User login
Outcomes of Treatment with Recombinant Tissue Plasminogen Activator in Patients Age 80 Years and Older Presenting with Acute Ischemic Stroke
From Summa Health System, Akron, OH.
Abstract
- Background: Ischemic stroke is a major cause of morbidity and mortality for patients ≥ 80 years old. The use of intravenous recombinant tissue plasminogen activator (tPA) in patients ≥ 80 years for treatment of ischemic stroke remains controversial.
- Objective: To examine outcomes in patients ≥ 80 years old who received tPA in our institution.
- Methods: This was a retrospective cohort study at a community-based certified acute stroke center. Individuals age ≥ 80 years evaluated emergently for acute neurologic changes consistent with ischemic stroke were included (n = 184). The comparison groups within this sample were patients who received tPA versus with those who did not because they came to the ED outside of the treatment window. Outcome measures included length of stay, symptomatic intracerebral hemorrhage (ICH), discharge disposition, and in-hospital death
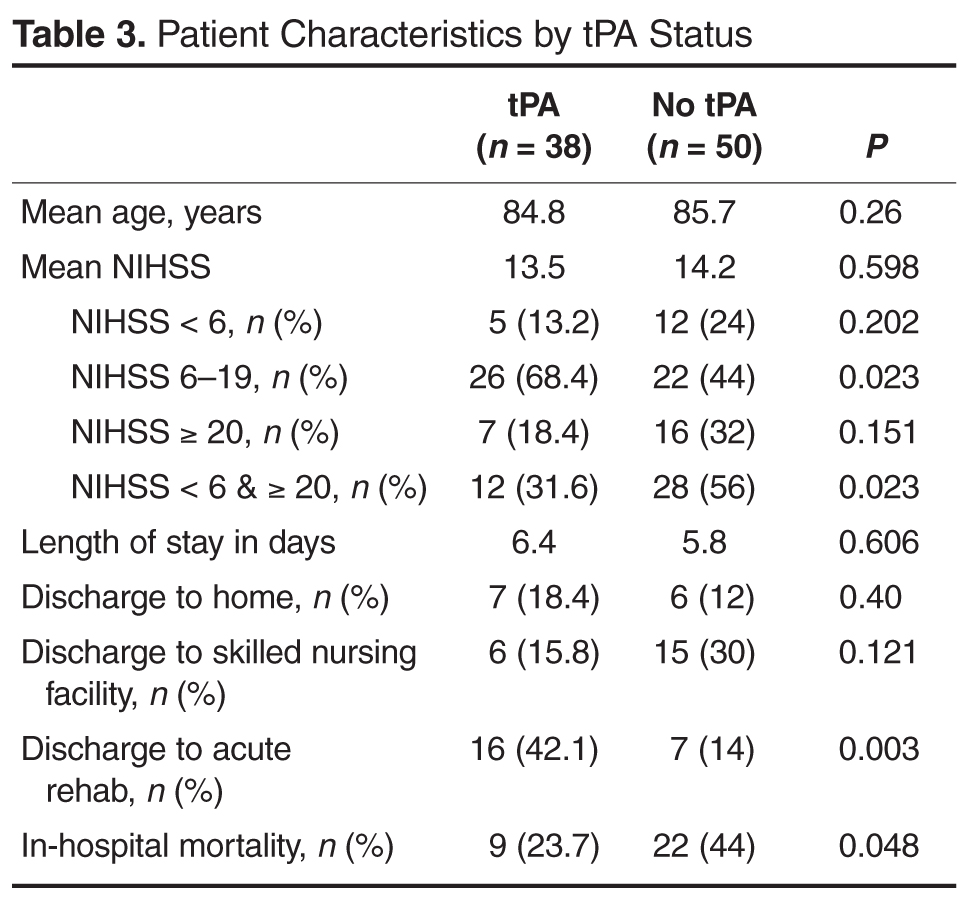
- Results: 38 patients (20.7%) received tPA. 50 patients (27.2%) presented outside of treatment windows and were included in comparative analysis. There was no difference between groups in age (P = 0.26) or initial National Institute of Health Stroke Scale (P = 0.598). One patient (2.6%) who received tPA developed symptomatic ICH. Those receiving tPA were more likely to be discharged to acute rehabilitation hospitals (P = 0.012) and less likely to experience in hospital death (P = 0.048).
- Conclusion: At this institution, the use of tPA in patients ≥ 80 years old is not associated with increased mortality or risk of symptomatic ICH. Those who received tPA were more likely to be discharged to acute rehabilitation hospitals, suggesting greater potential for functional recovery.
Acute ischemic stroke is a major cause of morbidity and mortality in patients 80 years or older. Though less than 5% of the United States population is over the age of 80 [1], studies have shown that up to one-third of patients presenting with ischemic stroke are ≥ 80 years old [2] and among first-time strokes, a third occur in those ≥ 80 [3]. Older adults present with worse symptoms associated with ischemic stroke as measured by the National Institutes of Health Stroke Scale (NIHSS) compared with younger (< 80 years) counterparts [4]. Older patients are more likely to be discharged to a location other than home [5]. Older age is associated with higher hospital, 30-day, and 1-year mortality [3,5,6]. Patients ≥ 80 are significantly more likely to die in the hospital compared to younger patients, 11.7% to 23.6% vs 5.1%, respectively [3,7].
The Food and Drug Administration (FDA) approved the use of intravenous recombinant tissue plasminogen activator (tPA) in 1996 for the treatment of ischemic stroke [8]. Studies evaluating the safety and efficacy of tPA in ischemic stroke excluded or underrepresented patients ≥ 80 [8,9]. The use of tPA in those ≥ 80 has not been shown consistently to improve outcomes [6,10,11]. Post-hoc analysis of the National Institute of Neurologic Disorders and Stroke (NINDS) study did not show worse outcomes or harms to older adults treated with tPA [12]. Likewise, data from the International Stroke Treatment (IST-3) collaborative group show that treatment with tPA up to 6 hours from the onset of symptoms improves outcomes in the elderly [13]. Use of tPA in the oldest adults remains controversial due to perceived higher risk of symptomatic intracerebral hemorrhage (ICH). Published data suggest overall ICH risk of 4.3% to 6.4% across all age-groups [9,14,15].Studies have failed to demonstrate an increased risk in the oldest adults [4,10,16,17], though they may have higher mortality rates associated with ICH [15]. Despite this, trends suggest increasing use of tPA in those ≥ 80 over the past decade [2]. Along with primary data from NINDS [12] and IST-3 [13], a meta-analysis conducted in 2014 suggests that regardless of age, patients have improved outcomes with tPA [18].With the increasing age of the population, effective treatment of strokes in patients ≥ 80 will continue to be an important clinical and research endeavor.
This study evaluates the outcomes of clinical use of tPA for treatment of patients ≥ 80 years old who present to a community-based certified stroke center with ischemic stroke.
Methods
The study setting was a 540-bed acute care hospital that is a community-based certified stroke center. This study was deemed nonhuman subjects research by the institutional review board as the goal was to evaluate processes and outcomes of this institution’s stroke team in treating a subgroup of patients according to clinically accepted practice (quality improvement initiative). All patients presenting to the emergency department (ED) between 1 January 2011 and 30 November 2013 with the onset of stroke-like neurological deficits underwent evaluation and treatment by a neurologist and/or specially trained stroke team. This team consists of the attending neurologist, ED physician, resident physicians, advanced practice nurses, and ED staff nurses and emergency medicine technicians. Team members involved in the evaluation and treatment of these patients undergo routine clinical education and testing to ensure standardization. Patients undergo emergent evaluation including the National Institutes of Health Stroke Scale (NIHSS) and obtain brain imaging with computed tomography (CT).
Patients ≥ 80 years were identified among all those who presented to the ED with ischemic stroke. Patients were included if they were subsequently diagnosed with ischemic stroke or transient ischemic attacks (TIA). They were excluded from analysis if neurological changes were due to primary hemorrhagic stroke, intracranial hemorrhage, subarachnoid hemorrhage, seizure, conversion disorder, or metabolic derangements. They were also excluded from analysis if the acute ischemic stroke treatment included intra-arterial administration of tPA or endovascular revascularization.
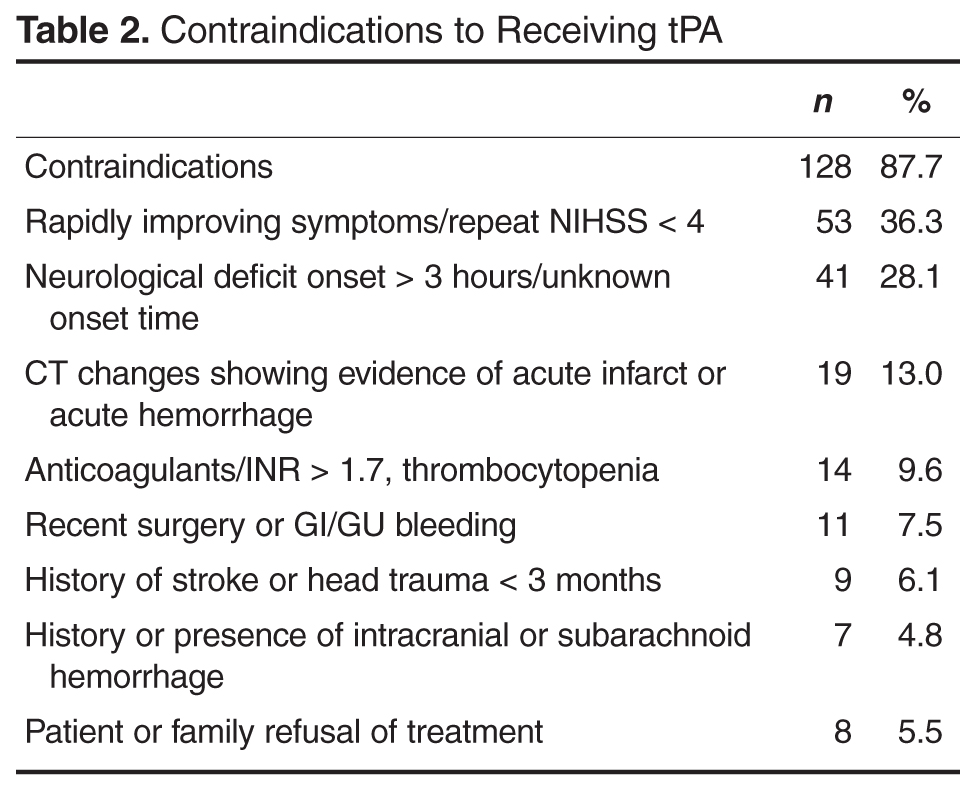
Patient data collected included age, NIHSS at presentation to ED, time to presentation at ED, treatment with tPA, contraindications to tPA, discharge disposition, length of stay and in-hospital mortality. Raw NIHSS values were collected at the time of presentation. NIHSS were categorized into mild symptoms (NIHSS < 6), moderate symptoms (NIHSS 6–19), or severe symptoms (NIHSS ≥ 20). Clinical indications for receiving tPA include NIHSS > 4, focal neurological deficit onset < 3 hours (for those ≥ 80 years old), and no evidence of acute hemorrhage or acute infarct on CT. Contraindications include rapidly improving symptoms (repeat NIHSS < 4), active or history of intracranial hemorrhage, history of stroke or head trauma in past 3 months, gastrointestinal or genitourinary hemorrhage within 21 days, major surgery within 14 days, arterial puncture at a noncompressible site in past 7 days, treatment with anticoagulation with therapeutic indices, systolic blood pressure > 185 mm Hg or diastolic blood pressure > 110 mm Hg and not responding to treatment, or platelet count < 100,000/mm3. Patients who were not eligible for tPA based on contraindications with the exception of being outside the treatment window (3 hours) were excluded from comparative analysis. Patient length of stay was rounded to nearest full day. Discharge disposition was categorized as home, acute rehabilitation hospital, skilled nursing facility, home or facility with hospice services, other hospital setting, or death.
Statistics were calculated using SPSS statistical software. Variables were reported as means and percentages. Group means were compared using t tests and differences in proportions were compared using the chi square test. Correlations were performed using Pearson’s correlation. A 2-tailed P < 0.05 was considered statistically significant.
Results

Discussion
Ischemic stroke remains a major cause of morbidity and mortality for very old patients. Though less than 5% of the United States population is over the age of 80 [1], at this community-based hospital 18% of those presenting to the ED with ischemic stroke were in this age-group. With a population of increasing age, more people in this age-group will present with ischemic stroke and need effective treatment to limit the associated morbidity and mortality. Being able to quickly and safely treat acute ischemic stroke may help very old adults maintain independence or prevent institutionalization. While the original studies demonstrating the effectiveness of tPA for acute ischemic stroke excluded or underrepresented those ≥ 80 years, retrospective analysis has not been conclusive regarding its use in very old patients [4–6,10,12,13].However, post-hoc analysis of NINDS and IST-3 data demonstrate efficacy and safety of treatment [12,13].
This study explored the use of tPA at a community-based certified stroke center. Similar to previous studies, it demonstrates the large proportion of patients presenting with acute neurological findings consistent with ischemic stroke are ≥ 80 years old [3,6]. Our incidence of acute ischemic stroke in the oldest patients may be slightly lower than reported elsewhere, which may reflect community differences, with higher rates of younger patients with multiple comorbidities presenting with stroke-like symptoms. Amongst this very old cohort, age was positively correlated with stroke severity. Mortality in patients ≥ 80 years old who present with acute ischemic stroke approaches 25%.
The majority of patients who did not receive tPA had documented contraindications to receiving the medication. The most common reason was rapidly improving symptoms with repeat NIHSS often ≤ 4. The second most common reason was presentation outside the treatment window of 3 hours. We compared those who either arrived too late to receive treatment with tPA or already had ischemic changes on CT to those who received tPA as this suggests the natural history of stroke progression and outcome without effective, early treatment. The outcomes at this institution support this trend. Very old patients who received tPA did not experience harm as evidenced by similar lengths of stay and rates of discharge to home. Also, rates of symptomatic ICH were lower than those reported in the literature. In fact, patients who received tPA were less likely to experience in-hospital death and more likely to be discharged to acute rehabilitation hospitals, suggesting more functional ability to tolerate aggressive recovery efforts.
Very few people who presented with acute ischemic stroke and were eligible for treatment with tPA failed to receive it. This suggests that despite the perceived increased risk to treating these patients with tPA, the specialized stroke team aggressively treats patients age ≥ 80 years who present with acute ischemic stroke. However, those who did not receive tPA were more likely to have presented with mild or severe strokes. This may suggest that treatment time frames are more strongly held, or that treatment teams are more likely to use time frames as a reason to not treat with tPA for patients with mild or severe strokes. Also, very few patients and families who were eligible to receive tPA declined treatment despite the associated risks. This suggests that patients and families are eager for aggressive treatment in attempt to prevent death or disability associated with ischemic stroke.
There are several limitations associated with this evaluation. First, this is a retrospective analysis of a single institution’s acute stroke procedures. Data was collected in an effort to evaluate the processes and outcomes of the specialized stroke team in evaluating and treating this very old cohort who present to a community-based hospital. It involved individualized clinical evaluation and decision making by multiple care providers who may offer different perspectives on the risk of treating patients ≥ 80 years old with tPA, which may result in selection bias. While comparing those who arrived outside treatment windows offers a comparison group who represents the natural course of untreated strokes, patient characteristics that prevented timely evaluation may also impact their outcomes including baseline mobility, care giving availability and underlying medical comorbidities. The similarity in mean presenting NIHSS scores of the two groups, however, argues against this possibility. Lastly, exclusion criteria to receiving tPA may represent intrinsic characteristics that impart higher risk of negative outcomes.
Conculsion
Although there have been no randomized controlled trials that evaluate the safety and efficacy of tPA in the treatment of acute ischemic stroke in very old patients, use at the community-based stroke center was not associated with worse outcomes including symptomatic ICH, hospital length of stay, and in-hospital mortality. In fact, there were trends towards better outcomes in older patients who received tPA, including a significant reduction in in-hospital mortality. This evaluation supports the benefits of using tPA to treat acute ischemic stroke as seen in prior randomized controlled trials that included the treatment of very old patients. Though ongoing research is needed, a growing body of evidence supports the use of tPA to treat acute ischemic stroke in patients ≥ 80 years.
Corresponding author: Jennifer C. Drost, DO, MPH, Summa Health System, 75 Arch St., Ste. G1, Akron, OH 44304, [email protected].
Financial disclosures: None.
Author contributions: Conception and design, JCD, SMB; analysis and interpretation of data, JCD, SMB; drafting of article, JCD; critical revision of the article, JCD, SMB; provision of study materials or patients, SMB; collection and assembly of data, JCD.
1. US Census Bureau. Annual estimates of the resident population for selected age groups by sex for the United States, States, Counties, and Puerto Rico Commonwealth and Municipios: April 1, 2010 to July 1, 2013. Accessed at www.census.gov/popest/index.html.
2. Fang MC, Cutler DM, Rosen AB. Trends in thrombolytic use for ischemic stroke in the United States. J Hosp Med 2010;5:406–9.
3. Marini C, Baldassarre M, Russo T, et al. Burden of first-ever ischemic stroke in the oldest old: evidence from a population-based study. Neurology 2004;62:77–81.
4. Sylaja PN, Cote R, Buchan AM, Hill MD. Thrombolysis in patients older than 80 years with acute ischaemic stroke: Canadian Alteplase for Stroke Effectiveness Study. J Neurol Neurosurg Psychiatry 2006;77:826–9.
5. Heitsch LE, Panagos PD. Treating the elderly stroke patient: complications, controversies, and best care metrics. Clin Geriatr Med 2013;29:231–55.
6. Engelter ST, Bonati LH, Lyrer PA. Intravenous thrombolysis in stroke patients of > or = 80 versus < 80 years of age--a systematic review across cohort studies. Age Ageing 2006;35:572–580.
7. Forti P, Maioli F, Procaccianti G, et al. Independent predictors of ischemic stroke in the elderly: prospective data from a stroke unit. Neurology 2013;80:29–38.
8. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTischemic stroke, ECASS, and NINDS rt-PA stroke trials. Lancet 2004;363:768–74.
9. Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med 1995;333:1581–7.
10. Sung PS, Chen CH, Hsieh HC, et al. Outcome of acute ischemic stroke in very elderly patients: is intravenous thrombolysis beneficial? Eur Neurol 2011;66:110–6.
11. Saposnik G, Guzik AK, Reeves M, et al. Stroke prognostication using age and NIH Stroke Scale: SPAN-100. Neurology 2013;80:21–8.
12. Generalized efficacy of t-PA for acute stroke. Subgroup analysis of the NINDS t-PA Stroke Trial. Stroke 1997;28:2119–25.
13. Sandercock P, Wardlaw JM, Lindley RI, et al. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet 2012;379:2352–63.
14. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA Stroke Study Group. Stroke 1997;28:2109–18.
15. Bray BD, Campbell J, Hoffman A, et al. Stroke thrombolysis in England: an age stratified analysis of practice and outcome. Age Ageing 2013;42:240–5.
16. Kono S, Deguchi K, Morimoto N, et al. Intravenous thrombolysis with neuroprotective therapy by edaravone for ischemic stroke patients older than 80 years of age. J Stroke Cerebrovasc Dis 2013;22:1175–83.
17. Berrouschot J, Rother J, Glahn J, et al. Outcome and severe hemorrhagic complications of intravenous thrombolysis with tissue plasminogen activator in very old (> or =80 years) stroke patients. Stroke 2005;36:2421–5.
18. Emberson J, Lees KR, Lyden P, et al; Stroke Thrombolysis Trialists' Collaborative Group. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet 2014;384:1929–35.
From Summa Health System, Akron, OH.
Abstract
- Background: Ischemic stroke is a major cause of morbidity and mortality for patients ≥ 80 years old. The use of intravenous recombinant tissue plasminogen activator (tPA) in patients ≥ 80 years for treatment of ischemic stroke remains controversial.
- Objective: To examine outcomes in patients ≥ 80 years old who received tPA in our institution.
- Methods: This was a retrospective cohort study at a community-based certified acute stroke center. Individuals age ≥ 80 years evaluated emergently for acute neurologic changes consistent with ischemic stroke were included (n = 184). The comparison groups within this sample were patients who received tPA versus with those who did not because they came to the ED outside of the treatment window. Outcome measures included length of stay, symptomatic intracerebral hemorrhage (ICH), discharge disposition, and in-hospital death
- Results: 38 patients (20.7%) received tPA. 50 patients (27.2%) presented outside of treatment windows and were included in comparative analysis. There was no difference between groups in age (P = 0.26) or initial National Institute of Health Stroke Scale (P = 0.598). One patient (2.6%) who received tPA developed symptomatic ICH. Those receiving tPA were more likely to be discharged to acute rehabilitation hospitals (P = 0.012) and less likely to experience in hospital death (P = 0.048).
- Conclusion: At this institution, the use of tPA in patients ≥ 80 years old is not associated with increased mortality or risk of symptomatic ICH. Those who received tPA were more likely to be discharged to acute rehabilitation hospitals, suggesting greater potential for functional recovery.
Acute ischemic stroke is a major cause of morbidity and mortality in patients 80 years or older. Though less than 5% of the United States population is over the age of 80 [1], studies have shown that up to one-third of patients presenting with ischemic stroke are ≥ 80 years old [2] and among first-time strokes, a third occur in those ≥ 80 [3]. Older adults present with worse symptoms associated with ischemic stroke as measured by the National Institutes of Health Stroke Scale (NIHSS) compared with younger (< 80 years) counterparts [4]. Older patients are more likely to be discharged to a location other than home [5]. Older age is associated with higher hospital, 30-day, and 1-year mortality [3,5,6]. Patients ≥ 80 are significantly more likely to die in the hospital compared to younger patients, 11.7% to 23.6% vs 5.1%, respectively [3,7].
The Food and Drug Administration (FDA) approved the use of intravenous recombinant tissue plasminogen activator (tPA) in 1996 for the treatment of ischemic stroke [8]. Studies evaluating the safety and efficacy of tPA in ischemic stroke excluded or underrepresented patients ≥ 80 [8,9]. The use of tPA in those ≥ 80 has not been shown consistently to improve outcomes [6,10,11]. Post-hoc analysis of the National Institute of Neurologic Disorders and Stroke (NINDS) study did not show worse outcomes or harms to older adults treated with tPA [12]. Likewise, data from the International Stroke Treatment (IST-3) collaborative group show that treatment with tPA up to 6 hours from the onset of symptoms improves outcomes in the elderly [13]. Use of tPA in the oldest adults remains controversial due to perceived higher risk of symptomatic intracerebral hemorrhage (ICH). Published data suggest overall ICH risk of 4.3% to 6.4% across all age-groups [9,14,15].Studies have failed to demonstrate an increased risk in the oldest adults [4,10,16,17], though they may have higher mortality rates associated with ICH [15]. Despite this, trends suggest increasing use of tPA in those ≥ 80 over the past decade [2]. Along with primary data from NINDS [12] and IST-3 [13], a meta-analysis conducted in 2014 suggests that regardless of age, patients have improved outcomes with tPA [18].With the increasing age of the population, effective treatment of strokes in patients ≥ 80 will continue to be an important clinical and research endeavor.
This study evaluates the outcomes of clinical use of tPA for treatment of patients ≥ 80 years old who present to a community-based certified stroke center with ischemic stroke.
Methods
The study setting was a 540-bed acute care hospital that is a community-based certified stroke center. This study was deemed nonhuman subjects research by the institutional review board as the goal was to evaluate processes and outcomes of this institution’s stroke team in treating a subgroup of patients according to clinically accepted practice (quality improvement initiative). All patients presenting to the emergency department (ED) between 1 January 2011 and 30 November 2013 with the onset of stroke-like neurological deficits underwent evaluation and treatment by a neurologist and/or specially trained stroke team. This team consists of the attending neurologist, ED physician, resident physicians, advanced practice nurses, and ED staff nurses and emergency medicine technicians. Team members involved in the evaluation and treatment of these patients undergo routine clinical education and testing to ensure standardization. Patients undergo emergent evaluation including the National Institutes of Health Stroke Scale (NIHSS) and obtain brain imaging with computed tomography (CT).
Patients ≥ 80 years were identified among all those who presented to the ED with ischemic stroke. Patients were included if they were subsequently diagnosed with ischemic stroke or transient ischemic attacks (TIA). They were excluded from analysis if neurological changes were due to primary hemorrhagic stroke, intracranial hemorrhage, subarachnoid hemorrhage, seizure, conversion disorder, or metabolic derangements. They were also excluded from analysis if the acute ischemic stroke treatment included intra-arterial administration of tPA or endovascular revascularization.
Patient data collected included age, NIHSS at presentation to ED, time to presentation at ED, treatment with tPA, contraindications to tPA, discharge disposition, length of stay and in-hospital mortality. Raw NIHSS values were collected at the time of presentation. NIHSS were categorized into mild symptoms (NIHSS < 6), moderate symptoms (NIHSS 6–19), or severe symptoms (NIHSS ≥ 20). Clinical indications for receiving tPA include NIHSS > 4, focal neurological deficit onset < 3 hours (for those ≥ 80 years old), and no evidence of acute hemorrhage or acute infarct on CT. Contraindications include rapidly improving symptoms (repeat NIHSS < 4), active or history of intracranial hemorrhage, history of stroke or head trauma in past 3 months, gastrointestinal or genitourinary hemorrhage within 21 days, major surgery within 14 days, arterial puncture at a noncompressible site in past 7 days, treatment with anticoagulation with therapeutic indices, systolic blood pressure > 185 mm Hg or diastolic blood pressure > 110 mm Hg and not responding to treatment, or platelet count < 100,000/mm3. Patients who were not eligible for tPA based on contraindications with the exception of being outside the treatment window (3 hours) were excluded from comparative analysis. Patient length of stay was rounded to nearest full day. Discharge disposition was categorized as home, acute rehabilitation hospital, skilled nursing facility, home or facility with hospice services, other hospital setting, or death.
Statistics were calculated using SPSS statistical software. Variables were reported as means and percentages. Group means were compared using t tests and differences in proportions were compared using the chi square test. Correlations were performed using Pearson’s correlation. A 2-tailed P < 0.05 was considered statistically significant.
Results
Discussion
Ischemic stroke remains a major cause of morbidity and mortality for very old patients. Though less than 5% of the United States population is over the age of 80 [1], at this community-based hospital 18% of those presenting to the ED with ischemic stroke were in this age-group. With a population of increasing age, more people in this age-group will present with ischemic stroke and need effective treatment to limit the associated morbidity and mortality. Being able to quickly and safely treat acute ischemic stroke may help very old adults maintain independence or prevent institutionalization. While the original studies demonstrating the effectiveness of tPA for acute ischemic stroke excluded or underrepresented those ≥ 80 years, retrospective analysis has not been conclusive regarding its use in very old patients [4–6,10,12,13].However, post-hoc analysis of NINDS and IST-3 data demonstrate efficacy and safety of treatment [12,13].
This study explored the use of tPA at a community-based certified stroke center. Similar to previous studies, it demonstrates the large proportion of patients presenting with acute neurological findings consistent with ischemic stroke are ≥ 80 years old [3,6]. Our incidence of acute ischemic stroke in the oldest patients may be slightly lower than reported elsewhere, which may reflect community differences, with higher rates of younger patients with multiple comorbidities presenting with stroke-like symptoms. Amongst this very old cohort, age was positively correlated with stroke severity. Mortality in patients ≥ 80 years old who present with acute ischemic stroke approaches 25%.
The majority of patients who did not receive tPA had documented contraindications to receiving the medication. The most common reason was rapidly improving symptoms with repeat NIHSS often ≤ 4. The second most common reason was presentation outside the treatment window of 3 hours. We compared those who either arrived too late to receive treatment with tPA or already had ischemic changes on CT to those who received tPA as this suggests the natural history of stroke progression and outcome without effective, early treatment. The outcomes at this institution support this trend. Very old patients who received tPA did not experience harm as evidenced by similar lengths of stay and rates of discharge to home. Also, rates of symptomatic ICH were lower than those reported in the literature. In fact, patients who received tPA were less likely to experience in-hospital death and more likely to be discharged to acute rehabilitation hospitals, suggesting more functional ability to tolerate aggressive recovery efforts.
Very few people who presented with acute ischemic stroke and were eligible for treatment with tPA failed to receive it. This suggests that despite the perceived increased risk to treating these patients with tPA, the specialized stroke team aggressively treats patients age ≥ 80 years who present with acute ischemic stroke. However, those who did not receive tPA were more likely to have presented with mild or severe strokes. This may suggest that treatment time frames are more strongly held, or that treatment teams are more likely to use time frames as a reason to not treat with tPA for patients with mild or severe strokes. Also, very few patients and families who were eligible to receive tPA declined treatment despite the associated risks. This suggests that patients and families are eager for aggressive treatment in attempt to prevent death or disability associated with ischemic stroke.
There are several limitations associated with this evaluation. First, this is a retrospective analysis of a single institution’s acute stroke procedures. Data was collected in an effort to evaluate the processes and outcomes of the specialized stroke team in evaluating and treating this very old cohort who present to a community-based hospital. It involved individualized clinical evaluation and decision making by multiple care providers who may offer different perspectives on the risk of treating patients ≥ 80 years old with tPA, which may result in selection bias. While comparing those who arrived outside treatment windows offers a comparison group who represents the natural course of untreated strokes, patient characteristics that prevented timely evaluation may also impact their outcomes including baseline mobility, care giving availability and underlying medical comorbidities. The similarity in mean presenting NIHSS scores of the two groups, however, argues against this possibility. Lastly, exclusion criteria to receiving tPA may represent intrinsic characteristics that impart higher risk of negative outcomes.
Conculsion
Although there have been no randomized controlled trials that evaluate the safety and efficacy of tPA in the treatment of acute ischemic stroke in very old patients, use at the community-based stroke center was not associated with worse outcomes including symptomatic ICH, hospital length of stay, and in-hospital mortality. In fact, there were trends towards better outcomes in older patients who received tPA, including a significant reduction in in-hospital mortality. This evaluation supports the benefits of using tPA to treat acute ischemic stroke as seen in prior randomized controlled trials that included the treatment of very old patients. Though ongoing research is needed, a growing body of evidence supports the use of tPA to treat acute ischemic stroke in patients ≥ 80 years.
Corresponding author: Jennifer C. Drost, DO, MPH, Summa Health System, 75 Arch St., Ste. G1, Akron, OH 44304, [email protected].
Financial disclosures: None.
Author contributions: Conception and design, JCD, SMB; analysis and interpretation of data, JCD, SMB; drafting of article, JCD; critical revision of the article, JCD, SMB; provision of study materials or patients, SMB; collection and assembly of data, JCD.
From Summa Health System, Akron, OH.
Abstract
- Background: Ischemic stroke is a major cause of morbidity and mortality for patients ≥ 80 years old. The use of intravenous recombinant tissue plasminogen activator (tPA) in patients ≥ 80 years for treatment of ischemic stroke remains controversial.
- Objective: To examine outcomes in patients ≥ 80 years old who received tPA in our institution.
- Methods: This was a retrospective cohort study at a community-based certified acute stroke center. Individuals age ≥ 80 years evaluated emergently for acute neurologic changes consistent with ischemic stroke were included (n = 184). The comparison groups within this sample were patients who received tPA versus with those who did not because they came to the ED outside of the treatment window. Outcome measures included length of stay, symptomatic intracerebral hemorrhage (ICH), discharge disposition, and in-hospital death
- Results: 38 patients (20.7%) received tPA. 50 patients (27.2%) presented outside of treatment windows and were included in comparative analysis. There was no difference between groups in age (P = 0.26) or initial National Institute of Health Stroke Scale (P = 0.598). One patient (2.6%) who received tPA developed symptomatic ICH. Those receiving tPA were more likely to be discharged to acute rehabilitation hospitals (P = 0.012) and less likely to experience in hospital death (P = 0.048).
- Conclusion: At this institution, the use of tPA in patients ≥ 80 years old is not associated with increased mortality or risk of symptomatic ICH. Those who received tPA were more likely to be discharged to acute rehabilitation hospitals, suggesting greater potential for functional recovery.
Acute ischemic stroke is a major cause of morbidity and mortality in patients 80 years or older. Though less than 5% of the United States population is over the age of 80 [1], studies have shown that up to one-third of patients presenting with ischemic stroke are ≥ 80 years old [2] and among first-time strokes, a third occur in those ≥ 80 [3]. Older adults present with worse symptoms associated with ischemic stroke as measured by the National Institutes of Health Stroke Scale (NIHSS) compared with younger (< 80 years) counterparts [4]. Older patients are more likely to be discharged to a location other than home [5]. Older age is associated with higher hospital, 30-day, and 1-year mortality [3,5,6]. Patients ≥ 80 are significantly more likely to die in the hospital compared to younger patients, 11.7% to 23.6% vs 5.1%, respectively [3,7].
The Food and Drug Administration (FDA) approved the use of intravenous recombinant tissue plasminogen activator (tPA) in 1996 for the treatment of ischemic stroke [8]. Studies evaluating the safety and efficacy of tPA in ischemic stroke excluded or underrepresented patients ≥ 80 [8,9]. The use of tPA in those ≥ 80 has not been shown consistently to improve outcomes [6,10,11]. Post-hoc analysis of the National Institute of Neurologic Disorders and Stroke (NINDS) study did not show worse outcomes or harms to older adults treated with tPA [12]. Likewise, data from the International Stroke Treatment (IST-3) collaborative group show that treatment with tPA up to 6 hours from the onset of symptoms improves outcomes in the elderly [13]. Use of tPA in the oldest adults remains controversial due to perceived higher risk of symptomatic intracerebral hemorrhage (ICH). Published data suggest overall ICH risk of 4.3% to 6.4% across all age-groups [9,14,15].Studies have failed to demonstrate an increased risk in the oldest adults [4,10,16,17], though they may have higher mortality rates associated with ICH [15]. Despite this, trends suggest increasing use of tPA in those ≥ 80 over the past decade [2]. Along with primary data from NINDS [12] and IST-3 [13], a meta-analysis conducted in 2014 suggests that regardless of age, patients have improved outcomes with tPA [18].With the increasing age of the population, effective treatment of strokes in patients ≥ 80 will continue to be an important clinical and research endeavor.
This study evaluates the outcomes of clinical use of tPA for treatment of patients ≥ 80 years old who present to a community-based certified stroke center with ischemic stroke.
Methods
The study setting was a 540-bed acute care hospital that is a community-based certified stroke center. This study was deemed nonhuman subjects research by the institutional review board as the goal was to evaluate processes and outcomes of this institution’s stroke team in treating a subgroup of patients according to clinically accepted practice (quality improvement initiative). All patients presenting to the emergency department (ED) between 1 January 2011 and 30 November 2013 with the onset of stroke-like neurological deficits underwent evaluation and treatment by a neurologist and/or specially trained stroke team. This team consists of the attending neurologist, ED physician, resident physicians, advanced practice nurses, and ED staff nurses and emergency medicine technicians. Team members involved in the evaluation and treatment of these patients undergo routine clinical education and testing to ensure standardization. Patients undergo emergent evaluation including the National Institutes of Health Stroke Scale (NIHSS) and obtain brain imaging with computed tomography (CT).
Patients ≥ 80 years were identified among all those who presented to the ED with ischemic stroke. Patients were included if they were subsequently diagnosed with ischemic stroke or transient ischemic attacks (TIA). They were excluded from analysis if neurological changes were due to primary hemorrhagic stroke, intracranial hemorrhage, subarachnoid hemorrhage, seizure, conversion disorder, or metabolic derangements. They were also excluded from analysis if the acute ischemic stroke treatment included intra-arterial administration of tPA or endovascular revascularization.
