User login
Computed tomography angiography after NCCT delays thrombectomy
BALTIMORE – Performing computed tomography angiography (CTA) following noncontrast computed tomography (NCCT) to obtain a high-resolution image of the large-vessel occlusion significantly delays the time to thrombectomy.
In clinical practice, omitting CTA in patients whose middle cerebral artery visualized on NCCT reveals the hyperdense sign may speed the time to thrombectomy and improve outcome. The findings of a retrospective cohort study of prospectively collected data were presented by Kunakorn Atchaneeyasakul, MD, of the University of Miami, as a poster and a brief oral presentation at the annual meeting of the American Neurological Association.

This study retrospectively compared the time from imaging to groin puncture, which is the first step in thrombectomy, in patients who received NCCT followed by CTA with those who received just NCCT for anterior circulation occlusion at the tertiary care University of Miami medical center. Of the 289 patients who received thrombectomy, 255 were excluded because of transfer from another hospital, occurrence of stroke while hospitalized, or use of other imaging prior to thrombectomy.
The remaining 34 patients were all evaluated with thin (0.625-mm) NCCT with automated image reconstruction. Fourteen received NCCT only, and 20 received CTA in addition to NCCT. The two groups were similar in mean age (64-71 years), gender (50% were female in each group), prevalence of hypertension (64% and 70% in the NCCT and NCCT + CTA group, respectively), and prevalence of diabetes, hyperlipidemia, atrial fibrillation, smoking, occlusion site, modified Rankin Scale score at discharge, and National Institutes of Health Stroke Scale scores at presentation and discharge. All 14 NCCT patients received intravenous tPA in contrast to 11 of the 20 (55%) NCCT + CTA patients (P = .003).
The middle cerebral artery was visualized on NCCT in about 85% of patients in each treatment group. Reperfusion was successful in 64% and 80% of patients receiving NCCT and NCCT + CTA, respectively (P = .31).
The total duration of imaging was 2 minutes (range, 1-6) in the NCCT group. The duration was significantly longer in the NCCT + CTA group (28 minutes; range, 23-65; P less than .001). The time from imaging to groin puncture was 68 minutes (range, 32-99) in the NCCT group. This was more than 30 minutes shorter than the NCCT + CTA group (104 minutes; range, 79-128; P = .030).
The times from emergency department admission to NCCT and from admission to groin puncture were similar in both groups.
“Avoiding advanced imaging in patients with anterior circulation large-vessel occlusion in whom thin-section NCCT with maximum-intensity projections reveals a hyperdense sign significantly shortens the imaging to groin puncture time,” concluded Dr. Atchaneeyasakul.
In the scenario, the detection of hyperdense middle cerebral artery would fast track the patient to the angiography suite, forgoing CTA. The result, according to Dr. Atchaneeyasakul, could alleviate a delay in thrombectomy, which could better preserve brain function.
Funding information was not provided.
BALTIMORE – Performing computed tomography angiography (CTA) following noncontrast computed tomography (NCCT) to obtain a high-resolution image of the large-vessel occlusion significantly delays the time to thrombectomy.
In clinical practice, omitting CTA in patients whose middle cerebral artery visualized on NCCT reveals the hyperdense sign may speed the time to thrombectomy and improve outcome. The findings of a retrospective cohort study of prospectively collected data were presented by Kunakorn Atchaneeyasakul, MD, of the University of Miami, as a poster and a brief oral presentation at the annual meeting of the American Neurological Association.
This study retrospectively compared the time from imaging to groin puncture, which is the first step in thrombectomy, in patients who received NCCT followed by CTA with those who received just NCCT for anterior circulation occlusion at the tertiary care University of Miami medical center. Of the 289 patients who received thrombectomy, 255 were excluded because of transfer from another hospital, occurrence of stroke while hospitalized, or use of other imaging prior to thrombectomy.
The remaining 34 patients were all evaluated with thin (0.625-mm) NCCT with automated image reconstruction. Fourteen received NCCT only, and 20 received CTA in addition to NCCT. The two groups were similar in mean age (64-71 years), gender (50% were female in each group), prevalence of hypertension (64% and 70% in the NCCT and NCCT + CTA group, respectively), and prevalence of diabetes, hyperlipidemia, atrial fibrillation, smoking, occlusion site, modified Rankin Scale score at discharge, and National Institutes of Health Stroke Scale scores at presentation and discharge. All 14 NCCT patients received intravenous tPA in contrast to 11 of the 20 (55%) NCCT + CTA patients (P = .003).
The middle cerebral artery was visualized on NCCT in about 85% of patients in each treatment group. Reperfusion was successful in 64% and 80% of patients receiving NCCT and NCCT + CTA, respectively (P = .31).
The total duration of imaging was 2 minutes (range, 1-6) in the NCCT group. The duration was significantly longer in the NCCT + CTA group (28 minutes; range, 23-65; P less than .001). The time from imaging to groin puncture was 68 minutes (range, 32-99) in the NCCT group. This was more than 30 minutes shorter than the NCCT + CTA group (104 minutes; range, 79-128; P = .030).
The times from emergency department admission to NCCT and from admission to groin puncture were similar in both groups.
“Avoiding advanced imaging in patients with anterior circulation large-vessel occlusion in whom thin-section NCCT with maximum-intensity projections reveals a hyperdense sign significantly shortens the imaging to groin puncture time,” concluded Dr. Atchaneeyasakul.
In the scenario, the detection of hyperdense middle cerebral artery would fast track the patient to the angiography suite, forgoing CTA. The result, according to Dr. Atchaneeyasakul, could alleviate a delay in thrombectomy, which could better preserve brain function.
Funding information was not provided.
BALTIMORE – Performing computed tomography angiography (CTA) following noncontrast computed tomography (NCCT) to obtain a high-resolution image of the large-vessel occlusion significantly delays the time to thrombectomy.
In clinical practice, omitting CTA in patients whose middle cerebral artery visualized on NCCT reveals the hyperdense sign may speed the time to thrombectomy and improve outcome. The findings of a retrospective cohort study of prospectively collected data were presented by Kunakorn Atchaneeyasakul, MD, of the University of Miami, as a poster and a brief oral presentation at the annual meeting of the American Neurological Association.
This study retrospectively compared the time from imaging to groin puncture, which is the first step in thrombectomy, in patients who received NCCT followed by CTA with those who received just NCCT for anterior circulation occlusion at the tertiary care University of Miami medical center. Of the 289 patients who received thrombectomy, 255 were excluded because of transfer from another hospital, occurrence of stroke while hospitalized, or use of other imaging prior to thrombectomy.
The remaining 34 patients were all evaluated with thin (0.625-mm) NCCT with automated image reconstruction. Fourteen received NCCT only, and 20 received CTA in addition to NCCT. The two groups were similar in mean age (64-71 years), gender (50% were female in each group), prevalence of hypertension (64% and 70% in the NCCT and NCCT + CTA group, respectively), and prevalence of diabetes, hyperlipidemia, atrial fibrillation, smoking, occlusion site, modified Rankin Scale score at discharge, and National Institutes of Health Stroke Scale scores at presentation and discharge. All 14 NCCT patients received intravenous tPA in contrast to 11 of the 20 (55%) NCCT + CTA patients (P = .003).
The middle cerebral artery was visualized on NCCT in about 85% of patients in each treatment group. Reperfusion was successful in 64% and 80% of patients receiving NCCT and NCCT + CTA, respectively (P = .31).
The total duration of imaging was 2 minutes (range, 1-6) in the NCCT group. The duration was significantly longer in the NCCT + CTA group (28 minutes; range, 23-65; P less than .001). The time from imaging to groin puncture was 68 minutes (range, 32-99) in the NCCT group. This was more than 30 minutes shorter than the NCCT + CTA group (104 minutes; range, 79-128; P = .030).
The times from emergency department admission to NCCT and from admission to groin puncture were similar in both groups.
“Avoiding advanced imaging in patients with anterior circulation large-vessel occlusion in whom thin-section NCCT with maximum-intensity projections reveals a hyperdense sign significantly shortens the imaging to groin puncture time,” concluded Dr. Atchaneeyasakul.
In the scenario, the detection of hyperdense middle cerebral artery would fast track the patient to the angiography suite, forgoing CTA. The result, according to Dr. Atchaneeyasakul, could alleviate a delay in thrombectomy, which could better preserve brain function.
Funding information was not provided.
AT ANA 2016
Key clinical point:
Major finding: Time from imaging to groin puncture was 68 minutes for NCCT vs. 104 minutes for NCCT + CTA.
Data source: Retrospective cohort study of prospectively collected data.
Disclosures: Dr. Atchaneeyasakul had no disclosures.
VIDEO: Pre–gastric bypass antibiotics alter gut microbiome
WASHINGTON – Antibiotics given in advance of gastric bypass surgery preferentially alter the microbiome, nudging it toward a more “lean” physiologic profile.
Given before a sleeve gastrectomy, vancomycin, which has little gut penetration, barely shifted the high ratio of Firmicutes to Bacteroidetes, a profile typically associated with obesity and insulin resistance. But cefazolin, which has much higher gut penetration, suppressed the presence of Firmicutes, which metabolize fat, and allowed the expansion of carbohydrate-loving Bacteroidetes – a profile generally seen in lean people.
Cyrus Jahansouz, MD, of the University of Minnesota, Minneapolis, and his colleagues wanted to examine whether a shift in preoperative antibiotics might affect the way the microbiome re-establishes itself in the wake of vertical sleeve gastrectomy. They enrolled 32 patients who were candidates for the procedure. None had undergone prior gastrointestinal surgery, and none had been exposed to antibiotics in the 3 months prior to bariatric surgery. They were similar in age, weight, body mass index, and fasting glucose. The mean HbA1c was about 6%.
Patients were randomized to three groups: maximal diet therapy (800 calories per day) without surgery; vertical sleeve gastrectomy with the usual preoperative antibiotic cefazolin and the postsurgical diet; and vertical sleeve gastrectomy with preoperative vancomycin and the postsurgical diet. All patients gave a fecal sample immediately before surgery and another one 6 days after surgery.
Preoperative cluster analysis of bacterial DNA showed that all of the samples had a similar composition, predominated by Firmicutes species (60%-70%). Bacteroidetes species made up about 20%-30%, with Proteobacteriae, Actinobacteriae, Verrucomicrobia, and other phyla comprising the remainder of the microbiome.
At the second sampling, the diet-only group showed no microbiome changes at all. The vancomycin group showed a very small but not significant expansion of Bacteroidetes and reduction of Firmicutes.
Patients in the cefazolin group showed a significant shift in the ratio – and it was quite striking, Dr. Jahansouz said. Among these patients, Firmicutes had decreased from 70% to 40% of the community. Bacteroidetes showed a corresponding shift, increasing from 20% of the community to 45%. The findings are quite surprising, he noted, considering that only one dose of antibiotic was associated with the changes and that they were evident within just a few days.
Although “a little hard to interpret” because of its small size and short follow-up, the study suggests that antibiotic choice might contribute to the success of weight-loss surgery, Dr. Jahansouz said at the annual clinical congress of the American College of Surgeons.
“There are still several factors in the perioperative period that we have to study to be able to identify what other things might have also influenced the shift,” he said in an interview. “But I do think that, in the future, these changes can be manipulated to benefit metabolic outcomes.”
Two phyla – Bacteroidetes and Firmicutes – dominate the human gut microbiome in a dynamic ratio that is highly associated with the way energy is extracted from food. Bacteroidetes species specialize in carbohydrate digestion and Firmicutes in fat digestion. “In a lean, insulin-sensitive state, Bacteroidetes dominates the human gut microbiome,” Dr. Jahansouz said. “With the progression of obesity and insulin resistance, there is a subsequent shift in the microbiome phenotype, favoring the growth of Firmicutes at the expense and reduction of Bacteroidetes. This is a significant change, because this obesity-associated phenotype has an increased capacity to harvest energy. It’s not the same for a lean person to consume 1,000 calories as it is for an obese person to consume them.”
Bariatric surgery has been shown to alter the gut microbiome, shifting it toward this more “lean” profile (Cell Metab. 2015 Aug 4;22[2]:228-38). This shift may be an important component of the still not fully elucidated mechanisms by which bariatric surgery causes weight loss and normalizes insulin signaling, Dr. Jahansouz said.
Dr. Jahansouz is following this group of patients to explore whether there are differences in weight loss and insulin signaling. He also will track whether the microbiome stabilizes at its early postsurgical profile, or continues to shift, either toward an even higher Bacteroidetes to Firmicutes ratio, or back to a more “obese” profile.
He and his colleagues are also investigating the effect of antibiotics and gastric bypass surgery in mouse models. “I can say that antibiotics seem to have a remarkable impact on the effect of mouse sleeve gastrectomy. We’re not quite there yet with humans,” but the data are compelling.
Dr. Jahansouz said that he had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @Alz_Gal
WASHINGTON – Antibiotics given in advance of gastric bypass surgery preferentially alter the microbiome, nudging it toward a more “lean” physiologic profile.
Given before a sleeve gastrectomy, vancomycin, which has little gut penetration, barely shifted the high ratio of Firmicutes to Bacteroidetes, a profile typically associated with obesity and insulin resistance. But cefazolin, which has much higher gut penetration, suppressed the presence of Firmicutes, which metabolize fat, and allowed the expansion of carbohydrate-loving Bacteroidetes – a profile generally seen in lean people.
Cyrus Jahansouz, MD, of the University of Minnesota, Minneapolis, and his colleagues wanted to examine whether a shift in preoperative antibiotics might affect the way the microbiome re-establishes itself in the wake of vertical sleeve gastrectomy. They enrolled 32 patients who were candidates for the procedure. None had undergone prior gastrointestinal surgery, and none had been exposed to antibiotics in the 3 months prior to bariatric surgery. They were similar in age, weight, body mass index, and fasting glucose. The mean HbA1c was about 6%.
Patients were randomized to three groups: maximal diet therapy (800 calories per day) without surgery; vertical sleeve gastrectomy with the usual preoperative antibiotic cefazolin and the postsurgical diet; and vertical sleeve gastrectomy with preoperative vancomycin and the postsurgical diet. All patients gave a fecal sample immediately before surgery and another one 6 days after surgery.
Preoperative cluster analysis of bacterial DNA showed that all of the samples had a similar composition, predominated by Firmicutes species (60%-70%). Bacteroidetes species made up about 20%-30%, with Proteobacteriae, Actinobacteriae, Verrucomicrobia, and other phyla comprising the remainder of the microbiome.
At the second sampling, the diet-only group showed no microbiome changes at all. The vancomycin group showed a very small but not significant expansion of Bacteroidetes and reduction of Firmicutes.
Patients in the cefazolin group showed a significant shift in the ratio – and it was quite striking, Dr. Jahansouz said. Among these patients, Firmicutes had decreased from 70% to 40% of the community. Bacteroidetes showed a corresponding shift, increasing from 20% of the community to 45%. The findings are quite surprising, he noted, considering that only one dose of antibiotic was associated with the changes and that they were evident within just a few days.
