User login
Risk Stratification for Cellulitis Versus Noncellulitic Conditions of the Lower Extremity: A Retrospective Review of the NEW HAvUN Criteria
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.

Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
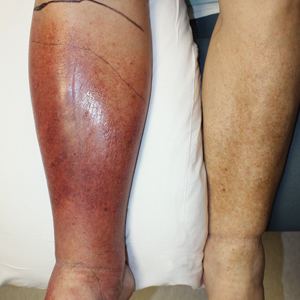
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7

Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
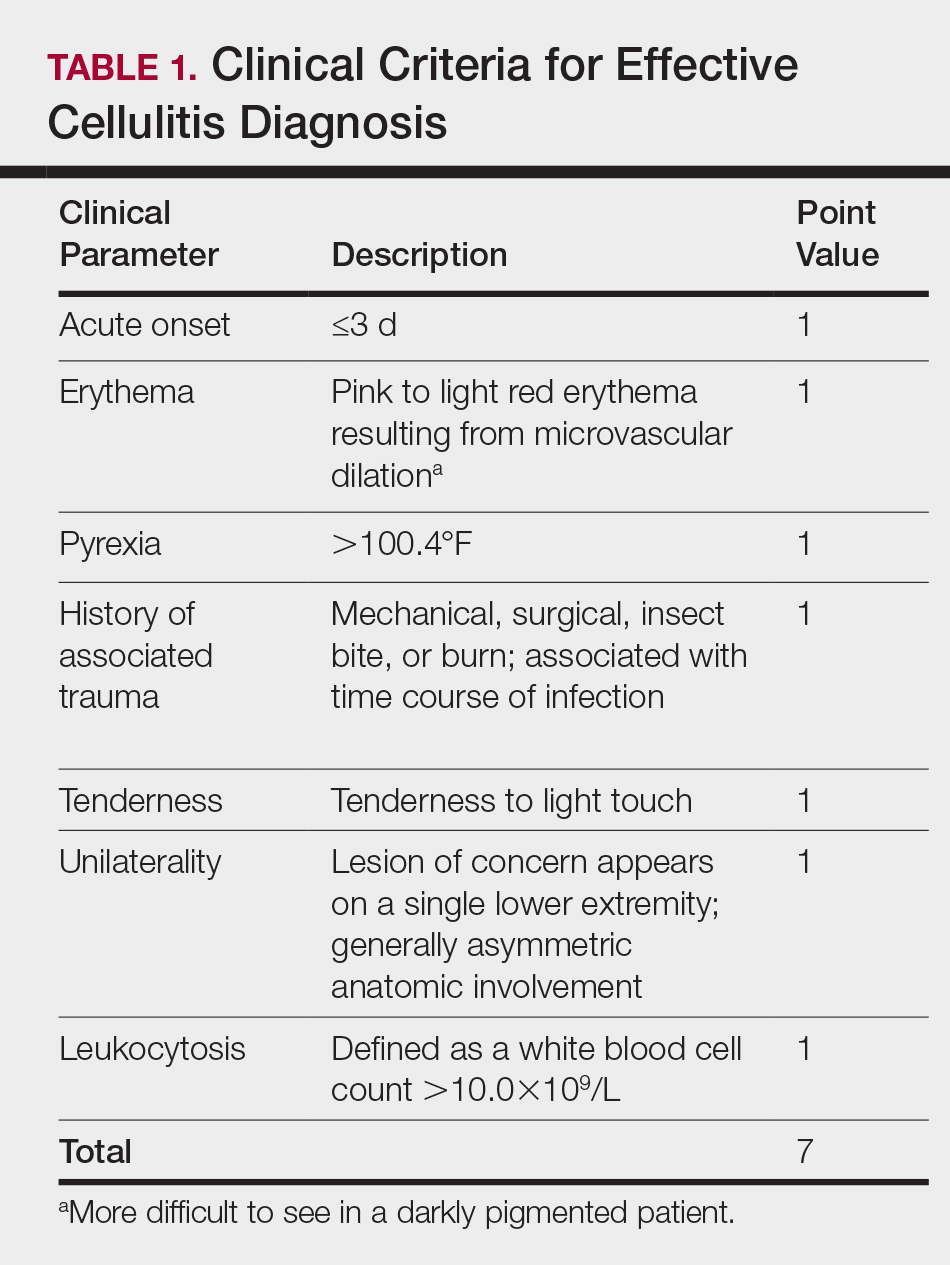
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).

Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).

A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
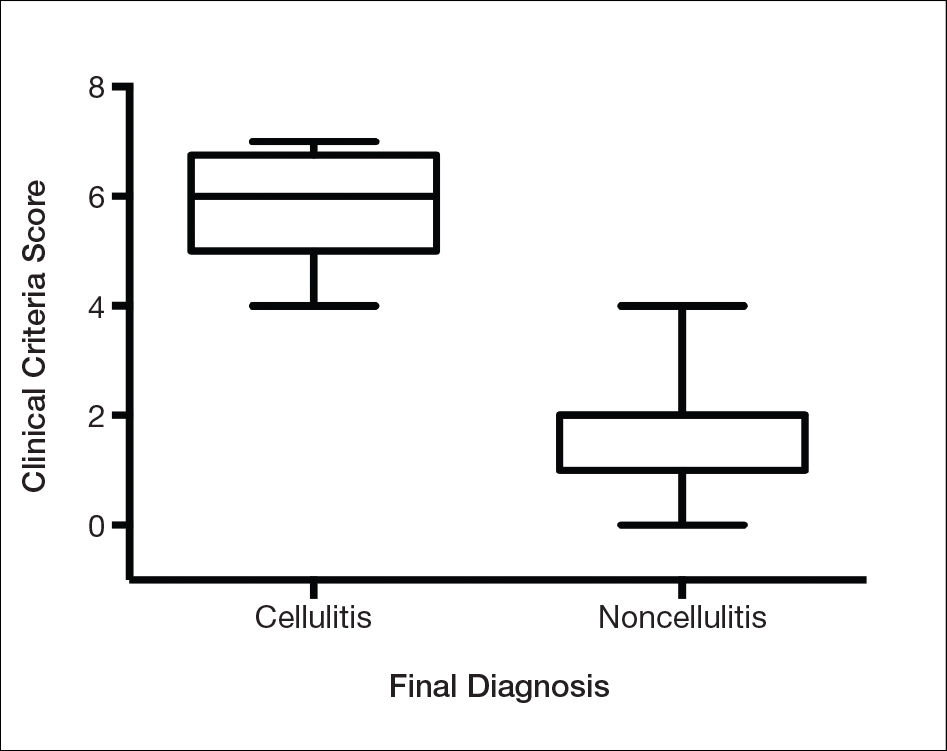
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.
Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7
Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).
Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).
A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
Cellulitis is defined as an acute or subacute, bacterial-induced inflammation of subcutaneous tissue that can extend superficially. The inciting incident often is assumed to be invasion of bacteria through loose connective tissue.1 Although cellulitis is bacterial in origin, it often is difficult to culture the offending microorganism from biopsy sites, swabs, or blood. Erythema, fever, induration, and tenderness are largely seen as clinical manifestations. Moderate and severe cases may be accompanied by fever, malaise, and leukocytosis. The lower extremity is the most common location of involvement (Figure 1), and usually a wound, ulcer, or interdigital superficial infection can be identified and implicated as the source of entry.
Effective treatment of cellulitis is necessary because complications such as abscesses, underlying fascia or muscle involvement, and septicemia can develop, leading to poor outcomes. Antibiotics should be administered intravenously in patients with suspected fascial involvement, septicemia, or dermal necrosis, or in those with an immunological comorbidity.2
The differential diagnosis of lower extremity cellulitis is wide due to the existence of several mimicking dermatologic conditions. These so-called pseudocellulitis conditions include stasis dermatitis, venous ulceration, acute lipodermatosclerosis, pigmented purpura, vasculopathy, contact dermatitis, adverse medication reaction, and arthropod bite. Stasis dermatitis and lipodermatosclerosis, both arising from venous insufficiency, are by far 2 of the most common skin conditions that imitate cellulitis.
Stasis dermatitis is a common condition in the United States and Europe, usually manifesting as a pigmented purpuric dermatosis on anterior tibial surfaces, around the ankle, or overlying dependent varicosities. Skin changes can include hyperpigmentation, edema, mild scaling, eczematous patches, and even ulceration.3
Lipodermatosclerosis is a disorder of progressive fibrosis of subcutaneous fat. It is more common in middle-aged women who have a high body mass index and a venous abnormality.4 This form of panniculitis typically affects the lower extremities bilaterally, manifesting as erythematous and indurated skin changes, sometimes described as inverted champagne bottles (Figure 2). At times, there can be accompanying painful ulceration on the erythematous areas, features that closely resemble cellulitis.5,6 Lipodermatosclerosis is commonly misdiagnosed as cellulitis, leading to inappropriate prescription of antibiotics.7
Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting. With a reported incidence of 24.6 per 100 person-years, cellulitis constitutes 1% to 14% of emergency department visits and 4% to 7% of hospital admissions.Therefore, prompt appropriate diagnosis and treatment can avoid life-threatening complications associated with infection such as sepsis, abscess, lymphangitis, and necrotizing fasciitis.8-11
It is estimated that 10% to 20% of patients who have been given a diagnosis of cellulitis do not actually have the disease.2,12 This discrepancy consumes a remarkable amount of hospital resources and can lead to inappropriate or excessive use of antibiotics.13 Although the true incidence of adverse antibiotic reactions is unknown, it is estimated that they are the cause of 3% to 6% of acute hospital admissions and occur in 10% to 15% of inpatients admitted for other primary reasons.14 These findings illustrate the potential for an increased risk for morbidity and increased length of stay for patients beginning an antibiotic regimen, especially when the agents are administered unnecessarily. In addition, inappropriate antibiotic use contributes to antibiotic resistance, which continues to be a major problem, especially in hospitalized patients.
There is a lack of consensus in the literature about methods to risk stratify patients who present with acute dermatologic conditions that include and resemble cellulitis. We sought to identify clinical features based on available clinical literature-derived variables. We tested our scheme in a series of patients with a known diagnosis of cellulitis or other dermatologic pathology of the lower extremity to assess the validity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Materials and Methods
This retrospective chart review was approved by the Yale University (New Haven, Connecticut) institutional review board (HIC#1409014533). Final diagnosis, demographic data, clinical manifestations, and relevant diagnostic laboratory values of 57 patients were obtained from a database in the dermatology department’s consultation log and electronic medical record database (December 2011 to December 2014). The presence of each clinical symptom—acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis—was assigned a score equal to 1; values were tallied to achieve a final score for each patient (Table 1). Patients who were seen initially as a consultation for possible cellulitis but given a final diagnosis of stasis dermatitis or lipodermatosclerosis were included (Table 2).
Clinical Criteria
The clinical criteria were developed based largely on clinical experience and relevant secondary literature.15-17 At the patient encounter, presence of each of the variables (Table 1) was assessed according to the following definitions:
- acute onset: within the prior 72 hours and more indicative of an acute infective process than a gradual and chronic consequence of venous stasis
- erythema: a subjective clinical marker for inflammation that can be associated with cellulitis, though darker, erythematous-appearing discolorations also can be seen in patients with chronic venous hypertension or valvular incompetence4,15
- pyrexia: body temperature greater than 100.4°F
- history of associated trauma: encompassing mechanical wounds, surgical incisions, burns, and insect bites that correlate closely to the time course of symptomatic development
- tenderness: tenderness to light touch, which may be more common in patients afflicted with cellulitis than in those with venous insufficiency
- unilaterality: a helpful distinguishing feature that points the diagnosis away from a dermatitislike clinical picture, especially because bilateral cellulitis is rare and regarded as a diagnostic pitfall18
- leukocytosis: white blood cell count greater than 10.0×109/L and is reasonably considered a cardinal metric of inflammatory processes, though it can be confounded by immunocompromise (low count) or steroid use (high count)
Statistical Analysis
Odds ratios (ORs) were calculated and χ2 analysis was performed for each presenting symptom using JMP 10.0 analytical software (SAS Institute Inc). Each patient was rated separately by means of the clinical feature–based scoring system for the calculation of a total score. After application of the score to the patient population, receiver operating characteristic curves were constructed to identify the optimal score threshold for discriminating cellulitis from dermatitis in this group. For each clinical feature, P<.05 was considered significant.
Results
Our cohort included 32 male and 25 female patients with a mean age of 63 and 61 years, respectively. The final clinical diagnosis of cellulitis was made in 20 patients (35%). An established diagnosis of cellulitis was assigned based on a dermatology evaluation located within our electronic medical record database (Table 2).
Each clinical parameter was evaluated separately for each patient; combined results are summarized in Table 3. Acute onset (≤3 days) was a clinical characteristic seen in 80% (16/20) of cellulitis cases and 22% (8/37) of noncellulitis cases (OR, 14.5; P<.001). Erythema had similar significance (OR, 10.3; prevalence, 95% [19/20] vs 65% [24/37]; P=.012). Pyrexia possessed an OR of 99.2 for cellulitis and was seen in 85% (17/20) of cellulitis cases and only 5% (2/37) of noncellulitis cases (P<.001).
A history of associated trauma had an OR of 36.0 for cellulitis, with 50% (10/20) and 3% (1/37) prevalence in cellulitis cases and noncellulitis cases, respectively (P<.001). Tenderness, documented in 90% (18/20) of cellulitis cases and 43% (16/37) of noncellulitis cases, had an OR of 11.8 (P<.001).
Unilaterality had 100% (20/20) prevalence in our cellulitis cohort and was the only characteristic within the algorithm that yielded an incalculable OR. Noncellulitis or stasis dermatitis of the lower extremity exhibited a unilateral lesion in 11 cases (30%), of which 1 case resulted from a unilateral tibial fracture. Leukocytosis was seen in 65% (13/20) of cellulitis cases and 8% (3/37) of noncellulitis cases, with an OR for cellulitis of 21.0 (P<.001).