Patient data collected included age, NIHSS at presentation to ED, time to presentation at ED, treatment with tPA, contraindications to tPA, discharge disposition, length of stay and in-hospital mortality. Raw NIHSS values were collected at the time of presentation. NIHSS were categorized into mild symptoms (NIHSS < 6), moderate symptoms (NIHSS 6–19), or severe symptoms (NIHSS ≥ 20). Clinical indications for receiving tPA include NIHSS > 4, focal neurological deficit onset < 3 hours (for those ≥ 80 years old), and no evidence of acute hemorrhage or acute infarct on CT. Contraindications include rapidly improving symptoms (repeat NIHSS < 4), active or history of intracranial hemorrhage, history of stroke or head trauma in past 3 months, gastrointestinal or genitourinary hemorrhage within 21 days, major surgery within 14 days, arterial puncture at a noncompressible site in past 7 days, treatment with anticoagulation with therapeutic indices, systolic blood pressure > 185 mm Hg or diastolic blood pressure > 110 mm Hg and not responding to treatment, or platelet count < 100,000/mm3. Patients who were not eligible for tPA based on contraindications with the exception of being outside the treatment window (3 hours) were excluded from comparative analysis. Patient length of stay was rounded to nearest full day. Discharge disposition was categorized as home, acute rehabilitation hospital, skilled nursing facility, home or facility with hospice services, other hospital setting, or death.
Statistics were calculated using SPSS statistical software. Variables were reported as means and percentages. Group means were compared using t tests and differences in proportions were compared using the chi square test. Correlations were performed using Pearson’s correlation. A 2-tailed P < 0.05 was considered statistically significant.
Results
Discussion
Ischemic stroke remains a major cause of morbidity and mortality for very old patients. Though less than 5% of the United States population is over the age of 80 [1], at this community-based hospital 18% of those presenting to the ED with ischemic stroke were in this age-group. With a population of increasing age, more people in this age-group will present with ischemic stroke and need effective treatment to limit the associated morbidity and mortality. Being able to quickly and safely treat acute ischemic stroke may help very old adults maintain independence or prevent institutionalization. While the original studies demonstrating the effectiveness of tPA for acute ischemic stroke excluded or underrepresented those ≥ 80 years, retrospective analysis has not been conclusive regarding its use in very old patients [4–6,10,12,13].However, post-hoc analysis of NINDS and IST-3 data demonstrate efficacy and safety of treatment [12,13].
This study explored the use of tPA at a community-based certified stroke center. Similar to previous studies, it demonstrates the large proportion of patients presenting with acute neurological findings consistent with ischemic stroke are ≥ 80 years old [3,6]. Our incidence of acute ischemic stroke in the oldest patients may be slightly lower than reported elsewhere, which may reflect community differences, with higher rates of younger patients with multiple comorbidities presenting with stroke-like symptoms. Amongst this very old cohort, age was positively correlated with stroke severity. Mortality in patients ≥ 80 years old who present with acute ischemic stroke approaches 25%.
The majority of patients who did not receive tPA had documented contraindications to receiving the medication. The most common reason was rapidly improving symptoms with repeat NIHSS often ≤ 4. The second most common reason was presentation outside the treatment window of 3 hours. We compared those who either arrived too late to receive treatment with tPA or already had ischemic changes on CT to those who received tPA as this suggests the natural history of stroke progression and outcome without effective, early treatment. The outcomes at this institution support this trend. Very old patients who received tPA did not experience harm as evidenced by similar lengths of stay and rates of discharge to home. Also, rates of symptomatic ICH were lower than those reported in the literature. In fact, patients who received tPA were less likely to experience in-hospital death and more likely to be discharged to acute rehabilitation hospitals, suggesting more functional ability to tolerate aggressive recovery efforts.
Very few people who presented with acute ischemic stroke and were eligible for treatment with tPA failed to receive it. This suggests that despite the perceived increased risk to treating these patients with tPA, the specialized stroke team aggressively treats patients age ≥ 80 years who present with acute ischemic stroke. However, those who did not receive tPA were more likely to have presented with mild or severe strokes. This may suggest that treatment time frames are more strongly held, or that treatment teams are more likely to use time frames as a reason to not treat with tPA for patients with mild or severe strokes. Also, very few patients and families who were eligible to receive tPA declined treatment despite the associated risks. This suggests that patients and families are eager for aggressive treatment in attempt to prevent death or disability associated with ischemic stroke.
There are several limitations associated with this evaluation. First, this is a retrospective analysis of a single institution’s acute stroke procedures. Data was collected in an effort to evaluate the processes and outcomes of the specialized stroke team in evaluating and treating this very old cohort who present to a community-based hospital. It involved individualized clinical evaluation and decision making by multiple care providers who may offer different perspectives on the risk of treating patients ≥ 80 years old with tPA, which may result in selection bias. While comparing those who arrived outside treatment windows offers a comparison group who represents the natural course of untreated strokes, patient characteristics that prevented timely evaluation may also impact their outcomes including baseline mobility, care giving availability and underlying medical comorbidities. The similarity in mean presenting NIHSS scores of the two groups, however, argues against this possibility. Lastly, exclusion criteria to receiving tPA may represent intrinsic characteristics that impart higher risk of negative outcomes.
Conculsion
Although there have been no randomized controlled trials that evaluate the safety and efficacy of tPA in the treatment of acute ischemic stroke in very old patients, use at the community-based stroke center was not associated with worse outcomes including symptomatic ICH, hospital length of stay, and in-hospital mortality. In fact, there were trends towards better outcomes in older patients who received tPA, including a significant reduction in in-hospital mortality. This evaluation supports the benefits of using tPA to treat acute ischemic stroke as seen in prior randomized controlled trials that included the treatment of very old patients. Though ongoing research is needed, a growing body of evidence supports the use of tPA to treat acute ischemic stroke in patients ≥ 80 years.
Corresponding author: Jennifer C. Drost, DO, MPH, Summa Health System, 75 Arch St., Ste. G1, Akron, OH 44304, [email protected].
Financial disclosures: None.
Author contributions: Conception and design, JCD, SMB; analysis and interpretation of data, JCD, SMB; drafting of article, JCD; critical revision of the article, JCD, SMB; provision of study materials or patients, SMB; collection and assembly of data, JCD.
1. US Census Bureau. Annual estimates of the resident population for selected age groups by sex for the United States, States, Counties, and Puerto Rico Commonwealth and Municipios: April 1, 2010 to July 1, 2013. Accessed at www.census.gov/popest/index.html.
2. Fang MC, Cutler DM, Rosen AB. Trends in thrombolytic use for ischemic stroke in the United States. J Hosp Med 2010;5:406–9.
3. Marini C, Baldassarre M, Russo T, et al. Burden of first-ever ischemic stroke in the oldest old: evidence from a population-based study. Neurology 2004;62:77–81.
4. Sylaja PN, Cote R, Buchan AM, Hill MD. Thrombolysis in patients older than 80 years with acute ischaemic stroke: Canadian Alteplase for Stroke Effectiveness Study. J Neurol Neurosurg Psychiatry 2006;77:826–9.
5. Heitsch LE, Panagos PD. Treating the elderly stroke patient: complications, controversies, and best care metrics. Clin Geriatr Med 2013;29:231–55.
6. Engelter ST, Bonati LH, Lyrer PA. Intravenous thrombolysis in stroke patients of > or = 80 versus < 80 years of age--a systematic review across cohort studies. Age Ageing 2006;35:572–580.
7. Forti P, Maioli F, Procaccianti G, et al. Independent predictors of ischemic stroke in the elderly: prospective data from a stroke unit. Neurology 2013;80:29–38.
8. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTischemic stroke, ECASS, and NINDS rt-PA stroke trials. Lancet 2004;363:768–74.
9. Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med 1995;333:1581–7.
10. Sung PS, Chen CH, Hsieh HC, et al. Outcome of acute ischemic stroke in very elderly patients: is intravenous thrombolysis beneficial? Eur Neurol 2011;66:110–6.
11. Saposnik G, Guzik AK, Reeves M, et al. Stroke prognostication using age and NIH Stroke Scale: SPAN-100. Neurology 2013;80:21–8.
12. Generalized efficacy of t-PA for acute stroke. Subgroup analysis of the NINDS t-PA Stroke Trial. Stroke 1997;28:2119–25.
13. Sandercock P, Wardlaw JM, Lindley RI, et al. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet 2012;379:2352–63.
14. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA Stroke Study Group. Stroke 1997;28:2109–18.
15. Bray BD, Campbell J, Hoffman A, et al. Stroke thrombolysis in England: an age stratified analysis of practice and outcome. Age Ageing 2013;42:240–5.
16. Kono S, Deguchi K, Morimoto N, et al. Intravenous thrombolysis with neuroprotective therapy by edaravone for ischemic stroke patients older than 80 years of age. J Stroke Cerebrovasc Dis 2013;22:1175–83.
17. Berrouschot J, Rother J, Glahn J, et al. Outcome and severe hemorrhagic complications of intravenous thrombolysis with tissue plasminogen activator in very old (> or =80 years) stroke patients. Stroke 2005;36:2421–5.
18. Emberson J, Lees KR, Lyden P, et al; Stroke Thrombolysis Trialists' Collaborative Group. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet 2014;384:1929–35.
1. US Census Bureau. Annual estimates of the resident population for selected age groups by sex for the United States, States, Counties, and Puerto Rico Commonwealth and Municipios: April 1, 2010 to July 1, 2013. Accessed at www.census.gov/popest/index.html.
2. Fang MC, Cutler DM, Rosen AB. Trends in thrombolytic use for ischemic stroke in the United States. J Hosp Med 2010;5:406–9.
3. Marini C, Baldassarre M, Russo T, et al. Burden of first-ever ischemic stroke in the oldest old: evidence from a population-based study. Neurology 2004;62:77–81.
4. Sylaja PN, Cote R, Buchan AM, Hill MD. Thrombolysis in patients older than 80 years with acute ischaemic stroke: Canadian Alteplase for Stroke Effectiveness Study. J Neurol Neurosurg Psychiatry 2006;77:826–9.
5. Heitsch LE, Panagos PD. Treating the elderly stroke patient: complications, controversies, and best care metrics. Clin Geriatr Med 2013;29:231–55.
6. Engelter ST, Bonati LH, Lyrer PA. Intravenous thrombolysis in stroke patients of > or = 80 versus < 80 years of age--a systematic review across cohort studies. Age Ageing 2006;35:572–580.
7. Forti P, Maioli F, Procaccianti G, et al. Independent predictors of ischemic stroke in the elderly: prospective data from a stroke unit. Neurology 2013;80:29–38.
8. Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTischemic stroke, ECASS, and NINDS rt-PA stroke trials. Lancet 2004;363:768–74.
9. Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med 1995;333:1581–7.
10. Sung PS, Chen CH, Hsieh HC, et al. Outcome of acute ischemic stroke in very elderly patients: is intravenous thrombolysis beneficial? Eur Neurol 2011;66:110–6.
11. Saposnik G, Guzik AK, Reeves M, et al. Stroke prognostication using age and NIH Stroke Scale: SPAN-100. Neurology 2013;80:21–8.
12. Generalized efficacy of t-PA for acute stroke. Subgroup analysis of the NINDS t-PA Stroke Trial. Stroke 1997;28:2119–25.
13. Sandercock P, Wardlaw JM, Lindley RI, et al. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet 2012;379:2352–63.
14. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA Stroke Study Group. Stroke 1997;28:2109–18.
15. Bray BD, Campbell J, Hoffman A, et al. Stroke thrombolysis in England: an age stratified analysis of practice and outcome. Age Ageing 2013;42:240–5.
16. Kono S, Deguchi K, Morimoto N, et al. Intravenous thrombolysis with neuroprotective therapy by edaravone for ischemic stroke patients older than 80 years of age. J Stroke Cerebrovasc Dis 2013;22:1175–83.
17. Berrouschot J, Rother J, Glahn J, et al. Outcome and severe hemorrhagic complications of intravenous thrombolysis with tissue plasminogen activator in very old (> or =80 years) stroke patients. Stroke 2005;36:2421–5.
18. Emberson J, Lees KR, Lyden P, et al; Stroke Thrombolysis Trialists' Collaborative Group. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet 2014;384:1929–35.
Comparison of Parent and Child versus Child-Only Weight Management Interventions in the Patient-Centered Medical Home
Study Overview
Objective. To determine the efficacy, both short and long term, of a behavioral intervention targeting overweight parents and their children simultaneously versus an intervention focused on weight management only for the child within the patient-centered medical home (PCMH).
Design. 4-center, 2-arm, randomized controlled trial.
Setting and participants. Study participants were recruited from 4 urban/suburban pediatric practices. Primary care providers (PCPs) recruited patients at the time of well or sick visits based on body mass index (BMI) flagged prior to the visit by Patient Enhancement Assistants (PEAs). 171 parent/child dyads were assessed for eligibility and 105 were randomized in blocks of 12 dyads using a random number generator and stratified by child’s gender. Pediatricians were blind to their patient’s group assignments. Inclusion criteria were as follows: children aged 2–5 with a BMI higher than the 85th percentile for both age and gender, and 1 parent with a BMI greater than 25. Exclusion criteria were limited to children who were small for gestational age and/or short stature, and child or parent inability to perform physical activity. Specific precautions were taken to prevent contamination between intervention and information control (IC) groups [1].
Intervention. Three PEAs who held a masters or bachelors degree in psychology, nutrition, exercise science, or equivalent, or were registered dietitians, were embedded within each PCMH practice. For both the intervention and IC groups, parents attended 13 one-hour group sessions led by a PEA over a 12-month period, followed by a 12-month follow-up period with 3 additional meetings. The PEA telephoned parents between scheduled meetings. Pediatricians reviewed child’s weight changes every 6 months during scheduled appointments and the PEA sent progress notes in between these visits [2]. Dietary, physical, and sedentary activity guidelines were given based on the recommendations of a national multi-organizational expert committee [3]. Parents were given specific goals for their child, including a 0.5- to 1-pound per week loss, 60 minutes per day of physical activity, and limiting TV and screen time to less than 2 hours per day.
In addition, the intervention group received parenting and behavior change strategies to promote both parent and child weight loss. Parents were instructed to weigh themselves and their child once per week and monitor physical activity and diet. They received individual meetings with the PEA before or after group meetings to review goal setting and food/physical activity diaries. Parents were also given a weight loss goal of 1 to 2 pounds per week and were advised to model physical activity by engaging in active play with their child for at least 10 minutes per day.
Main outcome measures. The main outcome measures were %0BMI and z-BMI. Percent 0BMI is defined as [(child’s BMI – 50th percentile BMI)/50th percentile BMI] x 100 [2]. The authors chose %0BMI as the primary outcome measure because z-BMI can diminish the effect of weight change in heavier children [4]. Both measures were expressed as mean ± standard error (SEM). Parent weight change was measured using BMI alone.
The child’s weight was measured at each session and height was measured at baseline, 3, 6, 12, 18, and 24 months. Parent weight was measured every session in the intervention group, but only at baseline, 6, 12, 18, and 24 months in the IC group. A standardized protocol was followed for all height and weight measurements. An intention to treat analysis (ITT) was conducted on all parent/child dyads, regardless of whether or not they completed the study (n = 96).
Results. Research assistants assessed 171 parent/child dyads for eligibility. 66 were excluded for either not meeting inclusion criteria (n = 24) or declining to participate (n = 42). 105 dyads were randomized, but 9 did not receive the allocated intervention because they did not start the study, resulting in a total of 96 dyads included in analysis: 46 in the intervention group and 50 in the IC. Twelve- and 24-month completion rates were 83% and 73% respectively; there was no difference in attrition between intervention and IC groups.
The mean child ages of the intervention and IC groups were 4.6 ± 0.2 and 4.4 ± 0.2 years, respectively. 33 of the 46 children in the intervention group and 37 of the 50 children in the IC group were identified as non-Hispanic white. The mean yearly income of all families was $65,729 ± $3068, with only 8.3% of families below $20,000.
The intervention group had greater decreases in child %0BMI from baseline to 6, 12, 18, and 24 months than the IC group. Similar trends were seen with child z-BMI. A slower increase in height was observed in the intervention group when compared with the IC at both 18 months (P < 0.001) and at 24 months (P < 0.02). Parents showed greater overall BMI reduction in the intervention group as opposed to the IC group at all time points (P < 0.001). BMI changes achieved at 6 months were maintained at 24 months. %0BMI and parent BMI changes were correlated from baseline to 12, 18, and 24 months. No significant baseline moderators were found among the children in either group.
Conclusion. This study demonstrated that within the PCMH model of pediatric primary care, an intervention focused on joint behavior change and weight modification treatment of parents and children led to better initial and sustained improvements in %0BMI and z-BMI (in children) and BMI (in parents) than a child-focused IC.
Commentary
Over one-third of children and adolescents are considered to be overweight or have obesity, a number that has doubled in the past 30 years [5]. Pediatrician and primary care physician visits are optimal places to identify overweight children who are at risk for obesity and begin prevention measures, although identifying overweight and obese younger children can be difficult [6]. This study used PEAs to aid physicians in identification, implementation, and delivery. With increasing evidence to support pediatrician involvement in intensive weight management in a primary versus specialty care setting, embedding PEAs within the PCMH model may be an important way to help deliver care for overweight/obese children [7].
Although many approaches have been considered to target childhood obesity, this study represents an important contribution to the literature because it demonstrates that a primary care–based intervention targeting parents as well as their young children is more efficacious for weight management than a more traditional, child-only focused intervention. In addition, the intervention included many different evidence-based components such as teaching behavior modification techniques to parents, consideration of parenting styles and techniques, and encouraging simultaneous parental weight modification. While the U.S. Preventive Services Task Force (USPSTF) recommends intensive interventions with 30 sessions over 2 years [8], this study was able to accomplish significant weight change in 13 sessions.
This intervention is unique in its integration of parenting techniques with other evidence-based strategies for child weight management. Although it has been shown in the literature that certain parenting styles can positively impact children’s health behaviors [9], namely the use of positive reinforcement and monitoring children’s health practices [10], only a few studies have looked at the impact of parenting interventions on childhood obesity. Mazzeo et al demonstrated a significant reduction in child BMI with a parenting-only intervention in the NOURISH trial [11], Slusser et al found a significant child BMI reduction using parent training for low-income, 2- to 4-year-old children [12], and Brotman et al conducted a longitudinal study demonstrating that a family intervention could decrease BMI and improve overall child health behaviors [13]. Despite these aforementioned studies, there is a lack of longitudinal data on the association between general parenting style and weight [14], and this study addresses this gap in literature by providing 2-year follow-up and demonstrating sustained impact on the intervention group.
