites faulty explanation: A person demands amputation of a leg because “it doesn’t belong to my body.”4 The cause of this strange and confusing disorder has been misinterpreted as a paraphilia, a desire by the affected person to achieve greater sexual satisfaction by having a stump. It was first reported in the September/October 1972 issue of the magazine Penthouse, where it was described as the motivation to heighten one’s sexual appeal because stumps can be sexually exciting to their partners.
It took many years of neurologic research to demonstrate that apotemnophilia is caused by pathology in the parietal lobe, where the physical representation of the body is located. Incomplete neurodevelopment of the parietal lobe can cause a person to fail to recognize a leg as a “legitimate” part of his body, and he (she) then desperately seeks amputation of the so-called alien limb (see the description of xenomelia below) that is attached to his body.
When an affected person is asked to delineate the borders of an alien limb, he draws a line on the skin at the precise border between the alien limb and the rest of his body—where the amputation should take place. Requests for surgical amputation were adamantly denied when the disorder was thought to be a weird sexual practice, but elective amputation in the context of neuropsychopathology is seriously debated now—and has, in fact, been reported.5 The term “body impaired integrity disorder” has been proposed, but neurologists consider the disorder an example of xenomelia.
Xenomelia (‘alien limb syndrome’). An odd neurologic disorder produced by brain pathology, in which a person has a sense of estrangement about 1 or more limbs.5 The disorder can be caused by a neurologic lesion such as tumor, Creutzfeldt-Jakob disease, hereditary diffuse leukoencephalopathy, demyelinating disease, progressive dementia, corpus callosotomy, intracerebral hemorrhage, or thalamic degeneration.6
So-called “alien hand syndrome,” or asomatognosia, is a widely recognized example of xenomelia, and is associated with medial frontal lobe damage.
Another variant of xenomelia is somatoparaphrenia, unawareness of a part of one’s body.7
Cotard syndrome. A nihilistic delusion of the nonexistence or dissolution of a body part; in extreme form, the delusion of being dead or nonexistent.8 The syndrome sometimes occurs in the setting of severe depression. Research has shown an association with atrophy of the insula,9 which is responsible for internal proprioception (interoception).
Delusional misidentification syndrome. A set of neuropsychiatric conditions in which a person misidentifies people, places, objects, or events10:
- Capgras syndrome (one perceives a familiar person as an imposter)
- Fregoli syndrome (one perceives that a familiar person is repeatedly disguised to change appearance)
- intermetamorphosis (one perceives that a person changes his external appearance and personality or identity)
- lycanthropy (one delusionally misidentifies one’s self as an animal—eg, a wolf, rabbit, or snake, and behaves accordingly)
- Ekbom syndrome (delusional belief of being infested with parasites )
- delusion of hermaphroditism (one has merged in the same body with another person of the opposite sex)
- delusion of sexual transformation (one has changed to the opposite sex)
- delusion of being the Antichrist.
Delusional misidentification syndrome can develop after the onset of focal or diffuse brain pathology, such as right hemispheric stroke, multiple sclerosis, hyperparathyroidism, traumatic brain injury, dementia, and schizophrenia. In several studies, researchers have reported an increased risk of violence in delusional misidentification syndromes.11
Neurological, not diabolical!
A disruption in brain anatomy, neurodevelopment, or circuitry/interconnectivity can produce odd beliefs and bizarre behavior that might prompt a lay observer to believe that a demon or an evil spirt is responsible for the incomprehensible symptoms. I have one response to the “blame-the-devil” proponents: It’s the brain pathology, stupid!
1. Irmak MK. Schizophrenia or possession? J Relig Health. 2014;53(3):773-777.
2. Goff DC, Brotman AW, Kindlon D, et al. The delusion of possession in chronically psychotic patients. J Nerv Ment Dis. 1991;179(9):567-571.
3. Nasrallah HA. Impaired mental proprioception in schizophrenia. Current Psychiatry. 2012;11(8):4-5.
4. Brang D, McGeoch PD, Ramachandran VS. Apotemnophilia: a neurological disorder. Neuroreport. 2008;19(13):1305-1306.
5. McGeoch PD, Brang D, Song T, et al. Xenomelia: a new right parietal lobe syndrome. J Neurol Neurosurg Psychiatry. 2011;82(12):1314-1319.
6. Graff-Radford J, Rubin MN, Jones DT, et al. The alien limb phenomenon. J Neurol. 2013;260(7):1880-1888.
7. Feinberg TE, Venneri A, Simone AM, et al. The neuroanatomy of asomatognosia and somatoparaphrenia. J Neurol Neurosurg Psychiatry. 2010;81(3):276-281.
