User login
Proteins may be therapeutic targets for AML subtype
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Image by Eric Smith
Preclinical research suggests a pair of histone-modifying proteins may be promising therapeutic targets for NPM1-mutated acute myeloid leukemia (AML).
Investigators found that these proteins—MLL and DOT1L—play key roles in NPM1-mutated AML.
Pharmacologic inhibition of either protein alone produced anti-leukemic activity in vitro and in vivo, but inhibiting both proteins together had a more profound effect.
Michael Kühn, MD, of the Mainz University Medical Center in Mainz, Germany, and his colleagues reported these findings in Cancer Discovery.
The investigators noted that nearly all NPM1-mutated AMLs are characterized by aberrant HOX expression, and FLT3 is concomitantly mutated in roughly 60% of these cases. However, it hasn’t been clear how mutant NPM1 cells maintain aberrant gene expression.
With this study, Dr Kühn and his colleagues showed that MLL1 and DOT1L control HOX and FLT3 expression and differentiation in NPM1-mutated AML.
The investigators were able to demonstrate that survival of NPM1-mutated AML cells depends on these 2 proteins. And NPM1-mutated AML is “exceptionally dependent” on the menin binding site in MLL1.
The team tested MI-503, a menin–MLL1 inhibitor, and the DOT1L inhibitor EPZ4777 in human and murine models of NPM1-mutated AML.
Each of the drugs reduced the activity of HOX genes in NPM1-mutated AML cells, but combining the drugs resulted in near-complete inactivation of HOX genes.
When given alone, EPZ4777 and MI-503 each reduced the proliferation and colony-forming potential of NPM1-mutated AML cells in vitro. And each of the drugs prolonged survival in mouse models of NPM1-mutated AML.
However, EPZ4777 and MI-503 given in combination significantly delayed the onset of leukemia and significantly prolonged survival when compared to either drug given alone.
The investigators said this suggests that inhibiting both DOT1L and menin–MLL1 affects leukemia-initiating cells, and this approach represents the first molecularly targeted treatment of NPM1-mutated AML that works by reversing a key mechanism of leukemogenesis.
They added that this research paves the way for trials assessing EPZ4777 and MI-503 in patients with NPM1-mutated AML. ![]()
Heart failure in Primary Care
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

NOACs show benefit in calciphylaxis
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.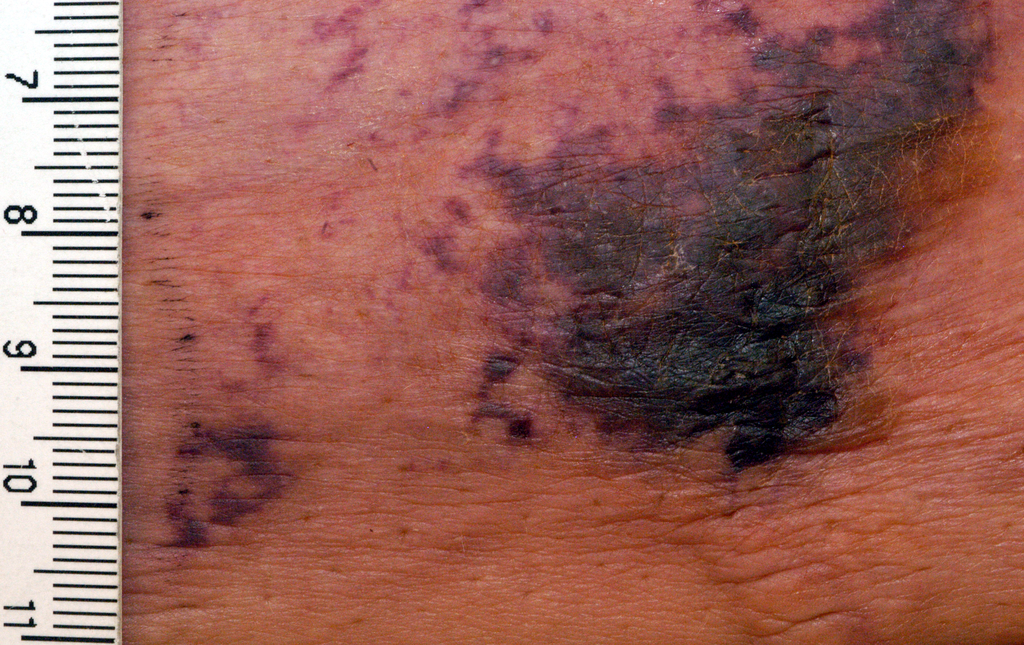
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
VIENNA – The novel oral anticoagulants may provide effective adjunctive therapy in patients with calciphylaxis, Brian J. King, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
He presented a retrospective case series of 16 patients with a confirmed diagnosis of calciphylaxis who were treated with NOACs at the Mayo Clinic in Rochester, Minn., where he is a dermatology resident. The results were impressive, particularly given that the estimated 1-year survival following diagnosis of calciphylaxis is only 45%.
At a mean followup of 418 days, 9 of 16 patients were still alive. More remarkably, five of those nine experienced complete resolution of their clinical lesions and remained alive at a mean followup of 775 days.
