User login
FDA approves use of assay to screen blood for Zika virus
![]()
Photo courtesy of UAB Hospital
The US Food and Drug Administration (FDA) has approved the use of a new assay to screen donated blood for the Zika virus.
The Procleix Zika Virus Assay is approved for use under an investigational new drug study protocol.
Blood banks can use the test to screen donated blood for the Zika virus in potentially endemic areas of the southern US. Testing may be extended to other areas of the US if the Zika virus continues to spread.
The Procleix Zika Virus Assay, which was co-developed by Hologic, Inc. and Grifols, is designed to run on the Procleix Panther System, an automated, nucleic acid technology blood screening platform. The system has received regulatory approvals in countries around the world and is currently in development for the US market.
“The American Red Cross is pleased to participate in the Procleix Zika Virus Assay investigational study, which will allow us to begin blood donor testing for Zika virus early this summer in areas most likely to have local mosquito transmission of the virus,” said Susan Stramer, PhD, vice-president of scientific affairs at the American Red Cross.
The FDA previously authorized use of another test, the cobas® Zika test (developed by Roche), to screen blood donations for Zika virus. The cobas Zika test can be used under an investigational new drug study protocol for screening donated blood in areas with active, mosquito-borne transmission of the Zika virus. ![]()
![]()
Photo courtesy of UAB Hospital
The US Food and Drug Administration (FDA) has approved the use of a new assay to screen donated blood for the Zika virus.
The Procleix Zika Virus Assay is approved for use under an investigational new drug study protocol.
Blood banks can use the test to screen donated blood for the Zika virus in potentially endemic areas of the southern US. Testing may be extended to other areas of the US if the Zika virus continues to spread.
The Procleix Zika Virus Assay, which was co-developed by Hologic, Inc. and Grifols, is designed to run on the Procleix Panther System, an automated, nucleic acid technology blood screening platform. The system has received regulatory approvals in countries around the world and is currently in development for the US market.
“The American Red Cross is pleased to participate in the Procleix Zika Virus Assay investigational study, which will allow us to begin blood donor testing for Zika virus early this summer in areas most likely to have local mosquito transmission of the virus,” said Susan Stramer, PhD, vice-president of scientific affairs at the American Red Cross.
The FDA previously authorized use of another test, the cobas® Zika test (developed by Roche), to screen blood donations for Zika virus. The cobas Zika test can be used under an investigational new drug study protocol for screening donated blood in areas with active, mosquito-borne transmission of the Zika virus. ![]()
![]()
Photo courtesy of UAB Hospital
The US Food and Drug Administration (FDA) has approved the use of a new assay to screen donated blood for the Zika virus.
The Procleix Zika Virus Assay is approved for use under an investigational new drug study protocol.
Blood banks can use the test to screen donated blood for the Zika virus in potentially endemic areas of the southern US. Testing may be extended to other areas of the US if the Zika virus continues to spread.
The Procleix Zika Virus Assay, which was co-developed by Hologic, Inc. and Grifols, is designed to run on the Procleix Panther System, an automated, nucleic acid technology blood screening platform. The system has received regulatory approvals in countries around the world and is currently in development for the US market.
“The American Red Cross is pleased to participate in the Procleix Zika Virus Assay investigational study, which will allow us to begin blood donor testing for Zika virus early this summer in areas most likely to have local mosquito transmission of the virus,” said Susan Stramer, PhD, vice-president of scientific affairs at the American Red Cross.
The FDA previously authorized use of another test, the cobas® Zika test (developed by Roche), to screen blood donations for Zika virus. The cobas Zika test can be used under an investigational new drug study protocol for screening donated blood in areas with active, mosquito-borne transmission of the Zika virus. ![]()
Study provides clues to AML survival after chemotherapy

Preclinical research suggests that some leukemia cells harvest energy resources from normal cells during chemotherapy, and this helps the leukemia cells survive and thrive after treatment.
Investigators found that acute myeloid leukemia (AML) cells are capable of stealing mitochondria from stromal cells, and these stolen mitochondria give an energy boost to surviving AML cells, which helps fuel the leukemia’s resurgence after chemotherapy.
“There are multiple mechanisms for resistance to chemotherapy, and it will be important to target them all in order to eliminate all leukemic cells,” said Jean-François Peyron, PhD, of the Centre Méditerranéen de Médecine Moléculaire (C3M) in Nice, France.
“Targeting this protective mitochondrial transfer could represent a new strategy to improve the efficacy of the current treatments for acute myeloid leukemia.”
Dr Peyron and his colleagues described their discovery of the mitochondrial transfer in Blood.
The team conducted their experiments using cell cultures and mouse models of AML. They found that nearly all AML cells died when exposed to chemotherapy drugs, but some survived. And these cells issued a “mayday” signal that “tricked” nearby non-cancerous cells into yielding their mitochondria to the AML cells, thus strengthening the leukemia cells.
“Mitochondria produce the energy that is vital for cell functions,” explained study author Emmanuel Griessinger, PhD, also of C3M.
“Through the uptake of mitochondria, chemotherapy-injured acute myeloid leukemia cells recover new energy to survive.”
The AML cells were found to increase their mitochondria mass by an average of 14%. This increase led to a 1.5-fold increase in energy production and significantly better survival rates. That is, the leukemia cells that had a high level of mitochondria were also more resistant to the chemotherapy.
The investigators observed the phenomenon in several types of leukemia cells, most notably leukemia-initiating cells. The team said this finding may explain why it can be difficult to treat AML and other cancers.
They also believe these findings offer new hope for developing better treatments for AML. If researchers can find a way to interfere with the transfer of mitochondria, that could reduce the risk of relapse.
The study may also shed light on other cancer types. Similar mechanisms may be at play in other hematologic malignancies and even solid tumors, according to the investigators.
For now, the team’s next step for this research is to identify the mechanism underlying the transfer of mitochondria in AML. ![]()
Preclinical research suggests that some leukemia cells harvest energy resources from normal cells during chemotherapy, and this helps the leukemia cells survive and thrive after treatment.
Investigators found that acute myeloid leukemia (AML) cells are capable of stealing mitochondria from stromal cells, and these stolen mitochondria give an energy boost to surviving AML cells, which helps fuel the leukemia’s resurgence after chemotherapy.
“There are multiple mechanisms for resistance to chemotherapy, and it will be important to target them all in order to eliminate all leukemic cells,” said Jean-François Peyron, PhD, of the Centre Méditerranéen de Médecine Moléculaire (C3M) in Nice, France.
“Targeting this protective mitochondrial transfer could represent a new strategy to improve the efficacy of the current treatments for acute myeloid leukemia.”
Dr Peyron and his colleagues described their discovery of the mitochondrial transfer in Blood.
The team conducted their experiments using cell cultures and mouse models of AML. They found that nearly all AML cells died when exposed to chemotherapy drugs, but some survived. And these cells issued a “mayday” signal that “tricked” nearby non-cancerous cells into yielding their mitochondria to the AML cells, thus strengthening the leukemia cells.
“Mitochondria produce the energy that is vital for cell functions,” explained study author Emmanuel Griessinger, PhD, also of C3M.
“Through the uptake of mitochondria, chemotherapy-injured acute myeloid leukemia cells recover new energy to survive.”
The AML cells were found to increase their mitochondria mass by an average of 14%. This increase led to a 1.5-fold increase in energy production and significantly better survival rates. That is, the leukemia cells that had a high level of mitochondria were also more resistant to the chemotherapy.
The investigators observed the phenomenon in several types of leukemia cells, most notably leukemia-initiating cells. The team said this finding may explain why it can be difficult to treat AML and other cancers.
They also believe these findings offer new hope for developing better treatments for AML. If researchers can find a way to interfere with the transfer of mitochondria, that could reduce the risk of relapse.
The study may also shed light on other cancer types. Similar mechanisms may be at play in other hematologic malignancies and even solid tumors, according to the investigators.
For now, the team’s next step for this research is to identify the mechanism underlying the transfer of mitochondria in AML. ![]()
Preclinical research suggests that some leukemia cells harvest energy resources from normal cells during chemotherapy, and this helps the leukemia cells survive and thrive after treatment.