Although “a little hard to interpret” because of its small size and short follow-up, the study suggests that antibiotic choice might contribute to the success of weight-loss surgery, Dr. Jahansouz said at the annual clinical congress of the American College of Surgeons.
“There are still several factors in the perioperative period that we have to study to be able to identify what other things might have also influenced the shift,” he said in an interview. “But I do think that, in the future, these changes can be manipulated to benefit metabolic outcomes.”
Two phyla – Bacteroidetes and Firmicutes – dominate the human gut microbiome in a dynamic ratio that is highly associated with the way energy is extracted from food. Bacteroidetes species specialize in carbohydrate digestion and Firmicutes in fat digestion. “In a lean, insulin-sensitive state, Bacteroidetes dominates the human gut microbiome,” Dr. Jahansouz said. “With the progression of obesity and insulin resistance, there is a subsequent shift in the microbiome phenotype, favoring the growth of Firmicutes at the expense and reduction of Bacteroidetes. This is a significant change, because this obesity-associated phenotype has an increased capacity to harvest energy. It’s not the same for a lean person to consume 1,000 calories as it is for an obese person to consume them.”
Bariatric surgery has been shown to alter the gut microbiome, shifting it toward this more “lean” profile (Cell Metab. 2015 Aug 4;22[2]:228-38). This shift may be an important component of the still not fully elucidated mechanisms by which bariatric surgery causes weight loss and normalizes insulin signaling, Dr. Jahansouz said.
Dr. Jahansouz is following this group of patients to explore whether there are differences in weight loss and insulin signaling. He also will track whether the microbiome stabilizes at its early postsurgical profile, or continues to shift, either toward an even higher Bacteroidetes to Firmicutes ratio, or back to a more “obese” profile.
He and his colleagues are also investigating the effect of antibiotics and gastric bypass surgery in mouse models. “I can say that antibiotics seem to have a remarkable impact on the effect of mouse sleeve gastrectomy. We’re not quite there yet with humans,” but the data are compelling.
Dr. Jahansouz said that he had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @Alz_Gal
WASHINGTON – Antibiotics given in advance of gastric bypass surgery preferentially alter the microbiome, nudging it toward a more “lean” physiologic profile.
Given before a sleeve gastrectomy, vancomycin, which has little gut penetration, barely shifted the high ratio of Firmicutes to Bacteroidetes, a profile typically associated with obesity and insulin resistance. But cefazolin, which has much higher gut penetration, suppressed the presence of Firmicutes, which metabolize fat, and allowed the expansion of carbohydrate-loving Bacteroidetes – a profile generally seen in lean people.
Cyrus Jahansouz, MD, of the University of Minnesota, Minneapolis, and his colleagues wanted to examine whether a shift in preoperative antibiotics might affect the way the microbiome re-establishes itself in the wake of vertical sleeve gastrectomy. They enrolled 32 patients who were candidates for the procedure. None had undergone prior gastrointestinal surgery, and none had been exposed to antibiotics in the 3 months prior to bariatric surgery. They were similar in age, weight, body mass index, and fasting glucose. The mean HbA1c was about 6%.
Patients were randomized to three groups: maximal diet therapy (800 calories per day) without surgery; vertical sleeve gastrectomy with the usual preoperative antibiotic cefazolin and the postsurgical diet; and vertical sleeve gastrectomy with preoperative vancomycin and the postsurgical diet. All patients gave a fecal sample immediately before surgery and another one 6 days after surgery.
Preoperative cluster analysis of bacterial DNA showed that all of the samples had a similar composition, predominated by Firmicutes species (60%-70%). Bacteroidetes species made up about 20%-30%, with Proteobacteriae, Actinobacteriae, Verrucomicrobia, and other phyla comprising the remainder of the microbiome.
At the second sampling, the diet-only group showed no microbiome changes at all. The vancomycin group showed a very small but not significant expansion of Bacteroidetes and reduction of Firmicutes.
Patients in the cefazolin group showed a significant shift in the ratio – and it was quite striking, Dr. Jahansouz said. Among these patients, Firmicutes had decreased from 70% to 40% of the community. Bacteroidetes showed a corresponding shift, increasing from 20% of the community to 45%. The findings are quite surprising, he noted, considering that only one dose of antibiotic was associated with the changes and that they were evident within just a few days.
Although “a little hard to interpret” because of its small size and short follow-up, the study suggests that antibiotic choice might contribute to the success of weight-loss surgery, Dr. Jahansouz said at the annual clinical congress of the American College of Surgeons.
“There are still several factors in the perioperative period that we have to study to be able to identify what other things might have also influenced the shift,” he said in an interview. “But I do think that, in the future, these changes can be manipulated to benefit metabolic outcomes.”
Two phyla – Bacteroidetes and Firmicutes – dominate the human gut microbiome in a dynamic ratio that is highly associated with the way energy is extracted from food. Bacteroidetes species specialize in carbohydrate digestion and Firmicutes in fat digestion. “In a lean, insulin-sensitive state, Bacteroidetes dominates the human gut microbiome,” Dr. Jahansouz said. “With the progression of obesity and insulin resistance, there is a subsequent shift in the microbiome phenotype, favoring the growth of Firmicutes at the expense and reduction of Bacteroidetes. This is a significant change, because this obesity-associated phenotype has an increased capacity to harvest energy. It’s not the same for a lean person to consume 1,000 calories as it is for an obese person to consume them.”
Bariatric surgery has been shown to alter the gut microbiome, shifting it toward this more “lean” profile (Cell Metab. 2015 Aug 4;22[2]:228-38). This shift may be an important component of the still not fully elucidated mechanisms by which bariatric surgery causes weight loss and normalizes insulin signaling, Dr. Jahansouz said.
Dr. Jahansouz is following this group of patients to explore whether there are differences in weight loss and insulin signaling. He also will track whether the microbiome stabilizes at its early postsurgical profile, or continues to shift, either toward an even higher Bacteroidetes to Firmicutes ratio, or back to a more “obese” profile.
He and his colleagues are also investigating the effect of antibiotics and gastric bypass surgery in mouse models. “I can say that antibiotics seem to have a remarkable impact on the effect of mouse sleeve gastrectomy. We’re not quite there yet with humans,” but the data are compelling.
Dr. Jahansouz said that he had no financial disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
[email protected]
On Twitter @Alz_Gal
EXPERT ANALYSIS FROM THE ACS CLINICAL CONGRESS

FBN-1 Overexpression Linked to Testicular Tumor Development
Researchers from Comenius University in Bratislava, Slovakia, have shown for the first time that fibrillin-1 (FBN-1) is overexpressed in testicular germ cell tumors (TGCTs). Their data suggest that progression of germ cell neoplasia in situ (GCNIS) to overt germ cell tumor is connected with decreased FBN-1 production. Fibrillin is a glycoprotein essential in forming the elastic fibers in connective tissue.
The researchers evaluated 350 tumor specimens and 85 adjacent nonneoplastic testicular tissues in 203 patients. They detected FBN-1 expression in 68 (97%) GCNIS, 21 (96%) yolk sac tumors, 85 (87%) seminomas, 78 (83%) embryonal carcinomas, 35 (71%) teratomas, 3 (18%) choriocarcinomas, and 1 (1%) nonneoplastic tissue. The highest FBN-1 positivity was found in GCNIS, with overexpression of FBN-1 in 77% of cases. The fact that the highest expression was observed in seminomas and the lowest in choriocarcinomas, the researchers say, suggests a reduction of FBN-1 expression with the process of differentiation.
Related: Is Chemotherapy a Good Choice for Neuroendocrine Tumors?
GCNIS represents the preinvasive stage of most TGCTs in adults. GCNIS cells are believed to be germ cells that have failed to mature normally, the researchers note. The appearance of FBN-1 expression in germ cells in the stage of GCNIS, they suggest, can be an early event in the process of malignant transformation.
Source:
Cierna Z, Mego M, Jurisica I, et al. BMC Cancer. 2016;16:597
doi: 10.1186/s12885-016-2644-z.
Researchers from Comenius University in Bratislava, Slovakia, have shown for the first time that fibrillin-1 (FBN-1) is overexpressed in testicular germ cell tumors (TGCTs). Their data suggest that progression of germ cell neoplasia in situ (GCNIS) to overt germ cell tumor is connected with decreased FBN-1 production. Fibrillin is a glycoprotein essential in forming the elastic fibers in connective tissue.
The researchers evaluated 350 tumor specimens and 85 adjacent nonneoplastic testicular tissues in 203 patients. They detected FBN-1 expression in 68 (97%) GCNIS, 21 (96%) yolk sac tumors, 85 (87%) seminomas, 78 (83%) embryonal carcinomas, 35 (71%) teratomas, 3 (18%) choriocarcinomas, and 1 (1%) nonneoplastic tissue. The highest FBN-1 positivity was found in GCNIS, with overexpression of FBN-1 in 77% of cases. The fact that the highest expression was observed in seminomas and the lowest in choriocarcinomas, the researchers say, suggests a reduction of FBN-1 expression with the process of differentiation.
Related: Is Chemotherapy a Good Choice for Neuroendocrine Tumors?
GCNIS represents the preinvasive stage of most TGCTs in adults. GCNIS cells are believed to be germ cells that have failed to mature normally, the researchers note. The appearance of FBN-1 expression in germ cells in the stage of GCNIS, they suggest, can be an early event in the process of malignant transformation.
Source:
Cierna Z, Mego M, Jurisica I, et al. BMC Cancer. 2016;16:597
doi: 10.1186/s12885-016-2644-z.
Researchers from Comenius University in Bratislava, Slovakia, have shown for the first time that fibrillin-1 (FBN-1) is overexpressed in testicular germ cell tumors (TGCTs). Their data suggest that progression of germ cell neoplasia in situ (GCNIS) to overt germ cell tumor is connected with decreased FBN-1 production. Fibrillin is a glycoprotein essential in forming the elastic fibers in connective tissue.
The researchers evaluated 350 tumor specimens and 85 adjacent nonneoplastic testicular tissues in 203 patients. They detected FBN-1 expression in 68 (97%) GCNIS, 21 (96%) yolk sac tumors, 85 (87%) seminomas, 78 (83%) embryonal carcinomas, 35 (71%) teratomas, 3 (18%) choriocarcinomas, and 1 (1%) nonneoplastic tissue. The highest FBN-1 positivity was found in GCNIS, with overexpression of FBN-1 in 77% of cases. The fact that the highest expression was observed in seminomas and the lowest in choriocarcinomas, the researchers say, suggests a reduction of FBN-1 expression with the process of differentiation.
Related: Is Chemotherapy a Good Choice for Neuroendocrine Tumors?
GCNIS represents the preinvasive stage of most TGCTs in adults. GCNIS cells are believed to be germ cells that have failed to mature normally, the researchers note. The appearance of FBN-1 expression in germ cells in the stage of GCNIS, they suggest, can be an early event in the process of malignant transformation.
Source:
Cierna Z, Mego M, Jurisica I, et al. BMC Cancer. 2016;16:597
doi: 10.1186/s12885-016-2644-z.
ACOS under-recognized, under-treated
Exacerbations in bronchodilator-responsive asthma–chronic obstructive pulmonary disease overlap syndrome (ACOS) were more frequent and severe than in COPD with emphysema, but only a minority of patients were treated to prevent them, in a review of 1,005 patients from the Annals of the American Thoracic Society.
All the subjects were current or former smokers culled from the COPDGene Study, a multicenter observational study looking for the genetic roots of COPD susceptibility; 385 patients met the investigators’ criteria for ACOS with bronchodilator response (ACOS-BDR), which included a history of asthma or hay fever, airway obstruction with significant bronchodilator responsiveness, and less then 15% emphysema on chest CT.
Another 620 subjects met criteria for COPD with emphysema, including airway obstruction without bronchodilator reversibility, and more than 15% emphysema on chest CT (Ann Am Thorac Soc. 2016 Sep;13(9):1483-9).
Although the ACOS patients had better lung function, they had similar severity and frequency of exacerbations, compared with the COPD group. After adjustment for forced expiratory volume in 1 second percent predicted and other factors, the patients with ACOS-BDR were actually more likely to have severe and frequent exacerbations. Possible explanations for this are that they were more likely to smoke and have gastroesophageal reflux disease and obstructive sleep apnea, all of which increase the risk of exacerbations.
Even so, ACOS-BDR patients were less likely to be on a long-acting beta-agonist (6.8% versus 13.9%); a long-acting muscarinic antagonist (20% versus 60.8%); or a combination long-acting beta-agonist/inhaled corticosteroid (29.9% versus 55.6%).
“Only a small percentage of them were being treated ... Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations,” said investigators led by James Cosentino, DO, of Temple University School of Medicine, Philadelphia. Patients with ACOS “are a particularly high-risk group.” They deserve “special attention, and practitioners need to be diligent in evaluation of them.”
ACOS is being increasingly recognized as a distinct clinical entity with perhaps a worse prognosis than either asthma or COPD alone. The goal of the study was to better characterize the disease.
To that end, the team found four features that seemed to distinguish ACOS-BDR from COPD with emphysema: ACOS-BDR patients were younger (60.6 versus 65.9 years old); heavier (body mass index 29.6 versus 25.1 kg/m2; more likely to be African American (26.8% versus 14.4%); and more likely to be current smokers (50.9% versus 20.7%).
It’s “likely that current smoking in subjects with ACOS, coupled with the long duration of asthma, leads to inflammation and small airway remodeling with development of symptoms earlier in the disease course than that seen in those with COPD with emphysema,” the investigators said.
“Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations. Recognition and treatment of comorbidities and aggressive smoking cessation may also play a key role in preventing exacerbations and alleviating the morbidity associated with ACOS; however, future studies on the treatment of ACOS are needed,” they said.
The majority of subjects with ACOS-BDR met criteria for Global Initiative for Chronic Obstructive Lung Disease grade B, indicating a high degree of symptoms despite less severe airflow obstruction.
The funding source wasn’t reported. Dr. Cosentino had no conflicts. Other authors disclosed personal fees from Concert Pharmaceuticals, CSA Medical, CSL Behring, Gala Therapeutics, and Novartis.
The importance of this study is that it used readily available metrics to define ACOS in a COPD population. Although diffusion capacity was not reported, quantification of emphysema on chest CT scans combined with history and spirometry provide a reasonable approach to distinguishing ACOS-BDR from COPD with emphysema.
Although subjects with COPD had smoked more heavily as measured by cigarette pack-years, subjects with ACOS were much more likely to be current smokers... Subjects with ACOS also had a higher prevalence of comorbidities such as sleep apnea, diabetes mellitus, hypertension, and hypercholesterolemia as compared with patients with COPD. Having a higher BMI, to near obesity, and a greater prevalence of gastroesophageal reflux disease raises questions related to diet, lifestyle, and nutrition as potential contributors to ACOS pathophysiology.
