User login
Carpal tunnel syndrome—try these diagnostic maneuvers
• Before considering surgery, offer patients with mild-to-moderate carpal tunnel syndrome (CTS) a trial of conservative therapy such as splinting or corticosteroids. A
• Order electrodiagnostic studies (EDS) as needed, to rule out other conditions with a similar presentation, confirm an uncertain diagnosis, and gauge the severity of CTS C, or when surgery is being considered. B
• Recommend carpal tunnel release for patients who have severe CTS or have failed to respond to nonsurgical t0reatment. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane K, 52, comes to see you because of discomfort in her right wrist and tingling in her hand. The symptoms began 3 months ago, but have been getting progressively worse, and have started to interfere with her sleep. Ms. K often awakens with “pins and needles” in her hand, and says that she often has the urge to “shake it out.” Her sister has carpal tunnel syndrome (CTS), and Ms. K suspects that she does, too. On exam, you find that Ms. K has a positive Phalen’s and Durkan’s compression test, but normal Tinel’s test. She has normal strength and sensation in her hands. Her neck and upper extremity exam is otherwise unremarkable. You note that her hypothyroidism is well controlled, with a recent thyroid-stimulating hormone level of 1.2 mIU/L.
The patient has tried acetaminophen and ibuprofen, with little relief. She has researched CTS on the Internet and read about cold laser therapy, and wants to know whether you think it will work. What should you tell her?
Carpal tunnel syndrome is one of the most common disorders of the upper extremities and the most prevalent compression neuropathy.1 About 3% of US adults are affected, typically those between the ages of 40 and 60 years.2 Women are almost 3 times more likely than men to develop CTS.1
Other risk factors include diabetes, hypothyroidism, rheumatoid arthritis, pregnancy, obesity, family history, and trauma. A history of hand-related repetitive motions also increases the risk.3-5 Evidence does not support a definite link between keyboard or mouse use and CTS; however, occupations that require use of hand-operated vibratory tools or repeated and forceful movements of the hand/wrist (such as assembly work and food processing or packaging) are associated with CTS.6
The optimal diagnostic approach incorporates history and physical exam findings, including the results of a number of provocative maneuvers, as well as electrodiagnostic studies (EDS) in some cases.7 While surgery is the definitive treatment for CTS, numerous nonsurgical options exist, including splinting, corticosteroids, and a variety of alternative therapies, some of which (eg, chiropractic and cold laser therapy) have little evidence to support them.
Because family physicians are often the first to see patients with symptoms associated with CTS, you need to know what to look for, when to test, and whether to provide treatment or a referral. Here’s what to keep in mind.
Clinical presentation of CTS
Increased pressure in the carpal tunnel compresses the median nerve, leading to numbness, tingling, or pain in the palmar aspect of the first 3 fingers and the radial half of the fourth (FIGURE). Symptoms vary widely, with pain or numbness localized to the hand or wrist in some cases and pain radiating into the forearm or shoulder in others.
Figure
Compressed median nerve leads to numbness and tingling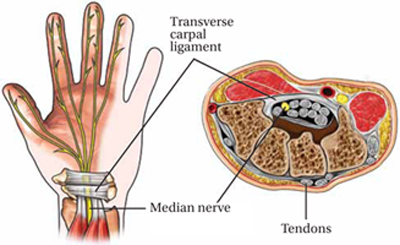
Early in the course of CTS, symptoms are often most bothersome at night. In a scenario like that reported by Ms. K, patients are often awakened by numbness or tingling and the desire to shake out the affected hand—a phenomenon known as the flick sign.8 Pain and numbness may occur intermittently at first, especially with repetitive wrist motion. Activities such as driving or holding a telephone often aggravate symptoms.
As CTS progresses, the intensity and duration of symptoms increase. Patients may complain of weakness in the hand and report that they often drop things. Paradoxically, patients with more severe CTS sometimes have less pain, rather than more, because of increasing sensory loss.9
Late in the course of CTS, physical exam findings typically include decreased sensation in the fingers innervated by the median nerve, sparing the thenar eminence. (A loss of sensation in the thenar eminence suggests the presence of a lesion proximal to the carpal tunnel, rather than CTS itself.10) In advanced cases, weakness of thumb abduction and opposition may occur, as well as atrophy of the thenar eminence.11
Sudden onset of severe symptoms with minimal trauma to the wrist should raise suspicion of a hematoma in the carpal tunnel—a particular risk for patients who have a clotting disorder or are being treated with newer anticoagulants such as dabigatran. Prompt surgical decompression is required to prevent permanent median nerve damage in such cases.12
Include these maneuvers in the physical exam
A thorough evaluation of the neck, shoulder, elbow, and wrist is crucial for all patients with signs and symptoms associated with CTS. Provocative maneuvers (TABLE 1)7,13 are also important as an aid to diagnosis. The results of the following tests should be viewed with caution, however, as studies have found wide variations in their sensitivity and specificity:
TABLE 1
Diagnosing carpal tunnel syndrome, using physical maneuvers7,13
| Test | Technique | Positive test | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|
| Phalen’s | Patient holds wrist flexed 90° with elbow in full extension | Pain or paresthesia ≤60 sec | 68 | 73 |
| Tinel’s | Clinician repetitively taps wrist over transverse carpal ligament | Pain or paresthesia | 50 | 77 |
| Median nerve compression* (MNC) | Clinician applies direct pressure over the transverse carpal ligament | Pain or paresthesia ≤30 sec | 64 | 83 |
| MNC + Phalen’s | Same as above | Same as above | 80 | 92 |
| *also known as Durkan’s test. | ||||
Phalen’s maneuver. The patient flexes his or her wrist with the elbow in full extension to increase pressure on the median nerve, and holds the position for 60 seconds. The onset of pain or paresthesia is a positive test. A meta-analysis found the sensitivity and specificity of a positive Phalen’s sign to be 68% and 73%, respectively.7
Tinel’s test. Tap the volar surface of the patient’s wrist just proximal to, or on top of, the carpal tunnel. Pain or paresthesia in the fingers innervated by the median nerve as a result of the percussion constitutes a positive result. Tinel’s test is less sensitive than the Phalen’s maneuver, but has a similar specificity.13
The median nerve (Durkan’s) compression test. Apply pressure over the transverse carpal ligament; the test is positive if pain or paresthesia develops within 30 seconds.7
The hand elevation test. The patient raises both hands overhead for 60 seconds; here, too, pain or paresthesia is a positive result.14
Combining results of provocative maneuvers may increase sensitivity and specificity. Positive results in both the Phalen’s and median nerve compression tests, for example, have a collective sensitivity and specificity of 80% and 92%, respectively.13
When (or whether) to order electrodiagnostic studies
While some clinicians consider EDS to be the gold standard in CTS diagnosis,6 evidence is limited. One issue is the lack of universally accepted reference standards; another is that most studies have been affected by “spectrum bias.”15 What’s more, EDS—which include nerve conduction studies (NCS) and electromyography (EMG)—do not always correlate directly with symptoms, and 16% to 34% of mild cases can be missed.16
EDS are useful in many instances, however. EMG can rule out other causes of CTS symptoms (TABLE 2 details the differential diagnosis),7,11 while NCS can aid in diagnosing CTS, gauging its severity, and arriving at a prognosis. Specifically, NCS can detect delayed distal latencies and slowed conduction velocities that can occur when the myelin sheath is damaged by prolonged compression of the median nerve.17 With more severe compression, axonal damage occurs, as evidenced by reduced action potential amplitudes on NCS. Results of the nerve conduction tests are compared to age-dependent normal values and to results from other nerves on either the same or the contralateral hand. In a 2002 systemic review, the sensitivity of NCS for CTS was 56% to 85% and the specificity was 94% to 99%.18
TABLE 2
Differential diagnosis for CTS7,11
| Condition | Characteristics |
|---|---|
| Carpometacarpal arthritis of thumb | Thumb is painful when in motion; radiographic findings |
| Cervical radiculopathy | Neck pain, nerve root distribution (eg, C6), positive Spurling’s test |
| DeQuervain’s tenosynovitis | Painful resisted thumb dorsiflexion, tender at base of thumb |
| Hypothyroidism | Fatigue, cold intolerance, dry skin, hair loss, abnormal thyroid function tests |
| Peripheral neuropathy | History of DM, lower extremity involvement |
| Pronator syndrome (median nerve compression at the elbow) | Tenderness at proximal forearm |
| Ulnar compressive neuropathy | Compression and positive Tinel’s sign: ulnar nerve at elbow or wrist produces pain or paresthesias in 4th and 5th fingers |
| Vibration white finger | History of use of power drill or other hand-held vibratory tool; symptoms of Raynaud’s syndrome |
| Wrist arthritis | Painful wrist ROM, radiographic findings |
| CTS, carpal tunnel syndrome; DM, diabetes mellitus; ROM, range of motion. | |
Before and after surgery. The American Academy of Orthopedic Surgeons (AAOS) recommends EDS when CTS surgery is being considered. 7 EDS may also be used after surgery, to verify neurologic improvement.
