User login
APOLLO: SLN360 clears first major hurdle, hammering Lp(a)
The short interfering RNA (siRNA) agent SLN360 was well tolerated and lowered lipoprotein(a) by up to 98% in volunteers without cardiovascular disease but with elevated Lp(a) in the small dose-ranging APOLLO trial.
Following a single subcutaneous dose of SLN360 (Silence Therapeutics), there was a dose-dependent reduction in Lp(a) plasma levels by a median of 46%, 86%, 96%, and 98% at about 45-60 days with 30-mg, 100-mg, 300-mg, and 600-mg doses, respectively.

Lp(a) levels at 150 days were 70% and 81% below baseline with the 300-and 600-mg doses.
In addition, for participants receiving the two highest doses, apolipoprotein B (apo B) was reduced was 21% and 24%, respectively, and LDL cholesterol (LDL-C), by 21% and 26%, respectively.
“The development of therapies targeting messenger RNA has made possible significant lowering of lipoprotein(a). Whether these reductions can impact on the incidence of ASCVD [atherosclerotic cardiovascular disease] or prevent progression of aortic stenosis remains to be determined but, we think, that optimism is warranted,” said principal investigator Steven E. Nissen, MD, Cleveland Clinic.
The results were presented in a late-breaking clinical trial session at the annual scientific sessions of the American College of Cardiology and published simultaneously in JAMA.
Elevated Lp(a) is a powerful genetic risk factor for ASCVD and aortic stenosis, which affects some 64 million Americans and 1.4 billion people globally. Although several experimental agents are under investigation, no currently approved drugs selectively lower Lp(a).
SLN360 is designed to lower Lp(a) production by using RNA interference to silence messenger RNA transcribed from the LPA gene in liver cells.
Testing vacuum
Dr. Nissen said in an interview that one of the big takeaways from the study is the need for greater testing of Lp(a). Automatic assays are available in almost every hospital, but two-unit systems (nmol/L and mg/dL) are used and thresholds for accelerated risk vary. The Cleveland Clinic currently tests all patients in its cardiac critical care unit and its prevention clinic.
“Someone comes in with an MI in their 40s and we measure it and it’s 100, 150 [mg/dL], clearly abnormal, and often these patients don’t have a lot of other risk factors,” Dr. Nissen said. “So the explanation very likely for their premature disease is this risk factor. We now have to educate everybody about the importance of getting it tested and finding out about it.”
During a media briefing, ACC 2022 program cochair Pamela B. Morris, MD, Medical University of South Carolina, Charleston, said testing for Lp(a) is not well reimbursed by insurance providers and that her patients will often cancel the test after learning it won’t be reimbursed because they don’t understand it.
“What Dr. Nissen is telling you: It should be measured in everyone at least once, we all believe that, but it hasn’t made it into the major guidelines,” she added. “I think what we’re going to have to do is have the guidelines mandate it and the insurers will follow.”
Guidelines currently list elevated Lp(a) as a “risk-enhancing factor,” which can help with at least recommending LDL-C treatment in patients with borderline risk and a sky-high Lp(a), noted Dr. Nissen. “But we need to go beyond that.”
Safety analyses
The first-in-human APOLLO trial evaluated 32 adults without known ASCVD and an Lp(a) concentration greater than 150 nmol/L (approximately 60 mg/dL) who received one of the four doses of SLN360 or placebo subcutaneously. Participants were monitored in a research unit for the first 24 hours and then followed periodically for up to 150 days. At baseline, their median Lp(a) level was 224 nmol/L, mean apo B level was 85 mg/dL, and mean LDL-C level was 108 mg/dL.
Treatment-emergent adverse events were generally mild, mostly grade 1 injection site reactions (83% at 30 mg, 100% at 100 mg, 67% at 300 mg, and 33% at 600 mg) and headache (33%, 17%, 0%, and 83%).
At the highest dose, C-reactive protein was increased in four patients and neutrophil counts in three. ALT and AST levels were elevated three times above the upper limit of normal in one patient at the lowest dose.
One participant in the lowest-dose group experienced two serious adverse events unrelated to SLN360 at day 45 after receiving a SARS-Co-V-2 vaccine.
Dr. Nissen noted that safety cannot be comprehensively assessed in a trial of this duration or size and that follow-up has been extended to 1 year in the two highest-dose groups.
Enrollment continues in the multiple-ascending dose portion of the study in patients with high Lp(a) and a history of stable ASCVD. A phase 2 study of SLN360 is also planned for the second half of 2022, pending regulatory discussions.
But will it reduce ASCVD events?
Study discussant Vera Bittner, MD, MSPH, University of Alabama at Birmingham, said that the development of Lp(a)-specific lowering agents has been a “holy grail” for years and congratulated the authors on a successful trial demonstrating very robust Lp(a) lowering.

She asked Dr. Nissen about the observation in proprotein convertase subtilisin/kexin type 9 inhibitor trials that absolute Lp(a) lowering is greater at higher baseline levels.
Dr. Nissen said this kind of analysis wasn’t possible because of the small sample size but “because these agents so effectively degrade messenger RNA, it’s very likely we will see robust suppression of plasma levels virtually regardless of the baseline level.”
Dr. Bittner also questioned if “LDL-C declined because of the cholesterol content in the lipoprotein(a) or is there some additional effect on LDL particles themselves?”
“It’s a really terrific question that will ultimately need to be answered,” Dr. Nissen replied. “There’s some controversy about the extent to which suppressing lipoprotein(a) will reduce LDL because the assays for LDL are measuring the LDL that’s in lipoprotein(a) and the LDL that is not. ... I think it’s probably a bystander effect, but it may also contribute to efficacy from a morbidity and mortality point of view, which is why we measured it.”
Dr. Bittner also called out the elevation in C-reactive protein and leukocytosis, which has not been seen in other siRNA studies. Dr. Nissen said the increases in C-reactive protein occurred in the first few days after administration and were gone after a week or so. “I don’t see it as a long-term limitation.”

In an accompanying editorial, Brian Ference, MD, MPhil, MSc, University of Cambridge (England), suggests that because circulating Lp(a) particles can progressively become trapped within the artery wall over time, it’s unlikely that lowering Lp(a) for only a few years starting later in life will eliminate the effect of lifelong exposure to Lp(a) and may only cut cardiovascular event risk by about 10%-15%.
He called for continued safety and efficacy evaluation of SLN360 and olpasiran, a similar siRNA agent in early development, and said further insights into whether large absolute reductions in Lp(a) can reduce the risk for major cardiovascular events will come from cardiovascular trials, such as the ongoing phase 3 Lp(a)HORIZON trial. It follows strong phase 2 results with the antisense agent AKCEA-APO(a)-LRx and has Dr. Nissen pulling double duty as study chair.
The study was funded by Silence Therapeutics. Dr. Nissen reported consulting for many pharmaceutical companies, which are directed to pay any renumeration directly to charity. Dr. Bittner reported consultant fees or honoraria from Pfizer; other from AstraZeneca, DalCor, Esperion, and Sanofi-Aventis; and research/research grants from Amgen and Novartis. Dr. Ference reported financial ties to Merck, Novartis, Amgen, Pfizer, Esperion Therapeutics, and numerous other companies.
A version of this article first appeared on Medscape.com.
The short interfering RNA (siRNA) agent SLN360 was well tolerated and lowered lipoprotein(a) by up to 98% in volunteers without cardiovascular disease but with elevated Lp(a) in the small dose-ranging APOLLO trial.
Following a single subcutaneous dose of SLN360 (Silence Therapeutics), there was a dose-dependent reduction in Lp(a) plasma levels by a median of 46%, 86%, 96%, and 98% at about 45-60 days with 30-mg, 100-mg, 300-mg, and 600-mg doses, respectively.
Lp(a) levels at 150 days were 70% and 81% below baseline with the 300-and 600-mg doses.
In addition, for participants receiving the two highest doses, apolipoprotein B (apo B) was reduced was 21% and 24%, respectively, and LDL cholesterol (LDL-C), by 21% and 26%, respectively.
“The development of therapies targeting messenger RNA has made possible significant lowering of lipoprotein(a). Whether these reductions can impact on the incidence of ASCVD [atherosclerotic cardiovascular disease] or prevent progression of aortic stenosis remains to be determined but, we think, that optimism is warranted,” said principal investigator Steven E. Nissen, MD, Cleveland Clinic.
The results were presented in a late-breaking clinical trial session at the annual scientific sessions of the American College of Cardiology and published simultaneously in JAMA.
Elevated Lp(a) is a powerful genetic risk factor for ASCVD and aortic stenosis, which affects some 64 million Americans and 1.4 billion people globally. Although several experimental agents are under investigation, no currently approved drugs selectively lower Lp(a).
SLN360 is designed to lower Lp(a) production by using RNA interference to silence messenger RNA transcribed from the LPA gene in liver cells.
Testing vacuum
Dr. Nissen said in an interview that one of the big takeaways from the study is the need for greater testing of Lp(a). Automatic assays are available in almost every hospital, but two-unit systems (nmol/L and mg/dL) are used and thresholds for accelerated risk vary. The Cleveland Clinic currently tests all patients in its cardiac critical care unit and its prevention clinic.
“Someone comes in with an MI in their 40s and we measure it and it’s 100, 150 [mg/dL], clearly abnormal, and often these patients don’t have a lot of other risk factors,” Dr. Nissen said. “So the explanation very likely for their premature disease is this risk factor. We now have to educate everybody about the importance of getting it tested and finding out about it.”
During a media briefing, ACC 2022 program cochair Pamela B. Morris, MD, Medical University of South Carolina, Charleston, said testing for Lp(a) is not well reimbursed by insurance providers and that her patients will often cancel the test after learning it won’t be reimbursed because they don’t understand it.
“What Dr. Nissen is telling you: It should be measured in everyone at least once, we all believe that, but it hasn’t made it into the major guidelines,” she added. “I think what we’re going to have to do is have the guidelines mandate it and the insurers will follow.”
Guidelines currently list elevated Lp(a) as a “risk-enhancing factor,” which can help with at least recommending LDL-C treatment in patients with borderline risk and a sky-high Lp(a), noted Dr. Nissen. “But we need to go beyond that.”
Safety analyses
The first-in-human APOLLO trial evaluated 32 adults without known ASCVD and an Lp(a) concentration greater than 150 nmol/L (approximately 60 mg/dL) who received one of the four doses of SLN360 or placebo subcutaneously. Participants were monitored in a research unit for the first 24 hours and then followed periodically for up to 150 days. At baseline, their median Lp(a) level was 224 nmol/L, mean apo B level was 85 mg/dL, and mean LDL-C level was 108 mg/dL.
Treatment-emergent adverse events were generally mild, mostly grade 1 injection site reactions (83% at 30 mg, 100% at 100 mg, 67% at 300 mg, and 33% at 600 mg) and headache (33%, 17%, 0%, and 83%).
At the highest dose, C-reactive protein was increased in four patients and neutrophil counts in three. ALT and AST levels were elevated three times above the upper limit of normal in one patient at the lowest dose.
One participant in the lowest-dose group experienced two serious adverse events unrelated to SLN360 at day 45 after receiving a SARS-Co-V-2 vaccine.
Dr. Nissen noted that safety cannot be comprehensively assessed in a trial of this duration or size and that follow-up has been extended to 1 year in the two highest-dose groups.
Enrollment continues in the multiple-ascending dose portion of the study in patients with high Lp(a) and a history of stable ASCVD. A phase 2 study of SLN360 is also planned for the second half of 2022, pending regulatory discussions.
But will it reduce ASCVD events?
Study discussant Vera Bittner, MD, MSPH, University of Alabama at Birmingham, said that the development of Lp(a)-specific lowering agents has been a “holy grail” for years and congratulated the authors on a successful trial demonstrating very robust Lp(a) lowering.
She asked Dr. Nissen about the observation in proprotein convertase subtilisin/kexin type 9 inhibitor trials that absolute Lp(a) lowering is greater at higher baseline levels.
Dr. Nissen said this kind of analysis wasn’t possible because of the small sample size but “because these agents so effectively degrade messenger RNA, it’s very likely we will see robust suppression of plasma levels virtually regardless of the baseline level.”
Dr. Bittner also questioned if “LDL-C declined because of the cholesterol content in the lipoprotein(a) or is there some additional effect on LDL particles themselves?”
“It’s a really terrific question that will ultimately need to be answered,” Dr. Nissen replied. “There’s some controversy about the extent to which suppressing lipoprotein(a) will reduce LDL because the assays for LDL are measuring the LDL that’s in lipoprotein(a) and the LDL that is not. ... I think it’s probably a bystander effect, but it may also contribute to efficacy from a morbidity and mortality point of view, which is why we measured it.”
Dr. Bittner also called out the elevation in C-reactive protein and leukocytosis, which has not been seen in other siRNA studies. Dr. Nissen said the increases in C-reactive protein occurred in the first few days after administration and were gone after a week or so. “I don’t see it as a long-term limitation.”
In an accompanying editorial, Brian Ference, MD, MPhil, MSc, University of Cambridge (England), suggests that because circulating Lp(a) particles can progressively become trapped within the artery wall over time, it’s unlikely that lowering Lp(a) for only a few years starting later in life will eliminate the effect of lifelong exposure to Lp(a) and may only cut cardiovascular event risk by about 10%-15%.
He called for continued safety and efficacy evaluation of SLN360 and olpasiran, a similar siRNA agent in early development, and said further insights into whether large absolute reductions in Lp(a) can reduce the risk for major cardiovascular events will come from cardiovascular trials, such as the ongoing phase 3 Lp(a)HORIZON trial. It follows strong phase 2 results with the antisense agent AKCEA-APO(a)-LRx and has Dr. Nissen pulling double duty as study chair.
The study was funded by Silence Therapeutics. Dr. Nissen reported consulting for many pharmaceutical companies, which are directed to pay any renumeration directly to charity. Dr. Bittner reported consultant fees or honoraria from Pfizer; other from AstraZeneca, DalCor, Esperion, and Sanofi-Aventis; and research/research grants from Amgen and Novartis. Dr. Ference reported financial ties to Merck, Novartis, Amgen, Pfizer, Esperion Therapeutics, and numerous other companies.
A version of this article first appeared on Medscape.com.
The short interfering RNA (siRNA) agent SLN360 was well tolerated and lowered lipoprotein(a) by up to 98% in volunteers without cardiovascular disease but with elevated Lp(a) in the small dose-ranging APOLLO trial.
Following a single subcutaneous dose of SLN360 (Silence Therapeutics), there was a dose-dependent reduction in Lp(a) plasma levels by a median of 46%, 86%, 96%, and 98% at about 45-60 days with 30-mg, 100-mg, 300-mg, and 600-mg doses, respectively.
Lp(a) levels at 150 days were 70% and 81% below baseline with the 300-and 600-mg doses.
In addition, for participants receiving the two highest doses, apolipoprotein B (apo B) was reduced was 21% and 24%, respectively, and LDL cholesterol (LDL-C), by 21% and 26%, respectively.
“The development of therapies targeting messenger RNA has made possible significant lowering of lipoprotein(a). Whether these reductions can impact on the incidence of ASCVD [atherosclerotic cardiovascular disease] or prevent progression of aortic stenosis remains to be determined but, we think, that optimism is warranted,” said principal investigator Steven E. Nissen, MD, Cleveland Clinic.
The results were presented in a late-breaking clinical trial session at the annual scientific sessions of the American College of Cardiology and published simultaneously in JAMA.
Elevated Lp(a) is a powerful genetic risk factor for ASCVD and aortic stenosis, which affects some 64 million Americans and 1.4 billion people globally. Although several experimental agents are under investigation, no currently approved drugs selectively lower Lp(a).
SLN360 is designed to lower Lp(a) production by using RNA interference to silence messenger RNA transcribed from the LPA gene in liver cells.
Testing vacuum
Dr. Nissen said in an interview that one of the big takeaways from the study is the need for greater testing of Lp(a). Automatic assays are available in almost every hospital, but two-unit systems (nmol/L and mg/dL) are used and thresholds for accelerated risk vary. The Cleveland Clinic currently tests all patients in its cardiac critical care unit and its prevention clinic.
“Someone comes in with an MI in their 40s and we measure it and it’s 100, 150 [mg/dL], clearly abnormal, and often these patients don’t have a lot of other risk factors,” Dr. Nissen said. “So the explanation very likely for their premature disease is this risk factor. We now have to educate everybody about the importance of getting it tested and finding out about it.”
During a media briefing, ACC 2022 program cochair Pamela B. Morris, MD, Medical University of South Carolina, Charleston, said testing for Lp(a) is not well reimbursed by insurance providers and that her patients will often cancel the test after learning it won’t be reimbursed because they don’t understand it.
“What Dr. Nissen is telling you: It should be measured in everyone at least once, we all believe that, but it hasn’t made it into the major guidelines,” she added. “I think what we’re going to have to do is have the guidelines mandate it and the insurers will follow.”
Guidelines currently list elevated Lp(a) as a “risk-enhancing factor,” which can help with at least recommending LDL-C treatment in patients with borderline risk and a sky-high Lp(a), noted Dr. Nissen. “But we need to go beyond that.”
Safety analyses
The first-in-human APOLLO trial evaluated 32 adults without known ASCVD and an Lp(a) concentration greater than 150 nmol/L (approximately 60 mg/dL) who received one of the four doses of SLN360 or placebo subcutaneously. Participants were monitored in a research unit for the first 24 hours and then followed periodically for up to 150 days. At baseline, their median Lp(a) level was 224 nmol/L, mean apo B level was 85 mg/dL, and mean LDL-C level was 108 mg/dL.
Treatment-emergent adverse events were generally mild, mostly grade 1 injection site reactions (83% at 30 mg, 100% at 100 mg, 67% at 300 mg, and 33% at 600 mg) and headache (33%, 17%, 0%, and 83%).
At the highest dose, C-reactive protein was increased in four patients and neutrophil counts in three. ALT and AST levels were elevated three times above the upper limit of normal in one patient at the lowest dose.
One participant in the lowest-dose group experienced two serious adverse events unrelated to SLN360 at day 45 after receiving a SARS-Co-V-2 vaccine.
Dr. Nissen noted that safety cannot be comprehensively assessed in a trial of this duration or size and that follow-up has been extended to 1 year in the two highest-dose groups.
Enrollment continues in the multiple-ascending dose portion of the study in patients with high Lp(a) and a history of stable ASCVD. A phase 2 study of SLN360 is also planned for the second half of 2022, pending regulatory discussions.
But will it reduce ASCVD events?
Study discussant Vera Bittner, MD, MSPH, University of Alabama at Birmingham, said that the development of Lp(a)-specific lowering agents has been a “holy grail” for years and congratulated the authors on a successful trial demonstrating very robust Lp(a) lowering.