In the future, the use of diagnostic terms such as “asthma,” “COPD,” and “ACOS,” will likely give way to the more unifying diagnosis of obstructive airway disease (OAD)... OAD would be further delineated on the basis of molecular phenotyping, genomic, and systems biology approaches, in combination with more traditional clinical and physiological parameters... This new mindset can help us solve the problem of obstructive airway disease taxonomy and develop not only better treatments, but eventually invent lasting cures – if we are so lucky.
Amir Zeki, MD , is an assistant professor in the Division of Pulmonary, Critical Care, and Sleep Medicine at the University of California, Davis. Nizar Jarjour, MD , is a professor of medicine and head of the allergy, pulmonary, and critical care division at the University of Wisconsin, Madison. Dr. Zeki had no disclosures. Dr. Jarjour reported consulting fees from AstraZeneca, Daiichi Sankyo, and Teva. They made their comments in an editorial ( Ann Am Thorac Soc. 2016 Sep;13(9):1440-2 ).
The importance of this study is that it used readily available metrics to define ACOS in a COPD population. Although diffusion capacity was not reported, quantification of emphysema on chest CT scans combined with history and spirometry provide a reasonable approach to distinguishing ACOS-BDR from COPD with emphysema.
Although subjects with COPD had smoked more heavily as measured by cigarette pack-years, subjects with ACOS were much more likely to be current smokers... Subjects with ACOS also had a higher prevalence of comorbidities such as sleep apnea, diabetes mellitus, hypertension, and hypercholesterolemia as compared with patients with COPD. Having a higher BMI, to near obesity, and a greater prevalence of gastroesophageal reflux disease raises questions related to diet, lifestyle, and nutrition as potential contributors to ACOS pathophysiology.
In the future, the use of diagnostic terms such as “asthma,” “COPD,” and “ACOS,” will likely give way to the more unifying diagnosis of obstructive airway disease (OAD)... OAD would be further delineated on the basis of molecular phenotyping, genomic, and systems biology approaches, in combination with more traditional clinical and physiological parameters... This new mindset can help us solve the problem of obstructive airway disease taxonomy and develop not only better treatments, but eventually invent lasting cures – if we are so lucky.
Amir Zeki, MD , is an assistant professor in the Division of Pulmonary, Critical Care, and Sleep Medicine at the University of California, Davis. Nizar Jarjour, MD , is a professor of medicine and head of the allergy, pulmonary, and critical care division at the University of Wisconsin, Madison. Dr. Zeki had no disclosures. Dr. Jarjour reported consulting fees from AstraZeneca, Daiichi Sankyo, and Teva. They made their comments in an editorial ( Ann Am Thorac Soc. 2016 Sep;13(9):1440-2 ).
The importance of this study is that it used readily available metrics to define ACOS in a COPD population. Although diffusion capacity was not reported, quantification of emphysema on chest CT scans combined with history and spirometry provide a reasonable approach to distinguishing ACOS-BDR from COPD with emphysema.
Although subjects with COPD had smoked more heavily as measured by cigarette pack-years, subjects with ACOS were much more likely to be current smokers... Subjects with ACOS also had a higher prevalence of comorbidities such as sleep apnea, diabetes mellitus, hypertension, and hypercholesterolemia as compared with patients with COPD. Having a higher BMI, to near obesity, and a greater prevalence of gastroesophageal reflux disease raises questions related to diet, lifestyle, and nutrition as potential contributors to ACOS pathophysiology.
In the future, the use of diagnostic terms such as “asthma,” “COPD,” and “ACOS,” will likely give way to the more unifying diagnosis of obstructive airway disease (OAD)... OAD would be further delineated on the basis of molecular phenotyping, genomic, and systems biology approaches, in combination with more traditional clinical and physiological parameters... This new mindset can help us solve the problem of obstructive airway disease taxonomy and develop not only better treatments, but eventually invent lasting cures – if we are so lucky.
Amir Zeki, MD , is an assistant professor in the Division of Pulmonary, Critical Care, and Sleep Medicine at the University of California, Davis. Nizar Jarjour, MD , is a professor of medicine and head of the allergy, pulmonary, and critical care division at the University of Wisconsin, Madison. Dr. Zeki had no disclosures. Dr. Jarjour reported consulting fees from AstraZeneca, Daiichi Sankyo, and Teva. They made their comments in an editorial ( Ann Am Thorac Soc. 2016 Sep;13(9):1440-2 ).
Exacerbations in bronchodilator-responsive asthma–chronic obstructive pulmonary disease overlap syndrome (ACOS) were more frequent and severe than in COPD with emphysema, but only a minority of patients were treated to prevent them, in a review of 1,005 patients from the Annals of the American Thoracic Society.
All the subjects were current or former smokers culled from the COPDGene Study, a multicenter observational study looking for the genetic roots of COPD susceptibility; 385 patients met the investigators’ criteria for ACOS with bronchodilator response (ACOS-BDR), which included a history of asthma or hay fever, airway obstruction with significant bronchodilator responsiveness, and less then 15% emphysema on chest CT.
Another 620 subjects met criteria for COPD with emphysema, including airway obstruction without bronchodilator reversibility, and more than 15% emphysema on chest CT (Ann Am Thorac Soc. 2016 Sep;13(9):1483-9).
Although the ACOS patients had better lung function, they had similar severity and frequency of exacerbations, compared with the COPD group. After adjustment for forced expiratory volume in 1 second percent predicted and other factors, the patients with ACOS-BDR were actually more likely to have severe and frequent exacerbations. Possible explanations for this are that they were more likely to smoke and have gastroesophageal reflux disease and obstructive sleep apnea, all of which increase the risk of exacerbations.
Even so, ACOS-BDR patients were less likely to be on a long-acting beta-agonist (6.8% versus 13.9%); a long-acting muscarinic antagonist (20% versus 60.8%); or a combination long-acting beta-agonist/inhaled corticosteroid (29.9% versus 55.6%).
“Only a small percentage of them were being treated ... Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations,” said investigators led by James Cosentino, DO, of Temple University School of Medicine, Philadelphia. Patients with ACOS “are a particularly high-risk group.” They deserve “special attention, and practitioners need to be diligent in evaluation of them.”
ACOS is being increasingly recognized as a distinct clinical entity with perhaps a worse prognosis than either asthma or COPD alone. The goal of the study was to better characterize the disease.
To that end, the team found four features that seemed to distinguish ACOS-BDR from COPD with emphysema: ACOS-BDR patients were younger (60.6 versus 65.9 years old); heavier (body mass index 29.6 versus 25.1 kg/m2; more likely to be African American (26.8% versus 14.4%); and more likely to be current smokers (50.9% versus 20.7%).
It’s “likely that current smoking in subjects with ACOS, coupled with the long duration of asthma, leads to inflammation and small airway remodeling with development of symptoms earlier in the disease course than that seen in those with COPD with emphysema,” the investigators said.
“Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations. Recognition and treatment of comorbidities and aggressive smoking cessation may also play a key role in preventing exacerbations and alleviating the morbidity associated with ACOS; however, future studies on the treatment of ACOS are needed,” they said.
The majority of subjects with ACOS-BDR met criteria for Global Initiative for Chronic Obstructive Lung Disease grade B, indicating a high degree of symptoms despite less severe airflow obstruction.
The funding source wasn’t reported. Dr. Cosentino had no conflicts. Other authors disclosed personal fees from Concert Pharmaceuticals, CSA Medical, CSL Behring, Gala Therapeutics, and Novartis.
Exacerbations in bronchodilator-responsive asthma–chronic obstructive pulmonary disease overlap syndrome (ACOS) were more frequent and severe than in COPD with emphysema, but only a minority of patients were treated to prevent them, in a review of 1,005 patients from the Annals of the American Thoracic Society.
All the subjects were current or former smokers culled from the COPDGene Study, a multicenter observational study looking for the genetic roots of COPD susceptibility; 385 patients met the investigators’ criteria for ACOS with bronchodilator response (ACOS-BDR), which included a history of asthma or hay fever, airway obstruction with significant bronchodilator responsiveness, and less then 15% emphysema on chest CT.
Another 620 subjects met criteria for COPD with emphysema, including airway obstruction without bronchodilator reversibility, and more than 15% emphysema on chest CT (Ann Am Thorac Soc. 2016 Sep;13(9):1483-9).
Although the ACOS patients had better lung function, they had similar severity and frequency of exacerbations, compared with the COPD group. After adjustment for forced expiratory volume in 1 second percent predicted and other factors, the patients with ACOS-BDR were actually more likely to have severe and frequent exacerbations. Possible explanations for this are that they were more likely to smoke and have gastroesophageal reflux disease and obstructive sleep apnea, all of which increase the risk of exacerbations.
Even so, ACOS-BDR patients were less likely to be on a long-acting beta-agonist (6.8% versus 13.9%); a long-acting muscarinic antagonist (20% versus 60.8%); or a combination long-acting beta-agonist/inhaled corticosteroid (29.9% versus 55.6%).
“Only a small percentage of them were being treated ... Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations,” said investigators led by James Cosentino, DO, of Temple University School of Medicine, Philadelphia. Patients with ACOS “are a particularly high-risk group.” They deserve “special attention, and practitioners need to be diligent in evaluation of them.”
ACOS is being increasingly recognized as a distinct clinical entity with perhaps a worse prognosis than either asthma or COPD alone. The goal of the study was to better characterize the disease.
To that end, the team found four features that seemed to distinguish ACOS-BDR from COPD with emphysema: ACOS-BDR patients were younger (60.6 versus 65.9 years old); heavier (body mass index 29.6 versus 25.1 kg/m2; more likely to be African American (26.8% versus 14.4%); and more likely to be current smokers (50.9% versus 20.7%).
It’s “likely that current smoking in subjects with ACOS, coupled with the long duration of asthma, leads to inflammation and small airway remodeling with development of symptoms earlier in the disease course than that seen in those with COPD with emphysema,” the investigators said.
“Early and aggressive treatment with combination therapy may help alleviate symptoms and decrease exacerbations. Recognition and treatment of comorbidities and aggressive smoking cessation may also play a key role in preventing exacerbations and alleviating the morbidity associated with ACOS; however, future studies on the treatment of ACOS are needed,” they said.
The majority of subjects with ACOS-BDR met criteria for Global Initiative for Chronic Obstructive Lung Disease grade B, indicating a high degree of symptoms despite less severe airflow obstruction.
The funding source wasn’t reported. Dr. Cosentino had no conflicts. Other authors disclosed personal fees from Concert Pharmaceuticals, CSA Medical, CSL Behring, Gala Therapeutics, and Novartis.
FROM THE ANNALS OF THE AMERICAN THORACIC SOCIETY
Key clinical point:
Major finding: ACOS-BDR patients were less likely to be on a long-acting beta-agonist (6.8% versus 13.9%); a long-acting muscarinic antagonist (20% versus 60.8%); or a combination long-acting beta-agonist/inhaled corticosteroid (29.9% versus 55.6%).
Data source: 1,005 patients from the COPDGene study.
Disclosures: The funding source wasn’t reported. Authors disclosed personal fees from Concert Pharmaceuticals, CSA Medical, CSL Behring, Gala Therapeutics, and Novartis.
Odanacatib reduced fractures but upped stroke risk
The novel oral osteoporosis drug odanacatib significantly reduced fractures in postmenopausal women but was also associated with a significantly higher risk for stroke, according to data from a 5-year extension of a large phase III clinical trial.
Based on an independent analysis and verification of the risk for stroke, the drug’s sponsor, Merck, has withdrawn odanacatib from review by the Food and Drug Administration. “We are disappointed that the overall benefit-risk profile for odanacatib does not support filing or further development,” Roger M. Perlmutter, MD, president of Merck Research Laboratories, said in a statement.

Compared with placebo, odanacatib resulted in relative risk reductions of 52% for vertebral fracture, 48% for hip fracture, 26% for nonvertebral fracture, and 67% for clinical vertebral fracture (all values, P less than .001). Lumbar spine bone density increased by a mean of 10.9%, as did total hip bone density by a mean of 10.3%, in the odanacatib group (for both, P less than .001), according to results presented at the meeting.
The Long-Term Odanacatib Fracture Trial (LOFT) was a randomized, double-blind placebo-controlled study that investigated the efficacy and safety of odanacatib, a cathepsin K inhibitor, as a treatment for osteoporosis and for fracture prevention in postmenopausal women. Odanacatib was taken as an oral, once-weekly 50-mg pill.
During the base period of the study, 16,071 postmenopausal women aged 65 and older were enrolled, and 12,290 completed the study. Women had to have total hip or femoral neck bone mineral density T scores less than or equal to –2.5, or a radiographically confirmed vertebral fracture and total hip or femoral neck T scores less than or equal to –1.5. Participants were demographically well matched between study arms; 6,092 odanacatib patients and 6,198 on placebo completed the base period of the study.
The original phase III study was stopped early because of robust efficacy data (Osteoporos Int. 2015 Feb;26[2]:699-712). Participants were eligible to continue in a preplanned 5-year double-blind, placebo-controlled extension period of the LOFT study; 3,432 odanacatib and 2,615 placebo participants completed the full 5 years.
An early, but statistically nonsignificant, signal for increased stroke and atrial fibrillation and flutter was seen at the end of the study’s base period. The trend continued and appeared to be amplified during the 5-year, double-blind, placebo-controlled extension period of the LOFT study.

In an interview, Dr. O’Donoghue said that the TIMI study group was brought in by Merck for a “second perspective,” to add rigor to an examination of events that had not been anticipated in the base period of the LOFT study. This was an important step, said Dr. O’Donoghue, because LOFT was not a dedicated cardiovascular safety trial.
The prespecified primary endpoints for TIMI’s safety analysis included new-onset atrial flutter or atrial fibrillation, as well as a composite endpoint of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or cardiovascular death.
Examination of the composite MACE endpoint showed a numeric, but not statistically significant, difference between the odanacatib and placebo arms of the extension study. However, in a preplanned subanalysis, when the 324 stroke events were isolated, a hazard ratio (HR) for stroke of 1.37 for odanacatib emerged (95% confidence interval [CI], 1.10-1.71; P = .005).
Atrial fibrillation and atrial flutter were more common in the odanacatib group, but the difference was not significant (HR, 1.22; 95% CI, 0.99-1.50; P = .06). Dr. O’Donoghue said that the individuals with supraventricular arrhythmias were not the same individuals who had strokes, although the strokes were almost entirely ischemic, rather than hemorrhagic, events.
Dr. O’Donoghue, a cardiovascular medicine specialist at Brigham and Women’s Hospital, noted in her presentation that “preclinical data suggested that inhibition of cathepsin K may reduce atherosclerosis progression and promote plaque stability.”
Odanacatib is a cathepsin K inhibitor, one of a member of a class of proteases. Cathepsin K targets kinins that are involved in bone resorption, but it is expressed in other tissues as well, and it may target other classes of kinins. However, exactly how odanacatib may have contributed to strokes in the study population remains a mystery.