Ultrasound. In patients with CTS, ultrasound reveals an increased cross-sectional area of the median nerve, a finding that has prompted studies of this modality as a diagnostic tool.19 Although evidence suggests that ultrasound’s sensitivity and specificity for CTS would be similar to that of EDS, the optimal cutoff for an abnormal test has not been defined,19 and ultrasound does not provide information on prognosis or alternate causes.
Thus, AAOS does not currently recommend ultrasound for CTS diagnosis.7 Magnetic resonance imaging is inappropriate for routine CTS diagnosis, as well.7
Treatment: Start conservatively
Multiple nonsurgical options are available, but the best evidence supports splinting, steroid injection, and oral steroids. Splinting or steroids alone may bring long-term relief for patients with mild to moderate cases;20 in fact, about a third of mild cases improve spontaneously.21
Conservative therapy can also provide relief for those who wish to avoid or delay surgery and for cases of transient CTS (pediatric patients, for example, or those whose condition is associated with pregnancy or hypothyroidism).18 A successful response to therapy can also help to confirm a CTS diagnosis.
Most conservative treatments begin providing relief within 2 to 6 weeks and reach the maximal benefit at 3 months.22 If there is no response after 6 weeks, it’s time to consider another approach.
In initiating splinting or corticosteroids, here’s a look at what to keep in mind:
Splinting. A splint can be used to maintain the wrist in a position with the least intracanal pressure, thereby limiting pressure on the median nerve. Splinting is equally effective whether used continually or only at night.23
Splinting can relieve symptoms and improve functional status within 2 weeks and the effects can last for 3 to 6 months, eliminating the need for surgery for some patients with mild CTS.19,20 Nerve gliding exercises, (see image at left), have been evaluated in combination with splinting. While evidence is limited, an at-home program involving these simple exercises may be a beneficial adjunctive treatment with minimal cost or harm.24,25
Local corticosteroid injection. A Cochrane meta-analysis found significant improvement in symptoms and function at one month among patients with CTS who were treated with steroid injection.26 In many cases, the effects last for many months.
A recent trial found that nearly half of patients with mild to moderate CTS who were treated with steroid injections had improved symptoms and EDS results at the 12-month follow-up.20 However, while patients with severe CTS experienced improvement at 4 weeks postinjection, most eventually required surgery.20
Evidence does not support one particular steroid dose or formulation over another, or one particular injection site.22 Injecting 4 cm proximal to the wrist flexion crease is as effective as a more distal injection.26,27
Caution is required, however, as risks associated with local injections include tendon rupture and median nerve injury. If a patient experiences intense pain or paresthesia in the median nerve distribution when the needle is inserted, redirect the needle away from the median nerve immediately. For patients who respond well to this treatment, one additional injection can be given after 6 months if symptoms recur.
Oral corticosteroids. Oral prednisone at a dose of 20 mg/d for 2 weeks improves symptoms and function in patients with CTS, but is less effective than steroid injections.28 Treatment for 2 weeks is as effective as treatment for 4 weeks; the effects tend to wane after 8 weeks in both cases.29 Nonsteroidal anti-inflammatory drugs, diuretics, and vitamin B6 have not been found to be effective.30
CASE Ms. K also asks about “those needle tests”—a reference to EDS—which her sister had to diagnose her CTS. You explain that these studies are not necessary at this time because her symptoms are mild and there is no need for other causes to be ruled out.
Instead, you offer her a neutral wrist splint for night-time use and recommend home-based nerve glide exercises. There is no evidence that cold laser therapy is effective, you explain to Ms. K, and it is expensive. She agrees to try the splint and the exercises, and you schedule a follow-up visit in 6 weeks.
A look at alternative therapies
There are many nontraditional treatments for CTS, with yoga, carpal bone mobilization, ergonomic keyboards, and ultrasound therapy among them. Some have limited evidence to suggest that they may have a therapeutic effect;30 others have little or no evidence to support them.
Yoga. Stretching and improved joint posture with specific yoga exercises may lead to decreased compression within the carpal tunnel and increased blood flow to the median nerve. One small study found that yoga was more effective than nocturnal wrist splinting for pain relief, and had similar improvement for nocturnal symptoms and grip strength.31
Carpal bone mobilization. One small study found this physical therapy technique, which involves movement of the bones in the wrist, to improve symptoms such as numbness and tingling after 3 weeks of therapy. Yet carpal bone mobilization did not relieve pain or help restore function.32
Ergonomic keyboard. Patients who use computers at work may find that an ergonomic keyboard helps to relieve pain associated with CTS, compared with a standard keyboard.33
Therapeutic ultrasound. A recent meta-analysis found that there is only poor-quality evidence for ultrasound as an effective treatment for CTS—a process in which a round-headed instrument applied to the skin delivers sound waves that are absorbed by underlying tissues in the carpal tunnel. And there is insufficient evidence for one type of ultrasound over another, or to suggest that ultrasound is more effective than other nonsurgical treatments.34 Notably, ultrasound takes several weeks to provide a therapeutic benefit.
What about acupuncture? A recent trial found that acupuncture was no more effective than sham acupuncture in relieving symptoms of CTS in patients wearing wrist splints.35 Magnet therapy, chiropractic, and cold laser therapy are not supported by evidence either.28
Is the patient a candidate for surgery?
Carpal tunnel release provides good long-term outcomes for 70% to 90% of patients and is a cost-effective treatment.36,37 Evidence supports a trial of conservative therapy, however, before considering surgery for patients with mild-to-moderate CTS.22 Future studies are needed to identify prognostic characteristics of patients most likely to respond to each type of intervention, and the optimal timing for surgical release.
Patients with severe CTS—with findings such as thenar atrophy, diminished hand function, and median nerve denervation—should be referred for surgery without delay. This recommendation is based on expert opinion, however, as most clinical trials comparing surgical vs nonsurgical treatment exclude those with severe CTS.38
3 surgical techniques, and a novel approach
Surgical techniques include open, endoscopic, and minimal incision carpal tunnel release, with benefits and drawbacks for each. Compared with open release, for example, patients who undergo endoscopic release have less postoperative pain at 12 weeks, quicker return to work, and fewer wound complications, but are more likely to require surgical revision. And minimal incision release is associated with improved symptoms and function compared with open release.38 However, there is no long-term evidence that any one of these 3 surgical approaches is more effective than another.39
Percutaneous carpal tunnel release is a novel approach that may be offered in outpatient settings, with local anesthesia and ultrasound guidance to avoid median nerve damage.40 Because studies of the safety and efficacy of percutaneous carpal tunnel release are limited, however, this approach is considered experimental.41 Percutaneous release is not a treatment recommended by the AAOS.38
What to tell patients about postop care
Regardless of the method used for carpal tunnel release, most complications are minor—eg, a painful or hypertrophic scar, stiffness, swelling, and pain or tenderness on either side of the incision—and resolve within a few months.42 Advise patients not to continue to wear a wrist splint after surgery; doing so can cause stiffness or adhesions and may compromise surgical outcomes.41 Postoperatively, patients should be instructed to do nerve gliding exercises and to massage their scars, both of which they can safely do at home.43
Patients can expect significant symptomatic improvement within 1 week of surgery, and most will be able to return to normal activities in 2 weeks.44 Those with severe CTS should be warned, however, that it could take up to a year to determine the extent of recovery.22 Evidence suggests that from 3% to 19% of patients may have persistent or recurrent symptoms even after carpal tunnel release, with up to 12% requiring surgical revision.45
CASE When Ms. K returns, she reports that while there has been some improvement, some activities—such as driving long distances and talking on the phone—still cause numbness and tingling. And, if she doesn’t wear the splint at night, she awakens with tingling in her hands. You discuss 2 options—continued conservative treatment with a local steroid injection, or EDS and surgical referral. The patient opts for the injection and continued use of the nocturnal wrist splint and exercises. When she returns in another 6 weeks, Ms. K reports significant improvement. You agree to stop the wrist splint and exercises and advise her to follow-up on an as-needed basis if the symptoms return.
CORRESPONDENCE Jennifer Wipperman, MD, MPH, Via Christi Family Medicine, 1121 S. Clifton, Wichita, KS 67218; [email protected]
1. Atroshi I, Gummesson C, Johnsson, et al. Prevalence of carpal tunnel syndrome in a general population. JAMA. 1999;282:153-158.
