User login
Introduction—Medicine’s future: Helping patients stay healthy at home
Home-based care will undoubtedly play an increasingly important role in the health care system as the United States seeks ways to provide cost-effective and compassionate care to a growing population of older adults with chronic illness. “Home health care,” a term that refers more specifically to visiting nurses, therapists, and related services, is currently the prominent home care model in this country.
Home health services were developed around the start of the 20th century to address the unmet health and social needs of vulnerable populations living in the shadows. Today, there are more than 10,000 home health agencies and visiting nurse organizations across the country that care for millions of homebound patients each year. With the onset of health reform and the increasing focus on value and “accountability,” there are many opportunities and challenges for home health providers and the physicians, hospitals, and facilities they work with to try to find the best ways to keep patients healthy at home and drive value for society.
There is a paucity of medical and health services literature to guide providers and policymakers’ decisions about the right types and approaches to care at home. Maybe this is because academic centers and American medicine became so focused on acute institutional care in the past half century that the home has been overlooked. However, that pendulum is likely swinging back as almost every sober analysis of our current health care environment suggests a need for better care for the chronically ill at home and in the community. It is important that research and academic enterprises emphasize scholarly efforts to understand and improve home and community care so that the anticipated shift in care to home is informed by the best possible evidence, ultimately ensuring that patients get the best possible care.
The articles in this online, CME-certified Cleveland Clinic Journal of Medicine supplement address contemporary topics in home health and other home-based care concepts. The authors have diverse backgrounds and discuss issues related to technology, palliative care, care transitions, heart failure, knee replacement, primary care, and health reform. Several articles share concepts and outcomes from innovative approaches being developed throughout the country to help patients succeed at home, especially when returning home from a hospitalization.
The articles should improve readers’ understanding of a wide range of initiatives and ideas for how home health and home care might look in the future delivery system. The authors also raise numerous yet-unanswered questions and opportunities for future study. The needs for further home care research from clinical, public health, and policy perspectives are evident. Health care is going home, and this transformation will be enhanced and possibly accelerated by thoughtful research and synthesis.
I am incredibly thankful to my fellow authors, and hope that we have produced a useful supplement that will help readers in their efforts to assist the most vulnerable patients and families in their efforts to remain independent at home.
Home-based care will undoubtedly play an increasingly important role in the health care system as the United States seeks ways to provide cost-effective and compassionate care to a growing population of older adults with chronic illness. “Home health care,” a term that refers more specifically to visiting nurses, therapists, and related services, is currently the prominent home care model in this country.
Home health services were developed around the start of the 20th century to address the unmet health and social needs of vulnerable populations living in the shadows. Today, there are more than 10,000 home health agencies and visiting nurse organizations across the country that care for millions of homebound patients each year. With the onset of health reform and the increasing focus on value and “accountability,” there are many opportunities and challenges for home health providers and the physicians, hospitals, and facilities they work with to try to find the best ways to keep patients healthy at home and drive value for society.
There is a paucity of medical and health services literature to guide providers and policymakers’ decisions about the right types and approaches to care at home. Maybe this is because academic centers and American medicine became so focused on acute institutional care in the past half century that the home has been overlooked. However, that pendulum is likely swinging back as almost every sober analysis of our current health care environment suggests a need for better care for the chronically ill at home and in the community. It is important that research and academic enterprises emphasize scholarly efforts to understand and improve home and community care so that the anticipated shift in care to home is informed by the best possible evidence, ultimately ensuring that patients get the best possible care.
The articles in this online, CME-certified Cleveland Clinic Journal of Medicine supplement address contemporary topics in home health and other home-based care concepts. The authors have diverse backgrounds and discuss issues related to technology, palliative care, care transitions, heart failure, knee replacement, primary care, and health reform. Several articles share concepts and outcomes from innovative approaches being developed throughout the country to help patients succeed at home, especially when returning home from a hospitalization.
The articles should improve readers’ understanding of a wide range of initiatives and ideas for how home health and home care might look in the future delivery system. The authors also raise numerous yet-unanswered questions and opportunities for future study. The needs for further home care research from clinical, public health, and policy perspectives are evident. Health care is going home, and this transformation will be enhanced and possibly accelerated by thoughtful research and synthesis.
I am incredibly thankful to my fellow authors, and hope that we have produced a useful supplement that will help readers in their efforts to assist the most vulnerable patients and families in their efforts to remain independent at home.
Home-based care will undoubtedly play an increasingly important role in the health care system as the United States seeks ways to provide cost-effective and compassionate care to a growing population of older adults with chronic illness. “Home health care,” a term that refers more specifically to visiting nurses, therapists, and related services, is currently the prominent home care model in this country.
Home health services were developed around the start of the 20th century to address the unmet health and social needs of vulnerable populations living in the shadows. Today, there are more than 10,000 home health agencies and visiting nurse organizations across the country that care for millions of homebound patients each year. With the onset of health reform and the increasing focus on value and “accountability,” there are many opportunities and challenges for home health providers and the physicians, hospitals, and facilities they work with to try to find the best ways to keep patients healthy at home and drive value for society.
There is a paucity of medical and health services literature to guide providers and policymakers’ decisions about the right types and approaches to care at home. Maybe this is because academic centers and American medicine became so focused on acute institutional care in the past half century that the home has been overlooked. However, that pendulum is likely swinging back as almost every sober analysis of our current health care environment suggests a need for better care for the chronically ill at home and in the community. It is important that research and academic enterprises emphasize scholarly efforts to understand and improve home and community care so that the anticipated shift in care to home is informed by the best possible evidence, ultimately ensuring that patients get the best possible care.
The articles in this online, CME-certified Cleveland Clinic Journal of Medicine supplement address contemporary topics in home health and other home-based care concepts. The authors have diverse backgrounds and discuss issues related to technology, palliative care, care transitions, heart failure, knee replacement, primary care, and health reform. Several articles share concepts and outcomes from innovative approaches being developed throughout the country to help patients succeed at home, especially when returning home from a hospitalization.
The articles should improve readers’ understanding of a wide range of initiatives and ideas for how home health and home care might look in the future delivery system. The authors also raise numerous yet-unanswered questions and opportunities for future study. The needs for further home care research from clinical, public health, and policy perspectives are evident. Health care is going home, and this transformation will be enhanced and possibly accelerated by thoughtful research and synthesis.
I am incredibly thankful to my fellow authors, and hope that we have produced a useful supplement that will help readers in their efforts to assist the most vulnerable patients and families in their efforts to remain independent at home.
Improving patient outcomes with better care transitions: The role for home health
The US health care system faces many challenges. Quality, cost, access, fragmentation, and misalignment of incentives are only a few. The most pressing dilemma is how this challenged system will handle the demographic wave of aging Americans. Our 21st-century population is living longer with a greater chronic disease burden than its predecessors, and has reasonable expectations of quality care. No setting portrays this challenge more clearly than that of transition: the transfer of a patient and his or her care from the hospital or facility setting to the home. Addressing this challenge requires that we adopt a set of proven effective interventions that can improve quality of care, meet the needs of the patients and families we serve, and lower the staggering economic and social burden of preventable hospital readmissions.
The Medicare system, designed in 1965, has not kept pace with the needs and challenges of the rapidly aging US population. Further, the system is not aligned with today’s—and tomorrow’s—needs. In 1965, average life expectancy for Americans was 70 years; by 2020, that average is predicted to be nearly 80 years.1 In 2000, one in eight Americans, or 12% of the US population, was aged 65 years or older.2 It is expected that by 2030, this group will represent 19% of the population. This means that in 2030, some 72 million Americans will be aged 65 or older—more than twice the number in this age group in 2000.2
The 1965 health care system focused on treating acute disease, but the health care system of the 21st century must effectively manage chronic disease. The burden of chronic disease is especially significant for aging patients, who are likely to be under the care of multiple providers and require multiple medications and ever-higher levels of professional care. The management and sequelae of chronic diseases frequently lead to impaired quality of life as well as significant expense for Medicare.
The discrepancy between our health care system and unmet needs is acutely obvious at the time of hospital discharge. In fact, the Medicare Payment Advisory Commission (MedPAC) has stated that this burden of unmet needs at hospital discharge is primarily driven by hospital admissions and readmissions.3 Thirty-day readmission rates among older Medicare beneficiaries range from 15% to 25%.4–6 Disagreement persists regarding what percentage of hospital readmissions within 30 days might be preventable. A systematic review of 34 studies has reported that, on average, 27% of readmissions were preventable.7
To address the challenge of avoidable readmissions, our home health and hospice care organization, Amedisys, Inc., developed a care transitions initiative designed to improve quality of life, improve patient outcomes, and prevent unnecessary hospital readmissions. This article, which includes an illustrative case study, describes the initiative and the outcomes observed during its first 12 months of testing.
CASE STUDY
Mrs. Smith is 84 years old and lives alone in her home. She suffers from mild to moderate dementia and heart failure (HF). Mrs. Smith’s daughter is her main caregiver, talking to Mrs. Smith multiple times a day and stopping by Mrs. Smith’s house at least two to three times a week.
Mrs. Smith was admitted to the hospital after her daughter brought her to the emergency department over the weekend because of shortness of breath. This was her third visit to the emergency department within the past year, with each visit resulting in a hospitalization. Because of questions regarding her homebound status, home health was not considered part of the care plan during either of Mrs. Smith’s previous discharges.
Hospitalists made rounds over the weekend and notified Mrs. Smith that she would be released on Tuesday morning; because of her weakness and disorientation, the hospitalist issued an order for home health and a prescription for a new HF medication. Upon hearing the news on Monday of the planned discharge, Mrs. Smith and her daughter selected the home health provider they wished to use and, within the next few hours, a care transitions coordinator (CTC) visited them in the hospital.
The CTC, a registered nurse, talked with Mrs. Smith about her illness, educating her on the impact of diet on her condition and the medications she takes, including the new medication prescribed by the hospitalist. Most importantly, the CTC talked to Mrs. Smith about her personal goals during her recovery. For example, Mrs. Smith loves to visit her granddaughter, where she spends hours at a time watching her great-grandchildren play. Mrs. Smith wants to control her HF so that she can continue these visits that bring her such joy.
Mrs. Smith’s daughter asked the CTC if she would make Mrs. Smith’s primary care physician aware of the change in medication and schedule an appointment within the next week. The CTC did so before Mrs. Smith left the hospital. She also completed a primary care discharge notification, which documented Mrs. Smith’s discharge diagnoses, discharge medications, important test results, and the date of the appointment, and e-faxed it to Mrs. Smith’s primary care physician. The CTC also communicated with the home health nurse who would care for Mrs. Smith following discharge, reviewing her clinical needs as well as her personal goals.
Mrs. Smith’s daughter was present when the home health nurse conducted the admission and in-home assessment. The home health nurse educated both Mrs. Smith and her daughter about foods that might exacerbate HF, reinforcing the education started in the hospital by the CTC. In the course of this conversation, Mrs. Smith’s daughter realized that her mother had been eating popcorn late at night when she could not sleep. The CTC helped both mother and daughter to understand that the salt in her popcorn could have an impact on Mrs. Smith’s illness that would likely result in rehospitalization and an increase in medication dosage; this educational process enhanced the patient’s understanding of her disease and likely reduced the chances of her emergency department–rehospitalization cycle continuing.
INTERVENTION
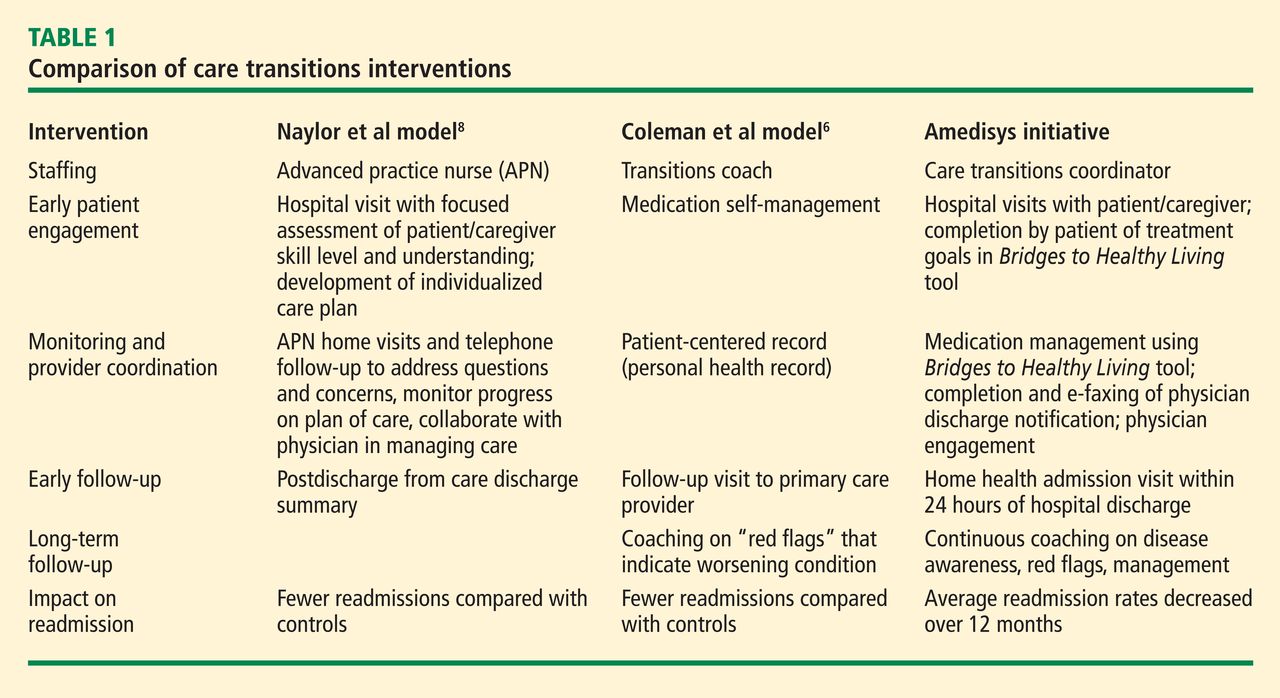
The design of the Amedisys care transitions initiative is based on work by Naylor et al8 and Coleman et al,6 who are recognized in the home health industry for their models of intervention at the time of hospital discharge. The Amedisys initiative’s objective is to prevent avoidable readmissions through patient and caregiver health coaching and care coordination, starting in the hospital and continuing through completion of the patient’s home health plan of care. Table 1 compares the essential interventions of the Naylor and Coleman models with those of the Amedisys initiative.
The Amedisys initiative includes these specific interventions:
- use of a CTC;
- early engagement of the patient, caregiver, and family with condition-specific coaching;
- careful medication management; and
- physician engagement with scheduling and reminders of physician visits early in the transition process.
Using a care transitions coordinator
Amedisys has placed CTCs in the acute care facilities that it serves. The CTC’s responsibility is to ensure that patients transition safely home from the acute care setting. With fragmentation of care, patients are most vulnerable during the initial few days postdischarge; this is particularly true for the frail elderly. Consequently, the CTC meets with the patient and caregiver as soon as possible upon his or her referral to Amedisys to plan the transition home from the facility and determine the resources needed once home. The CTC becomes the patient’s “touchpoint” for any questions or problems that arise between the time of discharge and the time when an Amedisys nurse visits the patient’s home.
Early engagement and coaching
The CTC uses a proprietary tool, Bridge to Healt0hy Living, to begin the process of early engagement, education, and coaching. This bound notebook is personalized for each patient with the CTC’s name and 24-hour phone contact information. The CTC records the patient’s diagnoses as well as social and economic barriers that may affect the patient’s outcomes. The diagnoses are written in the notebook along with a list of the patient’s medications that describes what each drug is for, its exact dosage, and instructions for taking it.
Coaching focuses on the patient’s diagnoses and capabilities, with discussion of diet and lifestyle needs and identification of “red flags” about each condition. The CTC asks the patient to describe his or her treatment goals and care plan. Ideally, the patient or a family member puts the goals and care plan in writing in the notebook in the patient’s own words; this strategy makes the goals and plan more meaningful and relevant to the patient. The CTC revisits this information at each encounter with the patient and caregiver.
Patient/family and caregiver engagement are crucial to the success of the initiative with frail, older patients.8,9 One 1998 study indicated that patient and caregiver satisfaction with home health services correlated with receiving information from the home health staff regarding medications, equipment and supplies, and self-care; further, the degree of caregiver burden was inversely related to receipt of information from the home health staff.10 The engagement required for the patient and caregiver to record the necessary information in the care transitions tool improves the likelihood of their understanding and adhering to lifestyle, behavioral, and medication recommendations.
At the time of hospital discharge, the CTC arranges the patient’s appointment with the primary care physician and records this in the patient’s notebook. The date and time for the patient’s first home nursing visit is also arranged and recorded so that the patient and caregiver know exactly when to expect that visit.
Medication management
The first home nursing visit typically occurs within 24 hours of hospital discharge. During this visit, the home health nurse reviews the Bridge to Healthy Living tool and uses it to guide care in partnership with the patient, enhancing adherence to the care plan. The nurse reviews the patient’s medications, checks them against the hospital discharge list, and then asks about other medications that might be in a cabinet or the refrigerator that the patient might be taking. At each subsequent visit, the nurse reviews the medication list and adjusts it as indicated if the patient’s physicians have changed any medication. If there has been a medication change, this is communicated by the home health nurse to all physicians caring for the patient.
The initial home nursing visit includes an environmental assessment with observation for hazards that could increase the risk for falls or other injury. The nurse also reinforces coaching on medications, red flags, and dietary or lifestyle issues that was begun by the CTC in the hospital.
Physician engagement
Physician engagement in the transition process is critical to reducing avoidable rehospitalizations. Coleman’s work has emphasized the need for the patient to follow up with his or her primary care physician within 1 week of discharge; but too frequently, the primary care physician is unaware that the patient was admitted to the hospital, and discharge summaries may take weeks to arrive. The care transitions initiative is a relationship-based, physician-led care delivery model in which the CTC serves as the funnel for information-sharing among all providers engaged with the patient. Although the CTC functions as the information manager, a successful transition requires an unprecedented level of cooperation among physicians and other health care providers. Health care is changing; outcomes must improve and costs must decrease. Therefore, this level of cooperation is no longer optional, but has become mandatory.
OUTCOMES
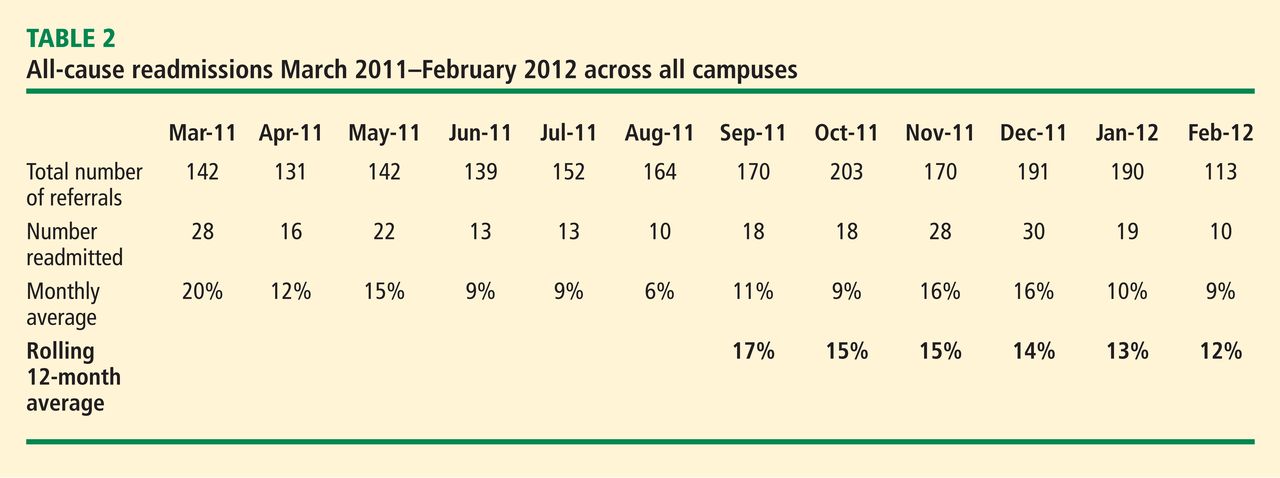
The primary outcome measure in the care transitions initiative was the rate of nonelective rehospitalization related to any cause, recurrence, or exacerbation of the index hospitalization diagnosis-related group, comorbid conditions, or new health problems. The Amedisys care transitions initiative was tested in three large, academic institutions in the northeast and southeast United States for 12 months. The 12-month average readmission rate (as calculated month by month) in the last 6 months of the study decreased from 17% to 12% (Table 2). During this period both patient and physician satisfaction were enhanced, according to internal survey data.
CALL TO ACTION
Americans want to live in their own homes as long as possible. In fact, when elderly Americans are admitted to a hospital, what is actually occurring is that they are being “discharged from their communities.”11 A health care delivery system that provides a true patient-centered approach to care recognizes that this situation often compounds issues of health care costs and quality. Adequate transitional care can provide simpler and more cost-effective options. If a CTC and follow-up care at home had been provided to Mrs. Smith and her daughter upon the first emergency room visit earlier in the year (see “Case study,” page e-S2), Mrs. Smith might have avoided multiple costly readmissions. Each member of the home health industry and its partners should be required to provide a basic set of evidence-based care transition elements to the patients they serve. By coordinating care at the time of discharge, some of the fragmentation that has become embedded in our system might be overcome.
- Life expectancy—United States. Data360 Web site. http://www.data360.org/dsg.aspx?Data_Set_Group_Id=195. Accessed August 15, 2012.
- Aging statistics. Administration on Aging Web site. http://www.aoa.gov/aoaroot/aging_statistics/index.aspx. Updated September 1, 2011. Accessed August 15, 2012.
- Report to the Congress. Medicare Payment Policy. Medicare Payment Advisory Commission Web site. http://medpac.gov/documents/Mar08_EntireReport.pdf. Published March 2008. Accessed August 15, 2012.
- Coleman EA, Min S, Chomiak A, Kramer AM. Post-hospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Quality initiatives—general information. Centers for Medicare & Medicaid Services Web site. http://www.cms.hhs.gov/QualityInitiativesGenInfo/15_MQMS.asp. Updated April 4, 2012. Accessed August 15, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review [published online ahead of print]. CMAJ 2011; 183:E391–E402. 10.1503/cmaj.101860
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Coleman EA, Smith JD, Frank JC, Min S, Parry C, Kramer AM. Preparing patients and caregivers to participate in care delivered across settings: the Care Transitions Intervention. J Am Geriatr Soc 2004; 52:1817–1825.
- Weaver FM, Perloff L, Waters T. Patients’ and caregivers’ transition from hospital to home: needs and recommendations. Home Health Care Serv Q 1998; 17:27–48.
- Fleming MO. The value of healthcare at home. Presented at: American Osteopathic Visiting Professorship, Louisiana State University Health System; April 12, 2012; New Orleans, LA.
The US health care system faces many challenges. Quality, cost, access, fragmentation, and misalignment of incentives are only a few. The most pressing dilemma is how this challenged system will handle the demographic wave of aging Americans. Our 21st-century population is living longer with a greater chronic disease burden than its predecessors, and has reasonable expectations of quality care. No setting portrays this challenge more clearly than that of transition: the transfer of a patient and his or her care from the hospital or facility setting to the home. Addressing this challenge requires that we adopt a set of proven effective interventions that can improve quality of care, meet the needs of the patients and families we serve, and lower the staggering economic and social burden of preventable hospital readmissions.
The Medicare system, designed in 1965, has not kept pace with the needs and challenges of the rapidly aging US population. Further, the system is not aligned with today’s—and tomorrow’s—needs. In 1965, average life expectancy for Americans was 70 years; by 2020, that average is predicted to be nearly 80 years.1 In 2000, one in eight Americans, or 12% of the US population, was aged 65 years or older.2 It is expected that by 2030, this group will represent 19% of the population. This means that in 2030, some 72 million Americans will be aged 65 or older—more than twice the number in this age group in 2000.2
The 1965 health care system focused on treating acute disease, but the health care system of the 21st century must effectively manage chronic disease. The burden of chronic disease is especially significant for aging patients, who are likely to be under the care of multiple providers and require multiple medications and ever-higher levels of professional care. The management and sequelae of chronic diseases frequently lead to impaired quality of life as well as significant expense for Medicare.
The discrepancy between our health care system and unmet needs is acutely obvious at the time of hospital discharge. In fact, the Medicare Payment Advisory Commission (MedPAC) has stated that this burden of unmet needs at hospital discharge is primarily driven by hospital admissions and readmissions.3 Thirty-day readmission rates among older Medicare beneficiaries range from 15% to 25%.4–6 Disagreement persists regarding what percentage of hospital readmissions within 30 days might be preventable. A systematic review of 34 studies has reported that, on average, 27% of readmissions were preventable.7
To address the challenge of avoidable readmissions, our home health and hospice care organization, Amedisys, Inc., developed a care transitions initiative designed to improve quality of life, improve patient outcomes, and prevent unnecessary hospital readmissions. This article, which includes an illustrative case study, describes the initiative and the outcomes observed during its first 12 months of testing.
CASE STUDY
Mrs. Smith is 84 years old and lives alone in her home. She suffers from mild to moderate dementia and heart failure (HF). Mrs. Smith’s daughter is her main caregiver, talking to Mrs. Smith multiple times a day and stopping by Mrs. Smith’s house at least two to three times a week.
Mrs. Smith was admitted to the hospital after her daughter brought her to the emergency department over the weekend because of shortness of breath. This was her third visit to the emergency department within the past year, with each visit resulting in a hospitalization. Because of questions regarding her homebound status, home health was not considered part of the care plan during either of Mrs. Smith’s previous discharges.
Hospitalists made rounds over the weekend and notified Mrs. Smith that she would be released on Tuesday morning; because of her weakness and disorientation, the hospitalist issued an order for home health and a prescription for a new HF medication. Upon hearing the news on Monday of the planned discharge, Mrs. Smith and her daughter selected the home health provider they wished to use and, within the next few hours, a care transitions coordinator (CTC) visited them in the hospital.
The CTC, a registered nurse, talked with Mrs. Smith about her illness, educating her on the impact of diet on her condition and the medications she takes, including the new medication prescribed by the hospitalist. Most importantly, the CTC talked to Mrs. Smith about her personal goals during her recovery. For example, Mrs. Smith loves to visit her granddaughter, where she spends hours at a time watching her great-grandchildren play. Mrs. Smith wants to control her HF so that she can continue these visits that bring her such joy.
Mrs. Smith’s daughter asked the CTC if she would make Mrs. Smith’s primary care physician aware of the change in medication and schedule an appointment within the next week. The CTC did so before Mrs. Smith left the hospital. She also completed a primary care discharge notification, which documented Mrs. Smith’s discharge diagnoses, discharge medications, important test results, and the date of the appointment, and e-faxed it to Mrs. Smith’s primary care physician. The CTC also communicated with the home health nurse who would care for Mrs. Smith following discharge, reviewing her clinical needs as well as her personal goals.
Mrs. Smith’s daughter was present when the home health nurse conducted the admission and in-home assessment. The home health nurse educated both Mrs. Smith and her daughter about foods that might exacerbate HF, reinforcing the education started in the hospital by the CTC. In the course of this conversation, Mrs. Smith’s daughter realized that her mother had been eating popcorn late at night when she could not sleep. The CTC helped both mother and daughter to understand that the salt in her popcorn could have an impact on Mrs. Smith’s illness that would likely result in rehospitalization and an increase in medication dosage; this educational process enhanced the patient’s understanding of her disease and likely reduced the chances of her emergency department–rehospitalization cycle continuing.
INTERVENTION
The design of the Amedisys care transitions initiative is based on work by Naylor et al8 and Coleman et al,6 who are recognized in the home health industry for their models of intervention at the time of hospital discharge. The Amedisys initiative’s objective is to prevent avoidable readmissions through patient and caregiver health coaching and care coordination, starting in the hospital and continuing through completion of the patient’s home health plan of care. Table 1 compares the essential interventions of the Naylor and Coleman models with those of the Amedisys initiative.
The Amedisys initiative includes these specific interventions:
- use of a CTC;
- early engagement of the patient, caregiver, and family with condition-specific coaching;
- careful medication management; and
- physician engagement with scheduling and reminders of physician visits early in the transition process.
Using a care transitions coordinator
Amedisys has placed CTCs in the acute care facilities that it serves. The CTC’s responsibility is to ensure that patients transition safely home from the acute care setting. With fragmentation of care, patients are most vulnerable during the initial few days postdischarge; this is particularly true for the frail elderly. Consequently, the CTC meets with the patient and caregiver as soon as possible upon his or her referral to Amedisys to plan the transition home from the facility and determine the resources needed once home. The CTC becomes the patient’s “touchpoint” for any questions or problems that arise between the time of discharge and the time when an Amedisys nurse visits the patient’s home.
Early engagement and coaching
The CTC uses a proprietary tool, Bridge to Healt0hy Living, to begin the process of early engagement, education, and coaching. This bound notebook is personalized for each patient with the CTC’s name and 24-hour phone contact information. The CTC records the patient’s diagnoses as well as social and economic barriers that may affect the patient’s outcomes. The diagnoses are written in the notebook along with a list of the patient’s medications that describes what each drug is for, its exact dosage, and instructions for taking it.
Coaching focuses on the patient’s diagnoses and capabilities, with discussion of diet and lifestyle needs and identification of “red flags” about each condition. The CTC asks the patient to describe his or her treatment goals and care plan. Ideally, the patient or a family member puts the goals and care plan in writing in the notebook in the patient’s own words; this strategy makes the goals and plan more meaningful and relevant to the patient. The CTC revisits this information at each encounter with the patient and caregiver.
Patient/family and caregiver engagement are crucial to the success of the initiative with frail, older patients.8,9 One 1998 study indicated that patient and caregiver satisfaction with home health services correlated with receiving information from the home health staff regarding medications, equipment and supplies, and self-care; further, the degree of caregiver burden was inversely related to receipt of information from the home health staff.10 The engagement required for the patient and caregiver to record the necessary information in the care transitions tool improves the likelihood of their understanding and adhering to lifestyle, behavioral, and medication recommendations.
At the time of hospital discharge, the CTC arranges the patient’s appointment with the primary care physician and records this in the patient’s notebook. The date and time for the patient’s first home nursing visit is also arranged and recorded so that the patient and caregiver know exactly when to expect that visit.
Medication management
The first home nursing visit typically occurs within 24 hours of hospital discharge. During this visit, the home health nurse reviews the Bridge to Healthy Living tool and uses it to guide care in partnership with the patient, enhancing adherence to the care plan. The nurse reviews the patient’s medications, checks them against the hospital discharge list, and then asks about other medications that might be in a cabinet or the refrigerator that the patient might be taking. At each subsequent visit, the nurse reviews the medication list and adjusts it as indicated if the patient’s physicians have changed any medication. If there has been a medication change, this is communicated by the home health nurse to all physicians caring for the patient.
The initial home nursing visit includes an environmental assessment with observation for hazards that could increase the risk for falls or other injury. The nurse also reinforces coaching on medications, red flags, and dietary or lifestyle issues that was begun by the CTC in the hospital.
Physician engagement
Physician engagement in the transition process is critical to reducing avoidable rehospitalizations. Coleman’s work has emphasized the need for the patient to follow up with his or her primary care physician within 1 week of discharge; but too frequently, the primary care physician is unaware that the patient was admitted to the hospital, and discharge summaries may take weeks to arrive. The care transitions initiative is a relationship-based, physician-led care delivery model in which the CTC serves as the funnel for information-sharing among all providers engaged with the patient. Although the CTC functions as the information manager, a successful transition requires an unprecedented level of cooperation among physicians and other health care providers. Health care is changing; outcomes must improve and costs must decrease. Therefore, this level of cooperation is no longer optional, but has become mandatory.
OUTCOMES
The primary outcome measure in the care transitions initiative was the rate of nonelective rehospitalization related to any cause, recurrence, or exacerbation of the index hospitalization diagnosis-related group, comorbid conditions, or new health problems. The Amedisys care transitions initiative was tested in three large, academic institutions in the northeast and southeast United States for 12 months. The 12-month average readmission rate (as calculated month by month) in the last 6 months of the study decreased from 17% to 12% (Table 2). During this period both patient and physician satisfaction were enhanced, according to internal survey data.
CALL TO ACTION
Americans want to live in their own homes as long as possible. In fact, when elderly Americans are admitted to a hospital, what is actually occurring is that they are being “discharged from their communities.”11 A health care delivery system that provides a true patient-centered approach to care recognizes that this situation often compounds issues of health care costs and quality. Adequate transitional care can provide simpler and more cost-effective options. If a CTC and follow-up care at home had been provided to Mrs. Smith and her daughter upon the first emergency room visit earlier in the year (see “Case study,” page e-S2), Mrs. Smith might have avoided multiple costly readmissions. Each member of the home health industry and its partners should be required to provide a basic set of evidence-based care transition elements to the patients they serve. By coordinating care at the time of discharge, some of the fragmentation that has become embedded in our system might be overcome.
The US health care system faces many challenges. Quality, cost, access, fragmentation, and misalignment of incentives are only a few. The most pressing dilemma is how this challenged system will handle the demographic wave of aging Americans. Our 21st-century population is living longer with a greater chronic disease burden than its predecessors, and has reasonable expectations of quality care. No setting portrays this challenge more clearly than that of transition: the transfer of a patient and his or her care from the hospital or facility setting to the home. Addressing this challenge requires that we adopt a set of proven effective interventions that can improve quality of care, meet the needs of the patients and families we serve, and lower the staggering economic and social burden of preventable hospital readmissions.
The Medicare system, designed in 1965, has not kept pace with the needs and challenges of the rapidly aging US population. Further, the system is not aligned with today’s—and tomorrow’s—needs. In 1965, average life expectancy for Americans was 70 years; by 2020, that average is predicted to be nearly 80 years.1 In 2000, one in eight Americans, or 12% of the US population, was aged 65 years or older.2 It is expected that by 2030, this group will represent 19% of the population. This means that in 2030, some 72 million Americans will be aged 65 or older—more than twice the number in this age group in 2000.2
The 1965 health care system focused on treating acute disease, but the health care system of the 21st century must effectively manage chronic disease. The burden of chronic disease is especially significant for aging patients, who are likely to be under the care of multiple providers and require multiple medications and ever-higher levels of professional care. The management and sequelae of chronic diseases frequently lead to impaired quality of life as well as significant expense for Medicare.
The discrepancy between our health care system and unmet needs is acutely obvious at the time of hospital discharge. In fact, the Medicare Payment Advisory Commission (MedPAC) has stated that this burden of unmet needs at hospital discharge is primarily driven by hospital admissions and readmissions.3 Thirty-day readmission rates among older Medicare beneficiaries range from 15% to 25%.4–6 Disagreement persists regarding what percentage of hospital readmissions within 30 days might be preventable. A systematic review of 34 studies has reported that, on average, 27% of readmissions were preventable.7
To address the challenge of avoidable readmissions, our home health and hospice care organization, Amedisys, Inc., developed a care transitions initiative designed to improve quality of life, improve patient outcomes, and prevent unnecessary hospital readmissions. This article, which includes an illustrative case study, describes the initiative and the outcomes observed during its first 12 months of testing.
CASE STUDY
Mrs. Smith is 84 years old and lives alone in her home. She suffers from mild to moderate dementia and heart failure (HF). Mrs. Smith’s daughter is her main caregiver, talking to Mrs. Smith multiple times a day and stopping by Mrs. Smith’s house at least two to three times a week.
Mrs. Smith was admitted to the hospital after her daughter brought her to the emergency department over the weekend because of shortness of breath. This was her third visit to the emergency department within the past year, with each visit resulting in a hospitalization. Because of questions regarding her homebound status, home health was not considered part of the care plan during either of Mrs. Smith’s previous discharges.
Hospitalists made rounds over the weekend and notified Mrs. Smith that she would be released on Tuesday morning; because of her weakness and disorientation, the hospitalist issued an order for home health and a prescription for a new HF medication. Upon hearing the news on Monday of the planned discharge, Mrs. Smith and her daughter selected the home health provider they wished to use and, within the next few hours, a care transitions coordinator (CTC) visited them in the hospital.
The CTC, a registered nurse, talked with Mrs. Smith about her illness, educating her on the impact of diet on her condition and the medications she takes, including the new medication prescribed by the hospitalist. Most importantly, the CTC talked to Mrs. Smith about her personal goals during her recovery. For example, Mrs. Smith loves to visit her granddaughter, where she spends hours at a time watching her great-grandchildren play. Mrs. Smith wants to control her HF so that she can continue these visits that bring her such joy.
Mrs. Smith’s daughter asked the CTC if she would make Mrs. Smith’s primary care physician aware of the change in medication and schedule an appointment within the next week. The CTC did so before Mrs. Smith left the hospital. She also completed a primary care discharge notification, which documented Mrs. Smith’s discharge diagnoses, discharge medications, important test results, and the date of the appointment, and e-faxed it to Mrs. Smith’s primary care physician. The CTC also communicated with the home health nurse who would care for Mrs. Smith following discharge, reviewing her clinical needs as well as her personal goals.
Mrs. Smith’s daughter was present when the home health nurse conducted the admission and in-home assessment. The home health nurse educated both Mrs. Smith and her daughter about foods that might exacerbate HF, reinforcing the education started in the hospital by the CTC. In the course of this conversation, Mrs. Smith’s daughter realized that her mother had been eating popcorn late at night when she could not sleep. The CTC helped both mother and daughter to understand that the salt in her popcorn could have an impact on Mrs. Smith’s illness that would likely result in rehospitalization and an increase in medication dosage; this educational process enhanced the patient’s understanding of her disease and likely reduced the chances of her emergency department–rehospitalization cycle continuing.
INTERVENTION
The design of the Amedisys care transitions initiative is based on work by Naylor et al8 and Coleman et al,6 who are recognized in the home health industry for their models of intervention at the time of hospital discharge. The Amedisys initiative’s objective is to prevent avoidable readmissions through patient and caregiver health coaching and care coordination, starting in the hospital and continuing through completion of the patient’s home health plan of care. Table 1 compares the essential interventions of the Naylor and Coleman models with those of the Amedisys initiative.
The Amedisys initiative includes these specific interventions:
- use of a CTC;
- early engagement of the patient, caregiver, and family with condition-specific coaching;
- careful medication management; and
- physician engagement with scheduling and reminders of physician visits early in the transition process.
Using a care transitions coordinator
Amedisys has placed CTCs in the acute care facilities that it serves. The CTC’s responsibility is to ensure that patients transition safely home from the acute care setting. With fragmentation of care, patients are most vulnerable during the initial few days postdischarge; this is particularly true for the frail elderly. Consequently, the CTC meets with the patient and caregiver as soon as possible upon his or her referral to Amedisys to plan the transition home from the facility and determine the resources needed once home. The CTC becomes the patient’s “touchpoint” for any questions or problems that arise between the time of discharge and the time when an Amedisys nurse visits the patient’s home.
Early engagement and coaching
The CTC uses a proprietary tool, Bridge to Healt0hy Living, to begin the process of early engagement, education, and coaching. This bound notebook is personalized for each patient with the CTC’s name and 24-hour phone contact information. The CTC records the patient’s diagnoses as well as social and economic barriers that may affect the patient’s outcomes. The diagnoses are written in the notebook along with a list of the patient’s medications that describes what each drug is for, its exact dosage, and instructions for taking it.
Coaching focuses on the patient’s diagnoses and capabilities, with discussion of diet and lifestyle needs and identification of “red flags” about each condition. The CTC asks the patient to describe his or her treatment goals and care plan. Ideally, the patient or a family member puts the goals and care plan in writing in the notebook in the patient’s own words; this strategy makes the goals and plan more meaningful and relevant to the patient. The CTC revisits this information at each encounter with the patient and caregiver.
Patient/family and caregiver engagement are crucial to the success of the initiative with frail, older patients.8,9 One 1998 study indicated that patient and caregiver satisfaction with home health services correlated with receiving information from the home health staff regarding medications, equipment and supplies, and self-care; further, the degree of caregiver burden was inversely related to receipt of information from the home health staff.10 The engagement required for the patient and caregiver to record the necessary information in the care transitions tool improves the likelihood of their understanding and adhering to lifestyle, behavioral, and medication recommendations.
At the time of hospital discharge, the CTC arranges the patient’s appointment with the primary care physician and records this in the patient’s notebook. The date and time for the patient’s first home nursing visit is also arranged and recorded so that the patient and caregiver know exactly when to expect that visit.
Medication management
The first home nursing visit typically occurs within 24 hours of hospital discharge. During this visit, the home health nurse reviews the Bridge to Healthy Living tool and uses it to guide care in partnership with the patient, enhancing adherence to the care plan. The nurse reviews the patient’s medications, checks them against the hospital discharge list, and then asks about other medications that might be in a cabinet or the refrigerator that the patient might be taking. At each subsequent visit, the nurse reviews the medication list and adjusts it as indicated if the patient’s physicians have changed any medication. If there has been a medication change, this is communicated by the home health nurse to all physicians caring for the patient.
The initial home nursing visit includes an environmental assessment with observation for hazards that could increase the risk for falls or other injury. The nurse also reinforces coaching on medications, red flags, and dietary or lifestyle issues that was begun by the CTC in the hospital.
Physician engagement
Physician engagement in the transition process is critical to reducing avoidable rehospitalizations. Coleman’s work has emphasized the need for the patient to follow up with his or her primary care physician within 1 week of discharge; but too frequently, the primary care physician is unaware that the patient was admitted to the hospital, and discharge summaries may take weeks to arrive. The care transitions initiative is a relationship-based, physician-led care delivery model in which the CTC serves as the funnel for information-sharing among all providers engaged with the patient. Although the CTC functions as the information manager, a successful transition requires an unprecedented level of cooperation among physicians and other health care providers. Health care is changing; outcomes must improve and costs must decrease. Therefore, this level of cooperation is no longer optional, but has become mandatory.
OUTCOMES
The primary outcome measure in the care transitions initiative was the rate of nonelective rehospitalization related to any cause, recurrence, or exacerbation of the index hospitalization diagnosis-related group, comorbid conditions, or new health problems. The Amedisys care transitions initiative was tested in three large, academic institutions in the northeast and southeast United States for 12 months. The 12-month average readmission rate (as calculated month by month) in the last 6 months of the study decreased from 17% to 12% (Table 2). During this period both patient and physician satisfaction were enhanced, according to internal survey data.
CALL TO ACTION
Americans want to live in their own homes as long as possible. In fact, when elderly Americans are admitted to a hospital, what is actually occurring is that they are being “discharged from their communities.”11 A health care delivery system that provides a true patient-centered approach to care recognizes that this situation often compounds issues of health care costs and quality. Adequate transitional care can provide simpler and more cost-effective options. If a CTC and follow-up care at home had been provided to Mrs. Smith and her daughter upon the first emergency room visit earlier in the year (see “Case study,” page e-S2), Mrs. Smith might have avoided multiple costly readmissions. Each member of the home health industry and its partners should be required to provide a basic set of evidence-based care transition elements to the patients they serve. By coordinating care at the time of discharge, some of the fragmentation that has become embedded in our system might be overcome.
- Life expectancy—United States. Data360 Web site. http://www.data360.org/dsg.aspx?Data_Set_Group_Id=195. Accessed August 15, 2012.
- Aging statistics. Administration on Aging Web site. http://www.aoa.gov/aoaroot/aging_statistics/index.aspx. Updated September 1, 2011. Accessed August 15, 2012.
- Report to the Congress. Medicare Payment Policy. Medicare Payment Advisory Commission Web site. http://medpac.gov/documents/Mar08_EntireReport.pdf. Published March 2008. Accessed August 15, 2012.
- Coleman EA, Min S, Chomiak A, Kramer AM. Post-hospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Quality initiatives—general information. Centers for Medicare & Medicaid Services Web site. http://www.cms.hhs.gov/QualityInitiativesGenInfo/15_MQMS.asp. Updated April 4, 2012. Accessed August 15, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review [published online ahead of print]. CMAJ 2011; 183:E391–E402. 10.1503/cmaj.101860
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Coleman EA, Smith JD, Frank JC, Min S, Parry C, Kramer AM. Preparing patients and caregivers to participate in care delivered across settings: the Care Transitions Intervention. J Am Geriatr Soc 2004; 52:1817–1825.
- Weaver FM, Perloff L, Waters T. Patients’ and caregivers’ transition from hospital to home: needs and recommendations. Home Health Care Serv Q 1998; 17:27–48.
- Fleming MO. The value of healthcare at home. Presented at: American Osteopathic Visiting Professorship, Louisiana State University Health System; April 12, 2012; New Orleans, LA.
- Life expectancy—United States. Data360 Web site. http://www.data360.org/dsg.aspx?Data_Set_Group_Id=195. Accessed August 15, 2012.
- Aging statistics. Administration on Aging Web site. http://www.aoa.gov/aoaroot/aging_statistics/index.aspx. Updated September 1, 2011. Accessed August 15, 2012.
- Report to the Congress. Medicare Payment Policy. Medicare Payment Advisory Commission Web site. http://medpac.gov/documents/Mar08_EntireReport.pdf. Published March 2008. Accessed August 15, 2012.
- Coleman EA, Min S, Chomiak A, Kramer AM. Post-hospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Quality initiatives—general information. Centers for Medicare & Medicaid Services Web site. http://www.cms.hhs.gov/QualityInitiativesGenInfo/15_MQMS.asp. Updated April 4, 2012. Accessed August 15, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- van Walraven C, Bennett C, Jennings A, Austin PC, Forster AJ. Proportion of hospital readmissions deemed avoidable: a systematic review [published online ahead of print]. CMAJ 2011; 183:E391–E402. 10.1503/cmaj.101860
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Coleman EA, Smith JD, Frank JC, Min S, Parry C, Kramer AM. Preparing patients and caregivers to participate in care delivered across settings: the Care Transitions Intervention. J Am Geriatr Soc 2004; 52:1817–1825.
- Weaver FM, Perloff L, Waters T. Patients’ and caregivers’ transition from hospital to home: needs and recommendations. Home Health Care Serv Q 1998; 17:27–48.
- Fleming MO. The value of healthcare at home. Presented at: American Osteopathic Visiting Professorship, Louisiana State University Health System; April 12, 2012; New Orleans, LA.
Improving outcomes and lowering costs by applying advanced models of in-home care
When it can be done safely, most people prefer to be treated and recover from illness at home.1,2 Home-based services have improved considerably since Brickner called the homebound aged “a medically unreached group.”3 Still, home care has not achieved its full potential and scientific investigation of home care models is scant compared with that of other therapeutic approaches.
The challenges of studying home care include variability in interventions, difficulty defining treatment and comparison groups, and high research costs. The care itself can be demanding, requiring providers to mobilize processes that have become institution-based and immobile, integrate care across insular settings, incorporate complex social issues into the care plan, and develop a viable home care financing model.
This article reviews evidence favoring investment in advanced home care and adds perspective from 3 decades’ experience at Virginia Commonwealth University (VCU), Richmond, Virginia.