“The mechanistic underpinnings to explain these findings is unknown and warrants investigation,” Dr. O’Donoghue said. “It’s a little bit more unsatisfying when you’re not able to provide a cause. … I share the disappointment of the endocrinologists in the community who were very hopeful for the cathepsin K class to be the next breakthrough in the management of osteoporosis.”
Dr. McClung reported financial relationships with Merck and several other pharmaceutical companies. Dr. O’Donoghue reported receiving grant support from several pharmaceutical companies. The LOFT trial and the TIMI study group analysis were sponsored by Merck.
[email protected]
On Twitter @karioakes
The novel oral osteoporosis drug odanacatib significantly reduced fractures in postmenopausal women but was also associated with a significantly higher risk for stroke, according to data from a 5-year extension of a large phase III clinical trial.
Based on an independent analysis and verification of the risk for stroke, the drug’s sponsor, Merck, has withdrawn odanacatib from review by the Food and Drug Administration. “We are disappointed that the overall benefit-risk profile for odanacatib does not support filing or further development,” Roger M. Perlmutter, MD, president of Merck Research Laboratories, said in a statement.
Compared with placebo, odanacatib resulted in relative risk reductions of 52% for vertebral fracture, 48% for hip fracture, 26% for nonvertebral fracture, and 67% for clinical vertebral fracture (all values, P less than .001). Lumbar spine bone density increased by a mean of 10.9%, as did total hip bone density by a mean of 10.3%, in the odanacatib group (for both, P less than .001), according to results presented at the meeting.
The Long-Term Odanacatib Fracture Trial (LOFT) was a randomized, double-blind placebo-controlled study that investigated the efficacy and safety of odanacatib, a cathepsin K inhibitor, as a treatment for osteoporosis and for fracture prevention in postmenopausal women. Odanacatib was taken as an oral, once-weekly 50-mg pill.
During the base period of the study, 16,071 postmenopausal women aged 65 and older were enrolled, and 12,290 completed the study. Women had to have total hip or femoral neck bone mineral density T scores less than or equal to –2.5, or a radiographically confirmed vertebral fracture and total hip or femoral neck T scores less than or equal to –1.5. Participants were demographically well matched between study arms; 6,092 odanacatib patients and 6,198 on placebo completed the base period of the study.
The original phase III study was stopped early because of robust efficacy data (Osteoporos Int. 2015 Feb;26[2]:699-712). Participants were eligible to continue in a preplanned 5-year double-blind, placebo-controlled extension period of the LOFT study; 3,432 odanacatib and 2,615 placebo participants completed the full 5 years.
An early, but statistically nonsignificant, signal for increased stroke and atrial fibrillation and flutter was seen at the end of the study’s base period. The trend continued and appeared to be amplified during the 5-year, double-blind, placebo-controlled extension period of the LOFT study.
In an interview, Dr. O’Donoghue said that the TIMI study group was brought in by Merck for a “second perspective,” to add rigor to an examination of events that had not been anticipated in the base period of the LOFT study. This was an important step, said Dr. O’Donoghue, because LOFT was not a dedicated cardiovascular safety trial.
The prespecified primary endpoints for TIMI’s safety analysis included new-onset atrial flutter or atrial fibrillation, as well as a composite endpoint of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or cardiovascular death.
Examination of the composite MACE endpoint showed a numeric, but not statistically significant, difference between the odanacatib and placebo arms of the extension study. However, in a preplanned subanalysis, when the 324 stroke events were isolated, a hazard ratio (HR) for stroke of 1.37 for odanacatib emerged (95% confidence interval [CI], 1.10-1.71; P = .005).
Atrial fibrillation and atrial flutter were more common in the odanacatib group, but the difference was not significant (HR, 1.22; 95% CI, 0.99-1.50; P = .06). Dr. O’Donoghue said that the individuals with supraventricular arrhythmias were not the same individuals who had strokes, although the strokes were almost entirely ischemic, rather than hemorrhagic, events.
Dr. O’Donoghue, a cardiovascular medicine specialist at Brigham and Women’s Hospital, noted in her presentation that “preclinical data suggested that inhibition of cathepsin K may reduce atherosclerosis progression and promote plaque stability.”
Odanacatib is a cathepsin K inhibitor, one of a member of a class of proteases. Cathepsin K targets kinins that are involved in bone resorption, but it is expressed in other tissues as well, and it may target other classes of kinins. However, exactly how odanacatib may have contributed to strokes in the study population remains a mystery.
“The mechanistic underpinnings to explain these findings is unknown and warrants investigation,” Dr. O’Donoghue said. “It’s a little bit more unsatisfying when you’re not able to provide a cause. … I share the disappointment of the endocrinologists in the community who were very hopeful for the cathepsin K class to be the next breakthrough in the management of osteoporosis.”
Dr. McClung reported financial relationships with Merck and several other pharmaceutical companies. Dr. O’Donoghue reported receiving grant support from several pharmaceutical companies. The LOFT trial and the TIMI study group analysis were sponsored by Merck.
[email protected]
On Twitter @karioakes
The novel oral osteoporosis drug odanacatib significantly reduced fractures in postmenopausal women but was also associated with a significantly higher risk for stroke, according to data from a 5-year extension of a large phase III clinical trial.
Based on an independent analysis and verification of the risk for stroke, the drug’s sponsor, Merck, has withdrawn odanacatib from review by the Food and Drug Administration. “We are disappointed that the overall benefit-risk profile for odanacatib does not support filing or further development,” Roger M. Perlmutter, MD, president of Merck Research Laboratories, said in a statement.
Compared with placebo, odanacatib resulted in relative risk reductions of 52% for vertebral fracture, 48% for hip fracture, 26% for nonvertebral fracture, and 67% for clinical vertebral fracture (all values, P less than .001). Lumbar spine bone density increased by a mean of 10.9%, as did total hip bone density by a mean of 10.3%, in the odanacatib group (for both, P less than .001), according to results presented at the meeting.
The Long-Term Odanacatib Fracture Trial (LOFT) was a randomized, double-blind placebo-controlled study that investigated the efficacy and safety of odanacatib, a cathepsin K inhibitor, as a treatment for osteoporosis and for fracture prevention in postmenopausal women. Odanacatib was taken as an oral, once-weekly 50-mg pill.
During the base period of the study, 16,071 postmenopausal women aged 65 and older were enrolled, and 12,290 completed the study. Women had to have total hip or femoral neck bone mineral density T scores less than or equal to –2.5, or a radiographically confirmed vertebral fracture and total hip or femoral neck T scores less than or equal to –1.5. Participants were demographically well matched between study arms; 6,092 odanacatib patients and 6,198 on placebo completed the base period of the study.
The original phase III study was stopped early because of robust efficacy data (Osteoporos Int. 2015 Feb;26[2]:699-712). Participants were eligible to continue in a preplanned 5-year double-blind, placebo-controlled extension period of the LOFT study; 3,432 odanacatib and 2,615 placebo participants completed the full 5 years.
An early, but statistically nonsignificant, signal for increased stroke and atrial fibrillation and flutter was seen at the end of the study’s base period. The trend continued and appeared to be amplified during the 5-year, double-blind, placebo-controlled extension period of the LOFT study.
In an interview, Dr. O’Donoghue said that the TIMI study group was brought in by Merck for a “second perspective,” to add rigor to an examination of events that had not been anticipated in the base period of the LOFT study. This was an important step, said Dr. O’Donoghue, because LOFT was not a dedicated cardiovascular safety trial.
The prespecified primary endpoints for TIMI’s safety analysis included new-onset atrial flutter or atrial fibrillation, as well as a composite endpoint of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or cardiovascular death.
Examination of the composite MACE endpoint showed a numeric, but not statistically significant, difference between the odanacatib and placebo arms of the extension study. However, in a preplanned subanalysis, when the 324 stroke events were isolated, a hazard ratio (HR) for stroke of 1.37 for odanacatib emerged (95% confidence interval [CI], 1.10-1.71; P = .005).
Atrial fibrillation and atrial flutter were more common in the odanacatib group, but the difference was not significant (HR, 1.22; 95% CI, 0.99-1.50; P = .06). Dr. O’Donoghue said that the individuals with supraventricular arrhythmias were not the same individuals who had strokes, although the strokes were almost entirely ischemic, rather than hemorrhagic, events.
Dr. O’Donoghue, a cardiovascular medicine specialist at Brigham and Women’s Hospital, noted in her presentation that “preclinical data suggested that inhibition of cathepsin K may reduce atherosclerosis progression and promote plaque stability.”
Odanacatib is a cathepsin K inhibitor, one of a member of a class of proteases. Cathepsin K targets kinins that are involved in bone resorption, but it is expressed in other tissues as well, and it may target other classes of kinins. However, exactly how odanacatib may have contributed to strokes in the study population remains a mystery.
“The mechanistic underpinnings to explain these findings is unknown and warrants investigation,” Dr. O’Donoghue said. “It’s a little bit more unsatisfying when you’re not able to provide a cause. … I share the disappointment of the endocrinologists in the community who were very hopeful for the cathepsin K class to be the next breakthrough in the management of osteoporosis.”
Dr. McClung reported financial relationships with Merck and several other pharmaceutical companies. Dr. O’Donoghue reported receiving grant support from several pharmaceutical companies. The LOFT trial and the TIMI study group analysis were sponsored by Merck.
[email protected]
On Twitter @karioakes
FROM THE NAMS 2016 ANNUAL MEETING
Abnormal Wound Healing Related to High-Dose Systemic Corticosteroid Therapy in a Patient With Ehlers-Danlos Syndrome Benign Hypermobility Type
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
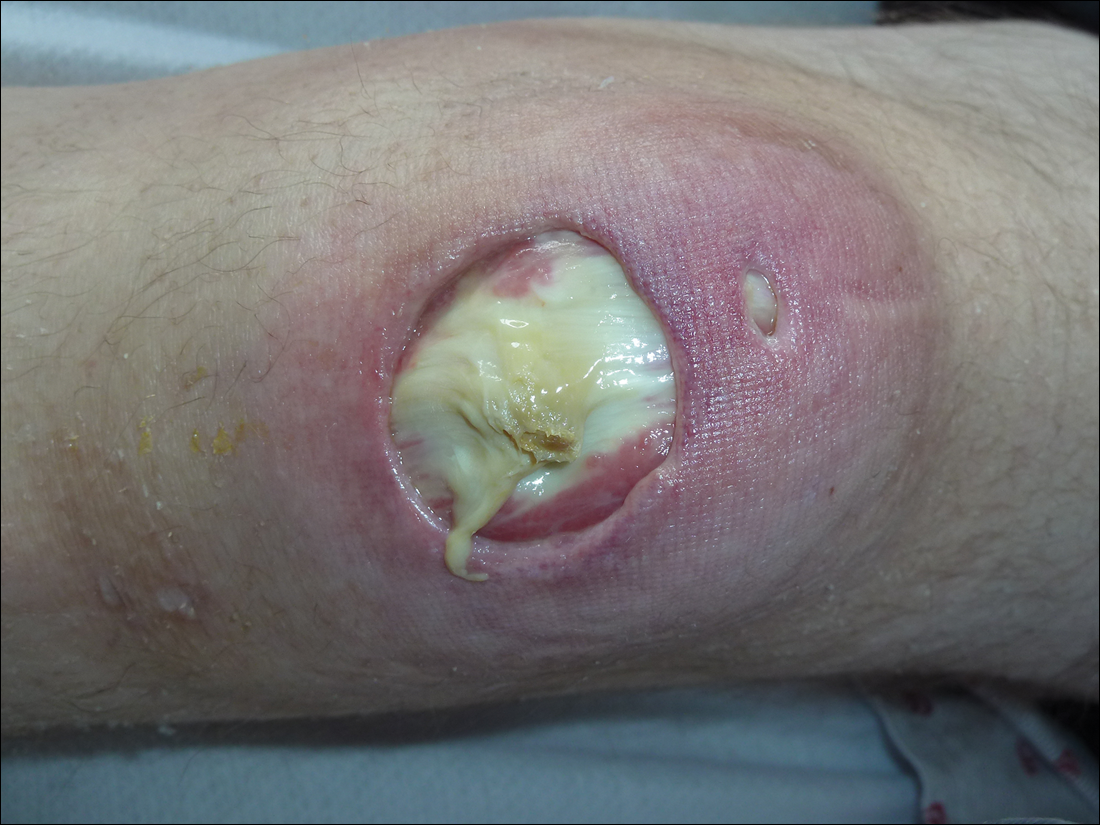
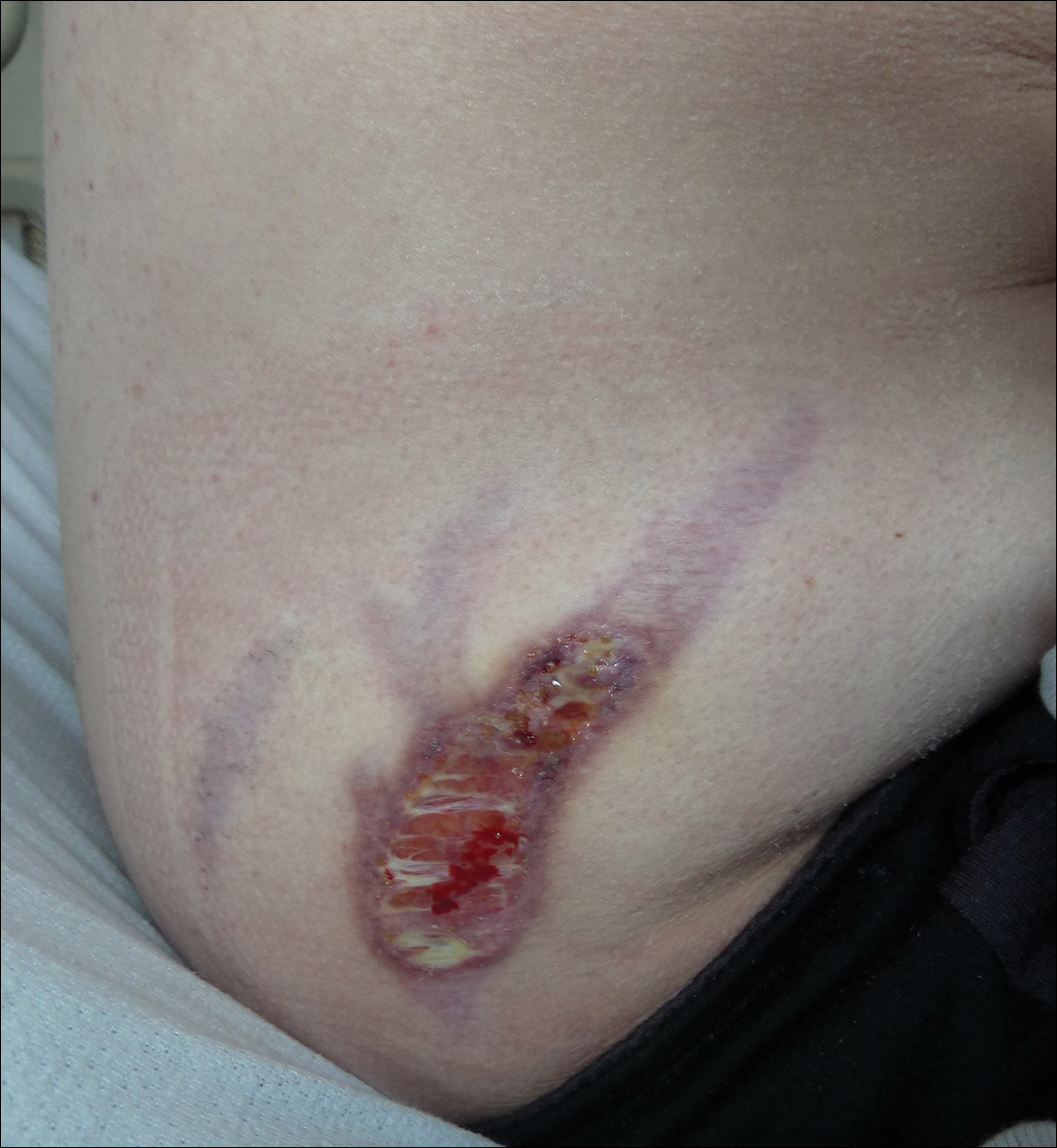
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
Practice Points
- Chronic corticosteroids have profound effects on the wound-healing process, and their detrimental effects may be amplified in patients with underlying connective tissue defects.