2. Luckhaupt SE, Dahlhamer JM, Ward BW, et al. Prevalence and work-relatedness of carpal tunnel syndrome in the working population, United States, 2010 national health interview survey. Am J Ind Med. 2012 April 12. [Epub ahead of print.]
3. van Dijk MA, Reitsma JB, Fischer JC, et al. Indications for requesting laboratory tests for concurrent diseases in patients with carpal tunnel syndrome: a systematic review. Clin Chem. 2003;49:1437-1444.
4. Padua L, Di Pasquale A, Pazzaglia C, et al. Systematic review of pregnancy-related carpal tunnel syndrome. Muscle Nerve. 2010;42:697-702.
5. Bland JD. The relationship of obesity, age, and carpal tunnel syndrome: more complex than was thought? Muscle Nerve. 2005;32:527-532.
6. Prime MS, Palmer J, Khan WS, et al. Is there light at the end of the tunnel? Controversies in the diagnosis and management of carpal tunnel syndrome. Hand. 2010;5:354-360.
7. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons Clinical Practice Guideline on diagnosis of carpal tunnel syndrome. J Bone Joint Surg Am. 2009;91:2478-2479.
8. Hansen PA, Micklesen P, Robinson LR. Clinical utility of the flick maneuver in diagnosing carpal tunnel syndrome. Am J Phys Med Rehabil. 2004;83:363-367.
9. Padua L, Padua R, Aprile I, et al. Carpal tunnel syndrome: relationship between clinical and patient-oriented assessment. Clin Orthop Relat Res. 2002;395:128-134.
10. Bland JD. Carpal tunnel syndrome. BMJ. 2007;335:343-346.
11. Wright PE. Carpal tunnel syndrome. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, Pa: Mosby, Inc; 2008:4285–4291.
12. Sibley PA, Mandel RJ. Atraumatic acute carpal tunnel syndrome in a patient taking dabigatran. Orthopedics. 2012;35:e1286-e1289.
13. MacDermid JC, Wessel J. Clinical diagnosis of carpal tunnel syndrome: a systematic review. J Hand Ther. 2004;17:309-319.
14. Ahn DS. Hand elevation: a new test for carpal tunnel syndrome. Ann Plastic Surg. 2001;46:120-124.
15. Boyer K, Wies J, Turkelson CM. Effects of bias on the results of diagnostic studies of carpal tunnel syndrome. J Hand Surg. 2009;34:1006-1013.
16. Witt JC, Hentz JG, Stevens JC. Carpal tunnel syndrome with normal nerve conduction studies. Muscle Nerve. 2004;29:515-522.
17. Graham B. The value added by electrodiagnostic testing in the diagnosis of carpal tunnel syndrome. J Bone Joint Surg. 2008;90:2587-2593.
18. Jablecki CK, Andary MT, Floeter MK, et al. Practice parameter: electrodiagnostic studies in carpal tunnel syndrome. Report of the American Association of Electrodiagnostic Medicine, American Academy of Neurology, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2002;58:1589-1592.
19. Descatha A, Huard L, Aubert F, et al. Meta-analysis on the performance of sonography for the diagnosis of carpal tunnel syndrome. Semin Arthritis Rheum. 2012;41:914-922.
20. Visser LH, Ngo Q, Groeneweg SJ, et al. Long term effect of local corticosteroid injection for carpal tunnel syndrome: a relation with electrodiagnostic severity. Clin Neurophysiol. 2012;123:838-841.
21. Padua L, Padua R, Aprile I, et al. Multiperspective follow-up of untreated carpal tunnel syndrome: a multicenter study. Neurology. 2001;56:1459-1466.
22. Shi Q, MacDermid JC. Is surgical intervention more effective than non-surgical treatment for carpal tunnel syndrome? A systematic review. J Orthop Surg Res. 2011;6:17.-
23. Walker WC, Metzler M, Cifu DX, et al. Neutral wrist splinting in carpal tunnel syndrome: a comparison of night-only versus full-time wear instructions. Arch Phys Med Rehabil. 2000;81:424-429.
24. Brininger TL, Rogers JC, Holm MB, et al. Efficacy of a fabricated customized splint and tendon and nerve gliding exercises for the treatment of carpal tunnel syndrome: a randomized controlled trial. Arch Phys Med Rehabil. 2007;88:1429-1435.
25. Schmid AB, Elliott JM, Strudwick MW, et al. Effect of splinting and exercise on intraneural edema of the median nerve in carpal tunnel syndrome-an MRI study to reveal therapeutic mechanisms. J Orthop Res. 2012;30:1343-1350.
26. Marshall S, Tardif G, Ashworth N. Local corticosteroid injection for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(2):CD001554.-
27. Kamanli A, Bezgincan M, Kaya A. Comparison of local steroid injection into carpal tunnel via proximal and distal approach in patients with carpal tunnel syndrome. Bratislavske Lek Listy. 2011;112:337-341.
28. Huisstede BM, Hoogvliet P, Randsdorp MS, et al. Carpal tunnel syndrome. Part I: effectiveness of nonsurgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:981-1004.
29. Chang MH, Ger LP, Hsieh PF, et al. A randomised clinical trial of oral steroids in the treatment of carpal tunnel syndrome: a long term follow up. J Neurol Neurosurg Psychiatry. 2002;73:710-714.
30. O’Connor D, Marshall S, Massy-Westropp N. Non-surgical treatment (other than steroid injection) for carpal tunnel syndrome. Cochrane Database Syst Rev. 2003;(1):CD003219.-
31. Garfinkel MS, Singhal A, Katz WA, et al. Yoga-based intervention for carpal tunnel syndrome: a randomized trial. JAMA. 1998;280:1601-1603.
32. Tal-Akabi A, Rushton A. An investigation to compare the effectiveness of carpal bone mobilisation and neurodynamic mobilisation as methods of treatment for carpal tunnel syndrome. Man Ther. 2000;5:214-222.
33. O’Connor D, Page MJ, Marshall SC, et al. Ergonomic positioning or equipment for treating carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009600.-
34. Page MJ, O’Connor D, Pitt V, et al. Therapeutic ultrasound for carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009601.-
35. Yao E, Gerritz PK, Henricson E, et al. Randomized controlled trial comparing acupuncture with placebo acupuncture for the treatment of carpal tunnel syndrome. PMR. 2012;4:367-373.
36. Pomerance J, Zurakowski D, Fine I. The cost-effectiveness of nonsurgical versus surgical treatment for carpal tunnel syndrome. J Hand Surg. 2009;34:1193-1200.
37. Turner A, Kimble F, Gulyas K, et al. Can the outcome of open carpal tunnel release be predicted? A review of the literature. ANZ J Surg. 2010;80:50-54.
38. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of carpal tunnel syndrome. J Bone Joint Surg. 2010;92:218-219.
39. Scholten RJ, Mink van der Molen A, Uitdehaag BM, et al. Surgical treatment options for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(4):CD003905.-
40. Nakamichi K, Tachibana S, Yamamoto S, et al. Percutaneous carpal tunnel release compared with mini-open release using ultrasonographic guidance for both techniques. J Hand Surg Am. 2010;35:437-445.
41. Huisstede BM, Randsdorp MS, Coert JH, et al. Carpal tunnel syndrome. Part II: effectiveness of surgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:1005-1024.
42. Ludlow KS, Merla JL, Cox JA, et al. Pillar pain as a postoperative complication of carpal tunnel release: a review of the literature. J Hand Ther. 1997;10:277-282.
43. Pomerance J, Fine I. Outcomes of carpal tunnel surgery with and without supervised postoperative therapy. J Hand Surg. 2007;32:1159-1163.
44. Acharya AD, Auchincloss JM. Return to functional hand use and work following open carpal tunnel surgery. J Hand Surg Br. 2005;30:607-610.
45. Dahlin LB, Salo M, Thomsen N, et al. Carpal tunnel syndrome and treatment of recurrent symptoms. Scand J Plast Reconstr Surg Hand Surg. 2010;44:4-11.
• Before considering surgery, offer patients with mild-to-moderate carpal tunnel syndrome (CTS) a trial of conservative therapy such as splinting or corticosteroids. A
• Order electrodiagnostic studies (EDS) as needed, to rule out other conditions with a similar presentation, confirm an uncertain diagnosis, and gauge the severity of CTS C, or when surgery is being considered. B
• Recommend carpal tunnel release for patients who have severe CTS or have failed to respond to nonsurgical t0reatment. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane K, 52, comes to see you because of discomfort in her right wrist and tingling in her hand. The symptoms began 3 months ago, but have been getting progressively worse, and have started to interfere with her sleep. Ms. K often awakens with “pins and needles” in her hand, and says that she often has the urge to “shake it out.” Her sister has carpal tunnel syndrome (CTS), and Ms. K suspects that she does, too. On exam, you find that Ms. K has a positive Phalen’s and Durkan’s compression test, but normal Tinel’s test. She has normal strength and sensation in her hands. Her neck and upper extremity exam is otherwise unremarkable. You note that her hypothyroidism is well controlled, with a recent thyroid-stimulating hormone level of 1.2 mIU/L.