8. Ramirez-Bermudez J, Aguilar-Venegas LC, Crail- Melendez D, et al. Cotard syndrome in neurological and psychiatric patients. J Neuropsychiatry Clin Neurosci. 2010;22(4):409-416.
9. Chatterjee SS, Mitra S. “I do not exist”-Cotard syndrome in insular cortex atrophy. Biol Psychiatry. 2015;77(11):e52-e53.
10. Cipriani G, Vedovello M, Ulivi M, et al. Delusional misidentification syndromes and dementia: a border zone between neurology and psychiatry. Am J Alzheimers Dis Other Demen. 2013;28(7):671-678.
11. Klein CA, Hirachan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
That expression is a residue of the absurd belief during the Middle Ages that mental illness is caused by evil spirits—that justified burning the afflicted person at the stake. (Remember Joan of Arc?)
This ignorant, even maliciously unscientific, portrayal of psychiatric symptoms is an appalling disservice to all our patients who struggle with a potentially disabling neuropsychiatric disorder. Regrettably, some religious entities still propagate the fallacy of possession by an evil spirit and call for exorcism of bizarre behaviors sometimes associated with psychosis.1 What is really needed is an exorcism of unscientific and harmful misconceptions that mental illness is the nefarious work of Beelzebub or Lucifer.
Strange manifestations beget weird explanations
I can understand how ignorance about the neurologic basis of unusual delusions and behavior can trigger absurd religious explanations for their cause. Sometimes, brain pathology can have strange clinical manifestations that are beyond the ken of the average layperson, which invites metaphysical, religious, or philosophical explanation. Here are examples of neuropsychiatric symptoms in the category of “very unusual” that might summon a demonic malfeasance.
Delusion of possession or alien control. Some people complain of being possessed2; delusional people who have a strong religious background might believe they are possessed by Satan himself. Some of my patients with psychotic depression believe this, expressing great guilt and anguish about being doomed to go to Hell.
Alternately, patients with schizophrenia often think they are under the control of an “alien force” that shapes their behavior, feelings, and thoughts (a Schneiderian first-rank symptom). In a 2012 editorial, I proposed that this “alien intruder” is the unintegrated right hemispheric consciousness,3 and that disintegration of the 200 million inter-hemispheric white matter fibers of the corpus callosum might be the cause of the loss of integration of the right hemisphere into the dominant left hemisphere.
Some people attribute external control on their lives to a government agency, a foreign country, or a spiteful neighbor; others believe it is the work of evil spirits. Whereas the foundation of the delusion is brain pathology, the content of the delusion is colored by the affected person’s cultural and religious background.
Apotemnophilia. A neurologic disorder that manifests in a bizarre clinical symptom that invites faulty explanation: A person demands amputation of a leg because “it doesn’t belong to my body.”4 The cause of this strange and confusing disorder has been misinterpreted as a paraphilia, a desire by the affected person to achieve greater sexual satisfaction by having a stump. It was first reported in the September/October 1972 issue of the magazine Penthouse, where it was described as the motivation to heighten one’s sexual appeal because stumps can be sexually exciting to their partners.
It took many years of neurologic research to demonstrate that apotemnophilia is caused by pathology in the parietal lobe, where the physical representation of the body is located. Incomplete neurodevelopment of the parietal lobe can cause a person to fail to recognize a leg as a “legitimate” part of his body, and he (she) then desperately seeks amputation of the so-called alien limb (see the description of xenomelia below) that is attached to his body.
When an affected person is asked to delineate the borders of an alien limb, he draws a line on the skin at the precise border between the alien limb and the rest of his body—where the amputation should take place. Requests for surgical amputation were adamantly denied when the disorder was thought to be a weird sexual practice, but elective amputation in the context of neuropsychopathology is seriously debated now—and has, in fact, been reported.5 The term “body impaired integrity disorder” has been proposed, but neurologists consider the disorder an example of xenomelia.
Xenomelia (‘alien limb syndrome’). An odd neurologic disorder produced by brain pathology, in which a person has a sense of estrangement about 1 or more limbs.5 The disorder can be caused by a neurologic lesion such as tumor, Creutzfeldt-Jakob disease, hereditary diffuse leukoencephalopathy, demyelinating disease, progressive dementia, corpus callosotomy, intracerebral hemorrhage, or thalamic degeneration.6
So-called “alien hand syndrome,” or asomatognosia, is a widely recognized example of xenomelia, and is associated with medial frontal lobe damage.
Another variant of xenomelia is somatoparaphrenia, unawareness of a part of one’s body.7
Cotard syndrome. A nihilistic delusion of the nonexistence or dissolution of a body part; in extreme form, the delusion of being dead or nonexistent.8 The syndrome sometimes occurs in the setting of severe depression. Research has shown an association with atrophy of the insula,9 which is responsible for internal proprioception (interoception).