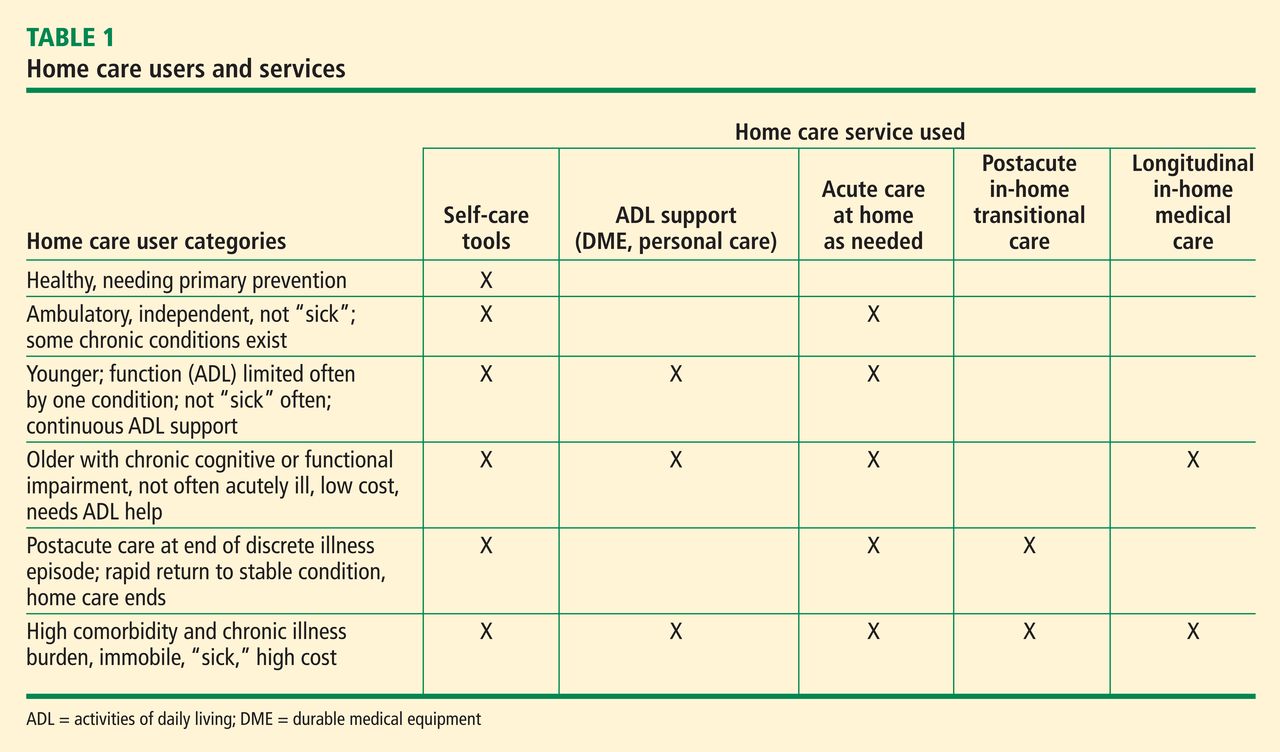
The term home care has a broad scope, ranging from basic support to highly technical care involving intravenous lines, ventilators, portable diagnostic tests, and remote monitors.4 Patients cared for at home range from those who are ambulatory to those who are permanently bedfast and seriously ill. The home care user population can be categorized based on the types of health care resources they consume (Table 1). Much attention has been paid to home-based care during recuperation after acute illness. The aim has been to foster recovery and prevent further need for institutional care. Lately the term transitional care has been used in this context.
TRANSITIONAL CARE
Transitional care has long been a priority for visiting nurse agencies. In 1965, Medicare Part A, building from the tradition of urban parish nursing services, created an interdisciplinary industry. Medicare now certifies more than 10,000 agencies with more than 250,000 professional staff.5 For several reasons, beginning in the 1970s, US physicians have become less integrated into in-home care. Despite this and the challenge of managing medically complex patients with minimal active physician involvement, home health agencies provide a vital service. Further, they have demonstrated improved outcomes and cost savings.
Transitional care refers to specialized, short-term care for selected high-risk patients after an acute illness. The original objective of transitional care was to reduce hospital readmissions. Tested models include an approach developed by Coleman et al,6 based on four pillars: assistance with medication self-management, patient-centered and -owned medical record, timely follow-up with primary or specialty care, and “red flags” that indicate a worsening condition. This model, which yielded one-third fewer hospital readmissions and a savings of about $500 per patient in 6 months, is being adopted in many locations nationally.
Naylor and colleagues7,8 collaborated with hospital-based nurse practitioners (NPs) for 2 decades on a more intensive model. In the Naylor model, the NPs form a health care bridge from hospital to home for 4 weeks after hospital care and add an active medical care component to the home care team. Naylor et al7 reported a 50% reduction in the rehospitalization rate and a cost savings of approximately $3,000 per patient over 24 weeks. Naylor’s team observed these results among frail, elderly patients with a variety of conditions and comorbidities. The 2010 federal health care reform law as well as state and private insurer initiatives now encourage use of this and other integrated care models.
In a national demonstration program using performance improvement methods and careful data collection, 73 US home health agencies improved targeted clinical outcomes and reduced hospitalizations from baseline rates by approximately 7% within 3 to 4 years.9 The study included approximately 158,000 patients in the intervention group and 249,000 in the comparison group. However, in general the success demonstrated in this study has not been reflected nationally, and home health agencies have been weakly integrated with the remainder of the health care delivery system.
Medicare home health agency care has evolved rapidly in the past 15 years, with reporting of numerous quality measures that has created direct accountability of physicians to the public. Until as recently as the 1990s, many important measures of quality in medicine were available only to physicians and physician and hospital organizations through governmental and, in some cases, legal routes. This new quality-based accountability, along with fiscal pressure to reduce lengths of stay and to limit visits under prospective payment, are among the changes that are transforming the home health industry.
THE VCU TRANSITIONAL CARE EXPERIENCE
The VCU Medical Center implemented a Naylor-model hospital-based transitional care program (TCP) 12 years ago that has served more than 500 patients. Targeted patients have histories similar to those observed by Naylor et al7: multiple hospitalizations, prolonged inpatient stays, many comorbidities and medications, complex care plans, and poor social support. Referrals come from physician teams, care coordinators, nurses, and social workers. The electronic medical record (EMR) has triggers for referrals.
Transitional care NPs meet patients in the hospital to ensure the appropriateness of their referral, introduce the program, and verify information. As shown in the Naylor model and later in the Coleman model,6 inpatient contact creates rapport with the patient and with family caregivers.
The first home visit is made on a weekday within 24 to 72 hours of discharge. At this initial visit, which takes a considerable amount of time, we attempt to reconcile medications, clarify social needs and resources, conduct physical assessments, modify medical regimens, educate the patient and his or her caregivers, and run diagnostic laboratory tests as needed. What we see in the home on this first visit often does not correspond with what was previously reported by hospital-based clinicians. For example, we have found that many patients are not taking medications as prescribed.
Typically, we visit homes weekly for 4 to 8 weeks. Some patients remain in transitional care for longer periods due to medical and social reasons. The NPs maintain close contact with home health agency staff via mobile phones. In some cases we conduct joint visits with home health agency staff in order to facilitate adjustments to medical care plans. Regular communication with primary care providers via the EMR, fax, and phone helps close the follow-up gap. The NP’s ability to observe the home setting, identify barriers to medical compliance (including literacy), and address social issues offers a clearer picture to care providers and fosters better outcomes. As patients improve and become more mobile, they return to the care of the primary provider.
Positive results with some limitations
We collected data between 2003 and 2006 on patients enrolled in the VCU Medical Center TCP. Our demographic results were similar to those reported by Naylor et al.7 Prevalent diseases included heart failure (HF), coronary artery disease, diabetes, and chronic obstructive pulmonary disease (COPD). The mean age was 71 years. The patient population was 63% female and 77% African American. About 73% of patients returned to the care of their primary physicians, 13% enrolled in the VCU House Calls program, 12% died, and 3% were admitted to nursing homes.10
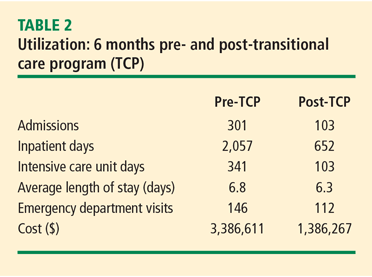
A comparison of utilization data for 199 patients 6 months before and after their enrollment in the TCP over a period of 4 years showed decreased use of hospital resources—ie, fewer inpatient days, shorter lengths of stay, and fewer intensive care unit days— after enrollment. Aggregate cost after TCP enrollment reduction was $2,251,34410 which is 38% less than the 6-month pre-enrollment baseline (Table 2). Regression to the mean played a role, but most patients had a sustained high-use pattern for 6 months before enrollment. The high rate of consumption of health care resources dropped quickly following implementation of the TCP and stayed down for many months.
We largely concur with Naylor’s description of transitional care implementation.11 However, we have found that many transitional care patients are unable return to the clinic after 2 months, as suggested by Naylor. In our system, these patients default to our House Calls program for continuing care. Thus, in our estimation, transitional care is an important but incomplete response to population-based health needs. Supporting this conclusion is the Congressional Budget Office report, which states that among high-cost Medicare patients in an index year (2001), those who lived for 5 years were high-cost patients on a month-by-month basis in 22 of the next 60 months, reflecting chronic illness and cyclical service use patterns.12
Extension of the TCP to outpatients
Because of the favorable effect observed in the hospital-based TCP, we created a role for transitional care in our outpatient geriatric practice. Transitional care NPs from the clinic practice have the option of making home visits in a variety of scenarios. In the least serious cases a single “diagnostic” home visit provides invaluable insight. For example, we evaluate support systems and compliance with medication instructions and put systems in place to help patients maintain independence and safety at home, including nutrition and fall prevention programs. Patients with poor social support benefit especially from home visits.
We find that high-risk patients recently discharged from facilities, including those outside our health system, benefit from NP visits. When a high-risk clinic patient is hospitalized, we maintain a connection with the inpatient team, follow the patient’s progress, and assist with discharge planning. Based on our relationship with the patient prior to admission, we are able to anticipate problems and to address them promptly after discharge. The NP functions as the “hub of the wheel” to coordinate the multidisciplinary plan among primary care providers; specialists; and support services such as home health, social work, and physical therapy.
We also initiate periodic NP home visits as chronic diseases progress and as clinic patients become increasingly frail. Interim visits are made to monitor the medical plan and perform follow-up blood testing. Once patients are no longer able to use the office practice, they transition into the House Calls program.
HOSPITAL AT HOME
The ultimate in substitutive, intensive home care occurs when one replaces acute care hospital admission with care delivered entirely at home. Robust research has shown comparable or better clinical outcomes with fewer complications and lower costs when home care is applied to common conditions such as pneumonia, COPD, cellulitis, and HF.13,14 Rapidly advancing technology now supports increasingly sophisticated care at home. For example, with low molecular weight heparin, the care of deep vein thrombosis and stable pulmonary embolism—which always required inpatient care 25 years ago—can now be delivered entirely at home in many cases. Soon, these conditions may be managed solely with oral medication.15,16 The range of conditions that are now being managed at home is extensive, and the transformation of health care by portable technology is just beginning.17
LONGITUDINAL IN-HOME PRIMARY CARE
In the United States, patients who are immobile and cannot easily access office-based care often suffer with suboptimal mobile primary care. This represents a major limitation in care access for these patients. There is good evidence that longitudinal medical care, primarily delivered at home for periods lasting many months to several years, is effective and that it makes clinical sense. In the home, providers can accurately assess the patient’s living situation, engender trust, and respond in a timely manner when a patient’s condition changes. The Geriatric Resources for Assessment and Care of Elders (GRACE) program and the Veterans Affairs (VA) home-based primary care model are two examples of the benefits of longitudinal in-home care.
In the GRACE model, patients receive comprehensive in-home assessment by NPs with quarterly follow-up, and recommendations are given to primary care providers. The program’s clinical trial demonstrated markedly improved treatment of a variety of common geriatric ailments and reduced costs in a high-risk subset of patients.18 GRACE was not designed for urgent care but the approach was linked to lower costs in high-risk cases, likely due to better care and improved access.
The VA home-based primary care model has grown rapidly in the past decade, now operating at more than 200 medical centers, each with a full interprofessional team. House calls by physicians and NPs are part of the model, although the frequency varies across sites. Every team includes actively engaged physicians. Medicoeconomic evaluation based on tens of thousands of patient-years has shown an overall reduction in health care costs of 15% to 25% compared with historical values and prospectively modeled dollars.19,20 Home-based primary care teams are emerging across the United States at many academic centers and in the private sector.
To fund comprehensive longitudinal home care services for patients with complex health problems, the Independence at Home21 demonstration program was created under section 3024 of the Patient Protection and Affordable Care Act, using robust gain-sharing from demonstrated cost savings to reward house call teams. This multisite 3-year program started in June 2012. Rapid growth of this model is likely as private insurers have also taken an active interest in mobile medical care designs, using a variety of reward structures.
TELEMEDICINE
A debate continues over the use of communication technology in home care. It seems intuitive that “virtual visits” would be more efficient than clinicians visiting patients at home. Yet, the challenges of improving care by telemedicine alone are underestimated. For example, a recent large randomized trial, in which 33 cardiology practice sites provided at-home postdis-charge telemonitoring for HF patients, demonstrated no difference in clinical outcomes compared with patients monitored in the hospital or clinic.22
Proponents of telemedicine cite integrated models where data are managed proactively by a physician-led team that is engaged in care. This view seems valid, but other than anecdotal reports from integrated health systems, the published evidence of reduced costs is sparse. Some combination of in-person care and telemedicine is likely to be the optimal design and will emerge in coming years.
PACE: SYSTEM-BASED HOME CARE
In the 1980s, health maintenance organization risk contracts seemed a likely context for developing advanced home care models, but this did not happen. However, the Program for All-Inclusive Care of the Elderly (PACE) was tested and became a defined federal benefit in 1997. There are now nearly 100 PACE centers nationwide. PACE offers comprehensive care for people aged 55 years and older who are nursing home–eligible. The program appears to effectively help people stay home.23
An interdisciplinary team (IDT) coordinates PACE medical and social services to promote independence and quality of life. The program has been referred to as “a nursing home without walls.” Services include primary and specialty care, adult day care, case management, nursing, home health care, assistance with activities of daily living (ADL), medications, social work, rehabilitation, hospitalization, nursing facility care, nutritional support, caregiver respite, and transportation to and from the PACE adult day health center (ADHC) and medical appointments. The ADHC is the cornerstone and coordinating center for most care provided to PACE participants. Home-based care is provided in several ways:
- Home nursing care may be provided by external agencies, including skilled care, personal care, and hospice care, under contract with PACE. In Richmond, the home care manager oversees care after it is approved by the IDT. Weekly hours of care are changed often according to the participant’s need (eg, increased hours after hospital discharge and decreased hours when a family member visits and can provide more care). Home care provides assistance with ADLs and instrumental ADLs; “sitter” services are provided at the ADHC.
- The program supports home modifications and provides durable medical equipment (DME). Assessment is done by one or more team members upon enrollment and then at least every 6 months. PACE provides all DME the participant needs to remain safely in the community. At disenrollment or death, some equipment can be returned to PACE after review by the rehabilitation department.
- Primary care, basic laboratory services, and medical specialty care can be provided to the participant at home if for any reason he or she is unable to travel to the ADHC. PACE physicians make house calls to better understand patients’ living situations and needs. On-call nurses make home visits after hours or on weekends for clinical assessments, point-of-care diagnostic testing, specimen collection (stool or urine), and participant and family education on proper use of medications or equipment.
- As PACE participants approach the end of life, they transition to a palliative care model. A decision is made by the family and the IDT to discontinue attendance at the ADHC and to focus on care at home,24 allowing the participant to spend the last days or weeks in the relative comfort of home. Nurses make home visits when needed and educate families on symptom palliation.
- Additional in-home respite services can be provided to decrease caregiver burden.
- Skilled rehabilitation services are delivered either at home or in the ADHC depending on the judgment of the rehabilitation department and the IDT. The PACE site offers advanced transportation and full onsite therapy services 5 days per week.
The PACE sites become the insurers, receive defined capitation payments from Medicare and Medicaid that are adjusted for patient complexity, and assume the risk for all health care costs. Because of a 5% withholding in the capitation amount relative to projected Medicare expenses, PACE should reduce governmental costs. PACE must provide or pay for all usual Medicare and Medicaid services, and it may provide other services deemed necessary by the PACE team. Within PACE, hospital use is markedly reduced compared with conventional Medicare,25 and home care is one of several strategies employed. The PACE experience shows that care can be safely shifted from hospitals to other settings.
IMPACT ON MEDICAL EDUCATION
Since 1984, several thousand medical students; internal medicine residents; geriatric fellows; and N P, social work, and pharmacy students have participated in the VCU House Calls program and have come to see home care as a viable care model. House calls have been mandatory in the VCU School of Medicine curriculum since 2002. Qualitative evidence from these encounters demonstrates that learners value the experience and gain a better understanding of health care as a result.
Medical students’ interest in geriatrics is low,26,27 but positive, intense, or unique experiences with elders, and interactions with positive role models may improve the outlook for the specialty. The home setting gives learners an opportunity to observe the care of medically complex patients in the community, exposes the students to the team of professionals needed for comprehensive care, and enhances learners’ awareness of the challenges in providing continuity of care for this population.
We previously reported on a qualitative study of comments of second-year medical students who participated in our House Calls program.28 Students frequently noted the apparent comfort and positive attitude of the patients; the dedication, patience, compassion, commitment, and hard work of the caregivers; and the personalized and comprehensive care provided. The students identified both the challenges and the rewards for the doctors and expressed increased interest in conducting house calls in the future.
The training of competent and caring physicians and other health professionals is the goal of medical education. Fourth-year medical students were surveyed nationally regarding the qualities of a humanistic doctor.29 The students noted the importance of role models and participatory experiences. House calls provide an opportunity for learners to see health care in the community. Such experiences can create a memorable lesson in care delivery and in doctor-patient-caregiver relationships.
PALLIATIVE CARE AND HOME CARE
Ideally, care plans would gradually shift in focus from curative therapy to palliative care as patients with significant chronic illness advance in age and debility. In our geriatric practice, palliation is always important throughout extended chronic illness. Care plans progress and palliation becomes the primary focus in the final months of life. This transition may take years. Hospice referral is frequently a final step because the payment system reimburses for comprehensive team-based hospice care only when life expectancy is less than 6 months. The reason for this is economic: comprehensive team-based care is costly, and lengthening the hospice benefit as it is now structured could be prohibitively expensive. Our patients may live for years in a state of advanced debility, yet need intensive team care only at intervals. Optimally, the care model, team intensity, and related payments should flex with clinical need. This is what we have experienced by making house calls the mode of longitudinal primary care delivery, supported by our institution. Our teams help patients and families shift focus and decide when to accept hospice care; this requires more art than science and usually involves a gradual process of adaptation.
Our approach is consistent with the definition of palliative care published by the Centers for Medicare & Medicaid Services in 2008: patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care addresses physical, intellectual, emotional, social, and spiritual needs and facilitates patient autonomy, access to information, and choice.30 Geriatric clinicians seek to help patients and families maximize quality of life and to maintain function by focusing on symptom management and clarification of patient and family goals rather than on specific diseases. This approach is applied without regard to patient age, condition, or stage of disease, and it can coexist with curative treatments. Thus it is distinguished in concept from “what we do when there is nothing more we can do.”31
In ways that are less clear when working in other care settings, home visits reveal patient goals, true rehabilitative potential, and family capacity for care-giving. Home visits take longer than office encounters, but make the provider’s job easier. By observing the patient at home, providers can better assess barriers to comfort and devise strategies to improve function, while also evaluating whether life is truly nearing the end. The home care clinician often engages in palliative care even if he or she did not initially intend to do so.
Furthermore, compared with the hospital or office setting, a home is more conducive to reasonably paced discussions about goals of care. Patients are more physically and emotionally comfortable and may talk more easily about potentially disturbing subjects. The clinician may be able to engage the patient by referring to pictures or mementos that help the patient to reflect on life values. And, a patient who is seen at home will more readily trust that the clinician places patients’ needs first. This opens the door to difficult discussions about code status, health care proxies, dialysis and ventilator support, or whether the patient would ever want to go to a hospital or a nursing home. Preferences change with time; patients ultimately feel less need to rely on ambulances and emergency care, given a timely response at home from a clinician who is familiar.32
Most dying patients are at home with their families during most of their final year of life; yet, despite studies showing that most patients prefer to die at home33–35 about 60% of all deaths still occur in the hospital.36 In our House Calls program’s experience, the percentage of patients who die at home is closer to 60.
Cherin and colleagues32 described a successful end-of-life home care program demonstrating a significant benefit to patients over usual care. The program integrated curative and palliative therapies. Similarly, Brumley and colleagues37 demonstrated that, compared with usual care, patients receiving in-home palliative care reported greater satisfaction, had fewer emergency department and hospital visits, and were more likely to die at home, with significantly lower overall costs. These findings conform to our experience. (Also see “Innovative models of home-based palliative care”)
CONCLUSION
Advanced home care with a strong medical component is an important part of the supportive and recuperative care options in the United States. For these programs to reach their full potential, we must expand on the successful in-home medical care models and create responsible financing methods that control overall costs while rewarding providers appropriately. We must broaden the application of portable and information technologies and develop an interdisciplinary workforce. These approaches will lead us toward our overall goals of optimal care at minimal cost.
- Levine SA, Boal J, Boling PA. Home care. JAMA 2003; 290:1203–1207.
- Boling PA. Home care: the first option. In:Cornwell T, Schwartz-berg JGeds. Medical Management of the Home Care Patient: Guidelines for Physicians. 4th ed. Chicago, IL: American Medical Association; 2012:1–14.
- Brickner PW, Duque Sister Teresita, Kaufman A, et al. The homebound aged: a medically unreached group. Ann Intern Med 1975; 82:1–6.
- Boling PA. Effects of policy, reimbursement, and regulation on home health care. In:Olson S. The Role of Human Factors in Home Health Care: Workshop Summary. Washington, DC: The National Academies Press; 2010:275–302.
- Basic statistics about home care. National Association for Home Care & Hospice Web site. http://www.nahc.org/facts/10HC_Stats.pdf. Updated 2010. Accessed October 11, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Shaughnessy PW, Hittle DF, Crisler KS, et al. Improving patient outcomes of home health care: findings from two demonstration trials of outcome-based quality improvement. J Am Geriatr Soc 2002; 50:1354–1364.
- Smigelski C, Hungate B, Holdren J, Goodloe L, Boling PA. Transitional model of care at VCU Medical Center—6 years’ experience. J Am Geriatr Soc 2008; 56 suppl 1:S197.
- Naylor MD. Advancing high value transitional care: the central role of nursing and its leadership. Nurs Adm Q 2012; 36:115–126.
- Holtz-Eakin D. High-cost medicare beneficiaries. Congressional Budget Office Web site. http://www.cbo.gov/sites/default/fles/cbofles/ftpdocs/63xx/doc6332/05-03-medispending.pdf. Published May 2005. Accessed October 11, 2012.
- Cryer L, Shannon SB, Van Amsterdam M, Leff B. Costs for “hospital at home” patients were 19 percent lower, with equal or better outcomes compared to similar inpatients. Health Aff (Millwood) 2012; 31:1237–1243.
- Leff B, Burton L, Mader SL, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Aujesky D, Roy PM, Verschuren F, et al. Outpatient versus inpatient treatment for patients with acute pulmonary embolism: an international, open-label, randomised, non-inferiority trial [published online ahead of print June 22, 2011]. Lancet 2011; 378 9785:41–48. 10.1016/S0140-6736 1160824–6
- Büller HR, Prins MH, Lensing AWA, et al the EINSTEIN– PE Investigators. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Landers SH. Why health care is going home [published online ahead of print October 20, 2010]. N Engl J Med 2010; 363:1690–1691. 10.1056/NEJMp1000401
- Counsell SR, Callahan CM, Clark DO, et al. Geriatric care management for low-income seniors: a randomized controlled trial. JAMA 2007; 298:2623–2633.
- Beales JL, Edes T. Veteran’s Affairs home based primary care. Clin Geriatr Med 2009; 25:149–154.
- Kinosian B, Edes T, Davis D, Hossain M. Financial savings of home based primary care for frail veterans with chronic disabling disease. J Am Geriatr Soc 2010; 589 (suppl s1):S3. Abstract P7.
- DeJonge KE, Taler G, Boling PA. Independence at home: community-based care for older adults with severe chronic illness. Clin Geriatr Med 2009; 25:155–169.
- Chaudhry SI, Mattera JA, Curtis JP, et al. Telemonitoring in patients with heart failure [published online ahead of print November 16, 2010]. N Engl J Med 2010; 363:2301–2309. 10.1056/NEJMoa1010029
- Wieland D, Boland R, Baskins J, Kinosian B. Five-year survival in a Program of All-inclusive Care for Elderly compared with alternative institutional and home- and community-based care [published online ahead of print March 30, 2010]. J Gerontol A Biol Sci Med Sci 2010; 65:721–726. 10.1093/gerona/glq040
- Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives [published online ahead of print May 30, 2006]. J Am Med Dir Assoc 2006; 7:339–344. 10.1016/j.jamda.2006.01.022
- Meret-Hanke LA. Effects of the Program of All-inclusive Care for the Elderly on hospital use [published online ahead of print July 6, 2011]. Gerontologist 2011; 51:774–785. 10.1093/geront/gnr040
- Fitzgerald JT, Wray LA, Halter JB, Williams BC, Supiano MA. Relating medical students’ knowledge, attitudes, and experience to an interest in geriatric medicine. Gerontologist 2003; 43:849–855.
- Voogt SJ, Mickus M, Santiago O, Herman SE. Attitudes, experiences, and interest in geriatrics of first-year allopathic and osteopathic medical students [published online ahead of print December 11, 2007]. J Am Geriatr Soc 2008; 56:339–344. 10.1111/j.1532-5415.2007.01541.
- Abbey L, Willett R, Selby-Penczak R, McKnight R. Social learning: medical student perceptions of geriatric house calls. Gerontol Geriatr Educ 2010; 31:149–162.
- Moyer CA, Arnold L, Quaintance J, et al. What factors create a humanistic doctor? A nationwide survey of fourth-year medical students. Acad Med 2010; 85:1800–1807.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid programs: hospice conditions of participation. Federal Register 2008; 73:32204–32220.
- Meier DE, Brawley OW. Palliative care and the quality of life [published online ahead of print June 13, 2011]. J Clin Oncol 2011; 29:2750–2752. 10.1200/JCO.2011.35.9729
- Cherin DA, Enguidanos SM, Jamison P. Physicians as medical center “extenders” in end-of-life care: physician home visits as the lynch pin in creating an end-of-life care system. Home Health Care Serv Q 2004; 23:41–53.
- Hays JC, Galanos AN, Palmer TA, McQuoid DR, Flint EP. Preference for place of death in a continuing care retirement community. Gerontologist 2001; 41:123–128.
- Karlsen S, Addington-Hall J. How do cancer patients who die at home differ from those who die elsewhere? Palliat Med 1998; 12:279–286.
- Townsend J, Frank AO, Fermont D, et al. Terminal cancer care and patients’ preference for place of death: a prospective study. BMJ 1990; 301:415–417.
- Weitzen S, Teno JM, Fennell M, Mor V. Factors associated with site of death: a national study of where people die. Med Care 2003; 41:323–335.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
When it can be done safely, most people prefer to be treated and recover from illness at home.1,2 Home-based services have improved considerably since Brickner called the homebound aged “a medically unreached group.”3 Still, home care has not achieved its full potential and scientific investigation of home care models is scant compared with that of other therapeutic approaches.
The challenges of studying home care include variability in interventions, difficulty defining treatment and comparison groups, and high research costs. The care itself can be demanding, requiring providers to mobilize processes that have become institution-based and immobile, integrate care across insular settings, incorporate complex social issues into the care plan, and develop a viable home care financing model.
This article reviews evidence favoring investment in advanced home care and adds perspective from 3 decades’ experience at Virginia Commonwealth University (VCU), Richmond, Virginia.
The term home care has a broad scope, ranging from basic support to highly technical care involving intravenous lines, ventilators, portable diagnostic tests, and remote monitors.4 Patients cared for at home range from those who are ambulatory to those who are permanently bedfast and seriously ill. The home care user population can be categorized based on the types of health care resources they consume (Table 1). Much attention has been paid to home-based care during recuperation after acute illness. The aim has been to foster recovery and prevent further need for institutional care. Lately the term transitional care has been used in this context.
TRANSITIONAL CARE
Transitional care has long been a priority for visiting nurse agencies. In 1965, Medicare Part A, building from the tradition of urban parish nursing services, created an interdisciplinary industry. Medicare now certifies more than 10,000 agencies with more than 250,000 professional staff.5 For several reasons, beginning in the 1970s, US physicians have become less integrated into in-home care. Despite this and the challenge of managing medically complex patients with minimal active physician involvement, home health agencies provide a vital service. Further, they have demonstrated improved outcomes and cost savings.
Transitional care refers to specialized, short-term care for selected high-risk patients after an acute illness. The original objective of transitional care was to reduce hospital readmissions. Tested models include an approach developed by Coleman et al,6 based on four pillars: assistance with medication self-management, patient-centered and -owned medical record, timely follow-up with primary or specialty care, and “red flags” that indicate a worsening condition. This model, which yielded one-third fewer hospital readmissions and a savings of about $500 per patient in 6 months, is being adopted in many locations nationally.
Naylor and colleagues7,8 collaborated with hospital-based nurse practitioners (NPs) for 2 decades on a more intensive model. In the Naylor model, the NPs form a health care bridge from hospital to home for 4 weeks after hospital care and add an active medical care component to the home care team. Naylor et al7 reported a 50% reduction in the rehospitalization rate and a cost savings of approximately $3,000 per patient over 24 weeks. Naylor’s team observed these results among frail, elderly patients with a variety of conditions and comorbidities. The 2010 federal health care reform law as well as state and private insurer initiatives now encourage use of this and other integrated care models.
In a national demonstration program using performance improvement methods and careful data collection, 73 US home health agencies improved targeted clinical outcomes and reduced hospitalizations from baseline rates by approximately 7% within 3 to 4 years.9 The study included approximately 158,000 patients in the intervention group and 249,000 in the comparison group. However, in general the success demonstrated in this study has not been reflected nationally, and home health agencies have been weakly integrated with the remainder of the health care delivery system.
Medicare home health agency care has evolved rapidly in the past 15 years, with reporting of numerous quality measures that has created direct accountability of physicians to the public. Until as recently as the 1990s, many important measures of quality in medicine were available only to physicians and physician and hospital organizations through governmental and, in some cases, legal routes. This new quality-based accountability, along with fiscal pressure to reduce lengths of stay and to limit visits under prospective payment, are among the changes that are transforming the home health industry.
THE VCU TRANSITIONAL CARE EXPERIENCE
The VCU Medical Center implemented a Naylor-model hospital-based transitional care program (TCP) 12 years ago that has served more than 500 patients. Targeted patients have histories similar to those observed by Naylor et al7: multiple hospitalizations, prolonged inpatient stays, many comorbidities and medications, complex care plans, and poor social support. Referrals come from physician teams, care coordinators, nurses, and social workers. The electronic medical record (EMR) has triggers for referrals.
Transitional care NPs meet patients in the hospital to ensure the appropriateness of their referral, introduce the program, and verify information. As shown in the Naylor model and later in the Coleman model,6 inpatient contact creates rapport with the patient and with family caregivers.
The first home visit is made on a weekday within 24 to 72 hours of discharge. At this initial visit, which takes a considerable amount of time, we attempt to reconcile medications, clarify social needs and resources, conduct physical assessments, modify medical regimens, educate the patient and his or her caregivers, and run diagnostic laboratory tests as needed. What we see in the home on this first visit often does not correspond with what was previously reported by hospital-based clinicians. For example, we have found that many patients are not taking medications as prescribed.
Typically, we visit homes weekly for 4 to 8 weeks. Some patients remain in transitional care for longer periods due to medical and social reasons. The NPs maintain close contact with home health agency staff via mobile phones. In some cases we conduct joint visits with home health agency staff in order to facilitate adjustments to medical care plans. Regular communication with primary care providers via the EMR, fax, and phone helps close the follow-up gap. The NP’s ability to observe the home setting, identify barriers to medical compliance (including literacy), and address social issues offers a clearer picture to care providers and fosters better outcomes. As patients improve and become more mobile, they return to the care of the primary provider.
Positive results with some limitations
We collected data between 2003 and 2006 on patients enrolled in the VCU Medical Center TCP. Our demographic results were similar to those reported by Naylor et al.7 Prevalent diseases included heart failure (HF), coronary artery disease, diabetes, and chronic obstructive pulmonary disease (COPD). The mean age was 71 years. The patient population was 63% female and 77% African American. About 73% of patients returned to the care of their primary physicians, 13% enrolled in the VCU House Calls program, 12% died, and 3% were admitted to nursing homes.10
A comparison of utilization data for 199 patients 6 months before and after their enrollment in the TCP over a period of 4 years showed decreased use of hospital resources—ie, fewer inpatient days, shorter lengths of stay, and fewer intensive care unit days— after enrollment. Aggregate cost after TCP enrollment reduction was $2,251,34410 which is 38% less than the 6-month pre-enrollment baseline (Table 2). Regression to the mean played a role, but most patients had a sustained high-use pattern for 6 months before enrollment. The high rate of consumption of health care resources dropped quickly following implementation of the TCP and stayed down for many months.
We largely concur with Naylor’s description of transitional care implementation.11 However, we have found that many transitional care patients are unable return to the clinic after 2 months, as suggested by Naylor. In our system, these patients default to our House Calls program for continuing care. Thus, in our estimation, transitional care is an important but incomplete response to population-based health needs. Supporting this conclusion is the Congressional Budget Office report, which states that among high-cost Medicare patients in an index year (2001), those who lived for 5 years were high-cost patients on a month-by-month basis in 22 of the next 60 months, reflecting chronic illness and cyclical service use patterns.12
Extension of the TCP to outpatients
Because of the favorable effect observed in the hospital-based TCP, we created a role for transitional care in our outpatient geriatric practice. Transitional care NPs from the clinic practice have the option of making home visits in a variety of scenarios. In the least serious cases a single “diagnostic” home visit provides invaluable insight. For example, we evaluate support systems and compliance with medication instructions and put systems in place to help patients maintain independence and safety at home, including nutrition and fall prevention programs. Patients with poor social support benefit especially from home visits.
We find that high-risk patients recently discharged from facilities, including those outside our health system, benefit from NP visits. When a high-risk clinic patient is hospitalized, we maintain a connection with the inpatient team, follow the patient’s progress, and assist with discharge planning. Based on our relationship with the patient prior to admission, we are able to anticipate problems and to address them promptly after discharge. The NP functions as the “hub of the wheel” to coordinate the multidisciplinary plan among primary care providers; specialists; and support services such as home health, social work, and physical therapy.
We also initiate periodic NP home visits as chronic diseases progress and as clinic patients become increasingly frail. Interim visits are made to monitor the medical plan and perform follow-up blood testing. Once patients are no longer able to use the office practice, they transition into the House Calls program.
HOSPITAL AT HOME
The ultimate in substitutive, intensive home care occurs when one replaces acute care hospital admission with care delivered entirely at home. Robust research has shown comparable or better clinical outcomes with fewer complications and lower costs when home care is applied to common conditions such as pneumonia, COPD, cellulitis, and HF.13,14 Rapidly advancing technology now supports increasingly sophisticated care at home. For example, with low molecular weight heparin, the care of deep vein thrombosis and stable pulmonary embolism—which always required inpatient care 25 years ago—can now be delivered entirely at home in many cases. Soon, these conditions may be managed solely with oral medication.15,16 The range of conditions that are now being managed at home is extensive, and the transformation of health care by portable technology is just beginning.17
LONGITUDINAL IN-HOME PRIMARY CARE
In the United States, patients who are immobile and cannot easily access office-based care often suffer with suboptimal mobile primary care. This represents a major limitation in care access for these patients. There is good evidence that longitudinal medical care, primarily delivered at home for periods lasting many months to several years, is effective and that it makes clinical sense. In the home, providers can accurately assess the patient’s living situation, engender trust, and respond in a timely manner when a patient’s condition changes. The Geriatric Resources for Assessment and Care of Elders (GRACE) program and the Veterans Affairs (VA) home-based primary care model are two examples of the benefits of longitudinal in-home care.
In the GRACE model, patients receive comprehensive in-home assessment by NPs with quarterly follow-up, and recommendations are given to primary care providers. The program’s clinical trial demonstrated markedly improved treatment of a variety of common geriatric ailments and reduced costs in a high-risk subset of patients.18 GRACE was not designed for urgent care but the approach was linked to lower costs in high-risk cases, likely due to better care and improved access.
The VA home-based primary care model has grown rapidly in the past decade, now operating at more than 200 medical centers, each with a full interprofessional team. House calls by physicians and NPs are part of the model, although the frequency varies across sites. Every team includes actively engaged physicians. Medicoeconomic evaluation based on tens of thousands of patient-years has shown an overall reduction in health care costs of 15% to 25% compared with historical values and prospectively modeled dollars.19,20 Home-based primary care teams are emerging across the United States at many academic centers and in the private sector.
To fund comprehensive longitudinal home care services for patients with complex health problems, the Independence at Home21 demonstration program was created under section 3024 of the Patient Protection and Affordable Care Act, using robust gain-sharing from demonstrated cost savings to reward house call teams. This multisite 3-year program started in June 2012. Rapid growth of this model is likely as private insurers have also taken an active interest in mobile medical care designs, using a variety of reward structures.
TELEMEDICINE
A debate continues over the use of communication technology in home care. It seems intuitive that “virtual visits” would be more efficient than clinicians visiting patients at home. Yet, the challenges of improving care by telemedicine alone are underestimated. For example, a recent large randomized trial, in which 33 cardiology practice sites provided at-home postdis-charge telemonitoring for HF patients, demonstrated no difference in clinical outcomes compared with patients monitored in the hospital or clinic.22
Proponents of telemedicine cite integrated models where data are managed proactively by a physician-led team that is engaged in care. This view seems valid, but other than anecdotal reports from integrated health systems, the published evidence of reduced costs is sparse. Some combination of in-person care and telemedicine is likely to be the optimal design and will emerge in coming years.
PACE: SYSTEM-BASED HOME CARE
In the 1980s, health maintenance organization risk contracts seemed a likely context for developing advanced home care models, but this did not happen. However, the Program for All-Inclusive Care of the Elderly (PACE) was tested and became a defined federal benefit in 1997. There are now nearly 100 PACE centers nationwide. PACE offers comprehensive care for people aged 55 years and older who are nursing home–eligible. The program appears to effectively help people stay home.23
An interdisciplinary team (IDT) coordinates PACE medical and social services to promote independence and quality of life. The program has been referred to as “a nursing home without walls.” Services include primary and specialty care, adult day care, case management, nursing, home health care, assistance with activities of daily living (ADL), medications, social work, rehabilitation, hospitalization, nursing facility care, nutritional support, caregiver respite, and transportation to and from the PACE adult day health center (ADHC) and medical appointments. The ADHC is the cornerstone and coordinating center for most care provided to PACE participants. Home-based care is provided in several ways:
- Home nursing care may be provided by external agencies, including skilled care, personal care, and hospice care, under contract with PACE. In Richmond, the home care manager oversees care after it is approved by the IDT. Weekly hours of care are changed often according to the participant’s need (eg, increased hours after hospital discharge and decreased hours when a family member visits and can provide more care). Home care provides assistance with ADLs and instrumental ADLs; “sitter” services are provided at the ADHC.
- The program supports home modifications and provides durable medical equipment (DME). Assessment is done by one or more team members upon enrollment and then at least every 6 months. PACE provides all DME the participant needs to remain safely in the community. At disenrollment or death, some equipment can be returned to PACE after review by the rehabilitation department.
- Primary care, basic laboratory services, and medical specialty care can be provided to the participant at home if for any reason he or she is unable to travel to the ADHC. PACE physicians make house calls to better understand patients’ living situations and needs. On-call nurses make home visits after hours or on weekends for clinical assessments, point-of-care diagnostic testing, specimen collection (stool or urine), and participant and family education on proper use of medications or equipment.
- As PACE participants approach the end of life, they transition to a palliative care model. A decision is made by the family and the IDT to discontinue attendance at the ADHC and to focus on care at home,24 allowing the participant to spend the last days or weeks in the relative comfort of home. Nurses make home visits when needed and educate families on symptom palliation.
- Additional in-home respite services can be provided to decrease caregiver burden.
- Skilled rehabilitation services are delivered either at home or in the ADHC depending on the judgment of the rehabilitation department and the IDT. The PACE site offers advanced transportation and full onsite therapy services 5 days per week.
The PACE sites become the insurers, receive defined capitation payments from Medicare and Medicaid that are adjusted for patient complexity, and assume the risk for all health care costs. Because of a 5% withholding in the capitation amount relative to projected Medicare expenses, PACE should reduce governmental costs. PACE must provide or pay for all usual Medicare and Medicaid services, and it may provide other services deemed necessary by the PACE team. Within PACE, hospital use is markedly reduced compared with conventional Medicare,25 and home care is one of several strategies employed. The PACE experience shows that care can be safely shifted from hospitals to other settings.
IMPACT ON MEDICAL EDUCATION
Since 1984, several thousand medical students; internal medicine residents; geriatric fellows; and N P, social work, and pharmacy students have participated in the VCU House Calls program and have come to see home care as a viable care model. House calls have been mandatory in the VCU School of Medicine curriculum since 2002. Qualitative evidence from these encounters demonstrates that learners value the experience and gain a better understanding of health care as a result.
Medical students’ interest in geriatrics is low,26,27 but positive, intense, or unique experiences with elders, and interactions with positive role models may improve the outlook for the specialty. The home setting gives learners an opportunity to observe the care of medically complex patients in the community, exposes the students to the team of professionals needed for comprehensive care, and enhances learners’ awareness of the challenges in providing continuity of care for this population.
We previously reported on a qualitative study of comments of second-year medical students who participated in our House Calls program.28 Students frequently noted the apparent comfort and positive attitude of the patients; the dedication, patience, compassion, commitment, and hard work of the caregivers; and the personalized and comprehensive care provided. The students identified both the challenges and the rewards for the doctors and expressed increased interest in conducting house calls in the future.
The training of competent and caring physicians and other health professionals is the goal of medical education. Fourth-year medical students were surveyed nationally regarding the qualities of a humanistic doctor.29 The students noted the importance of role models and participatory experiences. House calls provide an opportunity for learners to see health care in the community. Such experiences can create a memorable lesson in care delivery and in doctor-patient-caregiver relationships.
PALLIATIVE CARE AND HOME CARE
Ideally, care plans would gradually shift in focus from curative therapy to palliative care as patients with significant chronic illness advance in age and debility. In our geriatric practice, palliation is always important throughout extended chronic illness. Care plans progress and palliation becomes the primary focus in the final months of life. This transition may take years. Hospice referral is frequently a final step because the payment system reimburses for comprehensive team-based hospice care only when life expectancy is less than 6 months. The reason for this is economic: comprehensive team-based care is costly, and lengthening the hospice benefit as it is now structured could be prohibitively expensive. Our patients may live for years in a state of advanced debility, yet need intensive team care only at intervals. Optimally, the care model, team intensity, and related payments should flex with clinical need. This is what we have experienced by making house calls the mode of longitudinal primary care delivery, supported by our institution. Our teams help patients and families shift focus and decide when to accept hospice care; this requires more art than science and usually involves a gradual process of adaptation.
Our approach is consistent with the definition of palliative care published by the Centers for Medicare & Medicaid Services in 2008: patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care addresses physical, intellectual, emotional, social, and spiritual needs and facilitates patient autonomy, access to information, and choice.30 Geriatric clinicians seek to help patients and families maximize quality of life and to maintain function by focusing on symptom management and clarification of patient and family goals rather than on specific diseases. This approach is applied without regard to patient age, condition, or stage of disease, and it can coexist with curative treatments. Thus it is distinguished in concept from “what we do when there is nothing more we can do.”31
In ways that are less clear when working in other care settings, home visits reveal patient goals, true rehabilitative potential, and family capacity for care-giving. Home visits take longer than office encounters, but make the provider’s job easier. By observing the patient at home, providers can better assess barriers to comfort and devise strategies to improve function, while also evaluating whether life is truly nearing the end. The home care clinician often engages in palliative care even if he or she did not initially intend to do so.
Furthermore, compared with the hospital or office setting, a home is more conducive to reasonably paced discussions about goals of care. Patients are more physically and emotionally comfortable and may talk more easily about potentially disturbing subjects. The clinician may be able to engage the patient by referring to pictures or mementos that help the patient to reflect on life values. And, a patient who is seen at home will more readily trust that the clinician places patients’ needs first. This opens the door to difficult discussions about code status, health care proxies, dialysis and ventilator support, or whether the patient would ever want to go to a hospital or a nursing home. Preferences change with time; patients ultimately feel less need to rely on ambulances and emergency care, given a timely response at home from a clinician who is familiar.32
Most dying patients are at home with their families during most of their final year of life; yet, despite studies showing that most patients prefer to die at home33–35 about 60% of all deaths still occur in the hospital.36 In our House Calls program’s experience, the percentage of patients who die at home is closer to 60.
Cherin and colleagues32 described a successful end-of-life home care program demonstrating a significant benefit to patients over usual care. The program integrated curative and palliative therapies. Similarly, Brumley and colleagues37 demonstrated that, compared with usual care, patients receiving in-home palliative care reported greater satisfaction, had fewer emergency department and hospital visits, and were more likely to die at home, with significantly lower overall costs. These findings conform to our experience. (Also see “Innovative models of home-based palliative care”)
CONCLUSION
Advanced home care with a strong medical component is an important part of the supportive and recuperative care options in the United States. For these programs to reach their full potential, we must expand on the successful in-home medical care models and create responsible financing methods that control overall costs while rewarding providers appropriately. We must broaden the application of portable and information technologies and develop an interdisciplinary workforce. These approaches will lead us toward our overall goals of optimal care at minimal cost.
When it can be done safely, most people prefer to be treated and recover from illness at home.1,2 Home-based services have improved considerably since Brickner called the homebound aged “a medically unreached group.”3 Still, home care has not achieved its full potential and scientific investigation of home care models is scant compared with that of other therapeutic approaches.
The challenges of studying home care include variability in interventions, difficulty defining treatment and comparison groups, and high research costs. The care itself can be demanding, requiring providers to mobilize processes that have become institution-based and immobile, integrate care across insular settings, incorporate complex social issues into the care plan, and develop a viable home care financing model.
This article reviews evidence favoring investment in advanced home care and adds perspective from 3 decades’ experience at Virginia Commonwealth University (VCU), Richmond, Virginia.
The term home care has a broad scope, ranging from basic support to highly technical care involving intravenous lines, ventilators, portable diagnostic tests, and remote monitors.4 Patients cared for at home range from those who are ambulatory to those who are permanently bedfast and seriously ill. The home care user population can be categorized based on the types of health care resources they consume (Table 1). Much attention has been paid to home-based care during recuperation after acute illness. The aim has been to foster recovery and prevent further need for institutional care. Lately the term transitional care has been used in this context.
TRANSITIONAL CARE
Transitional care has long been a priority for visiting nurse agencies. In 1965, Medicare Part A, building from the tradition of urban parish nursing services, created an interdisciplinary industry. Medicare now certifies more than 10,000 agencies with more than 250,000 professional staff.5 For several reasons, beginning in the 1970s, US physicians have become less integrated into in-home care. Despite this and the challenge of managing medically complex patients with minimal active physician involvement, home health agencies provide a vital service. Further, they have demonstrated improved outcomes and cost savings.
Transitional care refers to specialized, short-term care for selected high-risk patients after an acute illness. The original objective of transitional care was to reduce hospital readmissions. Tested models include an approach developed by Coleman et al,6 based on four pillars: assistance with medication self-management, patient-centered and -owned medical record, timely follow-up with primary or specialty care, and “red flags” that indicate a worsening condition. This model, which yielded one-third fewer hospital readmissions and a savings of about $500 per patient in 6 months, is being adopted in many locations nationally.
Naylor and colleagues7,8 collaborated with hospital-based nurse practitioners (NPs) for 2 decades on a more intensive model. In the Naylor model, the NPs form a health care bridge from hospital to home for 4 weeks after hospital care and add an active medical care component to the home care team. Naylor et al7 reported a 50% reduction in the rehospitalization rate and a cost savings of approximately $3,000 per patient over 24 weeks. Naylor’s team observed these results among frail, elderly patients with a variety of conditions and comorbidities. The 2010 federal health care reform law as well as state and private insurer initiatives now encourage use of this and other integrated care models.
