User login
Mobile stroke units becoming more common despite cost effectiveness questions
HOUSTON – Mobile stroke units – specially equipped ambulances that bring a diagnostic CT scanner and therapeutic thrombolysis directly to patients in the field – have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost effectiveness.
The first U.S. mobile stroke unit (MSU) launched in Houston in early 2014 (following the world’s first in Berlin, which began running in early 2011), and by early 2017, at least eight other U.S. MSUs were in operation, most of them put into service during the prior 15 months. U.S. MSU locations now include Cleveland; Denver; Memphis; New York; Toledo, Ohio; Trenton, N.J., and Northwestern Medicine and Rush University Medical Center in the western Chicago region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to start thrombolytic therapy with tissue plasminogen activator (tPA) compared with that at a hospital, and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get a MSU on the road for the first time, and also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every U.S. MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr. Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged godfather of all U.S. MSUs.

The concept behind MSUs is simple. Each one carries a CT scanner on board so that, once the vehicle’s staff identifies a patient with clinical signs of a significant–acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.

Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr. Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr. Saver said during a session at the meeting.

Early data on effectiveness
Dr. Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data collected by the Berlin group and published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU with similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of TPA treatment did not begin until the patient reached a hospital.
The German analysis showed that, in the observational Pre-Hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference. However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 0-1 when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance (Lancet Neurol. 2016 Sept;15[10]:1035-43).
Dr. Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr. Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that, among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90 day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at emergency departments operated by the Cleveland Clinic, a statistically significant difference. However, this report included no data on clinical outcomes (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003786).
Running the financial numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality adjusted life years. We calculated a cost of $35,000 per quality adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.

Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years, enough to balance the operating cost, Dr. Grotta said.
A key part of controlling costs is having the neurologic consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital (Stroke. 2017 Jan;48[1]:222-4), said Dr. Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr. Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, something not formally examined in the study he reported.
However, telemedicine assessment of CT results gathered in a MSU has one major limitation: the time needed to transmit the huge amount of information in a CT angiogram.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT–angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CT angiogram are delivered on a memory stick to the attending hospital neurologist once the MSU returns.

Waiting for more data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially non-sustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr. Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March. “Even if MSUs meet an acceptable societal threshold for cost effectiveness, cost efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003833). They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 TPA treatments and 349 cancellations before arrival, noted Dr. Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr. Brandler, an emergency medicine physician at Stony Brook (N.Y.) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of U.S. patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver Houston, and Memphis. Although currently designed to enroll 697 patients, Dr. Grotta said he hopes to kick that up to 1,000 patients.
“We are following the healthcare use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr. Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
Dr. Southerland and Dr. Brandler suggested that “making MSUs multipurpose vehicles might also enhance cost-effectiveness,” an option that Dr. Grotta and his colleagues embrace. The MSUs on U.S. roads already also treat patients with intracranial hemorrhages using blood pressure reduction medications. Other neurologic diagnoses considered potential targets for MSU interventions include encephalopathy, seizures, central nervous system–tumors, and infections.
Stroke is a prime example of “a disease that is extremely time sensitive, and for the first time the field of stroke is ahead of the rest of the medical world in trying to speed treatment,” Dr. Grotta said. “We could add other diagnostic equipment monitored by telemedicine specialists. The MSU concept could be expanded to make it much more cost effective” and spur wider adoption by EMS, he suggested.
Dr. Grotta is a consultant to Frazer, a company that produces mobile stroke units, and to Stryker Corporation, and he has received research support from Genentech. Dr. Saver has been a consultant to and received research support from St. Jude. Dr. Audebert has received honoraria from Pfizer, Boehringer Ingelheim, Bristol-Myers Squibb, and Ever Pharma. He has been a consultant to ReNeuron, and he has served as an expert witness for Pfizer and for Lundbeck. Dr. Hussain has been a consultant to Pulsar. Dr. Alexandrov has been a speaker for Genentech. Dr. Nour, Dr. Wu, Dr. Bledsoe, Dr. Southerland, and Dr. Brandler had no disclosures.
[email protected]
On Twitter @mitchelzoler
HOUSTON – Mobile stroke units – specially equipped ambulances that bring a diagnostic CT scanner and therapeutic thrombolysis directly to patients in the field – have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost effectiveness.
The first U.S. mobile stroke unit (MSU) launched in Houston in early 2014 (following the world’s first in Berlin, which began running in early 2011), and by early 2017, at least eight other U.S. MSUs were in operation, most of them put into service during the prior 15 months. U.S. MSU locations now include Cleveland; Denver; Memphis; New York; Toledo, Ohio; Trenton, N.J., and Northwestern Medicine and Rush University Medical Center in the western Chicago region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to start thrombolytic therapy with tissue plasminogen activator (tPA) compared with that at a hospital, and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get a MSU on the road for the first time, and also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every U.S. MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr. Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged godfather of all U.S. MSUs.
The concept behind MSUs is simple. Each one carries a CT scanner on board so that, once the vehicle’s staff identifies a patient with clinical signs of a significant–acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.
Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr. Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr. Saver said during a session at the meeting.
Early data on effectiveness
Dr. Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data collected by the Berlin group and published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU with similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of TPA treatment did not begin until the patient reached a hospital.
The German analysis showed that, in the observational Pre-Hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference. However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 0-1 when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance (Lancet Neurol. 2016 Sept;15[10]:1035-43).
Dr. Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr. Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that, among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90 day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at emergency departments operated by the Cleveland Clinic, a statistically significant difference. However, this report included no data on clinical outcomes (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003786).
Running the financial numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality adjusted life years. We calculated a cost of $35,000 per quality adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.
Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years, enough to balance the operating cost, Dr. Grotta said.
A key part of controlling costs is having the neurologic consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital (Stroke. 2017 Jan;48[1]:222-4), said Dr. Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr. Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, something not formally examined in the study he reported.
However, telemedicine assessment of CT results gathered in a MSU has one major limitation: the time needed to transmit the huge amount of information in a CT angiogram.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT–angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CT angiogram are delivered on a memory stick to the attending hospital neurologist once the MSU returns.
Waiting for more data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially non-sustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr. Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March. “Even if MSUs meet an acceptable societal threshold for cost effectiveness, cost efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003833). They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 TPA treatments and 349 cancellations before arrival, noted Dr. Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr. Brandler, an emergency medicine physician at Stony Brook (N.Y.) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of U.S. patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver Houston, and Memphis. Although currently designed to enroll 697 patients, Dr. Grotta said he hopes to kick that up to 1,000 patients.
“We are following the healthcare use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr. Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
Dr. Southerland and Dr. Brandler suggested that “making MSUs multipurpose vehicles might also enhance cost-effectiveness,” an option that Dr. Grotta and his colleagues embrace. The MSUs on U.S. roads already also treat patients with intracranial hemorrhages using blood pressure reduction medications. Other neurologic diagnoses considered potential targets for MSU interventions include encephalopathy, seizures, central nervous system–tumors, and infections.
Stroke is a prime example of “a disease that is extremely time sensitive, and for the first time the field of stroke is ahead of the rest of the medical world in trying to speed treatment,” Dr. Grotta said. “We could add other diagnostic equipment monitored by telemedicine specialists. The MSU concept could be expanded to make it much more cost effective” and spur wider adoption by EMS, he suggested.
Dr. Grotta is a consultant to Frazer, a company that produces mobile stroke units, and to Stryker Corporation, and he has received research support from Genentech. Dr. Saver has been a consultant to and received research support from St. Jude. Dr. Audebert has received honoraria from Pfizer, Boehringer Ingelheim, Bristol-Myers Squibb, and Ever Pharma. He has been a consultant to ReNeuron, and he has served as an expert witness for Pfizer and for Lundbeck. Dr. Hussain has been a consultant to Pulsar. Dr. Alexandrov has been a speaker for Genentech. Dr. Nour, Dr. Wu, Dr. Bledsoe, Dr. Southerland, and Dr. Brandler had no disclosures.
[email protected]
On Twitter @mitchelzoler
HOUSTON – Mobile stroke units – specially equipped ambulances that bring a diagnostic CT scanner and therapeutic thrombolysis directly to patients in the field – have begun to proliferate across the United States, although they remain investigational, with no clear proof of their incremental clinical value or cost effectiveness.
The first U.S. mobile stroke unit (MSU) launched in Houston in early 2014 (following the world’s first in Berlin, which began running in early 2011), and by early 2017, at least eight other U.S. MSUs were in operation, most of them put into service during the prior 15 months. U.S. MSU locations now include Cleveland; Denver; Memphis; New York; Toledo, Ohio; Trenton, N.J., and Northwestern Medicine and Rush University Medical Center in the western Chicago region. A tenth MSU is slated to start operation at the University of California, Los Angeles later this year.
Early data collected at some of these sites show that initiating care of an acute ischemic stroke patient in an MSU shaves precious minutes off the time it takes to start thrombolytic therapy with tissue plasminogen activator (tPA) compared with that at a hospital, and findings from preliminary analyses suggest better functional outcomes for patients treated this way. However, leaders in the nascent field readily admit that the data needed to clearly prove the benefit patients receive from operating MSUs are still a few years off. This uncertainty about the added benefit to patients from MSUs couples with one clear fact: MSUs are expensive to start up, with a price tag of roughly $1 million to get a MSU on the road for the first time, and also expensive to operate, with one estimate for the annual cost of keeping an MSU on the street at about $500,000 per year for staffing, supplies, and other expenses.
“Every U.S. MSU I know of started with philanthropic gifts, but you need a business model” to keep the program running long-term, James C. Grotta, MD, said during a session focused on MSUs at the International Stroke Conference sponsored by the American Heart Association. “You can’t sustain an MSU with philanthropy,” said Dr. Grotta, professor of neurology at the University of Texas Health Science Center in Houston, director and founder of the Houston MSU, and acknowledged godfather of all U.S. MSUs.
The concept behind MSUs is simple. Each one carries a CT scanner on board so that, once the vehicle’s staff identifies a patient with clinical signs of a significant–acute ischemic stroke in the field and confirms that the timing of the stroke onset suggests eligibility for tPA treatment, a CT scan can immediately be run on site to finalize tPA eligibility. The MSU staff can then begin infusing the drug in the ambulance as it speeds the patient to an appropriate hospital.
Another advantage to MSUs, in addition to quicker initiation of thrombolysis, is “getting patients to where they need to go faster and more directly,” said Dr. Nour.
“Instead of bringing patients first to a hospital that’s unable to do thrombectomy and where treatment gets slowed down, with an MSU you can give tPA on the street and go straight to a thrombectomy center,” agreed Jeffrey L. Saver, MD, professor of neurology and director of the stroke unit at UCLA. “The MSU offers the tantalizing possibility that you can give tPA with no time hit because you can give it on the way directly to a comprehensive stroke center,” Dr. Saver said during a session at the meeting.
Early data on effectiveness
Dr. Nour reported some of the best evidence for the incremental clinical benefit of MSUs based on the reduced time for starting a tPA infusion. She used data collected by the Berlin group and published in September 2016 that compared the treatment courses and outcomes of patients managed with an MSU with similar patients managed by conventional ambulance transport for whom CT scan assessment and the start of TPA treatment did not begin until the patient reached a hospital.
The German analysis showed that, in the observational Pre-Hospital Acute Neurological Therapy and Optimization of Medical Care in Stroke Patients–Study (PHANTOM-S), among 353 patients treated by conventional transport, the median time from stroke onset to thrombolysis was 112 minutes, compared with a median of 73 minutes among 305 patients managed with an MSU, a statistically significant difference. However, the study found no significant difference for its primary endpoint: the percentage of patients with a modified Rankin Scale score of 0-1 when measured 90 days after their respective strokes. This outcome occurred in 47% of the control patients managed conventionally and in 53% of those managed by an MSU, a difference that fell short of statistical significance (Lancet Neurol. 2016 Sept;15[10]:1035-43).
Dr. Nour attributed the lack of statistical significance for this primary endpoint to the relatively small number of patients enrolled in PHANTOM-S. “The study was underpowered,” she said.
Dr. Nour presented an analysis at the meeting that extrapolated the results out to 1,000 hypothetical patients and tallied the benefits that a larger number of patients could expect to receive if their outcomes paralleled those seen in the published results. It showed that, among 1,000 stroke patients treated with an MSU, 58 were expected to be free from disability 90 days later, and an additional 124 patients would have some improvement in their 90 day clinical outcome based on their modified Rankin Scale scores when compared with patients undergoing conventional hospitalization.
“If this finding was confirmed in a larger, controlled study, it would suggest that MSU-based thrombolysis has substantial clinical benefit,” she concluded.
Another recent report looked at the first 100 stroke patients treated by the Cleveland MSU during 2014. Researchers at the Cleveland Clinic and Case Western Reserve University said that 16 of those 100 patients received tPA, and the median time from their emergency call to thrombolytic treatment was 38.5 minutes faster than for 53 stroke patients treated during the same period at emergency departments operated by the Cleveland Clinic, a statistically significant difference. However, this report included no data on clinical outcomes (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003786).
Running the financial numbers
Nailing down the incremental clinical benefit from MSUs is clearly a very important part of determining the value of this strategy, but another very practical concern is how much the service costs and whether it is financially sustainable.
“We did a cost-effectiveness analysis based on the PHANTOM-S data, and we were conservative by only looking at the benefit from early tPA treatment,” Heinrich J. Audebert, MD, professor of neurology at Charité Hospital in Berlin and head of the team running Berlin’s MSU, said during the MSU session at the meeting. “We did not take into account saving money by avoiding long-term stroke disability and just considered the cost of [immediate] care and the quality adjusted life years. We calculated a cost of $35,000 per quality adjusted life year, which is absolutely acceptable.”
He cautioned that this analysis was not based on actual outcomes but on the numbers needed to treat calculated from the PHANTOM-S results. “We need to now show this in controlled trials,” he admitted.
Income from transport reimbursement, currently $500 per trip, and reimbursements of $17,000 above costs for administering tPA and of roughly $40,000 above costs for performing thrombectomy are balancing these costs. Based on an estimated additional one thrombolysis case per month and one additional thrombectomy case per month, the MSU yields a potential incremental income to the hospital running the MSU of about $3.8 million over 5 years, enough to balance the operating cost, Dr. Grotta said.
A key part of controlling costs is having the neurologic consult done via a telemedicine link rather than by neurologist at the MSU. “Telemedicine reduces operational costs and improves efficiency,” noted M. Shazam Hussain, MD, interim director of the Cerebrovascular Center at the Cleveland Clinic. “Cost effectiveness is a very important part of the concept” of MSUs, he said at the session.
The Houston group reported results from a study that directly compared the diagnostic performance of an onboard neurologist with that of a telemedicine neurologist linked in remotely during MSU deployments for 174 patients. For these cases, the two neurologists each made an independent diagnosis that the researchers then compared. The two diagnoses concurred for 88% of the cases, Tzu-Ching Wu, MD, reported at the meeting. This rate of agreement matched the incidence of concordance between two neurologists who independently assessed the same patients at the hospital (Stroke. 2017 Jan;48[1]:222-4), said Dr. Wu, a vascular neurologist and director of the telemedicine program at the University of Texas Health Science Center in Houston.
“The results support using telemedicine as the primary means of assessment on the MSU,” said Dr. Wu. “This may enhance MSU efficiency and reduce costs.” His group’s next study of MSU telemedicine will compare the time needed to make a diagnostic decision using the two approaches, something not formally examined in the study he reported.
However, telemedicine assessment of CT results gathered in a MSU has one major limitation: the time needed to transmit the huge amount of information in a CT angiogram.
The MSU used by clinicians at the University of Tennessee, Memphis, incorporates an extremely powerful battery that enables “full CT scanner capability with a moving gantry,” said Andrei V. Alexandrov, MD, professor and chairman of neurology at the university. With this set up “we can do in-the-field multiphasic CT–angiography from the aortic arch up within 4 minutes. The challenge of doing this is simple. It’s 1.7 gigabytes of data,” which would take a prohibitively long time to transmit from a remote site, he explained. As a result, the complete set of images from the field CT angiogram are delivered on a memory stick to the attending hospital neurologist once the MSU returns.
Waiting for more data
Despite these advances and the steady recent growth of MSUs, significant skepticism remains. “While mobile stroke units seem like a good idea and there is genuine hope that they will improve outcomes for selected stroke patients, there is not yet any evidence that this is the case,” wrote Bryan Bledsoe, DO, in a January 2017 editorial in the Journal of Emergency Medical Services. “They are expensive and financially non-sustainable. Without widespread deployment, they stand to benefit few, if any, patients. The money spent on these devices would be better spent on improving the current EMS system including paramedic education, the availability of stroke centers, and on the early recognition of ELVO [emergent large vessel occlusion] strokes,” wrote Dr. Bledsoe, professor of emergency medicine at the University of Nevada in Las Vegas.
Two other experts voiced concerns about MSUs in an editorial that accompanied a Cleveland Clinic report in March. “Even if MSUs meet an acceptable societal threshold for cost effectiveness, cost efficiency may prove a taller order to achieve return on investment for individual health systems and communities,” wrote Andrew M. Southerland, MD, and Ethan S. Brandler, MD (Neurology. 2017 March 8. doi: 10.1212/WNL.0000000000003833). They cited the Cleveland report, which noted that the group’s first 100 MSU-treated patients came from a total of 317 MSU deployments and included 217 trips that were canceled prior to the MSU’s arrival at the patient’s location. In Berlin’s initial experience, more than 2,000 MSU deployments led to 200 TPA treatments and 349 cancellations before arrival, noted Dr. Southerland, a neurologist at the University of Virginia in Charlottesville, and Dr. Brandler, an emergency medicine physician at Stony Brook (N.Y.) University.
“Hope remains that future trials may demonstrate the ultimate potential of mobile stroke units to improve long-term outcomes for more patients by treating them more quickly and effectively. In the meantime, ongoing efforts are needed to streamline MSU cost and efficiency,” they wrote.
Proponents of MSUs agree that what’s needed now are more data to prove efficacy and cost effectiveness, as well as better integration into EMS programs. The first opportunity for documenting the clinical impact of MSUs on larger numbers of U.S. patients may be from the BEnefits of Stroke Treatment Delivered using a Mobile Stroke Unit Compared to Standard Management by Emergency Medical Services (BEST-MSU) Study, funded by the Patient-Centered Outcomes Research Institute. This study is collecting data from the MSU programs in Denver Houston, and Memphis. Although currently designed to enroll 697 patients, Dr. Grotta said he hopes to kick that up to 1,000 patients.
“We are following the healthcare use and its cost for every enrolled MSU and conventional patient for 1 year,” Dr. Grotta explained in an interview. He hopes these results will provide the data needed to move MSUs from investigational status to routine and reimbursable care.
Dr. Southerland and Dr. Brandler suggested that “making MSUs multipurpose vehicles might also enhance cost-effectiveness,” an option that Dr. Grotta and his colleagues embrace. The MSUs on U.S. roads already also treat patients with intracranial hemorrhages using blood pressure reduction medications. Other neurologic diagnoses considered potential targets for MSU interventions include encephalopathy, seizures, central nervous system–tumors, and infections.
Stroke is a prime example of “a disease that is extremely time sensitive, and for the first time the field of stroke is ahead of the rest of the medical world in trying to speed treatment,” Dr. Grotta said. “We could add other diagnostic equipment monitored by telemedicine specialists. The MSU concept could be expanded to make it much more cost effective” and spur wider adoption by EMS, he suggested.
Dr. Grotta is a consultant to Frazer, a company that produces mobile stroke units, and to Stryker Corporation, and he has received research support from Genentech. Dr. Saver has been a consultant to and received research support from St. Jude. Dr. Audebert has received honoraria from Pfizer, Boehringer Ingelheim, Bristol-Myers Squibb, and Ever Pharma. He has been a consultant to ReNeuron, and he has served as an expert witness for Pfizer and for Lundbeck. Dr. Hussain has been a consultant to Pulsar. Dr. Alexandrov has been a speaker for Genentech. Dr. Nour, Dr. Wu, Dr. Bledsoe, Dr. Southerland, and Dr. Brandler had no disclosures.
[email protected]
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE INTERNATIONAL STROKE CONFERENCE
Synthetic cannabinoid use linked to multiple risk factors
An estimated 1 in 10 high school students uses synthetic cannabinoids, which are linked to multiple other risk behaviors, and use is more likely among students with depressive symptoms, marijuana use, and alcohol use, investigators in two studies reported.
Synthetic cannabinoids are structurally similar to delta-9-tetrahydrocannabinol, but they may be more potent, with adverse health effects not seen with natural tetrahydrocannabinol in marijuana. These synthetic products are accessible to teens online, in convenience stores, and in smoke shops. Past research has suggested that adolescents aren’t aware of the possible negative effects of these products, such as tachycardia, hypertension, lethargy, nausea, vomiting, irritability, chest pain, hallucinations, confusion, and vertigo.
“Overall, we observed that ever use of synthetic cannabinoids was associated with the majority of health risk behaviors included in our study and that those associations tended to be more pronounced for ever use of synthetic cannabinoids than for ever use of marijuana only, particularly for substance use behaviors and sexual risk behaviors,” they wrote (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-2675).
Dr. Clayton’s team analyzed data from 15,624 students in grades 9-12 from the cross-sectional 2015 Youth Risk Behavior Survey, including all 50 states and Washington. The questions asked about use of marijuana, synthetic cannabinoids, or both. The question about synthetic cannabinoids included reference to street names of the drug, such as K2, Spice, fake weed, King Kong, Yucatan Fire, Skunk, or Moon Rocks. Another three dozen questions asked about risk behaviors related to substance use, violence and injury, mental health, and sexual health.
The results revealed that 29% of students had ever used only marijuana and 9% had ever used synthetic cannabinoids. Most of the students, 61%, had never used either. Although 23% of marijuana users had used synthetic cannabinoids, nearly all (98%) of the cannabinoids users had used marijuana.
Compared with those who had used only marijuana, adolescents who had ever used synthetic cannabinoids were considerably more likely to engage in substance use (adjusted prevalence ratio [aPR] = 4.85 for current alcohol use; aPR =151.90 for ever use of heroin) or sexual risk behaviors (had sexual intercourse with four or more persons during their life; aPR = 6.20). They also were more than twice as likely to have tried marijuana before age 13 years (aPR = 2.35) and were more likely to have used marijuana at least once in the past month (aPR = 1.36) and to have used marijuana 20 or more times in the past month (aPR = 1.88).
“Youth may progress from marijuana use only to the use of synthetic cannabinoids for a variety of reasons, such as ease of access, perception of safety, and ability to be undetected by many drug tests,” Dr. Clayton and her associates wrote.
The second study found similar associations between marijuana use and later use of synthetic cannabinoids. Andrew L. Ninnemann of the University of Missouri–Columbia, and his associates collected data twice over a 12-month period from 964 high school students at seven public schools in Southeast Texas (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-3009), to examine the relationship of synthetic cannabinoid use with anxiety, depression, impulsivity, and marijuana use.
