User login
The sword of Damocles
As Washington hurtled toward the fiscal cliff, I watched with increasing confidence that savvy politicians would announce a last-minute deal. However, I gained even more confidence that it would not actually deal with the problem. Far from forming a grand bargain, Congress just kicked the can a bit further down the road.
The next hurdle the federal government will face and crawl under will be raising the debt ceiling by the end of February 2013. I have no plans to write my next column on that subject. This column is not meant to be a fount for political analysis. But there are two important ways in which the fiscal cliff debacle impacts physicians. One is exemplified by the legend of the sword of Damocles. The other comes from an aphorism attributed to Mahatma Gandhi.
In 1997, Congress created a correction factor, based on a Sustainable Growth Rate (SGR), to control runaway increases in health care spending. Starting in 1999, Medicare fees were to be adjusted so that the rate of growth in Medicare spending was no larger than the growth of the gross domestic product (GDP). In short, if spending increased more than that, physician fees for a given service would be reduced by a proportionate amount. However, each year since then, an act of Congress has postponed, but not repealed, implementation of the correction factor. This has happened so repeatedly that it has acquired the nickname "the doc fix." In total, these accumulated corrections now exceed 27%. Once again, as part of the bill passed by the Senate on Jan. 1, 2013, a postponement was authorized. Physicians will not see a sudden 27% drop in Medicare fees in 2013. But the threat of such a reduction in fees for 2014 remains on the legislative books.
By legend, Damocles temporarily sat upon the throne of Dionysius, but could not enjoy its luxury because over his head was a large sword suspended by a single hair of a horse’s tail. For physicians, the sword of Damocles grows larger annually. As the size of the Medicare SGR correction has accumulated, fewer people believe it will ever be implemented. I am reminded of an adage that "experience allows us to repeat the same mistakes with increasing levels of confidence." The longer the sword remains over our heads, the less worried we become that it will actually fall. That may not be wise in a world of political brinkmanship.
The second take-home message from Washington’s paralysis is more cynical and insidious. Health care in the United States, particularly public health, has been very successful, adding 10 years to the average life expectancy over the past 50 years. But it has created a Faustian bargain with unsustainable cost increases. We’ve gone from 6% of the GDP to 17% spent on health care. Of course, if the average working person gets to live 10 years longer, he might be willing to pay for that with 11% of the GDP. But other countries have obtained the same benefit for half the price. The state of Oregon once tried to prioritize Medicaid spending, creating a list of which medical interventions would be covered and which were too extravagant. The process failed. The fiscal cliff debacle is further demonstration that our current form of government cannot handle these difficult choices over diverse ideals.
The financing of health care in the United States has fostered wasteful and futile care for the elderly while services for children, particularly dental and mental health, remained woefully underfunded. It appears to be irrational to continue to wait for government to create a just framework for allocating medical care. Inaction is collusion with this insanity.
Change is needed before the sword falls. Instead of relying on centralized planning, can we find salvation in the individual choices of physicians? What could you personally do to increase access to the most beneficial types of health care services rather than the most lucrative? As Gandhi suggested, "You must be the change you want to see in the world."
This column, "Beyond the White Coat," regularly appears in Pediatric News. Dr. Powell is associate professor of pediatrics at St. Louis University and a pediatric hospitalist at SSM Cardinal Glennon Children’s Medical Center in St. Louis. E-mail Dr. Powell at [email protected].
As Washington hurtled toward the fiscal cliff, I watched with increasing confidence that savvy politicians would announce a last-minute deal. However, I gained even more confidence that it would not actually deal with the problem. Far from forming a grand bargain, Congress just kicked the can a bit further down the road.
The next hurdle the federal government will face and crawl under will be raising the debt ceiling by the end of February 2013. I have no plans to write my next column on that subject. This column is not meant to be a fount for political analysis. But there are two important ways in which the fiscal cliff debacle impacts physicians. One is exemplified by the legend of the sword of Damocles. The other comes from an aphorism attributed to Mahatma Gandhi.
In 1997, Congress created a correction factor, based on a Sustainable Growth Rate (SGR), to control runaway increases in health care spending. Starting in 1999, Medicare fees were to be adjusted so that the rate of growth in Medicare spending was no larger than the growth of the gross domestic product (GDP). In short, if spending increased more than that, physician fees for a given service would be reduced by a proportionate amount. However, each year since then, an act of Congress has postponed, but not repealed, implementation of the correction factor. This has happened so repeatedly that it has acquired the nickname "the doc fix." In total, these accumulated corrections now exceed 27%. Once again, as part of the bill passed by the Senate on Jan. 1, 2013, a postponement was authorized. Physicians will not see a sudden 27% drop in Medicare fees in 2013. But the threat of such a reduction in fees for 2014 remains on the legislative books.
By legend, Damocles temporarily sat upon the throne of Dionysius, but could not enjoy its luxury because over his head was a large sword suspended by a single hair of a horse’s tail. For physicians, the sword of Damocles grows larger annually. As the size of the Medicare SGR correction has accumulated, fewer people believe it will ever be implemented. I am reminded of an adage that "experience allows us to repeat the same mistakes with increasing levels of confidence." The longer the sword remains over our heads, the less worried we become that it will actually fall. That may not be wise in a world of political brinkmanship.
The second take-home message from Washington’s paralysis is more cynical and insidious. Health care in the United States, particularly public health, has been very successful, adding 10 years to the average life expectancy over the past 50 years. But it has created a Faustian bargain with unsustainable cost increases. We’ve gone from 6% of the GDP to 17% spent on health care. Of course, if the average working person gets to live 10 years longer, he might be willing to pay for that with 11% of the GDP. But other countries have obtained the same benefit for half the price. The state of Oregon once tried to prioritize Medicaid spending, creating a list of which medical interventions would be covered and which were too extravagant. The process failed. The fiscal cliff debacle is further demonstration that our current form of government cannot handle these difficult choices over diverse ideals.
The financing of health care in the United States has fostered wasteful and futile care for the elderly while services for children, particularly dental and mental health, remained woefully underfunded. It appears to be irrational to continue to wait for government to create a just framework for allocating medical care. Inaction is collusion with this insanity.
Change is needed before the sword falls. Instead of relying on centralized planning, can we find salvation in the individual choices of physicians? What could you personally do to increase access to the most beneficial types of health care services rather than the most lucrative? As Gandhi suggested, "You must be the change you want to see in the world."
This column, "Beyond the White Coat," regularly appears in Pediatric News. Dr. Powell is associate professor of pediatrics at St. Louis University and a pediatric hospitalist at SSM Cardinal Glennon Children’s Medical Center in St. Louis. E-mail Dr. Powell at [email protected].
As Washington hurtled toward the fiscal cliff, I watched with increasing confidence that savvy politicians would announce a last-minute deal. However, I gained even more confidence that it would not actually deal with the problem. Far from forming a grand bargain, Congress just kicked the can a bit further down the road.
The next hurdle the federal government will face and crawl under will be raising the debt ceiling by the end of February 2013. I have no plans to write my next column on that subject. This column is not meant to be a fount for political analysis. But there are two important ways in which the fiscal cliff debacle impacts physicians. One is exemplified by the legend of the sword of Damocles. The other comes from an aphorism attributed to Mahatma Gandhi.
In 1997, Congress created a correction factor, based on a Sustainable Growth Rate (SGR), to control runaway increases in health care spending. Starting in 1999, Medicare fees were to be adjusted so that the rate of growth in Medicare spending was no larger than the growth of the gross domestic product (GDP). In short, if spending increased more than that, physician fees for a given service would be reduced by a proportionate amount. However, each year since then, an act of Congress has postponed, but not repealed, implementation of the correction factor. This has happened so repeatedly that it has acquired the nickname "the doc fix." In total, these accumulated corrections now exceed 27%. Once again, as part of the bill passed by the Senate on Jan. 1, 2013, a postponement was authorized. Physicians will not see a sudden 27% drop in Medicare fees in 2013. But the threat of such a reduction in fees for 2014 remains on the legislative books.
By legend, Damocles temporarily sat upon the throne of Dionysius, but could not enjoy its luxury because over his head was a large sword suspended by a single hair of a horse’s tail. For physicians, the sword of Damocles grows larger annually. As the size of the Medicare SGR correction has accumulated, fewer people believe it will ever be implemented. I am reminded of an adage that "experience allows us to repeat the same mistakes with increasing levels of confidence." The longer the sword remains over our heads, the less worried we become that it will actually fall. That may not be wise in a world of political brinkmanship.
The second take-home message from Washington’s paralysis is more cynical and insidious. Health care in the United States, particularly public health, has been very successful, adding 10 years to the average life expectancy over the past 50 years. But it has created a Faustian bargain with unsustainable cost increases. We’ve gone from 6% of the GDP to 17% spent on health care. Of course, if the average working person gets to live 10 years longer, he might be willing to pay for that with 11% of the GDP. But other countries have obtained the same benefit for half the price. The state of Oregon once tried to prioritize Medicaid spending, creating a list of which medical interventions would be covered and which were too extravagant. The process failed. The fiscal cliff debacle is further demonstration that our current form of government cannot handle these difficult choices over diverse ideals.
The financing of health care in the United States has fostered wasteful and futile care for the elderly while services for children, particularly dental and mental health, remained woefully underfunded. It appears to be irrational to continue to wait for government to create a just framework for allocating medical care. Inaction is collusion with this insanity.
Change is needed before the sword falls. Instead of relying on centralized planning, can we find salvation in the individual choices of physicians? What could you personally do to increase access to the most beneficial types of health care services rather than the most lucrative? As Gandhi suggested, "You must be the change you want to see in the world."
This column, "Beyond the White Coat," regularly appears in Pediatric News. Dr. Powell is associate professor of pediatrics at St. Louis University and a pediatric hospitalist at SSM Cardinal Glennon Children’s Medical Center in St. Louis. E-mail Dr. Powell at [email protected].

Evidence-based medicine depends on quality evidence
Efforts to improve the quality of health care often emphasize evidence-based medicine, but flaws in how research is designed, conducted, and reported make this "a great time for skeptics, in looking at clinical trials," according to Dr. J. Russell Hoverman.
Multiple studies in recent years suggest that increasing influence from industry and researchers’ desire to emphasize positive results, as well as other factors, may be distorting choices about which studies get done and how they get reported, said Dr. Hoverman, a medical oncologist and hematologist at Texas Oncology in Austin, Tex.

If researchers don’t improve the way they conduct and assess clinical trials, a lot of money could be wasted on misguided research, he said at a quality care symposium sponsored by the American Society of Clinical Oncology.
He’s not the only one making the case. Physicians at Yale recently argued for greater transparency in pharmaceutical industry–sponsored research to improve the integrity of medical research (Am. J. Public Health 2012;102:72-80).
Over the last three decades, sponsorship of breast, colon, and lung cancer studies by for-profit companies increased from 4% to 57%, Dr. Hoverman noted. Industry sponsorship was associated with trial results that endorsed the experimental agent, according to one study (J. Clin. Oncol. 2008;26:5458-64).
A separate study showed that abstracts of study results presented at major oncology meetings before final publication were discordant from the published article 63% of the time. In 10% of cases, the abstract and article presented substantially different conclusions (J. Clin. Oncol. 2009;3938-44).
One example of this was a trial of a cancer treatment regimen using gemcitabine, cisplatin, and bevacizumab. The investigators initially released an early abstract reporting an improvement in progression-free survival using the regimen. "That actually changed some [oncologists’] practices," he noted. But that was before the study reached its main outcome measure – overall survival – which, in the end, did not improve significantly with the new regimen.
Only half of phase II clinical trials with positive findings lead to positive phase III trials, another study found. For some reason, industry-sponsored trials are much more likely to report positive findings, compared with all other trials – 90% and 45%, respectively (J. Clin. Oncol. 2008;26:1511-8).
When reading or interpreting abstract summaries from a medical conference, "one needs to be a little careful," Dr. Hoverman advised.
Yet another study found that only 45% of randomized clinical trials were registered, even though trial registration has been required since 2005 by the International Committee of Medical Journal Editors in order for the results to be published in participating journals.
Among the registered studies, 31% showed discrepancies between what the investigators said they would be studying and the published outcomes. Half of the studies with discrepancies could be assessed to try to figure out why this was so; of those, 83% of the time it appeared that the investigators decided to favor statistically significant findings in the published article (JAMA 2009;302:977-84).
One set of experts from within industry and from Johns Hopkins University called for "transformational change" in how randomized clinical trials are conducted (Ann. Intern. Med. 2009;151:206-209).
"Without major changes in how we conceive, design, conduct, and analyze randomized controlled trials, the nation risks spending large sums of money inefficiently to answer the wrong questions, or the right questions too late," Dr. Hoverman said.
"In fact, we probably can’t do randomized clinical trials on everything we want to know about. It’s simply impossible. There’s not enough money, and many things involve competing industries or competing members within an industry," making it unlikely that some head-to-head comparisons will ever be done, he added. "So, we are challenged to make decisions based on evidence."
The broader challenge for clinicians and researchers will be to improve the quality and integrity of medical studies while maintaining a healthy skepticism about the available evidence. Medicine has always been an art and a science. Where the science behind medicine is lacking, the art takes over.
Dr. Hoverman reported having no financial disclosures.
-- Sherry Boschert
On Twitter @sherryboschert
Efforts to improve the quality of health care often emphasize evidence-based medicine, but flaws in how research is designed, conducted, and reported make this "a great time for skeptics, in looking at clinical trials," according to Dr. J. Russell Hoverman.
Multiple studies in recent years suggest that increasing influence from industry and researchers’ desire to emphasize positive results, as well as other factors, may be distorting choices about which studies get done and how they get reported, said Dr. Hoverman, a medical oncologist and hematologist at Texas Oncology in Austin, Tex.
If researchers don’t improve the way they conduct and assess clinical trials, a lot of money could be wasted on misguided research, he said at a quality care symposium sponsored by the American Society of Clinical Oncology.
He’s not the only one making the case. Physicians at Yale recently argued for greater transparency in pharmaceutical industry–sponsored research to improve the integrity of medical research (Am. J. Public Health 2012;102:72-80).
Over the last three decades, sponsorship of breast, colon, and lung cancer studies by for-profit companies increased from 4% to 57%, Dr. Hoverman noted. Industry sponsorship was associated with trial results that endorsed the experimental agent, according to one study (J. Clin. Oncol. 2008;26:5458-64).
A separate study showed that abstracts of study results presented at major oncology meetings before final publication were discordant from the published article 63% of the time. In 10% of cases, the abstract and article presented substantially different conclusions (J. Clin. Oncol. 2009;3938-44).
One example of this was a trial of a cancer treatment regimen using gemcitabine, cisplatin, and bevacizumab. The investigators initially released an early abstract reporting an improvement in progression-free survival using the regimen. "That actually changed some [oncologists’] practices," he noted. But that was before the study reached its main outcome measure – overall survival – which, in the end, did not improve significantly with the new regimen.
Only half of phase II clinical trials with positive findings lead to positive phase III trials, another study found. For some reason, industry-sponsored trials are much more likely to report positive findings, compared with all other trials – 90% and 45%, respectively (J. Clin. Oncol. 2008;26:1511-8).
When reading or interpreting abstract summaries from a medical conference, "one needs to be a little careful," Dr. Hoverman advised.
Yet another study found that only 45% of randomized clinical trials were registered, even though trial registration has been required since 2005 by the International Committee of Medical Journal Editors in order for the results to be published in participating journals.
Among the registered studies, 31% showed discrepancies between what the investigators said they would be studying and the published outcomes. Half of the studies with discrepancies could be assessed to try to figure out why this was so; of those, 83% of the time it appeared that the investigators decided to favor statistically significant findings in the published article (JAMA 2009;302:977-84).
One set of experts from within industry and from Johns Hopkins University called for "transformational change" in how randomized clinical trials are conducted (Ann. Intern. Med. 2009;151:206-209).
"Without major changes in how we conceive, design, conduct, and analyze randomized controlled trials, the nation risks spending large sums of money inefficiently to answer the wrong questions, or the right questions too late," Dr. Hoverman said.
"In fact, we probably can’t do randomized clinical trials on everything we want to know about. It’s simply impossible. There’s not enough money, and many things involve competing industries or competing members within an industry," making it unlikely that some head-to-head comparisons will ever be done, he added. "So, we are challenged to make decisions based on evidence."
The broader challenge for clinicians and researchers will be to improve the quality and integrity of medical studies while maintaining a healthy skepticism about the available evidence. Medicine has always been an art and a science. Where the science behind medicine is lacking, the art takes over.
Dr. Hoverman reported having no financial disclosures.
-- Sherry Boschert
On Twitter @sherryboschert
Efforts to improve the quality of health care often emphasize evidence-based medicine, but flaws in how research is designed, conducted, and reported make this "a great time for skeptics, in looking at clinical trials," according to Dr. J. Russell Hoverman.
Multiple studies in recent years suggest that increasing influence from industry and researchers’ desire to emphasize positive results, as well as other factors, may be distorting choices about which studies get done and how they get reported, said Dr. Hoverman, a medical oncologist and hematologist at Texas Oncology in Austin, Tex.
If researchers don’t improve the way they conduct and assess clinical trials, a lot of money could be wasted on misguided research, he said at a quality care symposium sponsored by the American Society of Clinical Oncology.
He’s not the only one making the case. Physicians at Yale recently argued for greater transparency in pharmaceutical industry–sponsored research to improve the integrity of medical research (Am. J. Public Health 2012;102:72-80).
Over the last three decades, sponsorship of breast, colon, and lung cancer studies by for-profit companies increased from 4% to 57%, Dr. Hoverman noted. Industry sponsorship was associated with trial results that endorsed the experimental agent, according to one study (J. Clin. Oncol. 2008;26:5458-64).