Delusional misidentification syndrome. A set of neuropsychiatric conditions in which a person misidentifies people, places, objects, or events10:
- Capgras syndrome (one perceives a familiar person as an imposter)
- Fregoli syndrome (one perceives that a familiar person is repeatedly disguised to change appearance)
- intermetamorphosis (one perceives that a person changes his external appearance and personality or identity)
- lycanthropy (one delusionally misidentifies one’s self as an animal—eg, a wolf, rabbit, or snake, and behaves accordingly)
- Ekbom syndrome (delusional belief of being infested with parasites )
- delusion of hermaphroditism (one has merged in the same body with another person of the opposite sex)
- delusion of sexual transformation (one has changed to the opposite sex)
- delusion of being the Antichrist.
Delusional misidentification syndrome can develop after the onset of focal or diffuse brain pathology, such as right hemispheric stroke, multiple sclerosis, hyperparathyroidism, traumatic brain injury, dementia, and schizophrenia. In several studies, researchers have reported an increased risk of violence in delusional misidentification syndromes.11
Neurological, not diabolical!
A disruption in brain anatomy, neurodevelopment, or circuitry/interconnectivity can produce odd beliefs and bizarre behavior that might prompt a lay observer to believe that a demon or an evil spirt is responsible for the incomprehensible symptoms. I have one response to the “blame-the-devil” proponents: It’s the brain pathology, stupid!
That expression is a residue of the absurd belief during the Middle Ages that mental illness is caused by evil spirits—that justified burning the afflicted person at the stake. (Remember Joan of Arc?)
This ignorant, even maliciously unscientific, portrayal of psychiatric symptoms is an appalling disservice to all our patients who struggle with a potentially disabling neuropsychiatric disorder. Regrettably, some religious entities still propagate the fallacy of possession by an evil spirit and call for exorcism of bizarre behaviors sometimes associated with psychosis.1 What is really needed is an exorcism of unscientific and harmful misconceptions that mental illness is the nefarious work of Beelzebub or Lucifer.
Strange manifestations beget weird explanations
I can understand how ignorance about the neurologic basis of unusual delusions and behavior can trigger absurd religious explanations for their cause. Sometimes, brain pathology can have strange clinical manifestations that are beyond the ken of the average layperson, which invites metaphysical, religious, or philosophical explanation. Here are examples of neuropsychiatric symptoms in the category of “very unusual” that might summon a demonic malfeasance.
Delusion of possession or alien control. Some people complain of being possessed2; delusional people who have a strong religious background might believe they are possessed by Satan himself. Some of my patients with psychotic depression believe this, expressing great guilt and anguish about being doomed to go to Hell.
Alternately, patients with schizophrenia often think they are under the control of an “alien force” that shapes their behavior, feelings, and thoughts (a Schneiderian first-rank symptom). In a 2012 editorial, I proposed that this “alien intruder” is the unintegrated right hemispheric consciousness,3 and that disintegration of the 200 million inter-hemispheric white matter fibers of the corpus callosum might be the cause of the loss of integration of the right hemisphere into the dominant left hemisphere.
Some people attribute external control on their lives to a government agency, a foreign country, or a spiteful neighbor; others believe it is the work of evil spirits. Whereas the foundation of the delusion is brain pathology, the content of the delusion is colored by the affected person’s cultural and religious background.
Apotemnophilia. A neurologic disorder that manifests in a bizarre clinical symptom that invites faulty explanation: A person demands amputation of a leg because “it doesn’t belong to my body.”4 The cause of this strange and confusing disorder has been misinterpreted as a paraphilia, a desire by the affected person to achieve greater sexual satisfaction by having a stump. It was first reported in the September/October 1972 issue of the magazine Penthouse, where it was described as the motivation to heighten one’s sexual appeal because stumps can be sexually exciting to their partners.
It took many years of neurologic research to demonstrate that apotemnophilia is caused by pathology in the parietal lobe, where the physical representation of the body is located. Incomplete neurodevelopment of the parietal lobe can cause a person to fail to recognize a leg as a “legitimate” part of his body, and he (she) then desperately seeks amputation of the so-called alien limb (see the description of xenomelia below) that is attached to his body.
When an affected person is asked to delineate the borders of an alien limb, he draws a line on the skin at the precise border between the alien limb and the rest of his body—where the amputation should take place. Requests for surgical amputation were adamantly denied when the disorder was thought to be a weird sexual practice, but elective amputation in the context of neuropsychopathology is seriously debated now—and has, in fact, been reported.5 The term “body impaired integrity disorder” has been proposed, but neurologists consider the disorder an example of xenomelia.