Calciphylaxis is a cutaneous manifestation of arteriolar thrombosis. It is classically associated with end-stage renal disease, hyperparathyroidism, a variety of hypercoagulable states, diabetes, and/or obesity. Fifteen of the 16 patients in the Mayo series were women. Fourteen patients had proximal involvement. The lesions occurred most often in fatty tissue on the hips, abdomen, thighs, breasts, and buttocks.
“It’s important to know that this is a deep, incredibly painful process and should not be confused with superficial crusted ulcerations,” Dr. King said.
A variety of treatments have been utilized for calciphylaxis, including sodium thiosulfate, debridement, advanced wound care, hyperbaric oxygen, and parathyroidectomy. But they are often ineffective.
Why not simply use warfarin instead of a costlier NOAC in addressing the problem? Because warfarin has actually been implicated as a cause of the vascular calcification that leads to thrombosis of dermal and pannicular arterioles. Indeed, 12 of the 16 patients in this series were on warfarin at the time of diagnosis of calciphylaxis, either for deep venous thrombosis, pulmonary embolism, or stroke prevention in atrial fibrillation. All were transitioned to a NOAC.
One group of Belgian investigators has provided evidence that strongly suggests the mechanism by which warfarin causes vascular calcification is via inhibition of vitamin K-dependent activation of matrix GLA 1, an enzyme which prevents calcification of vascular endothelial cells (BMC Nephrol. 2014 Sep 4;15:145).
“It is possible and even likely that the vessel calcification we see in patients on warfarin predisposes to thrombosis,” according to Dr. King.
The pathologic diagnostic criteria for calciphylaxis utilized at the Mayo Clinic require skin biopsy evidence of medial calcification and intimal fibroplasia of pannicular arterioles with cutaneous necrosis. Extravascular calcium deposition or thrombosis of pannicular or dermal arterioles is also typically present.
The major clinical criteria are necrotic cutaneous ulcers over indurated plaques, or indurated plaques without ulceration in adipose-rich tissue. The minor criteria are livedo racemosa, hemorrhagic bullae, or hemorrhagic plaques.
Asked if the NOACs can be used interchangeably for treatment of calciphylaxis, Dr. King said the direct factor Xa inhibitor apixaban is the NOAC of choice for this condition at the Mayo Clinic because unlike rivaroxaban (Xarelto) it doesn’t require dosing adjustment in the setting of renal impairment, which is extremely common in patients with calciphylaxis. The direct thrombin inhibitor dabigatran (Pradaxa) is contraindicated in chronic renal failure. Edoxaban (Savaysa) is not on the Mayo Clinic’s formulary, but it is contraindicated in patients with a creatinine clearance of 95 mL/min or more.
Dr. King said he and his coinvestigators recognize that a retrospective case series such as this must be considered hypothesis-generating and nondefinitive. They have already begun a larger prospective comparative outcomes study.
Dr. King reported having no financial conflicts of interest.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Nine of 16 patients with calciphylaxis who were placed on adjunctive therapy with a novel oral anticoagulant responded with significant improvement in their cutaneous disease, four experienced disease stabilization, and three had progressive disease.
Data source: This was a retrospective case study of 16 patients with biopsy-confirmed calciphylaxis.
Disclosures: The study presenter reported having no financial conflicts of interest.
Atopic dermatitis prevention strategies under study
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.

Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.
Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
VIENNA – Diverse strategies aimed at preventing childhood atopic dermatitis (AD) now under study include installation of home water softeners, daily use of emollients starting at birth, and maternal consumption of probiotics beginning late in pregnancy, Carsten Flohr, PhD, said at a joint program of the International Eczema Council and the International Psoriasis Council held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
To date there is no effective method for preventing AD. Preventive strategies are needed sorely because the prevalence of pediatric AD worldwide is expected to increase substantially. It appears to have stabilized at roughly 20% in many affluent countries, but the global burden of the disease will climb as low-income countries – where AD is historically uncommon – become more developed and urbanized. This trend has been well documented via the International Study of Asthma and Allergies in Childhood (ISAAC), which in several phases has studied nearly 2 million children in more than 100 countries, noted Dr. Flohr of St. John’s Institute of Dermatology at King’s College London.
Dr. Flohr and coinvestigators in the Enquiring About Tolerance (EAT) study recently documented a significant association between water hardness and the risk of infant-onset AD. The investigators took advantage of the considerable variation in the amount of bedrock limestone across England, which enabled them to study the relationship between domestic water calcium carbonate concentrations and the presence of AD in 1,303 babies at 3 monthd of age drawn from the general population across the country. Filaggrin skin barrier gene mutation status was determined in all infants.
Infants whose water supply contained a calcium carbonate level above the median value were at an adjusted 46% greater risk of having visible AD at age 3 months than those whose household water calcium carbonate level was below the median. The AD risk rose by 1% for each 1 mg/L increase in calcium carbonate concentration above the median. This increased risk was confined to infants with a filaggrin skin barrier gene mutation; hard water didn’t increase early AD risk in children with the normal, wild-type version of the filaggrin gene (J Allergy Clin Immunol. 2016 Aug;138[2]:509-16).
As a result of these findings, a UK prevention trial is underway in which home water softeners are provided to families at high risk of having a baby with AD in water districts with high calcium carbonate concentrations. An earlier UK study found that installation of home water softeners didn’t reduce AD severity in children with established disease (PLoS Med. 2011 Feb 15;8[2]:e1000395), but disease prevention may be another story.