A separate study showed that abstracts of study results presented at major oncology meetings before final publication were discordant from the published article 63% of the time. In 10% of cases, the abstract and article presented substantially different conclusions (J. Clin. Oncol. 2009;3938-44).
One example of this was a trial of a cancer treatment regimen using gemcitabine, cisplatin, and bevacizumab. The investigators initially released an early abstract reporting an improvement in progression-free survival using the regimen. "That actually changed some [oncologists’] practices," he noted. But that was before the study reached its main outcome measure – overall survival – which, in the end, did not improve significantly with the new regimen.
Only half of phase II clinical trials with positive findings lead to positive phase III trials, another study found. For some reason, industry-sponsored trials are much more likely to report positive findings, compared with all other trials – 90% and 45%, respectively (J. Clin. Oncol. 2008;26:1511-8).
When reading or interpreting abstract summaries from a medical conference, "one needs to be a little careful," Dr. Hoverman advised.
Yet another study found that only 45% of randomized clinical trials were registered, even though trial registration has been required since 2005 by the International Committee of Medical Journal Editors in order for the results to be published in participating journals.
Among the registered studies, 31% showed discrepancies between what the investigators said they would be studying and the published outcomes. Half of the studies with discrepancies could be assessed to try to figure out why this was so; of those, 83% of the time it appeared that the investigators decided to favor statistically significant findings in the published article (JAMA 2009;302:977-84).
One set of experts from within industry and from Johns Hopkins University called for "transformational change" in how randomized clinical trials are conducted (Ann. Intern. Med. 2009;151:206-209).
"Without major changes in how we conceive, design, conduct, and analyze randomized controlled trials, the nation risks spending large sums of money inefficiently to answer the wrong questions, or the right questions too late," Dr. Hoverman said.
"In fact, we probably can’t do randomized clinical trials on everything we want to know about. It’s simply impossible. There’s not enough money, and many things involve competing industries or competing members within an industry," making it unlikely that some head-to-head comparisons will ever be done, he added. "So, we are challenged to make decisions based on evidence."
The broader challenge for clinicians and researchers will be to improve the quality and integrity of medical studies while maintaining a healthy skepticism about the available evidence. Medicine has always been an art and a science. Where the science behind medicine is lacking, the art takes over.
Dr. Hoverman reported having no financial disclosures.
-- Sherry Boschert
On Twitter @sherryboschert

A Double‐Edged Sword
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.

Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
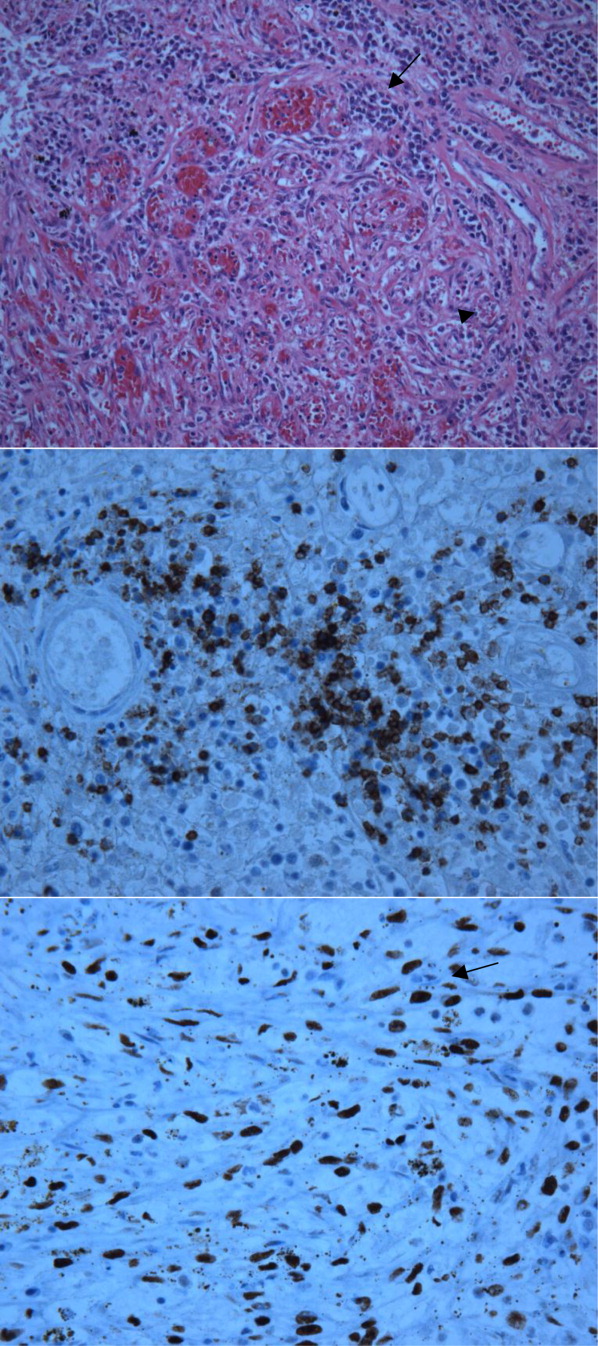
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
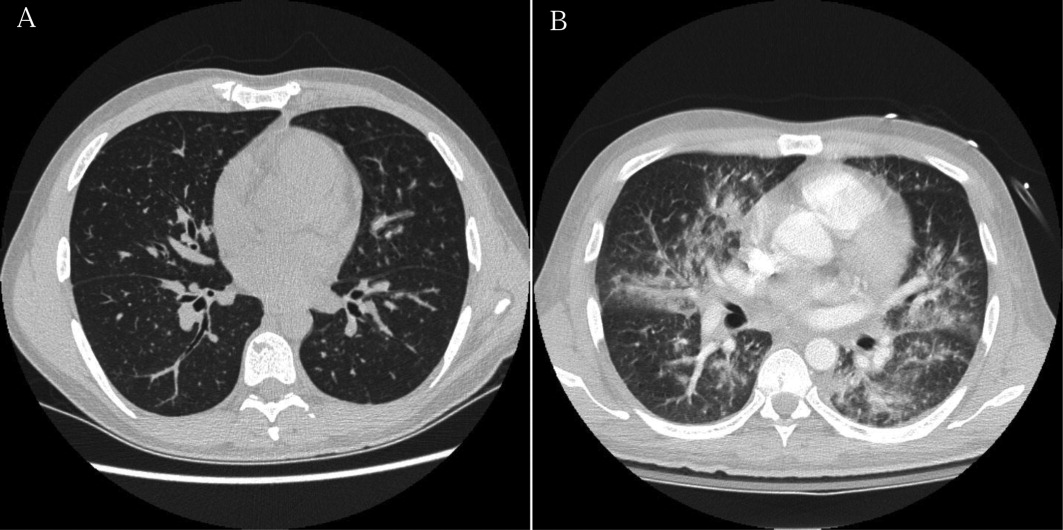
FDA approves apixaban for patients with NVAF
The FDA has approved the anticoagulant apixaban (Eliquis) as prophylaxis for stroke and systemic embolism in patients with nonvalvular atrial fibrillation (NVAF).
The agency’s decision is based on results of the ARISTOTLE trial, in which NVAF patients taking apixaban experienced fewer strokes than those on warfarin.
Apixaban use also resulted in lower rates of major bleeding and treatment discontinuation than warfarin.
The FDA had previously put off making a decision about apixaban, first last March and then in June, pending the receipt of additional data from the ARISTOTLE trial.
Though initial results of that study were promising, in other trials, apixaban produced mixed results.
In the APPRAISE-2 study, apixaban increased major bleeding in patients with acute coronary syndrome, without reducing recurrent ischemic events.
And although results of the ADVANCE-2 study suggested apixaban was superior to enoxaparin for the prevention of venous thromboembolism (VTE), the earlier ADVANCE study indicated the drugs were comparable in efficacy.
In the more recent AMPLIFY-EXTENSION study, apixaban reduced the incidence of VTE, VTE-related events, and death, when compared to placebo.
And in the AVERROES trial, apixaban proved superior to aspirin at preventing stroke in patients with atrial fibrillation who could not use vitamin K agonists.
Now, apixaban is approved for use in NVAF patients to reduce the risk of stroke and systemic embolism. The recommended dose for most patients is 5 mg orally twice daily.
But patients with at least 2 of the following characteristics—age of 80 years or older, weight of 60 kg or less, or serum creatinine of 1.5 mg/dL or greater—should receive 2.5 mg orally twice daily.
Patients with prosthetic heart valves should not take apixaban, nor should patients with atrial fibrillation caused by a heart valve problem. These patients have not been studied in clinical trials.
Bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban. Additionally, the drug’s anticoagulant effects are irreversible.
Apixaban has also been approved in Japan, Canada, and the European Union. The drug is manufactured by Bristol-Myers Squibb Company of Princeton, New Jersey, and marketed by BMS and Pfizer Inc., of New York.
For additional information on apixaban, see the package insert. ![]()
The FDA has approved the anticoagulant apixaban (Eliquis) as prophylaxis for stroke and systemic embolism in patients with nonvalvular atrial fibrillation (NVAF).
The agency’s decision is based on results of the ARISTOTLE trial, in which NVAF patients taking apixaban experienced fewer strokes than those on warfarin.
Apixaban use also resulted in lower rates of major bleeding and treatment discontinuation than warfarin.
The FDA had previously put off making a decision about apixaban, first last March and then in June, pending the receipt of additional data from the ARISTOTLE trial.
Though initial results of that study were promising, in other trials, apixaban produced mixed results.
In the APPRAISE-2 study, apixaban increased major bleeding in patients with acute coronary syndrome, without reducing recurrent ischemic events.
And although results of the ADVANCE-2 study suggested apixaban was superior to enoxaparin for the prevention of venous thromboembolism (VTE), the earlier ADVANCE study indicated the drugs were comparable in efficacy.
In the more recent AMPLIFY-EXTENSION study, apixaban reduced the incidence of VTE, VTE-related events, and death, when compared to placebo.
And in the AVERROES trial, apixaban proved superior to aspirin at preventing stroke in patients with atrial fibrillation who could not use vitamin K agonists.
Now, apixaban is approved for use in NVAF patients to reduce the risk of stroke and systemic embolism. The recommended dose for most patients is 5 mg orally twice daily.
But patients with at least 2 of the following characteristics—age of 80 years or older, weight of 60 kg or less, or serum creatinine of 1.5 mg/dL or greater—should receive 2.5 mg orally twice daily.
Patients with prosthetic heart valves should not take apixaban, nor should patients with atrial fibrillation caused by a heart valve problem. These patients have not been studied in clinical trials.
Bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban. Additionally, the drug’s anticoagulant effects are irreversible.
Apixaban has also been approved in Japan, Canada, and the European Union. The drug is manufactured by Bristol-Myers Squibb Company of Princeton, New Jersey, and marketed by BMS and Pfizer Inc., of New York.
For additional information on apixaban, see the package insert. ![]()
The FDA has approved the anticoagulant apixaban (Eliquis) as prophylaxis for stroke and systemic embolism in patients with nonvalvular atrial fibrillation (NVAF).
The agency’s decision is based on results of the ARISTOTLE trial, in which NVAF patients taking apixaban experienced fewer strokes than those on warfarin.
Apixaban use also resulted in lower rates of major bleeding and treatment discontinuation than warfarin.
The FDA had previously put off making a decision about apixaban, first last March and then in June, pending the receipt of additional data from the ARISTOTLE trial.
Though initial results of that study were promising, in other trials, apixaban produced mixed results.
In the APPRAISE-2 study, apixaban increased major bleeding in patients with acute coronary syndrome, without reducing recurrent ischemic events.
And although results of the ADVANCE-2 study suggested apixaban was superior to enoxaparin for the prevention of venous thromboembolism (VTE), the earlier ADVANCE study indicated the drugs were comparable in efficacy.
In the more recent AMPLIFY-EXTENSION study, apixaban reduced the incidence of VTE, VTE-related events, and death, when compared to placebo.
And in the AVERROES trial, apixaban proved superior to aspirin at preventing stroke in patients with atrial fibrillation who could not use vitamin K agonists.
Now, apixaban is approved for use in NVAF patients to reduce the risk of stroke and systemic embolism. The recommended dose for most patients is 5 mg orally twice daily.
But patients with at least 2 of the following characteristics—age of 80 years or older, weight of 60 kg or less, or serum creatinine of 1.5 mg/dL or greater—should receive 2.5 mg orally twice daily.
Patients with prosthetic heart valves should not take apixaban, nor should patients with atrial fibrillation caused by a heart valve problem. These patients have not been studied in clinical trials.
Bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban. Additionally, the drug’s anticoagulant effects are irreversible.
Apixaban has also been approved in Japan, Canada, and the European Union. The drug is manufactured by Bristol-Myers Squibb Company of Princeton, New Jersey, and marketed by BMS and Pfizer Inc., of New York.
For additional information on apixaban, see the package insert. ![]()
Hospitalist Care and Patient Satisfaction
Payers and policymakers are increasingly holding hospitals accountable for patients' experiences with their care. Since 2006, the Centers for Medicare and Medicaid Services (CMS) have collected data on patients' experiences with inpatient care using the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, a well‐validated and widely used tool. In 2008, these data on patient experience began to be publicly reported, and CMS now plans to base part of its payments to hospitals on HCAHPS performance scores. In this context, hospitals are looking for ways to improve patient satisfaction.
The effort to hold hospitals accountable for patient experience may conflict with another major trend in US hospitals: the increasing use of hospitalists.[1] Although hospitalists may have greater expertise in the day‐to‐day care of the hospitalized patient, they generally do not know the patient and cannot cater to patients' preferences in ways that the primary‐care provider might. Therefore, given that patients may prefer to be seen by their primary‐care provider,[2] greater use of hospitalists may actually lead to a decrease in patient satisfaction. Unfortunately, we are unaware of any national examination of the relationship between hospitalist use in an institution and that entity's performance on patient‐experience scores.
To better understand the relationship between greater hospitalist staffing and patient‐centered care, we examined the association between hospitalist staffing and patient satisfaction with both overall care and specific domains of patient‐centered care. We hypothesized that hospitals that used a high proportion of hospitalists would generally have lower patient‐experience scores. Further, we expected that the relationship would be monotonic (greater use of hospitalists associated with lower scores) and particularly pronounced in 2 domains: patient experience with discharge planning and patient experience with physician communication.
METHODS
Data
We sought to identify acute‐care hospitals with elderly medical patients cared for by hospitalists, non‐hospitalists, or some combination of the 2. To construct this cohort, we used 3 2009 Medicare files. The Beneficiary Summary File contains demographic information on Medicare beneficiaries and data on enrollment in managed‐care plans. To identify medical hospitalizations, we used the Medicare Provider Analysis and Review (MedPAR) 100% Files, which contain the clinical diagnoses and payments for all fee‐for‐service Medicare beneficiaries discharged from acute‐care hospitals. To identify hospitalists and non‐hospitalists, we used the 5% Carrier File, which contains physician billing data for a 5% random sample of fee‐for‐service Medicare beneficiaries. We also obtained information on hospital characteristics from the American Hospital Association (AHA) Annual Survey. We supplemented this with hospital‐level data on patient satisfaction from the HCAHPS survey conducted in 2009. The HCAHPS is a standard survey developed by the Agency for Healthcare Research and Quality (AHRQ) and administered by hospitals to a random sample of adult patients 48 hours to 6 weeks after discharge. The HCAHPS results are adjusted for patient mix and have been tested for nonresponse bias.[3] Details about the development and design of HCAHPS have been described previously.[4]
Patient and Hospital Sample
We started with 48,861,000 Medicare beneficiaries in the Beneficiary Summary File and excluded 38% either because their age was <65 years or they were members of an HMO. At the same time, from the 1,850,000 patients in the 5% Carrier File, we excluded 55% who had not been cared for by a general internist. Finally, we used the MedPAR File to identify 17,387,000 hospital admissions by fee‐for‐service Medicare beneficiaries. From MedPAR, we excluded admissions to a facility other than an acute‐care hospital (24%), surgical admissions identified by diagnosis‐related group (DRG) (29%), and admissions to hospitals with <5 medicine admissions in 2009 (<0.1%). After merging these 3 files (Beneficiary Summary, MedPAR, and 5% Carrier), we were left with 229,496 admissions among 180,399 patients at 3365 hospitals. We subsequently excluded readmissions and were left with 156,333 admissions at 3244 hospitals. Finally, we excluded those patients cared for by both hospitalists and non‐hospitalists during the same hospitalization, and those hospitals missing AHA or HCAHPS data, leaving us with 132,814 patients at 2843 hospitals.
Definition of Hospitalist
We used the claims‐based definition developed and validated by Kuo and Goodwin in earlier work.[1] Hospitalists are defined as those general internists (providers in general practice or internal medicine) who had 5 evaluation and management (E&M) billings (in a 5% sample of Medicare beneficiaries) in 2009 and generated >90% of their claims from the care of hospitalized patients in 2009.
Measures of Patient Satisfaction
There are 2 HCAHPS questions about overall satisfaction, one that asks patients to rate their experience on a scale of 0 to 10 and another that asks whether they would recommend the hospital. Not surprisingly, hospitals' performance on these 2 questions is highly correlated.[5] We measured overall patient experience using commonly used approaches: the proportion of patients who gave the hospital a 9 or 10 (on the 10‐point scale) or the proportion of patients who reported that they would definitely recommend the hospital. The HCAHPS also contains 24 questions, which are reported by CMS in 8 domains: communication with nurse, communication with physician, responsiveness of the staff, pain control, communication about medications, adequacy of discharge planning, cleanliness of the room, and quietness of the room. The patient‐satisfaction score for each of these domains represents the proportion of patients who answered always to each of the questions, or who answered yes to the question about discharge.