All parameters were significant by χ2 analysis (Table 3).
Comment
We found that testing positive for 4 of 7 clinical criteria for assessing cellulitis was highly specific (95%) and sensitive (100%) for a diagnosis of cellulitis among its range of mimics (Figure 3). These cellulitis criteria can be remembered, with some modification, using NEW HAvUN as a mnemonic device (New onset,
Consistent with the literature, pyrexia, history of associated trauma, and unilaterality also were predictors of cellulitis diagnosis. Unilaterality often is used as a diagnostic tool by dermatologist consultants when a patient lacks other criteria for cellulitis, so these findings are intuitive and consistent with our institutional experience. Interestingly, leukocytosis was seen in only 65% of cellulitis cases and 8% of noncellulitis cases and therefore might not serve as a sensitive independent predictor of a diagnosis of cellulitis, emphasizing the importance of the multifactorial scoring system we have put forward. Additionally, acuity of onset, erythema, and tenderness are not independently associated with cellulitis when assessing a patient because several of those findings are present in other dermatologic conditions of the lower extremity; when combined with the other criteria, however, these 3 findings can play a role in diagnosis.
Effective cellulitis diagnosis provides well-recognized challenges in the acute medical setting because many clinical mimics exist. The estimated rate of misdiagnosed cellulitis is certainly well-established: 30% to 75% in independent and multi-institutional studies. These studies also revealed that patients admitted for bilateral “cellulitis” overwhelmingly tended to be stasis clinical pictures.13,19
Cost implications from inappropriate diagnosis largely regard inappropriate antibiotic use and the potential for microbial resistance, with associated costs estimated to be more than $50 billion (2004 dollars).20,21 The true cost burden is extremely difficult to model or predict due to remarkable variations in the institutional misdiagnosis rate, prescribing pattern, and antibiotic cost and could represent avenues of further study. Misappropriation of antibiotics includes not only a monetary cost that encompasses all aspects of acute treatment and hospitalization but also an unquantifiable cost: human lives associated with the consequences of antibiotic resistance.
Conclusion
There is a lack of consensus or criteria for differentiating cellulitis from its most common clinical counterparts. Here, we propose a convenient clinical correlation system that we hope will lead to more efficient allocation of clinical resources, including antibiotics and hospital admissions, while lowering the incidence of adverse events and leading to better patient outcomes. We recognize that the small sample size of our study may limit broad application of these criteria, though we anticipate that further prospective studies can improve the diagnostic relevance and risk-assessment power of the NEW HAvUN criteria put forth here for assessing cellulitis in the acute medical setting.
Acknowledgement—Author H.H.E. recognizes the loving memory of Nadia Ezaldein for her profound influence on and motivation behind this research.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
- Lep
pard BJ, Seal DV, Colman G, et al. The value of bacteriology and serology in the diagnosis of cellulitis and erysipelas. Br J Dermatol. 1985;112:559-567. - Hep
burn MJ, Dooley DP, Skidmore PJ, et al. Comparison of short-course (5 days) and standard (10 days) treatment for uncomplicated cellulitis. Arch Int Med. 2004;164:1669-1674. - Bergan JJ, Schmid-Schönbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355:488-498.
- Bruc
e AJ, Bennett DD, Lohse CM, et al. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol. 2002;46:187-192. - Heym
ann WR. Lipodermatosclerosis. J Am Acad Dermatol. 2009;60:1022-1023. - Vesi
ć S, Vuković J, Medenica LJ, et al. Acute lipodermatosclerosis: an open clinical trial of stanozolol in patients unable to sustain compression therapy. Dermatol Online J. 2008;14:1. - Keller
EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-552. - Dong SL, Kelly KD, Oland RC, et al. ED management of cellulitis: a review of five urban centers. Am J Emerg Med. 2001;19:535-540.
- Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
- Manfredi R, Calza L, Chiodo F. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol. 2002;29:168-172.
- Pascarella L, Schonbein GW, Bergan JJ. Microcirculation and venous ulcers: a review. Ann Vasc Surg. 2005;19:921-927.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician. 2003;67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hay RJ, Adriaans BM. Bacterial infections. In: Thong BY, Tan TC. Epidemiology and risk factors for drug allergy. 8th ed. Br J Clin Pharmacol. 2011;71:684-700.
- Hay RJ, Adriaans BM. Bacterial infections. In: Burns T, Breathnach S, Cox N, et al. Rook’s Textbook of Dermatology. 8th ed. Hoboken, NJ: John Wiley & Sons, Inc; 2004:1345-1426.
- Wolff K, Goldsmith LA, Katz SI, et al. Fitzpatrick’s Dermatology In General Medicine. 7th ed. New York, NY: McGraw-Hill; 2003.
- Sommer LL, Reboli AC, Heymann WR. Bacterial infections. In: Bolognia J, Schaffer J, Cerroni L, et al. Dermatology. Vol 4. Philadelphia, PA: Elsevier Saunders; 2012:1462-1502.
- Cox NH. Management of lower leg cellulitis. Clin Med. 2002;2:23-27.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Pinder R, Sallis A, Berry D, et al. Behaviour change and antibiotic prescribing in healthcare settings: literature review and behavioural analysis. London, UK: Public Health England; February 2015. https://assets.publishing.service.gov.uk/government/
uploads/system/uploads/attachment_data/file/405031
/Behaviour_Change_for_Antibiotic_Prescribing_-_FINAL.pdf. Accessed May 7, 2018. - Smith R, Coast J. The true cost of antimicrobial resistance. BMJ. 2013;346:f1493.
Practice Points
- Distinguishing cellulitis from noncellulitic conditions of the lower extremity is paramount to effective patient management in the emergent setting, given that misdiagnosis consumes hospital resources and can lead to inappropriate or excessive use of antibiotics.
- We evaluated the specificity and sensitivity of the following 7 clinical criteria: acute onset, erythema, pyrexia, history of associated trauma, tenderness, unilaterality, and leukocytosis.
Gene assays reveal some ‘unknown primary’ cancers as RCC
Gene expression profiling and/or immunohistochemistry can identify occult renal cell carcinoma (RCC) in a subset of patients diagnosed with carcinoma of unknown primary (CUP), suggesting that these patients could benefit from RCC-specific targeted therapy or immunotherapy, investigators contend.
Of 539 patients presenting at a single center with CUP, a 92-gene reverse transcription polymerase chain reaction molecular cancer classifier assay (MCCA) performed on biopsy specimens identified 24 as having RCC. All of the patients had clinical characteristics typical of advanced RCC, but none had suspicious renal lesions on CT scans, reported F. Anthony Greco, MD and John D. Hainsworth, MD, of the Sarah Cannon Cancer Center and Research Institute in Nashville, Tenn.
“Although further experience is necessary, these patients responded to RCC-specific therapy in a manner consistent with advanced RCC. These patients are unlikely to benefit from treatment with empiric chemotherapy. The reliable identification of RCC patients within the heterogeneous CUP population is possible using MCCA, and has potentially important therapeutic implications,” they wrote. The report was published in Clinical Genitourinary Cancer.
They noted that previously the only therapeutic option for patients with CUP suspected of being renal in origin was ineffective systemic chemotherapy, making a specific diagnosis of more academic interest than clinical importance.
“This situation has now changed because of the introduction of several targeted agents and immune checkpoint blockers that improve survival in patients with advanced RCC. It is likely that these new RCC treatments are also more effective than empiric chemotherapy for patients with CUP who have an occult renal primary site. Therefore, recognition of the RCC subset of patients within the CUP population has practical therapeutic importance,” they wrote.
Dr. Greco and Dr. Hainsworth conducted a retrospective review of patients at their center with CUP from 2008 through 2013 who had RCC predicted by MCCA.
A total of 539 patients presented with CUP during the study period, and of this group, 24 (4.4%) had RCC identified by MCCA.
The patients, 18 men and 6 women, with a median age of 61 years, all had abdominal CT scans that failed to show renal lesions suggestive of a primary RCC. Nine of the 24 patients had baseline MCCA performed as part of a prospective phase 2 clinical trial; the other 15 were patients treated at the center who had MCCA performed later in the clinical course, usually during or after first-line empiric chemotherapy.
Sixteen patients had metastases in the retroperitoneum, 10 in the mediastinum, 6 in bone, 5 in the liver, and 5 in lungs and/or pleura.
Pathologic studies using light microscopy showed poorly differentiated carcinomas in eight patients, poorly differentiated adenocarcinomas in nine, and well or moderately differentiated adenocarcinomas in seven patients.
A pathologist identified RCC as the possible primary in only 4 of the 24 patients. Immunohistochemistry tests in these patients were consistent with a diagnosis of RCC. Only 5 of the 24 had focal features suggestive of RCC, including one clear-cell and four papillary histologies.
Sixteen of the 24 patients received first-line treatment for advanced RCC, including sunitinib, temsirolimus, bevacizumab, and/or interleukin 1. Four other patients received RCC-specific therapy following empiric chemotherapy (three patients who received RCC-specific therapies in the first line also received it in the second line).
Among the 16 patients who received first-line RCC-specific therapies there were 3 partial responses (PR), 10 cases of stable disease (SD), 2 of progressive disease (PD), and 1 patient was not evaluable. The median duration of both PR and SD was 8 months.
Of the eight patients who received first-line empiric chemotherapy, one had a PR, two had SD, and five had PD.
For the seven patients who received second-line RCC-specific therapy after either first-line chemotherapy or site-specific therapy, responses included one PR, two SD, two PD, and two not evaluable.
Median survival for all 24 patients was 12 months (range 2 to more than 43 months). Median survival of the 16 patients who received first-line RCC-specific treatment was 14 months (range 2-25 months).
Median survival for all 20 patients who received RCC-specific treatment at some time during their course was 16 months (range, 2 to more than 43 months).
The authors called for further prospective studies of this subset of patients with CUP.
SOURCE: Greco FA, Hainsworth JD. Clin Genitourin Cancer. 2018 Aug;16(4):e893-8.
Gene expression profiling and/or immunohistochemistry can identify occult renal cell carcinoma (RCC) in a subset of patients diagnosed with carcinoma of unknown primary (CUP), suggesting that these patients could benefit from RCC-specific targeted therapy or immunotherapy, investigators contend.
Of 539 patients presenting at a single center with CUP, a 92-gene reverse transcription polymerase chain reaction molecular cancer classifier assay (MCCA) performed on biopsy specimens identified 24 as having RCC. All of the patients had clinical characteristics typical of advanced RCC, but none had suspicious renal lesions on CT scans, reported F. Anthony Greco, MD and John D. Hainsworth, MD, of the Sarah Cannon Cancer Center and Research Institute in Nashville, Tenn.
“Although further experience is necessary, these patients responded to RCC-specific therapy in a manner consistent with advanced RCC. These patients are unlikely to benefit from treatment with empiric chemotherapy. The reliable identification of RCC patients within the heterogeneous CUP population is possible using MCCA, and has potentially important therapeutic implications,” they wrote. The report was published in Clinical Genitourinary Cancer.
They noted that previously the only therapeutic option for patients with CUP suspected of being renal in origin was ineffective systemic chemotherapy, making a specific diagnosis of more academic interest than clinical importance.
“This situation has now changed because of the introduction of several targeted agents and immune checkpoint blockers that improve survival in patients with advanced RCC. It is likely that these new RCC treatments are also more effective than empiric chemotherapy for patients with CUP who have an occult renal primary site. Therefore, recognition of the RCC subset of patients within the CUP population has practical therapeutic importance,” they wrote.
Dr. Greco and Dr. Hainsworth conducted a retrospective review of patients at their center with CUP from 2008 through 2013 who had RCC predicted by MCCA.
A total of 539 patients presented with CUP during the study period, and of this group, 24 (4.4%) had RCC identified by MCCA.
The patients, 18 men and 6 women, with a median age of 61 years, all had abdominal CT scans that failed to show renal lesions suggestive of a primary RCC. Nine of the 24 patients had baseline MCCA performed as part of a prospective phase 2 clinical trial; the other 15 were patients treated at the center who had MCCA performed later in the clinical course, usually during or after first-line empiric chemotherapy.
Sixteen patients had metastases in the retroperitoneum, 10 in the mediastinum, 6 in bone, 5 in the liver, and 5 in lungs and/or pleura.
Pathologic studies using light microscopy showed poorly differentiated carcinomas in eight patients, poorly differentiated adenocarcinomas in nine, and well or moderately differentiated adenocarcinomas in seven patients.
A pathologist identified RCC as the possible primary in only 4 of the 24 patients. Immunohistochemistry tests in these patients were consistent with a diagnosis of RCC. Only 5 of the 24 had focal features suggestive of RCC, including one clear-cell and four papillary histologies.
Sixteen of the 24 patients received first-line treatment for advanced RCC, including sunitinib, temsirolimus, bevacizumab, and/or interleukin 1. Four other patients received RCC-specific therapy following empiric chemotherapy (three patients who received RCC-specific therapies in the first line also received it in the second line).
Among the 16 patients who received first-line RCC-specific therapies there were 3 partial responses (PR), 10 cases of stable disease (SD), 2 of progressive disease (PD), and 1 patient was not evaluable. The median duration of both PR and SD was 8 months.
Of the eight patients who received first-line empiric chemotherapy, one had a PR, two had SD, and five had PD.
For the seven patients who received second-line RCC-specific therapy after either first-line chemotherapy or site-specific therapy, responses included one PR, two SD, two PD, and two not evaluable.
Median survival for all 24 patients was 12 months (range 2 to more than 43 months). Median survival of the 16 patients who received first-line RCC-specific treatment was 14 months (range 2-25 months).
Median survival for all 20 patients who received RCC-specific treatment at some time during their course was 16 months (range, 2 to more than 43 months).
The authors called for further prospective studies of this subset of patients with CUP.
SOURCE: Greco FA, Hainsworth JD. Clin Genitourin Cancer. 2018 Aug;16(4):e893-8.