The role of the gut microbiota in development of childhood AD is an active area of investigation. Dr. Flohr said “there is a signal” that maternal intake of probiotics including lactobacilli and bifidobacteria in the third trimester and postnatally may reduce a child’s risk of developing AD by encouraging establishment of a more diverse gut microflora. He cited a meta-analysis of 14 published studies of probiotics which provided evidence of a 21% reduction in the incidence of AD in young children (Epidemiology 2012 May;23[3]:402-14). The studies have methodologic shortcomings, so multiple research groups are continuing to pursue the signal of an AD preventive effect.
Dr. Flor reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Palliative care boosts heart failure patient outcomes
ORLANDO – Systematic introduction of palliative care interventions for patients with advanced heart failure improved patients’ quality of life and spurred their development of advanced-care preferences in a pair of independently performed, controlled, pilot studies.
But, despite demonstrating the ability of palliative-care interventions to help heart failure patients during their final months of life, the findings raised questions about the generalizability and reproducibility of palliative-care interventions that may depend on the skills and experience of the individual specialists who deliver the palliative care.

“Palliative care for patients with cardiovascular disease is in desperate need of good-quality evidence,” commented Larry A, Allen, MD, a heart failure cardiologist at the University of Colorado in Aurora and designated discussant for one of the two studies presented at the meeting. “We need large, randomized trials with clinical outcomes to look at patient outcomes from palliative-care interventions.”

The patients average 71 years old, about half were women, and about 40% were African Americans. They had been diagnosed with heart failure for an average of more than 5 years, all had advanced heart failure, about 60% spent at least half of their time awake immobilized in a bed or chair, and they had average NT-proBNP blood levels of greater than 10,000 pg/mL.
After 24 weeks of intervention, the palliative-care program produced both statistically significant and clinically meaningful improvements in two different measures of health-related quality of life, the Kansas City Cardiomyopathy Questionnaire (KCCQ) and the Functional Assessment of Chronic Illness Therapy – Palliative Care (FACIT-PAL). The KCCQ showed the palliative care intervention linked with an average rise of more than 9 points compared with patients in the control arm after adjustment for age and sex, a statistically significant increase on a scale where a 5-point rise is considered clinically meaningful. The FACIT-PAL showed an average, adjusted 11-point rise linked with the intervention, a statistically significant increase on a scale where an increase of at least 10 is judged clinically meaningful, reported Dr. Rogers, a heart failure cardiologist and professor of medicine at Duke University.
The palliative-care intervention also led to significant improvements in measures of spirituality, depression, and anxiety, but intervention had no impact on mortality.
“I like these endpoints and the idea that we can make quality-of-life better. These are very sick patients, with a predicted 6-month mortality of 50%. Patients reach a time when they don’t want to live longer but want better life quality for the days they still have,” he said in an interview.
The second report came from a single-center pilot study of 50 patients enrolled when they were hospitalized for acute decompensated heart failure and had at least one addition risk factor for poor prognosis such as age of at least 81 years, renal dysfunction, or a prior heart failure hospitalization within the past year. Patients randomized to the intervention arm underwent a structured evaluation based on the Serious Illness Conversation Guide and performed by a social worker experienced in palliative care and embedded in the heart failure clinical team. The primary endpoint of the SWAP-HF (Social Worker–Aided Palliative Care Intervention in High Risk Patients with Heart Failure) study was clinical-level documentation of advanced-care preferences by 6 months after the program began.

“Although more comprehensive, multidisciplinary palliative care interventions may also be effective, the focused approach [used in this study] may represent a cost-effective and scalable method for shepherding limited specialty resources to enhance the delivery of patient-centered care,” Dr. Desai said. In other words, a program with a social worker costs less than a two-person staff with a palliative-care physician and nurse practitioner.
Despite its relative simplicity, the SWAP-HF intervention had some unique aspects that make it generalizability uncertain, commented Dr. Allen. The embedding of a social worker on the heart failure team placed a professional with a “good understanding of social context” right on the scene with everyone else delivering care to the heart failure patient, a good strategy for minimizing fragmentation, he said. In addition, the place where the study was done, Brigham and Women’s Hospital, “is not your average hospital,” he noted,
In addition, the timing of the intervention studied during hospitalization may be problematic. Clinicians need to “be careful about patients making long-term decisions” about their care while they are hospitalized, a time when patients can be “ill, confused, and scared.” He cited recent findings from a study of hospital-based palliative-care interventions for family members of patients with chronic critical illness that did not reduce anxiety or depression symptoms among the treated family members and may have increased symptoms of posttraumatic stress disorder (JAMA. 2016 July 5;374[1]:51-62).
[email protected]
On Twitter @mitchelzoler
It’s very exciting to have these two studies presented at the Heart Failure Society of America’s annual meeting. Palliative-care research now receives funding from the National Institutes of Health, but consistently and successfully integrating palliative care into heart failure management still has a long way to go. In 2004, my colleagues and I published a set of consensus recommendations on how to apply palliative care methods to patients with advanced heart failure and what research needs existed for the field (J Card Fail. 2004 June;10[3]:200-9). Today, 12 years later, many of those research needs remain inadequately addressed.

Sarah J. Goodlin, MD , is chief of geriatrics at the Portland (Ore.) VA Medical Center. She had no disclosures. She made these comments as the designated discussant for Dr. Rogers’ report.