She asked Dr. Nissen about the observation in proprotein convertase subtilisin/kexin type 9 inhibitor trials that absolute Lp(a) lowering is greater at higher baseline levels.
Dr. Nissen said this kind of analysis wasn’t possible because of the small sample size but “because these agents so effectively degrade messenger RNA, it’s very likely we will see robust suppression of plasma levels virtually regardless of the baseline level.”
Dr. Bittner also questioned if “LDL-C declined because of the cholesterol content in the lipoprotein(a) or is there some additional effect on LDL particles themselves?”
“It’s a really terrific question that will ultimately need to be answered,” Dr. Nissen replied. “There’s some controversy about the extent to which suppressing lipoprotein(a) will reduce LDL because the assays for LDL are measuring the LDL that’s in lipoprotein(a) and the LDL that is not. ... I think it’s probably a bystander effect, but it may also contribute to efficacy from a morbidity and mortality point of view, which is why we measured it.”
Dr. Bittner also called out the elevation in C-reactive protein and leukocytosis, which has not been seen in other siRNA studies. Dr. Nissen said the increases in C-reactive protein occurred in the first few days after administration and were gone after a week or so. “I don’t see it as a long-term limitation.”
In an accompanying editorial, Brian Ference, MD, MPhil, MSc, University of Cambridge (England), suggests that because circulating Lp(a) particles can progressively become trapped within the artery wall over time, it’s unlikely that lowering Lp(a) for only a few years starting later in life will eliminate the effect of lifelong exposure to Lp(a) and may only cut cardiovascular event risk by about 10%-15%.
He called for continued safety and efficacy evaluation of SLN360 and olpasiran, a similar siRNA agent in early development, and said further insights into whether large absolute reductions in Lp(a) can reduce the risk for major cardiovascular events will come from cardiovascular trials, such as the ongoing phase 3 Lp(a)HORIZON trial. It follows strong phase 2 results with the antisense agent AKCEA-APO(a)-LRx and has Dr. Nissen pulling double duty as study chair.
The study was funded by Silence Therapeutics. Dr. Nissen reported consulting for many pharmaceutical companies, which are directed to pay any renumeration directly to charity. Dr. Bittner reported consultant fees or honoraria from Pfizer; other from AstraZeneca, DalCor, Esperion, and Sanofi-Aventis; and research/research grants from Amgen and Novartis. Dr. Ference reported financial ties to Merck, Novartis, Amgen, Pfizer, Esperion Therapeutics, and numerous other companies.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
New HF guidelines feature ‘quad’ therapy, tweaked terminology
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.

The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.

“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.
The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.
“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.
The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.
“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
Supermarket diet advice improves DASH adherence: SuperWIN
People who received personalized nutrition education in a series of sessions at their regular grocery store significantly improved adherence to a healthy diet, in a new “first-of-its-kind” study in which scientific researchers partnered with a large supermarket company.
In the SuperWIN study, participants were given individualized advice from supermarket-based dietitians using data on their own buying habits recorded on their supermarket loyalty cards. This was associated with an increased adherence to the DASH (Dietary Approaches to Stop Hypertension) diet, which emphasizes vegetables, fruits and whole grains while limiting foods that are high in saturated fat, sugar, and sodium and has been shown to lower blood pressure and LDL cholesterol.

One group of patients also received additional education about healthy eating and meal planning through online technologies, and this group showed even better adherence to the DASH diet.
The study was presented at the annual scientific sessions of the American College of Cardiology by Dylan Steen, MD, adjunct associate professor of medicine at the University of Cincinnati.
“The SuperWIN study provides evidence for the benefit of delivering healthy-eating interventions at modern supermarkets and retail-based clinics,” Dr. Steen said. “It demonstrates the efficacy of dietary interventions harnessing the physical environment of the supermarket, the retail-based dietitians working within the store, and the purchasing data captured on the store’s loyalty cards.”
The study was conducted in partnership with Kroger, the largest supermarket chain in the United States, which also operates a large chain of pharmacies and health clinics.
Dr. Steen said the study was addressing one of the biggest public health problems – unhealthy eating – with an innovative approach. “We need to think about how we can extend the reach of modern health care systems into communities and better deliver services right where people are; meet them where they live,” he said at an ACC press conference.
Commenting on the study at the press conference, Eileen Handberg, PhD, professor of medicine at University of Florida, Gainesville, and immediate past chair of the ACC Cardiovascular Care Team Council, said: “I am amazingly excited about this. There is so much potential here. We have never really taken advantage of the current explosion in retail-based health care before.”

Dr. Handberg suggested the study had major implications for the primary prevention of cardiovascular disease. “Little kids go shopping with their parents, so you have the ability here to change behavior from children on up if you can change the dynamic of the choices they make in the grocery store.”
In his presentation, Dr. Steen noted that, despite many longstanding guidelines on healthy eating, about 75% of Americans still have a poor-quality diet. This trial was conducted to see if a new approach could improve that situation. “If we change the environment in which we deliver dietary education, we can make a difference.”
The SuperWIN trial was conducted in 13 Kroger stores in Ohio and Kentucky. The study enrolled 267 people with at least one cardiovascular risk factor from a primary care network who regularly shopped at one of the study stores. All participants also had to be willing to follow the DASH diet, which was taught at each educational session in the trial.
All participants received one “enhanced” medical nutrition therapy that was guided by the individual’s own dietary intake analytics.
They were then randomly assigned to one of three arms. The control group received no further education. The strategy 1 group received six additional teaching sessions in the supermarket aisles over a 3-month period. Each session was guided by updated individualized purchasing data provided to the dietitian and the participant.
The strategy 2 group received the same six additional teaching sessions as strategy 1, but they also had some additional teaching on healthy eating and meal planning from a variety of online shopping tools, and nutrition and health care apps.
“The supermarket analytics were automatically collected so the dietitians could tell what each person liked to eat, how much of each product they were buying and how much they were spending,” Dr. Steen explained.
COVID hit halfway through the trial, and 20 participants were withdrawn for their own safety as they could no longer visit the stores, but the trial continued with the rest of the participants with enhanced safety precautions. The overall analysis cohort was 247 participants.
The average age of the participants was mid-50s, around 70% were female, and most did not have a history of cardiovascular disease.
Eating habits were assessed by three 24-hour dietary recalls assessed at the start of the study and at 3 and 6 months. The DASH score, which is a measure of adherence to the DASH diet, was calculated from this information. The score can range from 0 to 90, with an increased score showing increased adherence.
In one analysis, the researchers compared the DASH scores from the two intervention groups together with the control group, and in a second analysis they compared the scores in the strategy 2 group with those in the strategy 1 group.
Before the pandemic there was “near 100%” attendance for the six visits over the 3-month study period, which Dr. Steen said he thought was “remarkable.” During the pandemic, attendance came down to around 80%.
Results showed that the DASH score increased in all three groups at 3 months, with stepwise increases corresponding to the intensity of the intervention. DASH scores increased by 5.8 points in the control group, by 8.6 points in the strategy 1 group, and by 12.4 points in the strategy 2 group.
DASH scores significantly differed between the two intervention groups and the control group (P = .02). “This shows that purchasing data–guided in-store tours do increase the efficacy of dietary education,” Dr. Steen said.
The difference in scores between the strategy 1 and strategy 2 groups was also significant (P = .01). “This shows online enhancements increase adherence to the DASH diet even further,” Dr. Steen commented
By 6 months, the scores had dropped off a little but were still increased from baseline: by 4.4 points in the control group, 6.6 points in the strategy 1 group, and 8.4 points in the strategy 2 group. “There was again a stepwise increase as the intervention intensified, but there was no longer a significant difference between the interventions and control,” Dr. Steen noted.
Secondary endpoints included blood pressure and body mass index. Systolic blood pressure decreased slightly in all three groups: by 2.8 mm Hg in the control group, 6.6 mm Hg in the strategy 1 group, and 5.7 mm Hg in the strategy 2 group. Body mass index was reduced by 0.2, 0.4 and 0.8, respectively, but the between-group differences were not significant.
Dr. Steen said this is the first study of its kind to date in which scientific researchers collaborated with a large supermarket chain. He explained they also involved a primary care network so that health care utilization information will be available.
“We can the integrate retail-based health care information with traditional health care information. And we can start to look at downstream health care utilization and cost outcomes as well, which will be important as we start to think how to evolve the health care system,” he commented. “The hope is that we can get more scientists working with more retailers to really drive the evidence to shape the evolution of our health care system.”
Challenges ahead
Dr. Handberg pointed out there would be challenges in reaching the underserved population who do not shop at the major supermarkets. “We need to figure out how to get partnerships across the whole spectrum of grocery stores.”
She also noted that 3 months (the duration of the study intervention) was not much time to change the eating habits of a family. “Interventions may have to be a bit more intensive to get the change in blood pressure and weight that we would want to see.”
Dr Handberg hoped the major grocery store companies will see the opportunities in this approach. “Changing behavior is very complicated, and the key will be how to make people stick with the changes. But grocery stores are smart. They have got us going to their pharmacies, so getting us to see a dietitian is not that much of a stretch.”
Moderator of the ACC late-breaker session at which the study was presented, Pamela Morris, MD, from the Medical University of South Carolina, Charleston, who is also ACC annual scientific session chair, asked whether the approach could be sustained.
“I am thinking back to the barber shop study of blood pressure treatment and to my knowledge those PharmDs are no longer in those barbershops, taking blood pressures, counseling patients, and prescribing antihypertensives. So is Kroger maintaining a long-term commitment to providing this education, or how can this be financed over the long term?” she asked.
Dr. Steen replied that he believed sustainability to be one of the key strengths of this model. “Retail-based health care is exploding in the U.S. The number of retail outlets offering a comprehensive list of services is going up all the time. These programs exist regardless of whether this trial was conducted or not.”
But Dr. Steen stressed that having an evidence base will be critically important.
“Validation is an enormous part of this evolution in retail-based health care – not only to figure out what works but also to engage payors and others in the process of supporting these interventions. I think the sustainability is there – it is sort of baked into the model – but research will be a huge part of cementing this in and helping us to understand what we should do.”
The study was funded by Kroger. Dr. Steen is a consultant for Sanofi and CEO and cofounder of High Enroll.
A version of this article first appeared on Medscape.com.
People who received personalized nutrition education in a series of sessions at their regular grocery store significantly improved adherence to a healthy diet, in a new “first-of-its-kind” study in which scientific researchers partnered with a large supermarket company.
In the SuperWIN study, participants were given individualized advice from supermarket-based dietitians using data on their own buying habits recorded on their supermarket loyalty cards. This was associated with an increased adherence to the DASH (Dietary Approaches to Stop Hypertension) diet, which emphasizes vegetables, fruits and whole grains while limiting foods that are high in saturated fat, sugar, and sodium and has been shown to lower blood pressure and LDL cholesterol.
One group of patients also received additional education about healthy eating and meal planning through online technologies, and this group showed even better adherence to the DASH diet.
The study was presented at the annual scientific sessions of the American College of Cardiology by Dylan Steen, MD, adjunct associate professor of medicine at the University of Cincinnati.
“The SuperWIN study provides evidence for the benefit of delivering healthy-eating interventions at modern supermarkets and retail-based clinics,” Dr. Steen said. “It demonstrates the efficacy of dietary interventions harnessing the physical environment of the supermarket, the retail-based dietitians working within the store, and the purchasing data captured on the store’s loyalty cards.”
The study was conducted in partnership with Kroger, the largest supermarket chain in the United States, which also operates a large chain of pharmacies and health clinics.
Dr. Steen said the study was addressing one of the biggest public health problems – unhealthy eating – with an innovative approach. “We need to think about how we can extend the reach of modern health care systems into communities and better deliver services right where people are; meet them where they live,” he said at an ACC press conference.
Commenting on the study at the press conference, Eileen Handberg, PhD, professor of medicine at University of Florida, Gainesville, and immediate past chair of the ACC Cardiovascular Care Team Council, said: “I am amazingly excited about this. There is so much potential here. We have never really taken advantage of the current explosion in retail-based health care before.”
Dr. Handberg suggested the study had major implications for the primary prevention of cardiovascular disease. “Little kids go shopping with their parents, so you have the ability here to change behavior from children on up if you can change the dynamic of the choices they make in the grocery store.”
In his presentation, Dr. Steen noted that, despite many longstanding guidelines on healthy eating, about 75% of Americans still have a poor-quality diet. This trial was conducted to see if a new approach could improve that situation. “If we change the environment in which we deliver dietary education, we can make a difference.”
The SuperWIN trial was conducted in 13 Kroger stores in Ohio and Kentucky. The study enrolled 267 people with at least one cardiovascular risk factor from a primary care network who regularly shopped at one of the study stores. All participants also had to be willing to follow the DASH diet, which was taught at each educational session in the trial.
All participants received one “enhanced” medical nutrition therapy that was guided by the individual’s own dietary intake analytics.
They were then randomly assigned to one of three arms. The control group received no further education. The strategy 1 group received six additional teaching sessions in the supermarket aisles over a 3-month period. Each session was guided by updated individualized purchasing data provided to the dietitian and the participant.
The strategy 2 group received the same six additional teaching sessions as strategy 1, but they also had some additional teaching on healthy eating and meal planning from a variety of online shopping tools, and nutrition and health care apps.
“The supermarket analytics were automatically collected so the dietitians could tell what each person liked to eat, how much of each product they were buying and how much they were spending,” Dr. Steen explained.
COVID hit halfway through the trial, and 20 participants were withdrawn for their own safety as they could no longer visit the stores, but the trial continued with the rest of the participants with enhanced safety precautions. The overall analysis cohort was 247 participants.
The average age of the participants was mid-50s, around 70% were female, and most did not have a history of cardiovascular disease.
Eating habits were assessed by three 24-hour dietary recalls assessed at the start of the study and at 3 and 6 months. The DASH score, which is a measure of adherence to the DASH diet, was calculated from this information. The score can range from 0 to 90, with an increased score showing increased adherence.
In one analysis, the researchers compared the DASH scores from the two intervention groups together with the control group, and in a second analysis they compared the scores in the strategy 2 group with those in the strategy 1 group.
Before the pandemic there was “near 100%” attendance for the six visits over the 3-month study period, which Dr. Steen said he thought was “remarkable.” During the pandemic, attendance came down to around 80%.
Results showed that the DASH score increased in all three groups at 3 months, with stepwise increases corresponding to the intensity of the intervention. DASH scores increased by 5.8 points in the control group, by 8.6 points in the strategy 1 group, and by 12.4 points in the strategy 2 group.
DASH scores significantly differed between the two intervention groups and the control group (P = .02). “This shows that purchasing data–guided in-store tours do increase the efficacy of dietary education,” Dr. Steen said.
The difference in scores between the strategy 1 and strategy 2 groups was also significant (P = .01). “This shows online enhancements increase adherence to the DASH diet even further,” Dr. Steen commented
By 6 months, the scores had dropped off a little but were still increased from baseline: by 4.4 points in the control group, 6.6 points in the strategy 1 group, and 8.4 points in the strategy 2 group. “There was again a stepwise increase as the intervention intensified, but there was no longer a significant difference between the interventions and control,” Dr. Steen noted.
Secondary endpoints included blood pressure and body mass index. Systolic blood pressure decreased slightly in all three groups: by 2.8 mm Hg in the control group, 6.6 mm Hg in the strategy 1 group, and 5.7 mm Hg in the strategy 2 group. Body mass index was reduced by 0.2, 0.4 and 0.8, respectively, but the between-group differences were not significant.
Dr. Steen said this is the first study of its kind to date in which scientific researchers collaborated with a large supermarket chain. He explained they also involved a primary care network so that health care utilization information will be available.
“We can the integrate retail-based health care information with traditional health care information. And we can start to look at downstream health care utilization and cost outcomes as well, which will be important as we start to think how to evolve the health care system,” he commented. “The hope is that we can get more scientists working with more retailers to really drive the evidence to shape the evolution of our health care system.”
Challenges ahead
Dr. Handberg pointed out there would be challenges in reaching the underserved population who do not shop at the major supermarkets. “We need to figure out how to get partnerships across the whole spectrum of grocery stores.”
She also noted that 3 months (the duration of the study intervention) was not much time to change the eating habits of a family. “Interventions may have to be a bit more intensive to get the change in blood pressure and weight that we would want to see.”
Dr Handberg hoped the major grocery store companies will see the opportunities in this approach. “Changing behavior is very complicated, and the key will be how to make people stick with the changes. But grocery stores are smart. They have got us going to their pharmacies, so getting us to see a dietitian is not that much of a stretch.”
Moderator of the ACC late-breaker session at which the study was presented, Pamela Morris, MD, from the Medical University of South Carolina, Charleston, who is also ACC annual scientific session chair, asked whether the approach could be sustained.
“I am thinking back to the barber shop study of blood pressure treatment and to my knowledge those PharmDs are no longer in those barbershops, taking blood pressures, counseling patients, and prescribing antihypertensives. So is Kroger maintaining a long-term commitment to providing this education, or how can this be financed over the long term?” she asked.
Dr. Steen replied that he believed sustainability to be one of the key strengths of this model. “Retail-based health care is exploding in the U.S. The number of retail outlets offering a comprehensive list of services is going up all the time. These programs exist regardless of whether this trial was conducted or not.”
But Dr. Steen stressed that having an evidence base will be critically important.
“Validation is an enormous part of this evolution in retail-based health care – not only to figure out what works but also to engage payors and others in the process of supporting these interventions. I think the sustainability is there – it is sort of baked into the model – but research will be a huge part of cementing this in and helping us to understand what we should do.”
The study was funded by Kroger. Dr. Steen is a consultant for Sanofi and CEO and cofounder of High Enroll.
A version of this article first appeared on Medscape.com.
People who received personalized nutrition education in a series of sessions at their regular grocery store significantly improved adherence to a healthy diet, in a new “first-of-its-kind” study in which scientific researchers partnered with a large supermarket company.
In the SuperWIN study, participants were given individualized advice from supermarket-based dietitians using data on their own buying habits recorded on their supermarket loyalty cards. This was associated with an increased adherence to the DASH (Dietary Approaches to Stop Hypertension) diet, which emphasizes vegetables, fruits and whole grains while limiting foods that are high in saturated fat, sugar, and sodium and has been shown to lower blood pressure and LDL cholesterol.
One group of patients also received additional education about healthy eating and meal planning through online technologies, and this group showed even better adherence to the DASH diet.
The study was presented at the annual scientific sessions of the American College of Cardiology by Dylan Steen, MD, adjunct associate professor of medicine at the University of Cincinnati.