In a national demonstration program using performance improvement methods and careful data collection, 73 US home health agencies improved targeted clinical outcomes and reduced hospitalizations from baseline rates by approximately 7% within 3 to 4 years.9 The study included approximately 158,000 patients in the intervention group and 249,000 in the comparison group. However, in general the success demonstrated in this study has not been reflected nationally, and home health agencies have been weakly integrated with the remainder of the health care delivery system.
Medicare home health agency care has evolved rapidly in the past 15 years, with reporting of numerous quality measures that has created direct accountability of physicians to the public. Until as recently as the 1990s, many important measures of quality in medicine were available only to physicians and physician and hospital organizations through governmental and, in some cases, legal routes. This new quality-based accountability, along with fiscal pressure to reduce lengths of stay and to limit visits under prospective payment, are among the changes that are transforming the home health industry.
THE VCU TRANSITIONAL CARE EXPERIENCE
The VCU Medical Center implemented a Naylor-model hospital-based transitional care program (TCP) 12 years ago that has served more than 500 patients. Targeted patients have histories similar to those observed by Naylor et al7: multiple hospitalizations, prolonged inpatient stays, many comorbidities and medications, complex care plans, and poor social support. Referrals come from physician teams, care coordinators, nurses, and social workers. The electronic medical record (EMR) has triggers for referrals.
Transitional care NPs meet patients in the hospital to ensure the appropriateness of their referral, introduce the program, and verify information. As shown in the Naylor model and later in the Coleman model,6 inpatient contact creates rapport with the patient and with family caregivers.
The first home visit is made on a weekday within 24 to 72 hours of discharge. At this initial visit, which takes a considerable amount of time, we attempt to reconcile medications, clarify social needs and resources, conduct physical assessments, modify medical regimens, educate the patient and his or her caregivers, and run diagnostic laboratory tests as needed. What we see in the home on this first visit often does not correspond with what was previously reported by hospital-based clinicians. For example, we have found that many patients are not taking medications as prescribed.
Typically, we visit homes weekly for 4 to 8 weeks. Some patients remain in transitional care for longer periods due to medical and social reasons. The NPs maintain close contact with home health agency staff via mobile phones. In some cases we conduct joint visits with home health agency staff in order to facilitate adjustments to medical care plans. Regular communication with primary care providers via the EMR, fax, and phone helps close the follow-up gap. The NP’s ability to observe the home setting, identify barriers to medical compliance (including literacy), and address social issues offers a clearer picture to care providers and fosters better outcomes. As patients improve and become more mobile, they return to the care of the primary provider.
Positive results with some limitations
We collected data between 2003 and 2006 on patients enrolled in the VCU Medical Center TCP. Our demographic results were similar to those reported by Naylor et al.7 Prevalent diseases included heart failure (HF), coronary artery disease, diabetes, and chronic obstructive pulmonary disease (COPD). The mean age was 71 years. The patient population was 63% female and 77% African American. About 73% of patients returned to the care of their primary physicians, 13% enrolled in the VCU House Calls program, 12% died, and 3% were admitted to nursing homes.10
A comparison of utilization data for 199 patients 6 months before and after their enrollment in the TCP over a period of 4 years showed decreased use of hospital resources—ie, fewer inpatient days, shorter lengths of stay, and fewer intensive care unit days— after enrollment. Aggregate cost after TCP enrollment reduction was $2,251,34410 which is 38% less than the 6-month pre-enrollment baseline (Table 2). Regression to the mean played a role, but most patients had a sustained high-use pattern for 6 months before enrollment. The high rate of consumption of health care resources dropped quickly following implementation of the TCP and stayed down for many months.
We largely concur with Naylor’s description of transitional care implementation.11 However, we have found that many transitional care patients are unable return to the clinic after 2 months, as suggested by Naylor. In our system, these patients default to our House Calls program for continuing care. Thus, in our estimation, transitional care is an important but incomplete response to population-based health needs. Supporting this conclusion is the Congressional Budget Office report, which states that among high-cost Medicare patients in an index year (2001), those who lived for 5 years were high-cost patients on a month-by-month basis in 22 of the next 60 months, reflecting chronic illness and cyclical service use patterns.12
Extension of the TCP to outpatients
Because of the favorable effect observed in the hospital-based TCP, we created a role for transitional care in our outpatient geriatric practice. Transitional care NPs from the clinic practice have the option of making home visits in a variety of scenarios. In the least serious cases a single “diagnostic” home visit provides invaluable insight. For example, we evaluate support systems and compliance with medication instructions and put systems in place to help patients maintain independence and safety at home, including nutrition and fall prevention programs. Patients with poor social support benefit especially from home visits.
We find that high-risk patients recently discharged from facilities, including those outside our health system, benefit from NP visits. When a high-risk clinic patient is hospitalized, we maintain a connection with the inpatient team, follow the patient’s progress, and assist with discharge planning. Based on our relationship with the patient prior to admission, we are able to anticipate problems and to address them promptly after discharge. The NP functions as the “hub of the wheel” to coordinate the multidisciplinary plan among primary care providers; specialists; and support services such as home health, social work, and physical therapy.
We also initiate periodic NP home visits as chronic diseases progress and as clinic patients become increasingly frail. Interim visits are made to monitor the medical plan and perform follow-up blood testing. Once patients are no longer able to use the office practice, they transition into the House Calls program.
HOSPITAL AT HOME
The ultimate in substitutive, intensive home care occurs when one replaces acute care hospital admission with care delivered entirely at home. Robust research has shown comparable or better clinical outcomes with fewer complications and lower costs when home care is applied to common conditions such as pneumonia, COPD, cellulitis, and HF.13,14 Rapidly advancing technology now supports increasingly sophisticated care at home. For example, with low molecular weight heparin, the care of deep vein thrombosis and stable pulmonary embolism—which always required inpatient care 25 years ago—can now be delivered entirely at home in many cases. Soon, these conditions may be managed solely with oral medication.15,16 The range of conditions that are now being managed at home is extensive, and the transformation of health care by portable technology is just beginning.17
LONGITUDINAL IN-HOME PRIMARY CARE
In the United States, patients who are immobile and cannot easily access office-based care often suffer with suboptimal mobile primary care. This represents a major limitation in care access for these patients. There is good evidence that longitudinal medical care, primarily delivered at home for periods lasting many months to several years, is effective and that it makes clinical sense. In the home, providers can accurately assess the patient’s living situation, engender trust, and respond in a timely manner when a patient’s condition changes. The Geriatric Resources for Assessment and Care of Elders (GRACE) program and the Veterans Affairs (VA) home-based primary care model are two examples of the benefits of longitudinal in-home care.
In the GRACE model, patients receive comprehensive in-home assessment by NPs with quarterly follow-up, and recommendations are given to primary care providers. The program’s clinical trial demonstrated markedly improved treatment of a variety of common geriatric ailments and reduced costs in a high-risk subset of patients.18 GRACE was not designed for urgent care but the approach was linked to lower costs in high-risk cases, likely due to better care and improved access.
The VA home-based primary care model has grown rapidly in the past decade, now operating at more than 200 medical centers, each with a full interprofessional team. House calls by physicians and NPs are part of the model, although the frequency varies across sites. Every team includes actively engaged physicians. Medicoeconomic evaluation based on tens of thousands of patient-years has shown an overall reduction in health care costs of 15% to 25% compared with historical values and prospectively modeled dollars.19,20 Home-based primary care teams are emerging across the United States at many academic centers and in the private sector.
To fund comprehensive longitudinal home care services for patients with complex health problems, the Independence at Home21 demonstration program was created under section 3024 of the Patient Protection and Affordable Care Act, using robust gain-sharing from demonstrated cost savings to reward house call teams. This multisite 3-year program started in June 2012. Rapid growth of this model is likely as private insurers have also taken an active interest in mobile medical care designs, using a variety of reward structures.
TELEMEDICINE
A debate continues over the use of communication technology in home care. It seems intuitive that “virtual visits” would be more efficient than clinicians visiting patients at home. Yet, the challenges of improving care by telemedicine alone are underestimated. For example, a recent large randomized trial, in which 33 cardiology practice sites provided at-home postdis-charge telemonitoring for HF patients, demonstrated no difference in clinical outcomes compared with patients monitored in the hospital or clinic.22
Proponents of telemedicine cite integrated models where data are managed proactively by a physician-led team that is engaged in care. This view seems valid, but other than anecdotal reports from integrated health systems, the published evidence of reduced costs is sparse. Some combination of in-person care and telemedicine is likely to be the optimal design and will emerge in coming years.
PACE: SYSTEM-BASED HOME CARE
In the 1980s, health maintenance organization risk contracts seemed a likely context for developing advanced home care models, but this did not happen. However, the Program for All-Inclusive Care of the Elderly (PACE) was tested and became a defined federal benefit in 1997. There are now nearly 100 PACE centers nationwide. PACE offers comprehensive care for people aged 55 years and older who are nursing home–eligible. The program appears to effectively help people stay home.23
An interdisciplinary team (IDT) coordinates PACE medical and social services to promote independence and quality of life. The program has been referred to as “a nursing home without walls.” Services include primary and specialty care, adult day care, case management, nursing, home health care, assistance with activities of daily living (ADL), medications, social work, rehabilitation, hospitalization, nursing facility care, nutritional support, caregiver respite, and transportation to and from the PACE adult day health center (ADHC) and medical appointments. The ADHC is the cornerstone and coordinating center for most care provided to PACE participants. Home-based care is provided in several ways:
- Home nursing care may be provided by external agencies, including skilled care, personal care, and hospice care, under contract with PACE. In Richmond, the home care manager oversees care after it is approved by the IDT. Weekly hours of care are changed often according to the participant’s need (eg, increased hours after hospital discharge and decreased hours when a family member visits and can provide more care). Home care provides assistance with ADLs and instrumental ADLs; “sitter” services are provided at the ADHC.
- The program supports home modifications and provides durable medical equipment (DME). Assessment is done by one or more team members upon enrollment and then at least every 6 months. PACE provides all DME the participant needs to remain safely in the community. At disenrollment or death, some equipment can be returned to PACE after review by the rehabilitation department.
- Primary care, basic laboratory services, and medical specialty care can be provided to the participant at home if for any reason he or she is unable to travel to the ADHC. PACE physicians make house calls to better understand patients’ living situations and needs. On-call nurses make home visits after hours or on weekends for clinical assessments, point-of-care diagnostic testing, specimen collection (stool or urine), and participant and family education on proper use of medications or equipment.
- As PACE participants approach the end of life, they transition to a palliative care model. A decision is made by the family and the IDT to discontinue attendance at the ADHC and to focus on care at home,24 allowing the participant to spend the last days or weeks in the relative comfort of home. Nurses make home visits when needed and educate families on symptom palliation.
- Additional in-home respite services can be provided to decrease caregiver burden.
- Skilled rehabilitation services are delivered either at home or in the ADHC depending on the judgment of the rehabilitation department and the IDT. The PACE site offers advanced transportation and full onsite therapy services 5 days per week.
The PACE sites become the insurers, receive defined capitation payments from Medicare and Medicaid that are adjusted for patient complexity, and assume the risk for all health care costs. Because of a 5% withholding in the capitation amount relative to projected Medicare expenses, PACE should reduce governmental costs. PACE must provide or pay for all usual Medicare and Medicaid services, and it may provide other services deemed necessary by the PACE team. Within PACE, hospital use is markedly reduced compared with conventional Medicare,25 and home care is one of several strategies employed. The PACE experience shows that care can be safely shifted from hospitals to other settings.
IMPACT ON MEDICAL EDUCATION
Since 1984, several thousand medical students; internal medicine residents; geriatric fellows; and N P, social work, and pharmacy students have participated in the VCU House Calls program and have come to see home care as a viable care model. House calls have been mandatory in the VCU School of Medicine curriculum since 2002. Qualitative evidence from these encounters demonstrates that learners value the experience and gain a better understanding of health care as a result.
Medical students’ interest in geriatrics is low,26,27 but positive, intense, or unique experiences with elders, and interactions with positive role models may improve the outlook for the specialty. The home setting gives learners an opportunity to observe the care of medically complex patients in the community, exposes the students to the team of professionals needed for comprehensive care, and enhances learners’ awareness of the challenges in providing continuity of care for this population.
We previously reported on a qualitative study of comments of second-year medical students who participated in our House Calls program.28 Students frequently noted the apparent comfort and positive attitude of the patients; the dedication, patience, compassion, commitment, and hard work of the caregivers; and the personalized and comprehensive care provided. The students identified both the challenges and the rewards for the doctors and expressed increased interest in conducting house calls in the future.
The training of competent and caring physicians and other health professionals is the goal of medical education. Fourth-year medical students were surveyed nationally regarding the qualities of a humanistic doctor.29 The students noted the importance of role models and participatory experiences. House calls provide an opportunity for learners to see health care in the community. Such experiences can create a memorable lesson in care delivery and in doctor-patient-caregiver relationships.
PALLIATIVE CARE AND HOME CARE
Ideally, care plans would gradually shift in focus from curative therapy to palliative care as patients with significant chronic illness advance in age and debility. In our geriatric practice, palliation is always important throughout extended chronic illness. Care plans progress and palliation becomes the primary focus in the final months of life. This transition may take years. Hospice referral is frequently a final step because the payment system reimburses for comprehensive team-based hospice care only when life expectancy is less than 6 months. The reason for this is economic: comprehensive team-based care is costly, and lengthening the hospice benefit as it is now structured could be prohibitively expensive. Our patients may live for years in a state of advanced debility, yet need intensive team care only at intervals. Optimally, the care model, team intensity, and related payments should flex with clinical need. This is what we have experienced by making house calls the mode of longitudinal primary care delivery, supported by our institution. Our teams help patients and families shift focus and decide when to accept hospice care; this requires more art than science and usually involves a gradual process of adaptation.
Our approach is consistent with the definition of palliative care published by the Centers for Medicare & Medicaid Services in 2008: patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care addresses physical, intellectual, emotional, social, and spiritual needs and facilitates patient autonomy, access to information, and choice.30 Geriatric clinicians seek to help patients and families maximize quality of life and to maintain function by focusing on symptom management and clarification of patient and family goals rather than on specific diseases. This approach is applied without regard to patient age, condition, or stage of disease, and it can coexist with curative treatments. Thus it is distinguished in concept from “what we do when there is nothing more we can do.”31
In ways that are less clear when working in other care settings, home visits reveal patient goals, true rehabilitative potential, and family capacity for care-giving. Home visits take longer than office encounters, but make the provider’s job easier. By observing the patient at home, providers can better assess barriers to comfort and devise strategies to improve function, while also evaluating whether life is truly nearing the end. The home care clinician often engages in palliative care even if he or she did not initially intend to do so.
Furthermore, compared with the hospital or office setting, a home is more conducive to reasonably paced discussions about goals of care. Patients are more physically and emotionally comfortable and may talk more easily about potentially disturbing subjects. The clinician may be able to engage the patient by referring to pictures or mementos that help the patient to reflect on life values. And, a patient who is seen at home will more readily trust that the clinician places patients’ needs first. This opens the door to difficult discussions about code status, health care proxies, dialysis and ventilator support, or whether the patient would ever want to go to a hospital or a nursing home. Preferences change with time; patients ultimately feel less need to rely on ambulances and emergency care, given a timely response at home from a clinician who is familiar.32
Most dying patients are at home with their families during most of their final year of life; yet, despite studies showing that most patients prefer to die at home33–35 about 60% of all deaths still occur in the hospital.36 In our House Calls program’s experience, the percentage of patients who die at home is closer to 60.
Cherin and colleagues32 described a successful end-of-life home care program demonstrating a significant benefit to patients over usual care. The program integrated curative and palliative therapies. Similarly, Brumley and colleagues37 demonstrated that, compared with usual care, patients receiving in-home palliative care reported greater satisfaction, had fewer emergency department and hospital visits, and were more likely to die at home, with significantly lower overall costs. These findings conform to our experience. (Also see “Innovative models of home-based palliative care”)
CONCLUSION
Advanced home care with a strong medical component is an important part of the supportive and recuperative care options in the United States. For these programs to reach their full potential, we must expand on the successful in-home medical care models and create responsible financing methods that control overall costs while rewarding providers appropriately. We must broaden the application of portable and information technologies and develop an interdisciplinary workforce. These approaches will lead us toward our overall goals of optimal care at minimal cost.
- Levine SA, Boal J, Boling PA. Home care. JAMA 2003; 290:1203–1207.
- Boling PA. Home care: the first option. In:Cornwell T, Schwartz-berg JGeds. Medical Management of the Home Care Patient: Guidelines for Physicians. 4th ed. Chicago, IL: American Medical Association; 2012:1–14.
- Brickner PW, Duque Sister Teresita, Kaufman A, et al. The homebound aged: a medically unreached group. Ann Intern Med 1975; 82:1–6.
- Boling PA. Effects of policy, reimbursement, and regulation on home health care. In:Olson S. The Role of Human Factors in Home Health Care: Workshop Summary. Washington, DC: The National Academies Press; 2010:275–302.
- Basic statistics about home care. National Association for Home Care & Hospice Web site. http://www.nahc.org/facts/10HC_Stats.pdf. Updated 2010. Accessed October 11, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Shaughnessy PW, Hittle DF, Crisler KS, et al. Improving patient outcomes of home health care: findings from two demonstration trials of outcome-based quality improvement. J Am Geriatr Soc 2002; 50:1354–1364.
- Smigelski C, Hungate B, Holdren J, Goodloe L, Boling PA. Transitional model of care at VCU Medical Center—6 years’ experience. J Am Geriatr Soc 2008; 56 suppl 1:S197.
- Naylor MD. Advancing high value transitional care: the central role of nursing and its leadership. Nurs Adm Q 2012; 36:115–126.
- Holtz-Eakin D. High-cost medicare beneficiaries. Congressional Budget Office Web site. http://www.cbo.gov/sites/default/fles/cbofles/ftpdocs/63xx/doc6332/05-03-medispending.pdf. Published May 2005. Accessed October 11, 2012.
- Cryer L, Shannon SB, Van Amsterdam M, Leff B. Costs for “hospital at home” patients were 19 percent lower, with equal or better outcomes compared to similar inpatients. Health Aff (Millwood) 2012; 31:1237–1243.
- Leff B, Burton L, Mader SL, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Aujesky D, Roy PM, Verschuren F, et al. Outpatient versus inpatient treatment for patients with acute pulmonary embolism: an international, open-label, randomised, non-inferiority trial [published online ahead of print June 22, 2011]. Lancet 2011; 378 9785:41–48. 10.1016/S0140-6736 1160824–6
- Büller HR, Prins MH, Lensing AWA, et al the EINSTEIN– PE Investigators. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Landers SH. Why health care is going home [published online ahead of print October 20, 2010]. N Engl J Med 2010; 363:1690–1691. 10.1056/NEJMp1000401
- Counsell SR, Callahan CM, Clark DO, et al. Geriatric care management for low-income seniors: a randomized controlled trial. JAMA 2007; 298:2623–2633.
- Beales JL, Edes T. Veteran’s Affairs home based primary care. Clin Geriatr Med 2009; 25:149–154.
- Kinosian B, Edes T, Davis D, Hossain M. Financial savings of home based primary care for frail veterans with chronic disabling disease. J Am Geriatr Soc 2010; 589 (suppl s1):S3. Abstract P7.
- DeJonge KE, Taler G, Boling PA. Independence at home: community-based care for older adults with severe chronic illness. Clin Geriatr Med 2009; 25:155–169.
- Chaudhry SI, Mattera JA, Curtis JP, et al. Telemonitoring in patients with heart failure [published online ahead of print November 16, 2010]. N Engl J Med 2010; 363:2301–2309. 10.1056/NEJMoa1010029
- Wieland D, Boland R, Baskins J, Kinosian B. Five-year survival in a Program of All-inclusive Care for Elderly compared with alternative institutional and home- and community-based care [published online ahead of print March 30, 2010]. J Gerontol A Biol Sci Med Sci 2010; 65:721–726. 10.1093/gerona/glq040
- Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives [published online ahead of print May 30, 2006]. J Am Med Dir Assoc 2006; 7:339–344. 10.1016/j.jamda.2006.01.022
- Meret-Hanke LA. Effects of the Program of All-inclusive Care for the Elderly on hospital use [published online ahead of print July 6, 2011]. Gerontologist 2011; 51:774–785. 10.1093/geront/gnr040
- Fitzgerald JT, Wray LA, Halter JB, Williams BC, Supiano MA. Relating medical students’ knowledge, attitudes, and experience to an interest in geriatric medicine. Gerontologist 2003; 43:849–855.
- Voogt SJ, Mickus M, Santiago O, Herman SE. Attitudes, experiences, and interest in geriatrics of first-year allopathic and osteopathic medical students [published online ahead of print December 11, 2007]. J Am Geriatr Soc 2008; 56:339–344. 10.1111/j.1532-5415.2007.01541.
- Abbey L, Willett R, Selby-Penczak R, McKnight R. Social learning: medical student perceptions of geriatric house calls. Gerontol Geriatr Educ 2010; 31:149–162.
- Moyer CA, Arnold L, Quaintance J, et al. What factors create a humanistic doctor? A nationwide survey of fourth-year medical students. Acad Med 2010; 85:1800–1807.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid programs: hospice conditions of participation. Federal Register 2008; 73:32204–32220.
- Meier DE, Brawley OW. Palliative care and the quality of life [published online ahead of print June 13, 2011]. J Clin Oncol 2011; 29:2750–2752. 10.1200/JCO.2011.35.9729
- Cherin DA, Enguidanos SM, Jamison P. Physicians as medical center “extenders” in end-of-life care: physician home visits as the lynch pin in creating an end-of-life care system. Home Health Care Serv Q 2004; 23:41–53.
- Hays JC, Galanos AN, Palmer TA, McQuoid DR, Flint EP. Preference for place of death in a continuing care retirement community. Gerontologist 2001; 41:123–128.
- Karlsen S, Addington-Hall J. How do cancer patients who die at home differ from those who die elsewhere? Palliat Med 1998; 12:279–286.
- Townsend J, Frank AO, Fermont D, et al. Terminal cancer care and patients’ preference for place of death: a prospective study. BMJ 1990; 301:415–417.
- Weitzen S, Teno JM, Fennell M, Mor V. Factors associated with site of death: a national study of where people die. Med Care 2003; 41:323–335.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
- Levine SA, Boal J, Boling PA. Home care. JAMA 2003; 290:1203–1207.
- Boling PA. Home care: the first option. In:Cornwell T, Schwartz-berg JGeds. Medical Management of the Home Care Patient: Guidelines for Physicians. 4th ed. Chicago, IL: American Medical Association; 2012:1–14.
- Brickner PW, Duque Sister Teresita, Kaufman A, et al. The homebound aged: a medically unreached group. Ann Intern Med 1975; 82:1–6.
- Boling PA. Effects of policy, reimbursement, and regulation on home health care. In:Olson S. The Role of Human Factors in Home Health Care: Workshop Summary. Washington, DC: The National Academies Press; 2010:275–302.
- Basic statistics about home care. National Association for Home Care & Hospice Web site. http://www.nahc.org/facts/10HC_Stats.pdf. Updated 2010. Accessed October 11, 2012.
- Coleman EA, Parry C, Chalmers S, Min SJ. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten D, Campbell R, et al. Comprehensive discharge planning and home follow-up of hospitalized elders: a randomized clinical trial. JAMA 1999; 281:613–620.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Shaughnessy PW, Hittle DF, Crisler KS, et al. Improving patient outcomes of home health care: findings from two demonstration trials of outcome-based quality improvement. J Am Geriatr Soc 2002; 50:1354–1364.
- Smigelski C, Hungate B, Holdren J, Goodloe L, Boling PA. Transitional model of care at VCU Medical Center—6 years’ experience. J Am Geriatr Soc 2008; 56 suppl 1:S197.
- Naylor MD. Advancing high value transitional care: the central role of nursing and its leadership. Nurs Adm Q 2012; 36:115–126.
- Holtz-Eakin D. High-cost medicare beneficiaries. Congressional Budget Office Web site. http://www.cbo.gov/sites/default/fles/cbofles/ftpdocs/63xx/doc6332/05-03-medispending.pdf. Published May 2005. Accessed October 11, 2012.
- Cryer L, Shannon SB, Van Amsterdam M, Leff B. Costs for “hospital at home” patients were 19 percent lower, with equal or better outcomes compared to similar inpatients. Health Aff (Millwood) 2012; 31:1237–1243.
- Leff B, Burton L, Mader SL, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Aujesky D, Roy PM, Verschuren F, et al. Outpatient versus inpatient treatment for patients with acute pulmonary embolism: an international, open-label, randomised, non-inferiority trial [published online ahead of print June 22, 2011]. Lancet 2011; 378 9785:41–48. 10.1016/S0140-6736 1160824–6
- Büller HR, Prins MH, Lensing AWA, et al the EINSTEIN– PE Investigators. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Landers SH. Why health care is going home [published online ahead of print October 20, 2010]. N Engl J Med 2010; 363:1690–1691. 10.1056/NEJMp1000401
- Counsell SR, Callahan CM, Clark DO, et al. Geriatric care management for low-income seniors: a randomized controlled trial. JAMA 2007; 298:2623–2633.
- Beales JL, Edes T. Veteran’s Affairs home based primary care. Clin Geriatr Med 2009; 25:149–154.
- Kinosian B, Edes T, Davis D, Hossain M. Financial savings of home based primary care for frail veterans with chronic disabling disease. J Am Geriatr Soc 2010; 589 (suppl s1):S3. Abstract P7.
- DeJonge KE, Taler G, Boling PA. Independence at home: community-based care for older adults with severe chronic illness. Clin Geriatr Med 2009; 25:155–169.
- Chaudhry SI, Mattera JA, Curtis JP, et al. Telemonitoring in patients with heart failure [published online ahead of print November 16, 2010]. N Engl J Med 2010; 363:2301–2309. 10.1056/NEJMoa1010029
- Wieland D, Boland R, Baskins J, Kinosian B. Five-year survival in a Program of All-inclusive Care for Elderly compared with alternative institutional and home- and community-based care [published online ahead of print March 30, 2010]. J Gerontol A Biol Sci Med Sci 2010; 65:721–726. 10.1093/gerona/glq040
- Schamp R, Tenkku L. Managed death in a PACE: pathways in present and advance directives [published online ahead of print May 30, 2006]. J Am Med Dir Assoc 2006; 7:339–344. 10.1016/j.jamda.2006.01.022
- Meret-Hanke LA. Effects of the Program of All-inclusive Care for the Elderly on hospital use [published online ahead of print July 6, 2011]. Gerontologist 2011; 51:774–785. 10.1093/geront/gnr040
- Fitzgerald JT, Wray LA, Halter JB, Williams BC, Supiano MA. Relating medical students’ knowledge, attitudes, and experience to an interest in geriatric medicine. Gerontologist 2003; 43:849–855.
- Voogt SJ, Mickus M, Santiago O, Herman SE. Attitudes, experiences, and interest in geriatrics of first-year allopathic and osteopathic medical students [published online ahead of print December 11, 2007]. J Am Geriatr Soc 2008; 56:339–344. 10.1111/j.1532-5415.2007.01541.
- Abbey L, Willett R, Selby-Penczak R, McKnight R. Social learning: medical student perceptions of geriatric house calls. Gerontol Geriatr Educ 2010; 31:149–162.
- Moyer CA, Arnold L, Quaintance J, et al. What factors create a humanistic doctor? A nationwide survey of fourth-year medical students. Acad Med 2010; 85:1800–1807.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid programs: hospice conditions of participation. Federal Register 2008; 73:32204–32220.
- Meier DE, Brawley OW. Palliative care and the quality of life [published online ahead of print June 13, 2011]. J Clin Oncol 2011; 29:2750–2752. 10.1200/JCO.2011.35.9729
- Cherin DA, Enguidanos SM, Jamison P. Physicians as medical center “extenders” in end-of-life care: physician home visits as the lynch pin in creating an end-of-life care system. Home Health Care Serv Q 2004; 23:41–53.
- Hays JC, Galanos AN, Palmer TA, McQuoid DR, Flint EP. Preference for place of death in a continuing care retirement community. Gerontologist 2001; 41:123–128.
- Karlsen S, Addington-Hall J. How do cancer patients who die at home differ from those who die elsewhere? Palliat Med 1998; 12:279–286.
- Townsend J, Frank AO, Fermont D, et al. Terminal cancer care and patients’ preference for place of death: a prospective study. BMJ 1990; 301:415–417.
- Weitzen S, Teno JM, Fennell M, Mor V. Factors associated with site of death: a national study of where people die. Med Care 2003; 41:323–335.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
In-home care following total knee replacement
Total knee replacement (TKR) is a reliable treatment for end-stage arthritis of the knee, resulting in pain relief and return of function. While surgeons have historically focused on surgical technique and implant selection as important factors on the path to a successful outcome, additional care elements may play similarly important roles. As hospital length of stay continues to decrease, more of the patient’s postoperative care occurs in a postacute setting, with home care becoming a more important component of a well-designed care path. Early experience suggests that this shift toward home care has resulted in a more cost-effective approach with improved outcomes.1–4
Although TKR has traditionally been viewed as a surgical procedure, an important shift in thinking has increased recognition that TKR is best viewed as part of a spectrum of care required to obtain an end result. Viewing the procedure as an episode of care is gaining significant traction. In this approach, the surgical procedure and its attendant features and factors remain paramount and central in driving outcomes, but the care that precedes and follows the procedure can have a significant impact on important measures of success. From the patient’s perspective, this view is intuitive; ie, the outcome of the intervention can only be assessed when complete healing has occurred and the patient has returned to routine activities of daily living (ADL). As such, a more holistic or global view of the episode is warranted and is receiving increasing attention.5–8
INNOVATIVE PAYMENT METHODS AND RESOURCE ALLOCATION
Recently, the Center for Medicare & Medicaid Services (CMS) launched a call for innovative payment methods for episodes of care. Traditionally, CMS has paid for each component of care separately; the new approach, represented in this call for proposals and driven by the Patient Protection and Affordable Care Act (PPACA), is to pay for care based on defined episodes. This method of payment is sometimes referred to as “bundling,” in that the payment for a group of services is linked into a single payment. Although the details and definitions of the episodes may vary, the conceptual framework supports the integration of care along a continuum. By paying for care based on the entire episode, CMS believes it can encourage more rational allocation of resources along the care path.9
It is widely recognized that one area where care can be better managed is during the transitions that occur at many points along the care path—for example, transition from operating theater to postoperative unit and then to the acute care hospital setting, and transition from acute care hospital to a postacute setting.1,4,10
When a patient no longer requires hospital services but needs the benefits of continued care, the transition to postacute care must be managed carefully. Optimizing this transition and choosing among postacute care venues can significantly affect cost and outcomes of the procedure. In fact, there is increasing evidence that the transition from hospitalization to postacute care has been significantly undermanaged, with deferral of some important considerations until after the process has already begun.1,4,10 Neglecting this important transition results in unwarranted variation in process and outcomes. For example, physicians often delegate decisions regarding the location and intensity of postacute services to other team members. Patient preferences and, at times, misconceptions can drive the choices for postacute care, with patients erroneously believing that one venue is inherently better than another or that more is somehow better than less. Such patterns can lead to over- or underutilization, with care unmatched to individual need or circumstance. Careful scrutiny by an engaged team of the resources necessary for patients as they transition to the postacute component of the episode is likely to result in a more rational, cost-effective approach to care. It is also likely to increase patient satisfaction and improve patient outcome measures.5,10–13
MEETING THE CHALLENGE OF INCREASED DEMAND WITH HOME CARE
With the rising incidence of knee arthritis, the demand for TKR is expected to more than double in the coming years.14 This increased utilization is driven by an aging population that desires to remain active, as well as by evidence suggesting health benefits associated with increased activity levels. Along with these demographic and utilization trends, another evolution in joint replacement derives from patients’ expectation of continuously improving results. Patients measure the success of TKR not only by relative reduction in pain, but also by other outcome metrics, including, importantly, return to sport or work.5,7 The tandem challenge posed by increased demand for services and increased patient expectations regarding outcomes is testing health care providers as they consider the resources that will be required to meet the demand.
Health care systems, payers, and physicians are looking for ways to more efficiently meet this growing need for TKR services in the context of finite health care resources subject to competing demand from several clinical entities. Regardless of TKR’s record of clinical success, the resources applied to this orthopedic intervention come at the expense of the same resources being applied to other health care needs. As demand is unlikely to wane, the only rational approach is to redesign care delivery in favor of a more efficient model. In order to meet the demand with the available resources, several goals need to be achieved: fewer inpatient hospital and postacute bed days consumed by joint replacement services, better streamlined care paths, and improved engagement of the patient and his or her home-based support network. Key to this process is driving care to the home environment, provided that quality is at least comparable and cost is significantly less.3,15–17
Postoperative rehabilitation and physical therapy is essential to restoration of function after TKR. It is therefore no surprise that rehabilitation and physical therapy make up a significant proportion of the home care services for this patient population.8,17,18 Among its advantages, therapy in the home environment gives the therapist the opportunity to identify and address the patient’s unique needs in his or her own home. In addition, family and other support personnel often feel more comfortable assuming responsibility for assisting with care in a familiar setting. Tailored therapy in the home setting can improve safety and satisfaction and speed the resumption of ADL; it is increasingly seen as an essential component of the care path.4,11
Recently, care path designs have been subject to careful analyses that compare in-home rehabilitation outcomes with outcomes achieved in an inpatient environment. Observational, retrospective, and prospective study designs have confirmed that the in-home rehabilitation model of care delivery is not only viable, but in many circumstances preferable.5,10,12,17,19 The quality is comparable to inpatient care for most TKR patient populations and the cost and resource utilization intensity are considerably reduced. Such reports have lent credence to the movement to incorporate home care services into successful post–joint replacement care paths. The approach appears to have a large potential for benefit with very little risk. Strategies that aim to more rationally deliver needed rehabilitation services at home promise to keep TKR services within the reach of our strained health care resources.
THE HOME CARE CLINICAL PATH

The underlying principle of a home care clinical path is that the patient remains at the center of the program and shares in decisions about care strategies (Table). One of the greatest concerns patients have about a pending knee replacement is the duration of their expected recovery. To meet this concern, a Rapid Recovery Care Path has been developed that incorporates an integrated approach to acute and postacute care, with increased emphasis on discharging patients to their home environment as early as it appears safe to do so. The goals of a rapid recovery home-centered care program following routine TKR include reduced postoperative pain and early return to function.2,15,16 Meeting these goals minimizes the development of a vicious cycle of pain and stiffness that may lead to chronic pain and fibrosis. As a result, the patient can pursue more aggressive rehabilitation, which maintains joint range of motion, permits earlier hospital discharge and discharge to home rather than another health care facility, and improves patient satisfaction.
The Cleveland Clinic Total Knee Care Path effectively incorporates the rapid recovery approach, with home care taking the lead in discharge planning and transition of care management. Education is essential and should start early, at the time of informed consent; involve the patient and family; and continue throughout the care path.
The key to a successful outcome is patient engagement with agreed-upon principles of care, which form the basis for the care path. In the Cleveland Clinic program, patients are engaged to embrace the following goals:
- Shared decision-making
- A home care environment that includes support of family and friends
- Patient and family education to enhance shared decision-making
- Return to the home environment as soon as it is deemed safe
- Elimination of unnecessary or duplicative treatments, tests, or interventions
- Acceptance of multiple plans or paths in response to changing clinical conditions
All patients undergo a preoperative evaluation, during which they are introduced to and educated about the Rapid Recovery Total Knee Care Path. The Rapid Recovery Path accommodates planned interventions and contingencies depending on clinical course. Every patient envisions a safe return home as a primary goal, with as short an exposure to inpatient acute and postacute settings as is necessary. No fixed length of stay or discharge destination is mandated. Rather, patients are encouraged to articulate their goals, drive their discharge, and return home. Such shared decision-making empowers patients and improves satisfaction.
Factors that affect recovery are assessed through a detailed perioperative history and physical examination. The patient’s readiness for an intervention such as TKR is assessed in three phases:
- The preoperative history, physical examination, and radiographic parameters establish that appropriate indications exist in terms of diagnosis and level of disability.
- The assessment team identifies conditions that affect risk and devises plans for their perioperative management—for example, control of blood glucose or decolonization of methicillin-resistant Staphylococcus Taurus carriers. Plans are made for the perioperative as well as seamless postdischarge management of chronic conditions such as atrial fibrillation requiring anticoagulation or hypertension.
- Psychosocial factors are evaluated for their potential impact on discharge planning and postacute management. Patients must establish their ability to participate actively in their care and consider their access to family, friends, and neighbors who can assist with care management in the home. Successful management of the care episode depends on an effective and reliable advocate. If the patient is unable to perform this function, then a surrogate advocate must be identified. If this role cannot be filled, the patient will require transfer to an inpatient rehabilitation facility.
POSITIVE RESULTS, BUT REGULATORY CHALLENGES
Since our 2006 incorporation of an active postacute home care program into our rapid recovery protocol, we have observed several improved outcome metrics:
- Average acute care hospital length of stay has been reduced by an average of 0.9 days.
- Our discharge to home rate has risen from 32% to 74%. In fact, among surgeons who have fully embraced the rapid recovery protocol, the discharge to home rate is 74% compared with 45% among the remaining surgeons. The difference is statistically (P < .05) and clinically significant.
- The readmission rate for patients discharged to home using this protocol is significantly lower compared with the rate before the protocol was implemented and with the rate of a control cohort discharged to a skilled nursing facility. Patients discharged to home consume significantly fewer resources and cost the system about one-third as much as those sent to an inpatient postacute facility.
Despite these gains, the regulatory environment is not structured to reward good stewardship of health care resources. For example, current payment rules penalize institutions that achieve early discharge (less than 3 days) from an acute care hospital when the patient will be transferred to another care venue. In addition, requirements for home care can be stringent, limiting the beneficial application of therapy in the home if alternatives, such as outpatient or subacute care, exist. Fortunately, PPACA and the request for bundled pricing of episodes of care gives providers the opportunity to apply for exceptions to rules that hinder cost containment. As such, relief may be in sight.
OUTLOOK
The future is bright for care path development and incorporation of better methods to manage care episodes.20,21 Although the concept of outpatient joint replacement has been considered by some, questions remain regarding the lower limit of resources that should be applied to a given episode and how best to predict which patients can benefit from even less inpatient care. Predictive modeling based on patient-specific factors might assist in this, but prudence suggests that flexibility in care path management will always be the most important element of protection for patients. Specifically, early detection of significant clinical deviation requiring a change in venue is paramount and is routinely incorporated into any well-designed care path. The goal is not to minimize resource utilization, but rather to ensure appropriate and rational distribution of health care resources to meet the clinical needs of each patient. Refining our approaches to achieving this balance will require ongoing work and monitoring of metrics of success.
- Chimenti CE, Ingersoll G. Comparison of home health care physical therapy outcomes following total knee replacement with and without subacute rehabilitation. J Geriatr Phys Ther 2007; 30:102–108.
- Iyengar KP, Nadkarni JB, Ivanovic N, Mahale A. Targeted early rehabilitation at home after total hip and knee joint replacement: does it work? Disabil Rehabil 2007; 29:495–502
- Mitchell C, Walker J, Walters S, Morgan AB, Binns T, Mathers N. Costs and effectiveness of pre- and post-operative home physiotherapy for total knee replacement: randomized controlled trial. J Eval Clin Pract 2005; 11:283–292.
- Stevens M, van den Akker-Scheek I, Spriensma A, Boss NA, Diercks RL, van Horn JR. The Groningen Orthopedic Exit Strategy (GOES): a home-based support program for total hip and knee arthroplasty patients after shortened hospital stay. Patient Educ Couns 2004; 54:95–99.
- Tousignant M, Boissy P, Moffet H, et al. Patients’ satisfaction of healthcare services and perception with in-home telerehabilitation and physiotherapists’ satisfaction toward technology for post-knee arthroplasty: an embedded study in a randomized trial [published online ahead of print April 14, 2011]. Telemed J E Health 2011; 17:376–382. 10.1089/tmj.2010.0198
- Kramer JF, Speechley M, Bourne R, Rorabeck C, Vaz M. Comparison of clinic- and home-based rehabilitation programs after total knee arthroplasty. Clin Orthop Relat Res 2003; 410:225–234.
- Loft M, McWilliam C, Ward-Griffin C. Patient empowerment after total hip and knee replacement. Orthop Nurs 2003; 22:42–47.
- Harris MD, Candando P. The physical therapist as a member of the home healthcare team: caring for patients with replacements. Home Healthc Nurse 1998; 16:153–156.
- Collins T, Herness J, Martenas J, Roberson A. Medicare prospective payment before and after implementation: a review of visits and physical performance among Medicare home health patients after total knee replacements. Home Healthc Nurse 2007; 25:401–407.
- Mallinson TR, Bateman J, Tseng HY, et al. A comparison of discharge functional status after rehabilitation in skilled nursing, home health, and medical rehabilitation settings for patients after lower-extremity joint replacement surgery. Arch Phys Med Rehabil 2011; 92:712–720.
- Stineman MG, Chan L. Commentary on the comparative effectiveness of alternative settings for joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1257–1259.
- Lin CW, March L, Crosbie J, et al. Maximum recovery after knee replacement—the MARKER study rationale and protocol. BMC Musculoskelet Disord 2009; 10:69.
- Thomas G, Faisal M, Young S, Asson R, Ritson M, Bawale R. Early discharge after hip arthroplasty with home support: experience at a UK District General Hospital. Hip Int 2008; 18:294–300.
- Tian W, DeJong G, Brown M, Hsieh CH, Zamfirov ZP, Horn SD. Looking upstream: factors shaping the demand for postacute joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1260–1268.
- Bade MJ, Stevens-Lapsley JE. Early high-intensity rehabilitation following total knee arthroplasty improves outcomes [published online ahead of print September 30, 2011]. J Orthop Sports Phys Ther 2011; 41:932–941. 10.2519/jospt.2011.3734
- Doman DM, Gerlinger TL. Total joint arthroplasty cost savings with a rapid recovery protocol in a military medical center. Mil Med 2012; 177:64–69.
- Liebs TR, Herzberg W, Rüther W, Haasters J, Russlies M, Hassenpflug J, Multicenter Arthroplasty Aftercare Project. Multicenter randomized controlled trial comparing early versus late aquatic therapy after total hip or knee arthroplasty [published online ahead of print December 21, 2011]. Arch Phys Med Rehabil 2012; 93:192–199. 10.1016/j.apmr.2011.09.011
- Mahomed NN, Koo Seen Lin MJ, Levesque J, Lan S, Bogoch ER. Determinants and outcomes of inpatient versus home based rehabilitation following elective hip and knee replacement. J Rheumatol 2000; 27:1753–1758.
- Mahomed NN, Davis AM, Hawker G, et al. Inpatient compared with home-based rehabilitation following primary unilateral total hip or knee replacement: a randomized controlled trial. J Bone Joint Surg Am 2008; 90:1673–1680.
- Aprile I, Rizzo RS, Romanini E, et al. Group rehabilitation versus individual rehabilitation following knee and hip replacement: a pilot study with randomized, single-blind, cross-over design. Eur J Phys Rehabil Med 2011; 47:551–559.
- Russell TG, Buttrum P, Wootton R, Jull GA. Rehabilitation after total knee replacement via low-bandwidth telemedicine: the patient and therapist experience. J Telemed Telecare 2004; 10(suppl 1):85–87.
Total knee replacement (TKR) is a reliable treatment for end-stage arthritis of the knee, resulting in pain relief and return of function. While surgeons have historically focused on surgical technique and implant selection as important factors on the path to a successful outcome, additional care elements may play similarly important roles. As hospital length of stay continues to decrease, more of the patient’s postoperative care occurs in a postacute setting, with home care becoming a more important component of a well-designed care path. Early experience suggests that this shift toward home care has resulted in a more cost-effective approach with improved outcomes.1–4
Although TKR has traditionally been viewed as a surgical procedure, an important shift in thinking has increased recognition that TKR is best viewed as part of a spectrum of care required to obtain an end result. Viewing the procedure as an episode of care is gaining significant traction. In this approach, the surgical procedure and its attendant features and factors remain paramount and central in driving outcomes, but the care that precedes and follows the procedure can have a significant impact on important measures of success. From the patient’s perspective, this view is intuitive; ie, the outcome of the intervention can only be assessed when complete healing has occurred and the patient has returned to routine activities of daily living (ADL). As such, a more holistic or global view of the episode is warranted and is receiving increasing attention.5–8
INNOVATIVE PAYMENT METHODS AND RESOURCE ALLOCATION
Recently, the Center for Medicare & Medicaid Services (CMS) launched a call for innovative payment methods for episodes of care. Traditionally, CMS has paid for each component of care separately; the new approach, represented in this call for proposals and driven by the Patient Protection and Affordable Care Act (PPACA), is to pay for care based on defined episodes. This method of payment is sometimes referred to as “bundling,” in that the payment for a group of services is linked into a single payment. Although the details and definitions of the episodes may vary, the conceptual framework supports the integration of care along a continuum. By paying for care based on the entire episode, CMS believes it can encourage more rational allocation of resources along the care path.9
It is widely recognized that one area where care can be better managed is during the transitions that occur at many points along the care path—for example, transition from operating theater to postoperative unit and then to the acute care hospital setting, and transition from acute care hospital to a postacute setting.1,4,10
When a patient no longer requires hospital services but needs the benefits of continued care, the transition to postacute care must be managed carefully. Optimizing this transition and choosing among postacute care venues can significantly affect cost and outcomes of the procedure. In fact, there is increasing evidence that the transition from hospitalization to postacute care has been significantly undermanaged, with deferral of some important considerations until after the process has already begun.1,4,10 Neglecting this important transition results in unwarranted variation in process and outcomes. For example, physicians often delegate decisions regarding the location and intensity of postacute services to other team members. Patient preferences and, at times, misconceptions can drive the choices for postacute care, with patients erroneously believing that one venue is inherently better than another or that more is somehow better than less. Such patterns can lead to over- or underutilization, with care unmatched to individual need or circumstance. Careful scrutiny by an engaged team of the resources necessary for patients as they transition to the postacute component of the episode is likely to result in a more rational, cost-effective approach to care. It is also likely to increase patient satisfaction and improve patient outcome measures.5,10–13
MEETING THE CHALLENGE OF INCREASED DEMAND WITH HOME CARE
With the rising incidence of knee arthritis, the demand for TKR is expected to more than double in the coming years.14 This increased utilization is driven by an aging population that desires to remain active, as well as by evidence suggesting health benefits associated with increased activity levels. Along with these demographic and utilization trends, another evolution in joint replacement derives from patients’ expectation of continuously improving results. Patients measure the success of TKR not only by relative reduction in pain, but also by other outcome metrics, including, importantly, return to sport or work.5,7 The tandem challenge posed by increased demand for services and increased patient expectations regarding outcomes is testing health care providers as they consider the resources that will be required to meet the demand.
Health care systems, payers, and physicians are looking for ways to more efficiently meet this growing need for TKR services in the context of finite health care resources subject to competing demand from several clinical entities. Regardless of TKR’s record of clinical success, the resources applied to this orthopedic intervention come at the expense of the same resources being applied to other health care needs. As demand is unlikely to wane, the only rational approach is to redesign care delivery in favor of a more efficient model. In order to meet the demand with the available resources, several goals need to be achieved: fewer inpatient hospital and postacute bed days consumed by joint replacement services, better streamlined care paths, and improved engagement of the patient and his or her home-based support network. Key to this process is driving care to the home environment, provided that quality is at least comparable and cost is significantly less.3,15–17
Postoperative rehabilitation and physical therapy is essential to restoration of function after TKR. It is therefore no surprise that rehabilitation and physical therapy make up a significant proportion of the home care services for this patient population.8,17,18 Among its advantages, therapy in the home environment gives the therapist the opportunity to identify and address the patient’s unique needs in his or her own home. In addition, family and other support personnel often feel more comfortable assuming responsibility for assisting with care in a familiar setting. Tailored therapy in the home setting can improve safety and satisfaction and speed the resumption of ADL; it is increasingly seen as an essential component of the care path.4,11
Recently, care path designs have been subject to careful analyses that compare in-home rehabilitation outcomes with outcomes achieved in an inpatient environment. Observational, retrospective, and prospective study designs have confirmed that the in-home rehabilitation model of care delivery is not only viable, but in many circumstances preferable.5,10,12,17,19 The quality is comparable to inpatient care for most TKR patient populations and the cost and resource utilization intensity are considerably reduced. Such reports have lent credence to the movement to incorporate home care services into successful post–joint replacement care paths. The approach appears to have a large potential for benefit with very little risk. Strategies that aim to more rationally deliver needed rehabilitation services at home promise to keep TKR services within the reach of our strained health care resources.