Xenomelia (‘alien limb syndrome’). An odd neurologic disorder produced by brain pathology, in which a person has a sense of estrangement about 1 or more limbs.5 The disorder can be caused by a neurologic lesion such as tumor, Creutzfeldt-Jakob disease, hereditary diffuse leukoencephalopathy, demyelinating disease, progressive dementia, corpus callosotomy, intracerebral hemorrhage, or thalamic degeneration.6
So-called “alien hand syndrome,” or asomatognosia, is a widely recognized example of xenomelia, and is associated with medial frontal lobe damage.
Another variant of xenomelia is somatoparaphrenia, unawareness of a part of one’s body.7
Cotard syndrome. A nihilistic delusion of the nonexistence or dissolution of a body part; in extreme form, the delusion of being dead or nonexistent.8 The syndrome sometimes occurs in the setting of severe depression. Research has shown an association with atrophy of the insula,9 which is responsible for internal proprioception (interoception).
Delusional misidentification syndrome. A set of neuropsychiatric conditions in which a person misidentifies people, places, objects, or events10:
- Capgras syndrome (one perceives a familiar person as an imposter)
- Fregoli syndrome (one perceives that a familiar person is repeatedly disguised to change appearance)
- intermetamorphosis (one perceives that a person changes his external appearance and personality or identity)
- lycanthropy (one delusionally misidentifies one’s self as an animal—eg, a wolf, rabbit, or snake, and behaves accordingly)
- Ekbom syndrome (delusional belief of being infested with parasites )
- delusion of hermaphroditism (one has merged in the same body with another person of the opposite sex)
- delusion of sexual transformation (one has changed to the opposite sex)
- delusion of being the Antichrist.
Delusional misidentification syndrome can develop after the onset of focal or diffuse brain pathology, such as right hemispheric stroke, multiple sclerosis, hyperparathyroidism, traumatic brain injury, dementia, and schizophrenia. In several studies, researchers have reported an increased risk of violence in delusional misidentification syndromes.11
Neurological, not diabolical!
A disruption in brain anatomy, neurodevelopment, or circuitry/interconnectivity can produce odd beliefs and bizarre behavior that might prompt a lay observer to believe that a demon or an evil spirt is responsible for the incomprehensible symptoms. I have one response to the “blame-the-devil” proponents: It’s the brain pathology, stupid!
1. Irmak MK. Schizophrenia or possession? J Relig Health. 2014;53(3):773-777.
2. Goff DC, Brotman AW, Kindlon D, et al. The delusion of possession in chronically psychotic patients. J Nerv Ment Dis. 1991;179(9):567-571.
3. Nasrallah HA. Impaired mental proprioception in schizophrenia. Current Psychiatry. 2012;11(8):4-5.
4. Brang D, McGeoch PD, Ramachandran VS. Apotemnophilia: a neurological disorder. Neuroreport. 2008;19(13):1305-1306.
5. McGeoch PD, Brang D, Song T, et al. Xenomelia: a new right parietal lobe syndrome. J Neurol Neurosurg Psychiatry. 2011;82(12):1314-1319.
6. Graff-Radford J, Rubin MN, Jones DT, et al. The alien limb phenomenon. J Neurol. 2013;260(7):1880-1888.
7. Feinberg TE, Venneri A, Simone AM, et al. The neuroanatomy of asomatognosia and somatoparaphrenia. J Neurol Neurosurg Psychiatry. 2010;81(3):276-281.
8. Ramirez-Bermudez J, Aguilar-Venegas LC, Crail- Melendez D, et al. Cotard syndrome in neurological and psychiatric patients. J Neuropsychiatry Clin Neurosci. 2010;22(4):409-416.
9. Chatterjee SS, Mitra S. “I do not exist”-Cotard syndrome in insular cortex atrophy. Biol Psychiatry. 2015;77(11):e52-e53.
10. Cipriani G, Vedovello M, Ulivi M, et al. Delusional misidentification syndromes and dementia: a border zone between neurology and psychiatry. Am J Alzheimers Dis Other Demen. 2013;28(7):671-678.
11. Klein CA, Hirachan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
1. Irmak MK. Schizophrenia or possession? J Relig Health. 2014;53(3):773-777.
2. Goff DC, Brotman AW, Kindlon D, et al. The delusion of possession in chronically psychotic patients. J Nerv Ment Dis. 1991;179(9):567-571.
3. Nasrallah HA. Impaired mental proprioception in schizophrenia. Current Psychiatry. 2012;11(8):4-5.
4. Brang D, McGeoch PD, Ramachandran VS. Apotemnophilia: a neurological disorder. Neuroreport. 2008;19(13):1305-1306.
5. McGeoch PD, Brang D, Song T, et al. Xenomelia: a new right parietal lobe syndrome. J Neurol Neurosurg Psychiatry. 2011;82(12):1314-1319.
