User login
Building a better SHM
As we enter the holiday season, the Society of Hospital Medicine is preparing to unwrap a refreshed experience for all members and partners.
Next month, SHM will launch its new association management system (AMS), its new online community platform for the Hospital Medicine Exchange (HMX), and a brand new website to better serve the needs of its constituents.
While many may be unaware of the systems and platforms SHM currently uses, an AMS is essentially SHM’s EHR for its members. It houses each member’s information, so the more information SHM has, the more SHM can customize the types of information you receive. All systems will be integrated so you can quickly access information on the chapter, interest group, or committee to which you belong.
What does this mean to you?
• You’ll be prompted to create a new password for your SHM account. When you set up your new password, we urge you to update your profile to make sure your information is current and that you are receiving content that is most relevant to you.
• As you update your profile, you will have an opportunity to edit your email preferences. If you have previously opted out of SHM emails, we urge you to opt back in to receive information on your local chapter meetings and more targeted messages about SHM offerings tailored specifically to your interests.
• The SHM website, www.hospitalmedicine.org, will be optimized for your smartphone and tablet and have a fresh look and feel on all devices, complete with new, intuitive navigation and streamlined content – making it easier for you to find the information that is the most relevant for you in even less time.
• The Hospital Medicine Exchange (HMX) will move to an intuitive new platform to enhance your online discussions and group collaborations, including chapters, interest groups, committees, and more.
In addition to these technological enhancements, watch for a refreshed design of The Hospitalist, the Journal of Hospital Medicine, and the overall SHM brand to bring a refined, sleek look to all SHM-related products, programs, and communications.
We look forward to better serving the needs of our members and partners with these improvements and encourage you to share your thoughts at [email protected].
Mr. Radler is marketing communications manager at the Society of Hospital Medicine.
As we enter the holiday season, the Society of Hospital Medicine is preparing to unwrap a refreshed experience for all members and partners.
Next month, SHM will launch its new association management system (AMS), its new online community platform for the Hospital Medicine Exchange (HMX), and a brand new website to better serve the needs of its constituents.
While many may be unaware of the systems and platforms SHM currently uses, an AMS is essentially SHM’s EHR for its members. It houses each member’s information, so the more information SHM has, the more SHM can customize the types of information you receive. All systems will be integrated so you can quickly access information on the chapter, interest group, or committee to which you belong.
What does this mean to you?
• You’ll be prompted to create a new password for your SHM account. When you set up your new password, we urge you to update your profile to make sure your information is current and that you are receiving content that is most relevant to you.
• As you update your profile, you will have an opportunity to edit your email preferences. If you have previously opted out of SHM emails, we urge you to opt back in to receive information on your local chapter meetings and more targeted messages about SHM offerings tailored specifically to your interests.
• The SHM website, www.hospitalmedicine.org, will be optimized for your smartphone and tablet and have a fresh look and feel on all devices, complete with new, intuitive navigation and streamlined content – making it easier for you to find the information that is the most relevant for you in even less time.
• The Hospital Medicine Exchange (HMX) will move to an intuitive new platform to enhance your online discussions and group collaborations, including chapters, interest groups, committees, and more.
In addition to these technological enhancements, watch for a refreshed design of The Hospitalist, the Journal of Hospital Medicine, and the overall SHM brand to bring a refined, sleek look to all SHM-related products, programs, and communications.
We look forward to better serving the needs of our members and partners with these improvements and encourage you to share your thoughts at [email protected].
Mr. Radler is marketing communications manager at the Society of Hospital Medicine.
As we enter the holiday season, the Society of Hospital Medicine is preparing to unwrap a refreshed experience for all members and partners.
Next month, SHM will launch its new association management system (AMS), its new online community platform for the Hospital Medicine Exchange (HMX), and a brand new website to better serve the needs of its constituents.
While many may be unaware of the systems and platforms SHM currently uses, an AMS is essentially SHM’s EHR for its members. It houses each member’s information, so the more information SHM has, the more SHM can customize the types of information you receive. All systems will be integrated so you can quickly access information on the chapter, interest group, or committee to which you belong.
What does this mean to you?
• You’ll be prompted to create a new password for your SHM account. When you set up your new password, we urge you to update your profile to make sure your information is current and that you are receiving content that is most relevant to you.
• As you update your profile, you will have an opportunity to edit your email preferences. If you have previously opted out of SHM emails, we urge you to opt back in to receive information on your local chapter meetings and more targeted messages about SHM offerings tailored specifically to your interests.
• The SHM website, www.hospitalmedicine.org, will be optimized for your smartphone and tablet and have a fresh look and feel on all devices, complete with new, intuitive navigation and streamlined content – making it easier for you to find the information that is the most relevant for you in even less time.
• The Hospital Medicine Exchange (HMX) will move to an intuitive new platform to enhance your online discussions and group collaborations, including chapters, interest groups, committees, and more.
In addition to these technological enhancements, watch for a refreshed design of The Hospitalist, the Journal of Hospital Medicine, and the overall SHM brand to bring a refined, sleek look to all SHM-related products, programs, and communications.
We look forward to better serving the needs of our members and partners with these improvements and encourage you to share your thoughts at [email protected].
Mr. Radler is marketing communications manager at the Society of Hospital Medicine.
Chikungunya virus goes undetected despite chronic arthritis in 25% of patients after 20 months
Up to a quarter of patients infected with chikungunya virus who reported joint pain as one of their initial symptoms continue to have joint pain after 20 months of follow-up, and evidence suggests that the persistent joint symptoms are not related to the continued presence of the virus in synovial fluid, according to two studies of patients infected during the 2014-2015 Colombian epidemic.
In the first large-scale, cross-sectional follow-up of a prospective cohort from the Colombian epidemic, Aileen Chang, MD, of George Washington University, Washington, and her colleagues at multiple other institutions reported persistent joint pain and multiple swollen and/or tender joints after 20 months in 123 of 485 Colombian patients who initially had joint pain with their Chikungunya virus infection (CHIKV) diagnosis. In their report in Arthritis & Rheumatology, they said that increased initial viral load and severe initial joint pain were predictors of persistent arthritis, which is consistent with the work of other researchers.
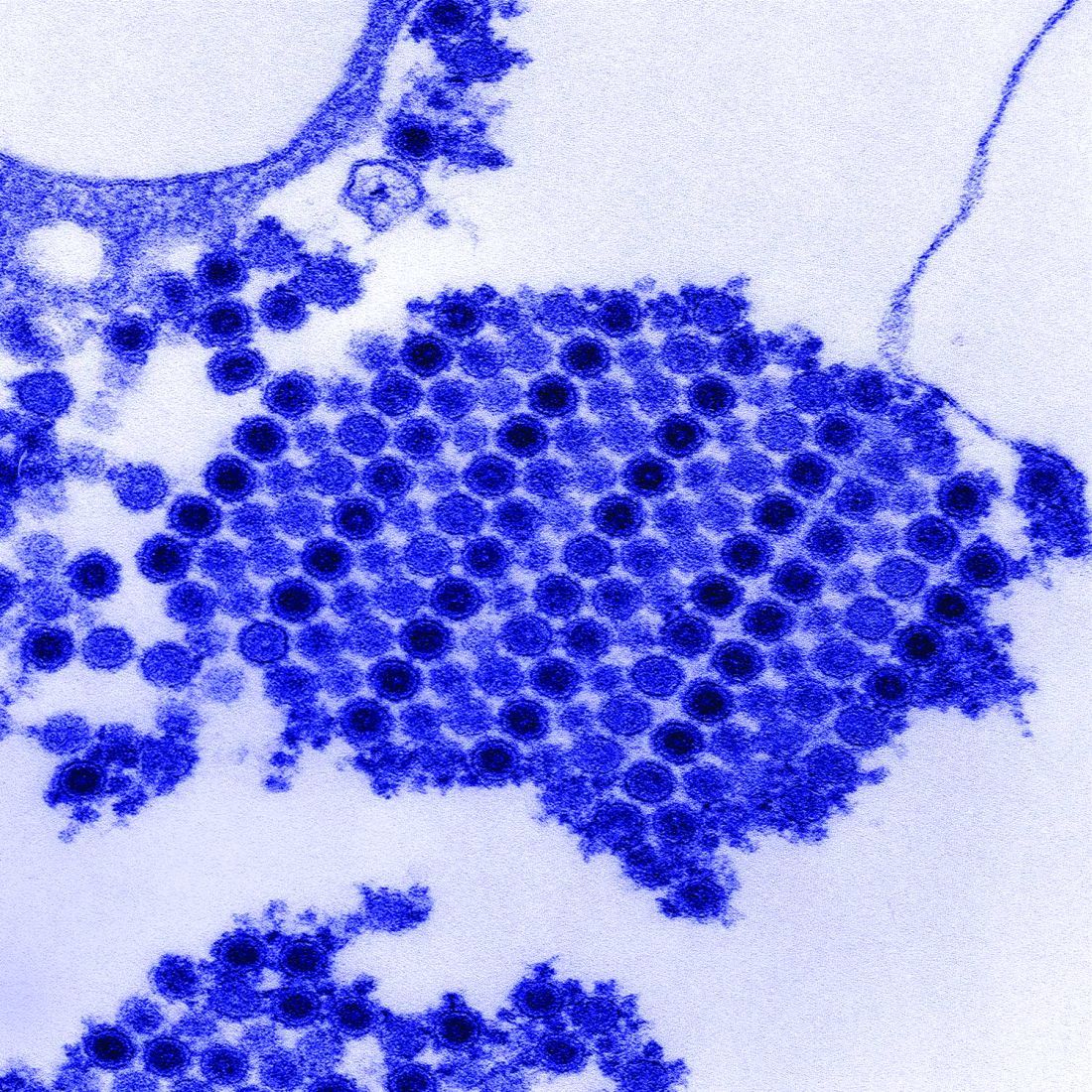
Dr. Chang worked with a variety of coinvestigators, some of whom were also involved in the larger symptom follow-up study, to conduct this Study of Chikungunya Arthritis Mechanisms in the Americas (CAMA). They collected synovial fluid and blood plasma from these 38 patients as well as 10 healthy controls who were serologically negative for CHIKV and never had arthritis, and analyzed the fluid and plasma for signs of CHIKV. They assessed viral RNA via quantitative reverse transcription polymerase chain reaction (qRT-PCR) testing, looked at viral proteins via mass spectrometry, and did viral cultures.
All samples from the 38 patients in the study were negative for CHIKV in two separate qRT-PCR assays. To determine if low-level viremia was present in synovial fluid samples, the samples were added to cell cultures to expand viral replication. No viral growth was found after three attempts and 10 days of culture. Conversely, controls with low quantities of virus (about 1 plaque-forming unit per well) yielded growth and detection of the virus.
Patients with CHIKV-associated arthritis also had no significant increase in rheumatoid arthritis markers or C-reactive protein. In fact, plasma markers for rheumatoid arthritis were found in only a fraction of patients with CHIKV arthritis: rheumatoid factor (RF) IgM antibody in 9%, RF IgG antibody in 12%, and anti–cyclic citrullinated peptide in 0%.
The more probable potential mechanisms through which CHIKV could cause persistent arthritis symptoms is through the presence of persistent CHIKV or viral antigens at low enough levels in the synovial tissue that it is undetectable in the synovial fluid, Dr. Chang and her associates suggested. There is also a possibility of epigenetic changes to the host DNA, altering host gene transcription. Other epigenetic changes, like epigenetic imprinting, could be possible in macrophages, leading to more aggressive cell behavior, they said. Unlikelier scenarios would be the presence of seronegative RA in these patients or, alternatively, the presence of seronegative RA indicating prior infection with CHIKV or other arthritogenic viruses.
Whatever mechanisms are causing CHIKV-associated arthritis, “these study findings may have important clinical relevance for CHIKV in the Americas. Since there is no current standard of care guidance for the treatment of CHIKV arthritis, some patients are currently being treated with immunosuppressant medications such as methotrexate, hydroxychloroquine, etanercept, adalimumab, sulfasalazine, fingolimod, abatacept, and tofacitinib,” Dr. Chang and her colleagues wrote. “This practice could be potentially harmful in the setting of replicating virus in the synovium as it could permit reemergence of a systemic viral infection.”
The CAMA study has several important limitations , the investigators said, the first being that during collection of synovial fluid, 0-20 mL of saline solution were used to flush the joints, which could have affected the ability to detect virus in the samples. In an attempt to mitigate this, the researchers cultured 0.5-1.5 mL of sampled synovial fluid to expand any replication-competent virus present in the sample, used two complementary PCR assays to detect nucleic acids, and a proteomic approach to look for viral proteins.
The researchers acknowledged that despite these measures, “proving the absence of a target is difficult, and we recognize that it is possible that our approach failed to detect low-level viral antigen; however, our orthogonal approach clearly demonstrates that if viral antigen exists in the synovial fluid, it is at extremely low levels.” They advised that future studies may want to use synovial biopsies rather than extracted fluid.
The investigators also did not include patients who had previously been infected by CHIKV without chronic arthritis. This issue was compounded by the lack of age- and sex-matched healthy controls.
All researchers involved in the studies reported no financial conflicts of interest. The studies were supported by various grants from the National Institutes of Health and the Rheumatology Research Foundation.
SOURCE: Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40383 and Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40384
Up to a quarter of patients infected with chikungunya virus who reported joint pain as one of their initial symptoms continue to have joint pain after 20 months of follow-up, and evidence suggests that the persistent joint symptoms are not related to the continued presence of the virus in synovial fluid, according to two studies of patients infected during the 2014-2015 Colombian epidemic.
In the first large-scale, cross-sectional follow-up of a prospective cohort from the Colombian epidemic, Aileen Chang, MD, of George Washington University, Washington, and her colleagues at multiple other institutions reported persistent joint pain and multiple swollen and/or tender joints after 20 months in 123 of 485 Colombian patients who initially had joint pain with their Chikungunya virus infection (CHIKV) diagnosis. In their report in Arthritis & Rheumatology, they said that increased initial viral load and severe initial joint pain were predictors of persistent arthritis, which is consistent with the work of other researchers.
Dr. Chang worked with a variety of coinvestigators, some of whom were also involved in the larger symptom follow-up study, to conduct this Study of Chikungunya Arthritis Mechanisms in the Americas (CAMA). They collected synovial fluid and blood plasma from these 38 patients as well as 10 healthy controls who were serologically negative for CHIKV and never had arthritis, and analyzed the fluid and plasma for signs of CHIKV. They assessed viral RNA via quantitative reverse transcription polymerase chain reaction (qRT-PCR) testing, looked at viral proteins via mass spectrometry, and did viral cultures.
All samples from the 38 patients in the study were negative for CHIKV in two separate qRT-PCR assays. To determine if low-level viremia was present in synovial fluid samples, the samples were added to cell cultures to expand viral replication. No viral growth was found after three attempts and 10 days of culture. Conversely, controls with low quantities of virus (about 1 plaque-forming unit per well) yielded growth and detection of the virus.
Patients with CHIKV-associated arthritis also had no significant increase in rheumatoid arthritis markers or C-reactive protein. In fact, plasma markers for rheumatoid arthritis were found in only a fraction of patients with CHIKV arthritis: rheumatoid factor (RF) IgM antibody in 9%, RF IgG antibody in 12%, and anti–cyclic citrullinated peptide in 0%.
The more probable potential mechanisms through which CHIKV could cause persistent arthritis symptoms is through the presence of persistent CHIKV or viral antigens at low enough levels in the synovial tissue that it is undetectable in the synovial fluid, Dr. Chang and her associates suggested. There is also a possibility of epigenetic changes to the host DNA, altering host gene transcription. Other epigenetic changes, like epigenetic imprinting, could be possible in macrophages, leading to more aggressive cell behavior, they said. Unlikelier scenarios would be the presence of seronegative RA in these patients or, alternatively, the presence of seronegative RA indicating prior infection with CHIKV or other arthritogenic viruses.
Whatever mechanisms are causing CHIKV-associated arthritis, “these study findings may have important clinical relevance for CHIKV in the Americas. Since there is no current standard of care guidance for the treatment of CHIKV arthritis, some patients are currently being treated with immunosuppressant medications such as methotrexate, hydroxychloroquine, etanercept, adalimumab, sulfasalazine, fingolimod, abatacept, and tofacitinib,” Dr. Chang and her colleagues wrote. “This practice could be potentially harmful in the setting of replicating virus in the synovium as it could permit reemergence of a systemic viral infection.”
The CAMA study has several important limitations , the investigators said, the first being that during collection of synovial fluid, 0-20 mL of saline solution were used to flush the joints, which could have affected the ability to detect virus in the samples. In an attempt to mitigate this, the researchers cultured 0.5-1.5 mL of sampled synovial fluid to expand any replication-competent virus present in the sample, used two complementary PCR assays to detect nucleic acids, and a proteomic approach to look for viral proteins.
The researchers acknowledged that despite these measures, “proving the absence of a target is difficult, and we recognize that it is possible that our approach failed to detect low-level viral antigen; however, our orthogonal approach clearly demonstrates that if viral antigen exists in the synovial fluid, it is at extremely low levels.” They advised that future studies may want to use synovial biopsies rather than extracted fluid.
The investigators also did not include patients who had previously been infected by CHIKV without chronic arthritis. This issue was compounded by the lack of age- and sex-matched healthy controls.
All researchers involved in the studies reported no financial conflicts of interest. The studies were supported by various grants from the National Institutes of Health and the Rheumatology Research Foundation.
SOURCE: Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40383 and Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40384
Up to a quarter of patients infected with chikungunya virus who reported joint pain as one of their initial symptoms continue to have joint pain after 20 months of follow-up, and evidence suggests that the persistent joint symptoms are not related to the continued presence of the virus in synovial fluid, according to two studies of patients infected during the 2014-2015 Colombian epidemic.
In the first large-scale, cross-sectional follow-up of a prospective cohort from the Colombian epidemic, Aileen Chang, MD, of George Washington University, Washington, and her colleagues at multiple other institutions reported persistent joint pain and multiple swollen and/or tender joints after 20 months in 123 of 485 Colombian patients who initially had joint pain with their Chikungunya virus infection (CHIKV) diagnosis. In their report in Arthritis & Rheumatology, they said that increased initial viral load and severe initial joint pain were predictors of persistent arthritis, which is consistent with the work of other researchers.
Dr. Chang worked with a variety of coinvestigators, some of whom were also involved in the larger symptom follow-up study, to conduct this Study of Chikungunya Arthritis Mechanisms in the Americas (CAMA). They collected synovial fluid and blood plasma from these 38 patients as well as 10 healthy controls who were serologically negative for CHIKV and never had arthritis, and analyzed the fluid and plasma for signs of CHIKV. They assessed viral RNA via quantitative reverse transcription polymerase chain reaction (qRT-PCR) testing, looked at viral proteins via mass spectrometry, and did viral cultures.
All samples from the 38 patients in the study were negative for CHIKV in two separate qRT-PCR assays. To determine if low-level viremia was present in synovial fluid samples, the samples were added to cell cultures to expand viral replication. No viral growth was found after three attempts and 10 days of culture. Conversely, controls with low quantities of virus (about 1 plaque-forming unit per well) yielded growth and detection of the virus.
Patients with CHIKV-associated arthritis also had no significant increase in rheumatoid arthritis markers or C-reactive protein. In fact, plasma markers for rheumatoid arthritis were found in only a fraction of patients with CHIKV arthritis: rheumatoid factor (RF) IgM antibody in 9%, RF IgG antibody in 12%, and anti–cyclic citrullinated peptide in 0%.
The more probable potential mechanisms through which CHIKV could cause persistent arthritis symptoms is through the presence of persistent CHIKV or viral antigens at low enough levels in the synovial tissue that it is undetectable in the synovial fluid, Dr. Chang and her associates suggested. There is also a possibility of epigenetic changes to the host DNA, altering host gene transcription. Other epigenetic changes, like epigenetic imprinting, could be possible in macrophages, leading to more aggressive cell behavior, they said. Unlikelier scenarios would be the presence of seronegative RA in these patients or, alternatively, the presence of seronegative RA indicating prior infection with CHIKV or other arthritogenic viruses.
Whatever mechanisms are causing CHIKV-associated arthritis, “these study findings may have important clinical relevance for CHIKV in the Americas. Since there is no current standard of care guidance for the treatment of CHIKV arthritis, some patients are currently being treated with immunosuppressant medications such as methotrexate, hydroxychloroquine, etanercept, adalimumab, sulfasalazine, fingolimod, abatacept, and tofacitinib,” Dr. Chang and her colleagues wrote. “This practice could be potentially harmful in the setting of replicating virus in the synovium as it could permit reemergence of a systemic viral infection.”
The CAMA study has several important limitations , the investigators said, the first being that during collection of synovial fluid, 0-20 mL of saline solution were used to flush the joints, which could have affected the ability to detect virus in the samples. In an attempt to mitigate this, the researchers cultured 0.5-1.5 mL of sampled synovial fluid to expand any replication-competent virus present in the sample, used two complementary PCR assays to detect nucleic acids, and a proteomic approach to look for viral proteins.
The researchers acknowledged that despite these measures, “proving the absence of a target is difficult, and we recognize that it is possible that our approach failed to detect low-level viral antigen; however, our orthogonal approach clearly demonstrates that if viral antigen exists in the synovial fluid, it is at extremely low levels.” They advised that future studies may want to use synovial biopsies rather than extracted fluid.
The investigators also did not include patients who had previously been infected by CHIKV without chronic arthritis. This issue was compounded by the lack of age- and sex-matched healthy controls.
All researchers involved in the studies reported no financial conflicts of interest. The studies were supported by various grants from the National Institutes of Health and the Rheumatology Research Foundation.
SOURCE: Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40383 and Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40384
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point:
Major finding: No signs of persistent CHIKV infection can be found in synovial fluid or blood plasma from patients with chronic arthritis after CHIKV.
Study details: Cross-sectional studies of 485 Colombian patients who had clinical CHIKV and associated arthritis symptoms in 2014-2015 and another 38 patients who underwent further synovial fluid and blood plasma testing after a median of 22 months.
Disclosures: All researchers involved in the studies reported no financial conflicts of interest. The studies were supported by various grants from the National Institutes of Health and the Rheumatology Research Foundation.
Source: Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40383 and Chang A et al. Arthritis Rheumatol. 2017 Dec 20. doi: 10.1002/art.40384.
DCIS risk signature is validated in SweDCIS population
SAN ANTONIO – A biological risk signature can help guide decisions about use of adjuvant radiation therapy in patients with ductal carcinoma in situ (DCIS), suggests a validation study reported at the San Antonio Breast Cancer Symposium.