Investigators found that acute myeloid leukemia (AML) cells are capable of stealing mitochondria from stromal cells, and these stolen mitochondria give an energy boost to surviving AML cells, which helps fuel the leukemia’s resurgence after chemotherapy.
“There are multiple mechanisms for resistance to chemotherapy, and it will be important to target them all in order to eliminate all leukemic cells,” said Jean-François Peyron, PhD, of the Centre Méditerranéen de Médecine Moléculaire (C3M) in Nice, France.
“Targeting this protective mitochondrial transfer could represent a new strategy to improve the efficacy of the current treatments for acute myeloid leukemia.”
Dr Peyron and his colleagues described their discovery of the mitochondrial transfer in Blood.
The team conducted their experiments using cell cultures and mouse models of AML. They found that nearly all AML cells died when exposed to chemotherapy drugs, but some survived. And these cells issued a “mayday” signal that “tricked” nearby non-cancerous cells into yielding their mitochondria to the AML cells, thus strengthening the leukemia cells.
“Mitochondria produce the energy that is vital for cell functions,” explained study author Emmanuel Griessinger, PhD, also of C3M.
“Through the uptake of mitochondria, chemotherapy-injured acute myeloid leukemia cells recover new energy to survive.”
The AML cells were found to increase their mitochondria mass by an average of 14%. This increase led to a 1.5-fold increase in energy production and significantly better survival rates. That is, the leukemia cells that had a high level of mitochondria were also more resistant to the chemotherapy.
The investigators observed the phenomenon in several types of leukemia cells, most notably leukemia-initiating cells. The team said this finding may explain why it can be difficult to treat AML and other cancers.
They also believe these findings offer new hope for developing better treatments for AML. If researchers can find a way to interfere with the transfer of mitochondria, that could reduce the risk of relapse.
The study may also shed light on other cancer types. Similar mechanisms may be at play in other hematologic malignancies and even solid tumors, according to the investigators.
For now, the team’s next step for this research is to identify the mechanism underlying the transfer of mitochondria in AML. ![]()
Long-term opioid use may not benefit SCD patients

and a normal one
Image by Betty Pace
Results of a small study suggest that long-term opioid treatment may not be the best option for pain management in adults with sickle cell disease (SCD).
The study showed that patients who received opioids long-term often fared worse in measures of pain, fatigue, and curtailed daily activities than patients who were not on long-term opioids.
Researchers reported these findings in the American Journal of Preventive Medicine.
“We need to be careful and skeptical about giving increasing doses of opioids to patients with sickle cell disease who are in chronic pain if it isn’t effective,” said study author C. Patrick Carroll, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Too little is known about the effects of long-term opioid management of chronic pain.”
For this study, Dr Carroll and his colleagues enrolled 83 SCD patients—57 women and 26 men. All were 18 and older, and their average age was 39.
Twenty-nine of the patients had been prescribed daily, long-acting opioids to manage their pain. The remaining 54 patients weren’t on long-term opioids.
The patients filled out daily electronic pain diaries for 90 days. Self-reported levels of pain, physical activity, fatigue, and pain-related daily activity interference were recorded, along with self-reported levels of pain relief and medication satisfaction on a scale of 0 to 100.
Crises, pain, and fatigue
The proportion of days in vaso-occlusive crisis was significantly higher for patients who were on long-term opioid therapy than for those who were not—29% and 11.9%, respectively (P<0.01).
Patients on long-term opioids also reported significantly higher levels of crisis pain—60.6 and 41.0, respectively (P<0.001)—and non-crisis pain—34.5 and 10.3, respectively (P<0.001).
Patients on long-term opioid therapy had higher levels of pain-related activity interference, both on non-crisis days—24.9 and 7.4, respectively (P<0.001)—and crisis days—56.7 and 37.7, respectively (P<0.01).
And patients on long-term opioid therapy had higher levels of fatigue on non-crisis days—49.7 and 27.0, respectively (P<0.001)—and crisis days—66.1 and 53.0, respectively (P<0.05).
Central sensitization
The researchers also performed some standard measures of pain processing on the test subjects, which measured and averaged variables such as how intensely participants experienced unpleasant heat and pressure.
The team was particularly interested in the phenomenon of central sensitization, in which the central nervous system amplifies painful sensations.
Central sensitization may be one way that opioids can increase pain sensitivity, and it also may play a role in how SCD causes chronic pain, Dr Carroll said.
For example, one such measure of central sensitization uses repeated pokes from a mildly painful stimulus in succession. In people who have this hypersensitization, each poke is perceived as more intense than the last because the nervous system becomes progressively more sensitive to the pain.
Combining the data from several measures of central sensitization, the researchers used a scoring system that sets a normal measurement at 0 and rates how abnormal something is by how far the values move away from 0. They calculated a central sensitization index for patients on long-term opioids and those not taking them.
Overall, patients on long-term opioid therapy showed higher levels of central sensitization, with an index of 0.34, than those who were not on opioids, with an index of -0.10.
In patients who were not on long-term opioid therapy, the level of central sensitization correlated with the level of non-crisis pain. However, in patients who were taking long-term opioid therapy and also had higher levels of central sensitization and clinical pain, the correlation essentially vanished.
Dr Carroll said this was surprising and suggests the mechanisms of pain in SCD patients on long-term opioid therapy may be different from patients who don’t take daily opioids for pain.
Dr Carroll cautioned that this work is preliminary and should not lead to the discontinuation of long-term opioid therapy in SCD patients.
“We need to better understand how long-term opioid use affects pain sensitization and determine if certain people are more sensitive to these effects so we can prescribe the best treatment option for each individual patient,” Dr Carroll said. “We also need to learn more about how sickle cell disease may sensitize the nervous system.” ![]()
and a normal one
Image by Betty Pace
Results of a small study suggest that long-term opioid treatment may not be the best option for pain management in adults with sickle cell disease (SCD).
The study showed that patients who received opioids long-term often fared worse in measures of pain, fatigue, and curtailed daily activities than patients who were not on long-term opioids.
Researchers reported these findings in the American Journal of Preventive Medicine.
“We need to be careful and skeptical about giving increasing doses of opioids to patients with sickle cell disease who are in chronic pain if it isn’t effective,” said study author C. Patrick Carroll, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Too little is known about the effects of long-term opioid management of chronic pain.”
For this study, Dr Carroll and his colleagues enrolled 83 SCD patients—57 women and 26 men. All were 18 and older, and their average age was 39.
Twenty-nine of the patients had been prescribed daily, long-acting opioids to manage their pain. The remaining 54 patients weren’t on long-term opioids.
The patients filled out daily electronic pain diaries for 90 days. Self-reported levels of pain, physical activity, fatigue, and pain-related daily activity interference were recorded, along with self-reported levels of pain relief and medication satisfaction on a scale of 0 to 100.
Crises, pain, and fatigue
The proportion of days in vaso-occlusive crisis was significantly higher for patients who were on long-term opioid therapy than for those who were not—29% and 11.9%, respectively (P<0.01).
Patients on long-term opioids also reported significantly higher levels of crisis pain—60.6 and 41.0, respectively (P<0.001)—and non-crisis pain—34.5 and 10.3, respectively (P<0.001).
Patients on long-term opioid therapy had higher levels of pain-related activity interference, both on non-crisis days—24.9 and 7.4, respectively (P<0.001)—and crisis days—56.7 and 37.7, respectively (P<0.01).
And patients on long-term opioid therapy had higher levels of fatigue on non-crisis days—49.7 and 27.0, respectively (P<0.001)—and crisis days—66.1 and 53.0, respectively (P<0.05).
Central sensitization
The researchers also performed some standard measures of pain processing on the test subjects, which measured and averaged variables such as how intensely participants experienced unpleasant heat and pressure.
The team was particularly interested in the phenomenon of central sensitization, in which the central nervous system amplifies painful sensations.
Central sensitization may be one way that opioids can increase pain sensitivity, and it also may play a role in how SCD causes chronic pain, Dr Carroll said.