6. Graff-Radford J, Rubin MN, Jones DT, et al. The alien limb phenomenon. J Neurol. 2013;260(7):1880-1888.
7. Feinberg TE, Venneri A, Simone AM, et al. The neuroanatomy of asomatognosia and somatoparaphrenia. J Neurol Neurosurg Psychiatry. 2010;81(3):276-281.
8. Ramirez-Bermudez J, Aguilar-Venegas LC, Crail- Melendez D, et al. Cotard syndrome in neurological and psychiatric patients. J Neuropsychiatry Clin Neurosci. 2010;22(4):409-416.
9. Chatterjee SS, Mitra S. “I do not exist”-Cotard syndrome in insular cortex atrophy. Biol Psychiatry. 2015;77(11):e52-e53.
10. Cipriani G, Vedovello M, Ulivi M, et al. Delusional misidentification syndromes and dementia: a border zone between neurology and psychiatry. Am J Alzheimers Dis Other Demen. 2013;28(7):671-678.
11. Klein CA, Hirachan S. The masks of identities: who’s who? Delusional misidentification syndromes. J Am Acad Psychiatry Law. 2014;42(3):369-378.
Quo vadis, Psychiatry?
Psychiatrists frequently complain about their lack of recognition by other specialties, stigmatization of mental illness and the practice of psychiatry, and diminishing sense of identity as a specialty. Although I share these concerns, there is another trend that worries me perhaps more: the deliberate abandonment of more and more areas of what has traditionally been and should be psychiatry’s area of expertise and skills. Not all of this is our own doing; the fact is that other clinicians would like to get “a piece of our pie”—a trend seen in other specialties as well (eg, parts of radiology taken over by cardiologists). However, I view our role in this process as larger than other specialties’ or disciplines’ efforts.
Many of us choose not to treat substance abuse patients and instead refer them to “specialists”; yet, don’t we have enough of our own trained specialists and don’t we fill our addiction psychiatry fellowship training positions? Similarly, many do not like to treat patients with comorbid psychiatric illness and substance abuse, although this occurs frequently in our practice. Cognitive disorders often are left to neurologists and our role in managing these patients is diminishing. Pulmonologists gradually are taking over sleep disorders; one wonders why. We do not like to ask our patients about their sexual history, not even talking about treating their sexual problems! Most psychiatrists are afraid of prescribing phosphodiesterase-5 inhibitors. We are leaving the entire field of human sexuality to gynecologists, urologists, and other specialists. Paraphilic disorders are something we do not want to manage and we would rather get the whole area out of our classification systems, with the implication that these are not really mental health problems.
Many of us prefer not to treat personality disorder patients—especially those with borderline personality disorder—because they are “difficult.” Some do not even feel comfortable managing adverse effects of psychotropics such as the metabolic syndrome, or use “unusual” augmentations such as thyroid hormone. We prescribe fewer and fewer older, yet efficacious, psychotropic medications; only a small fraction of psychiatrists still prescribes monoamine oxidase inhibitors. Other disciplines, eg, primary care and pain medicine, prescribe some tricyclic antidepressants more than we do. We irrationally avoid benzodiazepines and do not like prescribing lithium, because it requires ordering blood levels and lab tests. We seem comfortable only with newer antidepressants and antipsychotics. How is this way of prescribing different from what is done in primary care? Some of our leaders sneer at the idea of psychiatrists practicing psychotherapy, perhaps feeling that such a “lowly art” should be provided by psychologists and social workers. We do not address relational issues. Last but not least, I hear colleagues saying that they do not like to treat “difficult” patients.
What are we aspiring to be and to do? To treat schizophrenia, bipolar disorder, and maybe depression, with a limited medication armamentarium we feel comfortable with and no psychotherapy? I am sure that many will say I am exaggerating, but I think not. We have, as Pogo said, met the enemy and he is us. We should get off the slippery slope of selling out psychiatry piece-by-piece, and fully embrace—clinically and research-wise (funding!)—all of what has been part of psychiatry.
Psychiatrists frequently complain about their lack of recognition by other specialties, stigmatization of mental illness and the practice of psychiatry, and diminishing sense of identity as a specialty. Although I share these concerns, there is another trend that worries me perhaps more: the deliberate abandonment of more and more areas of what has traditionally been and should be psychiatry’s area of expertise and skills. Not all of this is our own doing; the fact is that other clinicians would like to get “a piece of our pie”—a trend seen in other specialties as well (eg, parts of radiology taken over by cardiologists). However, I view our role in this process as larger than other specialties’ or disciplines’ efforts.
