User login
First RCT evaluating benefits of colonoscopy screening rocks GI: NordICC
VIENNA – the 10-year follow-up of the large, multicenter, randomized Northern-European Initiative on Colorectal Cancer (NordICC) trial shows.
In effect, this means the number needed to invite to undergo screening to prevent one case of colorectal cancer is 455 (95% confidence interval, 270-1,429), the researchers determined.
The results were presented at the United European Gastroenterology Week 2022 meeting and were published simultaneously in The New England Journal of Medicine.
The results of the study, which was designed to be truly population based and to mimic national colorectal cancer screening programs, provide an estimate of the effect of screening colonoscopy in the general population.
The primary outcome was determined on an intention-to-screen basis. All persons who were invited to undergo colonoscopy screening were compared with people who received usual care (that is, received no invitation or screening). At UEG 2022, the researchers presented the interim 10-year colorectal cancer risk, which was found to be 0.98%, compared to 1.20%. This represents a risk reduction of 18% among colonoscopy invitees (risk ratio, 0.82; 95% CI, 0.70-0.93). During the study period, 259 cases of colorectal cancer were diagnosed in the invited group versus622 in the usual-care group.
The risk of death from colorectal cancer was 0.28% in the invited group and 0.31% in the usual-care group (RR, 0.90; 95% CI, 0.64-1.16). The risk of death from any cause was similar in both the invited group and the usual-care group, at 11.03% and 11.04%, respectively (RR, 0.99; 95% CI, 0.96-1.04).
The authors noted that the benefit would have been greater had more people undergone screening; only 42% of those who were invited actually underwent colonoscopy. In an adjusted analysis, had all those who had been invited to undergo screening undergone colonoscopy, the 10-year risk of colorectal cancer would have decreased from 1.22% to 0.84%, and the risk of colorectal cancer–related death would have fallen from 0.30% to 0.15%.
The researchers, led by gastroenterologist Michael Bretthauer, MD, from the department of medicine, gastrointestinal endoscopy, University of Oslo, who presented the data at UEG 2022 on behalf of the NordICC study group, acknowledged that, despite the “observed appreciable reductions in relative risks, the absolute risks of the risk of colorectal cancer and even more so of colorectal cancer–related death were lower than those in previous screening trials and lower than what we anticipated when the trial was planned.”
However, they add that “optimism related to the effects of screening on colorectal cancer–related death may be warranted in light of the 50% decrease observed in adjusted per-protocol analyses.”
With his coauthors, Dr. Bretthauer wrote that even their adjusted findings “probably underestimated the benefit because, as in most other large-scale trials of colorectal cancer screening, we could not adjust for all important confounders in all countries.”
Dr. Bretthauer also noted that results were similar to those achieved through sigmoidoscopy screening. By close comparison, sigmoidoscopy studies show the risk of colorectal cancer is reduced between 33% and 40%, according to per protocol analyses. “These results suggest that colonoscopy screening might not be substantially better in reducing the risk of colorectal cancer than sigmoidoscopy.”
Real-world, population-based study
NordICC is an ongoing, pragmatic study and is the first randomized trial to quantify the possible benefit of colonoscopy screening on risk of colorectal cancer and related death.
Researchers recruited healthy men and women from registries in Poland, Norway, Sweden, and the Netherlands between 2009 and 2014. Most participants came from Poland (54,258), followed by Norway (26,411) and Sweden (3,646). Data from the Netherlands could not be included owing to data protection law.
At baseline, 84,585 participants aged 55-64 years were randomly assigned in a 1:2 ratio either to receive an invitation to undergo a single screening colonoscopy (28,220; invited) or to undergo usual care in each participant country (56,365; no invitation or screening).
Any colorectal cancer lesions detected were removed, whenever possible. The primary endpoints were the risks of colorectal cancer and colorectal cancer–related death. The secondary endpoint was death from any cause.
‘Modest effectiveness,’ but longer follow-up to give fuller picture
In an editorial that accompanied publication of the study, Jason A. Dominitz, MD, from the division of gastroenterology, University of Washington, Seattle, and Douglas J. Robertson, MD, from White River Junction (Vt.) Veterans Affairs Medical Center, commented on the possible reasons for the low reduction in incident cancer and deaths seen in NordICC.
They pointed out that cohort studies suggest a 40%-69% decrease in the incidence of colorectal cancer and a 29%-88% decrease in the risk of death with colonoscopy. However, they noted that “cohort studies probably overestimate the real-world effectiveness of colonoscopy because of the inability to adjust for important factors such as incomplete adherence to testing and the tendency of healthier persons to seek preventive care.”
Referring to Dr. Bretthauer’s point about attendance to screening, Dr. Dominitz and Dr. Robertson added that, in the United States, colonoscopy is the predominant form of screening for colorectal cancer and that in countries where colonoscopy is less established, participation may be very different.
“The actual effectiveness of colonoscopy in populations that are more accepting of colonoscopy could more closely resemble the effectiveness shown in the per-protocol analysis in this trial,” they wrote.
The editorialists also pointed out that the benefits of screening colonoscopy take time to be realized “because the incidence of colorectal cancer is initially increased when presymptomatic cancers are identified.” A repeat and final analysis of the NordICC data is due at 15 years’ follow-up.
In addition, they noted that “colonoscopy is highly operator dependent” and that the adenoma detection rate is variable and affects cancer risk and related mortality.
Given the “modest effectiveness” of screening colonoscopy in the trial, they asserted that, “if the trial truly represents the real-world performance of population-based screening colonoscopy, it might be hard to justify the risk and expense of this form of screening when simpler, less-invasive strategies (e.g., sigmoidoscopy and FIT [fecal immunochemical test]) are available.”
However, they also noted that “additional analyses, including longer follow up and results from other ongoing comparative effectiveness trials, will help us to fully understand the benefits of this test.”
Also commenting on the study was Michiel Maas, MD, from the department of gastroenterology and hepatology, Radboud UMC, Nijmegen, the Netherlands, told this news organization that he agreed that the absolute effect on colorectal cancer risk or colorectal cancer–related death was not as high as expected and may be disappointing.
But Dr. Maas said that “around half of the patients in the study did not undergo colonoscopy, which may have negatively impacted the results.
“An additional factor, which can be influential in colonoscopy studies, is the potential variability in detection rates between operators/endoscopists,” he said.
Looking to the future, Dr. Maas noted that “AI [artificial intelligence] or computer-aided detection can level this playing field in detection rates.
“Nevertheless, this is a very interesting study, which sheds a new light on the efficacy on screening colonoscopies,” he said.
Dr. Bretthauer has relationships with Paion, Cybernet, and the Norwegian Council of Research. Dr. Dominitz is cochair of VA Cooperative Studies Program #577: “Colonoscopy vs. Fecal Immunochemical Test (FIT) in Reducing Mortality from Colorectal Cancer” (the CONFIRM Study), which is funded by the Department of Veterans Affairs. Dr. Robertson is national cochair (with Dr. Dominitz) of the CONFIRM trial and has received personal fees from Freenome outside of the submitted work. Dr. Maas reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
This study’s data show that colonoscopy is effective – if it is completed. Only 42% of patients randomized to colonoscopy completed the test; among patients who actually got a colonoscopy, results are much more impressive in colorectal cancer (CRC) prevention (31% decrease) and mortality (50% decrease). In this study, many endoscopists had ADRs below the 25% benchmark, and low ADRs are associated with a higher risk of postcolonoscopy CRC. Differences between the two groups may increase with longer follow-up, which is planned, because detection and removal of polyps via colonoscopy prevents future cancers.
Remind your patients that they shouldn’t let media headlines guide your health care decisions. You should also explain how colonoscopy can detect and remove polyps, which prevents those polyps from developing into cancer. Most of the patients in the Norway study skipped their colonoscopy, but the test can’t prevent cancers if it isn’t performed! Lastly, colonoscopy is effective in a U.S. population and can cut their risk of dying from CRC.
David Lieberman, MD, is a professor of medicine and chief of the division of gastroenterology and hepatology at Oregon Health and Science University, Portland. He disclosed being a consultant for Freenome and Check-Cap.
This study’s data show that colonoscopy is effective – if it is completed. Only 42% of patients randomized to colonoscopy completed the test; among patients who actually got a colonoscopy, results are much more impressive in colorectal cancer (CRC) prevention (31% decrease) and mortality (50% decrease). In this study, many endoscopists had ADRs below the 25% benchmark, and low ADRs are associated with a higher risk of postcolonoscopy CRC. Differences between the two groups may increase with longer follow-up, which is planned, because detection and removal of polyps via colonoscopy prevents future cancers.
Remind your patients that they shouldn’t let media headlines guide your health care decisions. You should also explain how colonoscopy can detect and remove polyps, which prevents those polyps from developing into cancer. Most of the patients in the Norway study skipped their colonoscopy, but the test can’t prevent cancers if it isn’t performed! Lastly, colonoscopy is effective in a U.S. population and can cut their risk of dying from CRC.
David Lieberman, MD, is a professor of medicine and chief of the division of gastroenterology and hepatology at Oregon Health and Science University, Portland. He disclosed being a consultant for Freenome and Check-Cap.
This study’s data show that colonoscopy is effective – if it is completed. Only 42% of patients randomized to colonoscopy completed the test; among patients who actually got a colonoscopy, results are much more impressive in colorectal cancer (CRC) prevention (31% decrease) and mortality (50% decrease). In this study, many endoscopists had ADRs below the 25% benchmark, and low ADRs are associated with a higher risk of postcolonoscopy CRC. Differences between the two groups may increase with longer follow-up, which is planned, because detection and removal of polyps via colonoscopy prevents future cancers.
Remind your patients that they shouldn’t let media headlines guide your health care decisions. You should also explain how colonoscopy can detect and remove polyps, which prevents those polyps from developing into cancer. Most of the patients in the Norway study skipped their colonoscopy, but the test can’t prevent cancers if it isn’t performed! Lastly, colonoscopy is effective in a U.S. population and can cut their risk of dying from CRC.
David Lieberman, MD, is a professor of medicine and chief of the division of gastroenterology and hepatology at Oregon Health and Science University, Portland. He disclosed being a consultant for Freenome and Check-Cap.
VIENNA – the 10-year follow-up of the large, multicenter, randomized Northern-European Initiative on Colorectal Cancer (NordICC) trial shows.
In effect, this means the number needed to invite to undergo screening to prevent one case of colorectal cancer is 455 (95% confidence interval, 270-1,429), the researchers determined.
The results were presented at the United European Gastroenterology Week 2022 meeting and were published simultaneously in The New England Journal of Medicine.
The results of the study, which was designed to be truly population based and to mimic national colorectal cancer screening programs, provide an estimate of the effect of screening colonoscopy in the general population.
The primary outcome was determined on an intention-to-screen basis. All persons who were invited to undergo colonoscopy screening were compared with people who received usual care (that is, received no invitation or screening). At UEG 2022, the researchers presented the interim 10-year colorectal cancer risk, which was found to be 0.98%, compared to 1.20%. This represents a risk reduction of 18% among colonoscopy invitees (risk ratio, 0.82; 95% CI, 0.70-0.93). During the study period, 259 cases of colorectal cancer were diagnosed in the invited group versus622 in the usual-care group.
The risk of death from colorectal cancer was 0.28% in the invited group and 0.31% in the usual-care group (RR, 0.90; 95% CI, 0.64-1.16). The risk of death from any cause was similar in both the invited group and the usual-care group, at 11.03% and 11.04%, respectively (RR, 0.99; 95% CI, 0.96-1.04).
The authors noted that the benefit would have been greater had more people undergone screening; only 42% of those who were invited actually underwent colonoscopy. In an adjusted analysis, had all those who had been invited to undergo screening undergone colonoscopy, the 10-year risk of colorectal cancer would have decreased from 1.22% to 0.84%, and the risk of colorectal cancer–related death would have fallen from 0.30% to 0.15%.
The researchers, led by gastroenterologist Michael Bretthauer, MD, from the department of medicine, gastrointestinal endoscopy, University of Oslo, who presented the data at UEG 2022 on behalf of the NordICC study group, acknowledged that, despite the “observed appreciable reductions in relative risks, the absolute risks of the risk of colorectal cancer and even more so of colorectal cancer–related death were lower than those in previous screening trials and lower than what we anticipated when the trial was planned.”
However, they add that “optimism related to the effects of screening on colorectal cancer–related death may be warranted in light of the 50% decrease observed in adjusted per-protocol analyses.”
With his coauthors, Dr. Bretthauer wrote that even their adjusted findings “probably underestimated the benefit because, as in most other large-scale trials of colorectal cancer screening, we could not adjust for all important confounders in all countries.”
Dr. Bretthauer also noted that results were similar to those achieved through sigmoidoscopy screening. By close comparison, sigmoidoscopy studies show the risk of colorectal cancer is reduced between 33% and 40%, according to per protocol analyses. “These results suggest that colonoscopy screening might not be substantially better in reducing the risk of colorectal cancer than sigmoidoscopy.”
Real-world, population-based study
NordICC is an ongoing, pragmatic study and is the first randomized trial to quantify the possible benefit of colonoscopy screening on risk of colorectal cancer and related death.
Researchers recruited healthy men and women from registries in Poland, Norway, Sweden, and the Netherlands between 2009 and 2014. Most participants came from Poland (54,258), followed by Norway (26,411) and Sweden (3,646). Data from the Netherlands could not be included owing to data protection law.
At baseline, 84,585 participants aged 55-64 years were randomly assigned in a 1:2 ratio either to receive an invitation to undergo a single screening colonoscopy (28,220; invited) or to undergo usual care in each participant country (56,365; no invitation or screening).
Any colorectal cancer lesions detected were removed, whenever possible. The primary endpoints were the risks of colorectal cancer and colorectal cancer–related death. The secondary endpoint was death from any cause.
‘Modest effectiveness,’ but longer follow-up to give fuller picture
In an editorial that accompanied publication of the study, Jason A. Dominitz, MD, from the division of gastroenterology, University of Washington, Seattle, and Douglas J. Robertson, MD, from White River Junction (Vt.) Veterans Affairs Medical Center, commented on the possible reasons for the low reduction in incident cancer and deaths seen in NordICC.
They pointed out that cohort studies suggest a 40%-69% decrease in the incidence of colorectal cancer and a 29%-88% decrease in the risk of death with colonoscopy. However, they noted that “cohort studies probably overestimate the real-world effectiveness of colonoscopy because of the inability to adjust for important factors such as incomplete adherence to testing and the tendency of healthier persons to seek preventive care.”
Referring to Dr. Bretthauer’s point about attendance to screening, Dr. Dominitz and Dr. Robertson added that, in the United States, colonoscopy is the predominant form of screening for colorectal cancer and that in countries where colonoscopy is less established, participation may be very different.
“The actual effectiveness of colonoscopy in populations that are more accepting of colonoscopy could more closely resemble the effectiveness shown in the per-protocol analysis in this trial,” they wrote.
The editorialists also pointed out that the benefits of screening colonoscopy take time to be realized “because the incidence of colorectal cancer is initially increased when presymptomatic cancers are identified.” A repeat and final analysis of the NordICC data is due at 15 years’ follow-up.
In addition, they noted that “colonoscopy is highly operator dependent” and that the adenoma detection rate is variable and affects cancer risk and related mortality.
Given the “modest effectiveness” of screening colonoscopy in the trial, they asserted that, “if the trial truly represents the real-world performance of population-based screening colonoscopy, it might be hard to justify the risk and expense of this form of screening when simpler, less-invasive strategies (e.g., sigmoidoscopy and FIT [fecal immunochemical test]) are available.”
However, they also noted that “additional analyses, including longer follow up and results from other ongoing comparative effectiveness trials, will help us to fully understand the benefits of this test.”