Potentially Confounding Variables
Because we were worried that hospitals with hospitalists would be different from hospitals without hospitalists, we identified a series of covariates for adjustment in a multivariable model. We extracted data from the AHA on hospitals' structural characteristics that we assumed might be associated both with having a hospitalist and with patient experience. These variables were size (number of beds), teaching status (membership in the Council of Teaching Hospitals vs no membership), location (urban vs rural), region (the 4 census regions), ownership (for profit, private nonprofit, or public), and presence of advanced clinical capabilities (as measured by having a medical, surgical, and/or cardiac intensive care unit [ICU]). We also used information about the patient population (proportion of patients with Medicare or with Medicaid) as well as nurse‐staffing level (ratio of full‐time equivalent registered nurses to total hospital beds).
Statistical Analyses
We first quantified hospital variation in the proportion of general‐medicine patients cared for by hospitalists, using basic descriptive statistics. Based on these analyses, we categorized hospitals into 3 groups: non‐hospitalist, mixed, and hospitalist (corresponding to lowest, middle, and highest tertile of hospitalist use respectively). We used bivariate techniques to describe the patient and hospital characteristics of hospitals in each group. Patient characteristics included number of comorbidities, which were calculated using software from the Healthcare Cost and Utilization Project (HCUP),[6] based on methods developed by Elixhauser et al.[7] We used the ‐square test to assess whether hospital or patient characteristics differed between hospitalist, mixed, and non‐hospitalist hospitals.
To examine the association between hospitalist care and patient satisfaction, we first constructed bivariate models for each measure of patient satisfaction. In these models, hospital type (hospitalist, mixed, and non‐hospitalist) was our predictor. We next constructed multivariable models, which adjusted for each of the hospital characteristics described above in order to assess the independent relationship between hospitalist care and HCAHPS performance.
In sensitivity analyses, we first examined hospitalist use as a continuous variable and had qualitatively very similar results. Those data are not presented. Additionally, we conducted a propensity score analysis, with results presented in the Appendix (see Supporting Information, Appendix 1, in the online version of this article). In our first‐stage logistic regression model, being a hospitalist hospital (defined as being in the top tertile of hospitalist use vs bottom 2 tertiles) was the outcome. Hospital structural factors were covariates. Based on this first‐stage model, each hospital was assigned a propensity of being a hospitalist hospital. We divided the hospitals into 3 groups (highest propensity tertile, middle propensity tertile, and lowest propensity tertile). In a second‐stage linear regression model, patient satisfaction score was the outcome. The predictors were hospital type (dichotomized, and defined as being in the top tertile of hospitalist use vs bottom 2 tertiles), and propensity of being a hospitalist hospital (3 categories, with low propensity as the reference).
All analyses were performed using SAS version 9.2. The project was reviewed by the Institutional Review Board at the University of Michigan and determined to be not regulated given our use of publicly available datasets.
RESULTS
Among all hospitals, the median proportion of general‐medicine admissions cared for by hospitalists was 41.2% (interquartile range [IQR], 11.5%67.4%). However, US hospitals varied widely in the proportion of general‐medicine patients cared for by hospitalists (Figure 1). Whereas 3.5% of hospitals had all of their general‐medicine patients cared for by hospitalists, 16.6% had none of their general‐medicine patients seen by hospitalists. For hospitals with at least some hospitalist care, the proportion of patients cared for by hospitalists was distributed fairly evenly across the range of possibilities (Figure 1).
Because hospitalist care varied widely among hospitals, we categorized hospitals into 3 groups (non‐hospitalist, mixed, and hospitalist). The median proportion of patients cared for by hospitalists in the 3 groups was 0%, 39.5%, and 76.5%, respectively (Table 1). The non‐hospitalist hospitals, when compared with mixed and hospitalist hospitals, were more likely to be small, nonteaching, for‐profit institutions located in the Midwestern United States. They also were less likely to have an ICU and had lower nurse‐to‐bed ratios.
| Hospital Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 943) | Mixed (N = 948) | Hospitalist (N = 952) | ||
| ||||
| GM admissions cared for by hospitalists, median (range), % | 0 (021) | 40 (2158) | 77 (58100) | <0.001 |
| Nurse‐to‐bed ratio | 1 | 1 | 2 | <0.001 |
| Presence of MICU, % | 79 | 84 | 85 | 0.001 |
| Medicaid patients, % | 19 | 18 | 18 | 0.06 |
| Hospital beds, % | <0.001 | |||
| Small (99) | 36 | 16 | 24 | |
| Medium (100399) | 59 | 64 | 58 | |
| Large (400) | 6 | 21 | 18 | |
| COTH membership, % | <0.001 | |||
| Yes | 3 | 13 | 11 | |
| No | 97 | 87 | 89 | |
| Urban, % | 0.10 | |||
| Yes | 88 | 89 | 91 | |
| No | 12 | 11 | 9 | |
| Profit status, % | <0.001 | |||
| For profit | 21 | 17 | 18 | |
| Not for profit, private | 62 | 71 | 67 | |
| Other | 18 | 12 | 15 | |
| Region, % | <0.001 | |||
| South | 41 | 42 | 42 | |
| Northeast | 14 | 21 | 16 | |
| Midwest | 30 | 22 | 18 | |
| West | 15 | 15 | 24 | |
The types of patients cared for at all 3 hospital types (non‐hospitalist, mixed, and hospitalist) were similar in age and day of admission (Table 2). Patients cared for at non‐hospitalist hospitals were slightly more likely to be female and non‐White, and less likely to be admitted from the emergency department or another hospital or healthcare facility.
| Patient Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 33,265) | Mixed (N = 52,844) | Hospitalist (N = 46,705) | ||
| ||||
| Age, y | 0.51 | |||
| 6574 | 27 | 27 | 27 | |
| 7584 | 39 | 39 | 39 | |
| 85 | 34 | 34 | 34 | |
| Sex | <0.001 | |||
| M | 35 | 35 | 36 | |
| F | 65 | 65 | 64 | |
| Race/ethnicity | <0.001 | |||
| White | 85 | 85 | 87 | |
| Black | 10 | 11 | 9 | |
| Other | 5 | 4 | 4 | |
| Unknown | 0 | 0 | 0 | |
| Comorbidities, % | <0.001 | |||
| 0 | 8 | 8 | 7 | |
| 1 | 23 | 23 | 22 | |
| 2+ | 69 | 69 | 71 | |
| Day of admission | 0.08 | |||
| Weekday | 73 | 73 | 73 | |
| Weekend | 27 | 27 | 27 | |
| Admission source | <0.001 | |||
| ED | 75 | 78 | 80 | |
| Another ACH | 1 | 2 | 3 | |
| Other healthcare facility | 4 | 4 | 4 | |
| Other | 20 | 17 | 13 | |
| ICU stay | <0.001 | |||
| Yes | 13 | 12 | 12 | |
| No | 87 | 88 | 88 | |
| Length of stay, d | <0.001 | |||
| Median (Q1, Q3) | 4 (3, 6) | 4 (2, 6) | 3 (2, 5) | |
| DRG | <0.001 | |||
| Septicemia or severe sepsis | 3 | 4 | 4 | |
| Esophagitis, gastroenteritis | 3 | 3 | 3 | |
| Kidney and urinary tract infections | 3 | 3 | 3 | |
| Syncope | 3 | 3 | 3 | |
| Pneumonia | 3 | 3 | 3 | |
When we examined unadjusted relationships between type of hospital and patient experience, we found that patients at hospitalist vs non‐hospitalist hospitals were more likely to recommend the hospital (69.4% vs 65.1%: P < 0.001), and report higher overall satisfaction (65.9% vs 63.6%: P < 0.001) ((see Supporting Information, Appendix, Table A1, in the online version of this article)). Care at hospitalist hospitals was associated with higher satisfaction with discharge, but lower satisfaction with room cleanliness and communication with doctors. These differences were statistically significant at the P < 0.05 level.
When we examined the relationship between having more hospitalists and patient experience using multivariable models that accounted for differences in hospital characteristics, we found largely similar results: The proportion of patients who were satisfied with their overall care was still higher at hospitalist compared with non‐hospitalist hospitals (65.6% vs 63.9%: P < 0.001) (Figure 2). Similarly, patients were more likely to definitely recommend their hospital if they had been cared for at a hospitalist vs non‐hospitalist hospital (66.0% vs 63.4%: P < 0.001).
To better understand which domains of care might be contributing to greater overall satisfaction, we also examined patient satisfaction with specific domains of care at hospitalist vs non‐hospitalist hospitals (Table 3) in our adjusted analyses. Among 8 domains, the largest difference in satisfaction between patients cared for at hospitalist vs non‐hospitalist hospitals occurred with discharge. At hospitalist hospitals, 80.3% of patients said they were satisfied with the quality of the discharge planning compared with 78.1% at non‐hospitalist hospitals (P < 0.001). Patients at hospitalist hospitals were more satisfied with most other domains of care as well. Patients cared for at hospitalist hospitals were slightly less likely to be satisfied with communication with doctors, but this difference was not statistically significant (P = 0.45). Results were qualitatively similar in propensity‐score analyses (see Supporting Information, Appendix, Table A2, in the online version of this article).
| Specific Domains of Care | Hospital Type, % Satisfied | Hospitalist vs Non‐Hospitalist | |||
|---|---|---|---|---|---|
| Non‐Hospitalist | Mixed | Hospitalist | Difference in % Satisfied | P Value | |
| |||||
| Discharge | 78.1 | 79.1 | 80.3 | 2.1 | <0.001 |
| Nursing services | 66.0 | 65.8 | 67.1 | 1.1 | <0.001 |
| Quiet | 63.3 | 63.1 | 64.4 | 1.1 | 0.001 |
| Communication, nurse | 76.7 | 76.7 | 77.7 | 1.0 | <0.001 |
| Pain control | 69.7 | 69.7 | 70.4 | 0.7 | 0.001 |
| Medications | 60.5 | 60.5 | 61.2 | 0.7 | 0.002 |
| Cleanliness | 72.7 | 72.1 | 72.9 | 0.2 | 0.56 |
| Communication, physician | 83.6 | 83.1 | 83.5 | 0.2 | 0.45 |
DISCUSSION
We found that in 2009, US hospitals varied widely in the proportion of general medicine patients cared for by hospitalists. Hospitals with higher levels of hospitalist care did better on most measures of patient satisfaction. Differences were largest in overall satisfaction and for discharge planning. In 5 other domains of care, differences were smaller, but hospitals with more hospitalist care consistently performed better than non‐hospitalist hospitals. Hospitalist care was not associated with patient satisfaction in 2 domains: communication with doctors and cleanliness of room.
Our findings of modestly higher patient satisfaction at hospitalist hospitals along most dimensions of care are surprising and reassuring. Indeed, when hospitalists first began caring for inpatients, some expressed concerns that hospitalist care would decrease patient satisfaction.[8, 9] Though this has been an ongoing concern, we found no evidence to support this contention. It may be that as a response to the concern, hospitals with hospitalists have paid particular attention to issues such as effective handoffs to primary‐care providers.[10, 11, 12, 13] Whether due to these efforts or other factors such as the 24/7 inpatient presence of hospitalists, we found that patients at hospitalist hospitals were more likely to be satisfied with their inpatient care, including their experience at discharge. In contrast, one area that may offer room for improvement for hospitalist hospitals is communication with physicians. It may be that patients cared for by hospitalists do not know their physicians as well as patients whose care is being orchestrated by their primary‐care provider, and thus the benefits of having an ever‐present hospitalist are diminished.
The magnitude of the associations that we found should also be placed in the context of existing research on patient satisfaction. Prior work has described baseline hospital performance, changes over time, and factors associated with greater inpatient satisfaction.[5, 14, 15] The associations that we found between hospitalist care and satisfaction with care at discharge were larger than those found for teaching (vs non‐teaching) hospitals.[5] However, compared with other hospital characteristics such as nurse staffing or profit status, hospitalist care was associated with smaller differences in patient satisfaction. In one study, hospitals in the highest quartile of nurse staffing had HCAHPS scores (ie, willingness to recommend measure) that were 6.7 points higher than those in the lowest quartile of nurse staffing, and similar differences existed between not‐for‐profit, public hospitals vs for‐profit hospitals.[5]
Taken together, our findings address an important gap in knowledge about hospitalist care. Prior research has documented growth in the use of hospitalist care[1] and described the association of hospitalist care with outcomes such as mortality and resource use, and receipt of recommended care.[16, 17, 18, 19] However, we are unaware of any national study that has examined the association of hospitalist care with patient satisfaction. One study surveyed patients in a single health system and found that patients were similarly satisfied with care provided by hospitalists and primary‐care physicians.[20] Our findings should be reassuring to clinical leaders and policymakers who have advocated greater use of hospitalists: the results suggest that there need be no tradeoff between greater use of hospitalist services and patient satisfaction. Indeed, patients appear to be even more satisfied in hospitals that have greater use of hospitalist physicians.
Our study has several limitations. First, it was a cross‐sectional study, and thus we cannot make any conclusions about causality. Although we adjusted for several potential confounders (eg, teaching status, advanced care capabilities, nurse staffing), it is possible that hospitalist care is a surrogate marker for features of hospitals that we could not measure but that directly influence patient experience. In addition, it is possible that patients cared for at hospitalist hospitals differ in unmeasured ways from patients cared for at other types of hospitals. Second, we constructed our primary predictor and outcome from different cohorts. Our primary predictor was derived from the proportion of general‐medicine patients cared for by hospitalists in Medicare claims data. In contrast, our primary outcome was based on HCAHPS responses from a random sampling of all hospital admissions. This misclassification likely would have biased us towards finding small or no associations. Therefore, we are likely underestimating the true association between hospitalist use and patient experience. Third, our findings may not be generalizable to hospitals that serve younger patients or have a large number of specialist hospitalists (who were not included in our definition of hospitalists). For example, compared with older patients with multiple comorbidities, relatively healthy younger patients may derive less benefit from an ever‐present hospitalist who can explain discharge plans or an attentive nurse.
In summary, we found that US hospitals varied widely in their use of hospitalist physicians, and those which a greater proportion of care was delivered by hospitalists generally had better scores on patient experience, especially on the global assessment of satisfaction and in discharge care. Our findings suggest that adoption of the hospitalist modelone of the strategies employed by US hospitals in the past 2 decades to provide efficient careshould not detract from achieving the goal of more patient‐centered care.
Disclosures
Dr. Chen's work is supported in part by the National Institutes of Health/National Institute on Aging (AG024824, University of Michigan Claude D. Pepper Older Americans Independence Center), and the National Institutes of Health/National Center for Research Resources (UL1‐RR024986, Michigan Institute for Clinical and Health Research). Dr. Chen is also supported by a Career Development Grant Award (K08HS020671) from the Agency for Healthcare Research and Quality.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , . How do patients view the role of the primary care physician in inpatient care? Dis Mon. 2002;48(4):230–238.
- , , , et al. Effects of survey mode, patient mix, and nonresponse on CAHPS hospital survey scores. Health Serv Res. 2009;44(2 pt 1):501–518.
- , , , , . Development, implementation, and public reporting of the HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- , , , . Patients' perception of hospital care in the United States. N Engl J Med. 2008;359(18):1921–1931.
- Agency for Healthcare Research and Quality. Healthcare Cost and Utilization Project (HCUP). HCUP Comorbidity Software. http://www.hcup‐us.ahrq.gov/toolssoftware/comorbidity/comorbidity.jsp Accessed November 12, 2012.
- , , , . Comorbidity measures for use with administrative data. Med Care. 1998;36(1):8–27.
- , , . Physician views on caring for hospitalized patients and the hospitalist model of inpatient care. J Gen Intern Med. 2001;16(2):116–119.
- , , , . How physicians perceive hospitalist services after implementation: anticipation vs reality. Arch Intern Med. 2003;163(19):2330–2336.
- , , , et al. Association of communication between hospital‐based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381–386.
- , , . Passing the clinical baton: 6 principles to guide the hospitalist. Dis Mon. 2002;48(4):260–266.
- , , , . Primary care physician attitudes regarding communication with hospitalists. Am J Med. 2001;111(9B):15S–20S.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- , , , et al. Hospital survey shows improvements in patient experience. Health Aff (Millwood). 2010;29(11):2061–2067.
- , , , , , . Characteristics of hospitals demonstrating superior performance in patient experience and clinical process measures of care. Med Care Res Rev. 2010;67(1):38–55.
- , . The impact of hospitalists on the cost and quality of inpatient care in the United States: a research synthesis. Med Care Res Rev. 2005;62(4):379–406.
- , , , , . Quality of care for patients hospitalized with heart failure: assessing the impact of hospitalists. Arch Intern Med. 2002;162(11):1251–1256.
- , , , , . Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389–1394.
- , , , et al. Quality of care for decompensated heart failure: comparable performance between academic hospitalists and non‐hospitalists. J Gen Intern Med. 2008;23(9):1399–1406.
- , , , et al. Patient satisfaction with hospital care provided by hospitalists and primary care physicians. J Hosp Med. 2012;7(2):131–136.
Payers and policymakers are increasingly holding hospitals accountable for patients' experiences with their care. Since 2006, the Centers for Medicare and Medicaid Services (CMS) have collected data on patients' experiences with inpatient care using the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, a well‐validated and widely used tool. In 2008, these data on patient experience began to be publicly reported, and CMS now plans to base part of its payments to hospitals on HCAHPS performance scores. In this context, hospitals are looking for ways to improve patient satisfaction.
The effort to hold hospitals accountable for patient experience may conflict with another major trend in US hospitals: the increasing use of hospitalists.[1] Although hospitalists may have greater expertise in the day‐to‐day care of the hospitalized patient, they generally do not know the patient and cannot cater to patients' preferences in ways that the primary‐care provider might. Therefore, given that patients may prefer to be seen by their primary‐care provider,[2] greater use of hospitalists may actually lead to a decrease in patient satisfaction. Unfortunately, we are unaware of any national examination of the relationship between hospitalist use in an institution and that entity's performance on patient‐experience scores.