The first assessment occurred in spring of 2011 and the second in spring of 2012. Most respondents (response rate, 62%) were sophomores (73%) or juniors (24%), with only 1% each of freshmen and seniors; 1% reported “other.” The sample included 31% African American students, 29% white students, 28% Hispanic students, and 12% of other ethnicities.
Males were more likely than females to use synthetic cannabinoids, and African American students were less likely to use them than teens of other ethnicities. Depression at baseline predicted use of synthetic cannabinoids a year later (adjusted odds ratio [aOR] = 1.42, P = .04), as did alcohol use (aOR = 1.85, P = .02), marijuana use (aOR = 2.47, P less than .001), and synthetic cannabinoid use at baseline (aOR = 2.36, P less than .001).
Students also were more likely to use marijuana at follow-up if they had used alcohol (aOR = 1.96, P less than .001) or marijuana (aOR = 4.52, P less than .001) at baseline. However, neither demographic variables nor anxiety, impulsivity, synthetic cannabinoid use, or other drug use significantly predicted marijuana use 1 year later.
Mr. Ninnemann’s study also found a slightly higher prevalence of synthetic cannabinoid use at baseline than the Clayton study did, with 13% of the Texas sample reporting use.
“The substantial risks associated with even a single episode of synthetic cannabinoid use emphasize the critical importance of identifying and targeting potential risk factors,” Mr. Ninnemann and his coauthors wrote. “Our findings indicate that prevention and intervention efforts may benefit from targeting depressive symptoms and alcohol and marijuana use to potentially reduce adolescent use of synthetic cannabinoids.”
Dr. Clayton and her colleagues mentioned a past study finding that 50% of elementary schools, 33% of middle schools, and 13% of high schools do not require instruction on alcohol or other drug use prevention. The U.S. trend of cannabis legalization also introduces uncertainty, the investigators noted.
“It is unclear what impact the legalization of marijuana will have on the use of synthetic cannabinoids,” Dr. Clayton’s team wrote. The evidence is contradictory on the likelihood of teens trying marijuana in these environments, but “there is a concern that if marijuana use increases, the use of synthetic marijuana may also increase,” they noted.
The Clayton study did not have external funding. The Ninnemann study received funding from the National Science Foundation, the National Institute of Child Health and Human Development, and the National Institute of Justice. The authors of both studies reported that they had no disclosures.
Synthetic cannabinoids are made in a lab to have marijuanalike properties, but often have more short-term medical and behavioral toxicities. We at the University of Florida McKnight Brain Institute in Gainesville have studied bath salts and synthetics in the laboratory, but there have been few human studies. The current studies by Clayton et al. and Ninnemann et al., following shortly after the American Academy of Pediatrics warning about the effects of cannabis smoking in adolescents (Pediatrics. 2017 10.1542/peds.2016-4069), are a grim reminder that adolescence is a period of extreme vulnerability to drugs of abuse. Synthetic cannabinoids, as addiction specialists will attest, produce some signs and symptoms of cannabis intoxication, but often with more acute problems, with greater intensity, and of longer duration. In the current two studies, it is clear that the reported consequences of synthetic cannabinoids are greater in terms of risk behaviors and depression than in marijuana smokers.

Mark S. Gold, MD, is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville. He also is chairman of the scientific advisory boards for RiverMend Health, Atlanta.
Synthetic cannabinoids are made in a lab to have marijuanalike properties, but often have more short-term medical and behavioral toxicities. We at the University of Florida McKnight Brain Institute in Gainesville have studied bath salts and synthetics in the laboratory, but there have been few human studies. The current studies by Clayton et al. and Ninnemann et al., following shortly after the American Academy of Pediatrics warning about the effects of cannabis smoking in adolescents (Pediatrics. 2017 10.1542/peds.2016-4069), are a grim reminder that adolescence is a period of extreme vulnerability to drugs of abuse. Synthetic cannabinoids, as addiction specialists will attest, produce some signs and symptoms of cannabis intoxication, but often with more acute problems, with greater intensity, and of longer duration. In the current two studies, it is clear that the reported consequences of synthetic cannabinoids are greater in terms of risk behaviors and depression than in marijuana smokers.
Mark S. Gold, MD, is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville. He also is chairman of the scientific advisory boards for RiverMend Health, Atlanta.
Synthetic cannabinoids are made in a lab to have marijuanalike properties, but often have more short-term medical and behavioral toxicities. We at the University of Florida McKnight Brain Institute in Gainesville have studied bath salts and synthetics in the laboratory, but there have been few human studies. The current studies by Clayton et al. and Ninnemann et al., following shortly after the American Academy of Pediatrics warning about the effects of cannabis smoking in adolescents (Pediatrics. 2017 10.1542/peds.2016-4069), are a grim reminder that adolescence is a period of extreme vulnerability to drugs of abuse. Synthetic cannabinoids, as addiction specialists will attest, produce some signs and symptoms of cannabis intoxication, but often with more acute problems, with greater intensity, and of longer duration. In the current two studies, it is clear that the reported consequences of synthetic cannabinoids are greater in terms of risk behaviors and depression than in marijuana smokers.
Mark S. Gold, MD, is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville. He also is chairman of the scientific advisory boards for RiverMend Health, Atlanta.
An estimated 1 in 10 high school students uses synthetic cannabinoids, which are linked to multiple other risk behaviors, and use is more likely among students with depressive symptoms, marijuana use, and alcohol use, investigators in two studies reported.
Synthetic cannabinoids are structurally similar to delta-9-tetrahydrocannabinol, but they may be more potent, with adverse health effects not seen with natural tetrahydrocannabinol in marijuana. These synthetic products are accessible to teens online, in convenience stores, and in smoke shops. Past research has suggested that adolescents aren’t aware of the possible negative effects of these products, such as tachycardia, hypertension, lethargy, nausea, vomiting, irritability, chest pain, hallucinations, confusion, and vertigo.
“Overall, we observed that ever use of synthetic cannabinoids was associated with the majority of health risk behaviors included in our study and that those associations tended to be more pronounced for ever use of synthetic cannabinoids than for ever use of marijuana only, particularly for substance use behaviors and sexual risk behaviors,” they wrote (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-2675).
Dr. Clayton’s team analyzed data from 15,624 students in grades 9-12 from the cross-sectional 2015 Youth Risk Behavior Survey, including all 50 states and Washington. The questions asked about use of marijuana, synthetic cannabinoids, or both. The question about synthetic cannabinoids included reference to street names of the drug, such as K2, Spice, fake weed, King Kong, Yucatan Fire, Skunk, or Moon Rocks. Another three dozen questions asked about risk behaviors related to substance use, violence and injury, mental health, and sexual health.
The results revealed that 29% of students had ever used only marijuana and 9% had ever used synthetic cannabinoids. Most of the students, 61%, had never used either. Although 23% of marijuana users had used synthetic cannabinoids, nearly all (98%) of the cannabinoids users had used marijuana.
Compared with those who had used only marijuana, adolescents who had ever used synthetic cannabinoids were considerably more likely to engage in substance use (adjusted prevalence ratio [aPR] = 4.85 for current alcohol use; aPR =151.90 for ever use of heroin) or sexual risk behaviors (had sexual intercourse with four or more persons during their life; aPR = 6.20). They also were more than twice as likely to have tried marijuana before age 13 years (aPR = 2.35) and were more likely to have used marijuana at least once in the past month (aPR = 1.36) and to have used marijuana 20 or more times in the past month (aPR = 1.88).
“Youth may progress from marijuana use only to the use of synthetic cannabinoids for a variety of reasons, such as ease of access, perception of safety, and ability to be undetected by many drug tests,” Dr. Clayton and her associates wrote.
The second study found similar associations between marijuana use and later use of synthetic cannabinoids. Andrew L. Ninnemann of the University of Missouri–Columbia, and his associates collected data twice over a 12-month period from 964 high school students at seven public schools in Southeast Texas (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-3009), to examine the relationship of synthetic cannabinoid use with anxiety, depression, impulsivity, and marijuana use.
The first assessment occurred in spring of 2011 and the second in spring of 2012. Most respondents (response rate, 62%) were sophomores (73%) or juniors (24%), with only 1% each of freshmen and seniors; 1% reported “other.” The sample included 31% African American students, 29% white students, 28% Hispanic students, and 12% of other ethnicities.
Males were more likely than females to use synthetic cannabinoids, and African American students were less likely to use them than teens of other ethnicities. Depression at baseline predicted use of synthetic cannabinoids a year later (adjusted odds ratio [aOR] = 1.42, P = .04), as did alcohol use (aOR = 1.85, P = .02), marijuana use (aOR = 2.47, P less than .001), and synthetic cannabinoid use at baseline (aOR = 2.36, P less than .001).
Students also were more likely to use marijuana at follow-up if they had used alcohol (aOR = 1.96, P less than .001) or marijuana (aOR = 4.52, P less than .001) at baseline. However, neither demographic variables nor anxiety, impulsivity, synthetic cannabinoid use, or other drug use significantly predicted marijuana use 1 year later.
Mr. Ninnemann’s study also found a slightly higher prevalence of synthetic cannabinoid use at baseline than the Clayton study did, with 13% of the Texas sample reporting use.
“The substantial risks associated with even a single episode of synthetic cannabinoid use emphasize the critical importance of identifying and targeting potential risk factors,” Mr. Ninnemann and his coauthors wrote. “Our findings indicate that prevention and intervention efforts may benefit from targeting depressive symptoms and alcohol and marijuana use to potentially reduce adolescent use of synthetic cannabinoids.”
Dr. Clayton and her colleagues mentioned a past study finding that 50% of elementary schools, 33% of middle schools, and 13% of high schools do not require instruction on alcohol or other drug use prevention. The U.S. trend of cannabis legalization also introduces uncertainty, the investigators noted.
“It is unclear what impact the legalization of marijuana will have on the use of synthetic cannabinoids,” Dr. Clayton’s team wrote. The evidence is contradictory on the likelihood of teens trying marijuana in these environments, but “there is a concern that if marijuana use increases, the use of synthetic marijuana may also increase,” they noted.
The Clayton study did not have external funding. The Ninnemann study received funding from the National Science Foundation, the National Institute of Child Health and Human Development, and the National Institute of Justice. The authors of both studies reported that they had no disclosures.
An estimated 1 in 10 high school students uses synthetic cannabinoids, which are linked to multiple other risk behaviors, and use is more likely among students with depressive symptoms, marijuana use, and alcohol use, investigators in two studies reported.
Synthetic cannabinoids are structurally similar to delta-9-tetrahydrocannabinol, but they may be more potent, with adverse health effects not seen with natural tetrahydrocannabinol in marijuana. These synthetic products are accessible to teens online, in convenience stores, and in smoke shops. Past research has suggested that adolescents aren’t aware of the possible negative effects of these products, such as tachycardia, hypertension, lethargy, nausea, vomiting, irritability, chest pain, hallucinations, confusion, and vertigo.
“Overall, we observed that ever use of synthetic cannabinoids was associated with the majority of health risk behaviors included in our study and that those associations tended to be more pronounced for ever use of synthetic cannabinoids than for ever use of marijuana only, particularly for substance use behaviors and sexual risk behaviors,” they wrote (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-2675).
Dr. Clayton’s team analyzed data from 15,624 students in grades 9-12 from the cross-sectional 2015 Youth Risk Behavior Survey, including all 50 states and Washington. The questions asked about use of marijuana, synthetic cannabinoids, or both. The question about synthetic cannabinoids included reference to street names of the drug, such as K2, Spice, fake weed, King Kong, Yucatan Fire, Skunk, or Moon Rocks. Another three dozen questions asked about risk behaviors related to substance use, violence and injury, mental health, and sexual health.
The results revealed that 29% of students had ever used only marijuana and 9% had ever used synthetic cannabinoids. Most of the students, 61%, had never used either. Although 23% of marijuana users had used synthetic cannabinoids, nearly all (98%) of the cannabinoids users had used marijuana.
Compared with those who had used only marijuana, adolescents who had ever used synthetic cannabinoids were considerably more likely to engage in substance use (adjusted prevalence ratio [aPR] = 4.85 for current alcohol use; aPR =151.90 for ever use of heroin) or sexual risk behaviors (had sexual intercourse with four or more persons during their life; aPR = 6.20). They also were more than twice as likely to have tried marijuana before age 13 years (aPR = 2.35) and were more likely to have used marijuana at least once in the past month (aPR = 1.36) and to have used marijuana 20 or more times in the past month (aPR = 1.88).
“Youth may progress from marijuana use only to the use of synthetic cannabinoids for a variety of reasons, such as ease of access, perception of safety, and ability to be undetected by many drug tests,” Dr. Clayton and her associates wrote.
The second study found similar associations between marijuana use and later use of synthetic cannabinoids. Andrew L. Ninnemann of the University of Missouri–Columbia, and his associates collected data twice over a 12-month period from 964 high school students at seven public schools in Southeast Texas (Pediatrics. 2017 March 13. doi: 10.1542/peds.2016-3009), to examine the relationship of synthetic cannabinoid use with anxiety, depression, impulsivity, and marijuana use.
The first assessment occurred in spring of 2011 and the second in spring of 2012. Most respondents (response rate, 62%) were sophomores (73%) or juniors (24%), with only 1% each of freshmen and seniors; 1% reported “other.” The sample included 31% African American students, 29% white students, 28% Hispanic students, and 12% of other ethnicities.
Males were more likely than females to use synthetic cannabinoids, and African American students were less likely to use them than teens of other ethnicities. Depression at baseline predicted use of synthetic cannabinoids a year later (adjusted odds ratio [aOR] = 1.42, P = .04), as did alcohol use (aOR = 1.85, P = .02), marijuana use (aOR = 2.47, P less than .001), and synthetic cannabinoid use at baseline (aOR = 2.36, P less than .001).
Students also were more likely to use marijuana at follow-up if they had used alcohol (aOR = 1.96, P less than .001) or marijuana (aOR = 4.52, P less than .001) at baseline. However, neither demographic variables nor anxiety, impulsivity, synthetic cannabinoid use, or other drug use significantly predicted marijuana use 1 year later.
Mr. Ninnemann’s study also found a slightly higher prevalence of synthetic cannabinoid use at baseline than the Clayton study did, with 13% of the Texas sample reporting use.
“The substantial risks associated with even a single episode of synthetic cannabinoid use emphasize the critical importance of identifying and targeting potential risk factors,” Mr. Ninnemann and his coauthors wrote. “Our findings indicate that prevention and intervention efforts may benefit from targeting depressive symptoms and alcohol and marijuana use to potentially reduce adolescent use of synthetic cannabinoids.”
Dr. Clayton and her colleagues mentioned a past study finding that 50% of elementary schools, 33% of middle schools, and 13% of high schools do not require instruction on alcohol or other drug use prevention. The U.S. trend of cannabis legalization also introduces uncertainty, the investigators noted.
“It is unclear what impact the legalization of marijuana will have on the use of synthetic cannabinoids,” Dr. Clayton’s team wrote. The evidence is contradictory on the likelihood of teens trying marijuana in these environments, but “there is a concern that if marijuana use increases, the use of synthetic marijuana may also increase,” they noted.
The Clayton study did not have external funding. The Ninnemann study received funding from the National Science Foundation, the National Institute of Child Health and Human Development, and the National Institute of Justice. The authors of both studies reported that they had no disclosures.
FROM PEDIATRICS
Key clinical point: Results of two studies found that 9%-13% of high school students have used synthetic cannabinoids.
Major finding: Depression, alcohol use, and marijuana use increase the likelihood of adolescents’ use of synthetic cannabinoids, which increases the risk of multiple substance use and risky sexual behavior.
Data source: Two studies, one surveying 15,624 high school students nationwide and one surveying 964 Texas public high school students twice over a period of 1 year.
Disclosures: The Clayton study had no external funding. The Ninnemann study received funding from the National Science Foundation, the National Institute of Child Health and Human Development, and the National Institute of Justice. The authors of both studies reported that they had no disclosures.
Ursolic acid
Ursolic acid (3beta-hydroxy-urs-12-en-28-oic acid) is a pentacyclic triterpenoid found naturally in apples, waxy berries, rosemary, oregano, and several other plants and herbs used in medicine and the diet.1,2 It is known to have significant antioxidant, anti-inflammatory, and antiproliferative properties, and has also been associated with a wider range of biologic activities, including anticancer, antimicrobial, antitumor, antiwrinkle, anti-HIV, cytotoxic, and hepatoprotective.3,4 In addition, ursolic acid is the focus of human clinical trials for potential uses in cancer and skin wrinkles.4 While this triterpenoid is known to suppress tumor formation and viability in various kinds of cancer, including skin cancer, several forms of cancer are resistant to ursolic acid.
Anti-inflammatory activity
In a 2013 study of the antibacterial and anti-inflammatory effects of Syzygium jambos on acne, Sharma et al. found that ursolic acid was one of the constituents of the leaf extracts that contributed to a significant suppression of the release of inflammatory cytokines interleukin (IL)-8 and tumor necrosis factor-alpha.5
In 2010, Yang et al. identified ursolic acid as a key constituent of Acanthopanax koreanum fruit, a popular fruit in Jeju Island, South Korea, extracts of which they found to exhibit significant anti-inflammatory activity and suitability as a topical agent.6
Yasukawa et al. conducted an in vivo two-stage carcinogenesis test in mice in 2009 in which extracts of the branches of Hippophae rhamnoides displayed significant antitumor activity after initiation with 7,12-dimethylbenz[a]anthracene (DMBA) and promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA). Ursolic acid and (-)-epigallocatechin were the constituents found to have the greatest inhibitory effects on TPA-induced inflammation.7

A 2002 study by Chattopadhyay et al. revealed that the ursolic acid present in Mallotus peltatus extract (long used in traditional folk medicine to treat skin infections and intestinal disorders) may partially account for the broad anti-inflammatory and antimicrobial activity of the plant.9
In 1997, Máñez et al. noted that ursolic acid was among two of the four selected natural triterpenoids tested and found to be significantly effective against inflammation in a TPA multiple-dose model of chronic skin inflammation.10
Anticancer activity
In 2015, Cho et al. reported on the inhibitory effects on skin tumor promotion from the topical application of ursolic acid, resveratrol, or the combination of the two prior to TPA treatment on mouse skin. The combination of the two botanical agents yielded the strongest suppression of TPA-induced epidermal hyperproliferation, skin inflammation, inflammatory gene expression, and skin tumor promotion.11
In another study that year buttressing the combination of the two botanical agents, Junco et al. demonstrated that chloroquine could be used to sensitize B16F10 metastatic mouse melanoma to the anticancer activities of ursolic acid and resveratrol. The investigators concluded that the combination of ursolic acid or resveratrol with chloroquine has potential for inclusion in melanoma treatment in humans.12 Previously, Junco et al. observed that the anti–skin cancer effects of ursolic acid are augmented by P-glycoprotein inhibitors, and that ursolic acid and the stilbene resveratrol, a potent antioxidant, work synergistically, although not by blocking P-glycoprotein. The investigators suggested that ursolic acid along with resveratrol and/or P-glycoprotein inhibitors have potential as effective anti–skin cancer regimens.
In 2014, Lee et al. showed that ursolic acid can differentially modulate apoptosis in cutaneous melanoma and retinal pigment epithelial cells exposed to ultraviolet to visible broadband radiation, exhibiting the potential to protect normal cells while sensitizing melanoma cells to the effects of UV radiation.13 These findings supported earlier work by the team showing that pretreatment of human cells derived from a malignant skin melanoma markedly enhanced the sensitivity of melanoma cells to UV radiation, while providing some photoprotection to retinal pigment epithelium.
Also that year, Soica et al. demonstrated, using in vitro tests and in vivo skin cancer models, that the mixture of oleanolic and ursolic acids and in complex with cyclodextrin rendered a synergistic antitumor activity.14
A year earlier, Kowalczyk et al. showed that the combined action of phytochemicals – dietary calcium D-glucarate and topical ursolic acid and resveratrol – was effective in suppressing the initiation (with 7,12-dimethylbenz[a]anthracene [DMBA]) and promotion (with TPA) of skin tumorigenesis in SENCAR mice. Ursolic acid alone or in combination with calcium D-glucarate significantly diminished epidermal hyperplasia when applied during promotion. All of the antipromotion protocols led to significant decreases in cyclooxygenase-2 and interleukin (IL)-6 expression. The researchers concluded that ursolic acid strongly inhibits skin tumor promotion and inflammatory signaling, and warrants attention as a potential preventive agent against skin and other epithelial cancers.15 Kowalczyk et al. had previously found that ursolic acid and other phytochemicals displayed significant in vitro and in vivo antioxidant and antitumorigenic activity, inhibiting murine skin carcinogenesis by blunting tumor initiation and tumor promotion/progression.16
In 2006, beta-ursolic acid isolated from Salvia officinalis was found by Jedinák et al. to be effective in suppressing lung colonization of beta16 mouse melanoma cells in vivo.17
Huang et al. showed in 1994 that extracts of the leaves of Rosmarinus officinalis (rosemary) were effective in suppressing tumor initiation and promotion in a two-stage skin tumorigenesis mouse model. Topically applied ursolic acid isolated from the leaves was found to hinder TPA-induced ear inflammation, ornithine decarboxylase activity, and tumor promotion. The number of tumors per mouse also declined significantly due to the topical application of ursolic acid concurrent with twice weekly application of the tumor-promoter TPA in DMBA-initiated mice.18
Antiaging and other activities
In 2015, Herndon et al. conducted an open-label clinical trial in 37 females (aged 35-60 years) to ascertain the effectiveness of an anti-aging moisturizer containing Astragalus membranaceus root extract, a peptide blend including palmitoyl tripeptide-38, standardized rosemary leaf extract (ursolic acid), tetrahexyldecyl ascorbate (THD ascorbate), and ubiquinone (coenzyme Q10). Subjects were instructed to apply the moisturizer once in the morning and once in the evening, and were assessed at baseline, and after 4, 8, and 12 weeks of twice daily application. Clinical evaluations after 8 weeks revealed a statistically significant improvement in all grading parameters (fine lines and wrinkles, clarity/brightness, visual roughness, tactile roughness, redness, hyperpigmentation, and overall appearance), with even more pronounced improvement at 12 weeks. The product was found to be mild and well tolerated, and digital photography reinforced clinical assessments and self-evaluations.19
Lee et al. reported in 2012 on in vitro results suggesting that ursolic acid was effective as an inhibitor of matrix metalloproteinase (MMP)-1 after UVB exposure and was more effective than retinoic acid.20
Based on studies with hairless mice, Lim et al. found in 2007 that ursolic and oleanolic acids can enhance the recovery of skin barrier function and, via peroxisome proliferator-activated receptor-alpha, spur epidermal keratinocyte differentiation. They concluded that both acids have potential for use as agents to promote epidermal permeability barrier function.21
In 2003, Soo et al. observed that pretreatment with ursolic acid inhibited UVA-induced oxidative stress and activation and expression of MMP-2 in HaCaT human keratinocytes. They concluded that ursolic acid may merit attention for the prevention of UVA-induced photoaging.22
Three years earlier, Yarosh et al. showed that liposomes containing ursolic acid augmented ceramide content in cultured normal human epidermal keratinocytes and collagen content in cultured normal human dermal fibroblasts. Over an 11-day period, clinical tests with the ursolic acid–containing liposome (Merotaine) revealed increases in the ceramide content in human skin.23 Two years later, many of the same researchers duplicated their results. This new study also demonstrated that ursolic acid liposomes raise ceramide levels in normal human epidermal keratinocytes, in contrast to the effects of retinoic acid, earlier shown to reduce such levels. They concluded that ursolic acid liposomes show promise for use alone or in combination to replenish or maintain cutaneous ceramide and collagen levels.24 Notably, ursolic acid is incorporated into topical oils and creams intended to confer rejuvenating effects to the skin.