Also commenting on the study was Michiel Maas, MD, from the department of gastroenterology and hepatology, Radboud UMC, Nijmegen, the Netherlands, told this news organization that he agreed that the absolute effect on colorectal cancer risk or colorectal cancer–related death was not as high as expected and may be disappointing.
But Dr. Maas said that “around half of the patients in the study did not undergo colonoscopy, which may have negatively impacted the results.
“An additional factor, which can be influential in colonoscopy studies, is the potential variability in detection rates between operators/endoscopists,” he said.
Looking to the future, Dr. Maas noted that “AI [artificial intelligence] or computer-aided detection can level this playing field in detection rates.
“Nevertheless, this is a very interesting study, which sheds a new light on the efficacy on screening colonoscopies,” he said.
Dr. Bretthauer has relationships with Paion, Cybernet, and the Norwegian Council of Research. Dr. Dominitz is cochair of VA Cooperative Studies Program #577: “Colonoscopy vs. Fecal Immunochemical Test (FIT) in Reducing Mortality from Colorectal Cancer” (the CONFIRM Study), which is funded by the Department of Veterans Affairs. Dr. Robertson is national cochair (with Dr. Dominitz) of the CONFIRM trial and has received personal fees from Freenome outside of the submitted work. Dr. Maas reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
VIENNA – the 10-year follow-up of the large, multicenter, randomized Northern-European Initiative on Colorectal Cancer (NordICC) trial shows.
In effect, this means the number needed to invite to undergo screening to prevent one case of colorectal cancer is 455 (95% confidence interval, 270-1,429), the researchers determined.
The results were presented at the United European Gastroenterology Week 2022 meeting and were published simultaneously in The New England Journal of Medicine.
The results of the study, which was designed to be truly population based and to mimic national colorectal cancer screening programs, provide an estimate of the effect of screening colonoscopy in the general population.
The primary outcome was determined on an intention-to-screen basis. All persons who were invited to undergo colonoscopy screening were compared with people who received usual care (that is, received no invitation or screening). At UEG 2022, the researchers presented the interim 10-year colorectal cancer risk, which was found to be 0.98%, compared to 1.20%. This represents a risk reduction of 18% among colonoscopy invitees (risk ratio, 0.82; 95% CI, 0.70-0.93). During the study period, 259 cases of colorectal cancer were diagnosed in the invited group versus622 in the usual-care group.
The risk of death from colorectal cancer was 0.28% in the invited group and 0.31% in the usual-care group (RR, 0.90; 95% CI, 0.64-1.16). The risk of death from any cause was similar in both the invited group and the usual-care group, at 11.03% and 11.04%, respectively (RR, 0.99; 95% CI, 0.96-1.04).
The authors noted that the benefit would have been greater had more people undergone screening; only 42% of those who were invited actually underwent colonoscopy. In an adjusted analysis, had all those who had been invited to undergo screening undergone colonoscopy, the 10-year risk of colorectal cancer would have decreased from 1.22% to 0.84%, and the risk of colorectal cancer–related death would have fallen from 0.30% to 0.15%.
The researchers, led by gastroenterologist Michael Bretthauer, MD, from the department of medicine, gastrointestinal endoscopy, University of Oslo, who presented the data at UEG 2022 on behalf of the NordICC study group, acknowledged that, despite the “observed appreciable reductions in relative risks, the absolute risks of the risk of colorectal cancer and even more so of colorectal cancer–related death were lower than those in previous screening trials and lower than what we anticipated when the trial was planned.”
However, they add that “optimism related to the effects of screening on colorectal cancer–related death may be warranted in light of the 50% decrease observed in adjusted per-protocol analyses.”
With his coauthors, Dr. Bretthauer wrote that even their adjusted findings “probably underestimated the benefit because, as in most other large-scale trials of colorectal cancer screening, we could not adjust for all important confounders in all countries.”
Dr. Bretthauer also noted that results were similar to those achieved through sigmoidoscopy screening. By close comparison, sigmoidoscopy studies show the risk of colorectal cancer is reduced between 33% and 40%, according to per protocol analyses. “These results suggest that colonoscopy screening might not be substantially better in reducing the risk of colorectal cancer than sigmoidoscopy.”
Real-world, population-based study
NordICC is an ongoing, pragmatic study and is the first randomized trial to quantify the possible benefit of colonoscopy screening on risk of colorectal cancer and related death.
Researchers recruited healthy men and women from registries in Poland, Norway, Sweden, and the Netherlands between 2009 and 2014. Most participants came from Poland (54,258), followed by Norway (26,411) and Sweden (3,646). Data from the Netherlands could not be included owing to data protection law.
At baseline, 84,585 participants aged 55-64 years were randomly assigned in a 1:2 ratio either to receive an invitation to undergo a single screening colonoscopy (28,220; invited) or to undergo usual care in each participant country (56,365; no invitation or screening).
Any colorectal cancer lesions detected were removed, whenever possible. The primary endpoints were the risks of colorectal cancer and colorectal cancer–related death. The secondary endpoint was death from any cause.
‘Modest effectiveness,’ but longer follow-up to give fuller picture
In an editorial that accompanied publication of the study, Jason A. Dominitz, MD, from the division of gastroenterology, University of Washington, Seattle, and Douglas J. Robertson, MD, from White River Junction (Vt.) Veterans Affairs Medical Center, commented on the possible reasons for the low reduction in incident cancer and deaths seen in NordICC.
They pointed out that cohort studies suggest a 40%-69% decrease in the incidence of colorectal cancer and a 29%-88% decrease in the risk of death with colonoscopy. However, they noted that “cohort studies probably overestimate the real-world effectiveness of colonoscopy because of the inability to adjust for important factors such as incomplete adherence to testing and the tendency of healthier persons to seek preventive care.”
Referring to Dr. Bretthauer’s point about attendance to screening, Dr. Dominitz and Dr. Robertson added that, in the United States, colonoscopy is the predominant form of screening for colorectal cancer and that in countries where colonoscopy is less established, participation may be very different.
“The actual effectiveness of colonoscopy in populations that are more accepting of colonoscopy could more closely resemble the effectiveness shown in the per-protocol analysis in this trial,” they wrote.
The editorialists also pointed out that the benefits of screening colonoscopy take time to be realized “because the incidence of colorectal cancer is initially increased when presymptomatic cancers are identified.” A repeat and final analysis of the NordICC data is due at 15 years’ follow-up.
In addition, they noted that “colonoscopy is highly operator dependent” and that the adenoma detection rate is variable and affects cancer risk and related mortality.
Given the “modest effectiveness” of screening colonoscopy in the trial, they asserted that, “if the trial truly represents the real-world performance of population-based screening colonoscopy, it might be hard to justify the risk and expense of this form of screening when simpler, less-invasive strategies (e.g., sigmoidoscopy and FIT [fecal immunochemical test]) are available.”
However, they also noted that “additional analyses, including longer follow up and results from other ongoing comparative effectiveness trials, will help us to fully understand the benefits of this test.”
Also commenting on the study was Michiel Maas, MD, from the department of gastroenterology and hepatology, Radboud UMC, Nijmegen, the Netherlands, told this news organization that he agreed that the absolute effect on colorectal cancer risk or colorectal cancer–related death was not as high as expected and may be disappointing.
But Dr. Maas said that “around half of the patients in the study did not undergo colonoscopy, which may have negatively impacted the results.
“An additional factor, which can be influential in colonoscopy studies, is the potential variability in detection rates between operators/endoscopists,” he said.
Looking to the future, Dr. Maas noted that “AI [artificial intelligence] or computer-aided detection can level this playing field in detection rates.
“Nevertheless, this is a very interesting study, which sheds a new light on the efficacy on screening colonoscopies,” he said.
Dr. Bretthauer has relationships with Paion, Cybernet, and the Norwegian Council of Research. Dr. Dominitz is cochair of VA Cooperative Studies Program #577: “Colonoscopy vs. Fecal Immunochemical Test (FIT) in Reducing Mortality from Colorectal Cancer” (the CONFIRM Study), which is funded by the Department of Veterans Affairs. Dr. Robertson is national cochair (with Dr. Dominitz) of the CONFIRM trial and has received personal fees from Freenome outside of the submitted work. Dr. Maas reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM UEG 2022
People of color more likely to be hospitalized for influenza, CDC report finds
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
Black Americans are 80% more likely to be hospitalized for the flu, compared with White Americans, according to new federal data.
Black, Hispanic, and American Indian/Alaska Native (AI/AN) adults in the United States also have had lower influenza vaccination rates, compared with their White counterparts, since 2010, researchers at the Centers for Disease Control and Prevention (CDC) revealed in a report.
The inequalities are the result of barriers to care, distrust of the medical system, and misinformation, the report said.
“We have many of the tools we need to address inequities and flu vaccination coverage and outcomes,” said CDC Acting Principal Deputy Director Debra Houry, MD, MPH, in a press call; “however, we must acknowledge that inequities in access to care continue to exist. To improve vaccine uptake, we must address the root causes of these ongoing disparities.”
The CDC has already reported early increases in flu activity in the United States, with the highest activity in the southeastern and south-central parts of the country. Experts also warn of a potentially more severe influenza season than in the previous 2 years. CDC officials emphasized that vaccination is the best protection against severe illness, hospitalization, and death from the flu. “Everyone should get vaccinated against flu today and encourage others and their community to get a flu vaccine for the best protection against flu this fall and winter,” Dr. Houry said.
In the recent report on disparities by community published October 18 in CDC Vital Signs, researchers looked at hospitalization rates from 2009 to 2022 and vaccination rates from 2010 to 2022 based on race and ethnicity using two national databases, the Influenza-Associated Hospitalization Surveillance Network and the Behavioral Risk Factor Surveillance System. All individuals included in the analysis were aged 18 years or older, and the 2020-2021 flu season was excluded from the analysis because of insufficient data.
Compared with those for White adults, hospitalization rates were 80% higher for Black adults, 30% higher for Hispanic adults, and 20% higher for AI/AN adults. While flu vaccination rates were similar in White and Asian adults (about 54%), coverage was lower in Black (42%), Hispanic (38%), AI/AN (41%), and other/multiracial (43%) adults. This disparity persisted even among individuals who had medical insurance, a personal health care provider, and a routine checkup within the last year.
Carla Black, PhD, MPH, an epidemiologist at the CDC’s Immunization Services Division, said during the press call. While flu vaccines may not always prevent infection, people who do get sick after being vaccinated tend to have better outcomes, she added. The report noted that building trust, increasing access to vaccination services, and combating misinformation are important steps to increasing vaccine coverage in minority groups.
While social distancing measures such as masking have made it difficult for the flu to spread, the relaxation of these safety measures could also lead to higher case counts. “We’ve had two mild flu seasons, and this means we might be ripe for a severe season,” Dr. Black said. “People haven’t had natural disease in 2 years, so there’s less natural immunity out there. People are going back to work. People are traveling again. All of these factors could contribute to us having a more severe flu season.”
A version of this article first appeared on Medscape.com.
An infant with a tender bump on her ear
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.

Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A 4-month-old female was referred to our clinic for evaluation of a bump on the right ear. The lesion was first noted at 2 months of age as a little pimple. She was evaluated by her pediatrician and was treated with topical and oral antibiotics without resolution of the lesion. The bump continued to grow and seemed tender to palpation, so she was referred to dermatology for evaluation.
She was born via normal vaginal delivery at 40 weeks. Her mother has no medical conditions and the pregnancy was uneventful. She has been growing and developing well. She takes vitamin D and is currently breast fed.
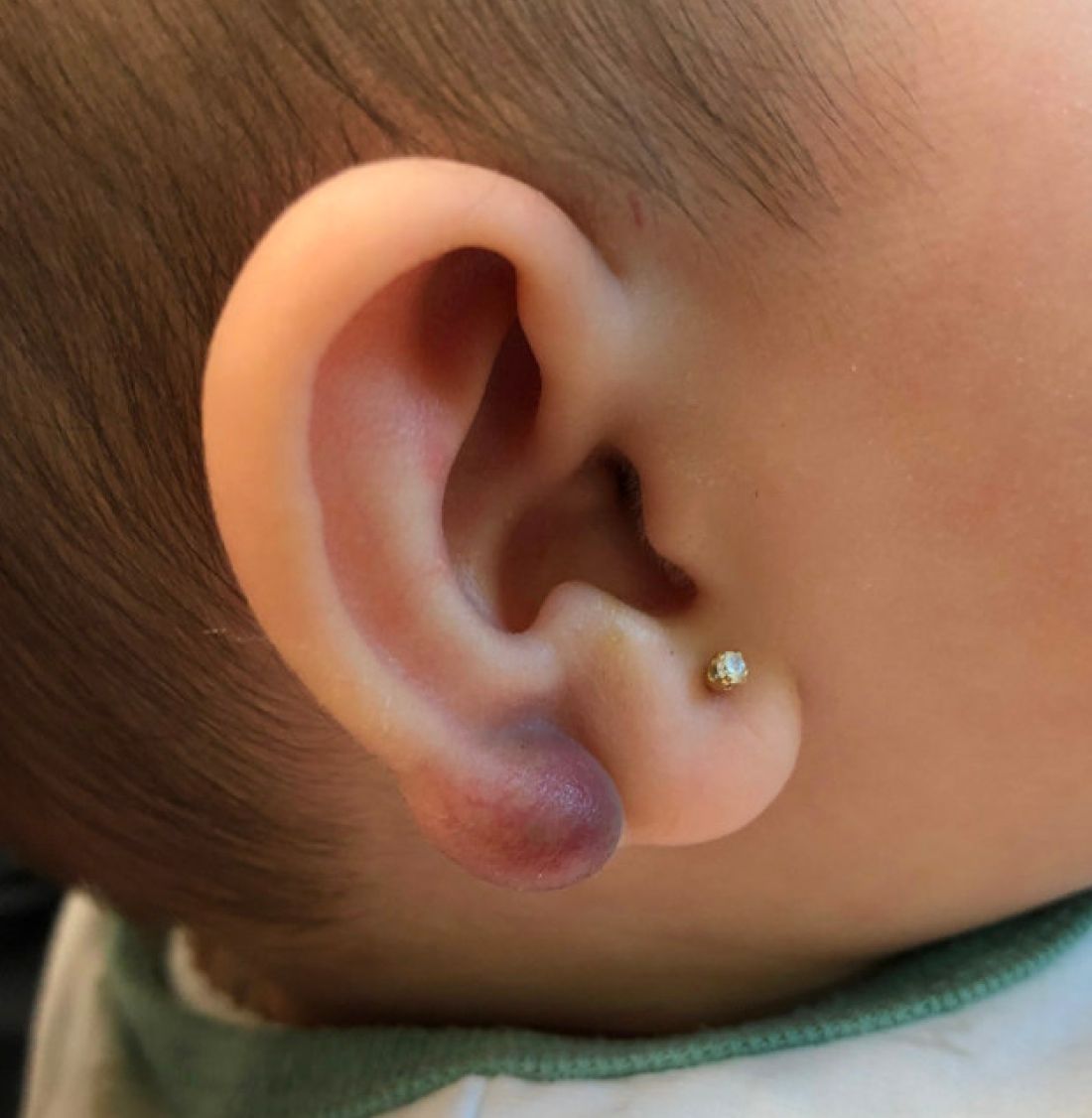
There have been no other family members with similar lesions. She had her ears pierced at a month of age without any complications.
On skin examination she has a firm red nodule on the right ear that appears slightly tender to touch. She has no other skin lesions of concern. She has normal muscle tone and there are no other abnormalities noted on the physical exam. She has no hepatomegaly, splenomegaly, or lymphadenopathy.
Bipolar risk and parental age: What’s the relationship?
