User login
Corticosteroids Show Benefit in Community-Acquired Pneumonia
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary care hospitals in Switzerland.
Synopsis: Seven hundred eighty-four patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33, 95% CI 1.15-1.50, P<0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin.
Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicenter, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511-1518.
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary care hospitals in Switzerland.
Synopsis: Seven hundred eighty-four patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33, 95% CI 1.15-1.50, P<0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin.
Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicenter, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511-1518.
Clinical question: Does corticosteroid treatment shorten systemic illness in patients admitted to the hospital for community-acquired pneumonia (CAP)?
Background: Pneumonia is the third-leading cause of death worldwide. Studies have yielded conflicting data about the benefit of adding systemic corticosteroids for treatment of CAP.
Study design: Double-blind, multicenter, randomized, placebo-controlled trial.
Setting: Seven tertiary care hospitals in Switzerland.
Synopsis: Seven hundred eighty-four patients hospitalized for CAP were randomized to receive either oral prednisone 50 mg daily for seven days or placebo, with the primary endpoint being time to stable vital signs. The intention-to-treat analysis found that the median time to clinical stability was 1.4 days earlier in the prednisone group (hazard ratio 1.33, 95% CI 1.15-1.50, P<0.0001) and that length of stay and IV antibiotics were reduced by one day; this effect was valid across all PSI classes and was not dependent on age. Pneumonia-associated complications in the two groups did not differ at 30 days, though the prednisone group had a higher incidence of hyperglycemia requiring insulin.
Because all study locations were in a single, fairly homogenous northern European country, care should be taken when hospitalists apply these findings to their patient population, and the risks of hyperglycemia requiring insulin should be taken into consideration.
Bottom line: Systemic steroids may reduce the time to clinical stability in patients with CAP.
Citation: Blum CA, Nigro N, Briel M, et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: a multicenter, double-blind, randomised, placebo-controlled trial. Lancet. 2015;385(9977):1511-1518.
Standard Discharge Communication Process Improves Verbal Handoffs between Hospitalists, PCPs
Clinical question: Can a standardized discharge communication process, coupled with an electronic health record (EHR) system, improve the proportion of completed verbal handoffs from in-hospital physicians to PCPs within 24 hours of patient discharge?
Background: Discharge from the hospital setting is known to be a transition of care fraught with patient safety risks, with more than half of discharged patients experiencing at least one error.1 Previous studies identified core elements that pediatric hospitalists and PCPs consider essential in discharge communication, which included:
- Pending laboratory or test results;
- Follow-up appointments;
- Discharge medications;
- Admission and discharge diagnoses;
- Dates of admission and discharge; and
- Suggested management plan.2
Rates of transmission and receipt of information have been found to be suboptimal after hospital discharge, and PCPs have been found to be less satisfied than hospitalists with communication.2,3 Additionally, PCPs and hospitalists have been found to have incongruent views on who should be responsible for pending labs, adverse events, or status changes, differences which can have safety implications.3 PCPs who refer to general hospitals have been found to report superior completeness of discharge communication compared to freestanding children’s hospitals, where resident physicians are generally responsible for discharge summary completion.4 Standardizing and promoting a process of verbal handoff after hospital discharge may address some of these safety concerns, although a relationship has not been established between aspects of discharge communication and associated adverse clinical outcomes.5
Study design: Quality improvement study using improvement science methods and run charts.
Setting: An urban, 598-bed, freestanding children’s hospital.
Synopsis: A 24/7 telephone operator service had been established at the investigators’ institution that was designed to facilitate communication between providers inside and outside the institution. At baseline, only 52% of hospital medicine (HM) provider discharges had a record of a discharge day call initiated to the PCP. A project team consisting of hospitalists, a chief resident, operator service administrators, and IT analysts identified system issues that led to unsuccessful communication, which facilitated identification of key drivers of improving communication and associated interventions (see Table 1).
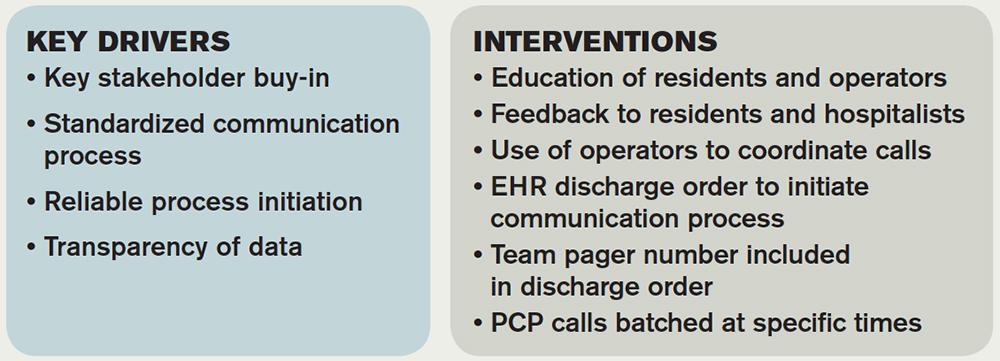
Discharging physicians, who were usually residents, were instructed to call the operator at the time of discharge. Operators would page the PCP, and PCPs were expected to return the page within 20 minutes. Discharging physicians were expected to return the call to the operator within two to four minutes. The EHR generated a message to the operator whenever a discharge order was placed for an HM patient, leading the operator to page the discharging physician to initiate the call.
Adaptations after project initiation included:
- Reassigning primary responsibility for discharge phone calls to the daily on-call resident, if the discharging physician was not available.
- Establishing a non-changing pager number on the automated discharge notification that would always reach the appropriate team member.
- Batching discharge phone calls at times of increased resident availability to minimize hold times for PCPs and work interruptions for discharging physicians.
Weekly failure data was generated and reviewed by the improvement team, and a call record was linked to the patient’s medical record. Team-specific and overall results for HM teams were posted weekly on a run-chart. The primary outcome measure was the percentage of completed calls between PCP and HM physician within 24 hours of discharge.
Over the approximately 32-month study period, the percentage of calls initiated improved from 50% to 97% after four interventions. After one year, data was collected to assess percentage of calls completed, a number that rose from 80% in the first eight weeks to a median of 93%, which was sustained for 18 months.
Bottom line: Utilizing improvement methods and reliability science, a process of improving verbal handoffs between hospital-based physicians and PCPs within 24 hours after discharge led to a sustained improvement, to above 90%, in successful verbal handoffs.
Citation: Mussman GM, Vossmeyer MT, Brady PW, Warrick DM, Simmons JM, White CM. Improving the reliability of verbal communication between primary care physicians and pediatric hospitalists at hospital discharge [published online ahead of print May 29, 2015]. J Hosp Med. doi: 10.1002/jhm.2392.

Clinical Shorts
MAJORITY OF NONOBSTRUCTING ASYMPTOMATIC RENAL STONES REMAIN ASYMPTOMATIC OVER TIME
Retrospective trial of active surveillance of asymptomatic nonobstructing renal calculi demonstrated that 28% of stones became symptomatic, with 17% requiring surgical intervention and 2% causing asymptomatic hydronephrosis over three years.
Citation: Dropkin BM, Moses RA, Sharma D, Pais VM Jr. The natural history of nonobstructing asymptomatic renal stones managed with active surveillance. J Urol. 2015;193(4):1265-1269.
CPR USE IS HIGH, YET OUTCOMES ARE POOR IN HEMODIALYSIS PATIENTS
In a national cohort of hemodialysis patients, receipt of in-hospital CPR was significantly higher (6.3% vs. 0.3%) than the general population, but post-discharge survival was substantially shorter (33 vs. five months).
Citation: Wong SY, Kreuter W, Curtis JR, Hall YN, O’Hare AM. Trends in in-hospital cardiopulmonary resuscitation and survival in adults receiving maintenance dialysis. JAMA Intern Med. 2015;175(6):1028-1035. doi:10.1001/jamainternmed.2015.0406.
ULTRASOUND GUIDANCE INCREASES RATE OF SUCCESSFUL FIRST ATTEMPT RADIAL ARTERY CANNULATION
Randomized controlled trial of 749 anesthesia trainees showed that ultrasound guidance increased rate of first attempt radial artery cannulation by 14% when compared to Doppler and palpation (95% CI 5-22%).
Citation: Ueda K, Bayman EO, Johnson C, Odum NJ, Lee JJ. A randomised controlled trial of radial artery cannulation guided by Doppler vs palpation vs ultrasound [published online ahead of print April 8, 2015]. Anaesthesia. doi: 10.1111/anae.13062.
NO DIFFERENCE BETWEEN EPIDURAL STEROID INJECTIONS AND GABAPENTIN FOR TREATMENT OF LUMBOSACRAL RADICULAR PAIN
Multicenter, randomized study found no difference in lumbosacral radicular pain at one and three months in patients treated with epidural steroid injection versus gabapentin.
Citation: Cohen SP, Hanling S, Bicket MC, et al. Epidural steroid injections compared with gabapentin for lumbosacral radicular pain: multicenter randomized double blind comparative efficacy study. BMJ. 2015;350:h1748 doi: 10.1136/bmj.h1748.
References
- Smith K. Effective communication with primary care providers. Pediatr Clin North Am. 2014;61(4):671-679.
- Coghlin DT, Leyenaar JK, Shen M, et al. Pediatric discharge content: a multisite assessment of physician preferences and experiences. Hosp Pediatr. 2014;4(1):9-15.
- Ruth JL, Geskey JM, Shaffer ML, Bramley HP, Paul IM. Evaluating communication between pediatric primary care physicians and hospitalists. Clin Pediatr (Phila). 2011;50(10):923-928.
- Leyenaar JK, Bergert L, Mallory LA, et al. Pediatric primary care providers’ perspectives regarding hospital discharge communication: a mixed methods analysis. Acad Pediatr. 2015;15(1):61-68.
- Bell CM, Schnipper JL, Auerbach AD, et al. Association of communication between hospital-based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381-386.
Clinical question: Can a standardized discharge communication process, coupled with an electronic health record (EHR) system, improve the proportion of completed verbal handoffs from in-hospital physicians to PCPs within 24 hours of patient discharge?
Background: Discharge from the hospital setting is known to be a transition of care fraught with patient safety risks, with more than half of discharged patients experiencing at least one error.1 Previous studies identified core elements that pediatric hospitalists and PCPs consider essential in discharge communication, which included:
- Pending laboratory or test results;
- Follow-up appointments;
- Discharge medications;
- Admission and discharge diagnoses;
- Dates of admission and discharge; and
- Suggested management plan.2
Rates of transmission and receipt of information have been found to be suboptimal after hospital discharge, and PCPs have been found to be less satisfied than hospitalists with communication.2,3 Additionally, PCPs and hospitalists have been found to have incongruent views on who should be responsible for pending labs, adverse events, or status changes, differences which can have safety implications.3 PCPs who refer to general hospitals have been found to report superior completeness of discharge communication compared to freestanding children’s hospitals, where resident physicians are generally responsible for discharge summary completion.4 Standardizing and promoting a process of verbal handoff after hospital discharge may address some of these safety concerns, although a relationship has not been established between aspects of discharge communication and associated adverse clinical outcomes.5
Study design: Quality improvement study using improvement science methods and run charts.
Setting: An urban, 598-bed, freestanding children’s hospital.
Synopsis: A 24/7 telephone operator service had been established at the investigators’ institution that was designed to facilitate communication between providers inside and outside the institution. At baseline, only 52% of hospital medicine (HM) provider discharges had a record of a discharge day call initiated to the PCP. A project team consisting of hospitalists, a chief resident, operator service administrators, and IT analysts identified system issues that led to unsuccessful communication, which facilitated identification of key drivers of improving communication and associated interventions (see Table 1).
Discharging physicians, who were usually residents, were instructed to call the operator at the time of discharge. Operators would page the PCP, and PCPs were expected to return the page within 20 minutes. Discharging physicians were expected to return the call to the operator within two to four minutes. The EHR generated a message to the operator whenever a discharge order was placed for an HM patient, leading the operator to page the discharging physician to initiate the call.
Adaptations after project initiation included:
- Reassigning primary responsibility for discharge phone calls to the daily on-call resident, if the discharging physician was not available.
- Establishing a non-changing pager number on the automated discharge notification that would always reach the appropriate team member.
- Batching discharge phone calls at times of increased resident availability to minimize hold times for PCPs and work interruptions for discharging physicians.
Weekly failure data was generated and reviewed by the improvement team, and a call record was linked to the patient’s medical record. Team-specific and overall results for HM teams were posted weekly on a run-chart. The primary outcome measure was the percentage of completed calls between PCP and HM physician within 24 hours of discharge.
Over the approximately 32-month study period, the percentage of calls initiated improved from 50% to 97% after four interventions. After one year, data was collected to assess percentage of calls completed, a number that rose from 80% in the first eight weeks to a median of 93%, which was sustained for 18 months.
Bottom line: Utilizing improvement methods and reliability science, a process of improving verbal handoffs between hospital-based physicians and PCPs within 24 hours after discharge led to a sustained improvement, to above 90%, in successful verbal handoffs.
Citation: Mussman GM, Vossmeyer MT, Brady PW, Warrick DM, Simmons JM, White CM. Improving the reliability of verbal communication between primary care physicians and pediatric hospitalists at hospital discharge [published online ahead of print May 29, 2015]. J Hosp Med. doi: 10.1002/jhm.2392.
Clinical Shorts
MAJORITY OF NONOBSTRUCTING ASYMPTOMATIC RENAL STONES REMAIN ASYMPTOMATIC OVER TIME
Retrospective trial of active surveillance of asymptomatic nonobstructing renal calculi demonstrated that 28% of stones became symptomatic, with 17% requiring surgical intervention and 2% causing asymptomatic hydronephrosis over three years.
Citation: Dropkin BM, Moses RA, Sharma D, Pais VM Jr. The natural history of nonobstructing asymptomatic renal stones managed with active surveillance. J Urol. 2015;193(4):1265-1269.
CPR USE IS HIGH, YET OUTCOMES ARE POOR IN HEMODIALYSIS PATIENTS
In a national cohort of hemodialysis patients, receipt of in-hospital CPR was significantly higher (6.3% vs. 0.3%) than the general population, but post-discharge survival was substantially shorter (33 vs. five months).
Citation: Wong SY, Kreuter W, Curtis JR, Hall YN, O’Hare AM. Trends in in-hospital cardiopulmonary resuscitation and survival in adults receiving maintenance dialysis. JAMA Intern Med. 2015;175(6):1028-1035. doi:10.1001/jamainternmed.2015.0406.
ULTRASOUND GUIDANCE INCREASES RATE OF SUCCESSFUL FIRST ATTEMPT RADIAL ARTERY CANNULATION
Randomized controlled trial of 749 anesthesia trainees showed that ultrasound guidance increased rate of first attempt radial artery cannulation by 14% when compared to Doppler and palpation (95% CI 5-22%).
Citation: Ueda K, Bayman EO, Johnson C, Odum NJ, Lee JJ. A randomised controlled trial of radial artery cannulation guided by Doppler vs palpation vs ultrasound [published online ahead of print April 8, 2015]. Anaesthesia. doi: 10.1111/anae.13062.
NO DIFFERENCE BETWEEN EPIDURAL STEROID INJECTIONS AND GABAPENTIN FOR TREATMENT OF LUMBOSACRAL RADICULAR PAIN
Multicenter, randomized study found no difference in lumbosacral radicular pain at one and three months in patients treated with epidural steroid injection versus gabapentin.