“The SuperWIN study provides evidence for the benefit of delivering healthy-eating interventions at modern supermarkets and retail-based clinics,” Dr. Steen said. “It demonstrates the efficacy of dietary interventions harnessing the physical environment of the supermarket, the retail-based dietitians working within the store, and the purchasing data captured on the store’s loyalty cards.”
The study was conducted in partnership with Kroger, the largest supermarket chain in the United States, which also operates a large chain of pharmacies and health clinics.
Dr. Steen said the study was addressing one of the biggest public health problems – unhealthy eating – with an innovative approach. “We need to think about how we can extend the reach of modern health care systems into communities and better deliver services right where people are; meet them where they live,” he said at an ACC press conference.
Commenting on the study at the press conference, Eileen Handberg, PhD, professor of medicine at University of Florida, Gainesville, and immediate past chair of the ACC Cardiovascular Care Team Council, said: “I am amazingly excited about this. There is so much potential here. We have never really taken advantage of the current explosion in retail-based health care before.”
Dr. Handberg suggested the study had major implications for the primary prevention of cardiovascular disease. “Little kids go shopping with their parents, so you have the ability here to change behavior from children on up if you can change the dynamic of the choices they make in the grocery store.”
In his presentation, Dr. Steen noted that, despite many longstanding guidelines on healthy eating, about 75% of Americans still have a poor-quality diet. This trial was conducted to see if a new approach could improve that situation. “If we change the environment in which we deliver dietary education, we can make a difference.”
The SuperWIN trial was conducted in 13 Kroger stores in Ohio and Kentucky. The study enrolled 267 people with at least one cardiovascular risk factor from a primary care network who regularly shopped at one of the study stores. All participants also had to be willing to follow the DASH diet, which was taught at each educational session in the trial.
All participants received one “enhanced” medical nutrition therapy that was guided by the individual’s own dietary intake analytics.
They were then randomly assigned to one of three arms. The control group received no further education. The strategy 1 group received six additional teaching sessions in the supermarket aisles over a 3-month period. Each session was guided by updated individualized purchasing data provided to the dietitian and the participant.
The strategy 2 group received the same six additional teaching sessions as strategy 1, but they also had some additional teaching on healthy eating and meal planning from a variety of online shopping tools, and nutrition and health care apps.
“The supermarket analytics were automatically collected so the dietitians could tell what each person liked to eat, how much of each product they were buying and how much they were spending,” Dr. Steen explained.
COVID hit halfway through the trial, and 20 participants were withdrawn for their own safety as they could no longer visit the stores, but the trial continued with the rest of the participants with enhanced safety precautions. The overall analysis cohort was 247 participants.
The average age of the participants was mid-50s, around 70% were female, and most did not have a history of cardiovascular disease.
Eating habits were assessed by three 24-hour dietary recalls assessed at the start of the study and at 3 and 6 months. The DASH score, which is a measure of adherence to the DASH diet, was calculated from this information. The score can range from 0 to 90, with an increased score showing increased adherence.
In one analysis, the researchers compared the DASH scores from the two intervention groups together with the control group, and in a second analysis they compared the scores in the strategy 2 group with those in the strategy 1 group.
Before the pandemic there was “near 100%” attendance for the six visits over the 3-month study period, which Dr. Steen said he thought was “remarkable.” During the pandemic, attendance came down to around 80%.
Results showed that the DASH score increased in all three groups at 3 months, with stepwise increases corresponding to the intensity of the intervention. DASH scores increased by 5.8 points in the control group, by 8.6 points in the strategy 1 group, and by 12.4 points in the strategy 2 group.
DASH scores significantly differed between the two intervention groups and the control group (P = .02). “This shows that purchasing data–guided in-store tours do increase the efficacy of dietary education,” Dr. Steen said.
The difference in scores between the strategy 1 and strategy 2 groups was also significant (P = .01). “This shows online enhancements increase adherence to the DASH diet even further,” Dr. Steen commented
By 6 months, the scores had dropped off a little but were still increased from baseline: by 4.4 points in the control group, 6.6 points in the strategy 1 group, and 8.4 points in the strategy 2 group. “There was again a stepwise increase as the intervention intensified, but there was no longer a significant difference between the interventions and control,” Dr. Steen noted.
Secondary endpoints included blood pressure and body mass index. Systolic blood pressure decreased slightly in all three groups: by 2.8 mm Hg in the control group, 6.6 mm Hg in the strategy 1 group, and 5.7 mm Hg in the strategy 2 group. Body mass index was reduced by 0.2, 0.4 and 0.8, respectively, but the between-group differences were not significant.
Dr. Steen said this is the first study of its kind to date in which scientific researchers collaborated with a large supermarket chain. He explained they also involved a primary care network so that health care utilization information will be available.
“We can the integrate retail-based health care information with traditional health care information. And we can start to look at downstream health care utilization and cost outcomes as well, which will be important as we start to think how to evolve the health care system,” he commented. “The hope is that we can get more scientists working with more retailers to really drive the evidence to shape the evolution of our health care system.”
Challenges ahead
Dr. Handberg pointed out there would be challenges in reaching the underserved population who do not shop at the major supermarkets. “We need to figure out how to get partnerships across the whole spectrum of grocery stores.”
She also noted that 3 months (the duration of the study intervention) was not much time to change the eating habits of a family. “Interventions may have to be a bit more intensive to get the change in blood pressure and weight that we would want to see.”
Dr Handberg hoped the major grocery store companies will see the opportunities in this approach. “Changing behavior is very complicated, and the key will be how to make people stick with the changes. But grocery stores are smart. They have got us going to their pharmacies, so getting us to see a dietitian is not that much of a stretch.”
Moderator of the ACC late-breaker session at which the study was presented, Pamela Morris, MD, from the Medical University of South Carolina, Charleston, who is also ACC annual scientific session chair, asked whether the approach could be sustained.
“I am thinking back to the barber shop study of blood pressure treatment and to my knowledge those PharmDs are no longer in those barbershops, taking blood pressures, counseling patients, and prescribing antihypertensives. So is Kroger maintaining a long-term commitment to providing this education, or how can this be financed over the long term?” she asked.
Dr. Steen replied that he believed sustainability to be one of the key strengths of this model. “Retail-based health care is exploding in the U.S. The number of retail outlets offering a comprehensive list of services is going up all the time. These programs exist regardless of whether this trial was conducted or not.”
But Dr. Steen stressed that having an evidence base will be critically important.
“Validation is an enormous part of this evolution in retail-based health care – not only to figure out what works but also to engage payors and others in the process of supporting these interventions. I think the sustainability is there – it is sort of baked into the model – but research will be a huge part of cementing this in and helping us to understand what we should do.”
The study was funded by Kroger. Dr. Steen is a consultant for Sanofi and CEO and cofounder of High Enroll.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
Mavacamten controlled hypertrophic cardiomyopathy for over 1 year
WASHINGTON – Treatment of patients with symptomatic obstructive hypertrophic cardiomyopathy who remained on treatment with the investigational agent mavacamten for a median of 62 weeks continued to show the same level of safe response to the drug as seen after the first 30 weeks on treatment in the pivotal trial for this agent.
The new findings from longer-term treatment bode well for mavacamten. That’s because if the drug is used in routine practice to avoid the need for surgery or an invasive intervention to reduce blockage of a patient’s left ventricular outflow tract, the duration of mavacamten treatment will likely need to continue for many years and even for decades, said Florian Rader, MD, who presented the results at the annual scientific sessions of the American College of Cardiology.

“In practice, mavacamten will probably be used for many, many years, especially as it replaces septal-reduction therapy, so we need long-term data,” noted Dr. Rader during a press briefing on his report. “I’m very happy with the long-term data” in the follow-up study.
The Food and Drug Administration is currently considering whether to approve mavacamten for routine marketing to treat patients with symptomatic obstructive hypertrophic cardiomyopathy (oHCM), with a decision expected by the end of April 2022.
The study Dr. Rader reported followed 231 patients with symptomatic oHCM who had completed the 30-week pivotal trial of mavacamten, EXPLORER-HCM, and opted to continue on open-label extended treatment with mavacamten, either continuing the treatment they started during the trial or crossing over to receive mavacamten after receiving placebo during the trial.
The major findings from EXPLORER-LTE (long-term extension) were that continued treatment for a median of about 62 weeks maintained the safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase, said Dr. Rader, codirector of the Clinic for Hypertrophic Cardiomyopathy and Aortopathies at Cedars-Sinai Medical Center in Los Angeles.
‘Almost revolutionary’
Mavacamten represents “an almost revolutionary change” for treating oHCM, commented Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis. “Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” said Dr. Guglin during the press briefing.

“All of us who care for patients with oHCM have looked forward to having a disease-specific therapy. It is encouraging to see that the safety and efficacy remained high with long-term follow-up,” commented Kyle W. Klarich, MD, a professor and cardiologist who specializes in treating patients with HCM at the Mayo Clinic in Rochester, Minn.
Mavacamten is a direct myosin inhibitor that reduces the excess number of myosin-actin cross bridges that form in patients with oHCM, and thereby directly targets the pathophysiology that underlies the disorder, explained Dr. Rader.
The patients on mavacamten included in the long-term extension reported by Dr. Rader averaged 60 years of age, and 61% were men. They averaged a 35.6–mm Hg drop in their resting left ventricular outflow tract (LVOT) gradient after 48 weeks on treatment, and a 32.8–mm Hg reduction after 84 weeks. When the investigators measured their LVOT gradient during a valsalva maneuver, their reductions from baseline averaged 45.3 mm Hg after 48 weeks and 46.4 mm Hg after 84 weeks.

Resting left ventricular ejection fraction also fell, by an average of 7.0 percentage points from baseline after 48 weeks, and by an average of 9.0 percentage points after 84 weeks. After 48 weeks on treatment, 68% of patients had at least a one-class improvement from baseline in their New York Heart Association functional class.
The safety results showed that most treatment-related adverse events were mild or moderate, and about 2% of patients had a serious drug-related adverse event. Ten of the 231 patients discontinued mavacamten because of a treated-related adverse event.
EXPLORER-HCM and EXPLORER-LTE were sponsored by MyoKardia, the company that is developing mavacamten and which is now owned by Bristol-Myers Squibb. Dr. Rader has been a consultant to MyoKardia as well as to Medtronic and ReCor. Dr. Guglin and Dr. Klarich had no disclosures.
WASHINGTON – Treatment of patients with symptomatic obstructive hypertrophic cardiomyopathy who remained on treatment with the investigational agent mavacamten for a median of 62 weeks continued to show the same level of safe response to the drug as seen after the first 30 weeks on treatment in the pivotal trial for this agent.
The new findings from longer-term treatment bode well for mavacamten. That’s because if the drug is used in routine practice to avoid the need for surgery or an invasive intervention to reduce blockage of a patient’s left ventricular outflow tract, the duration of mavacamten treatment will likely need to continue for many years and even for decades, said Florian Rader, MD, who presented the results at the annual scientific sessions of the American College of Cardiology.
“In practice, mavacamten will probably be used for many, many years, especially as it replaces septal-reduction therapy, so we need long-term data,” noted Dr. Rader during a press briefing on his report. “I’m very happy with the long-term data” in the follow-up study.
The Food and Drug Administration is currently considering whether to approve mavacamten for routine marketing to treat patients with symptomatic obstructive hypertrophic cardiomyopathy (oHCM), with a decision expected by the end of April 2022.
The study Dr. Rader reported followed 231 patients with symptomatic oHCM who had completed the 30-week pivotal trial of mavacamten, EXPLORER-HCM, and opted to continue on open-label extended treatment with mavacamten, either continuing the treatment they started during the trial or crossing over to receive mavacamten after receiving placebo during the trial.
The major findings from EXPLORER-LTE (long-term extension) were that continued treatment for a median of about 62 weeks maintained the safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase, said Dr. Rader, codirector of the Clinic for Hypertrophic Cardiomyopathy and Aortopathies at Cedars-Sinai Medical Center in Los Angeles.
‘Almost revolutionary’
Mavacamten represents “an almost revolutionary change” for treating oHCM, commented Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis. “Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” said Dr. Guglin during the press briefing.
“All of us who care for patients with oHCM have looked forward to having a disease-specific therapy. It is encouraging to see that the safety and efficacy remained high with long-term follow-up,” commented Kyle W. Klarich, MD, a professor and cardiologist who specializes in treating patients with HCM at the Mayo Clinic in Rochester, Minn.
Mavacamten is a direct myosin inhibitor that reduces the excess number of myosin-actin cross bridges that form in patients with oHCM, and thereby directly targets the pathophysiology that underlies the disorder, explained Dr. Rader.
The patients on mavacamten included in the long-term extension reported by Dr. Rader averaged 60 years of age, and 61% were men. They averaged a 35.6–mm Hg drop in their resting left ventricular outflow tract (LVOT) gradient after 48 weeks on treatment, and a 32.8–mm Hg reduction after 84 weeks. When the investigators measured their LVOT gradient during a valsalva maneuver, their reductions from baseline averaged 45.3 mm Hg after 48 weeks and 46.4 mm Hg after 84 weeks.
Resting left ventricular ejection fraction also fell, by an average of 7.0 percentage points from baseline after 48 weeks, and by an average of 9.0 percentage points after 84 weeks. After 48 weeks on treatment, 68% of patients had at least a one-class improvement from baseline in their New York Heart Association functional class.
The safety results showed that most treatment-related adverse events were mild or moderate, and about 2% of patients had a serious drug-related adverse event. Ten of the 231 patients discontinued mavacamten because of a treated-related adverse event.
EXPLORER-HCM and EXPLORER-LTE were sponsored by MyoKardia, the company that is developing mavacamten and which is now owned by Bristol-Myers Squibb. Dr. Rader has been a consultant to MyoKardia as well as to Medtronic and ReCor. Dr. Guglin and Dr. Klarich had no disclosures.
WASHINGTON – Treatment of patients with symptomatic obstructive hypertrophic cardiomyopathy who remained on treatment with the investigational agent mavacamten for a median of 62 weeks continued to show the same level of safe response to the drug as seen after the first 30 weeks on treatment in the pivotal trial for this agent.
The new findings from longer-term treatment bode well for mavacamten. That’s because if the drug is used in routine practice to avoid the need for surgery or an invasive intervention to reduce blockage of a patient’s left ventricular outflow tract, the duration of mavacamten treatment will likely need to continue for many years and even for decades, said Florian Rader, MD, who presented the results at the annual scientific sessions of the American College of Cardiology.
“In practice, mavacamten will probably be used for many, many years, especially as it replaces septal-reduction therapy, so we need long-term data,” noted Dr. Rader during a press briefing on his report. “I’m very happy with the long-term data” in the follow-up study.
The Food and Drug Administration is currently considering whether to approve mavacamten for routine marketing to treat patients with symptomatic obstructive hypertrophic cardiomyopathy (oHCM), with a decision expected by the end of April 2022.
The study Dr. Rader reported followed 231 patients with symptomatic oHCM who had completed the 30-week pivotal trial of mavacamten, EXPLORER-HCM, and opted to continue on open-label extended treatment with mavacamten, either continuing the treatment they started during the trial or crossing over to receive mavacamten after receiving placebo during the trial.
The major findings from EXPLORER-LTE (long-term extension) were that continued treatment for a median of about 62 weeks maintained the safety and efficacy findings seen at the end of the blinded, randomized, initial 30-week phase, said Dr. Rader, codirector of the Clinic for Hypertrophic Cardiomyopathy and Aortopathies at Cedars-Sinai Medical Center in Los Angeles.
‘Almost revolutionary’
Mavacamten represents “an almost revolutionary change” for treating oHCM, commented Maya E. Guglin, MD, professor of clinical medicine and an advanced heart failure physician at Indiana University, Indianapolis. “Until now, there was no good medical treatment for symptomatic oHCM. This will change the landscape, and without question it will change guidelines for treating oHCM,” said Dr. Guglin during the press briefing.
“All of us who care for patients with oHCM have looked forward to having a disease-specific therapy. It is encouraging to see that the safety and efficacy remained high with long-term follow-up,” commented Kyle W. Klarich, MD, a professor and cardiologist who specializes in treating patients with HCM at the Mayo Clinic in Rochester, Minn.
Mavacamten is a direct myosin inhibitor that reduces the excess number of myosin-actin cross bridges that form in patients with oHCM, and thereby directly targets the pathophysiology that underlies the disorder, explained Dr. Rader.
The patients on mavacamten included in the long-term extension reported by Dr. Rader averaged 60 years of age, and 61% were men. They averaged a 35.6–mm Hg drop in their resting left ventricular outflow tract (LVOT) gradient after 48 weeks on treatment, and a 32.8–mm Hg reduction after 84 weeks. When the investigators measured their LVOT gradient during a valsalva maneuver, their reductions from baseline averaged 45.3 mm Hg after 48 weeks and 46.4 mm Hg after 84 weeks.
Resting left ventricular ejection fraction also fell, by an average of 7.0 percentage points from baseline after 48 weeks, and by an average of 9.0 percentage points after 84 weeks. After 48 weeks on treatment, 68% of patients had at least a one-class improvement from baseline in their New York Heart Association functional class.
The safety results showed that most treatment-related adverse events were mild or moderate, and about 2% of patients had a serious drug-related adverse event. Ten of the 231 patients discontinued mavacamten because of a treated-related adverse event.
EXPLORER-HCM and EXPLORER-LTE were sponsored by MyoKardia, the company that is developing mavacamten and which is now owned by Bristol-Myers Squibb. Dr. Rader has been a consultant to MyoKardia as well as to Medtronic and ReCor. Dr. Guglin and Dr. Klarich had no disclosures.
AT ACC 2022
Novel cholesterol drug disappoints: TRANSLATE-TIMI 70
An investigational drug targeting a novel cholesterol pathway has shown disappointing results in the TRANSLATE-TIMI 70 phase 2b study.
Vupanorsen is an antisense oligonucleotide targeting hepatic angiopoietin-like protein 3 (ANGPTL3), a protein that inhibits enzymes involved in the metabolism of triglyceride and cholesterol. Inhibition of ANGPTL3 is one of several novel targets for lowering triglycerides and non-HDL cholesterol.

Results of the TRANSLATE-TIMI 70 study were presented at the annual scientific sessions of the American College of Cardiology by Brian Bergmark, MD, a cardiologist at Brigham and Women’s Hospital, Boston. They were also simultaneously published online in Circulation.
“While vupanorsen significantly reduced triglycerides and non-HDL cholesterol, the reduction in non-HDL cholesterol of 22%-27% was not to a degree that was clinically meaningful for cardiovascular risk reduction, and there were also some potentially important safety issues,” Dr. Bergmark said in an interview.