Conclusion
Ursolic acid is a compelling ingredient. I especially will be interested in the results of ongoing human clinical trials of this triterpenoid for treating cancer and skin wrinkles. As it is, ursolic acid is known to exert significant inhibitory activity against tumor formation and tumor cell viability in the laboratory. Given its wide range of biologic activity, and some promising cutaneous results, there is reason to believe that ursolic acid has the potential to play an increasingly useful role in topical skin care agents and dermatologic practice.
References
1. J Dermatol. 2007 Sep;34(9):625-34.
2. Folia Histochem Cytobiol. 2011;49(4):664-9.
3. J Cosmet Dermatol. 2004 Jan;3(1):26-34.
4. J Enzyme Inhib Med Chem. 2011 Oct;26(5):616-42.
5. BMC Complement Altern Med. 2013 Oct 29;13:292.
6. J Biomed Biotechnol. 2010;2010:715739.
7. Fitoterapia. 2009 Apr;80(3):164-7.
8. Biosci Biotechnol Biochem. 2004 Jan;68(1):85-90.
9. J Ethnopharmacol. 2002 Oct;82(2-3):229-37.
10. Eur J Pharmacol. 1997 Sep 3;334(1):103-5.
11. Cancer Prev Res (Phila). 2015 Sep;8(9):817-25.
12. Melanoma Res. 2015 Apr;25(2):103-12.
13. Apoptosis. 2014 May;19(5):816-28.
14. Molecules. 2014 Apr 17;19(4):4924-40.
15. Int J Oncol. 2013 Sep;43(3):911-8.
16. Carcinogenesis. 2009 Jun;30(6):1008-15.
17. Z Naturforsch C. 2006 Nov-Dec;61(11-12):777-82.
18. Cancer Res. 1994 Feb 1;54(3):701-8.
19. J Drugs Dermatol. 2015 Jul;14(7):699-704.
20. Bioorg Khim. 2012 May-Jun;38(3):374-81.
21. J Dermatol. 2007;34(9):625-34.
22. Eur J Pharmacol. 2003 Aug 29;476(3):173-8.
23. Horm Res. 2000;54(5-6):318-21.
24. Arch Dermatol Res. 2002 Jan;293(11):569-75.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
Ursolic acid (3beta-hydroxy-urs-12-en-28-oic acid) is a pentacyclic triterpenoid found naturally in apples, waxy berries, rosemary, oregano, and several other plants and herbs used in medicine and the diet.1,2 It is known to have significant antioxidant, anti-inflammatory, and antiproliferative properties, and has also been associated with a wider range of biologic activities, including anticancer, antimicrobial, antitumor, antiwrinkle, anti-HIV, cytotoxic, and hepatoprotective.3,4 In addition, ursolic acid is the focus of human clinical trials for potential uses in cancer and skin wrinkles.4 While this triterpenoid is known to suppress tumor formation and viability in various kinds of cancer, including skin cancer, several forms of cancer are resistant to ursolic acid.
Anti-inflammatory activity
In a 2013 study of the antibacterial and anti-inflammatory effects of Syzygium jambos on acne, Sharma et al. found that ursolic acid was one of the constituents of the leaf extracts that contributed to a significant suppression of the release of inflammatory cytokines interleukin (IL)-8 and tumor necrosis factor-alpha.5
In 2010, Yang et al. identified ursolic acid as a key constituent of Acanthopanax koreanum fruit, a popular fruit in Jeju Island, South Korea, extracts of which they found to exhibit significant anti-inflammatory activity and suitability as a topical agent.6
Yasukawa et al. conducted an in vivo two-stage carcinogenesis test in mice in 2009 in which extracts of the branches of Hippophae rhamnoides displayed significant antitumor activity after initiation with 7,12-dimethylbenz[a]anthracene (DMBA) and promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA). Ursolic acid and (-)-epigallocatechin were the constituents found to have the greatest inhibitory effects on TPA-induced inflammation.7
A 2002 study by Chattopadhyay et al. revealed that the ursolic acid present in Mallotus peltatus extract (long used in traditional folk medicine to treat skin infections and intestinal disorders) may partially account for the broad anti-inflammatory and antimicrobial activity of the plant.9
In 1997, Máñez et al. noted that ursolic acid was among two of the four selected natural triterpenoids tested and found to be significantly effective against inflammation in a TPA multiple-dose model of chronic skin inflammation.10
Anticancer activity
In 2015, Cho et al. reported on the inhibitory effects on skin tumor promotion from the topical application of ursolic acid, resveratrol, or the combination of the two prior to TPA treatment on mouse skin. The combination of the two botanical agents yielded the strongest suppression of TPA-induced epidermal hyperproliferation, skin inflammation, inflammatory gene expression, and skin tumor promotion.11
In another study that year buttressing the combination of the two botanical agents, Junco et al. demonstrated that chloroquine could be used to sensitize B16F10 metastatic mouse melanoma to the anticancer activities of ursolic acid and resveratrol. The investigators concluded that the combination of ursolic acid or resveratrol with chloroquine has potential for inclusion in melanoma treatment in humans.12 Previously, Junco et al. observed that the anti–skin cancer effects of ursolic acid are augmented by P-glycoprotein inhibitors, and that ursolic acid and the stilbene resveratrol, a potent antioxidant, work synergistically, although not by blocking P-glycoprotein. The investigators suggested that ursolic acid along with resveratrol and/or P-glycoprotein inhibitors have potential as effective anti–skin cancer regimens.
In 2014, Lee et al. showed that ursolic acid can differentially modulate apoptosis in cutaneous melanoma and retinal pigment epithelial cells exposed to ultraviolet to visible broadband radiation, exhibiting the potential to protect normal cells while sensitizing melanoma cells to the effects of UV radiation.13 These findings supported earlier work by the team showing that pretreatment of human cells derived from a malignant skin melanoma markedly enhanced the sensitivity of melanoma cells to UV radiation, while providing some photoprotection to retinal pigment epithelium.
Also that year, Soica et al. demonstrated, using in vitro tests and in vivo skin cancer models, that the mixture of oleanolic and ursolic acids and in complex with cyclodextrin rendered a synergistic antitumor activity.14
A year earlier, Kowalczyk et al. showed that the combined action of phytochemicals – dietary calcium D-glucarate and topical ursolic acid and resveratrol – was effective in suppressing the initiation (with 7,12-dimethylbenz[a]anthracene [DMBA]) and promotion (with TPA) of skin tumorigenesis in SENCAR mice. Ursolic acid alone or in combination with calcium D-glucarate significantly diminished epidermal hyperplasia when applied during promotion. All of the antipromotion protocols led to significant decreases in cyclooxygenase-2 and interleukin (IL)-6 expression. The researchers concluded that ursolic acid strongly inhibits skin tumor promotion and inflammatory signaling, and warrants attention as a potential preventive agent against skin and other epithelial cancers.15 Kowalczyk et al. had previously found that ursolic acid and other phytochemicals displayed significant in vitro and in vivo antioxidant and antitumorigenic activity, inhibiting murine skin carcinogenesis by blunting tumor initiation and tumor promotion/progression.16
In 2006, beta-ursolic acid isolated from Salvia officinalis was found by Jedinák et al. to be effective in suppressing lung colonization of beta16 mouse melanoma cells in vivo.17
Huang et al. showed in 1994 that extracts of the leaves of Rosmarinus officinalis (rosemary) were effective in suppressing tumor initiation and promotion in a two-stage skin tumorigenesis mouse model. Topically applied ursolic acid isolated from the leaves was found to hinder TPA-induced ear inflammation, ornithine decarboxylase activity, and tumor promotion. The number of tumors per mouse also declined significantly due to the topical application of ursolic acid concurrent with twice weekly application of the tumor-promoter TPA in DMBA-initiated mice.18
Antiaging and other activities
In 2015, Herndon et al. conducted an open-label clinical trial in 37 females (aged 35-60 years) to ascertain the effectiveness of an anti-aging moisturizer containing Astragalus membranaceus root extract, a peptide blend including palmitoyl tripeptide-38, standardized rosemary leaf extract (ursolic acid), tetrahexyldecyl ascorbate (THD ascorbate), and ubiquinone (coenzyme Q10). Subjects were instructed to apply the moisturizer once in the morning and once in the evening, and were assessed at baseline, and after 4, 8, and 12 weeks of twice daily application. Clinical evaluations after 8 weeks revealed a statistically significant improvement in all grading parameters (fine lines and wrinkles, clarity/brightness, visual roughness, tactile roughness, redness, hyperpigmentation, and overall appearance), with even more pronounced improvement at 12 weeks. The product was found to be mild and well tolerated, and digital photography reinforced clinical assessments and self-evaluations.19
Lee et al. reported in 2012 on in vitro results suggesting that ursolic acid was effective as an inhibitor of matrix metalloproteinase (MMP)-1 after UVB exposure and was more effective than retinoic acid.20
Based on studies with hairless mice, Lim et al. found in 2007 that ursolic and oleanolic acids can enhance the recovery of skin barrier function and, via peroxisome proliferator-activated receptor-alpha, spur epidermal keratinocyte differentiation. They concluded that both acids have potential for use as agents to promote epidermal permeability barrier function.21
In 2003, Soo et al. observed that pretreatment with ursolic acid inhibited UVA-induced oxidative stress and activation and expression of MMP-2 in HaCaT human keratinocytes. They concluded that ursolic acid may merit attention for the prevention of UVA-induced photoaging.22
Three years earlier, Yarosh et al. showed that liposomes containing ursolic acid augmented ceramide content in cultured normal human epidermal keratinocytes and collagen content in cultured normal human dermal fibroblasts. Over an 11-day period, clinical tests with the ursolic acid–containing liposome (Merotaine) revealed increases in the ceramide content in human skin.23 Two years later, many of the same researchers duplicated their results. This new study also demonstrated that ursolic acid liposomes raise ceramide levels in normal human epidermal keratinocytes, in contrast to the effects of retinoic acid, earlier shown to reduce such levels. They concluded that ursolic acid liposomes show promise for use alone or in combination to replenish or maintain cutaneous ceramide and collagen levels.24 Notably, ursolic acid is incorporated into topical oils and creams intended to confer rejuvenating effects to the skin.
Conclusion
Ursolic acid is a compelling ingredient. I especially will be interested in the results of ongoing human clinical trials of this triterpenoid for treating cancer and skin wrinkles. As it is, ursolic acid is known to exert significant inhibitory activity against tumor formation and tumor cell viability in the laboratory. Given its wide range of biologic activity, and some promising cutaneous results, there is reason to believe that ursolic acid has the potential to play an increasingly useful role in topical skin care agents and dermatologic practice.
References
1. J Dermatol. 2007 Sep;34(9):625-34.
2. Folia Histochem Cytobiol. 2011;49(4):664-9.
3. J Cosmet Dermatol. 2004 Jan;3(1):26-34.
4. J Enzyme Inhib Med Chem. 2011 Oct;26(5):616-42.
5. BMC Complement Altern Med. 2013 Oct 29;13:292.
6. J Biomed Biotechnol. 2010;2010:715739.
7. Fitoterapia. 2009 Apr;80(3):164-7.
8. Biosci Biotechnol Biochem. 2004 Jan;68(1):85-90.
9. J Ethnopharmacol. 2002 Oct;82(2-3):229-37.
10. Eur J Pharmacol. 1997 Sep 3;334(1):103-5.
11. Cancer Prev Res (Phila). 2015 Sep;8(9):817-25.
12. Melanoma Res. 2015 Apr;25(2):103-12.
13. Apoptosis. 2014 May;19(5):816-28.
14. Molecules. 2014 Apr 17;19(4):4924-40.
15. Int J Oncol. 2013 Sep;43(3):911-8.
16. Carcinogenesis. 2009 Jun;30(6):1008-15.
17. Z Naturforsch C. 2006 Nov-Dec;61(11-12):777-82.
18. Cancer Res. 1994 Feb 1;54(3):701-8.
19. J Drugs Dermatol. 2015 Jul;14(7):699-704.
20. Bioorg Khim. 2012 May-Jun;38(3):374-81.
21. J Dermatol. 2007;34(9):625-34.
22. Eur J Pharmacol. 2003 Aug 29;476(3):173-8.
23. Horm Res. 2000;54(5-6):318-21.
24. Arch Dermatol Res. 2002 Jan;293(11):569-75.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.
Ursolic acid (3beta-hydroxy-urs-12-en-28-oic acid) is a pentacyclic triterpenoid found naturally in apples, waxy berries, rosemary, oregano, and several other plants and herbs used in medicine and the diet.1,2 It is known to have significant antioxidant, anti-inflammatory, and antiproliferative properties, and has also been associated with a wider range of biologic activities, including anticancer, antimicrobial, antitumor, antiwrinkle, anti-HIV, cytotoxic, and hepatoprotective.3,4 In addition, ursolic acid is the focus of human clinical trials for potential uses in cancer and skin wrinkles.4 While this triterpenoid is known to suppress tumor formation and viability in various kinds of cancer, including skin cancer, several forms of cancer are resistant to ursolic acid.
Anti-inflammatory activity
In a 2013 study of the antibacterial and anti-inflammatory effects of Syzygium jambos on acne, Sharma et al. found that ursolic acid was one of the constituents of the leaf extracts that contributed to a significant suppression of the release of inflammatory cytokines interleukin (IL)-8 and tumor necrosis factor-alpha.5
In 2010, Yang et al. identified ursolic acid as a key constituent of Acanthopanax koreanum fruit, a popular fruit in Jeju Island, South Korea, extracts of which they found to exhibit significant anti-inflammatory activity and suitability as a topical agent.6
Yasukawa et al. conducted an in vivo two-stage carcinogenesis test in mice in 2009 in which extracts of the branches of Hippophae rhamnoides displayed significant antitumor activity after initiation with 7,12-dimethylbenz[a]anthracene (DMBA) and promotion with 12-O-tetradecanoylphorbol-13-acetate (TPA). Ursolic acid and (-)-epigallocatechin were the constituents found to have the greatest inhibitory effects on TPA-induced inflammation.7
A 2002 study by Chattopadhyay et al. revealed that the ursolic acid present in Mallotus peltatus extract (long used in traditional folk medicine to treat skin infections and intestinal disorders) may partially account for the broad anti-inflammatory and antimicrobial activity of the plant.9
In 1997, Máñez et al. noted that ursolic acid was among two of the four selected natural triterpenoids tested and found to be significantly effective against inflammation in a TPA multiple-dose model of chronic skin inflammation.10
Anticancer activity
In 2015, Cho et al. reported on the inhibitory effects on skin tumor promotion from the topical application of ursolic acid, resveratrol, or the combination of the two prior to TPA treatment on mouse skin. The combination of the two botanical agents yielded the strongest suppression of TPA-induced epidermal hyperproliferation, skin inflammation, inflammatory gene expression, and skin tumor promotion.11
In another study that year buttressing the combination of the two botanical agents, Junco et al. demonstrated that chloroquine could be used to sensitize B16F10 metastatic mouse melanoma to the anticancer activities of ursolic acid and resveratrol. The investigators concluded that the combination of ursolic acid or resveratrol with chloroquine has potential for inclusion in melanoma treatment in humans.12 Previously, Junco et al. observed that the anti–skin cancer effects of ursolic acid are augmented by P-glycoprotein inhibitors, and that ursolic acid and the stilbene resveratrol, a potent antioxidant, work synergistically, although not by blocking P-glycoprotein. The investigators suggested that ursolic acid along with resveratrol and/or P-glycoprotein inhibitors have potential as effective anti–skin cancer regimens.
In 2014, Lee et al. showed that ursolic acid can differentially modulate apoptosis in cutaneous melanoma and retinal pigment epithelial cells exposed to ultraviolet to visible broadband radiation, exhibiting the potential to protect normal cells while sensitizing melanoma cells to the effects of UV radiation.13 These findings supported earlier work by the team showing that pretreatment of human cells derived from a malignant skin melanoma markedly enhanced the sensitivity of melanoma cells to UV radiation, while providing some photoprotection to retinal pigment epithelium.
Also that year, Soica et al. demonstrated, using in vitro tests and in vivo skin cancer models, that the mixture of oleanolic and ursolic acids and in complex with cyclodextrin rendered a synergistic antitumor activity.14
A year earlier, Kowalczyk et al. showed that the combined action of phytochemicals – dietary calcium D-glucarate and topical ursolic acid and resveratrol – was effective in suppressing the initiation (with 7,12-dimethylbenz[a]anthracene [DMBA]) and promotion (with TPA) of skin tumorigenesis in SENCAR mice. Ursolic acid alone or in combination with calcium D-glucarate significantly diminished epidermal hyperplasia when applied during promotion. All of the antipromotion protocols led to significant decreases in cyclooxygenase-2 and interleukin (IL)-6 expression. The researchers concluded that ursolic acid strongly inhibits skin tumor promotion and inflammatory signaling, and warrants attention as a potential preventive agent against skin and other epithelial cancers.15 Kowalczyk et al. had previously found that ursolic acid and other phytochemicals displayed significant in vitro and in vivo antioxidant and antitumorigenic activity, inhibiting murine skin carcinogenesis by blunting tumor initiation and tumor promotion/progression.16
In 2006, beta-ursolic acid isolated from Salvia officinalis was found by Jedinák et al. to be effective in suppressing lung colonization of beta16 mouse melanoma cells in vivo.17
Huang et al. showed in 1994 that extracts of the leaves of Rosmarinus officinalis (rosemary) were effective in suppressing tumor initiation and promotion in a two-stage skin tumorigenesis mouse model. Topically applied ursolic acid isolated from the leaves was found to hinder TPA-induced ear inflammation, ornithine decarboxylase activity, and tumor promotion. The number of tumors per mouse also declined significantly due to the topical application of ursolic acid concurrent with twice weekly application of the tumor-promoter TPA in DMBA-initiated mice.18
Antiaging and other activities
In 2015, Herndon et al. conducted an open-label clinical trial in 37 females (aged 35-60 years) to ascertain the effectiveness of an anti-aging moisturizer containing Astragalus membranaceus root extract, a peptide blend including palmitoyl tripeptide-38, standardized rosemary leaf extract (ursolic acid), tetrahexyldecyl ascorbate (THD ascorbate), and ubiquinone (coenzyme Q10). Subjects were instructed to apply the moisturizer once in the morning and once in the evening, and were assessed at baseline, and after 4, 8, and 12 weeks of twice daily application. Clinical evaluations after 8 weeks revealed a statistically significant improvement in all grading parameters (fine lines and wrinkles, clarity/brightness, visual roughness, tactile roughness, redness, hyperpigmentation, and overall appearance), with even more pronounced improvement at 12 weeks. The product was found to be mild and well tolerated, and digital photography reinforced clinical assessments and self-evaluations.19
Lee et al. reported in 2012 on in vitro results suggesting that ursolic acid was effective as an inhibitor of matrix metalloproteinase (MMP)-1 after UVB exposure and was more effective than retinoic acid.20
Based on studies with hairless mice, Lim et al. found in 2007 that ursolic and oleanolic acids can enhance the recovery of skin barrier function and, via peroxisome proliferator-activated receptor-alpha, spur epidermal keratinocyte differentiation. They concluded that both acids have potential for use as agents to promote epidermal permeability barrier function.21
In 2003, Soo et al. observed that pretreatment with ursolic acid inhibited UVA-induced oxidative stress and activation and expression of MMP-2 in HaCaT human keratinocytes. They concluded that ursolic acid may merit attention for the prevention of UVA-induced photoaging.22
Three years earlier, Yarosh et al. showed that liposomes containing ursolic acid augmented ceramide content in cultured normal human epidermal keratinocytes and collagen content in cultured normal human dermal fibroblasts. Over an 11-day period, clinical tests with the ursolic acid–containing liposome (Merotaine) revealed increases in the ceramide content in human skin.23 Two years later, many of the same researchers duplicated their results. This new study also demonstrated that ursolic acid liposomes raise ceramide levels in normal human epidermal keratinocytes, in contrast to the effects of retinoic acid, earlier shown to reduce such levels. They concluded that ursolic acid liposomes show promise for use alone or in combination to replenish or maintain cutaneous ceramide and collagen levels.24 Notably, ursolic acid is incorporated into topical oils and creams intended to confer rejuvenating effects to the skin.
Conclusion
Ursolic acid is a compelling ingredient. I especially will be interested in the results of ongoing human clinical trials of this triterpenoid for treating cancer and skin wrinkles. As it is, ursolic acid is known to exert significant inhibitory activity against tumor formation and tumor cell viability in the laboratory. Given its wide range of biologic activity, and some promising cutaneous results, there is reason to believe that ursolic acid has the potential to play an increasingly useful role in topical skin care agents and dermatologic practice.
References
1. J Dermatol. 2007 Sep;34(9):625-34.
2. Folia Histochem Cytobiol. 2011;49(4):664-9.
3. J Cosmet Dermatol. 2004 Jan;3(1):26-34.
4. J Enzyme Inhib Med Chem. 2011 Oct;26(5):616-42.
5. BMC Complement Altern Med. 2013 Oct 29;13:292.
6. J Biomed Biotechnol. 2010;2010:715739.
7. Fitoterapia. 2009 Apr;80(3):164-7.
8. Biosci Biotechnol Biochem. 2004 Jan;68(1):85-90.
9. J Ethnopharmacol. 2002 Oct;82(2-3):229-37.
10. Eur J Pharmacol. 1997 Sep 3;334(1):103-5.
11. Cancer Prev Res (Phila). 2015 Sep;8(9):817-25.
12. Melanoma Res. 2015 Apr;25(2):103-12.
13. Apoptosis. 2014 May;19(5):816-28.
14. Molecules. 2014 Apr 17;19(4):4924-40.
15. Int J Oncol. 2013 Sep;43(3):911-8.
16. Carcinogenesis. 2009 Jun;30(6):1008-15.
17. Z Naturforsch C. 2006 Nov-Dec;61(11-12):777-82.
18. Cancer Res. 1994 Feb 1;54(3):701-8.
19. J Drugs Dermatol. 2015 Jul;14(7):699-704.
20. Bioorg Khim. 2012 May-Jun;38(3):374-81.
21. J Dermatol. 2007;34(9):625-34.
22. Eur J Pharmacol. 2003 Aug 29;476(3):173-8.
23. Horm Res. 2000;54(5-6):318-21.
24. Arch Dermatol Res. 2002 Jan;293(11):569-75.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. Dr. Baumann also developed and owns the Baumann Skin Type Solution skin typing systems and related products.