THE HOME CARE CLINICAL PATH
The underlying principle of a home care clinical path is that the patient remains at the center of the program and shares in decisions about care strategies (Table). One of the greatest concerns patients have about a pending knee replacement is the duration of their expected recovery. To meet this concern, a Rapid Recovery Care Path has been developed that incorporates an integrated approach to acute and postacute care, with increased emphasis on discharging patients to their home environment as early as it appears safe to do so. The goals of a rapid recovery home-centered care program following routine TKR include reduced postoperative pain and early return to function.2,15,16 Meeting these goals minimizes the development of a vicious cycle of pain and stiffness that may lead to chronic pain and fibrosis. As a result, the patient can pursue more aggressive rehabilitation, which maintains joint range of motion, permits earlier hospital discharge and discharge to home rather than another health care facility, and improves patient satisfaction.
The Cleveland Clinic Total Knee Care Path effectively incorporates the rapid recovery approach, with home care taking the lead in discharge planning and transition of care management. Education is essential and should start early, at the time of informed consent; involve the patient and family; and continue throughout the care path.
The key to a successful outcome is patient engagement with agreed-upon principles of care, which form the basis for the care path. In the Cleveland Clinic program, patients are engaged to embrace the following goals:
- Shared decision-making
- A home care environment that includes support of family and friends
- Patient and family education to enhance shared decision-making
- Return to the home environment as soon as it is deemed safe
- Elimination of unnecessary or duplicative treatments, tests, or interventions
- Acceptance of multiple plans or paths in response to changing clinical conditions
All patients undergo a preoperative evaluation, during which they are introduced to and educated about the Rapid Recovery Total Knee Care Path. The Rapid Recovery Path accommodates planned interventions and contingencies depending on clinical course. Every patient envisions a safe return home as a primary goal, with as short an exposure to inpatient acute and postacute settings as is necessary. No fixed length of stay or discharge destination is mandated. Rather, patients are encouraged to articulate their goals, drive their discharge, and return home. Such shared decision-making empowers patients and improves satisfaction.
Factors that affect recovery are assessed through a detailed perioperative history and physical examination. The patient’s readiness for an intervention such as TKR is assessed in three phases:
- The preoperative history, physical examination, and radiographic parameters establish that appropriate indications exist in terms of diagnosis and level of disability.
- The assessment team identifies conditions that affect risk and devises plans for their perioperative management—for example, control of blood glucose or decolonization of methicillin-resistant Staphylococcus Taurus carriers. Plans are made for the perioperative as well as seamless postdischarge management of chronic conditions such as atrial fibrillation requiring anticoagulation or hypertension.
- Psychosocial factors are evaluated for their potential impact on discharge planning and postacute management. Patients must establish their ability to participate actively in their care and consider their access to family, friends, and neighbors who can assist with care management in the home. Successful management of the care episode depends on an effective and reliable advocate. If the patient is unable to perform this function, then a surrogate advocate must be identified. If this role cannot be filled, the patient will require transfer to an inpatient rehabilitation facility.
POSITIVE RESULTS, BUT REGULATORY CHALLENGES
Since our 2006 incorporation of an active postacute home care program into our rapid recovery protocol, we have observed several improved outcome metrics:
- Average acute care hospital length of stay has been reduced by an average of 0.9 days.
- Our discharge to home rate has risen from 32% to 74%. In fact, among surgeons who have fully embraced the rapid recovery protocol, the discharge to home rate is 74% compared with 45% among the remaining surgeons. The difference is statistically (P < .05) and clinically significant.
- The readmission rate for patients discharged to home using this protocol is significantly lower compared with the rate before the protocol was implemented and with the rate of a control cohort discharged to a skilled nursing facility. Patients discharged to home consume significantly fewer resources and cost the system about one-third as much as those sent to an inpatient postacute facility.
Despite these gains, the regulatory environment is not structured to reward good stewardship of health care resources. For example, current payment rules penalize institutions that achieve early discharge (less than 3 days) from an acute care hospital when the patient will be transferred to another care venue. In addition, requirements for home care can be stringent, limiting the beneficial application of therapy in the home if alternatives, such as outpatient or subacute care, exist. Fortunately, PPACA and the request for bundled pricing of episodes of care gives providers the opportunity to apply for exceptions to rules that hinder cost containment. As such, relief may be in sight.
OUTLOOK
The future is bright for care path development and incorporation of better methods to manage care episodes.20,21 Although the concept of outpatient joint replacement has been considered by some, questions remain regarding the lower limit of resources that should be applied to a given episode and how best to predict which patients can benefit from even less inpatient care. Predictive modeling based on patient-specific factors might assist in this, but prudence suggests that flexibility in care path management will always be the most important element of protection for patients. Specifically, early detection of significant clinical deviation requiring a change in venue is paramount and is routinely incorporated into any well-designed care path. The goal is not to minimize resource utilization, but rather to ensure appropriate and rational distribution of health care resources to meet the clinical needs of each patient. Refining our approaches to achieving this balance will require ongoing work and monitoring of metrics of success.
Total knee replacement (TKR) is a reliable treatment for end-stage arthritis of the knee, resulting in pain relief and return of function. While surgeons have historically focused on surgical technique and implant selection as important factors on the path to a successful outcome, additional care elements may play similarly important roles. As hospital length of stay continues to decrease, more of the patient’s postoperative care occurs in a postacute setting, with home care becoming a more important component of a well-designed care path. Early experience suggests that this shift toward home care has resulted in a more cost-effective approach with improved outcomes.1–4
Although TKR has traditionally been viewed as a surgical procedure, an important shift in thinking has increased recognition that TKR is best viewed as part of a spectrum of care required to obtain an end result. Viewing the procedure as an episode of care is gaining significant traction. In this approach, the surgical procedure and its attendant features and factors remain paramount and central in driving outcomes, but the care that precedes and follows the procedure can have a significant impact on important measures of success. From the patient’s perspective, this view is intuitive; ie, the outcome of the intervention can only be assessed when complete healing has occurred and the patient has returned to routine activities of daily living (ADL). As such, a more holistic or global view of the episode is warranted and is receiving increasing attention.5–8
INNOVATIVE PAYMENT METHODS AND RESOURCE ALLOCATION
Recently, the Center for Medicare & Medicaid Services (CMS) launched a call for innovative payment methods for episodes of care. Traditionally, CMS has paid for each component of care separately; the new approach, represented in this call for proposals and driven by the Patient Protection and Affordable Care Act (PPACA), is to pay for care based on defined episodes. This method of payment is sometimes referred to as “bundling,” in that the payment for a group of services is linked into a single payment. Although the details and definitions of the episodes may vary, the conceptual framework supports the integration of care along a continuum. By paying for care based on the entire episode, CMS believes it can encourage more rational allocation of resources along the care path.9
It is widely recognized that one area where care can be better managed is during the transitions that occur at many points along the care path—for example, transition from operating theater to postoperative unit and then to the acute care hospital setting, and transition from acute care hospital to a postacute setting.1,4,10
When a patient no longer requires hospital services but needs the benefits of continued care, the transition to postacute care must be managed carefully. Optimizing this transition and choosing among postacute care venues can significantly affect cost and outcomes of the procedure. In fact, there is increasing evidence that the transition from hospitalization to postacute care has been significantly undermanaged, with deferral of some important considerations until after the process has already begun.1,4,10 Neglecting this important transition results in unwarranted variation in process and outcomes. For example, physicians often delegate decisions regarding the location and intensity of postacute services to other team members. Patient preferences and, at times, misconceptions can drive the choices for postacute care, with patients erroneously believing that one venue is inherently better than another or that more is somehow better than less. Such patterns can lead to over- or underutilization, with care unmatched to individual need or circumstance. Careful scrutiny by an engaged team of the resources necessary for patients as they transition to the postacute component of the episode is likely to result in a more rational, cost-effective approach to care. It is also likely to increase patient satisfaction and improve patient outcome measures.5,10–13
MEETING THE CHALLENGE OF INCREASED DEMAND WITH HOME CARE
With the rising incidence of knee arthritis, the demand for TKR is expected to more than double in the coming years.14 This increased utilization is driven by an aging population that desires to remain active, as well as by evidence suggesting health benefits associated with increased activity levels. Along with these demographic and utilization trends, another evolution in joint replacement derives from patients’ expectation of continuously improving results. Patients measure the success of TKR not only by relative reduction in pain, but also by other outcome metrics, including, importantly, return to sport or work.5,7 The tandem challenge posed by increased demand for services and increased patient expectations regarding outcomes is testing health care providers as they consider the resources that will be required to meet the demand.
Health care systems, payers, and physicians are looking for ways to more efficiently meet this growing need for TKR services in the context of finite health care resources subject to competing demand from several clinical entities. Regardless of TKR’s record of clinical success, the resources applied to this orthopedic intervention come at the expense of the same resources being applied to other health care needs. As demand is unlikely to wane, the only rational approach is to redesign care delivery in favor of a more efficient model. In order to meet the demand with the available resources, several goals need to be achieved: fewer inpatient hospital and postacute bed days consumed by joint replacement services, better streamlined care paths, and improved engagement of the patient and his or her home-based support network. Key to this process is driving care to the home environment, provided that quality is at least comparable and cost is significantly less.3,15–17
Postoperative rehabilitation and physical therapy is essential to restoration of function after TKR. It is therefore no surprise that rehabilitation and physical therapy make up a significant proportion of the home care services for this patient population.8,17,18 Among its advantages, therapy in the home environment gives the therapist the opportunity to identify and address the patient’s unique needs in his or her own home. In addition, family and other support personnel often feel more comfortable assuming responsibility for assisting with care in a familiar setting. Tailored therapy in the home setting can improve safety and satisfaction and speed the resumption of ADL; it is increasingly seen as an essential component of the care path.4,11
Recently, care path designs have been subject to careful analyses that compare in-home rehabilitation outcomes with outcomes achieved in an inpatient environment. Observational, retrospective, and prospective study designs have confirmed that the in-home rehabilitation model of care delivery is not only viable, but in many circumstances preferable.5,10,12,17,19 The quality is comparable to inpatient care for most TKR patient populations and the cost and resource utilization intensity are considerably reduced. Such reports have lent credence to the movement to incorporate home care services into successful post–joint replacement care paths. The approach appears to have a large potential for benefit with very little risk. Strategies that aim to more rationally deliver needed rehabilitation services at home promise to keep TKR services within the reach of our strained health care resources.
THE HOME CARE CLINICAL PATH
The underlying principle of a home care clinical path is that the patient remains at the center of the program and shares in decisions about care strategies (Table). One of the greatest concerns patients have about a pending knee replacement is the duration of their expected recovery. To meet this concern, a Rapid Recovery Care Path has been developed that incorporates an integrated approach to acute and postacute care, with increased emphasis on discharging patients to their home environment as early as it appears safe to do so. The goals of a rapid recovery home-centered care program following routine TKR include reduced postoperative pain and early return to function.2,15,16 Meeting these goals minimizes the development of a vicious cycle of pain and stiffness that may lead to chronic pain and fibrosis. As a result, the patient can pursue more aggressive rehabilitation, which maintains joint range of motion, permits earlier hospital discharge and discharge to home rather than another health care facility, and improves patient satisfaction.
The Cleveland Clinic Total Knee Care Path effectively incorporates the rapid recovery approach, with home care taking the lead in discharge planning and transition of care management. Education is essential and should start early, at the time of informed consent; involve the patient and family; and continue throughout the care path.
The key to a successful outcome is patient engagement with agreed-upon principles of care, which form the basis for the care path. In the Cleveland Clinic program, patients are engaged to embrace the following goals:
- Shared decision-making
- A home care environment that includes support of family and friends
- Patient and family education to enhance shared decision-making
- Return to the home environment as soon as it is deemed safe
- Elimination of unnecessary or duplicative treatments, tests, or interventions
- Acceptance of multiple plans or paths in response to changing clinical conditions
All patients undergo a preoperative evaluation, during which they are introduced to and educated about the Rapid Recovery Total Knee Care Path. The Rapid Recovery Path accommodates planned interventions and contingencies depending on clinical course. Every patient envisions a safe return home as a primary goal, with as short an exposure to inpatient acute and postacute settings as is necessary. No fixed length of stay or discharge destination is mandated. Rather, patients are encouraged to articulate their goals, drive their discharge, and return home. Such shared decision-making empowers patients and improves satisfaction.
Factors that affect recovery are assessed through a detailed perioperative history and physical examination. The patient’s readiness for an intervention such as TKR is assessed in three phases:
- The preoperative history, physical examination, and radiographic parameters establish that appropriate indications exist in terms of diagnosis and level of disability.
- The assessment team identifies conditions that affect risk and devises plans for their perioperative management—for example, control of blood glucose or decolonization of methicillin-resistant Staphylococcus Taurus carriers. Plans are made for the perioperative as well as seamless postdischarge management of chronic conditions such as atrial fibrillation requiring anticoagulation or hypertension.
- Psychosocial factors are evaluated for their potential impact on discharge planning and postacute management. Patients must establish their ability to participate actively in their care and consider their access to family, friends, and neighbors who can assist with care management in the home. Successful management of the care episode depends on an effective and reliable advocate. If the patient is unable to perform this function, then a surrogate advocate must be identified. If this role cannot be filled, the patient will require transfer to an inpatient rehabilitation facility.
POSITIVE RESULTS, BUT REGULATORY CHALLENGES
Since our 2006 incorporation of an active postacute home care program into our rapid recovery protocol, we have observed several improved outcome metrics:
- Average acute care hospital length of stay has been reduced by an average of 0.9 days.
- Our discharge to home rate has risen from 32% to 74%. In fact, among surgeons who have fully embraced the rapid recovery protocol, the discharge to home rate is 74% compared with 45% among the remaining surgeons. The difference is statistically (P < .05) and clinically significant.
- The readmission rate for patients discharged to home using this protocol is significantly lower compared with the rate before the protocol was implemented and with the rate of a control cohort discharged to a skilled nursing facility. Patients discharged to home consume significantly fewer resources and cost the system about one-third as much as those sent to an inpatient postacute facility.
Despite these gains, the regulatory environment is not structured to reward good stewardship of health care resources. For example, current payment rules penalize institutions that achieve early discharge (less than 3 days) from an acute care hospital when the patient will be transferred to another care venue. In addition, requirements for home care can be stringent, limiting the beneficial application of therapy in the home if alternatives, such as outpatient or subacute care, exist. Fortunately, PPACA and the request for bundled pricing of episodes of care gives providers the opportunity to apply for exceptions to rules that hinder cost containment. As such, relief may be in sight.
OUTLOOK
The future is bright for care path development and incorporation of better methods to manage care episodes.20,21 Although the concept of outpatient joint replacement has been considered by some, questions remain regarding the lower limit of resources that should be applied to a given episode and how best to predict which patients can benefit from even less inpatient care. Predictive modeling based on patient-specific factors might assist in this, but prudence suggests that flexibility in care path management will always be the most important element of protection for patients. Specifically, early detection of significant clinical deviation requiring a change in venue is paramount and is routinely incorporated into any well-designed care path. The goal is not to minimize resource utilization, but rather to ensure appropriate and rational distribution of health care resources to meet the clinical needs of each patient. Refining our approaches to achieving this balance will require ongoing work and monitoring of metrics of success.
- Chimenti CE, Ingersoll G. Comparison of home health care physical therapy outcomes following total knee replacement with and without subacute rehabilitation. J Geriatr Phys Ther 2007; 30:102–108.
- Iyengar KP, Nadkarni JB, Ivanovic N, Mahale A. Targeted early rehabilitation at home after total hip and knee joint replacement: does it work? Disabil Rehabil 2007; 29:495–502
- Mitchell C, Walker J, Walters S, Morgan AB, Binns T, Mathers N. Costs and effectiveness of pre- and post-operative home physiotherapy for total knee replacement: randomized controlled trial. J Eval Clin Pract 2005; 11:283–292.
- Stevens M, van den Akker-Scheek I, Spriensma A, Boss NA, Diercks RL, van Horn JR. The Groningen Orthopedic Exit Strategy (GOES): a home-based support program for total hip and knee arthroplasty patients after shortened hospital stay. Patient Educ Couns 2004; 54:95–99.
- Tousignant M, Boissy P, Moffet H, et al. Patients’ satisfaction of healthcare services and perception with in-home telerehabilitation and physiotherapists’ satisfaction toward technology for post-knee arthroplasty: an embedded study in a randomized trial [published online ahead of print April 14, 2011]. Telemed J E Health 2011; 17:376–382. 10.1089/tmj.2010.0198
- Kramer JF, Speechley M, Bourne R, Rorabeck C, Vaz M. Comparison of clinic- and home-based rehabilitation programs after total knee arthroplasty. Clin Orthop Relat Res 2003; 410:225–234.
- Loft M, McWilliam C, Ward-Griffin C. Patient empowerment after total hip and knee replacement. Orthop Nurs 2003; 22:42–47.
- Harris MD, Candando P. The physical therapist as a member of the home healthcare team: caring for patients with replacements. Home Healthc Nurse 1998; 16:153–156.
- Collins T, Herness J, Martenas J, Roberson A. Medicare prospective payment before and after implementation: a review of visits and physical performance among Medicare home health patients after total knee replacements. Home Healthc Nurse 2007; 25:401–407.
- Mallinson TR, Bateman J, Tseng HY, et al. A comparison of discharge functional status after rehabilitation in skilled nursing, home health, and medical rehabilitation settings for patients after lower-extremity joint replacement surgery. Arch Phys Med Rehabil 2011; 92:712–720.
- Stineman MG, Chan L. Commentary on the comparative effectiveness of alternative settings for joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1257–1259.
- Lin CW, March L, Crosbie J, et al. Maximum recovery after knee replacement—the MARKER study rationale and protocol. BMC Musculoskelet Disord 2009; 10:69.
- Thomas G, Faisal M, Young S, Asson R, Ritson M, Bawale R. Early discharge after hip arthroplasty with home support: experience at a UK District General Hospital. Hip Int 2008; 18:294–300.
- Tian W, DeJong G, Brown M, Hsieh CH, Zamfirov ZP, Horn SD. Looking upstream: factors shaping the demand for postacute joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1260–1268.
- Bade MJ, Stevens-Lapsley JE. Early high-intensity rehabilitation following total knee arthroplasty improves outcomes [published online ahead of print September 30, 2011]. J Orthop Sports Phys Ther 2011; 41:932–941. 10.2519/jospt.2011.3734
- Doman DM, Gerlinger TL. Total joint arthroplasty cost savings with a rapid recovery protocol in a military medical center. Mil Med 2012; 177:64–69.
- Liebs TR, Herzberg W, Rüther W, Haasters J, Russlies M, Hassenpflug J, Multicenter Arthroplasty Aftercare Project. Multicenter randomized controlled trial comparing early versus late aquatic therapy after total hip or knee arthroplasty [published online ahead of print December 21, 2011]. Arch Phys Med Rehabil 2012; 93:192–199. 10.1016/j.apmr.2011.09.011
- Mahomed NN, Koo Seen Lin MJ, Levesque J, Lan S, Bogoch ER. Determinants and outcomes of inpatient versus home based rehabilitation following elective hip and knee replacement. J Rheumatol 2000; 27:1753–1758.
- Mahomed NN, Davis AM, Hawker G, et al. Inpatient compared with home-based rehabilitation following primary unilateral total hip or knee replacement: a randomized controlled trial. J Bone Joint Surg Am 2008; 90:1673–1680.
- Aprile I, Rizzo RS, Romanini E, et al. Group rehabilitation versus individual rehabilitation following knee and hip replacement: a pilot study with randomized, single-blind, cross-over design. Eur J Phys Rehabil Med 2011; 47:551–559.
- Russell TG, Buttrum P, Wootton R, Jull GA. Rehabilitation after total knee replacement via low-bandwidth telemedicine: the patient and therapist experience. J Telemed Telecare 2004; 10(suppl 1):85–87.
- Chimenti CE, Ingersoll G. Comparison of home health care physical therapy outcomes following total knee replacement with and without subacute rehabilitation. J Geriatr Phys Ther 2007; 30:102–108.
- Iyengar KP, Nadkarni JB, Ivanovic N, Mahale A. Targeted early rehabilitation at home after total hip and knee joint replacement: does it work? Disabil Rehabil 2007; 29:495–502
- Mitchell C, Walker J, Walters S, Morgan AB, Binns T, Mathers N. Costs and effectiveness of pre- and post-operative home physiotherapy for total knee replacement: randomized controlled trial. J Eval Clin Pract 2005; 11:283–292.
- Stevens M, van den Akker-Scheek I, Spriensma A, Boss NA, Diercks RL, van Horn JR. The Groningen Orthopedic Exit Strategy (GOES): a home-based support program for total hip and knee arthroplasty patients after shortened hospital stay. Patient Educ Couns 2004; 54:95–99.
- Tousignant M, Boissy P, Moffet H, et al. Patients’ satisfaction of healthcare services and perception with in-home telerehabilitation and physiotherapists’ satisfaction toward technology for post-knee arthroplasty: an embedded study in a randomized trial [published online ahead of print April 14, 2011]. Telemed J E Health 2011; 17:376–382. 10.1089/tmj.2010.0198
- Kramer JF, Speechley M, Bourne R, Rorabeck C, Vaz M. Comparison of clinic- and home-based rehabilitation programs after total knee arthroplasty. Clin Orthop Relat Res 2003; 410:225–234.
- Loft M, McWilliam C, Ward-Griffin C. Patient empowerment after total hip and knee replacement. Orthop Nurs 2003; 22:42–47.
- Harris MD, Candando P. The physical therapist as a member of the home healthcare team: caring for patients with replacements. Home Healthc Nurse 1998; 16:153–156.
- Collins T, Herness J, Martenas J, Roberson A. Medicare prospective payment before and after implementation: a review of visits and physical performance among Medicare home health patients after total knee replacements. Home Healthc Nurse 2007; 25:401–407.
- Mallinson TR, Bateman J, Tseng HY, et al. A comparison of discharge functional status after rehabilitation in skilled nursing, home health, and medical rehabilitation settings for patients after lower-extremity joint replacement surgery. Arch Phys Med Rehabil 2011; 92:712–720.
- Stineman MG, Chan L. Commentary on the comparative effectiveness of alternative settings for joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1257–1259.
- Lin CW, March L, Crosbie J, et al. Maximum recovery after knee replacement—the MARKER study rationale and protocol. BMC Musculoskelet Disord 2009; 10:69.
- Thomas G, Faisal M, Young S, Asson R, Ritson M, Bawale R. Early discharge after hip arthroplasty with home support: experience at a UK District General Hospital. Hip Int 2008; 18:294–300.
- Tian W, DeJong G, Brown M, Hsieh CH, Zamfirov ZP, Horn SD. Looking upstream: factors shaping the demand for postacute joint replacement rehabilitation. Arch Phys Med Rehabil 2009; 90:1260–1268.
- Bade MJ, Stevens-Lapsley JE. Early high-intensity rehabilitation following total knee arthroplasty improves outcomes [published online ahead of print September 30, 2011]. J Orthop Sports Phys Ther 2011; 41:932–941. 10.2519/jospt.2011.3734
- Doman DM, Gerlinger TL. Total joint arthroplasty cost savings with a rapid recovery protocol in a military medical center. Mil Med 2012; 177:64–69.
- Liebs TR, Herzberg W, Rüther W, Haasters J, Russlies M, Hassenpflug J, Multicenter Arthroplasty Aftercare Project. Multicenter randomized controlled trial comparing early versus late aquatic therapy after total hip or knee arthroplasty [published online ahead of print December 21, 2011]. Arch Phys Med Rehabil 2012; 93:192–199. 10.1016/j.apmr.2011.09.011
- Mahomed NN, Koo Seen Lin MJ, Levesque J, Lan S, Bogoch ER. Determinants and outcomes of inpatient versus home based rehabilitation following elective hip and knee replacement. J Rheumatol 2000; 27:1753–1758.
- Mahomed NN, Davis AM, Hawker G, et al. Inpatient compared with home-based rehabilitation following primary unilateral total hip or knee replacement: a randomized controlled trial. J Bone Joint Surg Am 2008; 90:1673–1680.
- Aprile I, Rizzo RS, Romanini E, et al. Group rehabilitation versus individual rehabilitation following knee and hip replacement: a pilot study with randomized, single-blind, cross-over design. Eur J Phys Rehabil Med 2011; 47:551–559.
- Russell TG, Buttrum P, Wootton R, Jull GA. Rehabilitation after total knee replacement via low-bandwidth telemedicine: the patient and therapist experience. J Telemed Telecare 2004; 10(suppl 1):85–87.
Home-based care for heart failure: Cleveland Clinic’s “Heart Care at Home” transitional care program
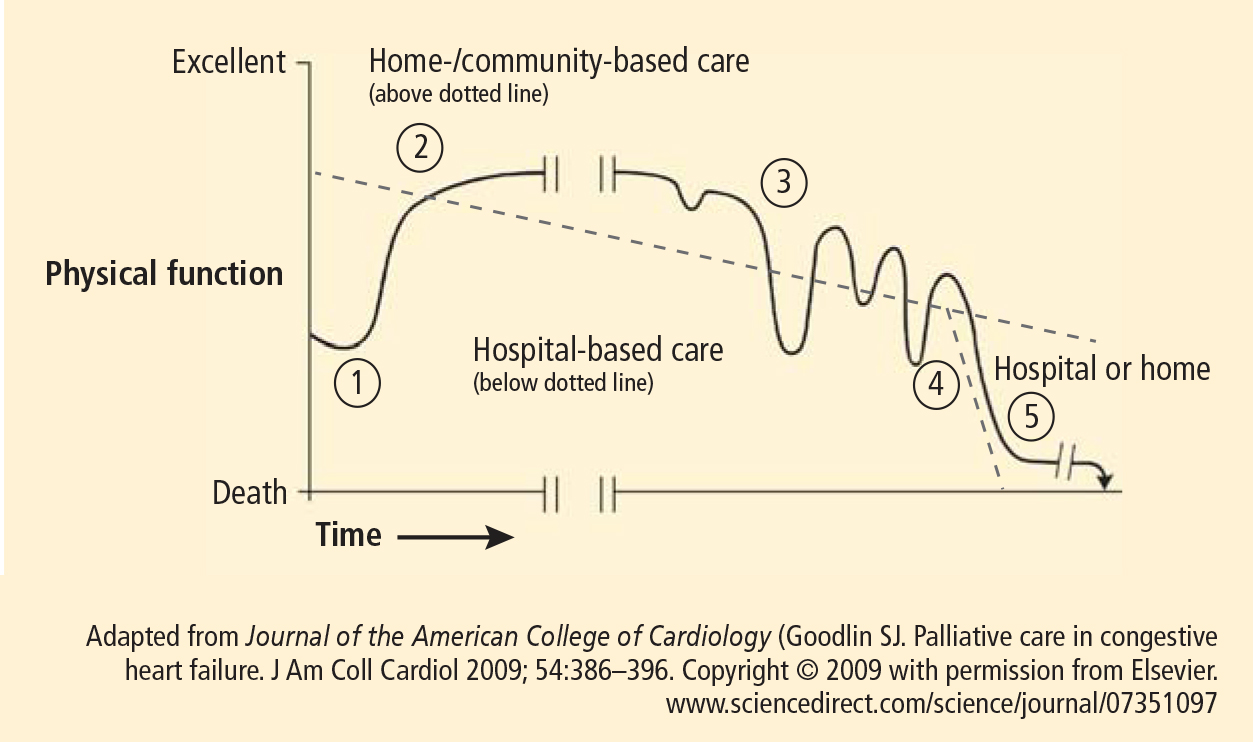
The home is the most important context of care for individuals with chronic heart failure and yet it is the least accessible to caregivers. Patients often struggle to manage a complex regimen of medications, follow an unfamiliar diet, monitor weight and vital signs, and work to coordinate care among various providers who, in some cases, fail to communicate effectively. Heart failure patients do all this while making difficult decisions about their livelihoods, social condition, and future direction. With progression of the disease and comorbidity, these patients often experience a downward cycle of repeat hospitalization and worsening functional capacity (Figure 1). Each subsequent transition from acute care to home becomes incrementally more difficult to manage.
According to the latest American College of Cardiology/American Heart Association Guidelines for the Diagnosis and Management of Heart Failure in Adults, appropriate care for patients with heart failure should include:
- Intensive patient education
- Encouragement of patients to be more aggressive participants in their care
- Close monitoring of patients through telephone follow-up or home nursing
- Careful review of medications to improve adherence to evidence-based guidelines
- Multidisciplinary care with nurse case management directed by a physician1
Beyond these general suggestions, recommendations about specific approaches and models of care in the home are lacking.
Contemporary research suggests that postacute, home-based care of heart failure patients may yield outcomes similar to those of clinic-based outpatient care. Results of the Which Heart Failure Intervention is Most Cost-Effective & Consumer-Friendly in Reducing Hospital Care (WHICH?) trial support this hypothesis. This multicenter, randomized clinical trial (n = 280) compared home- with clinic-based multidisciplinary management for postacute heart failure patients.2 Investigators compared outcomes in patients managed at a heart failure clinic with those managed at home. They found that postdischarge home visits by heart failure nurses did not significantly alter the primary composite end point of death or unplanned rehospitalization from any cause over 18 months (hazard ratio [HR] 0.97, 95% confidence interval [CI] 0.73–1.30, P = .8621). The rate of unplanned and total hospitalization was also similar in the two groups. However, the average length of hospital stay was significantly lower in the home care group (4 days) than in the clinic-based group (6 days); P = .004. A cost-effectiveness analysis is planned but has not yet been presented.
HEART CARE AT HOME
At Cleveland Clinic, our group of physicians (geriatrics and cardiology), nurses, nurse practitioners, and hospital administrators founded a primarily home-based postacute transitional care program in 2010 called “Heart Care at Home.” The design of our program was influenced by Coleman et al’s care transitions interventions program,3 Naylor et al’s transitional care intervention,4 and the contemporary remote monitoring literature.5 The program focuses primarily on older adults hospitalized for heart failure who are transitioning from hospital to home. In our model:
- Inpatient care advocates identify candidates during the index inpatient stay, introduce a model of care, and begin a coaching intervention.
- After discharge, home liaisons visit the patient at home, continue coaching intervention, and teach the patient to use the newly installed remote monitoring equipment.
- For 30 to 40 days after discharge, a team of telehealth nurses monitors the patient, makes contact with him or her weekly in order to reinforce coaching intervention, coordinates care, and tracks outcomes.
- Nurse practitioners experienced both in home care and heart failure provide clinical oversight and leadership and visit the highest-acuity patients at home.
To date, the program has provided care in more than 2,100 patient encounters, with approximately 50 to 80 patients actively enrolled at any time. We identified potential program candidates using a digital list tool embedded in Cleveland Clinic’s electronic medical record (EMR) system. This tool was developed by our team together with an internal business intelligence team. We have been approximately 65% successful in identifying eligible inpatients. Patients enrolled in our transitional care program tend to be older, have longer hospital stays, and have more comorbidities than other older adults hospitalized at Cleveland Clinic for similar reasons.
Following index hospital discharge, our home liaisons have been able to make an initial home visit after a median of 2 days (25th to 75th percentile: 1 to 3 days). Patients thought to be at higher risk for hospital readmissions have been seen at home by our nurse practitioners within the first week of discharge. The most common challenge that our at-home team members have faced relates to patients’ medications (for example, unfilled prescriptions and errors in utilization). On many occasions our at-home team has succeeded in transitioning patients not benefiting from care at home to nonhospital venues (skilled nursing facilities, chronic care facilities, inpatient hospice) or to higher levels of at-home care (at-home physician visits, home-care nursing and therapy, at-home hospice).
To date, patients have been enrolled in our program for a median of 30 days (25th to 75th percentile: 20 to 35 days). We have observed an increased level of patient satisfaction. Among heart failure patients enrolled in our program for the first time, we have observed a lower readmission rate compared with publicly reported Cleveland Clinic rates (24.5% vs 28.2%). However, there are several ongoing challenges in the care of heart failure patients in the home environment. These relate to longitudinal care across venues, cross training of providers, and home monitoring.
Longitudinal care across venues
Our program aims to address the lack of integrated care over time and between care venues. This problem lies at the intersection of health care reimbursement policy and clinical practice. Currently, the hospital reimbursement system does not encourage care coordination across settings. The system has, in fact, evolved into a string of disconnected care providers who act as “toll booths” providing services for a fee in isolation from other providers. Coleman and colleagues have documented the complexity of the transitions among these care providers for older patients with chronic disease, noting the implications for patient safety and cost.6
Hospitals receive a fixed payment for an inpatient admission, which increases the financial incentive to discharge patients faster to other venues of care. The study by Bueno et al of a Medicare population treated between 1993 and 2006 confirms that such a trend exists for heart failure patients.7 The authors found a steady decrease in the mean length of hospital stay from 8.81 days to 6.33 days over the study period (28% relative reduction, P < .001). During this same period, the 30-day all-cause readmission rate increased from 17.2% to 20.1% (a 17% relative increase, P < .001) with an associated 10% relative reduction in the proportion of patients discharged to home.7 Experience in other populations with heart failure, such as patients in the Veterans Affairs health care system, has shown similar trends in length of hospital stay and readmissions.8
During these transitions, information is often lost in the handoff from the discharging hospital to the next venue of care. Medication management is the most common problem area with the potential for patient noncompliance with prescriptions,9,10 which can have serious deleterious effects on quality and safety. Forster et al found that 66% of untoward outcomes in discharged patients were due to adverse drug events.11 Similarly, Gray et al identified adverse drug events in 20% of patients discharged from hospital to home with home health care services.12
In the Cleveland Clinic Health System, we are coupling our “Heart Care at Home” transitional care program with an aggressive plan to develop a more comprehensive cross-venue EMR. Connecting the hospital EMR with our health system–owned home health agency will enable a consistent medication record and communication system for patients transitioning from our hospitals to Cleveland Clinic home care services (nearly 20,000 patients per year).
Despite these issues, several care transition interventions have shown promising clinical and economic results. Coleman and colleagues conducted a randomized, controlled trial of a transition coaching model in which patients and caregivers were encouraged to take a more active role in care transitions. Results of this trial showed a significant decrease in 30- and 90-day rehospitalizations (the 90-day read-mission rate in the treatment group was 16.7 vs 22.5 in the control group, P = .04) with associated cost savings.3 Voss et al showed similar results in reduction of readmissions in a nonintegrated delivery system.13 Additionally, telephone-based chronic disease management programs have been shown to be cost-effective in chronically ill Medicare patients.14
When will the clinical evidence behind care transitions and financial incentives converge to create an atmosphere conducive to more optimal care coordination? Today, this question remains unanswered. Health care reform, with the passage of the Patient Protection and Affordable Care Act (PPACA) (http://housedocs.house.gov/energycommerce/ppacacon.pdf), may spur the creation of programs to increase incentives for care coordination. These include a move to episodic reimbursement that would bundle payments for acute and postacute care, thus creating more incentives for coordinating care across settings. The “Bundled Payments for Care Improvement” project run by the Center for Medicare & Medicaid Innovation will test different models and approaches to bundled payments (http://innovations.cms.gov/initiatives/bundled-payments). Additionally, beginning in fiscal year 2013, Medicare will penalize hospitals that have high readmission rates for heart failure, acute myocardial infarction, and pneumonia with a financial risk of up to 3% of total hospital Medicare payments by year 3 of the program.
The PPACA will have a significant effect on home-based care for older adults with chronic conditions. The PPACA reforms will likely lead to more patients being treated at home (the lower-cost care setting), ideally under the care of highly skilled teams. Payment reforms will also create new incentives for providers to better coordinate care, keep patients healthy at home, and avoid the “toll-booth” description entirely, enabling providers to focus on patient care. However, more research and experimentation are required to streamline the elements on the transitions spectrum in order to create the most value for specific patient populations. New infrastructure, use of technology, changing culture, and dedicated clinical teams will be necessary to deliver on the hopes of more integrated longitudinal care across venues.
Cross training of providers
Older community-dwelling adults with heart failure exhibit more health instability; take more medications; have more comorbidities; and receive more nursing, homemaking, and meal services than do other home care clients.15 Nurses thus have a unique opportunity to improve outcomes for home-based heart failure patients,16,17 but are often insufficiently trained to do so. Delaney et al administered a validated 20-item heart failure knowledge questionnaire to 94 home care nurses from four different home care agencies.18 The investigators found a 79% knowledge level in overall heart failure education principles, with lowest scores related to issues of asymptomatic hypotension (25% answered correctly), daily weight monitoring (27%), and transient dizziness (31%). Nurses with poorer heart failure–related knowledge may partially explain worse process and outcome measures among this patient population.19
The home-based nursing workforce of the future, and specifically nurses who care for heart failure patients at home, will need to be better trained and specialized in issues relating both to home-based nursing and medical heart failure. These “hybrid nurses” should be allowed a central clinical leadership role among their peers, as they will need to be empowered to make medical and care coordination decisions.
At our center, hybrid-trained home care/heart failure nurse practitioners make home visits for higher-acuity home-based patients and provide clinical leadership and support for other home care nurses. These nurse practitioners have been instrumental in identifying and correcting heart failure medication–related problems, as well as effectively coordinating care. Examples include: independently prescribing and coordinating administration of intravenous diuretics at home for patients who have difficulty managing volume overload, avoiding hospital readmissions by transitioning ill patients to a skilled nursing facility or an at-home hospice, and effectively educating patients and families about appropriate heart failure self-care.
Home monitoring
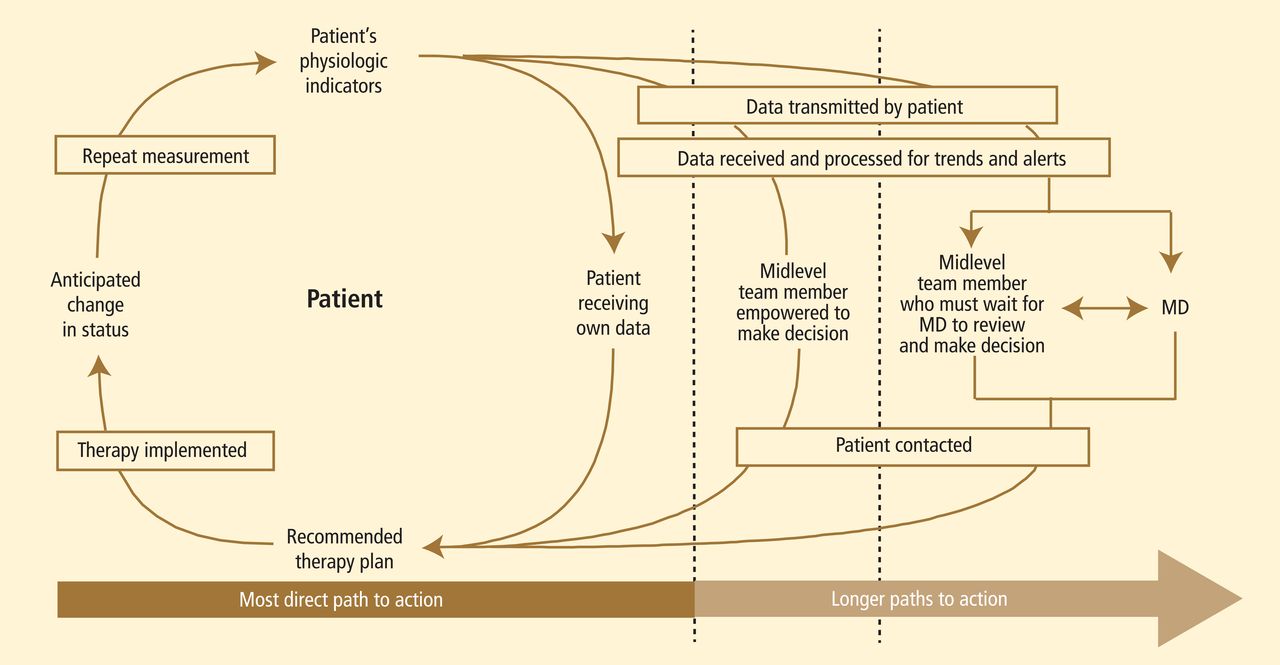
Home monitoring of selected physiologic parameters and patient-reported health status measures among heart failure patients may facilitate early detection of clinical deterioration and direct timely intervention to prevent adverse outcomes.20 Desai and Stevenson have previously proposed the “circle from home to heart-failure disease management,” a concept illustrating how home monitoring can be embedded in a comprehensive heart failure management approach (Figure 2).20 This concept emphasizes the following:
- Home monitoring should facilitate early detection of clinical deterioration.20
- Home monitoring data will most directly lead to action if the data can be used by the patient to improve self-care.
- In the setting of multidisciplinary care, data should be remotely transmitted to a midlevel team, preferably one empowered to make therapeutic decisions.
- Further engagement of physicians or other clinical providers may be beneficial but will delay the clinical response.
The most commonly monitored physiologic parameter of heart failure patients is daily weight. While nearly universally used, this parameter is in fact a poor surrogate for subclinical hemodynamic congestion and has poor diagnostic performance for clinical decompensation. Results are conflicting from studies evaluating the utility of daily body weight measurements in patients with heart failure who are being cared for in the home environment.
In one study, an increase in body weight of > 2 kg over 24 to 72 hours had a 9% sensitivity for detecting clinical deterioration.21 In another study, Chaudhry et al performed a nested case-control trial in 134 patients with heart failure and 134 matched controls referred to a home monitoring system by managed care organizations. The researchers found that increases in body weight were associated with hospitalization for heart failure and that the increases began at least 1 week before admission.22 However, they did not investigate whether the use of this information by clinicians altered outcomes. In a prior randomized clinical trial of symptom monitoring versus transtelephonic body weight monitoring in patients with symptomatic heart failure, the Weight Monitoring in Heart Failure trial (n = 280), weight monitoring did not result in improvement in the primary outcome of hospitalizations for heart failure over a 6-month period.23
The ideal monitoring parameters in heart failure patients may include direct hemodynamic measurements from the right ventricular outflow tract,24 pulmonary artery,25 or left atrium,26 using implantable devices. For example, the CHAMPION (CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Patients) trial (n = 550) was a randomized, single-blind, industry-sponsored trial of heart failure management guided by physiologic hemodynamic data derived from a percutaneously inserted pulmonary artery hemodynamic monitor (Champion HF Monitoring System; CardioMEMS, Atlanta, Georgia). The researchers found that monitoring these parameters was associated with a 28% reduction in heart failure–related hospitalizations during the first 6 months (rate 0.32 vs 0.44, HR 0.72, 95% CI 0.60–0.85, P = .0002) compared with usual care.25 At 6 months, the freedom from device- or system-related complications was 98.6%.
Despite success in the trial, the US Food and Drug Administration Circulatory System Devices Panel voted against approving the device. The panel was concerned that the e-mail–alert and care systems built into the intervention arm of the trial created bias in favor of the device, and that in a real-world situation it may not be as effective. This demonstrates the ongoing challenges and barriers to adoption of invasive hemodynamic monitoring.
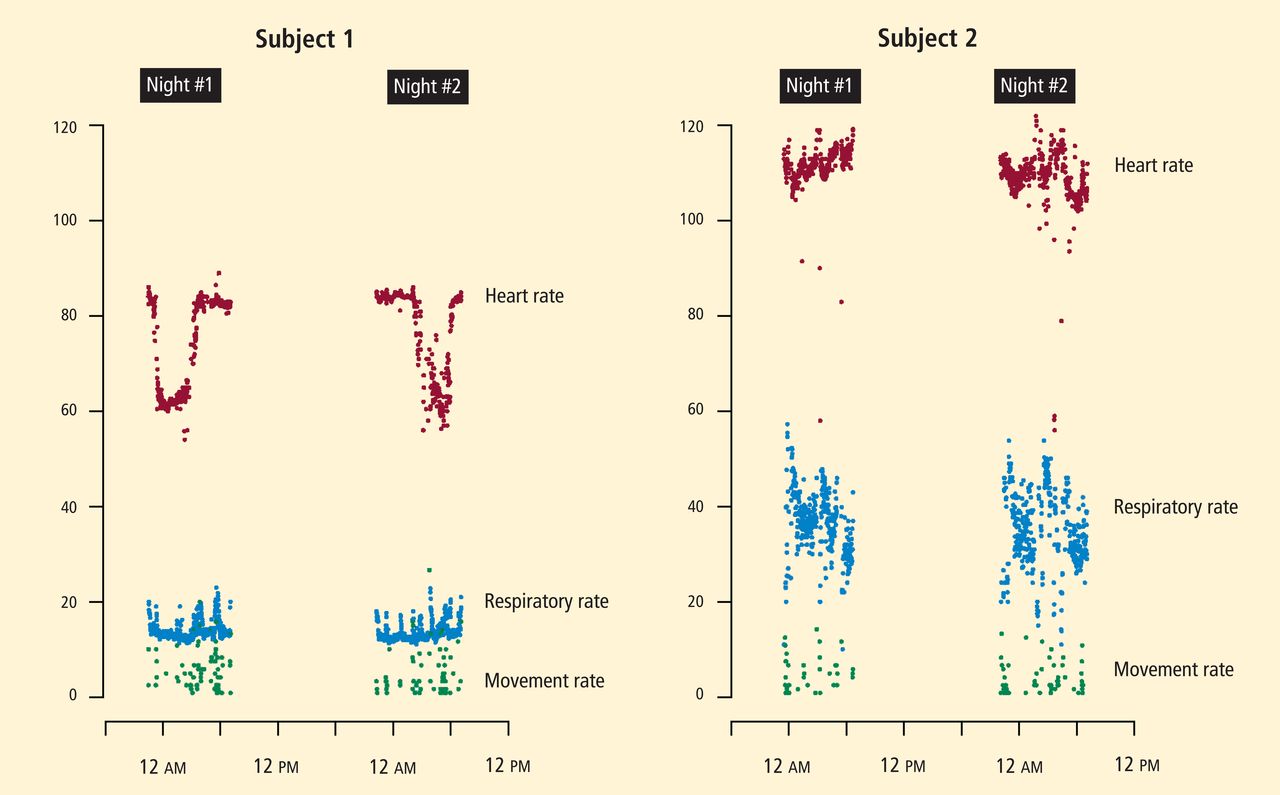
At our center, we are conducting an institutional review board–approved investigation of an entirely noninvasive under-the-mattress piezoelectric monitor in a cohort of postacute heart failure patients. Piezoelectricity is the charge that accumulates in certain solid materials in response to mechanical stress. Common applications of piezoelectricity include microphones, push-start propane barbecues, and cigarette lighters. The device under investigation (EverOn; EarlySense, Ramat-Gan, Israel) detects heart rate, respiratory rate, and movement rate through vibrations of the mattress. Case examples are shown in Figure 3. Whether such monitoring technology will play a future role in the home environment remains to be seen.
SUMMARY
At the time of this writing, the Supreme Court of the United States has reaffirmed the constitutionality of the PPACA, clearing the way for implementation of significant changes in the US health care delivery system. The implications for in-home care for older adults with chronic conditions, including heart failure, are significant. The home will become an increasingly common venue of postacute care. Today is the time to investigate beneficial models of care and optimal uses of technology, and to develop a specialized mobile workforce that will confidently care for individuals with heart failure at home, responsibly and at lower cost.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 119:e391–e479.