Gene expression profiling and/or immunohistochemistry can identify occult renal cell carcinoma (RCC) in a subset of patients diagnosed with carcinoma of unknown primary (CUP), suggesting that these patients could benefit from RCC-specific targeted therapy or immunotherapy, investigators contend.
Of 539 patients presenting at a single center with CUP, a 92-gene reverse transcription polymerase chain reaction molecular cancer classifier assay (MCCA) performed on biopsy specimens identified 24 as having RCC. All of the patients had clinical characteristics typical of advanced RCC, but none had suspicious renal lesions on CT scans, reported F. Anthony Greco, MD and John D. Hainsworth, MD, of the Sarah Cannon Cancer Center and Research Institute in Nashville, Tenn.
“Although further experience is necessary, these patients responded to RCC-specific therapy in a manner consistent with advanced RCC. These patients are unlikely to benefit from treatment with empiric chemotherapy. The reliable identification of RCC patients within the heterogeneous CUP population is possible using MCCA, and has potentially important therapeutic implications,” they wrote. The report was published in Clinical Genitourinary Cancer.
They noted that previously the only therapeutic option for patients with CUP suspected of being renal in origin was ineffective systemic chemotherapy, making a specific diagnosis of more academic interest than clinical importance.
“This situation has now changed because of the introduction of several targeted agents and immune checkpoint blockers that improve survival in patients with advanced RCC. It is likely that these new RCC treatments are also more effective than empiric chemotherapy for patients with CUP who have an occult renal primary site. Therefore, recognition of the RCC subset of patients within the CUP population has practical therapeutic importance,” they wrote.
Dr. Greco and Dr. Hainsworth conducted a retrospective review of patients at their center with CUP from 2008 through 2013 who had RCC predicted by MCCA.
A total of 539 patients presented with CUP during the study period, and of this group, 24 (4.4%) had RCC identified by MCCA.
The patients, 18 men and 6 women, with a median age of 61 years, all had abdominal CT scans that failed to show renal lesions suggestive of a primary RCC. Nine of the 24 patients had baseline MCCA performed as part of a prospective phase 2 clinical trial; the other 15 were patients treated at the center who had MCCA performed later in the clinical course, usually during or after first-line empiric chemotherapy.
Sixteen patients had metastases in the retroperitoneum, 10 in the mediastinum, 6 in bone, 5 in the liver, and 5 in lungs and/or pleura.
Pathologic studies using light microscopy showed poorly differentiated carcinomas in eight patients, poorly differentiated adenocarcinomas in nine, and well or moderately differentiated adenocarcinomas in seven patients.
A pathologist identified RCC as the possible primary in only 4 of the 24 patients. Immunohistochemistry tests in these patients were consistent with a diagnosis of RCC. Only 5 of the 24 had focal features suggestive of RCC, including one clear-cell and four papillary histologies.
Sixteen of the 24 patients received first-line treatment for advanced RCC, including sunitinib, temsirolimus, bevacizumab, and/or interleukin 1. Four other patients received RCC-specific therapy following empiric chemotherapy (three patients who received RCC-specific therapies in the first line also received it in the second line).
Among the 16 patients who received first-line RCC-specific therapies there were 3 partial responses (PR), 10 cases of stable disease (SD), 2 of progressive disease (PD), and 1 patient was not evaluable. The median duration of both PR and SD was 8 months.
Of the eight patients who received first-line empiric chemotherapy, one had a PR, two had SD, and five had PD.
For the seven patients who received second-line RCC-specific therapy after either first-line chemotherapy or site-specific therapy, responses included one PR, two SD, two PD, and two not evaluable.
Median survival for all 24 patients was 12 months (range 2 to more than 43 months). Median survival of the 16 patients who received first-line RCC-specific treatment was 14 months (range 2-25 months).
Median survival for all 20 patients who received RCC-specific treatment at some time during their course was 16 months (range, 2 to more than 43 months).
The authors called for further prospective studies of this subset of patients with CUP.
SOURCE: Greco FA, Hainsworth JD. Clin Genitourin Cancer. 2018 Aug;16(4):e893-8.
FROM CLINICAL GENITOURINARY CANCER
Key clinical point: Some carcinomas of unknown primary (CUP) can be identified as renal in origin by molecular assays and treated accordingly.
Major finding: Molecular assays identified RCC as the primary in 24 of 539 patients with CUP.
Study details: Retrospective review of patients with CUP presenting at a single center from 2008 through 2013.
Disclosures: The Minnie Pearl Cancer Research Foundation supported the study. Dr. Greco disclosed a consultant role and speakers bureau activities for bioTheranostics.
Source: Greco FA, Hainsworth JD. Clinical Genitourinary Cancer 16(4): e893-8.
Rituximab reduces risk of follicular lymphoma transformation
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
FROM THE LANCET HAEMATOLOGY
Key clinical point:
Major finding: The 10-year cumulative hazard of histologic transformation was 5.2% for patients who had received rituximab and 8.7% for those who had not.
Study details: Retrospective pooled analysis of 8,116 patients with FL, 509 of whom had transformation over a 10-year period.
Disclosures: The study was funded by Associazione Angela Serra per la Ricerca sul Cancro, European Lymphoma Institute, European Hematology Association Lymphoma Group, Fondazione Italiana Linfomi, and the Spanish Group of Lymphoma and Bone Marrow Transplantation. The researchers reported having no financial disclosures.
Source: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
ACS NSQIP project collected patient-reported data on surgery outcomes
ORLANDO – A pilot survey to generate had a high response rate and yielded clinically meaningful data, an investigator reported at the American College of Surgeons Quality and Safety Conference.
The 45-question electronic survey, conducted as part of the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) had 1,300 respondents with a response rate of 20%, according to Jason B. Liu, MD, an ACS Clinical Scholar-in-Residence and general surgery resident at the University of Chicago.
Results to date have demonstrated that in patients undergoing total knee arthroplasty (TKA), pain had a greater impact on daily activities than for other procedures, Dr. Liu said in a general session presentation the conference.
“Overall, the lesson learned is that in the current health care landscape, with its regulations and privacy issues, it is indeed both feasible and acceptable to electronically measure patient-reported outcomes using the ACS NSQIP platform,” Dr. Liu said at the meeting.
Eighteen hospitals in the United States and Canada participated in the pilot survey, which elicited responses from patients with a median age of 63 years, representing more than 340 types of operations.
The survey incorporates measurements from the PROMIS Pain Interference instrument, which measures how much pain hinders daily activities; PROMIS Global Health, which measures physical and mental health; and aspects of the Consumer Assessment of Healthcare Providers and Systems Surgical Care Survey (S-CAHPS), Dr. Liu said.
The TKA finding is just one example of the data obtained through the pilot, he said. Looking at PROMIS Pain Interference, pain had more impact in TKA patients compared with open GI, breast hernia, and laparoscopic GI procedures. Difference between means ranged from 3.2 to 9.4 for TKA, compared with those procedures.
Conducting the pilot has been an “uphill battle,” according to Dr. Liu, citing critics who wondered if the program would generate meaningful data, whether older patients would respond to an electronic survey, and whether patients would take time to fill out a 45-question survey.
In fact, the average completion time for the survey was just 6.4 minutes, and the median number of items missing was zero, meaning that patients who started the survey tended to finish it, he said.
“We really hope to expand what we’ve learned across all of the [ACS] quality programs so that we can begin to really incorporate the patients’ perspective in improving national surgical quality,” he said.
Dr. Liu had no disclosures to report.
ORLANDO – A pilot survey to generate had a high response rate and yielded clinically meaningful data, an investigator reported at the American College of Surgeons Quality and Safety Conference.
The 45-question electronic survey, conducted as part of the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) had 1,300 respondents with a response rate of 20%, according to Jason B. Liu, MD, an ACS Clinical Scholar-in-Residence and general surgery resident at the University of Chicago.
Results to date have demonstrated that in patients undergoing total knee arthroplasty (TKA), pain had a greater impact on daily activities than for other procedures, Dr. Liu said in a general session presentation the conference.
“Overall, the lesson learned is that in the current health care landscape, with its regulations and privacy issues, it is indeed both feasible and acceptable to electronically measure patient-reported outcomes using the ACS NSQIP platform,” Dr. Liu said at the meeting.
Eighteen hospitals in the United States and Canada participated in the pilot survey, which elicited responses from patients with a median age of 63 years, representing more than 340 types of operations.
The survey incorporates measurements from the PROMIS Pain Interference instrument, which measures how much pain hinders daily activities; PROMIS Global Health, which measures physical and mental health; and aspects of the Consumer Assessment of Healthcare Providers and Systems Surgical Care Survey (S-CAHPS), Dr. Liu said.
The TKA finding is just one example of the data obtained through the pilot, he said. Looking at PROMIS Pain Interference, pain had more impact in TKA patients compared with open GI, breast hernia, and laparoscopic GI procedures. Difference between means ranged from 3.2 to 9.4 for TKA, compared with those procedures.
Conducting the pilot has been an “uphill battle,” according to Dr. Liu, citing critics who wondered if the program would generate meaningful data, whether older patients would respond to an electronic survey, and whether patients would take time to fill out a 45-question survey.
In fact, the average completion time for the survey was just 6.4 minutes, and the median number of items missing was zero, meaning that patients who started the survey tended to finish it, he said.
“We really hope to expand what we’ve learned across all of the [ACS] quality programs so that we can begin to really incorporate the patients’ perspective in improving national surgical quality,” he said.
Dr. Liu had no disclosures to report.
ORLANDO – A pilot survey to generate had a high response rate and yielded clinically meaningful data, an investigator reported at the American College of Surgeons Quality and Safety Conference.
The 45-question electronic survey, conducted as part of the American College of Surgeons National Surgical Quality Improvement Program (ACS NSQIP) had 1,300 respondents with a response rate of 20%, according to Jason B. Liu, MD, an ACS Clinical Scholar-in-Residence and general surgery resident at the University of Chicago.
Results to date have demonstrated that in patients undergoing total knee arthroplasty (TKA), pain had a greater impact on daily activities than for other procedures, Dr. Liu said in a general session presentation the conference.
“Overall, the lesson learned is that in the current health care landscape, with its regulations and privacy issues, it is indeed both feasible and acceptable to electronically measure patient-reported outcomes using the ACS NSQIP platform,” Dr. Liu said at the meeting.
Eighteen hospitals in the United States and Canada participated in the pilot survey, which elicited responses from patients with a median age of 63 years, representing more than 340 types of operations.
The survey incorporates measurements from the PROMIS Pain Interference instrument, which measures how much pain hinders daily activities; PROMIS Global Health, which measures physical and mental health; and aspects of the Consumer Assessment of Healthcare Providers and Systems Surgical Care Survey (S-CAHPS), Dr. Liu said.
The TKA finding is just one example of the data obtained through the pilot, he said. Looking at PROMIS Pain Interference, pain had more impact in TKA patients compared with open GI, breast hernia, and laparoscopic GI procedures. Difference between means ranged from 3.2 to 9.4 for TKA, compared with those procedures.
Conducting the pilot has been an “uphill battle,” according to Dr. Liu, citing critics who wondered if the program would generate meaningful data, whether older patients would respond to an electronic survey, and whether patients would take time to fill out a 45-question survey.
In fact, the average completion time for the survey was just 6.4 minutes, and the median number of items missing was zero, meaning that patients who started the survey tended to finish it, he said.
“We really hope to expand what we’ve learned across all of the [ACS] quality programs so that we can begin to really incorporate the patients’ perspective in improving national surgical quality,” he said.
Dr. Liu had no disclosures to report.
REPORTING FROM ACSQSC 2018
Key clinical point: Clinically meaningful data on patient-reported outcomes can be obtained using the ACS NSQIP platform.
Major finding: The average completion time for the survey was 6.4 minutes, and the median number of items missing was zero.
Study details: A 45-question electronic survey of 1,300 patients treated at 18 hospitals for 340 different types of surgical procedures.
Disclosures: Dr. Liu had no disclosures to report.
Pseudotumor cerebri pediatric rates are rising
Pseudotumor cerebri, benign intracranial hypertension, and idiopathic intracranial hypertension are all terms to describe a syndrome of increased intracranial pressure, headaches, vision loss, or changes without an associated mass lesion.1 The condition was considered relatively rare, presenting most commonly in obese women in childbearing years. Surprisingly, 2

Obesity is the fastest growing morbidity among adolescents. The Centers for Disease Control and Prevention reported 32% of children 2-19 years were obese.1 This reality is impacting many areas of an adolescent’s health, but it also is changing the landscape of diseases that present in this age group. Although pediatric and adult pseudotumor cerebri always have had slightly varied features, many features were similar such as the papilledema, vision loss, headaches, and sixth nerve palsy. Obesity and female predominance tended to present more in the adult population, as many pediatric patients were not obese,2 and had fewer associated symptoms at the time of diagnosis, and the cause was thought to idiopathic.
Now, with the increase in obesity, more adolescents and more male patients are presenting with pseudotumor cerebri as a cause for their headache, and 57%-100% are obese, making it a compounding factor.3
Pediatric populations also are at risk of secondary pseudotumor cerebri, which is an increase in intracranial pressure from the use of medication, or other disease states such as anemia, kidney disease, or Down syndrome. Minocycline use is the most common medication cause and usually presents 1-2 months after normal use.4 Discontinuing the drug does lead to resolution. Retinoids, vitamin A products, growth hormone, and steroids also have been implicated. Given that acne is a common complaint amongst teens, knowledge of these side effects is important.4
In 2013, the criteria for diagnosis of pseudotumor cerebri was revised. Currently, the presence of papilledema, normal neurologic exam except for abnormal sixth cranial nerve, normal cerebral spinal fluid, elevated lumbar opening pressure, and normal imaging are needed for a definitive diagnosis. A probable diagnosis can be made if papilledema is not present but there abducens nerve palsy.2
In a routine physical exam, when I questioned a patient on any medication that was used daily, she replied she took ibuprofen daily for headaches and that she had been doing this for several months. Headaches were not in her chief complaints as she had learned to live with and ignore this symptom. Upon further evaluation, she was slightly overweight and has a questionable fundoscopic exam. After further evaluation by an ophthalmologist and a neurologist, pseudotumor cerebri was diagnosed.