It’s very exciting to have these two studies presented at the Heart Failure Society of America’s annual meeting. Palliative-care research now receives funding from the National Institutes of Health, but consistently and successfully integrating palliative care into heart failure management still has a long way to go. In 2004, my colleagues and I published a set of consensus recommendations on how to apply palliative care methods to patients with advanced heart failure and what research needs existed for the field (J Card Fail. 2004 June;10[3]:200-9). Today, 12 years later, many of those research needs remain inadequately addressed.
Sarah J. Goodlin, MD , is chief of geriatrics at the Portland (Ore.) VA Medical Center. She had no disclosures. She made these comments as the designated discussant for Dr. Rogers’ report.
It’s very exciting to have these two studies presented at the Heart Failure Society of America’s annual meeting. Palliative-care research now receives funding from the National Institutes of Health, but consistently and successfully integrating palliative care into heart failure management still has a long way to go. In 2004, my colleagues and I published a set of consensus recommendations on how to apply palliative care methods to patients with advanced heart failure and what research needs existed for the field (J Card Fail. 2004 June;10[3]:200-9). Today, 12 years later, many of those research needs remain inadequately addressed.
Sarah J. Goodlin, MD , is chief of geriatrics at the Portland (Ore.) VA Medical Center. She had no disclosures. She made these comments as the designated discussant for Dr. Rogers’ report.
ORLANDO – Systematic introduction of palliative care interventions for patients with advanced heart failure improved patients’ quality of life and spurred their development of advanced-care preferences in a pair of independently performed, controlled, pilot studies.
But, despite demonstrating the ability of palliative-care interventions to help heart failure patients during their final months of life, the findings raised questions about the generalizability and reproducibility of palliative-care interventions that may depend on the skills and experience of the individual specialists who deliver the palliative care.
“Palliative care for patients with cardiovascular disease is in desperate need of good-quality evidence,” commented Larry A, Allen, MD, a heart failure cardiologist at the University of Colorado in Aurora and designated discussant for one of the two studies presented at the meeting. “We need large, randomized trials with clinical outcomes to look at patient outcomes from palliative-care interventions.”
The patients average 71 years old, about half were women, and about 40% were African Americans. They had been diagnosed with heart failure for an average of more than 5 years, all had advanced heart failure, about 60% spent at least half of their time awake immobilized in a bed or chair, and they had average NT-proBNP blood levels of greater than 10,000 pg/mL.
After 24 weeks of intervention, the palliative-care program produced both statistically significant and clinically meaningful improvements in two different measures of health-related quality of life, the Kansas City Cardiomyopathy Questionnaire (KCCQ) and the Functional Assessment of Chronic Illness Therapy – Palliative Care (FACIT-PAL). The KCCQ showed the palliative care intervention linked with an average rise of more than 9 points compared with patients in the control arm after adjustment for age and sex, a statistically significant increase on a scale where a 5-point rise is considered clinically meaningful. The FACIT-PAL showed an average, adjusted 11-point rise linked with the intervention, a statistically significant increase on a scale where an increase of at least 10 is judged clinically meaningful, reported Dr. Rogers, a heart failure cardiologist and professor of medicine at Duke University.
The palliative-care intervention also led to significant improvements in measures of spirituality, depression, and anxiety, but intervention had no impact on mortality.
“I like these endpoints and the idea that we can make quality-of-life better. These are very sick patients, with a predicted 6-month mortality of 50%. Patients reach a time when they don’t want to live longer but want better life quality for the days they still have,” he said in an interview.
The second report came from a single-center pilot study of 50 patients enrolled when they were hospitalized for acute decompensated heart failure and had at least one addition risk factor for poor prognosis such as age of at least 81 years, renal dysfunction, or a prior heart failure hospitalization within the past year. Patients randomized to the intervention arm underwent a structured evaluation based on the Serious Illness Conversation Guide and performed by a social worker experienced in palliative care and embedded in the heart failure clinical team. The primary endpoint of the SWAP-HF (Social Worker–Aided Palliative Care Intervention in High Risk Patients with Heart Failure) study was clinical-level documentation of advanced-care preferences by 6 months after the program began.
“Although more comprehensive, multidisciplinary palliative care interventions may also be effective, the focused approach [used in this study] may represent a cost-effective and scalable method for shepherding limited specialty resources to enhance the delivery of patient-centered care,” Dr. Desai said. In other words, a program with a social worker costs less than a two-person staff with a palliative-care physician and nurse practitioner.
Despite its relative simplicity, the SWAP-HF intervention had some unique aspects that make it generalizability uncertain, commented Dr. Allen. The embedding of a social worker on the heart failure team placed a professional with a “good understanding of social context” right on the scene with everyone else delivering care to the heart failure patient, a good strategy for minimizing fragmentation, he said. In addition, the place where the study was done, Brigham and Women’s Hospital, “is not your average hospital,” he noted,
In addition, the timing of the intervention studied during hospitalization may be problematic. Clinicians need to “be careful about patients making long-term decisions” about their care while they are hospitalized, a time when patients can be “ill, confused, and scared.” He cited recent findings from a study of hospital-based palliative-care interventions for family members of patients with chronic critical illness that did not reduce anxiety or depression symptoms among the treated family members and may have increased symptoms of posttraumatic stress disorder (JAMA. 2016 July 5;374[1]:51-62).