To better understand the relationship between greater hospitalist staffing and patient‐centered care, we examined the association between hospitalist staffing and patient satisfaction with both overall care and specific domains of patient‐centered care. We hypothesized that hospitals that used a high proportion of hospitalists would generally have lower patient‐experience scores. Further, we expected that the relationship would be monotonic (greater use of hospitalists associated with lower scores) and particularly pronounced in 2 domains: patient experience with discharge planning and patient experience with physician communication.
METHODS
Data
We sought to identify acute‐care hospitals with elderly medical patients cared for by hospitalists, non‐hospitalists, or some combination of the 2. To construct this cohort, we used 3 2009 Medicare files. The Beneficiary Summary File contains demographic information on Medicare beneficiaries and data on enrollment in managed‐care plans. To identify medical hospitalizations, we used the Medicare Provider Analysis and Review (MedPAR) 100% Files, which contain the clinical diagnoses and payments for all fee‐for‐service Medicare beneficiaries discharged from acute‐care hospitals. To identify hospitalists and non‐hospitalists, we used the 5% Carrier File, which contains physician billing data for a 5% random sample of fee‐for‐service Medicare beneficiaries. We also obtained information on hospital characteristics from the American Hospital Association (AHA) Annual Survey. We supplemented this with hospital‐level data on patient satisfaction from the HCAHPS survey conducted in 2009. The HCAHPS is a standard survey developed by the Agency for Healthcare Research and Quality (AHRQ) and administered by hospitals to a random sample of adult patients 48 hours to 6 weeks after discharge. The HCAHPS results are adjusted for patient mix and have been tested for nonresponse bias.[3] Details about the development and design of HCAHPS have been described previously.[4]
Patient and Hospital Sample
We started with 48,861,000 Medicare beneficiaries in the Beneficiary Summary File and excluded 38% either because their age was <65 years or they were members of an HMO. At the same time, from the 1,850,000 patients in the 5% Carrier File, we excluded 55% who had not been cared for by a general internist. Finally, we used the MedPAR File to identify 17,387,000 hospital admissions by fee‐for‐service Medicare beneficiaries. From MedPAR, we excluded admissions to a facility other than an acute‐care hospital (24%), surgical admissions identified by diagnosis‐related group (DRG) (29%), and admissions to hospitals with <5 medicine admissions in 2009 (<0.1%). After merging these 3 files (Beneficiary Summary, MedPAR, and 5% Carrier), we were left with 229,496 admissions among 180,399 patients at 3365 hospitals. We subsequently excluded readmissions and were left with 156,333 admissions at 3244 hospitals. Finally, we excluded those patients cared for by both hospitalists and non‐hospitalists during the same hospitalization, and those hospitals missing AHA or HCAHPS data, leaving us with 132,814 patients at 2843 hospitals.
Definition of Hospitalist
We used the claims‐based definition developed and validated by Kuo and Goodwin in earlier work.[1] Hospitalists are defined as those general internists (providers in general practice or internal medicine) who had 5 evaluation and management (E&M) billings (in a 5% sample of Medicare beneficiaries) in 2009 and generated >90% of their claims from the care of hospitalized patients in 2009.
Measures of Patient Satisfaction
There are 2 HCAHPS questions about overall satisfaction, one that asks patients to rate their experience on a scale of 0 to 10 and another that asks whether they would recommend the hospital. Not surprisingly, hospitals' performance on these 2 questions is highly correlated.[5] We measured overall patient experience using commonly used approaches: the proportion of patients who gave the hospital a 9 or 10 (on the 10‐point scale) or the proportion of patients who reported that they would definitely recommend the hospital. The HCAHPS also contains 24 questions, which are reported by CMS in 8 domains: communication with nurse, communication with physician, responsiveness of the staff, pain control, communication about medications, adequacy of discharge planning, cleanliness of the room, and quietness of the room. The patient‐satisfaction score for each of these domains represents the proportion of patients who answered always to each of the questions, or who answered yes to the question about discharge.
Potentially Confounding Variables
Because we were worried that hospitals with hospitalists would be different from hospitals without hospitalists, we identified a series of covariates for adjustment in a multivariable model. We extracted data from the AHA on hospitals' structural characteristics that we assumed might be associated both with having a hospitalist and with patient experience. These variables were size (number of beds), teaching status (membership in the Council of Teaching Hospitals vs no membership), location (urban vs rural), region (the 4 census regions), ownership (for profit, private nonprofit, or public), and presence of advanced clinical capabilities (as measured by having a medical, surgical, and/or cardiac intensive care unit [ICU]). We also used information about the patient population (proportion of patients with Medicare or with Medicaid) as well as nurse‐staffing level (ratio of full‐time equivalent registered nurses to total hospital beds).
Statistical Analyses
We first quantified hospital variation in the proportion of general‐medicine patients cared for by hospitalists, using basic descriptive statistics. Based on these analyses, we categorized hospitals into 3 groups: non‐hospitalist, mixed, and hospitalist (corresponding to lowest, middle, and highest tertile of hospitalist use respectively). We used bivariate techniques to describe the patient and hospital characteristics of hospitals in each group. Patient characteristics included number of comorbidities, which were calculated using software from the Healthcare Cost and Utilization Project (HCUP),[6] based on methods developed by Elixhauser et al.[7] We used the ‐square test to assess whether hospital or patient characteristics differed between hospitalist, mixed, and non‐hospitalist hospitals.
To examine the association between hospitalist care and patient satisfaction, we first constructed bivariate models for each measure of patient satisfaction. In these models, hospital type (hospitalist, mixed, and non‐hospitalist) was our predictor. We next constructed multivariable models, which adjusted for each of the hospital characteristics described above in order to assess the independent relationship between hospitalist care and HCAHPS performance.
In sensitivity analyses, we first examined hospitalist use as a continuous variable and had qualitatively very similar results. Those data are not presented. Additionally, we conducted a propensity score analysis, with results presented in the Appendix (see Supporting Information, Appendix 1, in the online version of this article). In our first‐stage logistic regression model, being a hospitalist hospital (defined as being in the top tertile of hospitalist use vs bottom 2 tertiles) was the outcome. Hospital structural factors were covariates. Based on this first‐stage model, each hospital was assigned a propensity of being a hospitalist hospital. We divided the hospitals into 3 groups (highest propensity tertile, middle propensity tertile, and lowest propensity tertile). In a second‐stage linear regression model, patient satisfaction score was the outcome. The predictors were hospital type (dichotomized, and defined as being in the top tertile of hospitalist use vs bottom 2 tertiles), and propensity of being a hospitalist hospital (3 categories, with low propensity as the reference).
All analyses were performed using SAS version 9.2. The project was reviewed by the Institutional Review Board at the University of Michigan and determined to be not regulated given our use of publicly available datasets.
RESULTS
Among all hospitals, the median proportion of general‐medicine admissions cared for by hospitalists was 41.2% (interquartile range [IQR], 11.5%67.4%). However, US hospitals varied widely in the proportion of general‐medicine patients cared for by hospitalists (Figure 1). Whereas 3.5% of hospitals had all of their general‐medicine patients cared for by hospitalists, 16.6% had none of their general‐medicine patients seen by hospitalists. For hospitals with at least some hospitalist care, the proportion of patients cared for by hospitalists was distributed fairly evenly across the range of possibilities (Figure 1).
Because hospitalist care varied widely among hospitals, we categorized hospitals into 3 groups (non‐hospitalist, mixed, and hospitalist). The median proportion of patients cared for by hospitalists in the 3 groups was 0%, 39.5%, and 76.5%, respectively (Table 1). The non‐hospitalist hospitals, when compared with mixed and hospitalist hospitals, were more likely to be small, nonteaching, for‐profit institutions located in the Midwestern United States. They also were less likely to have an ICU and had lower nurse‐to‐bed ratios.
| Hospital Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 943) | Mixed (N = 948) | Hospitalist (N = 952) | ||
| ||||
| GM admissions cared for by hospitalists, median (range), % | 0 (021) | 40 (2158) | 77 (58100) | <0.001 |
| Nurse‐to‐bed ratio | 1 | 1 | 2 | <0.001 |
| Presence of MICU, % | 79 | 84 | 85 | 0.001 |
| Medicaid patients, % | 19 | 18 | 18 | 0.06 |
| Hospital beds, % | <0.001 | |||
| Small (99) | 36 | 16 | 24 | |
| Medium (100399) | 59 | 64 | 58 | |
| Large (400) | 6 | 21 | 18 | |
| COTH membership, % | <0.001 | |||
| Yes | 3 | 13 | 11 | |
| No | 97 | 87 | 89 | |
| Urban, % | 0.10 | |||
| Yes | 88 | 89 | 91 | |
| No | 12 | 11 | 9 | |
| Profit status, % | <0.001 | |||
| For profit | 21 | 17 | 18 | |
| Not for profit, private | 62 | 71 | 67 | |
| Other | 18 | 12 | 15 | |
| Region, % | <0.001 | |||
| South | 41 | 42 | 42 | |
| Northeast | 14 | 21 | 16 | |
| Midwest | 30 | 22 | 18 | |
| West | 15 | 15 | 24 | |
The types of patients cared for at all 3 hospital types (non‐hospitalist, mixed, and hospitalist) were similar in age and day of admission (Table 2). Patients cared for at non‐hospitalist hospitals were slightly more likely to be female and non‐White, and less likely to be admitted from the emergency department or another hospital or healthcare facility.
| Patient Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 33,265) | Mixed (N = 52,844) | Hospitalist (N = 46,705) | ||
| ||||
| Age, y | 0.51 | |||
| 6574 | 27 | 27 | 27 | |
| 7584 | 39 | 39 | 39 | |
| 85 | 34 | 34 | 34 | |
| Sex | <0.001 | |||
| M | 35 | 35 | 36 | |
| F | 65 | 65 | 64 | |
| Race/ethnicity | <0.001 | |||
| White | 85 | 85 | 87 | |
| Black | 10 | 11 | 9 | |
| Other | 5 | 4 | 4 | |
| Unknown | 0 | 0 | 0 | |
| Comorbidities, % | <0.001 | |||
| 0 | 8 | 8 | 7 | |
| 1 | 23 | 23 | 22 | |
| 2+ | 69 | 69 | 71 | |
| Day of admission | 0.08 | |||
| Weekday | 73 | 73 | 73 | |
| Weekend | 27 | 27 | 27 | |
| Admission source | <0.001 | |||
| ED | 75 | 78 | 80 | |
| Another ACH | 1 | 2 | 3 | |
| Other healthcare facility | 4 | 4 | 4 | |
| Other | 20 | 17 | 13 | |
| ICU stay | <0.001 | |||
| Yes | 13 | 12 | 12 | |
| No | 87 | 88 | 88 | |
| Length of stay, d | <0.001 | |||
| Median (Q1, Q3) | 4 (3, 6) | 4 (2, 6) | 3 (2, 5) | |
| DRG | <0.001 | |||
| Septicemia or severe sepsis | 3 | 4 | 4 | |
| Esophagitis, gastroenteritis | 3 | 3 | 3 | |
| Kidney and urinary tract infections | 3 | 3 | 3 | |
| Syncope | 3 | 3 | 3 | |
| Pneumonia | 3 | 3 | 3 | |
When we examined unadjusted relationships between type of hospital and patient experience, we found that patients at hospitalist vs non‐hospitalist hospitals were more likely to recommend the hospital (69.4% vs 65.1%: P < 0.001), and report higher overall satisfaction (65.9% vs 63.6%: P < 0.001) ((see Supporting Information, Appendix, Table A1, in the online version of this article)). Care at hospitalist hospitals was associated with higher satisfaction with discharge, but lower satisfaction with room cleanliness and communication with doctors. These differences were statistically significant at the P < 0.05 level.
When we examined the relationship between having more hospitalists and patient experience using multivariable models that accounted for differences in hospital characteristics, we found largely similar results: The proportion of patients who were satisfied with their overall care was still higher at hospitalist compared with non‐hospitalist hospitals (65.6% vs 63.9%: P < 0.001) (Figure 2). Similarly, patients were more likely to definitely recommend their hospital if they had been cared for at a hospitalist vs non‐hospitalist hospital (66.0% vs 63.4%: P < 0.001).
To better understand which domains of care might be contributing to greater overall satisfaction, we also examined patient satisfaction with specific domains of care at hospitalist vs non‐hospitalist hospitals (Table 3) in our adjusted analyses. Among 8 domains, the largest difference in satisfaction between patients cared for at hospitalist vs non‐hospitalist hospitals occurred with discharge. At hospitalist hospitals, 80.3% of patients said they were satisfied with the quality of the discharge planning compared with 78.1% at non‐hospitalist hospitals (P < 0.001). Patients at hospitalist hospitals were more satisfied with most other domains of care as well. Patients cared for at hospitalist hospitals were slightly less likely to be satisfied with communication with doctors, but this difference was not statistically significant (P = 0.45). Results were qualitatively similar in propensity‐score analyses (see Supporting Information, Appendix, Table A2, in the online version of this article).
| Specific Domains of Care | Hospital Type, % Satisfied | Hospitalist vs Non‐Hospitalist | |||
|---|---|---|---|---|---|
| Non‐Hospitalist | Mixed | Hospitalist | Difference in % Satisfied | P Value | |
| |||||
| Discharge | 78.1 | 79.1 | 80.3 | 2.1 | <0.001 |
| Nursing services | 66.0 | 65.8 | 67.1 | 1.1 | <0.001 |
| Quiet | 63.3 | 63.1 | 64.4 | 1.1 | 0.001 |
| Communication, nurse | 76.7 | 76.7 | 77.7 | 1.0 | <0.001 |
| Pain control | 69.7 | 69.7 | 70.4 | 0.7 | 0.001 |
| Medications | 60.5 | 60.5 | 61.2 | 0.7 | 0.002 |
| Cleanliness | 72.7 | 72.1 | 72.9 | 0.2 | 0.56 |
| Communication, physician | 83.6 | 83.1 | 83.5 | 0.2 | 0.45 |
DISCUSSION
We found that in 2009, US hospitals varied widely in the proportion of general medicine patients cared for by hospitalists. Hospitals with higher levels of hospitalist care did better on most measures of patient satisfaction. Differences were largest in overall satisfaction and for discharge planning. In 5 other domains of care, differences were smaller, but hospitals with more hospitalist care consistently performed better than non‐hospitalist hospitals. Hospitalist care was not associated with patient satisfaction in 2 domains: communication with doctors and cleanliness of room.
Our findings of modestly higher patient satisfaction at hospitalist hospitals along most dimensions of care are surprising and reassuring. Indeed, when hospitalists first began caring for inpatients, some expressed concerns that hospitalist care would decrease patient satisfaction.[8, 9] Though this has been an ongoing concern, we found no evidence to support this contention. It may be that as a response to the concern, hospitals with hospitalists have paid particular attention to issues such as effective handoffs to primary‐care providers.[10, 11, 12, 13] Whether due to these efforts or other factors such as the 24/7 inpatient presence of hospitalists, we found that patients at hospitalist hospitals were more likely to be satisfied with their inpatient care, including their experience at discharge. In contrast, one area that may offer room for improvement for hospitalist hospitals is communication with physicians. It may be that patients cared for by hospitalists do not know their physicians as well as patients whose care is being orchestrated by their primary‐care provider, and thus the benefits of having an ever‐present hospitalist are diminished.
The magnitude of the associations that we found should also be placed in the context of existing research on patient satisfaction. Prior work has described baseline hospital performance, changes over time, and factors associated with greater inpatient satisfaction.[5, 14, 15] The associations that we found between hospitalist care and satisfaction with care at discharge were larger than those found for teaching (vs non‐teaching) hospitals.[5] However, compared with other hospital characteristics such as nurse staffing or profit status, hospitalist care was associated with smaller differences in patient satisfaction. In one study, hospitals in the highest quartile of nurse staffing had HCAHPS scores (ie, willingness to recommend measure) that were 6.7 points higher than those in the lowest quartile of nurse staffing, and similar differences existed between not‐for‐profit, public hospitals vs for‐profit hospitals.[5]
Taken together, our findings address an important gap in knowledge about hospitalist care. Prior research has documented growth in the use of hospitalist care[1] and described the association of hospitalist care with outcomes such as mortality and resource use, and receipt of recommended care.[16, 17, 18, 19] However, we are unaware of any national study that has examined the association of hospitalist care with patient satisfaction. One study surveyed patients in a single health system and found that patients were similarly satisfied with care provided by hospitalists and primary‐care physicians.[20] Our findings should be reassuring to clinical leaders and policymakers who have advocated greater use of hospitalists: the results suggest that there need be no tradeoff between greater use of hospitalist services and patient satisfaction. Indeed, patients appear to be even more satisfied in hospitals that have greater use of hospitalist physicians.
Our study has several limitations. First, it was a cross‐sectional study, and thus we cannot make any conclusions about causality. Although we adjusted for several potential confounders (eg, teaching status, advanced care capabilities, nurse staffing), it is possible that hospitalist care is a surrogate marker for features of hospitals that we could not measure but that directly influence patient experience. In addition, it is possible that patients cared for at hospitalist hospitals differ in unmeasured ways from patients cared for at other types of hospitals. Second, we constructed our primary predictor and outcome from different cohorts. Our primary predictor was derived from the proportion of general‐medicine patients cared for by hospitalists in Medicare claims data. In contrast, our primary outcome was based on HCAHPS responses from a random sampling of all hospital admissions. This misclassification likely would have biased us towards finding small or no associations. Therefore, we are likely underestimating the true association between hospitalist use and patient experience. Third, our findings may not be generalizable to hospitals that serve younger patients or have a large number of specialist hospitalists (who were not included in our definition of hospitalists). For example, compared with older patients with multiple comorbidities, relatively healthy younger patients may derive less benefit from an ever‐present hospitalist who can explain discharge plans or an attentive nurse.