Citation: Cohen SP, Hanling S, Bicket MC, et al. Epidural steroid injections compared with gabapentin for lumbosacral radicular pain: multicenter randomized double blind comparative efficacy study. BMJ. 2015;350:h1748 doi: 10.1136/bmj.h1748.
References
- Smith K. Effective communication with primary care providers. Pediatr Clin North Am. 2014;61(4):671-679.
- Coghlin DT, Leyenaar JK, Shen M, et al. Pediatric discharge content: a multisite assessment of physician preferences and experiences. Hosp Pediatr. 2014;4(1):9-15.
- Ruth JL, Geskey JM, Shaffer ML, Bramley HP, Paul IM. Evaluating communication between pediatric primary care physicians and hospitalists. Clin Pediatr (Phila). 2011;50(10):923-928.
- Leyenaar JK, Bergert L, Mallory LA, et al. Pediatric primary care providers’ perspectives regarding hospital discharge communication: a mixed methods analysis. Acad Pediatr. 2015;15(1):61-68.
- Bell CM, Schnipper JL, Auerbach AD, et al. Association of communication between hospital-based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381-386.
Clinical question: Can a standardized discharge communication process, coupled with an electronic health record (EHR) system, improve the proportion of completed verbal handoffs from in-hospital physicians to PCPs within 24 hours of patient discharge?
Background: Discharge from the hospital setting is known to be a transition of care fraught with patient safety risks, with more than half of discharged patients experiencing at least one error.1 Previous studies identified core elements that pediatric hospitalists and PCPs consider essential in discharge communication, which included:
- Pending laboratory or test results;
- Follow-up appointments;
- Discharge medications;
- Admission and discharge diagnoses;
- Dates of admission and discharge; and
- Suggested management plan.2
Rates of transmission and receipt of information have been found to be suboptimal after hospital discharge, and PCPs have been found to be less satisfied than hospitalists with communication.2,3 Additionally, PCPs and hospitalists have been found to have incongruent views on who should be responsible for pending labs, adverse events, or status changes, differences which can have safety implications.3 PCPs who refer to general hospitals have been found to report superior completeness of discharge communication compared to freestanding children’s hospitals, where resident physicians are generally responsible for discharge summary completion.4 Standardizing and promoting a process of verbal handoff after hospital discharge may address some of these safety concerns, although a relationship has not been established between aspects of discharge communication and associated adverse clinical outcomes.5
Study design: Quality improvement study using improvement science methods and run charts.
Setting: An urban, 598-bed, freestanding children’s hospital.
Synopsis: A 24/7 telephone operator service had been established at the investigators’ institution that was designed to facilitate communication between providers inside and outside the institution. At baseline, only 52% of hospital medicine (HM) provider discharges had a record of a discharge day call initiated to the PCP. A project team consisting of hospitalists, a chief resident, operator service administrators, and IT analysts identified system issues that led to unsuccessful communication, which facilitated identification of key drivers of improving communication and associated interventions (see Table 1).
Discharging physicians, who were usually residents, were instructed to call the operator at the time of discharge. Operators would page the PCP, and PCPs were expected to return the page within 20 minutes. Discharging physicians were expected to return the call to the operator within two to four minutes. The EHR generated a message to the operator whenever a discharge order was placed for an HM patient, leading the operator to page the discharging physician to initiate the call.
Adaptations after project initiation included:
- Reassigning primary responsibility for discharge phone calls to the daily on-call resident, if the discharging physician was not available.
- Establishing a non-changing pager number on the automated discharge notification that would always reach the appropriate team member.
- Batching discharge phone calls at times of increased resident availability to minimize hold times for PCPs and work interruptions for discharging physicians.
Weekly failure data was generated and reviewed by the improvement team, and a call record was linked to the patient’s medical record. Team-specific and overall results for HM teams were posted weekly on a run-chart. The primary outcome measure was the percentage of completed calls between PCP and HM physician within 24 hours of discharge.
Over the approximately 32-month study period, the percentage of calls initiated improved from 50% to 97% after four interventions. After one year, data was collected to assess percentage of calls completed, a number that rose from 80% in the first eight weeks to a median of 93%, which was sustained for 18 months.
Bottom line: Utilizing improvement methods and reliability science, a process of improving verbal handoffs between hospital-based physicians and PCPs within 24 hours after discharge led to a sustained improvement, to above 90%, in successful verbal handoffs.
Citation: Mussman GM, Vossmeyer MT, Brady PW, Warrick DM, Simmons JM, White CM. Improving the reliability of verbal communication between primary care physicians and pediatric hospitalists at hospital discharge [published online ahead of print May 29, 2015]. J Hosp Med. doi: 10.1002/jhm.2392.
Clinical Shorts
MAJORITY OF NONOBSTRUCTING ASYMPTOMATIC RENAL STONES REMAIN ASYMPTOMATIC OVER TIME
Retrospective trial of active surveillance of asymptomatic nonobstructing renal calculi demonstrated that 28% of stones became symptomatic, with 17% requiring surgical intervention and 2% causing asymptomatic hydronephrosis over three years.
Citation: Dropkin BM, Moses RA, Sharma D, Pais VM Jr. The natural history of nonobstructing asymptomatic renal stones managed with active surveillance. J Urol. 2015;193(4):1265-1269.
CPR USE IS HIGH, YET OUTCOMES ARE POOR IN HEMODIALYSIS PATIENTS
In a national cohort of hemodialysis patients, receipt of in-hospital CPR was significantly higher (6.3% vs. 0.3%) than the general population, but post-discharge survival was substantially shorter (33 vs. five months).
Citation: Wong SY, Kreuter W, Curtis JR, Hall YN, O’Hare AM. Trends in in-hospital cardiopulmonary resuscitation and survival in adults receiving maintenance dialysis. JAMA Intern Med. 2015;175(6):1028-1035. doi:10.1001/jamainternmed.2015.0406.
ULTRASOUND GUIDANCE INCREASES RATE OF SUCCESSFUL FIRST ATTEMPT RADIAL ARTERY CANNULATION
Randomized controlled trial of 749 anesthesia trainees showed that ultrasound guidance increased rate of first attempt radial artery cannulation by 14% when compared to Doppler and palpation (95% CI 5-22%).
Citation: Ueda K, Bayman EO, Johnson C, Odum NJ, Lee JJ. A randomised controlled trial of radial artery cannulation guided by Doppler vs palpation vs ultrasound [published online ahead of print April 8, 2015]. Anaesthesia. doi: 10.1111/anae.13062.
NO DIFFERENCE BETWEEN EPIDURAL STEROID INJECTIONS AND GABAPENTIN FOR TREATMENT OF LUMBOSACRAL RADICULAR PAIN
Multicenter, randomized study found no difference in lumbosacral radicular pain at one and three months in patients treated with epidural steroid injection versus gabapentin.
Citation: Cohen SP, Hanling S, Bicket MC, et al. Epidural steroid injections compared with gabapentin for lumbosacral radicular pain: multicenter randomized double blind comparative efficacy study. BMJ. 2015;350:h1748 doi: 10.1136/bmj.h1748.
References
- Smith K. Effective communication with primary care providers. Pediatr Clin North Am. 2014;61(4):671-679.
- Coghlin DT, Leyenaar JK, Shen M, et al. Pediatric discharge content: a multisite assessment of physician preferences and experiences. Hosp Pediatr. 2014;4(1):9-15.
- Ruth JL, Geskey JM, Shaffer ML, Bramley HP, Paul IM. Evaluating communication between pediatric primary care physicians and hospitalists. Clin Pediatr (Phila). 2011;50(10):923-928.
- Leyenaar JK, Bergert L, Mallory LA, et al. Pediatric primary care providers’ perspectives regarding hospital discharge communication: a mixed methods analysis. Acad Pediatr. 2015;15(1):61-68.
- Bell CM, Schnipper JL, Auerbach AD, et al. Association of communication between hospital-based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381-386.
Nomogram Predicts Post-Operative Readmission
Clinical question: Can a nomogram accurately predict a patient’s risk of post-operative 30-day readmission?
Background: Medicare and Medicaid have implemented penalties for hospitals with high readmission rates. While this does not yet apply to post-operative readmissions, there is concern that it soon will. Algorithms for predicting readmission have been developed for medical patients; however, to date, no such tool has been developed for post-operative patients.
Study design: Retrospective review and prospective validation of a predictive nomogram.
Setting: Single academic hospital.
Synopsis: Using the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP) and hospital billing data, a retrospective analysis of 2,799 patients who had elective surgery between 2006 and 2011 was performed in order to develop a predictive nomogram for post-operative readmissions. Pre-operative, operative, and post-operative variables associated with readmission were evaluated, and the following variables were found to be independently associated with readmission:
- Bleeding disorder;
- Prolonged procedure length;
- In-hospital complications;
- Dependent functional status; and/or
- Higher care at discharge.
Using a linear regression model, a nomogram was developed that was prospectively validated in 255 patients from a single center. The nomogram accurately predicted the risk of post-operative readmission (C statistic=0.756) in the prospective analysis.
The nomogram has limited generalizability given the fact that it included patients from a single institution; it would benefit from external validation before widespread use.
Bottom line: The use of this predictive nomogram could aid in identifying patients at high risk of readmission.
Citation: Tevis SE, Weber SM, Kent KC, Kennedy GD. Nomogram to predict postoperative readmission in patients who undergo general surgery. JAMA Surg. 2015;150(6):505-510. doi: 10.1001/jamasurg.2014.4043.
Clinical question: Can a nomogram accurately predict a patient’s risk of post-operative 30-day readmission?
Background: Medicare and Medicaid have implemented penalties for hospitals with high readmission rates. While this does not yet apply to post-operative readmissions, there is concern that it soon will. Algorithms for predicting readmission have been developed for medical patients; however, to date, no such tool has been developed for post-operative patients.
Study design: Retrospective review and prospective validation of a predictive nomogram.
Setting: Single academic hospital.
Synopsis: Using the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP) and hospital billing data, a retrospective analysis of 2,799 patients who had elective surgery between 2006 and 2011 was performed in order to develop a predictive nomogram for post-operative readmissions. Pre-operative, operative, and post-operative variables associated with readmission were evaluated, and the following variables were found to be independently associated with readmission:
- Bleeding disorder;
- Prolonged procedure length;
- In-hospital complications;
- Dependent functional status; and/or
- Higher care at discharge.
Using a linear regression model, a nomogram was developed that was prospectively validated in 255 patients from a single center. The nomogram accurately predicted the risk of post-operative readmission (C statistic=0.756) in the prospective analysis.
The nomogram has limited generalizability given the fact that it included patients from a single institution; it would benefit from external validation before widespread use.
Bottom line: The use of this predictive nomogram could aid in identifying patients at high risk of readmission.
Citation: Tevis SE, Weber SM, Kent KC, Kennedy GD. Nomogram to predict postoperative readmission in patients who undergo general surgery. JAMA Surg. 2015;150(6):505-510. doi: 10.1001/jamasurg.2014.4043.
Clinical question: Can a nomogram accurately predict a patient’s risk of post-operative 30-day readmission?
Background: Medicare and Medicaid have implemented penalties for hospitals with high readmission rates. While this does not yet apply to post-operative readmissions, there is concern that it soon will. Algorithms for predicting readmission have been developed for medical patients; however, to date, no such tool has been developed for post-operative patients.
Study design: Retrospective review and prospective validation of a predictive nomogram.
Setting: Single academic hospital.
Synopsis: Using the American College of Surgeons’ National Surgical Quality Improvement Program (ACS NSQIP) and hospital billing data, a retrospective analysis of 2,799 patients who had elective surgery between 2006 and 2011 was performed in order to develop a predictive nomogram for post-operative readmissions. Pre-operative, operative, and post-operative variables associated with readmission were evaluated, and the following variables were found to be independently associated with readmission:
- Bleeding disorder;
- Prolonged procedure length;
- In-hospital complications;
- Dependent functional status; and/or
- Higher care at discharge.
Using a linear regression model, a nomogram was developed that was prospectively validated in 255 patients from a single center. The nomogram accurately predicted the risk of post-operative readmission (C statistic=0.756) in the prospective analysis.
The nomogram has limited generalizability given the fact that it included patients from a single institution; it would benefit from external validation before widespread use.
Bottom line: The use of this predictive nomogram could aid in identifying patients at high risk of readmission.
Citation: Tevis SE, Weber SM, Kent KC, Kennedy GD. Nomogram to predict postoperative readmission in patients who undergo general surgery. JAMA Surg. 2015;150(6):505-510. doi: 10.1001/jamasurg.2014.4043.
The Hospitalist Earns Pair of 2015 Awards for Publication Excellence for Health and Medical Writing

The Hospitalist has been honored with two 2015 Awards of Excellence in the Health and Medical Writing category from the Awards for Publication Excellence (APEX).
The annual awards, presented to corporate and nonprofit publications, received 1,851 total entries, including nearly 400 entries to the writing category. Only 16 Awards of Excellence were presented.
Freelance medical writer Bryn Nelson, PhD, was honored for his eight-page special report on ObamaCare.
Freelance writer Gretchen Henkel was honored for her cover story, “Deaf Hospitalist Focuses on Teaching, Co-Management, Patient-Centered Care,” which explores the inspiring and enlightening careers of hospitalists with hearing impairments.
Published by Wiley Inc., The Hospitalist is the official newsmagazine of the Society of Hospital Medicine. The Hospitalist has garnered nine APEX Awards in the past seven years, receiving the APEX Grand Award for Magazines, Journals, and Tabloids last year, as well as attaining finalist status for “Best Healthcare Business Publication” from Medical Marketing and Media in 2009.
“We have long known that The Hospitalist provides hospitalists with some of the most compelling and informative content in hospital medicine,” SHM President Bob Harrington, MD, SFHM, wrote in an email. “It is great to have that content recognized by APEX this year, as it has been in the past. We know that APEX judges received nearly 2,000 entries this year, so we are thrilled to see our newsmagazine rise to the top again."
“This year’s recognition is especially valuable, as The Hospitalist is now distributed to more than 32,000 hospitalists and other leaders in healthcare.”
Physician Editor Danielle Scheurer, MD, MSCR, SFHM, also commends The Hospitalist, emphasizing the importance of these awards.
“These awards exemplify how well written the content is for The Hospitalist,” she said. “This is a huge honor for the publication, as well as for all hospitalists, for which the publication is intended. I am personally honored to be the physician editor of such an exemplary publication!”
The Hospitalist has been honored with two 2015 Awards of Excellence in the Health and Medical Writing category from the Awards for Publication Excellence (APEX).
The annual awards, presented to corporate and nonprofit publications, received 1,851 total entries, including nearly 400 entries to the writing category. Only 16 Awards of Excellence were presented.
Freelance medical writer Bryn Nelson, PhD, was honored for his eight-page special report on ObamaCare.
Freelance writer Gretchen Henkel was honored for her cover story, “Deaf Hospitalist Focuses on Teaching, Co-Management, Patient-Centered Care,” which explores the inspiring and enlightening careers of hospitalists with hearing impairments.
Published by Wiley Inc., The Hospitalist is the official newsmagazine of the Society of Hospital Medicine. The Hospitalist has garnered nine APEX Awards in the past seven years, receiving the APEX Grand Award for Magazines, Journals, and Tabloids last year, as well as attaining finalist status for “Best Healthcare Business Publication” from Medical Marketing and Media in 2009.