CF patients live longer in Canada than in U.S.
People with cystic fibrosis (CF) survive an average of 10 years longer if they live in Canada than if they live in the United States, according to a report published online March 14 in Annals of Internal Medicine.
Differences between the two nations’ health care systems, including access to insurance, “may, in part, explain the Canadian survival advantage,” said Anne L. Stephenson, MD, PhD, of St. Michael’s Hospital, Toronto, and her associates.
Overall there were 9,654 U.S. deaths and 1,288 Canadian deaths during the study period, for nearly identical overall mortality between the two countries (21.2% and 21.7%, respectively). However, the median survival was 10 years longer in Canada (50.9 years) than in the United States (40.6 years), a gap that persisted across numerous analyses that adjusted for patient characteristics and clinical factors, including CF severity.
One particular difference between the two study populations was found to be key: Canada has single-payer universal health insurance, while the United States does not. When U.S. patients were categorized according to their insurance status, Canadians had a 44% lower risk of death than did U.S. patients receiving continuous Medicaid or Medicare (95% confidence interval, 0.45-0.71; P less than .001), a 36% lower risk than for U.S. patients receiving intermittent Medicaid or Medicare (95% CI, 0.51-0.80; P = .002), and a 77% lower risk of death than U.S. patients with no or unknown health insurance (95% CI, 0.14-0.37; P less than .001), the investigators said (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M16-0858). In contrast, there was no survival advantage for Canadian patients when compared with U.S. patients who had private health insurance. This “[raises] the question of whether a disparity exists in access to therapeutic approaches or health care delivery,” the researchers noted.
This study was supported by the U.S. Cystic Fibrosis Foundation, Cystic Fibrosis Canada, the National Institutes of Health, and the U.S. Food and Drug Administration. Dr. Stephenson reported grants from the Cystic Fibrosis Foundation and fees from Cystic Fibrosis Canada. Several of the study’s other authors reported receiving fees from various sources and one of those authors reported serving on the boards of pharmaceutical companies.
Stephenson et al. confirmed that there is a “marked” [survival] “advantage” for CF patients in Canada, compared with those in the United States.
A key finding of this study was the survival difference between the two countries disappeared when U.S. patients insured by Medicaid or Medicare and those with no health insurance were excluded from the analysis. The fundamental differences between the two nations’ health care systems seem to be driving this disparity in survival.
Median predicted survival for all Canadians is higher than that of U.S. citizens, and this difference has increased over the last 2 decades.
Patrick A. Flume, MD, is at the Medical University of South Carolina in Charleston. Donald R. VanDevanter, PhD, is at Case Western Reserve University in Cleveland. They both reported ties to the Cystic Fibrosis Foundation. Dr. Flume and Dr. VanDevanter made these remarks in an editorial accompanying Dr. Stephenson’s report (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M17-0564).
Stephenson et al. confirmed that there is a “marked” [survival] “advantage” for CF patients in Canada, compared with those in the United States.
A key finding of this study was the survival difference between the two countries disappeared when U.S. patients insured by Medicaid or Medicare and those with no health insurance were excluded from the analysis. The fundamental differences between the two nations’ health care systems seem to be driving this disparity in survival.
Median predicted survival for all Canadians is higher than that of U.S. citizens, and this difference has increased over the last 2 decades.
Patrick A. Flume, MD, is at the Medical University of South Carolina in Charleston. Donald R. VanDevanter, PhD, is at Case Western Reserve University in Cleveland. They both reported ties to the Cystic Fibrosis Foundation. Dr. Flume and Dr. VanDevanter made these remarks in an editorial accompanying Dr. Stephenson’s report (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M17-0564).
Stephenson et al. confirmed that there is a “marked” [survival] “advantage” for CF patients in Canada, compared with those in the United States.
A key finding of this study was the survival difference between the two countries disappeared when U.S. patients insured by Medicaid or Medicare and those with no health insurance were excluded from the analysis. The fundamental differences between the two nations’ health care systems seem to be driving this disparity in survival.
Median predicted survival for all Canadians is higher than that of U.S. citizens, and this difference has increased over the last 2 decades.
Patrick A. Flume, MD, is at the Medical University of South Carolina in Charleston. Donald R. VanDevanter, PhD, is at Case Western Reserve University in Cleveland. They both reported ties to the Cystic Fibrosis Foundation. Dr. Flume and Dr. VanDevanter made these remarks in an editorial accompanying Dr. Stephenson’s report (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M17-0564).
People with cystic fibrosis (CF) survive an average of 10 years longer if they live in Canada than if they live in the United States, according to a report published online March 14 in Annals of Internal Medicine.
Differences between the two nations’ health care systems, including access to insurance, “may, in part, explain the Canadian survival advantage,” said Anne L. Stephenson, MD, PhD, of St. Michael’s Hospital, Toronto, and her associates.
Overall there were 9,654 U.S. deaths and 1,288 Canadian deaths during the study period, for nearly identical overall mortality between the two countries (21.2% and 21.7%, respectively). However, the median survival was 10 years longer in Canada (50.9 years) than in the United States (40.6 years), a gap that persisted across numerous analyses that adjusted for patient characteristics and clinical factors, including CF severity.
One particular difference between the two study populations was found to be key: Canada has single-payer universal health insurance, while the United States does not. When U.S. patients were categorized according to their insurance status, Canadians had a 44% lower risk of death than did U.S. patients receiving continuous Medicaid or Medicare (95% confidence interval, 0.45-0.71; P less than .001), a 36% lower risk than for U.S. patients receiving intermittent Medicaid or Medicare (95% CI, 0.51-0.80; P = .002), and a 77% lower risk of death than U.S. patients with no or unknown health insurance (95% CI, 0.14-0.37; P less than .001), the investigators said (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M16-0858). In contrast, there was no survival advantage for Canadian patients when compared with U.S. patients who had private health insurance. This “[raises] the question of whether a disparity exists in access to therapeutic approaches or health care delivery,” the researchers noted.
This study was supported by the U.S. Cystic Fibrosis Foundation, Cystic Fibrosis Canada, the National Institutes of Health, and the U.S. Food and Drug Administration. Dr. Stephenson reported grants from the Cystic Fibrosis Foundation and fees from Cystic Fibrosis Canada. Several of the study’s other authors reported receiving fees from various sources and one of those authors reported serving on the boards of pharmaceutical companies.
People with cystic fibrosis (CF) survive an average of 10 years longer if they live in Canada than if they live in the United States, according to a report published online March 14 in Annals of Internal Medicine.
Differences between the two nations’ health care systems, including access to insurance, “may, in part, explain the Canadian survival advantage,” said Anne L. Stephenson, MD, PhD, of St. Michael’s Hospital, Toronto, and her associates.
Overall there were 9,654 U.S. deaths and 1,288 Canadian deaths during the study period, for nearly identical overall mortality between the two countries (21.2% and 21.7%, respectively). However, the median survival was 10 years longer in Canada (50.9 years) than in the United States (40.6 years), a gap that persisted across numerous analyses that adjusted for patient characteristics and clinical factors, including CF severity.
One particular difference between the two study populations was found to be key: Canada has single-payer universal health insurance, while the United States does not. When U.S. patients were categorized according to their insurance status, Canadians had a 44% lower risk of death than did U.S. patients receiving continuous Medicaid or Medicare (95% confidence interval, 0.45-0.71; P less than .001), a 36% lower risk than for U.S. patients receiving intermittent Medicaid or Medicare (95% CI, 0.51-0.80; P = .002), and a 77% lower risk of death than U.S. patients with no or unknown health insurance (95% CI, 0.14-0.37; P less than .001), the investigators said (Ann. Intern. Med. 2017 Mar 14. doi: 10.7326/M16-0858). In contrast, there was no survival advantage for Canadian patients when compared with U.S. patients who had private health insurance. This “[raises] the question of whether a disparity exists in access to therapeutic approaches or health care delivery,” the researchers noted.
This study was supported by the U.S. Cystic Fibrosis Foundation, Cystic Fibrosis Canada, the National Institutes of Health, and the U.S. Food and Drug Administration. Dr. Stephenson reported grants from the Cystic Fibrosis Foundation and fees from Cystic Fibrosis Canada. Several of the study’s other authors reported receiving fees from various sources and one of those authors reported serving on the boards of pharmaceutical companies.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point: People with cystic fibrosis survive an average of 10 years longer if they live in Canada than if they live in the United States.
Major finding: Canadians with CF had a 44% lower risk of death than U.S. patients receiving Medicaid or Medicare and a striking 77% lower risk of death than U.S. patients with no health insurance, but the same risk as U.S. patients with private insurance.
Data source: A population-based cohort study involving 45,448 patients in a U.S. registry and 5,941 in a Canadian registry in 1990-2013.
Disclosures: This study was supported by the U.S. Cystic Fibrosis Foundation, Cystic Fibrosis Canada, the National Institutes of Health, and the Food and Drug Administration. The authors’ financial disclosures are available at www.acponline.org
One Hundred Case Series of Vocal Cord Dysfunction in a Military Treatment Facility
Vocal cord dysfunction (VCD), also known as paradoxical vocal cord movement, is described as paroxysms of glottis obstruction due to true vocal cord adduction.1 Since VCD presents as a constellation of symptoms associated with dyspnea, it often is misdiagnosed as asthma.2 Vocal cord dysfunction often manifests as episodic dyspnea and wheezing, may occur with exercise, and may be minimally responsive to initial therapies. Flattened inspiratory curves may be noted on pulmonary function tests (PFTs), but direct laryngoscopy is the gold standard for diagnosis.3 A cohort of proven patients with VCD with a plateau in the inspiratory curve of PFTs also had a plateau on expiratory phase in 81% of cases.4
The differential diagnosis of patients presenting with upper airway symptoms is broad. It must include VCD, asthma, angioedema, laryngomalacia, vocal cord polyps, vocal cord tumors, and neurologic conditions such as brain stem compression or movement disorders. Essentially, all movement disorders of vocal cords must be considered, and organic causes of this movement disorder can be evaluated by visualization of the vocal cords. Triggers for VCD include exercise, airborne irritants, gastroesophageal reflux disease (GERD), allergic rhinitis, medications, and psychological conditions.5 Additionally, VCD can coexist with asthma, further complicating accurate diagnoses.6
Therapies are reported in case studies, but no large randomized controlled trials exist to evaluate current therapy options. Primary treatments of asthma therapy were largely ineffective, and ideal therapy includes a multidisciplinary approach, including speech therapy to optimize laryngeal control and treatment of all identified laryngeal irritants.6
The prevalence of VCD is unknown, with no prospective cohort studies completed to date and conflicting diagnostic criteria used in many case studies.7 A prevalence of 2.8% was noted in one particular cohort of 1,028 patients admitted to a rehabilitation center in a calendar year with the primary pulmonary diagnosis on admission.6 Females seemed to be affected at a higher ratio than were males, 2 to 3 females per 1 male diagnosis.7
In the military population, certain risk factors were noted in returning deployed members, including anxiety/high stress, exercise, and acute respiratory illnesses.8 In that particular cohort, 72% positive predictive value was noted for VCD if flattened inspiratory flow loops with negative methacholine challenge were present.
Diagnostic criteria are challenging, as symptoms such as dyspnea may be present acutely, last < 2 minutes, be self-limiting, and completely resolve outside of acute events. Stridor may be noted, primarily above the vocal cords, and less audible on chest auscultation.6 A goal of therapy, in addition to dedicated speech pathologist input, is optimizing comedical conditions, including GERD, allergic rhinitis, concomitant asthma, and any psychological diagnoses.9
Athletes are a particular subset of patients with VCD who are crucial to appropriately diagnose, including a detailed history and physical, PFTs, and proceeding to direct laryngoscopy to confirm diagnoses.10 Behavioral management includes rescue breathing techniques, and speech therapy programs focus on relaxation of the larynx and diaphragmatic breathing techniques, with the goal of establishing sense of control during acute events.10 Military service members are expected to operate at a high-intensity level similar to that of athletes, and treatments considered for athletes are applicable to military service members as well. Military strength and cardiovascular standards are measured by a combination of push-ups, sit-ups, and a run test, in addition to waist measurements. Some of the cohort were identified during physical fitness standard failures, usually in the run test, and ultimately received a pulmonology referral for wheezing or dyspnea with exertion. The objective of this retrospective cohort study was to evaluate 100 consecutively diagnosed cases of VCD in a military treatment facility.
Methods
The authors conducted a retrospective chart review of DoD military medical records of outpatient diagnoses in 100 consecutive diagnoses of VCD from January 2011 to February 2014. Institutional review board approval was obtained under Project RSM20130001E by the Exempt Determination Official at Eglin Air Force Base (AFB), Florida.
All cases were identified at time of VCD visualization and were diagnosed with video stroboscopy by speech therapy or by visual laryngoscopy by the otolaryngology or pulmonology departments via direct visualization.
Cases were collected chronologically, and all diagnosed cases at Eglin AFB hospital were included. Follow-up was scheduled with all patients diagnosed in Speech Therapy, and most patients were concurrently treated by Pulmonology or Allergy/Immunology. Pulmonary function tests were obtained in 98 of the 100 diagnosed cases. Patients eligible for care at Eglin AFB included active-duty and Reserve military members plus dependents and retirees.
The majority of patients diagnosed in this cohort were seen and diagnosed by Speech Therapy. Video stroboscopy is based on the principle that a movement of an object higher than a certain flicker rate appears to stand still to direct visualization, but with a rate of light exposure and imaging above the flicker rate by video, the true movement of the object can be identified.¹¹ Video stroboscopy is considered highly sensitive for organic disorders of vocal cords, but it is not specific for either organic or dysfunctional disorders.¹¹ It is still the gold standard above direct visualization, as it can detect abnormal movement of vocal cords above the critical rate that the human eye would perceive as not moving due to the frequency of movement (Figures 1 & 2).¹¹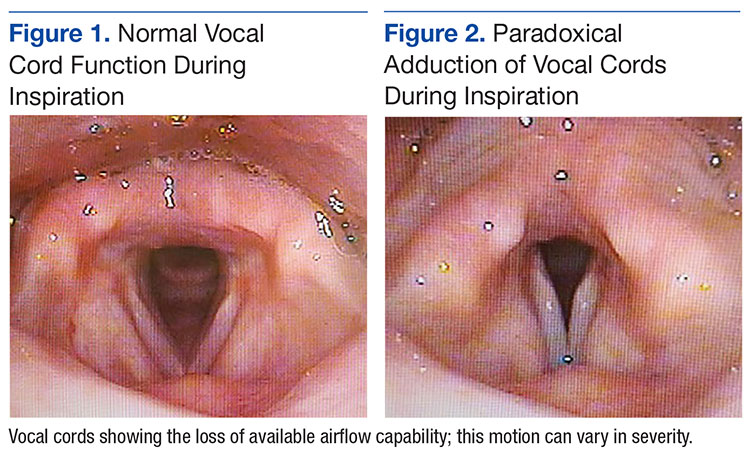
In an older study, laryngoscopy was able to diagnose 100% of patients with symptomatic paradoxical vocal cord movement and additional 60% asymptomatic patients with a constellation of symptoms consistent with paradoxical vocal cord movement.¹²
Speech Therapy; Ear, Nose, and Throat (ENT); and Pulmonology may not perform direct visualization in these patients at initial presentation due to other suspected diagnoses. A more common test is the PFT, especially if asthma or other airway tract diseases are suspected (Figure 3).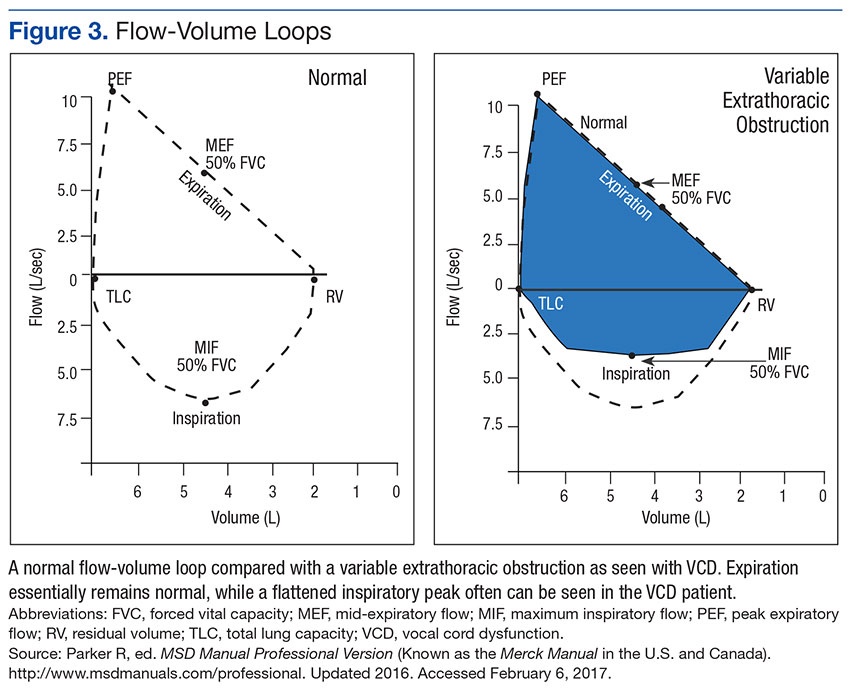
Patient Descriptions
Study patients were referred for a variety of reasons, often from primary care clinics for concerns for asthma, episodic dyspnea, wheezing, or decreased exercise tolerance thought to be related to pulmonary or allergy causes. Pulmonology worked closely with Speech Therapy and referred VCD cases for speech evaluation, including video stroboscopy. Notably, of the patients in this cohort, although some were suspected to have asthma, those patients were ruled out during part of the pulmonology evaluation, both with PFT testing and methacholine challenges. An asthma diagnosis is important in a military treatment facility, as asthma is often grounds for discharge.
Patients ranged in age from 13 to 68 years, with a median age at 31 years diagnosis. Thirty-nine females and 61 males comprised the total case series. Speech Therapy diagnosed 97 patients, 96 were diagnosed at Eglin AFB hospital via stroboscopy. One patient was diagnosed off-base by Speech Therapy via direct visualization, 1 patient was diagnosed by Pulmonology on-base via direct visualization, and 2 patients were diagnosed by ENT on-base via direct visualization. These patients had direct laryngoscopy completed, often to rule out other organic causes for upper airway disease processes, and were found to have visual paradoxical vocal cord movement. Ninety-eight patients completed PFTs. Several patients were lost to follow-up, as can be common in a military population with frequent moves or members leaving service.
On record review, patient symptoms were present in the range of 2 months to 20 years, with a median duration of symptomatic reports lasting 2 years prior to diagnosis. Common diagnoses prior to visual VCD diagnosis included asthma, exercise-induced asthma, anxiety, and episodic wheezing. Risk factors that were evaluated in this case series included age, sex, body mass index (BMI), GERD, allergic rhinitis, postnasal drip, active smoker, previous smoker, and mental health diagnoses (Figure 4).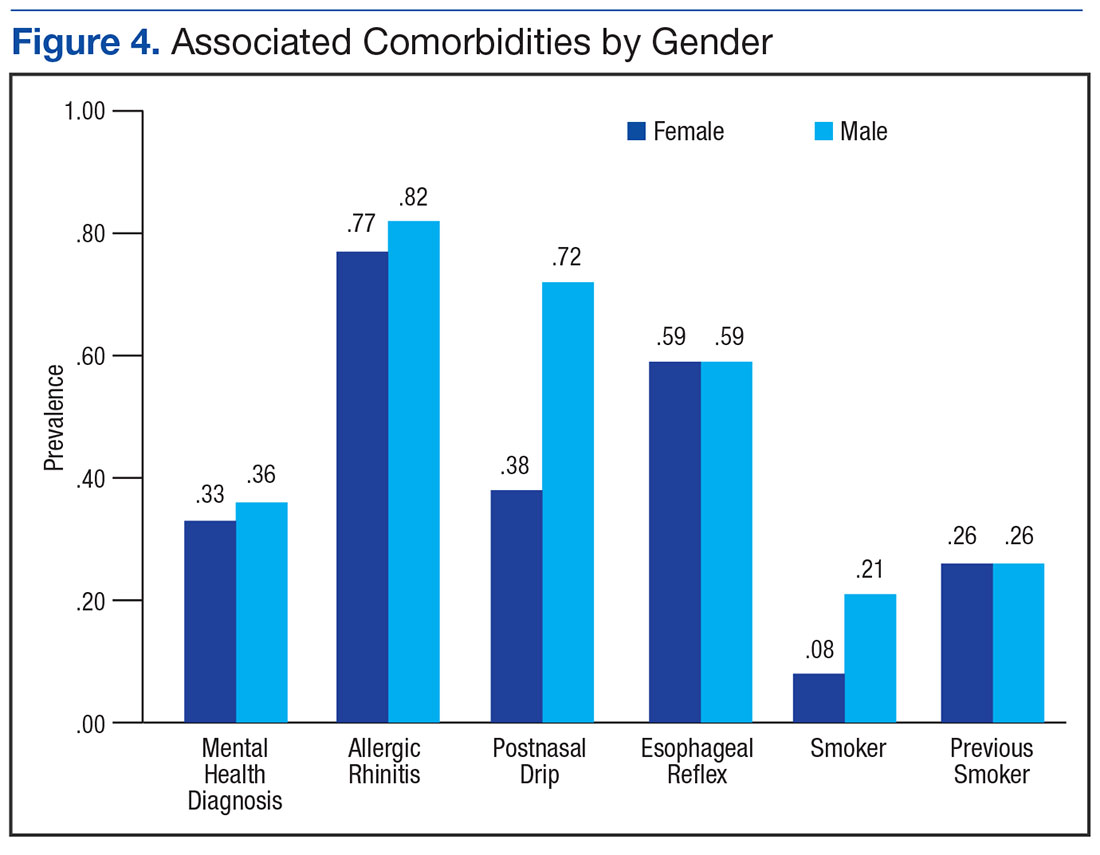
Pulmonary function test results were analyzed on 98 patients, including forced expiratory volume in 1 second (FEV1); forced vital capacity (FVC), FEV1/FVC ratio; peak inspiratory flow (PIF) and peak expiratory flow (PEF)—available in 97 studies; forced expiratory flow (FEF) at 25% to 75% of FVC (FEF 25%-75%)—available in 96 studies; and maximum voluntary ventilation (MVV) and MVV/FEV1 ratio—available in 60 of 98 PFTs.
Interventions
All patients diagnosed by Speech Therapy on-base were provided with laryngeal relaxation techniques, diaphragmatic breathing techniques, and controlled inhale/exhale techniques at time of diagnosis, with frequent follow-up scheduled with Speech Therapy and Pulmonology. All diagnoses potentially contributing to laryngeal irritation were treated, including GERD, allergic rhinitis, smoking cessation, weight loss, and exercise recommendations as needed.