In summary, we found that US hospitals varied widely in their use of hospitalist physicians, and those which a greater proportion of care was delivered by hospitalists generally had better scores on patient experience, especially on the global assessment of satisfaction and in discharge care. Our findings suggest that adoption of the hospitalist modelone of the strategies employed by US hospitals in the past 2 decades to provide efficient careshould not detract from achieving the goal of more patient‐centered care.
Disclosures
Dr. Chen's work is supported in part by the National Institutes of Health/National Institute on Aging (AG024824, University of Michigan Claude D. Pepper Older Americans Independence Center), and the National Institutes of Health/National Center for Research Resources (UL1‐RR024986, Michigan Institute for Clinical and Health Research). Dr. Chen is also supported by a Career Development Grant Award (K08HS020671) from the Agency for Healthcare Research and Quality.
Payers and policymakers are increasingly holding hospitals accountable for patients' experiences with their care. Since 2006, the Centers for Medicare and Medicaid Services (CMS) have collected data on patients' experiences with inpatient care using the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, a well‐validated and widely used tool. In 2008, these data on patient experience began to be publicly reported, and CMS now plans to base part of its payments to hospitals on HCAHPS performance scores. In this context, hospitals are looking for ways to improve patient satisfaction.
The effort to hold hospitals accountable for patient experience may conflict with another major trend in US hospitals: the increasing use of hospitalists.[1] Although hospitalists may have greater expertise in the day‐to‐day care of the hospitalized patient, they generally do not know the patient and cannot cater to patients' preferences in ways that the primary‐care provider might. Therefore, given that patients may prefer to be seen by their primary‐care provider,[2] greater use of hospitalists may actually lead to a decrease in patient satisfaction. Unfortunately, we are unaware of any national examination of the relationship between hospitalist use in an institution and that entity's performance on patient‐experience scores.
To better understand the relationship between greater hospitalist staffing and patient‐centered care, we examined the association between hospitalist staffing and patient satisfaction with both overall care and specific domains of patient‐centered care. We hypothesized that hospitals that used a high proportion of hospitalists would generally have lower patient‐experience scores. Further, we expected that the relationship would be monotonic (greater use of hospitalists associated with lower scores) and particularly pronounced in 2 domains: patient experience with discharge planning and patient experience with physician communication.
METHODS
Data
We sought to identify acute‐care hospitals with elderly medical patients cared for by hospitalists, non‐hospitalists, or some combination of the 2. To construct this cohort, we used 3 2009 Medicare files. The Beneficiary Summary File contains demographic information on Medicare beneficiaries and data on enrollment in managed‐care plans. To identify medical hospitalizations, we used the Medicare Provider Analysis and Review (MedPAR) 100% Files, which contain the clinical diagnoses and payments for all fee‐for‐service Medicare beneficiaries discharged from acute‐care hospitals. To identify hospitalists and non‐hospitalists, we used the 5% Carrier File, which contains physician billing data for a 5% random sample of fee‐for‐service Medicare beneficiaries. We also obtained information on hospital characteristics from the American Hospital Association (AHA) Annual Survey. We supplemented this with hospital‐level data on patient satisfaction from the HCAHPS survey conducted in 2009. The HCAHPS is a standard survey developed by the Agency for Healthcare Research and Quality (AHRQ) and administered by hospitals to a random sample of adult patients 48 hours to 6 weeks after discharge. The HCAHPS results are adjusted for patient mix and have been tested for nonresponse bias.[3] Details about the development and design of HCAHPS have been described previously.[4]
Patient and Hospital Sample
We started with 48,861,000 Medicare beneficiaries in the Beneficiary Summary File and excluded 38% either because their age was <65 years or they were members of an HMO. At the same time, from the 1,850,000 patients in the 5% Carrier File, we excluded 55% who had not been cared for by a general internist. Finally, we used the MedPAR File to identify 17,387,000 hospital admissions by fee‐for‐service Medicare beneficiaries. From MedPAR, we excluded admissions to a facility other than an acute‐care hospital (24%), surgical admissions identified by diagnosis‐related group (DRG) (29%), and admissions to hospitals with <5 medicine admissions in 2009 (<0.1%). After merging these 3 files (Beneficiary Summary, MedPAR, and 5% Carrier), we were left with 229,496 admissions among 180,399 patients at 3365 hospitals. We subsequently excluded readmissions and were left with 156,333 admissions at 3244 hospitals. Finally, we excluded those patients cared for by both hospitalists and non‐hospitalists during the same hospitalization, and those hospitals missing AHA or HCAHPS data, leaving us with 132,814 patients at 2843 hospitals.
Definition of Hospitalist
We used the claims‐based definition developed and validated by Kuo and Goodwin in earlier work.[1] Hospitalists are defined as those general internists (providers in general practice or internal medicine) who had 5 evaluation and management (E&M) billings (in a 5% sample of Medicare beneficiaries) in 2009 and generated >90% of their claims from the care of hospitalized patients in 2009.
Measures of Patient Satisfaction
There are 2 HCAHPS questions about overall satisfaction, one that asks patients to rate their experience on a scale of 0 to 10 and another that asks whether they would recommend the hospital. Not surprisingly, hospitals' performance on these 2 questions is highly correlated.[5] We measured overall patient experience using commonly used approaches: the proportion of patients who gave the hospital a 9 or 10 (on the 10‐point scale) or the proportion of patients who reported that they would definitely recommend the hospital. The HCAHPS also contains 24 questions, which are reported by CMS in 8 domains: communication with nurse, communication with physician, responsiveness of the staff, pain control, communication about medications, adequacy of discharge planning, cleanliness of the room, and quietness of the room. The patient‐satisfaction score for each of these domains represents the proportion of patients who answered always to each of the questions, or who answered yes to the question about discharge.
Potentially Confounding Variables
Because we were worried that hospitals with hospitalists would be different from hospitals without hospitalists, we identified a series of covariates for adjustment in a multivariable model. We extracted data from the AHA on hospitals' structural characteristics that we assumed might be associated both with having a hospitalist and with patient experience. These variables were size (number of beds), teaching status (membership in the Council of Teaching Hospitals vs no membership), location (urban vs rural), region (the 4 census regions), ownership (for profit, private nonprofit, or public), and presence of advanced clinical capabilities (as measured by having a medical, surgical, and/or cardiac intensive care unit [ICU]). We also used information about the patient population (proportion of patients with Medicare or with Medicaid) as well as nurse‐staffing level (ratio of full‐time equivalent registered nurses to total hospital beds).
Statistical Analyses
We first quantified hospital variation in the proportion of general‐medicine patients cared for by hospitalists, using basic descriptive statistics. Based on these analyses, we categorized hospitals into 3 groups: non‐hospitalist, mixed, and hospitalist (corresponding to lowest, middle, and highest tertile of hospitalist use respectively). We used bivariate techniques to describe the patient and hospital characteristics of hospitals in each group. Patient characteristics included number of comorbidities, which were calculated using software from the Healthcare Cost and Utilization Project (HCUP),[6] based on methods developed by Elixhauser et al.[7] We used the ‐square test to assess whether hospital or patient characteristics differed between hospitalist, mixed, and non‐hospitalist hospitals.
To examine the association between hospitalist care and patient satisfaction, we first constructed bivariate models for each measure of patient satisfaction. In these models, hospital type (hospitalist, mixed, and non‐hospitalist) was our predictor. We next constructed multivariable models, which adjusted for each of the hospital characteristics described above in order to assess the independent relationship between hospitalist care and HCAHPS performance.
In sensitivity analyses, we first examined hospitalist use as a continuous variable and had qualitatively very similar results. Those data are not presented. Additionally, we conducted a propensity score analysis, with results presented in the Appendix (see Supporting Information, Appendix 1, in the online version of this article). In our first‐stage logistic regression model, being a hospitalist hospital (defined as being in the top tertile of hospitalist use vs bottom 2 tertiles) was the outcome. Hospital structural factors were covariates. Based on this first‐stage model, each hospital was assigned a propensity of being a hospitalist hospital. We divided the hospitals into 3 groups (highest propensity tertile, middle propensity tertile, and lowest propensity tertile). In a second‐stage linear regression model, patient satisfaction score was the outcome. The predictors were hospital type (dichotomized, and defined as being in the top tertile of hospitalist use vs bottom 2 tertiles), and propensity of being a hospitalist hospital (3 categories, with low propensity as the reference).
All analyses were performed using SAS version 9.2. The project was reviewed by the Institutional Review Board at the University of Michigan and determined to be not regulated given our use of publicly available datasets.
RESULTS
Among all hospitals, the median proportion of general‐medicine admissions cared for by hospitalists was 41.2% (interquartile range [IQR], 11.5%67.4%). However, US hospitals varied widely in the proportion of general‐medicine patients cared for by hospitalists (Figure 1). Whereas 3.5% of hospitals had all of their general‐medicine patients cared for by hospitalists, 16.6% had none of their general‐medicine patients seen by hospitalists. For hospitals with at least some hospitalist care, the proportion of patients cared for by hospitalists was distributed fairly evenly across the range of possibilities (Figure 1).
Because hospitalist care varied widely among hospitals, we categorized hospitals into 3 groups (non‐hospitalist, mixed, and hospitalist). The median proportion of patients cared for by hospitalists in the 3 groups was 0%, 39.5%, and 76.5%, respectively (Table 1). The non‐hospitalist hospitals, when compared with mixed and hospitalist hospitals, were more likely to be small, nonteaching, for‐profit institutions located in the Midwestern United States. They also were less likely to have an ICU and had lower nurse‐to‐bed ratios.
| Hospital Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 943) | Mixed (N = 948) | Hospitalist (N = 952) | ||
| ||||
| GM admissions cared for by hospitalists, median (range), % | 0 (021) | 40 (2158) | 77 (58100) | <0.001 |
| Nurse‐to‐bed ratio | 1 | 1 | 2 | <0.001 |
| Presence of MICU, % | 79 | 84 | 85 | 0.001 |
| Medicaid patients, % | 19 | 18 | 18 | 0.06 |
| Hospital beds, % | <0.001 | |||
| Small (99) | 36 | 16 | 24 | |
| Medium (100399) | 59 | 64 | 58 | |
| Large (400) | 6 | 21 | 18 | |
| COTH membership, % | <0.001 | |||
| Yes | 3 | 13 | 11 | |
| No | 97 | 87 | 89 | |
| Urban, % | 0.10 | |||
| Yes | 88 | 89 | 91 | |
| No | 12 | 11 | 9 | |
| Profit status, % | <0.001 | |||
| For profit | 21 | 17 | 18 | |
| Not for profit, private | 62 | 71 | 67 | |
| Other | 18 | 12 | 15 | |
| Region, % | <0.001 | |||
| South | 41 | 42 | 42 | |
| Northeast | 14 | 21 | 16 | |
| Midwest | 30 | 22 | 18 | |
| West | 15 | 15 | 24 | |
The types of patients cared for at all 3 hospital types (non‐hospitalist, mixed, and hospitalist) were similar in age and day of admission (Table 2). Patients cared for at non‐hospitalist hospitals were slightly more likely to be female and non‐White, and less likely to be admitted from the emergency department or another hospital or healthcare facility.
| Patient Characteristics | Hospital Type | P Value | ||
|---|---|---|---|---|
| Non‐Hospitalist (N = 33,265) | Mixed (N = 52,844) | Hospitalist (N = 46,705) | ||
| ||||
| Age, y | 0.51 | |||
| 6574 | 27 | 27 | 27 | |
| 7584 | 39 | 39 | 39 | |
| 85 | 34 | 34 | 34 | |
| Sex | <0.001 | |||
| M | 35 | 35 | 36 | |
| F | 65 | 65 | 64 | |
| Race/ethnicity | <0.001 | |||
| White | 85 | 85 | 87 | |
| Black | 10 | 11 | 9 | |
| Other | 5 | 4 | 4 | |
| Unknown | 0 | 0 | 0 | |
| Comorbidities, % | <0.001 | |||
| 0 | 8 | 8 | 7 | |
| 1 | 23 | 23 | 22 | |
| 2+ | 69 | 69 | 71 | |
| Day of admission | 0.08 | |||
| Weekday | 73 | 73 | 73 | |
| Weekend | 27 | 27 | 27 | |
| Admission source | <0.001 | |||
| ED | 75 | 78 | 80 | |
| Another ACH | 1 | 2 | 3 | |
| Other healthcare facility | 4 | 4 | 4 | |
| Other | 20 | 17 | 13 | |
| ICU stay | <0.001 | |||
| Yes | 13 | 12 | 12 | |
| No | 87 | 88 | 88 | |
| Length of stay, d | <0.001 | |||
| Median (Q1, Q3) | 4 (3, 6) | 4 (2, 6) | 3 (2, 5) | |
| DRG | <0.001 | |||
| Septicemia or severe sepsis | 3 | 4 | 4 | |
| Esophagitis, gastroenteritis | 3 | 3 | 3 | |
| Kidney and urinary tract infections | 3 | 3 | 3 | |
| Syncope | 3 | 3 | 3 | |
| Pneumonia | 3 | 3 | 3 | |
When we examined unadjusted relationships between type of hospital and patient experience, we found that patients at hospitalist vs non‐hospitalist hospitals were more likely to recommend the hospital (69.4% vs 65.1%: P < 0.001), and report higher overall satisfaction (65.9% vs 63.6%: P < 0.001) ((see Supporting Information, Appendix, Table A1, in the online version of this article)). Care at hospitalist hospitals was associated with higher satisfaction with discharge, but lower satisfaction with room cleanliness and communication with doctors. These differences were statistically significant at the P < 0.05 level.
When we examined the relationship between having more hospitalists and patient experience using multivariable models that accounted for differences in hospital characteristics, we found largely similar results: The proportion of patients who were satisfied with their overall care was still higher at hospitalist compared with non‐hospitalist hospitals (65.6% vs 63.9%: P < 0.001) (Figure 2). Similarly, patients were more likely to definitely recommend their hospital if they had been cared for at a hospitalist vs non‐hospitalist hospital (66.0% vs 63.4%: P < 0.001).
To better understand which domains of care might be contributing to greater overall satisfaction, we also examined patient satisfaction with specific domains of care at hospitalist vs non‐hospitalist hospitals (Table 3) in our adjusted analyses. Among 8 domains, the largest difference in satisfaction between patients cared for at hospitalist vs non‐hospitalist hospitals occurred with discharge. At hospitalist hospitals, 80.3% of patients said they were satisfied with the quality of the discharge planning compared with 78.1% at non‐hospitalist hospitals (P < 0.001). Patients at hospitalist hospitals were more satisfied with most other domains of care as well. Patients cared for at hospitalist hospitals were slightly less likely to be satisfied with communication with doctors, but this difference was not statistically significant (P = 0.45). Results were qualitatively similar in propensity‐score analyses (see Supporting Information, Appendix, Table A2, in the online version of this article).
| Specific Domains of Care | Hospital Type, % Satisfied | Hospitalist vs Non‐Hospitalist | |||
|---|---|---|---|---|---|
| Non‐Hospitalist | Mixed | Hospitalist | Difference in % Satisfied | P Value | |
| |||||
| Discharge | 78.1 | 79.1 | 80.3 | 2.1 | <0.001 |
| Nursing services | 66.0 | 65.8 | 67.1 | 1.1 | <0.001 |
| Quiet | 63.3 | 63.1 | 64.4 | 1.1 | 0.001 |
| Communication, nurse | 76.7 | 76.7 | 77.7 | 1.0 | <0.001 |
| Pain control | 69.7 | 69.7 | 70.4 | 0.7 | 0.001 |
| Medications | 60.5 | 60.5 | 61.2 | 0.7 | 0.002 |
| Cleanliness | 72.7 | 72.1 | 72.9 | 0.2 | 0.56 |
| Communication, physician | 83.6 | 83.1 | 83.5 | 0.2 | 0.45 |
DISCUSSION
We found that in 2009, US hospitals varied widely in the proportion of general medicine patients cared for by hospitalists. Hospitals with higher levels of hospitalist care did better on most measures of patient satisfaction. Differences were largest in overall satisfaction and for discharge planning. In 5 other domains of care, differences were smaller, but hospitals with more hospitalist care consistently performed better than non‐hospitalist hospitals. Hospitalist care was not associated with patient satisfaction in 2 domains: communication with doctors and cleanliness of room.
Our findings of modestly higher patient satisfaction at hospitalist hospitals along most dimensions of care are surprising and reassuring. Indeed, when hospitalists first began caring for inpatients, some expressed concerns that hospitalist care would decrease patient satisfaction.[8, 9] Though this has been an ongoing concern, we found no evidence to support this contention. It may be that as a response to the concern, hospitals with hospitalists have paid particular attention to issues such as effective handoffs to primary‐care providers.[10, 11, 12, 13] Whether due to these efforts or other factors such as the 24/7 inpatient presence of hospitalists, we found that patients at hospitalist hospitals were more likely to be satisfied with their inpatient care, including their experience at discharge. In contrast, one area that may offer room for improvement for hospitalist hospitals is communication with physicians. It may be that patients cared for by hospitalists do not know their physicians as well as patients whose care is being orchestrated by their primary‐care provider, and thus the benefits of having an ever‐present hospitalist are diminished.