For example, one such measure of central sensitization uses repeated pokes from a mildly painful stimulus in succession. In people who have this hypersensitization, each poke is perceived as more intense than the last because the nervous system becomes progressively more sensitive to the pain.
Combining the data from several measures of central sensitization, the researchers used a scoring system that sets a normal measurement at 0 and rates how abnormal something is by how far the values move away from 0. They calculated a central sensitization index for patients on long-term opioids and those not taking them.
Overall, patients on long-term opioid therapy showed higher levels of central sensitization, with an index of 0.34, than those who were not on opioids, with an index of -0.10.
In patients who were not on long-term opioid therapy, the level of central sensitization correlated with the level of non-crisis pain. However, in patients who were taking long-term opioid therapy and also had higher levels of central sensitization and clinical pain, the correlation essentially vanished.
Dr Carroll said this was surprising and suggests the mechanisms of pain in SCD patients on long-term opioid therapy may be different from patients who don’t take daily opioids for pain.
Dr Carroll cautioned that this work is preliminary and should not lead to the discontinuation of long-term opioid therapy in SCD patients.
“We need to better understand how long-term opioid use affects pain sensitization and determine if certain people are more sensitive to these effects so we can prescribe the best treatment option for each individual patient,” Dr Carroll said. “We also need to learn more about how sickle cell disease may sensitize the nervous system.” ![]()
and a normal one
Image by Betty Pace
Results of a small study suggest that long-term opioid treatment may not be the best option for pain management in adults with sickle cell disease (SCD).
The study showed that patients who received opioids long-term often fared worse in measures of pain, fatigue, and curtailed daily activities than patients who were not on long-term opioids.
Researchers reported these findings in the American Journal of Preventive Medicine.
“We need to be careful and skeptical about giving increasing doses of opioids to patients with sickle cell disease who are in chronic pain if it isn’t effective,” said study author C. Patrick Carroll, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Too little is known about the effects of long-term opioid management of chronic pain.”
For this study, Dr Carroll and his colleagues enrolled 83 SCD patients—57 women and 26 men. All were 18 and older, and their average age was 39.
Twenty-nine of the patients had been prescribed daily, long-acting opioids to manage their pain. The remaining 54 patients weren’t on long-term opioids.
The patients filled out daily electronic pain diaries for 90 days. Self-reported levels of pain, physical activity, fatigue, and pain-related daily activity interference were recorded, along with self-reported levels of pain relief and medication satisfaction on a scale of 0 to 100.
Crises, pain, and fatigue
The proportion of days in vaso-occlusive crisis was significantly higher for patients who were on long-term opioid therapy than for those who were not—29% and 11.9%, respectively (P<0.01).
Patients on long-term opioids also reported significantly higher levels of crisis pain—60.6 and 41.0, respectively (P<0.001)—and non-crisis pain—34.5 and 10.3, respectively (P<0.001).
Patients on long-term opioid therapy had higher levels of pain-related activity interference, both on non-crisis days—24.9 and 7.4, respectively (P<0.001)—and crisis days—56.7 and 37.7, respectively (P<0.01).
And patients on long-term opioid therapy had higher levels of fatigue on non-crisis days—49.7 and 27.0, respectively (P<0.001)—and crisis days—66.1 and 53.0, respectively (P<0.05).
Central sensitization
The researchers also performed some standard measures of pain processing on the test subjects, which measured and averaged variables such as how intensely participants experienced unpleasant heat and pressure.
The team was particularly interested in the phenomenon of central sensitization, in which the central nervous system amplifies painful sensations.
Central sensitization may be one way that opioids can increase pain sensitivity, and it also may play a role in how SCD causes chronic pain, Dr Carroll said.
For example, one such measure of central sensitization uses repeated pokes from a mildly painful stimulus in succession. In people who have this hypersensitization, each poke is perceived as more intense than the last because the nervous system becomes progressively more sensitive to the pain.
Combining the data from several measures of central sensitization, the researchers used a scoring system that sets a normal measurement at 0 and rates how abnormal something is by how far the values move away from 0. They calculated a central sensitization index for patients on long-term opioids and those not taking them.
Overall, patients on long-term opioid therapy showed higher levels of central sensitization, with an index of 0.34, than those who were not on opioids, with an index of -0.10.
In patients who were not on long-term opioid therapy, the level of central sensitization correlated with the level of non-crisis pain. However, in patients who were taking long-term opioid therapy and also had higher levels of central sensitization and clinical pain, the correlation essentially vanished.
Dr Carroll said this was surprising and suggests the mechanisms of pain in SCD patients on long-term opioid therapy may be different from patients who don’t take daily opioids for pain.
Dr Carroll cautioned that this work is preliminary and should not lead to the discontinuation of long-term opioid therapy in SCD patients.
“We need to better understand how long-term opioid use affects pain sensitization and determine if certain people are more sensitive to these effects so we can prescribe the best treatment option for each individual patient,” Dr Carroll said. “We also need to learn more about how sickle cell disease may sensitize the nervous system.” ![]()
Health Canada shortens deferral for MSM blood donors
Photo by Marja Helander
Health Canada has decided to change its policy regarding blood donations from men who have sex with men (MSM).
The policy has been that MSMs can only donate blood if they have abstained from sexual contact with another man for a period of 5 years. As of August 15, that period will be shortened to 1 year.
The change is a result of proposals from Canada’s blood operators, Canadian Blood Services and Héma-Québec.
These proposals included scientific data indicating that decreasing the deferral period for MSMs would not affect the safety of the blood supply.
The change brings Canada in line with several other countries that have implemented a 1-year deferral period for MSM blood donors, including the US, Australia, New Zealand, England, Scotland, and France.
Health Canada said the country’s blood system continues to have rigorous scientific and screening processes in place to protect the safety of Canadians.
Canadian Blood Services and Héma-Québec will continue to screen all donations for HIV and other infections. As an extra precaution, Health Canada and the blood operators will monitor donations from new donors to see if there is an increase in HIV or other infection rates.
According to Health Canada, there has not been a single HIV infection from blood transfusion in Canada in 25 years.
However, the frequency of HIV infection remains higher among MSMs (10%) than among heterosexuals or lesbians (less than 1% for both). And it was because of this that Canada implemented the 5-year deferral period for MSM blood donors. Prior to the implementation of that policy, MSMs were completely barred from donating blood in Canada.
“It has been demonstrated that implementing a 5-year temporary exclusion in 2013 had no impact on the safety of the transfusion system,” said Marc Germain, vice-president of medical affairs at Héma-Québec.
“As a result of recent data concerning transfusion safety, the exclusion policy applied to men who have had sex with another man could be reviewed. [The new 1-year deferral period] is scientifically justified and will not endanger the very high level of safety of blood products.”
“This is an exciting, incremental step forward in updating our blood donation criteria based on the latest scientific evidence,” said Graham Sher, chief executive officer of Canadian Blood Services.
“Canadian Blood Services is dedicated to being as minimally restrictive as possible while also maintaining the safety of the blood supply.”
Canadian Blood Services said it is exploring the possibility of moving toward behavior-based screening of blood donors. The organization is working with researchers, the LGBTQ community, patient groups, and other stakeholders to determine how best to gather the scientific evidence required to determine future changes to donor eligibility criteria. ![]()
Photo by Marja Helander
Health Canada has decided to change its policy regarding blood donations from men who have sex with men (MSM).
The policy has been that MSMs can only donate blood if they have abstained from sexual contact with another man for a period of 5 years. As of August 15, that period will be shortened to 1 year.
The change is a result of proposals from Canada’s blood operators, Canadian Blood Services and Héma-Québec.
These proposals included scientific data indicating that decreasing the deferral period for MSMs would not affect the safety of the blood supply.
The change brings Canada in line with several other countries that have implemented a 1-year deferral period for MSM blood donors, including the US, Australia, New Zealand, England, Scotland, and France.
Health Canada said the country’s blood system continues to have rigorous scientific and screening processes in place to protect the safety of Canadians.
Canadian Blood Services and Héma-Québec will continue to screen all donations for HIV and other infections. As an extra precaution, Health Canada and the blood operators will monitor donations from new donors to see if there is an increase in HIV or other infection rates.