This study had many additional strengths, including randomized design, primary care physician blinding, use of intention to treat analysis, standardization of measurement tools, clear justification of sample size, long-term follow-up, and the use of child-appropriate BMI measures (eg, %0BMI vs. z-BMI as primary outcome measure). In addition, the intervention setting in a PCMH follows the trend of increasing interest in exploring this model of health care delivery [15,16]. It is also important to note that the intervention and IC groups received the same number of group visits and phone calls, the only difference being the content and the extra 1:1 PEA sessions received by the intervention group.
The few weaknesses include that the PEAs could not be blinded to treatment allocation, and generalizability is limited by the mostly non-Hispanic white population and that only 8.3% of the study population had an annual household income of less than $20,000. All parents included in this study were on the high end of the obese range (BMI 30–39.9), with baseline BMI values of 37.2 and 36.2 in the intervention and IC groups respectively. In addition, the age of the children included in the study were on the high end of the designated 2- to 5-year-old range: 4.6 years (IC) and 4.4 years (intervention). Although findings were promising within this specific population, further research in younger and more diverse populations is necessary [11].
Finally, it is unclear whether this intervention is scalable, and a cost-effectiveness analysis of this intervention is needed. This study was designed to limit the PCP’s role and simplify the process of identifying and intervening on overweight children and their parents, yet this required 3 part-time PEAs and a project coordinator responsible for delivering all of the group sessions and providing follow-up counseling to both intervention and IC groups.
Applications for Clinical Practice
This study demonstrates that in a mostly white, urban/suburban population, a parenting and behavior modification intervention focused on both parent and child leads to greater improvements in %0BMI and z-BMI in the child and BMI reduction in parents compared with an intervention focused on the child alone within pediatric PCMH practices. This intervention should be tested in more diverse populations. This study also suggests further exploration of the use of PEAs to help clinicians address obesity within the PCMH model of primary care.
—Natalie Berner, BA, and Melanie Jay, MD, MS
1. Quattrin T, Roemmich JN, Paluch R, et al. Efficacy of family-based weight control program for preschool children in primary care. Pediatrics 2012;130:660–6.
2. Paluch RA, Epstein LH, Roemmich JN. Comparison of methods to evaluate changes in relative body mass index in pediatric weight control. Am J Hum Biol 2007;19:487–94.
3. Barlow SE, for the Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120(suppl 4):S164–S192.
4. Cole TJ, Faith MS, Pietrobelli A, Heo M. What is the best measure of adiposity change in growing children: BMI, BMI %, BMI z-score or BMI centile? Eur J Clin Nutr 2005;59: 419–25.
5. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA 2014;311:806–14.
6. Miller JL, Silverstein JH. Management approaches for pediatric obesity. Nature Clinical Practice Endocrin Metab 2007;3:810–8.
7. Perrin EM, Finkle JP, Benjamin JT. Obesity prevention and the primary care pediatrician’s office. Curr Opin Pediatr 2007; 19:354–61.
8. Barton M; US Preventive Services Task Force. Screening for obesity in children and adolescents: US Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
9. Institute of Medicine. Early childhood obesity prevention policies. Washington, DC: National Academies Press; 2011.
10. Arredondo EM, Elder JP, Ayala GX,et al. Is parenting style related to children’s healthy eating and physical activity in Latino families? Health Educ Res 2006;21:862–71.
11. Mazzeo SE, Kelly NR, Stern M, et al. Parent skills training to enhance weight loss in overweight children: Evaluation of NOURISH. Eat Behav 2014;15:225–9.
12. Slusser W, Frankel F, Robison K, et al. Pediatric overweight prevention through a parent training program for 2-4 year old Latino children. Child Obesity 2012;8:52–9.
13. Brotman LM, Dawson-McClure S, Huang K, et al. Early childhood obesity family intervention and long-term obesity prevention among high-risk minority youth. Pediatrics 2012;129:e621–e628.
14. Ventura AK, Birch LL. Does parenting affect children’s eating and weight status? Int J Behav Nutr Phys Act 2008;5:15.
15. Rosenthal TC. The medical home: growing evidence to support a new approach to primary care. J Am Board Fam Med 200;21:427–40.
16. Jackson GL, Powers BJ, Chatterjee R, et al. The patient-centered medical home: a systematic review. Ann Intern Med 2013;158:169–78.
Study Overview
Objective. To determine the efficacy, both short and long term, of a behavioral intervention targeting overweight parents and their children simultaneously versus an intervention focused on weight management only for the child within the patient-centered medical home (PCMH).
Design. 4-center, 2-arm, randomized controlled trial.
Setting and participants. Study participants were recruited from 4 urban/suburban pediatric practices. Primary care providers (PCPs) recruited patients at the time of well or sick visits based on body mass index (BMI) flagged prior to the visit by Patient Enhancement Assistants (PEAs). 171 parent/child dyads were assessed for eligibility and 105 were randomized in blocks of 12 dyads using a random number generator and stratified by child’s gender. Pediatricians were blind to their patient’s group assignments. Inclusion criteria were as follows: children aged 2–5 with a BMI higher than the 85th percentile for both age and gender, and 1 parent with a BMI greater than 25. Exclusion criteria were limited to children who were small for gestational age and/or short stature, and child or parent inability to perform physical activity. Specific precautions were taken to prevent contamination between intervention and information control (IC) groups [1].
Intervention. Three PEAs who held a masters or bachelors degree in psychology, nutrition, exercise science, or equivalent, or were registered dietitians, were embedded within each PCMH practice. For both the intervention and IC groups, parents attended 13 one-hour group sessions led by a PEA over a 12-month period, followed by a 12-month follow-up period with 3 additional meetings. The PEA telephoned parents between scheduled meetings. Pediatricians reviewed child’s weight changes every 6 months during scheduled appointments and the PEA sent progress notes in between these visits [2]. Dietary, physical, and sedentary activity guidelines were given based on the recommendations of a national multi-organizational expert committee [3]. Parents were given specific goals for their child, including a 0.5- to 1-pound per week loss, 60 minutes per day of physical activity, and limiting TV and screen time to less than 2 hours per day.
In addition, the intervention group received parenting and behavior change strategies to promote both parent and child weight loss. Parents were instructed to weigh themselves and their child once per week and monitor physical activity and diet. They received individual meetings with the PEA before or after group meetings to review goal setting and food/physical activity diaries. Parents were also given a weight loss goal of 1 to 2 pounds per week and were advised to model physical activity by engaging in active play with their child for at least 10 minutes per day.
Main outcome measures. The main outcome measures were %0BMI and z-BMI. Percent 0BMI is defined as [(child’s BMI – 50th percentile BMI)/50th percentile BMI] x 100 [2]. The authors chose %0BMI as the primary outcome measure because z-BMI can diminish the effect of weight change in heavier children [4]. Both measures were expressed as mean ± standard error (SEM). Parent weight change was measured using BMI alone.
The child’s weight was measured at each session and height was measured at baseline, 3, 6, 12, 18, and 24 months. Parent weight was measured every session in the intervention group, but only at baseline, 6, 12, 18, and 24 months in the IC group. A standardized protocol was followed for all height and weight measurements. An intention to treat analysis (ITT) was conducted on all parent/child dyads, regardless of whether or not they completed the study (n = 96).
Results. Research assistants assessed 171 parent/child dyads for eligibility. 66 were excluded for either not meeting inclusion criteria (n = 24) or declining to participate (n = 42). 105 dyads were randomized, but 9 did not receive the allocated intervention because they did not start the study, resulting in a total of 96 dyads included in analysis: 46 in the intervention group and 50 in the IC. Twelve- and 24-month completion rates were 83% and 73% respectively; there was no difference in attrition between intervention and IC groups.
The mean child ages of the intervention and IC groups were 4.6 ± 0.2 and 4.4 ± 0.2 years, respectively. 33 of the 46 children in the intervention group and 37 of the 50 children in the IC group were identified as non-Hispanic white. The mean yearly income of all families was $65,729 ± $3068, with only 8.3% of families below $20,000.
The intervention group had greater decreases in child %0BMI from baseline to 6, 12, 18, and 24 months than the IC group. Similar trends were seen with child z-BMI. A slower increase in height was observed in the intervention group when compared with the IC at both 18 months (P < 0.001) and at 24 months (P < 0.02). Parents showed greater overall BMI reduction in the intervention group as opposed to the IC group at all time points (P < 0.001). BMI changes achieved at 6 months were maintained at 24 months. %0BMI and parent BMI changes were correlated from baseline to 12, 18, and 24 months. No significant baseline moderators were found among the children in either group.
Conclusion. This study demonstrated that within the PCMH model of pediatric primary care, an intervention focused on joint behavior change and weight modification treatment of parents and children led to better initial and sustained improvements in %0BMI and z-BMI (in children) and BMI (in parents) than a child-focused IC.
Commentary
Over one-third of children and adolescents are considered to be overweight or have obesity, a number that has doubled in the past 30 years [5]. Pediatrician and primary care physician visits are optimal places to identify overweight children who are at risk for obesity and begin prevention measures, although identifying overweight and obese younger children can be difficult [6]. This study used PEAs to aid physicians in identification, implementation, and delivery. With increasing evidence to support pediatrician involvement in intensive weight management in a primary versus specialty care setting, embedding PEAs within the PCMH model may be an important way to help deliver care for overweight/obese children [7].
Although many approaches have been considered to target childhood obesity, this study represents an important contribution to the literature because it demonstrates that a primary care–based intervention targeting parents as well as their young children is more efficacious for weight management than a more traditional, child-only focused intervention. In addition, the intervention included many different evidence-based components such as teaching behavior modification techniques to parents, consideration of parenting styles and techniques, and encouraging simultaneous parental weight modification. While the U.S. Preventive Services Task Force (USPSTF) recommends intensive interventions with 30 sessions over 2 years [8], this study was able to accomplish significant weight change in 13 sessions.
This intervention is unique in its integration of parenting techniques with other evidence-based strategies for child weight management. Although it has been shown in the literature that certain parenting styles can positively impact children’s health behaviors [9], namely the use of positive reinforcement and monitoring children’s health practices [10], only a few studies have looked at the impact of parenting interventions on childhood obesity. Mazzeo et al demonstrated a significant reduction in child BMI with a parenting-only intervention in the NOURISH trial [11], Slusser et al found a significant child BMI reduction using parent training for low-income, 2- to 4-year-old children [12], and Brotman et al conducted a longitudinal study demonstrating that a family intervention could decrease BMI and improve overall child health behaviors [13]. Despite these aforementioned studies, there is a lack of longitudinal data on the association between general parenting style and weight [14], and this study addresses this gap in literature by providing 2-year follow-up and demonstrating sustained impact on the intervention group.
This study had many additional strengths, including randomized design, primary care physician blinding, use of intention to treat analysis, standardization of measurement tools, clear justification of sample size, long-term follow-up, and the use of child-appropriate BMI measures (eg, %0BMI vs. z-BMI as primary outcome measure). In addition, the intervention setting in a PCMH follows the trend of increasing interest in exploring this model of health care delivery [15,16]. It is also important to note that the intervention and IC groups received the same number of group visits and phone calls, the only difference being the content and the extra 1:1 PEA sessions received by the intervention group.
The few weaknesses include that the PEAs could not be blinded to treatment allocation, and generalizability is limited by the mostly non-Hispanic white population and that only 8.3% of the study population had an annual household income of less than $20,000. All parents included in this study were on the high end of the obese range (BMI 30–39.9), with baseline BMI values of 37.2 and 36.2 in the intervention and IC groups respectively. In addition, the age of the children included in the study were on the high end of the designated 2- to 5-year-old range: 4.6 years (IC) and 4.4 years (intervention). Although findings were promising within this specific population, further research in younger and more diverse populations is necessary [11].
Finally, it is unclear whether this intervention is scalable, and a cost-effectiveness analysis of this intervention is needed. This study was designed to limit the PCP’s role and simplify the process of identifying and intervening on overweight children and their parents, yet this required 3 part-time PEAs and a project coordinator responsible for delivering all of the group sessions and providing follow-up counseling to both intervention and IC groups.
Applications for Clinical Practice
This study demonstrates that in a mostly white, urban/suburban population, a parenting and behavior modification intervention focused on both parent and child leads to greater improvements in %0BMI and z-BMI in the child and BMI reduction in parents compared with an intervention focused on the child alone within pediatric PCMH practices. This intervention should be tested in more diverse populations. This study also suggests further exploration of the use of PEAs to help clinicians address obesity within the PCMH model of primary care.
—Natalie Berner, BA, and Melanie Jay, MD, MS
Study Overview
Objective. To determine the efficacy, both short and long term, of a behavioral intervention targeting overweight parents and their children simultaneously versus an intervention focused on weight management only for the child within the patient-centered medical home (PCMH).
Design. 4-center, 2-arm, randomized controlled trial.
Setting and participants. Study participants were recruited from 4 urban/suburban pediatric practices. Primary care providers (PCPs) recruited patients at the time of well or sick visits based on body mass index (BMI) flagged prior to the visit by Patient Enhancement Assistants (PEAs). 171 parent/child dyads were assessed for eligibility and 105 were randomized in blocks of 12 dyads using a random number generator and stratified by child’s gender. Pediatricians were blind to their patient’s group assignments. Inclusion criteria were as follows: children aged 2–5 with a BMI higher than the 85th percentile for both age and gender, and 1 parent with a BMI greater than 25. Exclusion criteria were limited to children who were small for gestational age and/or short stature, and child or parent inability to perform physical activity. Specific precautions were taken to prevent contamination between intervention and information control (IC) groups [1].
Intervention. Three PEAs who held a masters or bachelors degree in psychology, nutrition, exercise science, or equivalent, or were registered dietitians, were embedded within each PCMH practice. For both the intervention and IC groups, parents attended 13 one-hour group sessions led by a PEA over a 12-month period, followed by a 12-month follow-up period with 3 additional meetings. The PEA telephoned parents between scheduled meetings. Pediatricians reviewed child’s weight changes every 6 months during scheduled appointments and the PEA sent progress notes in between these visits [2]. Dietary, physical, and sedentary activity guidelines were given based on the recommendations of a national multi-organizational expert committee [3]. Parents were given specific goals for their child, including a 0.5- to 1-pound per week loss, 60 minutes per day of physical activity, and limiting TV and screen time to less than 2 hours per day.
In addition, the intervention group received parenting and behavior change strategies to promote both parent and child weight loss. Parents were instructed to weigh themselves and their child once per week and monitor physical activity and diet. They received individual meetings with the PEA before or after group meetings to review goal setting and food/physical activity diaries. Parents were also given a weight loss goal of 1 to 2 pounds per week and were advised to model physical activity by engaging in active play with their child for at least 10 minutes per day.
Main outcome measures. The main outcome measures were %0BMI and z-BMI. Percent 0BMI is defined as [(child’s BMI – 50th percentile BMI)/50th percentile BMI] x 100 [2]. The authors chose %0BMI as the primary outcome measure because z-BMI can diminish the effect of weight change in heavier children [4]. Both measures were expressed as mean ± standard error (SEM). Parent weight change was measured using BMI alone.
The child’s weight was measured at each session and height was measured at baseline, 3, 6, 12, 18, and 24 months. Parent weight was measured every session in the intervention group, but only at baseline, 6, 12, 18, and 24 months in the IC group. A standardized protocol was followed for all height and weight measurements. An intention to treat analysis (ITT) was conducted on all parent/child dyads, regardless of whether or not they completed the study (n = 96).
Results. Research assistants assessed 171 parent/child dyads for eligibility. 66 were excluded for either not meeting inclusion criteria (n = 24) or declining to participate (n = 42). 105 dyads were randomized, but 9 did not receive the allocated intervention because they did not start the study, resulting in a total of 96 dyads included in analysis: 46 in the intervention group and 50 in the IC. Twelve- and 24-month completion rates were 83% and 73% respectively; there was no difference in attrition between intervention and IC groups.
The mean child ages of the intervention and IC groups were 4.6 ± 0.2 and 4.4 ± 0.2 years, respectively. 33 of the 46 children in the intervention group and 37 of the 50 children in the IC group were identified as non-Hispanic white. The mean yearly income of all families was $65,729 ± $3068, with only 8.3% of families below $20,000.
The intervention group had greater decreases in child %0BMI from baseline to 6, 12, 18, and 24 months than the IC group. Similar trends were seen with child z-BMI. A slower increase in height was observed in the intervention group when compared with the IC at both 18 months (P < 0.001) and at 24 months (P < 0.02). Parents showed greater overall BMI reduction in the intervention group as opposed to the IC group at all time points (P < 0.001). BMI changes achieved at 6 months were maintained at 24 months. %0BMI and parent BMI changes were correlated from baseline to 12, 18, and 24 months. No significant baseline moderators were found among the children in either group.
Conclusion. This study demonstrated that within the PCMH model of pediatric primary care, an intervention focused on joint behavior change and weight modification treatment of parents and children led to better initial and sustained improvements in %0BMI and z-BMI (in children) and BMI (in parents) than a child-focused IC.
Commentary
Over one-third of children and adolescents are considered to be overweight or have obesity, a number that has doubled in the past 30 years [5]. Pediatrician and primary care physician visits are optimal places to identify overweight children who are at risk for obesity and begin prevention measures, although identifying overweight and obese younger children can be difficult [6]. This study used PEAs to aid physicians in identification, implementation, and delivery. With increasing evidence to support pediatrician involvement in intensive weight management in a primary versus specialty care setting, embedding PEAs within the PCMH model may be an important way to help deliver care for overweight/obese children [7].
Although many approaches have been considered to target childhood obesity, this study represents an important contribution to the literature because it demonstrates that a primary care–based intervention targeting parents as well as their young children is more efficacious for weight management than a more traditional, child-only focused intervention. In addition, the intervention included many different evidence-based components such as teaching behavior modification techniques to parents, consideration of parenting styles and techniques, and encouraging simultaneous parental weight modification. While the U.S. Preventive Services Task Force (USPSTF) recommends intensive interventions with 30 sessions over 2 years [8], this study was able to accomplish significant weight change in 13 sessions.