Index of suspicion is key in correctly diagnosing patients, and understanding the changing landscape of medicine will lead to more thoughtful questioning during routine health exams and better outcomes for your patients.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Am J Ophthalmol. 2015 Feb;159(2):344-52.e1.
2. Horm Res Paediatr. 2014;81(4):217-25.
3. Clin Imaging. 2018 May 24. doi: 10.1016/j.clinimag.2018.05.020.
4. Am J Ophthalmol. 1998 Jul;126(1):116-21.
5. Glob Pediatr Health. 2018. doi:10.1177/2333794X18785550.
Pseudotumor cerebri, benign intracranial hypertension, and idiopathic intracranial hypertension are all terms to describe a syndrome of increased intracranial pressure, headaches, vision loss, or changes without an associated mass lesion.1 The condition was considered relatively rare, presenting most commonly in obese women in childbearing years. Surprisingly, 2
Obesity is the fastest growing morbidity among adolescents. The Centers for Disease Control and Prevention reported 32% of children 2-19 years were obese.1 This reality is impacting many areas of an adolescent’s health, but it also is changing the landscape of diseases that present in this age group. Although pediatric and adult pseudotumor cerebri always have had slightly varied features, many features were similar such as the papilledema, vision loss, headaches, and sixth nerve palsy. Obesity and female predominance tended to present more in the adult population, as many pediatric patients were not obese,2 and had fewer associated symptoms at the time of diagnosis, and the cause was thought to idiopathic.
Now, with the increase in obesity, more adolescents and more male patients are presenting with pseudotumor cerebri as a cause for their headache, and 57%-100% are obese, making it a compounding factor.3
Pediatric populations also are at risk of secondary pseudotumor cerebri, which is an increase in intracranial pressure from the use of medication, or other disease states such as anemia, kidney disease, or Down syndrome. Minocycline use is the most common medication cause and usually presents 1-2 months after normal use.4 Discontinuing the drug does lead to resolution. Retinoids, vitamin A products, growth hormone, and steroids also have been implicated. Given that acne is a common complaint amongst teens, knowledge of these side effects is important.4
In 2013, the criteria for diagnosis of pseudotumor cerebri was revised. Currently, the presence of papilledema, normal neurologic exam except for abnormal sixth cranial nerve, normal cerebral spinal fluid, elevated lumbar opening pressure, and normal imaging are needed for a definitive diagnosis. A probable diagnosis can be made if papilledema is not present but there abducens nerve palsy.2
In a routine physical exam, when I questioned a patient on any medication that was used daily, she replied she took ibuprofen daily for headaches and that she had been doing this for several months. Headaches were not in her chief complaints as she had learned to live with and ignore this symptom. Upon further evaluation, she was slightly overweight and has a questionable fundoscopic exam. After further evaluation by an ophthalmologist and a neurologist, pseudotumor cerebri was diagnosed.
Index of suspicion is key in correctly diagnosing patients, and understanding the changing landscape of medicine will lead to more thoughtful questioning during routine health exams and better outcomes for your patients.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Am J Ophthalmol. 2015 Feb;159(2):344-52.e1.
2. Horm Res Paediatr. 2014;81(4):217-25.
3. Clin Imaging. 2018 May 24. doi: 10.1016/j.clinimag.2018.05.020.
4. Am J Ophthalmol. 1998 Jul;126(1):116-21.
5. Glob Pediatr Health. 2018. doi:10.1177/2333794X18785550.
Pseudotumor cerebri, benign intracranial hypertension, and idiopathic intracranial hypertension are all terms to describe a syndrome of increased intracranial pressure, headaches, vision loss, or changes without an associated mass lesion.1 The condition was considered relatively rare, presenting most commonly in obese women in childbearing years. Surprisingly, 2
Obesity is the fastest growing morbidity among adolescents. The Centers for Disease Control and Prevention reported 32% of children 2-19 years were obese.1 This reality is impacting many areas of an adolescent’s health, but it also is changing the landscape of diseases that present in this age group. Although pediatric and adult pseudotumor cerebri always have had slightly varied features, many features were similar such as the papilledema, vision loss, headaches, and sixth nerve palsy. Obesity and female predominance tended to present more in the adult population, as many pediatric patients were not obese,2 and had fewer associated symptoms at the time of diagnosis, and the cause was thought to idiopathic.
Now, with the increase in obesity, more adolescents and more male patients are presenting with pseudotumor cerebri as a cause for their headache, and 57%-100% are obese, making it a compounding factor.3
Pediatric populations also are at risk of secondary pseudotumor cerebri, which is an increase in intracranial pressure from the use of medication, or other disease states such as anemia, kidney disease, or Down syndrome. Minocycline use is the most common medication cause and usually presents 1-2 months after normal use.4 Discontinuing the drug does lead to resolution. Retinoids, vitamin A products, growth hormone, and steroids also have been implicated. Given that acne is a common complaint amongst teens, knowledge of these side effects is important.4
In 2013, the criteria for diagnosis of pseudotumor cerebri was revised. Currently, the presence of papilledema, normal neurologic exam except for abnormal sixth cranial nerve, normal cerebral spinal fluid, elevated lumbar opening pressure, and normal imaging are needed for a definitive diagnosis. A probable diagnosis can be made if papilledema is not present but there abducens nerve palsy.2
In a routine physical exam, when I questioned a patient on any medication that was used daily, she replied she took ibuprofen daily for headaches and that she had been doing this for several months. Headaches were not in her chief complaints as she had learned to live with and ignore this symptom. Upon further evaluation, she was slightly overweight and has a questionable fundoscopic exam. After further evaluation by an ophthalmologist and a neurologist, pseudotumor cerebri was diagnosed.
Index of suspicion is key in correctly diagnosing patients, and understanding the changing landscape of medicine will lead to more thoughtful questioning during routine health exams and better outcomes for your patients.
Dr. Pearce is a pediatrician in Frankfort, Ill. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Am J Ophthalmol. 2015 Feb;159(2):344-52.e1.
2. Horm Res Paediatr. 2014;81(4):217-25.
3. Clin Imaging. 2018 May 24. doi: 10.1016/j.clinimag.2018.05.020.
4. Am J Ophthalmol. 1998 Jul;126(1):116-21.
5. Glob Pediatr Health. 2018. doi:10.1177/2333794X18785550.
New guidance offered for managing poorly controlled asthma in children
published in the Annals of Allergy, Asthma & Immunology.

“Although many children with asthma achieve symptom control with appropriate management, a substantial subset does not,” Bradley E. Chipps, MD, from the Capital Allergy & Respiratory Disease Center in Sacramento, Calif., and his colleagues wrote in the recommendations sponsored by the American College of Allergy, Asthma, and Immunology. “These children should undergo a step-up in care, but when and how to do that is not always straightforward. The Pediatric Asthma Yardstick is a practical resource for starting or adjusting controller therapy based on the options that are currently available for children, from infants to 18 years of age.”
In their recommendations, the authors grouped patients into age ranges of adolescent (12-18 years), school aged (6-11 years), and young children (5 years and under) as well as severity classifications.
Adolescents and school-aged children
For adolescents and school-aged children, step 1 was classified as intermittent asthma that can be controlled with low-dose inhaled corticosteroids (ICS) with short-acting beta2-agonist (SABA) for as-needed relief. Children considered for stepping up to the next therapy should show symptoms of mild persistent asthma that the authors recommended controlling with low-dose ICS, leukotriene receptor antagonist (LTRA), or low-dose theophylline with as-needed SABA.
In children 12-18 years with moderate persistent asthma (step 3), the authors recommended a combination of low-dose ICA and a long-acting beta2-agonist (LABA), while children 6-11 years should receive a medium dose of ICS; other considerations for school-aged children include a medium-high dose of ICS, a low-dose combination of ICS and LTRA, or low-dose ICS together with theophylline.
Adolescent or school-aged children with severe persistent asthma (step 4) should take a medium or high dose of ICS together with LABA, with the authors recommending adding tiotropium to a soft mist inhaler, combination high-dose ICS and LTRA, or a combination high-dose ICS and theophylline.
Dr. Chipps and his coauthors recommended children stepping up therapy beyond severe persistent asthma (step 5) should add on treatment such as low-dose oral corticosteroids, anti-immunoglobulin E therapy, and adding tiotropium to a soft-mist inhaler.
For adolescent and school-aged children going to steps 3-5, Dr. Chipps and his coauthors recommended prescribing as needed a short-acting beta2 agonist or low-dose ICS/LABA.
Children 5 years and younger

In children 5 years and younger, intermittent asthma (step 1) should be considered if the child has infrequent or viral wheezing but few or no symptoms in the interim that can be controlled with as-needed SABA. These young children who show symptoms of mild persistent asthma (step 2) can be treated with daily low-dose ICS, with other controller options of LTRA or intermittent ICS.
Stepping up therapy from mild to moderate persistent asthma (step 3), young children should receive double the daily dose of low-dose ICS from the previous step or use the low-dose ICS together with LTRA; if children show symptoms of severe persistent asthma (step 4), they should continue their daily controller and be referred to a specialist; other considerations for controllers at this step included adding LTRA, adding intermittent ICS, or increasing ICS frequency.
Other factors to consider

Inconsistencies in response to medication can occur because of comorbid conditions such as obesity, rhinosinusitis, respiratory infection or gastroesophageal reflux; suboptimal inhaled drug delivery; or failure to comply with treatment because of not wanting to take medication (common in adolescents), belief that even controller medicine can be taken intermittently, family stress, cost including lack of insurance or medication not covered by insurance. “Before adjusting therapy, it is important to ensure that the child’s change in symptoms is due to asthma and not to any of these factors that need to be addressed,” Dr. Chipps and his colleagues wrote.
Collaboration among children, their parents, and clinicians is needed to achieve good asthma control because of the “variable presentation within individuals and within the population of children affected” with asthma, they wrote.
The article summarizing the guidelines was sponsored by the American College of Allergy, Asthma, and Immunology. Most of the authors report various financial relationships with companies including AstraZeneca, Aerocrine, Aviragen, Boehringer Ingelheim, Cephalon, Circassia, Commense, Genentech, GlaxoSmithKline, Greer, Meda, Merck, Mylan, Novartis, Patara, Regeneron, Sanofi, TEVA, Theravance, and Vectura Group. Dr. Farrar and Dr. Szefler had no financial interests to disclose.
SOURCE: Chipps BE et al. Ann Allergy Asthma Immunol. 2018 Apr. doi: 1010.1016/j.anai.2018.04.002.
published in the Annals of Allergy, Asthma & Immunology.
“Although many children with asthma achieve symptom control with appropriate management, a substantial subset does not,” Bradley E. Chipps, MD, from the Capital Allergy & Respiratory Disease Center in Sacramento, Calif., and his colleagues wrote in the recommendations sponsored by the American College of Allergy, Asthma, and Immunology. “These children should undergo a step-up in care, but when and how to do that is not always straightforward. The Pediatric Asthma Yardstick is a practical resource for starting or adjusting controller therapy based on the options that are currently available for children, from infants to 18 years of age.”
In their recommendations, the authors grouped patients into age ranges of adolescent (12-18 years), school aged (6-11 years), and young children (5 years and under) as well as severity classifications.
Adolescents and school-aged children
For adolescents and school-aged children, step 1 was classified as intermittent asthma that can be controlled with low-dose inhaled corticosteroids (ICS) with short-acting beta2-agonist (SABA) for as-needed relief. Children considered for stepping up to the next therapy should show symptoms of mild persistent asthma that the authors recommended controlling with low-dose ICS, leukotriene receptor antagonist (LTRA), or low-dose theophylline with as-needed SABA.
In children 12-18 years with moderate persistent asthma (step 3), the authors recommended a combination of low-dose ICA and a long-acting beta2-agonist (LABA), while children 6-11 years should receive a medium dose of ICS; other considerations for school-aged children include a medium-high dose of ICS, a low-dose combination of ICS and LTRA, or low-dose ICS together with theophylline.
Adolescent or school-aged children with severe persistent asthma (step 4) should take a medium or high dose of ICS together with LABA, with the authors recommending adding tiotropium to a soft mist inhaler, combination high-dose ICS and LTRA, or a combination high-dose ICS and theophylline.
Dr. Chipps and his coauthors recommended children stepping up therapy beyond severe persistent asthma (step 5) should add on treatment such as low-dose oral corticosteroids, anti-immunoglobulin E therapy, and adding tiotropium to a soft-mist inhaler.
For adolescent and school-aged children going to steps 3-5, Dr. Chipps and his coauthors recommended prescribing as needed a short-acting beta2 agonist or low-dose ICS/LABA.
Children 5 years and younger
In children 5 years and younger, intermittent asthma (step 1) should be considered if the child has infrequent or viral wheezing but few or no symptoms in the interim that can be controlled with as-needed SABA. These young children who show symptoms of mild persistent asthma (step 2) can be treated with daily low-dose ICS, with other controller options of LTRA or intermittent ICS.
Stepping up therapy from mild to moderate persistent asthma (step 3), young children should receive double the daily dose of low-dose ICS from the previous step or use the low-dose ICS together with LTRA; if children show symptoms of severe persistent asthma (step 4), they should continue their daily controller and be referred to a specialist; other considerations for controllers at this step included adding LTRA, adding intermittent ICS, or increasing ICS frequency.
Other factors to consider
Inconsistencies in response to medication can occur because of comorbid conditions such as obesity, rhinosinusitis, respiratory infection or gastroesophageal reflux; suboptimal inhaled drug delivery; or failure to comply with treatment because of not wanting to take medication (common in adolescents), belief that even controller medicine can be taken intermittently, family stress, cost including lack of insurance or medication not covered by insurance. “Before adjusting therapy, it is important to ensure that the child’s change in symptoms is due to asthma and not to any of these factors that need to be addressed,” Dr. Chipps and his colleagues wrote.