Many of us choose not to treat substance abuse patients and instead refer them to “specialists”; yet, don’t we have enough of our own trained specialists and don’t we fill our addiction psychiatry fellowship training positions? Similarly, many do not like to treat patients with comorbid psychiatric illness and substance abuse, although this occurs frequently in our practice. Cognitive disorders often are left to neurologists and our role in managing these patients is diminishing. Pulmonologists gradually are taking over sleep disorders; one wonders why. We do not like to ask our patients about their sexual history, not even talking about treating their sexual problems! Most psychiatrists are afraid of prescribing phosphodiesterase-5 inhibitors. We are leaving the entire field of human sexuality to gynecologists, urologists, and other specialists. Paraphilic disorders are something we do not want to manage and we would rather get the whole area out of our classification systems, with the implication that these are not really mental health problems.
Many of us prefer not to treat personality disorder patients—especially those with borderline personality disorder—because they are “difficult.” Some do not even feel comfortable managing adverse effects of psychotropics such as the metabolic syndrome, or use “unusual” augmentations such as thyroid hormone. We prescribe fewer and fewer older, yet efficacious, psychotropic medications; only a small fraction of psychiatrists still prescribes monoamine oxidase inhibitors. Other disciplines, eg, primary care and pain medicine, prescribe some tricyclic antidepressants more than we do. We irrationally avoid benzodiazepines and do not like prescribing lithium, because it requires ordering blood levels and lab tests. We seem comfortable only with newer antidepressants and antipsychotics. How is this way of prescribing different from what is done in primary care? Some of our leaders sneer at the idea of psychiatrists practicing psychotherapy, perhaps feeling that such a “lowly art” should be provided by psychologists and social workers. We do not address relational issues. Last but not least, I hear colleagues saying that they do not like to treat “difficult” patients.
What are we aspiring to be and to do? To treat schizophrenia, bipolar disorder, and maybe depression, with a limited medication armamentarium we feel comfortable with and no psychotherapy? I am sure that many will say I am exaggerating, but I think not. We have, as Pogo said, met the enemy and he is us. We should get off the slippery slope of selling out psychiatry piece-by-piece, and fully embrace—clinically and research-wise (funding!)—all of what has been part of psychiatry.
Psychiatrists frequently complain about their lack of recognition by other specialties, stigmatization of mental illness and the practice of psychiatry, and diminishing sense of identity as a specialty. Although I share these concerns, there is another trend that worries me perhaps more: the deliberate abandonment of more and more areas of what has traditionally been and should be psychiatry’s area of expertise and skills. Not all of this is our own doing; the fact is that other clinicians would like to get “a piece of our pie”—a trend seen in other specialties as well (eg, parts of radiology taken over by cardiologists). However, I view our role in this process as larger than other specialties’ or disciplines’ efforts.
Many of us choose not to treat substance abuse patients and instead refer them to “specialists”; yet, don’t we have enough of our own trained specialists and don’t we fill our addiction psychiatry fellowship training positions? Similarly, many do not like to treat patients with comorbid psychiatric illness and substance abuse, although this occurs frequently in our practice. Cognitive disorders often are left to neurologists and our role in managing these patients is diminishing. Pulmonologists gradually are taking over sleep disorders; one wonders why. We do not like to ask our patients about their sexual history, not even talking about treating their sexual problems! Most psychiatrists are afraid of prescribing phosphodiesterase-5 inhibitors. We are leaving the entire field of human sexuality to gynecologists, urologists, and other specialists. Paraphilic disorders are something we do not want to manage and we would rather get the whole area out of our classification systems, with the implication that these are not really mental health problems.
Many of us prefer not to treat personality disorder patients—especially those with borderline personality disorder—because they are “difficult.” Some do not even feel comfortable managing adverse effects of psychotropics such as the metabolic syndrome, or use “unusual” augmentations such as thyroid hormone. We prescribe fewer and fewer older, yet efficacious, psychotropic medications; only a small fraction of psychiatrists still prescribes monoamine oxidase inhibitors. Other disciplines, eg, primary care and pain medicine, prescribe some tricyclic antidepressants more than we do. We irrationally avoid benzodiazepines and do not like prescribing lithium, because it requires ordering blood levels and lab tests. We seem comfortable only with newer antidepressants and antipsychotics. How is this way of prescribing different from what is done in primary care? Some of our leaders sneer at the idea of psychiatrists practicing psychotherapy, perhaps feeling that such a “lowly art” should be provided by psychologists and social workers. We do not address relational issues. Last but not least, I hear colleagues saying that they do not like to treat “difficult” patients.
What are we aspiring to be and to do? To treat schizophrenia, bipolar disorder, and maybe depression, with a limited medication armamentarium we feel comfortable with and no psychotherapy? I am sure that many will say I am exaggerating, but I think not. We have, as Pogo said, met the enemy and he is us. We should get off the slippery slope of selling out psychiatry piece-by-piece, and fully embrace—clinically and research-wise (funding!)—all of what has been part of psychiatry.