Radiation therapy reduces the 10-year risk of any ipsilateral recurrence in this population by about 50% as established in a large overview of trials (J Natl Cancer Inst Monogr. 2010;2010:162-77), noted lead investigator Fredrik Wärnberg, MD, PhD, of Uppsala Academic Hospital, Uppsala University, Sweden. But factors such as tumor size, grade, and margins have not been helpful in identifying patients most likely to benefit.

The risk signature incorporates four clinicopathologic factors and seven immunohistochemically assessed biomarkers of hormone receptor status, HER2 status, stress response, and proliferation. Possible scores range from 0 to 10, and are split into categories of low risk (0 to 3) and elevated risk (greater than 3). “To me, the magic of this signature is that it is nonlinear. Each factor can be dependent on the value of other factors in the model,” Dr. Wärnberg said.
Results of the validation study showed that among the 506 patients who had clear margins after surgery, radiation therapy significantly reduced the 10-year risk of invasive recurrence in those with an elevated risk score by more than three-fourths, but not in those with a low risk score.
“The biologic risk signature … correlated to risk. It’s prognostic, that’s nothing new,” he summarized. “More interestingly, it was also predictive for radiotherapy benefit. Not all patient groups had the same benefit from radiation therapy. In the low-risk group, there wasn’t any significant benefit from radiation therapy for invasive recurrences. But in the elevated risk group, the radiation therapy benefit was twice as high as expected – about a 76% relative risk reduction with radiotherapy for invasive recurrences.”
Study details
Main results from the SweDCIS trial, previously reported (J Clin Oncol. 2014;32:3613-8), showed that adjuvant radiation therapy reduced recurrences, yielding a 12% absolute reduction in risk of ipsilateral recurrence (10% for in situ recurrences and 2% for invasive recurrences).
For the validation study, Dr. Wärnberg and his colleagues were able to obtain tissue and biological signature results, blinded to patient outcome, for 56% of the original trial population. About half of patients each were determined to have low risk scores and elevated risk scores.
Among the 506 patients with clear margins, the score, analyzed as a continuous variable, was associated with risk of any (in situ or invasive) ipsilateral recurrence during follow-up (hazard ratio, 1.49 per 5-unit increase; P = .038).
In a multivariate model, receipt of radiation therapy was associated with a 52% relative reduction in 10-year risk of any ipsilateral recurrence for those with a low-risk score (HR, 0.48; P = .04) and a greater 69% relative reduction for those with an elevated risk score (HR, 0.31; P less than .001).
Radiation therapy did not significantly reduce the risk of ipsilateral invasive recurrence in the low risk group (HR, 0.84; P = .70), but it did in the elevated risk group (HR, 0.24; P = .012).
These findings essentially mirrored those of a 2015 validation study in a separate cohort of 526 patients from Uppsala University Hospital and the University of Massachusetts, according to Dr. Wärnberg. “We found highly consistent data with these two different sets,” he said.
Analyses additionally showed that radiation therapy reduced the 10-year risk of an invasive breast cancer recurrence by an absolute 1% for patients with low risk scores (not significant) but by an absolute 9% for patients with elevated risk scores (P = .012).
Dr. Wärnberg disclosed that he had no relevant conflicts of interest. The study was funded in part by PreludeDx.
SOURCE: Warnberg et al., SABCS Abstract GS5-08
SAN ANTONIO – A biological risk signature can help guide decisions about use of adjuvant radiation therapy in patients with ductal carcinoma in situ (DCIS), suggests a validation study reported at the San Antonio Breast Cancer Symposium.
Radiation therapy reduces the 10-year risk of any ipsilateral recurrence in this population by about 50% as established in a large overview of trials (J Natl Cancer Inst Monogr. 2010;2010:162-77), noted lead investigator Fredrik Wärnberg, MD, PhD, of Uppsala Academic Hospital, Uppsala University, Sweden. But factors such as tumor size, grade, and margins have not been helpful in identifying patients most likely to benefit.
The risk signature incorporates four clinicopathologic factors and seven immunohistochemically assessed biomarkers of hormone receptor status, HER2 status, stress response, and proliferation. Possible scores range from 0 to 10, and are split into categories of low risk (0 to 3) and elevated risk (greater than 3). “To me, the magic of this signature is that it is nonlinear. Each factor can be dependent on the value of other factors in the model,” Dr. Wärnberg said.
Results of the validation study showed that among the 506 patients who had clear margins after surgery, radiation therapy significantly reduced the 10-year risk of invasive recurrence in those with an elevated risk score by more than three-fourths, but not in those with a low risk score.
“The biologic risk signature … correlated to risk. It’s prognostic, that’s nothing new,” he summarized. “More interestingly, it was also predictive for radiotherapy benefit. Not all patient groups had the same benefit from radiation therapy. In the low-risk group, there wasn’t any significant benefit from radiation therapy for invasive recurrences. But in the elevated risk group, the radiation therapy benefit was twice as high as expected – about a 76% relative risk reduction with radiotherapy for invasive recurrences.”
Study details
Main results from the SweDCIS trial, previously reported (J Clin Oncol. 2014;32:3613-8), showed that adjuvant radiation therapy reduced recurrences, yielding a 12% absolute reduction in risk of ipsilateral recurrence (10% for in situ recurrences and 2% for invasive recurrences).
For the validation study, Dr. Wärnberg and his colleagues were able to obtain tissue and biological signature results, blinded to patient outcome, for 56% of the original trial population. About half of patients each were determined to have low risk scores and elevated risk scores.
Among the 506 patients with clear margins, the score, analyzed as a continuous variable, was associated with risk of any (in situ or invasive) ipsilateral recurrence during follow-up (hazard ratio, 1.49 per 5-unit increase; P = .038).
In a multivariate model, receipt of radiation therapy was associated with a 52% relative reduction in 10-year risk of any ipsilateral recurrence for those with a low-risk score (HR, 0.48; P = .04) and a greater 69% relative reduction for those with an elevated risk score (HR, 0.31; P less than .001).
Radiation therapy did not significantly reduce the risk of ipsilateral invasive recurrence in the low risk group (HR, 0.84; P = .70), but it did in the elevated risk group (HR, 0.24; P = .012).
These findings essentially mirrored those of a 2015 validation study in a separate cohort of 526 patients from Uppsala University Hospital and the University of Massachusetts, according to Dr. Wärnberg. “We found highly consistent data with these two different sets,” he said.
Analyses additionally showed that radiation therapy reduced the 10-year risk of an invasive breast cancer recurrence by an absolute 1% for patients with low risk scores (not significant) but by an absolute 9% for patients with elevated risk scores (P = .012).
Dr. Wärnberg disclosed that he had no relevant conflicts of interest. The study was funded in part by PreludeDx.
SOURCE: Warnberg et al., SABCS Abstract GS5-08
SAN ANTONIO – A biological risk signature can help guide decisions about use of adjuvant radiation therapy in patients with ductal carcinoma in situ (DCIS), suggests a validation study reported at the San Antonio Breast Cancer Symposium.
Radiation therapy reduces the 10-year risk of any ipsilateral recurrence in this population by about 50% as established in a large overview of trials (J Natl Cancer Inst Monogr. 2010;2010:162-77), noted lead investigator Fredrik Wärnberg, MD, PhD, of Uppsala Academic Hospital, Uppsala University, Sweden. But factors such as tumor size, grade, and margins have not been helpful in identifying patients most likely to benefit.
The risk signature incorporates four clinicopathologic factors and seven immunohistochemically assessed biomarkers of hormone receptor status, HER2 status, stress response, and proliferation. Possible scores range from 0 to 10, and are split into categories of low risk (0 to 3) and elevated risk (greater than 3). “To me, the magic of this signature is that it is nonlinear. Each factor can be dependent on the value of other factors in the model,” Dr. Wärnberg said.
Results of the validation study showed that among the 506 patients who had clear margins after surgery, radiation therapy significantly reduced the 10-year risk of invasive recurrence in those with an elevated risk score by more than three-fourths, but not in those with a low risk score.
“The biologic risk signature … correlated to risk. It’s prognostic, that’s nothing new,” he summarized. “More interestingly, it was also predictive for radiotherapy benefit. Not all patient groups had the same benefit from radiation therapy. In the low-risk group, there wasn’t any significant benefit from radiation therapy for invasive recurrences. But in the elevated risk group, the radiation therapy benefit was twice as high as expected – about a 76% relative risk reduction with radiotherapy for invasive recurrences.”
Study details
Main results from the SweDCIS trial, previously reported (J Clin Oncol. 2014;32:3613-8), showed that adjuvant radiation therapy reduced recurrences, yielding a 12% absolute reduction in risk of ipsilateral recurrence (10% for in situ recurrences and 2% for invasive recurrences).
For the validation study, Dr. Wärnberg and his colleagues were able to obtain tissue and biological signature results, blinded to patient outcome, for 56% of the original trial population. About half of patients each were determined to have low risk scores and elevated risk scores.
Among the 506 patients with clear margins, the score, analyzed as a continuous variable, was associated with risk of any (in situ or invasive) ipsilateral recurrence during follow-up (hazard ratio, 1.49 per 5-unit increase; P = .038).
In a multivariate model, receipt of radiation therapy was associated with a 52% relative reduction in 10-year risk of any ipsilateral recurrence for those with a low-risk score (HR, 0.48; P = .04) and a greater 69% relative reduction for those with an elevated risk score (HR, 0.31; P less than .001).
Radiation therapy did not significantly reduce the risk of ipsilateral invasive recurrence in the low risk group (HR, 0.84; P = .70), but it did in the elevated risk group (HR, 0.24; P = .012).
These findings essentially mirrored those of a 2015 validation study in a separate cohort of 526 patients from Uppsala University Hospital and the University of Massachusetts, according to Dr. Wärnberg. “We found highly consistent data with these two different sets,” he said.
Analyses additionally showed that radiation therapy reduced the 10-year risk of an invasive breast cancer recurrence by an absolute 1% for patients with low risk scores (not significant) but by an absolute 9% for patients with elevated risk scores (P = .012).
Dr. Wärnberg disclosed that he had no relevant conflicts of interest. The study was funded in part by PreludeDx.
SOURCE: Warnberg et al., SABCS Abstract GS5-08
REPORTING FROM SABCS 2017
Key clinical point:
Major finding: Among patients with clear margins, radiation therapy significantly reduced 10-year risk of invasive recurrence in those with an elevated risk score (hazard ratio, 0.24; P = .012) but not in those with a low risk score (HR, 0.84; P = .70).
Data source: A validation study in 584 patients with DCIS from a randomized trial of radiation therapy (SweDCIS).
Disclosures: Dr. Wärnberg disclosed that he had no relevant conflicts of interest. The study was funded in part by PreludeDx.
Source: Warnberg et al., SABCS Abstract GS5-08
Typhoid isn’t covered??!!
My wife and I decided to visit Morocco, to test the maxim that my fellow columnist Joe Eastern often cites: The words you won’t say on your deathbed are, “If only I had spent more time at the office.”
Though I’m not convinced he’s right about that – he’s never even seen my office – I thought I’d give being away a try. My office manager comes from near Marrakesh. While bound for Morocco, we could check out her hometown, even if there is no obvious tax angle.
As I contemplated exotic travel, the first things that came to mind of course were what rare diseases I might catch, which vaccines could prevent them, and how to get insurance to pay for getting immunized. Alexa helped me find CDC recommendations for immunizations for travel to Morocco, which included:
• Typhoid ... contaminated food or water.
• Hepatitis A ... contaminated food or water.
• Hepatitis B ... contaminated body fluids (sex, needles, etc.).
• Cholera ... contaminated food or water.
• Rabies ... infected animals.
• Influenza ... airborne droplets.
This trip was indeed starting to sound like an awful lot of fun.
My PCP called in several of the relevant vaccines to my local pharmacy, who informed me that typhoid vaccine is not covered by my health insurance. This spurred the following (somewhat embellished) dialogue with my insurer:
“Why is typhoid not covered?”
“Contractual exclusion. We don’t cover anything starting with “typ-,” including typhoid, typhus, typical, and typographic.”
“Do you cover bubonic plague?”
“Only for high-risk travel.”
“Such as?”
“Such as if you travel to Europe during the 14th century.”
“How about Hepatitis B and rabies?”
“That would depend.”
“On what?”
“On whether you plan to have sex with rabid bats, or rabid sex with placid bats.”
“I wouldn’t say I have plans. But, you know, in the moment ...”
“Sorry, not covered.”
“How about cholera?”
“Have you ever been threatened by cholera?
“Not exactly. But I did have a cranky uncle. When he was irritated, he often said, ‘May cholera grab you!’ ”
“You’re not covered. Your uncle might be.”
“We’ve decided on a side trip to Tanzania. As long as we’re already in Africa ...”
“Do you suffer from Sleeping Sickness?”
“Only at Grand Rounds.”
“We do cover eflornithine, but there is a problem ...”
“What problem?”
“Our only eflornithine manufacturing facility is in Bangladesh, where it takes up two floors of a factory that also makes designer jeans. That factory is closed for safety and child-labor violations.”
“For how long?”
“Indefinitely”
“Then what can I do?”
“You can apply eflornithine cream for your Sleeping Sickness and hope for the best.”
“Eflornithine cream?”
“Vaniqa. It may not help your sleeping symptoms, but you’ll need fewer haircuts.”
“Oh, thanks. What about River Blindness? Do you cover ivermectin?”
“Only if the preferred formulary alternatives have been exhausted.”
“What are those?”
“Metronidazole and azelaic acid.”
“Hold on! Are you looking at the page for onchocerciasis or the one for rosacea?”

“Yes. Did Montezuma ever make it to Morocco?”
“I don’t have that information. You’ll have to ask Alexa. Anything else?”
“No, I’m all set. Just remind me what you said about bats?”
In the end a family situation came up, and we had to cancel our trip. Instead, we watched the movie “Casablanca.” That is an excellent movie, with many pungent and memorable lines. Not only that but watching it does not cause jet lag.
As for the typhoid vaccine, in the end, it was not covered by insurance. Nevertheless, I haven’t had a bit of typhoid, so the vaccine seems to be working very well.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
My wife and I decided to visit Morocco, to test the maxim that my fellow columnist Joe Eastern often cites: The words you won’t say on your deathbed are, “If only I had spent more time at the office.”
Though I’m not convinced he’s right about that – he’s never even seen my office – I thought I’d give being away a try. My office manager comes from near Marrakesh. While bound for Morocco, we could check out her hometown, even if there is no obvious tax angle.
As I contemplated exotic travel, the first things that came to mind of course were what rare diseases I might catch, which vaccines could prevent them, and how to get insurance to pay for getting immunized. Alexa helped me find CDC recommendations for immunizations for travel to Morocco, which included:
• Typhoid ... contaminated food or water.
• Hepatitis A ... contaminated food or water.
• Hepatitis B ... contaminated body fluids (sex, needles, etc.).
• Cholera ... contaminated food or water.
• Rabies ... infected animals.
• Influenza ... airborne droplets.
This trip was indeed starting to sound like an awful lot of fun.
My PCP called in several of the relevant vaccines to my local pharmacy, who informed me that typhoid vaccine is not covered by my health insurance. This spurred the following (somewhat embellished) dialogue with my insurer:
“Why is typhoid not covered?”
“Contractual exclusion. We don’t cover anything starting with “typ-,” including typhoid, typhus, typical, and typographic.”
“Do you cover bubonic plague?”
“Only for high-risk travel.”
“Such as?”
“Such as if you travel to Europe during the 14th century.”
“How about Hepatitis B and rabies?”
“That would depend.”
“On what?”
“On whether you plan to have sex with rabid bats, or rabid sex with placid bats.”
“I wouldn’t say I have plans. But, you know, in the moment ...”
“Sorry, not covered.”
“How about cholera?”
“Have you ever been threatened by cholera?
“Not exactly. But I did have a cranky uncle. When he was irritated, he often said, ‘May cholera grab you!’ ”
“You’re not covered. Your uncle might be.”
“We’ve decided on a side trip to Tanzania. As long as we’re already in Africa ...”
“Do you suffer from Sleeping Sickness?”
“Only at Grand Rounds.”
“We do cover eflornithine, but there is a problem ...”
“What problem?”
“Our only eflornithine manufacturing facility is in Bangladesh, where it takes up two floors of a factory that also makes designer jeans. That factory is closed for safety and child-labor violations.”
“For how long?”
“Indefinitely”
“Then what can I do?”
“You can apply eflornithine cream for your Sleeping Sickness and hope for the best.”
“Eflornithine cream?”
“Vaniqa. It may not help your sleeping symptoms, but you’ll need fewer haircuts.”
“Oh, thanks. What about River Blindness? Do you cover ivermectin?”
“Only if the preferred formulary alternatives have been exhausted.”
“What are those?”
“Metronidazole and azelaic acid.”
“Hold on! Are you looking at the page for onchocerciasis or the one for rosacea?”
“Yes. Did Montezuma ever make it to Morocco?”
“I don’t have that information. You’ll have to ask Alexa. Anything else?”
“No, I’m all set. Just remind me what you said about bats?”
In the end a family situation came up, and we had to cancel our trip. Instead, we watched the movie “Casablanca.” That is an excellent movie, with many pungent and memorable lines. Not only that but watching it does not cause jet lag.
As for the typhoid vaccine, in the end, it was not covered by insurance. Nevertheless, I haven’t had a bit of typhoid, so the vaccine seems to be working very well.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
My wife and I decided to visit Morocco, to test the maxim that my fellow columnist Joe Eastern often cites: The words you won’t say on your deathbed are, “If only I had spent more time at the office.”
Though I’m not convinced he’s right about that – he’s never even seen my office – I thought I’d give being away a try. My office manager comes from near Marrakesh. While bound for Morocco, we could check out her hometown, even if there is no obvious tax angle.
As I contemplated exotic travel, the first things that came to mind of course were what rare diseases I might catch, which vaccines could prevent them, and how to get insurance to pay for getting immunized. Alexa helped me find CDC recommendations for immunizations for travel to Morocco, which included:
• Typhoid ... contaminated food or water.
• Hepatitis A ... contaminated food or water.
• Hepatitis B ... contaminated body fluids (sex, needles, etc.).
• Cholera ... contaminated food or water.
• Rabies ... infected animals.
• Influenza ... airborne droplets.
This trip was indeed starting to sound like an awful lot of fun.
My PCP called in several of the relevant vaccines to my local pharmacy, who informed me that typhoid vaccine is not covered by my health insurance. This spurred the following (somewhat embellished) dialogue with my insurer:
“Why is typhoid not covered?”
“Contractual exclusion. We don’t cover anything starting with “typ-,” including typhoid, typhus, typical, and typographic.”
“Do you cover bubonic plague?”
“Only for high-risk travel.”
“Such as?”
“Such as if you travel to Europe during the 14th century.”
“How about Hepatitis B and rabies?”
“That would depend.”
“On what?”
“On whether you plan to have sex with rabid bats, or rabid sex with placid bats.”
“I wouldn’t say I have plans. But, you know, in the moment ...”
“Sorry, not covered.”
“How about cholera?”
“Have you ever been threatened by cholera?
“Not exactly. But I did have a cranky uncle. When he was irritated, he often said, ‘May cholera grab you!’ ”
“You’re not covered. Your uncle might be.”
“We’ve decided on a side trip to Tanzania. As long as we’re already in Africa ...”
“Do you suffer from Sleeping Sickness?”
“Only at Grand Rounds.”
“We do cover eflornithine, but there is a problem ...”
“What problem?”
“Our only eflornithine manufacturing facility is in Bangladesh, where it takes up two floors of a factory that also makes designer jeans. That factory is closed for safety and child-labor violations.”
“For how long?”
“Indefinitely”
“Then what can I do?”
“You can apply eflornithine cream for your Sleeping Sickness and hope for the best.”
“Eflornithine cream?”
“Vaniqa. It may not help your sleeping symptoms, but you’ll need fewer haircuts.”
“Oh, thanks. What about River Blindness? Do you cover ivermectin?”
“Only if the preferred formulary alternatives have been exhausted.”
“What are those?”
“Metronidazole and azelaic acid.”
“Hold on! Are you looking at the page for onchocerciasis or the one for rosacea?”
“Yes. Did Montezuma ever make it to Morocco?”
“I don’t have that information. You’ll have to ask Alexa. Anything else?”
“No, I’m all set. Just remind me what you said about bats?”
In the end a family situation came up, and we had to cancel our trip. Instead, we watched the movie “Casablanca.” That is an excellent movie, with many pungent and memorable lines. Not only that but watching it does not cause jet lag.
As for the typhoid vaccine, in the end, it was not covered by insurance. Nevertheless, I haven’t had a bit of typhoid, so the vaccine seems to be working very well.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at [email protected].
Tau imaging predicts looming cognitive decline in cognitively normal elderly
BOSTON – Progressive tau accumulation in the temporal lobe of cognitively normal older adults was associated with cognitive decline over time in a prospective, longitudinal study presented at the Clinical Trials on Alzheimer’s Disease conference.
This track of cognitive impairment following tau pathology in a preclinical Alzheimer’s disease (AD) population suggests two roles for serial positron emission tomography (PET) scans with a tau binding agent, Bernard Hanseeuw, MD, PhD, said at the meeting. In the near future, they could be used to track therapeutic response in clinical trials. Farther out, if future validation studies confirm these preliminary results, they might be a useful clinical tool for predicting how fast an individual Alzheimer’s patient will progress, he said in an interview.

Serial tau scans, however, would, he said.
“Every patient with Alzheimer’s disease is different, with a different disease course. Amyloid scans can tell us if someone is on the wrong path, but tau scans could tell us how fast they are going. If you have Alzheimer’s, it’s important to know if you may not be able to live in your own home in a year. With tau PET, we could track the disease and predict how fast it might evolve. That is very clinically relevant,” said Dr. Hanseeuw.
Tau imaging remains investigational only. Several tau imaging agents are being developed, but none has yet been approved in the United States or in Europe.
To investigate the correlation of tau and cognitive decline in preclinical Alzheimer’s, Dr. Hanseeuw examined serial tau and amyloid PET scans conducted on 60 clinically normal older adults with a mean age of 75 years. About one-third of the cohort was positive for the APOE4 allele. All of them had a baseline Clinical Dementia Rating (CDR) score of 0 and a mean Mini-Mental State Exam score of at least 27. They also scored in the normal range on the Preclinical Alzheimer’s Cognitive Composite (PACC) test. This relatively new cognitive scale is an increasingly popular item in clinical trials. The PACC is a composite of the WAIS-R Digit Symbol Substitution Test, Mini-Mental State Exam, Free and Cued Selective Reminding Test, and Logical Memory IIA Delayed Recall, and correlates well with amyloid accumulation in the brain.