“We have long known that The Hospitalist provides hospitalists with some of the most compelling and informative content in hospital medicine,” SHM President Bob Harrington, MD, SFHM, wrote in an email. “It is great to have that content recognized by APEX this year, as it has been in the past. We know that APEX judges received nearly 2,000 entries this year, so we are thrilled to see our newsmagazine rise to the top again."
“This year’s recognition is especially valuable, as The Hospitalist is now distributed to more than 32,000 hospitalists and other leaders in healthcare.”
Physician Editor Danielle Scheurer, MD, MSCR, SFHM, also commends The Hospitalist, emphasizing the importance of these awards.
“These awards exemplify how well written the content is for The Hospitalist,” she said. “This is a huge honor for the publication, as well as for all hospitalists, for which the publication is intended. I am personally honored to be the physician editor of such an exemplary publication!”
The Hospitalist has been honored with two 2015 Awards of Excellence in the Health and Medical Writing category from the Awards for Publication Excellence (APEX).
The annual awards, presented to corporate and nonprofit publications, received 1,851 total entries, including nearly 400 entries to the writing category. Only 16 Awards of Excellence were presented.
Freelance medical writer Bryn Nelson, PhD, was honored for his eight-page special report on ObamaCare.
Freelance writer Gretchen Henkel was honored for her cover story, “Deaf Hospitalist Focuses on Teaching, Co-Management, Patient-Centered Care,” which explores the inspiring and enlightening careers of hospitalists with hearing impairments.
Published by Wiley Inc., The Hospitalist is the official newsmagazine of the Society of Hospital Medicine. The Hospitalist has garnered nine APEX Awards in the past seven years, receiving the APEX Grand Award for Magazines, Journals, and Tabloids last year, as well as attaining finalist status for “Best Healthcare Business Publication” from Medical Marketing and Media in 2009.
“We have long known that The Hospitalist provides hospitalists with some of the most compelling and informative content in hospital medicine,” SHM President Bob Harrington, MD, SFHM, wrote in an email. “It is great to have that content recognized by APEX this year, as it has been in the past. We know that APEX judges received nearly 2,000 entries this year, so we are thrilled to see our newsmagazine rise to the top again."
“This year’s recognition is especially valuable, as The Hospitalist is now distributed to more than 32,000 hospitalists and other leaders in healthcare.”
Physician Editor Danielle Scheurer, MD, MSCR, SFHM, also commends The Hospitalist, emphasizing the importance of these awards.
“These awards exemplify how well written the content is for The Hospitalist,” she said. “This is a huge honor for the publication, as well as for all hospitalists, for which the publication is intended. I am personally honored to be the physician editor of such an exemplary publication!”
Medical Professionalism: Its Evolution and What It Means to Hospitalists
Professionalism is an overused word in the medical industry. What exactly is meant by professionalism, and what should it mean to hospitalists? Wikipedia notes that a professional is one who earns a living from a specified activity that has standards of education and training that prepare members of the profession with the knowledge and skills necessary to perform the role required. In addition, professionals are subject to strict codes of conduct enshrining rigorous ethical and moral obligations.
Physicians are the consummate professionals; over centuries, we have been afforded the reputation of being one of the “highest-ranking” professionals within societies, along with divinity and law. We are held to very high standards, both in and out of the workplace, including arduous and rigorous standards of education and training. We are in one of the only professions that take the Hippocratic Oath at graduation. This oath requires us to swear to uphold specific ethical standards. Being a professional means we always act within our professional standards and advocate for our patients in all circumstances.1
Threats to Professionalism?
Over the past several decades, concern has been growing about a widespread decline in professionalism among physicians, a decline that extends beyond a single generation. There are many purported reasons for this erosion of professionalism; we need to first understand the threats to professionalism in order to guard against its erosion in ourselves and in the next generation of physicians.1
One major issue is that we do not have a common understanding of the nature of professionalism; the definition is both overused and misused. We often refer to professionalism by what it isn’t, rather than understanding what it is. For example, there are scores of definitions for “unprofessional” conduct in the medical industry, many of which refer to physician behaviors. These include actions that intimidate, berate, or bully others, regardless of the rationale or intent, and encompass any form of physical or psychological harm. The actions of the “disruptive physician” are often thought of as synonymous with unprofessional behavior. But professionalism is so much more than the absence of disruptive behavior. So part of the erosion of professionalism is an oversimplification of what it isn’t, rather than what it is.
Another issue with upholding professionalism over time is that many physicians forget their professional standards, because there are few “booster sessions” to remind us of why we practice medicine. Once we enter the workforce, we are confronted with so many obstacles to delivering good care to patients that we often feel overwhelmed or incapable of removing the real barriers to good care, and therefore incapable of fulfilling our mission. There are no regular “revivals” or checkpoints to refresh our memory of what we went into medicine to accomplish.
Although our ultimate goal is to take good care of patients, another threat to professionalism is that doing this often requires physicians to operate outside their “trained” knowledge and skill sets. It requires us to act on behalf of patients as an advocate in all aspects of their life, not just as a “diagnoser” or “prescriber.” As a result, maintaining the ideals of professionalism has become ever more complex, because the social determinants of health have a major impact on patient well-being and health, including access to food, housing, and transportation. Many times, diagnosing and prescribing have little impact on the patient’s outcome; these social determinants of disease take sole precedence. A patient’s education, income, and home environment have a much greater impact in determining their health outcomes than does access to prescription medications. This means that advocating for patient health and well-being extends far beyond the walls of a hospital or emergency room, a role in which most hospitalists are incapable and/or uncomfortable.1
Another major catalyst in the erosion of professionalism is the complex issue of money and income. Many physicians, including hospitalists, are “judged” by their relative value units, an indicator of the quantity and complexity of patients seen. Services not “billable” are generally delegated to others, or they go undone. Such services include communicating tirelessly with all the stakeholders in the patients’ care, including family members, primary care physicians, other physician specialists, and other disciplines. Untoward behaviors, such as “upcoding,” selecting funded patients for care, creating patient streams for highly lucrative services, and under-resourcing care provisions that “lose money”—regardless of the value to the patient—are inadvertently incentivized on individual and system levels to enhance revenue. Many hospitalists are strapped with student loans early in their careers, requiring them to earn enough to pay back these loans in a timely fashion. These perverse incentives can and often do confound our ability to act solely on behalf of our patients.2
How Do We Overcome These Threats?
The first step in reviving professionalism is to define it by what it is, not by what it isn’t. Professionalism is not the absence of bad behavior. Professionalism is the “commitment to carrying out professional responsibilities and an adherence to ethical principles.”3 Professionalism is the pursuit of the tenets of the Hippocratic Oath. As a litmus test, read and reread the oath, and honestly reflect upon your practice.
Another step is to continuously work in multidisciplinary teams, a skill that comes naturally to most hospitalists. In order to fulfill the oath, you should not work as a social worker, but you should advocate for your patients’ social work needs. You need a plethora of other disciplines to help you fulfill your role as a patient advocate. Know and respect the roles that your team members are playing, all of which are invaluable to you and your patients.
An additional step in helping you fulfill your role as a professional is to get the education and skills you need to function effectively within the complex systems in which we currently work. You should incorporate business and management education into your continuing medical education so that you can help patients traverse a system that is complex. You should know and understand the general concepts of value-based payment, insurance exchanges, federal-state-private insurances, and the basic tenets of health systems. You should know how to recognize and reduce waste and unnecessary variation in the system, and know how to measure and improve upon processes.
In the words of Emanuel Ezekiel, MD, PhD, “Learning clinical medicine is necessary for making patient well-being the physician’s primary obligation. But it is not sufficient. To promote professionalism and all that it entails (reducing errors; ensuring safe, consistent, high-quality, and convenient care; removing unnecessary services; and improving the efficiency in the delivery of services), physicians must develop better management skills … Becoming better managers will make physicians better medical professionals”.2
For those entering medical school, nine core competencies can predict success in medical school and later in practice; we should all commit to excellence in these, which go beyond clinical knowledge:
- ethical responsibility to self and others;
- reliability and dependability;
- service orientation;
- social skills;
- capacity for improvement;
- resilience and adaptability;
- cultural competence;
- oral communication; and
- teamwork.
Lastly, a critical step in preventing the erosion of professionalism in medicine is self-regulation. External regulation comes to those who refuse or are unwilling to regulate themselves. Professionalism is a set of skills that can be taught, learned, and modeled. As a new specialty, we all own the success or failure of the reputation of hospitalists as consummate professionals.1

Professionalism is an overused word in the medical industry. What exactly is meant by professionalism, and what should it mean to hospitalists? Wikipedia notes that a professional is one who earns a living from a specified activity that has standards of education and training that prepare members of the profession with the knowledge and skills necessary to perform the role required. In addition, professionals are subject to strict codes of conduct enshrining rigorous ethical and moral obligations.
Physicians are the consummate professionals; over centuries, we have been afforded the reputation of being one of the “highest-ranking” professionals within societies, along with divinity and law. We are held to very high standards, both in and out of the workplace, including arduous and rigorous standards of education and training. We are in one of the only professions that take the Hippocratic Oath at graduation. This oath requires us to swear to uphold specific ethical standards. Being a professional means we always act within our professional standards and advocate for our patients in all circumstances.1
Threats to Professionalism?
Over the past several decades, concern has been growing about a widespread decline in professionalism among physicians, a decline that extends beyond a single generation. There are many purported reasons for this erosion of professionalism; we need to first understand the threats to professionalism in order to guard against its erosion in ourselves and in the next generation of physicians.1
One major issue is that we do not have a common understanding of the nature of professionalism; the definition is both overused and misused. We often refer to professionalism by what it isn’t, rather than understanding what it is. For example, there are scores of definitions for “unprofessional” conduct in the medical industry, many of which refer to physician behaviors. These include actions that intimidate, berate, or bully others, regardless of the rationale or intent, and encompass any form of physical or psychological harm. The actions of the “disruptive physician” are often thought of as synonymous with unprofessional behavior. But professionalism is so much more than the absence of disruptive behavior. So part of the erosion of professionalism is an oversimplification of what it isn’t, rather than what it is.
Another issue with upholding professionalism over time is that many physicians forget their professional standards, because there are few “booster sessions” to remind us of why we practice medicine. Once we enter the workforce, we are confronted with so many obstacles to delivering good care to patients that we often feel overwhelmed or incapable of removing the real barriers to good care, and therefore incapable of fulfilling our mission. There are no regular “revivals” or checkpoints to refresh our memory of what we went into medicine to accomplish.
Although our ultimate goal is to take good care of patients, another threat to professionalism is that doing this often requires physicians to operate outside their “trained” knowledge and skill sets. It requires us to act on behalf of patients as an advocate in all aspects of their life, not just as a “diagnoser” or “prescriber.” As a result, maintaining the ideals of professionalism has become ever more complex, because the social determinants of health have a major impact on patient well-being and health, including access to food, housing, and transportation. Many times, diagnosing and prescribing have little impact on the patient’s outcome; these social determinants of disease take sole precedence. A patient’s education, income, and home environment have a much greater impact in determining their health outcomes than does access to prescription medications. This means that advocating for patient health and well-being extends far beyond the walls of a hospital or emergency room, a role in which most hospitalists are incapable and/or uncomfortable.1
Another major catalyst in the erosion of professionalism is the complex issue of money and income. Many physicians, including hospitalists, are “judged” by their relative value units, an indicator of the quantity and complexity of patients seen. Services not “billable” are generally delegated to others, or they go undone. Such services include communicating tirelessly with all the stakeholders in the patients’ care, including family members, primary care physicians, other physician specialists, and other disciplines. Untoward behaviors, such as “upcoding,” selecting funded patients for care, creating patient streams for highly lucrative services, and under-resourcing care provisions that “lose money”—regardless of the value to the patient—are inadvertently incentivized on individual and system levels to enhance revenue. Many hospitalists are strapped with student loans early in their careers, requiring them to earn enough to pay back these loans in a timely fashion. These perverse incentives can and often do confound our ability to act solely on behalf of our patients.2
How Do We Overcome These Threats?
The first step in reviving professionalism is to define it by what it is, not by what it isn’t. Professionalism is not the absence of bad behavior. Professionalism is the “commitment to carrying out professional responsibilities and an adherence to ethical principles.”3 Professionalism is the pursuit of the tenets of the Hippocratic Oath. As a litmus test, read and reread the oath, and honestly reflect upon your practice.
Another step is to continuously work in multidisciplinary teams, a skill that comes naturally to most hospitalists. In order to fulfill the oath, you should not work as a social worker, but you should advocate for your patients’ social work needs. You need a plethora of other disciplines to help you fulfill your role as a patient advocate. Know and respect the roles that your team members are playing, all of which are invaluable to you and your patients.
An additional step in helping you fulfill your role as a professional is to get the education and skills you need to function effectively within the complex systems in which we currently work. You should incorporate business and management education into your continuing medical education so that you can help patients traverse a system that is complex. You should know and understand the general concepts of value-based payment, insurance exchanges, federal-state-private insurances, and the basic tenets of health systems. You should know how to recognize and reduce waste and unnecessary variation in the system, and know how to measure and improve upon processes.
In the words of Emanuel Ezekiel, MD, PhD, “Learning clinical medicine is necessary for making patient well-being the physician’s primary obligation. But it is not sufficient. To promote professionalism and all that it entails (reducing errors; ensuring safe, consistent, high-quality, and convenient care; removing unnecessary services; and improving the efficiency in the delivery of services), physicians must develop better management skills … Becoming better managers will make physicians better medical professionals”.2
For those entering medical school, nine core competencies can predict success in medical school and later in practice; we should all commit to excellence in these, which go beyond clinical knowledge:
- ethical responsibility to self and others;
- reliability and dependability;
- service orientation;
- social skills;
- capacity for improvement;
- resilience and adaptability;
- cultural competence;
- oral communication; and
- teamwork.
Lastly, a critical step in preventing the erosion of professionalism in medicine is self-regulation. External regulation comes to those who refuse or are unwilling to regulate themselves. Professionalism is a set of skills that can be taught, learned, and modeled. As a new specialty, we all own the success or failure of the reputation of hospitalists as consummate professionals.1
Professionalism is an overused word in the medical industry. What exactly is meant by professionalism, and what should it mean to hospitalists? Wikipedia notes that a professional is one who earns a living from a specified activity that has standards of education and training that prepare members of the profession with the knowledge and skills necessary to perform the role required. In addition, professionals are subject to strict codes of conduct enshrining rigorous ethical and moral obligations.
Physicians are the consummate professionals; over centuries, we have been afforded the reputation of being one of the “highest-ranking” professionals within societies, along with divinity and law. We are held to very high standards, both in and out of the workplace, including arduous and rigorous standards of education and training. We are in one of the only professions that take the Hippocratic Oath at graduation. This oath requires us to swear to uphold specific ethical standards. Being a professional means we always act within our professional standards and advocate for our patients in all circumstances.1
Threats to Professionalism?
Over the past several decades, concern has been growing about a widespread decline in professionalism among physicians, a decline that extends beyond a single generation. There are many purported reasons for this erosion of professionalism; we need to first understand the threats to professionalism in order to guard against its erosion in ourselves and in the next generation of physicians.1
One major issue is that we do not have a common understanding of the nature of professionalism; the definition is both overused and misused. We often refer to professionalism by what it isn’t, rather than understanding what it is. For example, there are scores of definitions for “unprofessional” conduct in the medical industry, many of which refer to physician behaviors. These include actions that intimidate, berate, or bully others, regardless of the rationale or intent, and encompass any form of physical or psychological harm. The actions of the “disruptive physician” are often thought of as synonymous with unprofessional behavior. But professionalism is so much more than the absence of disruptive behavior. So part of the erosion of professionalism is an oversimplification of what it isn’t, rather than what it is.