Patients reported improvement on follow-up appointments with Speech Therapy in overall control of symptoms, subjectively categorized as poor improvement, partial improvement, and complete improvement. This was a subjective measurement of improvement and fully dependent on follow-up care and patient reporting for improvement. No predefined number of follow-ups was determined; patients were followed monthly until they declined further care, fully improved, moved out of the military treatment system, or were lost to follow-up.
Treatment included structured Speech Therapy sessions. Response to treatment was subjectively qualified by patient report. Fifteen patients reported complete resolution of symptoms, 57 reported partial improvement, 24 reported poor improvement, and 4 patients were lost to follow-up.
Results
Risk factors for the diagnosis of VCD included possible associations with GERD, allergic rhinitis, smoking, prior smoking, BMI, and mental health diagnoses. Body mass index ranged from 17 to 36 in the case series, with median BMI of 27. Mental health diagnoses were present in 35 patients and included diagnoses of anxiety, depression, and adjustment disorders. Gastroesophageal reflux disease diagnosis was present in 59 of the case series patients, 80 had the diagnosis of allergic rhinitis, 63 were diagnosed with postnasal drip. Sixteen case series patients were current smokers. An additional 26 were previous smokers (at least 100 cigarettes in lifetime) for a total of 42 patients that were current or prior smokers.
The chart review was completed to evaluate for the presence of these diagnoses, which included previous treatments; for example, proton pump inhibitors for GERD, antidepressants for depression, or intranasal steroids for allergic rhinitis. The diagnosis was counted as present if the patient was currently being treated for the particular diagnosis in question.
PFT Data
Data from PFTs were available for 98 of 100 cases diagnosed. Review of data across all 98 patients is noted for median FEV1 of 3.6, a median FVC of 4.5, with ratio of 0.80. 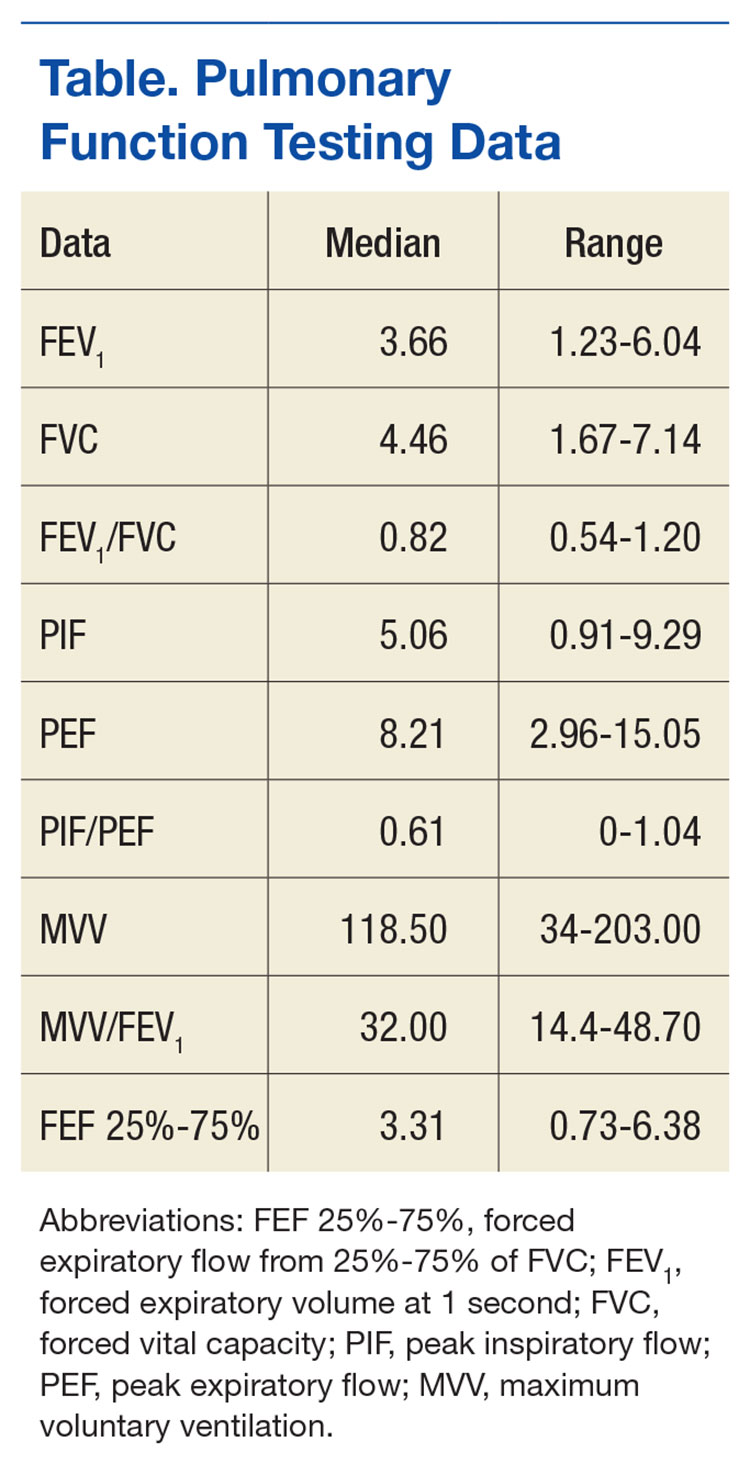
Since PFT values vary according to age, sex, and ethnicity, PFTs were analyzed for percent predicted values based on age, gender, and race. Notably, median values for FEV1, FVC, and PEF were all close to 100% of the predicted value. The MVV percent predicted was available in 60 cases and was 93% of predicted values. The most significant difference from expected values was FEF 25% to 75%, at 84% of expected results.
Flow-volume loop evaluations on the 97 PFTs available were completed, and 58 of the 97 were noted for variable extrathoracic airway obstruction consistent with inspiratory inhibition in the patient population. This is 60% of the available PFTs in this cohort study.
Discussion
This retrospective chart review of 100 consecutive VCD diagnoses in a military treatment facility reinforces many of the findings currently available in the literature. As illustrated in a Chest review article, the diagnosis of VCD on history, physical examination, or PFTs remains ellusive.1 The PFT evaluation contains some subjectivity regarding the flattening of inspiratory flow-volume loops and is not routinely reported in PFT results. In patients diagnosed with VCD, a clear consensus of treatment modalities remains lacking. Modification of risk factors (allergic rhinitis, GERD, smoking cessation, weight loss) assisted in self-reported patient improvement, as did focused speech therapy.
The median age of 31 years, likely reflected the younger military population served at Eglin AFB. Seventy-five of these patients were currently on active duty, 6 were retired from active duty (veterans), and 19 were dependents. The median time of symptoms to diagnosis was 2 years. Prior misdiagnosis with other diseases such as asthma was common. Also, referral to Pulmonology and Speech Therapy was usually completed after failed outpatient primary care management for the alternative diagnoses.
Improvement with therapy was mixed, and during the time of documented follow-up, 72 patients reported complete or partial improvement. Most active-duty patients in the partial improvement category based this subjective reporting on their ability to meet military physical fitness standards.
Previous data suggested a female predominance, but this study population was 61% male. Military populations are about 80% to 85% male, so an increase in male diagnosis is expected.
Many patients in the patient cohort arrived as a result of Pulmonology referrals with a presumptive diagnoses of asthma but were determined not to have asthma through PFT results inconsistent with asthma, no improvement with β-agonist therapies, and negative methacholine challenges (if performed). These results prompted evaluations for other conditions and eventually a VCD diagnosis. As noted, exclusion of asthma is of particular importance in a military population, as medical discharges often are pursued in service members with asthma whether controlled or uncontrolled. Lag time to referral also is possible in failures of military physical, which prompted medical evaluation once several failures had occurred over a 1- to 2-year time frame.
The PFT data evaluation was inconclusive for statistically significant changes when compared with age-matched normal PFT values. This also was noted in previous studies of VCD cases. Most notable was percent predicted values of FEF 25% to 75%, with 84% of expected values. The FEV1, FVC, and PEF all fell within predicted values of normal, despite wide ranges in age, sex, and ethnicity among the subjects. Inspiratory flattening consistent with extrathoracic obstruction was present in 58 of the 97 PFTs available for review at Eglin AFB.
Limitations
Limitations to this retrospective case series are illustrated here. Cases were found only when VCD was diagnosed and coded; and it is the authors’ suspicion that many have been misdiagnosed or improperly treated for asthma or other pulmonary/oropharynx conditions. If providers are not familiar with VCD or if PFT readings do not comment on inspiratory findings, diagnosis is less likely. Some of the authors’ colleagues already have determined that postdeployment prevalence of VCD seems to be elevated.8
This cohort was completed on all patients in a military treatment facility, with 75 active-duty personnel, 6 veterans, and 19 dependents of varying ages. This case series is retrospective and tabulates suspected risk factors; stronger and more informative studies could certainly be completed in prospective studies (although likely difficult with low prevalence) or in treatment comparison studies at the time of diagnosis.
Since the cohort had varied and lengthy time to diagnosis from onset of related symptoms, the treatment patients received prior to diagnosis differed extensively. Diagnosis was completed by numerous primary care managers or other subspecialties prior to arrival to Pulmonology and Speech Therapy at Eglin AFB. Once diagnosed in Speech Therapy, consistent treatment options were provided to patients in accordance with standard of care.
It is the authors’ suspicion that VCD may have a higher prevalence than previously reported in the literature. Military service members are tested annually or biannually on physical fitness standards and are evaluated for medical reasons for recurrent fitness standard failures. This selection of patients is more likely to have a VCD evaluation as part of a comprehensive evaluation than is a healthy adult in a civilian population. A prospective study in military service members would be more fruitful and possibly yield a higher prevalence postdeployment.
Conclusion
Vocal cord dysfunction remains a difficult diagnosis to treat, because multiple comorbidities likely contribute to the diagnosis. This retrospective case series attempted to compile common themes and noted that most of the patients had 2 or more risk factors of smoking, allergic rhinitis, GERD, or mental health diagnoses. A prospective trial would be ideal to evaluate VCD further. A focused trial in the particular communities of athletes or of military service members may be of increased benefit to better define VCD. It is notable that 100 cases were found in a relatively short period for a community hospital, and prevalence may be higher than previously reported.
1. Morris MJ, Christopher KL. Diagnostic criteria for the classification of vocal cord dysfunction. Chest. 2010;138(5):1213-1223.
2. National Heart, Lung, and Blood Institute. Expert panel report 3: guidelines for the diagnoses and management of asthma. Full report 2007. https://www.nhlbi.nih.gov/files/docs/guidelines/asthgdln .pdf. Published 2007.Accessed February 1, 2017.
3. Newman KB, Mason UG III, Schmaling KB. Clinical features of vocal cord dysfunction. Am J Respir Crit Care Med. 1995;152(4, pt 1):1382-1386.
4. Sanz Santiago V, López Neyra A, Almería Gil E, Villa Asensi JR. Spirometry patterns in vocal cord dysfunction [in Spanish]. An Pediatr (Barc). 2013;78(3):173-177.
5. Deckert J, Deckert L. Vocal cord dysfunction. Am Fam Physician. 2010;81(2):156-159.
6. Benninger C, Parsons JP, Mastronarde JG. Vocal cord dysfunction and asthma. Curr Opin Pulm Med. 2011;17(1):45-49.
7. Campainha S, Ribeiro C, Guimar M, Lima R. Vocal cord dysfunction: a frequently forgotten entity. Case Rep Pulmonol. 2012;2012:525493.
8. Morris MJ, Oleszewski RT, Sterner JB, Allan PF. Vocal cord dysfunction related to combat deployment. Mil Med. 2013;178(11):1208-1212.
9. Al-Alwan A, Kaminsky D. Vocal cord dysfunction in athletes: clinical presentation and review of the literature. Phys Sportsmed. 2012;40(2):22-27.
10. Kenn K, Schmitz M. Prevalence of vocal cord dysfunction in patients with dyspnea. First prospective clinical study. Am J Respir Crit Care Med. 1997;155:A965.
11. Wendler, J, Nawka, T, Verges, D. Instructional course: videolaryngo-stroboscopy and phonetography—basic tools for diagnostics and documentation in the voice clinic. Poster presented at: 15th European Congress of Oto-Rhino-Laryngology, Head and Neck Surgery; September 11-16, 2004; Rodos-Kos, Greece.
12. Ibrahim WH, Gheriani HA, Almohamed AA, Raza T. Paradoxical vocal cord motion disorder: past, present and future. Postgrad Med J. 2007;83(977):164-172.
Vocal cord dysfunction (VCD), also known as paradoxical vocal cord movement, is described as paroxysms of glottis obstruction due to true vocal cord adduction.1 Since VCD presents as a constellation of symptoms associated with dyspnea, it often is misdiagnosed as asthma.2 Vocal cord dysfunction often manifests as episodic dyspnea and wheezing, may occur with exercise, and may be minimally responsive to initial therapies. Flattened inspiratory curves may be noted on pulmonary function tests (PFTs), but direct laryngoscopy is the gold standard for diagnosis.3 A cohort of proven patients with VCD with a plateau in the inspiratory curve of PFTs also had a plateau on expiratory phase in 81% of cases.4
The differential diagnosis of patients presenting with upper airway symptoms is broad. It must include VCD, asthma, angioedema, laryngomalacia, vocal cord polyps, vocal cord tumors, and neurologic conditions such as brain stem compression or movement disorders. Essentially, all movement disorders of vocal cords must be considered, and organic causes of this movement disorder can be evaluated by visualization of the vocal cords. Triggers for VCD include exercise, airborne irritants, gastroesophageal reflux disease (GERD), allergic rhinitis, medications, and psychological conditions.5 Additionally, VCD can coexist with asthma, further complicating accurate diagnoses.6
Therapies are reported in case studies, but no large randomized controlled trials exist to evaluate current therapy options. Primary treatments of asthma therapy were largely ineffective, and ideal therapy includes a multidisciplinary approach, including speech therapy to optimize laryngeal control and treatment of all identified laryngeal irritants.6
The prevalence of VCD is unknown, with no prospective cohort studies completed to date and conflicting diagnostic criteria used in many case studies.7 A prevalence of 2.8% was noted in one particular cohort of 1,028 patients admitted to a rehabilitation center in a calendar year with the primary pulmonary diagnosis on admission.6 Females seemed to be affected at a higher ratio than were males, 2 to 3 females per 1 male diagnosis.7
In the military population, certain risk factors were noted in returning deployed members, including anxiety/high stress, exercise, and acute respiratory illnesses.8 In that particular cohort, 72% positive predictive value was noted for VCD if flattened inspiratory flow loops with negative methacholine challenge were present.
Diagnostic criteria are challenging, as symptoms such as dyspnea may be present acutely, last < 2 minutes, be self-limiting, and completely resolve outside of acute events. Stridor may be noted, primarily above the vocal cords, and less audible on chest auscultation.6 A goal of therapy, in addition to dedicated speech pathologist input, is optimizing comedical conditions, including GERD, allergic rhinitis, concomitant asthma, and any psychological diagnoses.9
Athletes are a particular subset of patients with VCD who are crucial to appropriately diagnose, including a detailed history and physical, PFTs, and proceeding to direct laryngoscopy to confirm diagnoses.10 Behavioral management includes rescue breathing techniques, and speech therapy programs focus on relaxation of the larynx and diaphragmatic breathing techniques, with the goal of establishing sense of control during acute events.10 Military service members are expected to operate at a high-intensity level similar to that of athletes, and treatments considered for athletes are applicable to military service members as well. Military strength and cardiovascular standards are measured by a combination of push-ups, sit-ups, and a run test, in addition to waist measurements. Some of the cohort were identified during physical fitness standard failures, usually in the run test, and ultimately received a pulmonology referral for wheezing or dyspnea with exertion. The objective of this retrospective cohort study was to evaluate 100 consecutively diagnosed cases of VCD in a military treatment facility.
Methods
The authors conducted a retrospective chart review of DoD military medical records of outpatient diagnoses in 100 consecutive diagnoses of VCD from January 2011 to February 2014. Institutional review board approval was obtained under Project RSM20130001E by the Exempt Determination Official at Eglin Air Force Base (AFB), Florida.
All cases were identified at time of VCD visualization and were diagnosed with video stroboscopy by speech therapy or by visual laryngoscopy by the otolaryngology or pulmonology departments via direct visualization.
Cases were collected chronologically, and all diagnosed cases at Eglin AFB hospital were included. Follow-up was scheduled with all patients diagnosed in Speech Therapy, and most patients were concurrently treated by Pulmonology or Allergy/Immunology. Pulmonary function tests were obtained in 98 of the 100 diagnosed cases. Patients eligible for care at Eglin AFB included active-duty and Reserve military members plus dependents and retirees.
The majority of patients diagnosed in this cohort were seen and diagnosed by Speech Therapy. Video stroboscopy is based on the principle that a movement of an object higher than a certain flicker rate appears to stand still to direct visualization, but with a rate of light exposure and imaging above the flicker rate by video, the true movement of the object can be identified.¹¹ Video stroboscopy is considered highly sensitive for organic disorders of vocal cords, but it is not specific for either organic or dysfunctional disorders.¹¹ It is still the gold standard above direct visualization, as it can detect abnormal movement of vocal cords above the critical rate that the human eye would perceive as not moving due to the frequency of movement (Figures 1 & 2).¹¹
In an older study, laryngoscopy was able to diagnose 100% of patients with symptomatic paradoxical vocal cord movement and additional 60% asymptomatic patients with a constellation of symptoms consistent with paradoxical vocal cord movement.¹²
Speech Therapy; Ear, Nose, and Throat (ENT); and Pulmonology may not perform direct visualization in these patients at initial presentation due to other suspected diagnoses. A more common test is the PFT, especially if asthma or other airway tract diseases are suspected (Figure 3).
Patient Descriptions
Study patients were referred for a variety of reasons, often from primary care clinics for concerns for asthma, episodic dyspnea, wheezing, or decreased exercise tolerance thought to be related to pulmonary or allergy causes. Pulmonology worked closely with Speech Therapy and referred VCD cases for speech evaluation, including video stroboscopy. Notably, of the patients in this cohort, although some were suspected to have asthma, those patients were ruled out during part of the pulmonology evaluation, both with PFT testing and methacholine challenges. An asthma diagnosis is important in a military treatment facility, as asthma is often grounds for discharge.
Patients ranged in age from 13 to 68 years, with a median age at 31 years diagnosis. Thirty-nine females and 61 males comprised the total case series. Speech Therapy diagnosed 97 patients, 96 were diagnosed at Eglin AFB hospital via stroboscopy. One patient was diagnosed off-base by Speech Therapy via direct visualization, 1 patient was diagnosed by Pulmonology on-base via direct visualization, and 2 patients were diagnosed by ENT on-base via direct visualization. These patients had direct laryngoscopy completed, often to rule out other organic causes for upper airway disease processes, and were found to have visual paradoxical vocal cord movement. Ninety-eight patients completed PFTs. Several patients were lost to follow-up, as can be common in a military population with frequent moves or members leaving service.
On record review, patient symptoms were present in the range of 2 months to 20 years, with a median duration of symptomatic reports lasting 2 years prior to diagnosis. Common diagnoses prior to visual VCD diagnosis included asthma, exercise-induced asthma, anxiety, and episodic wheezing. Risk factors that were evaluated in this case series included age, sex, body mass index (BMI), GERD, allergic rhinitis, postnasal drip, active smoker, previous smoker, and mental health diagnoses (Figure 4).
Pulmonary function test results were analyzed on 98 patients, including forced expiratory volume in 1 second (FEV1); forced vital capacity (FVC), FEV1/FVC ratio; peak inspiratory flow (PIF) and peak expiratory flow (PEF)—available in 97 studies; forced expiratory flow (FEF) at 25% to 75% of FVC (FEF 25%-75%)—available in 96 studies; and maximum voluntary ventilation (MVV) and MVV/FEV1 ratio—available in 60 of 98 PFTs.
Interventions
All patients diagnosed by Speech Therapy on-base were provided with laryngeal relaxation techniques, diaphragmatic breathing techniques, and controlled inhale/exhale techniques at time of diagnosis, with frequent follow-up scheduled with Speech Therapy and Pulmonology. All diagnoses potentially contributing to laryngeal irritation were treated, including GERD, allergic rhinitis, smoking cessation, weight loss, and exercise recommendations as needed.
Patients reported improvement on follow-up appointments with Speech Therapy in overall control of symptoms, subjectively categorized as poor improvement, partial improvement, and complete improvement. This was a subjective measurement of improvement and fully dependent on follow-up care and patient reporting for improvement. No predefined number of follow-ups was determined; patients were followed monthly until they declined further care, fully improved, moved out of the military treatment system, or were lost to follow-up.
Treatment included structured Speech Therapy sessions. Response to treatment was subjectively qualified by patient report. Fifteen patients reported complete resolution of symptoms, 57 reported partial improvement, 24 reported poor improvement, and 4 patients were lost to follow-up.
Results
Risk factors for the diagnosis of VCD included possible associations with GERD, allergic rhinitis, smoking, prior smoking, BMI, and mental health diagnoses. Body mass index ranged from 17 to 36 in the case series, with median BMI of 27. Mental health diagnoses were present in 35 patients and included diagnoses of anxiety, depression, and adjustment disorders. Gastroesophageal reflux disease diagnosis was present in 59 of the case series patients, 80 had the diagnosis of allergic rhinitis, 63 were diagnosed with postnasal drip. Sixteen case series patients were current smokers. An additional 26 were previous smokers (at least 100 cigarettes in lifetime) for a total of 42 patients that were current or prior smokers.
The chart review was completed to evaluate for the presence of these diagnoses, which included previous treatments; for example, proton pump inhibitors for GERD, antidepressants for depression, or intranasal steroids for allergic rhinitis. The diagnosis was counted as present if the patient was currently being treated for the particular diagnosis in question.
PFT Data
Data from PFTs were available for 98 of 100 cases diagnosed. Review of data across all 98 patients is noted for median FEV1 of 3.6, a median FVC of 4.5, with ratio of 0.80.
Since PFT values vary according to age, sex, and ethnicity, PFTs were analyzed for percent predicted values based on age, gender, and race. Notably, median values for FEV1, FVC, and PEF were all close to 100% of the predicted value. The MVV percent predicted was available in 60 cases and was 93% of predicted values. The most significant difference from expected values was FEF 25% to 75%, at 84% of expected results.
Flow-volume loop evaluations on the 97 PFTs available were completed, and 58 of the 97 were noted for variable extrathoracic airway obstruction consistent with inspiratory inhibition in the patient population. This is 60% of the available PFTs in this cohort study.