- Although genetic testing is available, the diagnosis of Ehlers-Danlos syndrome benign hypermobility type usually is made clinically.
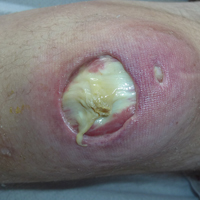
Low serum AMH predicted miscarriage after IVF
SALT LAKE CITY – Low serum levels of anti-Müllerian hormone predicted miscarriage after in vitro fertilization and embryo transfer, according to a single-center prospective study of more than 2,000 women.
This relationship remained statistically significant after the study controlled for age and response to controlled ovarian hyperstimulation, which suggests that AMH is a marker of the reproductive potential of oocytes, not just of oocyte quantity, Bruno Tarasconi, MD, said at the annual meeting of the American Society for Reproductive Medicine.
“Low” serum AMH was defined as less than 1.61 ng per mL, but specific levels vary by test type, so “this is not a definite cutoff,” Dr. Tarasconi emphasized. “What we need to know is that patients with low AMH are probably experiencing some problems in the ovary that affect oocyte quality.”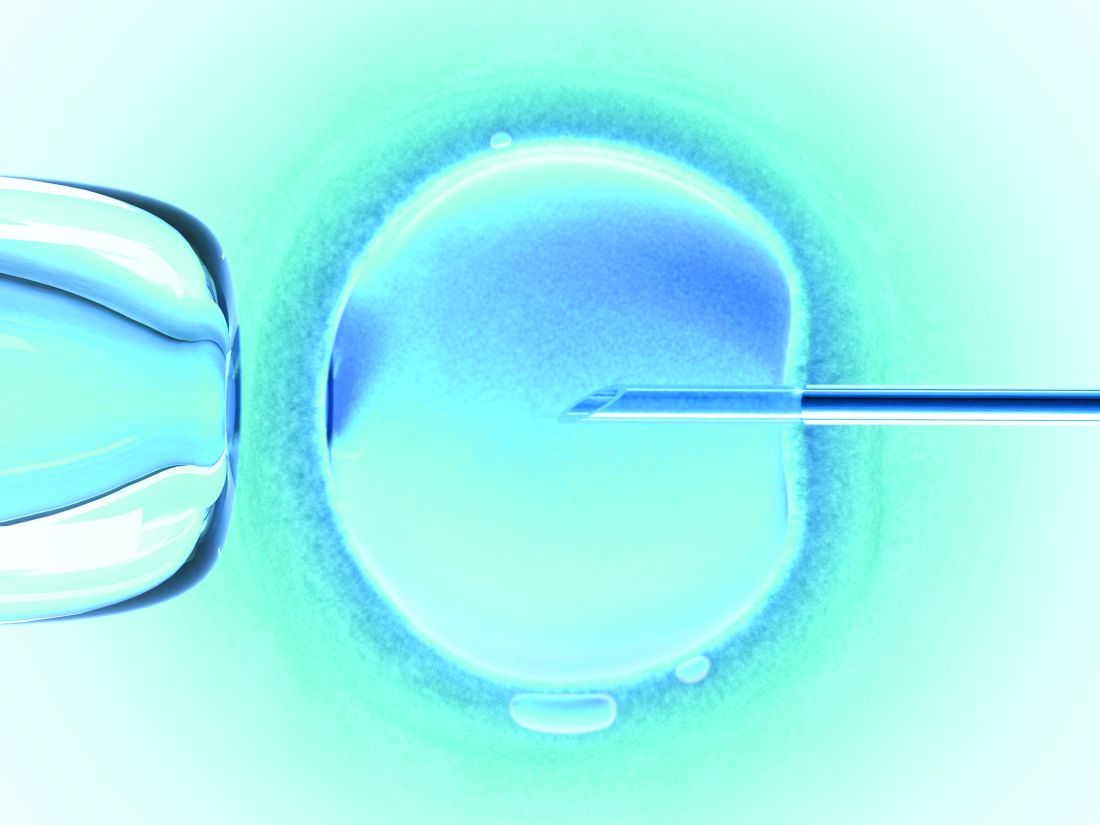
The study comprised 2,365 women undergoing 2,688 IVF cycles with fresh oocytes. Levels of AMH were measured by ELISA (AMH Gen II, Beckman Coulter) within 12 months before IVF on cycle days 2 and 4, said Dr. Tarasconi of University of Paris Ouest in France.
Women younger than 34 years old, women 34-36 years old, and women 37 years and older had the highest rates of miscarriage when they had low serum AMH levels. For the two older age groups, low AMH was associated with about a 33% miscarriage rate – about twice the rate of miscarriage among women whose AMH measured at least 5.6 ng per mL. This difference reached statistical significance for both age groups (P less than .05).
Among women younger than 34 years, low serum AMH was associated with a 22% rate of miscarriage, compared with about 13% among women with higher AMH levels. This difference did not reach significance because of a lack of power, Dr. Tarasconi said. “However, binary logistic regression of the whole population confirmed the association between low serum AMH and miscarriage, independent of patient’s age and oocyte yield, with the P value less than .001.”
Low circulating AMH can reflect an above-average prevalence of oocyte abnormalities, which are linked to miscarriage, Dr. Tarasconi noted. “The fact that oocyte yield failed to alter the relationship between AMH and miscarriage suggests that altered per-follicle AMH production, not oocyte or embryo availability, fosters pregnancy loss,” he added. However, the researchers did not test either preimplantation embryos or the expelled gestational sacs to directly link miscarriages to abnormalities such as aneuploidy, he noted.
Low AMH levels also were associated with lower rates of pregnancy after IVF, regardless of age, as has been previously reported. Patients in the study were of normal karyotype, with no history of recurrent pregnancy loss, uterine abnormalities, or family history of genetic or congenital disorders, Dr. Tarasconi noted.
He reported having no financial disclosures.
SALT LAKE CITY – Low serum levels of anti-Müllerian hormone predicted miscarriage after in vitro fertilization and embryo transfer, according to a single-center prospective study of more than 2,000 women.
This relationship remained statistically significant after the study controlled for age and response to controlled ovarian hyperstimulation, which suggests that AMH is a marker of the reproductive potential of oocytes, not just of oocyte quantity, Bruno Tarasconi, MD, said at the annual meeting of the American Society for Reproductive Medicine.
“Low” serum AMH was defined as less than 1.61 ng per mL, but specific levels vary by test type, so “this is not a definite cutoff,” Dr. Tarasconi emphasized. “What we need to know is that patients with low AMH are probably experiencing some problems in the ovary that affect oocyte quality.”
The study comprised 2,365 women undergoing 2,688 IVF cycles with fresh oocytes. Levels of AMH were measured by ELISA (AMH Gen II, Beckman Coulter) within 12 months before IVF on cycle days 2 and 4, said Dr. Tarasconi of University of Paris Ouest in France.
Women younger than 34 years old, women 34-36 years old, and women 37 years and older had the highest rates of miscarriage when they had low serum AMH levels. For the two older age groups, low AMH was associated with about a 33% miscarriage rate – about twice the rate of miscarriage among women whose AMH measured at least 5.6 ng per mL. This difference reached statistical significance for both age groups (P less than .05).
Among women younger than 34 years, low serum AMH was associated with a 22% rate of miscarriage, compared with about 13% among women with higher AMH levels. This difference did not reach significance because of a lack of power, Dr. Tarasconi said. “However, binary logistic regression of the whole population confirmed the association between low serum AMH and miscarriage, independent of patient’s age and oocyte yield, with the P value less than .001.”
Low circulating AMH can reflect an above-average prevalence of oocyte abnormalities, which are linked to miscarriage, Dr. Tarasconi noted. “The fact that oocyte yield failed to alter the relationship between AMH and miscarriage suggests that altered per-follicle AMH production, not oocyte or embryo availability, fosters pregnancy loss,” he added. However, the researchers did not test either preimplantation embryos or the expelled gestational sacs to directly link miscarriages to abnormalities such as aneuploidy, he noted.
Low AMH levels also were associated with lower rates of pregnancy after IVF, regardless of age, as has been previously reported. Patients in the study were of normal karyotype, with no history of recurrent pregnancy loss, uterine abnormalities, or family history of genetic or congenital disorders, Dr. Tarasconi noted.
He reported having no financial disclosures.
SALT LAKE CITY – Low serum levels of anti-Müllerian hormone predicted miscarriage after in vitro fertilization and embryo transfer, according to a single-center prospective study of more than 2,000 women.
This relationship remained statistically significant after the study controlled for age and response to controlled ovarian hyperstimulation, which suggests that AMH is a marker of the reproductive potential of oocytes, not just of oocyte quantity, Bruno Tarasconi, MD, said at the annual meeting of the American Society for Reproductive Medicine.
“Low” serum AMH was defined as less than 1.61 ng per mL, but specific levels vary by test type, so “this is not a definite cutoff,” Dr. Tarasconi emphasized. “What we need to know is that patients with low AMH are probably experiencing some problems in the ovary that affect oocyte quality.”
The study comprised 2,365 women undergoing 2,688 IVF cycles with fresh oocytes. Levels of AMH were measured by ELISA (AMH Gen II, Beckman Coulter) within 12 months before IVF on cycle days 2 and 4, said Dr. Tarasconi of University of Paris Ouest in France.
Women younger than 34 years old, women 34-36 years old, and women 37 years and older had the highest rates of miscarriage when they had low serum AMH levels. For the two older age groups, low AMH was associated with about a 33% miscarriage rate – about twice the rate of miscarriage among women whose AMH measured at least 5.6 ng per mL. This difference reached statistical significance for both age groups (P less than .05).
Among women younger than 34 years, low serum AMH was associated with a 22% rate of miscarriage, compared with about 13% among women with higher AMH levels. This difference did not reach significance because of a lack of power, Dr. Tarasconi said. “However, binary logistic regression of the whole population confirmed the association between low serum AMH and miscarriage, independent of patient’s age and oocyte yield, with the P value less than .001.”
Low circulating AMH can reflect an above-average prevalence of oocyte abnormalities, which are linked to miscarriage, Dr. Tarasconi noted. “The fact that oocyte yield failed to alter the relationship between AMH and miscarriage suggests that altered per-follicle AMH production, not oocyte or embryo availability, fosters pregnancy loss,” he added. However, the researchers did not test either preimplantation embryos or the expelled gestational sacs to directly link miscarriages to abnormalities such as aneuploidy, he noted.
Low AMH levels also were associated with lower rates of pregnancy after IVF, regardless of age, as has been previously reported. Patients in the study were of normal karyotype, with no history of recurrent pregnancy loss, uterine abnormalities, or family history of genetic or congenital disorders, Dr. Tarasconi noted.
He reported having no financial disclosures.
Key clinical point:
Major finding: The miscarriage rate was about 33% in women with low serum AMH levels after IVF, about twice that of women with high AMH levels.
Data source: A single-center prospective study of 2,365 women undergoing 2,688 IVF cycles with fresh oocytes.
Disclosures: Dr. Tarasconi reported having no financial disclosures.
Tildrakizumab for psoriasis scores high marks in phase III
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.

Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.
Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
VIENNA – The investigational interleukin-23 p-19 subunit inhibitor tildrakizumab achieved PASI 90 improvement rates approaching 60% in patients with moderate to severe plaque psoriasis at week 28 of the pivotal phase III reSURFACE 1 and reSURFACE 2 trials, Kristian Reich, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, the PASI 100 rate at week 28 in the two clinical trials was 24% with tildrakizumab, a humanized monoclonal antibody, at the 100-mg dose and 30% at 200 mg.
It was at these highest efficacy thresholds that the p-19 inhibitor really separated itself from etanercept (Enbrel) in reSURFACE 2, where the two biologics went head-to-head in randomized fashion. Patients on etanercept had a PASI 90 rate at week 28 of 31%, roughly only half that of tildrakizumab at the higher dose.
Guselkumab was the other IL-23 p-19 inhibitor that was a featured attraction at the EADV congress, with 48-week outcomes presented from the 837-patient, pivotal phase III VOYAGE 1 trial. Although caution is always warranted in comparing results across clinical trials because of differences in study populations, guselkumab achieved better top-end efficacy numbers than did tildrakizumab: a PASI 90 of 73.3% at 16 weeks and 80.2% at 24 weeks, along with PASI 100 responses of 34.4% at 16 weeks and 44.4% at 24 weeks.
“I believe there will be characteristics of the new drugs beyond efficacy that will come into play when making treatment decisions: Is dosing every 8 weeks or every 12 weeks? What is the price? What is the outcome after 1 year? I think it’s too early to close the book in trying to understand what these different drugs do, but these phase III results do give us the insight that IL-23 p-19 is actually the sweet spot in psoriasis. By targeting it we are able to keep the disease under control with drugs that are very convenient to use,” Dr. Reich said.
He added that his psoriasis patients really appreciate the convenience of quarterly as opposed to more frequent dosing of biologics, and he does, too.
reSURFACE 1 is a 64-week, randomized, phase III trial conducted in the United States, Canada, and Europe in which 772 patients were randomized 2:2:1 to tildrakizumab at 100 mg or 200 mg or to placebo. reSURFACE 2 is a 64-week trial in which 1,090 patients were randomized 2:2:1:2 to tildrakizumab at 100 mg or 200 mg, placebo, or etanercept at 50 mg twice weekly for the first 12 weeks and once weekly thereafter. At week 12 in both trials, patients on placebo were rerandomized to tildrakizumab at 100 or 200 mg for the duration. Participants averaged a baseline Psoriasis Area and Severity index score of 20, a body weight of 88 kg, and disease involvement over 31% of their body surface area.