Another issue with upholding professionalism over time is that many physicians forget their professional standards, because there are few “booster sessions” to remind us of why we practice medicine. Once we enter the workforce, we are confronted with so many obstacles to delivering good care to patients that we often feel overwhelmed or incapable of removing the real barriers to good care, and therefore incapable of fulfilling our mission. There are no regular “revivals” or checkpoints to refresh our memory of what we went into medicine to accomplish.
Although our ultimate goal is to take good care of patients, another threat to professionalism is that doing this often requires physicians to operate outside their “trained” knowledge and skill sets. It requires us to act on behalf of patients as an advocate in all aspects of their life, not just as a “diagnoser” or “prescriber.” As a result, maintaining the ideals of professionalism has become ever more complex, because the social determinants of health have a major impact on patient well-being and health, including access to food, housing, and transportation. Many times, diagnosing and prescribing have little impact on the patient’s outcome; these social determinants of disease take sole precedence. A patient’s education, income, and home environment have a much greater impact in determining their health outcomes than does access to prescription medications. This means that advocating for patient health and well-being extends far beyond the walls of a hospital or emergency room, a role in which most hospitalists are incapable and/or uncomfortable.1
Another major catalyst in the erosion of professionalism is the complex issue of money and income. Many physicians, including hospitalists, are “judged” by their relative value units, an indicator of the quantity and complexity of patients seen. Services not “billable” are generally delegated to others, or they go undone. Such services include communicating tirelessly with all the stakeholders in the patients’ care, including family members, primary care physicians, other physician specialists, and other disciplines. Untoward behaviors, such as “upcoding,” selecting funded patients for care, creating patient streams for highly lucrative services, and under-resourcing care provisions that “lose money”—regardless of the value to the patient—are inadvertently incentivized on individual and system levels to enhance revenue. Many hospitalists are strapped with student loans early in their careers, requiring them to earn enough to pay back these loans in a timely fashion. These perverse incentives can and often do confound our ability to act solely on behalf of our patients.2
How Do We Overcome These Threats?
The first step in reviving professionalism is to define it by what it is, not by what it isn’t. Professionalism is not the absence of bad behavior. Professionalism is the “commitment to carrying out professional responsibilities and an adherence to ethical principles.”3 Professionalism is the pursuit of the tenets of the Hippocratic Oath. As a litmus test, read and reread the oath, and honestly reflect upon your practice.
Another step is to continuously work in multidisciplinary teams, a skill that comes naturally to most hospitalists. In order to fulfill the oath, you should not work as a social worker, but you should advocate for your patients’ social work needs. You need a plethora of other disciplines to help you fulfill your role as a patient advocate. Know and respect the roles that your team members are playing, all of which are invaluable to you and your patients.
An additional step in helping you fulfill your role as a professional is to get the education and skills you need to function effectively within the complex systems in which we currently work. You should incorporate business and management education into your continuing medical education so that you can help patients traverse a system that is complex. You should know and understand the general concepts of value-based payment, insurance exchanges, federal-state-private insurances, and the basic tenets of health systems. You should know how to recognize and reduce waste and unnecessary variation in the system, and know how to measure and improve upon processes.
In the words of Emanuel Ezekiel, MD, PhD, “Learning clinical medicine is necessary for making patient well-being the physician’s primary obligation. But it is not sufficient. To promote professionalism and all that it entails (reducing errors; ensuring safe, consistent, high-quality, and convenient care; removing unnecessary services; and improving the efficiency in the delivery of services), physicians must develop better management skills … Becoming better managers will make physicians better medical professionals”.2
For those entering medical school, nine core competencies can predict success in medical school and later in practice; we should all commit to excellence in these, which go beyond clinical knowledge:
- ethical responsibility to self and others;
- reliability and dependability;
- service orientation;
- social skills;
- capacity for improvement;
- resilience and adaptability;
- cultural competence;
- oral communication; and
- teamwork.
Lastly, a critical step in preventing the erosion of professionalism in medicine is self-regulation. External regulation comes to those who refuse or are unwilling to regulate themselves. Professionalism is a set of skills that can be taught, learned, and modeled. As a new specialty, we all own the success or failure of the reputation of hospitalists as consummate professionals.1
Antibiotic Stewardship and Hospitalists: How to Educate Patients on Antibiotics
Editor’s note: This article originally appeared on SHM’s official blog, “The Hospital Leader,” in June 2015.
“Tell me what you know about antibiotics.”
That’s the discussion I start with hospitalized patients all the time, right after they ask me to prescribe antibiotics for their simple cough or other viral-like illness.
And, from their perspective, asking for antibiotics makes sense. After all, antibiotics have been the physician’s knee-jerk reaction to a number of patient symptoms for decades, especially for a cough or upper respiratory infection. We have inadvertently trained our patients that there is an easy solution to almost any common medical problem.
But patients often answer my question with something like “not much,” coupled with a little surprise that I haven’t already started ordering the prescription.
That’s when I talk about the potential harms of antibiotics. And that’s also when their eyebrows go up. I start with the easy harms, like the fact that many antibiotics can cause diarrhea, a symptom nobody wants to deal with along with their runny nose. Then I move on to the big ones: Use of antibiotics today could make the patient resistant to antibiotics later in life, when they might really need them, and using antibiotics can lead to other painful and even fatal conditions, like Clostridium difficile.
After that, every patient agrees with my recommendations that we hold off on antibiotics for certain, particularly viral-like, ailments.
Change the Conversation. Change the Approach.
It’s a longer conversation, but it’s worth it. Overuse of antibiotics affects not only the patient in front of me, but also entire communities. By creating antibiotic-resistant bacteria, we make everyone more vulnerable to the very diseases the antibiotics were originally intended to treat, like tuberculosis, staph infections, and numerous others.
That’s why the hospitalists in my hospital at Johns Hopkins Bayview teamed up with the infectious diseases division to improve our approach to cellulitis and antibiotic use.
In short, cellulitis is a bacterial skin infection. The most feared bacterial skin infection is MRSA (methicillin-resistant Staphylococcus aureus), a “super bug” that requires highly selective antibiotics like vancomycin; however, other more common and less virulent bacteria also cause cellulitis, and they don’t need super bug fighter medications. Some types of skin ailments, like those caused by poor circulation in the legs, are not infectious at all but can look like cellulitis, even to experienced doctors.
Thanks to the collaboration between infectious disease doctors and hospitalists, the hospitalists are much less likely to prescribe inappropriate antibiotics. That’s a triple-win: It reduces the length of stay for the patient, the incidence of C. diff, and costs.
The Front Line
This concern isn’t limited to a single hospital. There are now more than 44,000 hospitalists nationwide, and every one of us plays an important role in antibiotic stewardship. The bedside is the front line of the fight against antibiotic resistance.
tuberculosis, staph infections, and numerous others.
—Eric Howell, MD, SFHM

There are more than 5,000 hospitals across the country, and hospitalists in every one of them must take responsibility for the appropriate use of antibiotics for their patients.
Announcing SHM’s National Commitment to Antibiotic Stewardship
SHM was proud to join more than 150 major organizations at the White House Forum on Antibiotic Stewardship to announce commitments to implement changes over the next five years that will slow the emergence of antibiotic-resistant bacteria, detect resistant strains, preserve the efficacy of our existing antibiotics, and prevent the spread of resistant infections.
Specifically, SHM has committed to three national initiatives that are aligned with our organizational goal of providing the best possible care for the hospitalized patient and the federal government’s dedication to this important issue:
- Enhance hospitalists’ awareness of key antimicrobial stewardship best practices and ask them to formally commit to at least two behavior changes to reduce inappropriate antimicrobial use and antimicrobial resistance;
- Support national initiatives that advocate for the appropriate use of antimicrobials and promote strategies to reduce antimicrobial resistance; and
- Identify partnerships and other opportunities to support the development of a comprehensive program to implement antimicrobial stewardship best practices in America’s hospitals.
These commitments, which I shared with White House Forum participants, play to the strengths of hospitalists in healthcare: advocacy on behalf of patients and quality improvement and collaboration with others.
What Hospitalists Can Do Now
I also know, however, that you aren’t the kind of people to wait for official pronouncements and campaigns to start a program that will improve the care of hospitalized patients. That’s why SHM and I are recommending that all hospitalists begin to take steps immediately to address this national healthcare crisis:
- Start the conversation with your patients. It’s easy to prescribe antibiotics, but it can also be harmful. Talk with your patients about when antibiotics are medically appropriate and the potential harms they may cause.
- Prescribe antibiotics for specific diagnoses. Prescribing “just in case” is a prescription for antibiotic resistance. Make sure you understand the signs and symptoms of the conditions for which you’re prescribing antibiotics. As we learned at our hospital, cellulitis and venous insufficiency can look similar, but only one responds to antibiotic treatment.
- Work with your infectious disease colleagues. They can help you create systems and diagnose patients to help improve your hospital’s antibiotic stewardship.
After all, we are on the front lines, protecting our current and future patients. And we can’t afford to wait.
Dr. Howell is a veteran hospitalist at Johns Hopkins Bayview Hospital in Baltimore and a past president of SHM.
Editor’s note: This article originally appeared on SHM’s official blog, “The Hospital Leader,” in June 2015.
“Tell me what you know about antibiotics.”
That’s the discussion I start with hospitalized patients all the time, right after they ask me to prescribe antibiotics for their simple cough or other viral-like illness.
And, from their perspective, asking for antibiotics makes sense. After all, antibiotics have been the physician’s knee-jerk reaction to a number of patient symptoms for decades, especially for a cough or upper respiratory infection. We have inadvertently trained our patients that there is an easy solution to almost any common medical problem.
But patients often answer my question with something like “not much,” coupled with a little surprise that I haven’t already started ordering the prescription.
That’s when I talk about the potential harms of antibiotics. And that’s also when their eyebrows go up. I start with the easy harms, like the fact that many antibiotics can cause diarrhea, a symptom nobody wants to deal with along with their runny nose. Then I move on to the big ones: Use of antibiotics today could make the patient resistant to antibiotics later in life, when they might really need them, and using antibiotics can lead to other painful and even fatal conditions, like Clostridium difficile.
After that, every patient agrees with my recommendations that we hold off on antibiotics for certain, particularly viral-like, ailments.
Change the Conversation. Change the Approach.
It’s a longer conversation, but it’s worth it. Overuse of antibiotics affects not only the patient in front of me, but also entire communities. By creating antibiotic-resistant bacteria, we make everyone more vulnerable to the very diseases the antibiotics were originally intended to treat, like tuberculosis, staph infections, and numerous others.
That’s why the hospitalists in my hospital at Johns Hopkins Bayview teamed up with the infectious diseases division to improve our approach to cellulitis and antibiotic use.
In short, cellulitis is a bacterial skin infection. The most feared bacterial skin infection is MRSA (methicillin-resistant Staphylococcus aureus), a “super bug” that requires highly selective antibiotics like vancomycin; however, other more common and less virulent bacteria also cause cellulitis, and they don’t need super bug fighter medications. Some types of skin ailments, like those caused by poor circulation in the legs, are not infectious at all but can look like cellulitis, even to experienced doctors.
Thanks to the collaboration between infectious disease doctors and hospitalists, the hospitalists are much less likely to prescribe inappropriate antibiotics. That’s a triple-win: It reduces the length of stay for the patient, the incidence of C. diff, and costs.
The Front Line
This concern isn’t limited to a single hospital. There are now more than 44,000 hospitalists nationwide, and every one of us plays an important role in antibiotic stewardship. The bedside is the front line of the fight against antibiotic resistance.
tuberculosis, staph infections, and numerous others.
—Eric Howell, MD, SFHM
There are more than 5,000 hospitals across the country, and hospitalists in every one of them must take responsibility for the appropriate use of antibiotics for their patients.
Announcing SHM’s National Commitment to Antibiotic Stewardship
SHM was proud to join more than 150 major organizations at the White House Forum on Antibiotic Stewardship to announce commitments to implement changes over the next five years that will slow the emergence of antibiotic-resistant bacteria, detect resistant strains, preserve the efficacy of our existing antibiotics, and prevent the spread of resistant infections.
Specifically, SHM has committed to three national initiatives that are aligned with our organizational goal of providing the best possible care for the hospitalized patient and the federal government’s dedication to this important issue:
- Enhance hospitalists’ awareness of key antimicrobial stewardship best practices and ask them to formally commit to at least two behavior changes to reduce inappropriate antimicrobial use and antimicrobial resistance;
- Support national initiatives that advocate for the appropriate use of antimicrobials and promote strategies to reduce antimicrobial resistance; and
- Identify partnerships and other opportunities to support the development of a comprehensive program to implement antimicrobial stewardship best practices in America’s hospitals.
These commitments, which I shared with White House Forum participants, play to the strengths of hospitalists in healthcare: advocacy on behalf of patients and quality improvement and collaboration with others.
What Hospitalists Can Do Now
I also know, however, that you aren’t the kind of people to wait for official pronouncements and campaigns to start a program that will improve the care of hospitalized patients. That’s why SHM and I are recommending that all hospitalists begin to take steps immediately to address this national healthcare crisis:
- Start the conversation with your patients. It’s easy to prescribe antibiotics, but it can also be harmful. Talk with your patients about when antibiotics are medically appropriate and the potential harms they may cause.
- Prescribe antibiotics for specific diagnoses. Prescribing “just in case” is a prescription for antibiotic resistance. Make sure you understand the signs and symptoms of the conditions for which you’re prescribing antibiotics. As we learned at our hospital, cellulitis and venous insufficiency can look similar, but only one responds to antibiotic treatment.
- Work with your infectious disease colleagues. They can help you create systems and diagnose patients to help improve your hospital’s antibiotic stewardship.
After all, we are on the front lines, protecting our current and future patients. And we can’t afford to wait.
Dr. Howell is a veteran hospitalist at Johns Hopkins Bayview Hospital in Baltimore and a past president of SHM.
Editor’s note: This article originally appeared on SHM’s official blog, “The Hospital Leader,” in June 2015.
“Tell me what you know about antibiotics.”
That’s the discussion I start with hospitalized patients all the time, right after they ask me to prescribe antibiotics for their simple cough or other viral-like illness.
And, from their perspective, asking for antibiotics makes sense. After all, antibiotics have been the physician’s knee-jerk reaction to a number of patient symptoms for decades, especially for a cough or upper respiratory infection. We have inadvertently trained our patients that there is an easy solution to almost any common medical problem.
But patients often answer my question with something like “not much,” coupled with a little surprise that I haven’t already started ordering the prescription.
That’s when I talk about the potential harms of antibiotics. And that’s also when their eyebrows go up. I start with the easy harms, like the fact that many antibiotics can cause diarrhea, a symptom nobody wants to deal with along with their runny nose. Then I move on to the big ones: Use of antibiotics today could make the patient resistant to antibiotics later in life, when they might really need them, and using antibiotics can lead to other painful and even fatal conditions, like Clostridium difficile.
After that, every patient agrees with my recommendations that we hold off on antibiotics for certain, particularly viral-like, ailments.