The study included up to 4 years of data on cognition and amyloid PET imaging, and up to 3 years of tau PET imaging data. The investigators assessed amyloid as a whole-brain aggregate and tau in the bilateral inferior temporal neocortex. “This is where the change is most happening in patients, and it’s a place where relatively few normal elderly would have tau,” Dr. Hanseeuw said. All of the analyses controlled for age, sex, and years of education.
Baseline amyloid levels were low in 36 participants and high in 24. At least some tau was present in all of the subjects. This is not an unexpected finding, since tau accumulates with age, Dr. Hanseeuw said. Over the study period, six subjects progressed to a CDR of 0.5 – a rating consistent with mild cognitive impairment. At baseline, high tau and high amyloid levels were both associated with a progressive decline in PACC scores in the following years. However, the rate of change in tau predicted change in cognition better than did the baseline measurements. In contrast, the rate of change in amyloid was not associated with cognitive decline.
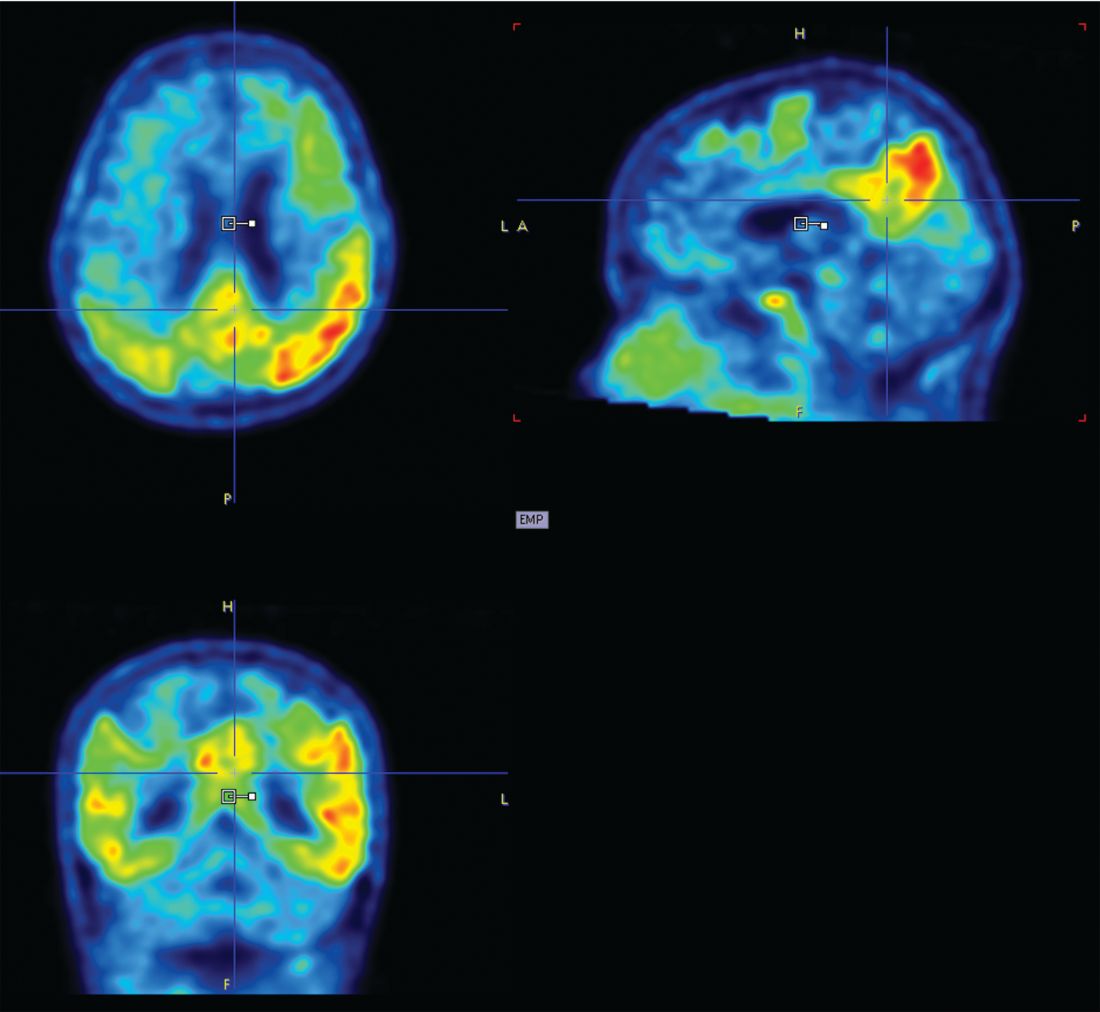
“What is interesting here is that tau changed four times faster than amyloid,” Dr. Hanseeuw said. “The average subject needed 5 years to change 1 standard deviation in tau, but would have needed 20 years to change 1 standard deviation in amyloid.”
Fast-changing outcomes are important to accelerate drug assessment in clinical trials. Currently, it takes 3-5 years to conduct most anti-AD trials, he added.
Dr. Hanseeuw had no relevant financial disclosures.
BOSTON – Progressive tau accumulation in the temporal lobe of cognitively normal older adults was associated with cognitive decline over time in a prospective, longitudinal study presented at the Clinical Trials on Alzheimer’s Disease conference.
This track of cognitive impairment following tau pathology in a preclinical Alzheimer’s disease (AD) population suggests two roles for serial positron emission tomography (PET) scans with a tau binding agent, Bernard Hanseeuw, MD, PhD, said at the meeting. In the near future, they could be used to track therapeutic response in clinical trials. Farther out, if future validation studies confirm these preliminary results, they might be a useful clinical tool for predicting how fast an individual Alzheimer’s patient will progress, he said in an interview.
Serial tau scans, however, would, he said.
“Every patient with Alzheimer’s disease is different, with a different disease course. Amyloid scans can tell us if someone is on the wrong path, but tau scans could tell us how fast they are going. If you have Alzheimer’s, it’s important to know if you may not be able to live in your own home in a year. With tau PET, we could track the disease and predict how fast it might evolve. That is very clinically relevant,” said Dr. Hanseeuw.
Tau imaging remains investigational only. Several tau imaging agents are being developed, but none has yet been approved in the United States or in Europe.
To investigate the correlation of tau and cognitive decline in preclinical Alzheimer’s, Dr. Hanseeuw examined serial tau and amyloid PET scans conducted on 60 clinically normal older adults with a mean age of 75 years. About one-third of the cohort was positive for the APOE4 allele. All of them had a baseline Clinical Dementia Rating (CDR) score of 0 and a mean Mini-Mental State Exam score of at least 27. They also scored in the normal range on the Preclinical Alzheimer’s Cognitive Composite (PACC) test. This relatively new cognitive scale is an increasingly popular item in clinical trials. The PACC is a composite of the WAIS-R Digit Symbol Substitution Test, Mini-Mental State Exam, Free and Cued Selective Reminding Test, and Logical Memory IIA Delayed Recall, and correlates well with amyloid accumulation in the brain.
The study included up to 4 years of data on cognition and amyloid PET imaging, and up to 3 years of tau PET imaging data. The investigators assessed amyloid as a whole-brain aggregate and tau in the bilateral inferior temporal neocortex. “This is where the change is most happening in patients, and it’s a place where relatively few normal elderly would have tau,” Dr. Hanseeuw said. All of the analyses controlled for age, sex, and years of education.
Baseline amyloid levels were low in 36 participants and high in 24. At least some tau was present in all of the subjects. This is not an unexpected finding, since tau accumulates with age, Dr. Hanseeuw said. Over the study period, six subjects progressed to a CDR of 0.5 – a rating consistent with mild cognitive impairment. At baseline, high tau and high amyloid levels were both associated with a progressive decline in PACC scores in the following years. However, the rate of change in tau predicted change in cognition better than did the baseline measurements. In contrast, the rate of change in amyloid was not associated with cognitive decline.
“What is interesting here is that tau changed four times faster than amyloid,” Dr. Hanseeuw said. “The average subject needed 5 years to change 1 standard deviation in tau, but would have needed 20 years to change 1 standard deviation in amyloid.”
Fast-changing outcomes are important to accelerate drug assessment in clinical trials. Currently, it takes 3-5 years to conduct most anti-AD trials, he added.
Dr. Hanseeuw had no relevant financial disclosures.
BOSTON – Progressive tau accumulation in the temporal lobe of cognitively normal older adults was associated with cognitive decline over time in a prospective, longitudinal study presented at the Clinical Trials on Alzheimer’s Disease conference.
This track of cognitive impairment following tau pathology in a preclinical Alzheimer’s disease (AD) population suggests two roles for serial positron emission tomography (PET) scans with a tau binding agent, Bernard Hanseeuw, MD, PhD, said at the meeting. In the near future, they could be used to track therapeutic response in clinical trials. Farther out, if future validation studies confirm these preliminary results, they might be a useful clinical tool for predicting how fast an individual Alzheimer’s patient will progress, he said in an interview.
Serial tau scans, however, would, he said.
“Every patient with Alzheimer’s disease is different, with a different disease course. Amyloid scans can tell us if someone is on the wrong path, but tau scans could tell us how fast they are going. If you have Alzheimer’s, it’s important to know if you may not be able to live in your own home in a year. With tau PET, we could track the disease and predict how fast it might evolve. That is very clinically relevant,” said Dr. Hanseeuw.
Tau imaging remains investigational only. Several tau imaging agents are being developed, but none has yet been approved in the United States or in Europe.
To investigate the correlation of tau and cognitive decline in preclinical Alzheimer’s, Dr. Hanseeuw examined serial tau and amyloid PET scans conducted on 60 clinically normal older adults with a mean age of 75 years. About one-third of the cohort was positive for the APOE4 allele. All of them had a baseline Clinical Dementia Rating (CDR) score of 0 and a mean Mini-Mental State Exam score of at least 27. They also scored in the normal range on the Preclinical Alzheimer’s Cognitive Composite (PACC) test. This relatively new cognitive scale is an increasingly popular item in clinical trials. The PACC is a composite of the WAIS-R Digit Symbol Substitution Test, Mini-Mental State Exam, Free and Cued Selective Reminding Test, and Logical Memory IIA Delayed Recall, and correlates well with amyloid accumulation in the brain.
The study included up to 4 years of data on cognition and amyloid PET imaging, and up to 3 years of tau PET imaging data. The investigators assessed amyloid as a whole-brain aggregate and tau in the bilateral inferior temporal neocortex. “This is where the change is most happening in patients, and it’s a place where relatively few normal elderly would have tau,” Dr. Hanseeuw said. All of the analyses controlled for age, sex, and years of education.
Baseline amyloid levels were low in 36 participants and high in 24. At least some tau was present in all of the subjects. This is not an unexpected finding, since tau accumulates with age, Dr. Hanseeuw said. Over the study period, six subjects progressed to a CDR of 0.5 – a rating consistent with mild cognitive impairment. At baseline, high tau and high amyloid levels were both associated with a progressive decline in PACC scores in the following years. However, the rate of change in tau predicted change in cognition better than did the baseline measurements. In contrast, the rate of change in amyloid was not associated with cognitive decline.
“What is interesting here is that tau changed four times faster than amyloid,” Dr. Hanseeuw said. “The average subject needed 5 years to change 1 standard deviation in tau, but would have needed 20 years to change 1 standard deviation in amyloid.”
Fast-changing outcomes are important to accelerate drug assessment in clinical trials. Currently, it takes 3-5 years to conduct most anti-AD trials, he added.
Dr. Hanseeuw had no relevant financial disclosures.
REPORTING FROM CTAD
Key clinical point:
Major finding: Tau levels changed twice as fast as cognition, suggesting that the protein is a significant marker of future cognitive change.
Data source: A prospective, longitudinal study of 60 cognitively normal subjects.
Disclosures: Dr. Hanseeuw had no relevant financial disclosures.
Source: Hanseeuw B et al. CTAD 2017 Abstract OC2.
Think before you Tweet: Social media guidelines for surgeons aim to prevent Internet regret
Think before you tweet. That’s what surgeons should remember before they express themselves on social media.
Anger and frustration can prompt ill-advised social media postings that have a big potential for blowback, Heather J. Logghe, MD, FACS, and her colleagues wrote in the Journal of the American College of Surgeons. But so can enthusiasm about posting about a new device or procedure, a fascination with a difficult case, the sense of relief that a patient made it though a harrowing period, or even just the simple joy of tossing back a beer or two with pals at the local watering hole (J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022).

“In a survey of 48 state medical boards, 44 (92%) reported online-related misbehavior with serious disciplinary consequences leading to license restriction, suspension, or revocation. A 2011 study of ‘Physicians on Twitter’ revealed that 10% of the physicians sampled had tweeted potential patient privacy violations. A 2014 study of publicly available Facebook profiles of 319 Midwest residents found 14% had ‘potentially unprofessional content’ and 12.2% had ‘clearly unprofessional’ content, the latter including references to binge drinking, sexually suggestive photos, and HIPAA violations.”
Dr. Logghe, of Thomas Jefferson University, Philadelphia, is a member of the American College of Surgeons’ (ACS’s) social media committee tasked with creating practice recommendations for clinicians’ use of social media. Conducting a literature review was the first step to creating a surgeon-specific document, and the team found seven online behavior guidelines directed at physicians. Groups authoring these papers included the American Medical Association, the Federation of State Medical Boards, the American Congress of Obstetricians and Gynecologists, and several international groups.
Dr. Logghe and her colleagues reviewed each one, synthesized the information, and created a practice recommendation statement specific to the ACS. While not encoded in any professional ethics requirements, “Best Practices for Surgeons’ Social Media Use: Statement of the Resident and Associate Society of the American College of Surgeons” does lay out some common, potentially problematic scenarios and offers some suggestions about how to avoid Internet regret.
Everything discussed in the paper revolves around maintaining a decorous public persona. Professionalism on and off the clock is a key tenet of the recommendations. Definitions of key terms like “professionalism” are an important basis for any practice guideline, but sometimes concepts are not easy to define, the team wrote. “Perhaps the limitation most difficult to address in any formalized guideline is the necessary subjectivity in interpreting what is ‘appropriate’ or ‘professional’ online – or in any other setting,” the authors wrote. The ACS Code of Professional Conduct does not explicitly define either of those terms or discuss the appearance of unprofessional behavior.
In the absence of a plain-and-simple definition, the authors attempted to couch the social media recommendations in terms of ACS’s commitment to maintaining the patient trust. It urges surgeons to “avoid even the appearance of impropriety.”
The practice recommendations touch on a number of areas that are potentially problematic for surgeons, including confidentiality, financial conflicts, collegial support, and general social responsibility.
Confidentiality
Maintaining privacy is more than a courtesy to patients: It’s a federally mandated law with serious punitive repercussions if violated. Blogs, YouTube, Twitter, and Facebook offer a vast potential for sharing information with and educating the public, but postings can also easily violate HIPPA standards, the team wrote.
“In general, most social media platforms are not HIPPA-compliant,” no matter how the privacy settings are adjusted. These modes of communication are never appropriate for patient-physician communication: They can’t be archived in an electronic health record, and it is ill advised to give any medical advice by using these channels.
Discussing a particular case online, even with the usual defining details omitted, can be a bad idea.“Simply de-identifying patient information may not be sufficient. When posting information online, one must be cognizant of the context of other information available online. Such information includes the poster’s place of employment, news media, and publicly available vital statistics. Therefore even when posting general comments about hospital events, surgical cases, or patients under one’s care, it is essential to consider the sum of information available to the reader, rather than simply the information shared in the isolated post.”
Employment
Most employers have social media guidelines and don’t take kindly to violations – which can affect both current and future job postings. “A strong social media presence can be of benefit to one’s employer, [but] content that portrays a surgeon in an unprofessional or controversial light can be detrimental and even career-damaging.”
This reaches beyond professional communications online and deep into a surgeon’s personal life, the team noted, so exercise caution when “friending.”
“While this practice is inevitable, surgeons should be aware of potential conflicts. Connecting with or accepting friend requests from some but not all coworkers or coresidents could be interpreted as favoritism and may create a problematic work relationship. … Surgeons should consider primarily connecting with coworkers on professional websites if they have little contact with them outside the workplace.”
As for friending patients – just don’t, for both your sake and theirs. “Accepting a patient’s Facebook friend request may allow them access to events, details, and commentary not traditionally appropriate for the patient-physician relationship. Accepting such requests is strongly discouraged. If concerned about appearing rude or rejecting a patient’s request to be Facebook friends, the patient can be referred to society guidelines or best practices such as these.” One helpful alternative to such a request may be to invite patients to follow a practice website or other professional page.
Conflicts of interest
Online friends might not require disclosures when a surgeon posts about an exciting procedure or piece of equipment, such as whether there is a financial interest in doing so, but it’s important to be proactive. “As always, it is the physician’s responsibility to avoid even the appearance of impropriety. If it is not feasible to include a relevant conflict of interest within a post, the post should not be made.”
Defamation
Irritated about a colleague? Keep it to yourself – especially if you’ve had a beer. “It is never appropriate to post derogatory comments about patients or colleagues. Surgeons should be careful not to post in anger or under the influence of any substance. Statements about a colleague’s abilities, experience, or outcomes intended in jest may be appropriate for the surgeon’s lounge, yet entirely inappropriate for public consumption. Again, the ‘pause-before-posting’ practice is likely to prevent regretful posts in this vein.”
Privacy and Permanence
The Internet goes everywhere and lasts forever. A snappy quote that’s funny at 2 a.m. might not seem so hilarious in the light of day – or even in the light of a day 5 years yet to come.
The delete key is a false friend, and that clever pseudonym you dreamed up is probably as crackable as the classic “Pa55word” password. “One should presume that all content posted online will remain there forever and may be seen by anyone. Again, ‘pause-before-posting’ is a recommended practice.”
Privacy settings should be viewed as an illusion, the team noted. In this era of face recognition and tagging, images carry just as much risk as words.
Collegial support
Maybe your mother was right when she said, “This is for your own good.” If a colleague’s postings are getting out of hand, a tactful heart-to-heart might be the best course of action. “As coined by Dr. Sarah Mansfield, ‘Looking after colleagues is an integral element of professional conduct.’ Surgeons who notice colleagues posting unprofessional content that could be damaging to both the colleague and the public’s trust in the profession should discreetly express their concern to the individual, who should then take any appropriate corrective actions. … If the action is in violation of the law or medical board regulations, it should be reported to the appropriate governing bodies.”
Physician, Google Thyself
The team acknowledged that an online presence is virtually a must for professional development. And even if you don’t create a web page, chances are your university or hospital has done it for you. The media is interested in your life, too, and may make mention of your activities – both positive or negative.
“To better understand and control this publicly accessible information, surgeons are encouraged to periodically self-audit themselves online and taking measures to ensure that the information present is accurate and professional.” Some professional service websites are more trustworthy than others. The team encouraged physicians to participate in the ACS professional pages, LinkedIn, Doximity, and ResearchGate.
Not rules – just recommendations
The team stressed that their recommendations aren’t meant to stifle personal expression. Instead, their aim is to prompt a more conscious use of what can be a very powerful tool for both self-expression and professional development.
“The authors recommend no punitive action based on a perceived ‘violation’ of these recommendations alone. While they refer to other guidelines, including laws such as HIPAA, that must be appropriately enforced, these best practices are intended to guide the practicing surgeon in the use of social media rather than act as regulations or encourage reprimand. Rather than encouraging a social media landscape as sterile as the operating theater, the authors hope these recommendations lead to conscious consideration of online behavior, to avoidance of preventable harm, and to recognition of others’ views of their posts.”
None of the authors reported any financial disclosures.
SOURCE: Logghe HJ et al. J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022.
As Editor of the ACS Communities, I am thrilled to see the RAS paper of social media recommendations. We who did not grow up with a keyboard in our hands can learn valuable and career-saving lessons from our younger colleagues who have had a lifetime of experience with social media.
There’s nothing like social media to get your thoughts “out there,” but the other side of the sword is excellently described in this article. I have seen or had to intervene on each of the subjects mentioned in it while reading through the thousands of posts that the ACS Communities’ users have generated over the last three-and-a-half years. When sitting in front of a screen, we can easily lose sight of the fact that our comments are going out into the real world and how rapidly they might reflect back on us and affect friends, relatives, employers, patients, foreign governments, cultures vastly different from our own, and other breathing, feeling human beings – in short, the entire universe hears regardless of whether the site is “password protected.”
I urge everyone using social media to read these guidelines, laminate them, and put them in their wallets, purses, or somewhere else that’s handy. Being self-aware and insightful in your posts can do a world of good, but a lack thereof can result in an avalanche of harm to yourself or others.
Tyler G. Hughes, MD, FACS, is a clinical professor in the department of surgery and the director of medical education at the Kansas University in Salina, Kan., as well as a Co-Editor of ACS Surgery News.
As Editor of the ACS Communities, I am thrilled to see the RAS paper of social media recommendations. We who did not grow up with a keyboard in our hands can learn valuable and career-saving lessons from our younger colleagues who have had a lifetime of experience with social media.
There’s nothing like social media to get your thoughts “out there,” but the other side of the sword is excellently described in this article. I have seen or had to intervene on each of the subjects mentioned in it while reading through the thousands of posts that the ACS Communities’ users have generated over the last three-and-a-half years. When sitting in front of a screen, we can easily lose sight of the fact that our comments are going out into the real world and how rapidly they might reflect back on us and affect friends, relatives, employers, patients, foreign governments, cultures vastly different from our own, and other breathing, feeling human beings – in short, the entire universe hears regardless of whether the site is “password protected.”
I urge everyone using social media to read these guidelines, laminate them, and put them in their wallets, purses, or somewhere else that’s handy. Being self-aware and insightful in your posts can do a world of good, but a lack thereof can result in an avalanche of harm to yourself or others.
Tyler G. Hughes, MD, FACS, is a clinical professor in the department of surgery and the director of medical education at the Kansas University in Salina, Kan., as well as a Co-Editor of ACS Surgery News.
As Editor of the ACS Communities, I am thrilled to see the RAS paper of social media recommendations. We who did not grow up with a keyboard in our hands can learn valuable and career-saving lessons from our younger colleagues who have had a lifetime of experience with social media.
There’s nothing like social media to get your thoughts “out there,” but the other side of the sword is excellently described in this article. I have seen or had to intervene on each of the subjects mentioned in it while reading through the thousands of posts that the ACS Communities’ users have generated over the last three-and-a-half years. When sitting in front of a screen, we can easily lose sight of the fact that our comments are going out into the real world and how rapidly they might reflect back on us and affect friends, relatives, employers, patients, foreign governments, cultures vastly different from our own, and other breathing, feeling human beings – in short, the entire universe hears regardless of whether the site is “password protected.”