Tildrakizumab was dosed in a regimen that’s the same as for ustekinumab (Stelara), which inhibits IL-12 as well as IL-23: one subcutaneous injection at baseline, another 1 month later, and every 12 weeks thereafter.
Dr. Reich presented results of the two pivotal trials through week 28. The coprimary efficacy endpoints in both studies were the PASI 75 response and proportion of subjects with a Physician’s Global Assessment (PGA) score of 0 or 1, meaning clear or minimal disease, compared with placebo at week 12. In hindsight, he said, those were not the best endpoints to have employed.
“We have here a drug that takes a little while to get to full throttle. The primary endpoint selected here at week 12 does not show efficacy data that really separates tildrakizumab from a drug like Stelara. But at week 28 you move toward levels of PASI 90 and PASI 100 response that we really want to see,” the dermatologist said.
Combining the results of reSURFACE 1 and 2, the PASI 75 response rate at week 12 – after only two doses – was 63% in the 100-mg arm and 64% at 200 mg. But the rates kept climbing thereafter such that by week 28 the PASI 75 rates were 77% and 78%.
Fifty-seven percent of patients on tildrakizumab at 100 mg had a PGA score of 0 or 1 at week 12, as did 6% of placebo-treated controls. By week 28, 66% of patients on the lower dose of the p-19 inhibitor had a PGA of 0 or 1. Rates in patients on tildrakizumab at 200 mg were 57% and 66% at 12 and 28 weeks, respectively.
Rates of adverse events of special interest in new biologic agents, including severe infections, malignancies, and major cardiovascular events, were similarly low across all study arms.
“My feeling is that looking at week 12 and week 28 safety data is of limited value. All I can say here is that through week 28 in these two studies I don’t see a safety signal. But for me, the real insight will have to come from larger studies with longer follow-up,” Dr. Reich said.
Asked why he thinks tildrakizumab is a slow starter, with only middling efficacy at the 12-week mark before subsequently picking up steam, he said it’s probably not a matter of the wrong doses being selected for reSURFACE 1 and 2, since the outcomes with 100 and 200 mg are fairly similar. More likely, the monoclonal antibody takes a bit longer to bind to its target and neutralize it than do some of the other biologics.
“It could be that if you dosed tildrakizumab at weeks 0, 2, and 8 as induction therapy you’d hit the mark at 12 weeks,” he added.
The reSURFACE trials are funded by Sun Pharma and Merck. Dr. Reich reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies interested in new treatments for inflammatory skin diseases.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Tildrakizumab dosed quarterly at 100 or 200 mg achieved PASI 75 response rates of 63%-64% at week 12 and 77%-78% at week 28.
Data source: reSURFACE 1 and 2: two pivotal, phase III, randomized clinical trials comprising 1,862 patients with moderate to severe plaque psoriasis.
Disclosures: The reSURFACE trials are funded by Sun Pharma and Merck. The presenter reported having received research grants from and serving as a consultant to Merck and numerous other pharmaceutical companies developing treatments for inflammatory skin diseases.
MACRA
Given the copious amount of printed and blog space that has been devoted in recent months to MACRA – the Medicare Access and CHIP Reauthorization Act of 2015 – I felt no particular obligation to add to the din. Then I was startled to read a recent poll from Deloitte that found that half of all private-practice physicians had never heard of MACRA. Furthermore, only 21% of solo or small-group physicians, and 9% of those employed by hospitals or larger groups, were even somewhat familiar with its financial implications.
Since yet another significant percentage of your Medicare reimbursements will be at risk under this new bureaucracy, an introduction is in order.

When the new system is implemented in 2019, physicians must choose between two payment tracks: the Merit-Based Incentive System (MIPS) or one of the so-called Alternate Payment Models (APM).
The MIPS track will use the four reporting programs just mentioned to compile a composite score between 0 and 100 each year for every practitioner, based on four performance metrics: quality measures listed in Qualified Clinical Data Registries, such as Approved Quality Improvement; total resources used by each practitioner, as measured by VBM; “improvement activities” (MOC); and MU, in some new, as-yet-undefined form. You can earn a bonus of 4% of reimbursement in 2019, rising to 5% in 2020, 7% in 2021, and 9% in 2022 – or you can be penalized those amounts (“negative adjustments”) if your performance doesn’t measure up.
The Centers for Medicare & Medicaid Services initially estimated that most physicians in groups of 24 or fewer on the MIPS track would incur a penalty in 2019; but the final MACRA regulations, issued in mid-October, allow a more gradual implementation that should decrease the penalty burden for small practices, at least initially. For example, you can avoid a penalty – but not qualify for a bonus – in 2019 by reporting your performance in only one quality-of-care or practice-improvement category, or by reporting for only a portion of the year. A decrease in penalties, however, means a smaller pot for bonuses – and reprieves will be temporary.
The alternative, APM, is difficult to discuss at present as very few models have been presented, or even defined, to date. Only Accountable Care Organizations (ACOs) have been introduced in any quantity, and most have failed miserably in real-world settings. The Episode of Care model, which pays providers a fixed amount for all services rendered in a bundle (“episode”) of care, has been discussed at some length, but remains untested, and in the end, may turn out to be just another variant of capitation.
So, which to choose? Long term, I strongly suggest that everyone prepare for the APM track as soon as better, more efficient APMs become available, as it appears that there will be more financial security, with less risk of penalties; but you will probably need to start in the MIPS program, as most projections indicate that the great majority of practitioners, particularly those in smaller operations, will do.
While some may be prompted to join a larger organization or network to decrease their risk of MIPS penalties and gain quicker access to the APM track – which may well be one of CMS’ surreptitious goals in introducing MACRA in the first place – there are steps that those individuals and small groups who choose to remain independent can take now to maximize their chances of landing on the bonus side of the MIPS ledger.
First, make sure your practice data is accurate on the Medicare Provider Enrollment, Chain, and Ownership System (PECOS) – where CMS will gather data for the VBM and Physician Feedback Reports. Study the quality benchmarks, and review your Quality Resource and Use Report (QRUR), which gathers information about each practice’s quality and performance rates for the VBM. (Both PECOS and QRUR can be downloaded at CMS.gov.) And, of course, report successfully for PQRS, which will avoid an automatic penalty of 4% 2 years hence.
If the alphabet soup above has your head swimming, join the club – you’re far from alone; but don’t be discouraged. CMS has already indicated its willingness to make changes aimed at decreasing the administrative burden and, in its words, “making the transition to MACRA as simple and as flexible as possible.”
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected] .
Given the copious amount of printed and blog space that has been devoted in recent months to MACRA – the Medicare Access and CHIP Reauthorization Act of 2015 – I felt no particular obligation to add to the din. Then I was startled to read a recent poll from Deloitte that found that half of all private-practice physicians had never heard of MACRA. Furthermore, only 21% of solo or small-group physicians, and 9% of those employed by hospitals or larger groups, were even somewhat familiar with its financial implications.
Since yet another significant percentage of your Medicare reimbursements will be at risk under this new bureaucracy, an introduction is in order.
When the new system is implemented in 2019, physicians must choose between two payment tracks: the Merit-Based Incentive System (MIPS) or one of the so-called Alternate Payment Models (APM).
The MIPS track will use the four reporting programs just mentioned to compile a composite score between 0 and 100 each year for every practitioner, based on four performance metrics: quality measures listed in Qualified Clinical Data Registries, such as Approved Quality Improvement; total resources used by each practitioner, as measured by VBM; “improvement activities” (MOC); and MU, in some new, as-yet-undefined form. You can earn a bonus of 4% of reimbursement in 2019, rising to 5% in 2020, 7% in 2021, and 9% in 2022 – or you can be penalized those amounts (“negative adjustments”) if your performance doesn’t measure up.
The Centers for Medicare & Medicaid Services initially estimated that most physicians in groups of 24 or fewer on the MIPS track would incur a penalty in 2019; but the final MACRA regulations, issued in mid-October, allow a more gradual implementation that should decrease the penalty burden for small practices, at least initially. For example, you can avoid a penalty – but not qualify for a bonus – in 2019 by reporting your performance in only one quality-of-care or practice-improvement category, or by reporting for only a portion of the year. A decrease in penalties, however, means a smaller pot for bonuses – and reprieves will be temporary.
The alternative, APM, is difficult to discuss at present as very few models have been presented, or even defined, to date. Only Accountable Care Organizations (ACOs) have been introduced in any quantity, and most have failed miserably in real-world settings. The Episode of Care model, which pays providers a fixed amount for all services rendered in a bundle (“episode”) of care, has been discussed at some length, but remains untested, and in the end, may turn out to be just another variant of capitation.
So, which to choose? Long term, I strongly suggest that everyone prepare for the APM track as soon as better, more efficient APMs become available, as it appears that there will be more financial security, with less risk of penalties; but you will probably need to start in the MIPS program, as most projections indicate that the great majority of practitioners, particularly those in smaller operations, will do.
While some may be prompted to join a larger organization or network to decrease their risk of MIPS penalties and gain quicker access to the APM track – which may well be one of CMS’ surreptitious goals in introducing MACRA in the first place – there are steps that those individuals and small groups who choose to remain independent can take now to maximize their chances of landing on the bonus side of the MIPS ledger.
First, make sure your practice data is accurate on the Medicare Provider Enrollment, Chain, and Ownership System (PECOS) – where CMS will gather data for the VBM and Physician Feedback Reports. Study the quality benchmarks, and review your Quality Resource and Use Report (QRUR), which gathers information about each practice’s quality and performance rates for the VBM. (Both PECOS and QRUR can be downloaded at CMS.gov.) And, of course, report successfully for PQRS, which will avoid an automatic penalty of 4% 2 years hence.
If the alphabet soup above has your head swimming, join the club – you’re far from alone; but don’t be discouraged. CMS has already indicated its willingness to make changes aimed at decreasing the administrative burden and, in its words, “making the transition to MACRA as simple and as flexible as possible.”
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected] .
Given the copious amount of printed and blog space that has been devoted in recent months to MACRA – the Medicare Access and CHIP Reauthorization Act of 2015 – I felt no particular obligation to add to the din. Then I was startled to read a recent poll from Deloitte that found that half of all private-practice physicians had never heard of MACRA. Furthermore, only 21% of solo or small-group physicians, and 9% of those employed by hospitals or larger groups, were even somewhat familiar with its financial implications.
Since yet another significant percentage of your Medicare reimbursements will be at risk under this new bureaucracy, an introduction is in order.
When the new system is implemented in 2019, physicians must choose between two payment tracks: the Merit-Based Incentive System (MIPS) or one of the so-called Alternate Payment Models (APM).
The MIPS track will use the four reporting programs just mentioned to compile a composite score between 0 and 100 each year for every practitioner, based on four performance metrics: quality measures listed in Qualified Clinical Data Registries, such as Approved Quality Improvement; total resources used by each practitioner, as measured by VBM; “improvement activities” (MOC); and MU, in some new, as-yet-undefined form. You can earn a bonus of 4% of reimbursement in 2019, rising to 5% in 2020, 7% in 2021, and 9% in 2022 – or you can be penalized those amounts (“negative adjustments”) if your performance doesn’t measure up.
The Centers for Medicare & Medicaid Services initially estimated that most physicians in groups of 24 or fewer on the MIPS track would incur a penalty in 2019; but the final MACRA regulations, issued in mid-October, allow a more gradual implementation that should decrease the penalty burden for small practices, at least initially. For example, you can avoid a penalty – but not qualify for a bonus – in 2019 by reporting your performance in only one quality-of-care or practice-improvement category, or by reporting for only a portion of the year. A decrease in penalties, however, means a smaller pot for bonuses – and reprieves will be temporary.
The alternative, APM, is difficult to discuss at present as very few models have been presented, or even defined, to date. Only Accountable Care Organizations (ACOs) have been introduced in any quantity, and most have failed miserably in real-world settings. The Episode of Care model, which pays providers a fixed amount for all services rendered in a bundle (“episode”) of care, has been discussed at some length, but remains untested, and in the end, may turn out to be just another variant of capitation.
So, which to choose? Long term, I strongly suggest that everyone prepare for the APM track as soon as better, more efficient APMs become available, as it appears that there will be more financial security, with less risk of penalties; but you will probably need to start in the MIPS program, as most projections indicate that the great majority of practitioners, particularly those in smaller operations, will do.
While some may be prompted to join a larger organization or network to decrease their risk of MIPS penalties and gain quicker access to the APM track – which may well be one of CMS’ surreptitious goals in introducing MACRA in the first place – there are steps that those individuals and small groups who choose to remain independent can take now to maximize their chances of landing on the bonus side of the MIPS ledger.
First, make sure your practice data is accurate on the Medicare Provider Enrollment, Chain, and Ownership System (PECOS) – where CMS will gather data for the VBM and Physician Feedback Reports. Study the quality benchmarks, and review your Quality Resource and Use Report (QRUR), which gathers information about each practice’s quality and performance rates for the VBM. (Both PECOS and QRUR can be downloaded at CMS.gov.) And, of course, report successfully for PQRS, which will avoid an automatic penalty of 4% 2 years hence.
If the alphabet soup above has your head swimming, join the club – you’re far from alone; but don’t be discouraged. CMS has already indicated its willingness to make changes aimed at decreasing the administrative burden and, in its words, “making the transition to MACRA as simple and as flexible as possible.”
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected] .

10 Things Hospitalists Need to Know about Palliative Care

In fact, according to the 2015 Palliative Care Report Card from the Center to Advance Palliative Care (CAPC), 67 percent of hospitals with 50 or more beds had a designated palliative care program.1
While core palliative care skills can be performed by frontline clinicians including hospitalists, specialty palliative care consults are the ones who are called in for complicated cases. The Hospitalist asked several palliative care experts for advice on how to clarify definitions, distinctions, and roles. This is what they told us:
Palliative care is not synonymous with end-of-life care.
Palliative care advocates call this the biggest misconception they struggle to overcome, with the potential to inhibit its contributions to patient care in the hospital. Palliative care, they say, is for any patient with a serious illness who is struggling to cope with the fallout from that illness in their lives.
“Our biggest impact can come earlier in the illness,” says Jeanie Youngwerth, MD, director of the University of Colorado Hospital’s Palliative Care Consult Service in Aurora. “We help people do the best they can for as long as they can. If you’re even considering a palliative-care consult, then do it sooner rather than later.”

Palliative care can offer more than just help with difficult conversations, adds Daniel Fischberg, MD, PhD, FAAHPM, medical director of the Pain and Palliative Care Department at The Queen’s Medical Center in Honolulu. For example, the palliative-care team can work with patients to clarify their expectations and goals for care, plan for what comes next, and address troubling symptoms—whether physical or emotional, Dr. Fischberg says.