Pfizer has announced that, after reviewing the results of this study, it is discontinuing development of vupanorsen and will return rights to Ionis, from which it licensed the investigational therapy in 2019.
In response to a question at an ACC press conference on whether there could be any future for the drug, Dr. Bergmark said that “the degree of lipid lowering was not as much as what had been suggested was potentially possible by acting on this pathway, and then there are the additional safety concerns. So, for the specific question of what we were looking at – cardiovascular risk reduction by impacting non-HDL cholesterol and apo [apolipoprotein] B – the modest efficacy paired with the safety concerns does not look favorable for future development of this drug.”
But he added: “Whether some other person or company wants to think about triglyceride lowering and try to find a dose that is a bit safer, that is not for me to say.”
In his ACC presentation, Dr. Bergmark explained that ANGPTL3 is a protein secreted by the liver that inhibits lipases, including lipoprotein lipase. Loss-of-function variants in ANGPTL3 are associated with lower levels of plasma lipids and a monoclonal antibody targeting ANGPTL3, evinacumab (Evkeeza, Regeneron), is approved as an intravenous infusion for the treatment of familial hypercholesterolemia. Vupanorsen is a second-generation antisense oligonucleotide targeting hepatic ANGPTL3 messenger RNA with a potential role for cardiovascular risk reduction.
A previous phase 2a study of vupanorsen in patients with hypertriglyceridemia, hepatic steatosis, and type 2 diabetes mellitus showed significant reductions in triglycerides at all doses studied, as well as reductions in non-HDL cholesterol at the highest doses (80 mg per month given by subcutaneous injection).
Dr. Bergmark noted that, because a potential cardiovascular benefit of vupanorsen would best be reflected by its effects on non-HDL cholesterol, the current TRANSLATE-TIMI 70 trial was designed to assess the effect of escalating doses of vupanorsen on non-HDL cholesterol levels in statin-treated adults with hyperlipidemia.
For the study, 286 adults with non-HDL cholesterol levels of 100 mg/dL or greater (median, 132 mg/dL) and triglyceride levels of 150-500 mg/dL (median, 216 mg/dL) who were receiving statin therapy were randomly assigned to placebo or one of seven vupanorsen dose regimens (80, 120, or 160 mg every 4 weeks or 60, 80, 120, or 160 mg every 2 weeks). All doses were given by subcutaneous injection.
The study population was said to reflect “a typical cohort intended for cardiovascular risk reduction, with type 2 diabetes in approximately one-half of patients and prevalent atherosclerotic cardiovascular disease in a substantial portion,” the researchers wrote in the published report.
The primary endpoint was placebo-adjusted percentage change from baseline in non-HDL cholesterol at 24 weeks. Secondary endpoints included placebo-adjusted percentage changes from baseline in triglycerides, LDL cholesterol, apo B, and ANGPTL3.
Vupanorsen resulted in significant decreases from baseline over placebo in non-HDL cholesterol ranging from 22.0% in the group receiving 60 mg every 2 weeks to 27.7% in the group receiving 80 mg every 2 weeks, but there did not appear to be a dose response.
Regarding additional lipid endpoints, vupanorsen reduced triglyceride levels in a dose-dependent manner, ranging from 41.3% in the group receiving 120 mg every 4 weeks to 56.8% in the group receiving 160 mg every 2 weeks.
The effects of vupanorsen on LDL cholesterol and apo B were more modest and without a clear dose response. Vupanorsen also lowered HDL cholesterol levels at all doses studied, and there was no significant change in high-sensitivity C-reactive protein at any dose.
Liver enzymes and hepatic fat increases of concern
In terms of safety, vupanorsen treatment was linked to liver enzyme elevations; more than three-times elevations of alanine aminotransferase or aspartate aminotransferase were more common at higher total monthly doses (up to 33.3% and 44.4%, respectively). Injection site reactions were also an issue, including recall reactions at sites of previous injections when subsequent injections were given. In addition, there was a dose-related increase (up to 76%) in hepatic fat fraction.
In the Circulation paper, the researchers say it is unclear whether the increases in hepatic fat fraction and liver enzymes reflect a metabolic effect of vupanorsen specifically or an off-target effect resulting from hepatic targeting of ANGPTL3. “Regardless, these are medically meaningful findings with important safety ramifications,” they wrote.
They pointed out that, whereas the reduction in ANGPTL3 levels increased with total monthly dose of vupanorsen, there was no clear dose-response reduction in LDL cholesterol, apo B, or non-HDL cholesterol.
In comparison, evinacumab, a monoclonal antibody against ANGPTL3 that is thought to cause near-total suppression of ANGPTL3 activity, reduces apo B levels by more than 40% in adults with refractory hypercholesterolemia or homozygous familial hypercholesterolemia.
Asked why vupanorsen showed less of an effect on non-HDL cholesterol than evinacumab, Dr. Bergmark suggested that the monoclonal antibody may achieve greater inhibition of ANGPTL3. “It may be that near complete suppression is needed to obtain clinically meaningful reductions in apo B and non-HDL cholesterol. That is a speculative and simplistic explanation,” he commented.
Conversely, reductions in triglycerides with vupanorsen showed a dose-response relationship, mirroring the reduction in ANGPTL3 and consistent with the expected increases in lipoprotein lipase activity, the researchers reported.
They note that the “relatively muted effect on apo B levels” suggests that vupanorsen is primarily decreasing the triglyceride and, to a lesser extent, cholesterol content of very low-density lipoprotein cholesterol particles rather than reducing the number of such particles.
“These observations have important implications for the potential ability of this mechanism to reduce lipid-mediated cardiovascular risk, which largely appears to be a function of the number of ApoB-containing lipoproteins,” they said.

Designated discussant of the study at the ACC late-breaking session, Pradeep Natarajan, MD, director of preventive cardiology at Massachusetts General Hospital in Boston, asked Dr. Bergmark what minimum degree of non-HDL cholesterol reduction would be compelling for a new drug to be considered for wide-scale use.
Dr. Bergmark replied there was no clear to answer to that question, as it would depend on many factors, including the risk of the population and the time horizon involved. But he added: “I think a minimum of at least a 30%-40% reduction in non-HDL cholesterol would be needed for a meaningful reduction in cardiovascular risk across a variety of settings.”
The TRANSLATE-TIMI 70 study was funded by Pfizer. Dr. Bergmark is a member of the TIMI Study Group, which has received institutional grant support through Brigham and Women’s Hospital from numerous pharmaceutical companies. Dr. Bergmark also reported receiving grant support through Brigham and Women’s Hospital from Pfizer, Ionis, AstraZeneca, and Abbott Vascular and consulting/personal fees from Abiomed, CSI, Philips, Abbott Vascular, Servier, DaiichiSankyo, Janssen, and Quark.
A version of this article first appeared on Medscape.com.
An investigational drug targeting a novel cholesterol pathway has shown disappointing results in the TRANSLATE-TIMI 70 phase 2b study.
Vupanorsen is an antisense oligonucleotide targeting hepatic angiopoietin-like protein 3 (ANGPTL3), a protein that inhibits enzymes involved in the metabolism of triglyceride and cholesterol. Inhibition of ANGPTL3 is one of several novel targets for lowering triglycerides and non-HDL cholesterol.
Results of the TRANSLATE-TIMI 70 study were presented at the annual scientific sessions of the American College of Cardiology by Brian Bergmark, MD, a cardiologist at Brigham and Women’s Hospital, Boston. They were also simultaneously published online in Circulation.
“While vupanorsen significantly reduced triglycerides and non-HDL cholesterol, the reduction in non-HDL cholesterol of 22%-27% was not to a degree that was clinically meaningful for cardiovascular risk reduction, and there were also some potentially important safety issues,” Dr. Bergmark said in an interview.
Pfizer has announced that, after reviewing the results of this study, it is discontinuing development of vupanorsen and will return rights to Ionis, from which it licensed the investigational therapy in 2019.
In response to a question at an ACC press conference on whether there could be any future for the drug, Dr. Bergmark said that “the degree of lipid lowering was not as much as what had been suggested was potentially possible by acting on this pathway, and then there are the additional safety concerns. So, for the specific question of what we were looking at – cardiovascular risk reduction by impacting non-HDL cholesterol and apo [apolipoprotein] B – the modest efficacy paired with the safety concerns does not look favorable for future development of this drug.”
But he added: “Whether some other person or company wants to think about triglyceride lowering and try to find a dose that is a bit safer, that is not for me to say.”
In his ACC presentation, Dr. Bergmark explained that ANGPTL3 is a protein secreted by the liver that inhibits lipases, including lipoprotein lipase. Loss-of-function variants in ANGPTL3 are associated with lower levels of plasma lipids and a monoclonal antibody targeting ANGPTL3, evinacumab (Evkeeza, Regeneron), is approved as an intravenous infusion for the treatment of familial hypercholesterolemia. Vupanorsen is a second-generation antisense oligonucleotide targeting hepatic ANGPTL3 messenger RNA with a potential role for cardiovascular risk reduction.
A previous phase 2a study of vupanorsen in patients with hypertriglyceridemia, hepatic steatosis, and type 2 diabetes mellitus showed significant reductions in triglycerides at all doses studied, as well as reductions in non-HDL cholesterol at the highest doses (80 mg per month given by subcutaneous injection).
Dr. Bergmark noted that, because a potential cardiovascular benefit of vupanorsen would best be reflected by its effects on non-HDL cholesterol, the current TRANSLATE-TIMI 70 trial was designed to assess the effect of escalating doses of vupanorsen on non-HDL cholesterol levels in statin-treated adults with hyperlipidemia.
For the study, 286 adults with non-HDL cholesterol levels of 100 mg/dL or greater (median, 132 mg/dL) and triglyceride levels of 150-500 mg/dL (median, 216 mg/dL) who were receiving statin therapy were randomly assigned to placebo or one of seven vupanorsen dose regimens (80, 120, or 160 mg every 4 weeks or 60, 80, 120, or 160 mg every 2 weeks). All doses were given by subcutaneous injection.
The study population was said to reflect “a typical cohort intended for cardiovascular risk reduction, with type 2 diabetes in approximately one-half of patients and prevalent atherosclerotic cardiovascular disease in a substantial portion,” the researchers wrote in the published report.
The primary endpoint was placebo-adjusted percentage change from baseline in non-HDL cholesterol at 24 weeks. Secondary endpoints included placebo-adjusted percentage changes from baseline in triglycerides, LDL cholesterol, apo B, and ANGPTL3.
Vupanorsen resulted in significant decreases from baseline over placebo in non-HDL cholesterol ranging from 22.0% in the group receiving 60 mg every 2 weeks to 27.7% in the group receiving 80 mg every 2 weeks, but there did not appear to be a dose response.
Regarding additional lipid endpoints, vupanorsen reduced triglyceride levels in a dose-dependent manner, ranging from 41.3% in the group receiving 120 mg every 4 weeks to 56.8% in the group receiving 160 mg every 2 weeks.
The effects of vupanorsen on LDL cholesterol and apo B were more modest and without a clear dose response. Vupanorsen also lowered HDL cholesterol levels at all doses studied, and there was no significant change in high-sensitivity C-reactive protein at any dose.
Liver enzymes and hepatic fat increases of concern
In terms of safety, vupanorsen treatment was linked to liver enzyme elevations; more than three-times elevations of alanine aminotransferase or aspartate aminotransferase were more common at higher total monthly doses (up to 33.3% and 44.4%, respectively). Injection site reactions were also an issue, including recall reactions at sites of previous injections when subsequent injections were given. In addition, there was a dose-related increase (up to 76%) in hepatic fat fraction.
In the Circulation paper, the researchers say it is unclear whether the increases in hepatic fat fraction and liver enzymes reflect a metabolic effect of vupanorsen specifically or an off-target effect resulting from hepatic targeting of ANGPTL3. “Regardless, these are medically meaningful findings with important safety ramifications,” they wrote.
They pointed out that, whereas the reduction in ANGPTL3 levels increased with total monthly dose of vupanorsen, there was no clear dose-response reduction in LDL cholesterol, apo B, or non-HDL cholesterol.
In comparison, evinacumab, a monoclonal antibody against ANGPTL3 that is thought to cause near-total suppression of ANGPTL3 activity, reduces apo B levels by more than 40% in adults with refractory hypercholesterolemia or homozygous familial hypercholesterolemia.
Asked why vupanorsen showed less of an effect on non-HDL cholesterol than evinacumab, Dr. Bergmark suggested that the monoclonal antibody may achieve greater inhibition of ANGPTL3. “It may be that near complete suppression is needed to obtain clinically meaningful reductions in apo B and non-HDL cholesterol. That is a speculative and simplistic explanation,” he commented.
Conversely, reductions in triglycerides with vupanorsen showed a dose-response relationship, mirroring the reduction in ANGPTL3 and consistent with the expected increases in lipoprotein lipase activity, the researchers reported.
They note that the “relatively muted effect on apo B levels” suggests that vupanorsen is primarily decreasing the triglyceride and, to a lesser extent, cholesterol content of very low-density lipoprotein cholesterol particles rather than reducing the number of such particles.
“These observations have important implications for the potential ability of this mechanism to reduce lipid-mediated cardiovascular risk, which largely appears to be a function of the number of ApoB-containing lipoproteins,” they said.
Designated discussant of the study at the ACC late-breaking session, Pradeep Natarajan, MD, director of preventive cardiology at Massachusetts General Hospital in Boston, asked Dr. Bergmark what minimum degree of non-HDL cholesterol reduction would be compelling for a new drug to be considered for wide-scale use.
Dr. Bergmark replied there was no clear to answer to that question, as it would depend on many factors, including the risk of the population and the time horizon involved. But he added: “I think a minimum of at least a 30%-40% reduction in non-HDL cholesterol would be needed for a meaningful reduction in cardiovascular risk across a variety of settings.”
The TRANSLATE-TIMI 70 study was funded by Pfizer. Dr. Bergmark is a member of the TIMI Study Group, which has received institutional grant support through Brigham and Women’s Hospital from numerous pharmaceutical companies. Dr. Bergmark also reported receiving grant support through Brigham and Women’s Hospital from Pfizer, Ionis, AstraZeneca, and Abbott Vascular and consulting/personal fees from Abiomed, CSI, Philips, Abbott Vascular, Servier, DaiichiSankyo, Janssen, and Quark.
A version of this article first appeared on Medscape.com.
An investigational drug targeting a novel cholesterol pathway has shown disappointing results in the TRANSLATE-TIMI 70 phase 2b study.
Vupanorsen is an antisense oligonucleotide targeting hepatic angiopoietin-like protein 3 (ANGPTL3), a protein that inhibits enzymes involved in the metabolism of triglyceride and cholesterol. Inhibition of ANGPTL3 is one of several novel targets for lowering triglycerides and non-HDL cholesterol.
Results of the TRANSLATE-TIMI 70 study were presented at the annual scientific sessions of the American College of Cardiology by Brian Bergmark, MD, a cardiologist at Brigham and Women’s Hospital, Boston. They were also simultaneously published online in Circulation.
“While vupanorsen significantly reduced triglycerides and non-HDL cholesterol, the reduction in non-HDL cholesterol of 22%-27% was not to a degree that was clinically meaningful for cardiovascular risk reduction, and there were also some potentially important safety issues,” Dr. Bergmark said in an interview.
Pfizer has announced that, after reviewing the results of this study, it is discontinuing development of vupanorsen and will return rights to Ionis, from which it licensed the investigational therapy in 2019.
In response to a question at an ACC press conference on whether there could be any future for the drug, Dr. Bergmark said that “the degree of lipid lowering was not as much as what had been suggested was potentially possible by acting on this pathway, and then there are the additional safety concerns. So, for the specific question of what we were looking at – cardiovascular risk reduction by impacting non-HDL cholesterol and apo [apolipoprotein] B – the modest efficacy paired with the safety concerns does not look favorable for future development of this drug.”
But he added: “Whether some other person or company wants to think about triglyceride lowering and try to find a dose that is a bit safer, that is not for me to say.”
In his ACC presentation, Dr. Bergmark explained that ANGPTL3 is a protein secreted by the liver that inhibits lipases, including lipoprotein lipase. Loss-of-function variants in ANGPTL3 are associated with lower levels of plasma lipids and a monoclonal antibody targeting ANGPTL3, evinacumab (Evkeeza, Regeneron), is approved as an intravenous infusion for the treatment of familial hypercholesterolemia. Vupanorsen is a second-generation antisense oligonucleotide targeting hepatic ANGPTL3 messenger RNA with a potential role for cardiovascular risk reduction.
A previous phase 2a study of vupanorsen in patients with hypertriglyceridemia, hepatic steatosis, and type 2 diabetes mellitus showed significant reductions in triglycerides at all doses studied, as well as reductions in non-HDL cholesterol at the highest doses (80 mg per month given by subcutaneous injection).
Dr. Bergmark noted that, because a potential cardiovascular benefit of vupanorsen would best be reflected by its effects on non-HDL cholesterol, the current TRANSLATE-TIMI 70 trial was designed to assess the effect of escalating doses of vupanorsen on non-HDL cholesterol levels in statin-treated adults with hyperlipidemia.
For the study, 286 adults with non-HDL cholesterol levels of 100 mg/dL or greater (median, 132 mg/dL) and triglyceride levels of 150-500 mg/dL (median, 216 mg/dL) who were receiving statin therapy were randomly assigned to placebo or one of seven vupanorsen dose regimens (80, 120, or 160 mg every 4 weeks or 60, 80, 120, or 160 mg every 2 weeks). All doses were given by subcutaneous injection.
The study population was said to reflect “a typical cohort intended for cardiovascular risk reduction, with type 2 diabetes in approximately one-half of patients and prevalent atherosclerotic cardiovascular disease in a substantial portion,” the researchers wrote in the published report.
The primary endpoint was placebo-adjusted percentage change from baseline in non-HDL cholesterol at 24 weeks. Secondary endpoints included placebo-adjusted percentage changes from baseline in triglycerides, LDL cholesterol, apo B, and ANGPTL3.
Vupanorsen resulted in significant decreases from baseline over placebo in non-HDL cholesterol ranging from 22.0% in the group receiving 60 mg every 2 weeks to 27.7% in the group receiving 80 mg every 2 weeks, but there did not appear to be a dose response.
Regarding additional lipid endpoints, vupanorsen reduced triglyceride levels in a dose-dependent manner, ranging from 41.3% in the group receiving 120 mg every 4 weeks to 56.8% in the group receiving 160 mg every 2 weeks.
The effects of vupanorsen on LDL cholesterol and apo B were more modest and without a clear dose response. Vupanorsen also lowered HDL cholesterol levels at all doses studied, and there was no significant change in high-sensitivity C-reactive protein at any dose.