The patient has tried acetaminophen and ibuprofen, with little relief. She has researched CTS on the Internet and read about cold laser therapy, and wants to know whether you think it will work. What should you tell her?
Carpal tunnel syndrome is one of the most common disorders of the upper extremities and the most prevalent compression neuropathy.1 About 3% of US adults are affected, typically those between the ages of 40 and 60 years.2 Women are almost 3 times more likely than men to develop CTS.1
Other risk factors include diabetes, hypothyroidism, rheumatoid arthritis, pregnancy, obesity, family history, and trauma. A history of hand-related repetitive motions also increases the risk.3-5 Evidence does not support a definite link between keyboard or mouse use and CTS; however, occupations that require use of hand-operated vibratory tools or repeated and forceful movements of the hand/wrist (such as assembly work and food processing or packaging) are associated with CTS.6
The optimal diagnostic approach incorporates history and physical exam findings, including the results of a number of provocative maneuvers, as well as electrodiagnostic studies (EDS) in some cases.7 While surgery is the definitive treatment for CTS, numerous nonsurgical options exist, including splinting, corticosteroids, and a variety of alternative therapies, some of which (eg, chiropractic and cold laser therapy) have little evidence to support them.
Because family physicians are often the first to see patients with symptoms associated with CTS, you need to know what to look for, when to test, and whether to provide treatment or a referral. Here’s what to keep in mind.
Clinical presentation of CTS
Increased pressure in the carpal tunnel compresses the median nerve, leading to numbness, tingling, or pain in the palmar aspect of the first 3 fingers and the radial half of the fourth (FIGURE). Symptoms vary widely, with pain or numbness localized to the hand or wrist in some cases and pain radiating into the forearm or shoulder in others.
Figure
Compressed median nerve leads to numbness and tingling
Early in the course of CTS, symptoms are often most bothersome at night. In a scenario like that reported by Ms. K, patients are often awakened by numbness or tingling and the desire to shake out the affected hand—a phenomenon known as the flick sign.8 Pain and numbness may occur intermittently at first, especially with repetitive wrist motion. Activities such as driving or holding a telephone often aggravate symptoms.
As CTS progresses, the intensity and duration of symptoms increase. Patients may complain of weakness in the hand and report that they often drop things. Paradoxically, patients with more severe CTS sometimes have less pain, rather than more, because of increasing sensory loss.9
Late in the course of CTS, physical exam findings typically include decreased sensation in the fingers innervated by the median nerve, sparing the thenar eminence. (A loss of sensation in the thenar eminence suggests the presence of a lesion proximal to the carpal tunnel, rather than CTS itself.10) In advanced cases, weakness of thumb abduction and opposition may occur, as well as atrophy of the thenar eminence.11
Sudden onset of severe symptoms with minimal trauma to the wrist should raise suspicion of a hematoma in the carpal tunnel—a particular risk for patients who have a clotting disorder or are being treated with newer anticoagulants such as dabigatran. Prompt surgical decompression is required to prevent permanent median nerve damage in such cases.12
Include these maneuvers in the physical exam
A thorough evaluation of the neck, shoulder, elbow, and wrist is crucial for all patients with signs and symptoms associated with CTS. Provocative maneuvers (TABLE 1)7,13 are also important as an aid to diagnosis. The results of the following tests should be viewed with caution, however, as studies have found wide variations in their sensitivity and specificity:
TABLE 1
Diagnosing carpal tunnel syndrome, using physical maneuvers7,13
| Test | Technique | Positive test | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|
| Phalen’s | Patient holds wrist flexed 90° with elbow in full extension | Pain or paresthesia ≤60 sec | 68 | 73 |
| Tinel’s | Clinician repetitively taps wrist over transverse carpal ligament | Pain or paresthesia | 50 | 77 |
| Median nerve compression* (MNC) | Clinician applies direct pressure over the transverse carpal ligament | Pain or paresthesia ≤30 sec | 64 | 83 |
| MNC + Phalen’s | Same as above | Same as above | 80 | 92 |
| *also known as Durkan’s test. | ||||
Phalen’s maneuver. The patient flexes his or her wrist with the elbow in full extension to increase pressure on the median nerve, and holds the position for 60 seconds. The onset of pain or paresthesia is a positive test. A meta-analysis found the sensitivity and specificity of a positive Phalen’s sign to be 68% and 73%, respectively.7
Tinel’s test. Tap the volar surface of the patient’s wrist just proximal to, or on top of, the carpal tunnel. Pain or paresthesia in the fingers innervated by the median nerve as a result of the percussion constitutes a positive result. Tinel’s test is less sensitive than the Phalen’s maneuver, but has a similar specificity.13
The median nerve (Durkan’s) compression test. Apply pressure over the transverse carpal ligament; the test is positive if pain or paresthesia develops within 30 seconds.7
The hand elevation test. The patient raises both hands overhead for 60 seconds; here, too, pain or paresthesia is a positive result.14
Combining results of provocative maneuvers may increase sensitivity and specificity. Positive results in both the Phalen’s and median nerve compression tests, for example, have a collective sensitivity and specificity of 80% and 92%, respectively.13
When (or whether) to order electrodiagnostic studies
While some clinicians consider EDS to be the gold standard in CTS diagnosis,6 evidence is limited. One issue is the lack of universally accepted reference standards; another is that most studies have been affected by “spectrum bias.”15 What’s more, EDS—which include nerve conduction studies (NCS) and electromyography (EMG)—do not always correlate directly with symptoms, and 16% to 34% of mild cases can be missed.16
EDS are useful in many instances, however. EMG can rule out other causes of CTS symptoms (TABLE 2 details the differential diagnosis),7,11 while NCS can aid in diagnosing CTS, gauging its severity, and arriving at a prognosis. Specifically, NCS can detect delayed distal latencies and slowed conduction velocities that can occur when the myelin sheath is damaged by prolonged compression of the median nerve.17 With more severe compression, axonal damage occurs, as evidenced by reduced action potential amplitudes on NCS. Results of the nerve conduction tests are compared to age-dependent normal values and to results from other nerves on either the same or the contralateral hand. In a 2002 systemic review, the sensitivity of NCS for CTS was 56% to 85% and the specificity was 94% to 99%.18
TABLE 2
Differential diagnosis for CTS7,11
| Condition | Characteristics |
|---|---|
| Carpometacarpal arthritis of thumb | Thumb is painful when in motion; radiographic findings |
| Cervical radiculopathy | Neck pain, nerve root distribution (eg, C6), positive Spurling’s test |
| DeQuervain’s tenosynovitis | Painful resisted thumb dorsiflexion, tender at base of thumb |
| Hypothyroidism | Fatigue, cold intolerance, dry skin, hair loss, abnormal thyroid function tests |
| Peripheral neuropathy | History of DM, lower extremity involvement |
| Pronator syndrome (median nerve compression at the elbow) | Tenderness at proximal forearm |
| Ulnar compressive neuropathy | Compression and positive Tinel’s sign: ulnar nerve at elbow or wrist produces pain or paresthesias in 4th and 5th fingers |
| Vibration white finger | History of use of power drill or other hand-held vibratory tool; symptoms of Raynaud’s syndrome |
| Wrist arthritis | Painful wrist ROM, radiographic findings |
| CTS, carpal tunnel syndrome; DM, diabetes mellitus; ROM, range of motion. | |
Before and after surgery. The American Academy of Orthopedic Surgeons (AAOS) recommends EDS when CTS surgery is being considered. 7 EDS may also be used after surgery, to verify neurologic improvement.
Ultrasound. In patients with CTS, ultrasound reveals an increased cross-sectional area of the median nerve, a finding that has prompted studies of this modality as a diagnostic tool.19 Although evidence suggests that ultrasound’s sensitivity and specificity for CTS would be similar to that of EDS, the optimal cutoff for an abnormal test has not been defined,19 and ultrasound does not provide information on prognosis or alternate causes.
Thus, AAOS does not currently recommend ultrasound for CTS diagnosis.7 Magnetic resonance imaging is inappropriate for routine CTS diagnosis, as well.7
Treatment: Start conservatively
Multiple nonsurgical options are available, but the best evidence supports splinting, steroid injection, and oral steroids. Splinting or steroids alone may bring long-term relief for patients with mild to moderate cases;20 in fact, about a third of mild cases improve spontaneously.21
Conservative therapy can also provide relief for those who wish to avoid or delay surgery and for cases of transient CTS (pediatric patients, for example, or those whose condition is associated with pregnancy or hypothyroidism).18 A successful response to therapy can also help to confirm a CTS diagnosis.