Change the Conversation. Change the Approach.
It’s a longer conversation, but it’s worth it. Overuse of antibiotics affects not only the patient in front of me, but also entire communities. By creating antibiotic-resistant bacteria, we make everyone more vulnerable to the very diseases the antibiotics were originally intended to treat, like tuberculosis, staph infections, and numerous others.
That’s why the hospitalists in my hospital at Johns Hopkins Bayview teamed up with the infectious diseases division to improve our approach to cellulitis and antibiotic use.
In short, cellulitis is a bacterial skin infection. The most feared bacterial skin infection is MRSA (methicillin-resistant Staphylococcus aureus), a “super bug” that requires highly selective antibiotics like vancomycin; however, other more common and less virulent bacteria also cause cellulitis, and they don’t need super bug fighter medications. Some types of skin ailments, like those caused by poor circulation in the legs, are not infectious at all but can look like cellulitis, even to experienced doctors.
Thanks to the collaboration between infectious disease doctors and hospitalists, the hospitalists are much less likely to prescribe inappropriate antibiotics. That’s a triple-win: It reduces the length of stay for the patient, the incidence of C. diff, and costs.
The Front Line
This concern isn’t limited to a single hospital. There are now more than 44,000 hospitalists nationwide, and every one of us plays an important role in antibiotic stewardship. The bedside is the front line of the fight against antibiotic resistance.
tuberculosis, staph infections, and numerous others.
—Eric Howell, MD, SFHM
There are more than 5,000 hospitals across the country, and hospitalists in every one of them must take responsibility for the appropriate use of antibiotics for their patients.
Announcing SHM’s National Commitment to Antibiotic Stewardship
SHM was proud to join more than 150 major organizations at the White House Forum on Antibiotic Stewardship to announce commitments to implement changes over the next five years that will slow the emergence of antibiotic-resistant bacteria, detect resistant strains, preserve the efficacy of our existing antibiotics, and prevent the spread of resistant infections.
Specifically, SHM has committed to three national initiatives that are aligned with our organizational goal of providing the best possible care for the hospitalized patient and the federal government’s dedication to this important issue:
- Enhance hospitalists’ awareness of key antimicrobial stewardship best practices and ask them to formally commit to at least two behavior changes to reduce inappropriate antimicrobial use and antimicrobial resistance;
- Support national initiatives that advocate for the appropriate use of antimicrobials and promote strategies to reduce antimicrobial resistance; and
- Identify partnerships and other opportunities to support the development of a comprehensive program to implement antimicrobial stewardship best practices in America’s hospitals.
These commitments, which I shared with White House Forum participants, play to the strengths of hospitalists in healthcare: advocacy on behalf of patients and quality improvement and collaboration with others.
What Hospitalists Can Do Now
I also know, however, that you aren’t the kind of people to wait for official pronouncements and campaigns to start a program that will improve the care of hospitalized patients. That’s why SHM and I are recommending that all hospitalists begin to take steps immediately to address this national healthcare crisis:
- Start the conversation with your patients. It’s easy to prescribe antibiotics, but it can also be harmful. Talk with your patients about when antibiotics are medically appropriate and the potential harms they may cause.
- Prescribe antibiotics for specific diagnoses. Prescribing “just in case” is a prescription for antibiotic resistance. Make sure you understand the signs and symptoms of the conditions for which you’re prescribing antibiotics. As we learned at our hospital, cellulitis and venous insufficiency can look similar, but only one responds to antibiotic treatment.
- Work with your infectious disease colleagues. They can help you create systems and diagnose patients to help improve your hospital’s antibiotic stewardship.
After all, we are on the front lines, protecting our current and future patients. And we can’t afford to wait.
Dr. Howell is a veteran hospitalist at Johns Hopkins Bayview Hospital in Baltimore and a past president of SHM.
The Three-Year Plan
Although 2019 may seem like a long way away, it isn’t too soon to start thinking about and preparing for the Merit-based Incentive Payment System (MIPS) or its (seemingly preferable) alternative, participation in an alternative payment model (APM) such as an ACO, a medical home, or a bundled payment program.
In April, Congress permanently repealed Medicare’s sustainable growth rate (SGR) formula for controlling physician payment. In yet another sign that we are in the midst of the biggest healthcare transformation in a generation, the 18-year-old SGR formula will be replaced by a far-reaching package of payment reforms. Here we will focus on the MIPS and its alternative, an APM, which involves assuming risk for financial loss or gain and measuring and reporting on quality.
The MIPS replaces three existing quality measurement programs that, to greater and lesser degrees, physicians have struggled with:
- Physician Quality Reporting System (PQRS);
- Value-based payment modifier; and
- Meaningful use of electronic health records.
MIPS will not totally eliminate these programs but will instead incorporate yet-to-be-defined elements of them and, presumably, though it is yet unclear, add new elements. For 2015-2018, the current payment system will remain intact. For 2019, physicians will have a choice. Either they must participate in MIPS, which will likely be complex and involve some administrative burden, or derive at least 25% of their practice revenue from an APM.
For those participating in MIPS, physician payment rates will be subject to an up or down adjustment based on performance in four categories: quality, meaningful use of EHRs, resource use, and clinical practice improvement.
There is an opportunity to avoid MIPS altogether, however. One of the most notable elements of the SGR fix is its push for physicians to participate in APMs such as ACOs, medical homes, bundled payment arrangements, and other payment models now being evaluated by the CMS Innovation Center. Physicians who gain a substantial portion—this means 25% in 2019 and 2020, and likely more thereafter—of their revenue through APMs like these will have the dual benefit of being exempt from MIPS participation and receiving a 5% annual bonus through 2024. After that, physicians in APMs will receive annual fee increases of 0.75%, while all other physicians will receive only a 0.25% increase.1
Strategic Thinking for Hospitalists: Enter an APM
If you’re asking yourself where you want your hospitalist practice to be in three years, I would suggest the answer is “in an alternative payment model of one kind or another.”
If you are an employed practice, strategic planning will involve assessing the APMs your hospital or health system is participating in and planning how your hospitalist practice can become a formal member of the arrangement.
If you are a freestanding practice, you should become a student of the APM policy coming from the CMS Innovation Center, and determine the best “insertion point” for your practice, such that you gain at least a quarter of your revenue through an APM within three years.

Reference
- Steinbrook R. The repeal of Medicare’s sustainable growth rate for physician payment. JAMA. 2015;313(20):2025-2026.
Obituary
Remembering Frank Michota, 1967-2015

Frank founded the “Update in Hospital Medicine” series at SHM annual meetings and in the Annals of Internal Medicine. He was a prolific speaker, writer, and teacher. More than that, Frank was an original kind of persona, one who collaborated often and led frequently, but rarely followed. Yet he had the humility to engage and contribute whenever he was asked.
I had the good fortune of working with him on a number of speaking and writing projects in the early days. I recall his charisma, irreverence, and larger-than-life presence whenever he addressed an audience.
Frank’s signature is etched forever on hospital medicine; our patients and hospitalists everywhere are the lucky beneficiaries of his work.
Although 2019 may seem like a long way away, it isn’t too soon to start thinking about and preparing for the Merit-based Incentive Payment System (MIPS) or its (seemingly preferable) alternative, participation in an alternative payment model (APM) such as an ACO, a medical home, or a bundled payment program.
In April, Congress permanently repealed Medicare’s sustainable growth rate (SGR) formula for controlling physician payment. In yet another sign that we are in the midst of the biggest healthcare transformation in a generation, the 18-year-old SGR formula will be replaced by a far-reaching package of payment reforms. Here we will focus on the MIPS and its alternative, an APM, which involves assuming risk for financial loss or gain and measuring and reporting on quality.
The MIPS replaces three existing quality measurement programs that, to greater and lesser degrees, physicians have struggled with:
- Physician Quality Reporting System (PQRS);
- Value-based payment modifier; and
- Meaningful use of electronic health records.
MIPS will not totally eliminate these programs but will instead incorporate yet-to-be-defined elements of them and, presumably, though it is yet unclear, add new elements. For 2015-2018, the current payment system will remain intact. For 2019, physicians will have a choice. Either they must participate in MIPS, which will likely be complex and involve some administrative burden, or derive at least 25% of their practice revenue from an APM.
For those participating in MIPS, physician payment rates will be subject to an up or down adjustment based on performance in four categories: quality, meaningful use of EHRs, resource use, and clinical practice improvement.
There is an opportunity to avoid MIPS altogether, however. One of the most notable elements of the SGR fix is its push for physicians to participate in APMs such as ACOs, medical homes, bundled payment arrangements, and other payment models now being evaluated by the CMS Innovation Center. Physicians who gain a substantial portion—this means 25% in 2019 and 2020, and likely more thereafter—of their revenue through APMs like these will have the dual benefit of being exempt from MIPS participation and receiving a 5% annual bonus through 2024. After that, physicians in APMs will receive annual fee increases of 0.75%, while all other physicians will receive only a 0.25% increase.1
Strategic Thinking for Hospitalists: Enter an APM
If you’re asking yourself where you want your hospitalist practice to be in three years, I would suggest the answer is “in an alternative payment model of one kind or another.”
If you are an employed practice, strategic planning will involve assessing the APMs your hospital or health system is participating in and planning how your hospitalist practice can become a formal member of the arrangement.
If you are a freestanding practice, you should become a student of the APM policy coming from the CMS Innovation Center, and determine the best “insertion point” for your practice, such that you gain at least a quarter of your revenue through an APM within three years.
Reference
- Steinbrook R. The repeal of Medicare’s sustainable growth rate for physician payment. JAMA. 2015;313(20):2025-2026.
Obituary
Remembering Frank Michota, 1967-2015
Frank founded the “Update in Hospital Medicine” series at SHM annual meetings and in the Annals of Internal Medicine. He was a prolific speaker, writer, and teacher. More than that, Frank was an original kind of persona, one who collaborated often and led frequently, but rarely followed. Yet he had the humility to engage and contribute whenever he was asked.
I had the good fortune of working with him on a number of speaking and writing projects in the early days. I recall his charisma, irreverence, and larger-than-life presence whenever he addressed an audience.
Frank’s signature is etched forever on hospital medicine; our patients and hospitalists everywhere are the lucky beneficiaries of his work.
Although 2019 may seem like a long way away, it isn’t too soon to start thinking about and preparing for the Merit-based Incentive Payment System (MIPS) or its (seemingly preferable) alternative, participation in an alternative payment model (APM) such as an ACO, a medical home, or a bundled payment program.
In April, Congress permanently repealed Medicare’s sustainable growth rate (SGR) formula for controlling physician payment. In yet another sign that we are in the midst of the biggest healthcare transformation in a generation, the 18-year-old SGR formula will be replaced by a far-reaching package of payment reforms. Here we will focus on the MIPS and its alternative, an APM, which involves assuming risk for financial loss or gain and measuring and reporting on quality.
The MIPS replaces three existing quality measurement programs that, to greater and lesser degrees, physicians have struggled with:
- Physician Quality Reporting System (PQRS);
- Value-based payment modifier; and
- Meaningful use of electronic health records.
MIPS will not totally eliminate these programs but will instead incorporate yet-to-be-defined elements of them and, presumably, though it is yet unclear, add new elements. For 2015-2018, the current payment system will remain intact. For 2019, physicians will have a choice. Either they must participate in MIPS, which will likely be complex and involve some administrative burden, or derive at least 25% of their practice revenue from an APM.
For those participating in MIPS, physician payment rates will be subject to an up or down adjustment based on performance in four categories: quality, meaningful use of EHRs, resource use, and clinical practice improvement.
There is an opportunity to avoid MIPS altogether, however. One of the most notable elements of the SGR fix is its push for physicians to participate in APMs such as ACOs, medical homes, bundled payment arrangements, and other payment models now being evaluated by the CMS Innovation Center. Physicians who gain a substantial portion—this means 25% in 2019 and 2020, and likely more thereafter—of their revenue through APMs like these will have the dual benefit of being exempt from MIPS participation and receiving a 5% annual bonus through 2024. After that, physicians in APMs will receive annual fee increases of 0.75%, while all other physicians will receive only a 0.25% increase.1
Strategic Thinking for Hospitalists: Enter an APM
If you’re asking yourself where you want your hospitalist practice to be in three years, I would suggest the answer is “in an alternative payment model of one kind or another.”
If you are an employed practice, strategic planning will involve assessing the APMs your hospital or health system is participating in and planning how your hospitalist practice can become a formal member of the arrangement.
If you are a freestanding practice, you should become a student of the APM policy coming from the CMS Innovation Center, and determine the best “insertion point” for your practice, such that you gain at least a quarter of your revenue through an APM within three years.
Reference
- Steinbrook R. The repeal of Medicare’s sustainable growth rate for physician payment. JAMA. 2015;313(20):2025-2026.
Obituary
Remembering Frank Michota, 1967-2015
Frank founded the “Update in Hospital Medicine” series at SHM annual meetings and in the Annals of Internal Medicine. He was a prolific speaker, writer, and teacher. More than that, Frank was an original kind of persona, one who collaborated often and led frequently, but rarely followed. Yet he had the humility to engage and contribute whenever he was asked.
I had the good fortune of working with him on a number of speaking and writing projects in the early days. I recall his charisma, irreverence, and larger-than-life presence whenever he addressed an audience.
Frank’s signature is etched forever on hospital medicine; our patients and hospitalists everywhere are the lucky beneficiaries of his work.
Institute of Medicine Report Prompts Debate Over Graduate Medical Education Funding, Oversight
Ever since 1997, when the federal Balanced Budget Act froze Medicare’s overall funding for graduate medical education, debates have flared regularly over whether and how the U.S. government should support medical resident training.
Discussions about the possible redistribution of billions of dollars are bound to make people nervous, but the controversy reached a fever pitch in 2014 when the Institute of Medicine released a report penned by a 21-member committee that recommended significant—and contentious—changes to the existing graduate medical education (GME) financing and governance structure to “address current deficiencies and better shape the physician workforce for the future.”
Should Medicare shake up the system to redistribute existing training slots to where they’re needed most, as the report recommends? Should it instead lift its funding cap to avert a potential bottleneck in the physician pipeline, as several medical associations have requested? One year later, the report has gained little traction amid a largely unchanged status quo that few experts believe is ultimately sustainable. The continuing debate, however, has prompted fresh questions about whether the current GME structure is adequately supporting the nation’s healthcare needs and has spurred widespread agreement on the need for greater transparency, accountability, and innovation.
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and one of the report’s co-authors, says she has seen firsthand the challenges arising from a lack of physicians in multiple specialties, especially in rural areas. “We believed that simply adding new money to a system that is outdated would not solve the issues in physician education and physician workforce,” she says.
Some HM leaders and other physicians’ groups have cautiously welcomed the report’s focus on better equipping doctors for a rapidly changing reality.
“It wasn’t wrong for them to look at this,” says Darlene B. Tad-y, MD, FHM, chair of the SHM Physicians in Training Committee and assistant professor of medicine at the University of Colorado in Denver. “And it’s probably not wrong for them to propose new ways to think about how we fund GME.”
In fact, she says, efforts to align such funding with healthcare funding in general could be timely in the face of added pressures like ensuring that new insurance beneficiaries have access to primary care.
Scott Sears, MD, FACP, MBA, chief clinical officer of Tacoma, Wash.-based hospitalist management firm Sound Physicians, says healthcare is also moving rapidly toward managing populations as part of team-based care that increases quality while lowering costs. So why not better align GME with innovative Medicare initiatives like bundled payments, he asks, and then use the savings to reward those training programs that accept the risk and achieve results?