VIENNA –
Results from a meta-analysis of more than 210,000 patients with bipolar disorder and over 13 million healthy individuals showed that children of mothers younger than 20 years had a 23% increased risk for bipolar disorder vs. those whose parents were aged 25-29 years. For participants whose mothers were aged 35-39 years, there was a 10% increased risk for bipolar disorder, which rose to 20% if the mother was aged 40 or older.
Having a father younger than 20 years conferred a 29% increased risk for bipolar disorder, which was the same increase in risk found in individuals whose fathers were aged 45 years or older.
These findings, which are an update of data published in the journal European Pharmacology, were presented at the 35th European College of Neuropsychopharmacology (ECNP) Congress.
Fourteen studies included
Previous studies have suggested that parental age at birth is a risk factor for several psychiatric disorders in offspring, including bipolar disorder, and that advanced parental age, specifically, is associated with earlier onset schizophrenia.
To investigate further, the current researchers conducted a systematic review and meta-analysis, searching the PubMed/MEDLINE, EMBASE, Scopus, and PsychINFO databases for relevant studies published to Dec. 1, 2021.
From 712 studies initially identified, 16 met all the inclusion criteria and 14 were included in the quantitative analysis.
Five studies reported only paternal age and risk for bipolar disorder in their offspring, one included just maternal age, and eight reported both maternal and paternal age in relation to the risk for offspring bipolar disorder.
Individuals with a history of any psychiatric disorders were excluded, leaving a total of 13.4 million individuals without bipolar disorder and 217,089 who had received a diagnosis for the disorder.
The investigators also corrected for both socioeconomic status and, when assessing the impact of maternal or paternal age at birth, corrected for the age of the other parent. However, they were unable to correct for the number of children in a family.
Results after stratifying maternal and paternal age showed that, compared with those born to parents aged 25-29 years, there was an increased risk for bipolar disorder in the offspring of both fathers and mothers younger than 20 years of age, with adjusted odds ratios of 1.29 (95% confidence interval, 1.13-1.48) and 1.23 (95% CI, 1.14-1.33), respectively.
Compared with those aged 25-29 years, there was also an increased risk for bipolar disorder in children born to mothers aged 35-39 years (adjusted OR, 1.1; 95% CI, 1.01-1.19) and aged 40 or older (OR, 1.2; 95% CI, 1.02-1.40).
Among fathers, there was increased risk for offspring bipolar disorder in those aged 45 or older vs. those aged 25-29 years (adjusted OR, 1.29; 95% CI, 1.15-1.46).
Several hypotheses
There are several hypotheses that could explain the results, lead study author Giovanna Fico, MD, bipolar and depressive disorders unit, Hospital Clínic Barcelona, told this news organization.
In older age, it may be “more related to genetic or epigenetic modification, especially in fathers,” Dr. Fico said. “Some studies have shown that there are de novo mutations in the germ lines, which increase the risk of several diseases, including schizophrenia.”
In younger individuals, there could be a “mixed effect between sociocultural factors, such as substance abuse, low educational status,” and other issues, Dr. Fico noted.
Moreover, as bipolar disorder onset can be as late as 30 years of age, the younger group could include “undiagnosed patients with bipolar disorder, which would increase the risk” of the disease in their offspring, she added.
Dr. Fico noted the investigators are now planning on studying the impact of environmental factors such as pollution, climate change, and urbanization on risk for bipolar disorder, with the aim of being better able to inform parents or to develop prevention strategies.
Psychoeducation is “very common for infertility, birth defects, and Down syndrome, but it’s not so common for psychiatric disorders because we need more data. But I think it’s important that parents know they have an increased risk,” she said.
Nevertheless, “We must stress that this risk is moderate, and it must be kept in perspective,” Dr. Fico said in a news release.
‘Exciting’ questions raised
The study “raises several exciting research questions, including the possibility of early prevention and intervention,” Maj Vinberg, MD, PhD, clinical professor, department of clinical medicine, University of Copenhagen, said in the release.
She said she agrees there are likely to be different factors at play at different ages, with the risk for bipolar disorder associated with younger-age parenthood more likely to be related to socioeconomic status.
For older parents, “there has been a lot of speculation around the father’s age especially, which everybody thought didn’t matter,” said Dr. Vinberg, who was not involved with the research.
“But you might have some epigenetic changes as you grow older that might transfer into the next generation,” given that there is 20 years of additional exposure to potential epigenetic changes between a man aged 25 years and one aged 45 years, she noted.
Dr. Vinberg also highlighted that there could be cases of undiagnosed bipolar disorder among the younger parents, and she noted that “men with bipolar disorder tend to have more children,” particularly during manic phases.
She explained that if someone were to get divorced at 35 years of age, then have a new manic episode at 45 “and have a new wife and children, I don’t know whether it’s possible to correct for that.”
The research is supported by a fellowship from “la Caixa” Foundation. The investigators have reported no relevant financial relationships. Dr. Vinberg reported having relationships with Lundbeck and Janssen.
A version of this article first appeared on Medscape.com.
VIENNA –
Results from a meta-analysis of more than 210,000 patients with bipolar disorder and over 13 million healthy individuals showed that children of mothers younger than 20 years had a 23% increased risk for bipolar disorder vs. those whose parents were aged 25-29 years. For participants whose mothers were aged 35-39 years, there was a 10% increased risk for bipolar disorder, which rose to 20% if the mother was aged 40 or older.
Having a father younger than 20 years conferred a 29% increased risk for bipolar disorder, which was the same increase in risk found in individuals whose fathers were aged 45 years or older.
These findings, which are an update of data published in the journal European Pharmacology, were presented at the 35th European College of Neuropsychopharmacology (ECNP) Congress.
Fourteen studies included
Previous studies have suggested that parental age at birth is a risk factor for several psychiatric disorders in offspring, including bipolar disorder, and that advanced parental age, specifically, is associated with earlier onset schizophrenia.
To investigate further, the current researchers conducted a systematic review and meta-analysis, searching the PubMed/MEDLINE, EMBASE, Scopus, and PsychINFO databases for relevant studies published to Dec. 1, 2021.
From 712 studies initially identified, 16 met all the inclusion criteria and 14 were included in the quantitative analysis.
Five studies reported only paternal age and risk for bipolar disorder in their offspring, one included just maternal age, and eight reported both maternal and paternal age in relation to the risk for offspring bipolar disorder.
Individuals with a history of any psychiatric disorders were excluded, leaving a total of 13.4 million individuals without bipolar disorder and 217,089 who had received a diagnosis for the disorder.
The investigators also corrected for both socioeconomic status and, when assessing the impact of maternal or paternal age at birth, corrected for the age of the other parent. However, they were unable to correct for the number of children in a family.
Results after stratifying maternal and paternal age showed that, compared with those born to parents aged 25-29 years, there was an increased risk for bipolar disorder in the offspring of both fathers and mothers younger than 20 years of age, with adjusted odds ratios of 1.29 (95% confidence interval, 1.13-1.48) and 1.23 (95% CI, 1.14-1.33), respectively.
Compared with those aged 25-29 years, there was also an increased risk for bipolar disorder in children born to mothers aged 35-39 years (adjusted OR, 1.1; 95% CI, 1.01-1.19) and aged 40 or older (OR, 1.2; 95% CI, 1.02-1.40).
Among fathers, there was increased risk for offspring bipolar disorder in those aged 45 or older vs. those aged 25-29 years (adjusted OR, 1.29; 95% CI, 1.15-1.46).
Several hypotheses
There are several hypotheses that could explain the results, lead study author Giovanna Fico, MD, bipolar and depressive disorders unit, Hospital Clínic Barcelona, told this news organization.
In older age, it may be “more related to genetic or epigenetic modification, especially in fathers,” Dr. Fico said. “Some studies have shown that there are de novo mutations in the germ lines, which increase the risk of several diseases, including schizophrenia.”
In younger individuals, there could be a “mixed effect between sociocultural factors, such as substance abuse, low educational status,” and other issues, Dr. Fico noted.
Moreover, as bipolar disorder onset can be as late as 30 years of age, the younger group could include “undiagnosed patients with bipolar disorder, which would increase the risk” of the disease in their offspring, she added.
Dr. Fico noted the investigators are now planning on studying the impact of environmental factors such as pollution, climate change, and urbanization on risk for bipolar disorder, with the aim of being better able to inform parents or to develop prevention strategies.
Psychoeducation is “very common for infertility, birth defects, and Down syndrome, but it’s not so common for psychiatric disorders because we need more data. But I think it’s important that parents know they have an increased risk,” she said.
Nevertheless, “We must stress that this risk is moderate, and it must be kept in perspective,” Dr. Fico said in a news release.
‘Exciting’ questions raised
The study “raises several exciting research questions, including the possibility of early prevention and intervention,” Maj Vinberg, MD, PhD, clinical professor, department of clinical medicine, University of Copenhagen, said in the release.
She said she agrees there are likely to be different factors at play at different ages, with the risk for bipolar disorder associated with younger-age parenthood more likely to be related to socioeconomic status.
For older parents, “there has been a lot of speculation around the father’s age especially, which everybody thought didn’t matter,” said Dr. Vinberg, who was not involved with the research.
“But you might have some epigenetic changes as you grow older that might transfer into the next generation,” given that there is 20 years of additional exposure to potential epigenetic changes between a man aged 25 years and one aged 45 years, she noted.
Dr. Vinberg also highlighted that there could be cases of undiagnosed bipolar disorder among the younger parents, and she noted that “men with bipolar disorder tend to have more children,” particularly during manic phases.
She explained that if someone were to get divorced at 35 years of age, then have a new manic episode at 45 “and have a new wife and children, I don’t know whether it’s possible to correct for that.”
The research is supported by a fellowship from “la Caixa” Foundation. The investigators have reported no relevant financial relationships. Dr. Vinberg reported having relationships with Lundbeck and Janssen.
A version of this article first appeared on Medscape.com.
VIENNA –
Results from a meta-analysis of more than 210,000 patients with bipolar disorder and over 13 million healthy individuals showed that children of mothers younger than 20 years had a 23% increased risk for bipolar disorder vs. those whose parents were aged 25-29 years. For participants whose mothers were aged 35-39 years, there was a 10% increased risk for bipolar disorder, which rose to 20% if the mother was aged 40 or older.
Having a father younger than 20 years conferred a 29% increased risk for bipolar disorder, which was the same increase in risk found in individuals whose fathers were aged 45 years or older.
These findings, which are an update of data published in the journal European Pharmacology, were presented at the 35th European College of Neuropsychopharmacology (ECNP) Congress.
Fourteen studies included
Previous studies have suggested that parental age at birth is a risk factor for several psychiatric disorders in offspring, including bipolar disorder, and that advanced parental age, specifically, is associated with earlier onset schizophrenia.
To investigate further, the current researchers conducted a systematic review and meta-analysis, searching the PubMed/MEDLINE, EMBASE, Scopus, and PsychINFO databases for relevant studies published to Dec. 1, 2021.
From 712 studies initially identified, 16 met all the inclusion criteria and 14 were included in the quantitative analysis.
Five studies reported only paternal age and risk for bipolar disorder in their offspring, one included just maternal age, and eight reported both maternal and paternal age in relation to the risk for offspring bipolar disorder.
Individuals with a history of any psychiatric disorders were excluded, leaving a total of 13.4 million individuals without bipolar disorder and 217,089 who had received a diagnosis for the disorder.
The investigators also corrected for both socioeconomic status and, when assessing the impact of maternal or paternal age at birth, corrected for the age of the other parent. However, they were unable to correct for the number of children in a family.
Results after stratifying maternal and paternal age showed that, compared with those born to parents aged 25-29 years, there was an increased risk for bipolar disorder in the offspring of both fathers and mothers younger than 20 years of age, with adjusted odds ratios of 1.29 (95% confidence interval, 1.13-1.48) and 1.23 (95% CI, 1.14-1.33), respectively.
Compared with those aged 25-29 years, there was also an increased risk for bipolar disorder in children born to mothers aged 35-39 years (adjusted OR, 1.1; 95% CI, 1.01-1.19) and aged 40 or older (OR, 1.2; 95% CI, 1.02-1.40).
Among fathers, there was increased risk for offspring bipolar disorder in those aged 45 or older vs. those aged 25-29 years (adjusted OR, 1.29; 95% CI, 1.15-1.46).
Several hypotheses
There are several hypotheses that could explain the results, lead study author Giovanna Fico, MD, bipolar and depressive disorders unit, Hospital Clínic Barcelona, told this news organization.
In older age, it may be “more related to genetic or epigenetic modification, especially in fathers,” Dr. Fico said. “Some studies have shown that there are de novo mutations in the germ lines, which increase the risk of several diseases, including schizophrenia.”
In younger individuals, there could be a “mixed effect between sociocultural factors, such as substance abuse, low educational status,” and other issues, Dr. Fico noted.
Moreover, as bipolar disorder onset can be as late as 30 years of age, the younger group could include “undiagnosed patients with bipolar disorder, which would increase the risk” of the disease in their offspring, she added.
Dr. Fico noted the investigators are now planning on studying the impact of environmental factors such as pollution, climate change, and urbanization on risk for bipolar disorder, with the aim of being better able to inform parents or to develop prevention strategies.
Psychoeducation is “very common for infertility, birth defects, and Down syndrome, but it’s not so common for psychiatric disorders because we need more data. But I think it’s important that parents know they have an increased risk,” she said.
Nevertheless, “We must stress that this risk is moderate, and it must be kept in perspective,” Dr. Fico said in a news release.
‘Exciting’ questions raised
The study “raises several exciting research questions, including the possibility of early prevention and intervention,” Maj Vinberg, MD, PhD, clinical professor, department of clinical medicine, University of Copenhagen, said in the release.
She said she agrees there are likely to be different factors at play at different ages, with the risk for bipolar disorder associated with younger-age parenthood more likely to be related to socioeconomic status.
For older parents, “there has been a lot of speculation around the father’s age especially, which everybody thought didn’t matter,” said Dr. Vinberg, who was not involved with the research.
“But you might have some epigenetic changes as you grow older that might transfer into the next generation,” given that there is 20 years of additional exposure to potential epigenetic changes between a man aged 25 years and one aged 45 years, she noted.
Dr. Vinberg also highlighted that there could be cases of undiagnosed bipolar disorder among the younger parents, and she noted that “men with bipolar disorder tend to have more children,” particularly during manic phases.
She explained that if someone were to get divorced at 35 years of age, then have a new manic episode at 45 “and have a new wife and children, I don’t know whether it’s possible to correct for that.”
The research is supported by a fellowship from “la Caixa” Foundation. The investigators have reported no relevant financial relationships. Dr. Vinberg reported having relationships with Lundbeck and Janssen.
A version of this article first appeared on Medscape.com.
AT ECNP CONGRESS 2022
Nonhormonal drug fezolinetant found safe for hot flashes in yearlong study
The drug fezolinetant, a selective neurokinin-3 receptor antagonist under investigation for treatment of menopausal vasomotor symptoms, showed acceptable long-term safety and tolerability during a 1-year phase 3 randomized controlled trial, according to data presented at the annual meeting of the North American Menopause Society. The study, called SKYLIGHT 4, examined fezolinetant treatment, especially in terms of endometrial health.
The findings mean that fezolinetant “may help bridge a gap in the management of vasomotor symptoms,” according to lead author Genevieve Neal-Perry, MD, PhD, chair of the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill.

This study was an important step in fezolinetant’s path toward potential approval by the Food and Drug Administration for vasomotor symptoms.
”Moderate and severe vasomotor symptoms can adversely affect quality of life of those affected and result in sleep disruption as well as increased risk for heart disease and other high-risk medical problems,” Dr. Neal-Perry said. “Although menopausal hormone therapy significantly improves vasomotor symptoms, it may not be desired or it may not be safe for some women,” resulting in gaps in care and a need for targeted, nonhormonal therapies for hot flashes. A planned study will also assess the safety of the drug in patients with a diagnosis of hormone-sensitive cancer and disorders that increase the risk for blood clots.