I urge everyone using social media to read these guidelines, laminate them, and put them in their wallets, purses, or somewhere else that’s handy. Being self-aware and insightful in your posts can do a world of good, but a lack thereof can result in an avalanche of harm to yourself or others.
Tyler G. Hughes, MD, FACS, is a clinical professor in the department of surgery and the director of medical education at the Kansas University in Salina, Kan., as well as a Co-Editor of ACS Surgery News.
Think before you tweet. That’s what surgeons should remember before they express themselves on social media.
Anger and frustration can prompt ill-advised social media postings that have a big potential for blowback, Heather J. Logghe, MD, FACS, and her colleagues wrote in the Journal of the American College of Surgeons. But so can enthusiasm about posting about a new device or procedure, a fascination with a difficult case, the sense of relief that a patient made it though a harrowing period, or even just the simple joy of tossing back a beer or two with pals at the local watering hole (J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022).
“In a survey of 48 state medical boards, 44 (92%) reported online-related misbehavior with serious disciplinary consequences leading to license restriction, suspension, or revocation. A 2011 study of ‘Physicians on Twitter’ revealed that 10% of the physicians sampled had tweeted potential patient privacy violations. A 2014 study of publicly available Facebook profiles of 319 Midwest residents found 14% had ‘potentially unprofessional content’ and 12.2% had ‘clearly unprofessional’ content, the latter including references to binge drinking, sexually suggestive photos, and HIPAA violations.”
Dr. Logghe, of Thomas Jefferson University, Philadelphia, is a member of the American College of Surgeons’ (ACS’s) social media committee tasked with creating practice recommendations for clinicians’ use of social media. Conducting a literature review was the first step to creating a surgeon-specific document, and the team found seven online behavior guidelines directed at physicians. Groups authoring these papers included the American Medical Association, the Federation of State Medical Boards, the American Congress of Obstetricians and Gynecologists, and several international groups.
Dr. Logghe and her colleagues reviewed each one, synthesized the information, and created a practice recommendation statement specific to the ACS. While not encoded in any professional ethics requirements, “Best Practices for Surgeons’ Social Media Use: Statement of the Resident and Associate Society of the American College of Surgeons” does lay out some common, potentially problematic scenarios and offers some suggestions about how to avoid Internet regret.
Everything discussed in the paper revolves around maintaining a decorous public persona. Professionalism on and off the clock is a key tenet of the recommendations. Definitions of key terms like “professionalism” are an important basis for any practice guideline, but sometimes concepts are not easy to define, the team wrote. “Perhaps the limitation most difficult to address in any formalized guideline is the necessary subjectivity in interpreting what is ‘appropriate’ or ‘professional’ online – or in any other setting,” the authors wrote. The ACS Code of Professional Conduct does not explicitly define either of those terms or discuss the appearance of unprofessional behavior.
In the absence of a plain-and-simple definition, the authors attempted to couch the social media recommendations in terms of ACS’s commitment to maintaining the patient trust. It urges surgeons to “avoid even the appearance of impropriety.”
The practice recommendations touch on a number of areas that are potentially problematic for surgeons, including confidentiality, financial conflicts, collegial support, and general social responsibility.
Confidentiality
Maintaining privacy is more than a courtesy to patients: It’s a federally mandated law with serious punitive repercussions if violated. Blogs, YouTube, Twitter, and Facebook offer a vast potential for sharing information with and educating the public, but postings can also easily violate HIPPA standards, the team wrote.
“In general, most social media platforms are not HIPPA-compliant,” no matter how the privacy settings are adjusted. These modes of communication are never appropriate for patient-physician communication: They can’t be archived in an electronic health record, and it is ill advised to give any medical advice by using these channels.
Discussing a particular case online, even with the usual defining details omitted, can be a bad idea.“Simply de-identifying patient information may not be sufficient. When posting information online, one must be cognizant of the context of other information available online. Such information includes the poster’s place of employment, news media, and publicly available vital statistics. Therefore even when posting general comments about hospital events, surgical cases, or patients under one’s care, it is essential to consider the sum of information available to the reader, rather than simply the information shared in the isolated post.”
Employment
Most employers have social media guidelines and don’t take kindly to violations – which can affect both current and future job postings. “A strong social media presence can be of benefit to one’s employer, [but] content that portrays a surgeon in an unprofessional or controversial light can be detrimental and even career-damaging.”
This reaches beyond professional communications online and deep into a surgeon’s personal life, the team noted, so exercise caution when “friending.”
“While this practice is inevitable, surgeons should be aware of potential conflicts. Connecting with or accepting friend requests from some but not all coworkers or coresidents could be interpreted as favoritism and may create a problematic work relationship. … Surgeons should consider primarily connecting with coworkers on professional websites if they have little contact with them outside the workplace.”
As for friending patients – just don’t, for both your sake and theirs. “Accepting a patient’s Facebook friend request may allow them access to events, details, and commentary not traditionally appropriate for the patient-physician relationship. Accepting such requests is strongly discouraged. If concerned about appearing rude or rejecting a patient’s request to be Facebook friends, the patient can be referred to society guidelines or best practices such as these.” One helpful alternative to such a request may be to invite patients to follow a practice website or other professional page.
Conflicts of interest
Online friends might not require disclosures when a surgeon posts about an exciting procedure or piece of equipment, such as whether there is a financial interest in doing so, but it’s important to be proactive. “As always, it is the physician’s responsibility to avoid even the appearance of impropriety. If it is not feasible to include a relevant conflict of interest within a post, the post should not be made.”
Defamation
Irritated about a colleague? Keep it to yourself – especially if you’ve had a beer. “It is never appropriate to post derogatory comments about patients or colleagues. Surgeons should be careful not to post in anger or under the influence of any substance. Statements about a colleague’s abilities, experience, or outcomes intended in jest may be appropriate for the surgeon’s lounge, yet entirely inappropriate for public consumption. Again, the ‘pause-before-posting’ practice is likely to prevent regretful posts in this vein.”
Privacy and Permanence
The Internet goes everywhere and lasts forever. A snappy quote that’s funny at 2 a.m. might not seem so hilarious in the light of day – or even in the light of a day 5 years yet to come.
The delete key is a false friend, and that clever pseudonym you dreamed up is probably as crackable as the classic “Pa55word” password. “One should presume that all content posted online will remain there forever and may be seen by anyone. Again, ‘pause-before-posting’ is a recommended practice.”
Privacy settings should be viewed as an illusion, the team noted. In this era of face recognition and tagging, images carry just as much risk as words.
Collegial support
Maybe your mother was right when she said, “This is for your own good.” If a colleague’s postings are getting out of hand, a tactful heart-to-heart might be the best course of action. “As coined by Dr. Sarah Mansfield, ‘Looking after colleagues is an integral element of professional conduct.’ Surgeons who notice colleagues posting unprofessional content that could be damaging to both the colleague and the public’s trust in the profession should discreetly express their concern to the individual, who should then take any appropriate corrective actions. … If the action is in violation of the law or medical board regulations, it should be reported to the appropriate governing bodies.”
Physician, Google Thyself
The team acknowledged that an online presence is virtually a must for professional development. And even if you don’t create a web page, chances are your university or hospital has done it for you. The media is interested in your life, too, and may make mention of your activities – both positive or negative.
“To better understand and control this publicly accessible information, surgeons are encouraged to periodically self-audit themselves online and taking measures to ensure that the information present is accurate and professional.” Some professional service websites are more trustworthy than others. The team encouraged physicians to participate in the ACS professional pages, LinkedIn, Doximity, and ResearchGate.
Not rules – just recommendations
The team stressed that their recommendations aren’t meant to stifle personal expression. Instead, their aim is to prompt a more conscious use of what can be a very powerful tool for both self-expression and professional development.
“The authors recommend no punitive action based on a perceived ‘violation’ of these recommendations alone. While they refer to other guidelines, including laws such as HIPAA, that must be appropriately enforced, these best practices are intended to guide the practicing surgeon in the use of social media rather than act as regulations or encourage reprimand. Rather than encouraging a social media landscape as sterile as the operating theater, the authors hope these recommendations lead to conscious consideration of online behavior, to avoidance of preventable harm, and to recognition of others’ views of their posts.”
None of the authors reported any financial disclosures.
SOURCE: Logghe HJ et al. J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022.
Think before you tweet. That’s what surgeons should remember before they express themselves on social media.
Anger and frustration can prompt ill-advised social media postings that have a big potential for blowback, Heather J. Logghe, MD, FACS, and her colleagues wrote in the Journal of the American College of Surgeons. But so can enthusiasm about posting about a new device or procedure, a fascination with a difficult case, the sense of relief that a patient made it though a harrowing period, or even just the simple joy of tossing back a beer or two with pals at the local watering hole (J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022).
“In a survey of 48 state medical boards, 44 (92%) reported online-related misbehavior with serious disciplinary consequences leading to license restriction, suspension, or revocation. A 2011 study of ‘Physicians on Twitter’ revealed that 10% of the physicians sampled had tweeted potential patient privacy violations. A 2014 study of publicly available Facebook profiles of 319 Midwest residents found 14% had ‘potentially unprofessional content’ and 12.2% had ‘clearly unprofessional’ content, the latter including references to binge drinking, sexually suggestive photos, and HIPAA violations.”
Dr. Logghe, of Thomas Jefferson University, Philadelphia, is a member of the American College of Surgeons’ (ACS’s) social media committee tasked with creating practice recommendations for clinicians’ use of social media. Conducting a literature review was the first step to creating a surgeon-specific document, and the team found seven online behavior guidelines directed at physicians. Groups authoring these papers included the American Medical Association, the Federation of State Medical Boards, the American Congress of Obstetricians and Gynecologists, and several international groups.
Dr. Logghe and her colleagues reviewed each one, synthesized the information, and created a practice recommendation statement specific to the ACS. While not encoded in any professional ethics requirements, “Best Practices for Surgeons’ Social Media Use: Statement of the Resident and Associate Society of the American College of Surgeons” does lay out some common, potentially problematic scenarios and offers some suggestions about how to avoid Internet regret.
Everything discussed in the paper revolves around maintaining a decorous public persona. Professionalism on and off the clock is a key tenet of the recommendations. Definitions of key terms like “professionalism” are an important basis for any practice guideline, but sometimes concepts are not easy to define, the team wrote. “Perhaps the limitation most difficult to address in any formalized guideline is the necessary subjectivity in interpreting what is ‘appropriate’ or ‘professional’ online – or in any other setting,” the authors wrote. The ACS Code of Professional Conduct does not explicitly define either of those terms or discuss the appearance of unprofessional behavior.
In the absence of a plain-and-simple definition, the authors attempted to couch the social media recommendations in terms of ACS’s commitment to maintaining the patient trust. It urges surgeons to “avoid even the appearance of impropriety.”
The practice recommendations touch on a number of areas that are potentially problematic for surgeons, including confidentiality, financial conflicts, collegial support, and general social responsibility.
Confidentiality
Maintaining privacy is more than a courtesy to patients: It’s a federally mandated law with serious punitive repercussions if violated. Blogs, YouTube, Twitter, and Facebook offer a vast potential for sharing information with and educating the public, but postings can also easily violate HIPPA standards, the team wrote.
“In general, most social media platforms are not HIPPA-compliant,” no matter how the privacy settings are adjusted. These modes of communication are never appropriate for patient-physician communication: They can’t be archived in an electronic health record, and it is ill advised to give any medical advice by using these channels.
Discussing a particular case online, even with the usual defining details omitted, can be a bad idea.“Simply de-identifying patient information may not be sufficient. When posting information online, one must be cognizant of the context of other information available online. Such information includes the poster’s place of employment, news media, and publicly available vital statistics. Therefore even when posting general comments about hospital events, surgical cases, or patients under one’s care, it is essential to consider the sum of information available to the reader, rather than simply the information shared in the isolated post.”
Employment
Most employers have social media guidelines and don’t take kindly to violations – which can affect both current and future job postings. “A strong social media presence can be of benefit to one’s employer, [but] content that portrays a surgeon in an unprofessional or controversial light can be detrimental and even career-damaging.”
This reaches beyond professional communications online and deep into a surgeon’s personal life, the team noted, so exercise caution when “friending.”
“While this practice is inevitable, surgeons should be aware of potential conflicts. Connecting with or accepting friend requests from some but not all coworkers or coresidents could be interpreted as favoritism and may create a problematic work relationship. … Surgeons should consider primarily connecting with coworkers on professional websites if they have little contact with them outside the workplace.”
As for friending patients – just don’t, for both your sake and theirs. “Accepting a patient’s Facebook friend request may allow them access to events, details, and commentary not traditionally appropriate for the patient-physician relationship. Accepting such requests is strongly discouraged. If concerned about appearing rude or rejecting a patient’s request to be Facebook friends, the patient can be referred to society guidelines or best practices such as these.” One helpful alternative to such a request may be to invite patients to follow a practice website or other professional page.
Conflicts of interest
Online friends might not require disclosures when a surgeon posts about an exciting procedure or piece of equipment, such as whether there is a financial interest in doing so, but it’s important to be proactive. “As always, it is the physician’s responsibility to avoid even the appearance of impropriety. If it is not feasible to include a relevant conflict of interest within a post, the post should not be made.”
Defamation
Irritated about a colleague? Keep it to yourself – especially if you’ve had a beer. “It is never appropriate to post derogatory comments about patients or colleagues. Surgeons should be careful not to post in anger or under the influence of any substance. Statements about a colleague’s abilities, experience, or outcomes intended in jest may be appropriate for the surgeon’s lounge, yet entirely inappropriate for public consumption. Again, the ‘pause-before-posting’ practice is likely to prevent regretful posts in this vein.”
Privacy and Permanence
The Internet goes everywhere and lasts forever. A snappy quote that’s funny at 2 a.m. might not seem so hilarious in the light of day – or even in the light of a day 5 years yet to come.
The delete key is a false friend, and that clever pseudonym you dreamed up is probably as crackable as the classic “Pa55word” password. “One should presume that all content posted online will remain there forever and may be seen by anyone. Again, ‘pause-before-posting’ is a recommended practice.”
Privacy settings should be viewed as an illusion, the team noted. In this era of face recognition and tagging, images carry just as much risk as words.
Collegial support
Maybe your mother was right when she said, “This is for your own good.” If a colleague’s postings are getting out of hand, a tactful heart-to-heart might be the best course of action. “As coined by Dr. Sarah Mansfield, ‘Looking after colleagues is an integral element of professional conduct.’ Surgeons who notice colleagues posting unprofessional content that could be damaging to both the colleague and the public’s trust in the profession should discreetly express their concern to the individual, who should then take any appropriate corrective actions. … If the action is in violation of the law or medical board regulations, it should be reported to the appropriate governing bodies.”
Physician, Google Thyself
The team acknowledged that an online presence is virtually a must for professional development. And even if you don’t create a web page, chances are your university or hospital has done it for you. The media is interested in your life, too, and may make mention of your activities – both positive or negative.
“To better understand and control this publicly accessible information, surgeons are encouraged to periodically self-audit themselves online and taking measures to ensure that the information present is accurate and professional.” Some professional service websites are more trustworthy than others. The team encouraged physicians to participate in the ACS professional pages, LinkedIn, Doximity, and ResearchGate.
Not rules – just recommendations
The team stressed that their recommendations aren’t meant to stifle personal expression. Instead, their aim is to prompt a more conscious use of what can be a very powerful tool for both self-expression and professional development.
“The authors recommend no punitive action based on a perceived ‘violation’ of these recommendations alone. While they refer to other guidelines, including laws such as HIPAA, that must be appropriately enforced, these best practices are intended to guide the practicing surgeon in the use of social media rather than act as regulations or encourage reprimand. Rather than encouraging a social media landscape as sterile as the operating theater, the authors hope these recommendations lead to conscious consideration of online behavior, to avoidance of preventable harm, and to recognition of others’ views of their posts.”
None of the authors reported any financial disclosures.
SOURCE: Logghe HJ et al. J Am Coll Surg. 2017. doi: 10.1016/j.jamcollsurg.2017.11.022.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF SURGEONS
MyPlate as effective as calorie counting after 12 months
MONTREAL – The high-satiety MyPlate approach to weight loss reduced waist circumference, improved mental health and quality of life, and increased satiety 12 months into a study that found the strategy as effective as a conventional calorie-counting approach.
After 12 months, waist circumference had decreased by about 2 cm in study participants, regardless of whether they used the MyPlate approach or counted calories. Body weight was unchanged after a year in both groups.
“The simpler MyPlate approach may be easier for primary care patients to sustain long-term than the calorie-counting approach and has equal waist circumference reduction outcomes, supporting the U.S. government’s recommendation to use the MyPlate approach,” wrote Lillian Gelberg, MD, and her collaborators.

MyPlate recommends eating more fruits and vegetables, making half of grain choices whole grain, replacing sugary drinks with water, limiting sodium intake.
Over the study period, neither group significantly increased their physical activity. “Increasing patient physical activity seems not to be necessary to effect sustained, desirable weight control,” the researchers wrote.
The comparative effectiveness study compared the two weight loss approaches in 261 primarily Hispanic patients at two federally qualified health center clinics.
The primary outcome in the patient-centered study was perceived satiety; secondary outcomes included waist circumference, weight, mental health, quality of life, intake of sugary drinks, water intake, and exercise.
Participants received 11 health coaching sessions from bilingual community health workers, dubbed “promotoras,” over a 6 month period. “Community health workers were effective change agents in coaching overweight patients on how to eat more high-satiety foods,” Dr. Gelberg and her colleagues noted in a poster presentation at the annual meeting of the North American Primary Care Research Group.
Two of the sessions were hour-long home-based sessions; the MyPlate group also received two hour-long cooking demonstrations. Both groups received two hour-long group education sessions, as well as a total of seven 20-minute telephone coaching sessions.
Most participants (56%) were in five sessions during the study period, with a quarter completing 10 of the 11 sessions. At 12 months, 80% of study members were still being followed.
Overall, 95% of the participants were female, and about half (n = 126) did not have a high school diploma or equivalent.
A total of 86% were Hispanic, and Spanish was the preferred language for about three quarters of all participants. Most patients (82%) were born outside the United States.
The mean age at enrollment was 41 years. Mean body mass index was 30 to less than 35 kg/m2 for about half (46%) of the patients; 5% had a BMI of 40 kg/m2 or more.
In an interview, Dr. Gelberg, professor of family medicine and public health and management at the University of California, Los Angeles, said that the investigators had hypothesized that the MyPlate approach would be easier for patients to follow, because it’s easier to understand and less cumbersome in terms of record keeping. The investigators did not think that one approach would result in greater reductions in body weight or waist circumference, a finding borne out by the data presented.
Both groups reduced their intake of sugary drinks and increased the amount of water they drank, but the differences were not statistically significant.
The study was funded by the Patient-Centered Outcome Research Institute. Dr. Gelberg reported no relevant disclosures.
SOURCE: Gelberg L, abstract P414.
This article was updated on 2/13/18
MONTREAL – The high-satiety MyPlate approach to weight loss reduced waist circumference, improved mental health and quality of life, and increased satiety 12 months into a study that found the strategy as effective as a conventional calorie-counting approach.
After 12 months, waist circumference had decreased by about 2 cm in study participants, regardless of whether they used the MyPlate approach or counted calories. Body weight was unchanged after a year in both groups.
“The simpler MyPlate approach may be easier for primary care patients to sustain long-term than the calorie-counting approach and has equal waist circumference reduction outcomes, supporting the U.S. government’s recommendation to use the MyPlate approach,” wrote Lillian Gelberg, MD, and her collaborators.
MyPlate recommends eating more fruits and vegetables, making half of grain choices whole grain, replacing sugary drinks with water, limiting sodium intake.
Over the study period, neither group significantly increased their physical activity. “Increasing patient physical activity seems not to be necessary to effect sustained, desirable weight control,” the researchers wrote.
The comparative effectiveness study compared the two weight loss approaches in 261 primarily Hispanic patients at two federally qualified health center clinics.
The primary outcome in the patient-centered study was perceived satiety; secondary outcomes included waist circumference, weight, mental health, quality of life, intake of sugary drinks, water intake, and exercise.
Participants received 11 health coaching sessions from bilingual community health workers, dubbed “promotoras,” over a 6 month period. “Community health workers were effective change agents in coaching overweight patients on how to eat more high-satiety foods,” Dr. Gelberg and her colleagues noted in a poster presentation at the annual meeting of the North American Primary Care Research Group.
Two of the sessions were hour-long home-based sessions; the MyPlate group also received two hour-long cooking demonstrations. Both groups received two hour-long group education sessions, as well as a total of seven 20-minute telephone coaching sessions.
Most participants (56%) were in five sessions during the study period, with a quarter completing 10 of the 11 sessions. At 12 months, 80% of study members were still being followed.
Overall, 95% of the participants were female, and about half (n = 126) did not have a high school diploma or equivalent.
A total of 86% were Hispanic, and Spanish was the preferred language for about three quarters of all participants. Most patients (82%) were born outside the United States.
The mean age at enrollment was 41 years. Mean body mass index was 30 to less than 35 kg/m2 for about half (46%) of the patients; 5% had a BMI of 40 kg/m2 or more.
In an interview, Dr. Gelberg, professor of family medicine and public health and management at the University of California, Los Angeles, said that the investigators had hypothesized that the MyPlate approach would be easier for patients to follow, because it’s easier to understand and less cumbersome in terms of record keeping. The investigators did not think that one approach would result in greater reductions in body weight or waist circumference, a finding borne out by the data presented.
Both groups reduced their intake of sugary drinks and increased the amount of water they drank, but the differences were not statistically significant.
The study was funded by the Patient-Centered Outcome Research Institute. Dr. Gelberg reported no relevant disclosures.
SOURCE: Gelberg L, abstract P414.
This article was updated on 2/13/18
MONTREAL – The high-satiety MyPlate approach to weight loss reduced waist circumference, improved mental health and quality of life, and increased satiety 12 months into a study that found the strategy as effective as a conventional calorie-counting approach.
After 12 months, waist circumference had decreased by about 2 cm in study participants, regardless of whether they used the MyPlate approach or counted calories. Body weight was unchanged after a year in both groups.
“The simpler MyPlate approach may be easier for primary care patients to sustain long-term than the calorie-counting approach and has equal waist circumference reduction outcomes, supporting the U.S. government’s recommendation to use the MyPlate approach,” wrote Lillian Gelberg, MD, and her collaborators.
MyPlate recommends eating more fruits and vegetables, making half of grain choices whole grain, replacing sugary drinks with water, limiting sodium intake.
Over the study period, neither group significantly increased their physical activity. “Increasing patient physical activity seems not to be necessary to effect sustained, desirable weight control,” the researchers wrote.
The comparative effectiveness study compared the two weight loss approaches in 261 primarily Hispanic patients at two federally qualified health center clinics.
