User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Drug interactions with tobacco smoke: Implications for patient care
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
Vitamin deficiencies and mental health: How are they linked?
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
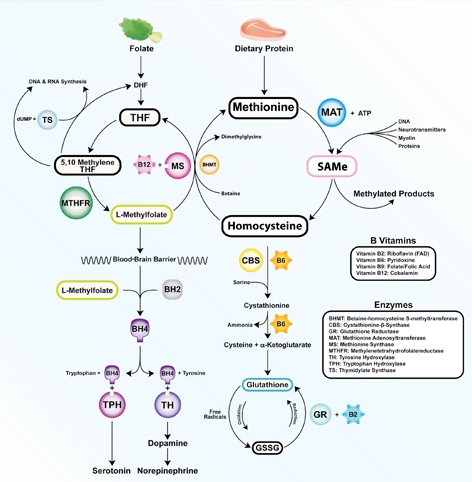
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients today often are overfed but undernourished. A growing body of literature links dietary choices to brain health and the risk of psychiatric illness. Vitamin deficiencies can affect psychiatric patients in several ways:
- deficiencies may play a causative role in mental illness and exacerbate symptoms
- psychiatric symptoms can result in poor nutrition
- vitamin insufficiency—defined as subclinical deficiency—may compromise patient recovery.
Additionally, genetic differences may compromise vitamin and essential nutrient pathways.
Vitamins are dietary components other than carbohydrates, fats, minerals, and proteins that are necessary for life. B vitamins are required for proper functioning of the methylation cycle, monoamine production, DNA synthesis, and maintenance of phospholipids such as myelin (Figure). Fat-soluble vitamins A, D, and E play important roles in genetic transcription, antioxidant recycling, and inflammatory regulation in the brain.
Figure: The methylation cycle
Vitamins B2, B6, B9, and B12 directly impact the functioning of the methylation cycle. Deficiencies pertain to brain function, as neurotransmitters, myelin, and active glutathione are dependent on one-carbon metabolism
Illustration: Mala Nimalasuriya with permission from DrewRamseyMD.com
To help clinicians recognize and treat vitamin deficiencies among psychiatric patients, this article reviews the role of the 6 essential water-soluble vitamins (B1, B2, B6, B9, B12, and C; Table 1,1) and 3 fat-soluble vitamins (A, D, and E; Table 2,1) in brain metabolism and psychiatric pathology. Because numerous sources address using supplements to treat vitamin deficiencies, this article emphasizes food sources, which for many patients are adequate to sustain nutrient status.
Table 1
Water-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| B1 (thiamine): Glycolysis, tricarboxylic acid cycle | ||||
| Rare; 7% in heart failure patients | 5% total, 12% of older women | Wernicke-Korsakoff syndrome, memory impairment, confusion, lack of coordination, paralysis | Older adults, malabsorptive conditions, heavy alcohol use. Those with diabetes are at risk because of increased clearance | Pork, fish, beans, lentils, nuts, rice, and wheat germ. Raw fish, tea, and betel nuts impair absorption |
| B2 (riboflavin): FMN, FAD cofactors in glycolysis and oxidative pathways. B6, folate, and glutathione synthesis | ||||
| 10% to 27% of older adults | <3%; 95% of adolescent girls (measured by EGRAC) | Fatigue, cracked lips, sore throat, bloodshot eyes | Older adults, low intake of animal and dairy products, heavy alcohol use | Dairy, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes |
| B6 (pyridoxal): Methylation cycle | ||||
| 11% to 24% (<5 ng/mL); 38% of heart failure patients | 14% total, 26% of adults | Dermatitis, glossitis, convulsions, migraine, chronic pain, depression | Older adults, women who use oral contraceptives, alcoholism. 33% to 49% of women age >51 have inadequate intake | Bananas, beans, potatoes, navy beans, salmon, steak, and whole grains |
| B9 (folate): Methylation cycle | ||||
| 0.5% total; up to 50% of depressed patients | 16% of adults, 19% of adolescent girls | Loss of appetite, weight loss, weakness, heart palpitations, behavioral disorders | Depression, pregnancy and lactation, alcoholism, dialysis, liver disease. Deficiency during pregnancy is linked to neural tube defects | Leafy green vegetables, fruits, dried beans, and peas |
| B12 (cobalamin): Methylation cycle (cofactor methionine synthase) | ||||
| 10% to 15% of older adults | <3% to 9% | Depression, irritability, anemia, fatigue, shortness of breath, high blood pressure | Vegetarian or vegan diet, achlorhydria, older adults. Deficiency more often due to poor absorption than low consumption | Meat, seafood, eggs, and dairy |
| C (ascorbic acid): Antioxidant | ||||
| 7.1% | 31% | Scurvy, fatigue, anemia, joint pain, petechia. Symptoms develop after 1 to 3 months of no dietary intake | Smokers, infants fed boiled or evaporated milk, limited dietary variation, patients with malabsorption, chronic illnesses | Citrus fruits, tomatoes and tomato juice, and potatoes |
| EGRAC: erythrocyte glutathione reductase activation coefficient; FAD: flavin adenine dinucleotide; FMN: flavin mononucleotide Source: Reference 1 | ||||
Table 2
Fat-soluble vitamins: Deficiency, insufficiency, symptoms, and dietary sources
| Deficiency | Insufficiency | Symptoms | At-risk patients | Dietary sources |
|---|---|---|---|---|
| A (retinol): Transcription regulation, vision | ||||
| <5% of U.S. population | 44% | Blindness, decreased immunity, corneal and retinal damage | Pregnant women, individuals with strict dietary restrictions, heavy alcohol use, chronic diarrhea, fat malabsorptive conditions | Beef liver, dairy products. Convertible beta-carotene sources: sweet potatoes, carrots, spinach, butternut squash, greens, broccoli, cantaloupe |
| D (cholecalciferol): Hormone, transcriptional regulation | ||||
| ≥50%, 90% of adults age >50 | 69% | Rickets, osteoporosis, muscle twitching | Breast-fed infants, older adults, limited sun exposure, pigmented skin, fat malabsorption, obesity. Older adults have an impaired ability to make vitamin D from the sun. SPF 15 reduces production by 99% | Fatty fish and fish liver oils, sun-dried mushrooms |
| E (tocopherols and tocotrienols): Antioxidant, PUFA protectant, gene regulation | ||||
| Rare | 93% | Anemia, neuropathy, myopathy, abnormal eye movements, weakness, retinal damage | Malabsorptive conditions, HIV, depression | Sunflower, wheat germ, and safflower oils; meats; fish; dairy; green vegetables |
| HIV: human immunodeficiency virus; PUFA: polyunsaturated fatty acids; SPF: sun protection factor Source: Reference 1 | ||||
Water-soluble vitamins
Vitamin B1 (thiamine) is essential for glucose metabolism. Pregnancy, lactation, and fever increase the need for thiamine, and tea, coffee, and shellfish can impair its absorption. Although rare, severe B1 deficiency can lead to beriberi, Wernicke’s encephalopathy (confusion, ataxia, nystagmus), and Korsakoff’s psychosis (confabulation, lack of insight, retrograde and anterograde amnesia, and apathy). Confusion and disorientation stem from the brain’s inability to oxidize glucose for energy because B1 is a critical cofactor in glycolysis and the tricarboxylic acid cycle. Deficiency leads to an increase in reactive oxygen species, proinflammatory cytokines, and blood-brain barrier dysfunction.2 Wernicke’s encephalopathy is most frequently encountered in patients with chronic alcoholism, diabetes, or eating disorders, and after bariatric surgery.3 Iatrogenic Wernicke’s encephalopathy may occur when depleted patients receive IV saline with dextrose without receiving thiamine. Top dietary sources of B1 include pork, fish, beans, lentils, nuts, rice, and wheat germ.
Vitamin B2 (riboflavin) is essential for oxidative pathways, monoamine synthesis, and the methylation cycle. B2 is needed to create the essential flavoprotein coenzymes for synthesis of L-methylfolate—the active form of folate—and for proper utilization of B6. Deficiency can occur after 4 months of inadequate intake.
Although generally B2 deficiency is rare, surveys in the United States have found that 10% to 27% of older adults (age ≥65) are deficient.4 Low intake of dairy products and meat and chronic, excessive alcohol intake are associated with deficiency. Marginal B2 levels are more prevalent in depressed patients, possibly because of B2’s role in the function of glutathione, an endogenous antioxidant.5 Top dietary sources of B2 are dairy products, meat and fish, eggs, mushrooms, almonds, leafy greens, and legumes.
Vitamin B6 refers to 3 distinct compounds: pyridoxine, pyridoxal, and pyridoxamine. B6 is essential to glycolysis, the methylation cycle, and recharging glutathione, an innate antioxidant in the brain. Higher levels of vitamin B6 are associated with a lower prevalence of depression in adolescents,6 and low dietary and plasma B6 increases the risk and severity of depression in geriatric patients7 and predicts depression in prospective trials.8 Deficiency is common (24% to 56%) among patients receiving hemodialysis.9 Women who take oral contraceptives are at increased risk of vitamin B6 deficiency.10 Top dietary sources are fish, beef, poultry, potatoes, legumes, and spinach.
Vitamin B9 (folate) is needed for proper one-carbon metabolism and thus requisite in synthesis of serotonin, norepinephrine, dopamine, and DNA and in phospholipid production. Low maternal folate status increases the risk of neural tube defects in newborns. Folate deficiency and insufficiency are common among patients with mood disorders and correlate with illness severity.11 In a study of 2,682 Finnish men, those in the lowest one-third of folate consumption had a 67% increased relative risk of depression.12 A meta-analysis of 11 studies of 15,315 persons found those who had low folate levels had a significant risk of depression.13 Patients without deficiency but with folate levels near the low end of the normal range also report low mood.14 Compared with controls, patients experiencing a first episode of psychosis have lower levels of folate, B12, and docosahexaenoic acid.15
Dietary folate must be converted to L-methylfolate for use in the brain. Patients with a methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism produce a less active form of the enzyme. The TT genotype is associated with major depression and bipolar disorder.16 Clinical trials have shown that several forms of folate can enhance antidepressant treatment.17 Augmentation with L-methylfolate, which bypasses the MTHFR enzyme, can be an effective strategy for treating depression in these patients.18
Leafy greens and legumes such as lentils are top dietary sources of folate; supplemental folic acid has been linked to an increased risk of cancer and overall mortality.19,20
Vitamin B12 (cobalamin). An essential cofactor in one-carbon metabolism, B12 is needed to produce monoamine neurotransmitters and maintain myelin. Deficiency is found in up to one-third of depressed patients11 and compromises antidepressant response,21 whereas higher vitamin B12 levels are associated with better treatment outcomes.22 B12 deficiency can cause depression, irritability, agitation, psychosis, and obsessive symptoms.23,24 Low B12 levels and elevated homocysteine increase the risk of cognitive decline and Alzheimer’s disease and are linked to a 5-fold increase in the rate of brain atrophy.26
B12 deficiencies may be seen in patients with gastrointestinal illness, older adults with achlorhydria, and vegans and vegetarians, in whom B12 intake can be low. Proton pump inhibitors such as omeprazole interfere with B12 absorption from food.
Psychiatric symptoms of B12 deficiency may present before hematologic findings.23 Folic acid supplementation may mask a B12 deficiency by delaying anemia but will not delay psychiatric symptoms. Ten percent of patients with an insufficiency (low normal levels of 200 to 400 pg/mL) have elevated homocysteine, which increases the risk of psychiatric disorders as well as comorbid illnesses such as cardiovascular disease. Top dietary sources include fish, mollusks (oysters, mussels, and clams), meat, and dairy products.