Liver enzymes and hepatic fat increases of concern
In terms of safety, vupanorsen treatment was linked to liver enzyme elevations; more than three-times elevations of alanine aminotransferase or aspartate aminotransferase were more common at higher total monthly doses (up to 33.3% and 44.4%, respectively). Injection site reactions were also an issue, including recall reactions at sites of previous injections when subsequent injections were given. In addition, there was a dose-related increase (up to 76%) in hepatic fat fraction.
In the Circulation paper, the researchers say it is unclear whether the increases in hepatic fat fraction and liver enzymes reflect a metabolic effect of vupanorsen specifically or an off-target effect resulting from hepatic targeting of ANGPTL3. “Regardless, these are medically meaningful findings with important safety ramifications,” they wrote.
They pointed out that, whereas the reduction in ANGPTL3 levels increased with total monthly dose of vupanorsen, there was no clear dose-response reduction in LDL cholesterol, apo B, or non-HDL cholesterol.
In comparison, evinacumab, a monoclonal antibody against ANGPTL3 that is thought to cause near-total suppression of ANGPTL3 activity, reduces apo B levels by more than 40% in adults with refractory hypercholesterolemia or homozygous familial hypercholesterolemia.
Asked why vupanorsen showed less of an effect on non-HDL cholesterol than evinacumab, Dr. Bergmark suggested that the monoclonal antibody may achieve greater inhibition of ANGPTL3. “It may be that near complete suppression is needed to obtain clinically meaningful reductions in apo B and non-HDL cholesterol. That is a speculative and simplistic explanation,” he commented.
Conversely, reductions in triglycerides with vupanorsen showed a dose-response relationship, mirroring the reduction in ANGPTL3 and consistent with the expected increases in lipoprotein lipase activity, the researchers reported.
They note that the “relatively muted effect on apo B levels” suggests that vupanorsen is primarily decreasing the triglyceride and, to a lesser extent, cholesterol content of very low-density lipoprotein cholesterol particles rather than reducing the number of such particles.
“These observations have important implications for the potential ability of this mechanism to reduce lipid-mediated cardiovascular risk, which largely appears to be a function of the number of ApoB-containing lipoproteins,” they said.
Designated discussant of the study at the ACC late-breaking session, Pradeep Natarajan, MD, director of preventive cardiology at Massachusetts General Hospital in Boston, asked Dr. Bergmark what minimum degree of non-HDL cholesterol reduction would be compelling for a new drug to be considered for wide-scale use.
Dr. Bergmark replied there was no clear to answer to that question, as it would depend on many factors, including the risk of the population and the time horizon involved. But he added: “I think a minimum of at least a 30%-40% reduction in non-HDL cholesterol would be needed for a meaningful reduction in cardiovascular risk across a variety of settings.”
The TRANSLATE-TIMI 70 study was funded by Pfizer. Dr. Bergmark is a member of the TIMI Study Group, which has received institutional grant support through Brigham and Women’s Hospital from numerous pharmaceutical companies. Dr. Bergmark also reported receiving grant support through Brigham and Women’s Hospital from Pfizer, Ionis, AstraZeneca, and Abbott Vascular and consulting/personal fees from Abiomed, CSI, Philips, Abbott Vascular, Servier, DaiichiSankyo, Janssen, and Quark.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
VALOR-HCM: Novel drug may delay, avert invasive therapy in OHCM
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
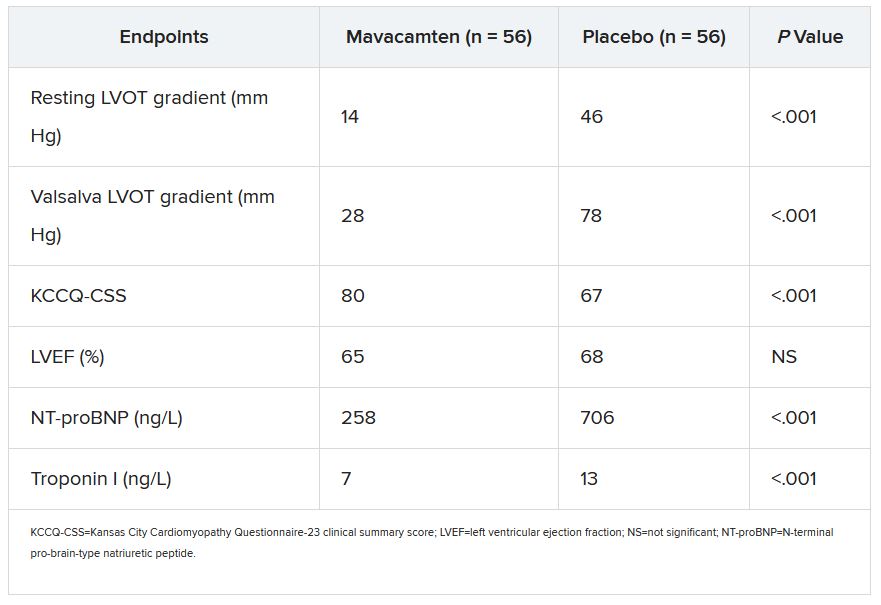
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
Treatment with a novel myosin-inhibiting agent may improve symptoms and hemodynamics enough in patients with obstructive hypertrophic cardiomyopathy (OHCM) so that they can avoid or at least delay septal reduction therapy (SRT), suggests a randomized trial of modest size and duration.
Of 112 patients with OHCM who were sick enough while receiving standard medications to qualify for SRT, those assigned to take mavacamten (MyoKardia) instead of placebo were far less likely to still be eligible for SRT 16 weeks later.
In other words, their OHCM had improved enough during therapy with mavacamten such that SRT, either surgical septal myectomy or transcatheter alcohol septal ablation, could no longer be recommended per guidelines.
Mavacamten, which lessens myocardial contractility by selective inhibition of cardiac myosin, is the first agent tested in prospective trials to appear as a viable medical option in patients with severe, symptomatic OHCM, observed principal investigator Milind Y. Desai, MD, MBA, of the Cleveland Clinic.
“There’s clearly an unmet need for noninvasive therapies, medical therapies, that work in OHCM,” he said in an interview. Mavacamten “adds to the armamentarium” of OHCM management options and may give patients with symptoms despite conventional medications an alternative to SRT, which is considered definitive but has drawbacks.
The goal of SRT is to alleviate obstruction of the left ventricular outflow tract (LVOT), but surgical SRT requires a sternotomy, with all the risks and recovery time that entails. Catheter-based alcohol septal ablation is a less common alternative for some patients with suitable anatomy, Dr. Desai noted.
But those procedures “are not uniformly available, and even when available, the outcomes are fairly heterogeneous,” he said. “The guidelines recommend that you should go to a center with a mortality rate of less than 1% with these procedures. Centers like that are very few across the world,” and procedural mortality can be much higher at centers with less SRT experience.
Dr. Desai presented the results of VALOR-HCM at the annual scientific sessions of the American College of Cardiology. Of the 56 patients assigned to mavacamten, 10 (17.9%) decided to undergo SRT by the end of the trial, or otherwise still met guideline-recommended criteria for receiving SRT, the primary endpoint. In comparison, 43 of the 56 patients (76.8%) in the control group (P < .0001) met that endpoint.
More patients receiving mavacamten improved by at least one New York Heart Association (NYHA) functional class during the trial’s 16 weeks: 63% versus 21% for those assigned to placebo. And 27% and 2%, respectively, improved by at least two NYHA classes, Dr. Desai said.
Guidelines recommend that SRT be reserved for patients in NYHA class III or IV heart failure with a resting or provoked LVOT gradient of at least 50 mm Hg.
Of note, Desai said, only two patients in each group elected to undergo SRT during the study. “The primary endpoint was driven by reduction in guideline eligibility for SRT, but 95% of patients in the study chose to continue with medical therapy.”
Speaking as a panelist after Dr. Desai’s presentation, Lynne W. Stevenson, MD, lauded the phase 3 trial’s “brave design,” which featured a highly unusual subjective primary endpoint and framed it as an advantage.
That the trial showed a significant mavacamten effect for that endpoint “answered, in one step, the question of what does this actually mean to the patient – which often takes much longer,” observed Dr. Stevenson, from Vanderbilt University, Nashville, Tenn.
Even so, she added, whether patients still qualified for SRT in the trial at least had to be supported by objective measures of LVOT gradient and NT-proBNP levels.
“My perspective is that of a cardiac surgeon who performs septal myectomies,” said John Cleveland, MD, University of Colorado at Denver, Aurora, who said he was impressed at how few patients receiving mavacamten went on to undergo SRT, while the rest were able to at least defer that decision.
Current recommendations are that patients who go to SRT “should be maximally medically treated and still symptomatic,” Dr. Cleveland observed at a press conference on VALOR-HCM. Should mavacamten be added to the list of agents to use before resorting to invasive therapy? “My answer would be yes,” he said, and patients who remain symptomatic even while receiving the myosin inhibitor and other medications should proceed to SRT.
The trial’s patients had documented OHCM, severe symptoms, and a resting or provoked LVOT gradient of at least 50 mm Hg despite maximally tolerated medications – which could include disopyramide, beta-blockers, and calcium channel blockers. About half the study population was female, and 89% were White. All had been referred for SRT.
Active therapy consisted of mavacamten initiated at 5 mg/day, with up-titrations at 8 and 12 weeks as tolerated, guided by echocardiographic left ventricular ejection fraction and LVOT gradient.
Most secondary endpoints improved significantly in patients receiving the drug, compared with placebo. They included measures of quality of life, symptom status, ventricular function, natriuretic peptides, and troponin I.
The secondary outcomes are consistent with what was observed in the EXPLORER-HCM trial, which in 2020 suggested that mavacamten could improve measures of quality of life, NYHA functional class, LVOT gradient, peak VO2, and other metrics in patients with OHCM.
Dr. Desai said mavacamten was well tolerated. “There were two patients who had a transient drop in ejection fraction to less than 50%, so the drug was temporarily discontinued, but resumed at a lower dose and they were able to complete the study.”
Dr. Stevenson commented on the “pretty quick” up-titration of mavacamten dosages in a study lasting only 4 months, which could have been a concern given the drug’s limited track record and its mechanism of action targeting contractility. “Fortunately, no serious safety signals” were observed.
Dr. Desai emphasized that mavacamten up-titrations were strictly guided by regular echocardiographic monitoring and assessment of LVOT gradients, in addition to clinical responses. And that, he said, is likely how up-titrations should be carried out if mavacamten is approved for OHCM.
VALOR-HCM was supported by MyoKardia. Dr. Desai disclosed receiving honoraria or consulting fees from Caristo Diagnostics, Medtronic, and MyoKardia. Dr. Stevenson disclosed receiving honoraria or consulting fees from Novartis; serving on a data safety monitoring board for Livanova; and other relationships with Abbott Medical, Biotronik, Boston Scientific, Bristol-Myers Squibb, Endotronic, Gore Medical, and Johnson & Johnson. Dr. Cleveland had no disclosures.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
FAME 3 subanalysis adds twist to negative primary results
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
A new subanalysis of the FAME 3 trial, which failed to show that percutaneous intervention (PCI) guided by fractional flow reserve (FFR) is noninferior to coronary artery bypass grafting (CABG) for treating three-vessel coronary artery disease, has associated PCI with early quality of life (QOL) advantages, according to findings presented at the annual scientific sessions of the American College of Cardiology.
Despite a modestly greater risk of major adverse cardiac events (MACE) at the end of 12 months’ follow-up among those treated with FFR-guided PCI, the greater QOL early after the procedure might be relevant to patients weighing these options, according to Frederik M. Zimmerman, MD, of Catharina Hospital in Eindhoven, the Netherlands.
“FFR-guided PCI results in a faster improvement in quality of life than CABG during the first year after revascularization, and it improved working status in patients younger than 65 years of age,” Dr. Zimmermann said.
The primary results of FAME 3 were presented at the 2021 Transcatheter Cardiovascular Therapeutics annual meeting by lead author William F. Fearon, MD, of Stanford (Calif.) University and published simultaneously in the New England Journal of Medicine.
Rather than confirming the hypothesis that FFR-guided PCI is comparable with CABG for the primary composite MACE outcome death from any cause, myocardial infarction, stroke, or revascularization, the incidence of MACE at 12 months was 10.6% in those randomized to PCI and 6.9% in the group assigned to CABG.
This translated into a hazard ratio for MACE of 1.5, signifying a 50% increase in risk for FFR-guided PCI relative to CABG for the primary outcome, a difference that negated the study definition of noninferiority (P = .35).
In this new health-related subanalysis, which was published simultaneously with his ACC presentation, the groups were compared over 12 months for QOL as measured with European Quality of Life–5 dimensions (EQ-5D) scale, angina as measured with the Canadian Cardiovascular Classification (CCC) system, and employment.
Outcomes data available in >85% of patients
Of the 1,500 patients enrolled and randomized in FAME 3 (757 to FFR-guided PCI and 743 to CABG), this health outcomes subanalysis was performed with complete data at 12 months from 89% of those in the PCI group and 88% of those in the CABG group.
Ultimately, the study did not show differences in any of these measures at the end of 12 months, but there were significant differences in QOL and employment at earlier time points. In particular, the significantly different (P < .001) trajectory for QOL improvement at 1 and 6 months favored FFR-guided PCI whether evaluated with the EQ-5D instrument or an EQ visual analog scale.
Rates of angina defined by as CCC class of at least 2 were low after revascularization in both arms of the study, negating any opportunity for differences, but patients aged younger than 65 years were almost twice as likely to have returned to full- or part-time work 1 month after revascularization (60.2% vs. 33.1%), and they remained at higher odds for working at 12 months (68.1% vs. 57.4%).
In patients aged older than 65 years, return-to-work rates did not differ significantly at any time point.
These results suggest potentially clinically meaningful early advantages for FFR-guided PCI, but some experts questioned the rationale for reporting positive secondary findings from a negative trial.
“This subanalysis is curious,” said Allen Jeremias, MD, director of interventional cardiology research, Saint Francis Hospital, Roslyn, N.Y. He pointed out that reporting these data is an anomaly.
Subanalyses uncommon in negative trials
“CABG was found to be better, so why look at QOL,” said Dr. Jeremias, who was an ACC-invited expert to discuss the results. However, he went on to say, “this could be an exception to the rule.”
The reason, according to Dr. Jeremias, is that the absolute difference at 12 months between FFR-guided PCI and CABG for the MACE events of greatest concern – death, MI, or stroke – was only about 2% greater in the FFR-guided PCI group (7.3% vs. 5.2%). The biggest contributor to the difference in MACE in FAME 3 at 12 months was the higher rate of repeat revascularization (5.9% vs. 3.9%).
Moreover, patients randomized to FFR-guided PCI had lower rates of many adverse events. This included risk of bleeding (1.6% vs. 3.8%; P = .009 as defined by type ≥3 Bleeding Academic Research Consortium , acute kidney injury (0.1% vs. 0.9%; P = .04), atrial fibrillation (2.4% vs. 14.1%; P < .001) and rehospitalization within 30 days (5.5% vs. 10.2%; P < .001).
In the context of a modest increase in risk of MACE and the lower rate of several important treatment-related adverse events, the QOL advantages identified in this subanalysis “might be a reasonable topic for patient-shared decision-making,” Dr. Jeremias suggested.
New data might inform patient decision-making
He granted the possibility that well-informed patients might accept the modestly increased risk of MACE for one or more of the outcomes, such as a higher likelihood of an early return to work, that favored FFR-guided PCI.
This is the point of this subanalysis, agreed Dr. Zimmermann.
“It is all about shared decision-making,” he said. Also emphasizing that the negative trial endpoint of FAME 3 “was driven largely by an increased risk of revascularization,” he believes that these new data might be a basis for discussions with patients weighing relative risks and benefits.
There are more data to come, according to Dr. Zimmermann, who said that follow-up of up to 5 years is planned. The 3-year data will be made available in 2023.
Dr. Zimmermann reported no potential conflicts of interest. Dr. Jeremias reported financial relationships with Abbott, ACIST, Boston Scientific, and Volcano. The investigator-initiated trial received research grants from Abbott Vascular and Medtronic.
FROM ACC 2021
POISE-3 backs wider use of tranexamic acid in noncardiac surgery
The antifibrinolytic tranexamic acid (TXA) reduced serious bleeding without a significant effect on major vascular outcomes in patients undergoing noncardiac surgery at risk for these complications in the POISE-3 trial.
TXA cut the primary efficacy outcome of life-threatening, major, and critical organ bleeding at 30 days by 24% compared with placebo (9.1% vs. 11.7%; hazard ratio [HR], 0.76; P < .0001).
The primary safety outcome of myocardial injury after noncardiac surgery (MINS), nonhemorrhagic stroke, peripheral arterial thrombosis, and symptomatic proximal venous thromboembolism (VTE) at 30 days occurred in 14.2% vs.. 13.9% of patients, respectively (HR, 1.023). This failed, however, to meet the study›s threshold to prove TXA noninferior to placebo (one-sided P = .044).
There was no increased risk for death or stroke with TXA, according to results published April 2 in the New England Journal of Medicine.
Principal investigator P.J. Devereaux, MD, PhD, Population Health Research Institute and McMaster University, Hamilton, Ontario, Canada, pointed out that there is only a 4.4% probability that the composite vascular outcome hazard ratio was above the noninferiority margin and that just 10 events separated the two groups (649 vs.. 639).
“Healthcare providers and patients will have to weigh a clear beneficial reduction in the composite bleeding outcome, which is an absolute difference of 2.7%, a result that was highly statistically significant, versus a low probability of a small increase in risk of the composite vascular endpoint, with an absolute difference of 0.3%,” a nonsignificant result, Dr. Devereaux said during the formal presentation of the results at the hybrid annual scientific sessions of the American College of Cardiology.
The findings, he said, should also be put in the context that 300 million adults have a major surgery each year worldwide and most don’t receive TXA. At the same time, there’s an annual global shortage of 30 million blood product units, and surgical bleeding accounts for up to 40% of all transfusions.
“POISE-3 identifies that use of TXA could avoid upwards of 8 million bleeding events resulting in transfusion on an annual basis, indicating potential for large public health and clinical benefit if TXA become standard practice in noncardiac surgery,” Dr. Devereaux said during the late-breaking trial session.
TXA is indicated for heavy menstrual bleeding and hemophilia and has been used in cardiac surgery, but it is increasingly being used in noncardiac surgeries. As previously reported, POISE showed that the beta-blocker metoprolol lowered the risk for myocardial infarction (MI) but increased the risk for severe stroke and overall death, whereas in POISE-2, perioperative low-dose aspirin lowered the risk for MI but was linked to more major bleeding.
The cumulative data have not shown an increased risk for thrombotic events in other settings, Dr. Devereaux told this news organization.