According to Health Canada, there has not been a single HIV infection from blood transfusion in Canada in 25 years.
However, the frequency of HIV infection remains higher among MSMs (10%) than among heterosexuals or lesbians (less than 1% for both). And it was because of this that Canada implemented the 5-year deferral period for MSM blood donors. Prior to the implementation of that policy, MSMs were completely barred from donating blood in Canada.
“It has been demonstrated that implementing a 5-year temporary exclusion in 2013 had no impact on the safety of the transfusion system,” said Marc Germain, vice-president of medical affairs at Héma-Québec.
“As a result of recent data concerning transfusion safety, the exclusion policy applied to men who have had sex with another man could be reviewed. [The new 1-year deferral period] is scientifically justified and will not endanger the very high level of safety of blood products.”
“This is an exciting, incremental step forward in updating our blood donation criteria based on the latest scientific evidence,” said Graham Sher, chief executive officer of Canadian Blood Services.
“Canadian Blood Services is dedicated to being as minimally restrictive as possible while also maintaining the safety of the blood supply.”
Canadian Blood Services said it is exploring the possibility of moving toward behavior-based screening of blood donors. The organization is working with researchers, the LGBTQ community, patient groups, and other stakeholders to determine how best to gather the scientific evidence required to determine future changes to donor eligibility criteria. ![]()
Photo by Marja Helander
Health Canada has decided to change its policy regarding blood donations from men who have sex with men (MSM).
The policy has been that MSMs can only donate blood if they have abstained from sexual contact with another man for a period of 5 years. As of August 15, that period will be shortened to 1 year.
The change is a result of proposals from Canada’s blood operators, Canadian Blood Services and Héma-Québec.
These proposals included scientific data indicating that decreasing the deferral period for MSMs would not affect the safety of the blood supply.
The change brings Canada in line with several other countries that have implemented a 1-year deferral period for MSM blood donors, including the US, Australia, New Zealand, England, Scotland, and France.
Health Canada said the country’s blood system continues to have rigorous scientific and screening processes in place to protect the safety of Canadians.
Canadian Blood Services and Héma-Québec will continue to screen all donations for HIV and other infections. As an extra precaution, Health Canada and the blood operators will monitor donations from new donors to see if there is an increase in HIV or other infection rates.
According to Health Canada, there has not been a single HIV infection from blood transfusion in Canada in 25 years.
However, the frequency of HIV infection remains higher among MSMs (10%) than among heterosexuals or lesbians (less than 1% for both). And it was because of this that Canada implemented the 5-year deferral period for MSM blood donors. Prior to the implementation of that policy, MSMs were completely barred from donating blood in Canada.
“It has been demonstrated that implementing a 5-year temporary exclusion in 2013 had no impact on the safety of the transfusion system,” said Marc Germain, vice-president of medical affairs at Héma-Québec.
“As a result of recent data concerning transfusion safety, the exclusion policy applied to men who have had sex with another man could be reviewed. [The new 1-year deferral period] is scientifically justified and will not endanger the very high level of safety of blood products.”
“This is an exciting, incremental step forward in updating our blood donation criteria based on the latest scientific evidence,” said Graham Sher, chief executive officer of Canadian Blood Services.
“Canadian Blood Services is dedicated to being as minimally restrictive as possible while also maintaining the safety of the blood supply.”
Canadian Blood Services said it is exploring the possibility of moving toward behavior-based screening of blood donors. The organization is working with researchers, the LGBTQ community, patient groups, and other stakeholders to determine how best to gather the scientific evidence required to determine future changes to donor eligibility criteria. ![]()
HIV research update: Early June 2016
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
Interventions that seek to promptly house homeless individuals might assist in maximizing the clinical and public health benefits of antiretroviral therapy among people living with HIV/AIDS, according to a study in AIDS Care.
MRSA colonization among HIV-infected youth is more closely related to living in a low-income or slum community than to HIV-related clinical factors, according to a study in the Pediatric Infectious Disease Journal.
Marijuana use was not associated with progression to significant liver fibrosis, in a large cohort study of HIV/hepatitis C virus (HCV) co-infected women. Alcohol use may better predict fibrosis progression in HIV/HCV co-infected women, the authors said.
More than 60% of deaths occurring in a rural South African community between 1992 and 2013 could be attributed directly or indirectly to HIV, according to a recent study. However, the authors noted that there has been an increasing level of non-HIV mortality, which has important implications for local health care provision.
A study in AIDS Care found that providers value same-day, electronic, patient-reported measures for use in clinical HIV care, with the condition that patient-reported outcomes are 1) tailored to be the most clinically relevant to their population; 2) well integrated into clinic flow; and 3) easy to interpret, highlighting chief patient concerns and changes over time.
Investigators found that nasal and salivary samples can be collected from HIV-infected patients in a standardized manner over repeated visits in both low and high resource settings, and these methods may be used in support of future HIV vaccine clinical trials.
A study in the Journal of Infectious Diseases found no intrinsic effect of viral subtype on the efficacy of tenofovir-containing regimens for treating HIV-1 subtype C infections. There were no differences between subtypes C and non-B/C in either univariate or multivariate analysis.
New research provides preliminary evidence that heavy drinking may increase a key inflammatory marker in HIV-infected individuals with suppressed infection.
The phase III Women AntiretroViral Efficacy and Safety study (WAVES) showed that clinical trials of antiretroviral regimens in global and diverse populations of treatment-naive women are possible, investigators concluded. They said their findings support guidelines recommending integrase inhibitor–based regimens in first-line antiretroviral therapy.
A hybrid mobile strategy for HIV testing of adults was leveraged successfully to reach adolescents for HIV treatment and prevention, according to a study in Kenya and Uganda.
With viral suppression induced by combination antiretroviral therapy, women have less reduction in key markers of inflammation and immune activation, compared with men, report the authors of a study in JAIDS.
The transfer of HIV-1-p17-specific T-cell receptors (TCRs) into T-cells is functional both in HIV-1–infected patients and in healthy blood donors, according to a study in the journal AIDS. The authors say TCR-transfer is a promising method to boost the immune system against HIV-1.
HIV-1 subtype C (HIV-1C) exhibits lower in vivo fitness, compared with HIV-1B, which allows successful treatment despite high baseline viral loads, a new study demonstrated. The lower fitness – and potentially lower virulence – together with high viral loads may underlie the heightened transmission potential of HIV-1C and its growing global spread.
Thailand has achieved World Health Organization targets for elimination of mother-to-child-transmission of HIV and can serve as a model for other countries, according to a report in MMWR.
Regular CD4 testing may be unnecessary for virally suppressed children aged 5-15 years with CD4 greater than or equal to 500 cells/mm3, according to a study published in the Journal of the Pediatric Infectious Diseases Society.
Investigators at Case Western Reserve University, Cleveland found that 24 weeks of 10 mg daily rosuvastatin decreases plasma coenzyme Q10 concentration and increases CoQ10/LDL ratio in HIV-infected patients on antiretroviral therapy.
Researchers from the University of California, San Francisco, and Yale University, New Haven, Conn., found that half of people newly infected with HIV experience neurologic issues that are generally not severe and usually resolve after starting antiretroviral therapy.
A study of HIV-infected individuals in metropolitan Washington identified high prevalence of transmitted drug resistance, regardless of gender. The authors said active surveillance for transmitted drug resistance is needed to guide antiretroviral usage and analyses of risk group contributions to HIV transmission and resistance.
Single-nucleotide polymorphisms in genes encoding the Toll-like receptor 4 (TLR4) and CD14 are independently associated with long-term CD4+ T-cell recovery in HIV-infected individuals after antiretroviral therapy, according to a study in the journal AIDS.
In HIV/HCV co-infected individuals, the crude incidence of hepatocellular carcinoma increased from 2001 to 2014, while other liver events declined, according to a recent study.
On Twitter @richpizzi
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
Interventions that seek to promptly house homeless individuals might assist in maximizing the clinical and public health benefits of antiretroviral therapy among people living with HIV/AIDS, according to a study in AIDS Care.