Vitamin C is vital for the synthesis of monoamines such as serotonin and norepinephrine. Vitamin C’s primary role in the brain is as an antioxidant. As a necessary cofactor, it keeps the copper and iron in metalloenzymes reduced, and also recycles vitamin E. Proper function of the methylation cycle depends on vitamin C, as does collagen synthesis and metabolism of xenobiotics by the liver. It is concentrated in cerebrospinal fluid.
Humans cannot manufacture vitamin C. Although the need for vitamin C (90 mg/d) is thought to be met by diet, studies have found that up to 13.7% of healthy, middle class patients in the United States are depleted.27 Older adults and patients with a poor diet due to drug or alcohol abuse, eating disorders, or affective symptoms are at risk.
Scurvy is caused by vitamin C deficiency and leads to bleeding gums and petechiae. Patients with insufficiency report irritability, loss of appetite, weight loss, and hypochondriasis. Vitamin C intake is significantly lower in older adults (age ≥60) with depression.28 Some research indicates patients with schizophrenia have decreased vitamin C levels and dysfunction of antioxidant defenses.29 Citrus, potatoes, and tomatoes are top dietary sources of vitamin C.
Fat-soluble vitamins
Vitamin A. Although vitamin A activity in the brain is poorly understood, retinol—the active form of vitamin A—is crucial for formation of opsins, which are the basis for vision. Childhood vitamin A deficiency may lead to blindness. Vitamin A also plays an important role in maintaining bone growth, reproduction, cell division, and immune system integrity.30 Animal sources such as beef liver, dairy products, and eggs provide retinol, and plant sources such as carrots, sweet potatoes, and leafy greens provide provitamin A carotenoids that humans convert into retinol.
Deficiency rarely is observed in the United States but remains a common problem for developing nations. In the United States, vitamin A deficiency is most often seen with excessive alcohol use, rigorous dietary restrictions, and gastrointestinal diseases accompanied by poor fat absorption.
Excess vitamin A ingestion may result in bone abnormalities, liver damage, birth defects, and depression. Isotretinoin—a form of vitamin A used to treat severe acne—carries an FDA “black-box” warning for psychiatric adverse effects, including aggression, depression, psychosis, and suicide.
Vitamin D is produced from cholesterol in the epidermis through exposure to sunlight, namely ultraviolet B radiation. After dermal synthesis or ingestion, vitamin D is converted through a series of steps into the active form of vitamin D, calcitriol, which also is known as 25(OH)D3.
Although vitamin D is known for its role in bone growth and mineralization,31 increasing evidence reveals vitamin D’s role in brain function and development.32 Both glial and neuronal cells possess vitamin D receptors in the hippocampus, prefrontal cortex, hypothalamus, thalamus, and substantia nigra—all regions theorized to be linked to depression pathophysiology.33 A review of the association of vitamin D deficiency and psychiatric illnesses will be published in a future issue of Current Psychiatry.
Vitamin D exists in food as either D2 or D3, from plant and animal sources, respectively. Concentrated sources include oily fish, sun-dried or “UVB-irradiated” mushrooms, and milk.
Vitamin E. There are 8 isoforms of vitamin E—4 tocopherols and 4 tocotrienols—that function as fat-soluble antioxidants and also promote innate antioxidant enzymes. Because vitamin E protects neuronal membranes from oxidation, low levels may affect the brain via increased inflammation. Alpha-tocopherol is the most common form of vitamin E in humans, but emerging evidence suggests tocotrienols mediate disease by modifying transcription factors in the brain, such as glutathione reductase, superoxide dismutase, and nuclear factor-kappaB.34 Low plasma vitamin E levels are found in depressed patients, although some data suggest this may be caused by factors other than dietary intake.35 Low vitamin status has been found in up to 70% of older adults.36 Although deficiency is rare, most of the U.S. population (93%) has inadequate dietary intake of vitamin E.1 The reasons for this discrepancy are unclear. Foods rich in vitamin E include almonds, sunflower seeds, leafy greens, and wheat germ.
Recommendations
Patients with depression, alcohol abuse, eating disorders, obsessive-compulsive disorder, or schizophrenia may neglect to care for themselves or adopt particular eating patterns. Deficiencies are more common among geriatric patients and those who are medically ill. Because dietary patterns are linked to the risk of psychiatric disorders, nutritional inquiry often identifies multiple modifiable risk factors, such as folate, vitamin B12, and vitamin D intake.37,38 Nutritional counseling offers clinicians an intervention with minimal side effect risks and the opportunity to modify a behavior that patients engage in 3 times a day.
Psychiatrists should assess patients’ dietary patterns and vitamin status, particularly older adults and those with:
- lower socioeconomic status or food insecurity
- a history of treatment resistance
- restrictive dietary patterns such as veganism
- alcohol abuse.
On initial assessment, test or obtain from other health care providers your patient’s blood levels of folate and vitamins D and B12. In some patients, assessing B2 and B6 levels may provide etiological guidance regarding onset of psychiatric symptoms or failure to respond to pharmacologic treatment. Because treating vitamin deficiencies often includes using supplements, evaluate recent reviews of specific deficiencies and consider consulting with the patient’s primary care provider.
Conduct a simple assessment of dietary patterns by asking patients about a typical breakfast, lunch, and dinner, their favorite snacks and foods, and specific dietary habits or restrictions (eg, not consuming seafood, dairy, meat, etc.). Rudimentary nutritional recommendations can be effective in changing a patient’s eating habits, particularly when provided by a physician. Encourage patients to eat nutrient-dense foods such as leafy greens, beans and legumes, seafood, whole grains, and a variety of vegetables and fruits. For more complex patients, consult with a clinical nutritionist.
Related Resources
- Institute of Medicine. Dietary Reference Intakes: Recommended intakes for individuals. SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf" target="_blank">www.iom.edu/Activities/Nutrition/
SummaryDRIs/~/media/Files
/Activity%20Files/Nutrition
/DRIs/5_Summary%20Table%20Tables%201-4.pdf. - The Farmacy: Vitamins. http://drewramseymd.com/index.php/resources/farmacy/category/vitamins.
- Office of Dietary Supplements. National Institutes of Health. Dietary supplements fact sheets. http://ods.od.nih.gov/factsheets/list-all.
- Oregon State University. Linus Pauling Institute. Micronutrient information center. http://lpi.oregonstate.edu/infocenter/vitamins.html.
Drug Brand Names
- Isotretinoin • Accutane
- L-methylfolate • Deplin
- Omeprazole • Prilosec
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
1. Moshfegh A, Goldman J, Cleveland L. United States Department of Agriculture, Agricultural Research Service. What we eat in America NHANES 2001-2002: Usual nutrient intakes from food compared to dietary reference intakes. http://www.ars.usda.gov/SP2UserFiles/Place/12355000/pdf/0102/usualintaketables2001-02.pdf. Published September 2005. Accessed November 27, 2012.
2. Page GL, Laight D, Cummings MH. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int J Clin Pract. 2011;65(6):684-690.
3. McCormick LM, Buchanan JR, Onwuameze OE, et al. Beyond alcoholism: Wernicke-Korsakoff syndrome in patients with psychiatric disorders. Cogn Behav Neurol. 2011;24(4):209-216.
4. Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77(6):1352-1360.
5. Naghashpour M, Amani R, Nutr R, et al. Riboflavin status and its association with serum hs-CRP levels among clinical nurses with depression. J Am Coll Nutr. 2011;30(5):340-347.
6. Murakami K, Miyake Y, Sasaki S, et al. Dietary folate, riboflavin, vitamin B-6, and vitamin B-12 and depressive symptoms in early adolescence: the Ryukyus Child Health Study. Psychosom Med. 2010;72(8):763-768.
7. Merete C, Falcon LM, Tucker KL. Vitamin B6 is associated with depressive symptomatology in Massachusetts elders. J Am Coll Nutr. 2008;27(3):421-427.
8. Skarupski KA, Tangney C, Li H, et al. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am J Clin Nutr. 2010;92(2):330-335.
9. Corken M, Porter J. Is vitamin B(6) deficiency an under-recognized risk in patients receiving haemodialysis? A systematic review: 2000-2010. Nephrology (Carlton). 2011;16(7):619-625.
10. Wilson SM, Bivins BN, Russell KA, et al. Oral contraceptive use: impact on folate, vitamin B6, and vitamin B12 status. Nutr Rev. 2011;69(10):572-583.
11. Coppen A, Bolander-Gouaille C. Treatment of depression: time to consider folic acid and vitamin B12. J Psychopharmacol. 2005;19(1):59-65.
12. Tolmunen T, Voutilainen S, Hintikka J, et al. Dietary folate and depressive symptoms are associated in middle-aged Finnish men. J Nutr. 2003;133(10):3233-3236.
13. Gilbody S, Lightfoot T, Sheldon T. Is low folate a risk factor for depression? A meta-analysis and exploration of heterogeneity. J Epidemiol Community Health. 2007;61(7):631-637.
14. Rösche J, Uhlmann C, Fröscher W. Low serum folate levels as a risk factor for depressive mood in patients with chronic epilepsy. J Neuropsychiatry Clin Neurosci. 2003;15(1):64-66.
15. Kale A, Naphade N, Sapkale S, et al. Reduced folic acid, vitamin B12 and docosahexaenoic acid and increased homocysteine and cortisol in never-medicated schizophrenia patients: implications for altered one-carbon metabolism. Psychiatry Res. 2010;175(1-2):47-53.
16. Gilbody S, Lewis S, Lightfoot T. Methylenetetrahydrofolate reductase (MTHFR) genetic polymorphisms and psychiatric disorders: a HuGE review. Am J Epidemiol. 2007;165(1):1-13.
17. Di Palma C, Urani R, Agricola R, et al. Is methylfolate effective in relieving major depression in chronic alcoholics? A hypothesis of treatment. Curr Ther Res Clin Exp. 1994;55(5):559-568.
18. Papakostas GI, Shelton RC, Zajecka JM, et al. l-Methylfolate as adjunctive therapy for ssri-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267-1274.
19. Baggott JE, Oster RA, Tamura T. Meta-analysis of cancer risk in folic acid supplementation trials. Cancer Epidemiol. 2012;36(1):78-81.
20. Figueiredo JC, Grau MV, Haile RW, et al. Folic acid and risk of prostate cancer: results from a randomized clinical trial. J Natl Cancer Inst. 2009;101(6):432-435.
21. Kate N, Grover S, Agarwal M. Does B12 deficiency lead to lack of treatment response to conventional antidepressants? Psychiatry (Edgmont). 2010;7(11):42-44.
22. Hintikka J, Tolmunen T, Tanskanen A, et al. High vitamin B12 level and good treatment outcome may be associated in major depressive disorder. BMC Psychiatry. 2003;3:17.-
23. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318(26):1720-1728.
24. Bar-Shai M, Gott D, Marmor S. Acute psychotic depression as a sole manifestation of vitamin B12 deficiency. Psychosomatics. 2011;52(4):384-386.
25. Sharma V, Biswas D. Cobalamin deficiency presenting as obsessive compulsive disorder: case report. Gen Hosp Psychiatry. 2012;34(5):578.e7-e8.
26. Vogiatzoglou A, Refsum H, Johnston C, et al. Vitamin B12 status and rate of brain volume loss in community-dwelling elderly. Neurology. 2008;71(11):826-832.