Collaboration among children, their parents, and clinicians is needed to achieve good asthma control because of the “variable presentation within individuals and within the population of children affected” with asthma, they wrote.
The article summarizing the guidelines was sponsored by the American College of Allergy, Asthma, and Immunology. Most of the authors report various financial relationships with companies including AstraZeneca, Aerocrine, Aviragen, Boehringer Ingelheim, Cephalon, Circassia, Commense, Genentech, GlaxoSmithKline, Greer, Meda, Merck, Mylan, Novartis, Patara, Regeneron, Sanofi, TEVA, Theravance, and Vectura Group. Dr. Farrar and Dr. Szefler had no financial interests to disclose.
SOURCE: Chipps BE et al. Ann Allergy Asthma Immunol. 2018 Apr. doi: 1010.1016/j.anai.2018.04.002.
published in the Annals of Allergy, Asthma & Immunology.
“Although many children with asthma achieve symptom control with appropriate management, a substantial subset does not,” Bradley E. Chipps, MD, from the Capital Allergy & Respiratory Disease Center in Sacramento, Calif., and his colleagues wrote in the recommendations sponsored by the American College of Allergy, Asthma, and Immunology. “These children should undergo a step-up in care, but when and how to do that is not always straightforward. The Pediatric Asthma Yardstick is a practical resource for starting or adjusting controller therapy based on the options that are currently available for children, from infants to 18 years of age.”
In their recommendations, the authors grouped patients into age ranges of adolescent (12-18 years), school aged (6-11 years), and young children (5 years and under) as well as severity classifications.
Adolescents and school-aged children
For adolescents and school-aged children, step 1 was classified as intermittent asthma that can be controlled with low-dose inhaled corticosteroids (ICS) with short-acting beta2-agonist (SABA) for as-needed relief. Children considered for stepping up to the next therapy should show symptoms of mild persistent asthma that the authors recommended controlling with low-dose ICS, leukotriene receptor antagonist (LTRA), or low-dose theophylline with as-needed SABA.
In children 12-18 years with moderate persistent asthma (step 3), the authors recommended a combination of low-dose ICA and a long-acting beta2-agonist (LABA), while children 6-11 years should receive a medium dose of ICS; other considerations for school-aged children include a medium-high dose of ICS, a low-dose combination of ICS and LTRA, or low-dose ICS together with theophylline.
Adolescent or school-aged children with severe persistent asthma (step 4) should take a medium or high dose of ICS together with LABA, with the authors recommending adding tiotropium to a soft mist inhaler, combination high-dose ICS and LTRA, or a combination high-dose ICS and theophylline.
Dr. Chipps and his coauthors recommended children stepping up therapy beyond severe persistent asthma (step 5) should add on treatment such as low-dose oral corticosteroids, anti-immunoglobulin E therapy, and adding tiotropium to a soft-mist inhaler.
For adolescent and school-aged children going to steps 3-5, Dr. Chipps and his coauthors recommended prescribing as needed a short-acting beta2 agonist or low-dose ICS/LABA.
Children 5 years and younger
In children 5 years and younger, intermittent asthma (step 1) should be considered if the child has infrequent or viral wheezing but few or no symptoms in the interim that can be controlled with as-needed SABA. These young children who show symptoms of mild persistent asthma (step 2) can be treated with daily low-dose ICS, with other controller options of LTRA or intermittent ICS.
Stepping up therapy from mild to moderate persistent asthma (step 3), young children should receive double the daily dose of low-dose ICS from the previous step or use the low-dose ICS together with LTRA; if children show symptoms of severe persistent asthma (step 4), they should continue their daily controller and be referred to a specialist; other considerations for controllers at this step included adding LTRA, adding intermittent ICS, or increasing ICS frequency.
Other factors to consider
Inconsistencies in response to medication can occur because of comorbid conditions such as obesity, rhinosinusitis, respiratory infection or gastroesophageal reflux; suboptimal inhaled drug delivery; or failure to comply with treatment because of not wanting to take medication (common in adolescents), belief that even controller medicine can be taken intermittently, family stress, cost including lack of insurance or medication not covered by insurance. “Before adjusting therapy, it is important to ensure that the child’s change in symptoms is due to asthma and not to any of these factors that need to be addressed,” Dr. Chipps and his colleagues wrote.
Collaboration among children, their parents, and clinicians is needed to achieve good asthma control because of the “variable presentation within individuals and within the population of children affected” with asthma, they wrote.
The article summarizing the guidelines was sponsored by the American College of Allergy, Asthma, and Immunology. Most of the authors report various financial relationships with companies including AstraZeneca, Aerocrine, Aviragen, Boehringer Ingelheim, Cephalon, Circassia, Commense, Genentech, GlaxoSmithKline, Greer, Meda, Merck, Mylan, Novartis, Patara, Regeneron, Sanofi, TEVA, Theravance, and Vectura Group. Dr. Farrar and Dr. Szefler had no financial interests to disclose.
SOURCE: Chipps BE et al. Ann Allergy Asthma Immunol. 2018 Apr. doi: 1010.1016/j.anai.2018.04.002.
FROM ANNALS OF ALLERGY, ASTHMA & IMMUNOLOGY
Brain connectivity in depression tied to poor sleep quality
An increase in functional connectivity in certain regions of the brains of people with depression could explain the link between the disease and poor sleep quality – and could have implications for the treatment of both conditions, the first research of its kind suggests.
Wei Cheng, PhD, of the Institute of Science and Technology for Brain-Inspired Intelligence at the Fudan University in Shanghai, China, and colleagues noted that many people with depression report poor sleep quality and sleep disturbance.
“Understanding the neural connectivity that underlies both conditions and mediates the association between them is likely to lead to better-directed treatments for depression and associated sleep problems,” they wrote in JAMA Psychiatry.
In the current study, the research team used data from 1,017 participants of the Human Connectome Project drawn from the general population in the United States (who were not selected for symptoms of depression). Subjects, whose ages ranged from 22 to 35 years, had completed the Adult Self-Report of Depressive Problems portion of the Achenbach Adult Self-Report for Ages 18-59 – a survey of self-reported sleep quality and resting-state functional MRI.
“Resting-state functional connectivity between brain areas, which reflects correlations of activity, is a fundamental tool in augmenting understanding of the brain regions with altered connectivity and function in mental disorders,” the study authors noted.
The researchers then cross-validated the sleep findings using a sample of 8,718 participants from with the United Kingdom Biobank data set.
In total, the research team identified 162 functional connectivity links involving areas associated with sleep, with 39 of these areas also associated with Depressive Problems Scores (P less than .001).
Overall, the brain areas with increased functional connectivity associated with the Pittsburgh Sleep Quality Index score and Depressive Problems scores included the lateral orbitofrontal cortex, dorsolateral prefrontal cortex, anterior and posterior cingulate cortices, insula, parahippocampal gyrus, hippocampus, amygdala, temporal cortex, and precuneus.
A mediation analysis conducted by the authors aimed at assessing the underlying mechanisms showed that “these functional connectivities underlie the association of depressive problems score with poor sleep quality (P less than .001).”
They observed “much smaller” associations in the reverse direction – in that the associations of sleep quality with depressive problems mediated by these links were less significant.
“ and this in turn has implications for treatment because of the brain areas identified,” the research team concluded.
Dr. Cheng and colleagues cited several limitations. One is that the Depressive Problems scores used were not reflective of a formal diagnosis. Nevertheless, they said, the current findings provided “strong support” for the role of the lateral orbitofrontal cortex in depression, particularly as the investigators observed relatively high correlations with functional connectivities in this area of the brains of 92 participants who had been diagnosed with a major depressive episode over their lifetime.
“The understanding that we developed in this study is consistent with areas of the brain involved in short-term memory (the dorsolateral prefrontal cortex), the self (precuneus), and negative emotion (the lateral orbitofrontal cortex) being highly connected in depression which results in increased ruminating thoughts that are at least part of the mechanism that impairs sleep quality,” they added.
The study was supported by several entities, including the Shanghai Science & Technology Innovation Plan and the National Natural Science Foundation of China. No conflicts of interest were reported.
SOURCE: Cheng W et al. JAMA Psychiatry. 2018 Jul 25. doi: 10.1001/jamapsychiatry.2018.1941.
An increase in functional connectivity in certain regions of the brains of people with depression could explain the link between the disease and poor sleep quality – and could have implications for the treatment of both conditions, the first research of its kind suggests.
Wei Cheng, PhD, of the Institute of Science and Technology for Brain-Inspired Intelligence at the Fudan University in Shanghai, China, and colleagues noted that many people with depression report poor sleep quality and sleep disturbance.
“Understanding the neural connectivity that underlies both conditions and mediates the association between them is likely to lead to better-directed treatments for depression and associated sleep problems,” they wrote in JAMA Psychiatry.
In the current study, the research team used data from 1,017 participants of the Human Connectome Project drawn from the general population in the United States (who were not selected for symptoms of depression). Subjects, whose ages ranged from 22 to 35 years, had completed the Adult Self-Report of Depressive Problems portion of the Achenbach Adult Self-Report for Ages 18-59 – a survey of self-reported sleep quality and resting-state functional MRI.
“Resting-state functional connectivity between brain areas, which reflects correlations of activity, is a fundamental tool in augmenting understanding of the brain regions with altered connectivity and function in mental disorders,” the study authors noted.
The researchers then cross-validated the sleep findings using a sample of 8,718 participants from with the United Kingdom Biobank data set.
In total, the research team identified 162 functional connectivity links involving areas associated with sleep, with 39 of these areas also associated with Depressive Problems Scores (P less than .001).
Overall, the brain areas with increased functional connectivity associated with the Pittsburgh Sleep Quality Index score and Depressive Problems scores included the lateral orbitofrontal cortex, dorsolateral prefrontal cortex, anterior and posterior cingulate cortices, insula, parahippocampal gyrus, hippocampus, amygdala, temporal cortex, and precuneus.
A mediation analysis conducted by the authors aimed at assessing the underlying mechanisms showed that “these functional connectivities underlie the association of depressive problems score with poor sleep quality (P less than .001).”
They observed “much smaller” associations in the reverse direction – in that the associations of sleep quality with depressive problems mediated by these links were less significant.
“ and this in turn has implications for treatment because of the brain areas identified,” the research team concluded.
Dr. Cheng and colleagues cited several limitations. One is that the Depressive Problems scores used were not reflective of a formal diagnosis. Nevertheless, they said, the current findings provided “strong support” for the role of the lateral orbitofrontal cortex in depression, particularly as the investigators observed relatively high correlations with functional connectivities in this area of the brains of 92 participants who had been diagnosed with a major depressive episode over their lifetime.
“The understanding that we developed in this study is consistent with areas of the brain involved in short-term memory (the dorsolateral prefrontal cortex), the self (precuneus), and negative emotion (the lateral orbitofrontal cortex) being highly connected in depression which results in increased ruminating thoughts that are at least part of the mechanism that impairs sleep quality,” they added.
The study was supported by several entities, including the Shanghai Science & Technology Innovation Plan and the National Natural Science Foundation of China. No conflicts of interest were reported.
SOURCE: Cheng W et al. JAMA Psychiatry. 2018 Jul 25. doi: 10.1001/jamapsychiatry.2018.1941.
An increase in functional connectivity in certain regions of the brains of people with depression could explain the link between the disease and poor sleep quality – and could have implications for the treatment of both conditions, the first research of its kind suggests.
Wei Cheng, PhD, of the Institute of Science and Technology for Brain-Inspired Intelligence at the Fudan University in Shanghai, China, and colleagues noted that many people with depression report poor sleep quality and sleep disturbance.
“Understanding the neural connectivity that underlies both conditions and mediates the association between them is likely to lead to better-directed treatments for depression and associated sleep problems,” they wrote in JAMA Psychiatry.
In the current study, the research team used data from 1,017 participants of the Human Connectome Project drawn from the general population in the United States (who were not selected for symptoms of depression). Subjects, whose ages ranged from 22 to 35 years, had completed the Adult Self-Report of Depressive Problems portion of the Achenbach Adult Self-Report for Ages 18-59 – a survey of self-reported sleep quality and resting-state functional MRI.
“Resting-state functional connectivity between brain areas, which reflects correlations of activity, is a fundamental tool in augmenting understanding of the brain regions with altered connectivity and function in mental disorders,” the study authors noted.
The researchers then cross-validated the sleep findings using a sample of 8,718 participants from with the United Kingdom Biobank data set.
In total, the research team identified 162 functional connectivity links involving areas associated with sleep, with 39 of these areas also associated with Depressive Problems Scores (P less than .001).
Overall, the brain areas with increased functional connectivity associated with the Pittsburgh Sleep Quality Index score and Depressive Problems scores included the lateral orbitofrontal cortex, dorsolateral prefrontal cortex, anterior and posterior cingulate cortices, insula, parahippocampal gyrus, hippocampus, amygdala, temporal cortex, and precuneus.
A mediation analysis conducted by the authors aimed at assessing the underlying mechanisms showed that “these functional connectivities underlie the association of depressive problems score with poor sleep quality (P less than .001).”
They observed “much smaller” associations in the reverse direction – in that the associations of sleep quality with depressive problems mediated by these links were less significant.
“ and this in turn has implications for treatment because of the brain areas identified,” the research team concluded.
Dr. Cheng and colleagues cited several limitations. One is that the Depressive Problems scores used were not reflective of a formal diagnosis. Nevertheless, they said, the current findings provided “strong support” for the role of the lateral orbitofrontal cortex in depression, particularly as the investigators observed relatively high correlations with functional connectivities in this area of the brains of 92 participants who had been diagnosed with a major depressive episode over their lifetime.
“The understanding that we developed in this study is consistent with areas of the brain involved in short-term memory (the dorsolateral prefrontal cortex), the self (precuneus), and negative emotion (the lateral orbitofrontal cortex) being highly connected in depression which results in increased ruminating thoughts that are at least part of the mechanism that impairs sleep quality,” they added.