MRSA colonization among HIV-infected youth is more closely related to living in a low-income or slum community than to HIV-related clinical factors, according to a study in the Pediatric Infectious Disease Journal.
Marijuana use was not associated with progression to significant liver fibrosis, in a large cohort study of HIV/hepatitis C virus (HCV) co-infected women. Alcohol use may better predict fibrosis progression in HIV/HCV co-infected women, the authors said.
More than 60% of deaths occurring in a rural South African community between 1992 and 2013 could be attributed directly or indirectly to HIV, according to a recent study. However, the authors noted that there has been an increasing level of non-HIV mortality, which has important implications for local health care provision.
A study in AIDS Care found that providers value same-day, electronic, patient-reported measures for use in clinical HIV care, with the condition that patient-reported outcomes are 1) tailored to be the most clinically relevant to their population; 2) well integrated into clinic flow; and 3) easy to interpret, highlighting chief patient concerns and changes over time.
Investigators found that nasal and salivary samples can be collected from HIV-infected patients in a standardized manner over repeated visits in both low and high resource settings, and these methods may be used in support of future HIV vaccine clinical trials.
A study in the Journal of Infectious Diseases found no intrinsic effect of viral subtype on the efficacy of tenofovir-containing regimens for treating HIV-1 subtype C infections. There were no differences between subtypes C and non-B/C in either univariate or multivariate analysis.
New research provides preliminary evidence that heavy drinking may increase a key inflammatory marker in HIV-infected individuals with suppressed infection.
The phase III Women AntiretroViral Efficacy and Safety study (WAVES) showed that clinical trials of antiretroviral regimens in global and diverse populations of treatment-naive women are possible, investigators concluded. They said their findings support guidelines recommending integrase inhibitor–based regimens in first-line antiretroviral therapy.
A hybrid mobile strategy for HIV testing of adults was leveraged successfully to reach adolescents for HIV treatment and prevention, according to a study in Kenya and Uganda.
With viral suppression induced by combination antiretroviral therapy, women have less reduction in key markers of inflammation and immune activation, compared with men, report the authors of a study in JAIDS.
The transfer of HIV-1-p17-specific T-cell receptors (TCRs) into T-cells is functional both in HIV-1–infected patients and in healthy blood donors, according to a study in the journal AIDS. The authors say TCR-transfer is a promising method to boost the immune system against HIV-1.
HIV-1 subtype C (HIV-1C) exhibits lower in vivo fitness, compared with HIV-1B, which allows successful treatment despite high baseline viral loads, a new study demonstrated. The lower fitness – and potentially lower virulence – together with high viral loads may underlie the heightened transmission potential of HIV-1C and its growing global spread.
Thailand has achieved World Health Organization targets for elimination of mother-to-child-transmission of HIV and can serve as a model for other countries, according to a report in MMWR.
Regular CD4 testing may be unnecessary for virally suppressed children aged 5-15 years with CD4 greater than or equal to 500 cells/mm3, according to a study published in the Journal of the Pediatric Infectious Diseases Society.
Investigators at Case Western Reserve University, Cleveland found that 24 weeks of 10 mg daily rosuvastatin decreases plasma coenzyme Q10 concentration and increases CoQ10/LDL ratio in HIV-infected patients on antiretroviral therapy.
Researchers from the University of California, San Francisco, and Yale University, New Haven, Conn., found that half of people newly infected with HIV experience neurologic issues that are generally not severe and usually resolve after starting antiretroviral therapy.
A study of HIV-infected individuals in metropolitan Washington identified high prevalence of transmitted drug resistance, regardless of gender. The authors said active surveillance for transmitted drug resistance is needed to guide antiretroviral usage and analyses of risk group contributions to HIV transmission and resistance.
Single-nucleotide polymorphisms in genes encoding the Toll-like receptor 4 (TLR4) and CD14 are independently associated with long-term CD4+ T-cell recovery in HIV-infected individuals after antiretroviral therapy, according to a study in the journal AIDS.
In HIV/HCV co-infected individuals, the crude incidence of hepatocellular carcinoma increased from 2001 to 2014, while other liver events declined, according to a recent study.
On Twitter @richpizzi
A great volume of HIV and AIDS research enters the medical literature every month. It’s difficult to monitor everything, so here’s a quick look at some notable news items and journal articles published over the past few weeks.
Interventions that seek to promptly house homeless individuals might assist in maximizing the clinical and public health benefits of antiretroviral therapy among people living with HIV/AIDS, according to a study in AIDS Care.
MRSA colonization among HIV-infected youth is more closely related to living in a low-income or slum community than to HIV-related clinical factors, according to a study in the Pediatric Infectious Disease Journal.
Marijuana use was not associated with progression to significant liver fibrosis, in a large cohort study of HIV/hepatitis C virus (HCV) co-infected women. Alcohol use may better predict fibrosis progression in HIV/HCV co-infected women, the authors said.
More than 60% of deaths occurring in a rural South African community between 1992 and 2013 could be attributed directly or indirectly to HIV, according to a recent study. However, the authors noted that there has been an increasing level of non-HIV mortality, which has important implications for local health care provision.
A study in AIDS Care found that providers value same-day, electronic, patient-reported measures for use in clinical HIV care, with the condition that patient-reported outcomes are 1) tailored to be the most clinically relevant to their population; 2) well integrated into clinic flow; and 3) easy to interpret, highlighting chief patient concerns and changes over time.
Investigators found that nasal and salivary samples can be collected from HIV-infected patients in a standardized manner over repeated visits in both low and high resource settings, and these methods may be used in support of future HIV vaccine clinical trials.
A study in the Journal of Infectious Diseases found no intrinsic effect of viral subtype on the efficacy of tenofovir-containing regimens for treating HIV-1 subtype C infections. There were no differences between subtypes C and non-B/C in either univariate or multivariate analysis.
New research provides preliminary evidence that heavy drinking may increase a key inflammatory marker in HIV-infected individuals with suppressed infection.
The phase III Women AntiretroViral Efficacy and Safety study (WAVES) showed that clinical trials of antiretroviral regimens in global and diverse populations of treatment-naive women are possible, investigators concluded. They said their findings support guidelines recommending integrase inhibitor–based regimens in first-line antiretroviral therapy.
A hybrid mobile strategy for HIV testing of adults was leveraged successfully to reach adolescents for HIV treatment and prevention, according to a study in Kenya and Uganda.
With viral suppression induced by combination antiretroviral therapy, women have less reduction in key markers of inflammation and immune activation, compared with men, report the authors of a study in JAIDS.
The transfer of HIV-1-p17-specific T-cell receptors (TCRs) into T-cells is functional both in HIV-1–infected patients and in healthy blood donors, according to a study in the journal AIDS. The authors say TCR-transfer is a promising method to boost the immune system against HIV-1.
HIV-1 subtype C (HIV-1C) exhibits lower in vivo fitness, compared with HIV-1B, which allows successful treatment despite high baseline viral loads, a new study demonstrated. The lower fitness – and potentially lower virulence – together with high viral loads may underlie the heightened transmission potential of HIV-1C and its growing global spread.
Thailand has achieved World Health Organization targets for elimination of mother-to-child-transmission of HIV and can serve as a model for other countries, according to a report in MMWR.
Regular CD4 testing may be unnecessary for virally suppressed children aged 5-15 years with CD4 greater than or equal to 500 cells/mm3, according to a study published in the Journal of the Pediatric Infectious Diseases Society.
Investigators at Case Western Reserve University, Cleveland found that 24 weeks of 10 mg daily rosuvastatin decreases plasma coenzyme Q10 concentration and increases CoQ10/LDL ratio in HIV-infected patients on antiretroviral therapy.
Researchers from the University of California, San Francisco, and Yale University, New Haven, Conn., found that half of people newly infected with HIV experience neurologic issues that are generally not severe and usually resolve after starting antiretroviral therapy.
A study of HIV-infected individuals in metropolitan Washington identified high prevalence of transmitted drug resistance, regardless of gender. The authors said active surveillance for transmitted drug resistance is needed to guide antiretroviral usage and analyses of risk group contributions to HIV transmission and resistance.