“We can really help patients and families who are facing unique and challenging needs,” he says.
The experts also say that palliative care is not synonymous with hospice care, which is a comprehensive service that provides specialized terminal care for patients with a prognosis of six months or fewer to live. Both, however, share many of the same principles and techniques of symptom management and psycho-social-spiritual support. But some patients and families may associate a palliative-care referral with hospice care or have other misconceptions and fears about it. Hospitalists are challenged to provide a consistent message clarifying that palliative care can be helpful for seriously ill patients regardless of prognosis or other medical treatments they’re receiving.
“It’s human nature not to want to deal with our mortality, and any word that gets associated with death and dying can turn people off,” says Joseph Rotella, MD, chief medical officer of the American Academy of Hospice and Palliative Medicine (AAHPM). “The best way to prevent this is to define it in terms of patient and family needs: ‘Let’s bring in our comfort specialists.’ Doctors should not apologize when referring to a service that has proven its value. We should be happy to recommend it often and early.”
Patients with serious illness can benefit from palliative care.
CAPC defines palliative care as “specialized medical care for people with serious illnesses.” It focuses on providing patients with relief from the symptoms and stress of a serious illness, regardless of their diagnosis, at any age and at any stage of a serious illness. This service is provided by a specially trained interdisciplinary palliative-care team of doctors, nurses, and other specialists who work together with patients’ other doctors. Their goal is to improve quality of life for both patients and their families with an extra layer of support.
Palliative care is also a medical specialty that involves specialty training, including year-long hospice and palliative medicine (HPM) fellowships now offered at 112 sites accredited by the Accreditation Council for Graduate Medical Education. Subspecialty board certification is also available through 10 collaborating medical specialty boards within the American Board of Medical Specialties as well as by the American Osteopathic Association. Palliative-care programs are now certified by The Joint Commission, with similar recognition under development by the Community Health Accreditation Partner.
Palliative care is intended for patients facing challenges.
Palliative care is intended for patients who might be expected to face stresses and challenges in any area of their lives as a result of serious illness. This may include, for example, patients who experience frequent emergency department visits, hospital readmissions, or prolonged ICU stays, as well as cancer patients who are admitted to the hospital solely to address out-of-control symptoms resulting from their disease and its treatment.
“We can help with the burdens of any challenging symptoms,” Dr. Fischberg says.
Other examples of appropriate palliative-care referrals are when next steps for patients’ treatment are not clear, when there are questions about patients’ real goals of care, and when unmet needs such as unrelieved symptoms have put their families in a state of distress, whether physical, emotional, social, or spiritual. Patients may need guidance about weighing their care options.
Palliative care is also available for children and their families.
The philosophy and organization of palliative care for delivering compassionate care for children with chronic, complex, or life-threatening conditions are much the same as for adults. In 2013, the American Academy of Pediatrics issued a Pediatric Palliative Care and Hospice Care policy statement,2 which outlined core commitments in such areas as respecting and partnering with patients and families and pursuing care that is high-quality, readily accessible, and equitable.
As with adults, a referral for palliative care typically is most helpful for the more complex cases, says Joanne Wolfe, MD, MPH, director of Pediatric Palliative Care at Boston Children’s Hospital. The palliative care team can offer emotional support to the parents of children with complex illnesses and help them understand confusing treatment options. The children, too, need a sounding board.
“If I were teaching a group of hospitalists, I would emphasize foundational principles of palliative care, starting with relationship and understanding patients’ and families’ goals of care,” Dr. Wolfe says. If the family is struggling to cope with the illness and the hospitalist doesn’t have good answers, that’s when to call palliative care, she adds.
Palliative care’s role is not to talk patients and their families out of treatments.
The palliative-care team tries to enter cases without an agenda, Dr. Fischberg says, rather than aiming to get patients to stop treatments or agree to a do-not-resuscitate (DNR) order.
“We’re interested in what the hospitalist thinks about what best care for this patient looks like but also in eliciting the patient’s values and preferences,” he says.
Palliative-care professionals are skilled at delicately communicating bad news and helping patients and families clarify what their goals of care really are, says Robert Crook, MD, FACP, associate medical director of Mount Carmel Hospice and Palliative Care in Columbus, Ohio.
“It’s more about helping to improve communication between the primary-care team and the patient and family—not talking somebody out of something but helping them understand each other better,” Dr. Crook says.
Palliative care can reduce costs of care on average, but it does not achieve this by rationing care or denying treatments.
“We’re not there to cut costs or to get patients discharged sooner or to steer them away from costly treatments,” explains Dr. Rotella. “The last thing a palliative-care team wants is to be viewed as a care rationer. But if the patient understands what’s really going on, they often won’t want treatments that don’t help. So, in that way, we are part of the solution.”
Dr. Rotella calls this cost-effectiveness a side effect of palliative care, when patients are empowered to make decisions about their own care. “That’s where you achieve the triple aim,” he says. “They feel better about it because they are in the driver’s seat. If a patient wants a treatment consistent with their values, we will advocate for it.”
One study found that patients at eight U.S. hospitals who received palliative care incurred significantly lower hospital costs than a matched group receiving usual care, with an average reduction in direct hospital costs of almost $1,700 for patients discharged alive and almost $5,000 per admission for patients who died in the hospital.3 Another study found that early palliative care interventions for cancer patients led to significant improvements in both quality of life and mood compared with patients receiving standard care, with less cost and fewer aggressive treatments at the end of life but longer survival.4
One of the main tools of the palliative care team is the family meeting.
Family meetings are scheduled to allow as many family members as possible to attend, and the primary-care team and relevant specialists typically are also invited. Many palliative-care teams use a standardized format that involves introductions, clarification of each participant’s understanding of the patient’s prognosis, and an effort to reconcile the patient’s hopes and values with medical realities and possibilities, Dr. Fischberg explains.
“That is such a critical component of our care, where we make sure the patient and family are fully informed and foster shared decision making that results in patients being more comfortable with care that better matches what they want,” says Dr. Rotella.
The palliative-care team typically becomes involved via a consultation request from a patient’s attending physician.
“A big part of our job is doing our homework,” Dr. Youngwerth says. “We’ll talk to the team about what’s going on. We want to get as much information as possible about the patient, about prognosis, about the perspectives of people caring for them. Don’t be surprised if the palliative-care team contacts you to get your input on the prognosis and other medical details in order to best inform their discussion with the patient and family.”
Then the palliative-care team will follow consult protocol in reporting back to the primary medical team.
Palliative-care teams can assist busy hospitalists with difficult patient conversations.
“When I’m on the hospitalist service, I’ll pull in the palliative-care team,” Dr. Youngwerth explains. “It’s not that I don’t have the skills; I don’t have the time.”
Conversations aimed at clarifying goals of care can take 90 minutes or more, but the palliative-care team will take as much time as needed to achieve clarification.
It’s important that hospitalists remain involved in these cases, says Christian Sinclair, MD, assistant professor in the division of palliative medicine at the University of Kansas Medical Center in Kansas City and president-elect of AAHPM.
“Just because you have access to palliative-care services, don’t miss the chance to enhance your own communication skills and your ability to address these issues head on,” Dr. Sinclair says.
American Academy of Hospice and Palliative Medicine is a participant in Choosing Wisely.
The Choosing Wisely program, initiated by the American Board of Internal Medicine Foundation, invites medical societies to identify five treatments that should be questioned by physicians and patients based on lack of supporting evidence in the research base. The Society of Hospital Medicine is also a participant in this initiative.
For AAHPM, one of its recommendations was: “Don’t delay palliative care for a patient with serious illness who has a physical, psychological, social, or spiritual distress because they are pursuing disease-directed treatment.” Other Choosing Wisely suggestions include not recommending feeding tubes for patients with advanced dementia, not leaving implantable cardioverter defibrillators (ICDs) activated when these are not consistent with patient/family goals of care, and not recommending more than a single fraction of palliative radiation for an uncomplicated painful bone metastasis.
Different palliative-care programs provide different services.
It is important for hospitalists to learn their local palliative-care programs and what they emphasize and are able to offer—or not, says Dr. Sinclair.
“There are so many different models,” he says. “Spend some time reaching out to them, outside of actual consults, and find out what their comfort level is on various issues. Hospitalists and palliative-care teams should get to know each other better.”
Access to palliative care and the comprehensiveness of the team and services can vary between hospitals, while access to community-based palliative care outside of the hospital is even more variable.
“Palliative-care teams often have a better sense of our partners in the community and access to community-based palliative care,” Dr. Fischberg says.
References
- Morrison RS, Meier DE. America’s Care of Serious Illness: 2015 State-by-State Report Card on Access to Palliative Care in Our Nation’s Hospitals. New York, NY: Center to Advance Palliative Care; 2015.
- American Academy of Pediatrics. Policy statement: pediatric palliative care and hospice care commitments, guidelines, and recommendations. Pediatr. 2013;132(5):966-972.
- Morrison RS, Penrod JD, Cassel JB, et al. Cost savings associated with US hospital palliative care consultation programs. Arch Intern Med. 2008;168(16):1783-1790.
- Temel JS, Greer JA, Muzikansky et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med. 2010;363(8):733-742.
In fact, according to the 2015 Palliative Care Report Card from the Center to Advance Palliative Care (CAPC), 67 percent of hospitals with 50 or more beds had a designated palliative care program.1
While core palliative care skills can be performed by frontline clinicians including hospitalists, specialty palliative care consults are the ones who are called in for complicated cases. The Hospitalist asked several palliative care experts for advice on how to clarify definitions, distinctions, and roles. This is what they told us:
Palliative care is not synonymous with end-of-life care.
Palliative care advocates call this the biggest misconception they struggle to overcome, with the potential to inhibit its contributions to patient care in the hospital. Palliative care, they say, is for any patient with a serious illness who is struggling to cope with the fallout from that illness in their lives.
“Our biggest impact can come earlier in the illness,” says Jeanie Youngwerth, MD, director of the University of Colorado Hospital’s Palliative Care Consult Service in Aurora. “We help people do the best they can for as long as they can. If you’re even considering a palliative-care consult, then do it sooner rather than later.”
Palliative care can offer more than just help with difficult conversations, adds Daniel Fischberg, MD, PhD, FAAHPM, medical director of the Pain and Palliative Care Department at The Queen’s Medical Center in Honolulu. For example, the palliative-care team can work with patients to clarify their expectations and goals for care, plan for what comes next, and address troubling symptoms—whether physical or emotional, Dr. Fischberg says.
“We can really help patients and families who are facing unique and challenging needs,” he says.
The experts also say that palliative care is not synonymous with hospice care, which is a comprehensive service that provides specialized terminal care for patients with a prognosis of six months or fewer to live. Both, however, share many of the same principles and techniques of symptom management and psycho-social-spiritual support. But some patients and families may associate a palliative-care referral with hospice care or have other misconceptions and fears about it. Hospitalists are challenged to provide a consistent message clarifying that palliative care can be helpful for seriously ill patients regardless of prognosis or other medical treatments they’re receiving.
“It’s human nature not to want to deal with our mortality, and any word that gets associated with death and dying can turn people off,” says Joseph Rotella, MD, chief medical officer of the American Academy of Hospice and Palliative Medicine (AAHPM). “The best way to prevent this is to define it in terms of patient and family needs: ‘Let’s bring in our comfort specialists.’ Doctors should not apologize when referring to a service that has proven its value. We should be happy to recommend it often and early.”
Patients with serious illness can benefit from palliative care.
CAPC defines palliative care as “specialized medical care for people with serious illnesses.” It focuses on providing patients with relief from the symptoms and stress of a serious illness, regardless of their diagnosis, at any age and at any stage of a serious illness. This service is provided by a specially trained interdisciplinary palliative-care team of doctors, nurses, and other specialists who work together with patients’ other doctors. Their goal is to improve quality of life for both patients and their families with an extra layer of support.
Palliative care is also a medical specialty that involves specialty training, including year-long hospice and palliative medicine (HPM) fellowships now offered at 112 sites accredited by the Accreditation Council for Graduate Medical Education. Subspecialty board certification is also available through 10 collaborating medical specialty boards within the American Board of Medical Specialties as well as by the American Osteopathic Association. Palliative-care programs are now certified by The Joint Commission, with similar recognition under development by the Community Health Accreditation Partner.
Palliative care is intended for patients facing challenges.
Palliative care is intended for patients who might be expected to face stresses and challenges in any area of their lives as a result of serious illness. This may include, for example, patients who experience frequent emergency department visits, hospital readmissions, or prolonged ICU stays, as well as cancer patients who are admitted to the hospital solely to address out-of-control symptoms resulting from their disease and its treatment.
“We can help with the burdens of any challenging symptoms,” Dr. Fischberg says.
Other examples of appropriate palliative-care referrals are when next steps for patients’ treatment are not clear, when there are questions about patients’ real goals of care, and when unmet needs such as unrelieved symptoms have put their families in a state of distress, whether physical, emotional, social, or spiritual. Patients may need guidance about weighing their care options.
Palliative care is also available for children and their families.
The philosophy and organization of palliative care for delivering compassionate care for children with chronic, complex, or life-threatening conditions are much the same as for adults. In 2013, the American Academy of Pediatrics issued a Pediatric Palliative Care and Hospice Care policy statement,2 which outlined core commitments in such areas as respecting and partnering with patients and families and pursuing care that is high-quality, readily accessible, and equitable.
As with adults, a referral for palliative care typically is most helpful for the more complex cases, says Joanne Wolfe, MD, MPH, director of Pediatric Palliative Care at Boston Children’s Hospital. The palliative care team can offer emotional support to the parents of children with complex illnesses and help them understand confusing treatment options. The children, too, need a sounding board.
“If I were teaching a group of hospitalists, I would emphasize foundational principles of palliative care, starting with relationship and understanding patients’ and families’ goals of care,” Dr. Wolfe says. If the family is struggling to cope with the illness and the hospitalist doesn’t have good answers, that’s when to call palliative care, she adds.
Palliative care’s role is not to talk patients and their families out of treatments.
The palliative-care team tries to enter cases without an agenda, Dr. Fischberg says, rather than aiming to get patients to stop treatments or agree to a do-not-resuscitate (DNR) order.
“We’re interested in what the hospitalist thinks about what best care for this patient looks like but also in eliciting the patient’s values and preferences,” he says.
Palliative-care professionals are skilled at delicately communicating bad news and helping patients and families clarify what their goals of care really are, says Robert Crook, MD, FACP, associate medical director of Mount Carmel Hospice and Palliative Care in Columbus, Ohio.
“It’s more about helping to improve communication between the primary-care team and the patient and family—not talking somebody out of something but helping them understand each other better,” Dr. Crook says.
Palliative care can reduce costs of care on average, but it does not achieve this by rationing care or denying treatments.
“We’re not there to cut costs or to get patients discharged sooner or to steer them away from costly treatments,” explains Dr. Rotella. “The last thing a palliative-care team wants is to be viewed as a care rationer. But if the patient understands what’s really going on, they often won’t want treatments that don’t help. So, in that way, we are part of the solution.”