27. Smith A, Di Primio G, Humphrey-Murto S. Scurvy in the developed world. CMAJ. 2011;183(11):E752-E725.
28. Payne ME, Steck SE, George RR, et al. Fruit, vegetable, and antioxidant intakes are lower in older adults with depression. J Acad Nutr Diet. 2012;112(12):2022-2027.
29. Dadheech G, Mishra S, Gautam S, et al. Oxidative stress, α-tocopherol, ascorbic acid and reduced glutathione status in schizophrenics. Indian J Clin Biochem. 2006;21(2):34-38.
30. Hinds TS, West WL, Knight EM. Carotenoids and retinoids: a review of research clinical, and public health applications. J Clin Pharmacol. 1997;37(7):551-558.
31. Thacher TD, Clarke BL. Vitamin D insufficiency. Mayo Clin Proc. 2011;86(1):50-60.
32. Berk M, Sanders KM, Pasco JA, et al. Vitamin D deficiency may play a role in depression. Med Hypotheses. 2007;69(6):1316-1319.
33. Eyles DW, Smith S, Kinobe R, et al. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29(1):21-30.
34. Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann N Y Acad Sci. 2004;1031:127-142.
35. Owen AJ, Batterham MJ, Probst YC, et al. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59(2):304-306.
36. Panemangalore M, Lee CJ. Evaluation of the indices of retinol and alpha-tocopherol status in free-living elderly. J Gerontol. 1992;47(3):B98-B104.
37. Sánchez-Villegas A, Delgado-Rodríguez M, Alonso A, et al. Association of the Mediterranean dietary pattern with the incidence of depression: the Seguimiento Universidad de Navarra/University of Navarra follow-up (SUN) cohort. Arch Gen Psychiatry. 2009;66(10):1090-1098.
38. Jacka FN, Pasco JA, Mykletun A, et al. Association of Western and traditional diets with depression and anxiety in women. Am J Psychiatry. 2010;167(3):305-311.
Options for treating antidepressant-induced sweating
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Excessive sweating—diaphoresis—affects up to 22% of patients who take antidepressants.1 Diaphoresis may interfere with social and occupational activities, which can lead to medication discontinuation and prevent effective treatment. Stopping, decreasing, or changing antidepressants are options, but patients may be reluctant if the current dose has relieved their depressive symptoms. Adding a medication to reduce diaphoresis may be appropriate.
Sympathetic division of the peripheral nervous system signals cholinergic neurons to stimulate sweat gland secretion. In the CNS, thermoregulation occurs in the hypothalamus through a balanced and complex interaction among serotonergic and dopaminergic neurons.1 Consequently, oral medications to decrease sweating target peripheral or CNS neurons. Although evidence is limited to case reports, consider cholinergic and serotonergic antagonists and dopamine partial agonists to relieve antidepressant-induced diaphoresis.
Pharmacologic options
Peripherally, the anticholinergic agent benztropine reduced or eliminated diaphoresis at doses ranging from 0.5 mg every other day to 1 mg/d.2,3 Dry mouth was the only reported side effect.
Centrally acting serotonin antagonists may decrease diaphoresis through the 5-HT2A receptor, which signals the hypothalamus to raise body temperature. Cyproheptadine is an antihistamine with serotonin receptor antagonism. In case reports, it reduced or eliminated sweating in doses of 4 mg once or twice daily.4 Mild sedation was the only noted adverse effect. The norepinephrine and serotonin antagonist mirtazapine reduced diaphoresis within 2 weeks of initiation at 15 mg/d with no adverse effects.5 Sweating resolved after mirtazapine was titrated to 60 mg/d.
In addition to excess serotonin activity, diaphoresis may result from decreased dopaminergic tone in the hypothalamus. Centrally acting dopamine agonists—even partial agonists—may restore homeostasis and decrease sweating. Aripiprazole, 10 to 20 mg/d, reduced sweating in 2 patients; no adverse effects were reported.6
Agents to avoid
Antiadrenergic medications such as clonidine have decreased or exacerbated diaphoresis in studies.1 Similarly, paroxetine may alleviate or cause sweating. It is difficult to attribute paroxetine’s occasional effectiveness in reducing sweating solely to its anticholinergic properties because improvement may be attributed to an initial anxiolytic effect or efficacy in treating the underlying anxiety disorder.1
Disclosure
Dr. Scarff reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
1. Marcy TR, Britton ML. Antidepressant-induced sweating. Ann Pharmacother. 2005;39(4):748-752.
2. Pierre JM, Guze BH. Benztropine for venlafaxine-induced night sweats. J Clin Psychopharmacol. 2000;20(2):269.-
3. Garber A, Gregory RJ. Benztropine in the treatment of venlafaxine-induced sweating. J Clin Psychiatry. 1997;58(4):176-177.
4. Ashton AK, Weinstein WL. Cyproheptadine for drug-induced sweating. Am J Psychiatry. 2002;159(5):874-875.
5. Buecking A, Vandeleur CL, Khazaal Y, et al. Mirtazapine in drug-induced excessive sweating. Eur J Clin Pharmacol. 2005;61(7):543-544.
6. Lu BY, Cullen CE, Eide CE, et al. Antidepressant-induced sweating alleviated by aripiprazole. J Clin Psychopharmacol. 2008;28(6):710-711.
Stiff person syndrome: What psychiatrists need to know
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Stiff person syndrome (SPS) is a rare autoimmune condition characterized by stiffness and rigidity in the lower limb muscles. Because SPS often is misdiagnosed as a psychiatric illness and psychiatric comorbidities are common in patients with this disorder,1 awareness and recognition of this unique condition is essential.
An insidious presentation
Patients with SPS present with:2
- axial muscle stiffness slowly progressing to proximal muscles
- unremarkable motor, sensory, and cranial nerve examinations with normal intellectual functioning
- normal muscle strength, although electromyography shows continuous motor activity
- spasms evoked by sudden movements, jarring noise, and emotional distress
- slow and cautious gait to avoid triggering spasms and falls.
Symptoms start slowly and insidiously. Axial muscle stiffness can result in spinal deformity. Involvement is asymmetrical, with a predilection for proximal lower limb and lumbar paraspinal muscles. Affected muscles reveal tight, hard, board-like rigidity. In later stages of SPS, mild atrophy and muscle weakness are likely.
Frequent misdiagnosis
Because facial muscle spasticity is prominent, SPS patients may be misdiagnosed with Parkinson’s disease, primary lateral sclerosis, or multiple sclerosis. Spasms affecting respiratory and thoracic paraspinal muscles (status spasticus) may be misdiagnosed as an anxiety-related condition. These spasms can be life-threatening and require IV diazepam and supportive measures.
More than 60% of SPS patients have a comorbid psychiatric disorder.3 Anxiety disorders—generalized anxiety disorder, agoraphobia, and panic disorder—major depression, and alcohol abuse are the most frequent psychiatric comorbidities seen in SPS patients.3
SPS patients who panic when in public may be misdiagnosed with agoraphobia.3 Emotional stimuli may cause muscle spasms leading to falls. Treating muscle spasticity with γ-aminobutyric acid (GABA) agonists and narcotics can lead to drug abuse and dependence. Muscle spasticity can fluctuate from hour to hour, abate with sleep, and get worse with emotional distress. These findings are why approximately 70% of SPS patients are initially misdiagnosed; conversion disorder is a frequent misdiagnosis.4 Mood disorder in SPS patients may be resistant to antidepressants until these patients are treated with immunotherapy.4
Treating SPS patients
Although early intervention can reduce long-term disability, approximately 50% of SPS patients eventually have to use a wheelchair as a result of pain and immobility.5
Antibodies to glutamic acid decarboxylase, which is the rate-limiting enzyme for GABA synthesis, are present in 85% of SPS patients.5 Therefore, treatment usually includes GABA-enhancing drugs, including sedative anxiolytics (clonazepam and diazepam), antiepileptics (gabapentin, levetiracetam, tiagabine, and vigabatrin), antispasticity drugs (baclofen, dantrolene, and tizanidine), and immunotherapy (corticosteroids, IV immunoglobulins, and rituximab).5 Antidepressants, biofeedback, and relaxation training also can offer relief. Psychotherapy and substance dependency interventions may be needed.
To achieve optimum outcomes in SPS patients, a close collaborative relationship among all treating clinicians—including primary care physicians, neurologists, anesthesiologists, and psychiatrists—is necessary.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
1. Tinsley JA, Barth EM, Black JL, et al. Psychiatric consultations in stiff-man syndrome. J Clin Psychiatry. 1997;58(10):444-449.
2. Egwuonwu S, Chedebeau F. Stiff-person syndrome: a case report and review of the literature. J Natl Med Assoc. 2010;102(12):1261-1263.
3. Black JL, Barth EM, Williams DE, et al. Stiff-man syndrome. Results of interviews and psychologic testing. Psychosomatics. 1998;39(1):38-44.
4. Culav-Sumić J, Bosnjak I, Pastar Z, et al. Anxious depression and the stiff-person plus syndrome. Cogn Behav Neurol. 2008;21(4):242-245.
5. Hadavi S, Noyce AJ, Leslie RD, et al. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282.
Treating thyroid disorders and depression: 3 case studies
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
Discuss this article at www.facebook.com/CurrentPsychiatry
Many endocrine disorders can manifest as depression, including relatively rare disorders such as Cushing’s syndrome (hypercortisolism) or Conn’s syndrome (primary hyperaldosteronism) as well as common ones such as diabetes mellitus. Most clinicians do not routinely screen for adrenal disorders when evaluating depressed patients because the yield is low, but do screen for thyroid disease because these disorders often mimic depression. The following 3 cases from my practice illustrate some nuances of screening and treating depressed patients with suspected thyroid abnormalities.
CASE 1: Feeling ‘like an 80-year-old’
Ms. A, age 25, has a gastrointestinal stromal tumor (GIST) and states that she feels “like an 80-year-old woman.” She is sore all over with facial swelling, abdominal cramping, and fatigue. This feeling has worsened since she started chemotherapy with sunitinib for the GIST. Her Patient Health Questionnaire-9 (PHQ-9) score is 14 out of 27, indicating moderate depression. As part of a workup for her depression, what general laboratory tests would be most helpful?
Because Ms. A is of menstruating age, check hemoglobin/hematocrit levels to evaluate for anemia. Monitoring electrolytes would allow you to assess for hypernatremia/hyponatremia, hyperkalemia/hypokalemia, and impaired renal function, all of which could cause depressive symptoms. Depending on Ms. A’s habitus or risk of metabolic syndrome, a fasting blood glucose or hemoglobin A1C test to screen for diabetes mellitus might be valuable because depression may be associated with diabetes.1 A1C is a preferred primary screening test for diabetes (≥6.5% constitutes a positive screen) based on revised clinical practice recommendations of the American Diabetes Association. A1C is available as an office-based test that requires just a drop of blood from a finger prick and does not require a fasting blood sample or a full laboratory analysis.
A popular test for a workup of depression is serum 25-hydroxyvitamin D [25(OH)D] (vitamin D), particularly for patients who live in areas with limited exposure to ultraviolet B radiation from sunlight.2 In a study of older adults, vitamin D levels were 14% lower in patients with minor depression and 14% lower in patients with major depressive disorder compared with controls. This study suggests that depression severity is associated with decreased serum vitamin D levels,3 but the association between depression and vitamin D insufficiency and deficiency is unknown. Checking sex hormones also may be helpful depending on the patient’s symptoms, because testosterone deficiency in men and dehydroepiandrosterone deficiency in women can have a direct impact on a patient’s libido and overall sense of well-being. If repleted, improved levels of sex hormones can lead to a dramatic improvement in mood as well.