The primary outcome in the patient-centered study was perceived satiety; secondary outcomes included waist circumference, weight, mental health, quality of life, intake of sugary drinks, water intake, and exercise.
Participants received 11 health coaching sessions from bilingual community health workers, dubbed “promotoras,” over a 6 month period. “Community health workers were effective change agents in coaching overweight patients on how to eat more high-satiety foods,” Dr. Gelberg and her colleagues noted in a poster presentation at the annual meeting of the North American Primary Care Research Group.
Two of the sessions were hour-long home-based sessions; the MyPlate group also received two hour-long cooking demonstrations. Both groups received two hour-long group education sessions, as well as a total of seven 20-minute telephone coaching sessions.
Most participants (56%) were in five sessions during the study period, with a quarter completing 10 of the 11 sessions. At 12 months, 80% of study members were still being followed.
Overall, 95% of the participants were female, and about half (n = 126) did not have a high school diploma or equivalent.
A total of 86% were Hispanic, and Spanish was the preferred language for about three quarters of all participants. Most patients (82%) were born outside the United States.
The mean age at enrollment was 41 years. Mean body mass index was 30 to less than 35 kg/m2 for about half (46%) of the patients; 5% had a BMI of 40 kg/m2 or more.
In an interview, Dr. Gelberg, professor of family medicine and public health and management at the University of California, Los Angeles, said that the investigators had hypothesized that the MyPlate approach would be easier for patients to follow, because it’s easier to understand and less cumbersome in terms of record keeping. The investigators did not think that one approach would result in greater reductions in body weight or waist circumference, a finding borne out by the data presented.
Both groups reduced their intake of sugary drinks and increased the amount of water they drank, but the differences were not statistically significant.
The study was funded by the Patient-Centered Outcome Research Institute. Dr. Gelberg reported no relevant disclosures.
SOURCE: Gelberg L, abstract P414.
This article was updated on 2/13/18
REPORTING FROM NAPCRG 2017
Key clinical point:
Major finding: At 12 months, both approaches reduced waist circumference by 2 cm but didn’t result in weight loss.
Data source: Prospective randomized comparative effectiveness trial of 261 patients.
Disclosures: Dr. Gelberg reported no conflicts of interest. The study was funded by the Patient-Centered Comparative Effectiveness Institute.
Source: Gelberg L, abstract P414.

FDA axes asthma drugs’ boxed warning
The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.

The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.
The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.
A Needs Review of Caregivers for Adults With Traumatic Brain Injury
Traumatic brain injury (TBI) is a health concern for the U.S. Military Health System (MHS) as well as the VHA. It occurs in both deployed and nondeployed settings; however, Operation Enduring Freedom (OEF) and Operation Iraqi Freedom (OIF) and improved reporting mechanisms have dramatically increased TBI diagnoses in active-duty service members. According to the Defense and Veterans Brain Injury Center (DVBIC), more than 370,000 service members have been diagnosed with a TBI since 2000 (Figure).1 
Background
The DoD and the VA are collaborating on clinical research studies to identify, understand, and treat the long-term effects of TBI that can affect patients and their families. Most TBIs are mild (mTBIs), also called concussions, and patients typically recover within a few weeks (Table 1). However, some individuals with mTBI experience symptoms that may persist for months or years. A meta-analysis by Perry and colleagues showed that the prevalence or risk of a neurologic disorder, depression, or other mental health issue following mTBI was 67% higher compared with that in uninjured controls.2 
Patients with any severity of TBI may require assistance with activities of daily living (ADLs), such as bathing, dressing, managing medications, and feeding. Patients also may need help with instrumental ADLs, such as meal preparation, grocery shopping, household chores, child care, getting to appointments or activities, coordination of educational and vocational services, financial and benefits management, and supportive listening.
Increased injuries have spurred the DoD and VA to coordinate health care to provide a seamless transition for patients between the 2 agencies. However, individuals who sustained a TBI may need various levels of caregiver assistance over time.
TBI and Caregivers
Despite better agency coordination for patients, caregivers can experience stress. Griffin and colleagues found that caregiving responsibilities can compete with other demands on the caregiver, such as work and family, and may negatively impact their health and finances.3,4
Lou and colleagues studied the factors associated with caring for chronically ill family members that may result in stress for the caregivers.5 Along with an unaccounted for economic contribution, caregivers may face lost work time and pay and limitations on work travel and work advancement. Additionally, lost time for leisure, travel, social activities, family obligations, and retirement could result in physical and mental drain on the caregiver. Stress may reach a level at which the caregivers risk psychological distress. The study also noted that families with perceived high stress experience disrupted family functioning. Some TBI caregiver studies sought to understand how best to evaluate and determine the level of caregiver burden, and other studies investigated appropriate interventions.6-9
Health care practitioners within the federal health care system may benefit from a greater awareness of caregiver needs and caregiver resources. Caregiver support can improve outcomes for both the caregiver and care recipient, and many organizations and resources already exist to assist the caregiver. This article reviews recent published literature on TBI caregivers of patients with TBI across civilian, military, and veteran populations and lists caregiver resources for additional information, assistance, and support.
Literature Review
The DVBIC defines the term caregiver as “any family or support person(s) relied upon by the service member or veteran with traumatic brain injury (TBI) who assumes primary responsibility for ensuring the needed level of care and overall well-being of that service member or veteran. A family or family caregiver may include spouse, parents, children, other extended family members, as well as significant others and friends.”3
In the following discussion, findings from military and veteran literature are separated from civilian population findings to highlight similarities and differences between these 2 bodies of research. Several of the studies in the military/veteran cohorts include polytrauma patients with comorbid physical and mental health issues not necessarily found in civilian literature.
Civilian Literature
A 2015 systematic review by Anderson and colleagues on coping and psychological adjustment in TBI caregivers indicated no Class I or Class II studies.10 Four Class III and 3 Class IV studies were found. The authors suggest that more rigorous studies (ie, Class I and II) are needed.
Despite these limitations, peer-reviewed literature indicates that the levels of stress and distress in TBI caregivers are consistent with reports for other diseases. In a civilian population, Carlozzi and colleagues found that TBI caregivers who reported stress, distress, anxiety, and feeling overwhelmed often had concerns for their social, emotional, physical, cognitive health, as well as feelings of loss.5 In addition, caregivers may need to take leaves of absence or leave the workforce entirely to provide for a family member or friend who had a TBI—often leading to financial strain (eg, depleting assets, accumulating debt). These challenges may occur during prime earning years, and the caregiver may lose the ability to resume work if the care receiver requires care for extended periods.11
Kratz and colleagues showed that caregivers of individuals with moderate-to-severe TBI: (1) felt overburdened with responsibilities; (2) lacked personal time and time for self-care; (3) felt their lives were interrupted or lost; (4) grieved the loss of the person with TBI; and (5) endorsed anger, guilt, anxiety, and sadness.12
Perceptions differed between caregiver parents and caregiver partners. Parents expressed feelings of grief and sadness related to the “loss of the person before the TBI.” Parents also reported a sense of guilt and responsibility for their child’s TBI and feelings of being tied down to the individual with TBI. Parents experienced a greater level of stress if the son or daughter with TBI still lived at home. Partners expressed frustration and despair related to their role as sole decision maker and care provider. Partners’ distress also related to the partner relationship and the relationship between children and the individual with TBI.
Verhasghe and colleagues found that partners experience a greater degree of stress than do parents.13 Young families with minimal social support for coping with financial, psychiatric, and medical problems were the most vulnerable to stress. A systematic review by Ennis and colleagues evaluated depression and anxiety in caregiver parents vs spouses.14 Although methods and quality differed in the studies, findings indicated high levels of distress regardless of the type of caregiver.
Anderson and colleagues used the Ways of Coping Questionnaire to evaluate the association between coping and psychological adjustment in caregivers of TBI individuals.10 The use of emotion-focused coping and problem solving was possibly associated with psychological adjustment in caregivers. Verhasghe and colleagues indicated that the nature of the injuries more than the severity of TBI determined the level of stress up to 15 years after the TBI.13 Gender and social and professional support also influenced coping. The review identified the need to develop models of long-term support and care.
An Australian cohort of 79 family caregivers participated in a study by Perlesz and colleagues.15 Participants’ caregiving responsibilities averaged 19.3 months posttrauma. The Family Satisfaction Scale, Beck Depression Inventory, State Anxiety Inventory, and Profile of Mood States were used in this analysis. Male caregivers reported distress in terms of anger and fatigue; female caregivers were at greatest risk of poor psychosocial outcomes. Although findings from primary caregivers indicated that 35% to 49% displayed enough distress to warrant clinical intervention, between 51% and 80% were not psychologically distressed and were satisfied with their families. Data supported previous reports suggesting caregivers are “not universally distressed.”15
Manskow and colleagues followed patients with severe TBI and assessed caregiver burden 1 year later. Using the Caregiver Burden Scale, caregivers reported the highest scores (N = 92) on the General Strain Index followed by the Disappointment Index.16 Bayen and colleagues also studied caregivers of severe TBI patients.17 Objective and subjective caregiver burden data 4 years later indicated 44% of caregivers (N = 98) reported multidimensional burden. Greater burden was associated in caring for individuals who had poorer Glasgow Outcome Scale Extended scores and more severe cognitive disorders.
Military and Veteran Literature
Griffin and colleagues conducted the Family and Caregiver Experience Study (FACES) with caregivers (N = 564) of service members who incurred a TBI.3 According to the caregivers, two-thirds of the patients lost consciousness for more than 30 minutes, which was followed by inpatient rehabilitation care at a VA polytrauma center between 2001 and 2009. The majority of caregivers of TBI patients were female (79%) and aged < 60 years (84%). Parents comprised 62% and spouses 32% of the cohort. Caregivers tended to have some level of education beyond high school (73%), were married (77%), either worked or were enrolled in school (55%), and earned less than $40,000 a year (70%). Common characteristics of the care receivers were male gender (95%), average age 30, high school educated (52%), married (almost 50%), and employed (50%). Forty-five percent of the care receivers were injured 4 to 6 years prior, and 12% were injured 7 or more years prior. The study determined the caregivers’ perception of intensity of care needed and indicated that families as well as clinicians need to plan for some level of long-term support and services.
In addition to the TBI-related caregiving needs, Griffin and colleagues found in a military population that other medical conditions impacted the level of caregiving and strained a marriage.18 Their study found that in a military population between 30% and 50% of marriages of patients with TBI dissolved within the first 10 years after injury. Caregivers may need to learn nursing activities, such as tube feedings, tracheostomy and stoma care, catheter care, wound care, and medication administration. Family stress with caregiving may interfere with the ability to understand information related to the care receivers’ medical care and may require multiple formats to explain care needs. Sander and colleagues associated better emotional functioning in caregivers with greater social integration and occupation outcomes in patients at the postacute rehabilitation program phase (within 6 months of injury).19 However, these outcomes did not continue more than 6 months postinjury.
Intervention and Research Studies
Powell and colleagues used a telephone-based, individualized TBI education intervention along with problem-solving mentoring (10 phone calls at 2-week intervals following patient discharge for moderate-to-severe TBI from a level 1 trauma center) to determine which programs, activities, and coping strategies could decrease caregiver challenges.20 The telephone interventions resulted in better caregiver outcomes than usual care as measured by composite scores on the Bakas Caregiving Outcomes Scale (BCOS) and the Brief Symptom Inventory (BSI-18) at 6 months post-TBI survivor discharge. Dyer and colleagues explored Internet approaches and mobile applications to provide support for caregivers. 18 In a small sample of 10 caregivers, Damianakis and colleagues conducted a 10-session pilot videoconferencing support-group intervention program led by a clinician. Results indicated that the intervention enhanced caregiver coping and problem-solving skills.7
Petranovich and colleagues examined the efficacy of counselor-assisted problem-solving interventions in improving long-term caregiver psychological functioning following TBI in adolescents.21 Their findings support the utility of online interventions in improving long-term caregiver psychological distress, particularly for lower income families. Although this study focused on adolescents, research may indicate merit in an adult population. In relatives of patients with severe TBI, Norup and colleagues associated improvements in health-related quality of life (HRQOL) with improvements in symptoms of anxiety and depression without specific intervention.22
Moriarty and colleagues conducted a randomized controlled trial for veterans who received care at a VA polytrauma center and their family members who participated in a veteran’s in-home program (VIP) intervention.9 The study aimed to evaluate how VIP affected family members’ caregiver burden, depressive symptoms, satisfaction with caregiving, and the program’s acceptability. Eighty-one veterans with a key family member were randomized. Of those, 63 veterans completed a follow-up interview. The intervention consisted of 6 home visits of 1 to 2 hours each and 2 telephone calls from an occupational therapist over 3 to 4 months. Family members were invited to participate during the home visits. The control group received usual clinic care with 2 telephone calls during the study period. All participants received the follow-up interview 3 to 4 months after baseline interviews. The severity of TBI was determined by a review of the electronic medical record using the VA/DoD Clinical Practice Guidelines. Findings of this study indicated that family members in the intervention group showed significantly lower depressive symptom scores and caregiver burden scores.9 Additionally, the veterans in the intervention group exhibited higher community integration and ability to manage their targeted outcomes. Further research may indicate that VIP could assist patients with TBI and caregivers in an active-duty population.
The DVBIC is the executive agent for a congressionally mandated 15-year longitudinal study on TBI incurred by members of the armed services in OEF and OIF. The John Warner National Defense Authorization Act for Fiscal Year 2007 outlined the study. An initial finding identified the need for an HRQOL outcomes assessment specific to TBI caregivers.23 Having these data will allow investigators to fully determine the comprehensive impact of caring for a person who sustained a mild, moderate, severe, or penetrating TBI and to evaluate the effectiveness of interventions designed to address caregivers’ needs. To date, the study has identified the following HRQOL themes generated among caregivers: social health, emotional health, physical/medical health, cognitive functioning, and feelings of loss (related to changing social roles). Carlozzi and colleagues noted that the study also aimed to identify a sensitive outcome measure to evaluate quality of life in the caregivers over time.7
Knowledge Gaps
Ongoing studies focus on caregiving for individuals with various illnesses and needs. Some of the information in each study may be beneficial to TBI caregivers who are not fully aware of resources and interventions. For example, Fortune and colleagues, Hirano and colleagues, and Grover and colleagues are studying caregiver activities involving other diseases to determine, more generally, which programs, activities, and coping strategies can decrease caregiver challenges.24-26 Further, understanding and addressing the needs of these families over many years will provide data that could inform policy, benefits, resources, and needed services (such as the Caregivers and Veterans Omnibus Health Services Act of 2010) and assist with family resilience efforts, including understanding and enhancing family protective and recovery factors.
As studies have indicated, some families do not report family distress when providing care to an individual with TBI. Understanding the factors that influence positive family adjustment is important to capture and perhaps replicate in future studies so that they can lead to effective treatment interventions. Although this review does not discuss caregiver needs for patients with TBI with disorders of consciousness that require more care than most caregivers can provide in the home setting, caregiving for this population deserves attention in future studies. Furthermore, an area that has not received much attention is the impact on children in the household. Children aged < 18 years can assist not only in the care of a disabled adult, but also of younger siblings; also they can help with household activities from housekeeping to meal preparation. Children also may provide physical and emotional support.
The impact of aging caregivers and subsequent needs for their own care as well as the person(s) they are providing care for has not been fully addressed. Areas requiring more research include both the aging caregiver taking care of an aging spouse or relative and the aging parent taking care of a young adult or child. Along with aging, the issue of long-term caregiving needs further development. For example, how do the differences between access to services between caregivers of adults with TBI in the military and those in the civilian sector impact the family/caregiver? Further research may answer questions such as:
- Which tools are most useful in evaluating and determining caregiver stress and burden?
- Are the needs of military and veteran caregivers unique?
- Do polytrauma patients with comorbid diagnoses have unique caregiver needs and trajectories?
- Do TBI caregiver stressors differ from stressors related to other medical conditions or chronic diseases?
- Is there a need for military and veteran TBI-specific caregiver programs?
- Which interventions best help caregivers and for how long?
- Should the approach to intervention depend on variables such as age and gender of the caregivers or relationship to the patient with a TBI (eg, spouse vs parent)?
Methods or processes to inform and update caregivers about available resources also are critically needed. Also, Sabab and colleagues noted the importance of research on the effects of denial as it relates to cognitive, emotional, social impact.27 Denial may impact delays in treatments.
Resource
Many national, state, local, and grassroots organizations provide information and support for persons with illness and/or disabilities. Most clinicians of neurologic, mental health, and cancer have developed various forms of support interventions for those with the disease and their caregivers (Table 2). Highlighted in this section are a few organizations that specifically provide resources for caregivers caring for active-duty service members or veterans with a TBI. 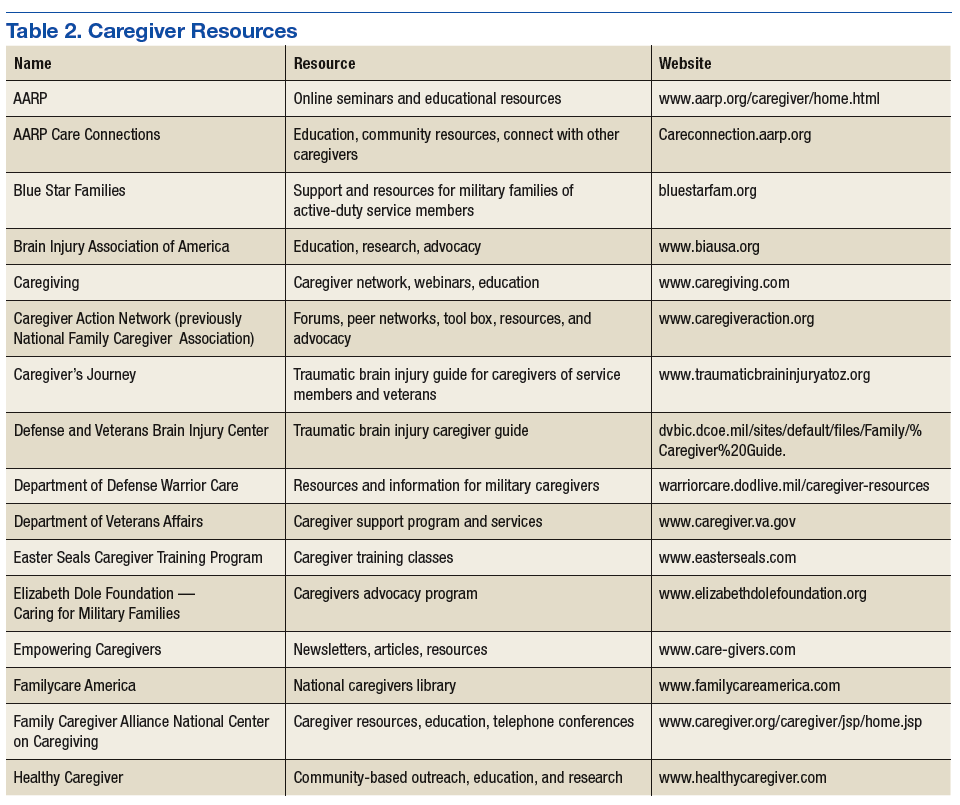
Although a caregiver generally does not receive money from an outside source for services, the DoD may consider the caregiver as a nonmedical attendant for an active-duty service member and provide a temporary stipend. The VA provides several support and service options for caregivers under the Caregiver Support Program, through which more than 300 VA health care professionals provide support to caregivers. The Caregivers and Veterans Omnibus Health Services Act of 2010 authorizes the VA to provide additional VA services for seriously injured post-9/11 veterans and their family caregivers through the Program of Comprehensive Assistance for Family Caregivers (VA Caregiver Support Program). After meeting eligibility criteria, primary caregivers of post-9/11 veterans may receive a monthly stipend (based on the level of care needed) as well as comprehensive caregiver training, referral services, access to health care insurance, mental health services, counseling, and respite care. The Caregiver Support Program offers a toll-free support line and a 24-hour crisis hot line. 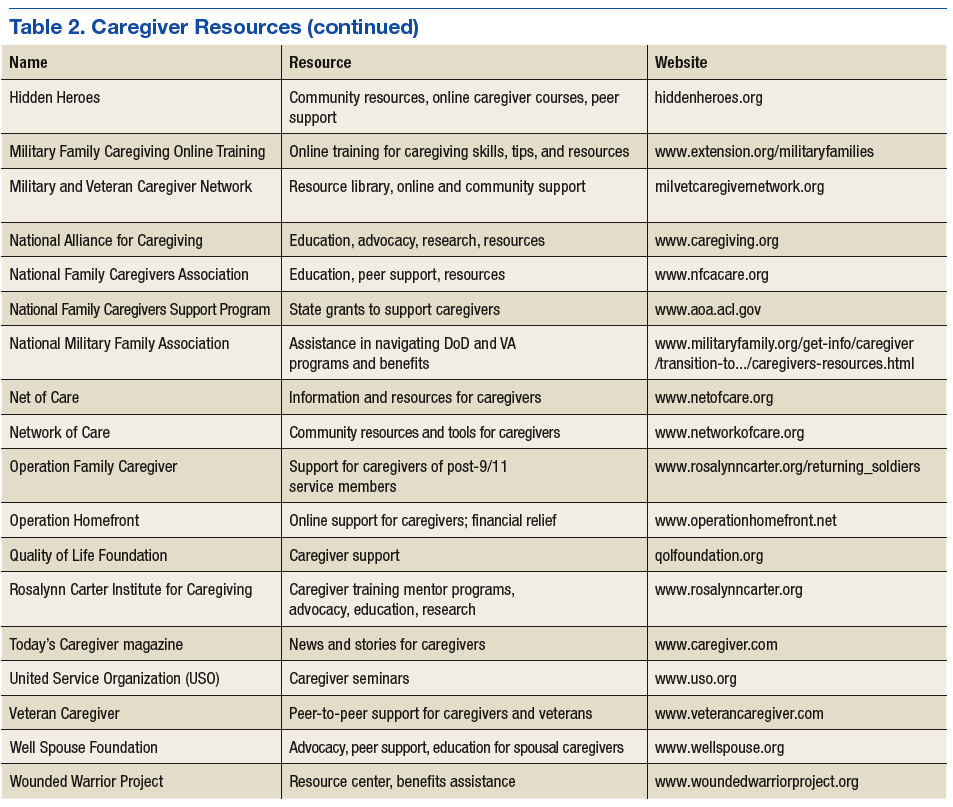
In 2014, the Government Accountability Office (GAO) outlined the VA health care improvements needed to manage the demand for the Caregiver Support Program, which are established at VA medical centers.28 The GAO reported that the “VA significantly underestimated caregivers’ demand for services… larger than expected workloads and …delays in approval determinations” with about 500 approved caregivers who are added to the program each month. Original estimates indicated that about 4,000 caregivers would be approved by September 2014; however, by May 2014 about 15,600 caregivers were approved.