”Fezolinetant has a low side effect profile, it is a nonhormonal option, and it is selective for the neurons that trigger and mediate hot flashes,” Dr. Neal-Perry said.
Hot flashes are caused by kisspeptin, neurokinin B, and dynorphin neurons located in the hypothalamus. Fezolinetant works by selectively blocking the neurokinin 3 receptor (NK3R), which regulates a person’s sense of temperature, Dr. Neal-Perry explained. Overactivation of NK3R, resulting from low estrogen levels, plays a role in the hot flashes and cold sweats women experience during menopause.
Drug development for hot flashes ”has been hampered by a lack of knowledge regarding the biological cause,” Dr. Neal-Perry said. “Now that we have a robust understanding of the basic biology of hot flashes, we can develop novel, highly effective, and targeted therapy.”
This safety study involved 1,830 women, ages 40-65, who were experiencing menopausal vasomotor symptoms and were randomly assigned to one of three arms for 52 weeks: 45 mg of fezolinetant, 30 mg of fezolinetant, or a placebo once daily.
The primary endpoints included the percentage of women with endometrial hyperplasia, the percentage of women with endometrial cancer, and the frequency and severity of treatment-emergent adverse events (TEAEs). To meet the primary safety endpoint, no more than 1% of participants could have hyperplasia or malignancy, with an upper confidence interval boundary not greater than 4%. Women who met prespecified criteria for their endometrial health to be assessed, underwent endometrial biopsies at baseline and at the end of the study. Three independent pathologists analyzed the tissue without knowledge of which study arm each sample came from. Among the 599 endometrial biopsy samples, 0.5% of the 203 participants taking 45 mg fezolinetant had hyperplasia while none of the women in the other two arms did. Among the 210 women taking 30 mg of fezolinetant, 0.5% had a malignancy; no malignancies occurred in the other two arms.
Overall adverse events were similar across all three arms, including rates of adverse events leading to discontinuation. The most common adverse events were headache and COVID-19. TEAEs related to the drug were 18.1% in the 45-mg arm, 15.4% in the 30-mg arm, and 17.4% in the placebo arm. Serious adverse events were similar across all three arms, and only 0.5% of participants in the 45-mg arm experienced drug-related serious adverse events, compared with none of the women in the 30-mg arm and 0.2% of women in the placebo group.
”The frequency of transaminase elevations was low, and these TEAEs were generally isolated, transient, and resolved on treatment or with discontinuation,” the authors reported.
The next steps for fezolinetant will be to assess its effect on mood and quality of life measures related to vasomotor symptoms, Dr. Neal-Perry said.

Samantha Dunham, MD, a NAMS-certified menopause practitioner and an associate professor of obstetrics and gynecology at New York University, suggested the drug’s safety in the study is encouraging.
”As a medication that treats menopausal symptoms, the study confirmed there are no issues with the endometrium, or lining of the uterus, not that one would expect issues given the mechanism of action,” Dr. Dunham, also codirector of NYU Langone’s Center for Midlife Health and Menopause, said in an interview. Dr. Dunham was not involved in the study.
”Earlier versions of medication in this class have caused liver enzyme elevation.” The trial of this medication showed that there were only transient elevations in liver enzymes, which resolved upon cessation of the medication. Dr. Dunham said. ”If the medicine proves to be safe over long periods of time in different populations, this will be a very significant medication for treating menopausal vasomotor symptoms.”
The research was funded by Astellas Pharma. Dr. Dunham had no disclosures. Dr. Neal-Perry is a scientific advisory board member for Astellas and Ferring Pharmaceuticals, and has received research funding from Merck and Overa.
The drug fezolinetant, a selective neurokinin-3 receptor antagonist under investigation for treatment of menopausal vasomotor symptoms, showed acceptable long-term safety and tolerability during a 1-year phase 3 randomized controlled trial, according to data presented at the annual meeting of the North American Menopause Society. The study, called SKYLIGHT 4, examined fezolinetant treatment, especially in terms of endometrial health.
The findings mean that fezolinetant “may help bridge a gap in the management of vasomotor symptoms,” according to lead author Genevieve Neal-Perry, MD, PhD, chair of the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill.
This study was an important step in fezolinetant’s path toward potential approval by the Food and Drug Administration for vasomotor symptoms.
”Moderate and severe vasomotor symptoms can adversely affect quality of life of those affected and result in sleep disruption as well as increased risk for heart disease and other high-risk medical problems,” Dr. Neal-Perry said. “Although menopausal hormone therapy significantly improves vasomotor symptoms, it may not be desired or it may not be safe for some women,” resulting in gaps in care and a need for targeted, nonhormonal therapies for hot flashes. A planned study will also assess the safety of the drug in patients with a diagnosis of hormone-sensitive cancer and disorders that increase the risk for blood clots.
”Fezolinetant has a low side effect profile, it is a nonhormonal option, and it is selective for the neurons that trigger and mediate hot flashes,” Dr. Neal-Perry said.
Hot flashes are caused by kisspeptin, neurokinin B, and dynorphin neurons located in the hypothalamus. Fezolinetant works by selectively blocking the neurokinin 3 receptor (NK3R), which regulates a person’s sense of temperature, Dr. Neal-Perry explained. Overactivation of NK3R, resulting from low estrogen levels, plays a role in the hot flashes and cold sweats women experience during menopause.
Drug development for hot flashes ”has been hampered by a lack of knowledge regarding the biological cause,” Dr. Neal-Perry said. “Now that we have a robust understanding of the basic biology of hot flashes, we can develop novel, highly effective, and targeted therapy.”
This safety study involved 1,830 women, ages 40-65, who were experiencing menopausal vasomotor symptoms and were randomly assigned to one of three arms for 52 weeks: 45 mg of fezolinetant, 30 mg of fezolinetant, or a placebo once daily.
The primary endpoints included the percentage of women with endometrial hyperplasia, the percentage of women with endometrial cancer, and the frequency and severity of treatment-emergent adverse events (TEAEs). To meet the primary safety endpoint, no more than 1% of participants could have hyperplasia or malignancy, with an upper confidence interval boundary not greater than 4%. Women who met prespecified criteria for their endometrial health to be assessed, underwent endometrial biopsies at baseline and at the end of the study. Three independent pathologists analyzed the tissue without knowledge of which study arm each sample came from. Among the 599 endometrial biopsy samples, 0.5% of the 203 participants taking 45 mg fezolinetant had hyperplasia while none of the women in the other two arms did. Among the 210 women taking 30 mg of fezolinetant, 0.5% had a malignancy; no malignancies occurred in the other two arms.
Overall adverse events were similar across all three arms, including rates of adverse events leading to discontinuation. The most common adverse events were headache and COVID-19. TEAEs related to the drug were 18.1% in the 45-mg arm, 15.4% in the 30-mg arm, and 17.4% in the placebo arm. Serious adverse events were similar across all three arms, and only 0.5% of participants in the 45-mg arm experienced drug-related serious adverse events, compared with none of the women in the 30-mg arm and 0.2% of women in the placebo group.
”The frequency of transaminase elevations was low, and these TEAEs were generally isolated, transient, and resolved on treatment or with discontinuation,” the authors reported.
The next steps for fezolinetant will be to assess its effect on mood and quality of life measures related to vasomotor symptoms, Dr. Neal-Perry said.
Samantha Dunham, MD, a NAMS-certified menopause practitioner and an associate professor of obstetrics and gynecology at New York University, suggested the drug’s safety in the study is encouraging.
”As a medication that treats menopausal symptoms, the study confirmed there are no issues with the endometrium, or lining of the uterus, not that one would expect issues given the mechanism of action,” Dr. Dunham, also codirector of NYU Langone’s Center for Midlife Health and Menopause, said in an interview. Dr. Dunham was not involved in the study.
”Earlier versions of medication in this class have caused liver enzyme elevation.” The trial of this medication showed that there were only transient elevations in liver enzymes, which resolved upon cessation of the medication. Dr. Dunham said. ”If the medicine proves to be safe over long periods of time in different populations, this will be a very significant medication for treating menopausal vasomotor symptoms.”
The research was funded by Astellas Pharma. Dr. Dunham had no disclosures. Dr. Neal-Perry is a scientific advisory board member for Astellas and Ferring Pharmaceuticals, and has received research funding from Merck and Overa.
The drug fezolinetant, a selective neurokinin-3 receptor antagonist under investigation for treatment of menopausal vasomotor symptoms, showed acceptable long-term safety and tolerability during a 1-year phase 3 randomized controlled trial, according to data presented at the annual meeting of the North American Menopause Society. The study, called SKYLIGHT 4, examined fezolinetant treatment, especially in terms of endometrial health.
The findings mean that fezolinetant “may help bridge a gap in the management of vasomotor symptoms,” according to lead author Genevieve Neal-Perry, MD, PhD, chair of the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill.
This study was an important step in fezolinetant’s path toward potential approval by the Food and Drug Administration for vasomotor symptoms.
”Moderate and severe vasomotor symptoms can adversely affect quality of life of those affected and result in sleep disruption as well as increased risk for heart disease and other high-risk medical problems,” Dr. Neal-Perry said. “Although menopausal hormone therapy significantly improves vasomotor symptoms, it may not be desired or it may not be safe for some women,” resulting in gaps in care and a need for targeted, nonhormonal therapies for hot flashes. A planned study will also assess the safety of the drug in patients with a diagnosis of hormone-sensitive cancer and disorders that increase the risk for blood clots.
”Fezolinetant has a low side effect profile, it is a nonhormonal option, and it is selective for the neurons that trigger and mediate hot flashes,” Dr. Neal-Perry said.
Hot flashes are caused by kisspeptin, neurokinin B, and dynorphin neurons located in the hypothalamus. Fezolinetant works by selectively blocking the neurokinin 3 receptor (NK3R), which regulates a person’s sense of temperature, Dr. Neal-Perry explained. Overactivation of NK3R, resulting from low estrogen levels, plays a role in the hot flashes and cold sweats women experience during menopause.
Drug development for hot flashes ”has been hampered by a lack of knowledge regarding the biological cause,” Dr. Neal-Perry said. “Now that we have a robust understanding of the basic biology of hot flashes, we can develop novel, highly effective, and targeted therapy.”
This safety study involved 1,830 women, ages 40-65, who were experiencing menopausal vasomotor symptoms and were randomly assigned to one of three arms for 52 weeks: 45 mg of fezolinetant, 30 mg of fezolinetant, or a placebo once daily.
The primary endpoints included the percentage of women with endometrial hyperplasia, the percentage of women with endometrial cancer, and the frequency and severity of treatment-emergent adverse events (TEAEs). To meet the primary safety endpoint, no more than 1% of participants could have hyperplasia or malignancy, with an upper confidence interval boundary not greater than 4%. Women who met prespecified criteria for their endometrial health to be assessed, underwent endometrial biopsies at baseline and at the end of the study. Three independent pathologists analyzed the tissue without knowledge of which study arm each sample came from. Among the 599 endometrial biopsy samples, 0.5% of the 203 participants taking 45 mg fezolinetant had hyperplasia while none of the women in the other two arms did. Among the 210 women taking 30 mg of fezolinetant, 0.5% had a malignancy; no malignancies occurred in the other two arms.
Overall adverse events were similar across all three arms, including rates of adverse events leading to discontinuation. The most common adverse events were headache and COVID-19. TEAEs related to the drug were 18.1% in the 45-mg arm, 15.4% in the 30-mg arm, and 17.4% in the placebo arm. Serious adverse events were similar across all three arms, and only 0.5% of participants in the 45-mg arm experienced drug-related serious adverse events, compared with none of the women in the 30-mg arm and 0.2% of women in the placebo group.
”The frequency of transaminase elevations was low, and these TEAEs were generally isolated, transient, and resolved on treatment or with discontinuation,” the authors reported.
The next steps for fezolinetant will be to assess its effect on mood and quality of life measures related to vasomotor symptoms, Dr. Neal-Perry said.
Samantha Dunham, MD, a NAMS-certified menopause practitioner and an associate professor of obstetrics and gynecology at New York University, suggested the drug’s safety in the study is encouraging.
”As a medication that treats menopausal symptoms, the study confirmed there are no issues with the endometrium, or lining of the uterus, not that one would expect issues given the mechanism of action,” Dr. Dunham, also codirector of NYU Langone’s Center for Midlife Health and Menopause, said in an interview. Dr. Dunham was not involved in the study.
”Earlier versions of medication in this class have caused liver enzyme elevation.” The trial of this medication showed that there were only transient elevations in liver enzymes, which resolved upon cessation of the medication. Dr. Dunham said. ”If the medicine proves to be safe over long periods of time in different populations, this will be a very significant medication for treating menopausal vasomotor symptoms.”
The research was funded by Astellas Pharma. Dr. Dunham had no disclosures. Dr. Neal-Perry is a scientific advisory board member for Astellas and Ferring Pharmaceuticals, and has received research funding from Merck and Overa.
FROM NAMS 2022
IM residents rate cardiology low on work-life balance
Both male and female internal medicine (IM) residents prioritized work-life balance, such as stable working hours and family friendliness, when considering career choices, and cardiology was perceived to fall short in this area, an updated survey revealed.
Originally conducted in 2010, the survey aimed to understand IM residents’ professional development preferences and perceptions of cardiology as a specialty. That survey demonstrated a discordance between what residents valued in making a career choice and their perceptions of a career in cardiology.
The discordance remained in 2020, with residents even more likely than their predecessors to report negative perceptions of cardiology.
Compared with residents surveyed in 2010, respondents in 2020 placed higher value on all aspects of work-life balance and of having role models who demonstrated a successful balance. The value change was particularly notable for men.
“While our survey does not elucidate why this is, speculation could be made that this value on work-life balance is generational and prominent in the youngest generations entering all professional fields, not just medicine,” lead author Meghan York, MD, of Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, told this news organization.
“There is also an interesting trend that dual-career couples are on the rise in the U.S.,” she said. “This may reflect that trend, [with] men in medical fields possibly taking on more domestic responsibility and requiring more work flexibility to do that.”
Regarding perceptions, she added, cardiology tends to show resident cardiologists who are working in inpatient services with “ballooning and unpredictable hours,” rather than those who are working in more time-controlled clinics. Therefore, “their prime exposure to physicians is not truly representative of the career.” The study was published online in JAMA Cardiology.
‘Lack of diversity’
The updated surveys were sent by various means to close to 30,000 residents, and were completed by 840 (mean age, 29; 50% male; 55% White). Cardiology was a favored subspecialty choice among men, with 46.5% reporting they were considering it vs. 29.7% of women. Women were more likely to report never having considered cardiology as a career choice (37.6%) compared with men (22.3%).
The survey incorporated a 5-point Likert scale of 1 (not important) to 5 (extremely important) for some of the questions.
The most important professional development preferences for respondents were positive role models (4.56), stimulating career (3.81), family friendly (3.78), patient focus (3.70), stable work hours (3.66), female or race friendly (3.33), professional challenges (3.21), and financial benefits (3.20).
The cardiology perception statements with the highest agreement were:
- Interferes with family life during training (3.93).
- Having met positive role models or having positive views of cardiovascular disease as a topic (3.85).
- Reasonable compensation (3.69).
- Adverse job conditions (3.16).
- Field lacks diversity (2.90).
Compared with the 2010 survey, the 2020 findings indicated increased importance on work-life balance components for both male and female residents, with a greater change among males.
In addition, 2020 respondents were more likely than their predecessors to report negative perceptions of cardiology, such as too much overnight or weekend call, challenging to have children during fellowship, and lack of diversity.
“The culture of the subspecialty of cardiology has not improved to become significantly more diverse or inclusive, whereas other specialties and subspecialties have, and residents interact with cardiologists frequently and can see that,” Dr. York noted.