Single-nucleotide polymorphisms in genes encoding the Toll-like receptor 4 (TLR4) and CD14 are independently associated with long-term CD4+ T-cell recovery in HIV-infected individuals after antiretroviral therapy, according to a study in the journal AIDS.
In HIV/HCV co-infected individuals, the crude incidence of hepatocellular carcinoma increased from 2001 to 2014, while other liver events declined, according to a recent study.
On Twitter @richpizzi
Study identifies changing trends in PBC incidence, mortality
A new study has identified a higher mortality rate among males with primary biliary cholangitis (PBC) and a lower sex ratio in disease prevalence than previously thought, according to findings published online in the journal Scientific Reports.
Investigators analyzed inpatient data from 2000 to 2009 for PBC patients in Denmark and in the Italian province of Lombardia. In the Lombardia cohort, 2,970 PBC patients were identified, with a female to male ratio of 2.3:1. In the Denmark population, 722 cases were identified, with a female to male ratio of 4.2:1, reported Dr. Ana Lleo from the Humanitas Clinical and Research Center in Rozzano, Italy, and her coauthors.

Among the Lombardia patients, survival at 1, 5, and 10 years was significantly higher for females (89%, 95% confidence interval, 88%-91%; 77%, 95% CI, 75%-78%; and 67%, 95% CI, 65%-70%, respectively) than for males (78%, 95% CI, 75%-80%; 55%, 95% CI, 52%-59%; and 47%, 95% CI, 43%-51%, respectively). Findings were similar in the Denmark cohort, with female patients having higher survival rates (survival at 1, 5, and 10 years of 90%, 87%-92%; 73%, 95% CI, 69%-77%; and 60%, 95% CI, 53%-67%, respectively) than males (72%, 95% CI, 63%-79%; 42%, 95% CI, 32%-51%; and 27%, 95% CI, 14%-43%, respectively), the authors reported.
The study findings question long-held beliefs on PBC, the authors said. Hepatologists should be aware that “male PBC patients have higher mortality than [do] their female counterparts, such that close clinical follow-up and checking adherence to therapy are strongly recommended,” Dr. Lleu and colleagues concluded.
Read the full article in Scientific Reports: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4872151.
A new study has identified a higher mortality rate among males with primary biliary cholangitis (PBC) and a lower sex ratio in disease prevalence than previously thought, according to findings published online in the journal Scientific Reports.
Investigators analyzed inpatient data from 2000 to 2009 for PBC patients in Denmark and in the Italian province of Lombardia. In the Lombardia cohort, 2,970 PBC patients were identified, with a female to male ratio of 2.3:1. In the Denmark population, 722 cases were identified, with a female to male ratio of 4.2:1, reported Dr. Ana Lleo from the Humanitas Clinical and Research Center in Rozzano, Italy, and her coauthors.
Among the Lombardia patients, survival at 1, 5, and 10 years was significantly higher for females (89%, 95% confidence interval, 88%-91%; 77%, 95% CI, 75%-78%; and 67%, 95% CI, 65%-70%, respectively) than for males (78%, 95% CI, 75%-80%; 55%, 95% CI, 52%-59%; and 47%, 95% CI, 43%-51%, respectively). Findings were similar in the Denmark cohort, with female patients having higher survival rates (survival at 1, 5, and 10 years of 90%, 87%-92%; 73%, 95% CI, 69%-77%; and 60%, 95% CI, 53%-67%, respectively) than males (72%, 95% CI, 63%-79%; 42%, 95% CI, 32%-51%; and 27%, 95% CI, 14%-43%, respectively), the authors reported.
The study findings question long-held beliefs on PBC, the authors said. Hepatologists should be aware that “male PBC patients have higher mortality than [do] their female counterparts, such that close clinical follow-up and checking adherence to therapy are strongly recommended,” Dr. Lleu and colleagues concluded.
Read the full article in Scientific Reports: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4872151.
A new study has identified a higher mortality rate among males with primary biliary cholangitis (PBC) and a lower sex ratio in disease prevalence than previously thought, according to findings published online in the journal Scientific Reports.
Investigators analyzed inpatient data from 2000 to 2009 for PBC patients in Denmark and in the Italian province of Lombardia. In the Lombardia cohort, 2,970 PBC patients were identified, with a female to male ratio of 2.3:1. In the Denmark population, 722 cases were identified, with a female to male ratio of 4.2:1, reported Dr. Ana Lleo from the Humanitas Clinical and Research Center in Rozzano, Italy, and her coauthors.
Among the Lombardia patients, survival at 1, 5, and 10 years was significantly higher for females (89%, 95% confidence interval, 88%-91%; 77%, 95% CI, 75%-78%; and 67%, 95% CI, 65%-70%, respectively) than for males (78%, 95% CI, 75%-80%; 55%, 95% CI, 52%-59%; and 47%, 95% CI, 43%-51%, respectively). Findings were similar in the Denmark cohort, with female patients having higher survival rates (survival at 1, 5, and 10 years of 90%, 87%-92%; 73%, 95% CI, 69%-77%; and 60%, 95% CI, 53%-67%, respectively) than males (72%, 95% CI, 63%-79%; 42%, 95% CI, 32%-51%; and 27%, 95% CI, 14%-43%, respectively), the authors reported.
The study findings question long-held beliefs on PBC, the authors said. Hepatologists should be aware that “male PBC patients have higher mortality than [do] their female counterparts, such that close clinical follow-up and checking adherence to therapy are strongly recommended,” Dr. Lleu and colleagues concluded.
Read the full article in Scientific Reports: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4872151.
FROM SCIENTIFIC REPORTS

Low paravalvular leak shown in real-world registry of Sapien 3 recipients
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.

The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.
The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
PARIS – The initial report from a large, prospective registry documenting outcomes in recipients of the Edwards Sapien 3 transcatheter aortic valve in real-world clinical practice confirm that the excellent results seen earlier in the rarified, randomized clinical trial setting are routinely reproducible in everyday practice, Dr. Olaf Wendler said at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
“The big thing with the Sapien 3 is the reduction in paravalvular leakage. Our rate of moderate or severe paravalvular leakage in the registry at 30 days is only 3.1%, with 73.6% of patients having no or only trace paravalvular leakage,” reported Dr. Wendler, professor of cardiothoracic surgery at King’s College Hospital, London.
The mean gradient improved from 43.8 mm Hg at baseline to 11.7 mm Hg at discharge, while the effective orifice area climbed from 0.72 to 1.67 cm2.
He presented 30-day outcomes from the SOURCE 3 registry for 1,947 recipients of the Edwards Sapien 3 valve during transcatheter aortic valve replacement at 80 European centers in 10 countries. Their average age was 81.6 years. Updates will continue during a planned 5 years of prospective follow-up.
“This will be a very rich dataset for subgroup analyses. We now have a number of procedural variables we can analyze over time. We will have good data to get to better outcomes in terms of how best to perform the procedure if you do it with a Sapien valve,” he explained.
Among the key findings: The lower profile of the Sapien 3 delivery system, compared with earlier iterations of the Sapien valve, enabled 87% of patients in the registry to undergo TAVR via transfemoral access. And, in these 1,695 patients, whose mean logistic EuroSCORE was 17.8, the 30-day rates of all-cause mortality and disabling stroke were just 1.9% and 0.5%, respectively.
“I think there are not many series that have shown better results than these,” Dr. Wendler commented.
The non–transfemoral-access group is a very different, higher surgical risk cohort with lots more comorbid conditions. Their mean logistic EuroSCORE was 21.8. Yet in this group, the 30-day all-cause mortality and disabling stroke rates were still only 4% and 0.8%.
The transfemoral access group had markedly lower rates of life-threatening bleeding (4%), new-onset atrial fibrillation (4.8%), extended ventilation (3.5%), stage 2-3 acute kidney injury (0.8%), plus a 2-day shorter mean hospital length of stay.