In addition to the VA Caregiver Support Program, a variety of state, local, and nonprofit organizations offer support for caregivers. Established in 2012, the Elizabeth Dole Foundation’s program Caring for Military Families “assists caregivers by raising awareness of the caregiver role, leveraging resources and partnerships to provide support, and identifying best practices and solutions to address the challenges caregivers face.” The foundation commissioned the RAND Corporation to “describe the magnitude of military caregiving in the United States, and to identify gaps in programs, policies, and services.” The 2014 RAND report estimated that among the 5.5 million military caregivers in the U.S., 1 million (19.6%) cared for post-9/11 veterans.29 The military caregivers consistently experienced poorer health outcomes, greater strains on family relationships, and more workplace problems than noncaregivers; post-9/11 military caregivers fared worse in those areas.
The Elizabeth Dole Foundation, Hidden Heroes Impact Council Forum advocates for caregiver empowerment, cultural competency awareness, and better policies, programs, and services. The council focuses its efforts on key impact: community support at home, education and training, employment and workplace support, financial and legal issues, interfaith action and ministry council, mental and physical health, and respite care. It aims to raise the money to build awareness and support for military and veterans’ caregivers. The Military and Veteran Caregiver Network is another Elizabeth Dole Foundation initiative. It is an online forum community, peer support group, and peer mentor program structure. A resource library for referrals to local services also is available.
A variety of other organizations, such as United Service Organizations; Easter Seals; Team Red, White and Blue; Operation Homefront; Blue Star Families; state Brain Injury Associations; and support groups for TBI at local hospitals and community centers provide resources to both patients and caregivers. Organizations for caregivers not exclusive to TBI patients include the Caregiver Action Network (formerly National Family Caregiver Association) and the Family Caregiver Alliance. The National Family Caregivers Support Program provides grants to states and territories to develop and provide supportive services to caregivers. Some training for caregivers could include long-term financial planning, legal issues, residential and educational planning, caregiver stress management, the benefits of utilizing support resources, and actions and behaviors that enhance coping strategies. In 2007, DVBIC developed The Traumatic Brain Injury Guide for Caregivers of Service Members and Veterans, which is intended for family caregivers assisting a service member or veteran who sustained a moderate or severe TBI.6 A recent assessment determined the need to update the guide. The Center of Excellence for Medical Multimedia is another source of information for caregivers.
Conclusion
The recent combat conflicts of OEF and OIF have resulted in a dramatic increase in the occurrence of TBI injuries in active-duty service members both in theater and stateside and have highlighted the need for some service members and veterans with a TBI to require ongoing assistance from a caregiver. The levels of assistance and length of time vary greatly, impacted by the severity of the TBI and psychosocial situations.
In response to elevated awareness, several programs and resources have been developed or enhanced to address the specific needs of caregivers. Certain programs and resources are specific for caregivers of military service members and veterans, whereas others benefit caregivers in general. Likewise, some programs are not specific to individuals with TBI.
Caregivers assume many roles in their efforts to support the person with a TBI. They may need to dramatically adjust their lives to serve as a caregiver. Providing adequate resources for the caregivers impacts their ability to continue providing care. Thus, awareness of and access to resources play a critical role in helping to reduce stress, distress, burden (eg, physical, emotional, and financial), and caregiver burnout. Programs and resources often change, making it difficult for health care practitioners to know which programs offer what or even whether they still exist. Therefore, the authors synthesized the current medical literature of the topic of TBI and their caregiver needs as well as current resources for additional information and support.
Ongoing research studies, such as the congressionally mandated 15-year longitudinal study, are examining the impact of caregiving in the military and veteran communities. Future research could identify specific needs of military caregivers, identify gaps in services or programs, and identify interventions that promote resilience. Moreover, research directed at military and veteran caregivers can promote change that will benefit the general population of caregivers. It will be important for health care practitioners to keep abreast of new findings and information to incorporate into care plans for their patients who have had a TBI and their families.
1. Defense and Veterans Brain Injury Center. DoD worldwide numbers for TBI. http://dvbic.dcoe.mil/dod-worldwide-numbers-tbi. Updated October 5, 2017. Accessed October 10, 2017.
2. Perry DC, Sturm VE, Peterson MJ, et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg. 2016;124(2):511-526.
3. Defense and Veterans Brain Injury Center. Traumatic brain injury: a guide for caregivers of service members and veterans. https://dvbic.dcoe.mil /sites/default/files/Family%20Caregiver%20Guide.All%20Modules_updated.pdf. Accessed October 10, 2017
4. Griffin JM, Friedemann-Sánchez G, Jensen AC, et al. The invisible side of war: families caring for US service members with traumatic brain injuries and polytrauma. J Head Trauma Rehabil. 2012;27(1):3-13.
5. Lou VW, Kwan CW, Chong ML, Chi I. Associations between secondary caregivers’ supportive behavior and psychological distress of primary spousal caregivers of cognitively intact and impaired elders. Gerontologist. 2015;55(4):584-594.
6. Carlozzi NE, Kratz AL, Sander AM, et al. Health-related quality of life in caregivers of individuals with traumatic brain injury: development of a conceptual model. Arch Phys Med Rehabil. 2015;96(1):105-113.
7. Damianakis T, Tough A, Marziali E, Dawson DR. Therapy online: a web-based video support group for family caregivers of survivors with traumatic brain injury. J Head Trauma Rehabil. 2016;31(4):E12-E20.
8. Dyer EA, Kansagara D, McInnes DK, Freeman M, Woods, S. Mobile applications and internet-based approaches for supporting non-professional caregivers: a systematic review. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0050675. Accessed October 10, 2017.
9. Moriarty H, Winter L, Robinson K, et al. A randomized controlled trial to evaluate the veterans’ in-home program for military veterans with traumatic brain injury and their families: report on impact for family members. PMR. 2016;8(6):495-509.
10. Anderson MI, Simpson GK, Daher M, Matheson L. Chapter 7 the relationship between coping and psychological adjustment in family caregivers of individuals with traumatic brain injury: a systematic review. Annu Rev Nurs Res. 2015;33:219-247.
11. Van Houtven CH, Friedemann-Sanchez G, Clothier B, et al. Is policy well-targeted to remedy financial strain among caregivers of severely injured U.S. service members? Inquiry. 2012-2013;49(4):339-351.
12. Kratz AL, Sander AM, Brickell TA, Lange RT, Carlozzi NE. Traumatic brain injury caregivers: a qualitative analysis of spouse and parent perspectives on quality of life. Neuropsychol Rehabil. 2017;27(1):16-37.13. Verhasghe S, Defloor T, Grypdonck M. Stress and coping among families of patients with traumatic brain injury: a review of the literature. J Clin Nurs. 2005;14(8):1004-1012.
14. Ennis N, Rosenbloom BN, Canzian S, Topolovec-Vranic J. Depression and anxiety in parent versus spouse caregivers of adult patients with traumatic brain injury: a systematic review. Neuropsychol Rehabil. 2013;23(1):1-18.
15. Perlesz A, Kinsella G, Crowe S. Psychological distress and family satisfaction following traumatic brain injury: injured individuals and their primary, secondary, and tertiary carers. J Head Trauma Rehabil. 2000;15(3):909-929.
16. Manskow US, Sigurdardottir S, Røe C, et al. Factors affecting caregiving burden 1 year after severe traumatic brain injury: a prospective nationwide multicenter study. J Head Trauma Rehabil. 2015;30(6):411-423.
17. Bayen E, Jourdan C, Ghout I, et al. Objective and subjective burden of informal caregivers 4 years after a severe traumatic brain injury: results from the Paris-TBI study. J Head Trauma Rehabil. 2016;31(5):E59-E67.
18. Griffin JM, Friedemann-Sanchez G, Hall C, Phelan S, van Ryn M. Families of patients with polytrauma: understanding the evidence and charting a new research agenda. J Rehabil Res Dev. 2009;46(6):879-892.
19. Sander AM, Maestas KL, Sherer M, Malac JF, Nakase-Richardson R. Relationship of caregiver and family functioning to participation outcomes after post-acute rehabilitation for traumatic brain injury: a multicenter investigation. Arch Phys Med Rehabil. 2012;93(5):842-848.
20. Powell JM, Fraser R, Brockway JA, Temkin N, Bell KR. A telehealth approach to caregiver self-management following traumatic brain injury: a randomized control trial. J Head Trauma Rehabil. 2015;31(3):180-190.
21. Petranovich CL, Wade SL, Taylor HG, et al. Long-term caregiver mental health outcomes following a predominately online intervention for adolescents with complicated mild to severe traumatic brain injury. J Pediatr Psychol, 2015;40(7):680-688.
22. Norup A, Kristensen KS, Poulsen I, Mortensen EL. Evaluating clinically significant changes in health-related quality of life: a sample of relatives of patients with severe traumatic brain injury. Neuropsychol Rehabil. 2017;27(2):196-215.
23. John Warner National Defense Authorization Act for Fiscal Year 2007, HR 5122, 109th Cong, 2nd Sess (2006).
24. Fortune DG, Rogan CR, Richards HL. A structured multicomponent group program for carers of people with acquired brain injury: effects on perceived criticism, strain, and psychological distress. Br J Health Psychol. 2016;21(1):224-243.
25. Hirano A, Umegaki H, Suzuki Y, Hayashi T, Kuzuya M. Effects of leisure activities at home on perceived care burden and the endocrine system of caregivers of dementia patients: a randomized controlled study. Int Psychogeriatr. 2016;28(2):261-268.
26. Grover S, Pradyumna, Chakrabarti S. Coping among caregivers of patients with schizophrenia. Ind Psychiatry J. 2015;24(1):5-11.
27. Saban KL, Hogan NS, Hogan TP, Pape TL. He looks normal but…challenges of family caregivers of veterans diagnosed with a traumatic brain injury. Rehabil Nurs. 2015;40(5):277-285.
28. Williamson RB; United States Government Accountability Office. VA health care improvements needed to manage higher-than-expected demand for the family caregiver program. http://www.gao.gov/assets/670/667275.pdf. Published December 3, 2014. Accessed October 10, 2017.
29. Ramchand R, Tanielian T, Fisher MP, et al. Hidden heroes America’s military caregivers. http://www.rand.org/content/dam/rand/pubs/research_reports/RR400/RR499/RAND_RR499.pdf. Published 2014. Accessed October 10, 2017.
Traumatic brain injury (TBI) is a health concern for the U.S. Military Health System (MHS) as well as the VHA. It occurs in both deployed and nondeployed settings; however, Operation Enduring Freedom (OEF) and Operation Iraqi Freedom (OIF) and improved reporting mechanisms have dramatically increased TBI diagnoses in active-duty service members. According to the Defense and Veterans Brain Injury Center (DVBIC), more than 370,000 service members have been diagnosed with a TBI since 2000 (Figure).1
Background
The DoD and the VA are collaborating on clinical research studies to identify, understand, and treat the long-term effects of TBI that can affect patients and their families. Most TBIs are mild (mTBIs), also called concussions, and patients typically recover within a few weeks (Table 1). However, some individuals with mTBI experience symptoms that may persist for months or years. A meta-analysis by Perry and colleagues showed that the prevalence or risk of a neurologic disorder, depression, or other mental health issue following mTBI was 67% higher compared with that in uninjured controls.2
Patients with any severity of TBI may require assistance with activities of daily living (ADLs), such as bathing, dressing, managing medications, and feeding. Patients also may need help with instrumental ADLs, such as meal preparation, grocery shopping, household chores, child care, getting to appointments or activities, coordination of educational and vocational services, financial and benefits management, and supportive listening.
Increased injuries have spurred the DoD and VA to coordinate health care to provide a seamless transition for patients between the 2 agencies. However, individuals who sustained a TBI may need various levels of caregiver assistance over time.
TBI and Caregivers
Despite better agency coordination for patients, caregivers can experience stress. Griffin and colleagues found that caregiving responsibilities can compete with other demands on the caregiver, such as work and family, and may negatively impact their health and finances.3,4
Lou and colleagues studied the factors associated with caring for chronically ill family members that may result in stress for the caregivers.5 Along with an unaccounted for economic contribution, caregivers may face lost work time and pay and limitations on work travel and work advancement. Additionally, lost time for leisure, travel, social activities, family obligations, and retirement could result in physical and mental drain on the caregiver. Stress may reach a level at which the caregivers risk psychological distress. The study also noted that families with perceived high stress experience disrupted family functioning. Some TBI caregiver studies sought to understand how best to evaluate and determine the level of caregiver burden, and other studies investigated appropriate interventions.6-9
Health care practitioners within the federal health care system may benefit from a greater awareness of caregiver needs and caregiver resources. Caregiver support can improve outcomes for both the caregiver and care recipient, and many organizations and resources already exist to assist the caregiver. This article reviews recent published literature on TBI caregivers of patients with TBI across civilian, military, and veteran populations and lists caregiver resources for additional information, assistance, and support.
Literature Review
The DVBIC defines the term caregiver as “any family or support person(s) relied upon by the service member or veteran with traumatic brain injury (TBI) who assumes primary responsibility for ensuring the needed level of care and overall well-being of that service member or veteran. A family or family caregiver may include spouse, parents, children, other extended family members, as well as significant others and friends.”3
In the following discussion, findings from military and veteran literature are separated from civilian population findings to highlight similarities and differences between these 2 bodies of research. Several of the studies in the military/veteran cohorts include polytrauma patients with comorbid physical and mental health issues not necessarily found in civilian literature.
Civilian Literature
A 2015 systematic review by Anderson and colleagues on coping and psychological adjustment in TBI caregivers indicated no Class I or Class II studies.10 Four Class III and 3 Class IV studies were found. The authors suggest that more rigorous studies (ie, Class I and II) are needed.
Despite these limitations, peer-reviewed literature indicates that the levels of stress and distress in TBI caregivers are consistent with reports for other diseases. In a civilian population, Carlozzi and colleagues found that TBI caregivers who reported stress, distress, anxiety, and feeling overwhelmed often had concerns for their social, emotional, physical, cognitive health, as well as feelings of loss.5 In addition, caregivers may need to take leaves of absence or leave the workforce entirely to provide for a family member or friend who had a TBI—often leading to financial strain (eg, depleting assets, accumulating debt). These challenges may occur during prime earning years, and the caregiver may lose the ability to resume work if the care receiver requires care for extended periods.11
Kratz and colleagues showed that caregivers of individuals with moderate-to-severe TBI: (1) felt overburdened with responsibilities; (2) lacked personal time and time for self-care; (3) felt their lives were interrupted or lost; (4) grieved the loss of the person with TBI; and (5) endorsed anger, guilt, anxiety, and sadness.12
Perceptions differed between caregiver parents and caregiver partners. Parents expressed feelings of grief and sadness related to the “loss of the person before the TBI.” Parents also reported a sense of guilt and responsibility for their child’s TBI and feelings of being tied down to the individual with TBI. Parents experienced a greater level of stress if the son or daughter with TBI still lived at home. Partners expressed frustration and despair related to their role as sole decision maker and care provider. Partners’ distress also related to the partner relationship and the relationship between children and the individual with TBI.
Verhasghe and colleagues found that partners experience a greater degree of stress than do parents.13 Young families with minimal social support for coping with financial, psychiatric, and medical problems were the most vulnerable to stress. A systematic review by Ennis and colleagues evaluated depression and anxiety in caregiver parents vs spouses.14 Although methods and quality differed in the studies, findings indicated high levels of distress regardless of the type of caregiver.
Anderson and colleagues used the Ways of Coping Questionnaire to evaluate the association between coping and psychological adjustment in caregivers of TBI individuals.10 The use of emotion-focused coping and problem solving was possibly associated with psychological adjustment in caregivers. Verhasghe and colleagues indicated that the nature of the injuries more than the severity of TBI determined the level of stress up to 15 years after the TBI.13 Gender and social and professional support also influenced coping. The review identified the need to develop models of long-term support and care.
An Australian cohort of 79 family caregivers participated in a study by Perlesz and colleagues.15 Participants’ caregiving responsibilities averaged 19.3 months posttrauma. The Family Satisfaction Scale, Beck Depression Inventory, State Anxiety Inventory, and Profile of Mood States were used in this analysis. Male caregivers reported distress in terms of anger and fatigue; female caregivers were at greatest risk of poor psychosocial outcomes. Although findings from primary caregivers indicated that 35% to 49% displayed enough distress to warrant clinical intervention, between 51% and 80% were not psychologically distressed and were satisfied with their families. Data supported previous reports suggesting caregivers are “not universally distressed.”15
Manskow and colleagues followed patients with severe TBI and assessed caregiver burden 1 year later. Using the Caregiver Burden Scale, caregivers reported the highest scores (N = 92) on the General Strain Index followed by the Disappointment Index.16 Bayen and colleagues also studied caregivers of severe TBI patients.17 Objective and subjective caregiver burden data 4 years later indicated 44% of caregivers (N = 98) reported multidimensional burden. Greater burden was associated in caring for individuals who had poorer Glasgow Outcome Scale Extended scores and more severe cognitive disorders.
Military and Veteran Literature
Griffin and colleagues conducted the Family and Caregiver Experience Study (FACES) with caregivers (N = 564) of service members who incurred a TBI.3 According to the caregivers, two-thirds of the patients lost consciousness for more than 30 minutes, which was followed by inpatient rehabilitation care at a VA polytrauma center between 2001 and 2009. The majority of caregivers of TBI patients were female (79%) and aged < 60 years (84%). Parents comprised 62% and spouses 32% of the cohort. Caregivers tended to have some level of education beyond high school (73%), were married (77%), either worked or were enrolled in school (55%), and earned less than $40,000 a year (70%). Common characteristics of the care receivers were male gender (95%), average age 30, high school educated (52%), married (almost 50%), and employed (50%). Forty-five percent of the care receivers were injured 4 to 6 years prior, and 12% were injured 7 or more years prior. The study determined the caregivers’ perception of intensity of care needed and indicated that families as well as clinicians need to plan for some level of long-term support and services.
In addition to the TBI-related caregiving needs, Griffin and colleagues found in a military population that other medical conditions impacted the level of caregiving and strained a marriage.18 Their study found that in a military population between 30% and 50% of marriages of patients with TBI dissolved within the first 10 years after injury. Caregivers may need to learn nursing activities, such as tube feedings, tracheostomy and stoma care, catheter care, wound care, and medication administration. Family stress with caregiving may interfere with the ability to understand information related to the care receivers’ medical care and may require multiple formats to explain care needs. Sander and colleagues associated better emotional functioning in caregivers with greater social integration and occupation outcomes in patients at the postacute rehabilitation program phase (within 6 months of injury).19 However, these outcomes did not continue more than 6 months postinjury.
Intervention and Research Studies
Powell and colleagues used a telephone-based, individualized TBI education intervention along with problem-solving mentoring (10 phone calls at 2-week intervals following patient discharge for moderate-to-severe TBI from a level 1 trauma center) to determine which programs, activities, and coping strategies could decrease caregiver challenges.20 The telephone interventions resulted in better caregiver outcomes than usual care as measured by composite scores on the Bakas Caregiving Outcomes Scale (BCOS) and the Brief Symptom Inventory (BSI-18) at 6 months post-TBI survivor discharge. Dyer and colleagues explored Internet approaches and mobile applications to provide support for caregivers. 18 In a small sample of 10 caregivers, Damianakis and colleagues conducted a 10-session pilot videoconferencing support-group intervention program led by a clinician. Results indicated that the intervention enhanced caregiver coping and problem-solving skills.7
Petranovich and colleagues examined the efficacy of counselor-assisted problem-solving interventions in improving long-term caregiver psychological functioning following TBI in adolescents.21 Their findings support the utility of online interventions in improving long-term caregiver psychological distress, particularly for lower income families. Although this study focused on adolescents, research may indicate merit in an adult population. In relatives of patients with severe TBI, Norup and colleagues associated improvements in health-related quality of life (HRQOL) with improvements in symptoms of anxiety and depression without specific intervention.22
Moriarty and colleagues conducted a randomized controlled trial for veterans who received care at a VA polytrauma center and their family members who participated in a veteran’s in-home program (VIP) intervention.9 The study aimed to evaluate how VIP affected family members’ caregiver burden, depressive symptoms, satisfaction with caregiving, and the program’s acceptability. Eighty-one veterans with a key family member were randomized. Of those, 63 veterans completed a follow-up interview. The intervention consisted of 6 home visits of 1 to 2 hours each and 2 telephone calls from an occupational therapist over 3 to 4 months. Family members were invited to participate during the home visits. The control group received usual clinic care with 2 telephone calls during the study period. All participants received the follow-up interview 3 to 4 months after baseline interviews. The severity of TBI was determined by a review of the electronic medical record using the VA/DoD Clinical Practice Guidelines. Findings of this study indicated that family members in the intervention group showed significantly lower depressive symptom scores and caregiver burden scores.9 Additionally, the veterans in the intervention group exhibited higher community integration and ability to manage their targeted outcomes. Further research may indicate that VIP could assist patients with TBI and caregivers in an active-duty population.
The DVBIC is the executive agent for a congressionally mandated 15-year longitudinal study on TBI incurred by members of the armed services in OEF and OIF. The John Warner National Defense Authorization Act for Fiscal Year 2007 outlined the study. An initial finding identified the need for an HRQOL outcomes assessment specific to TBI caregivers.23 Having these data will allow investigators to fully determine the comprehensive impact of caring for a person who sustained a mild, moderate, severe, or penetrating TBI and to evaluate the effectiveness of interventions designed to address caregivers’ needs. To date, the study has identified the following HRQOL themes generated among caregivers: social health, emotional health, physical/medical health, cognitive functioning, and feelings of loss (related to changing social roles). Carlozzi and colleagues noted that the study also aimed to identify a sensitive outcome measure to evaluate quality of life in the caregivers over time.7
Knowledge Gaps
Ongoing studies focus on caregiving for individuals with various illnesses and needs. Some of the information in each study may be beneficial to TBI caregivers who are not fully aware of resources and interventions. For example, Fortune and colleagues, Hirano and colleagues, and Grover and colleagues are studying caregiver activities involving other diseases to determine, more generally, which programs, activities, and coping strategies can decrease caregiver challenges.24-26 Further, understanding and addressing the needs of these families over many years will provide data that could inform policy, benefits, resources, and needed services (such as the Caregivers and Veterans Omnibus Health Services Act of 2010) and assist with family resilience efforts, including understanding and enhancing family protective and recovery factors.