Chronic pain and depression: Understanding 2 culprits in common
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).

Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
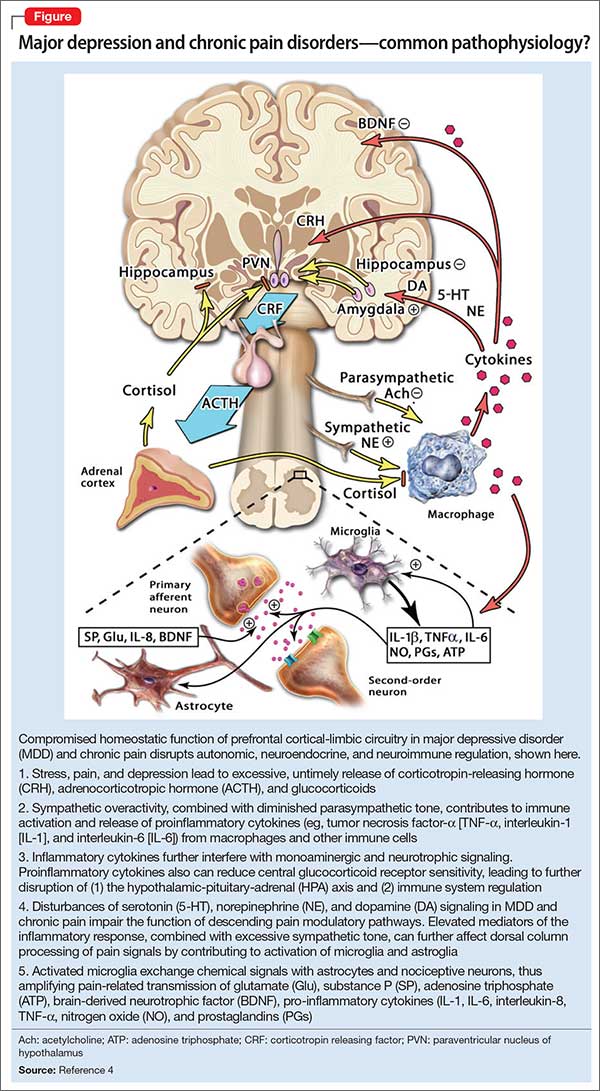
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).
Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).
Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.

Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
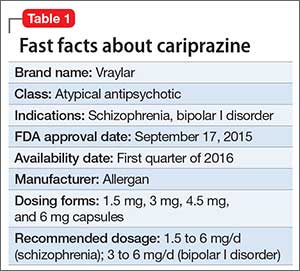
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
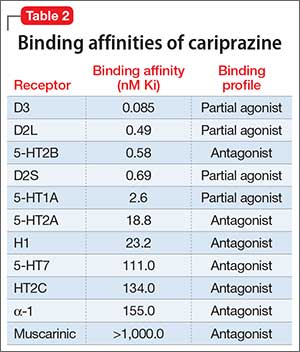
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
The view from my office: How psychiatry residency programs have changed
As I approach my twentieth year as Residency Program Coordinator in the Department of Psychiatry at Saint Louis University School of Medicine, I’ve been reflecting on the many changes that have occurred: within our residency program; in the requirements that all residency programs must meet to continue as an Accreditation Council for Graduate Medical Education (ACGME)-accredited program; and in the overall scope of psychiatry residency training.
What has changed
During my time as Residency Program Coordinator, I have assisted 5 program directors and 3 associate program directors with day-to-day details of residency training. Our residency program has had couples, and a father and son; some residents even married each other while still in training.
The Electronic Residency Application System was not available until 2001; before that, applicants interested in being invited for an interview with a psychiatry residency program had to mail in their applications for review. This was a time-consuming, tedious process. In addition, residency programs today are required to use the American Board of Psychiatry and Neurology (ABPN) PreCERT credentialing program to verify training—instead of (as in the past) simply submitting a letter to ABPN that detailed the rotations and clinical skills examinations completed.
Residency programs have gone from evaluating residents by using the 6 competencies to the Milestones requirement from ACGME, which is the newest system of measuring residents’ competencies. Every month, the program faculty meets to discuss the progress of 1 of the classes of residents and the residents who are completing an individual self-assessment. Milestone scores for each resident are then reported to ACGME.
At one time, a resident’s files could be stored in a 2-inch binder; now, we need a 4-inch binder to accommodate required documentation! I am relieved—as, I am sure, many other residency program coordinators are—that residency programs are no longer required to prepare a Program Information Form but, instead, perform a self-study and, every 10 years, have a site visit. Last, every academic year, the Residency Program Coordinator is required to enter the incoming residents’ information into the graduate medical education track, ACGME, and PreCERT Web site systems.