Because more than one-half of the estimated 27 million Americans with hyperthyroidism or hypothyroidism are undiagnosed, the American Thyroid Association recommends universal screening for thyroid dysfunction after age 35, with a recheck every 5 years.4 However, checking serum thyroid-stimulating hormone (TSH) levels this often may not be cost-effective. Typically, I do not follow this recommendation when assessing or treating asymptomatic individuals, but Ms. A has symptoms of hypothyroidism (Table 1) and is taking a medication—sunitinib—thought to be associated with hypothyroidism.5 Her serum TSH was very high (110 mIU/L; range 0.28 to 5.00) and her serum free T4 (FT4) was low (0.5 ng/dL; range 0.7 to 1.8). These values were consistent with overt hypothyroidism, defined as low FT4 and elevated TSH levels. This is in contrast to subclinical hypothyroidism (SH), which is defined as having an elevated serum TSH with normal thyroid hormone (T3 and T4) levels. SH presents in 5% of young patients (age <45) and increasingly is being diagnoses in older patients (age >55), who are most likely to suffer adverse effects in mood or cognition.6
Table 1
Hypothyroidism symptoms
| Psychiatric overlap |
| Fatigue |
| Hypersomnolence |
| Cognitive impairment (forgetfulness) |
| Difficulty concentrating or learning |
| Weight gain or fluid retention |
| Somatic signs and symptoms |
| Dry, itchy skin |
| Brittle hair and nails |
| Constipation |
| Myalgias |
| Heavy and/or irregular menstrual cycle |
| Increased rate of miscarriage |
| Sensitivity to cold |
CASE 1 CONTINUED: A classic case
Ms. A is started on a full levothyroxine replacement dose of 1.6 μg/kg/d. For hypothyroid patients who do not have cardiac symptoms, weight-based replacement is thought to be safe and more convenient than starting with a low dose and titrating up.7 Ms. A responds quickly. At 6-week follow-up—the recommended time interval for repeat thyroid lab testing after initiating thyroid replacement—her depressive symptoms are markedly improved and her PHQ-9 score is 6, indicating mild depression.
CASE 2: Chronic pain, low mood, and fatigue
Ms. B, age 62, has fibromyalgia and chronic back pain. She takes cyclobenzaprine, 5 mg 2 to 3 times daily, and oxycodone, 40 mg/d, and describes mild depressive symptoms when she presents for routine follow-up. Most of her complaints are related to chronic pain, but she has a history of low mood and fatigue. She says she was prescribed levothyroxine, but is unable to remember if she stopped taking it because of financial constraints or laboratory/clinical improvement. Her neurologist recently checked her serum TSH, which was elevated at 8.1 mIU/L. Is it best to restart thyroid replacement or wait 6 weeks and recheck her thyroid panel?
Mild SH typically is defined as TSH between 4.5 and 10 mIU/L. In contrast, TSH between 10 and 20 mIU/L is considered severe SH. Because Ms. B did not have prominent new symptoms, I felt it was reasonable to wait the recommended 6 weeks before rechecking her thyroid function. At follow-up, Ms. B’s TSH was 4.64 mIU/L and her FT4 was normal: 0.7 ng/dL. Thyroid replacement was not indicated because she did not have obvious symptoms and treating SH does not impact overall mood and cognition until TSH is ≥10 mIU/L.8,9
CASE 2 CONTINUED: Prominent symptoms emerge
Ms. B returns several months later. Another clinician prescribed duloxetine, titrated from 30 mg to 60 mg, for worsening fibromyalgia. Her depressive symptoms are more prominent at this visit, and her PHQ-9 score has risen from 7 to 14, indicating moderate depression. She says previously she failed or poorly tolerated several antidepressants—fluoxetine, sertraline, and citalopram—but was hoping for a pharmacologic adjustment. Most evidence-based augmentation algorithms for treating major depression start with adding a second “traditional” antidepressant such as bupropion, then move to lithium, second-generation antipsychotics, or lamotrigine.10 But what about thyroid hormone augmentation?
Thyroid hormone often is on the lower rungs of depression treatment algorithms despite Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial data. The data suggest triiodothyronine’s (T3) lower side effect burden and ease of use may offer an advantage over lithium augmentation for depressed patients who have failed several medication trials.11 Liothyronine sodium (triiodothyronine) is a relatively benign medication with potential for augmentation when started at 25 to 50 mcg/d concurrently with antidepressants such as sertraline.12 Unfortunately, most augmentation trials with T3 have been short-term—generally 4 to 8 weeks. In my practice, T3 has limited application; I use it mainly for patients with treatment-resistant depression who have failed several other treatments.
Lithium, the comparison medication to thyroid hormone in the third augmentation arm of the STAR*D trial, requires an annual check of thyroid function (TSH testing) to properly monitor for potential lithium-related hypothyroidism or thyroiditis. Hypothyroidism, for which thyroid replacement is required, with lithium therapy is common, affecting 8% to 27% of patients.13 Patients who rapidly gain weight at the beginning of lithium treatment seem to have a higher risk of developing hypothyroidism.13 However, the risk of developing lithium-induced hypothyroidism is tied to the length of treatment; the longer a patient has been treated with lithium, the greater the risk of developing lithium-induced hypothyroidism.
CASE 3: Unable to slow down
Mr. C, age 45, has a 20-year history of major depression controlled reasonably well with paroxetine, 40 mg. He presents with escalating anxiety, depression, and irritability. His wife is concerned about his overwhelming thoughts of death, especially because Mr. C’s father committed suicide 30 years ago under similar circumstances. Mr. C has been tremulous for the past month and has not been sleeping well. He feels like he is “in constant motion” and unable to slow down. He screens in the “highly likely” range for bipolar disorder on the Bipolar Spectrum Diagnostic Scale14 and is started on divalproex ER, 500 mg/d.
His thyroid function tests returns with a suppressed TSH of 0.03 mIU/L and an elevated FT4 of 3.26 ng/dL. Divalproex is discontinued and he is started on the beta blocker atenolol, 25 mg/d, to target his anxiety, tachycardia, and akathisia. TSH receptor antibody testing was positive, which, along with an abnormal radioactive iodine uptake scan, confirmed a diagnosis of Graves’ disease. He receives methimazole, 20 mg/d, as a temporizing measure. An endocrinologist completes a radioactive iodine (I-131) ablation procedure on Mr. C, which resolves his mood and anxiety symptoms.
Although hypothyroidism commonly is associated with depressive symptoms, hyperthyroidism also may present as depression. Most cases of overt hyperthyroidism are directly referred to an endocrinologist because when treating disorders such as Graves’ disease—the most common cause of hyperthyroidism, especially among women age 20 to 40—many nuclear medicine teams require the expert guidance of an endocrinologist before considering radioiodine ablation. Hyperthyroidism often is accompanied by psychiatric and somatic symptoms of an “overactive” nature (Table 2). However, older patients (age >65) with hyperthyroidism may develop apathetic hyperthyroidism, a subset that comprises approximately 10% to 15% of all hyperthyroidism cases in older adults.15 Rather than becoming nervous, jittery, and restless, patients with apathetic hyperthyroidism are depressed, lethargic, and weak, and may develop proximal myopathy or cardiomyopathy. It is essential to differentiate apathetic hyperthyroidism from typical hyperthyroidism because accurately diagnosing and treating apathetic hyperthyroidism will improve outcomes.15
Table 2
Hyperthyroidism symptoms
| Psychiatric overlap |
| Decrease or increase in appetite |
| Insomnia |
| Fatigue |
| Mood instability |
| Irritability |
| Anxiety, nervousness |
| Somatic signs and symptoms |
| Frequent bowel movement, eg, diarrhea |
| Heart palpitations |
| Heat intolerance |
| Increased sweating |
| Light or missed menstrual periods, fertility problems |
| Muscle weakness |
| Shortness of breath |
| Sudden paralysis |
| Tremor, shakiness, dizziness |
| Vision changes |
| Weight loss or gain |
| Thinning of hair |
| Itching and hives |
| Possible increase in blood sugar |
Using beta blockers to treat hyperthyroidism can help control tachycardia or palpitations, tremulousness, and anxiety that often are inherent in hyperthyroidism. But can beta blockers induce depressive symptoms? A 1-year prospective Dutch study of patients who had survived a myocardial infarction did not find evidence that beta blockers induced depressive symptoms.16 However, the long-term and high-dosage effects of beta blockers still are in question.16 In Mr. C’s case, beta blockers had only positive effects on his symptoms and did not exacerbate his depressive symptoms.
Related Resources
- National Women’s Health Resource Center, Inc. Thyroid disorders. www.healthywomen.org/condition/thyroid-disorders.
- American Thyroid Association. www.thyroid.org.
- American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/files/hypo-hyper.pdf.
Drug Brand Names
- Atenolol • Tenormin
- Bupropion • Wellbutrin, Zyban
- Citalopram • Celexa
- Cyclobenzaprine • Flexeril
- Divalproex ER • Depakote ER
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Lamotrigine • Lamictal
- Levothyroxine • Levoxyl, Synthroid
- Liothyronine sodium • Cytomel, Triostat
- Lithium • Eskalith, Lithobid
- Methimazole • Tapazole
- Oxycodone • OxyContin
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sunitinib • Sutent
Disclosure
Dr. Raj is a speaker for AstraZeneca and Merck.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
1. Campayo A, de Jonge P, Roy JF, et al. Depressive disorder and incident diabetes mellitus: the effect of characteristics of depression. Am J Psychiatry. 2010;167(5):580-588.
2. Gallagher JC, Sai AJ. Vitamin D insufficiency deficiency, and bone health. J Clin Endocrinol Metab. 2010;95(6):2630-2633.
3. Hoogendijk WJ, Lips P, Dik MG, et al. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008;65(5):508-512.
4. Ladenson PW, Singer PA, Ain KB, et al. American Thyroid Association guidelines for detection of thyroid dysfunction. Arch Intern Med. 2000;160(11):1573-1575.
5. Wolter P, Dumez H, Schöffski P. Sunitinib and hypothyroidism. N Engl J Med. 2007;356(15):1580; author reply 1580-1581.
6. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev. 2008;29(1):76-131.
7. Roos A, Linn-Rasker SP, van Domburg RT, et al. The starting dose of levothyroxine in primary hypothyroidism treatment: a prospective, randomized, double-blind trial. Arch Intern Med. 2005;165(15):1714-1720.
8. Raj YP. Subclinical hypothyroidism: merely monitor or time to treat? Current Psychiatry. 2009;8(2):47-48.
9. Samuels MH. Cognitive function in subclinical hypothyroidism. J Clin Endocrinol Metab. 2010;95(8):3611-3613.
10. Mann JJ. The medical management of depression. N Engl J Med. 2005;353(17):1819-1834.
11. Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T(3) augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1519-1530; quiz 1665.
12. Cooper-Kazaz R, Apter JT, Cohen R, et al. Combined treatment with sertraline and liothyronine in major depression: a randomized, double-blind, placebo-controlled trial. Arch Gen Psychiatry. 2007;64(6):679-688.
13. Henry C. Lithium side-effects and predictors of hypothyroidism in patients with bipolar disorder: sex differences. J Psychiatry Neurosci. 2002;27(2):104-107.
14. Ghaemi N, Pies R. The Bipolar Spectrum Diagnostic Scale. http://www.psycheducation.org/depression/BSDS.htm. Published October 2002. Updated June 2003. Accessed October 1 2012.