This intervention is unique in its integration of parenting techniques with other evidence-based strategies for child weight management. Although it has been shown in the literature that certain parenting styles can positively impact children’s health behaviors [9], namely the use of positive reinforcement and monitoring children’s health practices [10], only a few studies have looked at the impact of parenting interventions on childhood obesity. Mazzeo et al demonstrated a significant reduction in child BMI with a parenting-only intervention in the NOURISH trial [11], Slusser et al found a significant child BMI reduction using parent training for low-income, 2- to 4-year-old children [12], and Brotman et al conducted a longitudinal study demonstrating that a family intervention could decrease BMI and improve overall child health behaviors [13]. Despite these aforementioned studies, there is a lack of longitudinal data on the association between general parenting style and weight [14], and this study addresses this gap in literature by providing 2-year follow-up and demonstrating sustained impact on the intervention group.
This study had many additional strengths, including randomized design, primary care physician blinding, use of intention to treat analysis, standardization of measurement tools, clear justification of sample size, long-term follow-up, and the use of child-appropriate BMI measures (eg, %0BMI vs. z-BMI as primary outcome measure). In addition, the intervention setting in a PCMH follows the trend of increasing interest in exploring this model of health care delivery [15,16]. It is also important to note that the intervention and IC groups received the same number of group visits and phone calls, the only difference being the content and the extra 1:1 PEA sessions received by the intervention group.
The few weaknesses include that the PEAs could not be blinded to treatment allocation, and generalizability is limited by the mostly non-Hispanic white population and that only 8.3% of the study population had an annual household income of less than $20,000. All parents included in this study were on the high end of the obese range (BMI 30–39.9), with baseline BMI values of 37.2 and 36.2 in the intervention and IC groups respectively. In addition, the age of the children included in the study were on the high end of the designated 2- to 5-year-old range: 4.6 years (IC) and 4.4 years (intervention). Although findings were promising within this specific population, further research in younger and more diverse populations is necessary [11].
Finally, it is unclear whether this intervention is scalable, and a cost-effectiveness analysis of this intervention is needed. This study was designed to limit the PCP’s role and simplify the process of identifying and intervening on overweight children and their parents, yet this required 3 part-time PEAs and a project coordinator responsible for delivering all of the group sessions and providing follow-up counseling to both intervention and IC groups.
Applications for Clinical Practice
This study demonstrates that in a mostly white, urban/suburban population, a parenting and behavior modification intervention focused on both parent and child leads to greater improvements in %0BMI and z-BMI in the child and BMI reduction in parents compared with an intervention focused on the child alone within pediatric PCMH practices. This intervention should be tested in more diverse populations. This study also suggests further exploration of the use of PEAs to help clinicians address obesity within the PCMH model of primary care.
—Natalie Berner, BA, and Melanie Jay, MD, MS
1. Quattrin T, Roemmich JN, Paluch R, et al. Efficacy of family-based weight control program for preschool children in primary care. Pediatrics 2012;130:660–6.
2. Paluch RA, Epstein LH, Roemmich JN. Comparison of methods to evaluate changes in relative body mass index in pediatric weight control. Am J Hum Biol 2007;19:487–94.
3. Barlow SE, for the Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120(suppl 4):S164–S192.
4. Cole TJ, Faith MS, Pietrobelli A, Heo M. What is the best measure of adiposity change in growing children: BMI, BMI %, BMI z-score or BMI centile? Eur J Clin Nutr 2005;59: 419–25.
5. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA 2014;311:806–14.
6. Miller JL, Silverstein JH. Management approaches for pediatric obesity. Nature Clinical Practice Endocrin Metab 2007;3:810–8.
7. Perrin EM, Finkle JP, Benjamin JT. Obesity prevention and the primary care pediatrician’s office. Curr Opin Pediatr 2007; 19:354–61.
8. Barton M; US Preventive Services Task Force. Screening for obesity in children and adolescents: US Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
9. Institute of Medicine. Early childhood obesity prevention policies. Washington, DC: National Academies Press; 2011.
10. Arredondo EM, Elder JP, Ayala GX,et al. Is parenting style related to children’s healthy eating and physical activity in Latino families? Health Educ Res 2006;21:862–71.
11. Mazzeo SE, Kelly NR, Stern M, et al. Parent skills training to enhance weight loss in overweight children: Evaluation of NOURISH. Eat Behav 2014;15:225–9.
12. Slusser W, Frankel F, Robison K, et al. Pediatric overweight prevention through a parent training program for 2-4 year old Latino children. Child Obesity 2012;8:52–9.
13. Brotman LM, Dawson-McClure S, Huang K, et al. Early childhood obesity family intervention and long-term obesity prevention among high-risk minority youth. Pediatrics 2012;129:e621–e628.
14. Ventura AK, Birch LL. Does parenting affect children’s eating and weight status? Int J Behav Nutr Phys Act 2008;5:15.
15. Rosenthal TC. The medical home: growing evidence to support a new approach to primary care. J Am Board Fam Med 200;21:427–40.
16. Jackson GL, Powers BJ, Chatterjee R, et al. The patient-centered medical home: a systematic review. Ann Intern Med 2013;158:169–78.
1. Quattrin T, Roemmich JN, Paluch R, et al. Efficacy of family-based weight control program for preschool children in primary care. Pediatrics 2012;130:660–6.
2. Paluch RA, Epstein LH, Roemmich JN. Comparison of methods to evaluate changes in relative body mass index in pediatric weight control. Am J Hum Biol 2007;19:487–94.
3. Barlow SE, for the Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007;120(suppl 4):S164–S192.
4. Cole TJ, Faith MS, Pietrobelli A, Heo M. What is the best measure of adiposity change in growing children: BMI, BMI %, BMI z-score or BMI centile? Eur J Clin Nutr 2005;59: 419–25.
5. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA 2014;311:806–14.
6. Miller JL, Silverstein JH. Management approaches for pediatric obesity. Nature Clinical Practice Endocrin Metab 2007;3:810–8.
7. Perrin EM, Finkle JP, Benjamin JT. Obesity prevention and the primary care pediatrician’s office. Curr Opin Pediatr 2007; 19:354–61.
8. Barton M; US Preventive Services Task Force. Screening for obesity in children and adolescents: US Preventive Services Task Force recommendation statement. Pediatrics 2010;125:361–7.
9. Institute of Medicine. Early childhood obesity prevention policies. Washington, DC: National Academies Press; 2011.
10. Arredondo EM, Elder JP, Ayala GX,et al. Is parenting style related to children’s healthy eating and physical activity in Latino families? Health Educ Res 2006;21:862–71.
11. Mazzeo SE, Kelly NR, Stern M, et al. Parent skills training to enhance weight loss in overweight children: Evaluation of NOURISH. Eat Behav 2014;15:225–9.
12. Slusser W, Frankel F, Robison K, et al. Pediatric overweight prevention through a parent training program for 2-4 year old Latino children. Child Obesity 2012;8:52–9.
13. Brotman LM, Dawson-McClure S, Huang K, et al. Early childhood obesity family intervention and long-term obesity prevention among high-risk minority youth. Pediatrics 2012;129:e621–e628.
14. Ventura AK, Birch LL. Does parenting affect children’s eating and weight status? Int J Behav Nutr Phys Act 2008;5:15.
15. Rosenthal TC. The medical home: growing evidence to support a new approach to primary care. J Am Board Fam Med 200;21:427–40.
16. Jackson GL, Powers BJ, Chatterjee R, et al. The patient-centered medical home: a systematic review. Ann Intern Med 2013;158:169–78.
Mogamulizumab in PTCL: Europe vs Japan

SAN FRANCISCO—Two phase 2 studies testing mogamulizumab in peripheral T-cell lymphomas (PTCLs) suggest that higher response rates don’t necessarily translate to an improvement in progression-free survival (PFS).
The anti-CCR4 antibody produced a higher overall response rate (ORR) in a Japanese study than in a European study—34% and 11%, respectively.
However, median PFS times were similar—about 2 months in both studies.
This similarity is all the more interesting because the studies enrolled different types of patients and followed different dosing schedules, according to Pier Luigi Zinzani, MD, PhD, of the University of Bologna in Italy.
Dr Zinzani discussed details of the European experience testing mogamulizumab in PTCL, comparing it to the Japanese experience, in a presentation at the 7th Annual T-cell Lymphoma Forum.
Kensei Tobinai, MD, PhD, of the National Cancer Center Hospital in Tokyo, Japan, also reviewed the Japanese experience (TCLF 2013, JCO 2014) during the meeting’s keynote address and presented data from an ancillary analysis of this study (which is unpublished).
All of the research was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing mogamulizumab.
The Japanese experience
The Japanese study included 29 patients with PTCL and 8 with cutaneous T-cell lymphoma (CTCL). All patients had relapsed after their last chemotherapy regimen, and none had received an allogeneic stem cell transplant (allo-SCT). The PTCL patients had a median age of 67, and 69% were male.
All patients received mogamulizumab at 1.0 mg/kg/day weekly for 8 weeks. The ORR was 35%—34% for PTCL patients and 38% for CTCL patients.
Among PTCL patients, there were 5 complete responses (CRs) and 5 partial responses (PRs). Nine patients had stable disease (SD), and 10 progressed.
Of the 16 patients with PTCL-not otherwise specified (PTCL-NOS), 1 had a CR, 2 had a PR, 6 had SD, and 7 progressed. Of the 12 patients with angioimmunoblastic T-cell lymphoma (AITL), 3 had a CR, 3 had a PR, 3 had SD, and 3 progressed. The only patient with ALK- anaplastic large-cell lymphoma (ALCL) had an unconfirmed CR.
The ancillary analysis showed that tumor shrinkage of the target lesions occurred in 72% (21/29) of patients with PTCL. The patients’ median duration of response was 6.4 months, and the median time to response was 1.9 months.
Overall, the median PFS was 3.0 months—2.0 months in patients with PTCL and 3.4 months in patients with CTCL.
Common adverse events (for both PTCL and CTCL patients) included lymphopenia (81%), skin disorders (51%), leukopenia (43%), neutropenia (38%), thrombocytopenia (38%), pyrexia (30%), acute infusion reactions (24%), and anemia (14%).
Dr Tobinai noted that these results are not as favorable as those observed when patients with adult T-cell leukemia-lymphoma receive mogamulizumab.
“But compared to the efficacy rate of other approved agents—pralatrexate and romidepsin—this antibody has promising efficacy,” he said.
In fact, the results of this study prompted the December approval of mogamulizumab to treat PTCL and CTCL patients in Japan.
The European experience
The European study differed from the Japanese study in a few ways, Dr Zinzani pointed out. The European study only enrolled patients with PTCL. And it included patients with relapsed (49%) or refractory (51%) disease, whereas the Japanese study only included relapsed patients.
Furthermore, the Japanese study did not include any patients with an ECOG performance status of 2, while the European study did (39%). And the dosing schedule differed between the 2 studies.
In the European study, patients received mogamulizumab at 1 mg/kg once weekly for 4 weeks and then once every 2 weeks until they progressed or developed unacceptable toxicity.
There were 38 patients in the safety analysis. They had a median age of 58.5 years, and 61% were male.
Thirty-five of these patients were included in the efficacy analysis. They had a median of 2 prior treatments (range, 1-8), and 17 patients (49%) had responded to their last therapy.
The patients had PTCL-NOS (43%, 15/35), AITL (34%, 12), transformed mycosis fungoides (9%, 3), ALK- ALCL (11%, 4), and ALK+ ALCL (3%, 1).
The ORR was 11% (n=4), and 46% of patients (n=16) had SD or better. Two patients with PTCL-NOS responded, as did 2 with AITL.
Six patients with PTCL-NOS had SD, as did 3 with AITL, 1 with transformed mycosis fungoides, and 2 with ALK- ALCL.
The median duration of response (including SD) was 2.9 months. And the median PFS was 2.1 months. Two patients (1 with ALK- ALCL and 1 with PTCL-NOS) went on to allo-SCT.
The most frequent adverse events (occurring in at least 10% of patients) were drug eruption (n=12), pyrexia (n=9), pruritus (n=7), diarrhea (n=7), cough (n=6), vomiting (n=6), thrombocytopenia (n=6), hypotension (n=4), headache (n=4), peripheral edema (n=4), asthenia (n=4), nausea (n=4), anemia (n=4), and neutropenia (n=4).
“For the European experience, there were some differences from the Japanese experience,” Dr Zinzani said in closing. “It was worse in terms of overall response rate—only 11%—but roughly 50% of patients attained at least stable disease. And there was an acceptable safety profile in these really heavily pretreated, relapsed/refractory PTCL patients.” ![]()
SAN FRANCISCO—Two phase 2 studies testing mogamulizumab in peripheral T-cell lymphomas (PTCLs) suggest that higher response rates don’t necessarily translate to an improvement in progression-free survival (PFS).
The anti-CCR4 antibody produced a higher overall response rate (ORR) in a Japanese study than in a European study—34% and 11%, respectively.
However, median PFS times were similar—about 2 months in both studies.
This similarity is all the more interesting because the studies enrolled different types of patients and followed different dosing schedules, according to Pier Luigi Zinzani, MD, PhD, of the University of Bologna in Italy.
Dr Zinzani discussed details of the European experience testing mogamulizumab in PTCL, comparing it to the Japanese experience, in a presentation at the 7th Annual T-cell Lymphoma Forum.
Kensei Tobinai, MD, PhD, of the National Cancer Center Hospital in Tokyo, Japan, also reviewed the Japanese experience (TCLF 2013, JCO 2014) during the meeting’s keynote address and presented data from an ancillary analysis of this study (which is unpublished).
All of the research was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing mogamulizumab.
The Japanese experience
The Japanese study included 29 patients with PTCL and 8 with cutaneous T-cell lymphoma (CTCL). All patients had relapsed after their last chemotherapy regimen, and none had received an allogeneic stem cell transplant (allo-SCT). The PTCL patients had a median age of 67, and 69% were male.
All patients received mogamulizumab at 1.0 mg/kg/day weekly for 8 weeks. The ORR was 35%—34% for PTCL patients and 38% for CTCL patients.
Among PTCL patients, there were 5 complete responses (CRs) and 5 partial responses (PRs). Nine patients had stable disease (SD), and 10 progressed.
Of the 16 patients with PTCL-not otherwise specified (PTCL-NOS), 1 had a CR, 2 had a PR, 6 had SD, and 7 progressed. Of the 12 patients with angioimmunoblastic T-cell lymphoma (AITL), 3 had a CR, 3 had a PR, 3 had SD, and 3 progressed. The only patient with ALK- anaplastic large-cell lymphoma (ALCL) had an unconfirmed CR.
The ancillary analysis showed that tumor shrinkage of the target lesions occurred in 72% (21/29) of patients with PTCL. The patients’ median duration of response was 6.4 months, and the median time to response was 1.9 months.
Overall, the median PFS was 3.0 months—2.0 months in patients with PTCL and 3.4 months in patients with CTCL.
Common adverse events (for both PTCL and CTCL patients) included lymphopenia (81%), skin disorders (51%), leukopenia (43%), neutropenia (38%), thrombocytopenia (38%), pyrexia (30%), acute infusion reactions (24%), and anemia (14%).
Dr Tobinai noted that these results are not as favorable as those observed when patients with adult T-cell leukemia-lymphoma receive mogamulizumab.
“But compared to the efficacy rate of other approved agents—pralatrexate and romidepsin—this antibody has promising efficacy,” he said.
In fact, the results of this study prompted the December approval of mogamulizumab to treat PTCL and CTCL patients in Japan.
The European experience
The European study differed from the Japanese study in a few ways, Dr Zinzani pointed out. The European study only enrolled patients with PTCL. And it included patients with relapsed (49%) or refractory (51%) disease, whereas the Japanese study only included relapsed patients.
Furthermore, the Japanese study did not include any patients with an ECOG performance status of 2, while the European study did (39%). And the dosing schedule differed between the 2 studies.
In the European study, patients received mogamulizumab at 1 mg/kg once weekly for 4 weeks and then once every 2 weeks until they progressed or developed unacceptable toxicity.
There were 38 patients in the safety analysis. They had a median age of 58.5 years, and 61% were male.
Thirty-five of these patients were included in the efficacy analysis. They had a median of 2 prior treatments (range, 1-8), and 17 patients (49%) had responded to their last therapy.
The patients had PTCL-NOS (43%, 15/35), AITL (34%, 12), transformed mycosis fungoides (9%, 3), ALK- ALCL (11%, 4), and ALK+ ALCL (3%, 1).
The ORR was 11% (n=4), and 46% of patients (n=16) had SD or better. Two patients with PTCL-NOS responded, as did 2 with AITL.
Six patients with PTCL-NOS had SD, as did 3 with AITL, 1 with transformed mycosis fungoides, and 2 with ALK- ALCL.
The median duration of response (including SD) was 2.9 months. And the median PFS was 2.1 months. Two patients (1 with ALK- ALCL and 1 with PTCL-NOS) went on to allo-SCT.
The most frequent adverse events (occurring in at least 10% of patients) were drug eruption (n=12), pyrexia (n=9), pruritus (n=7), diarrhea (n=7), cough (n=6), vomiting (n=6), thrombocytopenia (n=6), hypotension (n=4), headache (n=4), peripheral edema (n=4), asthenia (n=4), nausea (n=4), anemia (n=4), and neutropenia (n=4).
“For the European experience, there were some differences from the Japanese experience,” Dr Zinzani said in closing. “It was worse in terms of overall response rate—only 11%—but roughly 50% of patients attained at least stable disease. And there was an acceptable safety profile in these really heavily pretreated, relapsed/refractory PTCL patients.” ![]()
SAN FRANCISCO—Two phase 2 studies testing mogamulizumab in peripheral T-cell lymphomas (PTCLs) suggest that higher response rates don’t necessarily translate to an improvement in progression-free survival (PFS).
The anti-CCR4 antibody produced a higher overall response rate (ORR) in a Japanese study than in a European study—34% and 11%, respectively.
However, median PFS times were similar—about 2 months in both studies.
This similarity is all the more interesting because the studies enrolled different types of patients and followed different dosing schedules, according to Pier Luigi Zinzani, MD, PhD, of the University of Bologna in Italy.