“As women now make up greater than 50% of medical students,” she said, “it is reasonable to focus on women in medical school and residency to bring them into the field of cardiology. But as racial and ethnic minority groups are also massively underrepresented in medical school, recruitment into medicine needs to start much earlier, in high school and college.
“Creating and supporting rotations that embed residents in the outpatient cardiology setting and exposure to more longitudinal experiences will provide a more realistic picture of the career,” she concluded.
ACC ‘at the forefront’
“Work-life balance looks different for each and every individual, but there are some themes that we need to think about,” Lisa Rose-Jones, MD, chair of the American College of Cardiology’s Program Directors and Graduate Medical Educators Section, said in her comments on the study. “The ACC is really at the forefront of this. They are putting together different work groups to focus on ‘how can we have some innovations?’ ”
The ACC is seeking mentors as part of its workforce diversity efforts among African American/Black, Hispanic/LatinX and Women’s IM cardiology programs, she noted. Furthermore, on Oct. 13, the organization released its 2022 health policy statement on career flexibility in cardiology, which calls for more leeway for cardiologists to deal with common life events without jeopardizing their careers.
Dr. Rose-Jones, director of the training program in cardiovascular disease at the University of North Carolina at Chapel Hill, said that because both male and female residents placed a high value on work-life balance, “we’ve got to think about how we can have flexibility in our work hours. That is critically important. Health systems need to be able to accommodate working families that may need to alter traditional 9 to 5 work hours to meet the demands of being a successful cardiologist and also being a parent.”
In addition, she said, “We need to have very clear policies at every institution on gender-related and parent-related discrimination. Data show that many female trainees are still being questioned on their family planning. That is absolutely not appropriate. It is none of our business. While we continue to do that, we continue to create stigma in our field.”
Like Dr. York, she noted generational differences in the doctors who are coming up now. “They’ve seen burnout firsthand and want to have a well-balanced life that includes medicine, but also life outside of the hospital,” Dr. Rose-Jones said. “So, those of us in cardiology really need to look deep inside and make changes. We need to be thoughtful about how we can be innovative.”
No commercial funding or conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
Both male and female internal medicine (IM) residents prioritized work-life balance, such as stable working hours and family friendliness, when considering career choices, and cardiology was perceived to fall short in this area, an updated survey revealed.
Originally conducted in 2010, the survey aimed to understand IM residents’ professional development preferences and perceptions of cardiology as a specialty. That survey demonstrated a discordance between what residents valued in making a career choice and their perceptions of a career in cardiology.
The discordance remained in 2020, with residents even more likely than their predecessors to report negative perceptions of cardiology.
Compared with residents surveyed in 2010, respondents in 2020 placed higher value on all aspects of work-life balance and of having role models who demonstrated a successful balance. The value change was particularly notable for men.
“While our survey does not elucidate why this is, speculation could be made that this value on work-life balance is generational and prominent in the youngest generations entering all professional fields, not just medicine,” lead author Meghan York, MD, of Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, told this news organization.
“There is also an interesting trend that dual-career couples are on the rise in the U.S.,” she said. “This may reflect that trend, [with] men in medical fields possibly taking on more domestic responsibility and requiring more work flexibility to do that.”
Regarding perceptions, she added, cardiology tends to show resident cardiologists who are working in inpatient services with “ballooning and unpredictable hours,” rather than those who are working in more time-controlled clinics. Therefore, “their prime exposure to physicians is not truly representative of the career.” The study was published online in JAMA Cardiology.
‘Lack of diversity’
The updated surveys were sent by various means to close to 30,000 residents, and were completed by 840 (mean age, 29; 50% male; 55% White). Cardiology was a favored subspecialty choice among men, with 46.5% reporting they were considering it vs. 29.7% of women. Women were more likely to report never having considered cardiology as a career choice (37.6%) compared with men (22.3%).
The survey incorporated a 5-point Likert scale of 1 (not important) to 5 (extremely important) for some of the questions.
The most important professional development preferences for respondents were positive role models (4.56), stimulating career (3.81), family friendly (3.78), patient focus (3.70), stable work hours (3.66), female or race friendly (3.33), professional challenges (3.21), and financial benefits (3.20).
The cardiology perception statements with the highest agreement were:
- Interferes with family life during training (3.93).
- Having met positive role models or having positive views of cardiovascular disease as a topic (3.85).
- Reasonable compensation (3.69).
- Adverse job conditions (3.16).
- Field lacks diversity (2.90).
Compared with the 2010 survey, the 2020 findings indicated increased importance on work-life balance components for both male and female residents, with a greater change among males.
In addition, 2020 respondents were more likely than their predecessors to report negative perceptions of cardiology, such as too much overnight or weekend call, challenging to have children during fellowship, and lack of diversity.
“The culture of the subspecialty of cardiology has not improved to become significantly more diverse or inclusive, whereas other specialties and subspecialties have, and residents interact with cardiologists frequently and can see that,” Dr. York noted.
“As women now make up greater than 50% of medical students,” she said, “it is reasonable to focus on women in medical school and residency to bring them into the field of cardiology. But as racial and ethnic minority groups are also massively underrepresented in medical school, recruitment into medicine needs to start much earlier, in high school and college.
“Creating and supporting rotations that embed residents in the outpatient cardiology setting and exposure to more longitudinal experiences will provide a more realistic picture of the career,” she concluded.
ACC ‘at the forefront’
“Work-life balance looks different for each and every individual, but there are some themes that we need to think about,” Lisa Rose-Jones, MD, chair of the American College of Cardiology’s Program Directors and Graduate Medical Educators Section, said in her comments on the study. “The ACC is really at the forefront of this. They are putting together different work groups to focus on ‘how can we have some innovations?’ ”
The ACC is seeking mentors as part of its workforce diversity efforts among African American/Black, Hispanic/LatinX and Women’s IM cardiology programs, she noted. Furthermore, on Oct. 13, the organization released its 2022 health policy statement on career flexibility in cardiology, which calls for more leeway for cardiologists to deal with common life events without jeopardizing their careers.
Dr. Rose-Jones, director of the training program in cardiovascular disease at the University of North Carolina at Chapel Hill, said that because both male and female residents placed a high value on work-life balance, “we’ve got to think about how we can have flexibility in our work hours. That is critically important. Health systems need to be able to accommodate working families that may need to alter traditional 9 to 5 work hours to meet the demands of being a successful cardiologist and also being a parent.”
In addition, she said, “We need to have very clear policies at every institution on gender-related and parent-related discrimination. Data show that many female trainees are still being questioned on their family planning. That is absolutely not appropriate. It is none of our business. While we continue to do that, we continue to create stigma in our field.”
Like Dr. York, she noted generational differences in the doctors who are coming up now. “They’ve seen burnout firsthand and want to have a well-balanced life that includes medicine, but also life outside of the hospital,” Dr. Rose-Jones said. “So, those of us in cardiology really need to look deep inside and make changes. We need to be thoughtful about how we can be innovative.”
No commercial funding or conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
Both male and female internal medicine (IM) residents prioritized work-life balance, such as stable working hours and family friendliness, when considering career choices, and cardiology was perceived to fall short in this area, an updated survey revealed.
Originally conducted in 2010, the survey aimed to understand IM residents’ professional development preferences and perceptions of cardiology as a specialty. That survey demonstrated a discordance between what residents valued in making a career choice and their perceptions of a career in cardiology.
The discordance remained in 2020, with residents even more likely than their predecessors to report negative perceptions of cardiology.
Compared with residents surveyed in 2010, respondents in 2020 placed higher value on all aspects of work-life balance and of having role models who demonstrated a successful balance. The value change was particularly notable for men.
“While our survey does not elucidate why this is, speculation could be made that this value on work-life balance is generational and prominent in the youngest generations entering all professional fields, not just medicine,” lead author Meghan York, MD, of Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, told this news organization.
“There is also an interesting trend that dual-career couples are on the rise in the U.S.,” she said. “This may reflect that trend, [with] men in medical fields possibly taking on more domestic responsibility and requiring more work flexibility to do that.”
Regarding perceptions, she added, cardiology tends to show resident cardiologists who are working in inpatient services with “ballooning and unpredictable hours,” rather than those who are working in more time-controlled clinics. Therefore, “their prime exposure to physicians is not truly representative of the career.” The study was published online in JAMA Cardiology.
‘Lack of diversity’
The updated surveys were sent by various means to close to 30,000 residents, and were completed by 840 (mean age, 29; 50% male; 55% White). Cardiology was a favored subspecialty choice among men, with 46.5% reporting they were considering it vs. 29.7% of women. Women were more likely to report never having considered cardiology as a career choice (37.6%) compared with men (22.3%).
The survey incorporated a 5-point Likert scale of 1 (not important) to 5 (extremely important) for some of the questions.
The most important professional development preferences for respondents were positive role models (4.56), stimulating career (3.81), family friendly (3.78), patient focus (3.70), stable work hours (3.66), female or race friendly (3.33), professional challenges (3.21), and financial benefits (3.20).
The cardiology perception statements with the highest agreement were:
- Interferes with family life during training (3.93).
- Having met positive role models or having positive views of cardiovascular disease as a topic (3.85).
- Reasonable compensation (3.69).
- Adverse job conditions (3.16).
- Field lacks diversity (2.90).
Compared with the 2010 survey, the 2020 findings indicated increased importance on work-life balance components for both male and female residents, with a greater change among males.
In addition, 2020 respondents were more likely than their predecessors to report negative perceptions of cardiology, such as too much overnight or weekend call, challenging to have children during fellowship, and lack of diversity.
“The culture of the subspecialty of cardiology has not improved to become significantly more diverse or inclusive, whereas other specialties and subspecialties have, and residents interact with cardiologists frequently and can see that,” Dr. York noted.
“As women now make up greater than 50% of medical students,” she said, “it is reasonable to focus on women in medical school and residency to bring them into the field of cardiology. But as racial and ethnic minority groups are also massively underrepresented in medical school, recruitment into medicine needs to start much earlier, in high school and college.
“Creating and supporting rotations that embed residents in the outpatient cardiology setting and exposure to more longitudinal experiences will provide a more realistic picture of the career,” she concluded.
ACC ‘at the forefront’
“Work-life balance looks different for each and every individual, but there are some themes that we need to think about,” Lisa Rose-Jones, MD, chair of the American College of Cardiology’s Program Directors and Graduate Medical Educators Section, said in her comments on the study. “The ACC is really at the forefront of this. They are putting together different work groups to focus on ‘how can we have some innovations?’ ”
The ACC is seeking mentors as part of its workforce diversity efforts among African American/Black, Hispanic/LatinX and Women’s IM cardiology programs, she noted. Furthermore, on Oct. 13, the organization released its 2022 health policy statement on career flexibility in cardiology, which calls for more leeway for cardiologists to deal with common life events without jeopardizing their careers.
Dr. Rose-Jones, director of the training program in cardiovascular disease at the University of North Carolina at Chapel Hill, said that because both male and female residents placed a high value on work-life balance, “we’ve got to think about how we can have flexibility in our work hours. That is critically important. Health systems need to be able to accommodate working families that may need to alter traditional 9 to 5 work hours to meet the demands of being a successful cardiologist and also being a parent.”
In addition, she said, “We need to have very clear policies at every institution on gender-related and parent-related discrimination. Data show that many female trainees are still being questioned on their family planning. That is absolutely not appropriate. It is none of our business. While we continue to do that, we continue to create stigma in our field.”
Like Dr. York, she noted generational differences in the doctors who are coming up now. “They’ve seen burnout firsthand and want to have a well-balanced life that includes medicine, but also life outside of the hospital,” Dr. Rose-Jones said. “So, those of us in cardiology really need to look deep inside and make changes. We need to be thoughtful about how we can be innovative.”
No commercial funding or conflicts of interest were declared.
A version of this article first appeared on Medscape.com.
FROM JAMA CARDIOLOGY
Islet transplants in type 1 diabetes durable up to 8 years
Transplantation of cadaveric pancreatic islet cells resulted in graft survival and function with acceptable safety for up to 8 years in selected individuals with type 1 diabetes, new research finds.
The study is a long-term follow-up of two phase 3 pivotal trials from the Clinical Islet Transplantation Consortium of a purified human pancreatic islet cell product for treating people with type 1 diabetes.
One trial involved islet transplantation in 48 people who experienced severe hypoglycemia and hypoglycemic unawareness, and the other trial included 24 people who also experienced those complications and were already receiving immunosuppression following kidney transplant. The trials, both registered with the U.S. Food and Drug Administration (FDA), met their primary efficacy and safety endpoints at 2- and 3-year timepoints.
The follow-up data have now been published in Diabetes Care by Michael Rickels, MD, and colleagues.
The procedure involved infusion through the hepatic portal vein of one or more purified human pancreatic islet products under standardized immunosuppression using methods that Dr. Rickels and colleagues have been developing since 2004. The approach involves multiple modalities to protect the islets prior to transplantation.
Among the 34 islet-alone and eight islet-after–kidney transplant recipients who entered the extended follow-up, durable graft survival allowing for achievement of glycemic targets occurred without severe hypoglycemia or adverse effects from immunosuppression.
The primary outcome, actuarial survival of graft islet function, was 56% at the maximum follow-up of 8.3 years for the islet-only transplantation group and 49% at 7.3 years for the islet-after–kidney transplantation group (P = .004).
The findings suggest that “in the long run, islet transplantation has efficacy, including among those who have had kidney transplants ... Most type 1 diabetes patients are improved tremendously with current insulin delivery systems ... but for those having the most difficulty controlling their blood sugar – and those whose diabetes has already been complicated by needing a kidney transplant – the outcomes we saw in this study are what we’ve been hoping to achieve for more than 20 years,” said Dr. Rickels in a statement from his institution, the University of Pennsylvania, Philadelphia.
In the initial trials at day 75 after the initial transplant, 87.5% of the islet-alone and 71% of the islet-after–kidney transplant group achieved hemoglobin A1c under 7%, and 85% and 54%, respectively, achieved A1c at or under 6.5%. At the end of maximal follow-up, 49% of islet-only transplant recipients maintained A1c under 7%, although none had A1c at or under 6.5%. For the islet-after–kidney transplant group, these proportions were 35% and 17%, respectively (P = .0017 for A1c under 7.0% and P < .0001 for A1c ≤ 6.5%, respectively, between the groups).
There were 12 severe hypoglycemic episodes in five patients (three islet-alone and two islet-after–kidney transplant group) during the initial trials, but no additional episodes occurred in either group during long-term follow-up.
Overall, 53 individuals – 37 in the islet-alone and 16 in the islet-after–kidney transplant group – or 74% of the total, achieved a period of insulin independence with A1c under 7%, ranging from 36 to 481 days. The range of time to achieving insulin independence reflects individuals who received one, two, or three islet infusions.
The fact that most patients achieved insulin independence following just one (n = 20) or two (n = 30) infusions and only three patients required three infusions was notable, Dr. Rickels said.
“Currently, around the world, there’s an expectation of two to three donor pancreases being needed. Here, it’s one, maybe two. It’s a much more efficient protocol and opens up access for more islet transplantation as a hoped-for alternative to pancreas transplants.”
Of those who achieved insulin independence, 30 (57%) remained insulin-independent throughout follow-up (20 of 37 islet-alone and 10 of 16 islet-after–kidney transplant patients), with no difference in duration of insulin independence between the groups.
There were no deaths during post-transplant follow-up. Rates of serious adverse events were 0.31 and 0.43 per patient-year for the islet-after–kidney and islet-alone transplant groups, respectively. Of a total of 104 serious adverse events, 65 occurred during the initial trials and had been previously reported. Of the additional 39 serious adverse events that occurred during long-term follow-up, 11 were possibly due to immunosuppression and 27 were deemed unrelated to the procedures.