As studies have indicated, some families do not report family distress when providing care to an individual with TBI. Understanding the factors that influence positive family adjustment is important to capture and perhaps replicate in future studies so that they can lead to effective treatment interventions. Although this review does not discuss caregiver needs for patients with TBI with disorders of consciousness that require more care than most caregivers can provide in the home setting, caregiving for this population deserves attention in future studies. Furthermore, an area that has not received much attention is the impact on children in the household. Children aged < 18 years can assist not only in the care of a disabled adult, but also of younger siblings; also they can help with household activities from housekeeping to meal preparation. Children also may provide physical and emotional support.
The impact of aging caregivers and subsequent needs for their own care as well as the person(s) they are providing care for has not been fully addressed. Areas requiring more research include both the aging caregiver taking care of an aging spouse or relative and the aging parent taking care of a young adult or child. Along with aging, the issue of long-term caregiving needs further development. For example, how do the differences between access to services between caregivers of adults with TBI in the military and those in the civilian sector impact the family/caregiver? Further research may answer questions such as:
- Which tools are most useful in evaluating and determining caregiver stress and burden?
- Are the needs of military and veteran caregivers unique?
- Do polytrauma patients with comorbid diagnoses have unique caregiver needs and trajectories?
- Do TBI caregiver stressors differ from stressors related to other medical conditions or chronic diseases?
- Is there a need for military and veteran TBI-specific caregiver programs?
- Which interventions best help caregivers and for how long?
- Should the approach to intervention depend on variables such as age and gender of the caregivers or relationship to the patient with a TBI (eg, spouse vs parent)?
Methods or processes to inform and update caregivers about available resources also are critically needed. Also, Sabab and colleagues noted the importance of research on the effects of denial as it relates to cognitive, emotional, social impact.27 Denial may impact delays in treatments.
Resource
Many national, state, local, and grassroots organizations provide information and support for persons with illness and/or disabilities. Most clinicians of neurologic, mental health, and cancer have developed various forms of support interventions for those with the disease and their caregivers (Table 2). Highlighted in this section are a few organizations that specifically provide resources for caregivers caring for active-duty service members or veterans with a TBI.
Although a caregiver generally does not receive money from an outside source for services, the DoD may consider the caregiver as a nonmedical attendant for an active-duty service member and provide a temporary stipend. The VA provides several support and service options for caregivers under the Caregiver Support Program, through which more than 300 VA health care professionals provide support to caregivers. The Caregivers and Veterans Omnibus Health Services Act of 2010 authorizes the VA to provide additional VA services for seriously injured post-9/11 veterans and their family caregivers through the Program of Comprehensive Assistance for Family Caregivers (VA Caregiver Support Program). After meeting eligibility criteria, primary caregivers of post-9/11 veterans may receive a monthly stipend (based on the level of care needed) as well as comprehensive caregiver training, referral services, access to health care insurance, mental health services, counseling, and respite care. The Caregiver Support Program offers a toll-free support line and a 24-hour crisis hot line.
In 2014, the Government Accountability Office (GAO) outlined the VA health care improvements needed to manage the demand for the Caregiver Support Program, which are established at VA medical centers.28 The GAO reported that the “VA significantly underestimated caregivers’ demand for services… larger than expected workloads and …delays in approval determinations” with about 500 approved caregivers who are added to the program each month. Original estimates indicated that about 4,000 caregivers would be approved by September 2014; however, by May 2014 about 15,600 caregivers were approved.
In addition to the VA Caregiver Support Program, a variety of state, local, and nonprofit organizations offer support for caregivers. Established in 2012, the Elizabeth Dole Foundation’s program Caring for Military Families “assists caregivers by raising awareness of the caregiver role, leveraging resources and partnerships to provide support, and identifying best practices and solutions to address the challenges caregivers face.” The foundation commissioned the RAND Corporation to “describe the magnitude of military caregiving in the United States, and to identify gaps in programs, policies, and services.” The 2014 RAND report estimated that among the 5.5 million military caregivers in the U.S., 1 million (19.6%) cared for post-9/11 veterans.29 The military caregivers consistently experienced poorer health outcomes, greater strains on family relationships, and more workplace problems than noncaregivers; post-9/11 military caregivers fared worse in those areas.
The Elizabeth Dole Foundation, Hidden Heroes Impact Council Forum advocates for caregiver empowerment, cultural competency awareness, and better policies, programs, and services. The council focuses its efforts on key impact: community support at home, education and training, employment and workplace support, financial and legal issues, interfaith action and ministry council, mental and physical health, and respite care. It aims to raise the money to build awareness and support for military and veterans’ caregivers. The Military and Veteran Caregiver Network is another Elizabeth Dole Foundation initiative. It is an online forum community, peer support group, and peer mentor program structure. A resource library for referrals to local services also is available.
A variety of other organizations, such as United Service Organizations; Easter Seals; Team Red, White and Blue; Operation Homefront; Blue Star Families; state Brain Injury Associations; and support groups for TBI at local hospitals and community centers provide resources to both patients and caregivers. Organizations for caregivers not exclusive to TBI patients include the Caregiver Action Network (formerly National Family Caregiver Association) and the Family Caregiver Alliance. The National Family Caregivers Support Program provides grants to states and territories to develop and provide supportive services to caregivers. Some training for caregivers could include long-term financial planning, legal issues, residential and educational planning, caregiver stress management, the benefits of utilizing support resources, and actions and behaviors that enhance coping strategies. In 2007, DVBIC developed The Traumatic Brain Injury Guide for Caregivers of Service Members and Veterans, which is intended for family caregivers assisting a service member or veteran who sustained a moderate or severe TBI.6 A recent assessment determined the need to update the guide. The Center of Excellence for Medical Multimedia is another source of information for caregivers.
Conclusion
The recent combat conflicts of OEF and OIF have resulted in a dramatic increase in the occurrence of TBI injuries in active-duty service members both in theater and stateside and have highlighted the need for some service members and veterans with a TBI to require ongoing assistance from a caregiver. The levels of assistance and length of time vary greatly, impacted by the severity of the TBI and psychosocial situations.
In response to elevated awareness, several programs and resources have been developed or enhanced to address the specific needs of caregivers. Certain programs and resources are specific for caregivers of military service members and veterans, whereas others benefit caregivers in general. Likewise, some programs are not specific to individuals with TBI.
Caregivers assume many roles in their efforts to support the person with a TBI. They may need to dramatically adjust their lives to serve as a caregiver. Providing adequate resources for the caregivers impacts their ability to continue providing care. Thus, awareness of and access to resources play a critical role in helping to reduce stress, distress, burden (eg, physical, emotional, and financial), and caregiver burnout. Programs and resources often change, making it difficult for health care practitioners to know which programs offer what or even whether they still exist. Therefore, the authors synthesized the current medical literature of the topic of TBI and their caregiver needs as well as current resources for additional information and support.
Ongoing research studies, such as the congressionally mandated 15-year longitudinal study, are examining the impact of caregiving in the military and veteran communities. Future research could identify specific needs of military caregivers, identify gaps in services or programs, and identify interventions that promote resilience. Moreover, research directed at military and veteran caregivers can promote change that will benefit the general population of caregivers. It will be important for health care practitioners to keep abreast of new findings and information to incorporate into care plans for their patients who have had a TBI and their families.
Traumatic brain injury (TBI) is a health concern for the U.S. Military Health System (MHS) as well as the VHA. It occurs in both deployed and nondeployed settings; however, Operation Enduring Freedom (OEF) and Operation Iraqi Freedom (OIF) and improved reporting mechanisms have dramatically increased TBI diagnoses in active-duty service members. According to the Defense and Veterans Brain Injury Center (DVBIC), more than 370,000 service members have been diagnosed with a TBI since 2000 (Figure).1
Background
The DoD and the VA are collaborating on clinical research studies to identify, understand, and treat the long-term effects of TBI that can affect patients and their families. Most TBIs are mild (mTBIs), also called concussions, and patients typically recover within a few weeks (Table 1). However, some individuals with mTBI experience symptoms that may persist for months or years. A meta-analysis by Perry and colleagues showed that the prevalence or risk of a neurologic disorder, depression, or other mental health issue following mTBI was 67% higher compared with that in uninjured controls.2
Patients with any severity of TBI may require assistance with activities of daily living (ADLs), such as bathing, dressing, managing medications, and feeding. Patients also may need help with instrumental ADLs, such as meal preparation, grocery shopping, household chores, child care, getting to appointments or activities, coordination of educational and vocational services, financial and benefits management, and supportive listening.
Increased injuries have spurred the DoD and VA to coordinate health care to provide a seamless transition for patients between the 2 agencies. However, individuals who sustained a TBI may need various levels of caregiver assistance over time.
TBI and Caregivers
Despite better agency coordination for patients, caregivers can experience stress. Griffin and colleagues found that caregiving responsibilities can compete with other demands on the caregiver, such as work and family, and may negatively impact their health and finances.3,4
Lou and colleagues studied the factors associated with caring for chronically ill family members that may result in stress for the caregivers.5 Along with an unaccounted for economic contribution, caregivers may face lost work time and pay and limitations on work travel and work advancement. Additionally, lost time for leisure, travel, social activities, family obligations, and retirement could result in physical and mental drain on the caregiver. Stress may reach a level at which the caregivers risk psychological distress. The study also noted that families with perceived high stress experience disrupted family functioning. Some TBI caregiver studies sought to understand how best to evaluate and determine the level of caregiver burden, and other studies investigated appropriate interventions.6-9
Health care practitioners within the federal health care system may benefit from a greater awareness of caregiver needs and caregiver resources. Caregiver support can improve outcomes for both the caregiver and care recipient, and many organizations and resources already exist to assist the caregiver. This article reviews recent published literature on TBI caregivers of patients with TBI across civilian, military, and veteran populations and lists caregiver resources for additional information, assistance, and support.
Literature Review
The DVBIC defines the term caregiver as “any family or support person(s) relied upon by the service member or veteran with traumatic brain injury (TBI) who assumes primary responsibility for ensuring the needed level of care and overall well-being of that service member or veteran. A family or family caregiver may include spouse, parents, children, other extended family members, as well as significant others and friends.”3
In the following discussion, findings from military and veteran literature are separated from civilian population findings to highlight similarities and differences between these 2 bodies of research. Several of the studies in the military/veteran cohorts include polytrauma patients with comorbid physical and mental health issues not necessarily found in civilian literature.
Civilian Literature
A 2015 systematic review by Anderson and colleagues on coping and psychological adjustment in TBI caregivers indicated no Class I or Class II studies.10 Four Class III and 3 Class IV studies were found. The authors suggest that more rigorous studies (ie, Class I and II) are needed.
Despite these limitations, peer-reviewed literature indicates that the levels of stress and distress in TBI caregivers are consistent with reports for other diseases. In a civilian population, Carlozzi and colleagues found that TBI caregivers who reported stress, distress, anxiety, and feeling overwhelmed often had concerns for their social, emotional, physical, cognitive health, as well as feelings of loss.5 In addition, caregivers may need to take leaves of absence or leave the workforce entirely to provide for a family member or friend who had a TBI—often leading to financial strain (eg, depleting assets, accumulating debt). These challenges may occur during prime earning years, and the caregiver may lose the ability to resume work if the care receiver requires care for extended periods.11
Kratz and colleagues showed that caregivers of individuals with moderate-to-severe TBI: (1) felt overburdened with responsibilities; (2) lacked personal time and time for self-care; (3) felt their lives were interrupted or lost; (4) grieved the loss of the person with TBI; and (5) endorsed anger, guilt, anxiety, and sadness.12
Perceptions differed between caregiver parents and caregiver partners. Parents expressed feelings of grief and sadness related to the “loss of the person before the TBI.” Parents also reported a sense of guilt and responsibility for their child’s TBI and feelings of being tied down to the individual with TBI. Parents experienced a greater level of stress if the son or daughter with TBI still lived at home. Partners expressed frustration and despair related to their role as sole decision maker and care provider. Partners’ distress also related to the partner relationship and the relationship between children and the individual with TBI.
Verhasghe and colleagues found that partners experience a greater degree of stress than do parents.13 Young families with minimal social support for coping with financial, psychiatric, and medical problems were the most vulnerable to stress. A systematic review by Ennis and colleagues evaluated depression and anxiety in caregiver parents vs spouses.14 Although methods and quality differed in the studies, findings indicated high levels of distress regardless of the type of caregiver.
Anderson and colleagues used the Ways of Coping Questionnaire to evaluate the association between coping and psychological adjustment in caregivers of TBI individuals.10 The use of emotion-focused coping and problem solving was possibly associated with psychological adjustment in caregivers. Verhasghe and colleagues indicated that the nature of the injuries more than the severity of TBI determined the level of stress up to 15 years after the TBI.13 Gender and social and professional support also influenced coping. The review identified the need to develop models of long-term support and care.
An Australian cohort of 79 family caregivers participated in a study by Perlesz and colleagues.15 Participants’ caregiving responsibilities averaged 19.3 months posttrauma. The Family Satisfaction Scale, Beck Depression Inventory, State Anxiety Inventory, and Profile of Mood States were used in this analysis. Male caregivers reported distress in terms of anger and fatigue; female caregivers were at greatest risk of poor psychosocial outcomes. Although findings from primary caregivers indicated that 35% to 49% displayed enough distress to warrant clinical intervention, between 51% and 80% were not psychologically distressed and were satisfied with their families. Data supported previous reports suggesting caregivers are “not universally distressed.”15
Manskow and colleagues followed patients with severe TBI and assessed caregiver burden 1 year later. Using the Caregiver Burden Scale, caregivers reported the highest scores (N = 92) on the General Strain Index followed by the Disappointment Index.16 Bayen and colleagues also studied caregivers of severe TBI patients.17 Objective and subjective caregiver burden data 4 years later indicated 44% of caregivers (N = 98) reported multidimensional burden. Greater burden was associated in caring for individuals who had poorer Glasgow Outcome Scale Extended scores and more severe cognitive disorders.
Military and Veteran Literature
Griffin and colleagues conducted the Family and Caregiver Experience Study (FACES) with caregivers (N = 564) of service members who incurred a TBI.3 According to the caregivers, two-thirds of the patients lost consciousness for more than 30 minutes, which was followed by inpatient rehabilitation care at a VA polytrauma center between 2001 and 2009. The majority of caregivers of TBI patients were female (79%) and aged < 60 years (84%). Parents comprised 62% and spouses 32% of the cohort. Caregivers tended to have some level of education beyond high school (73%), were married (77%), either worked or were enrolled in school (55%), and earned less than $40,000 a year (70%). Common characteristics of the care receivers were male gender (95%), average age 30, high school educated (52%), married (almost 50%), and employed (50%). Forty-five percent of the care receivers were injured 4 to 6 years prior, and 12% were injured 7 or more years prior. The study determined the caregivers’ perception of intensity of care needed and indicated that families as well as clinicians need to plan for some level of long-term support and services.
In addition to the TBI-related caregiving needs, Griffin and colleagues found in a military population that other medical conditions impacted the level of caregiving and strained a marriage.18 Their study found that in a military population between 30% and 50% of marriages of patients with TBI dissolved within the first 10 years after injury. Caregivers may need to learn nursing activities, such as tube feedings, tracheostomy and stoma care, catheter care, wound care, and medication administration. Family stress with caregiving may interfere with the ability to understand information related to the care receivers’ medical care and may require multiple formats to explain care needs. Sander and colleagues associated better emotional functioning in caregivers with greater social integration and occupation outcomes in patients at the postacute rehabilitation program phase (within 6 months of injury).19 However, these outcomes did not continue more than 6 months postinjury.
Intervention and Research Studies
Powell and colleagues used a telephone-based, individualized TBI education intervention along with problem-solving mentoring (10 phone calls at 2-week intervals following patient discharge for moderate-to-severe TBI from a level 1 trauma center) to determine which programs, activities, and coping strategies could decrease caregiver challenges.20 The telephone interventions resulted in better caregiver outcomes than usual care as measured by composite scores on the Bakas Caregiving Outcomes Scale (BCOS) and the Brief Symptom Inventory (BSI-18) at 6 months post-TBI survivor discharge. Dyer and colleagues explored Internet approaches and mobile applications to provide support for caregivers. 18 In a small sample of 10 caregivers, Damianakis and colleagues conducted a 10-session pilot videoconferencing support-group intervention program led by a clinician. Results indicated that the intervention enhanced caregiver coping and problem-solving skills.7
Petranovich and colleagues examined the efficacy of counselor-assisted problem-solving interventions in improving long-term caregiver psychological functioning following TBI in adolescents.21 Their findings support the utility of online interventions in improving long-term caregiver psychological distress, particularly for lower income families. Although this study focused on adolescents, research may indicate merit in an adult population. In relatives of patients with severe TBI, Norup and colleagues associated improvements in health-related quality of life (HRQOL) with improvements in symptoms of anxiety and depression without specific intervention.22
Moriarty and colleagues conducted a randomized controlled trial for veterans who received care at a VA polytrauma center and their family members who participated in a veteran’s in-home program (VIP) intervention.9 The study aimed to evaluate how VIP affected family members’ caregiver burden, depressive symptoms, satisfaction with caregiving, and the program’s acceptability. Eighty-one veterans with a key family member were randomized. Of those, 63 veterans completed a follow-up interview. The intervention consisted of 6 home visits of 1 to 2 hours each and 2 telephone calls from an occupational therapist over 3 to 4 months. Family members were invited to participate during the home visits. The control group received usual clinic care with 2 telephone calls during the study period. All participants received the follow-up interview 3 to 4 months after baseline interviews. The severity of TBI was determined by a review of the electronic medical record using the VA/DoD Clinical Practice Guidelines. Findings of this study indicated that family members in the intervention group showed significantly lower depressive symptom scores and caregiver burden scores.9 Additionally, the veterans in the intervention group exhibited higher community integration and ability to manage their targeted outcomes. Further research may indicate that VIP could assist patients with TBI and caregivers in an active-duty population.
The DVBIC is the executive agent for a congressionally mandated 15-year longitudinal study on TBI incurred by members of the armed services in OEF and OIF. The John Warner National Defense Authorization Act for Fiscal Year 2007 outlined the study. An initial finding identified the need for an HRQOL outcomes assessment specific to TBI caregivers.23 Having these data will allow investigators to fully determine the comprehensive impact of caring for a person who sustained a mild, moderate, severe, or penetrating TBI and to evaluate the effectiveness of interventions designed to address caregivers’ needs. To date, the study has identified the following HRQOL themes generated among caregivers: social health, emotional health, physical/medical health, cognitive functioning, and feelings of loss (related to changing social roles). Carlozzi and colleagues noted that the study also aimed to identify a sensitive outcome measure to evaluate quality of life in the caregivers over time.7
Knowledge Gaps
Ongoing studies focus on caregiving for individuals with various illnesses and needs. Some of the information in each study may be beneficial to TBI caregivers who are not fully aware of resources and interventions. For example, Fortune and colleagues, Hirano and colleagues, and Grover and colleagues are studying caregiver activities involving other diseases to determine, more generally, which programs, activities, and coping strategies can decrease caregiver challenges.24-26 Further, understanding and addressing the needs of these families over many years will provide data that could inform policy, benefits, resources, and needed services (such as the Caregivers and Veterans Omnibus Health Services Act of 2010) and assist with family resilience efforts, including understanding and enhancing family protective and recovery factors.
As studies have indicated, some families do not report family distress when providing care to an individual with TBI. Understanding the factors that influence positive family adjustment is important to capture and perhaps replicate in future studies so that they can lead to effective treatment interventions. Although this review does not discuss caregiver needs for patients with TBI with disorders of consciousness that require more care than most caregivers can provide in the home setting, caregiving for this population deserves attention in future studies. Furthermore, an area that has not received much attention is the impact on children in the household. Children aged < 18 years can assist not only in the care of a disabled adult, but also of younger siblings; also they can help with household activities from housekeeping to meal preparation. Children also may provide physical and emotional support.
The impact of aging caregivers and subsequent needs for their own care as well as the person(s) they are providing care for has not been fully addressed. Areas requiring more research include both the aging caregiver taking care of an aging spouse or relative and the aging parent taking care of a young adult or child. Along with aging, the issue of long-term caregiving needs further development. For example, how do the differences between access to services between caregivers of adults with TBI in the military and those in the civilian sector impact the family/caregiver? Further research may answer questions such as:
- Which tools are most useful in evaluating and determining caregiver stress and burden?
- Are the needs of military and veteran caregivers unique?
- Do polytrauma patients with comorbid diagnoses have unique caregiver needs and trajectories?
- Do TBI caregiver stressors differ from stressors related to other medical conditions or chronic diseases?
- Is there a need for military and veteran TBI-specific caregiver programs?
- Which interventions best help caregivers and for how long?
- Should the approach to intervention depend on variables such as age and gender of the caregivers or relationship to the patient with a TBI (eg, spouse vs parent)?
Methods or processes to inform and update caregivers about available resources also are critically needed. Also, Sabab and colleagues noted the importance of research on the effects of denial as it relates to cognitive, emotional, social impact.27 Denial may impact delays in treatments.
Resource
Many national, state, local, and grassroots organizations provide information and support for persons with illness and/or disabilities. Most clinicians of neurologic, mental health, and cancer have developed various forms of support interventions for those with the disease and their caregivers (Table 2). Highlighted in this section are a few organizations that specifically provide resources for caregivers caring for active-duty service members or veterans with a TBI.
Although a caregiver generally does not receive money from an outside source for services, the DoD may consider the caregiver as a nonmedical attendant for an active-duty service member and provide a temporary stipend. The VA provides several support and service options for caregivers under the Caregiver Support Program, through which more than 300 VA health care professionals provide support to caregivers. The Caregivers and Veterans Omnibus Health Services Act of 2010 authorizes the VA to provide additional VA services for seriously injured post-9/11 veterans and their family caregivers through the Program of Comprehensive Assistance for Family Caregivers (VA Caregiver Support Program). After meeting eligibility criteria, primary caregivers of post-9/11 veterans may receive a monthly stipend (based on the level of care needed) as well as comprehensive caregiver training, referral services, access to health care insurance, mental health services, counseling, and respite care. The Caregiver Support Program offers a toll-free support line and a 24-hour crisis hot line.
In 2014, the Government Accountability Office (GAO) outlined the VA health care improvements needed to manage the demand for the Caregiver Support Program, which are established at VA medical centers.28 The GAO reported that the “VA significantly underestimated caregivers’ demand for services… larger than expected workloads and …delays in approval determinations” with about 500 approved caregivers who are added to the program each month. Original estimates indicated that about 4,000 caregivers would be approved by September 2014; however, by May 2014 about 15,600 caregivers were approved.