15. Wu W, Sun Z, Yu J, et al. A clinical retrospective analysis of factors associated with apathetic hyperthyroidism. Pathobiology. 2010;77(1):46-51.
16. van Melle JP, Verbeek DE, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
A dismal situation
I thank Dr. Nasrallah for his November editorial (“Psychiatry’s ‘swords of Damocles,’” Current Psychiatry, November 2012, p. 4-5; http://bit.ly/1HY7Y33). The major problem is not the quality of patient care. We are no longer therapists; instead, we prescribe medications and see patients once a month for 15 minutes, while social workers and psychologists handle the interaction. Where is the president of the American Psychiatric Association (APA) in advocating for psychiatrists?
Also, psychiatrists can’t earn a decent living providing therapy to patients who are on insurance plans. I was struck by Edward M. Kennedy, Jr.’s lecture at the APA’s annual meeting in May. He spoke of psychiatrists as sacrificing for the good of humanity, meaning we earn far too little. I found that condescending.
I hope you can help in turning this dismal situation around.
Ruth Cohen, MD
Private Practice
New York, NY
I thank Dr. Nasrallah for his November editorial (“Psychiatry’s ‘swords of Damocles,’” Current Psychiatry, November 2012, p. 4-5; http://bit.ly/1HY7Y33). The major problem is not the quality of patient care. We are no longer therapists; instead, we prescribe medications and see patients once a month for 15 minutes, while social workers and psychologists handle the interaction. Where is the president of the American Psychiatric Association (APA) in advocating for psychiatrists?
Also, psychiatrists can’t earn a decent living providing therapy to patients who are on insurance plans. I was struck by Edward M. Kennedy, Jr.’s lecture at the APA’s annual meeting in May. He spoke of psychiatrists as sacrificing for the good of humanity, meaning we earn far too little. I found that condescending.
I hope you can help in turning this dismal situation around.
Ruth Cohen, MD
Private Practice
New York, NY
I thank Dr. Nasrallah for his November editorial (“Psychiatry’s ‘swords of Damocles,’” Current Psychiatry, November 2012, p. 4-5; http://bit.ly/1HY7Y33). The major problem is not the quality of patient care. We are no longer therapists; instead, we prescribe medications and see patients once a month for 15 minutes, while social workers and psychologists handle the interaction. Where is the president of the American Psychiatric Association (APA) in advocating for psychiatrists?
Also, psychiatrists can’t earn a decent living providing therapy to patients who are on insurance plans. I was struck by Edward M. Kennedy, Jr.’s lecture at the APA’s annual meeting in May. He spoke of psychiatrists as sacrificing for the good of humanity, meaning we earn far too little. I found that condescending.
I hope you can help in turning this dismal situation around.
Ruth Cohen, MD
Private Practice
New York, NY
The delirium/dementia treatment paradox
Delirium is the most common psychiatric disorder on medical/surgical units of general hospitals and is a major cause of consultations for consultation-liaison (C-L) psychiatrists in medical centers.
It primarily afflicts older patients and is characterized by a rapid onset of confusion with hallucinations, delusions, agitation (but sometimes hypoactivity), and fluctuating severity and outcomes. It can be triggered by multiple factors, including dehydration, electrolyte imbalance, upper respiratory infection, urinary tract infections, post-surgical state, and, very commonly, iatrogenic factors. Many medications, especially those with anticholinergic activity, can cause delirium in older patients. Delirium can lead to pronged hospital stays, functional decline, cognitive impairment, and accelerated mortality.1
So what is the evidence-based treatment for this common and serious neuropsychiatric brain disorder? None exists! Delirium is managed by identifying and addressing underlying factors, as well as supportive medical care (hydration, nutrition, sleep, monitoring vital signs, preventing aspiration, quiet rooms, orientation, and ambulation). Physical restraints are undesirable but may be hard to avoid for severely disinhibited and agitated patients who try to rip out their IV or endotracheal tube or assault staff or family members who try to calm them during their psychotic confusional state.
Doesn’t this sound similar to dementia patients in nursing homes who develop psychosis and agitation and pose management problems for their family and staff? These 2 neuropsychiatric conditions are clinically similar, and the use of antipsychotic agents is not FDA-approved for either of them. Antipsychotics have not been found efficacious for delirium2 or dementia with psychosis3 but their use continues. The paradox is that C-L psychiatrists use small doses of antipsychotics routinely for delirium and generally believe that such pharmacologic intervention works in many patients, although some older agents (eg, haloperidol) have been reported to be neurotoxic4 and associated with higher mortality in dementia patients with psychosis.5,6
The relationship of antipsychotic treatment with delirium and dementia is complex, paradoxical, and unsettled. Consider the following:
- Antipsychotics are not approved for the psychosis of dementia despite 17 large, placebo-controlled trials with 4 different second-generation agents.
- The FDA issued a “black-box” warning on all first- and second-generation antipsychotics because of a 1.6-times higher mortality rate among older patients with dementia.
- No placebo-controlled, FDA trials of delirium have been conducted with any antipsychotic agents.
- Clinicians frequently use first- and second-generation antipsychotics for delirium despite the lack of evidence.
- The FDA has not issued a “black-box” warning on antipsychotics for use in delirium as they did for dementia, although typically both populations are older and may be susceptible to the same complications observed in Alzheimer’s disease trials (eg, strokes, transient ischemic attack, and aspiration pneumonia). This may be because of the absence of safety data of antipsychotics in delirium based on industry-sponsored registration clinical trials. No data means no warning, although the risk factors may be present for millions of older patients who have suffered from delirium and have been treated with first-or second-generation antipsychotics.
The current convoluted state of pharmacotherapy for delirium and dementia is likely to continue: dementia patients with psychosis may or may not receive antipsychotics because of the FDA’s “black-box” warning while delirium patients readily receive various antipsychotics based on long-term practice, although not a single antipsychotic has been FDA-approved for delirium. Until a pharmaceutical company decides to conduct a large placebo-controlled trial for delirium, the current widespread, off-label use of antipsychotics in delirium will continue and “black-box” warning will apply only to 1 clinical population of older persons (those with dementia) but not another (those with delirium). Go figure!
1. Martins S, Fernandes L. Delirium in elderly people: a review. Front Neurol. 2012;3:101.-
2. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
3. Seitz DP, Gill SS, Herrmann N, et al. Pharmacological treatments for neuropsychiatric symptoms of dementia in long-term care: a systematic review. [published online October 19, 2012]. Int Psychogeriatr. 2012:1-19. doi: http://dx.doi.org/10.1017/S1041610212001627.
4. Nasrallah HA. Does the neurotoxicity of haloperidol explain the higher mortality in dementia patients compared with the second generation agents? Am J Psychiatry. 2012;169(6):663-664;author reply 664–665.
5. Nasrallah HA, White T, Nasrallah AT. Lower mortality in geriatric patients receiving risperidone and olanzapine versus haloperidol: preliminary analysis of retrospective data. Am J Geriatr Psychiatry. 2004;12(4):437-439.
6. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, visit http://www.facebook.com/CurrentPsychiatry, or click on the “Send Letters” link.
Delirium is the most common psychiatric disorder on medical/surgical units of general hospitals and is a major cause of consultations for consultation-liaison (C-L) psychiatrists in medical centers.
It primarily afflicts older patients and is characterized by a rapid onset of confusion with hallucinations, delusions, agitation (but sometimes hypoactivity), and fluctuating severity and outcomes. It can be triggered by multiple factors, including dehydration, electrolyte imbalance, upper respiratory infection, urinary tract infections, post-surgical state, and, very commonly, iatrogenic factors. Many medications, especially those with anticholinergic activity, can cause delirium in older patients. Delirium can lead to pronged hospital stays, functional decline, cognitive impairment, and accelerated mortality.1
So what is the evidence-based treatment for this common and serious neuropsychiatric brain disorder? None exists! Delirium is managed by identifying and addressing underlying factors, as well as supportive medical care (hydration, nutrition, sleep, monitoring vital signs, preventing aspiration, quiet rooms, orientation, and ambulation). Physical restraints are undesirable but may be hard to avoid for severely disinhibited and agitated patients who try to rip out their IV or endotracheal tube or assault staff or family members who try to calm them during their psychotic confusional state.
Doesn’t this sound similar to dementia patients in nursing homes who develop psychosis and agitation and pose management problems for their family and staff? These 2 neuropsychiatric conditions are clinically similar, and the use of antipsychotic agents is not FDA-approved for either of them. Antipsychotics have not been found efficacious for delirium2 or dementia with psychosis3 but their use continues. The paradox is that C-L psychiatrists use small doses of antipsychotics routinely for delirium and generally believe that such pharmacologic intervention works in many patients, although some older agents (eg, haloperidol) have been reported to be neurotoxic4 and associated with higher mortality in dementia patients with psychosis.5,6
The relationship of antipsychotic treatment with delirium and dementia is complex, paradoxical, and unsettled. Consider the following:
- Antipsychotics are not approved for the psychosis of dementia despite 17 large, placebo-controlled trials with 4 different second-generation agents.
- The FDA issued a “black-box” warning on all first- and second-generation antipsychotics because of a 1.6-times higher mortality rate among older patients with dementia.
- No placebo-controlled, FDA trials of delirium have been conducted with any antipsychotic agents.
- Clinicians frequently use first- and second-generation antipsychotics for delirium despite the lack of evidence.
- The FDA has not issued a “black-box” warning on antipsychotics for use in delirium as they did for dementia, although typically both populations are older and may be susceptible to the same complications observed in Alzheimer’s disease trials (eg, strokes, transient ischemic attack, and aspiration pneumonia). This may be because of the absence of safety data of antipsychotics in delirium based on industry-sponsored registration clinical trials. No data means no warning, although the risk factors may be present for millions of older patients who have suffered from delirium and have been treated with first-or second-generation antipsychotics.
The current convoluted state of pharmacotherapy for delirium and dementia is likely to continue: dementia patients with psychosis may or may not receive antipsychotics because of the FDA’s “black-box” warning while delirium patients readily receive various antipsychotics based on long-term practice, although not a single antipsychotic has been FDA-approved for delirium. Until a pharmaceutical company decides to conduct a large placebo-controlled trial for delirium, the current widespread, off-label use of antipsychotics in delirium will continue and “black-box” warning will apply only to 1 clinical population of older persons (those with dementia) but not another (those with delirium). Go figure!
Delirium is the most common psychiatric disorder on medical/surgical units of general hospitals and is a major cause of consultations for consultation-liaison (C-L) psychiatrists in medical centers.
It primarily afflicts older patients and is characterized by a rapid onset of confusion with hallucinations, delusions, agitation (but sometimes hypoactivity), and fluctuating severity and outcomes. It can be triggered by multiple factors, including dehydration, electrolyte imbalance, upper respiratory infection, urinary tract infections, post-surgical state, and, very commonly, iatrogenic factors. Many medications, especially those with anticholinergic activity, can cause delirium in older patients. Delirium can lead to pronged hospital stays, functional decline, cognitive impairment, and accelerated mortality.1
So what is the evidence-based treatment for this common and serious neuropsychiatric brain disorder? None exists! Delirium is managed by identifying and addressing underlying factors, as well as supportive medical care (hydration, nutrition, sleep, monitoring vital signs, preventing aspiration, quiet rooms, orientation, and ambulation). Physical restraints are undesirable but may be hard to avoid for severely disinhibited and agitated patients who try to rip out their IV or endotracheal tube or assault staff or family members who try to calm them during their psychotic confusional state.