Dr Zinzani discussed details of the European experience testing mogamulizumab in PTCL, comparing it to the Japanese experience, in a presentation at the 7th Annual T-cell Lymphoma Forum.
Kensei Tobinai, MD, PhD, of the National Cancer Center Hospital in Tokyo, Japan, also reviewed the Japanese experience (TCLF 2013, JCO 2014) during the meeting’s keynote address and presented data from an ancillary analysis of this study (which is unpublished).
All of the research was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing mogamulizumab.
The Japanese experience
The Japanese study included 29 patients with PTCL and 8 with cutaneous T-cell lymphoma (CTCL). All patients had relapsed after their last chemotherapy regimen, and none had received an allogeneic stem cell transplant (allo-SCT). The PTCL patients had a median age of 67, and 69% were male.
All patients received mogamulizumab at 1.0 mg/kg/day weekly for 8 weeks. The ORR was 35%—34% for PTCL patients and 38% for CTCL patients.
Among PTCL patients, there were 5 complete responses (CRs) and 5 partial responses (PRs). Nine patients had stable disease (SD), and 10 progressed.
Of the 16 patients with PTCL-not otherwise specified (PTCL-NOS), 1 had a CR, 2 had a PR, 6 had SD, and 7 progressed. Of the 12 patients with angioimmunoblastic T-cell lymphoma (AITL), 3 had a CR, 3 had a PR, 3 had SD, and 3 progressed. The only patient with ALK- anaplastic large-cell lymphoma (ALCL) had an unconfirmed CR.
The ancillary analysis showed that tumor shrinkage of the target lesions occurred in 72% (21/29) of patients with PTCL. The patients’ median duration of response was 6.4 months, and the median time to response was 1.9 months.
Overall, the median PFS was 3.0 months—2.0 months in patients with PTCL and 3.4 months in patients with CTCL.
Common adverse events (for both PTCL and CTCL patients) included lymphopenia (81%), skin disorders (51%), leukopenia (43%), neutropenia (38%), thrombocytopenia (38%), pyrexia (30%), acute infusion reactions (24%), and anemia (14%).
Dr Tobinai noted that these results are not as favorable as those observed when patients with adult T-cell leukemia-lymphoma receive mogamulizumab.
“But compared to the efficacy rate of other approved agents—pralatrexate and romidepsin—this antibody has promising efficacy,” he said.
In fact, the results of this study prompted the December approval of mogamulizumab to treat PTCL and CTCL patients in Japan.
The European experience
The European study differed from the Japanese study in a few ways, Dr Zinzani pointed out. The European study only enrolled patients with PTCL. And it included patients with relapsed (49%) or refractory (51%) disease, whereas the Japanese study only included relapsed patients.
Furthermore, the Japanese study did not include any patients with an ECOG performance status of 2, while the European study did (39%). And the dosing schedule differed between the 2 studies.
In the European study, patients received mogamulizumab at 1 mg/kg once weekly for 4 weeks and then once every 2 weeks until they progressed or developed unacceptable toxicity.
There were 38 patients in the safety analysis. They had a median age of 58.5 years, and 61% were male.
Thirty-five of these patients were included in the efficacy analysis. They had a median of 2 prior treatments (range, 1-8), and 17 patients (49%) had responded to their last therapy.
The patients had PTCL-NOS (43%, 15/35), AITL (34%, 12), transformed mycosis fungoides (9%, 3), ALK- ALCL (11%, 4), and ALK+ ALCL (3%, 1).
The ORR was 11% (n=4), and 46% of patients (n=16) had SD or better. Two patients with PTCL-NOS responded, as did 2 with AITL.
Six patients with PTCL-NOS had SD, as did 3 with AITL, 1 with transformed mycosis fungoides, and 2 with ALK- ALCL.
The median duration of response (including SD) was 2.9 months. And the median PFS was 2.1 months. Two patients (1 with ALK- ALCL and 1 with PTCL-NOS) went on to allo-SCT.
The most frequent adverse events (occurring in at least 10% of patients) were drug eruption (n=12), pyrexia (n=9), pruritus (n=7), diarrhea (n=7), cough (n=6), vomiting (n=6), thrombocytopenia (n=6), hypotension (n=4), headache (n=4), peripheral edema (n=4), asthenia (n=4), nausea (n=4), anemia (n=4), and neutropenia (n=4).
“For the European experience, there were some differences from the Japanese experience,” Dr Zinzani said in closing. “It was worse in terms of overall response rate—only 11%—but roughly 50% of patients attained at least stable disease. And there was an acceptable safety profile in these really heavily pretreated, relapsed/refractory PTCL patients.” ![]()
Though costly, blood cancer drugs appear cost-effective

Photo by Bill Branson
A new analysis indicates that certain high-cost therapies for hematologic malignancies provide reasonable value for money spent.
Most cost-effectiveness ratios were lower than thresholds commonly used to establish cost-effectiveness in the US—$50,000 or $100,000 per quality-adjusted life year (QALY) gained.
The median cost-effectiveness ratio was highest for chronic myeloid leukemia (CML), at $55,000/QALY, and lowest for non-Hodgkin lymphoma (NHL), at $21,500/QALY.
Researchers presented these data in Blood.
“Given the increased discussion about the high cost of these treatments, we were somewhat surprised to discover that their cost-effectiveness ratios were lower than expected,” said study author Peter J. Neumann, ScD, of Tufts Medical Center in Boston.
“Our analysis had a small sample size and included both industry- and non-industry-funded studies. In addition, cost-effectiveness ratios may have changed over time as associated costs or benefits have changed. However, the study underscores that debates in healthcare should consider the value of breakthrough drugs and not just costs.”
With that issue in mind, Dr Neumann and his colleagues had conducted a systematic review of studies published between 1996 and 2012 that examined the cost utility of agents for hematologic malignancies. The cost utility of a drug was depicted as a ratio of a drug’s total cost per patient QALY gained.
The researchers identified 29 studies, 22 of which were industry-funded. Nine studies were conducted from a US perspective, 6 from the UK, 3 from Norway, 3 from Sweden, 2 from France, 1 from Canada, 1 from Finland, and 4 from “other” countries.
The team grouped studies according to malignancy—CML, chronic lymphocytic leukemia (CLL), NHL, and multiple myeloma (MM)—as well as by treatment—α interferon, alemtuzumab, bendamustine, bortezomib, dasatinib, imatinib, lenalidomide, rituximab alone or in combination, and thalidomide.
The studies reported 44 cost-effectiveness ratios, most concerning interventions for NHL (41%) or CML (30%). Most ratios pertained to rituximab (43%), α interferon (18%), or imatinib (16%), and the most common intervention-disease combination was rituximab (alone or in combination) for NHL (36%).
The median cost-effectiveness ratios fluctuated over time, rising from $35,000/QALY (1996-2002) to $52,000/QALY (2003-2006), then falling to $22,000/QALY (2007-2012).
The median cost-effectiveness ratio reported by industry-funded studies was lower ($26,000/QALY) than for non-industry-funded studies ($33,000/QALY).
Four cost-effectiveness ratios, 1 from an industry-funded study, exceeded $100,000/QALY. This included 2 studies of bortezomib in MM, 1 of α interferon in CML, and 1 of imatinib in CML.
The researchers said these results suggest that many new treatments for hematologic malignancies may confer reasonable value for money spent. The distribution of cost-effectiveness ratios is comparable to those for cancers overall and for other healthcare fields, they said.
This study was funded by internal resources at the Center for the Evaluation of Value and Risk in Health. The center receives funding from federal, private foundation, and pharmaceutical industry sources. ![]()
Photo by Bill Branson
A new analysis indicates that certain high-cost therapies for hematologic malignancies provide reasonable value for money spent.
Most cost-effectiveness ratios were lower than thresholds commonly used to establish cost-effectiveness in the US—$50,000 or $100,000 per quality-adjusted life year (QALY) gained.
The median cost-effectiveness ratio was highest for chronic myeloid leukemia (CML), at $55,000/QALY, and lowest for non-Hodgkin lymphoma (NHL), at $21,500/QALY.
Researchers presented these data in Blood.
“Given the increased discussion about the high cost of these treatments, we were somewhat surprised to discover that their cost-effectiveness ratios were lower than expected,” said study author Peter J. Neumann, ScD, of Tufts Medical Center in Boston.
“Our analysis had a small sample size and included both industry- and non-industry-funded studies. In addition, cost-effectiveness ratios may have changed over time as associated costs or benefits have changed. However, the study underscores that debates in healthcare should consider the value of breakthrough drugs and not just costs.”
With that issue in mind, Dr Neumann and his colleagues had conducted a systematic review of studies published between 1996 and 2012 that examined the cost utility of agents for hematologic malignancies. The cost utility of a drug was depicted as a ratio of a drug’s total cost per patient QALY gained.
The researchers identified 29 studies, 22 of which were industry-funded. Nine studies were conducted from a US perspective, 6 from the UK, 3 from Norway, 3 from Sweden, 2 from France, 1 from Canada, 1 from Finland, and 4 from “other” countries.
The team grouped studies according to malignancy—CML, chronic lymphocytic leukemia (CLL), NHL, and multiple myeloma (MM)—as well as by treatment—α interferon, alemtuzumab, bendamustine, bortezomib, dasatinib, imatinib, lenalidomide, rituximab alone or in combination, and thalidomide.
The studies reported 44 cost-effectiveness ratios, most concerning interventions for NHL (41%) or CML (30%). Most ratios pertained to rituximab (43%), α interferon (18%), or imatinib (16%), and the most common intervention-disease combination was rituximab (alone or in combination) for NHL (36%).
The median cost-effectiveness ratios fluctuated over time, rising from $35,000/QALY (1996-2002) to $52,000/QALY (2003-2006), then falling to $22,000/QALY (2007-2012).
The median cost-effectiveness ratio reported by industry-funded studies was lower ($26,000/QALY) than for non-industry-funded studies ($33,000/QALY).
Four cost-effectiveness ratios, 1 from an industry-funded study, exceeded $100,000/QALY. This included 2 studies of bortezomib in MM, 1 of α interferon in CML, and 1 of imatinib in CML.
The researchers said these results suggest that many new treatments for hematologic malignancies may confer reasonable value for money spent. The distribution of cost-effectiveness ratios is comparable to those for cancers overall and for other healthcare fields, they said.
This study was funded by internal resources at the Center for the Evaluation of Value and Risk in Health. The center receives funding from federal, private foundation, and pharmaceutical industry sources. ![]()
Photo by Bill Branson
A new analysis indicates that certain high-cost therapies for hematologic malignancies provide reasonable value for money spent.
Most cost-effectiveness ratios were lower than thresholds commonly used to establish cost-effectiveness in the US—$50,000 or $100,000 per quality-adjusted life year (QALY) gained.
The median cost-effectiveness ratio was highest for chronic myeloid leukemia (CML), at $55,000/QALY, and lowest for non-Hodgkin lymphoma (NHL), at $21,500/QALY.
Researchers presented these data in Blood.
“Given the increased discussion about the high cost of these treatments, we were somewhat surprised to discover that their cost-effectiveness ratios were lower than expected,” said study author Peter J. Neumann, ScD, of Tufts Medical Center in Boston.
“Our analysis had a small sample size and included both industry- and non-industry-funded studies. In addition, cost-effectiveness ratios may have changed over time as associated costs or benefits have changed. However, the study underscores that debates in healthcare should consider the value of breakthrough drugs and not just costs.”
With that issue in mind, Dr Neumann and his colleagues had conducted a systematic review of studies published between 1996 and 2012 that examined the cost utility of agents for hematologic malignancies. The cost utility of a drug was depicted as a ratio of a drug’s total cost per patient QALY gained.
The researchers identified 29 studies, 22 of which were industry-funded. Nine studies were conducted from a US perspective, 6 from the UK, 3 from Norway, 3 from Sweden, 2 from France, 1 from Canada, 1 from Finland, and 4 from “other” countries.
The team grouped studies according to malignancy—CML, chronic lymphocytic leukemia (CLL), NHL, and multiple myeloma (MM)—as well as by treatment—α interferon, alemtuzumab, bendamustine, bortezomib, dasatinib, imatinib, lenalidomide, rituximab alone or in combination, and thalidomide.
The studies reported 44 cost-effectiveness ratios, most concerning interventions for NHL (41%) or CML (30%). Most ratios pertained to rituximab (43%), α interferon (18%), or imatinib (16%), and the most common intervention-disease combination was rituximab (alone or in combination) for NHL (36%).
The median cost-effectiveness ratios fluctuated over time, rising from $35,000/QALY (1996-2002) to $52,000/QALY (2003-2006), then falling to $22,000/QALY (2007-2012).
The median cost-effectiveness ratio reported by industry-funded studies was lower ($26,000/QALY) than for non-industry-funded studies ($33,000/QALY).
Four cost-effectiveness ratios, 1 from an industry-funded study, exceeded $100,000/QALY. This included 2 studies of bortezomib in MM, 1 of α interferon in CML, and 1 of imatinib in CML.
The researchers said these results suggest that many new treatments for hematologic malignancies may confer reasonable value for money spent. The distribution of cost-effectiveness ratios is comparable to those for cancers overall and for other healthcare fields, they said.
This study was funded by internal resources at the Center for the Evaluation of Value and Risk in Health. The center receives funding from federal, private foundation, and pharmaceutical industry sources. ![]()
Hand rejuvenation
The three most exposed areas of the body that give away a person’s age are the face, neck, and hands. Rejuvenation of the hands is an often simple and nice addition to facial and neck aesthetic rejuvenation.
When examining aging hands, the three most prominent features are decreased volume in the interosseous spaces (leading to increased crepiness of the skin and increased show of extensor tendons), lentigines, and prominent veins. Therefore, the treatment for hands is quite simple: Restore volume, treat the pigmented lesions, and if needed, treat the prominent veins.
The anatomy of the dorsal hand can be divided into three major compartments. First, the skin, which on the dorsal hand is quite pliable. Second, the subcutaneous tissue, which consists of a loose areolar tissue where the lymphatics and veins lie. Third, beneath the subcutaneous tissue is the dorsal fascia of the hand, which is contiguous with extensor tendons and underlying compartments. It is in the subcutaneous layer (or loose areolar tissue) where fillers or fat are placed to treat volume loss.
While several fillers are currently used off label for hand rejuvenation, the Food and Drug Administration is meeting in February to consider officially approving Radiesse for this indication. Currently, hyaluronic acid (HA) fillers, calcium- hydroxylapatite (Radiesse), poly-L-lactic acid, and autologous fat are all utilized. I tend to use HAs in this location because of the reversibility, if needed, and decreased risk of nodule formation. Several techniques exist, including injecting between each tendon space vs. a bolus technique. I tend to use a bolus technique, where one or two boluses are injected while tenting the skin up to ensure injection into the correct plane and to avoid the vessels. Subsequently, the boluses are massaged into place while the patient makes a fist.
Once the interosseous spaces have been treated, the veins often appear less prominent and often don’t require direct treatment. I typically do not treat the dorsal hand veins, but sclerotherapy can be performed. Lentigines may be treated with a variety of devices including intense pulse light, Q-switched lasers, and fractionated nonablative lasers. Chemical peels and topical antipigment agents also may help to a lesser degree or also may be used for maintenance to keep the lentigines away.
Dr. Talakoub and Dr. Wesley are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.
The three most exposed areas of the body that give away a person’s age are the face, neck, and hands. Rejuvenation of the hands is an often simple and nice addition to facial and neck aesthetic rejuvenation.
When examining aging hands, the three most prominent features are decreased volume in the interosseous spaces (leading to increased crepiness of the skin and increased show of extensor tendons), lentigines, and prominent veins. Therefore, the treatment for hands is quite simple: Restore volume, treat the pigmented lesions, and if needed, treat the prominent veins.
The anatomy of the dorsal hand can be divided into three major compartments. First, the skin, which on the dorsal hand is quite pliable. Second, the subcutaneous tissue, which consists of a loose areolar tissue where the lymphatics and veins lie. Third, beneath the subcutaneous tissue is the dorsal fascia of the hand, which is contiguous with extensor tendons and underlying compartments. It is in the subcutaneous layer (or loose areolar tissue) where fillers or fat are placed to treat volume loss.
While several fillers are currently used off label for hand rejuvenation, the Food and Drug Administration is meeting in February to consider officially approving Radiesse for this indication. Currently, hyaluronic acid (HA) fillers, calcium- hydroxylapatite (Radiesse), poly-L-lactic acid, and autologous fat are all utilized. I tend to use HAs in this location because of the reversibility, if needed, and decreased risk of nodule formation. Several techniques exist, including injecting between each tendon space vs. a bolus technique. I tend to use a bolus technique, where one or two boluses are injected while tenting the skin up to ensure injection into the correct plane and to avoid the vessels. Subsequently, the boluses are massaged into place while the patient makes a fist.
Once the interosseous spaces have been treated, the veins often appear less prominent and often don’t require direct treatment. I typically do not treat the dorsal hand veins, but sclerotherapy can be performed. Lentigines may be treated with a variety of devices including intense pulse light, Q-switched lasers, and fractionated nonablative lasers. Chemical peels and topical antipigment agents also may help to a lesser degree or also may be used for maintenance to keep the lentigines away.
Dr. Talakoub and Dr. Wesley are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.
The three most exposed areas of the body that give away a person’s age are the face, neck, and hands. Rejuvenation of the hands is an often simple and nice addition to facial and neck aesthetic rejuvenation.
When examining aging hands, the three most prominent features are decreased volume in the interosseous spaces (leading to increased crepiness of the skin and increased show of extensor tendons), lentigines, and prominent veins. Therefore, the treatment for hands is quite simple: Restore volume, treat the pigmented lesions, and if needed, treat the prominent veins.
The anatomy of the dorsal hand can be divided into three major compartments. First, the skin, which on the dorsal hand is quite pliable. Second, the subcutaneous tissue, which consists of a loose areolar tissue where the lymphatics and veins lie. Third, beneath the subcutaneous tissue is the dorsal fascia of the hand, which is contiguous with extensor tendons and underlying compartments. It is in the subcutaneous layer (or loose areolar tissue) where fillers or fat are placed to treat volume loss.