Most conservative treatments begin providing relief within 2 to 6 weeks and reach the maximal benefit at 3 months.22 If there is no response after 6 weeks, it’s time to consider another approach.
In initiating splinting or corticosteroids, here’s a look at what to keep in mind:
Splinting. A splint can be used to maintain the wrist in a position with the least intracanal pressure, thereby limiting pressure on the median nerve. Splinting is equally effective whether used continually or only at night.23
Splinting can relieve symptoms and improve functional status within 2 weeks and the effects can last for 3 to 6 months, eliminating the need for surgery for some patients with mild CTS.19,20 Nerve gliding exercises, (see image at left), have been evaluated in combination with splinting. While evidence is limited, an at-home program involving these simple exercises may be a beneficial adjunctive treatment with minimal cost or harm.24,25
Local corticosteroid injection. A Cochrane meta-analysis found significant improvement in symptoms and function at one month among patients with CTS who were treated with steroid injection.26 In many cases, the effects last for many months.
A recent trial found that nearly half of patients with mild to moderate CTS who were treated with steroid injections had improved symptoms and EDS results at the 12-month follow-up.20 However, while patients with severe CTS experienced improvement at 4 weeks postinjection, most eventually required surgery.20
Evidence does not support one particular steroid dose or formulation over another, or one particular injection site.22 Injecting 4 cm proximal to the wrist flexion crease is as effective as a more distal injection.26,27
Caution is required, however, as risks associated with local injections include tendon rupture and median nerve injury. If a patient experiences intense pain or paresthesia in the median nerve distribution when the needle is inserted, redirect the needle away from the median nerve immediately. For patients who respond well to this treatment, one additional injection can be given after 6 months if symptoms recur.
Oral corticosteroids. Oral prednisone at a dose of 20 mg/d for 2 weeks improves symptoms and function in patients with CTS, but is less effective than steroid injections.28 Treatment for 2 weeks is as effective as treatment for 4 weeks; the effects tend to wane after 8 weeks in both cases.29 Nonsteroidal anti-inflammatory drugs, diuretics, and vitamin B6 have not been found to be effective.30
CASE Ms. K also asks about “those needle tests”—a reference to EDS—which her sister had to diagnose her CTS. You explain that these studies are not necessary at this time because her symptoms are mild and there is no need for other causes to be ruled out.
Instead, you offer her a neutral wrist splint for night-time use and recommend home-based nerve glide exercises. There is no evidence that cold laser therapy is effective, you explain to Ms. K, and it is expensive. She agrees to try the splint and the exercises, and you schedule a follow-up visit in 6 weeks.
A look at alternative therapies
There are many nontraditional treatments for CTS, with yoga, carpal bone mobilization, ergonomic keyboards, and ultrasound therapy among them. Some have limited evidence to suggest that they may have a therapeutic effect;30 others have little or no evidence to support them.
Yoga. Stretching and improved joint posture with specific yoga exercises may lead to decreased compression within the carpal tunnel and increased blood flow to the median nerve. One small study found that yoga was more effective than nocturnal wrist splinting for pain relief, and had similar improvement for nocturnal symptoms and grip strength.31
Carpal bone mobilization. One small study found this physical therapy technique, which involves movement of the bones in the wrist, to improve symptoms such as numbness and tingling after 3 weeks of therapy. Yet carpal bone mobilization did not relieve pain or help restore function.32
Ergonomic keyboard. Patients who use computers at work may find that an ergonomic keyboard helps to relieve pain associated with CTS, compared with a standard keyboard.33
Therapeutic ultrasound. A recent meta-analysis found that there is only poor-quality evidence for ultrasound as an effective treatment for CTS—a process in which a round-headed instrument applied to the skin delivers sound waves that are absorbed by underlying tissues in the carpal tunnel. And there is insufficient evidence for one type of ultrasound over another, or to suggest that ultrasound is more effective than other nonsurgical treatments.34 Notably, ultrasound takes several weeks to provide a therapeutic benefit.
What about acupuncture? A recent trial found that acupuncture was no more effective than sham acupuncture in relieving symptoms of CTS in patients wearing wrist splints.35 Magnet therapy, chiropractic, and cold laser therapy are not supported by evidence either.28
Is the patient a candidate for surgery?
Carpal tunnel release provides good long-term outcomes for 70% to 90% of patients and is a cost-effective treatment.36,37 Evidence supports a trial of conservative therapy, however, before considering surgery for patients with mild-to-moderate CTS.22 Future studies are needed to identify prognostic characteristics of patients most likely to respond to each type of intervention, and the optimal timing for surgical release.
Patients with severe CTS—with findings such as thenar atrophy, diminished hand function, and median nerve denervation—should be referred for surgery without delay. This recommendation is based on expert opinion, however, as most clinical trials comparing surgical vs nonsurgical treatment exclude those with severe CTS.38
3 surgical techniques, and a novel approach
Surgical techniques include open, endoscopic, and minimal incision carpal tunnel release, with benefits and drawbacks for each. Compared with open release, for example, patients who undergo endoscopic release have less postoperative pain at 12 weeks, quicker return to work, and fewer wound complications, but are more likely to require surgical revision. And minimal incision release is associated with improved symptoms and function compared with open release.38 However, there is no long-term evidence that any one of these 3 surgical approaches is more effective than another.39
Percutaneous carpal tunnel release is a novel approach that may be offered in outpatient settings, with local anesthesia and ultrasound guidance to avoid median nerve damage.40 Because studies of the safety and efficacy of percutaneous carpal tunnel release are limited, however, this approach is considered experimental.41 Percutaneous release is not a treatment recommended by the AAOS.38
What to tell patients about postop care
Regardless of the method used for carpal tunnel release, most complications are minor—eg, a painful or hypertrophic scar, stiffness, swelling, and pain or tenderness on either side of the incision—and resolve within a few months.42 Advise patients not to continue to wear a wrist splint after surgery; doing so can cause stiffness or adhesions and may compromise surgical outcomes.41 Postoperatively, patients should be instructed to do nerve gliding exercises and to massage their scars, both of which they can safely do at home.43
Patients can expect significant symptomatic improvement within 1 week of surgery, and most will be able to return to normal activities in 2 weeks.44 Those with severe CTS should be warned, however, that it could take up to a year to determine the extent of recovery.22 Evidence suggests that from 3% to 19% of patients may have persistent or recurrent symptoms even after carpal tunnel release, with up to 12% requiring surgical revision.45
CASE When Ms. K returns, she reports that while there has been some improvement, some activities—such as driving long distances and talking on the phone—still cause numbness and tingling. And, if she doesn’t wear the splint at night, she awakens with tingling in her hands. You discuss 2 options—continued conservative treatment with a local steroid injection, or EDS and surgical referral. The patient opts for the injection and continued use of the nocturnal wrist splint and exercises. When she returns in another 6 weeks, Ms. K reports significant improvement. You agree to stop the wrist splint and exercises and advise her to follow-up on an as-needed basis if the symptoms return.
CORRESPONDENCE Jennifer Wipperman, MD, MPH, Via Christi Family Medicine, 1121 S. Clifton, Wichita, KS 67218; [email protected]
• Before considering surgery, offer patients with mild-to-moderate carpal tunnel syndrome (CTS) a trial of conservative therapy such as splinting or corticosteroids. A
• Order electrodiagnostic studies (EDS) as needed, to rule out other conditions with a similar presentation, confirm an uncertain diagnosis, and gauge the severity of CTS C, or when surgery is being considered. B
• Recommend carpal tunnel release for patients who have severe CTS or have failed to respond to nonsurgical t0reatment. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Jane K, 52, comes to see you because of discomfort in her right wrist and tingling in her hand. The symptoms began 3 months ago, but have been getting progressively worse, and have started to interfere with her sleep. Ms. K often awakens with “pins and needles” in her hand, and says that she often has the urge to “shake it out.” Her sister has carpal tunnel syndrome (CTS), and Ms. K suspects that she does, too. On exam, you find that Ms. K has a positive Phalen’s and Durkan’s compression test, but normal Tinel’s test. She has normal strength and sensation in her hands. Her neck and upper extremity exam is otherwise unremarkable. You note that her hypothyroidism is well controlled, with a recent thyroid-stimulating hormone level of 1.2 mIU/L.
The patient has tried acetaminophen and ibuprofen, with little relief. She has researched CTS on the Internet and read about cold laser therapy, and wants to know whether you think it will work. What should you tell her?