According to Dr. Rickels, “These are the most seriously affected patients, and you’d be expecting to see some hospitalizations in a population managed on immunosuppression therapy ... It’s important to note that none of the adverse events were related to the actual islet product. Also, kidney function remained stable during long-term follow-up in both cohorts, in fact, improving in those who had kidney transplants.”
Overall, he said, “This is a much less invasive procedure that opens itself up to significantly fewer complications than what many of these patients would otherwise require, a pancreas transplant, which involves major abdominal surgery.”
The investigators plan to submit these data as part of a biologic license application (BLA) to the FDA.
The research was supported by grants from JDRF, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Institute of Allergy and Infectious Diseases. Dr. Rickels has reported receiving consulting fees from Sernova and Vertex Pharmaceuticals.
A version of this article first appeared on Medscape.com.
Transplantation of cadaveric pancreatic islet cells resulted in graft survival and function with acceptable safety for up to 8 years in selected individuals with type 1 diabetes, new research finds.
The study is a long-term follow-up of two phase 3 pivotal trials from the Clinical Islet Transplantation Consortium of a purified human pancreatic islet cell product for treating people with type 1 diabetes.
One trial involved islet transplantation in 48 people who experienced severe hypoglycemia and hypoglycemic unawareness, and the other trial included 24 people who also experienced those complications and were already receiving immunosuppression following kidney transplant. The trials, both registered with the U.S. Food and Drug Administration (FDA), met their primary efficacy and safety endpoints at 2- and 3-year timepoints.
The follow-up data have now been published in Diabetes Care by Michael Rickels, MD, and colleagues.
The procedure involved infusion through the hepatic portal vein of one or more purified human pancreatic islet products under standardized immunosuppression using methods that Dr. Rickels and colleagues have been developing since 2004. The approach involves multiple modalities to protect the islets prior to transplantation.
Among the 34 islet-alone and eight islet-after–kidney transplant recipients who entered the extended follow-up, durable graft survival allowing for achievement of glycemic targets occurred without severe hypoglycemia or adverse effects from immunosuppression.
The primary outcome, actuarial survival of graft islet function, was 56% at the maximum follow-up of 8.3 years for the islet-only transplantation group and 49% at 7.3 years for the islet-after–kidney transplantation group (P = .004).
The findings suggest that “in the long run, islet transplantation has efficacy, including among those who have had kidney transplants ... Most type 1 diabetes patients are improved tremendously with current insulin delivery systems ... but for those having the most difficulty controlling their blood sugar – and those whose diabetes has already been complicated by needing a kidney transplant – the outcomes we saw in this study are what we’ve been hoping to achieve for more than 20 years,” said Dr. Rickels in a statement from his institution, the University of Pennsylvania, Philadelphia.
In the initial trials at day 75 after the initial transplant, 87.5% of the islet-alone and 71% of the islet-after–kidney transplant group achieved hemoglobin A1c under 7%, and 85% and 54%, respectively, achieved A1c at or under 6.5%. At the end of maximal follow-up, 49% of islet-only transplant recipients maintained A1c under 7%, although none had A1c at or under 6.5%. For the islet-after–kidney transplant group, these proportions were 35% and 17%, respectively (P = .0017 for A1c under 7.0% and P < .0001 for A1c ≤ 6.5%, respectively, between the groups).
There were 12 severe hypoglycemic episodes in five patients (three islet-alone and two islet-after–kidney transplant group) during the initial trials, but no additional episodes occurred in either group during long-term follow-up.
Overall, 53 individuals – 37 in the islet-alone and 16 in the islet-after–kidney transplant group – or 74% of the total, achieved a period of insulin independence with A1c under 7%, ranging from 36 to 481 days. The range of time to achieving insulin independence reflects individuals who received one, two, or three islet infusions.
The fact that most patients achieved insulin independence following just one (n = 20) or two (n = 30) infusions and only three patients required three infusions was notable, Dr. Rickels said.
“Currently, around the world, there’s an expectation of two to three donor pancreases being needed. Here, it’s one, maybe two. It’s a much more efficient protocol and opens up access for more islet transplantation as a hoped-for alternative to pancreas transplants.”
Of those who achieved insulin independence, 30 (57%) remained insulin-independent throughout follow-up (20 of 37 islet-alone and 10 of 16 islet-after–kidney transplant patients), with no difference in duration of insulin independence between the groups.
There were no deaths during post-transplant follow-up. Rates of serious adverse events were 0.31 and 0.43 per patient-year for the islet-after–kidney and islet-alone transplant groups, respectively. Of a total of 104 serious adverse events, 65 occurred during the initial trials and had been previously reported. Of the additional 39 serious adverse events that occurred during long-term follow-up, 11 were possibly due to immunosuppression and 27 were deemed unrelated to the procedures.
According to Dr. Rickels, “These are the most seriously affected patients, and you’d be expecting to see some hospitalizations in a population managed on immunosuppression therapy ... It’s important to note that none of the adverse events were related to the actual islet product. Also, kidney function remained stable during long-term follow-up in both cohorts, in fact, improving in those who had kidney transplants.”
Overall, he said, “This is a much less invasive procedure that opens itself up to significantly fewer complications than what many of these patients would otherwise require, a pancreas transplant, which involves major abdominal surgery.”
The investigators plan to submit these data as part of a biologic license application (BLA) to the FDA.
The research was supported by grants from JDRF, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Institute of Allergy and Infectious Diseases. Dr. Rickels has reported receiving consulting fees from Sernova and Vertex Pharmaceuticals.
A version of this article first appeared on Medscape.com.
Transplantation of cadaveric pancreatic islet cells resulted in graft survival and function with acceptable safety for up to 8 years in selected individuals with type 1 diabetes, new research finds.
The study is a long-term follow-up of two phase 3 pivotal trials from the Clinical Islet Transplantation Consortium of a purified human pancreatic islet cell product for treating people with type 1 diabetes.
One trial involved islet transplantation in 48 people who experienced severe hypoglycemia and hypoglycemic unawareness, and the other trial included 24 people who also experienced those complications and were already receiving immunosuppression following kidney transplant. The trials, both registered with the U.S. Food and Drug Administration (FDA), met their primary efficacy and safety endpoints at 2- and 3-year timepoints.
The follow-up data have now been published in Diabetes Care by Michael Rickels, MD, and colleagues.
The procedure involved infusion through the hepatic portal vein of one or more purified human pancreatic islet products under standardized immunosuppression using methods that Dr. Rickels and colleagues have been developing since 2004. The approach involves multiple modalities to protect the islets prior to transplantation.
Among the 34 islet-alone and eight islet-after–kidney transplant recipients who entered the extended follow-up, durable graft survival allowing for achievement of glycemic targets occurred without severe hypoglycemia or adverse effects from immunosuppression.
The primary outcome, actuarial survival of graft islet function, was 56% at the maximum follow-up of 8.3 years for the islet-only transplantation group and 49% at 7.3 years for the islet-after–kidney transplantation group (P = .004).
The findings suggest that “in the long run, islet transplantation has efficacy, including among those who have had kidney transplants ... Most type 1 diabetes patients are improved tremendously with current insulin delivery systems ... but for those having the most difficulty controlling their blood sugar – and those whose diabetes has already been complicated by needing a kidney transplant – the outcomes we saw in this study are what we’ve been hoping to achieve for more than 20 years,” said Dr. Rickels in a statement from his institution, the University of Pennsylvania, Philadelphia.
In the initial trials at day 75 after the initial transplant, 87.5% of the islet-alone and 71% of the islet-after–kidney transplant group achieved hemoglobin A1c under 7%, and 85% and 54%, respectively, achieved A1c at or under 6.5%. At the end of maximal follow-up, 49% of islet-only transplant recipients maintained A1c under 7%, although none had A1c at or under 6.5%. For the islet-after–kidney transplant group, these proportions were 35% and 17%, respectively (P = .0017 for A1c under 7.0% and P < .0001 for A1c ≤ 6.5%, respectively, between the groups).
There were 12 severe hypoglycemic episodes in five patients (three islet-alone and two islet-after–kidney transplant group) during the initial trials, but no additional episodes occurred in either group during long-term follow-up.
Overall, 53 individuals – 37 in the islet-alone and 16 in the islet-after–kidney transplant group – or 74% of the total, achieved a period of insulin independence with A1c under 7%, ranging from 36 to 481 days. The range of time to achieving insulin independence reflects individuals who received one, two, or three islet infusions.
The fact that most patients achieved insulin independence following just one (n = 20) or two (n = 30) infusions and only three patients required three infusions was notable, Dr. Rickels said.
“Currently, around the world, there’s an expectation of two to three donor pancreases being needed. Here, it’s one, maybe two. It’s a much more efficient protocol and opens up access for more islet transplantation as a hoped-for alternative to pancreas transplants.”
Of those who achieved insulin independence, 30 (57%) remained insulin-independent throughout follow-up (20 of 37 islet-alone and 10 of 16 islet-after–kidney transplant patients), with no difference in duration of insulin independence between the groups.
There were no deaths during post-transplant follow-up. Rates of serious adverse events were 0.31 and 0.43 per patient-year for the islet-after–kidney and islet-alone transplant groups, respectively. Of a total of 104 serious adverse events, 65 occurred during the initial trials and had been previously reported. Of the additional 39 serious adverse events that occurred during long-term follow-up, 11 were possibly due to immunosuppression and 27 were deemed unrelated to the procedures.
According to Dr. Rickels, “These are the most seriously affected patients, and you’d be expecting to see some hospitalizations in a population managed on immunosuppression therapy ... It’s important to note that none of the adverse events were related to the actual islet product. Also, kidney function remained stable during long-term follow-up in both cohorts, in fact, improving in those who had kidney transplants.”
Overall, he said, “This is a much less invasive procedure that opens itself up to significantly fewer complications than what many of these patients would otherwise require, a pancreas transplant, which involves major abdominal surgery.”
The investigators plan to submit these data as part of a biologic license application (BLA) to the FDA.
The research was supported by grants from JDRF, the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Institute of Allergy and Infectious Diseases. Dr. Rickels has reported receiving consulting fees from Sernova and Vertex Pharmaceuticals.
A version of this article first appeared on Medscape.com.
FROM DIABETES CARE
Menopause symptoms negatively affect women’s work
Symptoms of menopause can significantly disrupt a woman’s ability to work, according to a cross-sectional study presented at the annual meeting of the North American Menopause Society.
The study, by researchers at the Mayo Clinic, found that roughly one in eight women said issues stemming from menopause caused them to miss multiple days of work; reduce hours on the job; and even quit, retire, or be laid off.

“We were shocked to see the significant impact of menopause symptoms in the workplace,” Ekta Kapoor, MD, an associate professor of medicine at the Mayo Clinic in Rochester, Minn. said in an interview. “The potential economic impact of untreated menopause symptoms at the workplace is mind-boggling.”
The findings represent an opportunity to improve the treatment of menopause symptoms in working women and “draw attention to the need for creation of workplace policies that include education of employers, managers, and supervisors in order to support midlife women during this universal life stage transition,” Dr. Kapoor added.
Laurie Jeffers, DNP, certified menopause practitioner and codirector of the Center for Midlife Health and Menopause within the department of obstetrics & gynecology at New York University Langone Health, said the findings agree with the results of previous studies from the Netherlands and elsewhere.
“We know that across different studies up to 80% of women during the menopause transition and early post menopause will have high symptom burden, with vasomotor symptoms being the most common,” Dr. Jeffers said. “Psychological symptoms were notably significant in this study, which is also not surprising given that there can be an exacerbation of anxiety or depression during the menopausal transition due to the variability of hormonal activity during this time.”
4,400 women surveyed
Dr. Kapoor and colleagues analyzed data from 4,440 currently employed women, ages 45-60, who were enrolled in the Mayo Clinic Registry of Midlife Women and completed an online questionnaire between March and June 2021 about their menopause symptoms and the symptoms’ effects on their work. The participants all receive their primary care at one of four Mayo Clinic sites in Rochester; Scottsdale, Ariz.; Jacksonville, Fla.; and northwest Wisconsin.
The researchers defined an adverse outcome from a menopausal symptom as one that directly caused women to miss a day from work in the past year or, within the past 6 months, to cut back on work hours, to experience a layoff or job termination, or to quit, retire or change jobs.
Most of the respondents were White (95%), married (77%), and had at least a college degree (59%), and their average age was 54. Their overall average Menopause Rating Scale (MRS) score – including somatic, psychological, and urogenital domains – was 23.1, which indicated a severe level of menopause symptoms.
More than one in eight women (13%) reported having at least one adverse outcome because of menopause symptoms, most commonly missing work (11%).
The women reported missing an average 3 days of work because of menopause symptoms. About half as many (6%) reported cutting back on hours at work in the past 6 months. A small percentage reported being laid off in the past 6 months (0.3%), or quitting, retiring, or changing jobs in the past 6 months (1%) because of menopause symptoms.
Menopause symptoms may well be contributing to the gender wage gap, Dr. Kapoor said, in the same way that other factors affect women’s overall earnings, such as taking time off for having or raising a family, being responsible for a large share of housework, and taking on more mentoring or teaching roles that aren’t as highly valued at work.
“Women going through the menopause transition, and those who are postmenopausal, are at important stages of their careers,” Dr. Kapoor said. “They are often seeking, or already in leadership positions. Any impediments at this important stage in their professional lives can prove to be very costly, resulting in missed opportunities for promotion and leadership roles.”
Unsurprisingly, the higher a woman’s MRS score, the more likely she was to report an adverse work outcome, regardless of the symptom. For example, women whose symptom severity ranked in the top 25% overall were 15.6 times more likely to have an adverse work experience than those with the lowest level of symptoms (P < .001). Psychological symptoms had the greatest effect on work. Women whose psychological symptoms ranked in the top 25% in terms of severity were 21 times more likely to have an adverse work effect, compared with those with the lowest level of severity, according to the researchers.
The results echo findings from a recent survey from Carrot Fertility of 1,000 women, ages 40-55, about the effects of menopause on their careers. In that survey, 79% of respondents described working during menopause as more challenging than other common life stages and life experiences, including starting a new job, starting a family or getting a promotion.
Yet 77% of women felt uncomfortable talking with executives about the problem, and 63% didn’t feel comfortable talking to human resources about the issue. More than half (58%) didn’t want to discuss it with their immediate supervisor. Only 8% said their employer has offered significant support for menopause.
“Menopause symptoms continue to be undertreated for a variety of reasons [and] impact multiple aspects of a woman’s life, including her performance in the workplace,” Dr. Kapoor said. “In addition to focusing our attention on adequate treatment of menopause symptoms, we need advocacy for creation of workplace policies that can help women navigate this important and universal stage of their lives.”
Those policies might include education about menopause to increase knowledge and awareness among employers and managers, Dr. Kapoor said. She also noted the need to improve communication with women in discussing appropriate support and work adjustments during menopause.

"There is also evidence that less than 20%-30% of women seek help for their symptoms,” Dr. Jeffers said. “There are a variety of evidence-based hormonal and nonhormonal options available to ease these symptoms, and knowledgeable clinical management of these symptoms can favorably impact this transition. This study is interesting in that the population of women surveyed presumably had access to high-quality health resources and yet still had a high symptom burden.”
Dr. Kapoor cautioned that the data collection occurred in the midst of the COVID-19 pandemic, “which may have heightened the adverse experiences of women at the workplace. On the other hand, many of these women may have been working from home, which may have made their menopause experience more favorable than it would have been had they been working in actual offices,” thereby again underrepresenting the problem.
Dr. Kapoor added that the study population may not be representative since they all received treatment at a tertiary health care center and were almost all White women.