[email protected]
On Twitter @mitchelzoler
ORLANDO – Systematic introduction of palliative care interventions for patients with advanced heart failure improved patients’ quality of life and spurred their development of advanced-care preferences in a pair of independently performed, controlled, pilot studies.
But, despite demonstrating the ability of palliative-care interventions to help heart failure patients during their final months of life, the findings raised questions about the generalizability and reproducibility of palliative-care interventions that may depend on the skills and experience of the individual specialists who deliver the palliative care.
“Palliative care for patients with cardiovascular disease is in desperate need of good-quality evidence,” commented Larry A, Allen, MD, a heart failure cardiologist at the University of Colorado in Aurora and designated discussant for one of the two studies presented at the meeting. “We need large, randomized trials with clinical outcomes to look at patient outcomes from palliative-care interventions.”
The patients average 71 years old, about half were women, and about 40% were African Americans. They had been diagnosed with heart failure for an average of more than 5 years, all had advanced heart failure, about 60% spent at least half of their time awake immobilized in a bed or chair, and they had average NT-proBNP blood levels of greater than 10,000 pg/mL.
After 24 weeks of intervention, the palliative-care program produced both statistically significant and clinically meaningful improvements in two different measures of health-related quality of life, the Kansas City Cardiomyopathy Questionnaire (KCCQ) and the Functional Assessment of Chronic Illness Therapy – Palliative Care (FACIT-PAL). The KCCQ showed the palliative care intervention linked with an average rise of more than 9 points compared with patients in the control arm after adjustment for age and sex, a statistically significant increase on a scale where a 5-point rise is considered clinically meaningful. The FACIT-PAL showed an average, adjusted 11-point rise linked with the intervention, a statistically significant increase on a scale where an increase of at least 10 is judged clinically meaningful, reported Dr. Rogers, a heart failure cardiologist and professor of medicine at Duke University.
The palliative-care intervention also led to significant improvements in measures of spirituality, depression, and anxiety, but intervention had no impact on mortality.
“I like these endpoints and the idea that we can make quality-of-life better. These are very sick patients, with a predicted 6-month mortality of 50%. Patients reach a time when they don’t want to live longer but want better life quality for the days they still have,” he said in an interview.
The second report came from a single-center pilot study of 50 patients enrolled when they were hospitalized for acute decompensated heart failure and had at least one addition risk factor for poor prognosis such as age of at least 81 years, renal dysfunction, or a prior heart failure hospitalization within the past year. Patients randomized to the intervention arm underwent a structured evaluation based on the Serious Illness Conversation Guide and performed by a social worker experienced in palliative care and embedded in the heart failure clinical team. The primary endpoint of the SWAP-HF (Social Worker–Aided Palliative Care Intervention in High Risk Patients with Heart Failure) study was clinical-level documentation of advanced-care preferences by 6 months after the program began.
“Although more comprehensive, multidisciplinary palliative care interventions may also be effective, the focused approach [used in this study] may represent a cost-effective and scalable method for shepherding limited specialty resources to enhance the delivery of patient-centered care,” Dr. Desai said. In other words, a program with a social worker costs less than a two-person staff with a palliative-care physician and nurse practitioner.
Despite its relative simplicity, the SWAP-HF intervention had some unique aspects that make it generalizability uncertain, commented Dr. Allen. The embedding of a social worker on the heart failure team placed a professional with a “good understanding of social context” right on the scene with everyone else delivering care to the heart failure patient, a good strategy for minimizing fragmentation, he said. In addition, the place where the study was done, Brigham and Women’s Hospital, “is not your average hospital,” he noted,
In addition, the timing of the intervention studied during hospitalization may be problematic. Clinicians need to “be careful about patients making long-term decisions” about their care while they are hospitalized, a time when patients can be “ill, confused, and scared.” He cited recent findings from a study of hospital-based palliative-care interventions for family members of patients with chronic critical illness that did not reduce anxiety or depression symptoms among the treated family members and may have increased symptoms of posttraumatic stress disorder (JAMA. 2016 July 5;374[1]:51-62).
[email protected]
On Twitter @mitchelzoler
AT THE HFSA ANNUAL SCIENTIFIC MEETING
Key clinical point:
Major finding: Palliative care measures boosted patients’ Kansas City Cardiomyopathy Questionnaire score by an average of 9 points over that of controls.
Data source: PAL-HF, a single-center study with 150 randomized patients with heart failure and SWAP-HF, a single-center study with 50 randomized patients.
Disclosures: Dr. Rogers, Dr. Allen, and Dr. Desai had no relevant disclosures.
Help Improve Quality at Your Institution with SHM
October 16–22 is the National Association for Healthcare Quality’s “Healthcare Quality Week,” and SHM’s Center for Hospital Innovation & Improvement provides a variety of resources, tools, and programs to help address quality and patient safety issues at your institution. Find out how SHM can help you improve patient safety and outcomes through our Center for Hospital Innovation & Improvement at www.hospialmedicine.org/QI.
October 16–22 is the National Association for Healthcare Quality’s “Healthcare Quality Week,” and SHM’s Center for Hospital Innovation & Improvement provides a variety of resources, tools, and programs to help address quality and patient safety issues at your institution. Find out how SHM can help you improve patient safety and outcomes through our Center for Hospital Innovation & Improvement at www.hospialmedicine.org/QI.