Doesn’t this sound similar to dementia patients in nursing homes who develop psychosis and agitation and pose management problems for their family and staff? These 2 neuropsychiatric conditions are clinically similar, and the use of antipsychotic agents is not FDA-approved for either of them. Antipsychotics have not been found efficacious for delirium2 or dementia with psychosis3 but their use continues. The paradox is that C-L psychiatrists use small doses of antipsychotics routinely for delirium and generally believe that such pharmacologic intervention works in many patients, although some older agents (eg, haloperidol) have been reported to be neurotoxic4 and associated with higher mortality in dementia patients with psychosis.5,6
The relationship of antipsychotic treatment with delirium and dementia is complex, paradoxical, and unsettled. Consider the following:
- Antipsychotics are not approved for the psychosis of dementia despite 17 large, placebo-controlled trials with 4 different second-generation agents.
- The FDA issued a “black-box” warning on all first- and second-generation antipsychotics because of a 1.6-times higher mortality rate among older patients with dementia.
- No placebo-controlled, FDA trials of delirium have been conducted with any antipsychotic agents.
- Clinicians frequently use first- and second-generation antipsychotics for delirium despite the lack of evidence.
- The FDA has not issued a “black-box” warning on antipsychotics for use in delirium as they did for dementia, although typically both populations are older and may be susceptible to the same complications observed in Alzheimer’s disease trials (eg, strokes, transient ischemic attack, and aspiration pneumonia). This may be because of the absence of safety data of antipsychotics in delirium based on industry-sponsored registration clinical trials. No data means no warning, although the risk factors may be present for millions of older patients who have suffered from delirium and have been treated with first-or second-generation antipsychotics.
The current convoluted state of pharmacotherapy for delirium and dementia is likely to continue: dementia patients with psychosis may or may not receive antipsychotics because of the FDA’s “black-box” warning while delirium patients readily receive various antipsychotics based on long-term practice, although not a single antipsychotic has been FDA-approved for delirium. Until a pharmaceutical company decides to conduct a large placebo-controlled trial for delirium, the current widespread, off-label use of antipsychotics in delirium will continue and “black-box” warning will apply only to 1 clinical population of older persons (those with dementia) but not another (those with delirium). Go figure!
1. Martins S, Fernandes L. Delirium in elderly people: a review. Front Neurol. 2012;3:101.-
2. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
3. Seitz DP, Gill SS, Herrmann N, et al. Pharmacological treatments for neuropsychiatric symptoms of dementia in long-term care: a systematic review. [published online October 19, 2012]. Int Psychogeriatr. 2012:1-19. doi: http://dx.doi.org/10.1017/S1041610212001627.
4. Nasrallah HA. Does the neurotoxicity of haloperidol explain the higher mortality in dementia patients compared with the second generation agents? Am J Psychiatry. 2012;169(6):663-664;author reply 664–665.
5. Nasrallah HA, White T, Nasrallah AT. Lower mortality in geriatric patients receiving risperidone and olanzapine versus haloperidol: preliminary analysis of retrospective data. Am J Geriatr Psychiatry. 2004;12(4):437-439.
6. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.
1. Martins S, Fernandes L. Delirium in elderly people: a review. Front Neurol. 2012;3:101.-
2. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
3. Seitz DP, Gill SS, Herrmann N, et al. Pharmacological treatments for neuropsychiatric symptoms of dementia in long-term care: a systematic review. [published online October 19, 2012]. Int Psychogeriatr. 2012:1-19. doi: http://dx.doi.org/10.1017/S1041610212001627.
4. Nasrallah HA. Does the neurotoxicity of haloperidol explain the higher mortality in dementia patients compared with the second generation agents? Am J Psychiatry. 2012;169(6):663-664;author reply 664–665.
5. Nasrallah HA, White T, Nasrallah AT. Lower mortality in geriatric patients receiving risperidone and olanzapine versus haloperidol: preliminary analysis of retrospective data. Am J Geriatr Psychiatry. 2004;12(4):437-439.
6. Kales HC, Kim HM, Zivin K, et al. Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry. 2012;169(1):71-79.
Evolution of antipsychotic pharmacotherapy
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Stabilizing acute psychosis
Paranoia and slowed cognition
CASE: Behavioral changes
Mr. K, age 45, is brought to the emergency department (ED) by his wife for severe paranoia, combative behavior, confusion, and slowed cognition. Mr. K tells the ED staff that a chemical abrasion he sustained a few weeks earlier has spread to his penis, and insists that his penis is retracting into his body. He has tied a string around his penis to keep it from disappearing into his body. According to Mr. K’s wife, he went to an urgent care clinic 2 weeks ago after he sustained chemical abrasions from exposure to cleaning solution at home. The provider at the urgent care clinic started Mr. K on an unknown dose of oral prednisone.
Mr. K’s wife reports that her husband had a dysphoric episode approximately 6 months ago when his business was struggling but his mood improved without psychiatric care. Mr. K’s medical history includes episodic sarcoidosis of the eyes, skin, and lungs. In the past these symptoms remitted after he received oral prednisone.
ED clinicians consider neurosarcoidosis and substance-induced delirium in the differential diagnosis (Table).1 A CT scan of the head fails to show lesions suggestive of neurosarcoidosis. Chest radiography does not reveal lesions suggestive of lung sarcoids and Mr. K has no skin lesions.
Table
DSM-IV-TR criteria for substance-induced delirium
|
| Source: Reference 1 |
Mr. K is admitted to the psychiatric inpatient unit for acute stabilization, where he remains aggressive and combative. He throws chairs at his peers and staff on the unit and is placed in physical restraints. He requires several doses of IM haloperidol, 5 mg, lorazepam, 2 mg, and diphenhydramine, 50 mg, for severe agitation. Mr. K is guarded, perseverative, and selectively mute. He avoids eye contact and has poor grooming. He has slow thought processing and displays concrete thought process. Prednisone is discontinued and olanzapine, titrated to 30 mg/d, and mirtazapine, titrated to 30 mg/d, are started for psychosis and depression.
Mr. K’s mood and behavior eventually return to baseline but slowed cognition persists. He is discharged from our facility.
The authors’ observations
Cortisone was first used to treat rheumatoid arthritis in 1948 and corticosteroids have been linked to multiple neuropsychiatric complications that have been broadly defined as steroid psychosis. This syndrome includes reversible behavioral manifestations such as hypomania, irritability, mood reactivity, anxiety, and insomnia in addition to more severe symptoms such as depression, mania, and psychosis.2 Although mild cognitive deficits have been noted in patients taking corticosteroids, most published cases have focused on steroid-induced psychosis.
In 1984, Varney et al3 noted a phenomenon they called “steroid dementia” in 6 patients treated with corticosteroids. On first evaluation, these patients presented with symptoms similar to early Alzheimer’s dementia—impaired memory, attention, and concentration. Three patients initially were diagnosed first with Alzheimer’s dementia until their symptoms spontaneously improved when steroids were reduced or discontinued. Although their presentation resembled Alzheimer’s dementia, patients with steroid dementia had a specific cognitive presentation associated with corticosteroid use. Symptoms included impaired verbal memory and spatial thinking but normal procedural memory. These patients showed intact immediate recall but impaired delayed recall with difficulty tracking conversations and word finding. Overall, patients with steroid dementia showed a predominance of verbal declarative memory deficits out of proportion to other cognitive symptoms. These symptoms and recent corticosteroid exposure differentiated steroid dementia from other forms of dementia.
In a later article, Varney reviewed electroencephalography (EEG) and CT findings associated with steroid dementia, noting bilateral EEG abnormalities and acute cortical atrophy on CT.4 Steroid dementia largely was reversible, resolving 3 to 11 months after corticosteroid discontinuation. Additionally, Varney noted that patients who had psychosis and dementia had more severe and longer-lasting dementia.
TREATMENT: Progressive decline
Mr. K is college educated, has been married for 15 years, has 2 children, age 9 and 11, and owns a successful basketball coaching business. He has no history of substance abuse, legal issues, or violence. He reports a good childhood with normal developmental milestones and no history of trauma.
In the 6 months after his initial psychiatric admission, Mr. K sees various outpatient providers, who change his psychotropics multiple times. He also receives 4 courses of prednisone for ocular sarcoidosis. He is admitted twice to other psychiatric facilities. After he has paranoid interactions with colleagues and families of the youth he coaches, his business fails.
After his third psychiatric inpatient hospitalization, Mr. K becomes severely paranoid, believing his wife is having an affair. He becomes physically abusive to his wife, who obtains a restraining order and leaves with their children. Mr. K barely leaves his house and stops grooming. A friend notes that Mr. K’s home has become uninhabitable, and it goes into foreclosure. After Mr. K’s neighbors report combative behavior and paranoia, police bring him in on an involuntary hold for a fourth psychiatric hospitalization (the second in our facility).
During this hospitalization—6 months after the initial ED presentation—the neurology team conducts a repeat medical workup. EEG shows generalized slowing. Head CT and MRI show diffuse cortical atrophy that was not seen in previous imaging. Mr. K has ocular lesions characteristic of ocular sarcoidosis. His mental status examination is similar to his first presentation except that the psychosis and thought disorganization are considerably worse. His cognitive functioning also shows significant decline. Cognitive screening reveals intact remote memory with impaired recent memory. His thinking is concrete and his verbal memory is markedly impaired. His Mini-Mental State Examination score is 27/30, indicating functional capacity that is better than his clinical presentation. Because of difficulty with concentration and verbal processing, Mr. K is unable to complete the Minnesota Multiphasic Personality Inventory despite substantial assistance. On most days he cannot recall recent conversations with his wife, staff, or physicians. He is taking no medications at this time.
Mr. K is restarted on olanzapine, titrated to 30 mg/d, to control his psychosis; this medication was effective during his last stay in our facility. Oral prednisone is discontinued and methotrexate, 10 mg/week, is initiated for ocular sarcoidosis. Based on recommendations from a case series report,5 we start Mr. K on lithium, titrated to 600 mg twice a day, for steroid-induced mood symptoms, Mr. K’s psychosis and mood improve dramatically once he reaches a therapeutic lithium level; however, his cognition remains slowed and he is unable to care for his basic needs.
The authors’ observations
Steroid dementia may be the result of effects in the medial temporal lobe, specifically dorsolateral prefrontal cortex, which impairs working memory, and the parahippocampal gyrus.6,7 The cognitive presentation of steroid dementia Varney et al3 described has been replicated in healthy volunteers who received corticosteroids.3 Patients with Cushing’s syndrome also have been noted to have diminished hippocampal volume and similar cognitive deficits. Cognitive impairment experienced by patients treated with corticosteroids may be caused by neuronal death in the hippocampus and dorsolateral prefrontal cortex. The etiology of cell death is multifactorial and includes glutamate-mediated excitotoxicity, activation of proinflammatory pathways, inhibited utilization of glucose in the hippocampus, telomere shortening, and diminished cell repair by brain-derived neurotrophic factor. The net result is significant, widespread damage that in some cases is irreversible.8
Because of the severity of Mr. K’s psychosis and personality change from baseline, his cognitive symptoms were largely overlooked during his first psychiatric hospitalization. The affective flattening, delayed verbal response, and markedly concrete thought process were considered within the spectrum of resolving psychosis. After further hospitalizations and abnormal results on cognitive testing, Mr. K’s cognitive impairment was fully noted. His symptoms match those of previously documented cases of steroid dementia, including verbal deficits out of proportion to other impairment, acute cerebral atrophy on CT after corticosteroid treatment, and gradual improvement of symptoms when corticosteroids were discontinued.