“Shifting some of our education to match what Medicare is trying to drive out in the real world, I think, is long overdue,” Dr. Sears says.
Other groups, such as the Association of American Medical Colleges, however, contend that the report’s prescriptions are far less helpful than its diagnoses. “Politically, there’s just stuff in there for everybody to hate,” says Atul Grover, MD, PhD, FACP, FCCP, the AAMC’s chief public policy officer. “I think [the IOM report] did a decent job of pointing out some of the things that we want to improve moving forward, but I’m not sure that the answers are quite right.”
An Uneven Funding Landscape
The strong opinions engendered by the topic underscore the high stakes involved in GME. Every year, the federal government doles out about $15 billion for residency training, including about $10 billion from Medicare coffers. Medicare’s share is divided into two main funding streams that flow primarily to academic medical centers: direct graduate medical education (DGME) and indirect medical education (IME) payments. The first covers training expenses, while the second reimburses teaching hospitals that care for Medicare patients while training residents.
Some skeptics have questioned whether the government should be funding medical education at all, noting that the arrangement is utterly unique to the field. Advocates have countered that the funding concept was embedded in the original Medicare legislation and that it correctly recognized the added cost of offering GME training while providing more complex Medicare beneficiaries with specialty services.
Nearly everyone acknowledges that there are still enough residency slots for all U.S. graduates, but Dr. Grover says residency programs aren’t growing nearly fast enough to keep pace with medical school enrollment, creating a growing mismatch and a looming bottleneck in the supply chain. Compared to medical school numbers in 2002, for example, the AAMC says enrollment is on track to expand 29% by 2019, while osteopathic schools are set to expand by 162% over the same timeframe.
It wasn’t wrong for the [Institute of Medicine] to look at this. And it’s probably not wrong for them to propose new ways to think about how we fund GME. 
—Darlene B. Tad-y, MD, FHM, assistant professor of medicine, University of Colorado, Denver, chair, SHM Physicians in Training Committee
Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t. I think the challenge is that the devil’s in the details of how you do that. 
—Vikas Parekh, MD, FACP, SFHM, associate director, hospitalist program, University of Michigan, Ann Arbor, chair, SHM Academic Hospitalist Committee
Despite the continued freeze in Medicare funding, many large medical institutions continue to add residency spots.
“We’ve been hundreds of residency positions over our cap for a very long time,” says Vikas Parekh, MD, FACP, SFHM, associate director of the hospitalist program at the University of Michigan in Ann Arbor. “The hospital funds them through hospital operating margin because in the net, they still view the investment as worthwhile.”
Alternatively, some non-university-based training programs have secured money from other sources to fund their residency positions, potentially creating new funding models for the future if the programs can demonstrate both quality and stability.
One key rationale for the IOM report’s proposed overhaul, however, is the longstanding and sizeable geographical disparity in Medicare’s per capita GME spending, which has skewed heavily toward the Northeast. A 2013 study, in fact, found that one-fifth of all DGME funding in 2010—an estimated $2 billion—went to New York State alone.1 Florida, which recently overtook New York as the third most populous state, received only one-eighth as much money. And Mississippi—the state with the lowest doctor-to-patient ratio—received only $22 million, or about one-ninetieth as much.
The IOM report also suggests that the long-standing GME payment plan has yielded little data on whether it actually accomplishes what it was designed to do: help establish a well-prepared medical workforce in a cost-effective way. In response, one major IOM recommendation is to maintain the overall level of Medicare support but tie some of the payments to performance to ensure oversight and accountability, and provide new incentives for innovation in the content and financing of training programs.
As with other CMS initiatives, however, getting everyone to agree on which quality metrics to use in evaluating GME training could take awhile. For example, should Medicare judge the performance of the trainees, the GME programs, or even the sponsoring institutions? Despite the proliferation of performance-based carrots and sticks elsewhere in healthcare, Dr. Tad-y says, such incentives may work less well for GME.
“One thing that’s inherent with trainees is that they’re trainees,” she says. “They’re not as efficient or as effective as someone who’s an expert, right? That’s why it’s training.”
Dr. Parekh, who also serves as chair of the SHM Academic Hospitalist Committee, agrees that finding the right outcome measures could be tough. “It gets very dicey, because how do you define who’s a good doctor?” he says. Currently, residents often are assessed via the reputation and history of the training program. “People say, ‘I know that the people coming out of that program are good because they’ve always been good, and it’s a reputable program and has a big name.’ But it’s not objective data,” he says.
Dr. Sears, of Sound Physicians, notes that it’s also often difficult to attribute patients to specific providers.
“Many times in graduate medical education, patients are going in and out of the program or in and out of the hospital, and how do you attribute?” he says. “I think it becomes very complex.”
A New Take on Transformation
Another IOM recommendation would create a single GME fund with two subsidiaries: an operational fund for ongoing support and a transformation fund. The latter fund would finance four new initiatives to:
- Develop and evaluate innovative GME programs;
- Determine and validate appropriate performance measures;
- Establish pilot projects to test out alternative payment methods; and
- Award new training positions based on priority disciplines—such as primary care—and underserved geographic areas.
A related recommendation seeks to modernize the GME payment methodology. For example, the committee urged Medicare to combine the indirect and direct funding streams into one payment based on a national per-resident amount and adjusted according to each location. In addition, the report endorsed performance-based payments based on the results of pilots launched under the transformation fund.
Dr. Sears says he appreciates the report’s effort to address shortfalls in primary care providers relative to specialists. “That’s not to say that specialty medicine isn’t incredibly important, because it is,” he says. “But I think incentivizing or reallocating spots to ensure that we have adequate primary care physician coverage throughout the country will have tremendous impact on the ability to care for an aging population in the United States, at least.”
I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that. Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable. —Deborah Powell, MD, dean emerita, University of Minnesota Medical School, IOM committee member 
Shifting some of our [medical] education to match what Medicare is trying to drive out in the real world, I think, is long overdue. —Scott Sears, MD, FACP, MBA, chief clinical officer, Sound Physicians, Tacoma, Wash. 
Dr. Parekh agrees, at least in part.
“Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t,” he says. “I think the challenge is that the devil’s in the details of how you do that.”
A priority-based GME system, he continues, could potentially influence what type of physicians are trained.
“In my mind, it’s not irrational to think that if GME funding was more targeted around expanding slots in certain specialties and not expanding slots in other specialties, that there would be some ability to influence the workforce,” Dr. Parekh says. Influencing where residents go may be more difficult, though a growing mismatch between medical graduates and available residency slots might add a new wrinkle to that debate, as well.
Currently, U.S. medical graduates fill only about 60% of residency slots for specialties like internal medicine—a main conduit for hospital medicine—while foreign graduates make up the remainder.
“So who’s the first that’s going to be squeezed out? It will be foreign medical graduates,” Dr. Parekh says. Many of those graduates come to the U.S. on J-1 visas, which carry a payback requirement: practicing in underserved areas. “One worry is, will rural and underserved areas suffer from a physician shortage because U.S. grads won’t want to work there after you start squeezing out all of the foreign medical grads?” he asks.
Clear Line of Sight?
Dr. Parekh also supports efforts to establish a clearer connection between the funding’s intent and where the money actually goes. The IOM report’s proposal to do so, however, raises yet another controversy around the true purpose of IME funding. Teaching hospitals argue that the money should continue to be used to reimburse them for the added costs of providing comprehensive and specialized care like level I trauma centers to their more complex Medicare patient populations.
Number one, [the IOM] came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources. We found that problematic, given all the evidence we have of the growing, aging population. —Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer, Association of American Medical Colleges 
A big part of the problem here is that people are free agents. If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists. —Daniel Brotman, MD, FACP, SFHM, chair, SHM Education Committee, director, hospitalist program, Johns Hopkins Hospital, Baltimore 
Accordingly, the AAMC panned the report’s recommendation to replace separate IME funding with a single fund directed toward the GME sponsoring institution and subdivided instead into the operational and transformation funds. Dr. Grover says setting up a transformation fund with new money would make sense, but not as a carve-out from existing support.
“You’re removing those resources from the system and not replacing them, which is a challenge,” he says.
Medical schools are more inclined to want the money directed toward training goals, especially if they are to be held accountable for GME outcomes. “Right now, the hospital gets it, and it’s basically somewhere in the bottom line,” Dr. Parekh says. “No one really knows where that money goes. There’s very little accountability or clarity of purpose for that dollar.”
Amid the ongoing debate, the call for more transparency and accountability in GME seems to be gaining the most ground. “I don’t see tons of downside from it,” Dr. Parekh says. “I think it sheds light on the current funding environment and makes people have to justify a little bit more what they’re doing with that money.”
Dr. Tad-y puts it this way: “If you made your own budget at home, the first thing you’d do is try to figure out where all your money goes and what you’re spending your money on.” If Medicare is concerned that its GME money isn’t being spent wisely, then, the first step would be to do some accounting. “And that means a little bit of transparency,” she says. “I don’t think that’s a bad thing, to know exactly what we’re paying for; that makes sense. I mean, we do that for everything else.”
SHM and most other medical associations also agree on the necessary goal of increasing the nation’s primary care capacity, even if they differ on the details of how best to do so. In the long run, however, some observers say growing the workforce—whether that of primary care providers or of hospitalists—may depend less on the total number of residency spots and more on the enthusiasm of program leadership and the attractiveness of job conditions such as salary and workload.
“A big part of the problem here is that people are free agents,” says Daniel Brotman, MD, FACP, SFHM, chair of the SHM Education Committee and director of the hospitalist program at the Johns Hopkins Hospital in Baltimore. “If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists.”
Whatever happens, Dr. Parekh says hospitalists are well positioned to be integral parts of improving quality, accountability, and innovation in residency training programs.
“I think if more GME money is targeted toward the outcomes of the GME programs, hospitalists are going to be tapped to help with that work, in terms of training and broadening their skill sets,” he says. “So I think it’s a great opportunity.”
Bryn Nelson is a freelance medical writer in Seattle.
References
- Mullan F, Chen C, Steinmetz E. The geography of graduate medical education: imbalances signal need for new distribution policies. Health Aff. 2013;32(11):1914-1921.
Ever since 1997, when the federal Balanced Budget Act froze Medicare’s overall funding for graduate medical education, debates have flared regularly over whether and how the U.S. government should support medical resident training.
Discussions about the possible redistribution of billions of dollars are bound to make people nervous, but the controversy reached a fever pitch in 2014 when the Institute of Medicine released a report penned by a 21-member committee that recommended significant—and contentious—changes to the existing graduate medical education (GME) financing and governance structure to “address current deficiencies and better shape the physician workforce for the future.”
Should Medicare shake up the system to redistribute existing training slots to where they’re needed most, as the report recommends? Should it instead lift its funding cap to avert a potential bottleneck in the physician pipeline, as several medical associations have requested? One year later, the report has gained little traction amid a largely unchanged status quo that few experts believe is ultimately sustainable. The continuing debate, however, has prompted fresh questions about whether the current GME structure is adequately supporting the nation’s healthcare needs and has spurred widespread agreement on the need for greater transparency, accountability, and innovation.
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and one of the report’s co-authors, says she has seen firsthand the challenges arising from a lack of physicians in multiple specialties, especially in rural areas. “We believed that simply adding new money to a system that is outdated would not solve the issues in physician education and physician workforce,” she says.
Some HM leaders and other physicians’ groups have cautiously welcomed the report’s focus on better equipping doctors for a rapidly changing reality.
“It wasn’t wrong for them to look at this,” says Darlene B. Tad-y, MD, FHM, chair of the SHM Physicians in Training Committee and assistant professor of medicine at the University of Colorado in Denver. “And it’s probably not wrong for them to propose new ways to think about how we fund GME.”
In fact, she says, efforts to align such funding with healthcare funding in general could be timely in the face of added pressures like ensuring that new insurance beneficiaries have access to primary care.
Scott Sears, MD, FACP, MBA, chief clinical officer of Tacoma, Wash.-based hospitalist management firm Sound Physicians, says healthcare is also moving rapidly toward managing populations as part of team-based care that increases quality while lowering costs. So why not better align GME with innovative Medicare initiatives like bundled payments, he asks, and then use the savings to reward those training programs that accept the risk and achieve results?
“Shifting some of our education to match what Medicare is trying to drive out in the real world, I think, is long overdue,” Dr. Sears says.
Other groups, such as the Association of American Medical Colleges, however, contend that the report’s prescriptions are far less helpful than its diagnoses. “Politically, there’s just stuff in there for everybody to hate,” says Atul Grover, MD, PhD, FACP, FCCP, the AAMC’s chief public policy officer. “I think [the IOM report] did a decent job of pointing out some of the things that we want to improve moving forward, but I’m not sure that the answers are quite right.”
An Uneven Funding Landscape
The strong opinions engendered by the topic underscore the high stakes involved in GME. Every year, the federal government doles out about $15 billion for residency training, including about $10 billion from Medicare coffers. Medicare’s share is divided into two main funding streams that flow primarily to academic medical centers: direct graduate medical education (DGME) and indirect medical education (IME) payments. The first covers training expenses, while the second reimburses teaching hospitals that care for Medicare patients while training residents.
Some skeptics have questioned whether the government should be funding medical education at all, noting that the arrangement is utterly unique to the field. Advocates have countered that the funding concept was embedded in the original Medicare legislation and that it correctly recognized the added cost of offering GME training while providing more complex Medicare beneficiaries with specialty services.
Nearly everyone acknowledges that there are still enough residency slots for all U.S. graduates, but Dr. Grover says residency programs aren’t growing nearly fast enough to keep pace with medical school enrollment, creating a growing mismatch and a looming bottleneck in the supply chain. Compared to medical school numbers in 2002, for example, the AAMC says enrollment is on track to expand 29% by 2019, while osteopathic schools are set to expand by 162% over the same timeframe.
It wasn’t wrong for the [Institute of Medicine] to look at this. And it’s probably not wrong for them to propose new ways to think about how we fund GME. —Darlene B. Tad-y, MD, FHM, assistant professor of medicine, University of Colorado, Denver, chair, SHM Physicians in Training Committee
Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t. I think the challenge is that the devil’s in the details of how you do that. —Vikas Parekh, MD, FACP, SFHM, associate director, hospitalist program, University of Michigan, Ann Arbor, chair, SHM Academic Hospitalist Committee
Despite the continued freeze in Medicare funding, many large medical institutions continue to add residency spots.
“We’ve been hundreds of residency positions over our cap for a very long time,” says Vikas Parekh, MD, FACP, SFHM, associate director of the hospitalist program at the University of Michigan in Ann Arbor. “The hospital funds them through hospital operating margin because in the net, they still view the investment as worthwhile.”
Alternatively, some non-university-based training programs have secured money from other sources to fund their residency positions, potentially creating new funding models for the future if the programs can demonstrate both quality and stability.
One key rationale for the IOM report’s proposed overhaul, however, is the longstanding and sizeable geographical disparity in Medicare’s per capita GME spending, which has skewed heavily toward the Northeast. A 2013 study, in fact, found that one-fifth of all DGME funding in 2010—an estimated $2 billion—went to New York State alone.1 Florida, which recently overtook New York as the third most populous state, received only one-eighth as much money. And Mississippi—the state with the lowest doctor-to-patient ratio—received only $22 million, or about one-ninetieth as much.