The study was supported by several entities, including the Shanghai Science & Technology Innovation Plan and the National Natural Science Foundation of China. No conflicts of interest were reported.
SOURCE: Cheng W et al. JAMA Psychiatry. 2018 Jul 25. doi: 10.1001/jamapsychiatry.2018.1941.
FROM JAMA PSYCHIATRY
Key clinical point: The connections between depression and poor sleep quality hold implications for treating both conditions.
Major finding: Thirty-nine functional brain connectivity links involving sleep were associated with Depressive Problems Scores.
Study details: Depression and sleep data from 1,017 participants of the Human Connectome Project cross-validated with sleep findings from the United Kingdom Biobank data set using a sample of 8,718 participants.
Disclosures: The study was supported by several entities, including the Shanghai Science & Technology Innovation Plan and the National Natural Science Foundation of China. No conflicts of interest were reported.
Source: Cheng W et al. JAMA Psychiatry. 2018 Jul 25. doi: 10.1001/jamapsychiatry.2018.1941.
Weather changes trigger migraine
SAN FRANCISCO – Many migraineurs claim that changes in the weather can trigger their headache attacks. It took a headache specialist together with a meteorologist poring over surface weather maps to prove they are right.

“When patients tell you they can predict a headache from the weather, they really can,” Vincent T. Martin, MD, declared in presenting the evidence at the annual meeting of the American Headache Society.
Many physicians have been skeptical of patient self-reports of a weather/migraine connection because of mixed results in prior studies examining the impact of a single meteorologic factor at a time, such as barometric pressure, temperature, humidity, or wind speed.
“These studies, however, fail to account for the fact that weather events represent a confluence of meteorologic factors that occur in a specific temporal sequence. It may be necessary to model several variables together to achieve the optimal weather models,” explained Dr. Martin, a general internist, professor of medicine, and director of the Headache and Facial Pain Center at the University of Cincinnati.
He presented a retrospective cohort study of 218 patients with episodic migraine with a mean of 8.9 headache days per month who kept a daily electronic headache diary during two prior studies conducted in the St. Louis area. Their diary data were matched with hourly measurements of barometric pressure, temperature, relative humidity, and wind speed recorded at five St. Louis–area weather stations and archived at the National Climatic Data Center. Dr. Martin and his coinvestigators then created a series of models that predicted the weather conditions that were associated with each individual patient being in the top tertile for the presence of headache on a given day with no headache on the day before.
Preliminary analysis indicated that the most important predictor of new-onset headache in winter, spring, and fall was the barometric pressure differential between 2 consecutive days. These differentials were much smaller in the summer, so a separate model was created for that season. Multiple models were developed to identify binary cutpoints for each weather variable.
From fall through spring, during periods when barometric pressure was in the top tertile – that is, a high-pressure system was in play – a day-to-day difference in mean daily barometric pressure greater than 0.1 mm Hg was associated with a 4.9-fold increased risk of being in the top tertile for new-onset headache, and less than a 25% difference in minimal daily relative humidity was associated with a 4.6-fold increased risk.
In contrast, when barometric pressure was in the lowest tertile, a drop in mean daily barometric pressure of 0.05 mm Hg or less from one day to the next was associated with a 3.17-fold increased risk of entering the top tertile for new-onset headache, and a day-to-day increase in maximal wind speed of 7 mph or more was associated with a 2.64-fold increased risk.
In middle-tertile periods of barometric pressure, a drop in mean pressure of 0.05 mm Hg or less was associated with a 2.21-fold increase in new-onset headache, and a mean daily relative humidity of 79% or greater conferred a 4.43-fold relative risk.
“It’s very rare in epidemiologic studies to get magnitudes of association to those degrees,” Dr. Martin observed. “Our results provide strong evidence that weather is associated with days with a high probability of new-onset headache in persons with migraine.”
The mechanisms underlying this association aren’t known. Possibilities worthy of investigation include stimulation of hyperactivity of the sympathetic nervous system, increases in airborne environmental allergens or pollutants, or direct activation of trigeminal afferent nerve fibers, he said.
Dr. Martin reported having no financial conflicts of interest.
SAN FRANCISCO – Many migraineurs claim that changes in the weather can trigger their headache attacks. It took a headache specialist together with a meteorologist poring over surface weather maps to prove they are right.
“When patients tell you they can predict a headache from the weather, they really can,” Vincent T. Martin, MD, declared in presenting the evidence at the annual meeting of the American Headache Society.
Many physicians have been skeptical of patient self-reports of a weather/migraine connection because of mixed results in prior studies examining the impact of a single meteorologic factor at a time, such as barometric pressure, temperature, humidity, or wind speed.
“These studies, however, fail to account for the fact that weather events represent a confluence of meteorologic factors that occur in a specific temporal sequence. It may be necessary to model several variables together to achieve the optimal weather models,” explained Dr. Martin, a general internist, professor of medicine, and director of the Headache and Facial Pain Center at the University of Cincinnati.
He presented a retrospective cohort study of 218 patients with episodic migraine with a mean of 8.9 headache days per month who kept a daily electronic headache diary during two prior studies conducted in the St. Louis area. Their diary data were matched with hourly measurements of barometric pressure, temperature, relative humidity, and wind speed recorded at five St. Louis–area weather stations and archived at the National Climatic Data Center. Dr. Martin and his coinvestigators then created a series of models that predicted the weather conditions that were associated with each individual patient being in the top tertile for the presence of headache on a given day with no headache on the day before.
Preliminary analysis indicated that the most important predictor of new-onset headache in winter, spring, and fall was the barometric pressure differential between 2 consecutive days. These differentials were much smaller in the summer, so a separate model was created for that season. Multiple models were developed to identify binary cutpoints for each weather variable.
From fall through spring, during periods when barometric pressure was in the top tertile – that is, a high-pressure system was in play – a day-to-day difference in mean daily barometric pressure greater than 0.1 mm Hg was associated with a 4.9-fold increased risk of being in the top tertile for new-onset headache, and less than a 25% difference in minimal daily relative humidity was associated with a 4.6-fold increased risk.
In contrast, when barometric pressure was in the lowest tertile, a drop in mean daily barometric pressure of 0.05 mm Hg or less from one day to the next was associated with a 3.17-fold increased risk of entering the top tertile for new-onset headache, and a day-to-day increase in maximal wind speed of 7 mph or more was associated with a 2.64-fold increased risk.
In middle-tertile periods of barometric pressure, a drop in mean pressure of 0.05 mm Hg or less was associated with a 2.21-fold increase in new-onset headache, and a mean daily relative humidity of 79% or greater conferred a 4.43-fold relative risk.
“It’s very rare in epidemiologic studies to get magnitudes of association to those degrees,” Dr. Martin observed. “Our results provide strong evidence that weather is associated with days with a high probability of new-onset headache in persons with migraine.”
The mechanisms underlying this association aren’t known. Possibilities worthy of investigation include stimulation of hyperactivity of the sympathetic nervous system, increases in airborne environmental allergens or pollutants, or direct activation of trigeminal afferent nerve fibers, he said.
Dr. Martin reported having no financial conflicts of interest.
SAN FRANCISCO – Many migraineurs claim that changes in the weather can trigger their headache attacks. It took a headache specialist together with a meteorologist poring over surface weather maps to prove they are right.
“When patients tell you they can predict a headache from the weather, they really can,” Vincent T. Martin, MD, declared in presenting the evidence at the annual meeting of the American Headache Society.
Many physicians have been skeptical of patient self-reports of a weather/migraine connection because of mixed results in prior studies examining the impact of a single meteorologic factor at a time, such as barometric pressure, temperature, humidity, or wind speed.
“These studies, however, fail to account for the fact that weather events represent a confluence of meteorologic factors that occur in a specific temporal sequence. It may be necessary to model several variables together to achieve the optimal weather models,” explained Dr. Martin, a general internist, professor of medicine, and director of the Headache and Facial Pain Center at the University of Cincinnati.
He presented a retrospective cohort study of 218 patients with episodic migraine with a mean of 8.9 headache days per month who kept a daily electronic headache diary during two prior studies conducted in the St. Louis area. Their diary data were matched with hourly measurements of barometric pressure, temperature, relative humidity, and wind speed recorded at five St. Louis–area weather stations and archived at the National Climatic Data Center. Dr. Martin and his coinvestigators then created a series of models that predicted the weather conditions that were associated with each individual patient being in the top tertile for the presence of headache on a given day with no headache on the day before.
Preliminary analysis indicated that the most important predictor of new-onset headache in winter, spring, and fall was the barometric pressure differential between 2 consecutive days. These differentials were much smaller in the summer, so a separate model was created for that season. Multiple models were developed to identify binary cutpoints for each weather variable.
From fall through spring, during periods when barometric pressure was in the top tertile – that is, a high-pressure system was in play – a day-to-day difference in mean daily barometric pressure greater than 0.1 mm Hg was associated with a 4.9-fold increased risk of being in the top tertile for new-onset headache, and less than a 25% difference in minimal daily relative humidity was associated with a 4.6-fold increased risk.
In contrast, when barometric pressure was in the lowest tertile, a drop in mean daily barometric pressure of 0.05 mm Hg or less from one day to the next was associated with a 3.17-fold increased risk of entering the top tertile for new-onset headache, and a day-to-day increase in maximal wind speed of 7 mph or more was associated with a 2.64-fold increased risk.
In middle-tertile periods of barometric pressure, a drop in mean pressure of 0.05 mm Hg or less was associated with a 2.21-fold increase in new-onset headache, and a mean daily relative humidity of 79% or greater conferred a 4.43-fold relative risk.
“It’s very rare in epidemiologic studies to get magnitudes of association to those degrees,” Dr. Martin observed. “Our results provide strong evidence that weather is associated with days with a high probability of new-onset headache in persons with migraine.”
The mechanisms underlying this association aren’t known. Possibilities worthy of investigation include stimulation of hyperactivity of the sympathetic nervous system, increases in airborne environmental allergens or pollutants, or direct activation of trigeminal afferent nerve fibers, he said.
Dr. Martin reported having no financial conflicts of interest.
REPORTING FROM THE AHS ANNUAL MEETING
Key clinical point: Specific weather patterns can trigger migraine.
Major finding: During a low pressure front, a maximum wind speed of 7 mph or more on a given day was associated with a 2.6-fold increased relative risk of new-onset headache the next day.
Study details: This retrospective study of 218 episodic migraine patients linked their daily headache diary data to hourly measurements from local weather stations.
Disclosures: The presenter reported having no financial conflicts of interest.
Mild cognitive impairment risk slashed by 19% in SPRINT MIND
CHICAGO – Lowering systolic blood pressure to a target of 120 mm Hg or lower in people with cardiovascular risk factors reduced the risk of mild cognitive impairment by 19% and probable all-cause dementia by 17% relative to those who achieved a less intensive target of less than 140 mm Hg

Drug class didn’t matter. Cheap generics were just as effective as expensive name brands. It equally benefited men and women, whites, blacks, and Hispanics. And keeping systolic blood pressure at 120 mm Hg or lower prevented MCI just as well in those older than 75 as it did for younger subjects.
The stunning announcement came during a press briefing at the Alzheimer’s Association International Conference, as Jeff D. Williamson, MD, unveiled the results of the 4-year SPRINT MIND study. Strict blood pressure control for 3.2 years, with a systolic target of 120 mm Hg or lower, reduced the incidence of mild cognitive impairment by a magnitude of benefit that no amyloid-targeting investigational drug has ever approached.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during at the briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude. Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. People in the strict BP arm had 18% fewer white matter hyperintensities after 4 years of follow-up.
The news is an incredible step forward for the field that has stumbled repeatedly, clinicians agreed. Generic antihypertensives can be very inexpensive. They are almost globally available, and confer a host of other benefits, not only on cardiovascular health but on kidney health as well, said Dr. Williamson, chief of geriatric medicine at Wake Forest University, Winston-Salem, N.C.
“Hypertension is a highly prevalent condition, with 60%-70% having it. The 19% overall risk reduction for MCI will have a huge impact,” he said.
Maria Carrillo, PhD, chief scientific officer of the Alzheimer’s Association, was somewhat more guarded, but still very enthusiastic.
“I think the most we can say right now is we are able to reduce risk,” she said in an interview. “But the reality is that reducing the risk of MCI by 19% will have a huge impact on dementia overall. And slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, the I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, and standard care, a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 30% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 American Heart Association and American College of Cardiology high blood pressure clinical guidelines.
The SPRINT MIND substudy looked at whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume.
It comprised 9,361 SPRINT subjects who were 50 years or older (mean 68; 28% at least 75) and had at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but they were encouraged to use drugs with the strongest evidence of cardiovascular benefit: thiazide-type diuretics encouraged as first-line, and then loop diuretics and beta-adrenergic blockers. About 90% of the drugs used during the study were generics.
Subjects were seen monthly for the first 3 months, during which medications were adjusted to achieve the target, and then every 3 months after that. Medications could be adjusted monthly to keep on target.
At 1 year, the mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped in August 2015 due to the observed cardiovascular disease benefit, after a median follow up of 3.26 years, but cognitive assessment continued until the end of June (N Engl J Med. 2015 Nov 26; 373:2103-16).
The SPRINT MIND study did not meet its primary endpoint. Adjudicated cases of probable all-cause dementia developed in 175 of the standard care group and 147 of the intensive treatment group; the 17% risk reduction was not statistically significant (P = .10).
However, it did hit both secondary endpoints. Adjudicated cases of MCI developed in 348 of the standard treatment groups and 285 of the intensive treatment group: a statistically significant 19% risk reduction (P = .01). The combined secondary endpoint of MCI and probable dementia was a significant 15% risk reduction (P = .02), with 463 cases in the standard care group and 398 in the intensive care group.