Management recommendations
Educate patients taking steroids about possible side effects of mood changes, psychosis, and cognitive deficits. Close monitoring of patients on corticosteroids is paramount. If psychiatric or cognitive symptoms develop, gradually discontinue the corticosteroid and seek other treatments.
Randomized, placebo-controlled trials of lamotrigine and memantine have shown these medications are cognitively protective for patients taking prednisone.9
OUTCOME: Long-term deficits
After a 33-day stay in our adult inpatient psychiatric facility, the county places Mr. K in a permanent conservatorship for severe grave disability. He is discharged to a long-term psychiatric care locked facility for ongoing management. Mr. K spends 20 months in the long-term care facility while his family remains hopeful for his recovery and return home. He is admitted to our facility for acute stabilization of psychotic symptoms after he is released from the locked facility. Although no imaging studies are conducted, he remains significantly forgetful. Additionally, his paranoia persists.
Mr. K is poorly compliant with his psychotropics, which include divalproex, 1,000 mg/d, and olanzapine, 30 mg/d. Although he is discharged home with his family, his functional capacity is less than expected and he requires continuous support. Insisting that Mr. K abstain from steroids after the first psychiatric hospitalization might have prevented this seemingly irreversible dementia.
Related Resources
- Sacks O, Shulman M. Steroid dementia: an overlooked diagnosis? Neurology. 2005;64(4):707-709.
- Cipriani G, Picchi L, Vedovello M, et al. Reversible dementia from corticosteroid therapy. Clinical Geriatrics. 2012;20(7):38-41.
Drug Brand Names
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Memantine • Namenda
- Methotrexate • Rheumatrex, Trexall
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Prednisone • Deltasone, Meticorten, others
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Arlington VA: American Psychiatric Association; 2000.
2. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
3. Varney NR, Alexander B, MacIndoe JH. Reversible steroid dementia in patients without steroid psychosis. Am J Psychiatry. 1984;141(3):369-372.
4. Varney NR. A case of reversible steroid dementia. Arch Clin Neuropsychol. 1997;12(2):167-171.
5. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33.
6. Wolkowitz OM, Burke H, Epel ES, et al. Glucocorticoids: mood, memory, and mechanisms. Ann N Y Acad Sci. 2009;1179:19-40.
7. Lupien SJ, McEwen BS. The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Brain Res Rev. 1997;24(1):1-27.
8. Sapolsky RM. The physiological relevance of glucocorticoid endangerment of the hippocampus. Ann NY Acad Sci. 1994;746:294-304.
9. Brown ES. Effects of glucocorticoids on mood memory and the hippocampus. Treatment and preventative therapy. Ann N Y Acad Sci. 2009;1179:41-55.
CASE: Behavioral changes
Mr. K, age 45, is brought to the emergency department (ED) by his wife for severe paranoia, combative behavior, confusion, and slowed cognition. Mr. K tells the ED staff that a chemical abrasion he sustained a few weeks earlier has spread to his penis, and insists that his penis is retracting into his body. He has tied a string around his penis to keep it from disappearing into his body. According to Mr. K’s wife, he went to an urgent care clinic 2 weeks ago after he sustained chemical abrasions from exposure to cleaning solution at home. The provider at the urgent care clinic started Mr. K on an unknown dose of oral prednisone.
Mr. K’s wife reports that her husband had a dysphoric episode approximately 6 months ago when his business was struggling but his mood improved without psychiatric care. Mr. K’s medical history includes episodic sarcoidosis of the eyes, skin, and lungs. In the past these symptoms remitted after he received oral prednisone.
ED clinicians consider neurosarcoidosis and substance-induced delirium in the differential diagnosis (Table).1 A CT scan of the head fails to show lesions suggestive of neurosarcoidosis. Chest radiography does not reveal lesions suggestive of lung sarcoids and Mr. K has no skin lesions.
Table
DSM-IV-TR criteria for substance-induced delirium
|
| Source: Reference 1 |
Mr. K is admitted to the psychiatric inpatient unit for acute stabilization, where he remains aggressive and combative. He throws chairs at his peers and staff on the unit and is placed in physical restraints. He requires several doses of IM haloperidol, 5 mg, lorazepam, 2 mg, and diphenhydramine, 50 mg, for severe agitation. Mr. K is guarded, perseverative, and selectively mute. He avoids eye contact and has poor grooming. He has slow thought processing and displays concrete thought process. Prednisone is discontinued and olanzapine, titrated to 30 mg/d, and mirtazapine, titrated to 30 mg/d, are started for psychosis and depression.
Mr. K’s mood and behavior eventually return to baseline but slowed cognition persists. He is discharged from our facility.
The authors’ observations
Cortisone was first used to treat rheumatoid arthritis in 1948 and corticosteroids have been linked to multiple neuropsychiatric complications that have been broadly defined as steroid psychosis. This syndrome includes reversible behavioral manifestations such as hypomania, irritability, mood reactivity, anxiety, and insomnia in addition to more severe symptoms such as depression, mania, and psychosis.2 Although mild cognitive deficits have been noted in patients taking corticosteroids, most published cases have focused on steroid-induced psychosis.
In 1984, Varney et al3 noted a phenomenon they called “steroid dementia” in 6 patients treated with corticosteroids. On first evaluation, these patients presented with symptoms similar to early Alzheimer’s dementia—impaired memory, attention, and concentration. Three patients initially were diagnosed first with Alzheimer’s dementia until their symptoms spontaneously improved when steroids were reduced or discontinued. Although their presentation resembled Alzheimer’s dementia, patients with steroid dementia had a specific cognitive presentation associated with corticosteroid use. Symptoms included impaired verbal memory and spatial thinking but normal procedural memory. These patients showed intact immediate recall but impaired delayed recall with difficulty tracking conversations and word finding. Overall, patients with steroid dementia showed a predominance of verbal declarative memory deficits out of proportion to other cognitive symptoms. These symptoms and recent corticosteroid exposure differentiated steroid dementia from other forms of dementia.
In a later article, Varney reviewed electroencephalography (EEG) and CT findings associated with steroid dementia, noting bilateral EEG abnormalities and acute cortical atrophy on CT.4 Steroid dementia largely was reversible, resolving 3 to 11 months after corticosteroid discontinuation. Additionally, Varney noted that patients who had psychosis and dementia had more severe and longer-lasting dementia.
TREATMENT: Progressive decline
Mr. K is college educated, has been married for 15 years, has 2 children, age 9 and 11, and owns a successful basketball coaching business. He has no history of substance abuse, legal issues, or violence. He reports a good childhood with normal developmental milestones and no history of trauma.
In the 6 months after his initial psychiatric admission, Mr. K sees various outpatient providers, who change his psychotropics multiple times. He also receives 4 courses of prednisone for ocular sarcoidosis. He is admitted twice to other psychiatric facilities. After he has paranoid interactions with colleagues and families of the youth he coaches, his business fails.
After his third psychiatric inpatient hospitalization, Mr. K becomes severely paranoid, believing his wife is having an affair. He becomes physically abusive to his wife, who obtains a restraining order and leaves with their children. Mr. K barely leaves his house and stops grooming. A friend notes that Mr. K’s home has become uninhabitable, and it goes into foreclosure. After Mr. K’s neighbors report combative behavior and paranoia, police bring him in on an involuntary hold for a fourth psychiatric hospitalization (the second in our facility).
During this hospitalization—6 months after the initial ED presentation—the neurology team conducts a repeat medical workup. EEG shows generalized slowing. Head CT and MRI show diffuse cortical atrophy that was not seen in previous imaging. Mr. K has ocular lesions characteristic of ocular sarcoidosis. His mental status examination is similar to his first presentation except that the psychosis and thought disorganization are considerably worse. His cognitive functioning also shows significant decline. Cognitive screening reveals intact remote memory with impaired recent memory. His thinking is concrete and his verbal memory is markedly impaired. His Mini-Mental State Examination score is 27/30, indicating functional capacity that is better than his clinical presentation. Because of difficulty with concentration and verbal processing, Mr. K is unable to complete the Minnesota Multiphasic Personality Inventory despite substantial assistance. On most days he cannot recall recent conversations with his wife, staff, or physicians. He is taking no medications at this time.
Mr. K is restarted on olanzapine, titrated to 30 mg/d, to control his psychosis; this medication was effective during his last stay in our facility. Oral prednisone is discontinued and methotrexate, 10 mg/week, is initiated for ocular sarcoidosis. Based on recommendations from a case series report,5 we start Mr. K on lithium, titrated to 600 mg twice a day, for steroid-induced mood symptoms, Mr. K’s psychosis and mood improve dramatically once he reaches a therapeutic lithium level; however, his cognition remains slowed and he is unable to care for his basic needs.
The authors’ observations
Steroid dementia may be the result of effects in the medial temporal lobe, specifically dorsolateral prefrontal cortex, which impairs working memory, and the parahippocampal gyrus.6,7 The cognitive presentation of steroid dementia Varney et al3 described has been replicated in healthy volunteers who received corticosteroids.3 Patients with Cushing’s syndrome also have been noted to have diminished hippocampal volume and similar cognitive deficits. Cognitive impairment experienced by patients treated with corticosteroids may be caused by neuronal death in the hippocampus and dorsolateral prefrontal cortex. The etiology of cell death is multifactorial and includes glutamate-mediated excitotoxicity, activation of proinflammatory pathways, inhibited utilization of glucose in the hippocampus, telomere shortening, and diminished cell repair by brain-derived neurotrophic factor. The net result is significant, widespread damage that in some cases is irreversible.8
Because of the severity of Mr. K’s psychosis and personality change from baseline, his cognitive symptoms were largely overlooked during his first psychiatric hospitalization. The affective flattening, delayed verbal response, and markedly concrete thought process were considered within the spectrum of resolving psychosis. After further hospitalizations and abnormal results on cognitive testing, Mr. K’s cognitive impairment was fully noted. His symptoms match those of previously documented cases of steroid dementia, including verbal deficits out of proportion to other impairment, acute cerebral atrophy on CT after corticosteroid treatment, and gradual improvement of symptoms when corticosteroids were discontinued.
Management recommendations
Educate patients taking steroids about possible side effects of mood changes, psychosis, and cognitive deficits. Close monitoring of patients on corticosteroids is paramount. If psychiatric or cognitive symptoms develop, gradually discontinue the corticosteroid and seek other treatments.
Randomized, placebo-controlled trials of lamotrigine and memantine have shown these medications are cognitively protective for patients taking prednisone.9
OUTCOME: Long-term deficits
After a 33-day stay in our adult inpatient psychiatric facility, the county places Mr. K in a permanent conservatorship for severe grave disability. He is discharged to a long-term psychiatric care locked facility for ongoing management. Mr. K spends 20 months in the long-term care facility while his family remains hopeful for his recovery and return home. He is admitted to our facility for acute stabilization of psychotic symptoms after he is released from the locked facility. Although no imaging studies are conducted, he remains significantly forgetful. Additionally, his paranoia persists.
Mr. K is poorly compliant with his psychotropics, which include divalproex, 1,000 mg/d, and olanzapine, 30 mg/d. Although he is discharged home with his family, his functional capacity is less than expected and he requires continuous support. Insisting that Mr. K abstain from steroids after the first psychiatric hospitalization might have prevented this seemingly irreversible dementia.