“I’m a cardiologist, and I think that we’ve been guilty at times of always only focusing on the thrombotic side of the equation and ignoring that bleeding is a very important aspect of the circulatory system,” he said. “And I think this shows for the first time clear unequivocal evidence that there’s a cheap, very encouraging, safe way to prevent this.”
“An important point is that if you can give tranexamic acid and prevent bleeding in your cardiac patients having noncardiac surgery, then you can prevent the delay of reinitiating their anticoagulants and their antiplatelets after surgery and getting them back on the medications that are important for them to prevent their cardiovascular event,” Dr. Devereaux added.
Discussant Michael J. Mack, MD, commented that TXA, widely used in cardiac surgery, is an old, inexpensive drug that “should be more widely used in noncardiac surgery.” Dr. Mack, from Baylor Scott & White Health, Dallas, added that he would limit it to major noncardiac surgery.
International trial
PeriOperative ISchemic Evaluation-3 (POISE-3) investigators at 114 hospitals in 22 countries (including countries in North and South America, Europe, and Africa; Russia; India; and Australia) randomly assigned 9,535 patients, aged 45 years or older, with or at risk for cardiovascular and bleeding complications to receive a TXA 1-g intravenous bolus or placebo at the start and end of inpatient noncardiac surgery.
Patients taking at least one long-term antihypertensive medication were also randomly assigned to a perioperative hypotension- or hypertension-avoidance strategy, which differ in the use of antihypertensives on the morning of surgery and the first 2 days after surgery, and in the target mean arterial pressure during surgery. Results from these cohorts will be presented in a separate session on April 4.
The study had planned to enroll 10,000 patients but was stopped early by the steering committee because of financial constraints resulting from slow enrollment during the pandemic. The decision was made without knowledge of the trial results but with knowledge that aggregate composite bleeding and vascular outcomes were higher than originally estimated, Dr. Devereaux noted.
Among all participants, the mean age was 70 years, 56% were male, almost a third had coronary artery disease, 15% had peripheral artery disease, and 8% had a prior stroke. About 80% were undergoing major surgery. Adherence to the study medications was 96.3% in both groups.
Secondary bleeding outcomes were lower in the TXA and placebo groups, including bleeding independently associated with mortality after surgery (8.7% vs. 11.3%), life-threatening bleeding (1.6% vs. 1.7%), major bleeding (7.6% vs. 10.4%), and critical organ bleeding (0.3% vs. 0.4%).
Importantly, the TXA group had significantly lower rates of International Society on Thrombosis and Haemostasis major bleeding (6.6% vs. 8.7%; P = .0001) and the need for transfusion of 1 or more units of packed red blood cells (9.4% vs. 12.0%; P <.0001), Dr. Devereaux noted.
In terms of secondary vascular outcomes, there were no significant differences between the TXA and placebo groups in rates of MINS (12.8% vs. 12.6%), MINS not fulfilling definition of MI (both 11.5%), MI (1.4% vs. 1.1%), and the net risk-benefit outcome (a composite of vascular death and nonfatal life-threatening, major, or critical organ bleeding, MINS, stroke, peripheral arterial thrombosis, and symptomatic proximal VTE; 20.7% vs. 21.9%).
The two groups had similar rates of all-cause (1.1% vs. 1.2%) and vascular (0.5% vs. 0.6%) mortality.
There also were no significant differences in other tertiary outcomes, such as acute kidney injury (14.1% vs. 13.7%), rehospitalization for vascular reasons (1.8% vs. 1.6%), or seizures (0.2% vs. <0.1%). The latter has been a concern, with the risk reported to increase with higher doses.
Subgroup analyses
Preplanned subgroup analyses showed a benefit for TXA over placebo for the primary efficacy outcome in orthopedic and nonorthopedic surgery and in patients with hemoglobin level below 120 g/L or 120 g/L or higher, with an estimated glomerular filtration rate less than 45 mL/min/1.73 m 2 or 45 mL/min/1.73 m 2 or higher, or with an N-terminal pro– B-type natriuretic peptide level below 200 ng/L or 200 ng/L or higher.
For the primary safety outcome, the benefit favored placebo but the interaction was not statistically significant for any of the four subgroups.
A post hoc subgroup analysis also showed similar results across the major categories of surgery, including general, vascular, urologic, and gynecologic, Dr. Devereaux told this news organization.
Although TXA is commonly used in orthopedic procedures, Dr. Devereaux noted, in other types of surgeries, “it’s not used at all.” But because TXA “is so cheap, and we can apply it to a broad population, even at an economic level it looks like it’s a winner to give to almost all patients having noncardiac surgery.”
The team also recently published a risk prediction tool that can help estimate a patient’s baseline risk for bleeding.
“So just using a model, which will bring together the patient’s type of surgery and their risk factors, you can look to see, okay, this is enough risk of bleeding, I’m just going to give tranexamic acid,” he said. “We will also be doing economic analyses because blood is also not cheap.”
The study was funded by the Canadian Institutes of Health Research, National Health and Medical Research Council (Australia), and the Research Grant Council (Hong Kong). Dr. Devereaux reports research/research grants from Abbott Diagnostics, Philips Healthcare, Roche Diagnostics, and Siemens. Dr. Mack reports receiving research grants from Abbott Vascular, Edwards Lifesciences, and Medtronic.
A version of this article first appeared on Medscape.com.
The antifibrinolytic tranexamic acid (TXA) reduced serious bleeding without a significant effect on major vascular outcomes in patients undergoing noncardiac surgery at risk for these complications in the POISE-3 trial.
TXA cut the primary efficacy outcome of life-threatening, major, and critical organ bleeding at 30 days by 24% compared with placebo (9.1% vs. 11.7%; hazard ratio [HR], 0.76; P < .0001).
The primary safety outcome of myocardial injury after noncardiac surgery (MINS), nonhemorrhagic stroke, peripheral arterial thrombosis, and symptomatic proximal venous thromboembolism (VTE) at 30 days occurred in 14.2% vs.. 13.9% of patients, respectively (HR, 1.023). This failed, however, to meet the study›s threshold to prove TXA noninferior to placebo (one-sided P = .044).
There was no increased risk for death or stroke with TXA, according to results published April 2 in the New England Journal of Medicine.
Principal investigator P.J. Devereaux, MD, PhD, Population Health Research Institute and McMaster University, Hamilton, Ontario, Canada, pointed out that there is only a 4.4% probability that the composite vascular outcome hazard ratio was above the noninferiority margin and that just 10 events separated the two groups (649 vs.. 639).
“Healthcare providers and patients will have to weigh a clear beneficial reduction in the composite bleeding outcome, which is an absolute difference of 2.7%, a result that was highly statistically significant, versus a low probability of a small increase in risk of the composite vascular endpoint, with an absolute difference of 0.3%,” a nonsignificant result, Dr. Devereaux said during the formal presentation of the results at the hybrid annual scientific sessions of the American College of Cardiology.
The findings, he said, should also be put in the context that 300 million adults have a major surgery each year worldwide and most don’t receive TXA. At the same time, there’s an annual global shortage of 30 million blood product units, and surgical bleeding accounts for up to 40% of all transfusions.
“POISE-3 identifies that use of TXA could avoid upwards of 8 million bleeding events resulting in transfusion on an annual basis, indicating potential for large public health and clinical benefit if TXA become standard practice in noncardiac surgery,” Dr. Devereaux said during the late-breaking trial session.
TXA is indicated for heavy menstrual bleeding and hemophilia and has been used in cardiac surgery, but it is increasingly being used in noncardiac surgeries. As previously reported, POISE showed that the beta-blocker metoprolol lowered the risk for myocardial infarction (MI) but increased the risk for severe stroke and overall death, whereas in POISE-2, perioperative low-dose aspirin lowered the risk for MI but was linked to more major bleeding.
The cumulative data have not shown an increased risk for thrombotic events in other settings, Dr. Devereaux told this news organization.
“I’m a cardiologist, and I think that we’ve been guilty at times of always only focusing on the thrombotic side of the equation and ignoring that bleeding is a very important aspect of the circulatory system,” he said. “And I think this shows for the first time clear unequivocal evidence that there’s a cheap, very encouraging, safe way to prevent this.”
“An important point is that if you can give tranexamic acid and prevent bleeding in your cardiac patients having noncardiac surgery, then you can prevent the delay of reinitiating their anticoagulants and their antiplatelets after surgery and getting them back on the medications that are important for them to prevent their cardiovascular event,” Dr. Devereaux added.
Discussant Michael J. Mack, MD, commented that TXA, widely used in cardiac surgery, is an old, inexpensive drug that “should be more widely used in noncardiac surgery.” Dr. Mack, from Baylor Scott & White Health, Dallas, added that he would limit it to major noncardiac surgery.
International trial
PeriOperative ISchemic Evaluation-3 (POISE-3) investigators at 114 hospitals in 22 countries (including countries in North and South America, Europe, and Africa; Russia; India; and Australia) randomly assigned 9,535 patients, aged 45 years or older, with or at risk for cardiovascular and bleeding complications to receive a TXA 1-g intravenous bolus or placebo at the start and end of inpatient noncardiac surgery.
Patients taking at least one long-term antihypertensive medication were also randomly assigned to a perioperative hypotension- or hypertension-avoidance strategy, which differ in the use of antihypertensives on the morning of surgery and the first 2 days after surgery, and in the target mean arterial pressure during surgery. Results from these cohorts will be presented in a separate session on April 4.
The study had planned to enroll 10,000 patients but was stopped early by the steering committee because of financial constraints resulting from slow enrollment during the pandemic. The decision was made without knowledge of the trial results but with knowledge that aggregate composite bleeding and vascular outcomes were higher than originally estimated, Dr. Devereaux noted.
Among all participants, the mean age was 70 years, 56% were male, almost a third had coronary artery disease, 15% had peripheral artery disease, and 8% had a prior stroke. About 80% were undergoing major surgery. Adherence to the study medications was 96.3% in both groups.
Secondary bleeding outcomes were lower in the TXA and placebo groups, including bleeding independently associated with mortality after surgery (8.7% vs. 11.3%), life-threatening bleeding (1.6% vs. 1.7%), major bleeding (7.6% vs. 10.4%), and critical organ bleeding (0.3% vs. 0.4%).
Importantly, the TXA group had significantly lower rates of International Society on Thrombosis and Haemostasis major bleeding (6.6% vs. 8.7%; P = .0001) and the need for transfusion of 1 or more units of packed red blood cells (9.4% vs. 12.0%; P <.0001), Dr. Devereaux noted.
In terms of secondary vascular outcomes, there were no significant differences between the TXA and placebo groups in rates of MINS (12.8% vs. 12.6%), MINS not fulfilling definition of MI (both 11.5%), MI (1.4% vs. 1.1%), and the net risk-benefit outcome (a composite of vascular death and nonfatal life-threatening, major, or critical organ bleeding, MINS, stroke, peripheral arterial thrombosis, and symptomatic proximal VTE; 20.7% vs. 21.9%).
The two groups had similar rates of all-cause (1.1% vs. 1.2%) and vascular (0.5% vs. 0.6%) mortality.
There also were no significant differences in other tertiary outcomes, such as acute kidney injury (14.1% vs. 13.7%), rehospitalization for vascular reasons (1.8% vs. 1.6%), or seizures (0.2% vs. <0.1%). The latter has been a concern, with the risk reported to increase with higher doses.
Subgroup analyses
Preplanned subgroup analyses showed a benefit for TXA over placebo for the primary efficacy outcome in orthopedic and nonorthopedic surgery and in patients with hemoglobin level below 120 g/L or 120 g/L or higher, with an estimated glomerular filtration rate less than 45 mL/min/1.73 m 2 or 45 mL/min/1.73 m 2 or higher, or with an N-terminal pro– B-type natriuretic peptide level below 200 ng/L or 200 ng/L or higher.
For the primary safety outcome, the benefit favored placebo but the interaction was not statistically significant for any of the four subgroups.
A post hoc subgroup analysis also showed similar results across the major categories of surgery, including general, vascular, urologic, and gynecologic, Dr. Devereaux told this news organization.
Although TXA is commonly used in orthopedic procedures, Dr. Devereaux noted, in other types of surgeries, “it’s not used at all.” But because TXA “is so cheap, and we can apply it to a broad population, even at an economic level it looks like it’s a winner to give to almost all patients having noncardiac surgery.”
The team also recently published a risk prediction tool that can help estimate a patient’s baseline risk for bleeding.
“So just using a model, which will bring together the patient’s type of surgery and their risk factors, you can look to see, okay, this is enough risk of bleeding, I’m just going to give tranexamic acid,” he said. “We will also be doing economic analyses because blood is also not cheap.”
The study was funded by the Canadian Institutes of Health Research, National Health and Medical Research Council (Australia), and the Research Grant Council (Hong Kong). Dr. Devereaux reports research/research grants from Abbott Diagnostics, Philips Healthcare, Roche Diagnostics, and Siemens. Dr. Mack reports receiving research grants from Abbott Vascular, Edwards Lifesciences, and Medtronic.
A version of this article first appeared on Medscape.com.
The antifibrinolytic tranexamic acid (TXA) reduced serious bleeding without a significant effect on major vascular outcomes in patients undergoing noncardiac surgery at risk for these complications in the POISE-3 trial.
TXA cut the primary efficacy outcome of life-threatening, major, and critical organ bleeding at 30 days by 24% compared with placebo (9.1% vs. 11.7%; hazard ratio [HR], 0.76; P < .0001).
The primary safety outcome of myocardial injury after noncardiac surgery (MINS), nonhemorrhagic stroke, peripheral arterial thrombosis, and symptomatic proximal venous thromboembolism (VTE) at 30 days occurred in 14.2% vs.. 13.9% of patients, respectively (HR, 1.023). This failed, however, to meet the study›s threshold to prove TXA noninferior to placebo (one-sided P = .044).
There was no increased risk for death or stroke with TXA, according to results published April 2 in the New England Journal of Medicine.
Principal investigator P.J. Devereaux, MD, PhD, Population Health Research Institute and McMaster University, Hamilton, Ontario, Canada, pointed out that there is only a 4.4% probability that the composite vascular outcome hazard ratio was above the noninferiority margin and that just 10 events separated the two groups (649 vs.. 639).
“Healthcare providers and patients will have to weigh a clear beneficial reduction in the composite bleeding outcome, which is an absolute difference of 2.7%, a result that was highly statistically significant, versus a low probability of a small increase in risk of the composite vascular endpoint, with an absolute difference of 0.3%,” a nonsignificant result, Dr. Devereaux said during the formal presentation of the results at the hybrid annual scientific sessions of the American College of Cardiology.
The findings, he said, should also be put in the context that 300 million adults have a major surgery each year worldwide and most don’t receive TXA. At the same time, there’s an annual global shortage of 30 million blood product units, and surgical bleeding accounts for up to 40% of all transfusions.
“POISE-3 identifies that use of TXA could avoid upwards of 8 million bleeding events resulting in transfusion on an annual basis, indicating potential for large public health and clinical benefit if TXA become standard practice in noncardiac surgery,” Dr. Devereaux said during the late-breaking trial session.
TXA is indicated for heavy menstrual bleeding and hemophilia and has been used in cardiac surgery, but it is increasingly being used in noncardiac surgeries. As previously reported, POISE showed that the beta-blocker metoprolol lowered the risk for myocardial infarction (MI) but increased the risk for severe stroke and overall death, whereas in POISE-2, perioperative low-dose aspirin lowered the risk for MI but was linked to more major bleeding.
The cumulative data have not shown an increased risk for thrombotic events in other settings, Dr. Devereaux told this news organization.
“I’m a cardiologist, and I think that we’ve been guilty at times of always only focusing on the thrombotic side of the equation and ignoring that bleeding is a very important aspect of the circulatory system,” he said. “And I think this shows for the first time clear unequivocal evidence that there’s a cheap, very encouraging, safe way to prevent this.”
“An important point is that if you can give tranexamic acid and prevent bleeding in your cardiac patients having noncardiac surgery, then you can prevent the delay of reinitiating their anticoagulants and their antiplatelets after surgery and getting them back on the medications that are important for them to prevent their cardiovascular event,” Dr. Devereaux added.
Discussant Michael J. Mack, MD, commented that TXA, widely used in cardiac surgery, is an old, inexpensive drug that “should be more widely used in noncardiac surgery.” Dr. Mack, from Baylor Scott & White Health, Dallas, added that he would limit it to major noncardiac surgery.
International trial
PeriOperative ISchemic Evaluation-3 (POISE-3) investigators at 114 hospitals in 22 countries (including countries in North and South America, Europe, and Africa; Russia; India; and Australia) randomly assigned 9,535 patients, aged 45 years or older, with or at risk for cardiovascular and bleeding complications to receive a TXA 1-g intravenous bolus or placebo at the start and end of inpatient noncardiac surgery.
Patients taking at least one long-term antihypertensive medication were also randomly assigned to a perioperative hypotension- or hypertension-avoidance strategy, which differ in the use of antihypertensives on the morning of surgery and the first 2 days after surgery, and in the target mean arterial pressure during surgery. Results from these cohorts will be presented in a separate session on April 4.
The study had planned to enroll 10,000 patients but was stopped early by the steering committee because of financial constraints resulting from slow enrollment during the pandemic. The decision was made without knowledge of the trial results but with knowledge that aggregate composite bleeding and vascular outcomes were higher than originally estimated, Dr. Devereaux noted.
Among all participants, the mean age was 70 years, 56% were male, almost a third had coronary artery disease, 15% had peripheral artery disease, and 8% had a prior stroke. About 80% were undergoing major surgery. Adherence to the study medications was 96.3% in both groups.
Secondary bleeding outcomes were lower in the TXA and placebo groups, including bleeding independently associated with mortality after surgery (8.7% vs. 11.3%), life-threatening bleeding (1.6% vs. 1.7%), major bleeding (7.6% vs. 10.4%), and critical organ bleeding (0.3% vs. 0.4%).
Importantly, the TXA group had significantly lower rates of International Society on Thrombosis and Haemostasis major bleeding (6.6% vs. 8.7%; P = .0001) and the need for transfusion of 1 or more units of packed red blood cells (9.4% vs. 12.0%; P <.0001), Dr. Devereaux noted.
In terms of secondary vascular outcomes, there were no significant differences between the TXA and placebo groups in rates of MINS (12.8% vs. 12.6%), MINS not fulfilling definition of MI (both 11.5%), MI (1.4% vs. 1.1%), and the net risk-benefit outcome (a composite of vascular death and nonfatal life-threatening, major, or critical organ bleeding, MINS, stroke, peripheral arterial thrombosis, and symptomatic proximal VTE; 20.7% vs. 21.9%).