The imaging study comprised 454 subjects who had brain MRI at baseline and 4 years after randomization. There was no change in total brain volume, said Ilya Nasrallah, MD, of the University of Pennsylvania. But those in the intensively managed group had 18% lower white matter lesion load than those in the standard care group (P = .004).
White matter lesions often point to small vessel disease, which is conclusively linked to vascular dementia, and may also linked to Alzheimer’s disease. Most AD patients, in fact, have a mixed dementia that often includes a vascular component, Dr. Carillo said.
SPRINT MIND didn’t follow subjects past 4 years, and didn’t include any follow-up for amyloid or Alzheimer’s diagnosis. But preventing MCI is no trivial thing, according to David Knopman, MD, who moderated the session.
“There’s nothing that is benign about MCI,” said Dr. Knopman of the Mayo Clinic, Rochester, Minn. “it’s the first sign of overt cognitive dysfunction, and although the rate at which MCI progress to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think is going to change clinical practice for people in primary care and the benefits at the population level are going to be substantial.”
Dr. Williamson drove this point home in a later interview, suggesting that physicians may want to think about how the SPRINT MIND results might apply to even younger patients with hypertension, and even if they don’t have other cardiovascular risk factors.
“I can’t say as a scientist that we have evidence to do that, yet. But as a physician, and for my own self and my own patients, I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg, and certainly start treating people in their 50s, and probably in their 40s.”
***This article was updated 7/31/18.
SOURCE: Williamson et al. AAIC 2018 DT-0202 Nasrallah et al. AAIC 2018 DT-03-03
CHICAGO – Lowering systolic blood pressure to a target of 120 mm Hg or lower in people with cardiovascular risk factors reduced the risk of mild cognitive impairment by 19% and probable all-cause dementia by 17% relative to those who achieved a less intensive target of less than 140 mm Hg
Drug class didn’t matter. Cheap generics were just as effective as expensive name brands. It equally benefited men and women, whites, blacks, and Hispanics. And keeping systolic blood pressure at 120 mm Hg or lower prevented MCI just as well in those older than 75 as it did for younger subjects.
The stunning announcement came during a press briefing at the Alzheimer’s Association International Conference, as Jeff D. Williamson, MD, unveiled the results of the 4-year SPRINT MIND study. Strict blood pressure control for 3.2 years, with a systolic target of 120 mm Hg or lower, reduced the incidence of mild cognitive impairment by a magnitude of benefit that no amyloid-targeting investigational drug has ever approached.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during at the briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude. Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. People in the strict BP arm had 18% fewer white matter hyperintensities after 4 years of follow-up.
The news is an incredible step forward for the field that has stumbled repeatedly, clinicians agreed. Generic antihypertensives can be very inexpensive. They are almost globally available, and confer a host of other benefits, not only on cardiovascular health but on kidney health as well, said Dr. Williamson, chief of geriatric medicine at Wake Forest University, Winston-Salem, N.C.
“Hypertension is a highly prevalent condition, with 60%-70% having it. The 19% overall risk reduction for MCI will have a huge impact,” he said.
Maria Carrillo, PhD, chief scientific officer of the Alzheimer’s Association, was somewhat more guarded, but still very enthusiastic.
“I think the most we can say right now is we are able to reduce risk,” she said in an interview. “But the reality is that reducing the risk of MCI by 19% will have a huge impact on dementia overall. And slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, the I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, and standard care, a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 30% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 American Heart Association and American College of Cardiology high blood pressure clinical guidelines.
The SPRINT MIND substudy looked at whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume.
It comprised 9,361 SPRINT subjects who were 50 years or older (mean 68; 28% at least 75) and had at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but they were encouraged to use drugs with the strongest evidence of cardiovascular benefit: thiazide-type diuretics encouraged as first-line, and then loop diuretics and beta-adrenergic blockers. About 90% of the drugs used during the study were generics.
Subjects were seen monthly for the first 3 months, during which medications were adjusted to achieve the target, and then every 3 months after that. Medications could be adjusted monthly to keep on target.
At 1 year, the mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped in August 2015 due to the observed cardiovascular disease benefit, after a median follow up of 3.26 years, but cognitive assessment continued until the end of June (N Engl J Med. 2015 Nov 26; 373:2103-16).
The SPRINT MIND study did not meet its primary endpoint. Adjudicated cases of probable all-cause dementia developed in 175 of the standard care group and 147 of the intensive treatment group; the 17% risk reduction was not statistically significant (P = .10).
However, it did hit both secondary endpoints. Adjudicated cases of MCI developed in 348 of the standard treatment groups and 285 of the intensive treatment group: a statistically significant 19% risk reduction (P = .01). The combined secondary endpoint of MCI and probable dementia was a significant 15% risk reduction (P = .02), with 463 cases in the standard care group and 398 in the intensive care group.
The imaging study comprised 454 subjects who had brain MRI at baseline and 4 years after randomization. There was no change in total brain volume, said Ilya Nasrallah, MD, of the University of Pennsylvania. But those in the intensively managed group had 18% lower white matter lesion load than those in the standard care group (P = .004).
White matter lesions often point to small vessel disease, which is conclusively linked to vascular dementia, and may also linked to Alzheimer’s disease. Most AD patients, in fact, have a mixed dementia that often includes a vascular component, Dr. Carillo said.
SPRINT MIND didn’t follow subjects past 4 years, and didn’t include any follow-up for amyloid or Alzheimer’s diagnosis. But preventing MCI is no trivial thing, according to David Knopman, MD, who moderated the session.
“There’s nothing that is benign about MCI,” said Dr. Knopman of the Mayo Clinic, Rochester, Minn. “it’s the first sign of overt cognitive dysfunction, and although the rate at which MCI progress to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think is going to change clinical practice for people in primary care and the benefits at the population level are going to be substantial.”
Dr. Williamson drove this point home in a later interview, suggesting that physicians may want to think about how the SPRINT MIND results might apply to even younger patients with hypertension, and even if they don’t have other cardiovascular risk factors.
“I can’t say as a scientist that we have evidence to do that, yet. But as a physician, and for my own self and my own patients, I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg, and certainly start treating people in their 50s, and probably in their 40s.”
***This article was updated 7/31/18.
SOURCE: Williamson et al. AAIC 2018 DT-0202 Nasrallah et al. AAIC 2018 DT-03-03
CHICAGO – Lowering systolic blood pressure to a target of 120 mm Hg or lower in people with cardiovascular risk factors reduced the risk of mild cognitive impairment by 19% and probable all-cause dementia by 17% relative to those who achieved a less intensive target of less than 140 mm Hg
Drug class didn’t matter. Cheap generics were just as effective as expensive name brands. It equally benefited men and women, whites, blacks, and Hispanics. And keeping systolic blood pressure at 120 mm Hg or lower prevented MCI just as well in those older than 75 as it did for younger subjects.
The stunning announcement came during a press briefing at the Alzheimer’s Association International Conference, as Jeff D. Williamson, MD, unveiled the results of the 4-year SPRINT MIND study. Strict blood pressure control for 3.2 years, with a systolic target of 120 mm Hg or lower, reduced the incidence of mild cognitive impairment by a magnitude of benefit that no amyloid-targeting investigational drug has ever approached.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during at the briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude. Three years of treatment and 3.2 years of follow-up absolutely reduced the risk.”
Brain imaging underscored the clinical importance of this finding and showed its physiologic pathway. People in the strict BP arm had 18% fewer white matter hyperintensities after 4 years of follow-up.
The news is an incredible step forward for the field that has stumbled repeatedly, clinicians agreed. Generic antihypertensives can be very inexpensive. They are almost globally available, and confer a host of other benefits, not only on cardiovascular health but on kidney health as well, said Dr. Williamson, chief of geriatric medicine at Wake Forest University, Winston-Salem, N.C.
“Hypertension is a highly prevalent condition, with 60%-70% having it. The 19% overall risk reduction for MCI will have a huge impact,” he said.
Maria Carrillo, PhD, chief scientific officer of the Alzheimer’s Association, was somewhat more guarded, but still very enthusiastic.
“I think the most we can say right now is we are able to reduce risk,” she said in an interview. “But the reality is that reducing the risk of MCI by 19% will have a huge impact on dementia overall. And slowing down the disease progress is a disease modification, versus developing symptoms. So, if that is the definition we are using, the I would say yes, it is disease modifying,” for dementias arising from cerebrovascular pathology.
SPRINT MIND was a substudy of the Hypertension Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, and standard care, a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 30% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 American Heart Association and American College of Cardiology high blood pressure clinical guidelines.
The SPRINT MIND substudy looked at whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume.
It comprised 9,361 SPRINT subjects who were 50 years or older (mean 68; 28% at least 75) and had at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia.
In SPRINT, physicians could choose any appropriate antihypertensive regimen, but they were encouraged to use drugs with the strongest evidence of cardiovascular benefit: thiazide-type diuretics encouraged as first-line, and then loop diuretics and beta-adrenergic blockers. About 90% of the drugs used during the study were generics.
Subjects were seen monthly for the first 3 months, during which medications were adjusted to achieve the target, and then every 3 months after that. Medications could be adjusted monthly to keep on target.
At 1 year, the mean systolic blood pressure was 121.4 mm Hg in the intensive-treatment group and 136.2 mm Hg in the standard treatment group. Treatment was stopped in August 2015 due to the observed cardiovascular disease benefit, after a median follow up of 3.26 years, but cognitive assessment continued until the end of June (N Engl J Med. 2015 Nov 26; 373:2103-16).
The SPRINT MIND study did not meet its primary endpoint. Adjudicated cases of probable all-cause dementia developed in 175 of the standard care group and 147 of the intensive treatment group; the 17% risk reduction was not statistically significant (P = .10).
However, it did hit both secondary endpoints. Adjudicated cases of MCI developed in 348 of the standard treatment groups and 285 of the intensive treatment group: a statistically significant 19% risk reduction (P = .01). The combined secondary endpoint of MCI and probable dementia was a significant 15% risk reduction (P = .02), with 463 cases in the standard care group and 398 in the intensive care group.
The imaging study comprised 454 subjects who had brain MRI at baseline and 4 years after randomization. There was no change in total brain volume, said Ilya Nasrallah, MD, of the University of Pennsylvania. But those in the intensively managed group had 18% lower white matter lesion load than those in the standard care group (P = .004).
White matter lesions often point to small vessel disease, which is conclusively linked to vascular dementia, and may also linked to Alzheimer’s disease. Most AD patients, in fact, have a mixed dementia that often includes a vascular component, Dr. Carillo said.
SPRINT MIND didn’t follow subjects past 4 years, and didn’t include any follow-up for amyloid or Alzheimer’s diagnosis. But preventing MCI is no trivial thing, according to David Knopman, MD, who moderated the session.
“There’s nothing that is benign about MCI,” said Dr. Knopman of the Mayo Clinic, Rochester, Minn. “it’s the first sign of overt cognitive dysfunction, and although the rate at which MCI progress to dementia is slow, the appearance of it is just as important as the appearance of more severe dementia. To be able to see an effect in 3.2 years is quite remarkable. I think is going to change clinical practice for people in primary care and the benefits at the population level are going to be substantial.”
Dr. Williamson drove this point home in a later interview, suggesting that physicians may want to think about how the SPRINT MIND results might apply to even younger patients with hypertension, and even if they don’t have other cardiovascular risk factors.
“I can’t say as a scientist that we have evidence to do that, yet. But as a physician, and for my own self and my own patients, I will adhere to the guidelines we have and keep blood pressure at less than 130 mm Hg, and certainly start treating people in their 50s, and probably in their 40s.”
***This article was updated 7/31/18.
SOURCE: Williamson et al. AAIC 2018 DT-0202 Nasrallah et al. AAIC 2018 DT-03-03
AT AAIC 2018
Key clinical point: Keeping systolic blood pressure at less than 120 mm Hg reduced the risk of MCI and all-cause dementia more effectively than keeping it less than 140 mm Hg.
Major finding: After 3.2 years of treatment, there was a 19% lower risk of MCI in the intensively managed group relative to the standard of care group.
Study details: SPRINT MIND comprised more than 9,000 subjects treated for 3.2 years.
Disclosures: The study was funded by the National Institutes of Health. Neither presenter had any relevant financial disclosures.
Source: Williamson et al. AAIC 2018 DT-0202
Early-onset atopic dermatitis linked to elevated risk for seasonal allergies and asthma
LAKE TAHOE, CALIF. – results from a large, retrospective cohort study demonstrated.

“The atopic march is characterized by a progression from atopic dermatitis, usually early in childhood, to subsequent development of allergic rhinitis and asthma, lead study author Joy Wan, MD, said at the annual meeting of the Society for Pediatric Dermatology. “It is thought that the skin acts as the site of primary sensitization through a defective epithelial barrier, which then allows for allergic sensitization to occur in the airways. It is estimated that 30%-60% of AD patients go on to develop asthma and/or allergic rhinitis. However, not all patients complete the so-called atopic march, and this variation in the risk of asthma and allergic rhinitis among AD patients is not very well understood. Better ways to risk stratify these patients are needed.”
One possible explanation for this variation in the risk of atopy in AD patients could be the timing of their dermatitis onset. “We know that atopic dermatitis begins in infancy, but it can start at any age,” said Dr. Wan, who is a fellow in the section of pediatric dermatology at the Children’s Hospital of Philadelphia. “There has been a distinction between early-onset versus late-onset AD. Some past studies have also suggested that there is an increased risk of asthma and allergic rhinitis in children who have early-onset AD before the age of 1 or 2. This suggests that perhaps the model of the atopic march varies between early- and late-onset AD. However, past studies have had several limitations. They’ve often had short durations of follow-up, they’ve only examined narrow ranges of age of onset for AD, and most of them have been designed to primarily evaluate other exposures and outcomes, rather than looking at the timing of AD onset itself.”