The magnitude of the associations that we found should also be placed in the context of existing research on patient satisfaction. Prior work has described baseline hospital performance, changes over time, and factors associated with greater inpatient satisfaction.[5, 14, 15] The associations that we found between hospitalist care and satisfaction with care at discharge were larger than those found for teaching (vs non‐teaching) hospitals.[5] However, compared with other hospital characteristics such as nurse staffing or profit status, hospitalist care was associated with smaller differences in patient satisfaction. In one study, hospitals in the highest quartile of nurse staffing had HCAHPS scores (ie, willingness to recommend measure) that were 6.7 points higher than those in the lowest quartile of nurse staffing, and similar differences existed between not‐for‐profit, public hospitals vs for‐profit hospitals.[5]
Taken together, our findings address an important gap in knowledge about hospitalist care. Prior research has documented growth in the use of hospitalist care[1] and described the association of hospitalist care with outcomes such as mortality and resource use, and receipt of recommended care.[16, 17, 18, 19] However, we are unaware of any national study that has examined the association of hospitalist care with patient satisfaction. One study surveyed patients in a single health system and found that patients were similarly satisfied with care provided by hospitalists and primary‐care physicians.[20] Our findings should be reassuring to clinical leaders and policymakers who have advocated greater use of hospitalists: the results suggest that there need be no tradeoff between greater use of hospitalist services and patient satisfaction. Indeed, patients appear to be even more satisfied in hospitals that have greater use of hospitalist physicians.
Our study has several limitations. First, it was a cross‐sectional study, and thus we cannot make any conclusions about causality. Although we adjusted for several potential confounders (eg, teaching status, advanced care capabilities, nurse staffing), it is possible that hospitalist care is a surrogate marker for features of hospitals that we could not measure but that directly influence patient experience. In addition, it is possible that patients cared for at hospitalist hospitals differ in unmeasured ways from patients cared for at other types of hospitals. Second, we constructed our primary predictor and outcome from different cohorts. Our primary predictor was derived from the proportion of general‐medicine patients cared for by hospitalists in Medicare claims data. In contrast, our primary outcome was based on HCAHPS responses from a random sampling of all hospital admissions. This misclassification likely would have biased us towards finding small or no associations. Therefore, we are likely underestimating the true association between hospitalist use and patient experience. Third, our findings may not be generalizable to hospitals that serve younger patients or have a large number of specialist hospitalists (who were not included in our definition of hospitalists). For example, compared with older patients with multiple comorbidities, relatively healthy younger patients may derive less benefit from an ever‐present hospitalist who can explain discharge plans or an attentive nurse.
In summary, we found that US hospitals varied widely in their use of hospitalist physicians, and those which a greater proportion of care was delivered by hospitalists generally had better scores on patient experience, especially on the global assessment of satisfaction and in discharge care. Our findings suggest that adoption of the hospitalist modelone of the strategies employed by US hospitals in the past 2 decades to provide efficient careshould not detract from achieving the goal of more patient‐centered care.
Disclosures
Dr. Chen's work is supported in part by the National Institutes of Health/National Institute on Aging (AG024824, University of Michigan Claude D. Pepper Older Americans Independence Center), and the National Institutes of Health/National Center for Research Resources (UL1‐RR024986, Michigan Institute for Clinical and Health Research). Dr. Chen is also supported by a Career Development Grant Award (K08HS020671) from the Agency for Healthcare Research and Quality.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , . How do patients view the role of the primary care physician in inpatient care? Dis Mon. 2002;48(4):230–238.
- , , , et al. Effects of survey mode, patient mix, and nonresponse on CAHPS hospital survey scores. Health Serv Res. 2009;44(2 pt 1):501–518.
- , , , , . Development, implementation, and public reporting of the HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- , , , . Patients' perception of hospital care in the United States. N Engl J Med. 2008;359(18):1921–1931.
- Agency for Healthcare Research and Quality. Healthcare Cost and Utilization Project (HCUP). HCUP Comorbidity Software. http://www.hcup‐us.ahrq.gov/toolssoftware/comorbidity/comorbidity.jsp Accessed November 12, 2012.
- , , , . Comorbidity measures for use with administrative data. Med Care. 1998;36(1):8–27.
- , , . Physician views on caring for hospitalized patients and the hospitalist model of inpatient care. J Gen Intern Med. 2001;16(2):116–119.
- , , , . How physicians perceive hospitalist services after implementation: anticipation vs reality. Arch Intern Med. 2003;163(19):2330–2336.
- , , , et al. Association of communication between hospital‐based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381–386.
- , , . Passing the clinical baton: 6 principles to guide the hospitalist. Dis Mon. 2002;48(4):260–266.
- , , , . Primary care physician attitudes regarding communication with hospitalists. Am J Med. 2001;111(9B):15S–20S.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- , , , et al. Hospital survey shows improvements in patient experience. Health Aff (Millwood). 2010;29(11):2061–2067.
- , , , , , . Characteristics of hospitals demonstrating superior performance in patient experience and clinical process measures of care. Med Care Res Rev. 2010;67(1):38–55.
- , . The impact of hospitalists on the cost and quality of inpatient care in the United States: a research synthesis. Med Care Res Rev. 2005;62(4):379–406.
- , , , , . Quality of care for patients hospitalized with heart failure: assessing the impact of hospitalists. Arch Intern Med. 2002;162(11):1251–1256.
- , , , , . Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389–1394.
- , , , et al. Quality of care for decompensated heart failure: comparable performance between academic hospitalists and non‐hospitalists. J Gen Intern Med. 2008;23(9):1399–1406.
- , , , et al. Patient satisfaction with hospital care provided by hospitalists and primary care physicians. J Hosp Med. 2012;7(2):131–136.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , . How do patients view the role of the primary care physician in inpatient care? Dis Mon. 2002;48(4):230–238.
- , , , et al. Effects of survey mode, patient mix, and nonresponse on CAHPS hospital survey scores. Health Serv Res. 2009;44(2 pt 1):501–518.
- , , , , . Development, implementation, and public reporting of the HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- , , , . Patients' perception of hospital care in the United States. N Engl J Med. 2008;359(18):1921–1931.
- Agency for Healthcare Research and Quality. Healthcare Cost and Utilization Project (HCUP). HCUP Comorbidity Software. http://www.hcup‐us.ahrq.gov/toolssoftware/comorbidity/comorbidity.jsp Accessed November 12, 2012.
- , , , . Comorbidity measures for use with administrative data. Med Care. 1998;36(1):8–27.
- , , . Physician views on caring for hospitalized patients and the hospitalist model of inpatient care. J Gen Intern Med. 2001;16(2):116–119.
- , , , . How physicians perceive hospitalist services after implementation: anticipation vs reality. Arch Intern Med. 2003;163(19):2330–2336.
- , , , et al. Association of communication between hospital‐based physicians and primary care providers with patient outcomes. J Gen Intern Med. 2009;24(3):381–386.
- , , . Passing the clinical baton: 6 principles to guide the hospitalist. Dis Mon. 2002;48(4):260–266.
- , , , . Primary care physician attitudes regarding communication with hospitalists. Am J Med. 2001;111(9B):15S–20S.
- , , , et al. Transitions of Care Consensus policy statement: American College of Physicians, Society of General Internal Medicine, Society of Hospital Medicine, American Geriatrics Society, American College Of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364–370.
- , , , et al. Hospital survey shows improvements in patient experience. Health Aff (Millwood). 2010;29(11):2061–2067.
- , , , , , . Characteristics of hospitals demonstrating superior performance in patient experience and clinical process measures of care. Med Care Res Rev. 2010;67(1):38–55.
- , . The impact of hospitalists on the cost and quality of inpatient care in the United States: a research synthesis. Med Care Res Rev. 2005;62(4):379–406.
- , , , , . Quality of care for patients hospitalized with heart failure: assessing the impact of hospitalists. Arch Intern Med. 2002;162(11):1251–1256.
- , , , , . Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389–1394.
- , , , et al. Quality of care for decompensated heart failure: comparable performance between academic hospitalists and non‐hospitalists. J Gen Intern Med. 2008;23(9):1399–1406.
- , , , et al. Patient satisfaction with hospital care provided by hospitalists and primary care physicians. J Hosp Med. 2012;7(2):131–136.
Copyright © 2012 Society of Hospital Medicine
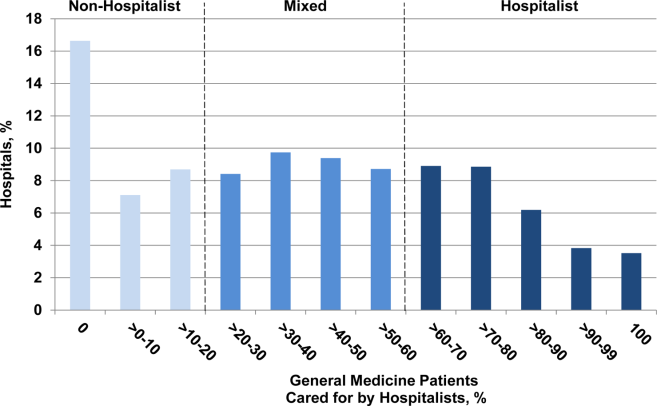
Cut to the Quick
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
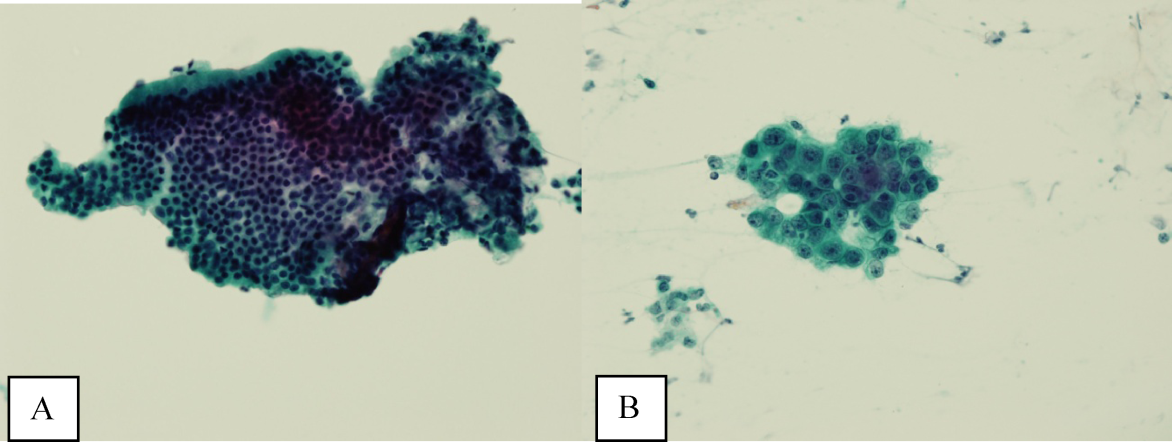
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
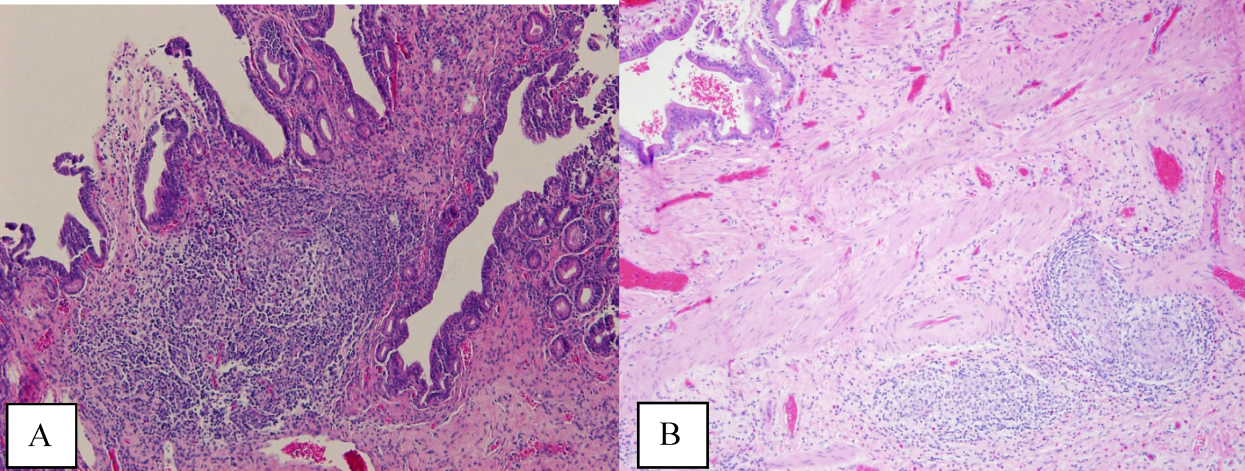
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
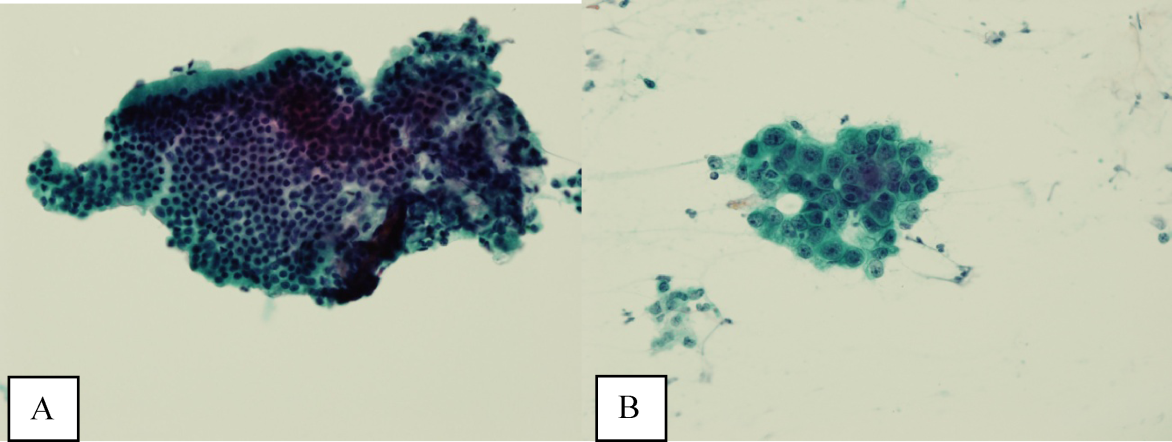
Teamwork Key to Effective Interdisciplinary Rounds
A new study in the Journal of Hospital Medicine is among the first to assess and characterize the effectiveness of teamwork in interdisciplinary rounds (IDR). The upshot: Varied performance on rounds suggests a need to improve the consistency of teamwork.
The report, "Assessment of Teamwork During Structured Interdisciplinary Rounds on Medical Units," adapted the Observational Teamwork Assessment for Surgery (OTAS) behavioral rating scale tool to evaluate and characterize teamwork of hospitalists. Mark Williams, MD, FACP, MHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, says the review shows that mere implementation of IDR is not enough. Physician leaders must occasionally check how the rounds operate to ensure against such roadblocks as a team member who dominates discussions, or the formation of hierarchal relationships that not everyone is comfortable participating in, he says.
"You can't just say, 'Oh, we're practicing teamwork, we have structured interdisciplinary rounds,'" says Dr. Williams, who credits the research to lead author Kevin O'Leary, MD, MS, also of Feinberg. "You need to ensure that it’s occurring."
The paper fills a gap in research, the authors write, as much of the prior work on IDR has focused on patient outcomes, cost, and length of stay. But Dr. Williams says he doesn't expect community hospital medicine groups to conduct similar research because of their busy schedules. Still, he hopes group leaders and administrators consider the research an impetus to periodically check those rounds.
"Even in an institution [like Northwestern] that has strong buy-in to this [teamwork], you need to go back and check," Dr. Williams adds. "We saw variation in performance and we realized we needed to do some retraining."
Visit our website for more information about interdisciplinary rounds.
A new study in the Journal of Hospital Medicine is among the first to assess and characterize the effectiveness of teamwork in interdisciplinary rounds (IDR). The upshot: Varied performance on rounds suggests a need to improve the consistency of teamwork.
The report, "Assessment of Teamwork During Structured Interdisciplinary Rounds on Medical Units," adapted the Observational Teamwork Assessment for Surgery (OTAS) behavioral rating scale tool to evaluate and characterize teamwork of hospitalists. Mark Williams, MD, FACP, MHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, says the review shows that mere implementation of IDR is not enough. Physician leaders must occasionally check how the rounds operate to ensure against such roadblocks as a team member who dominates discussions, or the formation of hierarchal relationships that not everyone is comfortable participating in, he says.
"You can't just say, 'Oh, we're practicing teamwork, we have structured interdisciplinary rounds,'" says Dr. Williams, who credits the research to lead author Kevin O'Leary, MD, MS, also of Feinberg. "You need to ensure that it’s occurring."
The paper fills a gap in research, the authors write, as much of the prior work on IDR has focused on patient outcomes, cost, and length of stay. But Dr. Williams says he doesn't expect community hospital medicine groups to conduct similar research because of their busy schedules. Still, he hopes group leaders and administrators consider the research an impetus to periodically check those rounds.
"Even in an institution [like Northwestern] that has strong buy-in to this [teamwork], you need to go back and check," Dr. Williams adds. "We saw variation in performance and we realized we needed to do some retraining."
Visit our website for more information about interdisciplinary rounds.
A new study in the Journal of Hospital Medicine is among the first to assess and characterize the effectiveness of teamwork in interdisciplinary rounds (IDR). The upshot: Varied performance on rounds suggests a need to improve the consistency of teamwork.
The report, "Assessment of Teamwork During Structured Interdisciplinary Rounds on Medical Units," adapted the Observational Teamwork Assessment for Surgery (OTAS) behavioral rating scale tool to evaluate and characterize teamwork of hospitalists. Mark Williams, MD, FACP, MHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, says the review shows that mere implementation of IDR is not enough. Physician leaders must occasionally check how the rounds operate to ensure against such roadblocks as a team member who dominates discussions, or the formation of hierarchal relationships that not everyone is comfortable participating in, he says.
"You can't just say, 'Oh, we're practicing teamwork, we have structured interdisciplinary rounds,'" says Dr. Williams, who credits the research to lead author Kevin O'Leary, MD, MS, also of Feinberg. "You need to ensure that it’s occurring."