Carpal tunnel syndrome is one of the most common disorders of the upper extremities and the most prevalent compression neuropathy.1 About 3% of US adults are affected, typically those between the ages of 40 and 60 years.2 Women are almost 3 times more likely than men to develop CTS.1
Other risk factors include diabetes, hypothyroidism, rheumatoid arthritis, pregnancy, obesity, family history, and trauma. A history of hand-related repetitive motions also increases the risk.3-5 Evidence does not support a definite link between keyboard or mouse use and CTS; however, occupations that require use of hand-operated vibratory tools or repeated and forceful movements of the hand/wrist (such as assembly work and food processing or packaging) are associated with CTS.6
The optimal diagnostic approach incorporates history and physical exam findings, including the results of a number of provocative maneuvers, as well as electrodiagnostic studies (EDS) in some cases.7 While surgery is the definitive treatment for CTS, numerous nonsurgical options exist, including splinting, corticosteroids, and a variety of alternative therapies, some of which (eg, chiropractic and cold laser therapy) have little evidence to support them.
Because family physicians are often the first to see patients with symptoms associated with CTS, you need to know what to look for, when to test, and whether to provide treatment or a referral. Here’s what to keep in mind.
Clinical presentation of CTS
Increased pressure in the carpal tunnel compresses the median nerve, leading to numbness, tingling, or pain in the palmar aspect of the first 3 fingers and the radial half of the fourth (FIGURE). Symptoms vary widely, with pain or numbness localized to the hand or wrist in some cases and pain radiating into the forearm or shoulder in others.
Figure
Compressed median nerve leads to numbness and tingling
Early in the course of CTS, symptoms are often most bothersome at night. In a scenario like that reported by Ms. K, patients are often awakened by numbness or tingling and the desire to shake out the affected hand—a phenomenon known as the flick sign.8 Pain and numbness may occur intermittently at first, especially with repetitive wrist motion. Activities such as driving or holding a telephone often aggravate symptoms.
As CTS progresses, the intensity and duration of symptoms increase. Patients may complain of weakness in the hand and report that they often drop things. Paradoxically, patients with more severe CTS sometimes have less pain, rather than more, because of increasing sensory loss.9
Late in the course of CTS, physical exam findings typically include decreased sensation in the fingers innervated by the median nerve, sparing the thenar eminence. (A loss of sensation in the thenar eminence suggests the presence of a lesion proximal to the carpal tunnel, rather than CTS itself.10) In advanced cases, weakness of thumb abduction and opposition may occur, as well as atrophy of the thenar eminence.11
Sudden onset of severe symptoms with minimal trauma to the wrist should raise suspicion of a hematoma in the carpal tunnel—a particular risk for patients who have a clotting disorder or are being treated with newer anticoagulants such as dabigatran. Prompt surgical decompression is required to prevent permanent median nerve damage in such cases.12
Include these maneuvers in the physical exam
A thorough evaluation of the neck, shoulder, elbow, and wrist is crucial for all patients with signs and symptoms associated with CTS. Provocative maneuvers (TABLE 1)7,13 are also important as an aid to diagnosis. The results of the following tests should be viewed with caution, however, as studies have found wide variations in their sensitivity and specificity:
TABLE 1
Diagnosing carpal tunnel syndrome, using physical maneuvers7,13
| Test | Technique | Positive test | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|
| Phalen’s | Patient holds wrist flexed 90° with elbow in full extension | Pain or paresthesia ≤60 sec | 68 | 73 |
| Tinel’s | Clinician repetitively taps wrist over transverse carpal ligament | Pain or paresthesia | 50 | 77 |
| Median nerve compression* (MNC) | Clinician applies direct pressure over the transverse carpal ligament | Pain or paresthesia ≤30 sec | 64 | 83 |
| MNC + Phalen’s | Same as above | Same as above | 80 | 92 |
| *also known as Durkan’s test. | ||||
Phalen’s maneuver. The patient flexes his or her wrist with the elbow in full extension to increase pressure on the median nerve, and holds the position for 60 seconds. The onset of pain or paresthesia is a positive test. A meta-analysis found the sensitivity and specificity of a positive Phalen’s sign to be 68% and 73%, respectively.7
Tinel’s test. Tap the volar surface of the patient’s wrist just proximal to, or on top of, the carpal tunnel. Pain or paresthesia in the fingers innervated by the median nerve as a result of the percussion constitutes a positive result. Tinel’s test is less sensitive than the Phalen’s maneuver, but has a similar specificity.13
The median nerve (Durkan’s) compression test. Apply pressure over the transverse carpal ligament; the test is positive if pain or paresthesia develops within 30 seconds.7
The hand elevation test. The patient raises both hands overhead for 60 seconds; here, too, pain or paresthesia is a positive result.14
Combining results of provocative maneuvers may increase sensitivity and specificity. Positive results in both the Phalen’s and median nerve compression tests, for example, have a collective sensitivity and specificity of 80% and 92%, respectively.13
When (or whether) to order electrodiagnostic studies
While some clinicians consider EDS to be the gold standard in CTS diagnosis,6 evidence is limited. One issue is the lack of universally accepted reference standards; another is that most studies have been affected by “spectrum bias.”15 What’s more, EDS—which include nerve conduction studies (NCS) and electromyography (EMG)—do not always correlate directly with symptoms, and 16% to 34% of mild cases can be missed.16
EDS are useful in many instances, however. EMG can rule out other causes of CTS symptoms (TABLE 2 details the differential diagnosis),7,11 while NCS can aid in diagnosing CTS, gauging its severity, and arriving at a prognosis. Specifically, NCS can detect delayed distal latencies and slowed conduction velocities that can occur when the myelin sheath is damaged by prolonged compression of the median nerve.17 With more severe compression, axonal damage occurs, as evidenced by reduced action potential amplitudes on NCS. Results of the nerve conduction tests are compared to age-dependent normal values and to results from other nerves on either the same or the contralateral hand. In a 2002 systemic review, the sensitivity of NCS for CTS was 56% to 85% and the specificity was 94% to 99%.18
TABLE 2
Differential diagnosis for CTS7,11
| Condition | Characteristics |
|---|---|
| Carpometacarpal arthritis of thumb | Thumb is painful when in motion; radiographic findings |
| Cervical radiculopathy | Neck pain, nerve root distribution (eg, C6), positive Spurling’s test |
| DeQuervain’s tenosynovitis | Painful resisted thumb dorsiflexion, tender at base of thumb |
| Hypothyroidism | Fatigue, cold intolerance, dry skin, hair loss, abnormal thyroid function tests |
| Peripheral neuropathy | History of DM, lower extremity involvement |
| Pronator syndrome (median nerve compression at the elbow) | Tenderness at proximal forearm |
| Ulnar compressive neuropathy | Compression and positive Tinel’s sign: ulnar nerve at elbow or wrist produces pain or paresthesias in 4th and 5th fingers |
| Vibration white finger | History of use of power drill or other hand-held vibratory tool; symptoms of Raynaud’s syndrome |
| Wrist arthritis | Painful wrist ROM, radiographic findings |
| CTS, carpal tunnel syndrome; DM, diabetes mellitus; ROM, range of motion. | |
Before and after surgery. The American Academy of Orthopedic Surgeons (AAOS) recommends EDS when CTS surgery is being considered. 7 EDS may also be used after surgery, to verify neurologic improvement.
Ultrasound. In patients with CTS, ultrasound reveals an increased cross-sectional area of the median nerve, a finding that has prompted studies of this modality as a diagnostic tool.19 Although evidence suggests that ultrasound’s sensitivity and specificity for CTS would be similar to that of EDS, the optimal cutoff for an abnormal test has not been defined,19 and ultrasound does not provide information on prognosis or alternate causes.
Thus, AAOS does not currently recommend ultrasound for CTS diagnosis.7 Magnetic resonance imaging is inappropriate for routine CTS diagnosis, as well.7
Treatment: Start conservatively
Multiple nonsurgical options are available, but the best evidence supports splinting, steroid injection, and oral steroids. Splinting or steroids alone may bring long-term relief for patients with mild to moderate cases;20 in fact, about a third of mild cases improve spontaneously.21
Conservative therapy can also provide relief for those who wish to avoid or delay surgery and for cases of transient CTS (pediatric patients, for example, or those whose condition is associated with pregnancy or hypothyroidism).18 A successful response to therapy can also help to confirm a CTS diagnosis.
Most conservative treatments begin providing relief within 2 to 6 weeks and reach the maximal benefit at 3 months.22 If there is no response after 6 weeks, it’s time to consider another approach.