Discussion
This retrospective chart review of 100 consecutive VCD diagnoses in a military treatment facility reinforces many of the findings currently available in the literature. As illustrated in a Chest review article, the diagnosis of VCD on history, physical examination, or PFTs remains ellusive.1 The PFT evaluation contains some subjectivity regarding the flattening of inspiratory flow-volume loops and is not routinely reported in PFT results. In patients diagnosed with VCD, a clear consensus of treatment modalities remains lacking. Modification of risk factors (allergic rhinitis, GERD, smoking cessation, weight loss) assisted in self-reported patient improvement, as did focused speech therapy.
The median age of 31 years, likely reflected the younger military population served at Eglin AFB. Seventy-five of these patients were currently on active duty, 6 were retired from active duty (veterans), and 19 were dependents. The median time of symptoms to diagnosis was 2 years. Prior misdiagnosis with other diseases such as asthma was common. Also, referral to Pulmonology and Speech Therapy was usually completed after failed outpatient primary care management for the alternative diagnoses.
Improvement with therapy was mixed, and during the time of documented follow-up, 72 patients reported complete or partial improvement. Most active-duty patients in the partial improvement category based this subjective reporting on their ability to meet military physical fitness standards.
Previous data suggested a female predominance, but this study population was 61% male. Military populations are about 80% to 85% male, so an increase in male diagnosis is expected.
Many patients in the patient cohort arrived as a result of Pulmonology referrals with a presumptive diagnoses of asthma but were determined not to have asthma through PFT results inconsistent with asthma, no improvement with β-agonist therapies, and negative methacholine challenges (if performed). These results prompted evaluations for other conditions and eventually a VCD diagnosis. As noted, exclusion of asthma is of particular importance in a military population, as medical discharges often are pursued in service members with asthma whether controlled or uncontrolled. Lag time to referral also is possible in failures of military physical, which prompted medical evaluation once several failures had occurred over a 1- to 2-year time frame.
The PFT data evaluation was inconclusive for statistically significant changes when compared with age-matched normal PFT values. This also was noted in previous studies of VCD cases. Most notable was percent predicted values of FEF 25% to 75%, with 84% of expected values. The FEV1, FVC, and PEF all fell within predicted values of normal, despite wide ranges in age, sex, and ethnicity among the subjects. Inspiratory flattening consistent with extrathoracic obstruction was present in 58 of the 97 PFTs available for review at Eglin AFB.
Limitations
Limitations to this retrospective case series are illustrated here. Cases were found only when VCD was diagnosed and coded; and it is the authors’ suspicion that many have been misdiagnosed or improperly treated for asthma or other pulmonary/oropharynx conditions. If providers are not familiar with VCD or if PFT readings do not comment on inspiratory findings, diagnosis is less likely. Some of the authors’ colleagues already have determined that postdeployment prevalence of VCD seems to be elevated.8
This cohort was completed on all patients in a military treatment facility, with 75 active-duty personnel, 6 veterans, and 19 dependents of varying ages. This case series is retrospective and tabulates suspected risk factors; stronger and more informative studies could certainly be completed in prospective studies (although likely difficult with low prevalence) or in treatment comparison studies at the time of diagnosis.
Since the cohort had varied and lengthy time to diagnosis from onset of related symptoms, the treatment patients received prior to diagnosis differed extensively. Diagnosis was completed by numerous primary care managers or other subspecialties prior to arrival to Pulmonology and Speech Therapy at Eglin AFB. Once diagnosed in Speech Therapy, consistent treatment options were provided to patients in accordance with standard of care.
It is the authors’ suspicion that VCD may have a higher prevalence than previously reported in the literature. Military service members are tested annually or biannually on physical fitness standards and are evaluated for medical reasons for recurrent fitness standard failures. This selection of patients is more likely to have a VCD evaluation as part of a comprehensive evaluation than is a healthy adult in a civilian population. A prospective study in military service members would be more fruitful and possibly yield a higher prevalence postdeployment.
Conclusion
Vocal cord dysfunction remains a difficult diagnosis to treat, because multiple comorbidities likely contribute to the diagnosis. This retrospective case series attempted to compile common themes and noted that most of the patients had 2 or more risk factors of smoking, allergic rhinitis, GERD, or mental health diagnoses. A prospective trial would be ideal to evaluate VCD further. A focused trial in the particular communities of athletes or of military service members may be of increased benefit to better define VCD. It is notable that 100 cases were found in a relatively short period for a community hospital, and prevalence may be higher than previously reported.
Vocal cord dysfunction (VCD), also known as paradoxical vocal cord movement, is described as paroxysms of glottis obstruction due to true vocal cord adduction.1 Since VCD presents as a constellation of symptoms associated with dyspnea, it often is misdiagnosed as asthma.2 Vocal cord dysfunction often manifests as episodic dyspnea and wheezing, may occur with exercise, and may be minimally responsive to initial therapies. Flattened inspiratory curves may be noted on pulmonary function tests (PFTs), but direct laryngoscopy is the gold standard for diagnosis.3 A cohort of proven patients with VCD with a plateau in the inspiratory curve of PFTs also had a plateau on expiratory phase in 81% of cases.4
The differential diagnosis of patients presenting with upper airway symptoms is broad. It must include VCD, asthma, angioedema, laryngomalacia, vocal cord polyps, vocal cord tumors, and neurologic conditions such as brain stem compression or movement disorders. Essentially, all movement disorders of vocal cords must be considered, and organic causes of this movement disorder can be evaluated by visualization of the vocal cords. Triggers for VCD include exercise, airborne irritants, gastroesophageal reflux disease (GERD), allergic rhinitis, medications, and psychological conditions.5 Additionally, VCD can coexist with asthma, further complicating accurate diagnoses.6
Therapies are reported in case studies, but no large randomized controlled trials exist to evaluate current therapy options. Primary treatments of asthma therapy were largely ineffective, and ideal therapy includes a multidisciplinary approach, including speech therapy to optimize laryngeal control and treatment of all identified laryngeal irritants.6
The prevalence of VCD is unknown, with no prospective cohort studies completed to date and conflicting diagnostic criteria used in many case studies.7 A prevalence of 2.8% was noted in one particular cohort of 1,028 patients admitted to a rehabilitation center in a calendar year with the primary pulmonary diagnosis on admission.6 Females seemed to be affected at a higher ratio than were males, 2 to 3 females per 1 male diagnosis.7
In the military population, certain risk factors were noted in returning deployed members, including anxiety/high stress, exercise, and acute respiratory illnesses.8 In that particular cohort, 72% positive predictive value was noted for VCD if flattened inspiratory flow loops with negative methacholine challenge were present.
Diagnostic criteria are challenging, as symptoms such as dyspnea may be present acutely, last < 2 minutes, be self-limiting, and completely resolve outside of acute events. Stridor may be noted, primarily above the vocal cords, and less audible on chest auscultation.6 A goal of therapy, in addition to dedicated speech pathologist input, is optimizing comedical conditions, including GERD, allergic rhinitis, concomitant asthma, and any psychological diagnoses.9
Athletes are a particular subset of patients with VCD who are crucial to appropriately diagnose, including a detailed history and physical, PFTs, and proceeding to direct laryngoscopy to confirm diagnoses.10 Behavioral management includes rescue breathing techniques, and speech therapy programs focus on relaxation of the larynx and diaphragmatic breathing techniques, with the goal of establishing sense of control during acute events.10 Military service members are expected to operate at a high-intensity level similar to that of athletes, and treatments considered for athletes are applicable to military service members as well. Military strength and cardiovascular standards are measured by a combination of push-ups, sit-ups, and a run test, in addition to waist measurements. Some of the cohort were identified during physical fitness standard failures, usually in the run test, and ultimately received a pulmonology referral for wheezing or dyspnea with exertion. The objective of this retrospective cohort study was to evaluate 100 consecutively diagnosed cases of VCD in a military treatment facility.
Methods
The authors conducted a retrospective chart review of DoD military medical records of outpatient diagnoses in 100 consecutive diagnoses of VCD from January 2011 to February 2014. Institutional review board approval was obtained under Project RSM20130001E by the Exempt Determination Official at Eglin Air Force Base (AFB), Florida.
All cases were identified at time of VCD visualization and were diagnosed with video stroboscopy by speech therapy or by visual laryngoscopy by the otolaryngology or pulmonology departments via direct visualization.
Cases were collected chronologically, and all diagnosed cases at Eglin AFB hospital were included. Follow-up was scheduled with all patients diagnosed in Speech Therapy, and most patients were concurrently treated by Pulmonology or Allergy/Immunology. Pulmonary function tests were obtained in 98 of the 100 diagnosed cases. Patients eligible for care at Eglin AFB included active-duty and Reserve military members plus dependents and retirees.
The majority of patients diagnosed in this cohort were seen and diagnosed by Speech Therapy. Video stroboscopy is based on the principle that a movement of an object higher than a certain flicker rate appears to stand still to direct visualization, but with a rate of light exposure and imaging above the flicker rate by video, the true movement of the object can be identified.¹¹ Video stroboscopy is considered highly sensitive for organic disorders of vocal cords, but it is not specific for either organic or dysfunctional disorders.¹¹ It is still the gold standard above direct visualization, as it can detect abnormal movement of vocal cords above the critical rate that the human eye would perceive as not moving due to the frequency of movement (Figures 1 & 2).¹¹
In an older study, laryngoscopy was able to diagnose 100% of patients with symptomatic paradoxical vocal cord movement and additional 60% asymptomatic patients with a constellation of symptoms consistent with paradoxical vocal cord movement.¹²
Speech Therapy; Ear, Nose, and Throat (ENT); and Pulmonology may not perform direct visualization in these patients at initial presentation due to other suspected diagnoses. A more common test is the PFT, especially if asthma or other airway tract diseases are suspected (Figure 3).
Patient Descriptions
Study patients were referred for a variety of reasons, often from primary care clinics for concerns for asthma, episodic dyspnea, wheezing, or decreased exercise tolerance thought to be related to pulmonary or allergy causes. Pulmonology worked closely with Speech Therapy and referred VCD cases for speech evaluation, including video stroboscopy. Notably, of the patients in this cohort, although some were suspected to have asthma, those patients were ruled out during part of the pulmonology evaluation, both with PFT testing and methacholine challenges. An asthma diagnosis is important in a military treatment facility, as asthma is often grounds for discharge.
Patients ranged in age from 13 to 68 years, with a median age at 31 years diagnosis. Thirty-nine females and 61 males comprised the total case series. Speech Therapy diagnosed 97 patients, 96 were diagnosed at Eglin AFB hospital via stroboscopy. One patient was diagnosed off-base by Speech Therapy via direct visualization, 1 patient was diagnosed by Pulmonology on-base via direct visualization, and 2 patients were diagnosed by ENT on-base via direct visualization. These patients had direct laryngoscopy completed, often to rule out other organic causes for upper airway disease processes, and were found to have visual paradoxical vocal cord movement. Ninety-eight patients completed PFTs. Several patients were lost to follow-up, as can be common in a military population with frequent moves or members leaving service.
On record review, patient symptoms were present in the range of 2 months to 20 years, with a median duration of symptomatic reports lasting 2 years prior to diagnosis. Common diagnoses prior to visual VCD diagnosis included asthma, exercise-induced asthma, anxiety, and episodic wheezing. Risk factors that were evaluated in this case series included age, sex, body mass index (BMI), GERD, allergic rhinitis, postnasal drip, active smoker, previous smoker, and mental health diagnoses (Figure 4).
Pulmonary function test results were analyzed on 98 patients, including forced expiratory volume in 1 second (FEV1); forced vital capacity (FVC), FEV1/FVC ratio; peak inspiratory flow (PIF) and peak expiratory flow (PEF)—available in 97 studies; forced expiratory flow (FEF) at 25% to 75% of FVC (FEF 25%-75%)—available in 96 studies; and maximum voluntary ventilation (MVV) and MVV/FEV1 ratio—available in 60 of 98 PFTs.
Interventions
All patients diagnosed by Speech Therapy on-base were provided with laryngeal relaxation techniques, diaphragmatic breathing techniques, and controlled inhale/exhale techniques at time of diagnosis, with frequent follow-up scheduled with Speech Therapy and Pulmonology. All diagnoses potentially contributing to laryngeal irritation were treated, including GERD, allergic rhinitis, smoking cessation, weight loss, and exercise recommendations as needed.
Patients reported improvement on follow-up appointments with Speech Therapy in overall control of symptoms, subjectively categorized as poor improvement, partial improvement, and complete improvement. This was a subjective measurement of improvement and fully dependent on follow-up care and patient reporting for improvement. No predefined number of follow-ups was determined; patients were followed monthly until they declined further care, fully improved, moved out of the military treatment system, or were lost to follow-up.
Treatment included structured Speech Therapy sessions. Response to treatment was subjectively qualified by patient report. Fifteen patients reported complete resolution of symptoms, 57 reported partial improvement, 24 reported poor improvement, and 4 patients were lost to follow-up.
Results
Risk factors for the diagnosis of VCD included possible associations with GERD, allergic rhinitis, smoking, prior smoking, BMI, and mental health diagnoses. Body mass index ranged from 17 to 36 in the case series, with median BMI of 27. Mental health diagnoses were present in 35 patients and included diagnoses of anxiety, depression, and adjustment disorders. Gastroesophageal reflux disease diagnosis was present in 59 of the case series patients, 80 had the diagnosis of allergic rhinitis, 63 were diagnosed with postnasal drip. Sixteen case series patients were current smokers. An additional 26 were previous smokers (at least 100 cigarettes in lifetime) for a total of 42 patients that were current or prior smokers.
The chart review was completed to evaluate for the presence of these diagnoses, which included previous treatments; for example, proton pump inhibitors for GERD, antidepressants for depression, or intranasal steroids for allergic rhinitis. The diagnosis was counted as present if the patient was currently being treated for the particular diagnosis in question.
PFT Data
Data from PFTs were available for 98 of 100 cases diagnosed. Review of data across all 98 patients is noted for median FEV1 of 3.6, a median FVC of 4.5, with ratio of 0.80.
Since PFT values vary according to age, sex, and ethnicity, PFTs were analyzed for percent predicted values based on age, gender, and race. Notably, median values for FEV1, FVC, and PEF were all close to 100% of the predicted value. The MVV percent predicted was available in 60 cases and was 93% of predicted values. The most significant difference from expected values was FEF 25% to 75%, at 84% of expected results.
Flow-volume loop evaluations on the 97 PFTs available were completed, and 58 of the 97 were noted for variable extrathoracic airway obstruction consistent with inspiratory inhibition in the patient population. This is 60% of the available PFTs in this cohort study.
Discussion
This retrospective chart review of 100 consecutive VCD diagnoses in a military treatment facility reinforces many of the findings currently available in the literature. As illustrated in a Chest review article, the diagnosis of VCD on history, physical examination, or PFTs remains ellusive.1 The PFT evaluation contains some subjectivity regarding the flattening of inspiratory flow-volume loops and is not routinely reported in PFT results. In patients diagnosed with VCD, a clear consensus of treatment modalities remains lacking. Modification of risk factors (allergic rhinitis, GERD, smoking cessation, weight loss) assisted in self-reported patient improvement, as did focused speech therapy.
The median age of 31 years, likely reflected the younger military population served at Eglin AFB. Seventy-five of these patients were currently on active duty, 6 were retired from active duty (veterans), and 19 were dependents. The median time of symptoms to diagnosis was 2 years. Prior misdiagnosis with other diseases such as asthma was common. Also, referral to Pulmonology and Speech Therapy was usually completed after failed outpatient primary care management for the alternative diagnoses.
Improvement with therapy was mixed, and during the time of documented follow-up, 72 patients reported complete or partial improvement. Most active-duty patients in the partial improvement category based this subjective reporting on their ability to meet military physical fitness standards.
Previous data suggested a female predominance, but this study population was 61% male. Military populations are about 80% to 85% male, so an increase in male diagnosis is expected.
Many patients in the patient cohort arrived as a result of Pulmonology referrals with a presumptive diagnoses of asthma but were determined not to have asthma through PFT results inconsistent with asthma, no improvement with β-agonist therapies, and negative methacholine challenges (if performed). These results prompted evaluations for other conditions and eventually a VCD diagnosis. As noted, exclusion of asthma is of particular importance in a military population, as medical discharges often are pursued in service members with asthma whether controlled or uncontrolled. Lag time to referral also is possible in failures of military physical, which prompted medical evaluation once several failures had occurred over a 1- to 2-year time frame.
The PFT data evaluation was inconclusive for statistically significant changes when compared with age-matched normal PFT values. This also was noted in previous studies of VCD cases. Most notable was percent predicted values of FEF 25% to 75%, with 84% of expected values. The FEV1, FVC, and PEF all fell within predicted values of normal, despite wide ranges in age, sex, and ethnicity among the subjects. Inspiratory flattening consistent with extrathoracic obstruction was present in 58 of the 97 PFTs available for review at Eglin AFB.
Limitations
Limitations to this retrospective case series are illustrated here. Cases were found only when VCD was diagnosed and coded; and it is the authors’ suspicion that many have been misdiagnosed or improperly treated for asthma or other pulmonary/oropharynx conditions. If providers are not familiar with VCD or if PFT readings do not comment on inspiratory findings, diagnosis is less likely. Some of the authors’ colleagues already have determined that postdeployment prevalence of VCD seems to be elevated.8
This cohort was completed on all patients in a military treatment facility, with 75 active-duty personnel, 6 veterans, and 19 dependents of varying ages. This case series is retrospective and tabulates suspected risk factors; stronger and more informative studies could certainly be completed in prospective studies (although likely difficult with low prevalence) or in treatment comparison studies at the time of diagnosis.
Since the cohort had varied and lengthy time to diagnosis from onset of related symptoms, the treatment patients received prior to diagnosis differed extensively. Diagnosis was completed by numerous primary care managers or other subspecialties prior to arrival to Pulmonology and Speech Therapy at Eglin AFB. Once diagnosed in Speech Therapy, consistent treatment options were provided to patients in accordance with standard of care.
It is the authors’ suspicion that VCD may have a higher prevalence than previously reported in the literature. Military service members are tested annually or biannually on physical fitness standards and are evaluated for medical reasons for recurrent fitness standard failures. This selection of patients is more likely to have a VCD evaluation as part of a comprehensive evaluation than is a healthy adult in a civilian population. A prospective study in military service members would be more fruitful and possibly yield a higher prevalence postdeployment.
Conclusion
Vocal cord dysfunction remains a difficult diagnosis to treat, because multiple comorbidities likely contribute to the diagnosis. This retrospective case series attempted to compile common themes and noted that most of the patients had 2 or more risk factors of smoking, allergic rhinitis, GERD, or mental health diagnoses. A prospective trial would be ideal to evaluate VCD further. A focused trial in the particular communities of athletes or of military service members may be of increased benefit to better define VCD. It is notable that 100 cases were found in a relatively short period for a community hospital, and prevalence may be higher than previously reported.
1. Morris MJ, Christopher KL. Diagnostic criteria for the classification of vocal cord dysfunction. Chest. 2010;138(5):1213-1223.
2. National Heart, Lung, and Blood Institute. Expert panel report 3: guidelines for the diagnoses and management of asthma. Full report 2007. https://www.nhlbi.nih.gov/files/docs/guidelines/asthgdln .pdf. Published 2007.Accessed February 1, 2017.
3. Newman KB, Mason UG III, Schmaling KB. Clinical features of vocal cord dysfunction. Am J Respir Crit Care Med. 1995;152(4, pt 1):1382-1386.
4. Sanz Santiago V, López Neyra A, Almería Gil E, Villa Asensi JR. Spirometry patterns in vocal cord dysfunction [in Spanish]. An Pediatr (Barc). 2013;78(3):173-177.
5. Deckert J, Deckert L. Vocal cord dysfunction. Am Fam Physician. 2010;81(2):156-159.
6. Benninger C, Parsons JP, Mastronarde JG. Vocal cord dysfunction and asthma. Curr Opin Pulm Med. 2011;17(1):45-49.
7. Campainha S, Ribeiro C, Guimar M, Lima R. Vocal cord dysfunction: a frequently forgotten entity. Case Rep Pulmonol. 2012;2012:525493.
8. Morris MJ, Oleszewski RT, Sterner JB, Allan PF. Vocal cord dysfunction related to combat deployment. Mil Med. 2013;178(11):1208-1212.
9. Al-Alwan A, Kaminsky D. Vocal cord dysfunction in athletes: clinical presentation and review of the literature. Phys Sportsmed. 2012;40(2):22-27.
10. Kenn K, Schmitz M. Prevalence of vocal cord dysfunction in patients with dyspnea. First prospective clinical study. Am J Respir Crit Care Med. 1997;155:A965.
11. Wendler, J, Nawka, T, Verges, D. Instructional course: videolaryngo-stroboscopy and phonetography—basic tools for diagnostics and documentation in the voice clinic. Poster presented at: 15th European Congress of Oto-Rhino-Laryngology, Head and Neck Surgery; September 11-16, 2004; Rodos-Kos, Greece.
12. Ibrahim WH, Gheriani HA, Almohamed AA, Raza T. Paradoxical vocal cord motion disorder: past, present and future. Postgrad Med J. 2007;83(977):164-172.
1. Morris MJ, Christopher KL. Diagnostic criteria for the classification of vocal cord dysfunction. Chest. 2010;138(5):1213-1223.
2. National Heart, Lung, and Blood Institute. Expert panel report 3: guidelines for the diagnoses and management of asthma. Full report 2007. https://www.nhlbi.nih.gov/files/docs/guidelines/asthgdln .pdf. Published 2007.Accessed February 1, 2017.
3. Newman KB, Mason UG III, Schmaling KB. Clinical features of vocal cord dysfunction. Am J Respir Crit Care Med. 1995;152(4, pt 1):1382-1386.
4. Sanz Santiago V, López Neyra A, Almería Gil E, Villa Asensi JR. Spirometry patterns in vocal cord dysfunction [in Spanish]. An Pediatr (Barc). 2013;78(3):173-177.
5. Deckert J, Deckert L. Vocal cord dysfunction. Am Fam Physician. 2010;81(2):156-159.
6. Benninger C, Parsons JP, Mastronarde JG. Vocal cord dysfunction and asthma. Curr Opin Pulm Med. 2011;17(1):45-49.
7. Campainha S, Ribeiro C, Guimar M, Lima R. Vocal cord dysfunction: a frequently forgotten entity. Case Rep Pulmonol. 2012;2012:525493.
8. Morris MJ, Oleszewski RT, Sterner JB, Allan PF. Vocal cord dysfunction related to combat deployment. Mil Med. 2013;178(11):1208-1212.
9. Al-Alwan A, Kaminsky D. Vocal cord dysfunction in athletes: clinical presentation and review of the literature. Phys Sportsmed. 2012;40(2):22-27.
10. Kenn K, Schmitz M. Prevalence of vocal cord dysfunction in patients with dyspnea. First prospective clinical study. Am J Respir Crit Care Med. 1997;155:A965.
11. Wendler, J, Nawka, T, Verges, D. Instructional course: videolaryngo-stroboscopy and phonetography—basic tools for diagnostics and documentation in the voice clinic. Poster presented at: 15th European Congress of Oto-Rhino-Laryngology, Head and Neck Surgery; September 11-16, 2004; Rodos-Kos, Greece.