The paper fills a gap in research, the authors write, as much of the prior work on IDR has focused on patient outcomes, cost, and length of stay. But Dr. Williams says he doesn't expect community hospital medicine groups to conduct similar research because of their busy schedules. Still, he hopes group leaders and administrators consider the research an impetus to periodically check those rounds.
"Even in an institution [like Northwestern] that has strong buy-in to this [teamwork], you need to go back and check," Dr. Williams adds. "We saw variation in performance and we realized we needed to do some retraining."
Visit our website for more information about interdisciplinary rounds.
ITL: Physician Reviews of HM-Relevant Research
Clinical question: Is there a difference between aspirin and warfarin in preventing thromboembolic complications and risk of bleeding in patients with chronic kidney disease (CKD) and nonvalvular atrial fibrillation (NVAF)?
Background: Data are lacking on risks and benefits of aspirin and warfarin in CKD, as this group of patients largely has been excluded from anticoagulation therapy trials for NVAF. This study examined the risks and benefits of aspirin and warfarin in patients with CKD with NVAF.
Study design: Retrospective, observational cohort study.
Setting: Danish National Registries.
Synopsis: Of 132,372 patients with NVAF, 2.7% had CKD and 0.7% had end-stage renal disease (ESRD). Compared to patients with no CKD, there was increased risk of stroke or systemic thromboembolism in patients with ESRD (HR, 1.83; 95% CI, 1.57-2.14) and with non-end-stage CKD (HR 1.49; 95% CI 1.38-1.59).
In patients with CKD, warfarin significantly reduced stroke risk (HR, 0.76; 95% CI, 0.64-0.91) and significantly increased bleeding risk (HR, 1.33; 95% CI, 1.16-1.53); aspirin significantly increased bleeding risk (HR, 1.17; 95% CI, 1.02-1.34), with no reduction in stroke risk.
Bottom line: CKD was associated with an increased risk of stroke among NVAF patients. While both aspirin and warfarin were associated with increased risk of bleeding, there was a reduction in the risk of stroke with warfarin, but not with aspirin.
Citation: Olesen JB, Lip GY, Kamper AL, et al. Stroke and bleeding in atrial fibrillation with chronic kidney disease. N Engl J Med. 2012;367(7):625-635.
Click here for more physician reviews of HM-relevant literature.
Clinical question: Is there a difference between aspirin and warfarin in preventing thromboembolic complications and risk of bleeding in patients with chronic kidney disease (CKD) and nonvalvular atrial fibrillation (NVAF)?
Background: Data are lacking on risks and benefits of aspirin and warfarin in CKD, as this group of patients largely has been excluded from anticoagulation therapy trials for NVAF. This study examined the risks and benefits of aspirin and warfarin in patients with CKD with NVAF.
Study design: Retrospective, observational cohort study.
Setting: Danish National Registries.
Synopsis: Of 132,372 patients with NVAF, 2.7% had CKD and 0.7% had end-stage renal disease (ESRD). Compared to patients with no CKD, there was increased risk of stroke or systemic thromboembolism in patients with ESRD (HR, 1.83; 95% CI, 1.57-2.14) and with non-end-stage CKD (HR 1.49; 95% CI 1.38-1.59).
In patients with CKD, warfarin significantly reduced stroke risk (HR, 0.76; 95% CI, 0.64-0.91) and significantly increased bleeding risk (HR, 1.33; 95% CI, 1.16-1.53); aspirin significantly increased bleeding risk (HR, 1.17; 95% CI, 1.02-1.34), with no reduction in stroke risk.
Bottom line: CKD was associated with an increased risk of stroke among NVAF patients. While both aspirin and warfarin were associated with increased risk of bleeding, there was a reduction in the risk of stroke with warfarin, but not with aspirin.
Citation: Olesen JB, Lip GY, Kamper AL, et al. Stroke and bleeding in atrial fibrillation with chronic kidney disease. N Engl J Med. 2012;367(7):625-635.
Click here for more physician reviews of HM-relevant literature.
Clinical question: Is there a difference between aspirin and warfarin in preventing thromboembolic complications and risk of bleeding in patients with chronic kidney disease (CKD) and nonvalvular atrial fibrillation (NVAF)?
Background: Data are lacking on risks and benefits of aspirin and warfarin in CKD, as this group of patients largely has been excluded from anticoagulation therapy trials for NVAF. This study examined the risks and benefits of aspirin and warfarin in patients with CKD with NVAF.
Study design: Retrospective, observational cohort study.
Setting: Danish National Registries.
Synopsis: Of 132,372 patients with NVAF, 2.7% had CKD and 0.7% had end-stage renal disease (ESRD). Compared to patients with no CKD, there was increased risk of stroke or systemic thromboembolism in patients with ESRD (HR, 1.83; 95% CI, 1.57-2.14) and with non-end-stage CKD (HR 1.49; 95% CI 1.38-1.59).
In patients with CKD, warfarin significantly reduced stroke risk (HR, 0.76; 95% CI, 0.64-0.91) and significantly increased bleeding risk (HR, 1.33; 95% CI, 1.16-1.53); aspirin significantly increased bleeding risk (HR, 1.17; 95% CI, 1.02-1.34), with no reduction in stroke risk.
Bottom line: CKD was associated with an increased risk of stroke among NVAF patients. While both aspirin and warfarin were associated with increased risk of bleeding, there was a reduction in the risk of stroke with warfarin, but not with aspirin.
Citation: Olesen JB, Lip GY, Kamper AL, et al. Stroke and bleeding in atrial fibrillation with chronic kidney disease. N Engl J Med. 2012;367(7):625-635.
Click here for more physician reviews of HM-relevant literature.
Quality Improvement Project Helps Hospital Patients Get Needed Prescriptions
A quality-improvement (QI) project to give high-risk patients ready access to prescribed medications at the time of hospital discharge achieved an 86% success rate, according to an abstract poster presented at HM12 in San Diego last April.1
Lead author Elizabeth Le, MD, then a resident at the University of California at San Francisco Medical Center (UCSF) and now a practicing hospitalist at the Veterans Administration Medical Center in Palo Alto, Calif., says the multidisciplinary “brown bag medications” project involved training house staff to recognize patients at risk. Staff meetings and rounds were used to identify appropriate candidates—those with limited mobility or cognitive issues, lacking insurance coverage or financial resources, a history of medication noncompliance, or leaving the hospital against medical advice—as well as those prescribed medications with a greater urgency for administration on schedule, such as anticoagulants or antibiotics.
About one-quarter of patients on the unit where this approach was first tested were found to need the service, which involved faxing prescriptions to an outpatient pharmacy across the street from the hospital for either pick-up by the family or delivery to the patient’s hospital room. For those with financial impediments, hospital social workers and case managers explored other options, including the social work department’s discretionary use fund, to pay for the drugs.
Dr. Le believes the project could be replicated in other facilities that lack access to in-house pharmacy services at discharge. She recommends involving social workers and case managers in the planning.
At UCSF, recent EHR implementation has automated the ordering of medications, but the challenge of recognizing who could benefit from extra help in obtaining their discharge medications remains a critical issue for hospitals trying to bring readmissions under control.
For more information about the brown bag medications program, contact Dr. Le at [email protected].
References
A quality-improvement (QI) project to give high-risk patients ready access to prescribed medications at the time of hospital discharge achieved an 86% success rate, according to an abstract poster presented at HM12 in San Diego last April.1
Lead author Elizabeth Le, MD, then a resident at the University of California at San Francisco Medical Center (UCSF) and now a practicing hospitalist at the Veterans Administration Medical Center in Palo Alto, Calif., says the multidisciplinary “brown bag medications” project involved training house staff to recognize patients at risk. Staff meetings and rounds were used to identify appropriate candidates—those with limited mobility or cognitive issues, lacking insurance coverage or financial resources, a history of medication noncompliance, or leaving the hospital against medical advice—as well as those prescribed medications with a greater urgency for administration on schedule, such as anticoagulants or antibiotics.
About one-quarter of patients on the unit where this approach was first tested were found to need the service, which involved faxing prescriptions to an outpatient pharmacy across the street from the hospital for either pick-up by the family or delivery to the patient’s hospital room. For those with financial impediments, hospital social workers and case managers explored other options, including the social work department’s discretionary use fund, to pay for the drugs.
Dr. Le believes the project could be replicated in other facilities that lack access to in-house pharmacy services at discharge. She recommends involving social workers and case managers in the planning.
At UCSF, recent EHR implementation has automated the ordering of medications, but the challenge of recognizing who could benefit from extra help in obtaining their discharge medications remains a critical issue for hospitals trying to bring readmissions under control.
For more information about the brown bag medications program, contact Dr. Le at [email protected].
References
A quality-improvement (QI) project to give high-risk patients ready access to prescribed medications at the time of hospital discharge achieved an 86% success rate, according to an abstract poster presented at HM12 in San Diego last April.1
Lead author Elizabeth Le, MD, then a resident at the University of California at San Francisco Medical Center (UCSF) and now a practicing hospitalist at the Veterans Administration Medical Center in Palo Alto, Calif., says the multidisciplinary “brown bag medications” project involved training house staff to recognize patients at risk. Staff meetings and rounds were used to identify appropriate candidates—those with limited mobility or cognitive issues, lacking insurance coverage or financial resources, a history of medication noncompliance, or leaving the hospital against medical advice—as well as those prescribed medications with a greater urgency for administration on schedule, such as anticoagulants or antibiotics.
About one-quarter of patients on the unit where this approach was first tested were found to need the service, which involved faxing prescriptions to an outpatient pharmacy across the street from the hospital for either pick-up by the family or delivery to the patient’s hospital room. For those with financial impediments, hospital social workers and case managers explored other options, including the social work department’s discretionary use fund, to pay for the drugs.
Dr. Le believes the project could be replicated in other facilities that lack access to in-house pharmacy services at discharge. She recommends involving social workers and case managers in the planning.
At UCSF, recent EHR implementation has automated the ordering of medications, but the challenge of recognizing who could benefit from extra help in obtaining their discharge medications remains a critical issue for hospitals trying to bring readmissions under control.
For more information about the brown bag medications program, contact Dr. Le at [email protected].
References
12 Things Hospitalists Need to Know About Billing and Coding
Documentation, CPT codes, modifiers—it’s not glamorous, but it’s an integral part of a 21st-century physician’s job description. The Hospitalist queried more than a handful of billing and coding experts about the advice they would dispense to clinicians navigating the reimbursement maze.
“Physicians often do more than what is reflected in the documentation,” says Barb Pierce, CCS-P, ACS-EM, a national coding consultant based in West Des Moines, Iowa, and CODE-H faculty. “They can’t always bill for everything they do, but they certainly can document and code to obtain the appropriate levels of service.”
Meanwhile, hospitalists have to be careful they aren’t excessive in their billing practices. “The name of the game isn’t just to bill higher,” Pierce adds, “but to make sure that your documentation supports the service being billed, and Medicare is watching. They’re doing a lot of focused audits.”
Some hospitalists might opt for a lower level of service, suspecting they’re less likely to be audited. Other hospitalists might seek reimbursement for more of their time and efforts.
“You have both ends of the spectrum,” says Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education for AAPC, formerly known as the American Academy of Professional Coders. “There are a lot of factors that would go into why a provider would code something incorrectly.”
Here’s how to land somewhere in the middle.
1 Be thorough in documenting the initial hospital visit.
When selecting the level of service for an initial hospital visit, the documentation consists of three key components: history, physical examination, and medical decision-making. The history includes the chief complaint as well as the review of systems. This is “an inventory of the patient’s organ systems.” Both the complaint and the systems review are often incorporated in the history of present illness, says Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist in the Department of Medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
A patient’s family history is commonly overlooked in a hospitalist’s notes, primarily when they know the patient from previous admissions for chronic diseases and when the family history will likely not have an impact on treatment. “If they do not document a complete review of systems or miss one of the histories, the service will definitely be down-coded,” Mulholland says, “no matter how complete the exam and medical decision-making documentation.”
2 Familiarize yourself with Medicare reimbursement rules in the state where you practice.
In some states, Medicare contractors require providers to document the status of each organ system reviewed individually. In other states, it’s acceptable to document a system review with pertinent findings, “whether positive or negative,” and the statement of “all other systems negative,” Mulholland says.
The auditor will give credit for the review based on the number of organ systems documented. “If you miss one system review, it will take down what otherwise would be a Level Three hospital admission to a Level One,” she says. “So there would be a significant financial impact.”
Medicare reimbursement for a Level Three initial visit in Mulholland’s area of practice—Philadelphia County in Pennsylvania—is $206.57, compared with $104.69 for a Level One. During this visit, each of the key components—history, exam, and medical decision-making—need to be documented completely for the provider to receive the highest level of reimbursement.
3 Ask about a patient’s social history.
Social history can be obtained by querying the patient about smoking, drug and alcohol use, his or her occupation, marital status, and type of living arrangement.
“Knowing the social history helps the hospitalist understand the home situation or social circumstances that may have contributed to the hospitalization or may complicate the discharge plan,” Mulholland says.
This is particularly important in decision-making that involves elderly patients. The clinician should “think down the road” as to where the patient will be discharged and if a social worker’s assistance will be needed. It’s about “seeing the whole patient,” she says, “not just the disease.”
4 Remember to include the actual diagnosis.
“As coders, we can see all the clinical indicators of a particular diagnosis,” says Kathryn DeVault, RHIA, CCS, CCS-P, a director at HIM Solutions at the American Health Management Association. However, “unless [physicians] write down the diagnosis, we can’t code it.”
Documents without a diagnosis are more common than one would expect. For example, if a patient has pain when urinating, the hospitalist typically orders a culture. If the result is positive, the hospitalist prescribes an antibiotic for the infection, and too often “the story ends there.” From experience, DeVault can decipher that the patient is being treated for a urinary tract infection, but she can’t assign a code without querying the physician. Hospitalists, she suggests, should try to “close the loop in their documentation.”
5 Be specific in your written assessment of the patient’s condition.
“The main thing that we see is missing documentation,” says Angie Comfort, RHIT, CCS, a director at HIM Solutions. For instance, if a hospitalist documents congestive heart failure, it’s important to indicate whether the condition is chronic or acute and systolic or diastolic.
In the case of a diabetic patient, the notes should specify the type of diabetes. Not doing so “could be a reimbursement-changer,” Comfort says. In contrast, documenting such specifics could result in higher reimbursement, especially if a patient has complications from Type 1 diabetes.
6 Note the severity of the patient’s case.
Hospitalists’ documentation doesn’t always capture everything they’re evaluating for patients. “I’ve seen notes to the extent of ‘patient doing well; waiting on test results,’” the AAPC’s Jimenez says. “If they’re doing certain tests, why are they doing them? What are they trying to diagnose for the patient? What treatment are they considering?”
The reasons for the tests need to be explained. When a provider is monitoring someone in the hospital, the documentation should elaborate on the patient’s response to a treatment, and whether the patient’s condition is better, stable, or worse. This information helps put the severity in perspective.
“A diabetic could be a diabetic out of control. It could be a diabetic who’s not responding or who has comorbidities,” Jimenez says. “No one diagnosis is the same for every patient.”

—Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist, department of medicine, University of Pennsylvania, Philadelphia
7 Indicate which aspect of the patient’s condition you are treating.
When multiple providers are involved in a hospitalized patient’s care, it’s important to document your specific role apart from the services rendered by specialists, Jimenez says. The codes billed must be supported by the documentation for each service. Many providers contribute to the inpatient documentation, so it must be clear what each clinician personally performs.
Only report the diagnosis you are treating or the diagnoses that affect the ones you are managing. If a specialist has been brought in to take over treatment for a specific condition, a hospitalist would not bill for that diagnosis code.

—Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education, AAPC Salt Lake City
8 Note your personal review of medical records and reports from other clinicians.
Hospitalists should document their review of lab data or radiology reports, discussion of the case with other providers, or collection of the history from someone other than the patient. It’s also helpful to document your personal review of any images, such as a chest X-ray or MRI. Examining the images yourself might lead to higher reimbursement, Mulholland says.
Providers also should note when they request or review old records, and they should include a short synopsis of the information obtained and how it contributed to the current treatment plan.
9 Learn the correct coding for patients being transferred.
A transfer can occur either from a different facility or from a hospital floor to a rehabilitation unit. Either way, the patient is seen twice in one day, with each visit covered by the same hospitalist practice.
“Both physicians often report a separate independent visit. However, because these services occurred on the same day, it is not appropriate to bill for two separate subsequent or initial hospital codes,” says Sherri Dumford, MBA, CHBME, director of operations and past president of the Healthcare Billing and Management Association. “Often what will happen is both services will be reported and get through the billing system. The second claim is just written off as a denied service, when, in fact, you could combine the elements of service of both visits and possibly bill for a single higher level of visit.”
10 Consider delegating to a coding expert.
While smaller hospitalist groups can turn to a coding consultant on an as-needed basis, larger groups might consider bringing a certified coder on staff. This person would inform physicians about proper coding, review their documentation, and “give real-time feedback,” Pierce says.
An internal audit would show if the documentation meets selected evaluation management codes. Also, it usually takes a coding professional to determine whether prolonged services are an option for the team on any given date of service. Someone would need to internally “add together” multiple services on one date to see if there is sufficient time documented to allow billing for these add-on codes, Pierce says. Similarly, critical-care time needs to be accumulated during a date of service.

—Barb Pierce, CCS-P, ACS-EM, national coding consultant, West Des Moines, Iowa
11 Indicate the number of minutes spent arranging for a patient’s discharge.
Discharging a patient involves various steps, says Peter Thompson, MD, chief of clinical operations at the Phoenix headquarters of Apogee Physicians, a hospitalist management company that employs about 750 hospitalists across the country. Hospitalists discuss the hospital stay with the patient and family members, prescribe medications, issue discharge recommendations, set up follow-up care, and coordinate with the case manager, specialists, and primary-care physician.