In initiating splinting or corticosteroids, here’s a look at what to keep in mind:
Splinting. A splint can be used to maintain the wrist in a position with the least intracanal pressure, thereby limiting pressure on the median nerve. Splinting is equally effective whether used continually or only at night.23
Splinting can relieve symptoms and improve functional status within 2 weeks and the effects can last for 3 to 6 months, eliminating the need for surgery for some patients with mild CTS.19,20 Nerve gliding exercises, (see image at left), have been evaluated in combination with splinting. While evidence is limited, an at-home program involving these simple exercises may be a beneficial adjunctive treatment with minimal cost or harm.24,25
Local corticosteroid injection. A Cochrane meta-analysis found significant improvement in symptoms and function at one month among patients with CTS who were treated with steroid injection.26 In many cases, the effects last for many months.
A recent trial found that nearly half of patients with mild to moderate CTS who were treated with steroid injections had improved symptoms and EDS results at the 12-month follow-up.20 However, while patients with severe CTS experienced improvement at 4 weeks postinjection, most eventually required surgery.20
Evidence does not support one particular steroid dose or formulation over another, or one particular injection site.22 Injecting 4 cm proximal to the wrist flexion crease is as effective as a more distal injection.26,27
Caution is required, however, as risks associated with local injections include tendon rupture and median nerve injury. If a patient experiences intense pain or paresthesia in the median nerve distribution when the needle is inserted, redirect the needle away from the median nerve immediately. For patients who respond well to this treatment, one additional injection can be given after 6 months if symptoms recur.
Oral corticosteroids. Oral prednisone at a dose of 20 mg/d for 2 weeks improves symptoms and function in patients with CTS, but is less effective than steroid injections.28 Treatment for 2 weeks is as effective as treatment for 4 weeks; the effects tend to wane after 8 weeks in both cases.29 Nonsteroidal anti-inflammatory drugs, diuretics, and vitamin B6 have not been found to be effective.30
CASE Ms. K also asks about “those needle tests”—a reference to EDS—which her sister had to diagnose her CTS. You explain that these studies are not necessary at this time because her symptoms are mild and there is no need for other causes to be ruled out.
Instead, you offer her a neutral wrist splint for night-time use and recommend home-based nerve glide exercises. There is no evidence that cold laser therapy is effective, you explain to Ms. K, and it is expensive. She agrees to try the splint and the exercises, and you schedule a follow-up visit in 6 weeks.
A look at alternative therapies
There are many nontraditional treatments for CTS, with yoga, carpal bone mobilization, ergonomic keyboards, and ultrasound therapy among them. Some have limited evidence to suggest that they may have a therapeutic effect;30 others have little or no evidence to support them.
Yoga. Stretching and improved joint posture with specific yoga exercises may lead to decreased compression within the carpal tunnel and increased blood flow to the median nerve. One small study found that yoga was more effective than nocturnal wrist splinting for pain relief, and had similar improvement for nocturnal symptoms and grip strength.31
Carpal bone mobilization. One small study found this physical therapy technique, which involves movement of the bones in the wrist, to improve symptoms such as numbness and tingling after 3 weeks of therapy. Yet carpal bone mobilization did not relieve pain or help restore function.32
Ergonomic keyboard. Patients who use computers at work may find that an ergonomic keyboard helps to relieve pain associated with CTS, compared with a standard keyboard.33
Therapeutic ultrasound. A recent meta-analysis found that there is only poor-quality evidence for ultrasound as an effective treatment for CTS—a process in which a round-headed instrument applied to the skin delivers sound waves that are absorbed by underlying tissues in the carpal tunnel. And there is insufficient evidence for one type of ultrasound over another, or to suggest that ultrasound is more effective than other nonsurgical treatments.34 Notably, ultrasound takes several weeks to provide a therapeutic benefit.
What about acupuncture? A recent trial found that acupuncture was no more effective than sham acupuncture in relieving symptoms of CTS in patients wearing wrist splints.35 Magnet therapy, chiropractic, and cold laser therapy are not supported by evidence either.28
Is the patient a candidate for surgery?
Carpal tunnel release provides good long-term outcomes for 70% to 90% of patients and is a cost-effective treatment.36,37 Evidence supports a trial of conservative therapy, however, before considering surgery for patients with mild-to-moderate CTS.22 Future studies are needed to identify prognostic characteristics of patients most likely to respond to each type of intervention, and the optimal timing for surgical release.
Patients with severe CTS—with findings such as thenar atrophy, diminished hand function, and median nerve denervation—should be referred for surgery without delay. This recommendation is based on expert opinion, however, as most clinical trials comparing surgical vs nonsurgical treatment exclude those with severe CTS.38
3 surgical techniques, and a novel approach
Surgical techniques include open, endoscopic, and minimal incision carpal tunnel release, with benefits and drawbacks for each. Compared with open release, for example, patients who undergo endoscopic release have less postoperative pain at 12 weeks, quicker return to work, and fewer wound complications, but are more likely to require surgical revision. And minimal incision release is associated with improved symptoms and function compared with open release.38 However, there is no long-term evidence that any one of these 3 surgical approaches is more effective than another.39
Percutaneous carpal tunnel release is a novel approach that may be offered in outpatient settings, with local anesthesia and ultrasound guidance to avoid median nerve damage.40 Because studies of the safety and efficacy of percutaneous carpal tunnel release are limited, however, this approach is considered experimental.41 Percutaneous release is not a treatment recommended by the AAOS.38
What to tell patients about postop care
Regardless of the method used for carpal tunnel release, most complications are minor—eg, a painful or hypertrophic scar, stiffness, swelling, and pain or tenderness on either side of the incision—and resolve within a few months.42 Advise patients not to continue to wear a wrist splint after surgery; doing so can cause stiffness or adhesions and may compromise surgical outcomes.41 Postoperatively, patients should be instructed to do nerve gliding exercises and to massage their scars, both of which they can safely do at home.43
Patients can expect significant symptomatic improvement within 1 week of surgery, and most will be able to return to normal activities in 2 weeks.44 Those with severe CTS should be warned, however, that it could take up to a year to determine the extent of recovery.22 Evidence suggests that from 3% to 19% of patients may have persistent or recurrent symptoms even after carpal tunnel release, with up to 12% requiring surgical revision.45
CASE When Ms. K returns, she reports that while there has been some improvement, some activities—such as driving long distances and talking on the phone—still cause numbness and tingling. And, if she doesn’t wear the splint at night, she awakens with tingling in her hands. You discuss 2 options—continued conservative treatment with a local steroid injection, or EDS and surgical referral. The patient opts for the injection and continued use of the nocturnal wrist splint and exercises. When she returns in another 6 weeks, Ms. K reports significant improvement. You agree to stop the wrist splint and exercises and advise her to follow-up on an as-needed basis if the symptoms return.
CORRESPONDENCE Jennifer Wipperman, MD, MPH, Via Christi Family Medicine, 1121 S. Clifton, Wichita, KS 67218; [email protected]
1. Atroshi I, Gummesson C, Johnsson, et al. Prevalence of carpal tunnel syndrome in a general population. JAMA. 1999;282:153-158.
2. Luckhaupt SE, Dahlhamer JM, Ward BW, et al. Prevalence and work-relatedness of carpal tunnel syndrome in the working population, United States, 2010 national health interview survey. Am J Ind Med. 2012 April 12. [Epub ahead of print.]
3. van Dijk MA, Reitsma JB, Fischer JC, et al. Indications for requesting laboratory tests for concurrent diseases in patients with carpal tunnel syndrome: a systematic review. Clin Chem. 2003;49:1437-1444.
4. Padua L, Di Pasquale A, Pazzaglia C, et al. Systematic review of pregnancy-related carpal tunnel syndrome. Muscle Nerve. 2010;42:697-702.
5. Bland JD. The relationship of obesity, age, and carpal tunnel syndrome: more complex than was thought? Muscle Nerve. 2005;32:527-532.
6. Prime MS, Palmer J, Khan WS, et al. Is there light at the end of the tunnel? Controversies in the diagnosis and management of carpal tunnel syndrome. Hand. 2010;5:354-360.
7. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons Clinical Practice Guideline on diagnosis of carpal tunnel syndrome. J Bone Joint Surg Am. 2009;91:2478-2479.
8. Hansen PA, Micklesen P, Robinson LR. Clinical utility of the flick maneuver in diagnosing carpal tunnel syndrome. Am J Phys Med Rehabil. 2004;83:363-367.
9. Padua L, Padua R, Aprile I, et al. Carpal tunnel syndrome: relationship between clinical and patient-oriented assessment. Clin Orthop Relat Res. 2002;395:128-134.
10. Bland JD. Carpal tunnel syndrome. BMJ. 2007;335:343-346.
11. Wright PE. Carpal tunnel syndrome. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, Pa: Mosby, Inc; 2008:4285–4291.