The IOM report also suggests that the long-standing GME payment plan has yielded little data on whether it actually accomplishes what it was designed to do: help establish a well-prepared medical workforce in a cost-effective way. In response, one major IOM recommendation is to maintain the overall level of Medicare support but tie some of the payments to performance to ensure oversight and accountability, and provide new incentives for innovation in the content and financing of training programs.
As with other CMS initiatives, however, getting everyone to agree on which quality metrics to use in evaluating GME training could take awhile. For example, should Medicare judge the performance of the trainees, the GME programs, or even the sponsoring institutions? Despite the proliferation of performance-based carrots and sticks elsewhere in healthcare, Dr. Tad-y says, such incentives may work less well for GME.
“One thing that’s inherent with trainees is that they’re trainees,” she says. “They’re not as efficient or as effective as someone who’s an expert, right? That’s why it’s training.”
Dr. Parekh, who also serves as chair of the SHM Academic Hospitalist Committee, agrees that finding the right outcome measures could be tough. “It gets very dicey, because how do you define who’s a good doctor?” he says. Currently, residents often are assessed via the reputation and history of the training program. “People say, ‘I know that the people coming out of that program are good because they’ve always been good, and it’s a reputable program and has a big name.’ But it’s not objective data,” he says.
Dr. Sears, of Sound Physicians, notes that it’s also often difficult to attribute patients to specific providers.
“Many times in graduate medical education, patients are going in and out of the program or in and out of the hospital, and how do you attribute?” he says. “I think it becomes very complex.”
A New Take on Transformation
Another IOM recommendation would create a single GME fund with two subsidiaries: an operational fund for ongoing support and a transformation fund. The latter fund would finance four new initiatives to:
- Develop and evaluate innovative GME programs;
- Determine and validate appropriate performance measures;
- Establish pilot projects to test out alternative payment methods; and
- Award new training positions based on priority disciplines—such as primary care—and underserved geographic areas.
A related recommendation seeks to modernize the GME payment methodology. For example, the committee urged Medicare to combine the indirect and direct funding streams into one payment based on a national per-resident amount and adjusted according to each location. In addition, the report endorsed performance-based payments based on the results of pilots launched under the transformation fund.
Dr. Sears says he appreciates the report’s effort to address shortfalls in primary care providers relative to specialists. “That’s not to say that specialty medicine isn’t incredibly important, because it is,” he says. “But I think incentivizing or reallocating spots to ensure that we have adequate primary care physician coverage throughout the country will have tremendous impact on the ability to care for an aging population in the United States, at least.”
I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that. Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable. —Deborah Powell, MD, dean emerita, University of Minnesota Medical School, IOM committee member
Shifting some of our [medical] education to match what Medicare is trying to drive out in the real world, I think, is long overdue. —Scott Sears, MD, FACP, MBA, chief clinical officer, Sound Physicians, Tacoma, Wash.
Dr. Parekh agrees, at least in part.
“Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t,” he says. “I think the challenge is that the devil’s in the details of how you do that.”
A priority-based GME system, he continues, could potentially influence what type of physicians are trained.
“In my mind, it’s not irrational to think that if GME funding was more targeted around expanding slots in certain specialties and not expanding slots in other specialties, that there would be some ability to influence the workforce,” Dr. Parekh says. Influencing where residents go may be more difficult, though a growing mismatch between medical graduates and available residency slots might add a new wrinkle to that debate, as well.
Currently, U.S. medical graduates fill only about 60% of residency slots for specialties like internal medicine—a main conduit for hospital medicine—while foreign graduates make up the remainder.
“So who’s the first that’s going to be squeezed out? It will be foreign medical graduates,” Dr. Parekh says. Many of those graduates come to the U.S. on J-1 visas, which carry a payback requirement: practicing in underserved areas. “One worry is, will rural and underserved areas suffer from a physician shortage because U.S. grads won’t want to work there after you start squeezing out all of the foreign medical grads?” he asks.
Clear Line of Sight?
Dr. Parekh also supports efforts to establish a clearer connection between the funding’s intent and where the money actually goes. The IOM report’s proposal to do so, however, raises yet another controversy around the true purpose of IME funding. Teaching hospitals argue that the money should continue to be used to reimburse them for the added costs of providing comprehensive and specialized care like level I trauma centers to their more complex Medicare patient populations.
Number one, [the IOM] came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources. We found that problematic, given all the evidence we have of the growing, aging population. —Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer, Association of American Medical Colleges
A big part of the problem here is that people are free agents. If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists. —Daniel Brotman, MD, FACP, SFHM, chair, SHM Education Committee, director, hospitalist program, Johns Hopkins Hospital, Baltimore
Accordingly, the AAMC panned the report’s recommendation to replace separate IME funding with a single fund directed toward the GME sponsoring institution and subdivided instead into the operational and transformation funds. Dr. Grover says setting up a transformation fund with new money would make sense, but not as a carve-out from existing support.
“You’re removing those resources from the system and not replacing them, which is a challenge,” he says.
Medical schools are more inclined to want the money directed toward training goals, especially if they are to be held accountable for GME outcomes. “Right now, the hospital gets it, and it’s basically somewhere in the bottom line,” Dr. Parekh says. “No one really knows where that money goes. There’s very little accountability or clarity of purpose for that dollar.”
Amid the ongoing debate, the call for more transparency and accountability in GME seems to be gaining the most ground. “I don’t see tons of downside from it,” Dr. Parekh says. “I think it sheds light on the current funding environment and makes people have to justify a little bit more what they’re doing with that money.”
Dr. Tad-y puts it this way: “If you made your own budget at home, the first thing you’d do is try to figure out where all your money goes and what you’re spending your money on.” If Medicare is concerned that its GME money isn’t being spent wisely, then, the first step would be to do some accounting. “And that means a little bit of transparency,” she says. “I don’t think that’s a bad thing, to know exactly what we’re paying for; that makes sense. I mean, we do that for everything else.”
SHM and most other medical associations also agree on the necessary goal of increasing the nation’s primary care capacity, even if they differ on the details of how best to do so. In the long run, however, some observers say growing the workforce—whether that of primary care providers or of hospitalists—may depend less on the total number of residency spots and more on the enthusiasm of program leadership and the attractiveness of job conditions such as salary and workload.
“A big part of the problem here is that people are free agents,” says Daniel Brotman, MD, FACP, SFHM, chair of the SHM Education Committee and director of the hospitalist program at the Johns Hopkins Hospital in Baltimore. “If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists.”
Whatever happens, Dr. Parekh says hospitalists are well positioned to be integral parts of improving quality, accountability, and innovation in residency training programs.
“I think if more GME money is targeted toward the outcomes of the GME programs, hospitalists are going to be tapped to help with that work, in terms of training and broadening their skill sets,” he says. “So I think it’s a great opportunity.”
Bryn Nelson is a freelance medical writer in Seattle.
References
- Mullan F, Chen C, Steinmetz E. The geography of graduate medical education: imbalances signal need for new distribution policies. Health Aff. 2013;32(11):1914-1921.
Ever since 1997, when the federal Balanced Budget Act froze Medicare’s overall funding for graduate medical education, debates have flared regularly over whether and how the U.S. government should support medical resident training.
Discussions about the possible redistribution of billions of dollars are bound to make people nervous, but the controversy reached a fever pitch in 2014 when the Institute of Medicine released a report penned by a 21-member committee that recommended significant—and contentious—changes to the existing graduate medical education (GME) financing and governance structure to “address current deficiencies and better shape the physician workforce for the future.”
Should Medicare shake up the system to redistribute existing training slots to where they’re needed most, as the report recommends? Should it instead lift its funding cap to avert a potential bottleneck in the physician pipeline, as several medical associations have requested? One year later, the report has gained little traction amid a largely unchanged status quo that few experts believe is ultimately sustainable. The continuing debate, however, has prompted fresh questions about whether the current GME structure is adequately supporting the nation’s healthcare needs and has spurred widespread agreement on the need for greater transparency, accountability, and innovation.
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and one of the report’s co-authors, says she has seen firsthand the challenges arising from a lack of physicians in multiple specialties, especially in rural areas. “We believed that simply adding new money to a system that is outdated would not solve the issues in physician education and physician workforce,” she says.
Some HM leaders and other physicians’ groups have cautiously welcomed the report’s focus on better equipping doctors for a rapidly changing reality.
“It wasn’t wrong for them to look at this,” says Darlene B. Tad-y, MD, FHM, chair of the SHM Physicians in Training Committee and assistant professor of medicine at the University of Colorado in Denver. “And it’s probably not wrong for them to propose new ways to think about how we fund GME.”
In fact, she says, efforts to align such funding with healthcare funding in general could be timely in the face of added pressures like ensuring that new insurance beneficiaries have access to primary care.
Scott Sears, MD, FACP, MBA, chief clinical officer of Tacoma, Wash.-based hospitalist management firm Sound Physicians, says healthcare is also moving rapidly toward managing populations as part of team-based care that increases quality while lowering costs. So why not better align GME with innovative Medicare initiatives like bundled payments, he asks, and then use the savings to reward those training programs that accept the risk and achieve results?
“Shifting some of our education to match what Medicare is trying to drive out in the real world, I think, is long overdue,” Dr. Sears says.
Other groups, such as the Association of American Medical Colleges, however, contend that the report’s prescriptions are far less helpful than its diagnoses. “Politically, there’s just stuff in there for everybody to hate,” says Atul Grover, MD, PhD, FACP, FCCP, the AAMC’s chief public policy officer. “I think [the IOM report] did a decent job of pointing out some of the things that we want to improve moving forward, but I’m not sure that the answers are quite right.”
An Uneven Funding Landscape
The strong opinions engendered by the topic underscore the high stakes involved in GME. Every year, the federal government doles out about $15 billion for residency training, including about $10 billion from Medicare coffers. Medicare’s share is divided into two main funding streams that flow primarily to academic medical centers: direct graduate medical education (DGME) and indirect medical education (IME) payments. The first covers training expenses, while the second reimburses teaching hospitals that care for Medicare patients while training residents.
Some skeptics have questioned whether the government should be funding medical education at all, noting that the arrangement is utterly unique to the field. Advocates have countered that the funding concept was embedded in the original Medicare legislation and that it correctly recognized the added cost of offering GME training while providing more complex Medicare beneficiaries with specialty services.
Nearly everyone acknowledges that there are still enough residency slots for all U.S. graduates, but Dr. Grover says residency programs aren’t growing nearly fast enough to keep pace with medical school enrollment, creating a growing mismatch and a looming bottleneck in the supply chain. Compared to medical school numbers in 2002, for example, the AAMC says enrollment is on track to expand 29% by 2019, while osteopathic schools are set to expand by 162% over the same timeframe.
It wasn’t wrong for the [Institute of Medicine] to look at this. And it’s probably not wrong for them to propose new ways to think about how we fund GME. —Darlene B. Tad-y, MD, FHM, assistant professor of medicine, University of Colorado, Denver, chair, SHM Physicians in Training Committee
Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t. I think the challenge is that the devil’s in the details of how you do that. —Vikas Parekh, MD, FACP, SFHM, associate director, hospitalist program, University of Michigan, Ann Arbor, chair, SHM Academic Hospitalist Committee
Despite the continued freeze in Medicare funding, many large medical institutions continue to add residency spots.
“We’ve been hundreds of residency positions over our cap for a very long time,” says Vikas Parekh, MD, FACP, SFHM, associate director of the hospitalist program at the University of Michigan in Ann Arbor. “The hospital funds them through hospital operating margin because in the net, they still view the investment as worthwhile.”
Alternatively, some non-university-based training programs have secured money from other sources to fund their residency positions, potentially creating new funding models for the future if the programs can demonstrate both quality and stability.
One key rationale for the IOM report’s proposed overhaul, however, is the longstanding and sizeable geographical disparity in Medicare’s per capita GME spending, which has skewed heavily toward the Northeast. A 2013 study, in fact, found that one-fifth of all DGME funding in 2010—an estimated $2 billion—went to New York State alone.1 Florida, which recently overtook New York as the third most populous state, received only one-eighth as much money. And Mississippi—the state with the lowest doctor-to-patient ratio—received only $22 million, or about one-ninetieth as much.
The IOM report also suggests that the long-standing GME payment plan has yielded little data on whether it actually accomplishes what it was designed to do: help establish a well-prepared medical workforce in a cost-effective way. In response, one major IOM recommendation is to maintain the overall level of Medicare support but tie some of the payments to performance to ensure oversight and accountability, and provide new incentives for innovation in the content and financing of training programs.
As with other CMS initiatives, however, getting everyone to agree on which quality metrics to use in evaluating GME training could take awhile. For example, should Medicare judge the performance of the trainees, the GME programs, or even the sponsoring institutions? Despite the proliferation of performance-based carrots and sticks elsewhere in healthcare, Dr. Tad-y says, such incentives may work less well for GME.
“One thing that’s inherent with trainees is that they’re trainees,” she says. “They’re not as efficient or as effective as someone who’s an expert, right? That’s why it’s training.”
Dr. Parekh, who also serves as chair of the SHM Academic Hospitalist Committee, agrees that finding the right outcome measures could be tough. “It gets very dicey, because how do you define who’s a good doctor?” he says. Currently, residents often are assessed via the reputation and history of the training program. “People say, ‘I know that the people coming out of that program are good because they’ve always been good, and it’s a reputable program and has a big name.’ But it’s not objective data,” he says.
Dr. Sears, of Sound Physicians, notes that it’s also often difficult to attribute patients to specific providers.
“Many times in graduate medical education, patients are going in and out of the program or in and out of the hospital, and how do you attribute?” he says. “I think it becomes very complex.”
A New Take on Transformation
Another IOM recommendation would create a single GME fund with two subsidiaries: an operational fund for ongoing support and a transformation fund. The latter fund would finance four new initiatives to:
- Develop and evaluate innovative GME programs;
- Determine and validate appropriate performance measures;
- Establish pilot projects to test out alternative payment methods; and
- Award new training positions based on priority disciplines—such as primary care—and underserved geographic areas.
A related recommendation seeks to modernize the GME payment methodology. For example, the committee urged Medicare to combine the indirect and direct funding streams into one payment based on a national per-resident amount and adjusted according to each location. In addition, the report endorsed performance-based payments based on the results of pilots launched under the transformation fund.
Dr. Sears says he appreciates the report’s effort to address shortfalls in primary care providers relative to specialists. “That’s not to say that specialty medicine isn’t incredibly important, because it is,” he says. “But I think incentivizing or reallocating spots to ensure that we have adequate primary care physician coverage throughout the country will have tremendous impact on the ability to care for an aging population in the United States, at least.”
I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that. Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable. —Deborah Powell, MD, dean emerita, University of Minnesota Medical School, IOM committee member
Shifting some of our [medical] education to match what Medicare is trying to drive out in the real world, I think, is long overdue. —Scott Sears, MD, FACP, MBA, chief clinical officer, Sound Physicians, Tacoma, Wash.
Dr. Parekh agrees, at least in part.
“Fundamentally, the idea is not a bad one, to say that programs that were more aligned with national needs and priorities in terms of how they train physicians would get more funding, and those that did not wouldn’t,” he says. “I think the challenge is that the devil’s in the details of how you do that.”
A priority-based GME system, he continues, could potentially influence what type of physicians are trained.