- Stewart S, Carrington MJ, Marwick TH, et al. Impact of home versus clinic-based management of chronic heart failure: the WHICH? (Which Heart Failure Intervention Is Most Cost-Effective and Consumer Friendly in Reducing Hospital Care) multicenter, randomized trial. J Am Coll Cardiol 2012; 60:1239–1248.
- Coleman EA, Parry C, Chalmers S, Min S-J. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Klersy C, De Silvestri A, Gabutti G, Regoli F, Auricchio A. A meta-analysis of remote monitoring of heart failure patients. J Am Coll Cardiol 2009; 54:1683–1694.
- Coleman EA, Min S-J, Chomiak A, Kramer AM. Posthospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993–2006. JAMA 2010; 303:2141–2147.
- Heidenreich PA, Sahay A, Kapoor JR, Pham MX, Massie B. Divergent trends in survival and readmission following a hospitalization for heart failure in the Veterans Affairs health care system 2002 to 2006. J Am Coll Cardiol 2010; 56:362–368.
- Moore C, Wisnivesky J, Williams S, McGinn T. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med 2003; 18:646–651.
- Coleman EA, Smith JD, Raha D, Min S-J. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Gray SL, Mahoney JE, Blough DK. Adverse drug events in elderly patients receiving home health services following hospital discharge. Ann Pharmacother 1999; 33:1147–1153.
- Voss R, Gardner R, Baier R, Butterfeld K, Lehrman S, Gravenstein S. The care transitions intervention: translating from efficacy to effectiveness. Arch Intern Med 2011; 171:1232–1237.
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare benefciaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Foebel AD, Hirdes JP, Heckman GA, Tyas SL, Tjam EY. A profile of older community-dwelling home care clients with heart failure in Ontario. Chronic Dis Can 2011; 31:49–57.
- Krumholz HM, Amatruda J, Smith GL, et al. Randomized trial of an education and support intervention to prevent readmission of patients with heart failure. J Am Coll Cardiol 2002; 39:83–89.
- González B, Lupón J, Herreros J, et al. Patient’s education by nurse: what we really do achieve? Eur J Cardiovasc Nurs 2005; 4:107–111.
- Delaney C, Apostolidis B, Lachapelle L, Fortinsky R. Home care nurses’ knowledge of evidence-based education topics for management of heart failure. Heart Lung 2011; 40:285–292.
- Foebel AD, Heckman GA, Hirdes JP, et al. Clinical, demographic and functional characteristics associated with pharmacotherapy for heart failure in older home care clients: a retrospective, population-level, cross-sectional study. Drugs Aging 2011; 28:561–573.
- Desai AS, Stevenson LW. Connecting the circle from home to heart-failure disease management. N Engl J Med 2010; 363:2364–2367.
- Lewin J, Ledwidge M, O’Loughlin C, McNally C, McDonald K. Clinical deterioration in established heart failure: what is the value of BNP and weight gain in aiding diagnosis? Eur J Heart Fail 2005; 7:953–957.
- Chaudhry SI, Wang Y, Concato J, Gill TM, Krumholz HM. Patterns of weight change preceding hospitalization for heart failure. Circulation 2007; 116:1549–1554.
- Goldberg LR, Piette JD, Walsh MN, et al. Randomized trial of a daily electronic home monitoring system in patients with advanced heart failure: the Weight Monitoring in Heart Failure (WHARF) trial. Am Heart J 2003; 146:705–712.
- Bourge RC, Abraham WT, Adamson PB, et al. Randomized controlled trial of an implantable continuous hemodynamic monitor in patients with advanced heart failure: the Compass-HF study. J Am Coll Cardiol 2008; 51:1073–1079.
- Abraham WT, Adamson PB, Bourge RC, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet 2011; 377:658–666.
- Ritzema J, Troughton R, Melton I, et al. Physician-directed patient self-management of left atrial pressure in advanced chronic heart failure. Circulation 2010; 121:1086–1095.
The home is the most important context of care for individuals with chronic heart failure and yet it is the least accessible to caregivers. Patients often struggle to manage a complex regimen of medications, follow an unfamiliar diet, monitor weight and vital signs, and work to coordinate care among various providers who, in some cases, fail to communicate effectively. Heart failure patients do all this while making difficult decisions about their livelihoods, social condition, and future direction. With progression of the disease and comorbidity, these patients often experience a downward cycle of repeat hospitalization and worsening functional capacity (Figure 1). Each subsequent transition from acute care to home becomes incrementally more difficult to manage.
According to the latest American College of Cardiology/American Heart Association Guidelines for the Diagnosis and Management of Heart Failure in Adults, appropriate care for patients with heart failure should include:
- Intensive patient education
- Encouragement of patients to be more aggressive participants in their care
- Close monitoring of patients through telephone follow-up or home nursing
- Careful review of medications to improve adherence to evidence-based guidelines
- Multidisciplinary care with nurse case management directed by a physician1
Beyond these general suggestions, recommendations about specific approaches and models of care in the home are lacking.
Contemporary research suggests that postacute, home-based care of heart failure patients may yield outcomes similar to those of clinic-based outpatient care. Results of the Which Heart Failure Intervention is Most Cost-Effective & Consumer-Friendly in Reducing Hospital Care (WHICH?) trial support this hypothesis. This multicenter, randomized clinical trial (n = 280) compared home- with clinic-based multidisciplinary management for postacute heart failure patients.2 Investigators compared outcomes in patients managed at a heart failure clinic with those managed at home. They found that postdischarge home visits by heart failure nurses did not significantly alter the primary composite end point of death or unplanned rehospitalization from any cause over 18 months (hazard ratio [HR] 0.97, 95% confidence interval [CI] 0.73–1.30, P = .8621). The rate of unplanned and total hospitalization was also similar in the two groups. However, the average length of hospital stay was significantly lower in the home care group (4 days) than in the clinic-based group (6 days); P = .004. A cost-effectiveness analysis is planned but has not yet been presented.
HEART CARE AT HOME
At Cleveland Clinic, our group of physicians (geriatrics and cardiology), nurses, nurse practitioners, and hospital administrators founded a primarily home-based postacute transitional care program in 2010 called “Heart Care at Home.” The design of our program was influenced by Coleman et al’s care transitions interventions program,3 Naylor et al’s transitional care intervention,4 and the contemporary remote monitoring literature.5 The program focuses primarily on older adults hospitalized for heart failure who are transitioning from hospital to home. In our model:
- Inpatient care advocates identify candidates during the index inpatient stay, introduce a model of care, and begin a coaching intervention.
- After discharge, home liaisons visit the patient at home, continue coaching intervention, and teach the patient to use the newly installed remote monitoring equipment.
- For 30 to 40 days after discharge, a team of telehealth nurses monitors the patient, makes contact with him or her weekly in order to reinforce coaching intervention, coordinates care, and tracks outcomes.
- Nurse practitioners experienced both in home care and heart failure provide clinical oversight and leadership and visit the highest-acuity patients at home.
To date, the program has provided care in more than 2,100 patient encounters, with approximately 50 to 80 patients actively enrolled at any time. We identified potential program candidates using a digital list tool embedded in Cleveland Clinic’s electronic medical record (EMR) system. This tool was developed by our team together with an internal business intelligence team. We have been approximately 65% successful in identifying eligible inpatients. Patients enrolled in our transitional care program tend to be older, have longer hospital stays, and have more comorbidities than other older adults hospitalized at Cleveland Clinic for similar reasons.
Following index hospital discharge, our home liaisons have been able to make an initial home visit after a median of 2 days (25th to 75th percentile: 1 to 3 days). Patients thought to be at higher risk for hospital readmissions have been seen at home by our nurse practitioners within the first week of discharge. The most common challenge that our at-home team members have faced relates to patients’ medications (for example, unfilled prescriptions and errors in utilization). On many occasions our at-home team has succeeded in transitioning patients not benefiting from care at home to nonhospital venues (skilled nursing facilities, chronic care facilities, inpatient hospice) or to higher levels of at-home care (at-home physician visits, home-care nursing and therapy, at-home hospice).
To date, patients have been enrolled in our program for a median of 30 days (25th to 75th percentile: 20 to 35 days). We have observed an increased level of patient satisfaction. Among heart failure patients enrolled in our program for the first time, we have observed a lower readmission rate compared with publicly reported Cleveland Clinic rates (24.5% vs 28.2%). However, there are several ongoing challenges in the care of heart failure patients in the home environment. These relate to longitudinal care across venues, cross training of providers, and home monitoring.
Longitudinal care across venues
Our program aims to address the lack of integrated care over time and between care venues. This problem lies at the intersection of health care reimbursement policy and clinical practice. Currently, the hospital reimbursement system does not encourage care coordination across settings. The system has, in fact, evolved into a string of disconnected care providers who act as “toll booths” providing services for a fee in isolation from other providers. Coleman and colleagues have documented the complexity of the transitions among these care providers for older patients with chronic disease, noting the implications for patient safety and cost.6
Hospitals receive a fixed payment for an inpatient admission, which increases the financial incentive to discharge patients faster to other venues of care. The study by Bueno et al of a Medicare population treated between 1993 and 2006 confirms that such a trend exists for heart failure patients.7 The authors found a steady decrease in the mean length of hospital stay from 8.81 days to 6.33 days over the study period (28% relative reduction, P < .001). During this same period, the 30-day all-cause readmission rate increased from 17.2% to 20.1% (a 17% relative increase, P < .001) with an associated 10% relative reduction in the proportion of patients discharged to home.7 Experience in other populations with heart failure, such as patients in the Veterans Affairs health care system, has shown similar trends in length of hospital stay and readmissions.8
During these transitions, information is often lost in the handoff from the discharging hospital to the next venue of care. Medication management is the most common problem area with the potential for patient noncompliance with prescriptions,9,10 which can have serious deleterious effects on quality and safety. Forster et al found that 66% of untoward outcomes in discharged patients were due to adverse drug events.11 Similarly, Gray et al identified adverse drug events in 20% of patients discharged from hospital to home with home health care services.12
In the Cleveland Clinic Health System, we are coupling our “Heart Care at Home” transitional care program with an aggressive plan to develop a more comprehensive cross-venue EMR. Connecting the hospital EMR with our health system–owned home health agency will enable a consistent medication record and communication system for patients transitioning from our hospitals to Cleveland Clinic home care services (nearly 20,000 patients per year).
Despite these issues, several care transition interventions have shown promising clinical and economic results. Coleman and colleagues conducted a randomized, controlled trial of a transition coaching model in which patients and caregivers were encouraged to take a more active role in care transitions. Results of this trial showed a significant decrease in 30- and 90-day rehospitalizations (the 90-day read-mission rate in the treatment group was 16.7 vs 22.5 in the control group, P = .04) with associated cost savings.3 Voss et al showed similar results in reduction of readmissions in a nonintegrated delivery system.13 Additionally, telephone-based chronic disease management programs have been shown to be cost-effective in chronically ill Medicare patients.14
When will the clinical evidence behind care transitions and financial incentives converge to create an atmosphere conducive to more optimal care coordination? Today, this question remains unanswered. Health care reform, with the passage of the Patient Protection and Affordable Care Act (PPACA) (http://housedocs.house.gov/energycommerce/ppacacon.pdf), may spur the creation of programs to increase incentives for care coordination. These include a move to episodic reimbursement that would bundle payments for acute and postacute care, thus creating more incentives for coordinating care across settings. The “Bundled Payments for Care Improvement” project run by the Center for Medicare & Medicaid Innovation will test different models and approaches to bundled payments (http://innovations.cms.gov/initiatives/bundled-payments). Additionally, beginning in fiscal year 2013, Medicare will penalize hospitals that have high readmission rates for heart failure, acute myocardial infarction, and pneumonia with a financial risk of up to 3% of total hospital Medicare payments by year 3 of the program.
The PPACA will have a significant effect on home-based care for older adults with chronic conditions. The PPACA reforms will likely lead to more patients being treated at home (the lower-cost care setting), ideally under the care of highly skilled teams. Payment reforms will also create new incentives for providers to better coordinate care, keep patients healthy at home, and avoid the “toll-booth” description entirely, enabling providers to focus on patient care. However, more research and experimentation are required to streamline the elements on the transitions spectrum in order to create the most value for specific patient populations. New infrastructure, use of technology, changing culture, and dedicated clinical teams will be necessary to deliver on the hopes of more integrated longitudinal care across venues.
Cross training of providers
Older community-dwelling adults with heart failure exhibit more health instability; take more medications; have more comorbidities; and receive more nursing, homemaking, and meal services than do other home care clients.15 Nurses thus have a unique opportunity to improve outcomes for home-based heart failure patients,16,17 but are often insufficiently trained to do so. Delaney et al administered a validated 20-item heart failure knowledge questionnaire to 94 home care nurses from four different home care agencies.18 The investigators found a 79% knowledge level in overall heart failure education principles, with lowest scores related to issues of asymptomatic hypotension (25% answered correctly), daily weight monitoring (27%), and transient dizziness (31%). Nurses with poorer heart failure–related knowledge may partially explain worse process and outcome measures among this patient population.19
The home-based nursing workforce of the future, and specifically nurses who care for heart failure patients at home, will need to be better trained and specialized in issues relating both to home-based nursing and medical heart failure. These “hybrid nurses” should be allowed a central clinical leadership role among their peers, as they will need to be empowered to make medical and care coordination decisions.
At our center, hybrid-trained home care/heart failure nurse practitioners make home visits for higher-acuity home-based patients and provide clinical leadership and support for other home care nurses. These nurse practitioners have been instrumental in identifying and correcting heart failure medication–related problems, as well as effectively coordinating care. Examples include: independently prescribing and coordinating administration of intravenous diuretics at home for patients who have difficulty managing volume overload, avoiding hospital readmissions by transitioning ill patients to a skilled nursing facility or an at-home hospice, and effectively educating patients and families about appropriate heart failure self-care.
Home monitoring
Home monitoring of selected physiologic parameters and patient-reported health status measures among heart failure patients may facilitate early detection of clinical deterioration and direct timely intervention to prevent adverse outcomes.20 Desai and Stevenson have previously proposed the “circle from home to heart-failure disease management,” a concept illustrating how home monitoring can be embedded in a comprehensive heart failure management approach (Figure 2).20 This concept emphasizes the following:
- Home monitoring should facilitate early detection of clinical deterioration.20
- Home monitoring data will most directly lead to action if the data can be used by the patient to improve self-care.
- In the setting of multidisciplinary care, data should be remotely transmitted to a midlevel team, preferably one empowered to make therapeutic decisions.
- Further engagement of physicians or other clinical providers may be beneficial but will delay the clinical response.
The most commonly monitored physiologic parameter of heart failure patients is daily weight. While nearly universally used, this parameter is in fact a poor surrogate for subclinical hemodynamic congestion and has poor diagnostic performance for clinical decompensation. Results are conflicting from studies evaluating the utility of daily body weight measurements in patients with heart failure who are being cared for in the home environment.
In one study, an increase in body weight of > 2 kg over 24 to 72 hours had a 9% sensitivity for detecting clinical deterioration.21 In another study, Chaudhry et al performed a nested case-control trial in 134 patients with heart failure and 134 matched controls referred to a home monitoring system by managed care organizations. The researchers found that increases in body weight were associated with hospitalization for heart failure and that the increases began at least 1 week before admission.22 However, they did not investigate whether the use of this information by clinicians altered outcomes. In a prior randomized clinical trial of symptom monitoring versus transtelephonic body weight monitoring in patients with symptomatic heart failure, the Weight Monitoring in Heart Failure trial (n = 280), weight monitoring did not result in improvement in the primary outcome of hospitalizations for heart failure over a 6-month period.23
The ideal monitoring parameters in heart failure patients may include direct hemodynamic measurements from the right ventricular outflow tract,24 pulmonary artery,25 or left atrium,26 using implantable devices. For example, the CHAMPION (CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Patients) trial (n = 550) was a randomized, single-blind, industry-sponsored trial of heart failure management guided by physiologic hemodynamic data derived from a percutaneously inserted pulmonary artery hemodynamic monitor (Champion HF Monitoring System; CardioMEMS, Atlanta, Georgia). The researchers found that monitoring these parameters was associated with a 28% reduction in heart failure–related hospitalizations during the first 6 months (rate 0.32 vs 0.44, HR 0.72, 95% CI 0.60–0.85, P = .0002) compared with usual care.25 At 6 months, the freedom from device- or system-related complications was 98.6%.
Despite success in the trial, the US Food and Drug Administration Circulatory System Devices Panel voted against approving the device. The panel was concerned that the e-mail–alert and care systems built into the intervention arm of the trial created bias in favor of the device, and that in a real-world situation it may not be as effective. This demonstrates the ongoing challenges and barriers to adoption of invasive hemodynamic monitoring.
At our center, we are conducting an institutional review board–approved investigation of an entirely noninvasive under-the-mattress piezoelectric monitor in a cohort of postacute heart failure patients. Piezoelectricity is the charge that accumulates in certain solid materials in response to mechanical stress. Common applications of piezoelectricity include microphones, push-start propane barbecues, and cigarette lighters. The device under investigation (EverOn; EarlySense, Ramat-Gan, Israel) detects heart rate, respiratory rate, and movement rate through vibrations of the mattress. Case examples are shown in Figure 3. Whether such monitoring technology will play a future role in the home environment remains to be seen.
SUMMARY
At the time of this writing, the Supreme Court of the United States has reaffirmed the constitutionality of the PPACA, clearing the way for implementation of significant changes in the US health care delivery system. The implications for in-home care for older adults with chronic conditions, including heart failure, are significant. The home will become an increasingly common venue of postacute care. Today is the time to investigate beneficial models of care and optimal uses of technology, and to develop a specialized mobile workforce that will confidently care for individuals with heart failure at home, responsibly and at lower cost.
The home is the most important context of care for individuals with chronic heart failure and yet it is the least accessible to caregivers. Patients often struggle to manage a complex regimen of medications, follow an unfamiliar diet, monitor weight and vital signs, and work to coordinate care among various providers who, in some cases, fail to communicate effectively. Heart failure patients do all this while making difficult decisions about their livelihoods, social condition, and future direction. With progression of the disease and comorbidity, these patients often experience a downward cycle of repeat hospitalization and worsening functional capacity (Figure 1). Each subsequent transition from acute care to home becomes incrementally more difficult to manage.
According to the latest American College of Cardiology/American Heart Association Guidelines for the Diagnosis and Management of Heart Failure in Adults, appropriate care for patients with heart failure should include:
- Intensive patient education
- Encouragement of patients to be more aggressive participants in their care
- Close monitoring of patients through telephone follow-up or home nursing
- Careful review of medications to improve adherence to evidence-based guidelines
- Multidisciplinary care with nurse case management directed by a physician1
Beyond these general suggestions, recommendations about specific approaches and models of care in the home are lacking.
Contemporary research suggests that postacute, home-based care of heart failure patients may yield outcomes similar to those of clinic-based outpatient care. Results of the Which Heart Failure Intervention is Most Cost-Effective & Consumer-Friendly in Reducing Hospital Care (WHICH?) trial support this hypothesis. This multicenter, randomized clinical trial (n = 280) compared home- with clinic-based multidisciplinary management for postacute heart failure patients.2 Investigators compared outcomes in patients managed at a heart failure clinic with those managed at home. They found that postdischarge home visits by heart failure nurses did not significantly alter the primary composite end point of death or unplanned rehospitalization from any cause over 18 months (hazard ratio [HR] 0.97, 95% confidence interval [CI] 0.73–1.30, P = .8621). The rate of unplanned and total hospitalization was also similar in the two groups. However, the average length of hospital stay was significantly lower in the home care group (4 days) than in the clinic-based group (6 days); P = .004. A cost-effectiveness analysis is planned but has not yet been presented.
HEART CARE AT HOME
At Cleveland Clinic, our group of physicians (geriatrics and cardiology), nurses, nurse practitioners, and hospital administrators founded a primarily home-based postacute transitional care program in 2010 called “Heart Care at Home.” The design of our program was influenced by Coleman et al’s care transitions interventions program,3 Naylor et al’s transitional care intervention,4 and the contemporary remote monitoring literature.5 The program focuses primarily on older adults hospitalized for heart failure who are transitioning from hospital to home. In our model:
- Inpatient care advocates identify candidates during the index inpatient stay, introduce a model of care, and begin a coaching intervention.
- After discharge, home liaisons visit the patient at home, continue coaching intervention, and teach the patient to use the newly installed remote monitoring equipment.
- For 30 to 40 days after discharge, a team of telehealth nurses monitors the patient, makes contact with him or her weekly in order to reinforce coaching intervention, coordinates care, and tracks outcomes.
- Nurse practitioners experienced both in home care and heart failure provide clinical oversight and leadership and visit the highest-acuity patients at home.
To date, the program has provided care in more than 2,100 patient encounters, with approximately 50 to 80 patients actively enrolled at any time. We identified potential program candidates using a digital list tool embedded in Cleveland Clinic’s electronic medical record (EMR) system. This tool was developed by our team together with an internal business intelligence team. We have been approximately 65% successful in identifying eligible inpatients. Patients enrolled in our transitional care program tend to be older, have longer hospital stays, and have more comorbidities than other older adults hospitalized at Cleveland Clinic for similar reasons.
Following index hospital discharge, our home liaisons have been able to make an initial home visit after a median of 2 days (25th to 75th percentile: 1 to 3 days). Patients thought to be at higher risk for hospital readmissions have been seen at home by our nurse practitioners within the first week of discharge. The most common challenge that our at-home team members have faced relates to patients’ medications (for example, unfilled prescriptions and errors in utilization). On many occasions our at-home team has succeeded in transitioning patients not benefiting from care at home to nonhospital venues (skilled nursing facilities, chronic care facilities, inpatient hospice) or to higher levels of at-home care (at-home physician visits, home-care nursing and therapy, at-home hospice).
To date, patients have been enrolled in our program for a median of 30 days (25th to 75th percentile: 20 to 35 days). We have observed an increased level of patient satisfaction. Among heart failure patients enrolled in our program for the first time, we have observed a lower readmission rate compared with publicly reported Cleveland Clinic rates (24.5% vs 28.2%). However, there are several ongoing challenges in the care of heart failure patients in the home environment. These relate to longitudinal care across venues, cross training of providers, and home monitoring.
Longitudinal care across venues
Our program aims to address the lack of integrated care over time and between care venues. This problem lies at the intersection of health care reimbursement policy and clinical practice. Currently, the hospital reimbursement system does not encourage care coordination across settings. The system has, in fact, evolved into a string of disconnected care providers who act as “toll booths” providing services for a fee in isolation from other providers. Coleman and colleagues have documented the complexity of the transitions among these care providers for older patients with chronic disease, noting the implications for patient safety and cost.6
Hospitals receive a fixed payment for an inpatient admission, which increases the financial incentive to discharge patients faster to other venues of care. The study by Bueno et al of a Medicare population treated between 1993 and 2006 confirms that such a trend exists for heart failure patients.7 The authors found a steady decrease in the mean length of hospital stay from 8.81 days to 6.33 days over the study period (28% relative reduction, P < .001). During this same period, the 30-day all-cause readmission rate increased from 17.2% to 20.1% (a 17% relative increase, P < .001) with an associated 10% relative reduction in the proportion of patients discharged to home.7 Experience in other populations with heart failure, such as patients in the Veterans Affairs health care system, has shown similar trends in length of hospital stay and readmissions.8
During these transitions, information is often lost in the handoff from the discharging hospital to the next venue of care. Medication management is the most common problem area with the potential for patient noncompliance with prescriptions,9,10 which can have serious deleterious effects on quality and safety. Forster et al found that 66% of untoward outcomes in discharged patients were due to adverse drug events.11 Similarly, Gray et al identified adverse drug events in 20% of patients discharged from hospital to home with home health care services.12
In the Cleveland Clinic Health System, we are coupling our “Heart Care at Home” transitional care program with an aggressive plan to develop a more comprehensive cross-venue EMR. Connecting the hospital EMR with our health system–owned home health agency will enable a consistent medication record and communication system for patients transitioning from our hospitals to Cleveland Clinic home care services (nearly 20,000 patients per year).
Despite these issues, several care transition interventions have shown promising clinical and economic results. Coleman and colleagues conducted a randomized, controlled trial of a transition coaching model in which patients and caregivers were encouraged to take a more active role in care transitions. Results of this trial showed a significant decrease in 30- and 90-day rehospitalizations (the 90-day read-mission rate in the treatment group was 16.7 vs 22.5 in the control group, P = .04) with associated cost savings.3 Voss et al showed similar results in reduction of readmissions in a nonintegrated delivery system.13 Additionally, telephone-based chronic disease management programs have been shown to be cost-effective in chronically ill Medicare patients.14
When will the clinical evidence behind care transitions and financial incentives converge to create an atmosphere conducive to more optimal care coordination? Today, this question remains unanswered. Health care reform, with the passage of the Patient Protection and Affordable Care Act (PPACA) (http://housedocs.house.gov/energycommerce/ppacacon.pdf), may spur the creation of programs to increase incentives for care coordination. These include a move to episodic reimbursement that would bundle payments for acute and postacute care, thus creating more incentives for coordinating care across settings. The “Bundled Payments for Care Improvement” project run by the Center for Medicare & Medicaid Innovation will test different models and approaches to bundled payments (http://innovations.cms.gov/initiatives/bundled-payments). Additionally, beginning in fiscal year 2013, Medicare will penalize hospitals that have high readmission rates for heart failure, acute myocardial infarction, and pneumonia with a financial risk of up to 3% of total hospital Medicare payments by year 3 of the program.
The PPACA will have a significant effect on home-based care for older adults with chronic conditions. The PPACA reforms will likely lead to more patients being treated at home (the lower-cost care setting), ideally under the care of highly skilled teams. Payment reforms will also create new incentives for providers to better coordinate care, keep patients healthy at home, and avoid the “toll-booth” description entirely, enabling providers to focus on patient care. However, more research and experimentation are required to streamline the elements on the transitions spectrum in order to create the most value for specific patient populations. New infrastructure, use of technology, changing culture, and dedicated clinical teams will be necessary to deliver on the hopes of more integrated longitudinal care across venues.
Cross training of providers
Older community-dwelling adults with heart failure exhibit more health instability; take more medications; have more comorbidities; and receive more nursing, homemaking, and meal services than do other home care clients.15 Nurses thus have a unique opportunity to improve outcomes for home-based heart failure patients,16,17 but are often insufficiently trained to do so. Delaney et al administered a validated 20-item heart failure knowledge questionnaire to 94 home care nurses from four different home care agencies.18 The investigators found a 79% knowledge level in overall heart failure education principles, with lowest scores related to issues of asymptomatic hypotension (25% answered correctly), daily weight monitoring (27%), and transient dizziness (31%). Nurses with poorer heart failure–related knowledge may partially explain worse process and outcome measures among this patient population.19
The home-based nursing workforce of the future, and specifically nurses who care for heart failure patients at home, will need to be better trained and specialized in issues relating both to home-based nursing and medical heart failure. These “hybrid nurses” should be allowed a central clinical leadership role among their peers, as they will need to be empowered to make medical and care coordination decisions.
At our center, hybrid-trained home care/heart failure nurse practitioners make home visits for higher-acuity home-based patients and provide clinical leadership and support for other home care nurses. These nurse practitioners have been instrumental in identifying and correcting heart failure medication–related problems, as well as effectively coordinating care. Examples include: independently prescribing and coordinating administration of intravenous diuretics at home for patients who have difficulty managing volume overload, avoiding hospital readmissions by transitioning ill patients to a skilled nursing facility or an at-home hospice, and effectively educating patients and families about appropriate heart failure self-care.
Home monitoring
Home monitoring of selected physiologic parameters and patient-reported health status measures among heart failure patients may facilitate early detection of clinical deterioration and direct timely intervention to prevent adverse outcomes.20 Desai and Stevenson have previously proposed the “circle from home to heart-failure disease management,” a concept illustrating how home monitoring can be embedded in a comprehensive heart failure management approach (Figure 2).20 This concept emphasizes the following:
- Home monitoring should facilitate early detection of clinical deterioration.20
- Home monitoring data will most directly lead to action if the data can be used by the patient to improve self-care.
- In the setting of multidisciplinary care, data should be remotely transmitted to a midlevel team, preferably one empowered to make therapeutic decisions.
- Further engagement of physicians or other clinical providers may be beneficial but will delay the clinical response.
The most commonly monitored physiologic parameter of heart failure patients is daily weight. While nearly universally used, this parameter is in fact a poor surrogate for subclinical hemodynamic congestion and has poor diagnostic performance for clinical decompensation. Results are conflicting from studies evaluating the utility of daily body weight measurements in patients with heart failure who are being cared for in the home environment.
In one study, an increase in body weight of > 2 kg over 24 to 72 hours had a 9% sensitivity for detecting clinical deterioration.21 In another study, Chaudhry et al performed a nested case-control trial in 134 patients with heart failure and 134 matched controls referred to a home monitoring system by managed care organizations. The researchers found that increases in body weight were associated with hospitalization for heart failure and that the increases began at least 1 week before admission.22 However, they did not investigate whether the use of this information by clinicians altered outcomes. In a prior randomized clinical trial of symptom monitoring versus transtelephonic body weight monitoring in patients with symptomatic heart failure, the Weight Monitoring in Heart Failure trial (n = 280), weight monitoring did not result in improvement in the primary outcome of hospitalizations for heart failure over a 6-month period.23
The ideal monitoring parameters in heart failure patients may include direct hemodynamic measurements from the right ventricular outflow tract,24 pulmonary artery,25 or left atrium,26 using implantable devices. For example, the CHAMPION (CardioMEMS Heart Sensor Allows Monitoring of Pressure to Improve Outcomes in NYHA Class III Patients) trial (n = 550) was a randomized, single-blind, industry-sponsored trial of heart failure management guided by physiologic hemodynamic data derived from a percutaneously inserted pulmonary artery hemodynamic monitor (Champion HF Monitoring System; CardioMEMS, Atlanta, Georgia). The researchers found that monitoring these parameters was associated with a 28% reduction in heart failure–related hospitalizations during the first 6 months (rate 0.32 vs 0.44, HR 0.72, 95% CI 0.60–0.85, P = .0002) compared with usual care.25 At 6 months, the freedom from device- or system-related complications was 98.6%.
Despite success in the trial, the US Food and Drug Administration Circulatory System Devices Panel voted against approving the device. The panel was concerned that the e-mail–alert and care systems built into the intervention arm of the trial created bias in favor of the device, and that in a real-world situation it may not be as effective. This demonstrates the ongoing challenges and barriers to adoption of invasive hemodynamic monitoring.
At our center, we are conducting an institutional review board–approved investigation of an entirely noninvasive under-the-mattress piezoelectric monitor in a cohort of postacute heart failure patients. Piezoelectricity is the charge that accumulates in certain solid materials in response to mechanical stress. Common applications of piezoelectricity include microphones, push-start propane barbecues, and cigarette lighters. The device under investigation (EverOn; EarlySense, Ramat-Gan, Israel) detects heart rate, respiratory rate, and movement rate through vibrations of the mattress. Case examples are shown in Figure 3. Whether such monitoring technology will play a future role in the home environment remains to be seen.
SUMMARY
At the time of this writing, the Supreme Court of the United States has reaffirmed the constitutionality of the PPACA, clearing the way for implementation of significant changes in the US health care delivery system. The implications for in-home care for older adults with chronic conditions, including heart failure, are significant. The home will become an increasingly common venue of postacute care. Today is the time to investigate beneficial models of care and optimal uses of technology, and to develop a specialized mobile workforce that will confidently care for individuals with heart failure at home, responsibly and at lower cost.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 119:e391–e479.
- Stewart S, Carrington MJ, Marwick TH, et al. Impact of home versus clinic-based management of chronic heart failure: the WHICH? (Which Heart Failure Intervention Is Most Cost-Effective and Consumer Friendly in Reducing Hospital Care) multicenter, randomized trial. J Am Coll Cardiol 2012; 60:1239–1248.
- Coleman EA, Parry C, Chalmers S, Min S-J. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Klersy C, De Silvestri A, Gabutti G, Regoli F, Auricchio A. A meta-analysis of remote monitoring of heart failure patients. J Am Coll Cardiol 2009; 54:1683–1694.
- Coleman EA, Min S-J, Chomiak A, Kramer AM. Posthospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993–2006. JAMA 2010; 303:2141–2147.
- Heidenreich PA, Sahay A, Kapoor JR, Pham MX, Massie B. Divergent trends in survival and readmission following a hospitalization for heart failure in the Veterans Affairs health care system 2002 to 2006. J Am Coll Cardiol 2010; 56:362–368.
- Moore C, Wisnivesky J, Williams S, McGinn T. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med 2003; 18:646–651.
- Coleman EA, Smith JD, Raha D, Min S-J. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Gray SL, Mahoney JE, Blough DK. Adverse drug events in elderly patients receiving home health services following hospital discharge. Ann Pharmacother 1999; 33:1147–1153.
- Voss R, Gardner R, Baier R, Butterfeld K, Lehrman S, Gravenstein S. The care transitions intervention: translating from efficacy to effectiveness. Arch Intern Med 2011; 171:1232–1237.
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare benefciaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Foebel AD, Hirdes JP, Heckman GA, Tyas SL, Tjam EY. A profile of older community-dwelling home care clients with heart failure in Ontario. Chronic Dis Can 2011; 31:49–57.
- Krumholz HM, Amatruda J, Smith GL, et al. Randomized trial of an education and support intervention to prevent readmission of patients with heart failure. J Am Coll Cardiol 2002; 39:83–89.
- González B, Lupón J, Herreros J, et al. Patient’s education by nurse: what we really do achieve? Eur J Cardiovasc Nurs 2005; 4:107–111.
- Delaney C, Apostolidis B, Lachapelle L, Fortinsky R. Home care nurses’ knowledge of evidence-based education topics for management of heart failure. Heart Lung 2011; 40:285–292.
- Foebel AD, Heckman GA, Hirdes JP, et al. Clinical, demographic and functional characteristics associated with pharmacotherapy for heart failure in older home care clients: a retrospective, population-level, cross-sectional study. Drugs Aging 2011; 28:561–573.
- Desai AS, Stevenson LW. Connecting the circle from home to heart-failure disease management. N Engl J Med 2010; 363:2364–2367.
- Lewin J, Ledwidge M, O’Loughlin C, McNally C, McDonald K. Clinical deterioration in established heart failure: what is the value of BNP and weight gain in aiding diagnosis? Eur J Heart Fail 2005; 7:953–957.
- Chaudhry SI, Wang Y, Concato J, Gill TM, Krumholz HM. Patterns of weight change preceding hospitalization for heart failure. Circulation 2007; 116:1549–1554.
- Goldberg LR, Piette JD, Walsh MN, et al. Randomized trial of a daily electronic home monitoring system in patients with advanced heart failure: the Weight Monitoring in Heart Failure (WHARF) trial. Am Heart J 2003; 146:705–712.
- Bourge RC, Abraham WT, Adamson PB, et al. Randomized controlled trial of an implantable continuous hemodynamic monitor in patients with advanced heart failure: the Compass-HF study. J Am Coll Cardiol 2008; 51:1073–1079.
- Abraham WT, Adamson PB, Bourge RC, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet 2011; 377:658–666.
- Ritzema J, Troughton R, Melton I, et al. Physician-directed patient self-management of left atrial pressure in advanced chronic heart failure. Circulation 2010; 121:1086–1095.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009; 119:e391–e479.
- Stewart S, Carrington MJ, Marwick TH, et al. Impact of home versus clinic-based management of chronic heart failure: the WHICH? (Which Heart Failure Intervention Is Most Cost-Effective and Consumer Friendly in Reducing Hospital Care) multicenter, randomized trial. J Am Coll Cardiol 2012; 60:1239–1248.
- Coleman EA, Parry C, Chalmers S, Min S-J. The care transitions intervention: results of a randomized controlled trial. Arch Intern Med 2006; 166:1822–1828.
- Naylor MD, Brooten DA, Campbell RL, Maislin G, McCauley KM, Schwartz JS. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc 2004; 52:675–684.
- Klersy C, De Silvestri A, Gabutti G, Regoli F, Auricchio A. A meta-analysis of remote monitoring of heart failure patients. J Am Coll Cardiol 2009; 54:1683–1694.
- Coleman EA, Min S-J, Chomiak A, Kramer AM. Posthospital care transitions: patterns, complications, and risk identification. Health Serv Res 2004; 39:1449–1465.
- Bueno H, Ross JS, Wang Y, et al. Trends in length of stay and short-term outcomes among Medicare patients hospitalized for heart failure, 1993–2006. JAMA 2010; 303:2141–2147.
- Heidenreich PA, Sahay A, Kapoor JR, Pham MX, Massie B. Divergent trends in survival and readmission following a hospitalization for heart failure in the Veterans Affairs health care system 2002 to 2006. J Am Coll Cardiol 2010; 56:362–368.
- Moore C, Wisnivesky J, Williams S, McGinn T. Medical errors related to discontinuity of care from an inpatient to an outpatient setting. J Gen Intern Med 2003; 18:646–651.
- Coleman EA, Smith JD, Raha D, Min S-J. Posthospital medication discrepancies: prevalence and contributing factors. Arch Intern Med 2005; 165:1842–1847.
- Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med 2003; 138:161–167.
- Gray SL, Mahoney JE, Blough DK. Adverse drug events in elderly patients receiving home health services following hospital discharge. Ann Pharmacother 1999; 33:1147–1153.
- Voss R, Gardner R, Baier R, Butterfeld K, Lehrman S, Gravenstein S. The care transitions intervention: translating from efficacy to effectiveness. Arch Intern Med 2011; 171:1232–1237.
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare benefciaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Foebel AD, Hirdes JP, Heckman GA, Tyas SL, Tjam EY. A profile of older community-dwelling home care clients with heart failure in Ontario. Chronic Dis Can 2011; 31:49–57.
- Krumholz HM, Amatruda J, Smith GL, et al. Randomized trial of an education and support intervention to prevent readmission of patients with heart failure. J Am Coll Cardiol 2002; 39:83–89.
- González B, Lupón J, Herreros J, et al. Patient’s education by nurse: what we really do achieve? Eur J Cardiovasc Nurs 2005; 4:107–111.
- Delaney C, Apostolidis B, Lachapelle L, Fortinsky R. Home care nurses’ knowledge of evidence-based education topics for management of heart failure. Heart Lung 2011; 40:285–292.
- Foebel AD, Heckman GA, Hirdes JP, et al. Clinical, demographic and functional characteristics associated with pharmacotherapy for heart failure in older home care clients: a retrospective, population-level, cross-sectional study. Drugs Aging 2011; 28:561–573.
- Desai AS, Stevenson LW. Connecting the circle from home to heart-failure disease management. N Engl J Med 2010; 363:2364–2367.
- Lewin J, Ledwidge M, O’Loughlin C, McNally C, McDonald K. Clinical deterioration in established heart failure: what is the value of BNP and weight gain in aiding diagnosis? Eur J Heart Fail 2005; 7:953–957.
- Chaudhry SI, Wang Y, Concato J, Gill TM, Krumholz HM. Patterns of weight change preceding hospitalization for heart failure. Circulation 2007; 116:1549–1554.
- Goldberg LR, Piette JD, Walsh MN, et al. Randomized trial of a daily electronic home monitoring system in patients with advanced heart failure: the Weight Monitoring in Heart Failure (WHARF) trial. Am Heart J 2003; 146:705–712.
- Bourge RC, Abraham WT, Adamson PB, et al. Randomized controlled trial of an implantable continuous hemodynamic monitor in patients with advanced heart failure: the Compass-HF study. J Am Coll Cardiol 2008; 51:1073–1079.
- Abraham WT, Adamson PB, Bourge RC, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet 2011; 377:658–666.
- Ritzema J, Troughton R, Melton I, et al. Physician-directed patient self-management of left atrial pressure in advanced chronic heart failure. Circulation 2010; 121:1086–1095.
The case for "connected health" at home
Many technologies have emerged to monitor, interact with, and support patients at home and change home health care delivery.1–5 This trend coincides with the explosion of consumer digital and mobile products such as “smartphones” and has brought with it many different names, such as telehealth, telemedicine, e-medicine, remote monitoring, “virtual” care, digital health, mobile medicine, interactive health, and distance health. Many of these terms and concepts raise concerns for those who value traditional expressions of caring, physical diagnosis, touch, and presence in health care. However, these new technologies may present opportunities to find ways to enhance humanism in home health care. This potential may be most evident among patients with serious chronic illness and their families, who often struggle 168 hours a week but find their access to help limited to brief visits at times convenient for the provider.
While our health care system offers heroic acute-care treatments for hundreds of life-threatening maladies, we seem to fall short in helping those with serious ongoing needs whose care must be coordinated over time and across health care venues. Thinking in terms of “connected health” may provide a more holistic nomenclature that suggests the bond between technology and the opportunity for closer personal relationships.6–8
OPPORTUNITIES
Can technology better connect our home health patients and families to care during the “white space,”9 between our visits? Can we use new mobile and digital technologies to improve care for the seriously chronically ill? We have the technology to turn many challenges into opportunities in the next decade. For example:
- Can we change our visit-based model of home health care to a model that provides 24/7 “inbound” multichannel access to home health care teams along with proactive “outbound” support between visits in the form of multimedia health education and virtual encounters? Can this free up time for longer visits targeted toward higher-risk and higher-complexity scenarios that require extensive team leadership and care coordination?
- Can “smart” home monitoring be integrated into home-based long-term care for patients who have dementia, fall risks, other safety issues, or unaddressed limitations in activities of daily living to increase independence and quality of life and reduce institutionalization while decreasing cost of care and accommodating workforce constraints?10
- How do we apply clinician-to-clinician and clinician-to-patient videoconferencing and other connected health approaches to increase home health patient access to specialized, but hard-to-find, clinicians for consultative and direct-care services?
- Can emerging technologies accelerate the shift in care whereby most acute care for exacerbations of chronic illness and other common acute scenarios move from hospitals into home-based models of acute care, such as “Hospital at home”?11
- To what extent can apps and other technologies provide self-management support to truly deliver the home health care version of the automatic teller machine? For example, diabetes self-management support tools provide patients feedback about their disease based on information input into mobile devices.12 Can this be expanded in a way that dramatically increases access, especially for vulnerable groups that have been hard to reach, while also decreasing costs?
- Can we improve the home health care experience by using connected health concepts to improve transparency, minimize common scheduling delays and annoyances, and empower patients while they are receiving care?
REAL-WORLD BARRIERS
Despite the opportunities, barriers remain for innovative providers. With few exceptions, there is no direct third-party reimbursement for care that comes through a device rather than the front door. Medicare does not reimburse home health providers for services outside of a visit, but specific guidance has been issued that clarifies some of the opportunities:
An HHA (Home Health Agency) may adopt telehealth technologies that it believes promote efficiencies or improve quality of care…. An HHA may not substitute telehealth services for Medicare-covered services ordered by a physician. However, if an HHA has telehealth services available to its clients, a doctor may take their availability into account when he or she prepares a plan…. If a physician intends that telehealth services be furnished while a patient is under a home health plan of care, the services should be recorded in the plan of care along with the Medicare covered home health services to be furnished.13
Thus, there is no reimbursement for telehealth services, but if telehealth is part of a physician-directed plan of care, it may be included if it promotes home health quality and efficiency. Beyond reimbursement, there are other regulatory barriers. If monitoring or other digital or virtual services are provided across state lines, the clinicians involved in a regional or national “command center” likely must meet the licensure requirements (or obtain waivers) for every jurisdiction in which their patients reside. Providers should seek counsel regarding the extent to which new devices and software need to be approved by the US Food and Drug Administration before being deployed. And, as with all health-related communication, it is essential that information transmitted in nontraditional ways be secure, private, and compliant with all mandated standards for privacy. Finally, if the technology or service is rolled out in a fashion that could be construed as a “gift” or “freebie” for marketing purposes rather than a tool to improve clinical outcomes and health care value, then there may be a risk that the approach runs afoul of laws to prevent undue inducements.
In addition to reimbursement and regulatory concerns, there are technical barriers to fully realizing the connected health opportunities in home care. Even if patients are provided with devices, there is variability in internet connectivity or bandwidth in any given home. Providing devices with built-in cellular capabilities can reduce these barriers, but cellular data coverage varies across different geographies. High-quality health care videoconferencing tends to require more bandwidth than that provided in the typical “3G” connection. Use of existing cable television connections, which are almost ubiquitous, is another option, but it typically requires a more customized set-up than consumer mobile devices with cellular and wireless capabilities. If the services were delivered or coordinated by the cable provider, some of these inconveniences might be resolved.
As with most innovation, there is no “cookbook,” and there is limited and conflicting evidence in the clinical sciences literature to guide best practices. Organizations that commit to using technology to improve the quality and efficiency of care will experience fits and starts before they find the right types and “doses” of technology in their new care models. The home health community should beware of these frustrations leading to undue skepticism, like that of Newsweek author Clifford Stoll, who in 1995 infamously wrote about the developing internet:
…today, I’m uneasy about this [trend]…. Visionaries see a future of telecommuting workers, interactive libraries and multimedia classrooms. They speak of electronic town meetings and virtual communities. Commerce and business will shift from offices and malls to networks and modems. And the freedom of digital networks will make government more democratic. Baloney. Do our computer pundits lack all common sense? The truth is no online database will replace your daily newspaper … no computer network will change the way government works.14
Like the internet of 15 years ago, mobile and digital technologies are now changing how people live and relate to one another and how businesses function. It is unlikely that the impact of these technologies on health care will be fully elucidated by controlled trials that consider incremental changes to existing care models and workflows. Rather, innovative providers and the next generation of clinicians that “grew up,” with mobile devices as part of their lives will create new home care workflows and care realities. Home health providers can use these technologies to better connect their patients and find new ways to reduce suffering, increase health and independence, and improve the care experience while lowering costs and increasing value. The individuals and organizations that seize the moment and “answer” these key questions in connected health with successful new approaches to care will be the winners of the future. There is such an opportunity to make a difference.
- Chen HF, Kalish MC, Pagan JA. Telehealth and hospitalizations for Medicare home healthcare patients. Am J Manag Care 2011; 17 (6 Spec No.):e224–e230.
- Gellis ZD, Kenaley B, McGinty J, Bardelli E, Davitt J, Ten Have T Outcomes of a telehealth intervention for homebound older adults with heart or chronic respiratory failure: a randomized controlled trial [published online ahead of print January 11, 2012]. Gerontologist 2012; 52:541–552. 10.1093/geront/gnr134
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare beneficiaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Franko OI, Bhola S. iPad apps for orthopedic surgeons. Orthopedics 2011; 34:978–981.
- The smartphone will see you now: “apps” and devices are turning cell phones into tools for health. Harv Heart Lett 2011; 22:3.
- Barr PJ, McElnay JC, Hughes CM. Connected health care: the future of health care and the role of the pharmacist [published online ahead of print August 4, 2010]. J Eval Clin Pract 2012; 18:56–62. 10.1111/j.1365-2753.2010.01522x
- O’Neill SA, Nugent CD, Donnelly MP, McCullagh P, McLaughlin J. Evaluation of connected health technology. Technol Health Care 2012; 20:151–167.
- Ziebland S, Wyke S. Health and illness in a connected world: how might sharing experiences on the internet affect people’s health? Milbank Q 2012; 90:219–249.
- Dobson A. Personal communication. 2011.
- Dreyfus D. Smart-home technology for persons with disabilities. Am Fam Physician 2009; 80:233.
- Leff B, Burton L, Mader SL, Naughton B, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Quinn CC, Shardell MD, Terrin ML, et al. Cluster-randomized trial of a mobile phone personalized behavioral intervention for blood glucose control [published online ahead of print July 25, 2011]. Diabetes Care 2011; 34:1934–1942. 10.2337/dc11-0366
- Medicare Home Health Agency Manual. Section 201.13, Tele-health. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R298HHA.pdf. Published January 22, 2002. Accessed September 18, 2012.
- Stoll C. The Internet? Bah! Newsweek 1995; 125 February 27:41.