Dr. Rotella calls this cost-effectiveness a side effect of palliative care, when patients are empowered to make decisions about their own care. “That’s where you achieve the triple aim,” he says. “They feel better about it because they are in the driver’s seat. If a patient wants a treatment consistent with their values, we will advocate for it.”
One study found that patients at eight U.S. hospitals who received palliative care incurred significantly lower hospital costs than a matched group receiving usual care, with an average reduction in direct hospital costs of almost $1,700 for patients discharged alive and almost $5,000 per admission for patients who died in the hospital.3 Another study found that early palliative care interventions for cancer patients led to significant improvements in both quality of life and mood compared with patients receiving standard care, with less cost and fewer aggressive treatments at the end of life but longer survival.4
One of the main tools of the palliative care team is the family meeting.
Family meetings are scheduled to allow as many family members as possible to attend, and the primary-care team and relevant specialists typically are also invited. Many palliative-care teams use a standardized format that involves introductions, clarification of each participant’s understanding of the patient’s prognosis, and an effort to reconcile the patient’s hopes and values with medical realities and possibilities, Dr. Fischberg explains.
“That is such a critical component of our care, where we make sure the patient and family are fully informed and foster shared decision making that results in patients being more comfortable with care that better matches what they want,” says Dr. Rotella.
The palliative-care team typically becomes involved via a consultation request from a patient’s attending physician.
“A big part of our job is doing our homework,” Dr. Youngwerth says. “We’ll talk to the team about what’s going on. We want to get as much information as possible about the patient, about prognosis, about the perspectives of people caring for them. Don’t be surprised if the palliative-care team contacts you to get your input on the prognosis and other medical details in order to best inform their discussion with the patient and family.”
Then the palliative-care team will follow consult protocol in reporting back to the primary medical team.
Palliative-care teams can assist busy hospitalists with difficult patient conversations.
“When I’m on the hospitalist service, I’ll pull in the palliative-care team,” Dr. Youngwerth explains. “It’s not that I don’t have the skills; I don’t have the time.”
Conversations aimed at clarifying goals of care can take 90 minutes or more, but the palliative-care team will take as much time as needed to achieve clarification.
It’s important that hospitalists remain involved in these cases, says Christian Sinclair, MD, assistant professor in the division of palliative medicine at the University of Kansas Medical Center in Kansas City and president-elect of AAHPM.
“Just because you have access to palliative-care services, don’t miss the chance to enhance your own communication skills and your ability to address these issues head on,” Dr. Sinclair says.
American Academy of Hospice and Palliative Medicine is a participant in Choosing Wisely.
The Choosing Wisely program, initiated by the American Board of Internal Medicine Foundation, invites medical societies to identify five treatments that should be questioned by physicians and patients based on lack of supporting evidence in the research base. The Society of Hospital Medicine is also a participant in this initiative.
For AAHPM, one of its recommendations was: “Don’t delay palliative care for a patient with serious illness who has a physical, psychological, social, or spiritual distress because they are pursuing disease-directed treatment.” Other Choosing Wisely suggestions include not recommending feeding tubes for patients with advanced dementia, not leaving implantable cardioverter defibrillators (ICDs) activated when these are not consistent with patient/family goals of care, and not recommending more than a single fraction of palliative radiation for an uncomplicated painful bone metastasis.
Different palliative-care programs provide different services.
It is important for hospitalists to learn their local palliative-care programs and what they emphasize and are able to offer—or not, says Dr. Sinclair.
“There are so many different models,” he says. “Spend some time reaching out to them, outside of actual consults, and find out what their comfort level is on various issues. Hospitalists and palliative-care teams should get to know each other better.”
Access to palliative care and the comprehensiveness of the team and services can vary between hospitals, while access to community-based palliative care outside of the hospital is even more variable.
“Palliative-care teams often have a better sense of our partners in the community and access to community-based palliative care,” Dr. Fischberg says.
References
- Morrison RS, Meier DE. America’s Care of Serious Illness: 2015 State-by-State Report Card on Access to Palliative Care in Our Nation’s Hospitals. New York, NY: Center to Advance Palliative Care; 2015.
- American Academy of Pediatrics. Policy statement: pediatric palliative care and hospice care commitments, guidelines, and recommendations. Pediatr. 2013;132(5):966-972.
- Morrison RS, Penrod JD, Cassel JB, et al. Cost savings associated with US hospital palliative care consultation programs. Arch Intern Med. 2008;168(16):1783-1790.
- Temel JS, Greer JA, Muzikansky et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med. 2010;363(8):733-742.
In fact, according to the 2015 Palliative Care Report Card from the Center to Advance Palliative Care (CAPC), 67 percent of hospitals with 50 or more beds had a designated palliative care program.1
While core palliative care skills can be performed by frontline clinicians including hospitalists, specialty palliative care consults are the ones who are called in for complicated cases. The Hospitalist asked several palliative care experts for advice on how to clarify definitions, distinctions, and roles. This is what they told us:
Palliative care is not synonymous with end-of-life care.
Palliative care advocates call this the biggest misconception they struggle to overcome, with the potential to inhibit its contributions to patient care in the hospital. Palliative care, they say, is for any patient with a serious illness who is struggling to cope with the fallout from that illness in their lives.
“Our biggest impact can come earlier in the illness,” says Jeanie Youngwerth, MD, director of the University of Colorado Hospital’s Palliative Care Consult Service in Aurora. “We help people do the best they can for as long as they can. If you’re even considering a palliative-care consult, then do it sooner rather than later.”
Palliative care can offer more than just help with difficult conversations, adds Daniel Fischberg, MD, PhD, FAAHPM, medical director of the Pain and Palliative Care Department at The Queen’s Medical Center in Honolulu. For example, the palliative-care team can work with patients to clarify their expectations and goals for care, plan for what comes next, and address troubling symptoms—whether physical or emotional, Dr. Fischberg says.
“We can really help patients and families who are facing unique and challenging needs,” he says.
The experts also say that palliative care is not synonymous with hospice care, which is a comprehensive service that provides specialized terminal care for patients with a prognosis of six months or fewer to live. Both, however, share many of the same principles and techniques of symptom management and psycho-social-spiritual support. But some patients and families may associate a palliative-care referral with hospice care or have other misconceptions and fears about it. Hospitalists are challenged to provide a consistent message clarifying that palliative care can be helpful for seriously ill patients regardless of prognosis or other medical treatments they’re receiving.
“It’s human nature not to want to deal with our mortality, and any word that gets associated with death and dying can turn people off,” says Joseph Rotella, MD, chief medical officer of the American Academy of Hospice and Palliative Medicine (AAHPM). “The best way to prevent this is to define it in terms of patient and family needs: ‘Let’s bring in our comfort specialists.’ Doctors should not apologize when referring to a service that has proven its value. We should be happy to recommend it often and early.”
Patients with serious illness can benefit from palliative care.
CAPC defines palliative care as “specialized medical care for people with serious illnesses.” It focuses on providing patients with relief from the symptoms and stress of a serious illness, regardless of their diagnosis, at any age and at any stage of a serious illness. This service is provided by a specially trained interdisciplinary palliative-care team of doctors, nurses, and other specialists who work together with patients’ other doctors. Their goal is to improve quality of life for both patients and their families with an extra layer of support.
Palliative care is also a medical specialty that involves specialty training, including year-long hospice and palliative medicine (HPM) fellowships now offered at 112 sites accredited by the Accreditation Council for Graduate Medical Education. Subspecialty board certification is also available through 10 collaborating medical specialty boards within the American Board of Medical Specialties as well as by the American Osteopathic Association. Palliative-care programs are now certified by The Joint Commission, with similar recognition under development by the Community Health Accreditation Partner.
Palliative care is intended for patients facing challenges.
Palliative care is intended for patients who might be expected to face stresses and challenges in any area of their lives as a result of serious illness. This may include, for example, patients who experience frequent emergency department visits, hospital readmissions, or prolonged ICU stays, as well as cancer patients who are admitted to the hospital solely to address out-of-control symptoms resulting from their disease and its treatment.
“We can help with the burdens of any challenging symptoms,” Dr. Fischberg says.
Other examples of appropriate palliative-care referrals are when next steps for patients’ treatment are not clear, when there are questions about patients’ real goals of care, and when unmet needs such as unrelieved symptoms have put their families in a state of distress, whether physical, emotional, social, or spiritual. Patients may need guidance about weighing their care options.
Palliative care is also available for children and their families.
The philosophy and organization of palliative care for delivering compassionate care for children with chronic, complex, or life-threatening conditions are much the same as for adults. In 2013, the American Academy of Pediatrics issued a Pediatric Palliative Care and Hospice Care policy statement,2 which outlined core commitments in such areas as respecting and partnering with patients and families and pursuing care that is high-quality, readily accessible, and equitable.
As with adults, a referral for palliative care typically is most helpful for the more complex cases, says Joanne Wolfe, MD, MPH, director of Pediatric Palliative Care at Boston Children’s Hospital. The palliative care team can offer emotional support to the parents of children with complex illnesses and help them understand confusing treatment options. The children, too, need a sounding board.
“If I were teaching a group of hospitalists, I would emphasize foundational principles of palliative care, starting with relationship and understanding patients’ and families’ goals of care,” Dr. Wolfe says. If the family is struggling to cope with the illness and the hospitalist doesn’t have good answers, that’s when to call palliative care, she adds.
Palliative care’s role is not to talk patients and their families out of treatments.
The palliative-care team tries to enter cases without an agenda, Dr. Fischberg says, rather than aiming to get patients to stop treatments or agree to a do-not-resuscitate (DNR) order.
“We’re interested in what the hospitalist thinks about what best care for this patient looks like but also in eliciting the patient’s values and preferences,” he says.
Palliative-care professionals are skilled at delicately communicating bad news and helping patients and families clarify what their goals of care really are, says Robert Crook, MD, FACP, associate medical director of Mount Carmel Hospice and Palliative Care in Columbus, Ohio.
“It’s more about helping to improve communication between the primary-care team and the patient and family—not talking somebody out of something but helping them understand each other better,” Dr. Crook says.
Palliative care can reduce costs of care on average, but it does not achieve this by rationing care or denying treatments.
“We’re not there to cut costs or to get patients discharged sooner or to steer them away from costly treatments,” explains Dr. Rotella. “The last thing a palliative-care team wants is to be viewed as a care rationer. But if the patient understands what’s really going on, they often won’t want treatments that don’t help. So, in that way, we are part of the solution.”
Dr. Rotella calls this cost-effectiveness a side effect of palliative care, when patients are empowered to make decisions about their own care. “That’s where you achieve the triple aim,” he says. “They feel better about it because they are in the driver’s seat. If a patient wants a treatment consistent with their values, we will advocate for it.”
One study found that patients at eight U.S. hospitals who received palliative care incurred significantly lower hospital costs than a matched group receiving usual care, with an average reduction in direct hospital costs of almost $1,700 for patients discharged alive and almost $5,000 per admission for patients who died in the hospital.3 Another study found that early palliative care interventions for cancer patients led to significant improvements in both quality of life and mood compared with patients receiving standard care, with less cost and fewer aggressive treatments at the end of life but longer survival.4
One of the main tools of the palliative care team is the family meeting.
Family meetings are scheduled to allow as many family members as possible to attend, and the primary-care team and relevant specialists typically are also invited. Many palliative-care teams use a standardized format that involves introductions, clarification of each participant’s understanding of the patient’s prognosis, and an effort to reconcile the patient’s hopes and values with medical realities and possibilities, Dr. Fischberg explains.
“That is such a critical component of our care, where we make sure the patient and family are fully informed and foster shared decision making that results in patients being more comfortable with care that better matches what they want,” says Dr. Rotella.
The palliative-care team typically becomes involved via a consultation request from a patient’s attending physician.
“A big part of our job is doing our homework,” Dr. Youngwerth says. “We’ll talk to the team about what’s going on. We want to get as much information as possible about the patient, about prognosis, about the perspectives of people caring for them. Don’t be surprised if the palliative-care team contacts you to get your input on the prognosis and other medical details in order to best inform their discussion with the patient and family.”
Then the palliative-care team will follow consult protocol in reporting back to the primary medical team.
Palliative-care teams can assist busy hospitalists with difficult patient conversations.
“When I’m on the hospitalist service, I’ll pull in the palliative-care team,” Dr. Youngwerth explains. “It’s not that I don’t have the skills; I don’t have the time.”
Conversations aimed at clarifying goals of care can take 90 minutes or more, but the palliative-care team will take as much time as needed to achieve clarification.
It’s important that hospitalists remain involved in these cases, says Christian Sinclair, MD, assistant professor in the division of palliative medicine at the University of Kansas Medical Center in Kansas City and president-elect of AAHPM.
“Just because you have access to palliative-care services, don’t miss the chance to enhance your own communication skills and your ability to address these issues head on,” Dr. Sinclair says.
American Academy of Hospice and Palliative Medicine is a participant in Choosing Wisely.
The Choosing Wisely program, initiated by the American Board of Internal Medicine Foundation, invites medical societies to identify five treatments that should be questioned by physicians and patients based on lack of supporting evidence in the research base. The Society of Hospital Medicine is also a participant in this initiative.
For AAHPM, one of its recommendations was: “Don’t delay palliative care for a patient with serious illness who has a physical, psychological, social, or spiritual distress because they are pursuing disease-directed treatment.” Other Choosing Wisely suggestions include not recommending feeding tubes for patients with advanced dementia, not leaving implantable cardioverter defibrillators (ICDs) activated when these are not consistent with patient/family goals of care, and not recommending more than a single fraction of palliative radiation for an uncomplicated painful bone metastasis.
Different palliative-care programs provide different services.
It is important for hospitalists to learn their local palliative-care programs and what they emphasize and are able to offer—or not, says Dr. Sinclair.
“There are so many different models,” he says. “Spend some time reaching out to them, outside of actual consults, and find out what their comfort level is on various issues. Hospitalists and palliative-care teams should get to know each other better.”
Access to palliative care and the comprehensiveness of the team and services can vary between hospitals, while access to community-based palliative care outside of the hospital is even more variable.
“Palliative-care teams often have a better sense of our partners in the community and access to community-based palliative care,” Dr. Fischberg says.
References
- Morrison RS, Meier DE. America’s Care of Serious Illness: 2015 State-by-State Report Card on Access to Palliative Care in Our Nation’s Hospitals. New York, NY: Center to Advance Palliative Care; 2015.
- American Academy of Pediatrics. Policy statement: pediatric palliative care and hospice care commitments, guidelines, and recommendations. Pediatr. 2013;132(5):966-972.
- Morrison RS, Penrod JD, Cassel JB, et al. Cost savings associated with US hospital palliative care consultation programs. Arch Intern Med. 2008;168(16):1783-1790.
- Temel JS, Greer JA, Muzikansky et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med. 2010;363(8):733-742.