Rewards of my position
As Residency Program Coordinator, I’ve had the rewarding experience of meeting physicians from all over the world without having to travel to other countries. Because I have a 3- or 4-year relationship with residents, I serve them in various roles: mentor, mother, confidante, motivator, and friend. As much as the job is rewarding, being the Residency Program Coordinator can, on some days, be overwhelming, particularly because I need to think “out of the box” to streamline decisions and thus avoid conflicts with program rotations and didactic schedules.
As I approach my twentieth year as Residency Program Coordinator in the Department of Psychiatry at Saint Louis University School of Medicine, I’ve been reflecting on the many changes that have occurred: within our residency program; in the requirements that all residency programs must meet to continue as an Accreditation Council for Graduate Medical Education (ACGME)-accredited program; and in the overall scope of psychiatry residency training.
What has changed
During my time as Residency Program Coordinator, I have assisted 5 program directors and 3 associate program directors with day-to-day details of residency training. Our residency program has had couples, and a father and son; some residents even married each other while still in training.
The Electronic Residency Application System was not available until 2001; before that, applicants interested in being invited for an interview with a psychiatry residency program had to mail in their applications for review. This was a time-consuming, tedious process. In addition, residency programs today are required to use the American Board of Psychiatry and Neurology (ABPN) PreCERT credentialing program to verify training—instead of (as in the past) simply submitting a letter to ABPN that detailed the rotations and clinical skills examinations completed.
Residency programs have gone from evaluating residents by using the 6 competencies to the Milestones requirement from ACGME, which is the newest system of measuring residents’ competencies. Every month, the program faculty meets to discuss the progress of 1 of the classes of residents and the residents who are completing an individual self-assessment. Milestone scores for each resident are then reported to ACGME.
At one time, a resident’s files could be stored in a 2-inch binder; now, we need a 4-inch binder to accommodate required documentation! I am relieved—as, I am sure, many other residency program coordinators are—that residency programs are no longer required to prepare a Program Information Form but, instead, perform a self-study and, every 10 years, have a site visit. Last, every academic year, the Residency Program Coordinator is required to enter the incoming residents’ information into the graduate medical education track, ACGME, and PreCERT Web site systems.
Rewards of my position
As Residency Program Coordinator, I’ve had the rewarding experience of meeting physicians from all over the world without having to travel to other countries. Because I have a 3- or 4-year relationship with residents, I serve them in various roles: mentor, mother, confidante, motivator, and friend. As much as the job is rewarding, being the Residency Program Coordinator can, on some days, be overwhelming, particularly because I need to think “out of the box” to streamline decisions and thus avoid conflicts with program rotations and didactic schedules.
As I approach my twentieth year as Residency Program Coordinator in the Department of Psychiatry at Saint Louis University School of Medicine, I’ve been reflecting on the many changes that have occurred: within our residency program; in the requirements that all residency programs must meet to continue as an Accreditation Council for Graduate Medical Education (ACGME)-accredited program; and in the overall scope of psychiatry residency training.
What has changed
During my time as Residency Program Coordinator, I have assisted 5 program directors and 3 associate program directors with day-to-day details of residency training. Our residency program has had couples, and a father and son; some residents even married each other while still in training.
The Electronic Residency Application System was not available until 2001; before that, applicants interested in being invited for an interview with a psychiatry residency program had to mail in their applications for review. This was a time-consuming, tedious process. In addition, residency programs today are required to use the American Board of Psychiatry and Neurology (ABPN) PreCERT credentialing program to verify training—instead of (as in the past) simply submitting a letter to ABPN that detailed the rotations and clinical skills examinations completed.
Residency programs have gone from evaluating residents by using the 6 competencies to the Milestones requirement from ACGME, which is the newest system of measuring residents’ competencies. Every month, the program faculty meets to discuss the progress of 1 of the classes of residents and the residents who are completing an individual self-assessment. Milestone scores for each resident are then reported to ACGME.
At one time, a resident’s files could be stored in a 2-inch binder; now, we need a 4-inch binder to accommodate required documentation! I am relieved—as, I am sure, many other residency program coordinators are—that residency programs are no longer required to prepare a Program Information Form but, instead, perform a self-study and, every 10 years, have a site visit. Last, every academic year, the Residency Program Coordinator is required to enter the incoming residents’ information into the graduate medical education track, ACGME, and PreCERT Web site systems.
Rewards of my position
As Residency Program Coordinator, I’ve had the rewarding experience of meeting physicians from all over the world without having to travel to other countries. Because I have a 3- or 4-year relationship with residents, I serve them in various roles: mentor, mother, confidante, motivator, and friend. As much as the job is rewarding, being the Residency Program Coordinator can, on some days, be overwhelming, particularly because I need to think “out of the box” to streamline decisions and thus avoid conflicts with program rotations and didactic schedules.