In addition to the VA Caregiver Support Program, a variety of state, local, and nonprofit organizations offer support for caregivers. Established in 2012, the Elizabeth Dole Foundation’s program Caring for Military Families “assists caregivers by raising awareness of the caregiver role, leveraging resources and partnerships to provide support, and identifying best practices and solutions to address the challenges caregivers face.” The foundation commissioned the RAND Corporation to “describe the magnitude of military caregiving in the United States, and to identify gaps in programs, policies, and services.” The 2014 RAND report estimated that among the 5.5 million military caregivers in the U.S., 1 million (19.6%) cared for post-9/11 veterans.29 The military caregivers consistently experienced poorer health outcomes, greater strains on family relationships, and more workplace problems than noncaregivers; post-9/11 military caregivers fared worse in those areas.
The Elizabeth Dole Foundation, Hidden Heroes Impact Council Forum advocates for caregiver empowerment, cultural competency awareness, and better policies, programs, and services. The council focuses its efforts on key impact: community support at home, education and training, employment and workplace support, financial and legal issues, interfaith action and ministry council, mental and physical health, and respite care. It aims to raise the money to build awareness and support for military and veterans’ caregivers. The Military and Veteran Caregiver Network is another Elizabeth Dole Foundation initiative. It is an online forum community, peer support group, and peer mentor program structure. A resource library for referrals to local services also is available.
A variety of other organizations, such as United Service Organizations; Easter Seals; Team Red, White and Blue; Operation Homefront; Blue Star Families; state Brain Injury Associations; and support groups for TBI at local hospitals and community centers provide resources to both patients and caregivers. Organizations for caregivers not exclusive to TBI patients include the Caregiver Action Network (formerly National Family Caregiver Association) and the Family Caregiver Alliance. The National Family Caregivers Support Program provides grants to states and territories to develop and provide supportive services to caregivers. Some training for caregivers could include long-term financial planning, legal issues, residential and educational planning, caregiver stress management, the benefits of utilizing support resources, and actions and behaviors that enhance coping strategies. In 2007, DVBIC developed The Traumatic Brain Injury Guide for Caregivers of Service Members and Veterans, which is intended for family caregivers assisting a service member or veteran who sustained a moderate or severe TBI.6 A recent assessment determined the need to update the guide. The Center of Excellence for Medical Multimedia is another source of information for caregivers.
Conclusion
The recent combat conflicts of OEF and OIF have resulted in a dramatic increase in the occurrence of TBI injuries in active-duty service members both in theater and stateside and have highlighted the need for some service members and veterans with a TBI to require ongoing assistance from a caregiver. The levels of assistance and length of time vary greatly, impacted by the severity of the TBI and psychosocial situations.
In response to elevated awareness, several programs and resources have been developed or enhanced to address the specific needs of caregivers. Certain programs and resources are specific for caregivers of military service members and veterans, whereas others benefit caregivers in general. Likewise, some programs are not specific to individuals with TBI.
Caregivers assume many roles in their efforts to support the person with a TBI. They may need to dramatically adjust their lives to serve as a caregiver. Providing adequate resources for the caregivers impacts their ability to continue providing care. Thus, awareness of and access to resources play a critical role in helping to reduce stress, distress, burden (eg, physical, emotional, and financial), and caregiver burnout. Programs and resources often change, making it difficult for health care practitioners to know which programs offer what or even whether they still exist. Therefore, the authors synthesized the current medical literature of the topic of TBI and their caregiver needs as well as current resources for additional information and support.
Ongoing research studies, such as the congressionally mandated 15-year longitudinal study, are examining the impact of caregiving in the military and veteran communities. Future research could identify specific needs of military caregivers, identify gaps in services or programs, and identify interventions that promote resilience. Moreover, research directed at military and veteran caregivers can promote change that will benefit the general population of caregivers. It will be important for health care practitioners to keep abreast of new findings and information to incorporate into care plans for their patients who have had a TBI and their families.
1. Defense and Veterans Brain Injury Center. DoD worldwide numbers for TBI. http://dvbic.dcoe.mil/dod-worldwide-numbers-tbi. Updated October 5, 2017. Accessed October 10, 2017.
2. Perry DC, Sturm VE, Peterson MJ, et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg. 2016;124(2):511-526.
3. Defense and Veterans Brain Injury Center. Traumatic brain injury: a guide for caregivers of service members and veterans. https://dvbic.dcoe.mil /sites/default/files/Family%20Caregiver%20Guide.All%20Modules_updated.pdf. Accessed October 10, 2017
4. Griffin JM, Friedemann-Sánchez G, Jensen AC, et al. The invisible side of war: families caring for US service members with traumatic brain injuries and polytrauma. J Head Trauma Rehabil. 2012;27(1):3-13.
5. Lou VW, Kwan CW, Chong ML, Chi I. Associations between secondary caregivers’ supportive behavior and psychological distress of primary spousal caregivers of cognitively intact and impaired elders. Gerontologist. 2015;55(4):584-594.
6. Carlozzi NE, Kratz AL, Sander AM, et al. Health-related quality of life in caregivers of individuals with traumatic brain injury: development of a conceptual model. Arch Phys Med Rehabil. 2015;96(1):105-113.
7. Damianakis T, Tough A, Marziali E, Dawson DR. Therapy online: a web-based video support group for family caregivers of survivors with traumatic brain injury. J Head Trauma Rehabil. 2016;31(4):E12-E20.
8. Dyer EA, Kansagara D, McInnes DK, Freeman M, Woods, S. Mobile applications and internet-based approaches for supporting non-professional caregivers: a systematic review. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0050675. Accessed October 10, 2017.
9. Moriarty H, Winter L, Robinson K, et al. A randomized controlled trial to evaluate the veterans’ in-home program for military veterans with traumatic brain injury and their families: report on impact for family members. PMR. 2016;8(6):495-509.
10. Anderson MI, Simpson GK, Daher M, Matheson L. Chapter 7 the relationship between coping and psychological adjustment in family caregivers of individuals with traumatic brain injury: a systematic review. Annu Rev Nurs Res. 2015;33:219-247.
11. Van Houtven CH, Friedemann-Sanchez G, Clothier B, et al. Is policy well-targeted to remedy financial strain among caregivers of severely injured U.S. service members? Inquiry. 2012-2013;49(4):339-351.
12. Kratz AL, Sander AM, Brickell TA, Lange RT, Carlozzi NE. Traumatic brain injury caregivers: a qualitative analysis of spouse and parent perspectives on quality of life. Neuropsychol Rehabil. 2017;27(1):16-37.13. Verhasghe S, Defloor T, Grypdonck M. Stress and coping among families of patients with traumatic brain injury: a review of the literature. J Clin Nurs. 2005;14(8):1004-1012.
14. Ennis N, Rosenbloom BN, Canzian S, Topolovec-Vranic J. Depression and anxiety in parent versus spouse caregivers of adult patients with traumatic brain injury: a systematic review. Neuropsychol Rehabil. 2013;23(1):1-18.
15. Perlesz A, Kinsella G, Crowe S. Psychological distress and family satisfaction following traumatic brain injury: injured individuals and their primary, secondary, and tertiary carers. J Head Trauma Rehabil. 2000;15(3):909-929.
16. Manskow US, Sigurdardottir S, Røe C, et al. Factors affecting caregiving burden 1 year after severe traumatic brain injury: a prospective nationwide multicenter study. J Head Trauma Rehabil. 2015;30(6):411-423.
17. Bayen E, Jourdan C, Ghout I, et al. Objective and subjective burden of informal caregivers 4 years after a severe traumatic brain injury: results from the Paris-TBI study. J Head Trauma Rehabil. 2016;31(5):E59-E67.
18. Griffin JM, Friedemann-Sanchez G, Hall C, Phelan S, van Ryn M. Families of patients with polytrauma: understanding the evidence and charting a new research agenda. J Rehabil Res Dev. 2009;46(6):879-892.
19. Sander AM, Maestas KL, Sherer M, Malac JF, Nakase-Richardson R. Relationship of caregiver and family functioning to participation outcomes after post-acute rehabilitation for traumatic brain injury: a multicenter investigation. Arch Phys Med Rehabil. 2012;93(5):842-848.
20. Powell JM, Fraser R, Brockway JA, Temkin N, Bell KR. A telehealth approach to caregiver self-management following traumatic brain injury: a randomized control trial. J Head Trauma Rehabil. 2015;31(3):180-190.
21. Petranovich CL, Wade SL, Taylor HG, et al. Long-term caregiver mental health outcomes following a predominately online intervention for adolescents with complicated mild to severe traumatic brain injury. J Pediatr Psychol, 2015;40(7):680-688.
22. Norup A, Kristensen KS, Poulsen I, Mortensen EL. Evaluating clinically significant changes in health-related quality of life: a sample of relatives of patients with severe traumatic brain injury. Neuropsychol Rehabil. 2017;27(2):196-215.
23. John Warner National Defense Authorization Act for Fiscal Year 2007, HR 5122, 109th Cong, 2nd Sess (2006).
24. Fortune DG, Rogan CR, Richards HL. A structured multicomponent group program for carers of people with acquired brain injury: effects on perceived criticism, strain, and psychological distress. Br J Health Psychol. 2016;21(1):224-243.
25. Hirano A, Umegaki H, Suzuki Y, Hayashi T, Kuzuya M. Effects of leisure activities at home on perceived care burden and the endocrine system of caregivers of dementia patients: a randomized controlled study. Int Psychogeriatr. 2016;28(2):261-268.
26. Grover S, Pradyumna, Chakrabarti S. Coping among caregivers of patients with schizophrenia. Ind Psychiatry J. 2015;24(1):5-11.
27. Saban KL, Hogan NS, Hogan TP, Pape TL. He looks normal but…challenges of family caregivers of veterans diagnosed with a traumatic brain injury. Rehabil Nurs. 2015;40(5):277-285.
28. Williamson RB; United States Government Accountability Office. VA health care improvements needed to manage higher-than-expected demand for the family caregiver program. http://www.gao.gov/assets/670/667275.pdf. Published December 3, 2014. Accessed October 10, 2017.
29. Ramchand R, Tanielian T, Fisher MP, et al. Hidden heroes America’s military caregivers. http://www.rand.org/content/dam/rand/pubs/research_reports/RR400/RR499/RAND_RR499.pdf. Published 2014. Accessed October 10, 2017.
1. Defense and Veterans Brain Injury Center. DoD worldwide numbers for TBI. http://dvbic.dcoe.mil/dod-worldwide-numbers-tbi. Updated October 5, 2017. Accessed October 10, 2017.
2. Perry DC, Sturm VE, Peterson MJ, et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg. 2016;124(2):511-526.
3. Defense and Veterans Brain Injury Center. Traumatic brain injury: a guide for caregivers of service members and veterans. https://dvbic.dcoe.mil /sites/default/files/Family%20Caregiver%20Guide.All%20Modules_updated.pdf. Accessed October 10, 2017
4. Griffin JM, Friedemann-Sánchez G, Jensen AC, et al. The invisible side of war: families caring for US service members with traumatic brain injuries and polytrauma. J Head Trauma Rehabil. 2012;27(1):3-13.
5. Lou VW, Kwan CW, Chong ML, Chi I. Associations between secondary caregivers’ supportive behavior and psychological distress of primary spousal caregivers of cognitively intact and impaired elders. Gerontologist. 2015;55(4):584-594.
6. Carlozzi NE, Kratz AL, Sander AM, et al. Health-related quality of life in caregivers of individuals with traumatic brain injury: development of a conceptual model. Arch Phys Med Rehabil. 2015;96(1):105-113.
7. Damianakis T, Tough A, Marziali E, Dawson DR. Therapy online: a web-based video support group for family caregivers of survivors with traumatic brain injury. J Head Trauma Rehabil. 2016;31(4):E12-E20.
8. Dyer EA, Kansagara D, McInnes DK, Freeman M, Woods, S. Mobile applications and internet-based approaches for supporting non-professional caregivers: a systematic review. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0050675. Accessed October 10, 2017.
9. Moriarty H, Winter L, Robinson K, et al. A randomized controlled trial to evaluate the veterans’ in-home program for military veterans with traumatic brain injury and their families: report on impact for family members. PMR. 2016;8(6):495-509.
10. Anderson MI, Simpson GK, Daher M, Matheson L. Chapter 7 the relationship between coping and psychological adjustment in family caregivers of individuals with traumatic brain injury: a systematic review. Annu Rev Nurs Res. 2015;33:219-247.
11. Van Houtven CH, Friedemann-Sanchez G, Clothier B, et al. Is policy well-targeted to remedy financial strain among caregivers of severely injured U.S. service members? Inquiry. 2012-2013;49(4):339-351.
12. Kratz AL, Sander AM, Brickell TA, Lange RT, Carlozzi NE. Traumatic brain injury caregivers: a qualitative analysis of spouse and parent perspectives on quality of life. Neuropsychol Rehabil. 2017;27(1):16-37.13. Verhasghe S, Defloor T, Grypdonck M. Stress and coping among families of patients with traumatic brain injury: a review of the literature. J Clin Nurs. 2005;14(8):1004-1012.
14. Ennis N, Rosenbloom BN, Canzian S, Topolovec-Vranic J. Depression and anxiety in parent versus spouse caregivers of adult patients with traumatic brain injury: a systematic review. Neuropsychol Rehabil. 2013;23(1):1-18.
15. Perlesz A, Kinsella G, Crowe S. Psychological distress and family satisfaction following traumatic brain injury: injured individuals and their primary, secondary, and tertiary carers. J Head Trauma Rehabil. 2000;15(3):909-929.
16. Manskow US, Sigurdardottir S, Røe C, et al. Factors affecting caregiving burden 1 year after severe traumatic brain injury: a prospective nationwide multicenter study. J Head Trauma Rehabil. 2015;30(6):411-423.
17. Bayen E, Jourdan C, Ghout I, et al. Objective and subjective burden of informal caregivers 4 years after a severe traumatic brain injury: results from the Paris-TBI study. J Head Trauma Rehabil. 2016;31(5):E59-E67.
18. Griffin JM, Friedemann-Sanchez G, Hall C, Phelan S, van Ryn M. Families of patients with polytrauma: understanding the evidence and charting a new research agenda. J Rehabil Res Dev. 2009;46(6):879-892.
19. Sander AM, Maestas KL, Sherer M, Malac JF, Nakase-Richardson R. Relationship of caregiver and family functioning to participation outcomes after post-acute rehabilitation for traumatic brain injury: a multicenter investigation. Arch Phys Med Rehabil. 2012;93(5):842-848.
20. Powell JM, Fraser R, Brockway JA, Temkin N, Bell KR. A telehealth approach to caregiver self-management following traumatic brain injury: a randomized control trial. J Head Trauma Rehabil. 2015;31(3):180-190.
21. Petranovich CL, Wade SL, Taylor HG, et al. Long-term caregiver mental health outcomes following a predominately online intervention for adolescents with complicated mild to severe traumatic brain injury. J Pediatr Psychol, 2015;40(7):680-688.
22. Norup A, Kristensen KS, Poulsen I, Mortensen EL. Evaluating clinically significant changes in health-related quality of life: a sample of relatives of patients with severe traumatic brain injury. Neuropsychol Rehabil. 2017;27(2):196-215.
23. John Warner National Defense Authorization Act for Fiscal Year 2007, HR 5122, 109th Cong, 2nd Sess (2006).
24. Fortune DG, Rogan CR, Richards HL. A structured multicomponent group program for carers of people with acquired brain injury: effects on perceived criticism, strain, and psychological distress. Br J Health Psychol. 2016;21(1):224-243.
25. Hirano A, Umegaki H, Suzuki Y, Hayashi T, Kuzuya M. Effects of leisure activities at home on perceived care burden and the endocrine system of caregivers of dementia patients: a randomized controlled study. Int Psychogeriatr. 2016;28(2):261-268.
26. Grover S, Pradyumna, Chakrabarti S. Coping among caregivers of patients with schizophrenia. Ind Psychiatry J. 2015;24(1):5-11.
27. Saban KL, Hogan NS, Hogan TP, Pape TL. He looks normal but…challenges of family caregivers of veterans diagnosed with a traumatic brain injury. Rehabil Nurs. 2015;40(5):277-285.
28. Williamson RB; United States Government Accountability Office. VA health care improvements needed to manage higher-than-expected demand for the family caregiver program. http://www.gao.gov/assets/670/667275.pdf. Published December 3, 2014. Accessed October 10, 2017.
29. Ramchand R, Tanielian T, Fisher MP, et al. Hidden heroes America’s military caregivers. http://www.rand.org/content/dam/rand/pubs/research_reports/RR400/RR499/RAND_RR499.pdf. Published 2014. Accessed October 10, 2017.
bb2121 induces durable, deepening responses in MM patients

ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121. ![]()
*Data presented differ from the abstract.
ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121. ![]()
*Data presented differ from the abstract.
ATLANTA—Updated results from a phase 1 trial have shown that bb2121, a chimeric antigen receptor (CAR) T-cell product, can induce durable, deepening responses in patients with relapsed/refractory multiple myeloma (MM).
Responses continue to improve from very good partial responses to complete responses (CRs), even 15 months after infusion.
In 5 months, the CR rate increased from 27% to 56%, and ongoing responses have now surpassed 1 year.
The overall response rate (ORR) stands at 94%, and the median progression-free survival (PFS) has not been reached with a follow-up of 40 weeks.
bb2121 is a second-generation CAR T-cell product that targets the B-cell maturation antigen (BCMA).
BCMA is expressed nearly universally on MM cells, and its expression is largely restricted to plasma cells and some mature B cells, making it “an attractive target for immunotherapies,” said James N. Kochenderfer, MD, of the National Cancer Institute/National Institutes of Health in Bethesda, Maryland.
Dr Kochenderfer reported results from the phase 1 study of bb2121 (NCT02658929) at the 2017 ASH Annual Meeting (abstract 740*).
Study sponsors and collaborators were bluebird bio and Celgene Corporation. Dr Kochenderfer disclosed that he has multiple patents in the CAR field and has received research funding from bluebird bio and Kite Pharma.
Study design
Patients with relapsed or refractory MM who had 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug, or who were double refractory were eligible for the dose-escalation cohort of the study. They had to have measurable disease, 50% or more BCMA expression, and adequate bone marrow, renal, and hepatic function.
BCMA expression was not required for the dose-expansion cohort. For this cohort, patients must have received daratumumab and have been refractory to their last line of therapy.
The dose-escalation cohort was a standard 3 + 3 design and included CAR T-cell doses of 50 x 106, 150 x 106, 450 x 106, and 800 x 106.
Patients were screened, underwent leukapheresis, and waited for the manufacture of their CAR T cells. One centralized manufacturing site produced the T-cell products for the 9 US clinical study sites.
“We had a manufacturing success rate of 100%,” Dr Kochenderfer noted, and the manufacturing took 10 days.
Five days prior to bb2121 infusion, patients received lymphodepletion with fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2).
Patient characteristics
Investigators dosed 21 patients as of the data cut-off of October 2.
Their median age was 58 (range, 37–74), 62% were male, and they had a median time since diagnosis of 4 years.
All patients had an ECOG performance status of 0 or 1, and 43% had high-risk cytogenetics, defined as del17p, t(4;14), and t(14;16).
“One of the most impressive things about our study was how heavily pretreated the patients were,” Dr Kochenderfer noted. “These patients had a median of 7 prior lines of therapy, and 100% of the patients had a prior autologous stem cell transplant.”
All patients were exposed to bortezomib and lenalidomide, and 67% and 86%, respectively, were refractory to those agents. Patients were also exposed to carfilzomib (91%), pomalidomide (91%), and daratumumab (71%) and had varying degrees of refractoriness to those agents.
Safety
“In general, the treatment was very well tolerated,” Dr Kochenderfer said. “[It was] well tolerated compared with other T-cell products I’ve had experience with.”
The investigators observed no dose-limiting toxicities.
Cytokine release syndrome (CRS) of all grades occurred in 15 (71%) patients, and grade 3 or higher CRS occurred in 2 (10%) patients. The latter resolved within 24 hours.
Five (24%) patients experienced neurotoxicity, none grade 3 or higher.
Dr Kochenderfer described 1 case of delayed-onset, grade 4, reversible neurotoxicity that was associated with tumor lysis syndrome (TLS) and CRS.
The patient had no toxicity until day 10. By day 12, magnetic resonance imaging showed cerebral edema.
The patient was transferred to the intensive care unit and required intubation. She was treated with high-dose methylprednisolone and tocilizumab. She also received hemodialysis for TLS.
“By day 17, she dramatically improved,” Dr Kochenderfer said.
Her mental status cleared, TLS resolved, she was extubated, and she was doing much better, he reported.
On day 30, the patient was out of the intensive care unit.
“So the whole course was fairly brief,” Dr Kochenderfer said. “And, today, she’s doing well. She’s actually asymptomatic.”
Cytopenias—neutropenia, thrombocytopenia, and anemia—were primarily related to the lymphodepleting drugs, and patients recovered to grade 3 or lower by month 2 after the infusion.
Fourteen patients experienced 1 or more serious adverse events. Four had grade 1-2 CRS that required hospitalization per protocol, and 2 had pyrexia.
Five patients died, 3 due to disease progression, all who received treatment at the lowest dose.
Two patients treated at active doses were in CR when they died. One had a cardiac arrest, and the other had myelodysplastic syndrome following discontinuation.
Efficacy
In addition to the high ORR (94%) and CR rate (56%) in this study, 9 of 10 patients evaluated for minimal residual disease were negative.
The median time to first response was 1.02 months, and median time to best response was 3.74 months. The median time to CR was 3.84 months.
The median duration of response and PFS have not been reached. The PFS rate was 81% at 6 months and 71% at 9 months.
“We found that all the doses between 150 million and 450 million were effective,” Dr Kochenderfer noted. “We didn’t see a clear difference in efficacy between those doses, so we’ve chosen to use the 150 – 300 million dose range for the follow-up study.”
The investigators observed robust expansion of bb2121, which peaked in the first week after the infusion. Six of 13 patients had evident CAR T cells at 6 months. One patient has persistence over 12 months.
The investigators also observed a robust decrease in M protein and rapid clearance of serum-free light chains and serum BCMA. They noted that the activity of the CAR-positive T cells was not inhibited by high baseline serum BCMA.
Four patients progressed. The investigators analyzed the patients’ tumor burden, bb2121 dose, best response, time to progression, BCMA expression, grades of CRS, and bb2121 persistence. And progression was independent of these factors.
“So we can’t pick out a very good factor of why they progressed,” Dr Kochenderfer said.
However, he noted that the patients are eligible for re-treatment.
Investigators have opened a global trial of bb2121 (NCT03361748) given at doses ranging from 150 – 300 million CAR T cells.
The US Food and Drug Administration and the European Medicines Agency recently fast-tracked bb2121. ![]()
*Data presented differ from the abstract.