While several fillers are currently used off label for hand rejuvenation, the Food and Drug Administration is meeting in February to consider officially approving Radiesse for this indication. Currently, hyaluronic acid (HA) fillers, calcium- hydroxylapatite (Radiesse), poly-L-lactic acid, and autologous fat are all utilized. I tend to use HAs in this location because of the reversibility, if needed, and decreased risk of nodule formation. Several techniques exist, including injecting between each tendon space vs. a bolus technique. I tend to use a bolus technique, where one or two boluses are injected while tenting the skin up to ensure injection into the correct plane and to avoid the vessels. Subsequently, the boluses are massaged into place while the patient makes a fist.
Once the interosseous spaces have been treated, the veins often appear less prominent and often don’t require direct treatment. I typically do not treat the dorsal hand veins, but sclerotherapy can be performed. Lentigines may be treated with a variety of devices including intense pulse light, Q-switched lasers, and fractionated nonablative lasers. Chemical peels and topical antipigment agents also may help to a lesser degree or also may be used for maintenance to keep the lentigines away.
Dr. Talakoub and Dr. Wesley are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.

EC approves bortezomib for MCL
Photo courtesy of Millennium
The European Commission (EC) has approved bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) in whom hematopoietic stem cell transplant (HSCT) is considered unsuitable.
Now, bortezomib can be marketed for this indication in all 28 countries of the European Union (EU).
Bortezomib is already approved in the EU to treat multiple myeloma (MM), either as monotherapy or in combination with other agents.
The EC’s approval of bortezomib in MCL is based on data from a phase 3 study known as LYM-3002.
This randomized trial included 487 patients with newly diagnosed MCL who were ineligible, or not considered, for HSCT. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AEs) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85% of patients, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3% of patients, respectively.
About bortezomib
Bortezomib works by reversibly interrupting the normal working of cell proteasomes, inducing cancerous cells to stop growing and die.
In addition to the new MCL indication, the drug is approved in the EU to treat various stages of MM. It’s approved in combination with melphalan and prednisone to treat previously untreated adults with MM who are unsuitable for high-dose chemotherapy with HSCT.
Bortezomib is also approved in combination with dexamethasone, or with dexamethasone plus thalidomide, to treat previously untreated MM patients set to receive high-dose chemotherapy followed by HSCT.
And the drug is approved as monotherapy or in combination with pegylated liposomal doxorubicin or dexamethasone to treat adults with MM whose disease has progressed after at least one other treatment and who have already had, or cannot undergo, HSCT.
Bortezomib is approved in more than 90 countries and has been used to treat more than 550,000 patients worldwide.
The product is co-developed by Millennium, the Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and Janssen Pharmaceutical Companies.
Millennium is responsible for commercialization in the US. Janssen Pharmaceutical Companies are responsible for commercialization in Europe and the rest of the world. Takeda Pharmaceutical Company Limited and Janssen Pharmaceutical K.K. co-promote the drug in Japan. ![]()
Photo courtesy of Millennium
The European Commission (EC) has approved bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) in whom hematopoietic stem cell transplant (HSCT) is considered unsuitable.
Now, bortezomib can be marketed for this indication in all 28 countries of the European Union (EU).
Bortezomib is already approved in the EU to treat multiple myeloma (MM), either as monotherapy or in combination with other agents.
The EC’s approval of bortezomib in MCL is based on data from a phase 3 study known as LYM-3002.
This randomized trial included 487 patients with newly diagnosed MCL who were ineligible, or not considered, for HSCT. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AEs) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85% of patients, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3% of patients, respectively.
About bortezomib
Bortezomib works by reversibly interrupting the normal working of cell proteasomes, inducing cancerous cells to stop growing and die.
In addition to the new MCL indication, the drug is approved in the EU to treat various stages of MM. It’s approved in combination with melphalan and prednisone to treat previously untreated adults with MM who are unsuitable for high-dose chemotherapy with HSCT.
Bortezomib is also approved in combination with dexamethasone, or with dexamethasone plus thalidomide, to treat previously untreated MM patients set to receive high-dose chemotherapy followed by HSCT.
And the drug is approved as monotherapy or in combination with pegylated liposomal doxorubicin or dexamethasone to treat adults with MM whose disease has progressed after at least one other treatment and who have already had, or cannot undergo, HSCT.
Bortezomib is approved in more than 90 countries and has been used to treat more than 550,000 patients worldwide.
The product is co-developed by Millennium, the Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and Janssen Pharmaceutical Companies.
Millennium is responsible for commercialization in the US. Janssen Pharmaceutical Companies are responsible for commercialization in Europe and the rest of the world. Takeda Pharmaceutical Company Limited and Janssen Pharmaceutical K.K. co-promote the drug in Japan. ![]()
Photo courtesy of Millennium
The European Commission (EC) has approved bortezomib (Velcade) in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to treat adults with previously untreated mantle cell lymphoma (MCL) in whom hematopoietic stem cell transplant (HSCT) is considered unsuitable.
Now, bortezomib can be marketed for this indication in all 28 countries of the European Union (EU).
Bortezomib is already approved in the EU to treat multiple myeloma (MM), either as monotherapy or in combination with other agents.
The EC’s approval of bortezomib in MCL is based on data from a phase 3 study known as LYM-3002.
This randomized trial included 487 patients with newly diagnosed MCL who were ineligible, or not considered, for HSCT. Patients were randomized to receive VR-CAP or R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
The VR-CAP regimen significantly improved progression-free survival (PFS), the primary endpoint, when compared to R-CHOP.
According to an independent review committee, there was a 59% improvement in PFS for the VR-CAP arm compared to the R-CHOP arm, with median times of 24.7 months and 14.4 months, respectively (hazard ratio=0.63; P<0.001).
Study investigators reported a 96% increase in PFS with VR-CAP compared to R-CHOP, with median times of 30.7 months and 16.1 months, respectively (hazard ratio=0.51, P<0.001).
VR-CAP was associated with additional, but manageable, toxicity when compared to R-CHOP. Serious adverse events (AEs) were reported in 38% and 30% of patients, respectively. And grade 3 or higher AEs were reported in 93% and 85% of patients, respectively.
Treatment discontinuation due to AEs occurred in 9% of patients in the VR-CAP arm and 7% in the R-CHOP arm. On-treatment, drug-related deaths occurred in 2% and 3% of patients, respectively.
About bortezomib
Bortezomib works by reversibly interrupting the normal working of cell proteasomes, inducing cancerous cells to stop growing and die.
In addition to the new MCL indication, the drug is approved in the EU to treat various stages of MM. It’s approved in combination with melphalan and prednisone to treat previously untreated adults with MM who are unsuitable for high-dose chemotherapy with HSCT.
Bortezomib is also approved in combination with dexamethasone, or with dexamethasone plus thalidomide, to treat previously untreated MM patients set to receive high-dose chemotherapy followed by HSCT.
And the drug is approved as monotherapy or in combination with pegylated liposomal doxorubicin or dexamethasone to treat adults with MM whose disease has progressed after at least one other treatment and who have already had, or cannot undergo, HSCT.
Bortezomib is approved in more than 90 countries and has been used to treat more than 550,000 patients worldwide.
The product is co-developed by Millennium, the Takeda Oncology Company, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and Janssen Pharmaceutical Companies.
Millennium is responsible for commercialization in the US. Janssen Pharmaceutical Companies are responsible for commercialization in Europe and the rest of the world. Takeda Pharmaceutical Company Limited and Janssen Pharmaceutical K.K. co-promote the drug in Japan. ![]()
AUDIO: Training broadens psychotherapy in primary care
NEW YORK– The REACH Institute trains primary care clinicians to include more mental health assessment and management in their practices, Dr. Lawrence V. Amsel said during an interview at the psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
Many primary care clinicians don’t feel adequately trained to interview patients, guage their mental status, and then act on the findings by treatment or referral. But over the past decade, psychiatrists have developed and validated several tools that are appropriate for a primary care practice, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York, and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. The program also tries to make clinicians comfortable prescribing psychiatric medications and links them with psychiatrists who can provide consultations when needed.
“It’s kind of like a psychiatrist extender,” when a psychiatrist consults with several primary care clinicians, which allows for improved psychiatric care of many more patients, he said.
Dr. Amsel is on the faculty of the REACH Institute.
On Twitter@mitchelzoler
NEW YORK– The REACH Institute trains primary care clinicians to include more mental health assessment and management in their practices, Dr. Lawrence V. Amsel said during an interview at the psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
Many primary care clinicians don’t feel adequately trained to interview patients, guage their mental status, and then act on the findings by treatment or referral. But over the past decade, psychiatrists have developed and validated several tools that are appropriate for a primary care practice, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York, and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. The program also tries to make clinicians comfortable prescribing psychiatric medications and links them with psychiatrists who can provide consultations when needed.
“It’s kind of like a psychiatrist extender,” when a psychiatrist consults with several primary care clinicians, which allows for improved psychiatric care of many more patients, he said.
Dr. Amsel is on the faculty of the REACH Institute.
On Twitter@mitchelzoler
NEW YORK– The REACH Institute trains primary care clinicians to include more mental health assessment and management in their practices, Dr. Lawrence V. Amsel said during an interview at the psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
Many primary care clinicians don’t feel adequately trained to interview patients, guage their mental status, and then act on the findings by treatment or referral. But over the past decade, psychiatrists have developed and validated several tools that are appropriate for a primary care practice, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York, and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. The program also tries to make clinicians comfortable prescribing psychiatric medications and links them with psychiatrists who can provide consultations when needed.
“It’s kind of like a psychiatrist extender,” when a psychiatrist consults with several primary care clinicians, which allows for improved psychiatric care of many more patients, he said.
Dr. Amsel is on the faculty of the REACH Institute.
On Twitter@mitchelzoler
EXPERT ANALYSIS FROM THE PSYCHOPHARMACOLOGY UPDATE INSTITUTE
Treating VTE in patients with gynecologic malignancies
Rudolph Virchow clearly demonstrated the association between malignancy and venous thromboembolic events. VTE – deep vein thrombosis and pulmonary embolism – affect between 15% and 38% of patients with gynecologic malignancies after surgery.
The rate of pulmonary embolism (PE) in this patient population can be as high as 6.8%, with the case fatality rate being 11%-12% (Obstet. Gynecol. 2012;119:155-67). Other factors associated with the development of VTE include prior VTE, older age, African American race, prolonged operative time, obesity, and prior radiation therapy (Obstet. Gynecol. 1987;69:146-50). The risk of VTE in women undergoing gynecologic surgery is quadrupled in the presence of malignancy(Obstet. Gynecol. 2006;107:666-71) and these patients are twice as likely to die from a VTE compared to matched controls (Gynecol. Oncol. 2007;106:439-45).
Additionally, cancer patients are typically older, have longer and more complex surgeries, and the presence of a pelvic mass further contributes to venous stasis (Obstet. Gynecol. 2012;119:155-67).
Although the treatment of VTE is fairly similar between patients with malignancy and those without cancer, treatment of a VTE in patients with cancer can be further complicated by higher VTE recurrence rates and increased risk of bleeding. Furthermore, issues related to the malignant disease process such as prognosis, presence and location of metastasis, and life expectancy should be taken into consideration when managing VTE in this patient population.
Generally, in the setting of an acute or recurrent VTE, initial therapy with a parenteral anticoagulant (heparin or low-molecular-weight heparins [LMWH]) should be immediately instituted in patients with a gynecologic malignancy, unless there is evidence of active bleeding or any other contraindication for the use of an anticoagulant.
Other factors associated with cancer such as immobilization, the presence of metastases, and impaired renal function with a creatinine clearance less than 30 mL/min, may increase the risk of bleeding complications but are not absolute contraindications to anticoagulation (Thromb. Haemost. 2008;100:435-9). The initial treatment phase, which last for 5-10 days, is then followed by a longer treatment phase lasting 3-6 months.
In the majority of cases, LMWH is the preferred agent for both the initial and prolonged treatment phase assuming adequate renal function. Based on evidence from a meta-analysis of 16 randomized controlled trials in cancer patients receiving initial anticoagulation for VTE, LMWH is associated with a 30% reduction in mortality without an increased risk of bleeding in comparison to unfractionated heparin (Cochrane Database. Syst. Rev. 2014;6:CD006649).
When compared with the vitamin K antagonist warfarin, LMWH appears to be associated with a significantly reduced rate of recurrent VTE (hazard ratio, 0.47; 95% confidence interval 0.32-0.71). However, this was not associated with a survival advantage (N. Engl. J. Med. 2003;349:146-53).
There are no trials comparing the different formulations of LMWH. In our practice, we routinely use the LMWH enoxaparin dosed at 1 mg/kg subcutaneously twice daily. Other well-studied LMWHs include dalteparin and tinzaparin.
LMWHs are primarily renally excreted, thus, in patients with compromised renal function, the biological half-life of the medication may be prolonged, leading to potential bleeding complications. The majority of LMWH trials excluded patients with creatinine clearance less than 30 mL/min, therefore, in patients with compromised renal function, one option would be to decrease the daily dose by as much as 50% and closely monitor antifactor XA levels. Alternatively, the use of unfractionated heparin in the acute setting followed by warfarin with close monitoring of the patient’s international normalized ratio could prove less cumbersome and ultimately safer for these patients. However, given the limitations of the currently available data we would not recommend the routine use of newer oral anticoagulation agents.
Patients with a malignancy are at increased risk for the development of a recurrent VTE even in the setting of anticoagulation. Some of the risks factors for this phenomenon include presence of central venous catheters, interruption of therapy for procedures, and immobilization. In cases of recurrent VTE, consideration should be given to extending the duration of treatment beyond the initial planned 3-6 months. Other patients that may benefit from extended therapy include those with continued immobility or active cancer burden.
LMWH is also the preferred agent for extended therapy based on very limited evidence from experimental studies suggesting that LMWH may have antineoplastic effects and thus a survival advantage. However, in the setting of a recurrent VTE, there is very limited data on which to base the choice of extended treatment. Options include switching the therapeutic agent, increasing the dose or frequency of administration, or placement of an inferior vena cava filter. Consultation with a hematologist may also be warranted in this and more complicated scenarios.
Ultimately, LMWH appears to be the best available therapy for patients with a gynecologic malignancy. However, the decision to anticoagulate should be carefully planned out, taking into consideration the individual patient’s disease burden and associated comorbidities in order to select the most appropriate treatment option.
Dr. Roque is a fellow in the gynecologic oncology program at the University of North Carolina at Chapel Hill. Dr. Clarke-Pearson is the chair and the Robert A. Ross Distinguished Professor of Obstetrics and Gynecology and a professor in the division of gynecologic oncology at the university. Dr. Roque and Dr. Clarke-Pearson said they had no relevant financial disclosures.
Rudolph Virchow clearly demonstrated the association between malignancy and venous thromboembolic events. VTE – deep vein thrombosis and pulmonary embolism – affect between 15% and 38% of patients with gynecologic malignancies after surgery.
The rate of pulmonary embolism (PE) in this patient population can be as high as 6.8%, with the case fatality rate being 11%-12% (Obstet. Gynecol. 2012;119:155-67). Other factors associated with the development of VTE include prior VTE, older age, African American race, prolonged operative time, obesity, and prior radiation therapy (Obstet. Gynecol. 1987;69:146-50). The risk of VTE in women undergoing gynecologic surgery is quadrupled in the presence of malignancy(Obstet. Gynecol. 2006;107:666-71) and these patients are twice as likely to die from a VTE compared to matched controls (Gynecol. Oncol. 2007;106:439-45).
Additionally, cancer patients are typically older, have longer and more complex surgeries, and the presence of a pelvic mass further contributes to venous stasis (Obstet. Gynecol. 2012;119:155-67).
Although the treatment of VTE is fairly similar between patients with malignancy and those without cancer, treatment of a VTE in patients with cancer can be further complicated by higher VTE recurrence rates and increased risk of bleeding. Furthermore, issues related to the malignant disease process such as prognosis, presence and location of metastasis, and life expectancy should be taken into consideration when managing VTE in this patient population.
Generally, in the setting of an acute or recurrent VTE, initial therapy with a parenteral anticoagulant (heparin or low-molecular-weight heparins [LMWH]) should be immediately instituted in patients with a gynecologic malignancy, unless there is evidence of active bleeding or any other contraindication for the use of an anticoagulant.
Other factors associated with cancer such as immobilization, the presence of metastases, and impaired renal function with a creatinine clearance less than 30 mL/min, may increase the risk of bleeding complications but are not absolute contraindications to anticoagulation (Thromb. Haemost. 2008;100:435-9). The initial treatment phase, which last for 5-10 days, is then followed by a longer treatment phase lasting 3-6 months.
In the majority of cases, LMWH is the preferred agent for both the initial and prolonged treatment phase assuming adequate renal function. Based on evidence from a meta-analysis of 16 randomized controlled trials in cancer patients receiving initial anticoagulation for VTE, LMWH is associated with a 30% reduction in mortality without an increased risk of bleeding in comparison to unfractionated heparin (Cochrane Database. Syst. Rev. 2014;6:CD006649).
When compared with the vitamin K antagonist warfarin, LMWH appears to be associated with a significantly reduced rate of recurrent VTE (hazard ratio, 0.47; 95% confidence interval 0.32-0.71). However, this was not associated with a survival advantage (N. Engl. J. Med. 2003;349:146-53).
There are no trials comparing the different formulations of LMWH. In our practice, we routinely use the LMWH enoxaparin dosed at 1 mg/kg subcutaneously twice daily. Other well-studied LMWHs include dalteparin and tinzaparin.
LMWHs are primarily renally excreted, thus, in patients with compromised renal function, the biological half-life of the medication may be prolonged, leading to potential bleeding complications. The majority of LMWH trials excluded patients with creatinine clearance less than 30 mL/min, therefore, in patients with compromised renal function, one option would be to decrease the daily dose by as much as 50% and closely monitor antifactor XA levels. Alternatively, the use of unfractionated heparin in the acute setting followed by warfarin with close monitoring of the patient’s international normalized ratio could prove less cumbersome and ultimately safer for these patients. However, given the limitations of the currently available data we would not recommend the routine use of newer oral anticoagulation agents.