Related Resources
- Sacks O, Shulman M. Steroid dementia: an overlooked diagnosis? Neurology. 2005;64(4):707-709.
- Cipriani G, Picchi L, Vedovello M, et al. Reversible dementia from corticosteroid therapy. Clinical Geriatrics. 2012;20(7):38-41.
Drug Brand Names
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Memantine • Namenda
- Methotrexate • Rheumatrex, Trexall
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Prednisone • Deltasone, Meticorten, others
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Behavioral changes
Mr. K, age 45, is brought to the emergency department (ED) by his wife for severe paranoia, combative behavior, confusion, and slowed cognition. Mr. K tells the ED staff that a chemical abrasion he sustained a few weeks earlier has spread to his penis, and insists that his penis is retracting into his body. He has tied a string around his penis to keep it from disappearing into his body. According to Mr. K’s wife, he went to an urgent care clinic 2 weeks ago after he sustained chemical abrasions from exposure to cleaning solution at home. The provider at the urgent care clinic started Mr. K on an unknown dose of oral prednisone.
Mr. K’s wife reports that her husband had a dysphoric episode approximately 6 months ago when his business was struggling but his mood improved without psychiatric care. Mr. K’s medical history includes episodic sarcoidosis of the eyes, skin, and lungs. In the past these symptoms remitted after he received oral prednisone.
ED clinicians consider neurosarcoidosis and substance-induced delirium in the differential diagnosis (Table).1 A CT scan of the head fails to show lesions suggestive of neurosarcoidosis. Chest radiography does not reveal lesions suggestive of lung sarcoids and Mr. K has no skin lesions.
Table
DSM-IV-TR criteria for substance-induced delirium
|
| Source: Reference 1 |
Mr. K is admitted to the psychiatric inpatient unit for acute stabilization, where he remains aggressive and combative. He throws chairs at his peers and staff on the unit and is placed in physical restraints. He requires several doses of IM haloperidol, 5 mg, lorazepam, 2 mg, and diphenhydramine, 50 mg, for severe agitation. Mr. K is guarded, perseverative, and selectively mute. He avoids eye contact and has poor grooming. He has slow thought processing and displays concrete thought process. Prednisone is discontinued and olanzapine, titrated to 30 mg/d, and mirtazapine, titrated to 30 mg/d, are started for psychosis and depression.
Mr. K’s mood and behavior eventually return to baseline but slowed cognition persists. He is discharged from our facility.
The authors’ observations
Cortisone was first used to treat rheumatoid arthritis in 1948 and corticosteroids have been linked to multiple neuropsychiatric complications that have been broadly defined as steroid psychosis. This syndrome includes reversible behavioral manifestations such as hypomania, irritability, mood reactivity, anxiety, and insomnia in addition to more severe symptoms such as depression, mania, and psychosis.2 Although mild cognitive deficits have been noted in patients taking corticosteroids, most published cases have focused on steroid-induced psychosis.
In 1984, Varney et al3 noted a phenomenon they called “steroid dementia” in 6 patients treated with corticosteroids. On first evaluation, these patients presented with symptoms similar to early Alzheimer’s dementia—impaired memory, attention, and concentration. Three patients initially were diagnosed first with Alzheimer’s dementia until their symptoms spontaneously improved when steroids were reduced or discontinued. Although their presentation resembled Alzheimer’s dementia, patients with steroid dementia had a specific cognitive presentation associated with corticosteroid use. Symptoms included impaired verbal memory and spatial thinking but normal procedural memory. These patients showed intact immediate recall but impaired delayed recall with difficulty tracking conversations and word finding. Overall, patients with steroid dementia showed a predominance of verbal declarative memory deficits out of proportion to other cognitive symptoms. These symptoms and recent corticosteroid exposure differentiated steroid dementia from other forms of dementia.
In a later article, Varney reviewed electroencephalography (EEG) and CT findings associated with steroid dementia, noting bilateral EEG abnormalities and acute cortical atrophy on CT.4 Steroid dementia largely was reversible, resolving 3 to 11 months after corticosteroid discontinuation. Additionally, Varney noted that patients who had psychosis and dementia had more severe and longer-lasting dementia.
TREATMENT: Progressive decline
Mr. K is college educated, has been married for 15 years, has 2 children, age 9 and 11, and owns a successful basketball coaching business. He has no history of substance abuse, legal issues, or violence. He reports a good childhood with normal developmental milestones and no history of trauma.
In the 6 months after his initial psychiatric admission, Mr. K sees various outpatient providers, who change his psychotropics multiple times. He also receives 4 courses of prednisone for ocular sarcoidosis. He is admitted twice to other psychiatric facilities. After he has paranoid interactions with colleagues and families of the youth he coaches, his business fails.
After his third psychiatric inpatient hospitalization, Mr. K becomes severely paranoid, believing his wife is having an affair. He becomes physically abusive to his wife, who obtains a restraining order and leaves with their children. Mr. K barely leaves his house and stops grooming. A friend notes that Mr. K’s home has become uninhabitable, and it goes into foreclosure. After Mr. K’s neighbors report combative behavior and paranoia, police bring him in on an involuntary hold for a fourth psychiatric hospitalization (the second in our facility).
During this hospitalization—6 months after the initial ED presentation—the neurology team conducts a repeat medical workup. EEG shows generalized slowing. Head CT and MRI show diffuse cortical atrophy that was not seen in previous imaging. Mr. K has ocular lesions characteristic of ocular sarcoidosis. His mental status examination is similar to his first presentation except that the psychosis and thought disorganization are considerably worse. His cognitive functioning also shows significant decline. Cognitive screening reveals intact remote memory with impaired recent memory. His thinking is concrete and his verbal memory is markedly impaired. His Mini-Mental State Examination score is 27/30, indicating functional capacity that is better than his clinical presentation. Because of difficulty with concentration and verbal processing, Mr. K is unable to complete the Minnesota Multiphasic Personality Inventory despite substantial assistance. On most days he cannot recall recent conversations with his wife, staff, or physicians. He is taking no medications at this time.
Mr. K is restarted on olanzapine, titrated to 30 mg/d, to control his psychosis; this medication was effective during his last stay in our facility. Oral prednisone is discontinued and methotrexate, 10 mg/week, is initiated for ocular sarcoidosis. Based on recommendations from a case series report,5 we start Mr. K on lithium, titrated to 600 mg twice a day, for steroid-induced mood symptoms, Mr. K’s psychosis and mood improve dramatically once he reaches a therapeutic lithium level; however, his cognition remains slowed and he is unable to care for his basic needs.
The authors’ observations
Steroid dementia may be the result of effects in the medial temporal lobe, specifically dorsolateral prefrontal cortex, which impairs working memory, and the parahippocampal gyrus.6,7 The cognitive presentation of steroid dementia Varney et al3 described has been replicated in healthy volunteers who received corticosteroids.3 Patients with Cushing’s syndrome also have been noted to have diminished hippocampal volume and similar cognitive deficits. Cognitive impairment experienced by patients treated with corticosteroids may be caused by neuronal death in the hippocampus and dorsolateral prefrontal cortex. The etiology of cell death is multifactorial and includes glutamate-mediated excitotoxicity, activation of proinflammatory pathways, inhibited utilization of glucose in the hippocampus, telomere shortening, and diminished cell repair by brain-derived neurotrophic factor. The net result is significant, widespread damage that in some cases is irreversible.8
Because of the severity of Mr. K’s psychosis and personality change from baseline, his cognitive symptoms were largely overlooked during his first psychiatric hospitalization. The affective flattening, delayed verbal response, and markedly concrete thought process were considered within the spectrum of resolving psychosis. After further hospitalizations and abnormal results on cognitive testing, Mr. K’s cognitive impairment was fully noted. His symptoms match those of previously documented cases of steroid dementia, including verbal deficits out of proportion to other impairment, acute cerebral atrophy on CT after corticosteroid treatment, and gradual improvement of symptoms when corticosteroids were discontinued.
Management recommendations
Educate patients taking steroids about possible side effects of mood changes, psychosis, and cognitive deficits. Close monitoring of patients on corticosteroids is paramount. If psychiatric or cognitive symptoms develop, gradually discontinue the corticosteroid and seek other treatments.
Randomized, placebo-controlled trials of lamotrigine and memantine have shown these medications are cognitively protective for patients taking prednisone.9
OUTCOME: Long-term deficits
After a 33-day stay in our adult inpatient psychiatric facility, the county places Mr. K in a permanent conservatorship for severe grave disability. He is discharged to a long-term psychiatric care locked facility for ongoing management. Mr. K spends 20 months in the long-term care facility while his family remains hopeful for his recovery and return home. He is admitted to our facility for acute stabilization of psychotic symptoms after he is released from the locked facility. Although no imaging studies are conducted, he remains significantly forgetful. Additionally, his paranoia persists.
Mr. K is poorly compliant with his psychotropics, which include divalproex, 1,000 mg/d, and olanzapine, 30 mg/d. Although he is discharged home with his family, his functional capacity is less than expected and he requires continuous support. Insisting that Mr. K abstain from steroids after the first psychiatric hospitalization might have prevented this seemingly irreversible dementia.
Related Resources
- Sacks O, Shulman M. Steroid dementia: an overlooked diagnosis? Neurology. 2005;64(4):707-709.
- Cipriani G, Picchi L, Vedovello M, et al. Reversible dementia from corticosteroid therapy. Clinical Geriatrics. 2012;20(7):38-41.
Drug Brand Names
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Lorazepam • Ativan
- Memantine • Namenda
- Methotrexate • Rheumatrex, Trexall
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Prednisone • Deltasone, Meticorten, others
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Arlington VA: American Psychiatric Association; 2000.
2. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
3. Varney NR, Alexander B, MacIndoe JH. Reversible steroid dementia in patients without steroid psychosis. Am J Psychiatry. 1984;141(3):369-372.
4. Varney NR. A case of reversible steroid dementia. Arch Clin Neuropsychol. 1997;12(2):167-171.
5. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33.
6. Wolkowitz OM, Burke H, Epel ES, et al. Glucocorticoids: mood, memory, and mechanisms. Ann N Y Acad Sci. 2009;1179:19-40.
7. Lupien SJ, McEwen BS. The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Brain Res Rev. 1997;24(1):1-27.
8. Sapolsky RM. The physiological relevance of glucocorticoid endangerment of the hippocampus. Ann NY Acad Sci. 1994;746:294-304.
9. Brown ES. Effects of glucocorticoids on mood memory and the hippocampus. Treatment and preventative therapy. Ann N Y Acad Sci. 2009;1179:41-55.
1. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Arlington VA: American Psychiatric Association; 2000.
2. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
3. Varney NR, Alexander B, MacIndoe JH. Reversible steroid dementia in patients without steroid psychosis. Am J Psychiatry. 1984;141(3):369-372.
4. Varney NR. A case of reversible steroid dementia. Arch Clin Neuropsychol. 1997;12(2):167-171.
5. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25(1):27-33.
6. Wolkowitz OM, Burke H, Epel ES, et al. Glucocorticoids: mood, memory, and mechanisms. Ann N Y Acad Sci. 2009;1179:19-40.
7. Lupien SJ, McEwen BS. The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Brain Res Rev. 1997;24(1):1-27.
8. Sapolsky RM. The physiological relevance of glucocorticoid endangerment of the hippocampus. Ann NY Acad Sci. 1994;746:294-304.
9. Brown ES. Effects of glucocorticoids on mood memory and the hippocampus. Treatment and preventative therapy. Ann N Y Acad Sci. 2009;1179:41-55.