Many technologies have emerged to monitor, interact with, and support patients at home and change home health care delivery.1–5 This trend coincides with the explosion of consumer digital and mobile products such as “smartphones” and has brought with it many different names, such as telehealth, telemedicine, e-medicine, remote monitoring, “virtual” care, digital health, mobile medicine, interactive health, and distance health. Many of these terms and concepts raise concerns for those who value traditional expressions of caring, physical diagnosis, touch, and presence in health care. However, these new technologies may present opportunities to find ways to enhance humanism in home health care. This potential may be most evident among patients with serious chronic illness and their families, who often struggle 168 hours a week but find their access to help limited to brief visits at times convenient for the provider.
While our health care system offers heroic acute-care treatments for hundreds of life-threatening maladies, we seem to fall short in helping those with serious ongoing needs whose care must be coordinated over time and across health care venues. Thinking in terms of “connected health” may provide a more holistic nomenclature that suggests the bond between technology and the opportunity for closer personal relationships.6–8
OPPORTUNITIES
Can technology better connect our home health patients and families to care during the “white space,”9 between our visits? Can we use new mobile and digital technologies to improve care for the seriously chronically ill? We have the technology to turn many challenges into opportunities in the next decade. For example:
- Can we change our visit-based model of home health care to a model that provides 24/7 “inbound” multichannel access to home health care teams along with proactive “outbound” support between visits in the form of multimedia health education and virtual encounters? Can this free up time for longer visits targeted toward higher-risk and higher-complexity scenarios that require extensive team leadership and care coordination?
- Can “smart” home monitoring be integrated into home-based long-term care for patients who have dementia, fall risks, other safety issues, or unaddressed limitations in activities of daily living to increase independence and quality of life and reduce institutionalization while decreasing cost of care and accommodating workforce constraints?10
- How do we apply clinician-to-clinician and clinician-to-patient videoconferencing and other connected health approaches to increase home health patient access to specialized, but hard-to-find, clinicians for consultative and direct-care services?
- Can emerging technologies accelerate the shift in care whereby most acute care for exacerbations of chronic illness and other common acute scenarios move from hospitals into home-based models of acute care, such as “Hospital at home”?11
- To what extent can apps and other technologies provide self-management support to truly deliver the home health care version of the automatic teller machine? For example, diabetes self-management support tools provide patients feedback about their disease based on information input into mobile devices.12 Can this be expanded in a way that dramatically increases access, especially for vulnerable groups that have been hard to reach, while also decreasing costs?
- Can we improve the home health care experience by using connected health concepts to improve transparency, minimize common scheduling delays and annoyances, and empower patients while they are receiving care?
REAL-WORLD BARRIERS
Despite the opportunities, barriers remain for innovative providers. With few exceptions, there is no direct third-party reimbursement for care that comes through a device rather than the front door. Medicare does not reimburse home health providers for services outside of a visit, but specific guidance has been issued that clarifies some of the opportunities:
An HHA (Home Health Agency) may adopt telehealth technologies that it believes promote efficiencies or improve quality of care…. An HHA may not substitute telehealth services for Medicare-covered services ordered by a physician. However, if an HHA has telehealth services available to its clients, a doctor may take their availability into account when he or she prepares a plan…. If a physician intends that telehealth services be furnished while a patient is under a home health plan of care, the services should be recorded in the plan of care along with the Medicare covered home health services to be furnished.13
Thus, there is no reimbursement for telehealth services, but if telehealth is part of a physician-directed plan of care, it may be included if it promotes home health quality and efficiency. Beyond reimbursement, there are other regulatory barriers. If monitoring or other digital or virtual services are provided across state lines, the clinicians involved in a regional or national “command center” likely must meet the licensure requirements (or obtain waivers) for every jurisdiction in which their patients reside. Providers should seek counsel regarding the extent to which new devices and software need to be approved by the US Food and Drug Administration before being deployed. And, as with all health-related communication, it is essential that information transmitted in nontraditional ways be secure, private, and compliant with all mandated standards for privacy. Finally, if the technology or service is rolled out in a fashion that could be construed as a “gift” or “freebie” for marketing purposes rather than a tool to improve clinical outcomes and health care value, then there may be a risk that the approach runs afoul of laws to prevent undue inducements.
In addition to reimbursement and regulatory concerns, there are technical barriers to fully realizing the connected health opportunities in home care. Even if patients are provided with devices, there is variability in internet connectivity or bandwidth in any given home. Providing devices with built-in cellular capabilities can reduce these barriers, but cellular data coverage varies across different geographies. High-quality health care videoconferencing tends to require more bandwidth than that provided in the typical “3G” connection. Use of existing cable television connections, which are almost ubiquitous, is another option, but it typically requires a more customized set-up than consumer mobile devices with cellular and wireless capabilities. If the services were delivered or coordinated by the cable provider, some of these inconveniences might be resolved.
As with most innovation, there is no “cookbook,” and there is limited and conflicting evidence in the clinical sciences literature to guide best practices. Organizations that commit to using technology to improve the quality and efficiency of care will experience fits and starts before they find the right types and “doses” of technology in their new care models. The home health community should beware of these frustrations leading to undue skepticism, like that of Newsweek author Clifford Stoll, who in 1995 infamously wrote about the developing internet:
…today, I’m uneasy about this [trend]…. Visionaries see a future of telecommuting workers, interactive libraries and multimedia classrooms. They speak of electronic town meetings and virtual communities. Commerce and business will shift from offices and malls to networks and modems. And the freedom of digital networks will make government more democratic. Baloney. Do our computer pundits lack all common sense? The truth is no online database will replace your daily newspaper … no computer network will change the way government works.14
Like the internet of 15 years ago, mobile and digital technologies are now changing how people live and relate to one another and how businesses function. It is unlikely that the impact of these technologies on health care will be fully elucidated by controlled trials that consider incremental changes to existing care models and workflows. Rather, innovative providers and the next generation of clinicians that “grew up,” with mobile devices as part of their lives will create new home care workflows and care realities. Home health providers can use these technologies to better connect their patients and find new ways to reduce suffering, increase health and independence, and improve the care experience while lowering costs and increasing value. The individuals and organizations that seize the moment and “answer” these key questions in connected health with successful new approaches to care will be the winners of the future. There is such an opportunity to make a difference.
Many technologies have emerged to monitor, interact with, and support patients at home and change home health care delivery.1–5 This trend coincides with the explosion of consumer digital and mobile products such as “smartphones” and has brought with it many different names, such as telehealth, telemedicine, e-medicine, remote monitoring, “virtual” care, digital health, mobile medicine, interactive health, and distance health. Many of these terms and concepts raise concerns for those who value traditional expressions of caring, physical diagnosis, touch, and presence in health care. However, these new technologies may present opportunities to find ways to enhance humanism in home health care. This potential may be most evident among patients with serious chronic illness and their families, who often struggle 168 hours a week but find their access to help limited to brief visits at times convenient for the provider.
While our health care system offers heroic acute-care treatments for hundreds of life-threatening maladies, we seem to fall short in helping those with serious ongoing needs whose care must be coordinated over time and across health care venues. Thinking in terms of “connected health” may provide a more holistic nomenclature that suggests the bond between technology and the opportunity for closer personal relationships.6–8
OPPORTUNITIES
Can technology better connect our home health patients and families to care during the “white space,”9 between our visits? Can we use new mobile and digital technologies to improve care for the seriously chronically ill? We have the technology to turn many challenges into opportunities in the next decade. For example:
- Can we change our visit-based model of home health care to a model that provides 24/7 “inbound” multichannel access to home health care teams along with proactive “outbound” support between visits in the form of multimedia health education and virtual encounters? Can this free up time for longer visits targeted toward higher-risk and higher-complexity scenarios that require extensive team leadership and care coordination?
- Can “smart” home monitoring be integrated into home-based long-term care for patients who have dementia, fall risks, other safety issues, or unaddressed limitations in activities of daily living to increase independence and quality of life and reduce institutionalization while decreasing cost of care and accommodating workforce constraints?10
- How do we apply clinician-to-clinician and clinician-to-patient videoconferencing and other connected health approaches to increase home health patient access to specialized, but hard-to-find, clinicians for consultative and direct-care services?
- Can emerging technologies accelerate the shift in care whereby most acute care for exacerbations of chronic illness and other common acute scenarios move from hospitals into home-based models of acute care, such as “Hospital at home”?11
- To what extent can apps and other technologies provide self-management support to truly deliver the home health care version of the automatic teller machine? For example, diabetes self-management support tools provide patients feedback about their disease based on information input into mobile devices.12 Can this be expanded in a way that dramatically increases access, especially for vulnerable groups that have been hard to reach, while also decreasing costs?
- Can we improve the home health care experience by using connected health concepts to improve transparency, minimize common scheduling delays and annoyances, and empower patients while they are receiving care?
REAL-WORLD BARRIERS
Despite the opportunities, barriers remain for innovative providers. With few exceptions, there is no direct third-party reimbursement for care that comes through a device rather than the front door. Medicare does not reimburse home health providers for services outside of a visit, but specific guidance has been issued that clarifies some of the opportunities:
An HHA (Home Health Agency) may adopt telehealth technologies that it believes promote efficiencies or improve quality of care…. An HHA may not substitute telehealth services for Medicare-covered services ordered by a physician. However, if an HHA has telehealth services available to its clients, a doctor may take their availability into account when he or she prepares a plan…. If a physician intends that telehealth services be furnished while a patient is under a home health plan of care, the services should be recorded in the plan of care along with the Medicare covered home health services to be furnished.13
Thus, there is no reimbursement for telehealth services, but if telehealth is part of a physician-directed plan of care, it may be included if it promotes home health quality and efficiency. Beyond reimbursement, there are other regulatory barriers. If monitoring or other digital or virtual services are provided across state lines, the clinicians involved in a regional or national “command center” likely must meet the licensure requirements (or obtain waivers) for every jurisdiction in which their patients reside. Providers should seek counsel regarding the extent to which new devices and software need to be approved by the US Food and Drug Administration before being deployed. And, as with all health-related communication, it is essential that information transmitted in nontraditional ways be secure, private, and compliant with all mandated standards for privacy. Finally, if the technology or service is rolled out in a fashion that could be construed as a “gift” or “freebie” for marketing purposes rather than a tool to improve clinical outcomes and health care value, then there may be a risk that the approach runs afoul of laws to prevent undue inducements.
In addition to reimbursement and regulatory concerns, there are technical barriers to fully realizing the connected health opportunities in home care. Even if patients are provided with devices, there is variability in internet connectivity or bandwidth in any given home. Providing devices with built-in cellular capabilities can reduce these barriers, but cellular data coverage varies across different geographies. High-quality health care videoconferencing tends to require more bandwidth than that provided in the typical “3G” connection. Use of existing cable television connections, which are almost ubiquitous, is another option, but it typically requires a more customized set-up than consumer mobile devices with cellular and wireless capabilities. If the services were delivered or coordinated by the cable provider, some of these inconveniences might be resolved.
As with most innovation, there is no “cookbook,” and there is limited and conflicting evidence in the clinical sciences literature to guide best practices. Organizations that commit to using technology to improve the quality and efficiency of care will experience fits and starts before they find the right types and “doses” of technology in their new care models. The home health community should beware of these frustrations leading to undue skepticism, like that of Newsweek author Clifford Stoll, who in 1995 infamously wrote about the developing internet:
…today, I’m uneasy about this [trend]…. Visionaries see a future of telecommuting workers, interactive libraries and multimedia classrooms. They speak of electronic town meetings and virtual communities. Commerce and business will shift from offices and malls to networks and modems. And the freedom of digital networks will make government more democratic. Baloney. Do our computer pundits lack all common sense? The truth is no online database will replace your daily newspaper … no computer network will change the way government works.14
Like the internet of 15 years ago, mobile and digital technologies are now changing how people live and relate to one another and how businesses function. It is unlikely that the impact of these technologies on health care will be fully elucidated by controlled trials that consider incremental changes to existing care models and workflows. Rather, innovative providers and the next generation of clinicians that “grew up,” with mobile devices as part of their lives will create new home care workflows and care realities. Home health providers can use these technologies to better connect their patients and find new ways to reduce suffering, increase health and independence, and improve the care experience while lowering costs and increasing value. The individuals and organizations that seize the moment and “answer” these key questions in connected health with successful new approaches to care will be the winners of the future. There is such an opportunity to make a difference.
- Chen HF, Kalish MC, Pagan JA. Telehealth and hospitalizations for Medicare home healthcare patients. Am J Manag Care 2011; 17 (6 Spec No.):e224–e230.
- Gellis ZD, Kenaley B, McGinty J, Bardelli E, Davitt J, Ten Have T Outcomes of a telehealth intervention for homebound older adults with heart or chronic respiratory failure: a randomized controlled trial [published online ahead of print January 11, 2012]. Gerontologist 2012; 52:541–552. 10.1093/geront/gnr134
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare beneficiaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Franko OI, Bhola S. iPad apps for orthopedic surgeons. Orthopedics 2011; 34:978–981.
- The smartphone will see you now: “apps” and devices are turning cell phones into tools for health. Harv Heart Lett 2011; 22:3.
- Barr PJ, McElnay JC, Hughes CM. Connected health care: the future of health care and the role of the pharmacist [published online ahead of print August 4, 2010]. J Eval Clin Pract 2012; 18:56–62. 10.1111/j.1365-2753.2010.01522x
- O’Neill SA, Nugent CD, Donnelly MP, McCullagh P, McLaughlin J. Evaluation of connected health technology. Technol Health Care 2012; 20:151–167.
- Ziebland S, Wyke S. Health and illness in a connected world: how might sharing experiences on the internet affect people’s health? Milbank Q 2012; 90:219–249.
- Dobson A. Personal communication. 2011.
- Dreyfus D. Smart-home technology for persons with disabilities. Am Fam Physician 2009; 80:233.
- Leff B, Burton L, Mader SL, Naughton B, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Quinn CC, Shardell MD, Terrin ML, et al. Cluster-randomized trial of a mobile phone personalized behavioral intervention for blood glucose control [published online ahead of print July 25, 2011]. Diabetes Care 2011; 34:1934–1942. 10.2337/dc11-0366
- Medicare Home Health Agency Manual. Section 201.13, Tele-health. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R298HHA.pdf. Published January 22, 2002. Accessed September 18, 2012.
- Stoll C. The Internet? Bah! Newsweek 1995; 125 February 27:41.
- Chen HF, Kalish MC, Pagan JA. Telehealth and hospitalizations for Medicare home healthcare patients. Am J Manag Care 2011; 17 (6 Spec No.):e224–e230.
- Gellis ZD, Kenaley B, McGinty J, Bardelli E, Davitt J, Ten Have T Outcomes of a telehealth intervention for homebound older adults with heart or chronic respiratory failure: a randomized controlled trial [published online ahead of print January 11, 2012]. Gerontologist 2012; 52:541–552. 10.1093/geront/gnr134
- Baker LC, Johnson SJ, Macaulay D, Birnbaum H. Integrated telehealth and care management program for Medicare beneficiaries with chronic disease linked to savings. Health Aff (Millwood) 2011; 30:1689–1697.
- Franko OI, Bhola S. iPad apps for orthopedic surgeons. Orthopedics 2011; 34:978–981.
- The smartphone will see you now: “apps” and devices are turning cell phones into tools for health. Harv Heart Lett 2011; 22:3.
- Barr PJ, McElnay JC, Hughes CM. Connected health care: the future of health care and the role of the pharmacist [published online ahead of print August 4, 2010]. J Eval Clin Pract 2012; 18:56–62. 10.1111/j.1365-2753.2010.01522x
- O’Neill SA, Nugent CD, Donnelly MP, McCullagh P, McLaughlin J. Evaluation of connected health technology. Technol Health Care 2012; 20:151–167.
- Ziebland S, Wyke S. Health and illness in a connected world: how might sharing experiences on the internet affect people’s health? Milbank Q 2012; 90:219–249.
- Dobson A. Personal communication. 2011.
- Dreyfus D. Smart-home technology for persons with disabilities. Am Fam Physician 2009; 80:233.
- Leff B, Burton L, Mader SL, Naughton B, et al. Hospital at home: feasibility and outcomes of a program to provide hospital-level care at home for acutely ill older patients. Ann Intern Med 2005; 143:798–808.
- Quinn CC, Shardell MD, Terrin ML, et al. Cluster-randomized trial of a mobile phone personalized behavioral intervention for blood glucose control [published online ahead of print July 25, 2011]. Diabetes Care 2011; 34:1934–1942. 10.2337/dc11-0366
- Medicare Home Health Agency Manual. Section 201.13, Tele-health. Centers for Medicare & Medicaid Services Web site. http://www.cms.gov/Regulations-and-Guidance/Guidance/Transmittals/downloads/R298HHA.pdf. Published January 22, 2002. Accessed September 18, 2012.
- Stoll C. The Internet? Bah! Newsweek 1995; 125 February 27:41.
Innovative models of home-based palliative care
As the prevalence of serious illness among the elderly population has increased, interest in palliative care has grown as an approach to care management that is patient-centered and focused on quality of life. Case management that employs palliative care has the potential to alleviate unnecessary pain and suffering for patients while they concurrently pursue life-prolonging therapy. Palliative care can be provided across the continuum of care, involving multiple health care providers and practitioners.
Home health care, while often used as a postacute care provider, also can provide longitudinal care to elderly patients without a preceding hospitalization. Home health providers often act as central liaisons to coordinate care while patients are at home, particularly chronically ill patients with multiple physician providers, complex medication regimens, and ongoing concerns with independence and safety in the home.
Home health care can play a critical role in providing palliative care and, through innovative programs, can improve access to it. This article provides context and background on the provision of palliative care and explores how home health can work seamlessly in coordination with other health care stakeholders in providing palliative care.
WHAT IS PALLIATIVE CARE?
Palliative care means patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care throughout the continuum of illness involves addressing physical, intellectual, emotional, social, and spiritual needs and [facilitating] patient autonomy, access to information, and choice.1
At its core, palliative care is a field of medicine aimed at alleviating the suffering of patients. As a “philosophy of care,” palliative care is appropriate for various sites of care at various stages of disease and all ages of patients. While hospice care is defined by the provision of palliative care for patients at the end of life, not all palliative care is hospice care. Rather, palliative care is an approach to care for any patient diagnosed with a serious illness that leverages expertise from multidisciplinary teams of health professionals and addresses pain and symptoms.
Palliative care addresses suffering by incorporating psychosocial and spiritual care with consideration of patient and family needs, preferences, values, beliefs and cultures. Palliative care can be provided throughout the continuum of care for patients with chronic, serious, and even life-threatening illnesses.1 To a degree, all aspects of health care can potentially address some palliative issues in that health care providers ideally combine a desire to cure the patient with a need to alleviate the patient’s pain and suffering.

Although the Medicare program recognizes the potential breadth of palliative care, the hospice benefit is relatively narrow. Consistent with the depiction in the Figure,2 the Medicare hospice benefit is limited to care that is focused on “comfort, not on curing an illness”3 (emphasis added). The Medicare hospice benefit is available to Medicare beneficiaries who: (1) are eligible for Medicare Part A; (2) have a doctor and hospice medical director certifying that they are terminally ill and have 6 months or less to live if their illness runs its normal course; (3) sign a statement choosing hospice care instead of other Medicare-covered benefits to treat their terminal illness (although Medicare will still pay for covered benefits for any health problems that are not related to the terminal illness); and (4) get care from a Medicare-certified hospice program.3
There are, however, clear benefits to providing palliative care outside of the Medicare hospice benefit. In particular, patients with serious illnesses may have more than 6 months to live if their illness runs its normal course. Patients who may die within 1 year due to serious illness can benefit from palliative care. Furthermore, some patients would like to continue to pursue curative treatment of their illnesses, but would benefit from a palliative care approach. By providing palliative care in the context of a plan of care with the patient’s physician, the patient and family can comprehensively make decisions and obtain support that enables access to appropriate treatments while allowing enhanced quality of life through symptom management.
WHO CAN PROVIDE PALLIATIVE CARE?
Palliative care can be provided in any care setting that has been accredited or certified to provide care, including those that are upstream from hospice along the continuum of care. Hospitals, nursing homes, and home health agencies can provide palliative care.
The Joint Commission, a nonprofit accrediting organization, currently accredits or certifies more than 17,000 organizations or programs across the care continuum, including hospitals, nursing homes, home health agencies, and hospices. Within the scope of the home care accreditation program, hospices and home health agencies are evaluated by certified field representatives to determine the extent to which their services meet the standards established by The Joint Commission. These standards are developed with input from health care professionals, providers, subject matter experts, consumers, government agencies (including the Centers for Medicare & Medicaid Services [CMS]) and employers. They are informed by scientific literature and expert consensus and approved by the board of commissioners.
The Joint Commission also has a certification program for palliative care services provided in hospitals and has certified 21 palliative care programs at various hospitals in the United States.
The Joint Commission’s Advanced Certification Program for Palliative Care recognizes hospital inpatient programs that demonstrate exceptional patient-and family-centered care and optimize quality of life for patients (both adult and pediatric) with serious illness. Certification standards emphasize:
- A formal, organized, palliative care program led by an interdisciplinary team whose members are experts in palliative care
- Leadership endorsement and support of the program’s goals for providing care, treatment and services
- Special focus on patient and family engagement
- Processes that support the coordination of care and communication among all care settings and providers
- The use of evidence-based national guidelines or expert consensus to guide patient care
The certification standards cover program management, provision of care, information management, and performance improvement. The standards are built on the National Consensus Project’s Clinical Practice Guidelines for Quality Palliative Care2 and the National Quality Forum’s National Framework and Preferred Practices for Palliative and Hospice Care Quality.4 Many of the concepts contained in the standards for inpatient palliative care have their origins in hospice care.
In addition to palliative care accreditation programs, certification in palliative care for clinicians is also possible. The American Board of Medical Specialties approved the creation of hospice and palliative medicine as a subspecialty in 2006. The National Board of Certification of Hospice and Palliative Nurses offers specialty certification for all levels of hospice and palliative care nursing. The National Association of Social Workers also offers an advanced certified hospice and palliative social worker (ACHP-SW) certifcation for MSW-level clinicians. These certification programs establish qualifications and standards for the members of a palliative care team.
Subject to federal and state requirements that regulate the way health care is provided, hospitals, nursing homes, home health agencies, and hospices are able to provide palliative care to patients who need such care.5,6
WHAT IS HOME HEALTH’S ROLE IN PROVIDING PALLIATIVE CARE?
Many Medicare-certified home health agencies also operate Medicare-approved hospice programs. Home health agencies have a heightened perspective on patients’ palliative care needs. Because of the limited nature of the Medicare hospice benefit, home health agencies have built palliative care programs to fill unmet patient needs. Home health agencies often provide palliative care to patients who may be ineligible for the hospice benefit or have chosen not to enroll in it. These programs are particularly attractive to patients who would like to pursue curative treatment for their serious illnesses or who are expected to live longer than 6 months.
Home health patients with advancing or serious illness or chronic illness are candidates for a palliative care service. For these patients, the burden of their illness continues to grow as distressing symptoms begin to more regularly impact their quality of life. As they continue curative treatment of their illness, they would benefit from palliative care services that provide greater relief of their symptoms and support advanced care planning. Palliative care interventions become an integrated part of the care plan for these patients. Home health agencies serving patients with chronic or advancing illnesses will see care benefits from incorporating palliative care into their team’s skill set.
Two innovative examples of home health–based programs that include a palliative care component have been reported in peer-reviewed literature to date: Kaiser Permanente’s In-Home Palliative Care program and Sutter Health’s Advanced Illness Management (AIM) program.7–10
Kaiser Permanente’s In-Home Palliative Care Program
Kaiser Permanente (KP) established the TriCentral Palliative Care Program in 1998 to achieve balance for seriously ill patients facing the end of life who were caught between “the extremes of too little care and too much.”11 KP began the program after discovering that patients were underusing their existing hospice program. The TriCentral Palliative Care program is an outpatient service, housed in the KP home health department and modeled after the KP hospice program with three key modifications designed to encourage timely referrals to the program:
- Physicians are asked to refer a patient if they “would not be surprised if this patient died in the next year.” Palliative care patients with a prognosis of 12 months or less to live are accepted into the program.
- Improved pain control and symptom management are emphasized, but patients do not need to forgo curative care as they do in hospice programs.
- Patients are assigned a palliative care physician who coordinates care from a variety of health care providers, preventing fragmentation.
The program has five core components that are geared toward enhanced quality of care and patient quality of life. These core components are:
- An interdisciplinary team approach, focused on patient and family, with care provided by a core team consisting of a physician, nurse, and social worker, all with expertise in pain control, other symptom management, and psychosocial intervention
- Home visits by all team members, including physicians, to provide medical care, support, and education as needed by patients and their caregivers
- Ongoing care management to fill gaps in care and ensure that the patient’s medical, social, and spiritual needs are being met
- Telephone support via a toll-free number and after-hours home visits available 24 hours a day, 7 days a week as needed by the patient
- Advanced-care planning that empowers patients and their families to make informed decisions and choices about end-of-life care11
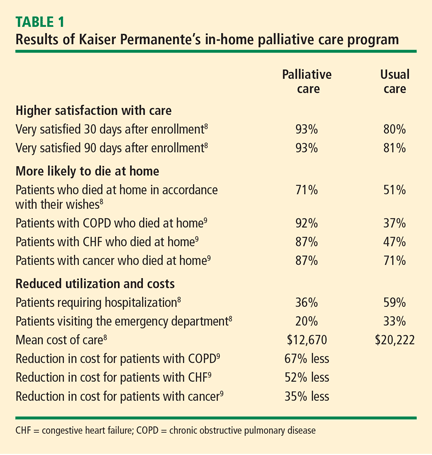
Assessments of the program’s results in a randomized controlled trial8 and a comparative study9 showed that patient satisfaction increased; patients were more likely to die at home in accordance with their wishes; and emergency department (ED) visits, inpatient admissions, and costs were reduced (Table 1).
Sutter Health AIM Program
Sutter Health in northern California, in collaboration with its home care and hospice affiliate, Sutter Care at Home, initiated a home health–based program, Advanced Illness Management (AIM), in 2000 in response to the growing population of patients with advanced illness who needed enhanced care planning and symptom management. This program served patients who met the Medicare eligibility criteria for home health, had a prognosis of 1 year or less, and were continuing to seek treatment or cure for their illness. These patients frequently lacked awareness of their health status, particularly as it related to choices and decisions connected to the progression and management of their conditions. They also were frequently receiving uncoordinated care through various health channels, resulting in substandard symptom management. As a result, patients tended to experience more acute episodes that required frequent use of “unwanted and inappropriate care at the end of life, and they, their families, and their providers were dissatisfied.”12
As the AIM program matured, it incorporated a broader care management model, including principles of patient/caregiver engagement and goal setting, self-management techniques, ongoing advanced care planning, symptom management, and other evidence-based practices related to care transitions and care management. The program connects with the patient’s network of care providers and coordinates the exchange of realtime information about the current status of care plans and medication, as well as the patient’s defined goals. This more comprehensive model of care for persons with advanced illness has achieved improved adherence to patient wishes and goals, reductions in unnecessary hospital and ED utilization, and higher patient/caregiver and provider satisfaction than usual care.
Today, AIM is not primarily a palliative care program. Rather, it provides a comprehensive approach to care management that moves the focus of care for advanced illness out of the hospital and into the home/community setting. AIM achieves this through integrating the patient’s “health system.”
This integration occurs through formation of an interdisciplinary team comprised of the home care team, representative clinicians connected to the hospital, and providers of care for the patient. This expanded team, then, becomes the AIM care management team that is trained on the principles of AIM and its interventions. With this enhanced level of care coordination and unified focus on supporting the patient’s personal health goals, the AIM program serves as a “health system integrator” for the vulnerable and costly population of people with advanced chronic illness.
Inpatient palliative care is a separate and distinct systemwide priority at Sutter Health and, because of this, AIM collaborates closely with the inpatient palliative care teams to ensure that patients experience a seamless transition from hospital to home. There, AIM staff work with patients and families over time to clarify and document their personal values and goals, then use these to develop and drive the care plan. Armed with clearer appreciation of the natural progression of illness, both clinically and practically, coupled with improved understanding of available options for care, most choose to stay in the safety and comfort of their homes and out of the hospital. These avoided hospitalizations are the primary source of AIM’s considerable cost savings.
Patients eligible for AIM are those with clinical, functional, or nutritional decline; with multiple hospitalizations, ED visits, or both within the past 12 months; and who are clinically eligible for hospice but have chosen to continue treatment or have not otherwise made the decision to use a hospice model of care. Once the patient is enrolled, the AIM team works with the patient, the family, and the physician on a preference-driven plan of care. That plan is shared with all providers supporting the patient and is regularly updated to reflect changes in the patient’s evolving choices as illness advances. This tracking of goals and preferences over time as illness progresses has been a critical factor in improving outcomes, especially those related to adherence or honoring a patient’s personal goals.
The AIM program started as a symptom management and care planning intervention for Medicare-eligible home health patients. The program has evolved over time into a pivotal fulcrum by which to engage or create an interdisciplinary focus and skill set across sites and providers of care in an effort to improve the overall outcomes for patients with advancing illness. In 2009, the AIM program began geographically expanding its home health–based AIM teams across 12 counties surrounding the San Francisco Bay area and the greater Sacramento region in northern California. The program now coordinates care with more than 17 hospitals and all of the large Sutter-affiliated medical groups, and it serves approximately 800 patients per day.

The AIM program has yielded significant results in terms of both quality of care and cost savings. Preliminary data on more than 300 AIM patients surveyed from November 2009 through September 2010 showed significant reductions in unnecessary hospitalizations and inpatient direct care costs (Table 2).12 Survey data also showed significant improvements in patient, family, and physician satisfaction when late-stage patients were served through AIM rather than through home care by itself.12
The Sutter Health AIM program recently received a Health Care Innovation Award from the Center for Medicare & Medicaid Innovation (CMMI) because of the program’s ability to “improve care and patient quality of life, increase physician, caregiver, and patient satisfaction, and reduce Medicare costs associated with avoidable hospital stays, ED visits, and days spent in intensive care units and skilled nursing facilities.”13 The $13 million CMMI grant will help expand AIM to the entire Sutter Health system. It is estimated that the program will save $29,388,894 over 3 years.13
CONCLUSION: CHALLENGES AND OPPORTUNITIES FOR THE US HEALTH CARE SYSTEM
The basic objective of AIM and programs like it is to move the focus of care for people with advanced illness out of the hospital and into home and community. This fulfills the Triple Aim vision set forth in 2008 by former CMS Administrator Don Berwick14:
- Improving health by reducing inpatient care that does not achieve person-centered goals or reduce overall mortality
- Improving care by basing it on the values and goals of people dealing with serious chronic illness
- Reducing costs by preventing unwanted hospital care
Sutter Health, a system that is on its way to becoming fully clinically integrated, was a logical choice for launching AIM because its hospitals are forming relationships with physician groups and home care providers. This integration process is supported nationally by CMS and CMMI, which are promoting new models of care and reimbursement such as accountable care organizations (ACOs) and bundled payments.
Nonintegrated hospitals and other provider groups can move in this same direction. AIM establishes key care coordination roles in each setting of care such as in hospitals and physician offices, as well as in the home care–based team and providers. The AIM care model emphasizes close coordination of clinical activities and communications, and integrates these with hospital and medical group operations. These provider groups can move strategically toward becoming “virtual ACOs” by coordinating care for people with advanced illness, who comprise the most vulnerable and costly segment of the US population and increasingly impact Medicare expenditures.
Changes in federal policy will be needed to facilitate national implementation of AIM-like programs. If ACOs and bundled payments were to be implemented overnight, the person-centered, cost-saving advantages of AIM would be obvious. However, until shared risk/shared savings models replace fee-for-service reimbursement, new payment policies will be needed on an interim basis to cover the costs of currently nonreimbursed care management services. This could be arranged through a per-enrollee-per-month payment or shared savings models tied to specific quality and utilization outcomes.
Simplification of regulatory requirements to better serve persons with advancing illness and to reduce the burden on providers operating such programs would be valuable. The pattern or progression of advancing chronic illness requires ongoing coordination in order to maintain a higher quality of life and symptom management. Current regulations and requirements foster an episodic focus in the home, as well in the hospital and physician’s office, which is not in alignment with the experience of persons living with advancing illness.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid program: conditions of participation. Federal Register 2008; 73:32088–32219.
- Clinical Practice Guidelines for Quality Palliative Care. 2nd ed. Pittsburgh, PA: National Consensus Project for Quality Palliative Care. National Consensus Project Web site. http:www.nationalconsensusproject.org. Published 2009. Accessed December 12, 2012.
- Medicare hospice benefits. Centers for Medicare & Medicaid Services Web site. http://www.medicare.gov/publications/pubs/pdf/02154.pdf. Revised August 2012. Accessed November 14, 2012.
- A national framework and preferred practices for palliative and hospice care quality. A consensus report. National Quality Forum Web site. http://www.qualityforum.org/Publications/2006/12/A_National_Framework_and_Preferred_Practices_for_Palliative_and_Hospice_Care_Quality.aspx. Published 2006. Accessed December 12, 2012.
- Michal MH, Pekarske MSL. Palliative care checklist: selected regulatory and risk management considerations. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/fles/public/palliativecare/pcchecklist.pdf. Published April 17, 2006. Accessed November 14, 2012.
- Raffa CA. Palliative care: the legal and regulatory requirements. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/files/public/palliativecare/legal_regulatorypart2.pdf. Published December 22, 2003. Accessed November 14, 2012.
- In-home palliative care allows more patients to die at home, leading to higher satisfaction and lower acute care utilization and costs. Agency for Healthcare Research and Quality Health Care Innovations Exchange Web site. http://www.innovations.ahrq.gov/content.aspx?id=2366. Published March 2, 2009. Updated November 07, 2012. Accessed November 14, 2012.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
- Enguidanos SM, Cherin D, Brumley R. Home-based palliative care study: site of death, and costs of medical care for patients with congestive heart failure, chronic obstructive pulmonary disease, and cancer. J Soc Work End Life Palliat Care 2005; 1:37–56.
- Brumley RD, Enguidanos S, Cherin DA. Effectiveness of a home-based palliative care program for end-of-life. J Palliat Med 2003; 6:715–724.
- Brumley RD, Hillary K. The TriCentral Palliative Care Program Toolkit. 1st ed. MyWhatever Web site. http://www.mywhatever.com/cifwriter/content/22/fles/sorostoolkitfnal120902.doc. Published 2002. Accessed November 14, 2012.
- Meyer H. Innovation profile: changing the conversation in California about care near the end of life. Health Aff 2011; 30:390–393.
- Health Care Innovation Awards. Center for Medicare & Medicaid Innovation Web site. http://innovations.cms.gov/initiatives/Innovation-Awards/california.html. Accessed November 14, 2012.
- Berwick DJ, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff 2008; 27:759–769.
As the prevalence of serious illness among the elderly population has increased, interest in palliative care has grown as an approach to care management that is patient-centered and focused on quality of life. Case management that employs palliative care has the potential to alleviate unnecessary pain and suffering for patients while they concurrently pursue life-prolonging therapy. Palliative care can be provided across the continuum of care, involving multiple health care providers and practitioners.
Home health care, while often used as a postacute care provider, also can provide longitudinal care to elderly patients without a preceding hospitalization. Home health providers often act as central liaisons to coordinate care while patients are at home, particularly chronically ill patients with multiple physician providers, complex medication regimens, and ongoing concerns with independence and safety in the home.
Home health care can play a critical role in providing palliative care and, through innovative programs, can improve access to it. This article provides context and background on the provision of palliative care and explores how home health can work seamlessly in coordination with other health care stakeholders in providing palliative care.
WHAT IS PALLIATIVE CARE?
Palliative care means patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care throughout the continuum of illness involves addressing physical, intellectual, emotional, social, and spiritual needs and [facilitating] patient autonomy, access to information, and choice.1
At its core, palliative care is a field of medicine aimed at alleviating the suffering of patients. As a “philosophy of care,” palliative care is appropriate for various sites of care at various stages of disease and all ages of patients. While hospice care is defined by the provision of palliative care for patients at the end of life, not all palliative care is hospice care. Rather, palliative care is an approach to care for any patient diagnosed with a serious illness that leverages expertise from multidisciplinary teams of health professionals and addresses pain and symptoms.
Palliative care addresses suffering by incorporating psychosocial and spiritual care with consideration of patient and family needs, preferences, values, beliefs and cultures. Palliative care can be provided throughout the continuum of care for patients with chronic, serious, and even life-threatening illnesses.1 To a degree, all aspects of health care can potentially address some palliative issues in that health care providers ideally combine a desire to cure the patient with a need to alleviate the patient’s pain and suffering.
Although the Medicare program recognizes the potential breadth of palliative care, the hospice benefit is relatively narrow. Consistent with the depiction in the Figure,2 the Medicare hospice benefit is limited to care that is focused on “comfort, not on curing an illness”3 (emphasis added). The Medicare hospice benefit is available to Medicare beneficiaries who: (1) are eligible for Medicare Part A; (2) have a doctor and hospice medical director certifying that they are terminally ill and have 6 months or less to live if their illness runs its normal course; (3) sign a statement choosing hospice care instead of other Medicare-covered benefits to treat their terminal illness (although Medicare will still pay for covered benefits for any health problems that are not related to the terminal illness); and (4) get care from a Medicare-certified hospice program.3
There are, however, clear benefits to providing palliative care outside of the Medicare hospice benefit. In particular, patients with serious illnesses may have more than 6 months to live if their illness runs its normal course. Patients who may die within 1 year due to serious illness can benefit from palliative care. Furthermore, some patients would like to continue to pursue curative treatment of their illnesses, but would benefit from a palliative care approach. By providing palliative care in the context of a plan of care with the patient’s physician, the patient and family can comprehensively make decisions and obtain support that enables access to appropriate treatments while allowing enhanced quality of life through symptom management.
WHO CAN PROVIDE PALLIATIVE CARE?
Palliative care can be provided in any care setting that has been accredited or certified to provide care, including those that are upstream from hospice along the continuum of care. Hospitals, nursing homes, and home health agencies can provide palliative care.
The Joint Commission, a nonprofit accrediting organization, currently accredits or certifies more than 17,000 organizations or programs across the care continuum, including hospitals, nursing homes, home health agencies, and hospices. Within the scope of the home care accreditation program, hospices and home health agencies are evaluated by certified field representatives to determine the extent to which their services meet the standards established by The Joint Commission. These standards are developed with input from health care professionals, providers, subject matter experts, consumers, government agencies (including the Centers for Medicare & Medicaid Services [CMS]) and employers. They are informed by scientific literature and expert consensus and approved by the board of commissioners.
The Joint Commission also has a certification program for palliative care services provided in hospitals and has certified 21 palliative care programs at various hospitals in the United States.
The Joint Commission’s Advanced Certification Program for Palliative Care recognizes hospital inpatient programs that demonstrate exceptional patient-and family-centered care and optimize quality of life for patients (both adult and pediatric) with serious illness. Certification standards emphasize:
- A formal, organized, palliative care program led by an interdisciplinary team whose members are experts in palliative care
- Leadership endorsement and support of the program’s goals for providing care, treatment and services
- Special focus on patient and family engagement
- Processes that support the coordination of care and communication among all care settings and providers
- The use of evidence-based national guidelines or expert consensus to guide patient care
The certification standards cover program management, provision of care, information management, and performance improvement. The standards are built on the National Consensus Project’s Clinical Practice Guidelines for Quality Palliative Care2 and the National Quality Forum’s National Framework and Preferred Practices for Palliative and Hospice Care Quality.4 Many of the concepts contained in the standards for inpatient palliative care have their origins in hospice care.
In addition to palliative care accreditation programs, certification in palliative care for clinicians is also possible. The American Board of Medical Specialties approved the creation of hospice and palliative medicine as a subspecialty in 2006. The National Board of Certification of Hospice and Palliative Nurses offers specialty certification for all levels of hospice and palliative care nursing. The National Association of Social Workers also offers an advanced certified hospice and palliative social worker (ACHP-SW) certifcation for MSW-level clinicians. These certification programs establish qualifications and standards for the members of a palliative care team.
Subject to federal and state requirements that regulate the way health care is provided, hospitals, nursing homes, home health agencies, and hospices are able to provide palliative care to patients who need such care.5,6
WHAT IS HOME HEALTH’S ROLE IN PROVIDING PALLIATIVE CARE?
Many Medicare-certified home health agencies also operate Medicare-approved hospice programs. Home health agencies have a heightened perspective on patients’ palliative care needs. Because of the limited nature of the Medicare hospice benefit, home health agencies have built palliative care programs to fill unmet patient needs. Home health agencies often provide palliative care to patients who may be ineligible for the hospice benefit or have chosen not to enroll in it. These programs are particularly attractive to patients who would like to pursue curative treatment for their serious illnesses or who are expected to live longer than 6 months.
Home health patients with advancing or serious illness or chronic illness are candidates for a palliative care service. For these patients, the burden of their illness continues to grow as distressing symptoms begin to more regularly impact their quality of life. As they continue curative treatment of their illness, they would benefit from palliative care services that provide greater relief of their symptoms and support advanced care planning. Palliative care interventions become an integrated part of the care plan for these patients. Home health agencies serving patients with chronic or advancing illnesses will see care benefits from incorporating palliative care into their team’s skill set.
Two innovative examples of home health–based programs that include a palliative care component have been reported in peer-reviewed literature to date: Kaiser Permanente’s In-Home Palliative Care program and Sutter Health’s Advanced Illness Management (AIM) program.7–10
Kaiser Permanente’s In-Home Palliative Care Program
Kaiser Permanente (KP) established the TriCentral Palliative Care Program in 1998 to achieve balance for seriously ill patients facing the end of life who were caught between “the extremes of too little care and too much.”11 KP began the program after discovering that patients were underusing their existing hospice program. The TriCentral Palliative Care program is an outpatient service, housed in the KP home health department and modeled after the KP hospice program with three key modifications designed to encourage timely referrals to the program:
- Physicians are asked to refer a patient if they “would not be surprised if this patient died in the next year.” Palliative care patients with a prognosis of 12 months or less to live are accepted into the program.
- Improved pain control and symptom management are emphasized, but patients do not need to forgo curative care as they do in hospice programs.
- Patients are assigned a palliative care physician who coordinates care from a variety of health care providers, preventing fragmentation.
The program has five core components that are geared toward enhanced quality of care and patient quality of life. These core components are:
- An interdisciplinary team approach, focused on patient and family, with care provided by a core team consisting of a physician, nurse, and social worker, all with expertise in pain control, other symptom management, and psychosocial intervention
- Home visits by all team members, including physicians, to provide medical care, support, and education as needed by patients and their caregivers
- Ongoing care management to fill gaps in care and ensure that the patient’s medical, social, and spiritual needs are being met
- Telephone support via a toll-free number and after-hours home visits available 24 hours a day, 7 days a week as needed by the patient
- Advanced-care planning that empowers patients and their families to make informed decisions and choices about end-of-life care11
Assessments of the program’s results in a randomized controlled trial8 and a comparative study9 showed that patient satisfaction increased; patients were more likely to die at home in accordance with their wishes; and emergency department (ED) visits, inpatient admissions, and costs were reduced (Table 1).
Sutter Health AIM Program
Sutter Health in northern California, in collaboration with its home care and hospice affiliate, Sutter Care at Home, initiated a home health–based program, Advanced Illness Management (AIM), in 2000 in response to the growing population of patients with advanced illness who needed enhanced care planning and symptom management. This program served patients who met the Medicare eligibility criteria for home health, had a prognosis of 1 year or less, and were continuing to seek treatment or cure for their illness. These patients frequently lacked awareness of their health status, particularly as it related to choices and decisions connected to the progression and management of their conditions. They also were frequently receiving uncoordinated care through various health channels, resulting in substandard symptom management. As a result, patients tended to experience more acute episodes that required frequent use of “unwanted and inappropriate care at the end of life, and they, their families, and their providers were dissatisfied.”12
As the AIM program matured, it incorporated a broader care management model, including principles of patient/caregiver engagement and goal setting, self-management techniques, ongoing advanced care planning, symptom management, and other evidence-based practices related to care transitions and care management. The program connects with the patient’s network of care providers and coordinates the exchange of realtime information about the current status of care plans and medication, as well as the patient’s defined goals. This more comprehensive model of care for persons with advanced illness has achieved improved adherence to patient wishes and goals, reductions in unnecessary hospital and ED utilization, and higher patient/caregiver and provider satisfaction than usual care.
Today, AIM is not primarily a palliative care program. Rather, it provides a comprehensive approach to care management that moves the focus of care for advanced illness out of the hospital and into the home/community setting. AIM achieves this through integrating the patient’s “health system.”
This integration occurs through formation of an interdisciplinary team comprised of the home care team, representative clinicians connected to the hospital, and providers of care for the patient. This expanded team, then, becomes the AIM care management team that is trained on the principles of AIM and its interventions. With this enhanced level of care coordination and unified focus on supporting the patient’s personal health goals, the AIM program serves as a “health system integrator” for the vulnerable and costly population of people with advanced chronic illness.
Inpatient palliative care is a separate and distinct systemwide priority at Sutter Health and, because of this, AIM collaborates closely with the inpatient palliative care teams to ensure that patients experience a seamless transition from hospital to home. There, AIM staff work with patients and families over time to clarify and document their personal values and goals, then use these to develop and drive the care plan. Armed with clearer appreciation of the natural progression of illness, both clinically and practically, coupled with improved understanding of available options for care, most choose to stay in the safety and comfort of their homes and out of the hospital. These avoided hospitalizations are the primary source of AIM’s considerable cost savings.
Patients eligible for AIM are those with clinical, functional, or nutritional decline; with multiple hospitalizations, ED visits, or both within the past 12 months; and who are clinically eligible for hospice but have chosen to continue treatment or have not otherwise made the decision to use a hospice model of care. Once the patient is enrolled, the AIM team works with the patient, the family, and the physician on a preference-driven plan of care. That plan is shared with all providers supporting the patient and is regularly updated to reflect changes in the patient’s evolving choices as illness advances. This tracking of goals and preferences over time as illness progresses has been a critical factor in improving outcomes, especially those related to adherence or honoring a patient’s personal goals.
The AIM program started as a symptom management and care planning intervention for Medicare-eligible home health patients. The program has evolved over time into a pivotal fulcrum by which to engage or create an interdisciplinary focus and skill set across sites and providers of care in an effort to improve the overall outcomes for patients with advancing illness. In 2009, the AIM program began geographically expanding its home health–based AIM teams across 12 counties surrounding the San Francisco Bay area and the greater Sacramento region in northern California. The program now coordinates care with more than 17 hospitals and all of the large Sutter-affiliated medical groups, and it serves approximately 800 patients per day.
The AIM program has yielded significant results in terms of both quality of care and cost savings. Preliminary data on more than 300 AIM patients surveyed from November 2009 through September 2010 showed significant reductions in unnecessary hospitalizations and inpatient direct care costs (Table 2).12 Survey data also showed significant improvements in patient, family, and physician satisfaction when late-stage patients were served through AIM rather than through home care by itself.12
The Sutter Health AIM program recently received a Health Care Innovation Award from the Center for Medicare & Medicaid Innovation (CMMI) because of the program’s ability to “improve care and patient quality of life, increase physician, caregiver, and patient satisfaction, and reduce Medicare costs associated with avoidable hospital stays, ED visits, and days spent in intensive care units and skilled nursing facilities.”13 The $13 million CMMI grant will help expand AIM to the entire Sutter Health system. It is estimated that the program will save $29,388,894 over 3 years.13
CONCLUSION: CHALLENGES AND OPPORTUNITIES FOR THE US HEALTH CARE SYSTEM
The basic objective of AIM and programs like it is to move the focus of care for people with advanced illness out of the hospital and into home and community. This fulfills the Triple Aim vision set forth in 2008 by former CMS Administrator Don Berwick14:
- Improving health by reducing inpatient care that does not achieve person-centered goals or reduce overall mortality
- Improving care by basing it on the values and goals of people dealing with serious chronic illness
- Reducing costs by preventing unwanted hospital care
Sutter Health, a system that is on its way to becoming fully clinically integrated, was a logical choice for launching AIM because its hospitals are forming relationships with physician groups and home care providers. This integration process is supported nationally by CMS and CMMI, which are promoting new models of care and reimbursement such as accountable care organizations (ACOs) and bundled payments.