The two groups had similar rates of all-cause (1.1% vs. 1.2%) and vascular (0.5% vs. 0.6%) mortality.
There also were no significant differences in other tertiary outcomes, such as acute kidney injury (14.1% vs. 13.7%), rehospitalization for vascular reasons (1.8% vs. 1.6%), or seizures (0.2% vs. <0.1%). The latter has been a concern, with the risk reported to increase with higher doses.
Subgroup analyses
Preplanned subgroup analyses showed a benefit for TXA over placebo for the primary efficacy outcome in orthopedic and nonorthopedic surgery and in patients with hemoglobin level below 120 g/L or 120 g/L or higher, with an estimated glomerular filtration rate less than 45 mL/min/1.73 m 2 or 45 mL/min/1.73 m 2 or higher, or with an N-terminal pro– B-type natriuretic peptide level below 200 ng/L or 200 ng/L or higher.
For the primary safety outcome, the benefit favored placebo but the interaction was not statistically significant for any of the four subgroups.
A post hoc subgroup analysis also showed similar results across the major categories of surgery, including general, vascular, urologic, and gynecologic, Dr. Devereaux told this news organization.
Although TXA is commonly used in orthopedic procedures, Dr. Devereaux noted, in other types of surgeries, “it’s not used at all.” But because TXA “is so cheap, and we can apply it to a broad population, even at an economic level it looks like it’s a winner to give to almost all patients having noncardiac surgery.”
The team also recently published a risk prediction tool that can help estimate a patient’s baseline risk for bleeding.
“So just using a model, which will bring together the patient’s type of surgery and their risk factors, you can look to see, okay, this is enough risk of bleeding, I’m just going to give tranexamic acid,” he said. “We will also be doing economic analyses because blood is also not cheap.”
The study was funded by the Canadian Institutes of Health Research, National Health and Medical Research Council (Australia), and the Research Grant Council (Hong Kong). Dr. Devereaux reports research/research grants from Abbott Diagnostics, Philips Healthcare, Roche Diagnostics, and Siemens. Dr. Mack reports receiving research grants from Abbott Vascular, Edwards Lifesciences, and Medtronic.
A version of this article first appeared on Medscape.com.
FROM ACC 2022
SCORED: Sotagliflozin shows robust MACE benefit
WASHINGTON – Results from new analyses further fleshed out the potent effect by the investigational SGLT1&2 inhibitor sotagliflozin on major cardiovascular adverse events in patients with type 2 diabetes, chronic kidney disease, and at high risk for cardiovascular disease in the SCORED trial that randomized more than 10,000 patients.
In prespecified, secondary analyses of the SCORED results, treatment with sotagliflozin during a median of 16 months was linked to a significant 21% risk reduction relative to placebo for the combined incidence of total major adverse cardiovascular events (MACE), which included cardiovascular death, first and recurrent episodes of nonfatal MI, and nonfatal stroke among the 5,144 randomized patients who entered the trial with a history of cardiovascular disease (CVD), Deepak L. Bhatt, MD, said at the annual scientific sessions of the American College of Cardiology.

Among the 5,440 patients in the study who did not have a history of CVD (although they did have at least one major risk factor or at least two minor risk factors), treatment with sotagliflozin was linked to a significant 26% relative risk reduction in total MACE events.
Part of these overall MACE benefits resulted from similar improvements from sotagliflozin treatment on the individual outcomes of total nonfatal MI and total nonfatal strokes. Treatment with sotagliflozin cut these MIs by a significant 31% in patients with a history of CVD relative to patients who received placebo, and by a relative 34% in those without a CVD event in their history, a difference compared with placebo that fell short of significance, said Dr. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Health, both in Boston.
Treatment with sotagliflozin also cut total nonfatal strokes by 31% relative to placebo in patients with a history of CVD, and by a relative 38% in those without a CVD history. Both differences fell short of significance.
An early MACE benefit and a stroke benefit
“This stroke benefit has not been clearly seen” with any agent from the closely related sodium-glucose cotransport-2 (SGLT2) inhibitor class, and “the MACE benefit appeared very early,” within 3 months from the start of sotagliflozin treatment, “which may be because of the SGLT1 inhibition,” Dr. Bhatt said during his report.
The SGLT1 receptor is the primary mechanism cells in the gut use to absorb glucose and galactose in the human gastrointestinal tract, Dr. Bhatt explained, while the SGLT2 receptor appears on kidney cells and is the major player in the reabsorption of filtered glucose. The SGLT2 inhibitor class includes the agents canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance), while sotagliflozin inhibits both SGLT1 and SGLT2.
Main results from SCORED appeared in a report first released in late 2020, and showed that for the study’s primary endpoint treatment with sotagliflozin linked with a significant 26% relative risk reduction for the composite of cardiovascular deaths, hospitalizations for heart failure, and urgent visits for heart failure (N Engl J Med. 2021 Jan 14;384[2]:129-39). Patient follow-up in SCORED was not as long as originally planned when the study stopped early due to a loss of funding from a sponsor that was triggered by the COVID-19 pandemic.
MACE results ‘heterogeneous’ from SGLT2 inhibitors
Sotagliflozin and agents from the SGLT2 inhibitor class “have been consistent” in their benefits for reducing cardiovascular death and hospitalization for heart failure, but for MACE, the results from the SGLT2 inhibitors “have been more heterogeneous,” and the effect of sotagliflozin on MACE “were different in SCORED,” commented Michelle L. O’Donoghue, MD, MPH, a cardiologist at Brigham and Women’s Hospital in Boston who was not involved with this work.

“The results suggest a benefit [from sotagliflozin] on atherosclerotic events, which could be a potential advantage” compared with the SGLT2 inhibitors, “but the heterogeneity of this effect” among these agents means that more confirmatory data are needed for sotagliflozin, Dr. O’Donoghue said in an interview.
“There is a lot of enthusiasm for the concept” of combined inhibition of the SGLT1 and 2 receptors, and if more evidence for unique benefits of this effect accumulate “it may lead to increased enthusiasm for sotagliflozin,” she said. “A lot will also depend on pricing decisions” for sotagliflozin, if it receives U.S. marketing approval from the Food and Drug Administration. Decisions about which agent from the SGLT2 inhibitor class to prescribe “are often being made based on price right now,” Dr. O’Donoghue said.
Lexicon Pharmaceuticals, the company developing sotagliflozin, has announced plans to resubmit its new drug application for sotagliflozin to the FDA later in 2022, with the agency’s approval decision likely occurring late in 2022 or sometime during 2023. In February, the company withdrew its December 2021 application to correct a “technical issue” it had found.
An additional analysis reported by Dr. Bhatt used combined data from SCORED as well as several additional randomized trials of sotagliflozin involving a total of more than 20,000 patients that showed a significant 21% reduction in the incidence of MACE compared with placebo.
During his talk, Dr. Bhatt said that sotagliflozin was potentially superior to the agents that inhibit only SGLT2. In an interview, he based this tentative assessment on at least four attributes of sotagliflozin that have emerged from trial results:
- The drug’s ability to significantly reduce MACE and to have this effect apparent within a few months of treatment onset;
- The significantly reduced rate of stroke with sotagliflozin (when patients are not subdivided into those with or without a history of CVD) that has not yet been seen with any SGLT2 inhibitor;
- The ability of sotagliflozin to reduce hemoglobin A1c levels in patients with type 2 diabetes even when their estimated glomerular filtration rate is less than 30 mL/min per 1.73 m2, an effect not seen with SGLT2 inhibitors and possibly explained by sotagliflozin having an effect on gut absorption of glucose in addition to its SGLT2 inhibitory effect in the kidney;
- And the proven ability of sotagliflozin to be safe and effective when initiated in patients hospitalized for heart failure, a property that so far has only also been shown for the SGLT2 inhibitor empagliflozin in the EMPULSE trial (Nature Med. 2022 Mar;28: 568-74).
SCORED was sponsored by Sanofi and Lexicon Pharmaceuticals, the companies originally developing sotagliflozin, although with the withdrawal of Sanofi’s support, further development is now sponsored entirely by Lexicon. Dr. Bhatt received research funding from Sanofi and Lexicon that was paid to Brigham and Women’s Health, and he has been an advisor to numerous companies. Dr. O’Donoghue has been a consultant to Amgen, Janssen, and Novartis, and has received research funding from Amgen, AZ MedImmune, Intarcia, Janssen, Merck, and Novartis.
WASHINGTON – Results from new analyses further fleshed out the potent effect by the investigational SGLT1&2 inhibitor sotagliflozin on major cardiovascular adverse events in patients with type 2 diabetes, chronic kidney disease, and at high risk for cardiovascular disease in the SCORED trial that randomized more than 10,000 patients.
In prespecified, secondary analyses of the SCORED results, treatment with sotagliflozin during a median of 16 months was linked to a significant 21% risk reduction relative to placebo for the combined incidence of total major adverse cardiovascular events (MACE), which included cardiovascular death, first and recurrent episodes of nonfatal MI, and nonfatal stroke among the 5,144 randomized patients who entered the trial with a history of cardiovascular disease (CVD), Deepak L. Bhatt, MD, said at the annual scientific sessions of the American College of Cardiology.
Among the 5,440 patients in the study who did not have a history of CVD (although they did have at least one major risk factor or at least two minor risk factors), treatment with sotagliflozin was linked to a significant 26% relative risk reduction in total MACE events.
Part of these overall MACE benefits resulted from similar improvements from sotagliflozin treatment on the individual outcomes of total nonfatal MI and total nonfatal strokes. Treatment with sotagliflozin cut these MIs by a significant 31% in patients with a history of CVD relative to patients who received placebo, and by a relative 34% in those without a CVD event in their history, a difference compared with placebo that fell short of significance, said Dr. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Health, both in Boston.
Treatment with sotagliflozin also cut total nonfatal strokes by 31% relative to placebo in patients with a history of CVD, and by a relative 38% in those without a CVD history. Both differences fell short of significance.
An early MACE benefit and a stroke benefit
“This stroke benefit has not been clearly seen” with any agent from the closely related sodium-glucose cotransport-2 (SGLT2) inhibitor class, and “the MACE benefit appeared very early,” within 3 months from the start of sotagliflozin treatment, “which may be because of the SGLT1 inhibition,” Dr. Bhatt said during his report.
The SGLT1 receptor is the primary mechanism cells in the gut use to absorb glucose and galactose in the human gastrointestinal tract, Dr. Bhatt explained, while the SGLT2 receptor appears on kidney cells and is the major player in the reabsorption of filtered glucose. The SGLT2 inhibitor class includes the agents canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance), while sotagliflozin inhibits both SGLT1 and SGLT2.
Main results from SCORED appeared in a report first released in late 2020, and showed that for the study’s primary endpoint treatment with sotagliflozin linked with a significant 26% relative risk reduction for the composite of cardiovascular deaths, hospitalizations for heart failure, and urgent visits for heart failure (N Engl J Med. 2021 Jan 14;384[2]:129-39). Patient follow-up in SCORED was not as long as originally planned when the study stopped early due to a loss of funding from a sponsor that was triggered by the COVID-19 pandemic.
MACE results ‘heterogeneous’ from SGLT2 inhibitors
Sotagliflozin and agents from the SGLT2 inhibitor class “have been consistent” in their benefits for reducing cardiovascular death and hospitalization for heart failure, but for MACE, the results from the SGLT2 inhibitors “have been more heterogeneous,” and the effect of sotagliflozin on MACE “were different in SCORED,” commented Michelle L. O’Donoghue, MD, MPH, a cardiologist at Brigham and Women’s Hospital in Boston who was not involved with this work.
“The results suggest a benefit [from sotagliflozin] on atherosclerotic events, which could be a potential advantage” compared with the SGLT2 inhibitors, “but the heterogeneity of this effect” among these agents means that more confirmatory data are needed for sotagliflozin, Dr. O’Donoghue said in an interview.
“There is a lot of enthusiasm for the concept” of combined inhibition of the SGLT1 and 2 receptors, and if more evidence for unique benefits of this effect accumulate “it may lead to increased enthusiasm for sotagliflozin,” she said. “A lot will also depend on pricing decisions” for sotagliflozin, if it receives U.S. marketing approval from the Food and Drug Administration. Decisions about which agent from the SGLT2 inhibitor class to prescribe “are often being made based on price right now,” Dr. O’Donoghue said.
Lexicon Pharmaceuticals, the company developing sotagliflozin, has announced plans to resubmit its new drug application for sotagliflozin to the FDA later in 2022, with the agency’s approval decision likely occurring late in 2022 or sometime during 2023. In February, the company withdrew its December 2021 application to correct a “technical issue” it had found.
An additional analysis reported by Dr. Bhatt used combined data from SCORED as well as several additional randomized trials of sotagliflozin involving a total of more than 20,000 patients that showed a significant 21% reduction in the incidence of MACE compared with placebo.
During his talk, Dr. Bhatt said that sotagliflozin was potentially superior to the agents that inhibit only SGLT2. In an interview, he based this tentative assessment on at least four attributes of sotagliflozin that have emerged from trial results:
- The drug’s ability to significantly reduce MACE and to have this effect apparent within a few months of treatment onset;
- The significantly reduced rate of stroke with sotagliflozin (when patients are not subdivided into those with or without a history of CVD) that has not yet been seen with any SGLT2 inhibitor;
- The ability of sotagliflozin to reduce hemoglobin A1c levels in patients with type 2 diabetes even when their estimated glomerular filtration rate is less than 30 mL/min per 1.73 m2, an effect not seen with SGLT2 inhibitors and possibly explained by sotagliflozin having an effect on gut absorption of glucose in addition to its SGLT2 inhibitory effect in the kidney;
- And the proven ability of sotagliflozin to be safe and effective when initiated in patients hospitalized for heart failure, a property that so far has only also been shown for the SGLT2 inhibitor empagliflozin in the EMPULSE trial (Nature Med. 2022 Mar;28: 568-74).
SCORED was sponsored by Sanofi and Lexicon Pharmaceuticals, the companies originally developing sotagliflozin, although with the withdrawal of Sanofi’s support, further development is now sponsored entirely by Lexicon. Dr. Bhatt received research funding from Sanofi and Lexicon that was paid to Brigham and Women’s Health, and he has been an advisor to numerous companies. Dr. O’Donoghue has been a consultant to Amgen, Janssen, and Novartis, and has received research funding from Amgen, AZ MedImmune, Intarcia, Janssen, Merck, and Novartis.
WASHINGTON – Results from new analyses further fleshed out the potent effect by the investigational SGLT1&2 inhibitor sotagliflozin on major cardiovascular adverse events in patients with type 2 diabetes, chronic kidney disease, and at high risk for cardiovascular disease in the SCORED trial that randomized more than 10,000 patients.
In prespecified, secondary analyses of the SCORED results, treatment with sotagliflozin during a median of 16 months was linked to a significant 21% risk reduction relative to placebo for the combined incidence of total major adverse cardiovascular events (MACE), which included cardiovascular death, first and recurrent episodes of nonfatal MI, and nonfatal stroke among the 5,144 randomized patients who entered the trial with a history of cardiovascular disease (CVD), Deepak L. Bhatt, MD, said at the annual scientific sessions of the American College of Cardiology.
Among the 5,440 patients in the study who did not have a history of CVD (although they did have at least one major risk factor or at least two minor risk factors), treatment with sotagliflozin was linked to a significant 26% relative risk reduction in total MACE events.
Part of these overall MACE benefits resulted from similar improvements from sotagliflozin treatment on the individual outcomes of total nonfatal MI and total nonfatal strokes. Treatment with sotagliflozin cut these MIs by a significant 31% in patients with a history of CVD relative to patients who received placebo, and by a relative 34% in those without a CVD event in their history, a difference compared with placebo that fell short of significance, said Dr. Bhatt, professor of medicine at Harvard Medical School and executive director of interventional cardiovascular programs at Brigham and Women’s Health, both in Boston.
Treatment with sotagliflozin also cut total nonfatal strokes by 31% relative to placebo in patients with a history of CVD, and by a relative 38% in those without a CVD history. Both differences fell short of significance.
An early MACE benefit and a stroke benefit
“This stroke benefit has not been clearly seen” with any agent from the closely related sodium-glucose cotransport-2 (SGLT2) inhibitor class, and “the MACE benefit appeared very early,” within 3 months from the start of sotagliflozin treatment, “which may be because of the SGLT1 inhibition,” Dr. Bhatt said during his report.
The SGLT1 receptor is the primary mechanism cells in the gut use to absorb glucose and galactose in the human gastrointestinal tract, Dr. Bhatt explained, while the SGLT2 receptor appears on kidney cells and is the major player in the reabsorption of filtered glucose. The SGLT2 inhibitor class includes the agents canagliflozin (Invokana), dapagliflozin (Farxiga), and empagliflozin (Jardiance), while sotagliflozin inhibits both SGLT1 and SGLT2.
Main results from SCORED appeared in a report first released in late 2020, and showed that for the study’s primary endpoint treatment with sotagliflozin linked with a significant 26% relative risk reduction for the composite of cardiovascular deaths, hospitalizations for heart failure, and urgent visits for heart failure (N Engl J Med. 2021 Jan 14;384[2]:129-39). Patient follow-up in SCORED was not as long as originally planned when the study stopped early due to a loss of funding from a sponsor that was triggered by the COVID-19 pandemic.
MACE results ‘heterogeneous’ from SGLT2 inhibitors
Sotagliflozin and agents from the SGLT2 inhibitor class “have been consistent” in their benefits for reducing cardiovascular death and hospitalization for heart failure, but for MACE, the results from the SGLT2 inhibitors “have been more heterogeneous,” and the effect of sotagliflozin on MACE “were different in SCORED,” commented Michelle L. O’Donoghue, MD, MPH, a cardiologist at Brigham and Women’s Hospital in Boston who was not involved with this work.
“The results suggest a benefit [from sotagliflozin] on atherosclerotic events, which could be a potential advantage” compared with the SGLT2 inhibitors, “but the heterogeneity of this effect” among these agents means that more confirmatory data are needed for sotagliflozin, Dr. O’Donoghue said in an interview.
“There is a lot of enthusiasm for the concept” of combined inhibition of the SGLT1 and 2 receptors, and if more evidence for unique benefits of this effect accumulate “it may lead to increased enthusiasm for sotagliflozin,” she said. “A lot will also depend on pricing decisions” for sotagliflozin, if it receives U.S. marketing approval from the Food and Drug Administration. Decisions about which agent from the SGLT2 inhibitor class to prescribe “are often being made based on price right now,” Dr. O’Donoghue said.
Lexicon Pharmaceuticals, the company developing sotagliflozin, has announced plans to resubmit its new drug application for sotagliflozin to the FDA later in 2022, with the agency’s approval decision likely occurring late in 2022 or sometime during 2023. In February, the company withdrew its December 2021 application to correct a “technical issue” it had found.