October 16–22 is the National Association for Healthcare Quality’s “Healthcare Quality Week,” and SHM’s Center for Hospital Innovation & Improvement provides a variety of resources, tools, and programs to help address quality and patient safety issues at your institution. Find out how SHM can help you improve patient safety and outcomes through our Center for Hospital Innovation & Improvement at www.hospialmedicine.org/QI.
Strengthen Your Role as a Practice Administrator with SHM’s Mentor Program

- Model 1: Mentors/Mentees. Less experienced administrators will be paired with seasoned professionals to gain more experience or exposure.
- Model 2: Buddy System. Administrators at any level of expertise or experience will be paired with a peer so they both can learn from each other.
Interested in being a mentor or mentee for the 2017 program? Complete the online form at www.hospitalmedicine.org/pamentor used to match you up to other individuals who have similar needs for improvement.
- Model 1: Mentors/Mentees. Less experienced administrators will be paired with seasoned professionals to gain more experience or exposure.
- Model 2: Buddy System. Administrators at any level of expertise or experience will be paired with a peer so they both can learn from each other.
Interested in being a mentor or mentee for the 2017 program? Complete the online form at www.hospitalmedicine.org/pamentor used to match you up to other individuals who have similar needs for improvement.
- Model 1: Mentors/Mentees. Less experienced administrators will be paired with seasoned professionals to gain more experience or exposure.
- Model 2: Buddy System. Administrators at any level of expertise or experience will be paired with a peer so they both can learn from each other.
Interested in being a mentor or mentee for the 2017 program? Complete the online form at www.hospitalmedicine.org/pamentor used to match you up to other individuals who have similar needs for improvement.

Become a Fellow in Hospital Medicine

The application is open through November 30, with a decision on or before December 31, 2016. Apply now and learn how you can join other hospitalists who have earned this exclusive designation and recognition at www.hospitalmedicine.org/fellows.
The application is open through November 30, with a decision on or before December 31, 2016. Apply now and learn how you can join other hospitalists who have earned this exclusive designation and recognition at www.hospitalmedicine.org/fellows.
The application is open through November 30, with a decision on or before December 31, 2016. Apply now and learn how you can join other hospitalists who have earned this exclusive designation and recognition at www.hospitalmedicine.org/fellows.

Lebrikizumab opens new door in atopic dermatitis therapy
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.

Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.
Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
VIENNA – The success of the interleukin-13 blocker lebrikizumab in the TREBLE trial provides a new avenue in the treatment of moderate-to-severe atopic dermatitis.
“This is another promising molecule in atopic dermatitis,” Eric L. Simpson, MD, declared in presenting the phase II TREBLE data at the annual congress of the European Academy of Dermatology and Venereology.
Interleukin-13 is known to be an especially potent promoter of type 1, IgE-mediated inflammation. For this reason, lebrikizumab is also under investigation in the treatment of severe asthma, where large phase III trials have been completed, as well as in idiopathic pulmonary fibrosis.
In mouse models of AD, topical anti-IL-13 therapy markedly reduces skin inflammation. This observation helped provide the rationale for investigating lebrikizumab as a novel therapy for AD.
TREBLE was a double-blind, dose-ranging study involving 209 adults with moderate-to-severe AD despite intensive topical corticosteroid therapy. Indeed, enrollment was restricted to patients who still had moderate or severe disease after 2 weeks of triamcinolone 0.1% cream BID. Continuation of that twice-daily topical steroid regimen was mandatory for the full 12-week study period that followed. Patients were randomized 1:1:1:1 to triamcinolone 0.1% BID plus either a single 125-mg subcutaneous dose of lebrikizumab at week 0, a single 250-mg dose of the biologic, 125 mg every 4 weeks, or placebo injections.
At baseline, after their 2 weeks of topical steroid monotherapy, patients had a median Eczema Area and Severity Index (EASI) score of 22 and a median SCORing Atopic Dermatitis (SCORAD) of about 56. They averaged 36 years of age. Slightly over 40% of their body surface area was affected.
The primary endpoint in TREBLE was the percentage of patients who achieved at least a 50% reduction from baseline on the EASI, or EASI 50. A dose-response effect was apparent: the EASI 50 rate was 62.3% in patients on placebo plus daily topical steroids, 69.2% with a single 125-mg dose of lebrikizumab, 69.8% with a single 250-mg dose, and 82.4% with 125 mg of lebrikizumab at weeks 0, 4, 8, and 12. Only the group with monthly dosing of the biologic plus daily triamcinolone 0.1% BID had an EASI 50 response rate significantly better than the controls on placebo plus topical steroid therapy.
A key secondary endpoint was the SCORAD 50 response. The rate was 26.4% in the control group, 34.6% in patients who received one 125-mg dose of lebrikizumab, 47.2% in those who got a single 250-mg dose, and 51% with monthly dosing. The SCORAD 50 rate was significantly greater than placebo in the latter two lebrikizumab arms.
The EASI 75 rate was significantly greater than placebo plus triamcinolone only in the group on monthly lebrikizumab plus topical steroids.