“It generally is one sequential event after the other,” lasting between 20 and 40 minutes and leading up to discharge, Thompson says. Reimbursement for a high-level discharge constitutes more than 30 minutes. However, without proper documentation, he cautions, the claim could be downgraded or denied.
12 Don’t forget to sign, date, and time your progress note.
Last but not least, when it comes to reimbursement, your signature really does matter.
“For an illegible signature, Medicare and the insurance companies have the option of not paying for the service,” Mulholland says. “They’re trying to establish or authenticate who provided the service.”
And they want to know when the hospitalist saw the patient, so it’s a good idea to indicate the exact time of your visit.
Susan Kreimer is a freelance medical writer in New York.
Documentation, CPT codes, modifiers—it’s not glamorous, but it’s an integral part of a 21st-century physician’s job description. The Hospitalist queried more than a handful of billing and coding experts about the advice they would dispense to clinicians navigating the reimbursement maze.
“Physicians often do more than what is reflected in the documentation,” says Barb Pierce, CCS-P, ACS-EM, a national coding consultant based in West Des Moines, Iowa, and CODE-H faculty. “They can’t always bill for everything they do, but they certainly can document and code to obtain the appropriate levels of service.”
Meanwhile, hospitalists have to be careful they aren’t excessive in their billing practices. “The name of the game isn’t just to bill higher,” Pierce adds, “but to make sure that your documentation supports the service being billed, and Medicare is watching. They’re doing a lot of focused audits.”
Some hospitalists might opt for a lower level of service, suspecting they’re less likely to be audited. Other hospitalists might seek reimbursement for more of their time and efforts.
“You have both ends of the spectrum,” says Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education for AAPC, formerly known as the American Academy of Professional Coders. “There are a lot of factors that would go into why a provider would code something incorrectly.”
Here’s how to land somewhere in the middle.
1 Be thorough in documenting the initial hospital visit.
When selecting the level of service for an initial hospital visit, the documentation consists of three key components: history, physical examination, and medical decision-making. The history includes the chief complaint as well as the review of systems. This is “an inventory of the patient’s organ systems.” Both the complaint and the systems review are often incorporated in the history of present illness, says Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist in the Department of Medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
A patient’s family history is commonly overlooked in a hospitalist’s notes, primarily when they know the patient from previous admissions for chronic diseases and when the family history will likely not have an impact on treatment. “If they do not document a complete review of systems or miss one of the histories, the service will definitely be down-coded,” Mulholland says, “no matter how complete the exam and medical decision-making documentation.”
2 Familiarize yourself with Medicare reimbursement rules in the state where you practice.
In some states, Medicare contractors require providers to document the status of each organ system reviewed individually. In other states, it’s acceptable to document a system review with pertinent findings, “whether positive or negative,” and the statement of “all other systems negative,” Mulholland says.
The auditor will give credit for the review based on the number of organ systems documented. “If you miss one system review, it will take down what otherwise would be a Level Three hospital admission to a Level One,” she says. “So there would be a significant financial impact.”
Medicare reimbursement for a Level Three initial visit in Mulholland’s area of practice—Philadelphia County in Pennsylvania—is $206.57, compared with $104.69 for a Level One. During this visit, each of the key components—history, exam, and medical decision-making—need to be documented completely for the provider to receive the highest level of reimbursement.
3 Ask about a patient’s social history.
Social history can be obtained by querying the patient about smoking, drug and alcohol use, his or her occupation, marital status, and type of living arrangement.
“Knowing the social history helps the hospitalist understand the home situation or social circumstances that may have contributed to the hospitalization or may complicate the discharge plan,” Mulholland says.
This is particularly important in decision-making that involves elderly patients. The clinician should “think down the road” as to where the patient will be discharged and if a social worker’s assistance will be needed. It’s about “seeing the whole patient,” she says, “not just the disease.”
4 Remember to include the actual diagnosis.
“As coders, we can see all the clinical indicators of a particular diagnosis,” says Kathryn DeVault, RHIA, CCS, CCS-P, a director at HIM Solutions at the American Health Management Association. However, “unless [physicians] write down the diagnosis, we can’t code it.”
Documents without a diagnosis are more common than one would expect. For example, if a patient has pain when urinating, the hospitalist typically orders a culture. If the result is positive, the hospitalist prescribes an antibiotic for the infection, and too often “the story ends there.” From experience, DeVault can decipher that the patient is being treated for a urinary tract infection, but she can’t assign a code without querying the physician. Hospitalists, she suggests, should try to “close the loop in their documentation.”
5 Be specific in your written assessment of the patient’s condition.
“The main thing that we see is missing documentation,” says Angie Comfort, RHIT, CCS, a director at HIM Solutions. For instance, if a hospitalist documents congestive heart failure, it’s important to indicate whether the condition is chronic or acute and systolic or diastolic.
In the case of a diabetic patient, the notes should specify the type of diabetes. Not doing so “could be a reimbursement-changer,” Comfort says. In contrast, documenting such specifics could result in higher reimbursement, especially if a patient has complications from Type 1 diabetes.
6 Note the severity of the patient’s case.
Hospitalists’ documentation doesn’t always capture everything they’re evaluating for patients. “I’ve seen notes to the extent of ‘patient doing well; waiting on test results,’” the AAPC’s Jimenez says. “If they’re doing certain tests, why are they doing them? What are they trying to diagnose for the patient? What treatment are they considering?”
The reasons for the tests need to be explained. When a provider is monitoring someone in the hospital, the documentation should elaborate on the patient’s response to a treatment, and whether the patient’s condition is better, stable, or worse. This information helps put the severity in perspective.
“A diabetic could be a diabetic out of control. It could be a diabetic who’s not responding or who has comorbidities,” Jimenez says. “No one diagnosis is the same for every patient.”
—Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist, department of medicine, University of Pennsylvania, Philadelphia
7 Indicate which aspect of the patient’s condition you are treating.
When multiple providers are involved in a hospitalized patient’s care, it’s important to document your specific role apart from the services rendered by specialists, Jimenez says. The codes billed must be supported by the documentation for each service. Many providers contribute to the inpatient documentation, so it must be clear what each clinician personally performs.
Only report the diagnosis you are treating or the diagnoses that affect the ones you are managing. If a specialist has been brought in to take over treatment for a specific condition, a hospitalist would not bill for that diagnosis code.
—Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education, AAPC Salt Lake City
8 Note your personal review of medical records and reports from other clinicians.
Hospitalists should document their review of lab data or radiology reports, discussion of the case with other providers, or collection of the history from someone other than the patient. It’s also helpful to document your personal review of any images, such as a chest X-ray or MRI. Examining the images yourself might lead to higher reimbursement, Mulholland says.
Providers also should note when they request or review old records, and they should include a short synopsis of the information obtained and how it contributed to the current treatment plan.
9 Learn the correct coding for patients being transferred.
A transfer can occur either from a different facility or from a hospital floor to a rehabilitation unit. Either way, the patient is seen twice in one day, with each visit covered by the same hospitalist practice.
“Both physicians often report a separate independent visit. However, because these services occurred on the same day, it is not appropriate to bill for two separate subsequent or initial hospital codes,” says Sherri Dumford, MBA, CHBME, director of operations and past president of the Healthcare Billing and Management Association. “Often what will happen is both services will be reported and get through the billing system. The second claim is just written off as a denied service, when, in fact, you could combine the elements of service of both visits and possibly bill for a single higher level of visit.”
10 Consider delegating to a coding expert.
While smaller hospitalist groups can turn to a coding consultant on an as-needed basis, larger groups might consider bringing a certified coder on staff. This person would inform physicians about proper coding, review their documentation, and “give real-time feedback,” Pierce says.
An internal audit would show if the documentation meets selected evaluation management codes. Also, it usually takes a coding professional to determine whether prolonged services are an option for the team on any given date of service. Someone would need to internally “add together” multiple services on one date to see if there is sufficient time documented to allow billing for these add-on codes, Pierce says. Similarly, critical-care time needs to be accumulated during a date of service.
—Barb Pierce, CCS-P, ACS-EM, national coding consultant, West Des Moines, Iowa
11 Indicate the number of minutes spent arranging for a patient’s discharge.
Discharging a patient involves various steps, says Peter Thompson, MD, chief of clinical operations at the Phoenix headquarters of Apogee Physicians, a hospitalist management company that employs about 750 hospitalists across the country. Hospitalists discuss the hospital stay with the patient and family members, prescribe medications, issue discharge recommendations, set up follow-up care, and coordinate with the case manager, specialists, and primary-care physician.
“It generally is one sequential event after the other,” lasting between 20 and 40 minutes and leading up to discharge, Thompson says. Reimbursement for a high-level discharge constitutes more than 30 minutes. However, without proper documentation, he cautions, the claim could be downgraded or denied.
12 Don’t forget to sign, date, and time your progress note.
Last but not least, when it comes to reimbursement, your signature really does matter.
“For an illegible signature, Medicare and the insurance companies have the option of not paying for the service,” Mulholland says. “They’re trying to establish or authenticate who provided the service.”
And they want to know when the hospitalist saw the patient, so it’s a good idea to indicate the exact time of your visit.
Susan Kreimer is a freelance medical writer in New York.
Documentation, CPT codes, modifiers—it’s not glamorous, but it’s an integral part of a 21st-century physician’s job description. The Hospitalist queried more than a handful of billing and coding experts about the advice they would dispense to clinicians navigating the reimbursement maze.
“Physicians often do more than what is reflected in the documentation,” says Barb Pierce, CCS-P, ACS-EM, a national coding consultant based in West Des Moines, Iowa, and CODE-H faculty. “They can’t always bill for everything they do, but they certainly can document and code to obtain the appropriate levels of service.”
Meanwhile, hospitalists have to be careful they aren’t excessive in their billing practices. “The name of the game isn’t just to bill higher,” Pierce adds, “but to make sure that your documentation supports the service being billed, and Medicare is watching. They’re doing a lot of focused audits.”
Some hospitalists might opt for a lower level of service, suspecting they’re less likely to be audited. Other hospitalists might seek reimbursement for more of their time and efforts.
“You have both ends of the spectrum,” says Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education for AAPC, formerly known as the American Academy of Professional Coders. “There are a lot of factors that would go into why a provider would code something incorrectly.”
Here’s how to land somewhere in the middle.
1 Be thorough in documenting the initial hospital visit.
When selecting the level of service for an initial hospital visit, the documentation consists of three key components: history, physical examination, and medical decision-making. The history includes the chief complaint as well as the review of systems. This is “an inventory of the patient’s organ systems.” Both the complaint and the systems review are often incorporated in the history of present illness, says Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist in the Department of Medicine at the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
A patient’s family history is commonly overlooked in a hospitalist’s notes, primarily when they know the patient from previous admissions for chronic diseases and when the family history will likely not have an impact on treatment. “If they do not document a complete review of systems or miss one of the histories, the service will definitely be down-coded,” Mulholland says, “no matter how complete the exam and medical decision-making documentation.”
2 Familiarize yourself with Medicare reimbursement rules in the state where you practice.
In some states, Medicare contractors require providers to document the status of each organ system reviewed individually. In other states, it’s acceptable to document a system review with pertinent findings, “whether positive or negative,” and the statement of “all other systems negative,” Mulholland says.
The auditor will give credit for the review based on the number of organ systems documented. “If you miss one system review, it will take down what otherwise would be a Level Three hospital admission to a Level One,” she says. “So there would be a significant financial impact.”
Medicare reimbursement for a Level Three initial visit in Mulholland’s area of practice—Philadelphia County in Pennsylvania—is $206.57, compared with $104.69 for a Level One. During this visit, each of the key components—history, exam, and medical decision-making—need to be documented completely for the provider to receive the highest level of reimbursement.
3 Ask about a patient’s social history.
Social history can be obtained by querying the patient about smoking, drug and alcohol use, his or her occupation, marital status, and type of living arrangement.
“Knowing the social history helps the hospitalist understand the home situation or social circumstances that may have contributed to the hospitalization or may complicate the discharge plan,” Mulholland says.
This is particularly important in decision-making that involves elderly patients. The clinician should “think down the road” as to where the patient will be discharged and if a social worker’s assistance will be needed. It’s about “seeing the whole patient,” she says, “not just the disease.”
4 Remember to include the actual diagnosis.
“As coders, we can see all the clinical indicators of a particular diagnosis,” says Kathryn DeVault, RHIA, CCS, CCS-P, a director at HIM Solutions at the American Health Management Association. However, “unless [physicians] write down the diagnosis, we can’t code it.”
Documents without a diagnosis are more common than one would expect. For example, if a patient has pain when urinating, the hospitalist typically orders a culture. If the result is positive, the hospitalist prescribes an antibiotic for the infection, and too often “the story ends there.” From experience, DeVault can decipher that the patient is being treated for a urinary tract infection, but she can’t assign a code without querying the physician. Hospitalists, she suggests, should try to “close the loop in their documentation.”
5 Be specific in your written assessment of the patient’s condition.
“The main thing that we see is missing documentation,” says Angie Comfort, RHIT, CCS, a director at HIM Solutions. For instance, if a hospitalist documents congestive heart failure, it’s important to indicate whether the condition is chronic or acute and systolic or diastolic.
In the case of a diabetic patient, the notes should specify the type of diabetes. Not doing so “could be a reimbursement-changer,” Comfort says. In contrast, documenting such specifics could result in higher reimbursement, especially if a patient has complications from Type 1 diabetes.
6 Note the severity of the patient’s case.
Hospitalists’ documentation doesn’t always capture everything they’re evaluating for patients. “I’ve seen notes to the extent of ‘patient doing well; waiting on test results,’” the AAPC’s Jimenez says. “If they’re doing certain tests, why are they doing them? What are they trying to diagnose for the patient? What treatment are they considering?”
The reasons for the tests need to be explained. When a provider is monitoring someone in the hospital, the documentation should elaborate on the patient’s response to a treatment, and whether the patient’s condition is better, stable, or worse. This information helps put the severity in perspective.
“A diabetic could be a diabetic out of control. It could be a diabetic who’s not responding or who has comorbidities,” Jimenez says. “No one diagnosis is the same for every patient.”
—Mary Mulholland, MHA, BSN, RN, CPC, senior coding and education specialist, department of medicine, University of Pennsylvania, Philadelphia
7 Indicate which aspect of the patient’s condition you are treating.
When multiple providers are involved in a hospitalized patient’s care, it’s important to document your specific role apart from the services rendered by specialists, Jimenez says. The codes billed must be supported by the documentation for each service. Many providers contribute to the inpatient documentation, so it must be clear what each clinician personally performs.
Only report the diagnosis you are treating or the diagnoses that affect the ones you are managing. If a specialist has been brought in to take over treatment for a specific condition, a hospitalist would not bill for that diagnosis code.
—Raemarie Jimenez, CPC, CPMA, CPC-I, CANPC, CRHC, director of education, AAPC Salt Lake City
8 Note your personal review of medical records and reports from other clinicians.
Hospitalists should document their review of lab data or radiology reports, discussion of the case with other providers, or collection of the history from someone other than the patient. It’s also helpful to document your personal review of any images, such as a chest X-ray or MRI. Examining the images yourself might lead to higher reimbursement, Mulholland says.
Providers also should note when they request or review old records, and they should include a short synopsis of the information obtained and how it contributed to the current treatment plan.
9 Learn the correct coding for patients being transferred.
A transfer can occur either from a different facility or from a hospital floor to a rehabilitation unit. Either way, the patient is seen twice in one day, with each visit covered by the same hospitalist practice.
“Both physicians often report a separate independent visit. However, because these services occurred on the same day, it is not appropriate to bill for two separate subsequent or initial hospital codes,” says Sherri Dumford, MBA, CHBME, director of operations and past president of the Healthcare Billing and Management Association. “Often what will happen is both services will be reported and get through the billing system. The second claim is just written off as a denied service, when, in fact, you could combine the elements of service of both visits and possibly bill for a single higher level of visit.”
10 Consider delegating to a coding expert.
While smaller hospitalist groups can turn to a coding consultant on an as-needed basis, larger groups might consider bringing a certified coder on staff. This person would inform physicians about proper coding, review their documentation, and “give real-time feedback,” Pierce says.
An internal audit would show if the documentation meets selected evaluation management codes. Also, it usually takes a coding professional to determine whether prolonged services are an option for the team on any given date of service. Someone would need to internally “add together” multiple services on one date to see if there is sufficient time documented to allow billing for these add-on codes, Pierce says. Similarly, critical-care time needs to be accumulated during a date of service.
—Barb Pierce, CCS-P, ACS-EM, national coding consultant, West Des Moines, Iowa
11 Indicate the number of minutes spent arranging for a patient’s discharge.
Discharging a patient involves various steps, says Peter Thompson, MD, chief of clinical operations at the Phoenix headquarters of Apogee Physicians, a hospitalist management company that employs about 750 hospitalists across the country. Hospitalists discuss the hospital stay with the patient and family members, prescribe medications, issue discharge recommendations, set up follow-up care, and coordinate with the case manager, specialists, and primary-care physician.
“It generally is one sequential event after the other,” lasting between 20 and 40 minutes and leading up to discharge, Thompson says. Reimbursement for a high-level discharge constitutes more than 30 minutes. However, without proper documentation, he cautions, the claim could be downgraded or denied.
12 Don’t forget to sign, date, and time your progress note.
Last but not least, when it comes to reimbursement, your signature really does matter.
“For an illegible signature, Medicare and the insurance companies have the option of not paying for the service,” Mulholland says. “They’re trying to establish or authenticate who provided the service.”
And they want to know when the hospitalist saw the patient, so it’s a good idea to indicate the exact time of your visit.
Susan Kreimer is a freelance medical writer in New York.