“In my mind, it’s not irrational to think that if GME funding was more targeted around expanding slots in certain specialties and not expanding slots in other specialties, that there would be some ability to influence the workforce,” Dr. Parekh says. Influencing where residents go may be more difficult, though a growing mismatch between medical graduates and available residency slots might add a new wrinkle to that debate, as well.
Currently, U.S. medical graduates fill only about 60% of residency slots for specialties like internal medicine—a main conduit for hospital medicine—while foreign graduates make up the remainder.
“So who’s the first that’s going to be squeezed out? It will be foreign medical graduates,” Dr. Parekh says. Many of those graduates come to the U.S. on J-1 visas, which carry a payback requirement: practicing in underserved areas. “One worry is, will rural and underserved areas suffer from a physician shortage because U.S. grads won’t want to work there after you start squeezing out all of the foreign medical grads?” he asks.
Clear Line of Sight?
Dr. Parekh also supports efforts to establish a clearer connection between the funding’s intent and where the money actually goes. The IOM report’s proposal to do so, however, raises yet another controversy around the true purpose of IME funding. Teaching hospitals argue that the money should continue to be used to reimburse them for the added costs of providing comprehensive and specialized care like level I trauma centers to their more complex Medicare patient populations.
Number one, [the IOM] came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources. We found that problematic, given all the evidence we have of the growing, aging population. —Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer, Association of American Medical Colleges
A big part of the problem here is that people are free agents. If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists. —Daniel Brotman, MD, FACP, SFHM, chair, SHM Education Committee, director, hospitalist program, Johns Hopkins Hospital, Baltimore
Accordingly, the AAMC panned the report’s recommendation to replace separate IME funding with a single fund directed toward the GME sponsoring institution and subdivided instead into the operational and transformation funds. Dr. Grover says setting up a transformation fund with new money would make sense, but not as a carve-out from existing support.
“You’re removing those resources from the system and not replacing them, which is a challenge,” he says.
Medical schools are more inclined to want the money directed toward training goals, especially if they are to be held accountable for GME outcomes. “Right now, the hospital gets it, and it’s basically somewhere in the bottom line,” Dr. Parekh says. “No one really knows where that money goes. There’s very little accountability or clarity of purpose for that dollar.”
Amid the ongoing debate, the call for more transparency and accountability in GME seems to be gaining the most ground. “I don’t see tons of downside from it,” Dr. Parekh says. “I think it sheds light on the current funding environment and makes people have to justify a little bit more what they’re doing with that money.”
Dr. Tad-y puts it this way: “If you made your own budget at home, the first thing you’d do is try to figure out where all your money goes and what you’re spending your money on.” If Medicare is concerned that its GME money isn’t being spent wisely, then, the first step would be to do some accounting. “And that means a little bit of transparency,” she says. “I don’t think that’s a bad thing, to know exactly what we’re paying for; that makes sense. I mean, we do that for everything else.”
SHM and most other medical associations also agree on the necessary goal of increasing the nation’s primary care capacity, even if they differ on the details of how best to do so. In the long run, however, some observers say growing the workforce—whether that of primary care providers or of hospitalists—may depend less on the total number of residency spots and more on the enthusiasm of program leadership and the attractiveness of job conditions such as salary and workload.
“A big part of the problem here is that people are free agents,” says Daniel Brotman, MD, FACP, SFHM, chair of the SHM Education Committee and director of the hospitalist program at the Johns Hopkins Hospital in Baltimore. “If you make more residency spots, but the economics are such that people decide to become cardiologists because cardiologists make twice or more what hospitalists make, then you may have increased residency spots but [added only] a very small increment in the number of hospitalists.”
Whatever happens, Dr. Parekh says hospitalists are well positioned to be integral parts of improving quality, accountability, and innovation in residency training programs.
“I think if more GME money is targeted toward the outcomes of the GME programs, hospitalists are going to be tapped to help with that work, in terms of training and broadening their skill sets,” he says. “So I think it’s a great opportunity.”
Bryn Nelson is a freelance medical writer in Seattle.
References
- Mullan F, Chen C, Steinmetz E. The geography of graduate medical education: imbalances signal need for new distribution policies. Health Aff. 2013;32(11):1914-1921.
The Difficulty of Predicting Physician Shortages

“Number one, they came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources,” says Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer for the Association of American Medical Colleges. “So we found that problematic, given all the evidence we have of the growing, aging population.”
Census figures indeed suggest a rapidly growing population of seniors: By 2030, one in five U.S. residents will be at least 65 years old. The estimated size of a future doctor shortage, however, has proven far more contentious.
Vikas I. Parekh, MD, FACP, SFHM, chair of the SHM Academic Hospitalist Committee, says most observers agree on a few basic points: that the pool of U.S. physicians leans more toward specialty than primary care and that significant workforce gaps exist in certain geographic locations—both in primary care and in other specialties.
Dr. Grover says the uneven distribution and an overall shortfall are both problematic; the AAMC has projected a shortage of up to 90,000 doctors by 2025. But Dr. Parekh says predicting future workforce numbers has always been a challenge.
–Gail Wilensky, PhD
“Historically, the projections of what the shortages might be have not been reliable or accurate,” he says.
In a recent outlook published by the National Institute of Healthcare Management, IOM committee co-chair Gail Wilensky, PhD, a senior fellow at Project HOPE and former Medicare administrator, goes a step farther. “We concluded that attempts to forecast physician supply and demand, both in the aggregate and by broad specialty types, have been singularly unsuccessful in the past,” she writes. “In fact, past projections have not always been even directionally correct.”
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and another IOM committee member, likewise defended the report’s analysis.
“I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that,” she writes in an e-mail to The Hospitalist. “Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable.
“However, we were agreed, and stated multiple times, that there were and are striking physician shortages by geography and specialty in multiple areas of the country, and we suggested a specific system change aimed at beginning to address these geographic and specialty shortages.”
The committee members decided against the “one size fits all” solution of simply expanding the current system, she says, because they believed the existing structure had contributed to the disparities in the first place.
“Number one, they came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources,” says Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer for the Association of American Medical Colleges. “So we found that problematic, given all the evidence we have of the growing, aging population.”
Census figures indeed suggest a rapidly growing population of seniors: By 2030, one in five U.S. residents will be at least 65 years old. The estimated size of a future doctor shortage, however, has proven far more contentious.
Vikas I. Parekh, MD, FACP, SFHM, chair of the SHM Academic Hospitalist Committee, says most observers agree on a few basic points: that the pool of U.S. physicians leans more toward specialty than primary care and that significant workforce gaps exist in certain geographic locations—both in primary care and in other specialties.
Dr. Grover says the uneven distribution and an overall shortfall are both problematic; the AAMC has projected a shortage of up to 90,000 doctors by 2025. But Dr. Parekh says predicting future workforce numbers has always been a challenge.
–Gail Wilensky, PhD
“Historically, the projections of what the shortages might be have not been reliable or accurate,” he says.
In a recent outlook published by the National Institute of Healthcare Management, IOM committee co-chair Gail Wilensky, PhD, a senior fellow at Project HOPE and former Medicare administrator, goes a step farther. “We concluded that attempts to forecast physician supply and demand, both in the aggregate and by broad specialty types, have been singularly unsuccessful in the past,” she writes. “In fact, past projections have not always been even directionally correct.”
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and another IOM committee member, likewise defended the report’s analysis.
“I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that,” she writes in an e-mail to The Hospitalist. “Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable.
“However, we were agreed, and stated multiple times, that there were and are striking physician shortages by geography and specialty in multiple areas of the country, and we suggested a specific system change aimed at beginning to address these geographic and specialty shortages.”
The committee members decided against the “one size fits all” solution of simply expanding the current system, she says, because they believed the existing structure had contributed to the disparities in the first place.
“Number one, they came out and said, ‘We don’t know that there’s a shortage of physicians and we’re, if anything, going to remove money from the training system rather than putting in additional resources,” says Atul Grover, MD, PhD, FACP, FCCP, chief public policy officer for the Association of American Medical Colleges. “So we found that problematic, given all the evidence we have of the growing, aging population.”
Census figures indeed suggest a rapidly growing population of seniors: By 2030, one in five U.S. residents will be at least 65 years old. The estimated size of a future doctor shortage, however, has proven far more contentious.
Vikas I. Parekh, MD, FACP, SFHM, chair of the SHM Academic Hospitalist Committee, says most observers agree on a few basic points: that the pool of U.S. physicians leans more toward specialty than primary care and that significant workforce gaps exist in certain geographic locations—both in primary care and in other specialties.
Dr. Grover says the uneven distribution and an overall shortfall are both problematic; the AAMC has projected a shortage of up to 90,000 doctors by 2025. But Dr. Parekh says predicting future workforce numbers has always been a challenge.
–Gail Wilensky, PhD
“Historically, the projections of what the shortages might be have not been reliable or accurate,” he says.
In a recent outlook published by the National Institute of Healthcare Management, IOM committee co-chair Gail Wilensky, PhD, a senior fellow at Project HOPE and former Medicare administrator, goes a step farther. “We concluded that attempts to forecast physician supply and demand, both in the aggregate and by broad specialty types, have been singularly unsuccessful in the past,” she writes. “In fact, past projections have not always been even directionally correct.”
Deborah Powell, MD, dean emerita of the University of Minnesota Medical School and another IOM committee member, likewise defended the report’s analysis.
“I have had physicians tell me that they do not understand why our report said that there was not a physician shortage, and I try to point out that we did NOT say that,” she writes in an e-mail to The Hospitalist. “Rather, the report [and the committee] said that we could not find compelling evidence of an impending physician shortage and that physician workforce projections had been and are quite unreliable.
“However, we were agreed, and stated multiple times, that there were and are striking physician shortages by geography and specialty in multiple areas of the country, and we suggested a specific system change aimed at beginning to address these geographic and specialty shortages.”
The committee members decided against the “one size fits all” solution of simply expanding the current system, she says, because they believed the existing structure had contributed to the disparities in the first place.
Antiepileptic Drugs Reduce Risk of Recurrent Unprovoked Seizures
Clinical question: What are the updated recommendations for treating first unprovoked seizure in adults?
Background: Approximately 150,000 adults present with an unprovoked first seizure in the U.S. annually, and these events are associated with physical and psychological trauma. Prior guidelines discussed evaluation of unprovoked first seizures in adults but did not address management. This publication aims to analyze existing evidence regarding prognosis and therapy with antiepileptic drugs (AEDs).
Study design: Evidence-based appraisal of a systematic review.
Setting: Literature published from 1966 to 2013 on MEDLINE, EMBASE, and Cochrane Central Register of Controlled Trials.
Synopsis: Ten prognostic studies describing risk of recurrence were found. Generalized tonic-clonic seizures were the major seizure type. Cumulative incidence of recurrent seizure increased over time, with the majority occurring within the first two years, regardless of treatment with AED; however, there were treatment differences among these studies and wide variation in recurrence rates.
Recurrence risk was lower with AED therapy, though patients were not randomized. Increased risk of recurrence was associated with prior brain lesion causing the seizure, EEG with epileptiform abnormalities, imaging abnormality, and nocturnal seizure.
Five studies were reviewed for prognosis following immediate AED therapy. Immediate AED treatment reduced risk of recurrence by 35% over the first two years. Among studies, “immediate” ranged from within one week to up to three months. Two studies described long-term prognosis, concluding that immediate AED treatment was unlikely to improve the chance of sustained seizure remission.
Five studies were used to describe adverse events in patients treated with AED. Adverse event incidence varied from 7% to 31%, and the incidents that occurred were largely mild and were reversible.
Bottom line: In adults presenting with unprovoked first seizure, the risk of recurrence is highest in the first two years and can be reduced with immediate AED therapy, though AED therapy was not shown to improve long-term prognosis.
Citation: Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: management of an unprovoked first seizure in adults. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2015;84(16):1705-1713.
Clinical question: What are the updated recommendations for treating first unprovoked seizure in adults?
Background: Approximately 150,000 adults present with an unprovoked first seizure in the U.S. annually, and these events are associated with physical and psychological trauma. Prior guidelines discussed evaluation of unprovoked first seizures in adults but did not address management. This publication aims to analyze existing evidence regarding prognosis and therapy with antiepileptic drugs (AEDs).
Study design: Evidence-based appraisal of a systematic review.
Setting: Literature published from 1966 to 2013 on MEDLINE, EMBASE, and Cochrane Central Register of Controlled Trials.
Synopsis: Ten prognostic studies describing risk of recurrence were found. Generalized tonic-clonic seizures were the major seizure type. Cumulative incidence of recurrent seizure increased over time, with the majority occurring within the first two years, regardless of treatment with AED; however, there were treatment differences among these studies and wide variation in recurrence rates.
Recurrence risk was lower with AED therapy, though patients were not randomized. Increased risk of recurrence was associated with prior brain lesion causing the seizure, EEG with epileptiform abnormalities, imaging abnormality, and nocturnal seizure.
Five studies were reviewed for prognosis following immediate AED therapy. Immediate AED treatment reduced risk of recurrence by 35% over the first two years. Among studies, “immediate” ranged from within one week to up to three months. Two studies described long-term prognosis, concluding that immediate AED treatment was unlikely to improve the chance of sustained seizure remission.
Five studies were used to describe adverse events in patients treated with AED. Adverse event incidence varied from 7% to 31%, and the incidents that occurred were largely mild and were reversible.
Bottom line: In adults presenting with unprovoked first seizure, the risk of recurrence is highest in the first two years and can be reduced with immediate AED therapy, though AED therapy was not shown to improve long-term prognosis.
Citation: Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: management of an unprovoked first seizure in adults. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2015;84(16):1705-1713.
Clinical question: What are the updated recommendations for treating first unprovoked seizure in adults?
Background: Approximately 150,000 adults present with an unprovoked first seizure in the U.S. annually, and these events are associated with physical and psychological trauma. Prior guidelines discussed evaluation of unprovoked first seizures in adults but did not address management. This publication aims to analyze existing evidence regarding prognosis and therapy with antiepileptic drugs (AEDs).
Study design: Evidence-based appraisal of a systematic review.
Setting: Literature published from 1966 to 2013 on MEDLINE, EMBASE, and Cochrane Central Register of Controlled Trials.
Synopsis: Ten prognostic studies describing risk of recurrence were found. Generalized tonic-clonic seizures were the major seizure type. Cumulative incidence of recurrent seizure increased over time, with the majority occurring within the first two years, regardless of treatment with AED; however, there were treatment differences among these studies and wide variation in recurrence rates.
Recurrence risk was lower with AED therapy, though patients were not randomized. Increased risk of recurrence was associated with prior brain lesion causing the seizure, EEG with epileptiform abnormalities, imaging abnormality, and nocturnal seizure.
Five studies were reviewed for prognosis following immediate AED therapy. Immediate AED treatment reduced risk of recurrence by 35% over the first two years. Among studies, “immediate” ranged from within one week to up to three months. Two studies described long-term prognosis, concluding that immediate AED treatment was unlikely to improve the chance of sustained seizure remission.
Five studies were used to describe adverse events in patients treated with AED. Adverse event incidence varied from 7% to 31%, and the incidents that occurred were largely mild and were reversible.
Bottom line: In adults presenting with unprovoked first seizure, the risk of recurrence is highest in the first two years and can be reduced with immediate AED therapy, though AED therapy was not shown to improve long-term prognosis.
Citation: Krumholz A, Wiebe S, Gronseth GS, et al. Evidence-based guideline: management of an unprovoked first seizure in adults. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2015;84(16):1705-1713.