Nonintegrated hospitals and other provider groups can move in this same direction. AIM establishes key care coordination roles in each setting of care such as in hospitals and physician offices, as well as in the home care–based team and providers. The AIM care model emphasizes close coordination of clinical activities and communications, and integrates these with hospital and medical group operations. These provider groups can move strategically toward becoming “virtual ACOs” by coordinating care for people with advanced illness, who comprise the most vulnerable and costly segment of the US population and increasingly impact Medicare expenditures.
Changes in federal policy will be needed to facilitate national implementation of AIM-like programs. If ACOs and bundled payments were to be implemented overnight, the person-centered, cost-saving advantages of AIM would be obvious. However, until shared risk/shared savings models replace fee-for-service reimbursement, new payment policies will be needed on an interim basis to cover the costs of currently nonreimbursed care management services. This could be arranged through a per-enrollee-per-month payment or shared savings models tied to specific quality and utilization outcomes.
Simplification of regulatory requirements to better serve persons with advancing illness and to reduce the burden on providers operating such programs would be valuable. The pattern or progression of advancing chronic illness requires ongoing coordination in order to maintain a higher quality of life and symptom management. Current regulations and requirements foster an episodic focus in the home, as well in the hospital and physician’s office, which is not in alignment with the experience of persons living with advancing illness.
As the prevalence of serious illness among the elderly population has increased, interest in palliative care has grown as an approach to care management that is patient-centered and focused on quality of life. Case management that employs palliative care has the potential to alleviate unnecessary pain and suffering for patients while they concurrently pursue life-prolonging therapy. Palliative care can be provided across the continuum of care, involving multiple health care providers and practitioners.
Home health care, while often used as a postacute care provider, also can provide longitudinal care to elderly patients without a preceding hospitalization. Home health providers often act as central liaisons to coordinate care while patients are at home, particularly chronically ill patients with multiple physician providers, complex medication regimens, and ongoing concerns with independence and safety in the home.
Home health care can play a critical role in providing palliative care and, through innovative programs, can improve access to it. This article provides context and background on the provision of palliative care and explores how home health can work seamlessly in coordination with other health care stakeholders in providing palliative care.
WHAT IS PALLIATIVE CARE?
Palliative care means patient- and family-centered care that optimizes quality of life by anticipating, preventing, and treating suffering. Palliative care throughout the continuum of illness involves addressing physical, intellectual, emotional, social, and spiritual needs and [facilitating] patient autonomy, access to information, and choice.1
At its core, palliative care is a field of medicine aimed at alleviating the suffering of patients. As a “philosophy of care,” palliative care is appropriate for various sites of care at various stages of disease and all ages of patients. While hospice care is defined by the provision of palliative care for patients at the end of life, not all palliative care is hospice care. Rather, palliative care is an approach to care for any patient diagnosed with a serious illness that leverages expertise from multidisciplinary teams of health professionals and addresses pain and symptoms.
Palliative care addresses suffering by incorporating psychosocial and spiritual care with consideration of patient and family needs, preferences, values, beliefs and cultures. Palliative care can be provided throughout the continuum of care for patients with chronic, serious, and even life-threatening illnesses.1 To a degree, all aspects of health care can potentially address some palliative issues in that health care providers ideally combine a desire to cure the patient with a need to alleviate the patient’s pain and suffering.
Although the Medicare program recognizes the potential breadth of palliative care, the hospice benefit is relatively narrow. Consistent with the depiction in the Figure,2 the Medicare hospice benefit is limited to care that is focused on “comfort, not on curing an illness”3 (emphasis added). The Medicare hospice benefit is available to Medicare beneficiaries who: (1) are eligible for Medicare Part A; (2) have a doctor and hospice medical director certifying that they are terminally ill and have 6 months or less to live if their illness runs its normal course; (3) sign a statement choosing hospice care instead of other Medicare-covered benefits to treat their terminal illness (although Medicare will still pay for covered benefits for any health problems that are not related to the terminal illness); and (4) get care from a Medicare-certified hospice program.3
There are, however, clear benefits to providing palliative care outside of the Medicare hospice benefit. In particular, patients with serious illnesses may have more than 6 months to live if their illness runs its normal course. Patients who may die within 1 year due to serious illness can benefit from palliative care. Furthermore, some patients would like to continue to pursue curative treatment of their illnesses, but would benefit from a palliative care approach. By providing palliative care in the context of a plan of care with the patient’s physician, the patient and family can comprehensively make decisions and obtain support that enables access to appropriate treatments while allowing enhanced quality of life through symptom management.
WHO CAN PROVIDE PALLIATIVE CARE?
Palliative care can be provided in any care setting that has been accredited or certified to provide care, including those that are upstream from hospice along the continuum of care. Hospitals, nursing homes, and home health agencies can provide palliative care.
The Joint Commission, a nonprofit accrediting organization, currently accredits or certifies more than 17,000 organizations or programs across the care continuum, including hospitals, nursing homes, home health agencies, and hospices. Within the scope of the home care accreditation program, hospices and home health agencies are evaluated by certified field representatives to determine the extent to which their services meet the standards established by The Joint Commission. These standards are developed with input from health care professionals, providers, subject matter experts, consumers, government agencies (including the Centers for Medicare & Medicaid Services [CMS]) and employers. They are informed by scientific literature and expert consensus and approved by the board of commissioners.
The Joint Commission also has a certification program for palliative care services provided in hospitals and has certified 21 palliative care programs at various hospitals in the United States.
The Joint Commission’s Advanced Certification Program for Palliative Care recognizes hospital inpatient programs that demonstrate exceptional patient-and family-centered care and optimize quality of life for patients (both adult and pediatric) with serious illness. Certification standards emphasize:
- A formal, organized, palliative care program led by an interdisciplinary team whose members are experts in palliative care
- Leadership endorsement and support of the program’s goals for providing care, treatment and services
- Special focus on patient and family engagement
- Processes that support the coordination of care and communication among all care settings and providers
- The use of evidence-based national guidelines or expert consensus to guide patient care
The certification standards cover program management, provision of care, information management, and performance improvement. The standards are built on the National Consensus Project’s Clinical Practice Guidelines for Quality Palliative Care2 and the National Quality Forum’s National Framework and Preferred Practices for Palliative and Hospice Care Quality.4 Many of the concepts contained in the standards for inpatient palliative care have their origins in hospice care.
In addition to palliative care accreditation programs, certification in palliative care for clinicians is also possible. The American Board of Medical Specialties approved the creation of hospice and palliative medicine as a subspecialty in 2006. The National Board of Certification of Hospice and Palliative Nurses offers specialty certification for all levels of hospice and palliative care nursing. The National Association of Social Workers also offers an advanced certified hospice and palliative social worker (ACHP-SW) certifcation for MSW-level clinicians. These certification programs establish qualifications and standards for the members of a palliative care team.
Subject to federal and state requirements that regulate the way health care is provided, hospitals, nursing homes, home health agencies, and hospices are able to provide palliative care to patients who need such care.5,6
WHAT IS HOME HEALTH’S ROLE IN PROVIDING PALLIATIVE CARE?
Many Medicare-certified home health agencies also operate Medicare-approved hospice programs. Home health agencies have a heightened perspective on patients’ palliative care needs. Because of the limited nature of the Medicare hospice benefit, home health agencies have built palliative care programs to fill unmet patient needs. Home health agencies often provide palliative care to patients who may be ineligible for the hospice benefit or have chosen not to enroll in it. These programs are particularly attractive to patients who would like to pursue curative treatment for their serious illnesses or who are expected to live longer than 6 months.
Home health patients with advancing or serious illness or chronic illness are candidates for a palliative care service. For these patients, the burden of their illness continues to grow as distressing symptoms begin to more regularly impact their quality of life. As they continue curative treatment of their illness, they would benefit from palliative care services that provide greater relief of their symptoms and support advanced care planning. Palliative care interventions become an integrated part of the care plan for these patients. Home health agencies serving patients with chronic or advancing illnesses will see care benefits from incorporating palliative care into their team’s skill set.
Two innovative examples of home health–based programs that include a palliative care component have been reported in peer-reviewed literature to date: Kaiser Permanente’s In-Home Palliative Care program and Sutter Health’s Advanced Illness Management (AIM) program.7–10
Kaiser Permanente’s In-Home Palliative Care Program
Kaiser Permanente (KP) established the TriCentral Palliative Care Program in 1998 to achieve balance for seriously ill patients facing the end of life who were caught between “the extremes of too little care and too much.”11 KP began the program after discovering that patients were underusing their existing hospice program. The TriCentral Palliative Care program is an outpatient service, housed in the KP home health department and modeled after the KP hospice program with three key modifications designed to encourage timely referrals to the program:
- Physicians are asked to refer a patient if they “would not be surprised if this patient died in the next year.” Palliative care patients with a prognosis of 12 months or less to live are accepted into the program.
- Improved pain control and symptom management are emphasized, but patients do not need to forgo curative care as they do in hospice programs.
- Patients are assigned a palliative care physician who coordinates care from a variety of health care providers, preventing fragmentation.
The program has five core components that are geared toward enhanced quality of care and patient quality of life. These core components are:
- An interdisciplinary team approach, focused on patient and family, with care provided by a core team consisting of a physician, nurse, and social worker, all with expertise in pain control, other symptom management, and psychosocial intervention
- Home visits by all team members, including physicians, to provide medical care, support, and education as needed by patients and their caregivers
- Ongoing care management to fill gaps in care and ensure that the patient’s medical, social, and spiritual needs are being met
- Telephone support via a toll-free number and after-hours home visits available 24 hours a day, 7 days a week as needed by the patient
- Advanced-care planning that empowers patients and their families to make informed decisions and choices about end-of-life care11
Assessments of the program’s results in a randomized controlled trial8 and a comparative study9 showed that patient satisfaction increased; patients were more likely to die at home in accordance with their wishes; and emergency department (ED) visits, inpatient admissions, and costs were reduced (Table 1).
Sutter Health AIM Program
Sutter Health in northern California, in collaboration with its home care and hospice affiliate, Sutter Care at Home, initiated a home health–based program, Advanced Illness Management (AIM), in 2000 in response to the growing population of patients with advanced illness who needed enhanced care planning and symptom management. This program served patients who met the Medicare eligibility criteria for home health, had a prognosis of 1 year or less, and were continuing to seek treatment or cure for their illness. These patients frequently lacked awareness of their health status, particularly as it related to choices and decisions connected to the progression and management of their conditions. They also were frequently receiving uncoordinated care through various health channels, resulting in substandard symptom management. As a result, patients tended to experience more acute episodes that required frequent use of “unwanted and inappropriate care at the end of life, and they, their families, and their providers were dissatisfied.”12
As the AIM program matured, it incorporated a broader care management model, including principles of patient/caregiver engagement and goal setting, self-management techniques, ongoing advanced care planning, symptom management, and other evidence-based practices related to care transitions and care management. The program connects with the patient’s network of care providers and coordinates the exchange of realtime information about the current status of care plans and medication, as well as the patient’s defined goals. This more comprehensive model of care for persons with advanced illness has achieved improved adherence to patient wishes and goals, reductions in unnecessary hospital and ED utilization, and higher patient/caregiver and provider satisfaction than usual care.
Today, AIM is not primarily a palliative care program. Rather, it provides a comprehensive approach to care management that moves the focus of care for advanced illness out of the hospital and into the home/community setting. AIM achieves this through integrating the patient’s “health system.”
This integration occurs through formation of an interdisciplinary team comprised of the home care team, representative clinicians connected to the hospital, and providers of care for the patient. This expanded team, then, becomes the AIM care management team that is trained on the principles of AIM and its interventions. With this enhanced level of care coordination and unified focus on supporting the patient’s personal health goals, the AIM program serves as a “health system integrator” for the vulnerable and costly population of people with advanced chronic illness.
Inpatient palliative care is a separate and distinct systemwide priority at Sutter Health and, because of this, AIM collaborates closely with the inpatient palliative care teams to ensure that patients experience a seamless transition from hospital to home. There, AIM staff work with patients and families over time to clarify and document their personal values and goals, then use these to develop and drive the care plan. Armed with clearer appreciation of the natural progression of illness, both clinically and practically, coupled with improved understanding of available options for care, most choose to stay in the safety and comfort of their homes and out of the hospital. These avoided hospitalizations are the primary source of AIM’s considerable cost savings.
Patients eligible for AIM are those with clinical, functional, or nutritional decline; with multiple hospitalizations, ED visits, or both within the past 12 months; and who are clinically eligible for hospice but have chosen to continue treatment or have not otherwise made the decision to use a hospice model of care. Once the patient is enrolled, the AIM team works with the patient, the family, and the physician on a preference-driven plan of care. That plan is shared with all providers supporting the patient and is regularly updated to reflect changes in the patient’s evolving choices as illness advances. This tracking of goals and preferences over time as illness progresses has been a critical factor in improving outcomes, especially those related to adherence or honoring a patient’s personal goals.
The AIM program started as a symptom management and care planning intervention for Medicare-eligible home health patients. The program has evolved over time into a pivotal fulcrum by which to engage or create an interdisciplinary focus and skill set across sites and providers of care in an effort to improve the overall outcomes for patients with advancing illness. In 2009, the AIM program began geographically expanding its home health–based AIM teams across 12 counties surrounding the San Francisco Bay area and the greater Sacramento region in northern California. The program now coordinates care with more than 17 hospitals and all of the large Sutter-affiliated medical groups, and it serves approximately 800 patients per day.
The AIM program has yielded significant results in terms of both quality of care and cost savings. Preliminary data on more than 300 AIM patients surveyed from November 2009 through September 2010 showed significant reductions in unnecessary hospitalizations and inpatient direct care costs (Table 2).12 Survey data also showed significant improvements in patient, family, and physician satisfaction when late-stage patients were served through AIM rather than through home care by itself.12
The Sutter Health AIM program recently received a Health Care Innovation Award from the Center for Medicare & Medicaid Innovation (CMMI) because of the program’s ability to “improve care and patient quality of life, increase physician, caregiver, and patient satisfaction, and reduce Medicare costs associated with avoidable hospital stays, ED visits, and days spent in intensive care units and skilled nursing facilities.”13 The $13 million CMMI grant will help expand AIM to the entire Sutter Health system. It is estimated that the program will save $29,388,894 over 3 years.13
CONCLUSION: CHALLENGES AND OPPORTUNITIES FOR THE US HEALTH CARE SYSTEM
The basic objective of AIM and programs like it is to move the focus of care for people with advanced illness out of the hospital and into home and community. This fulfills the Triple Aim vision set forth in 2008 by former CMS Administrator Don Berwick14:
- Improving health by reducing inpatient care that does not achieve person-centered goals or reduce overall mortality
- Improving care by basing it on the values and goals of people dealing with serious chronic illness
- Reducing costs by preventing unwanted hospital care
Sutter Health, a system that is on its way to becoming fully clinically integrated, was a logical choice for launching AIM because its hospitals are forming relationships with physician groups and home care providers. This integration process is supported nationally by CMS and CMMI, which are promoting new models of care and reimbursement such as accountable care organizations (ACOs) and bundled payments.
Nonintegrated hospitals and other provider groups can move in this same direction. AIM establishes key care coordination roles in each setting of care such as in hospitals and physician offices, as well as in the home care–based team and providers. The AIM care model emphasizes close coordination of clinical activities and communications, and integrates these with hospital and medical group operations. These provider groups can move strategically toward becoming “virtual ACOs” by coordinating care for people with advanced illness, who comprise the most vulnerable and costly segment of the US population and increasingly impact Medicare expenditures.
Changes in federal policy will be needed to facilitate national implementation of AIM-like programs. If ACOs and bundled payments were to be implemented overnight, the person-centered, cost-saving advantages of AIM would be obvious. However, until shared risk/shared savings models replace fee-for-service reimbursement, new payment policies will be needed on an interim basis to cover the costs of currently nonreimbursed care management services. This could be arranged through a per-enrollee-per-month payment or shared savings models tied to specific quality and utilization outcomes.
Simplification of regulatory requirements to better serve persons with advancing illness and to reduce the burden on providers operating such programs would be valuable. The pattern or progression of advancing chronic illness requires ongoing coordination in order to maintain a higher quality of life and symptom management. Current regulations and requirements foster an episodic focus in the home, as well in the hospital and physician’s office, which is not in alignment with the experience of persons living with advancing illness.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid program: conditions of participation. Federal Register 2008; 73:32088–32219.
- Clinical Practice Guidelines for Quality Palliative Care. 2nd ed. Pittsburgh, PA: National Consensus Project for Quality Palliative Care. National Consensus Project Web site. http:www.nationalconsensusproject.org. Published 2009. Accessed December 12, 2012.
- Medicare hospice benefits. Centers for Medicare & Medicaid Services Web site. http://www.medicare.gov/publications/pubs/pdf/02154.pdf. Revised August 2012. Accessed November 14, 2012.
- A national framework and preferred practices for palliative and hospice care quality. A consensus report. National Quality Forum Web site. http://www.qualityforum.org/Publications/2006/12/A_National_Framework_and_Preferred_Practices_for_Palliative_and_Hospice_Care_Quality.aspx. Published 2006. Accessed December 12, 2012.
- Michal MH, Pekarske MSL. Palliative care checklist: selected regulatory and risk management considerations. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/fles/public/palliativecare/pcchecklist.pdf. Published April 17, 2006. Accessed November 14, 2012.
- Raffa CA. Palliative care: the legal and regulatory requirements. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/files/public/palliativecare/legal_regulatorypart2.pdf. Published December 22, 2003. Accessed November 14, 2012.
- In-home palliative care allows more patients to die at home, leading to higher satisfaction and lower acute care utilization and costs. Agency for Healthcare Research and Quality Health Care Innovations Exchange Web site. http://www.innovations.ahrq.gov/content.aspx?id=2366. Published March 2, 2009. Updated November 07, 2012. Accessed November 14, 2012.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
- Enguidanos SM, Cherin D, Brumley R. Home-based palliative care study: site of death, and costs of medical care for patients with congestive heart failure, chronic obstructive pulmonary disease, and cancer. J Soc Work End Life Palliat Care 2005; 1:37–56.
- Brumley RD, Enguidanos S, Cherin DA. Effectiveness of a home-based palliative care program for end-of-life. J Palliat Med 2003; 6:715–724.
- Brumley RD, Hillary K. The TriCentral Palliative Care Program Toolkit. 1st ed. MyWhatever Web site. http://www.mywhatever.com/cifwriter/content/22/fles/sorostoolkitfnal120902.doc. Published 2002. Accessed November 14, 2012.
- Meyer H. Innovation profile: changing the conversation in California about care near the end of life. Health Aff 2011; 30:390–393.
- Health Care Innovation Awards. Center for Medicare & Medicaid Innovation Web site. http://innovations.cms.gov/initiatives/Innovation-Awards/california.html. Accessed November 14, 2012.
- Berwick DJ, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff 2008; 27:759–769.
- Centers for Medicare & Medicaid Services. Medicare and Medicaid program: conditions of participation. Federal Register 2008; 73:32088–32219.
- Clinical Practice Guidelines for Quality Palliative Care. 2nd ed. Pittsburgh, PA: National Consensus Project for Quality Palliative Care. National Consensus Project Web site. http:www.nationalconsensusproject.org. Published 2009. Accessed December 12, 2012.
- Medicare hospice benefits. Centers for Medicare & Medicaid Services Web site. http://www.medicare.gov/publications/pubs/pdf/02154.pdf. Revised August 2012. Accessed November 14, 2012.
- A national framework and preferred practices for palliative and hospice care quality. A consensus report. National Quality Forum Web site. http://www.qualityforum.org/Publications/2006/12/A_National_Framework_and_Preferred_Practices_for_Palliative_and_Hospice_Care_Quality.aspx. Published 2006. Accessed December 12, 2012.
- Michal MH, Pekarske MSL. Palliative care checklist: selected regulatory and risk management considerations. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/fles/public/palliativecare/pcchecklist.pdf. Published April 17, 2006. Accessed November 14, 2012.
- Raffa CA. Palliative care: the legal and regulatory requirements. National Hospice and Palliative Care Organization Web site. http://www.nhpco.org/files/public/palliativecare/legal_regulatorypart2.pdf. Published December 22, 2003. Accessed November 14, 2012.
- In-home palliative care allows more patients to die at home, leading to higher satisfaction and lower acute care utilization and costs. Agency for Healthcare Research and Quality Health Care Innovations Exchange Web site. http://www.innovations.ahrq.gov/content.aspx?id=2366. Published March 2, 2009. Updated November 07, 2012. Accessed November 14, 2012.
- Brumley R, Enguidanos S, Jamison P, et al. Increased satisfaction with care and lower costs: results of a randomized trial of in-home palliative care. J Am Geriatr Soc 2007; 55:993–1000.
- Enguidanos SM, Cherin D, Brumley R. Home-based palliative care study: site of death, and costs of medical care for patients with congestive heart failure, chronic obstructive pulmonary disease, and cancer. J Soc Work End Life Palliat Care 2005; 1:37–56.
- Brumley RD, Enguidanos S, Cherin DA. Effectiveness of a home-based palliative care program for end-of-life. J Palliat Med 2003; 6:715–724.
- Brumley RD, Hillary K. The TriCentral Palliative Care Program Toolkit. 1st ed. MyWhatever Web site. http://www.mywhatever.com/cifwriter/content/22/fles/sorostoolkitfnal120902.doc. Published 2002. Accessed November 14, 2012.
- Meyer H. Innovation profile: changing the conversation in California about care near the end of life. Health Aff 2011; 30:390–393.
- Health Care Innovation Awards. Center for Medicare & Medicaid Innovation Web site. http://innovations.cms.gov/initiatives/Innovation-Awards/california.html. Accessed November 14, 2012.
- Berwick DJ, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff 2008; 27:759–769.
Accountable care and patient-centered medical homes: Implications for office-based practice
The passage of the Patient Protection and Affordable Care Act will profoundly affect the way physicians—particularly those engaged in primary care—practice medicine. Clinicians and their colleagues will be obliged to meet government-mandated performance quality measures while achieving cost efficiencies. Two concepts are central to the implementation of reform in the US health care system: accountable care organizations (ACOs) and the patient-centered medical home (PCMH). To get some perspective on what these changes mean for the practicing clinician, Cleveland Clinic Journal of Medicine (CCJM) interviewed David Longworth, MD, who chairs the Cleveland Clinic Medicine Institute and directs strategy and implementation of Cleveland Clinic ACO-related activities.
CCJM: Please explain briefly the concept of PCMH.
Dr. Longworth: PCMH is not a new concept; first advanced by the American Academy of Pediatrics in 1967,1 it represents a model of care in which an individual patient has a primary relationship with one provider who manages and coordinates the different aspects of the patient’s health care. The provider collaborates with a team of health care professionals. The concept caught on about a decade ago when a consortium of family medicine organizations and ultimately industry, including IBM, endorsed the concept. IBM and others created the Primary Care Consortium and began to drive the concept of PCMH.
Increasingly, care delivered through PCMH is team-based. The team coordinates the patient’s care and, when appropriate, enlists specialists or subspecialists to provide necessary components of care, all while maintaining responsibility for care coordination across the continuum of care. The medical home model provides an opportunity for enhanced access and care coordination utilizing care outside of the office walls, such as through retail clinics, eVisits, online diagnostic services, phone and electronic communication, and house call services.
Patient-centered medical homes are springing up across the country. In 2008, the National Committee for Quality Assurance (NCQA) developed criteria for recognition of PCMHs.2 It scored the sophistication of medical homes at three levels, level 1 being the lowest and level 3 the highest. Between 2008 and the end of 2010, NCQA had recognized more than 1,500 PCMHs. According to the latest figures, more than 3,000 practices have now earned PCMH recognition from the NCQA.3

At Cleveland Clinic, pilot projects at three family health centers that cover 60,000 persons have recently been rolled out with the goal of determining the model of team care that yields the highest value, with value defined by the equation of quality over cost. Ideally, higher quality is delivered at lower cost to increase value.
CCJM: What are the goals of ACOs?
Dr. Longworth: The term “accountable care,” first used in 2006 by Elliot Fisher, Dartmouth Institute of Health Policy and Clinical Practice,6 expresses the idea that health care organizations be accountable for the care they deliver, with the three-part aim of better health for populations, better care for individuals, and reduced cost inefficiencies without compromised care.
With accountable care, institutions take on risk with the expectation that they will improve quality but reduce costs, and if they reduce costs and achieve certain quality targets for populations of patients, they will share in the savings accrued. The Affordable Care Act laid the groundwork for creation of ACOs. The regulation for ACOs released by the Centers for Medicare & Medicaid Services (CMS) became effective in January 2012.7,8 Many health care organizations opposed the rule for reasons related to complexity, prescriptiveness, onerous detail around governance and marketing, and shared savings arrangements, among others. The final rule addressed many of these concerns and enabled the creation of the first wave of ACOs.8 At present, 153 ACOs have been approved by CMS.9 Other ACOs funded by commercial payers are also being formed in many locations.
For ACOs to be effective, I believe that the cornerstone of management has to be PCMHs.
CCJM: You mentioned that institutions will take on risk. What kind of risk are you referring to?
Dr. Longworth: Added value must be rewarded with sustainable payment models. There are two payment models in the final ACO rule from CMS. Both models require 3-year commitments and both require involvement of primary care physicians. One model for organizations that want to stick a toe in the water has no downside risk and modest potential for gain if they hit certain quality and cost targets. For those organizations that are further along and want to assume risk, the second option is a shared savings/risk payment model, which creates greater incentives for efficiency and quality. In the shared savings/risk model, the ACO can retain a portion of savings if it meets performance and expenditure benchmarks based on its performance during the previous 3 years. It is also at risk for loss if expenditures are greater than a certain amount compared with benchmark expenditures. Ultimately, the final destination for ACOs will be a risk of loss if they don’t perform.
CCJM: How can these two structures—PCMHs and ACOs—optimize the use of home health?
Dr. Longworth: Home health, which is part of the postacute care continuum, will be vitally important for managing individuals and populations of patients as we move toward PCMHs and ACOs. Coordination of care will require communication between home health services and the primary care physicians who are integral to PCMHs. There will have to be an emphasis on transitions of care, from the hospital to home, from skilled nursing facilities to home, and so forth.
Accountable care organizations are responsible for a population of patients, and ACOs receive a fixed amount of money per year to cover an individual life in that population. Thus, managing quality and controlling cost is the name of the game no matter where the patient is in the health care continuum— the office, the emergency room, the hospital, a skilled nursing facility, or a home health setting. For some chronic diseases, managing patients in the home health setting may be vitally important to prevent unnecessary trips to the emergency room and hospital readmissions, thereby reducing expenditures while providing quality care.
CCJM: Do you expect an increase in the number of PCMHs and ACOs to increase the demand for home health services?
Dr. Longworth: Given the necessity of optimizing quality at lower cost, I anticipate a push to deliver as much care as we can in the least expensive “right” setting, which might be the home in some situations. Certainly, we don’t want to send patients home prematurely only to have them return to emergency departments or hospitals, but I think the demand for home health will increase as we try to decrease the number of days in skilled nursing facilities, which are expensive, and to move care from skilled nursing facilities to the home setting.
CCJM: Is there evidence that integrated delivery models such as PCMHs deliver value?
Dr. Longworth: The Patient-Centered Primary Care Collaborative demonstrated quality improvements in selected outcomes domains while also realizing savings through reductions in admissions, emergency department visits, skilled nursing facility days, and pharmacy costs.10
CCJM: What challenges do PCMHs and ACOs present to home health agencies and the way they provide services, and how will these challenges affect patients and clinicians?
Dr. Longworth: One challenge will be communication between home health services and primary care providers during transitions of care. A second will be managing costs for home health, which entails leveraging new technologies such as in-home devices and telemedicine to provide optimal and ideal monitoring of patients at the lowest potential cost. Home health, like other players along the care continuum, will face increasing scrutiny regarding quality metrics. Home health agencies will likely need to distinguish themselves from one another on the basis of performance measures such as emergency department utilization, unnecessary hospital readmissions, medication errors, and quality of service to patients as well as to primary care providers.
CCJM: How does personalized health care fit into the PCMH model?
Dr. Longworth: Personalized health care, which includes the use of genetic testing in certain situations, is an emerging field that is still in its infancy. Like PCMHs, personalized health care is proactive rather than reactive. Application of personalized health care can help deliver value with better prediction of disease and appropriate use of targeted therapies to improve outcomes for certain individuals. Such individualized treatment not only enables higher quality of care but wiser use of resources. For instance, genetic markers can be used to predict drug metabolism and adverse drug events for certain medications. In the field of oncology, the expression of genetic mutations in certain tumor types can help identify patients most likely to respond to specific targeted therapies. In these ways, personalized health care is patient-centered health care. As part of its proactive nature, personalized health care, beyond genetic testing, also implies advance planning of appointments with a focus on chronic care and keeping patients in the care system.
CCJM: How does participation in a PCMH or an ACO benefit the primary care provider? Are there any disadvantages to participation?
Dr. Longworth: In the current fee-for-service world, primary care physicians and all providers are paid on a widget-by-widget basis. Some primary care physicians and other specialists fear moving to this new world in which they will ultimately be accountable for quality and cost. Not everyone has embraced the concept, but I do think it is inevitable. Primary care physicians especially will be under increasing pressure to care for populations as opposed to individual patients. They will need to redesign the care delivery model to provide team-based, proactive care focusing on the highest-risk patients to try to keep them out of the emergency department and hospital. There will also be a greater emphasis on wellness moving forward, in an attempt to prevent the development of chronic diseases such as diabetes and obesity in individual patients and populations. All of these changes represent a different paradigm for the delivery of care, compared with the present model.
The benefit of participation for a primary care physician depends on the structure of an ACO, particularly the amount of personal financial liability an individual practitioner might have. In a staff-model, fixed-salary institution, primary care physicians would probably be more immune to financial liability than they would in other markets or other compensation models in which salary can fluctuate.
CCJM: What are some of the barriers to ACO implementation that are relevant to office-based practice, and how can they be overcome?
Dr. Longworth: There are a number of barriers to ACOs and true PCMHs. The barriers revolve around redefining workflows and moving away from reactive care—a physician-centric model in which a patient comes into the office with a problem and the physician reacts—to proactive care with the goal being to recognize how the patient is doing over time to prevent unnecessary trips to the emergency department and, ultimately, hospitalization. It is a fundamentally different mindset that involves proactive outreach targeted at high-risk patients whose chronic diseases are managed through a team-based approach. An essential feature of primary care practice will be care coordinators who will manage and proactively anticipate the needs of medically complex, high-risk patients who use a disproportionately large share of services.
In addition, a greater emphasis on wellness will be necessary to prevent the development of chronic diseases such as diabetes, obesity, and hypertension in the large segment of the population that is reasonably healthy.
CCJM: What steps can a clinician take to prepare his or her practice for ACO implementation?
Dr. Longworth: Small practices will be challenged. It is difficult to imagine accountable care without an electronic health record. To understand the population, the practitioner will need to do continuous performance management, which can’t be done without access to data from a population of patients. An increasing number of physicians are aligning with organizations that have the necessary infrastructure to provide the myriad data required to measure quality, to enable continuous improvement in performance, and to enhance the patient experience. Small practices may not have the resources to complete the administrative work necessary to become part of an ACO.
There are ways to align with an ACO that do not constitute full employment; for example, the Cleveland (Ohio) Quality Alliance has aligned with community-based physicians to provide informatics support. Linking with larger organizations that have the resources to provide quality measurement and contracting support will permit smaller community-based physicians’ practices to be part of the game.
CCJM: What steps should PCMHs and ACOs take to leverage and optimize home health services among other parts of the medical neighborhood?
Dr. Longworth: Frankly, the postacute continuum is a challenge for most systems across the country because postacute care is fragmented. Our strategy at Cleveland Clinic is to identify and align with preferred providers of home health services. The criteria that I look for are commitment to quality and transparency, service that is oriented to both patients and PCMHs, and openness to innovation for leveraging health care technology to deliver care at the best value. Home health providers need to think about how to best accomplish these results to position themselves to partner with ACOs.
CCJM: How do PCMHs and ACOs apply to special patient populations and their needs? Is there a population that’s best suited for the medical home model?
Dr. Longworth: Certain populations of higher-risk patients are ideally suited to home health coupled with chronic disease management using care coordinators. Some examples are children with asthma and children with intellectual and developmental disabilities (eg, autism) who have high utilization of emergency services. Another population is patients with heart failure who are often in and out of the emergency department and hospital; there has been a concerted effort to reduce 30-day readmission rates, which are as high as 30%, for this group. (Also see “Home-based care for heart failure: Cleveland Clinic’s ‘Heart Care at Home’ transitional care program”)
CCJM: What are the specific expectations for patient involvement in the PCMH setting?
Dr. Longworth: Our challenge lies in how best to motivate patients and engage them in their own care, especially patients who have chronic diseases. We all struggle to resolve the engagement question. Coaching and patient engagement are functions of PCMHs and at every point along the care continuum. Home health providers can serve as health coaches to promote adherence to medications, healthy lifestyles, and follow-up visits with patients’ doctors—these all need to happen to better engage patients. How to engage patients and motivate them to be more involved in their health is a basic challenge.
CCJM: Along similar lines, how can home health providers work with physicians to achieve patient-centered care?
Dr. Longworth: They can communicate early when they think that things are amiss, serve as health coaches, create technologic solutions that enhance efficiency of communication, and anticipate care needs of patients in the home setting.
CCJM: How might bundling affect the financial picture of PCMHs and patient care?
Dr. Longworth: When one talks about bundling, the devil is in the definition. In bundling, one gets paid for an episode of service. So, for example, a total knee replacement might be compensated by a 30-day bundle that covers only the surgery and the immediate postoperative period. Or it might be a 90-day bundle that includes hospitalization and perhaps some days in skilled nursing facility, but ideally transitioning from hospital to home. In the latter example, the bundle, or the total payment, will be split between the hospital and the home care services. If home health is included in a bundle, there will be tremendous pressure on the home health service to prevent readmission and emergency room visits and to eliminate waste of care. Home health’s vulnerability will depend upon how a bundle is defined for specific service.
CCJM: Who defines the terms of the bundle?
Dr. Longworth: Whoever is applying for the bundle—usually, a health care system, hospital, or ACO. It may be that home health services will subcontract for a fat fee in order to immunize themselves against risk, and shift all of the risk to the contracting organization. If I were a home health provider, I might try to minimize my own risk, but still offer my services at a price that is financially viable.
- Sia C, Tonniges TF, Osterhus E, Taba S. History of the medical home concept. Pediatrics 2004; 113 suppl 5:1473–1478.
- National Committee for Quality Assurance. Standards and Guidelines for Physician Practice Connections ®—Patient-Centered Medical Home (PPC-PCMH™). http://www.ncqa.org/Portals/0/Programs/Recognition/PCMH_Overview_Apr01.pdfPublished 2008. Accessed September 17, 2012.
- White paper. NCQA ’s Patient-centered medical home (PCMH) 2011. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Newsroom/PCMH%202011%20White%20Paper_4.6.12.pdf. Published 2011. Accessed September 17, 2012.
- 2011 annual report. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Publications/Resource%20Library/Annual%20Report/2011_Annual_Report.pdf. Published 2011. Accessed September 17, 2012.
- National Committee for Quality Assurance patient-centered medical home 2011. National Committee for Quality Assurance Web site http://www.ncqa.org/Portals/0/PCMH2011%20withCAHPSInsert.pdf. Published 2011. Accessed September 17, 2012.
- Fisher ES, Staiger DO, Bynum JP, Gottlieb DJ. Creating accountable care organizations: the extended hospital medical staff [published online ahead of print December 5, 2006]. Health Aff (Millwood) 2007; 26:w44–w57. 10.1377/hlthaff.26.1.w44
- Accountable care organizations: improving care coordination for people with Medicare. A U.S. Department of Health & Human Services Web site. www.HealthCare.gov/news/factsheets/accountablecare03312011a.html. Published March 31, 2011. Updated March 12, 2012. Accessed November 20, 2012.
- Centers for Medicare & Medicaid Services (CMS), HHS. Medicare program; Medicare shared savings program: accountable care organizations. Final rule. Fed Regist 2011; 76 212:67802–67990.
- Fact Sheets. CMS names 88 new Medicare shared savings accountable care organizations. A Centers for Medicare & Medicaid Services (CMS) Web site. http://www.cms.gov/apps/media/press/factsheet.asp?Counter=4405&intNumPerPage=10&checkDate=1&checkKey=&srchType=1&numDays=90&srchOpt=0&srchData=&keywordType=All&chkNewsType=6&intPage=&showAll=1&pYear=1&year=2012&desc=&cboOrder=date. Published July 9, 2012. Accessed November 20, 2012.
- Grumbach K, Grundy P. Outcomes of implementing patient centered medical home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative Web site. http://www.pcpcc.net/fles/evidence_outcomes_in_pcmh.pdf. Published November 16, 2010. Accessed November 20, 2012.
The passage of the Patient Protection and Affordable Care Act will profoundly affect the way physicians—particularly those engaged in primary care—practice medicine. Clinicians and their colleagues will be obliged to meet government-mandated performance quality measures while achieving cost efficiencies. Two concepts are central to the implementation of reform in the US health care system: accountable care organizations (ACOs) and the patient-centered medical home (PCMH). To get some perspective on what these changes mean for the practicing clinician, Cleveland Clinic Journal of Medicine (CCJM) interviewed David Longworth, MD, who chairs the Cleveland Clinic Medicine Institute and directs strategy and implementation of Cleveland Clinic ACO-related activities.
CCJM: Please explain briefly the concept of PCMH.
Dr. Longworth: PCMH is not a new concept; first advanced by the American Academy of Pediatrics in 1967,1 it represents a model of care in which an individual patient has a primary relationship with one provider who manages and coordinates the different aspects of the patient’s health care. The provider collaborates with a team of health care professionals. The concept caught on about a decade ago when a consortium of family medicine organizations and ultimately industry, including IBM, endorsed the concept. IBM and others created the Primary Care Consortium and began to drive the concept of PCMH.
Increasingly, care delivered through PCMH is team-based. The team coordinates the patient’s care and, when appropriate, enlists specialists or subspecialists to provide necessary components of care, all while maintaining responsibility for care coordination across the continuum of care. The medical home model provides an opportunity for enhanced access and care coordination utilizing care outside of the office walls, such as through retail clinics, eVisits, online diagnostic services, phone and electronic communication, and house call services.
Patient-centered medical homes are springing up across the country. In 2008, the National Committee for Quality Assurance (NCQA) developed criteria for recognition of PCMHs.2 It scored the sophistication of medical homes at three levels, level 1 being the lowest and level 3 the highest. Between 2008 and the end of 2010, NCQA had recognized more than 1,500 PCMHs. According to the latest figures, more than 3,000 practices have now earned PCMH recognition from the NCQA.3
At Cleveland Clinic, pilot projects at three family health centers that cover 60,000 persons have recently been rolled out with the goal of determining the model of team care that yields the highest value, with value defined by the equation of quality over cost. Ideally, higher quality is delivered at lower cost to increase value.
CCJM: What are the goals of ACOs?
Dr. Longworth: The term “accountable care,” first used in 2006 by Elliot Fisher, Dartmouth Institute of Health Policy and Clinical Practice,6 expresses the idea that health care organizations be accountable for the care they deliver, with the three-part aim of better health for populations, better care for individuals, and reduced cost inefficiencies without compromised care.
With accountable care, institutions take on risk with the expectation that they will improve quality but reduce costs, and if they reduce costs and achieve certain quality targets for populations of patients, they will share in the savings accrued. The Affordable Care Act laid the groundwork for creation of ACOs. The regulation for ACOs released by the Centers for Medicare & Medicaid Services (CMS) became effective in January 2012.7,8 Many health care organizations opposed the rule for reasons related to complexity, prescriptiveness, onerous detail around governance and marketing, and shared savings arrangements, among others. The final rule addressed many of these concerns and enabled the creation of the first wave of ACOs.8 At present, 153 ACOs have been approved by CMS.9 Other ACOs funded by commercial payers are also being formed in many locations.
For ACOs to be effective, I believe that the cornerstone of management has to be PCMHs.
CCJM: You mentioned that institutions will take on risk. What kind of risk are you referring to?
Dr. Longworth: Added value must be rewarded with sustainable payment models. There are two payment models in the final ACO rule from CMS. Both models require 3-year commitments and both require involvement of primary care physicians. One model for organizations that want to stick a toe in the water has no downside risk and modest potential for gain if they hit certain quality and cost targets. For those organizations that are further along and want to assume risk, the second option is a shared savings/risk payment model, which creates greater incentives for efficiency and quality. In the shared savings/risk model, the ACO can retain a portion of savings if it meets performance and expenditure benchmarks based on its performance during the previous 3 years. It is also at risk for loss if expenditures are greater than a certain amount compared with benchmark expenditures. Ultimately, the final destination for ACOs will be a risk of loss if they don’t perform.
CCJM: How can these two structures—PCMHs and ACOs—optimize the use of home health?
Dr. Longworth: Home health, which is part of the postacute care continuum, will be vitally important for managing individuals and populations of patients as we move toward PCMHs and ACOs. Coordination of care will require communication between home health services and the primary care physicians who are integral to PCMHs. There will have to be an emphasis on transitions of care, from the hospital to home, from skilled nursing facilities to home, and so forth.
Accountable care organizations are responsible for a population of patients, and ACOs receive a fixed amount of money per year to cover an individual life in that population. Thus, managing quality and controlling cost is the name of the game no matter where the patient is in the health care continuum— the office, the emergency room, the hospital, a skilled nursing facility, or a home health setting. For some chronic diseases, managing patients in the home health setting may be vitally important to prevent unnecessary trips to the emergency room and hospital readmissions, thereby reducing expenditures while providing quality care.
CCJM: Do you expect an increase in the number of PCMHs and ACOs to increase the demand for home health services?
Dr. Longworth: Given the necessity of optimizing quality at lower cost, I anticipate a push to deliver as much care as we can in the least expensive “right” setting, which might be the home in some situations. Certainly, we don’t want to send patients home prematurely only to have them return to emergency departments or hospitals, but I think the demand for home health will increase as we try to decrease the number of days in skilled nursing facilities, which are expensive, and to move care from skilled nursing facilities to the home setting.
CCJM: Is there evidence that integrated delivery models such as PCMHs deliver value?
Dr. Longworth: The Patient-Centered Primary Care Collaborative demonstrated quality improvements in selected outcomes domains while also realizing savings through reductions in admissions, emergency department visits, skilled nursing facility days, and pharmacy costs.10
CCJM: What challenges do PCMHs and ACOs present to home health agencies and the way they provide services, and how will these challenges affect patients and clinicians?
Dr. Longworth: One challenge will be communication between home health services and primary care providers during transitions of care. A second will be managing costs for home health, which entails leveraging new technologies such as in-home devices and telemedicine to provide optimal and ideal monitoring of patients at the lowest potential cost. Home health, like other players along the care continuum, will face increasing scrutiny regarding quality metrics. Home health agencies will likely need to distinguish themselves from one another on the basis of performance measures such as emergency department utilization, unnecessary hospital readmissions, medication errors, and quality of service to patients as well as to primary care providers.
CCJM: How does personalized health care fit into the PCMH model?
Dr. Longworth: Personalized health care, which includes the use of genetic testing in certain situations, is an emerging field that is still in its infancy. Like PCMHs, personalized health care is proactive rather than reactive. Application of personalized health care can help deliver value with better prediction of disease and appropriate use of targeted therapies to improve outcomes for certain individuals. Such individualized treatment not only enables higher quality of care but wiser use of resources. For instance, genetic markers can be used to predict drug metabolism and adverse drug events for certain medications. In the field of oncology, the expression of genetic mutations in certain tumor types can help identify patients most likely to respond to specific targeted therapies. In these ways, personalized health care is patient-centered health care. As part of its proactive nature, personalized health care, beyond genetic testing, also implies advance planning of appointments with a focus on chronic care and keeping patients in the care system.
CCJM: How does participation in a PCMH or an ACO benefit the primary care provider? Are there any disadvantages to participation?
Dr. Longworth: In the current fee-for-service world, primary care physicians and all providers are paid on a widget-by-widget basis. Some primary care physicians and other specialists fear moving to this new world in which they will ultimately be accountable for quality and cost. Not everyone has embraced the concept, but I do think it is inevitable. Primary care physicians especially will be under increasing pressure to care for populations as opposed to individual patients. They will need to redesign the care delivery model to provide team-based, proactive care focusing on the highest-risk patients to try to keep them out of the emergency department and hospital. There will also be a greater emphasis on wellness moving forward, in an attempt to prevent the development of chronic diseases such as diabetes and obesity in individual patients and populations. All of these changes represent a different paradigm for the delivery of care, compared with the present model.
The benefit of participation for a primary care physician depends on the structure of an ACO, particularly the amount of personal financial liability an individual practitioner might have. In a staff-model, fixed-salary institution, primary care physicians would probably be more immune to financial liability than they would in other markets or other compensation models in which salary can fluctuate.
CCJM: What are some of the barriers to ACO implementation that are relevant to office-based practice, and how can they be overcome?
Dr. Longworth: There are a number of barriers to ACOs and true PCMHs. The barriers revolve around redefining workflows and moving away from reactive care—a physician-centric model in which a patient comes into the office with a problem and the physician reacts—to proactive care with the goal being to recognize how the patient is doing over time to prevent unnecessary trips to the emergency department and, ultimately, hospitalization. It is a fundamentally different mindset that involves proactive outreach targeted at high-risk patients whose chronic diseases are managed through a team-based approach. An essential feature of primary care practice will be care coordinators who will manage and proactively anticipate the needs of medically complex, high-risk patients who use a disproportionately large share of services.
In addition, a greater emphasis on wellness will be necessary to prevent the development of chronic diseases such as diabetes, obesity, and hypertension in the large segment of the population that is reasonably healthy.
CCJM: What steps can a clinician take to prepare his or her practice for ACO implementation?
Dr. Longworth: Small practices will be challenged. It is difficult to imagine accountable care without an electronic health record. To understand the population, the practitioner will need to do continuous performance management, which can’t be done without access to data from a population of patients. An increasing number of physicians are aligning with organizations that have the necessary infrastructure to provide the myriad data required to measure quality, to enable continuous improvement in performance, and to enhance the patient experience. Small practices may not have the resources to complete the administrative work necessary to become part of an ACO.
There are ways to align with an ACO that do not constitute full employment; for example, the Cleveland (Ohio) Quality Alliance has aligned with community-based physicians to provide informatics support. Linking with larger organizations that have the resources to provide quality measurement and contracting support will permit smaller community-based physicians’ practices to be part of the game.
CCJM: What steps should PCMHs and ACOs take to leverage and optimize home health services among other parts of the medical neighborhood?
Dr. Longworth: Frankly, the postacute continuum is a challenge for most systems across the country because postacute care is fragmented. Our strategy at Cleveland Clinic is to identify and align with preferred providers of home health services. The criteria that I look for are commitment to quality and transparency, service that is oriented to both patients and PCMHs, and openness to innovation for leveraging health care technology to deliver care at the best value. Home health providers need to think about how to best accomplish these results to position themselves to partner with ACOs.
CCJM: How do PCMHs and ACOs apply to special patient populations and their needs? Is there a population that’s best suited for the medical home model?
Dr. Longworth: Certain populations of higher-risk patients are ideally suited to home health coupled with chronic disease management using care coordinators. Some examples are children with asthma and children with intellectual and developmental disabilities (eg, autism) who have high utilization of emergency services. Another population is patients with heart failure who are often in and out of the emergency department and hospital; there has been a concerted effort to reduce 30-day readmission rates, which are as high as 30%, for this group. (Also see “Home-based care for heart failure: Cleveland Clinic’s ‘Heart Care at Home’ transitional care program”)
CCJM: What are the specific expectations for patient involvement in the PCMH setting?