Sixty percent of transfemoral access patients had their TAVR done under conscious sedation. These 1,018 patients constitute the largest dataset ever treated using conscious sedation with one valve system. The conscious sedation group had significantly lower baseline rates of carotid artery disease, prior coronary artery bypass graft surgery, and heart failure than did patients who received general anesthesia. At 30 days post-TAVR, the conscious sedation group had significantly lower rates of extended ventilation and postdilatation, and they received less contrast volume than did the general anesthesia group. However, they had a significantly higher incidence of stroke: 1.7%, compared with 0.6% for the general anesthesia group.
“Why that is the case we don’t know at the moment. We need to look into this more in the future,” the surgeon said.
A particularly impressive finding was how well Sapien 3 valve recipients with a low left ventricular ejection fraction have done. For example, 30-day all-cause mortality in the 100 patients with a baseline LVEF below 30% was 3%, not significantly different from the 1.8% rate in the 1,198 subjects with an LVEF greater than 50% or the 2.6% in patients with an LVEF of 30%-50%.
“Three percent all-cause mortality with an ejection fraction below 30% is just remarkable from my point of view,” Dr. Wendler said.
The 778 who didn’t undergo balloon aortic valvuloplasty prior to Sapien 3 implantation did not fare any worse than did those who did in terms of 30-day all-cause mortality or all strokes, but they did have significantly higher rates of life-threatening bleeding and major vascular complications.
The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.
AT EUROPCR 2016
Key clinical point: Only 3.1% of patients had moderate or severe paravalvular leak at 30 days after TAVR with the Sapien 3.
Major finding: The 30-day, all-cause mortality and disabling stroke rates were 1.9% and 0.5% in Sapien 3 valve recipients whose transcatheter aortic valve replacement was done by the transfemoral route.
Data source: A prospective multicenter European registry which includes 1,947 patients who underwent transcatheter aortic valve replacement with the Edwards Sapien 3 valve in real-world commercial settings.
Disclosures: The SOURCE 3 registry is sponsored by Edwards Lifesciences. Dr. Wendler serves as a consultant to the company.

VIDEO: Depression worsens newly diagnosed juvenile idiopathic arthritis
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.

|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.
|
|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
Prior study results showed that about a third of adult patients with rheumatoid arthritis are depressed. There seems to be a vulnerability to depression among patients diagnosed with inflammatory arthritis. It makes sense that chronic arthritis can cause depression as it is a painful, debilitating, and long-term disease that is often nonremitting.
|
|
| Mitchel L. Zoler/Frontline Medical News Susan Barlett, Ph.D. |
Good evidence also suggests that people who are depressed are more vulnerable to develop rheumatoid arthritis or other autoimmune diseases.
The important new findings reported by Dr. Ioannou and his associates underscore the importance of providing psychological support to adolescents newly diagnosed with juvenile idiopathic arthritis. Among its many effects, depression is one of the few robust predictors of nonadherence to medical treatments by patients. Patients who are depressed are less likely to take their medications as prescribed. Depressed patients are also more likely to smoke because tobacco smoking can produce some depression relief. But smoking also contributes to the development and worsening of rheumatoid arthritis and likely other forms of inflammatory arthritis.
Susan Bartlett, Ph.D., is a psychologist and clinical epidemiologist at McGill University, Montreal, who specializes in chronic diseases including arthritis. She had no disclosures. She made these comments during a press conference.
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
LONDON – Depression is relatively common among teenagers newly diagnosed with juvenile idiopathic arthritis, and adolescents with both disorders appeared to have a less complete response to their treatment in a study of 102 patients.
Juvenile idiopathic arthritis (JIA) that first manifests when a patient is a teenager comes at a “vulnerable time” that can drive the development and worsening of depression, and depression can potentially exacerbate inflammation and also interfere with treatment compliance, Dr. John Ioannou said at the European Congress of Rheumatology,
Depression and JIA can produce a “vicious cycle in which depression exacerbates the disease and the disease exacerbates depression,” explained Dr. Ioannou, a rheumatologist at University College Hospital in London.
Although no study results have yet identified an effective intervention for depression identified in teenagers with newly diagnosed JIA, the immediate message from these new findings is that clinicians must assess the psychological health of adolescents with JIA both when they are first diagnosed as well as at subsequent visits, and if depression is found it requires some sort of intervention, Dr. Ioannou said in an interview.
He and his associates studied 102 patients from the United Kingdom, who were newly diagnosed with JIA and were 11-16 years old at baseline and enrolled in the Childhood Arthritis Prospective Study (CAPS), a nationwide cohort of patients with childhood-onset arthritis of various types. The average age of the group they studied was just under 13 years old, 57% were girls, 52% had persistent oligoarticular arthritis, 30% had polyarticular arthritis, and 18% had enthesitis-related arthritis. All patients underwent assessment at baseline for depression using the Mood and Feelings Questionnaire and 15 (15%) had a score that flagged them as having “probable” depression.
This depression prevalence is about three- to fourfold higher than for an otherwise healthy group of similarly aged adolescents, Dr. Ioannou said.
At baseline, the subgroup of teens with depression had a significantly higher number of inflamed joints, restricted joints, and also more overall pain and disability as measured on the Childhood Health Assessment Questionnaire.
The 102 teens with JIA underwent follow-up assessment 1-3 years later, after they had received ongoing treatment for their JIA. At follow-up, standard JIA treatment had largely resulted in resolution of joint inflammation and movement restriction among all patients, including those with depression at baseline. However the adolescents who had both JIA and depression at entry continued to have significantly more pain and disability at follow-up than did the nondepressed JIA patients, suggesting a link between depression and refractory pain and disability in JIA patients, the researchers reported.
“We need to ensure that psychological assessments and support are available to all young people diagnosed with JIA, and that this is fully integrated into routine care” for newly diagnosed JIA patients, Dr. Ioannou said. He had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
AT THE EULAR 2016 CONGRESS
Key clinical point: Pain and disability often persist despite effective antirheumatic treatment in depressed adolescents with juvenile idiopathic arthritis.
Major finding: Disability and pain levels remained significantly elevated among JIA teens with depression, compared with JIA teens without baseline depression.
Data source: The 102 adolescents enrolled in the Childhood Arthritis Prospective Study with juvenile idiopathic arthritis, including 15 patients with depression at baseline.
Disclosures: Dr. Ioannou had no disclosures.

Artificial pancreas can improve inpatient glycemic control in type 2 diabetes
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.

Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.
Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
NEW ORLEANS – Having people with type 2 diabetes mellitus use an artificial pancreas during hospitalization has the potential to improve control of their glycemia when compared with conventional insulin therapy, based on the results of a small study of inpatients in the United Kingdom.
“This is the first study to show that automated subcutaneous closed-loop insulin delivery without meal-time insulin is feasible and safe in patients with insulin-treated type 2 diabetes in the general wards,” Dr. Hood Thabit of the University of Cambridge (England) reported at the ADA annual scientific sessions. “Closed-loop [delivery] increased time in target, with reduced glucose variability and reduced time spent hyperglycemic without actually increasing time spent hypoglycemic,” he said.
The study involved 40 general ward inpatients evenly assigned to the closed-loop system and conventional insulin therapy for 72 hours.
Dr. Thabit said hyperglycemia in hospital patients is a common problem that’s poorly managed. “There’s an unmet need for an effective and safe glucose control, specifically in the underserved and understudied population of type 2 diabetes in the general wards,” he said. The use of the closed-loop system in inpatients with type 2 diabetes “remains untested until now,” Dr. Thabit added.
The 20 patients randomized to the closed-loop system spent an average of 61% of the whole study period within the sensor glucose target vs. 38% of those on conventional insulin therapy. The closed-loop patients also used comparable insulin daily on average: 62.6 U (±36.3 U) vs. 66.0 U (±39.6 U), Dr. Thabit said.
He noted that those on the closed-loop system did not have to announce meals to the control algorithm, or give any meal-time insulin – “we didn’t want to trouble our nurses with this, due to the increasing workload that health care professionals in the hospital currently face,” he said – and showed “significantly improved” nighttime control of glucose while “simultaneously reducing the risk of nocturnal hypoglycemia”. “The closed loop may potentially be an effective and safe tool to manage hospital inpatient hyperglycemia in this particularly underserved population of patients whilst easing the burden of health care professionals in hospital,” he said.