“Perhaps the impact of menopause symptoms in the minority populations and the community is even greater,” Dr. Kapoor said. “Our data might be underrepresenting the extent of the problem.”
The research did not use external funding. Dr. Kapoor has received grant support from Mithra Pharmaceuticals and consulted for Astellas, Mithra Pharmaceuticals, Scynexis, and Womaness. Dr. Jeffers had no disclosures.
*This story was updated on Nov. 28, 2022.
Symptoms of menopause can significantly disrupt a woman’s ability to work, according to a cross-sectional study presented at the annual meeting of the North American Menopause Society.
The study, by researchers at the Mayo Clinic, found that roughly one in eight women said issues stemming from menopause caused them to miss multiple days of work; reduce hours on the job; and even quit, retire, or be laid off.
“We were shocked to see the significant impact of menopause symptoms in the workplace,” Ekta Kapoor, MD, an associate professor of medicine at the Mayo Clinic in Rochester, Minn. said in an interview. “The potential economic impact of untreated menopause symptoms at the workplace is mind-boggling.”
The findings represent an opportunity to improve the treatment of menopause symptoms in working women and “draw attention to the need for creation of workplace policies that include education of employers, managers, and supervisors in order to support midlife women during this universal life stage transition,” Dr. Kapoor added.
Laurie Jeffers, DNP, certified menopause practitioner and codirector of the Center for Midlife Health and Menopause within the department of obstetrics & gynecology at New York University Langone Health, said the findings agree with the results of previous studies from the Netherlands and elsewhere.
“We know that across different studies up to 80% of women during the menopause transition and early post menopause will have high symptom burden, with vasomotor symptoms being the most common,” Dr. Jeffers said. “Psychological symptoms were notably significant in this study, which is also not surprising given that there can be an exacerbation of anxiety or depression during the menopausal transition due to the variability of hormonal activity during this time.”
4,400 women surveyed
Dr. Kapoor and colleagues analyzed data from 4,440 currently employed women, ages 45-60, who were enrolled in the Mayo Clinic Registry of Midlife Women and completed an online questionnaire between March and June 2021 about their menopause symptoms and the symptoms’ effects on their work. The participants all receive their primary care at one of four Mayo Clinic sites in Rochester; Scottsdale, Ariz.; Jacksonville, Fla.; and northwest Wisconsin.
The researchers defined an adverse outcome from a menopausal symptom as one that directly caused women to miss a day from work in the past year or, within the past 6 months, to cut back on work hours, to experience a layoff or job termination, or to quit, retire or change jobs.
Most of the respondents were White (95%), married (77%), and had at least a college degree (59%), and their average age was 54. Their overall average Menopause Rating Scale (MRS) score – including somatic, psychological, and urogenital domains – was 23.1, which indicated a severe level of menopause symptoms.
More than one in eight women (13%) reported having at least one adverse outcome because of menopause symptoms, most commonly missing work (11%).
The women reported missing an average 3 days of work because of menopause symptoms. About half as many (6%) reported cutting back on hours at work in the past 6 months. A small percentage reported being laid off in the past 6 months (0.3%), or quitting, retiring, or changing jobs in the past 6 months (1%) because of menopause symptoms.
Menopause symptoms may well be contributing to the gender wage gap, Dr. Kapoor said, in the same way that other factors affect women’s overall earnings, such as taking time off for having or raising a family, being responsible for a large share of housework, and taking on more mentoring or teaching roles that aren’t as highly valued at work.
“Women going through the menopause transition, and those who are postmenopausal, are at important stages of their careers,” Dr. Kapoor said. “They are often seeking, or already in leadership positions. Any impediments at this important stage in their professional lives can prove to be very costly, resulting in missed opportunities for promotion and leadership roles.”
Unsurprisingly, the higher a woman’s MRS score, the more likely she was to report an adverse work outcome, regardless of the symptom. For example, women whose symptom severity ranked in the top 25% overall were 15.6 times more likely to have an adverse work experience than those with the lowest level of symptoms (P < .001). Psychological symptoms had the greatest effect on work. Women whose psychological symptoms ranked in the top 25% in terms of severity were 21 times more likely to have an adverse work effect, compared with those with the lowest level of severity, according to the researchers.
The results echo findings from a recent survey from Carrot Fertility of 1,000 women, ages 40-55, about the effects of menopause on their careers. In that survey, 79% of respondents described working during menopause as more challenging than other common life stages and life experiences, including starting a new job, starting a family or getting a promotion.
Yet 77% of women felt uncomfortable talking with executives about the problem, and 63% didn’t feel comfortable talking to human resources about the issue. More than half (58%) didn’t want to discuss it with their immediate supervisor. Only 8% said their employer has offered significant support for menopause.
“Menopause symptoms continue to be undertreated for a variety of reasons [and] impact multiple aspects of a woman’s life, including her performance in the workplace,” Dr. Kapoor said. “In addition to focusing our attention on adequate treatment of menopause symptoms, we need advocacy for creation of workplace policies that can help women navigate this important and universal stage of their lives.”
Those policies might include education about menopause to increase knowledge and awareness among employers and managers, Dr. Kapoor said. She also noted the need to improve communication with women in discussing appropriate support and work adjustments during menopause.
"There is also evidence that less than 20%-30% of women seek help for their symptoms,” Dr. Jeffers said. “There are a variety of evidence-based hormonal and nonhormonal options available to ease these symptoms, and knowledgeable clinical management of these symptoms can favorably impact this transition. This study is interesting in that the population of women surveyed presumably had access to high-quality health resources and yet still had a high symptom burden.”
Dr. Kapoor cautioned that the data collection occurred in the midst of the COVID-19 pandemic, “which may have heightened the adverse experiences of women at the workplace. On the other hand, many of these women may have been working from home, which may have made their menopause experience more favorable than it would have been had they been working in actual offices,” thereby again underrepresenting the problem.
Dr. Kapoor added that the study population may not be representative since they all received treatment at a tertiary health care center and were almost all White women.
“Perhaps the impact of menopause symptoms in the minority populations and the community is even greater,” Dr. Kapoor said. “Our data might be underrepresenting the extent of the problem.”
The research did not use external funding. Dr. Kapoor has received grant support from Mithra Pharmaceuticals and consulted for Astellas, Mithra Pharmaceuticals, Scynexis, and Womaness. Dr. Jeffers had no disclosures.
*This story was updated on Nov. 28, 2022.
Symptoms of menopause can significantly disrupt a woman’s ability to work, according to a cross-sectional study presented at the annual meeting of the North American Menopause Society.
The study, by researchers at the Mayo Clinic, found that roughly one in eight women said issues stemming from menopause caused them to miss multiple days of work; reduce hours on the job; and even quit, retire, or be laid off.
“We were shocked to see the significant impact of menopause symptoms in the workplace,” Ekta Kapoor, MD, an associate professor of medicine at the Mayo Clinic in Rochester, Minn. said in an interview. “The potential economic impact of untreated menopause symptoms at the workplace is mind-boggling.”
The findings represent an opportunity to improve the treatment of menopause symptoms in working women and “draw attention to the need for creation of workplace policies that include education of employers, managers, and supervisors in order to support midlife women during this universal life stage transition,” Dr. Kapoor added.
Laurie Jeffers, DNP, certified menopause practitioner and codirector of the Center for Midlife Health and Menopause within the department of obstetrics & gynecology at New York University Langone Health, said the findings agree with the results of previous studies from the Netherlands and elsewhere.
“We know that across different studies up to 80% of women during the menopause transition and early post menopause will have high symptom burden, with vasomotor symptoms being the most common,” Dr. Jeffers said. “Psychological symptoms were notably significant in this study, which is also not surprising given that there can be an exacerbation of anxiety or depression during the menopausal transition due to the variability of hormonal activity during this time.”
4,400 women surveyed
Dr. Kapoor and colleagues analyzed data from 4,440 currently employed women, ages 45-60, who were enrolled in the Mayo Clinic Registry of Midlife Women and completed an online questionnaire between March and June 2021 about their menopause symptoms and the symptoms’ effects on their work. The participants all receive their primary care at one of four Mayo Clinic sites in Rochester; Scottsdale, Ariz.; Jacksonville, Fla.; and northwest Wisconsin.
The researchers defined an adverse outcome from a menopausal symptom as one that directly caused women to miss a day from work in the past year or, within the past 6 months, to cut back on work hours, to experience a layoff or job termination, or to quit, retire or change jobs.
Most of the respondents were White (95%), married (77%), and had at least a college degree (59%), and their average age was 54. Their overall average Menopause Rating Scale (MRS) score – including somatic, psychological, and urogenital domains – was 23.1, which indicated a severe level of menopause symptoms.
More than one in eight women (13%) reported having at least one adverse outcome because of menopause symptoms, most commonly missing work (11%).
The women reported missing an average 3 days of work because of menopause symptoms. About half as many (6%) reported cutting back on hours at work in the past 6 months. A small percentage reported being laid off in the past 6 months (0.3%), or quitting, retiring, or changing jobs in the past 6 months (1%) because of menopause symptoms.
Menopause symptoms may well be contributing to the gender wage gap, Dr. Kapoor said, in the same way that other factors affect women’s overall earnings, such as taking time off for having or raising a family, being responsible for a large share of housework, and taking on more mentoring or teaching roles that aren’t as highly valued at work.
“Women going through the menopause transition, and those who are postmenopausal, are at important stages of their careers,” Dr. Kapoor said. “They are often seeking, or already in leadership positions. Any impediments at this important stage in their professional lives can prove to be very costly, resulting in missed opportunities for promotion and leadership roles.”
Unsurprisingly, the higher a woman’s MRS score, the more likely she was to report an adverse work outcome, regardless of the symptom. For example, women whose symptom severity ranked in the top 25% overall were 15.6 times more likely to have an adverse work experience than those with the lowest level of symptoms (P < .001). Psychological symptoms had the greatest effect on work. Women whose psychological symptoms ranked in the top 25% in terms of severity were 21 times more likely to have an adverse work effect, compared with those with the lowest level of severity, according to the researchers.
The results echo findings from a recent survey from Carrot Fertility of 1,000 women, ages 40-55, about the effects of menopause on their careers. In that survey, 79% of respondents described working during menopause as more challenging than other common life stages and life experiences, including starting a new job, starting a family or getting a promotion.
Yet 77% of women felt uncomfortable talking with executives about the problem, and 63% didn’t feel comfortable talking to human resources about the issue. More than half (58%) didn’t want to discuss it with their immediate supervisor. Only 8% said their employer has offered significant support for menopause.
“Menopause symptoms continue to be undertreated for a variety of reasons [and] impact multiple aspects of a woman’s life, including her performance in the workplace,” Dr. Kapoor said. “In addition to focusing our attention on adequate treatment of menopause symptoms, we need advocacy for creation of workplace policies that can help women navigate this important and universal stage of their lives.”
Those policies might include education about menopause to increase knowledge and awareness among employers and managers, Dr. Kapoor said. She also noted the need to improve communication with women in discussing appropriate support and work adjustments during menopause.
"There is also evidence that less than 20%-30% of women seek help for their symptoms,” Dr. Jeffers said. “There are a variety of evidence-based hormonal and nonhormonal options available to ease these symptoms, and knowledgeable clinical management of these symptoms can favorably impact this transition. This study is interesting in that the population of women surveyed presumably had access to high-quality health resources and yet still had a high symptom burden.”
Dr. Kapoor cautioned that the data collection occurred in the midst of the COVID-19 pandemic, “which may have heightened the adverse experiences of women at the workplace. On the other hand, many of these women may have been working from home, which may have made their menopause experience more favorable than it would have been had they been working in actual offices,” thereby again underrepresenting the problem.
Dr. Kapoor added that the study population may not be representative since they all received treatment at a tertiary health care center and were almost all White women.
“Perhaps the impact of menopause symptoms in the minority populations and the community is even greater,” Dr. Kapoor said. “Our data might be underrepresenting the extent of the problem.”
The research did not use external funding. Dr. Kapoor has received grant support from Mithra Pharmaceuticals and consulted for Astellas, Mithra Pharmaceuticals, Scynexis, and Womaness. Dr. Jeffers had no disclosures.
*This story was updated on Nov. 28, 2022.
FROM NAMS 2022
VTE prophylaxis overused in low-risk hospitalized patients
A majority of hospitalized patients at low risk for venous thromboembolism were unnecessarily treated with medication, based on data from more than 400 individuals.
Prevention of venous thromboembolism (VTE) is important, and current guidelines from the American College of Chest Physicians suggest that patients with high or moderate risk for VTE be treated with mechanical prophylaxis, and that pharmacological prophylaxis is not recommended for patients at high risk for bleeding, said Hui Chong Lau, MD, in a presentation at the annual meeting of the American College of Chest Physicians (CHEST).
However, the nature of VTE prophylaxis using a risk assessment score has not been explored, said Dr. Lau, a third-year resident in internal medicine at Crozer-Chester Medical Center, Upland, Penn.
Low-molecular-weight heparin (LWMH) and intermittent pneumatic compression are often used to reduce VTE risk during hospitalization, but for patients with low VTE risk, prophylaxis is not necessarily recommended, he said. In fact, overuse of chemical prophylaxis in low-risk patients can increase bleeding risk and contribute to patient discomfort in the form of additional needle sticks while hospitalized, Dr. Lau said in the presentation.
“We wanted to see how well physicians in the hospital used a risk assessment model to stratify patients,” and how well the patients were assigned to the correct prophylaxis, he explained.
Dr. Lau and colleagues reviewed data from 469 adult patients hospitalized at a single medical center who were hospitalized between January 2021 and June 2021. The researchers retrospectively performed risk assessment using the Padua prediction score. A score of less than 4 was considered low risk for VTE, and a score of 4 or higher was considered high risk.
In the study population, 180 patients were identified as low risk and 289 were considered high risk.
Based on the Padua score, 95% of the patients at high risk were on the correct prophylaxis, Dr. Lau said.
A total of 193 high-risk patients were on heparin. However, many of these patients had good kidney function, and could have been treated with enoxaparin instead; “this would have spared them two needle sticks per day,” Dr. Lau noted.
Of the 180 low-risk patients, 168 (93.3%) were on chemical prophylaxis, and should have been on mechanical prophylaxis, he said. Only 10 patients (5%) who were considered low risk were placed on mechanical prophylaxis.
Overall, 3.6% of all patients who received chemical VTE prophylaxis developed bleeding.
The results were limited by the retrospective design and use of data from a single center. However, the findings emphasize the need for better attention to VTE risk when considering prophylaxis, said Dr. Lau. “We have to have risk assessment every day,” during a hospital stay, and adjust treatment accordingly, he said.
he concluded.
Additional research is needed to better understand the potential consequences of overusing chemical VTE, including not only bleeding risk, but also financial costs and patient discomfort, he said.
The study received no outside funding. The researchers had no financial conflicts to disclose.
A majority of hospitalized patients at low risk for venous thromboembolism were unnecessarily treated with medication, based on data from more than 400 individuals.
Prevention of venous thromboembolism (VTE) is important, and current guidelines from the American College of Chest Physicians suggest that patients with high or moderate risk for VTE be treated with mechanical prophylaxis, and that pharmacological prophylaxis is not recommended for patients at high risk for bleeding, said Hui Chong Lau, MD, in a presentation at the annual meeting of the American College of Chest Physicians (CHEST).
However, the nature of VTE prophylaxis using a risk assessment score has not been explored, said Dr. Lau, a third-year resident in internal medicine at Crozer-Chester Medical Center, Upland, Penn.
Low-molecular-weight heparin (LWMH) and intermittent pneumatic compression are often used to reduce VTE risk during hospitalization, but for patients with low VTE risk, prophylaxis is not necessarily recommended, he said. In fact, overuse of chemical prophylaxis in low-risk patients can increase bleeding risk and contribute to patient discomfort in the form of additional needle sticks while hospitalized, Dr. Lau said in the presentation.