For the current study, Dr. Wan and her associates set out to examine the risk of seasonal allergies and asthma among children with AD with respect to the age of AD onset. They used data from the Pediatric Eczema Elective Registry (PEER), an ongoing, prospective U.S. cohort of more than 7,700 children with physician-confirmed AD (JAMA Dermatol. 2014 Jun;150:593-600). All registry participants had used pimecrolimus cream in the past, but children with lymphoproliferative disease were excluded from the registry, as were those with malignancy or those who required the use of systemic immunosuppression.
The researchers evaluated 3,966 subjects in PEER with at least 3 years of follow-up. The exposure of interest was age of AD onset, and they divided patients into three broad age categories: early onset (age 2 years or younger), mid onset (3-7 years), and late onset (8-17 years). Primary outcomes were prevalent seasonal allergies and asthma at the time of registry enrollment, and incident seasonal allergies and asthma during follow-up, assessed via patient surveys every 3 years.
The study population included high proportions of white and black children, and there was a slight predominance of females. The median age at PEER enrollment increased with advancing age of AD onset (5.2 years in the early-onset group vs. 8.2 years in the mid-onset group and 13.1 years in the late-onset group), while the duration of follow-up was fairly similar across the three groups (a median of about 8.3 months). Family history of AD was common across all three groups, while patients in the late-onset group tended to have better control of their AD, compared with their younger counterparts.
At baseline, the prevalence of seasonal allergies was highest among the early-onset group at 74.6%, compared with 69.9% among the mid-onset group and 70.1% among the late-onset group. After adjusting for sex, race, and age at registry enrollment, the relative risk for prevalent seasonal allergies was 9% lower in the mid-onset group (0.91) and 18% lower in the late-onset group (0.82), compared with those in the early-onset group. Next, Dr. Wan and her associates calculated the incidence of seasonal allergies among 1,054 patients who did not have allergies at baseline. The cumulative incidence was highest among the early-onset group (56.1%), followed by the mid-onset group (46.8%), and the late-onset group (30.6%). On adjusted analysis, the relative risk for seasonal allergies among patients who had no allergies at baseline was 18% lower in the mid-onset group (0.82) and 36% lower in the late-onset group (0.64), compared with those in the early-onset group.
In the analysis of asthma risk by age of AD onset, prevalence was highest among patients in the early-onset group at 51.5%, compared with 44.7% among the mid-onset age group and 43% among the late-onset age group. On adjusted analysis, the relative risk for asthma was 15% lower in the mid-onset group (0.85) and 29% lower in the late-onset group (0.71), compared with those in the early-onset group. Meanwhile, the cumulative incidence of asthma among patients without asthma at baseline was also highest in the early-onset group (39.2%), compared with 31.9% in the mid-onset group and 29.9% in the late-onset group.
On adjusted analysis, the relative risk for asthma among this subset of patients was 4% lower in the mid-onset group (0.96) and 8% lower in the late-onset group (0.92), compared with those in the early-onset group, a difference that was not statistically significant. “One possible explanation for this is that asthma tends to develop soon after AD does, and the rates of developing asthma later on, as detected by our study, are nondifferential,” Dr. Wan said. “Another possibility is that the impact of early-onset versus late-onset AD is just different for asthma than it is for seasonal allergies.”
She acknowledged certain limitations of the study, including the risk of misclassification bias and limitations in recall with self-reported data, and the fact that the findings may not be generalizable to all patients with AD.
“Future studies with longer follow-up and studies of adult-onset AD will help extend our findings,” she concluded. “Nevertheless, our findings may inform how we risk stratify patients for AD treatment or atopic march prevention efforts in the future.”
PEER is funded by a grant from Valeant Pharmaceuticals, but Valeant had no role in this study. Dr. Wan reported having no financial disclosures. The study won an award at the meeting for best research presented by a dermatology resident or fellow.
LAKE TAHOE, CALIF. – results from a large, retrospective cohort study demonstrated.
“The atopic march is characterized by a progression from atopic dermatitis, usually early in childhood, to subsequent development of allergic rhinitis and asthma, lead study author Joy Wan, MD, said at the annual meeting of the Society for Pediatric Dermatology. “It is thought that the skin acts as the site of primary sensitization through a defective epithelial barrier, which then allows for allergic sensitization to occur in the airways. It is estimated that 30%-60% of AD patients go on to develop asthma and/or allergic rhinitis. However, not all patients complete the so-called atopic march, and this variation in the risk of asthma and allergic rhinitis among AD patients is not very well understood. Better ways to risk stratify these patients are needed.”
One possible explanation for this variation in the risk of atopy in AD patients could be the timing of their dermatitis onset. “We know that atopic dermatitis begins in infancy, but it can start at any age,” said Dr. Wan, who is a fellow in the section of pediatric dermatology at the Children’s Hospital of Philadelphia. “There has been a distinction between early-onset versus late-onset AD. Some past studies have also suggested that there is an increased risk of asthma and allergic rhinitis in children who have early-onset AD before the age of 1 or 2. This suggests that perhaps the model of the atopic march varies between early- and late-onset AD. However, past studies have had several limitations. They’ve often had short durations of follow-up, they’ve only examined narrow ranges of age of onset for AD, and most of them have been designed to primarily evaluate other exposures and outcomes, rather than looking at the timing of AD onset itself.”
For the current study, Dr. Wan and her associates set out to examine the risk of seasonal allergies and asthma among children with AD with respect to the age of AD onset. They used data from the Pediatric Eczema Elective Registry (PEER), an ongoing, prospective U.S. cohort of more than 7,700 children with physician-confirmed AD (JAMA Dermatol. 2014 Jun;150:593-600). All registry participants had used pimecrolimus cream in the past, but children with lymphoproliferative disease were excluded from the registry, as were those with malignancy or those who required the use of systemic immunosuppression.
The researchers evaluated 3,966 subjects in PEER with at least 3 years of follow-up. The exposure of interest was age of AD onset, and they divided patients into three broad age categories: early onset (age 2 years or younger), mid onset (3-7 years), and late onset (8-17 years). Primary outcomes were prevalent seasonal allergies and asthma at the time of registry enrollment, and incident seasonal allergies and asthma during follow-up, assessed via patient surveys every 3 years.
The study population included high proportions of white and black children, and there was a slight predominance of females. The median age at PEER enrollment increased with advancing age of AD onset (5.2 years in the early-onset group vs. 8.2 years in the mid-onset group and 13.1 years in the late-onset group), while the duration of follow-up was fairly similar across the three groups (a median of about 8.3 months). Family history of AD was common across all three groups, while patients in the late-onset group tended to have better control of their AD, compared with their younger counterparts.
At baseline, the prevalence of seasonal allergies was highest among the early-onset group at 74.6%, compared with 69.9% among the mid-onset group and 70.1% among the late-onset group. After adjusting for sex, race, and age at registry enrollment, the relative risk for prevalent seasonal allergies was 9% lower in the mid-onset group (0.91) and 18% lower in the late-onset group (0.82), compared with those in the early-onset group. Next, Dr. Wan and her associates calculated the incidence of seasonal allergies among 1,054 patients who did not have allergies at baseline. The cumulative incidence was highest among the early-onset group (56.1%), followed by the mid-onset group (46.8%), and the late-onset group (30.6%). On adjusted analysis, the relative risk for seasonal allergies among patients who had no allergies at baseline was 18% lower in the mid-onset group (0.82) and 36% lower in the late-onset group (0.64), compared with those in the early-onset group.
In the analysis of asthma risk by age of AD onset, prevalence was highest among patients in the early-onset group at 51.5%, compared with 44.7% among the mid-onset age group and 43% among the late-onset age group. On adjusted analysis, the relative risk for asthma was 15% lower in the mid-onset group (0.85) and 29% lower in the late-onset group (0.71), compared with those in the early-onset group. Meanwhile, the cumulative incidence of asthma among patients without asthma at baseline was also highest in the early-onset group (39.2%), compared with 31.9% in the mid-onset group and 29.9% in the late-onset group.
On adjusted analysis, the relative risk for asthma among this subset of patients was 4% lower in the mid-onset group (0.96) and 8% lower in the late-onset group (0.92), compared with those in the early-onset group, a difference that was not statistically significant. “One possible explanation for this is that asthma tends to develop soon after AD does, and the rates of developing asthma later on, as detected by our study, are nondifferential,” Dr. Wan said. “Another possibility is that the impact of early-onset versus late-onset AD is just different for asthma than it is for seasonal allergies.”
She acknowledged certain limitations of the study, including the risk of misclassification bias and limitations in recall with self-reported data, and the fact that the findings may not be generalizable to all patients with AD.
“Future studies with longer follow-up and studies of adult-onset AD will help extend our findings,” she concluded. “Nevertheless, our findings may inform how we risk stratify patients for AD treatment or atopic march prevention efforts in the future.”
PEER is funded by a grant from Valeant Pharmaceuticals, but Valeant had no role in this study. Dr. Wan reported having no financial disclosures. The study won an award at the meeting for best research presented by a dermatology resident or fellow.
LAKE TAHOE, CALIF. – results from a large, retrospective cohort study demonstrated.
“The atopic march is characterized by a progression from atopic dermatitis, usually early in childhood, to subsequent development of allergic rhinitis and asthma, lead study author Joy Wan, MD, said at the annual meeting of the Society for Pediatric Dermatology. “It is thought that the skin acts as the site of primary sensitization through a defective epithelial barrier, which then allows for allergic sensitization to occur in the airways. It is estimated that 30%-60% of AD patients go on to develop asthma and/or allergic rhinitis. However, not all patients complete the so-called atopic march, and this variation in the risk of asthma and allergic rhinitis among AD patients is not very well understood. Better ways to risk stratify these patients are needed.”
One possible explanation for this variation in the risk of atopy in AD patients could be the timing of their dermatitis onset. “We know that atopic dermatitis begins in infancy, but it can start at any age,” said Dr. Wan, who is a fellow in the section of pediatric dermatology at the Children’s Hospital of Philadelphia. “There has been a distinction between early-onset versus late-onset AD. Some past studies have also suggested that there is an increased risk of asthma and allergic rhinitis in children who have early-onset AD before the age of 1 or 2. This suggests that perhaps the model of the atopic march varies between early- and late-onset AD. However, past studies have had several limitations. They’ve often had short durations of follow-up, they’ve only examined narrow ranges of age of onset for AD, and most of them have been designed to primarily evaluate other exposures and outcomes, rather than looking at the timing of AD onset itself.”
For the current study, Dr. Wan and her associates set out to examine the risk of seasonal allergies and asthma among children with AD with respect to the age of AD onset. They used data from the Pediatric Eczema Elective Registry (PEER), an ongoing, prospective U.S. cohort of more than 7,700 children with physician-confirmed AD (JAMA Dermatol. 2014 Jun;150:593-600). All registry participants had used pimecrolimus cream in the past, but children with lymphoproliferative disease were excluded from the registry, as were those with malignancy or those who required the use of systemic immunosuppression.
The researchers evaluated 3,966 subjects in PEER with at least 3 years of follow-up. The exposure of interest was age of AD onset, and they divided patients into three broad age categories: early onset (age 2 years or younger), mid onset (3-7 years), and late onset (8-17 years). Primary outcomes were prevalent seasonal allergies and asthma at the time of registry enrollment, and incident seasonal allergies and asthma during follow-up, assessed via patient surveys every 3 years.
The study population included high proportions of white and black children, and there was a slight predominance of females. The median age at PEER enrollment increased with advancing age of AD onset (5.2 years in the early-onset group vs. 8.2 years in the mid-onset group and 13.1 years in the late-onset group), while the duration of follow-up was fairly similar across the three groups (a median of about 8.3 months). Family history of AD was common across all three groups, while patients in the late-onset group tended to have better control of their AD, compared with their younger counterparts.
At baseline, the prevalence of seasonal allergies was highest among the early-onset group at 74.6%, compared with 69.9% among the mid-onset group and 70.1% among the late-onset group. After adjusting for sex, race, and age at registry enrollment, the relative risk for prevalent seasonal allergies was 9% lower in the mid-onset group (0.91) and 18% lower in the late-onset group (0.82), compared with those in the early-onset group. Next, Dr. Wan and her associates calculated the incidence of seasonal allergies among 1,054 patients who did not have allergies at baseline. The cumulative incidence was highest among the early-onset group (56.1%), followed by the mid-onset group (46.8%), and the late-onset group (30.6%). On adjusted analysis, the relative risk for seasonal allergies among patients who had no allergies at baseline was 18% lower in the mid-onset group (0.82) and 36% lower in the late-onset group (0.64), compared with those in the early-onset group.
In the analysis of asthma risk by age of AD onset, prevalence was highest among patients in the early-onset group at 51.5%, compared with 44.7% among the mid-onset age group and 43% among the late-onset age group. On adjusted analysis, the relative risk for asthma was 15% lower in the mid-onset group (0.85) and 29% lower in the late-onset group (0.71), compared with those in the early-onset group. Meanwhile, the cumulative incidence of asthma among patients without asthma at baseline was also highest in the early-onset group (39.2%), compared with 31.9% in the mid-onset group and 29.9% in the late-onset group.
On adjusted analysis, the relative risk for asthma among this subset of patients was 4% lower in the mid-onset group (0.96) and 8% lower in the late-onset group (0.92), compared with those in the early-onset group, a difference that was not statistically significant. “One possible explanation for this is that asthma tends to develop soon after AD does, and the rates of developing asthma later on, as detected by our study, are nondifferential,” Dr. Wan said. “Another possibility is that the impact of early-onset versus late-onset AD is just different for asthma than it is for seasonal allergies.”
She acknowledged certain limitations of the study, including the risk of misclassification bias and limitations in recall with self-reported data, and the fact that the findings may not be generalizable to all patients with AD.
“Future studies with longer follow-up and studies of adult-onset AD will help extend our findings,” she concluded. “Nevertheless, our findings may inform how we risk stratify patients for AD treatment or atopic march prevention efforts in the future.”
PEER is funded by a grant from Valeant Pharmaceuticals, but Valeant had no role in this study. Dr. Wan reported having no financial disclosures. The study won an award at the meeting for best research presented by a dermatology resident or fellow.
AT SPD 2018