An additional analysis reported by Dr. Bhatt used combined data from SCORED as well as several additional randomized trials of sotagliflozin involving a total of more than 20,000 patients that showed a significant 21% reduction in the incidence of MACE compared with placebo.
During his talk, Dr. Bhatt said that sotagliflozin was potentially superior to the agents that inhibit only SGLT2. In an interview, he based this tentative assessment on at least four attributes of sotagliflozin that have emerged from trial results:
- The drug’s ability to significantly reduce MACE and to have this effect apparent within a few months of treatment onset;
- The significantly reduced rate of stroke with sotagliflozin (when patients are not subdivided into those with or without a history of CVD) that has not yet been seen with any SGLT2 inhibitor;
- The ability of sotagliflozin to reduce hemoglobin A1c levels in patients with type 2 diabetes even when their estimated glomerular filtration rate is less than 30 mL/min per 1.73 m2, an effect not seen with SGLT2 inhibitors and possibly explained by sotagliflozin having an effect on gut absorption of glucose in addition to its SGLT2 inhibitory effect in the kidney;
- And the proven ability of sotagliflozin to be safe and effective when initiated in patients hospitalized for heart failure, a property that so far has only also been shown for the SGLT2 inhibitor empagliflozin in the EMPULSE trial (Nature Med. 2022 Mar;28: 568-74).
SCORED was sponsored by Sanofi and Lexicon Pharmaceuticals, the companies originally developing sotagliflozin, although with the withdrawal of Sanofi’s support, further development is now sponsored entirely by Lexicon. Dr. Bhatt received research funding from Sanofi and Lexicon that was paid to Brigham and Women’s Health, and he has been an advisor to numerous companies. Dr. O’Donoghue has been a consultant to Amgen, Janssen, and Novartis, and has received research funding from Amgen, AZ MedImmune, Intarcia, Janssen, Merck, and Novartis.
AT ACC 2022
Hypertension control during pregnancy validated in major trial
Pregnant women with even mild hypertension should receive blood pressure–lowering medications to reduce the likelihood of adverse outcomes for the mother and the child, according to a large, open-label, randomized trial.
“Treating to the blood pressure goal in this study reduced the risk of adverse events associated with pregnancy but did not impair fetal growth,” Alan T. Tita, MD, PhD, associate dean for Global and Women’s Health, University of Alabama, Birmingham, reported at the annual scientific sessions of the American College of Cardiology.
The question of whether to treat chronic hypertension during pregnancy has been “an international controversy for decades,” said Dr. Tita, who led the investigator-initiated Chronic Hypertension and Pregnancy (CHAP) trial.
For the composite primary outcome of severe preeclampsia, medically indicated preterm birth at less than 35 weeks of gestation, placental abruption, or fetal/neonatal death, the treatment of hypertension versus no treatment showed a relative risk reduction of 18% (30.2% vs. 37%, (hazard ratio, 0.82; P < .001).
Small for gestational age is primary safety endpoint
An increase in preeclampsia risk in women whose fetus was small for gestational age (SGA), a theoretical consequence of reductions in arterial pressure, was not seen. The rate of SGA, defined as below the 10th percentile, was slightly higher in the treatment group (11.2% vs. 10.4%), but the difference did not approach significance (P = 0.76).
By answering this long-pending question, the CHAP data are “practice changing,” declared an ACC-invited commentator, Athena Poppas, MD, chief of cardiology and director of the Lifespan Cardiovascular Institute, Providence, R.I. She agreed that the need for treatment of mild chronic hypertension has been a dilemma for clinicians that is now acceptably resolved.
In this trial, 2,408 pregnant women with chronic mild hypertension defined as a blood pressure of 160/90 mm Hg were randomized to treatment with a goal blood pressure of less than 140/90 mm Hg or no treatment unless the blood pressure rose to at least 160/105. All women had singleton pregnancies. Enrollment before 23 weeks of gestation was required. Severe hypertension (at least 160/105 mm Hg) was an exclusion criterion, as were several comorbidities, such as kidney disease.
Combination therapy accepted for <140/90 mm Hg goal
The beta-blocker labetalol or the calcium channel blocker nifedipine as single agents were the preferred antihypertensive medications in the protocol, but other medications were permitted. To reach the blood pressure goal, the single-agent therapy was titrated to the maximum dose before starting a second agent.
After randomization the systolic and diastolic blood pressures fell in both groups, but they fell more and remained consistently lower in the active treatment group, particularly during the first 20 weeks after randomization, according to graphs displayed by Dr. Tita. Over the course of the study, the mean diastolic blood pressures were 129.5 and 132.6 mm Hg in the active treatment and control groups, respectively, while the systolic pressures were 79.1 vs. 81.5 mm Hg.
When the components of the primary outcome were evaluated separately, the greatest advantage of treatment was the reduction in the rate of severe eclampsia (23.3% vs. 29.1%; HR, 0.80: 95% confidence interval, 0.70-0.92) and preterm birth (12.2% vs. 16.7%; HR, 0.73: 95% CI, 0.60-0.89).
Across a large array of subgroups, including those with or without diabetes and those treated before or after 14 weeks of gestation, there was a consistent advantage for treatment, even if not statistically different. It is notable that 48% of patients were Black and 35% had a body mass index of at least 40. The active treatment was favored across all groups stratified by these characteristics.
Although the incidences of placental abruption (1.7% on treatment vs. 1.9% without) and fetal or neonatal death (3.5% vs. 4.3%) were lower in the active treatment group, they were uncommon events in both arms of the study. The differences did not reach statistical significance.
Maternal morbidity rates lower on treatment
Severe SGA, which was defined as below the 5th percentile, was also numerically but not significantly higher in the control arm than in the group receiving treatment (5.1% vs. 5.5%), but the incidence of composite adverse maternal events was numerically lower (2.1% vs. 2.8%). The incidences of all components of maternal morbidity, such as maternal death (0.1% vs. 0.2%) pulmonary edema (0.4% vs. 0.9%), heart failure (0.1% vs. 0.1%), and acute kidney injury (0.8% vs. 1.2%), were either lower or the same on active treatment versus no treatment.
According to Dr. Tita, who called CHAP one of the largest and most diverse studies to address the value of treating mild hypertension in pregnancy, the American College of Obstetricians and Gynecologists (ACOG) is evaluating these data for changing their current guidelines for managing hypertension during pregnancy.
“The rate of chronic hypertension during pregnancy has been rising in the United States due to the increase in the average age of pregnant women and the rising rates of obesity,” Dr. Tita commented.
“We definitely needed these data,” said Mary Norine Walsh, MD, medical director, Ascension Saint Vincent Cardiovascular Research Institute, Indianapolis. Not only has the value of treating mild hypertension been unresolved, but Dr. Walsh pointed out that the rates of maternal mortality in the United States are rising and now generally exceed those of many other developed countries.
There are several features in the design of this trial that make the results even more salient to clinical practice, according to Dr. Walsh. This includes the fact that about half of patients enrolled were on Medicaid. As a result, the study confirmed benefit in what Dr. Walsh characterized as a “vulnerable” population.
“We will be busy now to make sure that our [pregnant] patients are achieving these target blood pressures,” Dr. Walsh said. She indicated that CHAP validates the treatment target of 140/90 mm Hg as a standard of care.
The results were published in the New England Journal of Medicine simultaneously with its ACC presentation.
The trial was funded by the National Heart, Lung, and Blood Institute. Dr. Tita reports research grants from Pfizer. Dr. Walsh reports a financial relationship with EBR Systems. Dr. Poppas reports no potential conflicts of interest.
Pregnant women with even mild hypertension should receive blood pressure–lowering medications to reduce the likelihood of adverse outcomes for the mother and the child, according to a large, open-label, randomized trial.
“Treating to the blood pressure goal in this study reduced the risk of adverse events associated with pregnancy but did not impair fetal growth,” Alan T. Tita, MD, PhD, associate dean for Global and Women’s Health, University of Alabama, Birmingham, reported at the annual scientific sessions of the American College of Cardiology.
The question of whether to treat chronic hypertension during pregnancy has been “an international controversy for decades,” said Dr. Tita, who led the investigator-initiated Chronic Hypertension and Pregnancy (CHAP) trial.
For the composite primary outcome of severe preeclampsia, medically indicated preterm birth at less than 35 weeks of gestation, placental abruption, or fetal/neonatal death, the treatment of hypertension versus no treatment showed a relative risk reduction of 18% (30.2% vs. 37%, (hazard ratio, 0.82; P < .001).
Small for gestational age is primary safety endpoint
An increase in preeclampsia risk in women whose fetus was small for gestational age (SGA), a theoretical consequence of reductions in arterial pressure, was not seen. The rate of SGA, defined as below the 10th percentile, was slightly higher in the treatment group (11.2% vs. 10.4%), but the difference did not approach significance (P = 0.76).
By answering this long-pending question, the CHAP data are “practice changing,” declared an ACC-invited commentator, Athena Poppas, MD, chief of cardiology and director of the Lifespan Cardiovascular Institute, Providence, R.I. She agreed that the need for treatment of mild chronic hypertension has been a dilemma for clinicians that is now acceptably resolved.
In this trial, 2,408 pregnant women with chronic mild hypertension defined as a blood pressure of 160/90 mm Hg were randomized to treatment with a goal blood pressure of less than 140/90 mm Hg or no treatment unless the blood pressure rose to at least 160/105. All women had singleton pregnancies. Enrollment before 23 weeks of gestation was required. Severe hypertension (at least 160/105 mm Hg) was an exclusion criterion, as were several comorbidities, such as kidney disease.
Combination therapy accepted for <140/90 mm Hg goal
The beta-blocker labetalol or the calcium channel blocker nifedipine as single agents were the preferred antihypertensive medications in the protocol, but other medications were permitted. To reach the blood pressure goal, the single-agent therapy was titrated to the maximum dose before starting a second agent.
After randomization the systolic and diastolic blood pressures fell in both groups, but they fell more and remained consistently lower in the active treatment group, particularly during the first 20 weeks after randomization, according to graphs displayed by Dr. Tita. Over the course of the study, the mean diastolic blood pressures were 129.5 and 132.6 mm Hg in the active treatment and control groups, respectively, while the systolic pressures were 79.1 vs. 81.5 mm Hg.
When the components of the primary outcome were evaluated separately, the greatest advantage of treatment was the reduction in the rate of severe eclampsia (23.3% vs. 29.1%; HR, 0.80: 95% confidence interval, 0.70-0.92) and preterm birth (12.2% vs. 16.7%; HR, 0.73: 95% CI, 0.60-0.89).
Across a large array of subgroups, including those with or without diabetes and those treated before or after 14 weeks of gestation, there was a consistent advantage for treatment, even if not statistically different. It is notable that 48% of patients were Black and 35% had a body mass index of at least 40. The active treatment was favored across all groups stratified by these characteristics.
Although the incidences of placental abruption (1.7% on treatment vs. 1.9% without) and fetal or neonatal death (3.5% vs. 4.3%) were lower in the active treatment group, they were uncommon events in both arms of the study. The differences did not reach statistical significance.
Maternal morbidity rates lower on treatment
Severe SGA, which was defined as below the 5th percentile, was also numerically but not significantly higher in the control arm than in the group receiving treatment (5.1% vs. 5.5%), but the incidence of composite adverse maternal events was numerically lower (2.1% vs. 2.8%). The incidences of all components of maternal morbidity, such as maternal death (0.1% vs. 0.2%) pulmonary edema (0.4% vs. 0.9%), heart failure (0.1% vs. 0.1%), and acute kidney injury (0.8% vs. 1.2%), were either lower or the same on active treatment versus no treatment.
According to Dr. Tita, who called CHAP one of the largest and most diverse studies to address the value of treating mild hypertension in pregnancy, the American College of Obstetricians and Gynecologists (ACOG) is evaluating these data for changing their current guidelines for managing hypertension during pregnancy.
“The rate of chronic hypertension during pregnancy has been rising in the United States due to the increase in the average age of pregnant women and the rising rates of obesity,” Dr. Tita commented.
“We definitely needed these data,” said Mary Norine Walsh, MD, medical director, Ascension Saint Vincent Cardiovascular Research Institute, Indianapolis. Not only has the value of treating mild hypertension been unresolved, but Dr. Walsh pointed out that the rates of maternal mortality in the United States are rising and now generally exceed those of many other developed countries.
There are several features in the design of this trial that make the results even more salient to clinical practice, according to Dr. Walsh. This includes the fact that about half of patients enrolled were on Medicaid. As a result, the study confirmed benefit in what Dr. Walsh characterized as a “vulnerable” population.
“We will be busy now to make sure that our [pregnant] patients are achieving these target blood pressures,” Dr. Walsh said. She indicated that CHAP validates the treatment target of 140/90 mm Hg as a standard of care.
The results were published in the New England Journal of Medicine simultaneously with its ACC presentation.
The trial was funded by the National Heart, Lung, and Blood Institute. Dr. Tita reports research grants from Pfizer. Dr. Walsh reports a financial relationship with EBR Systems. Dr. Poppas reports no potential conflicts of interest.
Pregnant women with even mild hypertension should receive blood pressure–lowering medications to reduce the likelihood of adverse outcomes for the mother and the child, according to a large, open-label, randomized trial.
“Treating to the blood pressure goal in this study reduced the risk of adverse events associated with pregnancy but did not impair fetal growth,” Alan T. Tita, MD, PhD, associate dean for Global and Women’s Health, University of Alabama, Birmingham, reported at the annual scientific sessions of the American College of Cardiology.
The question of whether to treat chronic hypertension during pregnancy has been “an international controversy for decades,” said Dr. Tita, who led the investigator-initiated Chronic Hypertension and Pregnancy (CHAP) trial.
For the composite primary outcome of severe preeclampsia, medically indicated preterm birth at less than 35 weeks of gestation, placental abruption, or fetal/neonatal death, the treatment of hypertension versus no treatment showed a relative risk reduction of 18% (30.2% vs. 37%, (hazard ratio, 0.82; P < .001).
Small for gestational age is primary safety endpoint
An increase in preeclampsia risk in women whose fetus was small for gestational age (SGA), a theoretical consequence of reductions in arterial pressure, was not seen. The rate of SGA, defined as below the 10th percentile, was slightly higher in the treatment group (11.2% vs. 10.4%), but the difference did not approach significance (P = 0.76).
By answering this long-pending question, the CHAP data are “practice changing,” declared an ACC-invited commentator, Athena Poppas, MD, chief of cardiology and director of the Lifespan Cardiovascular Institute, Providence, R.I. She agreed that the need for treatment of mild chronic hypertension has been a dilemma for clinicians that is now acceptably resolved.
In this trial, 2,408 pregnant women with chronic mild hypertension defined as a blood pressure of 160/90 mm Hg were randomized to treatment with a goal blood pressure of less than 140/90 mm Hg or no treatment unless the blood pressure rose to at least 160/105. All women had singleton pregnancies. Enrollment before 23 weeks of gestation was required. Severe hypertension (at least 160/105 mm Hg) was an exclusion criterion, as were several comorbidities, such as kidney disease.
Combination therapy accepted for <140/90 mm Hg goal
The beta-blocker labetalol or the calcium channel blocker nifedipine as single agents were the preferred antihypertensive medications in the protocol, but other medications were permitted. To reach the blood pressure goal, the single-agent therapy was titrated to the maximum dose before starting a second agent.
After randomization the systolic and diastolic blood pressures fell in both groups, but they fell more and remained consistently lower in the active treatment group, particularly during the first 20 weeks after randomization, according to graphs displayed by Dr. Tita. Over the course of the study, the mean diastolic blood pressures were 129.5 and 132.6 mm Hg in the active treatment and control groups, respectively, while the systolic pressures were 79.1 vs. 81.5 mm Hg.
When the components of the primary outcome were evaluated separately, the greatest advantage of treatment was the reduction in the rate of severe eclampsia (23.3% vs. 29.1%; HR, 0.80: 95% confidence interval, 0.70-0.92) and preterm birth (12.2% vs. 16.7%; HR, 0.73: 95% CI, 0.60-0.89).
Across a large array of subgroups, including those with or without diabetes and those treated before or after 14 weeks of gestation, there was a consistent advantage for treatment, even if not statistically different. It is notable that 48% of patients were Black and 35% had a body mass index of at least 40. The active treatment was favored across all groups stratified by these characteristics.
Although the incidences of placental abruption (1.7% on treatment vs. 1.9% without) and fetal or neonatal death (3.5% vs. 4.3%) were lower in the active treatment group, they were uncommon events in both arms of the study. The differences did not reach statistical significance.
Maternal morbidity rates lower on treatment
Severe SGA, which was defined as below the 5th percentile, was also numerically but not significantly higher in the control arm than in the group receiving treatment (5.1% vs. 5.5%), but the incidence of composite adverse maternal events was numerically lower (2.1% vs. 2.8%). The incidences of all components of maternal morbidity, such as maternal death (0.1% vs. 0.2%) pulmonary edema (0.4% vs. 0.9%), heart failure (0.1% vs. 0.1%), and acute kidney injury (0.8% vs. 1.2%), were either lower or the same on active treatment versus no treatment.
According to Dr. Tita, who called CHAP one of the largest and most diverse studies to address the value of treating mild hypertension in pregnancy, the American College of Obstetricians and Gynecologists (ACOG) is evaluating these data for changing their current guidelines for managing hypertension during pregnancy.
“The rate of chronic hypertension during pregnancy has been rising in the United States due to the increase in the average age of pregnant women and the rising rates of obesity,” Dr. Tita commented.
“We definitely needed these data,” said Mary Norine Walsh, MD, medical director, Ascension Saint Vincent Cardiovascular Research Institute, Indianapolis. Not only has the value of treating mild hypertension been unresolved, but Dr. Walsh pointed out that the rates of maternal mortality in the United States are rising and now generally exceed those of many other developed countries.
There are several features in the design of this trial that make the results even more salient to clinical practice, according to Dr. Walsh. This includes the fact that about half of patients enrolled were on Medicaid. As a result, the study confirmed benefit in what Dr. Walsh characterized as a “vulnerable” population.
“We will be busy now to make sure that our [pregnant] patients are achieving these target blood pressures,” Dr. Walsh said. She indicated that CHAP validates the treatment target of 140/90 mm Hg as a standard of care.
The results were published in the New England Journal of Medicine simultaneously with its ACC presentation.
The trial was funded by the National Heart, Lung, and Blood Institute. Dr. Tita reports research grants from Pfizer. Dr. Walsh reports a financial relationship with EBR Systems. Dr. Poppas reports no potential conflicts of interest.
FROM ACC 2022