The cost is not insignificant. The pump and sensor devices together with related consumables can cost up to £6,000 (about $8,600), but he did note the artificial pancreas device itself is reusable. The cost of the automated closed-loop glucose control system can also potentially be offset by the reduced time of health care professionals spent managing inpatient hyperglycemia safely. The study investigators are in the process of planning a larger trial, Dr. Thabit said.
Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Key clinical point:Hospitalized patients in the general ward with type 2 diabetes mellitus can maintain better glycemic control by using an artificial pancreas than by taking conventional therapy.
Major finding: Patients on the closed-loop system spent an average of 61% of the study period within the sensor glucose target vs. 38% for those on conventional insulin therapy.
Data source: A single-center study of 40 patients randomized to wear the artificial pancreas or take conventional therapy.
Disclosures: Dr. Thabit had no financial disclosures. Some coauthors disclosed relationships with Novo Nordisk; Medtronic MiniMed; Becton, Dickinson and Co.; Abbott Diabetes Care; Roche Pharmaceuticals; Cell Novo; Animas; Eli Lilly; B. Braun Melsungen; Sanofi-Aventis Deutschland; and Profil Institute for Clinical Research.

AAP, NASPAG issue joint guidance on menstruation management in teens with disabilities
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”

A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
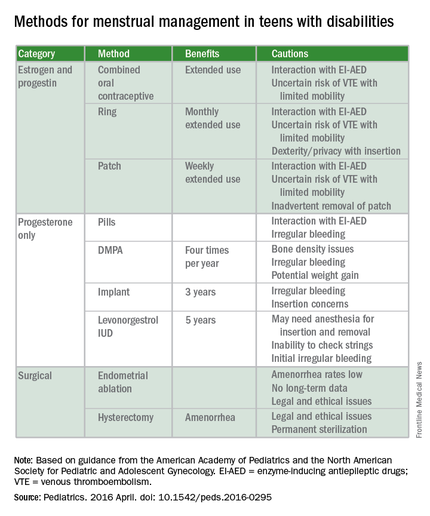
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.

|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.
|
|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
Although this guideline focuses on menstrual management and the guidance for you to help teens with disabilities through the pubertal transition, it’s very important to put this topic also into the context of sexuality. I think you have a great opportunity to do this because, often, you already have developed long-term relationships with these teenagers and their families, so the trust is already there. You should be the one to ensure all patients have appropriate sex education and help families with this.
|
|
Dr. Elisabeth Quint |
For some of these teens who are cognitively impaired, the initial conversations about sex may focus more on safety and abuse prevention. For example, which parts of their body should not be touched by other people. You can help the families really be the educators. Parents can be the ones to teach their kids how to protect themselves by rehearsing the answers to questions like, “What do you do if someone touches you? Who do you tell? Where do you go? What if it happens at school?” As part of the safety aspects, you also can help families assess whether the patient will be able to have a consensual sexual relationship. It’s the teens who have mild cognitive impairment that I worry about most, because often they are friendly and open to people, and can be taken advantage of. You just want to make sure they have the right information at their appropriate level.
Adolescents with physical disabilities are going to be just as interested in sex as any other teens and should be helped with any potential issues that they may have around that issue. They will likely get sex education in schools, but are still often viewed as not interested in sex or sexually active, and they may not get the usual confidential teen questions or appropriate screenings. Menstrual management and sexuality education both are important aspects of reproductive health care for teens with disabilities.
Dr. Elisabeth Quint, lead author of the AAP clinical report “Menstrual Management for Adolescents,” is a clinical professor of obstetrics and gynecology at the University of Michigan, Ann Arbor. She is also a past president of the North American Society for Pediatric and Adolescent Gynecology.
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”
A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
For the first time, the American Academy of Pediatrics is offering guidance on managing menstruation and sexuality education in adolescents with disabilities.
Written jointly with the North American Society for Pediatric and Adolescent Gynecology, the clinical report offers guidance on options for menstrual management, sexual education and expression, and protection from sexual abuse. The report gives guidance regarding the care of adolescents with physical and/or intellectual abilities, but not for those with psychiatric illnesses (Pediatrics. 2016 April. doi: 10.1542/peds.2016-0295).
“Taking care of teens with disabilities and figuring out what to do with menstrual management has been all over the map,” Dr. Cora Collette Breuner, chair of the AAP’s committee on adolescence, said in an interview. “So, we tried to clarify it and help clinicians know what to do and when.”
A particular concern for the two groups was the threat of adverse events. “We wanted to cover what’s safe and what’s not in menstruation management, especially around bone health and thromboembolic events,” said Dr. Breuner, a professor of pediatrics and adolescent medicine at the University of Washington, Seattle.
Some of the information will not surprise clinicians, but there are some data that will perhaps come as news, said Dr. Breuner, including the recommendation that long-acting reversible contraception (LARC), such as the levonorgestrel intrauterine device or the progesterone implant rod should be considered first-line management therapies. “We point out that a number of studies show that these are safe.”
The report emphasizes offering anticipatory guidance before menses begins, noting that most teens with disabilities mature at the same rate as teens without disabilities. The report does not recommend premenarchal suppression in these patients, because doing so can interfere with normal bone growth. Such suppression also prevents patients and their families and caregivers from discovering that coping with the onset of menses is perhaps not as difficult as they might fear, the report states.
Although combined oral contraceptives are not contraindicated in teens with mobility issues, to guard against the threat of thromboembolic events in teens who use wheelchairs, the report recommends taking a thorough family history to rule out inherited thrombophilia. Otherwise, the recommendation is to prescribe the lowest-dose estrogen with a first- or second-generation progestin, as these are associated with lower rates of venous thrombotic events.
The guidance states that if cycles are creating difficulties in the patient’s life, “as determined by health care providers, patients, and families,” then menstrual management is appropriate. Even though it may take up to 3 years before a menstrual cycle becomes regular, the report cites irregularities caused by certain medications can be reason enough for menstrual management. Specific drugs noted include those affecting the dopaminergic system, valproic acid, and medications that elevate prolactin. Teens with obesity, seizure disorders, and polycystic ovary syndrome also can experience higher rates of irregularity.
The report also warns against the assumption that teens with disabilities are asexual or uninterested in sex. When appropriate, they should be offered the same confidential conversations about sexuality as are recommended for all teenagers by the AAP and the American College of Obstetricians and Gynecologists. “Teenagers with physical disabilities are just as likely to be sexually active as their peers and have a higher incidence of sexual abuse,” the report states. It is typically when a patient is cognitively impaired that consent to confidential services may require “discussion about legal guardianship or medical power of attorney status for families,” according to the report.
The report’s comprehensive review of four main menstrual management techniques – estrogen-containing, progestin-only, nonhormonal methods, and surgical requests and options – begins with the caveat that regardless of the method used, the threat of abuse or sexually transmitted infections remain. When a patient’s family or caregivers request suppression of menarche in a patient, stating fears of abuse or pregnancy, further investigation into the patient’s circumstances is warranted, the report states.
“It’s always worth reminding physicians that in this cohort, endometrial ablation can have legal implications, and it’s not recommended in this age group,” Dr. Breuner said.
On average, 1.5 hormonal methods are tried before achieving management goals, according to the report. Data cited in the study showed that at 42%, oral contraception is the preferred method of menstrual suppression, followed by the patch at 20%. Expectant management was third at 15%, followed by DMPA (depot medroxyprogesterone acetate) at 12%. The least utilized method was the levonorgestrel intrauterine device at 3%. No data were provided for the implantable contraceptive rod.
The clinical report is a companion document to another AAP clinical report, “Sexuality of Children and Adolescents with Developmental Disabilities” (Pediatrics. 2006. doi: 10.1542/peds.2006-1115).
AAP guidance on these matters in teens with psychiatric illnesses is expected to be issued within a few years, Dr. Breuner said.
There was no external funding and the authors have no relevant financial disclosures.
On Twitter @whitneymcknight
FROM PEDIATRICS