Dr. Longworth: Our challenge lies in how best to motivate patients and engage them in their own care, especially patients who have chronic diseases. We all struggle to resolve the engagement question. Coaching and patient engagement are functions of PCMHs and at every point along the care continuum. Home health providers can serve as health coaches to promote adherence to medications, healthy lifestyles, and follow-up visits with patients’ doctors—these all need to happen to better engage patients. How to engage patients and motivate them to be more involved in their health is a basic challenge.
CCJM: Along similar lines, how can home health providers work with physicians to achieve patient-centered care?
Dr. Longworth: They can communicate early when they think that things are amiss, serve as health coaches, create technologic solutions that enhance efficiency of communication, and anticipate care needs of patients in the home setting.
CCJM: How might bundling affect the financial picture of PCMHs and patient care?
Dr. Longworth: When one talks about bundling, the devil is in the definition. In bundling, one gets paid for an episode of service. So, for example, a total knee replacement might be compensated by a 30-day bundle that covers only the surgery and the immediate postoperative period. Or it might be a 90-day bundle that includes hospitalization and perhaps some days in skilled nursing facility, but ideally transitioning from hospital to home. In the latter example, the bundle, or the total payment, will be split between the hospital and the home care services. If home health is included in a bundle, there will be tremendous pressure on the home health service to prevent readmission and emergency room visits and to eliminate waste of care. Home health’s vulnerability will depend upon how a bundle is defined for specific service.
CCJM: Who defines the terms of the bundle?
Dr. Longworth: Whoever is applying for the bundle—usually, a health care system, hospital, or ACO. It may be that home health services will subcontract for a fat fee in order to immunize themselves against risk, and shift all of the risk to the contracting organization. If I were a home health provider, I might try to minimize my own risk, but still offer my services at a price that is financially viable.
The passage of the Patient Protection and Affordable Care Act will profoundly affect the way physicians—particularly those engaged in primary care—practice medicine. Clinicians and their colleagues will be obliged to meet government-mandated performance quality measures while achieving cost efficiencies. Two concepts are central to the implementation of reform in the US health care system: accountable care organizations (ACOs) and the patient-centered medical home (PCMH). To get some perspective on what these changes mean for the practicing clinician, Cleveland Clinic Journal of Medicine (CCJM) interviewed David Longworth, MD, who chairs the Cleveland Clinic Medicine Institute and directs strategy and implementation of Cleveland Clinic ACO-related activities.
CCJM: Please explain briefly the concept of PCMH.
Dr. Longworth: PCMH is not a new concept; first advanced by the American Academy of Pediatrics in 1967,1 it represents a model of care in which an individual patient has a primary relationship with one provider who manages and coordinates the different aspects of the patient’s health care. The provider collaborates with a team of health care professionals. The concept caught on about a decade ago when a consortium of family medicine organizations and ultimately industry, including IBM, endorsed the concept. IBM and others created the Primary Care Consortium and began to drive the concept of PCMH.
Increasingly, care delivered through PCMH is team-based. The team coordinates the patient’s care and, when appropriate, enlists specialists or subspecialists to provide necessary components of care, all while maintaining responsibility for care coordination across the continuum of care. The medical home model provides an opportunity for enhanced access and care coordination utilizing care outside of the office walls, such as through retail clinics, eVisits, online diagnostic services, phone and electronic communication, and house call services.
Patient-centered medical homes are springing up across the country. In 2008, the National Committee for Quality Assurance (NCQA) developed criteria for recognition of PCMHs.2 It scored the sophistication of medical homes at three levels, level 1 being the lowest and level 3 the highest. Between 2008 and the end of 2010, NCQA had recognized more than 1,500 PCMHs. According to the latest figures, more than 3,000 practices have now earned PCMH recognition from the NCQA.3
At Cleveland Clinic, pilot projects at three family health centers that cover 60,000 persons have recently been rolled out with the goal of determining the model of team care that yields the highest value, with value defined by the equation of quality over cost. Ideally, higher quality is delivered at lower cost to increase value.
CCJM: What are the goals of ACOs?
Dr. Longworth: The term “accountable care,” first used in 2006 by Elliot Fisher, Dartmouth Institute of Health Policy and Clinical Practice,6 expresses the idea that health care organizations be accountable for the care they deliver, with the three-part aim of better health for populations, better care for individuals, and reduced cost inefficiencies without compromised care.
With accountable care, institutions take on risk with the expectation that they will improve quality but reduce costs, and if they reduce costs and achieve certain quality targets for populations of patients, they will share in the savings accrued. The Affordable Care Act laid the groundwork for creation of ACOs. The regulation for ACOs released by the Centers for Medicare & Medicaid Services (CMS) became effective in January 2012.7,8 Many health care organizations opposed the rule for reasons related to complexity, prescriptiveness, onerous detail around governance and marketing, and shared savings arrangements, among others. The final rule addressed many of these concerns and enabled the creation of the first wave of ACOs.8 At present, 153 ACOs have been approved by CMS.9 Other ACOs funded by commercial payers are also being formed in many locations.
For ACOs to be effective, I believe that the cornerstone of management has to be PCMHs.
CCJM: You mentioned that institutions will take on risk. What kind of risk are you referring to?
Dr. Longworth: Added value must be rewarded with sustainable payment models. There are two payment models in the final ACO rule from CMS. Both models require 3-year commitments and both require involvement of primary care physicians. One model for organizations that want to stick a toe in the water has no downside risk and modest potential for gain if they hit certain quality and cost targets. For those organizations that are further along and want to assume risk, the second option is a shared savings/risk payment model, which creates greater incentives for efficiency and quality. In the shared savings/risk model, the ACO can retain a portion of savings if it meets performance and expenditure benchmarks based on its performance during the previous 3 years. It is also at risk for loss if expenditures are greater than a certain amount compared with benchmark expenditures. Ultimately, the final destination for ACOs will be a risk of loss if they don’t perform.
CCJM: How can these two structures—PCMHs and ACOs—optimize the use of home health?
Dr. Longworth: Home health, which is part of the postacute care continuum, will be vitally important for managing individuals and populations of patients as we move toward PCMHs and ACOs. Coordination of care will require communication between home health services and the primary care physicians who are integral to PCMHs. There will have to be an emphasis on transitions of care, from the hospital to home, from skilled nursing facilities to home, and so forth.
Accountable care organizations are responsible for a population of patients, and ACOs receive a fixed amount of money per year to cover an individual life in that population. Thus, managing quality and controlling cost is the name of the game no matter where the patient is in the health care continuum— the office, the emergency room, the hospital, a skilled nursing facility, or a home health setting. For some chronic diseases, managing patients in the home health setting may be vitally important to prevent unnecessary trips to the emergency room and hospital readmissions, thereby reducing expenditures while providing quality care.
CCJM: Do you expect an increase in the number of PCMHs and ACOs to increase the demand for home health services?
Dr. Longworth: Given the necessity of optimizing quality at lower cost, I anticipate a push to deliver as much care as we can in the least expensive “right” setting, which might be the home in some situations. Certainly, we don’t want to send patients home prematurely only to have them return to emergency departments or hospitals, but I think the demand for home health will increase as we try to decrease the number of days in skilled nursing facilities, which are expensive, and to move care from skilled nursing facilities to the home setting.
CCJM: Is there evidence that integrated delivery models such as PCMHs deliver value?
Dr. Longworth: The Patient-Centered Primary Care Collaborative demonstrated quality improvements in selected outcomes domains while also realizing savings through reductions in admissions, emergency department visits, skilled nursing facility days, and pharmacy costs.10
CCJM: What challenges do PCMHs and ACOs present to home health agencies and the way they provide services, and how will these challenges affect patients and clinicians?
Dr. Longworth: One challenge will be communication between home health services and primary care providers during transitions of care. A second will be managing costs for home health, which entails leveraging new technologies such as in-home devices and telemedicine to provide optimal and ideal monitoring of patients at the lowest potential cost. Home health, like other players along the care continuum, will face increasing scrutiny regarding quality metrics. Home health agencies will likely need to distinguish themselves from one another on the basis of performance measures such as emergency department utilization, unnecessary hospital readmissions, medication errors, and quality of service to patients as well as to primary care providers.
CCJM: How does personalized health care fit into the PCMH model?
Dr. Longworth: Personalized health care, which includes the use of genetic testing in certain situations, is an emerging field that is still in its infancy. Like PCMHs, personalized health care is proactive rather than reactive. Application of personalized health care can help deliver value with better prediction of disease and appropriate use of targeted therapies to improve outcomes for certain individuals. Such individualized treatment not only enables higher quality of care but wiser use of resources. For instance, genetic markers can be used to predict drug metabolism and adverse drug events for certain medications. In the field of oncology, the expression of genetic mutations in certain tumor types can help identify patients most likely to respond to specific targeted therapies. In these ways, personalized health care is patient-centered health care. As part of its proactive nature, personalized health care, beyond genetic testing, also implies advance planning of appointments with a focus on chronic care and keeping patients in the care system.
CCJM: How does participation in a PCMH or an ACO benefit the primary care provider? Are there any disadvantages to participation?
Dr. Longworth: In the current fee-for-service world, primary care physicians and all providers are paid on a widget-by-widget basis. Some primary care physicians and other specialists fear moving to this new world in which they will ultimately be accountable for quality and cost. Not everyone has embraced the concept, but I do think it is inevitable. Primary care physicians especially will be under increasing pressure to care for populations as opposed to individual patients. They will need to redesign the care delivery model to provide team-based, proactive care focusing on the highest-risk patients to try to keep them out of the emergency department and hospital. There will also be a greater emphasis on wellness moving forward, in an attempt to prevent the development of chronic diseases such as diabetes and obesity in individual patients and populations. All of these changes represent a different paradigm for the delivery of care, compared with the present model.
The benefit of participation for a primary care physician depends on the structure of an ACO, particularly the amount of personal financial liability an individual practitioner might have. In a staff-model, fixed-salary institution, primary care physicians would probably be more immune to financial liability than they would in other markets or other compensation models in which salary can fluctuate.
CCJM: What are some of the barriers to ACO implementation that are relevant to office-based practice, and how can they be overcome?
Dr. Longworth: There are a number of barriers to ACOs and true PCMHs. The barriers revolve around redefining workflows and moving away from reactive care—a physician-centric model in which a patient comes into the office with a problem and the physician reacts—to proactive care with the goal being to recognize how the patient is doing over time to prevent unnecessary trips to the emergency department and, ultimately, hospitalization. It is a fundamentally different mindset that involves proactive outreach targeted at high-risk patients whose chronic diseases are managed through a team-based approach. An essential feature of primary care practice will be care coordinators who will manage and proactively anticipate the needs of medically complex, high-risk patients who use a disproportionately large share of services.
In addition, a greater emphasis on wellness will be necessary to prevent the development of chronic diseases such as diabetes, obesity, and hypertension in the large segment of the population that is reasonably healthy.
CCJM: What steps can a clinician take to prepare his or her practice for ACO implementation?
Dr. Longworth: Small practices will be challenged. It is difficult to imagine accountable care without an electronic health record. To understand the population, the practitioner will need to do continuous performance management, which can’t be done without access to data from a population of patients. An increasing number of physicians are aligning with organizations that have the necessary infrastructure to provide the myriad data required to measure quality, to enable continuous improvement in performance, and to enhance the patient experience. Small practices may not have the resources to complete the administrative work necessary to become part of an ACO.
There are ways to align with an ACO that do not constitute full employment; for example, the Cleveland (Ohio) Quality Alliance has aligned with community-based physicians to provide informatics support. Linking with larger organizations that have the resources to provide quality measurement and contracting support will permit smaller community-based physicians’ practices to be part of the game.
CCJM: What steps should PCMHs and ACOs take to leverage and optimize home health services among other parts of the medical neighborhood?
Dr. Longworth: Frankly, the postacute continuum is a challenge for most systems across the country because postacute care is fragmented. Our strategy at Cleveland Clinic is to identify and align with preferred providers of home health services. The criteria that I look for are commitment to quality and transparency, service that is oriented to both patients and PCMHs, and openness to innovation for leveraging health care technology to deliver care at the best value. Home health providers need to think about how to best accomplish these results to position themselves to partner with ACOs.
CCJM: How do PCMHs and ACOs apply to special patient populations and their needs? Is there a population that’s best suited for the medical home model?
Dr. Longworth: Certain populations of higher-risk patients are ideally suited to home health coupled with chronic disease management using care coordinators. Some examples are children with asthma and children with intellectual and developmental disabilities (eg, autism) who have high utilization of emergency services. Another population is patients with heart failure who are often in and out of the emergency department and hospital; there has been a concerted effort to reduce 30-day readmission rates, which are as high as 30%, for this group. (Also see “Home-based care for heart failure: Cleveland Clinic’s ‘Heart Care at Home’ transitional care program”)
CCJM: What are the specific expectations for patient involvement in the PCMH setting?
Dr. Longworth: Our challenge lies in how best to motivate patients and engage them in their own care, especially patients who have chronic diseases. We all struggle to resolve the engagement question. Coaching and patient engagement are functions of PCMHs and at every point along the care continuum. Home health providers can serve as health coaches to promote adherence to medications, healthy lifestyles, and follow-up visits with patients’ doctors—these all need to happen to better engage patients. How to engage patients and motivate them to be more involved in their health is a basic challenge.
CCJM: Along similar lines, how can home health providers work with physicians to achieve patient-centered care?
Dr. Longworth: They can communicate early when they think that things are amiss, serve as health coaches, create technologic solutions that enhance efficiency of communication, and anticipate care needs of patients in the home setting.
CCJM: How might bundling affect the financial picture of PCMHs and patient care?
Dr. Longworth: When one talks about bundling, the devil is in the definition. In bundling, one gets paid for an episode of service. So, for example, a total knee replacement might be compensated by a 30-day bundle that covers only the surgery and the immediate postoperative period. Or it might be a 90-day bundle that includes hospitalization and perhaps some days in skilled nursing facility, but ideally transitioning from hospital to home. In the latter example, the bundle, or the total payment, will be split between the hospital and the home care services. If home health is included in a bundle, there will be tremendous pressure on the home health service to prevent readmission and emergency room visits and to eliminate waste of care. Home health’s vulnerability will depend upon how a bundle is defined for specific service.
CCJM: Who defines the terms of the bundle?
Dr. Longworth: Whoever is applying for the bundle—usually, a health care system, hospital, or ACO. It may be that home health services will subcontract for a fat fee in order to immunize themselves against risk, and shift all of the risk to the contracting organization. If I were a home health provider, I might try to minimize my own risk, but still offer my services at a price that is financially viable.
- Sia C, Tonniges TF, Osterhus E, Taba S. History of the medical home concept. Pediatrics 2004; 113 suppl 5:1473–1478.
- National Committee for Quality Assurance. Standards and Guidelines for Physician Practice Connections ®—Patient-Centered Medical Home (PPC-PCMH™). http://www.ncqa.org/Portals/0/Programs/Recognition/PCMH_Overview_Apr01.pdfPublished 2008. Accessed September 17, 2012.
- White paper. NCQA ’s Patient-centered medical home (PCMH) 2011. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Newsroom/PCMH%202011%20White%20Paper_4.6.12.pdf. Published 2011. Accessed September 17, 2012.
- 2011 annual report. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Publications/Resource%20Library/Annual%20Report/2011_Annual_Report.pdf. Published 2011. Accessed September 17, 2012.
- National Committee for Quality Assurance patient-centered medical home 2011. National Committee for Quality Assurance Web site http://www.ncqa.org/Portals/0/PCMH2011%20withCAHPSInsert.pdf. Published 2011. Accessed September 17, 2012.
- Fisher ES, Staiger DO, Bynum JP, Gottlieb DJ. Creating accountable care organizations: the extended hospital medical staff [published online ahead of print December 5, 2006]. Health Aff (Millwood) 2007; 26:w44–w57. 10.1377/hlthaff.26.1.w44
- Accountable care organizations: improving care coordination for people with Medicare. A U.S. Department of Health & Human Services Web site. www.HealthCare.gov/news/factsheets/accountablecare03312011a.html. Published March 31, 2011. Updated March 12, 2012. Accessed November 20, 2012.
- Centers for Medicare & Medicaid Services (CMS), HHS. Medicare program; Medicare shared savings program: accountable care organizations. Final rule. Fed Regist 2011; 76 212:67802–67990.
- Fact Sheets. CMS names 88 new Medicare shared savings accountable care organizations. A Centers for Medicare & Medicaid Services (CMS) Web site. http://www.cms.gov/apps/media/press/factsheet.asp?Counter=4405&intNumPerPage=10&checkDate=1&checkKey=&srchType=1&numDays=90&srchOpt=0&srchData=&keywordType=All&chkNewsType=6&intPage=&showAll=1&pYear=1&year=2012&desc=&cboOrder=date. Published July 9, 2012. Accessed November 20, 2012.
- Grumbach K, Grundy P. Outcomes of implementing patient centered medical home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative Web site. http://www.pcpcc.net/fles/evidence_outcomes_in_pcmh.pdf. Published November 16, 2010. Accessed November 20, 2012.
- Sia C, Tonniges TF, Osterhus E, Taba S. History of the medical home concept. Pediatrics 2004; 113 suppl 5:1473–1478.
- National Committee for Quality Assurance. Standards and Guidelines for Physician Practice Connections ®—Patient-Centered Medical Home (PPC-PCMH™). http://www.ncqa.org/Portals/0/Programs/Recognition/PCMH_Overview_Apr01.pdfPublished 2008. Accessed September 17, 2012.
- White paper. NCQA ’s Patient-centered medical home (PCMH) 2011. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Newsroom/PCMH%202011%20White%20Paper_4.6.12.pdf. Published 2011. Accessed September 17, 2012.
- 2011 annual report. National Committee for Quality Assurance Web site. http://www.ncqa.org/Portals/0/Publications/Resource%20Library/Annual%20Report/2011_Annual_Report.pdf. Published 2011. Accessed September 17, 2012.
- National Committee for Quality Assurance patient-centered medical home 2011. National Committee for Quality Assurance Web site http://www.ncqa.org/Portals/0/PCMH2011%20withCAHPSInsert.pdf. Published 2011. Accessed September 17, 2012.
- Fisher ES, Staiger DO, Bynum JP, Gottlieb DJ. Creating accountable care organizations: the extended hospital medical staff [published online ahead of print December 5, 2006]. Health Aff (Millwood) 2007; 26:w44–w57. 10.1377/hlthaff.26.1.w44
- Accountable care organizations: improving care coordination for people with Medicare. A U.S. Department of Health & Human Services Web site. www.HealthCare.gov/news/factsheets/accountablecare03312011a.html. Published March 31, 2011. Updated March 12, 2012. Accessed November 20, 2012.
- Centers for Medicare & Medicaid Services (CMS), HHS. Medicare program; Medicare shared savings program: accountable care organizations. Final rule. Fed Regist 2011; 76 212:67802–67990.
- Fact Sheets. CMS names 88 new Medicare shared savings accountable care organizations. A Centers for Medicare & Medicaid Services (CMS) Web site. http://www.cms.gov/apps/media/press/factsheet.asp?Counter=4405&intNumPerPage=10&checkDate=1&checkKey=&srchType=1&numDays=90&srchOpt=0&srchData=&keywordType=All&chkNewsType=6&intPage=&showAll=1&pYear=1&year=2012&desc=&cboOrder=date. Published July 9, 2012. Accessed November 20, 2012.
- Grumbach K, Grundy P. Outcomes of implementing patient centered medical home interventions: a review of the evidence from prospective evaluation studies in the United States. Patient-Centered Primary Care Collaborative Web site. http://www.pcpcc.net/fles/evidence_outcomes_in_pcmh.pdf. Published November 16, 2010. Accessed November 20, 2012.
AGA releases new medical position statement on constipation
Assessment of colonic transit in a patient presenting with constipation is recommended only after excluding a defecatory disorder and after treatment with laxatives and first-line pharmacologic agents fails, or after pelvic floor training in those with a defecatory disorder fails, according to a new medical position statement from the American Gastroenterological Association.
This recommendation is in contrast to the previous AGA medical position statement on constipation, which called for earlier assessment for colonic transit.
The change is one of only three substantive changes to the statement, which is published in the January issue of Gastroenterology; the others are the use of GRADE (Grading of Recommendations Assessment, Development, and Evaluation), which rates for each recommendation, its strength and quality of evidence, and the inclusion of newer agents; and deletion of certain older agents in treatment recommendations.
The colonic transit assessment recommendation is based in part on concerns about potential long-term side effects associated with newer agents that might be prescribed in patients with slow colonic transit.
"At present, the medical approaches used for managing normal and slow-transit constipation are similar. However, the major pharmacological trials in chronic constipation did not assess if the response to therapy is influenced by colonic transit. While newer agents may also be considered without assessing colonic transit, the long-term side effects, if any, of these agents are unknown and exposure to such potential risks might be more appropriate in patients with the more severe forms of constipation associated with slow transit," according to the statement.
Also, up to 50% of all patients with defecatory disorders have slow colonic transit as well, thus slow colonic transit does not exclude a defecatory disorder – and it also does not alter the management of defecatory disorders.
As for the approach to assessing for slow transit once a defecatory disorder is excluded, the statement says, "consideration should be given to assessing colonic transit by radiopaque markers, scintigraphy, or a wireless motility capsule in patients with persistent symptoms on laxatives."
Identifying slow colonic transit can reassure patients about the pathophysiology of their symptoms and also can serve as an objective marker for documenting response to treatment and provide physicians with the appropriate rationale for prescribing newer, often more expensive treatments.
Recommendations in the AGA statement that address the initial clinical assessment of constipation include the following:
• When feasible, medications that can cause constipation should be discontinued before further testing is initiated. This is a "strong" recommendation based on low-quality evidence.
• A careful digital rectal examination, including assessment of pelvic floor motion during simulated evacuation, is preferable to a cursory examination without these maneuvers and should be performed prior to referral for anorectal manometry. A normal exam, however, does not exclude defecatory disorders. This is a "strong" recommendation based on moderate-quality evidence.
The recommendations also address testing to assess medical causes of constipation. In addition to colonic transit testing, after ruling out a defecatory disorder, other recommended tests to assess for medical causes of constipation include a complete red blood count. Metabolic tests such as glucose, calcium, and sensitive thyroid-stimulating hormone are necessary only when other clinical features warrant these tests, and a colonoscopy and an imaging procedure for colonic lesions is only necessary in the presence of "alarm features," including blood in the stool, anemia, and weight loss, for medically refractory constipation or when age-appropriate colon cancer screening has not been performed. Anorectal manometry and a rectal balloon expulsion are indicated in those who fail to respond to laxatives but defecography only when anorectal manometry and a rectal balloon expulsion are inconclusive for defecatory disorders. All of these are "strong" recommendations based on low- or moderate-quality evidence.
Initial medical management, according to the statement, should include:
• A therapeutic trial of fiber supplementation and/or osmotic or stimulant laxatives after discontinuing medications that can cause constipation and after performing blood and other tests as guided by clinical features, but before anorectal testing.
• Use of long-term laxatives for normal and slow-transit constipation.
• Anorectal testing in patients who do not respond to these measures.
• Pelvic floor retraining by biofeedback therapy rather than laxatives in those with defecatory disorders.
These are all "strong" recommendations based on moderate- or high-quality evidence.
As for treatments to consider in patients who fail to respond to initial approaches, the AGA says that newer agents, such as lubiprostone and linaclotide, should be considered in those with normal or slow transit constipation who fail to respond to simple laxatives. Based on the GRADE ratings, this is a "weak" recommendation (implying that benefits, risks, and the burden of intervention are balanced among several legitimate management options or that appreciable uncertainty exists, and is based on moderate-quality evidence).
Also, when symptoms persist despite an adequate trial of biofeedback therapy – which improves symptoms in more than 70% of patients with defecatory disorders – anorectal tests and colonic transit should be reevaluated. This is a "strong" recommendation based on low-quality evidence.
Subtotal colectomy, as opposed to chronic laxative therapy, should be considered in those with symptomatic slow-transit constipation without a defecatory disorder, and colonic intraluminal testing should be considered to document colonic motor dysfunction prior to colectomy. These are weak recommendations based on moderate-quality evidence.
Finally, suppositories or enemas, rather than oral laxatives alone, should be considered in those with refractory pelvic floor dysfunction. This is a weak recommendation based on low-quality evidence.
These recommendations, drafted by a medical position panel and ultimately approved by the AGA Institute Governing Board, were published in conjunction with a technical review, which provides the rationale for the recommendations included in the statement.
AGA Institute Medical Position Panel members listed the following disclosures: Dr. Anthony Lembo reported serving as a consultant to, and serving as an advisory board member for Ironwood Pharmaceuticals and Forest Laboratories; Dr. Spencer D. Dorn reported serving as a consultant to Ironwood Pharmaceuticals and Forest Laboratories, and receiving research support from these companies, as well as from Synergy Pharmaceutical and Takeda Pharmaceuticals; Dr. A. E. Bharucha reported having a financial interest in a new technology related to anal manometry and serving a consultant for Helsin Therapeutics and Asubio Pharmaceuticals.
Assessment of colonic transit in a patient presenting with constipation is recommended only after excluding a defecatory disorder and after treatment with laxatives and first-line pharmacologic agents fails, or after pelvic floor training in those with a defecatory disorder fails, according to a new medical position statement from the American Gastroenterological Association.
This recommendation is in contrast to the previous AGA medical position statement on constipation, which called for earlier assessment for colonic transit.
The change is one of only three substantive changes to the statement, which is published in the January issue of Gastroenterology; the others are the use of GRADE (Grading of Recommendations Assessment, Development, and Evaluation), which rates for each recommendation, its strength and quality of evidence, and the inclusion of newer agents; and deletion of certain older agents in treatment recommendations.
The colonic transit assessment recommendation is based in part on concerns about potential long-term side effects associated with newer agents that might be prescribed in patients with slow colonic transit.
"At present, the medical approaches used for managing normal and slow-transit constipation are similar. However, the major pharmacological trials in chronic constipation did not assess if the response to therapy is influenced by colonic transit. While newer agents may also be considered without assessing colonic transit, the long-term side effects, if any, of these agents are unknown and exposure to such potential risks might be more appropriate in patients with the more severe forms of constipation associated with slow transit," according to the statement.
Also, up to 50% of all patients with defecatory disorders have slow colonic transit as well, thus slow colonic transit does not exclude a defecatory disorder – and it also does not alter the management of defecatory disorders.
As for the approach to assessing for slow transit once a defecatory disorder is excluded, the statement says, "consideration should be given to assessing colonic transit by radiopaque markers, scintigraphy, or a wireless motility capsule in patients with persistent symptoms on laxatives."
Identifying slow colonic transit can reassure patients about the pathophysiology of their symptoms and also can serve as an objective marker for documenting response to treatment and provide physicians with the appropriate rationale for prescribing newer, often more expensive treatments.
Recommendations in the AGA statement that address the initial clinical assessment of constipation include the following:
• When feasible, medications that can cause constipation should be discontinued before further testing is initiated. This is a "strong" recommendation based on low-quality evidence.
• A careful digital rectal examination, including assessment of pelvic floor motion during simulated evacuation, is preferable to a cursory examination without these maneuvers and should be performed prior to referral for anorectal manometry. A normal exam, however, does not exclude defecatory disorders. This is a "strong" recommendation based on moderate-quality evidence.
The recommendations also address testing to assess medical causes of constipation. In addition to colonic transit testing, after ruling out a defecatory disorder, other recommended tests to assess for medical causes of constipation include a complete red blood count. Metabolic tests such as glucose, calcium, and sensitive thyroid-stimulating hormone are necessary only when other clinical features warrant these tests, and a colonoscopy and an imaging procedure for colonic lesions is only necessary in the presence of "alarm features," including blood in the stool, anemia, and weight loss, for medically refractory constipation or when age-appropriate colon cancer screening has not been performed. Anorectal manometry and a rectal balloon expulsion are indicated in those who fail to respond to laxatives but defecography only when anorectal manometry and a rectal balloon expulsion are inconclusive for defecatory disorders. All of these are "strong" recommendations based on low- or moderate-quality evidence.
Initial medical management, according to the statement, should include:
• A therapeutic trial of fiber supplementation and/or osmotic or stimulant laxatives after discontinuing medications that can cause constipation and after performing blood and other tests as guided by clinical features, but before anorectal testing.
• Use of long-term laxatives for normal and slow-transit constipation.
• Anorectal testing in patients who do not respond to these measures.
• Pelvic floor retraining by biofeedback therapy rather than laxatives in those with defecatory disorders.
These are all "strong" recommendations based on moderate- or high-quality evidence.
As for treatments to consider in patients who fail to respond to initial approaches, the AGA says that newer agents, such as lubiprostone and linaclotide, should be considered in those with normal or slow transit constipation who fail to respond to simple laxatives. Based on the GRADE ratings, this is a "weak" recommendation (implying that benefits, risks, and the burden of intervention are balanced among several legitimate management options or that appreciable uncertainty exists, and is based on moderate-quality evidence).
Also, when symptoms persist despite an adequate trial of biofeedback therapy – which improves symptoms in more than 70% of patients with defecatory disorders – anorectal tests and colonic transit should be reevaluated. This is a "strong" recommendation based on low-quality evidence.
Subtotal colectomy, as opposed to chronic laxative therapy, should be considered in those with symptomatic slow-transit constipation without a defecatory disorder, and colonic intraluminal testing should be considered to document colonic motor dysfunction prior to colectomy. These are weak recommendations based on moderate-quality evidence.
Finally, suppositories or enemas, rather than oral laxatives alone, should be considered in those with refractory pelvic floor dysfunction. This is a weak recommendation based on low-quality evidence.
These recommendations, drafted by a medical position panel and ultimately approved by the AGA Institute Governing Board, were published in conjunction with a technical review, which provides the rationale for the recommendations included in the statement.
AGA Institute Medical Position Panel members listed the following disclosures: Dr. Anthony Lembo reported serving as a consultant to, and serving as an advisory board member for Ironwood Pharmaceuticals and Forest Laboratories; Dr. Spencer D. Dorn reported serving as a consultant to Ironwood Pharmaceuticals and Forest Laboratories, and receiving research support from these companies, as well as from Synergy Pharmaceutical and Takeda Pharmaceuticals; Dr. A. E. Bharucha reported having a financial interest in a new technology related to anal manometry and serving a consultant for Helsin Therapeutics and Asubio Pharmaceuticals.
Assessment of colonic transit in a patient presenting with constipation is recommended only after excluding a defecatory disorder and after treatment with laxatives and first-line pharmacologic agents fails, or after pelvic floor training in those with a defecatory disorder fails, according to a new medical position statement from the American Gastroenterological Association.
This recommendation is in contrast to the previous AGA medical position statement on constipation, which called for earlier assessment for colonic transit.
The change is one of only three substantive changes to the statement, which is published in the January issue of Gastroenterology; the others are the use of GRADE (Grading of Recommendations Assessment, Development, and Evaluation), which rates for each recommendation, its strength and quality of evidence, and the inclusion of newer agents; and deletion of certain older agents in treatment recommendations.
The colonic transit assessment recommendation is based in part on concerns about potential long-term side effects associated with newer agents that might be prescribed in patients with slow colonic transit.
"At present, the medical approaches used for managing normal and slow-transit constipation are similar. However, the major pharmacological trials in chronic constipation did not assess if the response to therapy is influenced by colonic transit. While newer agents may also be considered without assessing colonic transit, the long-term side effects, if any, of these agents are unknown and exposure to such potential risks might be more appropriate in patients with the more severe forms of constipation associated with slow transit," according to the statement.
Also, up to 50% of all patients with defecatory disorders have slow colonic transit as well, thus slow colonic transit does not exclude a defecatory disorder – and it also does not alter the management of defecatory disorders.
As for the approach to assessing for slow transit once a defecatory disorder is excluded, the statement says, "consideration should be given to assessing colonic transit by radiopaque markers, scintigraphy, or a wireless motility capsule in patients with persistent symptoms on laxatives."
Identifying slow colonic transit can reassure patients about the pathophysiology of their symptoms and also can serve as an objective marker for documenting response to treatment and provide physicians with the appropriate rationale for prescribing newer, often more expensive treatments.
Recommendations in the AGA statement that address the initial clinical assessment of constipation include the following:
• When feasible, medications that can cause constipation should be discontinued before further testing is initiated. This is a "strong" recommendation based on low-quality evidence.
• A careful digital rectal examination, including assessment of pelvic floor motion during simulated evacuation, is preferable to a cursory examination without these maneuvers and should be performed prior to referral for anorectal manometry. A normal exam, however, does not exclude defecatory disorders. This is a "strong" recommendation based on moderate-quality evidence.
The recommendations also address testing to assess medical causes of constipation. In addition to colonic transit testing, after ruling out a defecatory disorder, other recommended tests to assess for medical causes of constipation include a complete red blood count. Metabolic tests such as glucose, calcium, and sensitive thyroid-stimulating hormone are necessary only when other clinical features warrant these tests, and a colonoscopy and an imaging procedure for colonic lesions is only necessary in the presence of "alarm features," including blood in the stool, anemia, and weight loss, for medically refractory constipation or when age-appropriate colon cancer screening has not been performed. Anorectal manometry and a rectal balloon expulsion are indicated in those who fail to respond to laxatives but defecography only when anorectal manometry and a rectal balloon expulsion are inconclusive for defecatory disorders. All of these are "strong" recommendations based on low- or moderate-quality evidence.
Initial medical management, according to the statement, should include:
• A therapeutic trial of fiber supplementation and/or osmotic or stimulant laxatives after discontinuing medications that can cause constipation and after performing blood and other tests as guided by clinical features, but before anorectal testing.
• Use of long-term laxatives for normal and slow-transit constipation.
• Anorectal testing in patients who do not respond to these measures.
• Pelvic floor retraining by biofeedback therapy rather than laxatives in those with defecatory disorders.
These are all "strong" recommendations based on moderate- or high-quality evidence.
As for treatments to consider in patients who fail to respond to initial approaches, the AGA says that newer agents, such as lubiprostone and linaclotide, should be considered in those with normal or slow transit constipation who fail to respond to simple laxatives. Based on the GRADE ratings, this is a "weak" recommendation (implying that benefits, risks, and the burden of intervention are balanced among several legitimate management options or that appreciable uncertainty exists, and is based on moderate-quality evidence).
Also, when symptoms persist despite an adequate trial of biofeedback therapy – which improves symptoms in more than 70% of patients with defecatory disorders – anorectal tests and colonic transit should be reevaluated. This is a "strong" recommendation based on low-quality evidence.
Subtotal colectomy, as opposed to chronic laxative therapy, should be considered in those with symptomatic slow-transit constipation without a defecatory disorder, and colonic intraluminal testing should be considered to document colonic motor dysfunction prior to colectomy. These are weak recommendations based on moderate-quality evidence.
Finally, suppositories or enemas, rather than oral laxatives alone, should be considered in those with refractory pelvic floor dysfunction. This is a weak recommendation based on low-quality evidence.
These recommendations, drafted by a medical position panel and ultimately approved by the AGA Institute Governing Board, were published in conjunction with a technical review, which provides the rationale for the recommendations included in the statement.
AGA Institute Medical Position Panel members listed the following disclosures: Dr. Anthony Lembo reported serving as a consultant to, and serving as an advisory board member for Ironwood Pharmaceuticals and Forest Laboratories; Dr. Spencer D. Dorn reported serving as a consultant to Ironwood Pharmaceuticals and Forest Laboratories, and receiving research support from these companies, as well as from Synergy Pharmaceutical and Takeda Pharmaceuticals; Dr. A. E. Bharucha reported having a financial interest in a new technology related to anal manometry and serving a consultant for Helsin Therapeutics and Asubio Pharmaceuticals.
Depressive symptoms doubled risk of Crohn's disease in women
Depressive symptoms were associated with a doubling of the risk of Crohn’s disease in two large prospective cohorts of women, Dr. Ashwin N. Ananthakrishnan and his colleagues reported in the January issue of Clinical Gastroenterology and Hepatology (2013;11:57-62).
For women with recent and past episodes of depressive symptoms, the associations with the development of Crohn’s disease were significant, and were stronger for those with recent depression. The effect sizes were "in the same range we found for current smoking, oral contraceptive use, and NSAID use," all of which are known risk factors for Crohn’s disease, said Dr. Ananthakrishnan of Massachusetts General Hospital and Harvard Medical School, both in Boston, and his associates.
Video Source: American Gastroenterological Association YouTube channe
"Our findings support the potential importance of a biopsychosocial model in the pathogenesis of Crohn’s disease, and suggest the need for further studies on the effect of depression and stress on immune function and regulation," they noted.
Depression and life stress long have been thought to contribute to immune dysfunction and to influence both the risk for and the course of immune-mediated disorders such as Crohn’s. But until now, few studies have examined the role of mood in the onset of Crohn’s disease and ulcerative colitis, and those that did so were retrospective, failed to adjust for possible confounders, and assessed only the occurrence of major life stressors rather than the presence of depressive symptoms.
To address these shortcomings, the investigators examined the link between depressive symptoms and later onset of Crohn’s disease and ulcerative colitis using data from the prospective Nurses Health Study I and II. NHS I enrolled 121,700 female registered nurses who were 30-55 years old at baseline in 1976, and NHS II enrolled 116,686 female RNs aged 25-42 years at baseline in 1989.
For this study, Dr. Ananthakrishnan and his colleagues analyzed data for 152,461 of these participants in the two NHS cohorts. A total of 170 developed incident Crohn’s disease and 203 developed incident ulcerative colitis during follow-up.
Depressive symptoms were assessed several times in both the NHS I and II subjects using the five-question Mental Health Index (MHI-5), a subscale of the Short Form 36 health status survey. The participants received scores ranging from 1 to 100; 16,986 women (11% of the study population) received scores of 0-52, indicating the presence of depressive symptoms.
Subjects who scored from 86 to 100 composed the reference group.
The data were adjusted to account for numerous covariates that might influence risk for Crohn’s disease and ulcerative colitis, including the subjects’ race/ethnicity; smoking status; weight; menopausal status; and use of oral contraceptives, hormone therapy, and aspirin or NSAIDs.
There was a significant and linear increase in the risk of developing Crohn’s disease as MHI-5 scores decreased. Compared with the reference group, women with an MHI-5 score of 76-85 had a hazard ratio for Crohn’s disease of 1.38, those with an MHI-5 score of 53-75 had an HR of 1.59, and women with depressive symptoms and a score of 0-52 had an HR of 2.36.
No such association was seen between depressive symptoms and ulcerative colitis.
To account for the possibility that the study subjects’ depressed mood might be due to as-yet undiagnosed GI symptoms, the researchers performed a "lag" analysis excluding all cases of Crohn’s and ulcerative colitis that developed within 2 years of MHI-5 assessment. The results were unchanged in this analysis, they said.
The investigators also performed a sensitivity analysis restricted only to cases of Crohn’s disease that developed after 1996, when the NHS subjects first were routinely questioned about their use of antidepressant medications. Adjusting for the use of these drugs only slightly attenuated the strong association between depressive symptoms and Crohn’s disease.
"Preliminary animal and human studies suggest that treating depression through administration of antidepressants, or through improvement in coping mechanisms, could reduce risk of disease relapse. Whether similar interventions can also influence risk of disease onset, particularly among individuals with genetic susceptibility for Crohn’s disease or ulcerative colitis, merits further study," Dr. Ananthakrishnan and his associates said.
This study was supported by the American Gastroenterological Association, the Crohn’s and Colitis Foundation of America, the Broad Foundation, and the National Institutes of Health. Dr. Ananthakrishnan reported no potential financial conflicts of interest; his associates reported ties to Policy Analysis, Bayer HealthCare, Millennium Pharmaceuticals, and Pfizer.
Depressive symptoms were associated with a doubling of the risk of Crohn’s disease in two large prospective cohorts of women, Dr. Ashwin N. Ananthakrishnan and his colleagues reported in the January issue of Clinical Gastroenterology and Hepatology (2013;11:57-62).
For women with recent and past episodes of depressive symptoms, the associations with the development of Crohn’s disease were significant, and were stronger for those with recent depression. The effect sizes were "in the same range we found for current smoking, oral contraceptive use, and NSAID use," all of which are known risk factors for Crohn’s disease, said Dr. Ananthakrishnan of Massachusetts General Hospital and Harvard Medical School, both in Boston, and his associates.
Video Source: American Gastroenterological Association YouTube channe
"Our findings support the potential importance of a biopsychosocial model in the pathogenesis of Crohn’s disease, and suggest the need for further studies on the effect of depression and stress on immune function and regulation," they noted.
Depression and life stress long have been thought to contribute to immune dysfunction and to influence both the risk for and the course of immune-mediated disorders such as Crohn’s. But until now, few studies have examined the role of mood in the onset of Crohn’s disease and ulcerative colitis, and those that did so were retrospective, failed to adjust for possible confounders, and assessed only the occurrence of major life stressors rather than the presence of depressive symptoms.
To address these shortcomings, the investigators examined the link between depressive symptoms and later onset of Crohn’s disease and ulcerative colitis using data from the prospective Nurses Health Study I and II. NHS I enrolled 121,700 female registered nurses who were 30-55 years old at baseline in 1976, and NHS II enrolled 116,686 female RNs aged 25-42 years at baseline in 1989.
For this study, Dr. Ananthakrishnan and his colleagues analyzed data for 152,461 of these participants in the two NHS cohorts. A total of 170 developed incident Crohn’s disease and 203 developed incident ulcerative colitis during follow-up.
Depressive symptoms were assessed several times in both the NHS I and II subjects using the five-question Mental Health Index (MHI-5), a subscale of the Short Form 36 health status survey. The participants received scores ranging from 1 to 100; 16,986 women (11% of the study population) received scores of 0-52, indicating the presence of depressive symptoms.
Subjects who scored from 86 to 100 composed the reference group.
The data were adjusted to account for numerous covariates that might influence risk for Crohn’s disease and ulcerative colitis, including the subjects’ race/ethnicity; smoking status; weight; menopausal status; and use of oral contraceptives, hormone therapy, and aspirin or NSAIDs.
There was a significant and linear increase in the risk of developing Crohn’s disease as MHI-5 scores decreased. Compared with the reference group, women with an MHI-5 score of 76-85 had a hazard ratio for Crohn’s disease of 1.38, those with an MHI-5 score of 53-75 had an HR of 1.59, and women with depressive symptoms and a score of 0-52 had an HR of 2.36.
No such association was seen between depressive symptoms and ulcerative colitis.
To account for the possibility that the study subjects’ depressed mood might be due to as-yet undiagnosed GI symptoms, the researchers performed a "lag" analysis excluding all cases of Crohn’s and ulcerative colitis that developed within 2 years of MHI-5 assessment. The results were unchanged in this analysis, they said.
The investigators also performed a sensitivity analysis restricted only to cases of Crohn’s disease that developed after 1996, when the NHS subjects first were routinely questioned about their use of antidepressant medications. Adjusting for the use of these drugs only slightly attenuated the strong association between depressive symptoms and Crohn’s disease.
"Preliminary animal and human studies suggest that treating depression through administration of antidepressants, or through improvement in coping mechanisms, could reduce risk of disease relapse. Whether similar interventions can also influence risk of disease onset, particularly among individuals with genetic susceptibility for Crohn’s disease or ulcerative colitis, merits further study," Dr. Ananthakrishnan and his associates said.
This study was supported by the American Gastroenterological Association, the Crohn’s and Colitis Foundation of America, the Broad Foundation, and the National Institutes of Health. Dr. Ananthakrishnan reported no potential financial conflicts of interest; his associates reported ties to Policy Analysis, Bayer HealthCare, Millennium Pharmaceuticals, and Pfizer.
Depressive symptoms were associated with a doubling of the risk of Crohn’s disease in two large prospective cohorts of women, Dr. Ashwin N. Ananthakrishnan and his colleagues reported in the January issue of Clinical Gastroenterology and Hepatology (2013;11:57-62).
For women with recent and past episodes of depressive symptoms, the associations with the development of Crohn’s disease were significant, and were stronger for those with recent depression. The effect sizes were "in the same range we found for current smoking, oral contraceptive use, and NSAID use," all of which are known risk factors for Crohn’s disease, said Dr. Ananthakrishnan of Massachusetts General Hospital and Harvard Medical School, both in Boston, and his associates.
Video Source: American Gastroenterological Association YouTube channe
"Our findings support the potential importance of a biopsychosocial model in the pathogenesis of Crohn’s disease, and suggest the need for further studies on the effect of depression and stress on immune function and regulation," they noted.
Depression and life stress long have been thought to contribute to immune dysfunction and to influence both the risk for and the course of immune-mediated disorders such as Crohn’s. But until now, few studies have examined the role of mood in the onset of Crohn’s disease and ulcerative colitis, and those that did so were retrospective, failed to adjust for possible confounders, and assessed only the occurrence of major life stressors rather than the presence of depressive symptoms.
To address these shortcomings, the investigators examined the link between depressive symptoms and later onset of Crohn’s disease and ulcerative colitis using data from the prospective Nurses Health Study I and II. NHS I enrolled 121,700 female registered nurses who were 30-55 years old at baseline in 1976, and NHS II enrolled 116,686 female RNs aged 25-42 years at baseline in 1989.
For this study, Dr. Ananthakrishnan and his colleagues analyzed data for 152,461 of these participants in the two NHS cohorts. A total of 170 developed incident Crohn’s disease and 203 developed incident ulcerative colitis during follow-up.
Depressive symptoms were assessed several times in both the NHS I and II subjects using the five-question Mental Health Index (MHI-5), a subscale of the Short Form 36 health status survey. The participants received scores ranging from 1 to 100; 16,986 women (11% of the study population) received scores of 0-52, indicating the presence of depressive symptoms.
Subjects who scored from 86 to 100 composed the reference group.
The data were adjusted to account for numerous covariates that might influence risk for Crohn’s disease and ulcerative colitis, including the subjects’ race/ethnicity; smoking status; weight; menopausal status; and use of oral contraceptives, hormone therapy, and aspirin or NSAIDs.
There was a significant and linear increase in the risk of developing Crohn’s disease as MHI-5 scores decreased. Compared with the reference group, women with an MHI-5 score of 76-85 had a hazard ratio for Crohn’s disease of 1.38, those with an MHI-5 score of 53-75 had an HR of 1.59, and women with depressive symptoms and a score of 0-52 had an HR of 2.36.
No such association was seen between depressive symptoms and ulcerative colitis.
To account for the possibility that the study subjects’ depressed mood might be due to as-yet undiagnosed GI symptoms, the researchers performed a "lag" analysis excluding all cases of Crohn’s and ulcerative colitis that developed within 2 years of MHI-5 assessment. The results were unchanged in this analysis, they said.
The investigators also performed a sensitivity analysis restricted only to cases of Crohn’s disease that developed after 1996, when the NHS subjects first were routinely questioned about their use of antidepressant medications. Adjusting for the use of these drugs only slightly attenuated the strong association between depressive symptoms and Crohn’s disease.
"Preliminary animal and human studies suggest that treating depression through administration of antidepressants, or through improvement in coping mechanisms, could reduce risk of disease relapse. Whether similar interventions can also influence risk of disease onset, particularly among individuals with genetic susceptibility for Crohn’s disease or ulcerative colitis, merits further study," Dr. Ananthakrishnan and his associates said.
This study was supported by the American Gastroenterological Association, the Crohn’s and Colitis Foundation of America, the Broad Foundation, and the National Institutes of Health. Dr. Ananthakrishnan reported no potential financial conflicts of interest; his associates reported ties to Policy Analysis, Bayer HealthCare, Millennium Pharmaceuticals, and Pfizer.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Major Finding: Women with depressive symptoms and an MHI-5 score of 0-52 had a hazard ratio of 2.36 for Crohn’s disease.
Data Source: An analysis of data on 152,461 subjects in the Nurses Health Study I and II.
Disclosures: This study was supported by the American Gastroenterological Association, the Crohn’s and Colitis Foundation of America, the Broad Foundation, and the National Institutes of Health. Dr. Ananthakrishnan reported no potential financial conflicts of interest; his associates reported ties to Policy Analysis, Bayer HealthCare, Millennium Pharmaceuticals, and Pfizer.