Patients with a malignancy are at increased risk for the development of a recurrent VTE even in the setting of anticoagulation. Some of the risks factors for this phenomenon include presence of central venous catheters, interruption of therapy for procedures, and immobilization. In cases of recurrent VTE, consideration should be given to extending the duration of treatment beyond the initial planned 3-6 months. Other patients that may benefit from extended therapy include those with continued immobility or active cancer burden.
LMWH is also the preferred agent for extended therapy based on very limited evidence from experimental studies suggesting that LMWH may have antineoplastic effects and thus a survival advantage. However, in the setting of a recurrent VTE, there is very limited data on which to base the choice of extended treatment. Options include switching the therapeutic agent, increasing the dose or frequency of administration, or placement of an inferior vena cava filter. Consultation with a hematologist may also be warranted in this and more complicated scenarios.
Ultimately, LMWH appears to be the best available therapy for patients with a gynecologic malignancy. However, the decision to anticoagulate should be carefully planned out, taking into consideration the individual patient’s disease burden and associated comorbidities in order to select the most appropriate treatment option.
Dr. Roque is a fellow in the gynecologic oncology program at the University of North Carolina at Chapel Hill. Dr. Clarke-Pearson is the chair and the Robert A. Ross Distinguished Professor of Obstetrics and Gynecology and a professor in the division of gynecologic oncology at the university. Dr. Roque and Dr. Clarke-Pearson said they had no relevant financial disclosures.
Rudolph Virchow clearly demonstrated the association between malignancy and venous thromboembolic events. VTE – deep vein thrombosis and pulmonary embolism – affect between 15% and 38% of patients with gynecologic malignancies after surgery.
The rate of pulmonary embolism (PE) in this patient population can be as high as 6.8%, with the case fatality rate being 11%-12% (Obstet. Gynecol. 2012;119:155-67). Other factors associated with the development of VTE include prior VTE, older age, African American race, prolonged operative time, obesity, and prior radiation therapy (Obstet. Gynecol. 1987;69:146-50). The risk of VTE in women undergoing gynecologic surgery is quadrupled in the presence of malignancy(Obstet. Gynecol. 2006;107:666-71) and these patients are twice as likely to die from a VTE compared to matched controls (Gynecol. Oncol. 2007;106:439-45).
Additionally, cancer patients are typically older, have longer and more complex surgeries, and the presence of a pelvic mass further contributes to venous stasis (Obstet. Gynecol. 2012;119:155-67).
Although the treatment of VTE is fairly similar between patients with malignancy and those without cancer, treatment of a VTE in patients with cancer can be further complicated by higher VTE recurrence rates and increased risk of bleeding. Furthermore, issues related to the malignant disease process such as prognosis, presence and location of metastasis, and life expectancy should be taken into consideration when managing VTE in this patient population.
Generally, in the setting of an acute or recurrent VTE, initial therapy with a parenteral anticoagulant (heparin or low-molecular-weight heparins [LMWH]) should be immediately instituted in patients with a gynecologic malignancy, unless there is evidence of active bleeding or any other contraindication for the use of an anticoagulant.
Other factors associated with cancer such as immobilization, the presence of metastases, and impaired renal function with a creatinine clearance less than 30 mL/min, may increase the risk of bleeding complications but are not absolute contraindications to anticoagulation (Thromb. Haemost. 2008;100:435-9). The initial treatment phase, which last for 5-10 days, is then followed by a longer treatment phase lasting 3-6 months.
In the majority of cases, LMWH is the preferred agent for both the initial and prolonged treatment phase assuming adequate renal function. Based on evidence from a meta-analysis of 16 randomized controlled trials in cancer patients receiving initial anticoagulation for VTE, LMWH is associated with a 30% reduction in mortality without an increased risk of bleeding in comparison to unfractionated heparin (Cochrane Database. Syst. Rev. 2014;6:CD006649).
When compared with the vitamin K antagonist warfarin, LMWH appears to be associated with a significantly reduced rate of recurrent VTE (hazard ratio, 0.47; 95% confidence interval 0.32-0.71). However, this was not associated with a survival advantage (N. Engl. J. Med. 2003;349:146-53).
There are no trials comparing the different formulations of LMWH. In our practice, we routinely use the LMWH enoxaparin dosed at 1 mg/kg subcutaneously twice daily. Other well-studied LMWHs include dalteparin and tinzaparin.
LMWHs are primarily renally excreted, thus, in patients with compromised renal function, the biological half-life of the medication may be prolonged, leading to potential bleeding complications. The majority of LMWH trials excluded patients with creatinine clearance less than 30 mL/min, therefore, in patients with compromised renal function, one option would be to decrease the daily dose by as much as 50% and closely monitor antifactor XA levels. Alternatively, the use of unfractionated heparin in the acute setting followed by warfarin with close monitoring of the patient’s international normalized ratio could prove less cumbersome and ultimately safer for these patients. However, given the limitations of the currently available data we would not recommend the routine use of newer oral anticoagulation agents.
Patients with a malignancy are at increased risk for the development of a recurrent VTE even in the setting of anticoagulation. Some of the risks factors for this phenomenon include presence of central venous catheters, interruption of therapy for procedures, and immobilization. In cases of recurrent VTE, consideration should be given to extending the duration of treatment beyond the initial planned 3-6 months. Other patients that may benefit from extended therapy include those with continued immobility or active cancer burden.
LMWH is also the preferred agent for extended therapy based on very limited evidence from experimental studies suggesting that LMWH may have antineoplastic effects and thus a survival advantage. However, in the setting of a recurrent VTE, there is very limited data on which to base the choice of extended treatment. Options include switching the therapeutic agent, increasing the dose or frequency of administration, or placement of an inferior vena cava filter. Consultation with a hematologist may also be warranted in this and more complicated scenarios.
Ultimately, LMWH appears to be the best available therapy for patients with a gynecologic malignancy. However, the decision to anticoagulate should be carefully planned out, taking into consideration the individual patient’s disease burden and associated comorbidities in order to select the most appropriate treatment option.
Dr. Roque is a fellow in the gynecologic oncology program at the University of North Carolina at Chapel Hill. Dr. Clarke-Pearson is the chair and the Robert A. Ross Distinguished Professor of Obstetrics and Gynecology and a professor in the division of gynecologic oncology at the university. Dr. Roque and Dr. Clarke-Pearson said they had no relevant financial disclosures.

Program fosters psychotherapy in primary care practices
NEW YORK – Making primary care clinicians comfortable performing basic mental health diagnoses and management is vital for adequately treating U.S. patients with psychiatric disorders, Dr. Lawrence V. Amsel said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
In many parts of the United States there are “far fewer mental health practitioners than are needed.” Training primary care clinicians so that they are willing to do more mental health work can help address this issue, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. “It’s like producing psychiatric extenders.” By consulting with a broad range of primary care clinicians, a psychiatrist can take care of a lot more kids than usual in a single psychiatric practice,” Dr. Amsel said.

But encouraging primary care providers to become more active in mental health diagnosis and management is not easy. “Most prescriptions for mental health indications are now written by primary care clinicians, but they often describe themselves as uncomfortable prescribing these medications and not adequately trained,” Dr. Amsel said in an interview.
“Their main anxiety comes from making the wrong diagnosis and then doing harm” as a consequence of their error, he explained during his talk at the meeting. Training by the REACH curriculum highlights the role of well-validated tools now available for refining assessment of a patient and boosting confidence in the diagnosis. This includes instruments like the Pediatric Symptom Checklist and the Mental Status Exam. “Reliable and validated tools are available to improve identification and assessment of mental health problems that can be used efficiently in clinical practice,” Dr. Amsel said.
Another aspect to mental health management that often troubles primary care clinicians is doubt about their knowledge and ability to safely and effectively prescribe psychiatric medications. The REACH Institute curriculum tells clinicians to focus on each patient’s primary diagnosis and treat that first, and whenever possible to use medications that are evidence based, with good supporting documentation from double-blind, randomized, controlled trials.
“We recommend that clinicians get a summary slide of the evidence that they can show to patients or family members if necessary to make clear that there is a scientific basis for the treatment and that it is based on facts and data rather than on opinion,” he said.
Training for primary care clinicians also emphasizes that management goes beyond drug treatment and also must include a psychosocial plan for each patient.
Members of the health care system have begun to “recognize that mental health is responsible for much if not most disability. Until now, this importance had not been recognized. Now that it is being recognized, I think people will develop systems that increase the capacity for identifying children with mental health issues and provide them with improved care,” Dr. Amsel said.
Dr. Amsel had no disclosures aside from his work for the REACH Institute.
On Twitter @mitchelzoler
NEW YORK – Making primary care clinicians comfortable performing basic mental health diagnoses and management is vital for adequately treating U.S. patients with psychiatric disorders, Dr. Lawrence V. Amsel said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
In many parts of the United States there are “far fewer mental health practitioners than are needed.” Training primary care clinicians so that they are willing to do more mental health work can help address this issue, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. “It’s like producing psychiatric extenders.” By consulting with a broad range of primary care clinicians, a psychiatrist can take care of a lot more kids than usual in a single psychiatric practice,” Dr. Amsel said.
But encouraging primary care providers to become more active in mental health diagnosis and management is not easy. “Most prescriptions for mental health indications are now written by primary care clinicians, but they often describe themselves as uncomfortable prescribing these medications and not adequately trained,” Dr. Amsel said in an interview.
“Their main anxiety comes from making the wrong diagnosis and then doing harm” as a consequence of their error, he explained during his talk at the meeting. Training by the REACH curriculum highlights the role of well-validated tools now available for refining assessment of a patient and boosting confidence in the diagnosis. This includes instruments like the Pediatric Symptom Checklist and the Mental Status Exam. “Reliable and validated tools are available to improve identification and assessment of mental health problems that can be used efficiently in clinical practice,” Dr. Amsel said.
Another aspect to mental health management that often troubles primary care clinicians is doubt about their knowledge and ability to safely and effectively prescribe psychiatric medications. The REACH Institute curriculum tells clinicians to focus on each patient’s primary diagnosis and treat that first, and whenever possible to use medications that are evidence based, with good supporting documentation from double-blind, randomized, controlled trials.
“We recommend that clinicians get a summary slide of the evidence that they can show to patients or family members if necessary to make clear that there is a scientific basis for the treatment and that it is based on facts and data rather than on opinion,” he said.
Training for primary care clinicians also emphasizes that management goes beyond drug treatment and also must include a psychosocial plan for each patient.
Members of the health care system have begun to “recognize that mental health is responsible for much if not most disability. Until now, this importance had not been recognized. Now that it is being recognized, I think people will develop systems that increase the capacity for identifying children with mental health issues and provide them with improved care,” Dr. Amsel said.
Dr. Amsel had no disclosures aside from his work for the REACH Institute.
On Twitter @mitchelzoler
NEW YORK – Making primary care clinicians comfortable performing basic mental health diagnoses and management is vital for adequately treating U.S. patients with psychiatric disorders, Dr. Lawrence V. Amsel said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
In many parts of the United States there are “far fewer mental health practitioners than are needed.” Training primary care clinicians so that they are willing to do more mental health work can help address this issue, said Dr. Amsel, a clinical psychiatrist at Columbia University in New York and a faculty member of the REACH Institute, a New York–based nonprofit focused on disseminating mental health skills to primary care clinicians, teachers, parents, and others. “It’s like producing psychiatric extenders.” By consulting with a broad range of primary care clinicians, a psychiatrist can take care of a lot more kids than usual in a single psychiatric practice,” Dr. Amsel said.
But encouraging primary care providers to become more active in mental health diagnosis and management is not easy. “Most prescriptions for mental health indications are now written by primary care clinicians, but they often describe themselves as uncomfortable prescribing these medications and not adequately trained,” Dr. Amsel said in an interview.
“Their main anxiety comes from making the wrong diagnosis and then doing harm” as a consequence of their error, he explained during his talk at the meeting. Training by the REACH curriculum highlights the role of well-validated tools now available for refining assessment of a patient and boosting confidence in the diagnosis. This includes instruments like the Pediatric Symptom Checklist and the Mental Status Exam. “Reliable and validated tools are available to improve identification and assessment of mental health problems that can be used efficiently in clinical practice,” Dr. Amsel said.
Another aspect to mental health management that often troubles primary care clinicians is doubt about their knowledge and ability to safely and effectively prescribe psychiatric medications. The REACH Institute curriculum tells clinicians to focus on each patient’s primary diagnosis and treat that first, and whenever possible to use medications that are evidence based, with good supporting documentation from double-blind, randomized, controlled trials.
“We recommend that clinicians get a summary slide of the evidence that they can show to patients or family members if necessary to make clear that there is a scientific basis for the treatment and that it is based on facts and data rather than on opinion,” he said.
Training for primary care clinicians also emphasizes that management goes beyond drug treatment and also must include a psychosocial plan for each patient.
Members of the health care system have begun to “recognize that mental health is responsible for much if not most disability. Until now, this importance had not been recognized. Now that it is being recognized, I think people will develop systems that increase the capacity for identifying children with mental health issues and provide them with improved care,” Dr. Amsel said.
Dr. Amsel had no disclosures aside from his work for the REACH Institute.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM THE PSYCHOPHARMACOLOGY UPDATE INSTITUTE

Aggressiveness of CLL linked to genetic variability

The genetic variability of chronic lymphocytic leukemia (CLL) appears to predict its aggressiveness, according to a study published in Genome Medicine.
Investigators found evidence suggesting that greater variability in gene expression is associated with more aggressive disease.
The team analyzed gene expression in two cohorts of CLL patients—those with IgVH mutations and a good prognosis and those with unmutated CLL who have more aggressive disease.
The researchers examined 70 mutated and 52 unmutated CLL samples, as well as 20 control samples taken from healthy individuals.
Unmutated, aggressive CLL showed increased gene expression variability across individuals, whereas gene expression variability was lower in less aggressive, mutated CLL.
The investigators validated these observations by comparing them against a second sample group consisting of 24 mutated and 36 unmutated CLL samples.
The results suggested that CLL aggressiveness is specifically determined by a set of 500 genes showing increased expression variability across individuals. The genes are involved in processes such as adaptation to the environment, cell death, tumor growth, and drug resistance.
“Our conclusion is that the coefficient of variation for gene expression in CLL efficiently predicts its aggressiveness,” said study author Alfonso Valencia, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain.
“More importantly, if further research confirms these findings, a classifier based on the measurement of gene expression variability could be created to predict the disease subtype of CLL patients.”
The researchers said their next step is to discover the mechanisms responsible for the high levels of expression variability for a given gene across individuals.
Understanding the mechanisms underlying this phenomenon is of crucial relevance for oncology research, the investigators said, as it is linked to tumor heterogeneity, a key feature of cancer progression and drug resistance.
The greater the genetic variability in a tumor, the better it will adapt to its environment, and the more probabilities for this tumor to spread, develop resistance to cancer therapies, and metastasize to distant organs. ![]()
The genetic variability of chronic lymphocytic leukemia (CLL) appears to predict its aggressiveness, according to a study published in Genome Medicine.
Investigators found evidence suggesting that greater variability in gene expression is associated with more aggressive disease.
The team analyzed gene expression in two cohorts of CLL patients—those with IgVH mutations and a good prognosis and those with unmutated CLL who have more aggressive disease.
The researchers examined 70 mutated and 52 unmutated CLL samples, as well as 20 control samples taken from healthy individuals.
Unmutated, aggressive CLL showed increased gene expression variability across individuals, whereas gene expression variability was lower in less aggressive, mutated CLL.
The investigators validated these observations by comparing them against a second sample group consisting of 24 mutated and 36 unmutated CLL samples.
The results suggested that CLL aggressiveness is specifically determined by a set of 500 genes showing increased expression variability across individuals. The genes are involved in processes such as adaptation to the environment, cell death, tumor growth, and drug resistance.
“Our conclusion is that the coefficient of variation for gene expression in CLL efficiently predicts its aggressiveness,” said study author Alfonso Valencia, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain.
“More importantly, if further research confirms these findings, a classifier based on the measurement of gene expression variability could be created to predict the disease subtype of CLL patients.”
The researchers said their next step is to discover the mechanisms responsible for the high levels of expression variability for a given gene across individuals.
Understanding the mechanisms underlying this phenomenon is of crucial relevance for oncology research, the investigators said, as it is linked to tumor heterogeneity, a key feature of cancer progression and drug resistance.
The greater the genetic variability in a tumor, the better it will adapt to its environment, and the more probabilities for this tumor to spread, develop resistance to cancer therapies, and metastasize to distant organs. ![]()
The genetic variability of chronic lymphocytic leukemia (CLL) appears to predict its aggressiveness, according to a study published in Genome Medicine.
Investigators found evidence suggesting that greater variability in gene expression is associated with more aggressive disease.
The team analyzed gene expression in two cohorts of CLL patients—those with IgVH mutations and a good prognosis and those with unmutated CLL who have more aggressive disease.
The researchers examined 70 mutated and 52 unmutated CLL samples, as well as 20 control samples taken from healthy individuals.
Unmutated, aggressive CLL showed increased gene expression variability across individuals, whereas gene expression variability was lower in less aggressive, mutated CLL.
The investigators validated these observations by comparing them against a second sample group consisting of 24 mutated and 36 unmutated CLL samples.
The results suggested that CLL aggressiveness is specifically determined by a set of 500 genes showing increased expression variability across individuals. The genes are involved in processes such as adaptation to the environment, cell death, tumor growth, and drug resistance.
“Our conclusion is that the coefficient of variation for gene expression in CLL efficiently predicts its aggressiveness,” said study author Alfonso Valencia, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain.
“More importantly, if further research confirms these findings, a classifier based on the measurement of gene expression variability could be created to predict the disease subtype of CLL patients.”
The researchers said their next step is to discover the mechanisms responsible for the high levels of expression variability for a given gene across individuals.
Understanding the mechanisms underlying this phenomenon is of crucial relevance for oncology research, the investigators said, as it is linked to tumor heterogeneity, a key feature of cancer progression and drug resistance.
The greater the genetic variability in a tumor, the better it will adapt to its environment, and the more probabilities for this tumor to spread, develop resistance to cancer therapies, and metastasize to distant organs. ![]()