“We wanted to see how well physicians in the hospital used a risk assessment model to stratify patients,” and how well the patients were assigned to the correct prophylaxis, he explained.
Dr. Lau and colleagues reviewed data from 469 adult patients hospitalized at a single medical center who were hospitalized between January 2021 and June 2021. The researchers retrospectively performed risk assessment using the Padua prediction score. A score of less than 4 was considered low risk for VTE, and a score of 4 or higher was considered high risk.
In the study population, 180 patients were identified as low risk and 289 were considered high risk.
Based on the Padua score, 95% of the patients at high risk were on the correct prophylaxis, Dr. Lau said.
A total of 193 high-risk patients were on heparin. However, many of these patients had good kidney function, and could have been treated with enoxaparin instead; “this would have spared them two needle sticks per day,” Dr. Lau noted.
Of the 180 low-risk patients, 168 (93.3%) were on chemical prophylaxis, and should have been on mechanical prophylaxis, he said. Only 10 patients (5%) who were considered low risk were placed on mechanical prophylaxis.
Overall, 3.6% of all patients who received chemical VTE prophylaxis developed bleeding.
The results were limited by the retrospective design and use of data from a single center. However, the findings emphasize the need for better attention to VTE risk when considering prophylaxis, said Dr. Lau. “We have to have risk assessment every day,” during a hospital stay, and adjust treatment accordingly, he said.
he concluded.
Additional research is needed to better understand the potential consequences of overusing chemical VTE, including not only bleeding risk, but also financial costs and patient discomfort, he said.
The study received no outside funding. The researchers had no financial conflicts to disclose.
A majority of hospitalized patients at low risk for venous thromboembolism were unnecessarily treated with medication, based on data from more than 400 individuals.
Prevention of venous thromboembolism (VTE) is important, and current guidelines from the American College of Chest Physicians suggest that patients with high or moderate risk for VTE be treated with mechanical prophylaxis, and that pharmacological prophylaxis is not recommended for patients at high risk for bleeding, said Hui Chong Lau, MD, in a presentation at the annual meeting of the American College of Chest Physicians (CHEST).
However, the nature of VTE prophylaxis using a risk assessment score has not been explored, said Dr. Lau, a third-year resident in internal medicine at Crozer-Chester Medical Center, Upland, Penn.
Low-molecular-weight heparin (LWMH) and intermittent pneumatic compression are often used to reduce VTE risk during hospitalization, but for patients with low VTE risk, prophylaxis is not necessarily recommended, he said. In fact, overuse of chemical prophylaxis in low-risk patients can increase bleeding risk and contribute to patient discomfort in the form of additional needle sticks while hospitalized, Dr. Lau said in the presentation.
“We wanted to see how well physicians in the hospital used a risk assessment model to stratify patients,” and how well the patients were assigned to the correct prophylaxis, he explained.
Dr. Lau and colleagues reviewed data from 469 adult patients hospitalized at a single medical center who were hospitalized between January 2021 and June 2021. The researchers retrospectively performed risk assessment using the Padua prediction score. A score of less than 4 was considered low risk for VTE, and a score of 4 or higher was considered high risk.
In the study population, 180 patients were identified as low risk and 289 were considered high risk.
Based on the Padua score, 95% of the patients at high risk were on the correct prophylaxis, Dr. Lau said.
A total of 193 high-risk patients were on heparin. However, many of these patients had good kidney function, and could have been treated with enoxaparin instead; “this would have spared them two needle sticks per day,” Dr. Lau noted.
Of the 180 low-risk patients, 168 (93.3%) were on chemical prophylaxis, and should have been on mechanical prophylaxis, he said. Only 10 patients (5%) who were considered low risk were placed on mechanical prophylaxis.
Overall, 3.6% of all patients who received chemical VTE prophylaxis developed bleeding.
The results were limited by the retrospective design and use of data from a single center. However, the findings emphasize the need for better attention to VTE risk when considering prophylaxis, said Dr. Lau. “We have to have risk assessment every day,” during a hospital stay, and adjust treatment accordingly, he said.
he concluded.
Additional research is needed to better understand the potential consequences of overusing chemical VTE, including not only bleeding risk, but also financial costs and patient discomfort, he said.
The study received no outside funding. The researchers had no financial conflicts to disclose.
FROM CHEST 2022
Rapid point-of-care test could help avoid inappropriate antibiotic prescribing
The fingerstick test, FebriDx, works by detecting myxovirus resistance protein A, which the body generates in response to viral infections, and C-reactive protein (CRP), which is associated with systemic bacterial or viral infection.
In a study of 520 adults and children with symptoms of acute respiratory illness who were treated in outpatient settings, the test correctly classified bacterial infections 93.2% of the time (95% confidence interval [CI], 84.9-97.0). The negative predictive value (NPV), or probability that a person with a negative test result was truly free of a bacterial infection, was 98.7% (95% CI, 96.9-99.4).
The findings of the study, which was sponsored by the test’s manufacturer, were published in JAMA Network Open).
The ability to rule out a bacterial cause “may provide clinicians with reassurance to withhold antibiotics when supported by the clinical assessment,” the researchers wrote.
They added that the ability to identify infections that may benefit from antibiotics and confidently rule out those that will not “is essential to optimizing clinical management and addressing global antimicrobial resistance.”
FDA concerned about false negative viral infection results
FebriDx has been cleared for sale in the United Kingdom, Europe, Canada, United Arab Emirates, Brazil, and Australia, according to the manufacturer, Australia-based Lumos Diagnostics.
However, the product is not available in the United States, where the Food and Drug Administration denied marketing clearance in July. In a news release, Lumos said the FDA determined that FebriDx did not demonstrate “substantial equivalence” to a predicate device and expressed concern that false negative viral infection results could lead to missed cases of COVID-19.
In the newly published study, FebriDx identified individuals with viral infections 70.3% of the time (95% CI, 64.8-75.2). The probability that a person who tested negative for a viral infection was truly negative was 66.7% (95%CI, 60.8-72.1).
The study included patients with respiratory symptoms and recent fever who were enrolled from October 2019 to April 2021 at nine emergency departments, six urgent care clinics, and five primary care clinics in the United States. All patients were tested with FebriDx and underwent separate laboratory testing to determine a final diagnosis.
In addition, researchers recruited a control group of 120 individuals without symptoms.
Among 496 symptomatic individuals who had a final diagnosis, 73 (14.7%) were classified as having a response associated with a bacterial infection, 296 (59.7%) as having a viral-associated response, and 127 (25.6%) as negative.
FebriDx correctly ruled out a bacterial infection 88.4% of the time (95% CI, 85.0-91.1). The probability that a patient with a positive result for bacterial infection actually had a bacterial infection was 58.1% (95%CI, 49.1-66.7).
The findings bolster those of a previous study on the same test. This research included 220 patients who reported having a fever within the prior 3 days or had a measurable fever at the time of enrollment. In that study, the test correctly identified bacterial infections 85% of the time and correctly ruled out bacterial infection 93% of the time, with a NPV of 97%.
Too early to say test will be useful in practice
The idea of a test to guide the prescribing of antibiotics isn’t new, according to an expert who was not involved in FebriDx research.
Noah Ivers, MD, PhD, a family physician and associate professor at the University of Toronto who studies strategies to optimize primary care delivery, said, “many such point-of-care tests have been tried” to detect biomarkers such as CRP or procalcitonin, which is associated with bacterial infections.

Such tests have looked good in initial studies, he said, but when trialed in urgent care clinics, primary care clinics, or emergency departments, “they tend run into implementation challenges or simply lack of effects, or both.
“So, while I am happy at the news of this result, it’s too early to say with any certainty that it will prove useful in practice,” he added.
Meanwhile, Dr. Ivers said it’s “crucial that people understand that most illnesses are likely to be viral” and therefore not helped by antibiotics. When antibiotics are needed for outpatients, he said, “5 days is usually ample.”
The study was funded by Lumos Diagnostics. Among the 15 study authors, 6 had conflicts of interest disclosures, reporting ties to Inflammatix, Medical College of Wisconsin, Siemens, Technomics Research, and Lumos Diagnostics. Dr. Ivers reported no relevant financial interests.
The fingerstick test, FebriDx, works by detecting myxovirus resistance protein A, which the body generates in response to viral infections, and C-reactive protein (CRP), which is associated with systemic bacterial or viral infection.
In a study of 520 adults and children with symptoms of acute respiratory illness who were treated in outpatient settings, the test correctly classified bacterial infections 93.2% of the time (95% confidence interval [CI], 84.9-97.0). The negative predictive value (NPV), or probability that a person with a negative test result was truly free of a bacterial infection, was 98.7% (95% CI, 96.9-99.4).
The findings of the study, which was sponsored by the test’s manufacturer, were published in JAMA Network Open).
The ability to rule out a bacterial cause “may provide clinicians with reassurance to withhold antibiotics when supported by the clinical assessment,” the researchers wrote.
They added that the ability to identify infections that may benefit from antibiotics and confidently rule out those that will not “is essential to optimizing clinical management and addressing global antimicrobial resistance.”
FDA concerned about false negative viral infection results
FebriDx has been cleared for sale in the United Kingdom, Europe, Canada, United Arab Emirates, Brazil, and Australia, according to the manufacturer, Australia-based Lumos Diagnostics.
However, the product is not available in the United States, where the Food and Drug Administration denied marketing clearance in July. In a news release, Lumos said the FDA determined that FebriDx did not demonstrate “substantial equivalence” to a predicate device and expressed concern that false negative viral infection results could lead to missed cases of COVID-19.
In the newly published study, FebriDx identified individuals with viral infections 70.3% of the time (95% CI, 64.8-75.2). The probability that a person who tested negative for a viral infection was truly negative was 66.7% (95%CI, 60.8-72.1).
The study included patients with respiratory symptoms and recent fever who were enrolled from October 2019 to April 2021 at nine emergency departments, six urgent care clinics, and five primary care clinics in the United States. All patients were tested with FebriDx and underwent separate laboratory testing to determine a final diagnosis.
In addition, researchers recruited a control group of 120 individuals without symptoms.
Among 496 symptomatic individuals who had a final diagnosis, 73 (14.7%) were classified as having a response associated with a bacterial infection, 296 (59.7%) as having a viral-associated response, and 127 (25.6%) as negative.
FebriDx correctly ruled out a bacterial infection 88.4% of the time (95% CI, 85.0-91.1). The probability that a patient with a positive result for bacterial infection actually had a bacterial infection was 58.1% (95%CI, 49.1-66.7).
The findings bolster those of a previous study on the same test. This research included 220 patients who reported having a fever within the prior 3 days or had a measurable fever at the time of enrollment. In that study, the test correctly identified bacterial infections 85% of the time and correctly ruled out bacterial infection 93% of the time, with a NPV of 97%.
Too early to say test will be useful in practice
The idea of a test to guide the prescribing of antibiotics isn’t new, according to an expert who was not involved in FebriDx research.
Noah Ivers, MD, PhD, a family physician and associate professor at the University of Toronto who studies strategies to optimize primary care delivery, said, “many such point-of-care tests have been tried” to detect biomarkers such as CRP or procalcitonin, which is associated with bacterial infections.
Such tests have looked good in initial studies, he said, but when trialed in urgent care clinics, primary care clinics, or emergency departments, “they tend run into implementation challenges or simply lack of effects, or both.
“So, while I am happy at the news of this result, it’s too early to say with any certainty that it will prove useful in practice,” he added.
Meanwhile, Dr. Ivers said it’s “crucial that people understand that most illnesses are likely to be viral” and therefore not helped by antibiotics. When antibiotics are needed for outpatients, he said, “5 days is usually ample.”
The study was funded by Lumos Diagnostics. Among the 15 study authors, 6 had conflicts of interest disclosures, reporting ties to Inflammatix, Medical College of Wisconsin, Siemens, Technomics Research, and Lumos Diagnostics. Dr. Ivers reported no relevant financial interests.
The fingerstick test, FebriDx, works by detecting myxovirus resistance protein A, which the body generates in response to viral infections, and C-reactive protein (CRP), which is associated with systemic bacterial or viral infection.
In a study of 520 adults and children with symptoms of acute respiratory illness who were treated in outpatient settings, the test correctly classified bacterial infections 93.2% of the time (95% confidence interval [CI], 84.9-97.0). The negative predictive value (NPV), or probability that a person with a negative test result was truly free of a bacterial infection, was 98.7% (95% CI, 96.9-99.4).
The findings of the study, which was sponsored by the test’s manufacturer, were published in JAMA Network Open).
The ability to rule out a bacterial cause “may provide clinicians with reassurance to withhold antibiotics when supported by the clinical assessment,” the researchers wrote.
They added that the ability to identify infections that may benefit from antibiotics and confidently rule out those that will not “is essential to optimizing clinical management and addressing global antimicrobial resistance.”
FDA concerned about false negative viral infection results
FebriDx has been cleared for sale in the United Kingdom, Europe, Canada, United Arab Emirates, Brazil, and Australia, according to the manufacturer, Australia-based Lumos Diagnostics.
However, the product is not available in the United States, where the Food and Drug Administration denied marketing clearance in July. In a news release, Lumos said the FDA determined that FebriDx did not demonstrate “substantial equivalence” to a predicate device and expressed concern that false negative viral infection results could lead to missed cases of COVID-19.
In the newly published study, FebriDx identified individuals with viral infections 70.3% of the time (95% CI, 64.8-75.2). The probability that a person who tested negative for a viral infection was truly negative was 66.7% (95%CI, 60.8-72.1).
The study included patients with respiratory symptoms and recent fever who were enrolled from October 2019 to April 2021 at nine emergency departments, six urgent care clinics, and five primary care clinics in the United States. All patients were tested with FebriDx and underwent separate laboratory testing to determine a final diagnosis.
In addition, researchers recruited a control group of 120 individuals without symptoms.
Among 496 symptomatic individuals who had a final diagnosis, 73 (14.7%) were classified as having a response associated with a bacterial infection, 296 (59.7%) as having a viral-associated response, and 127 (25.6%) as negative.
FebriDx correctly ruled out a bacterial infection 88.4% of the time (95% CI, 85.0-91.1). The probability that a patient with a positive result for bacterial infection actually had a bacterial infection was 58.1% (95%CI, 49.1-66.7).
The findings bolster those of a previous study on the same test. This research included 220 patients who reported having a fever within the prior 3 days or had a measurable fever at the time of enrollment. In that study, the test correctly identified bacterial infections 85% of the time and correctly ruled out bacterial infection 93% of the time, with a NPV of 97%.
Too early to say test will be useful in practice
The idea of a test to guide the prescribing of antibiotics isn’t new, according to an expert who was not involved in FebriDx research.
Noah Ivers, MD, PhD, a family physician and associate professor at the University of Toronto who studies strategies to optimize primary care delivery, said, “many such point-of-care tests have been tried” to detect biomarkers such as CRP or procalcitonin, which is associated with bacterial infections.
Such tests have looked good in initial studies, he said, but when trialed in urgent care clinics, primary care clinics, or emergency departments, “they tend run into implementation challenges or simply lack of effects, or both.
“So, while I am happy at the news of this result, it’s too early to say with any certainty that it will prove useful in practice,” he added.
Meanwhile, Dr. Ivers said it’s “crucial that people understand that most illnesses are likely to be viral” and therefore not helped by antibiotics. When antibiotics are needed for outpatients, he said, “5 days is usually ample.”
The study was funded by Lumos Diagnostics. Among the 15 study authors, 6 had conflicts of interest disclosures, reporting ties to Inflammatix, Medical College of Wisconsin, Siemens, Technomics Research, and Lumos Diagnostics. Dr. Ivers reported no relevant financial interests.
FROM JAMA NETWORK OPEN