12. Ibrahim WH, Gheriani HA, Almohamed AA, Raza T. Paradoxical vocal cord motion disorder: past, present and future. Postgrad Med J. 2007;83(977):164-172.
Family history impacts risk of second cancer after HL

A new study suggests Hodgkin lymphoma (HL) survivors have a high risk of developing a second malignancy, particularly if they have a family history of that malignancy.
The research showed that HL survivors in Sweden were roughly 2.4 times more likely than individuals in the country’s general population to develop a second cancer.
The risk for HL survivors remained high 30 years after treatment, and the risk was even greater in HL survivors who had a family history of specific cancers.
“The vast majority of patients with Hodgkin lymphoma are cured with a combination of chemotherapy and radiotherapy,” said study author Amit Sud, MBChB, of The Institute of Cancer Research, London in the UK.
“Our research has shown that these patients are at substantially increased risk of a second cancer later in life and particularly if they have a family history of cancer.”
Dr Sud and his colleagues described this research in the Journal of Clinical Oncology.
The team analyzed data from the Swedish Family-Cancer Project Database. They identified 9522 HL patients diagnosed between 1965 and 2013. During a median follow-up of 12.6 years, there were 1215 second cancers in 1121 HL patients (12%).
Compared to the general population, the HL patients had a significantly higher risk of all second malignancies, with a standardized incident ratio (SIR) of 2.39 and an absolute excess risk of 71.2 cases per 10,000 person-years.
Cancer types
HL patients had a significantly increased risk of several malignancies. The overall SIRs were as follows:
- NHL—7.99
- Leukemia—6.46
- Connective tissue cancer—5.73
- Thyroid cancer—5.13
- Squamous cell carcinoma—4.44
- Lung cancer—3.61
- Pharyngeal cancer—3.52
- Esophageal cancer—2.62
- Brain cancer—2.58
- Breast cancer—2.52
- Colon cancer—2.21
- Pancreatic cancer—2.09
- Melanoma—2.08
- Colorectal cancer—1.85
- Stomach cancer—1.78
- Bladder cancer—1.57
- Prostate cancer—1.21.
The researchers calculated SIRs over time and found the risk for many of the cancers remained high over 30 years following HL treatment.
Family history
The researchers identified 28,277 first-degree relatives of the HL survivors. Thirty percent of HL survivors (n=2785) had 1 or more first-degree relatives with a family history of cancer.
The SIR for cancers was 1.02 in the relatives. The SIR for second cancers was 2.83 for HL survivors who had first-degree relatives with cancer and 2.16 for HL survivors who did not have any first-degree relatives with cancer.
The researchers said the increased risk of second malignancy was correlated with the number of first-degree relatives with cancer.
The SIR was 2.67 for HL patients who had a single first-degree relative with cancer and 3.40 for HL patients who had 2 or more first-degree relatives with cancer.
The SIRs for different cancer types (for HL patients with at least 1 first-degree relative with cancer and no first-degree relatives with cancer, respectively) were as follows:
- NHL—14.43 vs 7.83
- Leukemia—14.31 vs 6.37
- Squamous cell carcinoma—10.85 vs 4.30
- Lung cancer—11.24 vs 3.39
- Breast cancer—4.36 vs 2.36
- Colorectal cancer—3.71 vs 1.76.
Sex and age
The researchers found significant differences in the SIRs for second cancers between HL patients diagnosed before the age of 35 and those diagnosed after age 35.
For men, the SIRs were:
- All cancers—4.26 for <35, 2.08 for ≥ 35
- Colorectal cancer—4.07 for < 35, 1.73 for ≥35
- Lung cancer—6.16 for < 35, 3.20 for ≥35
- Breast cancer—12.60 for < 35, 4.58 for ≥35
- Squamous cell carcinoma—5.89 for < 35, 3.96 for ≥35
- NHL—15.9 for < 35, 6.93 for ≥35
- Leukemia—12.15 for < 35, 5.57 for ≥35.
For women, the SIRs were:
- All cancers—4.61 for <35, 1.73 for ≥ 35
- Colorectal cancer—1.31 for < 35, 1.65 for ≥35
- Lung cancer—8.84 for < 35, 2.50 for ≥35
- Breast cancer—6.00 for < 35, 1.14 for ≥35
- Squamous cell carcinoma—6.37 for < 35, 4.87 for ≥35
- NHL—6.23 for < 35, 6.55 for ≥35
- Leukemia—10.36 for < 35, 4.51 for ≥35.
“Younger women who have been treated with radiotherapy to the chest for Hodgkin lymphoma are already screened for breast cancer, but our study suggests that we should be looking at ways of monitoring survivors for other forms of cancer too, and potentially offering preventative interventions,” Dr Sud said.
“After patients are cured, they no longer encounter oncologists, so it’s important that other healthcare providers are aware of the increased risk to Hodgkin lymphoma survivors to improve early diagnosis of second cancers.” ![]()
A new study suggests Hodgkin lymphoma (HL) survivors have a high risk of developing a second malignancy, particularly if they have a family history of that malignancy.
The research showed that HL survivors in Sweden were roughly 2.4 times more likely than individuals in the country’s general population to develop a second cancer.
The risk for HL survivors remained high 30 years after treatment, and the risk was even greater in HL survivors who had a family history of specific cancers.
“The vast majority of patients with Hodgkin lymphoma are cured with a combination of chemotherapy and radiotherapy,” said study author Amit Sud, MBChB, of The Institute of Cancer Research, London in the UK.
“Our research has shown that these patients are at substantially increased risk of a second cancer later in life and particularly if they have a family history of cancer.”
Dr Sud and his colleagues described this research in the Journal of Clinical Oncology.
The team analyzed data from the Swedish Family-Cancer Project Database. They identified 9522 HL patients diagnosed between 1965 and 2013. During a median follow-up of 12.6 years, there were 1215 second cancers in 1121 HL patients (12%).
Compared to the general population, the HL patients had a significantly higher risk of all second malignancies, with a standardized incident ratio (SIR) of 2.39 and an absolute excess risk of 71.2 cases per 10,000 person-years.
Cancer types
HL patients had a significantly increased risk of several malignancies. The overall SIRs were as follows:
- NHL—7.99
- Leukemia—6.46
- Connective tissue cancer—5.73
- Thyroid cancer—5.13
- Squamous cell carcinoma—4.44
- Lung cancer—3.61
- Pharyngeal cancer—3.52
- Esophageal cancer—2.62
- Brain cancer—2.58
- Breast cancer—2.52
- Colon cancer—2.21
- Pancreatic cancer—2.09
- Melanoma—2.08
- Colorectal cancer—1.85
- Stomach cancer—1.78
- Bladder cancer—1.57
- Prostate cancer—1.21.
The researchers calculated SIRs over time and found the risk for many of the cancers remained high over 30 years following HL treatment.
Family history
The researchers identified 28,277 first-degree relatives of the HL survivors. Thirty percent of HL survivors (n=2785) had 1 or more first-degree relatives with a family history of cancer.
The SIR for cancers was 1.02 in the relatives. The SIR for second cancers was 2.83 for HL survivors who had first-degree relatives with cancer and 2.16 for HL survivors who did not have any first-degree relatives with cancer.
The researchers said the increased risk of second malignancy was correlated with the number of first-degree relatives with cancer.
The SIR was 2.67 for HL patients who had a single first-degree relative with cancer and 3.40 for HL patients who had 2 or more first-degree relatives with cancer.
The SIRs for different cancer types (for HL patients with at least 1 first-degree relative with cancer and no first-degree relatives with cancer, respectively) were as follows:
- NHL—14.43 vs 7.83
- Leukemia—14.31 vs 6.37
- Squamous cell carcinoma—10.85 vs 4.30
- Lung cancer—11.24 vs 3.39
- Breast cancer—4.36 vs 2.36
- Colorectal cancer—3.71 vs 1.76.
Sex and age
The researchers found significant differences in the SIRs for second cancers between HL patients diagnosed before the age of 35 and those diagnosed after age 35.
For men, the SIRs were:
- All cancers—4.26 for <35, 2.08 for ≥ 35
- Colorectal cancer—4.07 for < 35, 1.73 for ≥35
- Lung cancer—6.16 for < 35, 3.20 for ≥35
- Breast cancer—12.60 for < 35, 4.58 for ≥35
- Squamous cell carcinoma—5.89 for < 35, 3.96 for ≥35
- NHL—15.9 for < 35, 6.93 for ≥35
- Leukemia—12.15 for < 35, 5.57 for ≥35.
For women, the SIRs were:
- All cancers—4.61 for <35, 1.73 for ≥ 35
- Colorectal cancer—1.31 for < 35, 1.65 for ≥35
- Lung cancer—8.84 for < 35, 2.50 for ≥35
- Breast cancer—6.00 for < 35, 1.14 for ≥35
- Squamous cell carcinoma—6.37 for < 35, 4.87 for ≥35
- NHL—6.23 for < 35, 6.55 for ≥35
- Leukemia—10.36 for < 35, 4.51 for ≥35.
“Younger women who have been treated with radiotherapy to the chest for Hodgkin lymphoma are already screened for breast cancer, but our study suggests that we should be looking at ways of monitoring survivors for other forms of cancer too, and potentially offering preventative interventions,” Dr Sud said.
“After patients are cured, they no longer encounter oncologists, so it’s important that other healthcare providers are aware of the increased risk to Hodgkin lymphoma survivors to improve early diagnosis of second cancers.” ![]()
A new study suggests Hodgkin lymphoma (HL) survivors have a high risk of developing a second malignancy, particularly if they have a family history of that malignancy.
The research showed that HL survivors in Sweden were roughly 2.4 times more likely than individuals in the country’s general population to develop a second cancer.
The risk for HL survivors remained high 30 years after treatment, and the risk was even greater in HL survivors who had a family history of specific cancers.
“The vast majority of patients with Hodgkin lymphoma are cured with a combination of chemotherapy and radiotherapy,” said study author Amit Sud, MBChB, of The Institute of Cancer Research, London in the UK.
“Our research has shown that these patients are at substantially increased risk of a second cancer later in life and particularly if they have a family history of cancer.”
Dr Sud and his colleagues described this research in the Journal of Clinical Oncology.
The team analyzed data from the Swedish Family-Cancer Project Database. They identified 9522 HL patients diagnosed between 1965 and 2013. During a median follow-up of 12.6 years, there were 1215 second cancers in 1121 HL patients (12%).
Compared to the general population, the HL patients had a significantly higher risk of all second malignancies, with a standardized incident ratio (SIR) of 2.39 and an absolute excess risk of 71.2 cases per 10,000 person-years.
Cancer types
HL patients had a significantly increased risk of several malignancies. The overall SIRs were as follows:
- NHL—7.99
- Leukemia—6.46
- Connective tissue cancer—5.73
- Thyroid cancer—5.13
- Squamous cell carcinoma—4.44
- Lung cancer—3.61
- Pharyngeal cancer—3.52
- Esophageal cancer—2.62
- Brain cancer—2.58
- Breast cancer—2.52
- Colon cancer—2.21
- Pancreatic cancer—2.09
- Melanoma—2.08
- Colorectal cancer—1.85
- Stomach cancer—1.78
- Bladder cancer—1.57
- Prostate cancer—1.21.
The researchers calculated SIRs over time and found the risk for many of the cancers remained high over 30 years following HL treatment.
Family history
The researchers identified 28,277 first-degree relatives of the HL survivors. Thirty percent of HL survivors (n=2785) had 1 or more first-degree relatives with a family history of cancer.
The SIR for cancers was 1.02 in the relatives. The SIR for second cancers was 2.83 for HL survivors who had first-degree relatives with cancer and 2.16 for HL survivors who did not have any first-degree relatives with cancer.
The researchers said the increased risk of second malignancy was correlated with the number of first-degree relatives with cancer.
The SIR was 2.67 for HL patients who had a single first-degree relative with cancer and 3.40 for HL patients who had 2 or more first-degree relatives with cancer.
The SIRs for different cancer types (for HL patients with at least 1 first-degree relative with cancer and no first-degree relatives with cancer, respectively) were as follows:
- NHL—14.43 vs 7.83
- Leukemia—14.31 vs 6.37
- Squamous cell carcinoma—10.85 vs 4.30
- Lung cancer—11.24 vs 3.39
- Breast cancer—4.36 vs 2.36
- Colorectal cancer—3.71 vs 1.76.
Sex and age
The researchers found significant differences in the SIRs for second cancers between HL patients diagnosed before the age of 35 and those diagnosed after age 35.
For men, the SIRs were:
- All cancers—4.26 for <35, 2.08 for ≥ 35
- Colorectal cancer—4.07 for < 35, 1.73 for ≥35
- Lung cancer—6.16 for < 35, 3.20 for ≥35
- Breast cancer—12.60 for < 35, 4.58 for ≥35
- Squamous cell carcinoma—5.89 for < 35, 3.96 for ≥35
- NHL—15.9 for < 35, 6.93 for ≥35
- Leukemia—12.15 for < 35, 5.57 for ≥35.
For women, the SIRs were:
- All cancers—4.61 for <35, 1.73 for ≥ 35
- Colorectal cancer—1.31 for < 35, 1.65 for ≥35
- Lung cancer—8.84 for < 35, 2.50 for ≥35
- Breast cancer—6.00 for < 35, 1.14 for ≥35
- Squamous cell carcinoma—6.37 for < 35, 4.87 for ≥35
- NHL—6.23 for < 35, 6.55 for ≥35
- Leukemia—10.36 for < 35, 4.51 for ≥35.
“Younger women who have been treated with radiotherapy to the chest for Hodgkin lymphoma are already screened for breast cancer, but our study suggests that we should be looking at ways of monitoring survivors for other forms of cancer too, and potentially offering preventative interventions,” Dr Sud said.
“After patients are cured, they no longer encounter oncologists, so it’s important that other healthcare providers are aware of the increased risk to Hodgkin lymphoma survivors to improve early diagnosis of second cancers.” ![]()
Selinexor trials placed on partial hold

The US Food and Drug Administration (FDA) has placed a partial clinical hold on all trials of selinexor (KPT-330).
Selinexor is an inhibitor being evaluated in multiple trials of patients with relapsed and/or refractory hematologic and solid tumor malignancies.
While the partial clinical hold remains in effect, patients with stable disease or better may remain on selinexor.
However, no new patients may be enrolled in selinexor trials until the hold is lifted.
The FDA has indicated that the partial clinical hold is due to incomplete information in the existing version of the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, said it has amended the brochure, updated the informed consent documents accordingly, and submitted the documents to the FDA as requested.
As of March 10, Karyopharm had provided all requested materials to the FDA believed to be required to lift the partial clinical hold. By regulation, the FDA has 30 days from the receipt of Karyopharm’s submission to notify the company whether the partial clinical hold is lifted.
Karyopharm said it is working with the FDA to seek the release of the hold and resume enrollment in its selinexor trials as expeditiously as possible. The company believes its previously disclosed enrollment rates and timelines for its ongoing trials will remain materially unchanged.
About selinexor
Selinexor is a selective inhibitor of nuclear export (SINE) XPO1 antagonist. The drug binds with and inhibits XPO1, leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to induce apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor. The drug is currently being evaluated in several trials across multiple cancer indications.
One of these is the phase 2 SOPRA trial, in which selinexor is being compared to investigator’s choice of therapy (1 of 3 potential salvage therapies). The trial is enrolling patients 60 years of age or older with relapsed or refractory acute myeloid leukemia who are ineligible for standard intensive chemotherapy and/or transplant.
The SADAL study is a phase 2b trial comparing high and low doses of selinexor in patients with relapsed and/or refractory de novo diffuse large B-cell lymphoma who have no therapeutic options of demonstrated clinical benefit.
STORM is a phase 2b trial evaluating selinexor and low-dose dexamethasone in patients with heavily pretreated multiple myeloma (MM). And STOMP is a phase 1b/2 study evaluating selinexor in combination with existing therapies across the broader population in MM.
Karyopharm is also planning a randomized, phase 3 study known as BOSTON. In this trial, researchers will compare selinexor plus bortezomib and low-dose dexamethasone to bortezomib and low-dose dexamethasone in MM patients who have had 1 to 3 prior lines of therapy.
Additional phase 1, 2, and 3 studies are ongoing or currently planned.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on all trials of selinexor (KPT-330).
Selinexor is an inhibitor being evaluated in multiple trials of patients with relapsed and/or refractory hematologic and solid tumor malignancies.
While the partial clinical hold remains in effect, patients with stable disease or better may remain on selinexor.
However, no new patients may be enrolled in selinexor trials until the hold is lifted.
The FDA has indicated that the partial clinical hold is due to incomplete information in the existing version of the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, said it has amended the brochure, updated the informed consent documents accordingly, and submitted the documents to the FDA as requested.
As of March 10, Karyopharm had provided all requested materials to the FDA believed to be required to lift the partial clinical hold. By regulation, the FDA has 30 days from the receipt of Karyopharm’s submission to notify the company whether the partial clinical hold is lifted.
Karyopharm said it is working with the FDA to seek the release of the hold and resume enrollment in its selinexor trials as expeditiously as possible. The company believes its previously disclosed enrollment rates and timelines for its ongoing trials will remain materially unchanged.
About selinexor
Selinexor is a selective inhibitor of nuclear export (SINE) XPO1 antagonist. The drug binds with and inhibits XPO1, leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to induce apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor. The drug is currently being evaluated in several trials across multiple cancer indications.
One of these is the phase 2 SOPRA trial, in which selinexor is being compared to investigator’s choice of therapy (1 of 3 potential salvage therapies). The trial is enrolling patients 60 years of age or older with relapsed or refractory acute myeloid leukemia who are ineligible for standard intensive chemotherapy and/or transplant.
The SADAL study is a phase 2b trial comparing high and low doses of selinexor in patients with relapsed and/or refractory de novo diffuse large B-cell lymphoma who have no therapeutic options of demonstrated clinical benefit.
STORM is a phase 2b trial evaluating selinexor and low-dose dexamethasone in patients with heavily pretreated multiple myeloma (MM). And STOMP is a phase 1b/2 study evaluating selinexor in combination with existing therapies across the broader population in MM.
Karyopharm is also planning a randomized, phase 3 study known as BOSTON. In this trial, researchers will compare selinexor plus bortezomib and low-dose dexamethasone to bortezomib and low-dose dexamethasone in MM patients who have had 1 to 3 prior lines of therapy.
Additional phase 1, 2, and 3 studies are ongoing or currently planned.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on all trials of selinexor (KPT-330).
Selinexor is an inhibitor being evaluated in multiple trials of patients with relapsed and/or refractory hematologic and solid tumor malignancies.
While the partial clinical hold remains in effect, patients with stable disease or better may remain on selinexor.
However, no new patients may be enrolled in selinexor trials until the hold is lifted.
The FDA has indicated that the partial clinical hold is due to incomplete information in the existing version of the investigator’s brochure, including an incomplete list of serious adverse events associated with selinexor.
Karyopharm Therapeutics Inc., the company developing selinexor, said it has amended the brochure, updated the informed consent documents accordingly, and submitted the documents to the FDA as requested.
As of March 10, Karyopharm had provided all requested materials to the FDA believed to be required to lift the partial clinical hold. By regulation, the FDA has 30 days from the receipt of Karyopharm’s submission to notify the company whether the partial clinical hold is lifted.
Karyopharm said it is working with the FDA to seek the release of the hold and resume enrollment in its selinexor trials as expeditiously as possible. The company believes its previously disclosed enrollment rates and timelines for its ongoing trials will remain materially unchanged.
About selinexor
Selinexor is a selective inhibitor of nuclear export (SINE) XPO1 antagonist. The drug binds with and inhibits XPO1, leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to induce apoptosis in cancer cells while largely sparing normal cells.
To date, more than 1900 patients have been treated with selinexor. The drug is currently being evaluated in several trials across multiple cancer indications.
One of these is the phase 2 SOPRA trial, in which selinexor is being compared to investigator’s choice of therapy (1 of 3 potential salvage therapies). The trial is enrolling patients 60 years of age or older with relapsed or refractory acute myeloid leukemia who are ineligible for standard intensive chemotherapy and/or transplant.
The SADAL study is a phase 2b trial comparing high and low doses of selinexor in patients with relapsed and/or refractory de novo diffuse large B-cell lymphoma who have no therapeutic options of demonstrated clinical benefit.
STORM is a phase 2b trial evaluating selinexor and low-dose dexamethasone in patients with heavily pretreated multiple myeloma (MM). And STOMP is a phase 1b/2 study evaluating selinexor in combination with existing therapies across the broader population in MM.
Karyopharm is also planning a randomized, phase 3 study known as BOSTON. In this trial, researchers will compare selinexor plus bortezomib and low-dose dexamethasone to bortezomib and low-dose dexamethasone in MM patients who have had 1 to 3 prior lines of therapy.
Additional phase 1, 2, and 3 studies are ongoing or currently planned.
Sickle cell trait linked to end-stage renal disease

Black individuals with sickle cell trait (SCT) have an increased risk of developing end-stage renal disease (ESRD), according to new research.
The study indicates that having SCT actually doubles the risk of ESRD.
And the trait confers a similar degree of risk as APOL1 gene variants, which are currently the most widely recognized genetic contributors to kidney disease in blacks.
Researchers believe this finding may have important public policy implications for genetic counseling for individuals with SCT.
Rakhi P. Naik, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues reported this finding in the Journal of the American Society of Nephrology.
Previous research suggested there is an association between SCT and chronic kidney disease, but it hasn’t been clear if that extends to ESRD. Studies have also suggested a possible association between kidney disease and hemoglobin C trait, but the link has not been confirmed.
So Dr Naik and her colleagues decided to investigate these potential links. To do so, the researchers analyzed data from a large, population-based study, the REasons for Geographic and Racial Differences in Stroke (REGARDS) study.
The team evaluated information on 9909 black individuals, 739 of whom had SCT and 243 of whom had hemoglobin C trait.
The data indicate that individuals with SCT have a 2-fold higher risk of developing ESRD when compared to those without SCT. But there is no association between hemoglobin C trait and ESRD.
At a median follow-up of 6.5 years, the incidence of ESRD was 5.4% (40/739) in participants with SCT, 2.5% (6/243) in subjects with hemoglobin C trait, and 2.6% (234/8927) in individuals without either trait.
The incidence rate for ESRD was 8.5 per 1000 person-years for participants with SCT, 3.9 per 1000 person-years for subjects with hemoglobin C trait, and 4.0 per 1000 person-years for individuals without either trait.
The researchers noted that SCT conferred a similar degree of ESRD risk as APOL1 gene variants. The hazard ratio for subjects with SCT was 2.03, and the hazard ratio for those with APOL1 high-risk genotypes was 1.77.
“Although you cannot change the genes you are born with, doctors can use this information to start screening for kidney disease earlier and to aggressively treat any other risk factors you may have, such as diabetes or high blood pressure,” Dr Naik said.