The EASI 50 and 75 response rates in the group on 125 mg of lebrikizumab every 4 weeks was still climbing with no plateau evident when the trial ended at 12 weeks. This suggests greater benefit might be achieved with longer treatment duration and/or a higher dosage, Dr. Simpson observed.
Turning to safety issues, he pointed out that the total number of adverse events and serious adverse events were similar across all four treatment arms. Herpes infections occurred in 2%-6% of patients who received lebrikizumab but in no controls. There was also a nonsignificant trend for more cases of conjunctivitis in the groups that received the biologic.
Audience member Andrew Blauvelt, MD, rose to take issue with the study design.
“This study is almost an advertisement for not using topical steroids in a biologic trial for atopic dermatitis. I think it just confuses everything. It especially doesn’t make sense when one of the inclusion criteria is inadequate control with topical steroids. Shouldn’t we be doing monotherapy trials in the new era of biologics for atopic dermatitis?” commented Dr. Blauvelt, president of the Oregon Medical Research Center in Portland.
Dr. Simpson replied that when TREBLE was designed, the thinking was that in real-world clinical practice, physicians often prescribe a biologic for AD on top of topical therapy. The goal was to conduct a trial that mimics that situation. That being said, he agreed with Dr. Blauvelt’s critique.
“I think we’re realizing that topical steroids have a very strong effect, especially if you use them all the way through a trial or ad lib,” he said. “Topical steroids bring too many confounders. If you can remove potent topical steroid therapy I think you’re going to have a clearer result.”
“We’re not where psoriasis is,” Dr. Simpson added. “We don’t have any standardized methodologies in atopic dermatitis clinical trials yet, but I think we’re getting there.”
The TREBLE trial was sponsored by Genentech/Roche. Dr. Simpson reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Of atopic dermatitis patients randomized to 125 mg of lebrikizumab every 4 weeks on top of twice-daily potent topical steroids, 82% experienced at least a 50% reduction in Eczema Area and Severity Index scores at 12 weeks, compared with 62% on the same topical steroid regimen plus placebo injections.
Data source: The TREBLE trial was a phase II, double-blind, placebo-controlled, 12-week study including 209 adults with moderate-to-severe atopic dermatitis inadequately controlled by intensive topical steroid therapy.
Disclosures: The study was sponsored by Genentech/Roche. The presenter reported serving as an investigator for and consultant to that organization and numerous other pharmaceutical companies.
ADT may increase dementia risk in prostate CA
Prostate cancer patients treated with androgen deprivation therapy are more than twice as likely as those who were not to develop dementia, according to a review of 9,272 prostate cancer cases at Stanford (Calif.) University.
Almost 8% of the 1,826 men treated with androgen deprivation therapy (ADT), a mainstay against prostate cancer, were diagnosed with dementia at 5 years, versus 3.5% of the 7,446 men not treated with ADT (JAMA Oncol. 2016 Oct 13. doi: 10.1001/jamaoncol.2016.3662).
“Our study extends previous work supporting an association between use of ADT and Alzheimer disease and suggests that ADT may more broadly affect neurocognitive function,” said the investigators, led by Kevin Nead, MD, formerly at Stanford but now a radiation oncology resident at the University of Pennsylvania, Philadelphia.
New-onset senile dementia, vascular dementia, frontotemporal dementia, and Alzheimer dementia were linked to ADT in both a propensity score-matched analysis (HR, 2.17; 95% CI, 1.58-2.99; P < .001) and multivariate analysis (adjusted HR, 2.21; 95% CI, 1.72-2.83; P < .001). The results held up when Alzheimer disease, just 30% of the 314 dementia cases, was excluded.
Men with at least 12 months of ADT had the greatest increased risk of dementia (HR, 2.36; 95% CI, 1.64-3.38; P < .001), suggesting a dose-response relationship; men on ADT who were at least 70 years old had the lowest cumulative probability of not developing dementia.
The link, the team said, is biologically plausible. Androgens have a role in neuron health and growth, and testosterone analogues have neuroprotective effects. Testosterone may be converted to estrogen, which is also neuroprotective. Low testosterone levels and ADT, meanwhile, have been shown to increase the risk of cardiometabolic diseases, which increase the risk of dementia.
The analyses controlled for age at prostate cancer diagnosis; race; smoking status; use of antiplatelet, anticoagulant, antihypertensive, and statin medications; and histories of cardiovascular disease, type 1 or 2 diabetes, stroke, and malignant neoplasms.
The men were treated at Stanford from 1994-2013. ADT patients tended to be older than their peers (70 versus 66 years), less likely to white (54% versus 60%), more likely to have smoked (44% versus 38%), and more likely to have other health problems.
More than 20 medications in the study were used for ADT, including leuprolide, goserelin, and triptorelin. Data were collectedd from electronic health records, and included diagnostic codes, medication lists, and clinical notes.
The work was funded by the National Institutes of Health. The senior author, Nigam Shah, PhD, has patents pending on the data-mining methods used in the study.
Prostate cancer patients treated with androgen deprivation therapy are more than twice as likely as those who were not to develop dementia, according to a review of 9,272 prostate cancer cases at Stanford (Calif.) University.
Almost 8% of the 1,826 men treated with androgen deprivation therapy (ADT), a mainstay against prostate cancer, were diagnosed with dementia at 5 years, versus 3.5% of the 7,446 men not treated with ADT (JAMA Oncol. 2016 Oct 13. doi: 10.1001/jamaoncol.2016.3662).
“Our study extends previous work supporting an association between use of ADT and Alzheimer disease and suggests that ADT may more broadly affect neurocognitive function,” said the investigators, led by Kevin Nead, MD, formerly at Stanford but now a radiation oncology resident at the University of Pennsylvania, Philadelphia.
New-onset senile dementia, vascular dementia, frontotemporal dementia, and Alzheimer dementia were linked to ADT in both a propensity score-matched analysis (HR, 2.17; 95% CI, 1.58-2.99; P < .001) and multivariate analysis (adjusted HR, 2.21; 95% CI, 1.72-2.83; P < .001). The results held up when Alzheimer disease, just 30% of the 314 dementia cases, was excluded.
Men with at least 12 months of ADT had the greatest increased risk of dementia (HR, 2.36; 95% CI, 1.64-3.38; P < .001), suggesting a dose-response relationship; men on ADT who were at least 70 years old had the lowest cumulative probability of not developing dementia.
The link, the team said, is biologically plausible. Androgens have a role in neuron health and growth, and testosterone analogues have neuroprotective effects. Testosterone may be converted to estrogen, which is also neuroprotective. Low testosterone levels and ADT, meanwhile, have been shown to increase the risk of cardiometabolic diseases, which increase the risk of dementia.
The analyses controlled for age at prostate cancer diagnosis; race; smoking status; use of antiplatelet, anticoagulant, antihypertensive, and statin medications; and histories of cardiovascular disease, type 1 or 2 diabetes, stroke, and malignant neoplasms.
The men were treated at Stanford from 1994-2013. ADT patients tended to be older than their peers (70 versus 66 years), less likely to white (54% versus 60%), more likely to have smoked (44% versus 38%), and more likely to have other health problems.
More than 20 medications in the study were used for ADT, including leuprolide, goserelin, and triptorelin. Data were collectedd from electronic health records, and included diagnostic codes, medication lists, and clinical notes.
The work was funded by the National Institutes of Health. The senior author, Nigam Shah, PhD, has patents pending on the data-mining methods used in the study.
Prostate cancer patients treated with androgen deprivation therapy are more than twice as likely as those who were not to develop dementia, according to a review of 9,272 prostate cancer cases at Stanford (Calif.) University.
Almost 8% of the 1,826 men treated with androgen deprivation therapy (ADT), a mainstay against prostate cancer, were diagnosed with dementia at 5 years, versus 3.5% of the 7,446 men not treated with ADT (JAMA Oncol. 2016 Oct 13. doi: 10.1001/jamaoncol.2016.3662).
“Our study extends previous work supporting an association between use of ADT and Alzheimer disease and suggests that ADT may more broadly affect neurocognitive function,” said the investigators, led by Kevin Nead, MD, formerly at Stanford but now a radiation oncology resident at the University of Pennsylvania, Philadelphia.
New-onset senile dementia, vascular dementia, frontotemporal dementia, and Alzheimer dementia were linked to ADT in both a propensity score-matched analysis (HR, 2.17; 95% CI, 1.58-2.99; P < .001) and multivariate analysis (adjusted HR, 2.21; 95% CI, 1.72-2.83; P < .001). The results held up when Alzheimer disease, just 30% of the 314 dementia cases, was excluded.
Men with at least 12 months of ADT had the greatest increased risk of dementia (HR, 2.36; 95% CI, 1.64-3.38; P < .001), suggesting a dose-response relationship; men on ADT who were at least 70 years old had the lowest cumulative probability of not developing dementia.
The link, the team said, is biologically plausible. Androgens have a role in neuron health and growth, and testosterone analogues have neuroprotective effects. Testosterone may be converted to estrogen, which is also neuroprotective. Low testosterone levels and ADT, meanwhile, have been shown to increase the risk of cardiometabolic diseases, which increase the risk of dementia.
The analyses controlled for age at prostate cancer diagnosis; race; smoking status; use of antiplatelet, anticoagulant, antihypertensive, and statin medications; and histories of cardiovascular disease, type 1 or 2 diabetes, stroke, and malignant neoplasms.
The men were treated at Stanford from 1994-2013. ADT patients tended to be older than their peers (70 versus 66 years), less likely to white (54% versus 60%), more likely to have smoked (44% versus 38%), and more likely to have other health problems.
More than 20 medications in the study were used for ADT, including leuprolide, goserelin, and triptorelin. Data were collectedd from electronic health records, and included diagnostic codes, medication lists, and clinical notes.
The work was funded by the National Institutes of Health. The senior author, Nigam Shah, PhD, has patents pending on the data-mining methods used in the study.
FROM JAMA ONCOLOGY
Key clinical point:
Major finding: New-onset senile dementia, vascular dementia, frontotemporal dementia, and Alzheimer dementia were linked to ADT in both a propensity score-matched analysis (HR, 2.17; 95% CI, 1.58-2.99; P < .001) and multivariate analysis (adjusted HR, 2.21; 95% CI, 1.72-2.83; P < .001).
Data source: Review of 9,272 prostate cancer cases at Stanford (Calif.) University.
Disclosures: The work was funded by the National Institutes of Health. The senior author, Nigam Shah, PhD, has patents pending on the data-mining methods used in the study.