“We still need more studies to determine if there are other treatments that can be used to slow the progression of kidney disease, specifically in individuals with sickle cell trait.” ![]()
Black individuals with sickle cell trait (SCT) have an increased risk of developing end-stage renal disease (ESRD), according to new research.
The study indicates that having SCT actually doubles the risk of ESRD.
And the trait confers a similar degree of risk as APOL1 gene variants, which are currently the most widely recognized genetic contributors to kidney disease in blacks.
Researchers believe this finding may have important public policy implications for genetic counseling for individuals with SCT.
Rakhi P. Naik, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues reported this finding in the Journal of the American Society of Nephrology.
Previous research suggested there is an association between SCT and chronic kidney disease, but it hasn’t been clear if that extends to ESRD. Studies have also suggested a possible association between kidney disease and hemoglobin C trait, but the link has not been confirmed.
So Dr Naik and her colleagues decided to investigate these potential links. To do so, the researchers analyzed data from a large, population-based study, the REasons for Geographic and Racial Differences in Stroke (REGARDS) study.
The team evaluated information on 9909 black individuals, 739 of whom had SCT and 243 of whom had hemoglobin C trait.
The data indicate that individuals with SCT have a 2-fold higher risk of developing ESRD when compared to those without SCT. But there is no association between hemoglobin C trait and ESRD.
At a median follow-up of 6.5 years, the incidence of ESRD was 5.4% (40/739) in participants with SCT, 2.5% (6/243) in subjects with hemoglobin C trait, and 2.6% (234/8927) in individuals without either trait.
The incidence rate for ESRD was 8.5 per 1000 person-years for participants with SCT, 3.9 per 1000 person-years for subjects with hemoglobin C trait, and 4.0 per 1000 person-years for individuals without either trait.
The researchers noted that SCT conferred a similar degree of ESRD risk as APOL1 gene variants. The hazard ratio for subjects with SCT was 2.03, and the hazard ratio for those with APOL1 high-risk genotypes was 1.77.
“Although you cannot change the genes you are born with, doctors can use this information to start screening for kidney disease earlier and to aggressively treat any other risk factors you may have, such as diabetes or high blood pressure,” Dr Naik said.
“We still need more studies to determine if there are other treatments that can be used to slow the progression of kidney disease, specifically in individuals with sickle cell trait.” ![]()
Black individuals with sickle cell trait (SCT) have an increased risk of developing end-stage renal disease (ESRD), according to new research.
The study indicates that having SCT actually doubles the risk of ESRD.
And the trait confers a similar degree of risk as APOL1 gene variants, which are currently the most widely recognized genetic contributors to kidney disease in blacks.
Researchers believe this finding may have important public policy implications for genetic counseling for individuals with SCT.
Rakhi P. Naik, MD, of Johns Hopkins University School of Medicine in Baltimore, Maryland, and her colleagues reported this finding in the Journal of the American Society of Nephrology.
Previous research suggested there is an association between SCT and chronic kidney disease, but it hasn’t been clear if that extends to ESRD. Studies have also suggested a possible association between kidney disease and hemoglobin C trait, but the link has not been confirmed.
So Dr Naik and her colleagues decided to investigate these potential links. To do so, the researchers analyzed data from a large, population-based study, the REasons for Geographic and Racial Differences in Stroke (REGARDS) study.
The team evaluated information on 9909 black individuals, 739 of whom had SCT and 243 of whom had hemoglobin C trait.
The data indicate that individuals with SCT have a 2-fold higher risk of developing ESRD when compared to those without SCT. But there is no association between hemoglobin C trait and ESRD.
At a median follow-up of 6.5 years, the incidence of ESRD was 5.4% (40/739) in participants with SCT, 2.5% (6/243) in subjects with hemoglobin C trait, and 2.6% (234/8927) in individuals without either trait.
The incidence rate for ESRD was 8.5 per 1000 person-years for participants with SCT, 3.9 per 1000 person-years for subjects with hemoglobin C trait, and 4.0 per 1000 person-years for individuals without either trait.
The researchers noted that SCT conferred a similar degree of ESRD risk as APOL1 gene variants. The hazard ratio for subjects with SCT was 2.03, and the hazard ratio for those with APOL1 high-risk genotypes was 1.77.
“Although you cannot change the genes you are born with, doctors can use this information to start screening for kidney disease earlier and to aggressively treat any other risk factors you may have, such as diabetes or high blood pressure,” Dr Naik said.
“We still need more studies to determine if there are other treatments that can be used to slow the progression of kidney disease, specifically in individuals with sickle cell trait.” ![]()
PRAC recommends suspending gadolinium agents
The European Medicines Agency’s (EMA) Pharmacovigilance and Risk Assessment Committee (PRAC) has recommended suspending marketing authorizations for 4 linear gadolinium contrast agents because of evidence that small amounts of the gadolinium they contain are deposited in the brain.
The agents concerned are intravenous injections of gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide, which are given to patients to enhance images from magnetic resonance imaging (MRI) body scans.
The PRAC’s review of gadolinium agents found “convincing evidence” of accumulation of gadolinium in the brain from studies directly measuring gadolinium in brain tissues and areas of increased signal intensity seen on MRI scan images many months after the last injection of a gadolinium contrast agent.
Although no symptoms or diseases linked to gadolinium in the brain have been reported, the PRAC took a precautionary approach, noting that data on the long-term effects in the brain are limited.
Companies concerned by this review have the right to request that the PRAC re-examine its recommendations.
The PRAC’s recommendations will be sent to the Committee for Medicinal Products for Human Use (CHMP) for its opinion. Further details will be published at the time of the CHMP opinion.
The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all European Union member states.
Details of the review, recommendations
The PRAC’s review was initiated on March 17, 2016, at the request of the European Commission, under Article 31 of Directive 2001/83/EC.
The review covers agents containing the following active substances: gadobenic acid, gadobutrol, gadodiamide, gadopentetic acid, gadoteric acid, gadoteridol, gadoversetamide, and gadoxetic acid.
The 4 agents recommended for suspension (gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide) are linear agents. They have a structure more likely to release gadolinium, which can build up in body tissues.
Other agents, known as macrocyclic agents, are more stable and have a much lower propensity to release gadolinium.
The PRAC recommends that macrocyclic agents (gadobutrol, gadoteric acid, and gadoteridol) be used at the lowest dose that enhances images sufficiently to make diagnoses and only when unenhanced body scans are not suitable.
The PRAC has recommended that some linear agents remain available. The committee said that gadoxetic acid, a linear agent used at a low dose for liver scans, can remain on the market as it meets an important diagnostic need in patients with few alternatives.
In addition, a formulation of gadopentetic acid injected directly into joints should remain available because its gadolinium concentration is very low—around 200 times lower than those of intravenous products.
Both agents should be used at the lowest dose that enhances images sufficiently to make diagnoses and only if unenhanced scans are not suitable.
For those marketing authorizations recommended for suspension, the suspensions can be lifted if the respective companies provide evidence of new benefits in an identified patient group that outweigh its risks or show that their product (modified or not) does not release gadolinium significantly or lead to its retention in tissues. ![]()
The European Medicines Agency’s (EMA) Pharmacovigilance and Risk Assessment Committee (PRAC) has recommended suspending marketing authorizations for 4 linear gadolinium contrast agents because of evidence that small amounts of the gadolinium they contain are deposited in the brain.
The agents concerned are intravenous injections of gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide, which are given to patients to enhance images from magnetic resonance imaging (MRI) body scans.
The PRAC’s review of gadolinium agents found “convincing evidence” of accumulation of gadolinium in the brain from studies directly measuring gadolinium in brain tissues and areas of increased signal intensity seen on MRI scan images many months after the last injection of a gadolinium contrast agent.
Although no symptoms or diseases linked to gadolinium in the brain have been reported, the PRAC took a precautionary approach, noting that data on the long-term effects in the brain are limited.
Companies concerned by this review have the right to request that the PRAC re-examine its recommendations.
The PRAC’s recommendations will be sent to the Committee for Medicinal Products for Human Use (CHMP) for its opinion. Further details will be published at the time of the CHMP opinion.
The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all European Union member states.
Details of the review, recommendations
The PRAC’s review was initiated on March 17, 2016, at the request of the European Commission, under Article 31 of Directive 2001/83/EC.
The review covers agents containing the following active substances: gadobenic acid, gadobutrol, gadodiamide, gadopentetic acid, gadoteric acid, gadoteridol, gadoversetamide, and gadoxetic acid.
The 4 agents recommended for suspension (gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide) are linear agents. They have a structure more likely to release gadolinium, which can build up in body tissues.
Other agents, known as macrocyclic agents, are more stable and have a much lower propensity to release gadolinium.
The PRAC recommends that macrocyclic agents (gadobutrol, gadoteric acid, and gadoteridol) be used at the lowest dose that enhances images sufficiently to make diagnoses and only when unenhanced body scans are not suitable.
The PRAC has recommended that some linear agents remain available. The committee said that gadoxetic acid, a linear agent used at a low dose for liver scans, can remain on the market as it meets an important diagnostic need in patients with few alternatives.
In addition, a formulation of gadopentetic acid injected directly into joints should remain available because its gadolinium concentration is very low—around 200 times lower than those of intravenous products.
Both agents should be used at the lowest dose that enhances images sufficiently to make diagnoses and only if unenhanced scans are not suitable.
For those marketing authorizations recommended for suspension, the suspensions can be lifted if the respective companies provide evidence of new benefits in an identified patient group that outweigh its risks or show that their product (modified or not) does not release gadolinium significantly or lead to its retention in tissues. ![]()
The European Medicines Agency’s (EMA) Pharmacovigilance and Risk Assessment Committee (PRAC) has recommended suspending marketing authorizations for 4 linear gadolinium contrast agents because of evidence that small amounts of the gadolinium they contain are deposited in the brain.
The agents concerned are intravenous injections of gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide, which are given to patients to enhance images from magnetic resonance imaging (MRI) body scans.
The PRAC’s review of gadolinium agents found “convincing evidence” of accumulation of gadolinium in the brain from studies directly measuring gadolinium in brain tissues and areas of increased signal intensity seen on MRI scan images many months after the last injection of a gadolinium contrast agent.
Although no symptoms or diseases linked to gadolinium in the brain have been reported, the PRAC took a precautionary approach, noting that data on the long-term effects in the brain are limited.
Companies concerned by this review have the right to request that the PRAC re-examine its recommendations.
The PRAC’s recommendations will be sent to the Committee for Medicinal Products for Human Use (CHMP) for its opinion. Further details will be published at the time of the CHMP opinion.
The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all European Union member states.
Details of the review, recommendations
The PRAC’s review was initiated on March 17, 2016, at the request of the European Commission, under Article 31 of Directive 2001/83/EC.
The review covers agents containing the following active substances: gadobenic acid, gadobutrol, gadodiamide, gadopentetic acid, gadoteric acid, gadoteridol, gadoversetamide, and gadoxetic acid.
The 4 agents recommended for suspension (gadobenic acid, gadodiamide, gadopentetic acid, and gadoversetamide) are linear agents. They have a structure more likely to release gadolinium, which can build up in body tissues.
Other agents, known as macrocyclic agents, are more stable and have a much lower propensity to release gadolinium.
The PRAC recommends that macrocyclic agents (gadobutrol, gadoteric acid, and gadoteridol) be used at the lowest dose that enhances images sufficiently to make diagnoses and only when unenhanced body scans are not suitable.
The PRAC has recommended that some linear agents remain available. The committee said that gadoxetic acid, a linear agent used at a low dose for liver scans, can remain on the market as it meets an important diagnostic need in patients with few alternatives.
In addition, a formulation of gadopentetic acid injected directly into joints should remain available because its gadolinium concentration is very low—around 200 times lower than those of intravenous products.
Both agents should be used at the lowest dose that enhances images sufficiently to make diagnoses and only if unenhanced scans are not suitable.
For those marketing authorizations recommended for suspension, the suspensions can be lifted if the respective companies provide evidence of new benefits in an identified patient group that outweigh its risks or show that their product (modified or not) does not release gadolinium significantly or lead to its retention in tissues. ![]()
Is Vitamin D Beneficial for MS Patients?
Q) What is the role of vitamin D in multiple sclerosis? Is it beneficial?
The exact etiology and pathophysiology of multiple sclerosis (MS) is still not fully understood. Research strongly suggests that there are two major causative factors: one genetic, and the other, environmental. From an environmental standpoint, multiple studies have shown that living farther from the equator, not being exposed to sunlight, and having a low vitamin D level are all correlated with increased risk for MS and MS relapse.1
Our bodies need sunlight to successfully synthesize vitamin D in the skin. Research has found that individuals with lightly pigmented skin are five times more efficient at synthesizing vitamin D in the presence of sunlight than those with darker skin.2 However, the ability to absorb sunlight is also correlated with the earth’s latitude; worse absorption occurs in areas beyond the 40th parallel (in either hemisphere), where UVB levels are too low to synthesize vitamin D four to six months out of the year.2
When exposed to UVB rays, our bodies start to synthesize vitamin D; it undergoes a transformation in the liver and then the kidneys and ultimately becomes the hormonally active form of vitamin D, 1,25-dihydroxyvitamin D3 (calcitriol).2 Calcitriol is recognized by multiple tissues throughout the body that contain vitamin D receptors. Specifically, in the central nervous system, receptors are located on microglia, activated monocytes, and B and T lymphocytes.1 In MS, myelin (the coating around the nerves) is destroyed by an immune-mediated inflammatory process involving the microglia and B and T lymphocytes. Vitamin D quiets down this inflammation, thereby reducing disability accumulation and relapse risk and resulting in fewer changes on MRI.
Vitamin D is also believed to shift the immune response to an anti-inflammatory state by focusing the response on the cytotoxic T cells often found in MS lesions, which attack neurons and oligodendrocytes.2 This theory was tested by Munger and colleagues, who used a pooled cohort of 187,000 women from the Nurses’ Health Study and Nurses’ Health Study II to assess vitamin D intake and risk for MS. Compared to women with lower vitamin D intake, those who took 700 IU/d had a 41% lower incidence of MS. Women who took ≥ 400 IU/d had a 33% lower risk for MS, compared to nonusers.3 In another evaluation of 7 million US military personnel, individuals with a serum vitamin D level of 40 ng/mL were 62% less likely to develop MS.4
In light of the anti-inflammatory effects of vitamin D and its purported reduction of MS risk, it is possible that patients with MS should begin vitamin D supplementation early to obtain maximum anti-inflammatory effects. While an optimal vitamin D goal has not been established in the literature, some studies suggest 30 to 55 ng/mL as a target range for serum vitamin D level.1
While vitamin D has been found to be well-tolerated, patients should be cautioned that very high doses can cause fatigue, abdominal cramps, nausea, vomiting, kidney damage, hypertension, hypercalcemia, and oth
Lisa Marie Fox, MSPAS, PA-C
Division of Multiple Sclerosis, Department of Neurology, Johns Hopkins Hosptial, Baltimore
1. Waubant E, Mowry E, Bowling A. The role of vitamin D in multiple sclerosis pathology and treatment: answers and opportunities. Int J MS Care. 2015;17(2):1-24.
2. Pierrot-Deseilligny C. Clinical implications of a possible role of vitamin D in multiple sclerosis. J Neurol. 2009;256(9):1468-1478.3. Munger KL, Zhang SM, O’Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65.
4. Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006; 296(23):2832-2838.
5. Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53-58.
Clinician Reviews in partnership with

MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month’s responses were authored by Bryan Walker and Lisa Marie Fox, MSPAS, PA-C, who is in the Division of Multiple Sclerosis, Department of Neurology, at Johns Hopkins Hospital in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month’s responses were authored by Bryan Walker and Lisa Marie Fox, MSPAS, PA-C, who is in the Division of Multiple Sclerosis, Department of Neurology, at Johns Hopkins Hospital in Baltimore.
Clinician Reviews in partnership with
MS Consult is edited by Colleen J. Harris, MN, NP, MSCN, Nurse Practitioner/Manager of the Multiple Sclerosis Clinic at Foothills Medical Centre in Calgary, Alberta, Canada, and Bryan Walker, MHS, PA-C, who is in the Department of Neurology, Division of MS and Neuroimmunology, at Duke University Medical Center in Durham, North Carolina. This month’s responses were authored by Bryan Walker and Lisa Marie Fox, MSPAS, PA-C, who is in the Division of Multiple Sclerosis, Department of Neurology, at Johns Hopkins Hospital in Baltimore.
Q) What is the role of vitamin D in multiple sclerosis? Is it beneficial?
The exact etiology and pathophysiology of multiple sclerosis (MS) is still not fully understood. Research strongly suggests that there are two major causative factors: one genetic, and the other, environmental. From an environmental standpoint, multiple studies have shown that living farther from the equator, not being exposed to sunlight, and having a low vitamin D level are all correlated with increased risk for MS and MS relapse.1
Our bodies need sunlight to successfully synthesize vitamin D in the skin. Research has found that individuals with lightly pigmented skin are five times more efficient at synthesizing vitamin D in the presence of sunlight than those with darker skin.2 However, the ability to absorb sunlight is also correlated with the earth’s latitude; worse absorption occurs in areas beyond the 40th parallel (in either hemisphere), where UVB levels are too low to synthesize vitamin D four to six months out of the year.2
When exposed to UVB rays, our bodies start to synthesize vitamin D; it undergoes a transformation in the liver and then the kidneys and ultimately becomes the hormonally active form of vitamin D, 1,25-dihydroxyvitamin D3 (calcitriol).2 Calcitriol is recognized by multiple tissues throughout the body that contain vitamin D receptors. Specifically, in the central nervous system, receptors are located on microglia, activated monocytes, and B and T lymphocytes.1 In MS, myelin (the coating around the nerves) is destroyed by an immune-mediated inflammatory process involving the microglia and B and T lymphocytes. Vitamin D quiets down this inflammation, thereby reducing disability accumulation and relapse risk and resulting in fewer changes on MRI.
Vitamin D is also believed to shift the immune response to an anti-inflammatory state by focusing the response on the cytotoxic T cells often found in MS lesions, which attack neurons and oligodendrocytes.2 This theory was tested by Munger and colleagues, who used a pooled cohort of 187,000 women from the Nurses’ Health Study and Nurses’ Health Study II to assess vitamin D intake and risk for MS. Compared to women with lower vitamin D intake, those who took 700 IU/d had a 41% lower incidence of MS. Women who took ≥ 400 IU/d had a 33% lower risk for MS, compared to nonusers.3 In another evaluation of 7 million US military personnel, individuals with a serum vitamin D level of 40 ng/mL were 62% less likely to develop MS.4
In light of the anti-inflammatory effects of vitamin D and its purported reduction of MS risk, it is possible that patients with MS should begin vitamin D supplementation early to obtain maximum anti-inflammatory effects. While an optimal vitamin D goal has not been established in the literature, some studies suggest 30 to 55 ng/mL as a target range for serum vitamin D level.1
While vitamin D has been found to be well-tolerated, patients should be cautioned that very high doses can cause fatigue, abdominal cramps, nausea, vomiting, kidney damage, hypertension, hypercalcemia, and oth
Lisa Marie Fox, MSPAS, PA-C
Division of Multiple Sclerosis, Department of Neurology, Johns Hopkins Hosptial, Baltimore
Q) What is the role of vitamin D in multiple sclerosis? Is it beneficial?
The exact etiology and pathophysiology of multiple sclerosis (MS) is still not fully understood. Research strongly suggests that there are two major causative factors: one genetic, and the other, environmental. From an environmental standpoint, multiple studies have shown that living farther from the equator, not being exposed to sunlight, and having a low vitamin D level are all correlated with increased risk for MS and MS relapse.1
Our bodies need sunlight to successfully synthesize vitamin D in the skin. Research has found that individuals with lightly pigmented skin are five times more efficient at synthesizing vitamin D in the presence of sunlight than those with darker skin.2 However, the ability to absorb sunlight is also correlated with the earth’s latitude; worse absorption occurs in areas beyond the 40th parallel (in either hemisphere), where UVB levels are too low to synthesize vitamin D four to six months out of the year.2
When exposed to UVB rays, our bodies start to synthesize vitamin D; it undergoes a transformation in the liver and then the kidneys and ultimately becomes the hormonally active form of vitamin D, 1,25-dihydroxyvitamin D3 (calcitriol).2 Calcitriol is recognized by multiple tissues throughout the body that contain vitamin D receptors. Specifically, in the central nervous system, receptors are located on microglia, activated monocytes, and B and T lymphocytes.1 In MS, myelin (the coating around the nerves) is destroyed by an immune-mediated inflammatory process involving the microglia and B and T lymphocytes. Vitamin D quiets down this inflammation, thereby reducing disability accumulation and relapse risk and resulting in fewer changes on MRI.
Vitamin D is also believed to shift the immune response to an anti-inflammatory state by focusing the response on the cytotoxic T cells often found in MS lesions, which attack neurons and oligodendrocytes.2 This theory was tested by Munger and colleagues, who used a pooled cohort of 187,000 women from the Nurses’ Health Study and Nurses’ Health Study II to assess vitamin D intake and risk for MS. Compared to women with lower vitamin D intake, those who took 700 IU/d had a 41% lower incidence of MS. Women who took ≥ 400 IU/d had a 33% lower risk for MS, compared to nonusers.3 In another evaluation of 7 million US military personnel, individuals with a serum vitamin D level of 40 ng/mL were 62% less likely to develop MS.4
In light of the anti-inflammatory effects of vitamin D and its purported reduction of MS risk, it is possible that patients with MS should begin vitamin D supplementation early to obtain maximum anti-inflammatory effects. While an optimal vitamin D goal has not been established in the literature, some studies suggest 30 to 55 ng/mL as a target range for serum vitamin D level.1
While vitamin D has been found to be well-tolerated, patients should be cautioned that very high doses can cause fatigue, abdominal cramps, nausea, vomiting, kidney damage, hypertension, hypercalcemia, and oth
Lisa Marie Fox, MSPAS, PA-C
Division of Multiple Sclerosis, Department of Neurology, Johns Hopkins Hosptial, Baltimore
1. Waubant E, Mowry E, Bowling A. The role of vitamin D in multiple sclerosis pathology and treatment: answers and opportunities. Int J MS Care. 2015;17(2):1-24.
2. Pierrot-Deseilligny C. Clinical implications of a possible role of vitamin D in multiple sclerosis. J Neurol. 2009;256(9):1468-1478.3. Munger KL, Zhang SM, O’Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65.
4. Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006; 296(23):2832-2838.
5. Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53-58.
1. Waubant E, Mowry E, Bowling A. The role of vitamin D in multiple sclerosis pathology and treatment: answers and opportunities. Int J MS Care. 2015;17(2):1-24.
2. Pierrot-Deseilligny C. Clinical implications of a possible role of vitamin D in multiple sclerosis. J Neurol. 2009;256(9):1468-1478.3. Munger KL, Zhang SM, O’Reilly E, et al. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62(1):60-65.
4. Munger KL, Levin LI, Hollis BW, et al. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006; 296(23):2832-2838.
5. Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53-58.