12. Sibley PA, Mandel RJ. Atraumatic acute carpal tunnel syndrome in a patient taking dabigatran. Orthopedics. 2012;35:e1286-e1289.
13. MacDermid JC, Wessel J. Clinical diagnosis of carpal tunnel syndrome: a systematic review. J Hand Ther. 2004;17:309-319.
14. Ahn DS. Hand elevation: a new test for carpal tunnel syndrome. Ann Plastic Surg. 2001;46:120-124.
15. Boyer K, Wies J, Turkelson CM. Effects of bias on the results of diagnostic studies of carpal tunnel syndrome. J Hand Surg. 2009;34:1006-1013.
16. Witt JC, Hentz JG, Stevens JC. Carpal tunnel syndrome with normal nerve conduction studies. Muscle Nerve. 2004;29:515-522.
17. Graham B. The value added by electrodiagnostic testing in the diagnosis of carpal tunnel syndrome. J Bone Joint Surg. 2008;90:2587-2593.
18. Jablecki CK, Andary MT, Floeter MK, et al. Practice parameter: electrodiagnostic studies in carpal tunnel syndrome. Report of the American Association of Electrodiagnostic Medicine, American Academy of Neurology, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2002;58:1589-1592.
19. Descatha A, Huard L, Aubert F, et al. Meta-analysis on the performance of sonography for the diagnosis of carpal tunnel syndrome. Semin Arthritis Rheum. 2012;41:914-922.
20. Visser LH, Ngo Q, Groeneweg SJ, et al. Long term effect of local corticosteroid injection for carpal tunnel syndrome: a relation with electrodiagnostic severity. Clin Neurophysiol. 2012;123:838-841.
21. Padua L, Padua R, Aprile I, et al. Multiperspective follow-up of untreated carpal tunnel syndrome: a multicenter study. Neurology. 2001;56:1459-1466.
22. Shi Q, MacDermid JC. Is surgical intervention more effective than non-surgical treatment for carpal tunnel syndrome? A systematic review. J Orthop Surg Res. 2011;6:17.-
23. Walker WC, Metzler M, Cifu DX, et al. Neutral wrist splinting in carpal tunnel syndrome: a comparison of night-only versus full-time wear instructions. Arch Phys Med Rehabil. 2000;81:424-429.
24. Brininger TL, Rogers JC, Holm MB, et al. Efficacy of a fabricated customized splint and tendon and nerve gliding exercises for the treatment of carpal tunnel syndrome: a randomized controlled trial. Arch Phys Med Rehabil. 2007;88:1429-1435.
25. Schmid AB, Elliott JM, Strudwick MW, et al. Effect of splinting and exercise on intraneural edema of the median nerve in carpal tunnel syndrome-an MRI study to reveal therapeutic mechanisms. J Orthop Res. 2012;30:1343-1350.
26. Marshall S, Tardif G, Ashworth N. Local corticosteroid injection for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(2):CD001554.-
27. Kamanli A, Bezgincan M, Kaya A. Comparison of local steroid injection into carpal tunnel via proximal and distal approach in patients with carpal tunnel syndrome. Bratislavske Lek Listy. 2011;112:337-341.
28. Huisstede BM, Hoogvliet P, Randsdorp MS, et al. Carpal tunnel syndrome. Part I: effectiveness of nonsurgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:981-1004.
29. Chang MH, Ger LP, Hsieh PF, et al. A randomised clinical trial of oral steroids in the treatment of carpal tunnel syndrome: a long term follow up. J Neurol Neurosurg Psychiatry. 2002;73:710-714.
30. O’Connor D, Marshall S, Massy-Westropp N. Non-surgical treatment (other than steroid injection) for carpal tunnel syndrome. Cochrane Database Syst Rev. 2003;(1):CD003219.-
31. Garfinkel MS, Singhal A, Katz WA, et al. Yoga-based intervention for carpal tunnel syndrome: a randomized trial. JAMA. 1998;280:1601-1603.
32. Tal-Akabi A, Rushton A. An investigation to compare the effectiveness of carpal bone mobilisation and neurodynamic mobilisation as methods of treatment for carpal tunnel syndrome. Man Ther. 2000;5:214-222.
33. O’Connor D, Page MJ, Marshall SC, et al. Ergonomic positioning or equipment for treating carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009600.-
34. Page MJ, O’Connor D, Pitt V, et al. Therapeutic ultrasound for carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009601.-
35. Yao E, Gerritz PK, Henricson E, et al. Randomized controlled trial comparing acupuncture with placebo acupuncture for the treatment of carpal tunnel syndrome. PMR. 2012;4:367-373.
36. Pomerance J, Zurakowski D, Fine I. The cost-effectiveness of nonsurgical versus surgical treatment for carpal tunnel syndrome. J Hand Surg. 2009;34:1193-1200.
37. Turner A, Kimble F, Gulyas K, et al. Can the outcome of open carpal tunnel release be predicted? A review of the literature. ANZ J Surg. 2010;80:50-54.
38. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of carpal tunnel syndrome. J Bone Joint Surg. 2010;92:218-219.
39. Scholten RJ, Mink van der Molen A, Uitdehaag BM, et al. Surgical treatment options for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(4):CD003905.-
40. Nakamichi K, Tachibana S, Yamamoto S, et al. Percutaneous carpal tunnel release compared with mini-open release using ultrasonographic guidance for both techniques. J Hand Surg Am. 2010;35:437-445.
41. Huisstede BM, Randsdorp MS, Coert JH, et al. Carpal tunnel syndrome. Part II: effectiveness of surgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:1005-1024.
42. Ludlow KS, Merla JL, Cox JA, et al. Pillar pain as a postoperative complication of carpal tunnel release: a review of the literature. J Hand Ther. 1997;10:277-282.
43. Pomerance J, Fine I. Outcomes of carpal tunnel surgery with and without supervised postoperative therapy. J Hand Surg. 2007;32:1159-1163.
44. Acharya AD, Auchincloss JM. Return to functional hand use and work following open carpal tunnel surgery. J Hand Surg Br. 2005;30:607-610.
45. Dahlin LB, Salo M, Thomsen N, et al. Carpal tunnel syndrome and treatment of recurrent symptoms. Scand J Plast Reconstr Surg Hand Surg. 2010;44:4-11.
1. Atroshi I, Gummesson C, Johnsson, et al. Prevalence of carpal tunnel syndrome in a general population. JAMA. 1999;282:153-158.
2. Luckhaupt SE, Dahlhamer JM, Ward BW, et al. Prevalence and work-relatedness of carpal tunnel syndrome in the working population, United States, 2010 national health interview survey. Am J Ind Med. 2012 April 12. [Epub ahead of print.]
3. van Dijk MA, Reitsma JB, Fischer JC, et al. Indications for requesting laboratory tests for concurrent diseases in patients with carpal tunnel syndrome: a systematic review. Clin Chem. 2003;49:1437-1444.
4. Padua L, Di Pasquale A, Pazzaglia C, et al. Systematic review of pregnancy-related carpal tunnel syndrome. Muscle Nerve. 2010;42:697-702.
5. Bland JD. The relationship of obesity, age, and carpal tunnel syndrome: more complex than was thought? Muscle Nerve. 2005;32:527-532.
6. Prime MS, Palmer J, Khan WS, et al. Is there light at the end of the tunnel? Controversies in the diagnosis and management of carpal tunnel syndrome. Hand. 2010;5:354-360.
7. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons Clinical Practice Guideline on diagnosis of carpal tunnel syndrome. J Bone Joint Surg Am. 2009;91:2478-2479.
8. Hansen PA, Micklesen P, Robinson LR. Clinical utility of the flick maneuver in diagnosing carpal tunnel syndrome. Am J Phys Med Rehabil. 2004;83:363-367.
9. Padua L, Padua R, Aprile I, et al. Carpal tunnel syndrome: relationship between clinical and patient-oriented assessment. Clin Orthop Relat Res. 2002;395:128-134.
10. Bland JD. Carpal tunnel syndrome. BMJ. 2007;335:343-346.
11. Wright PE. Carpal tunnel syndrome. In: Canale ST, Beaty JH, eds. Campbell’s Operative Orthopaedics. 11th ed. Philadelphia, Pa: Mosby, Inc; 2008:4285–4291.
12. Sibley PA, Mandel RJ. Atraumatic acute carpal tunnel syndrome in a patient taking dabigatran. Orthopedics. 2012;35:e1286-e1289.
13. MacDermid JC, Wessel J. Clinical diagnosis of carpal tunnel syndrome: a systematic review. J Hand Ther. 2004;17:309-319.
14. Ahn DS. Hand elevation: a new test for carpal tunnel syndrome. Ann Plastic Surg. 2001;46:120-124.
15. Boyer K, Wies J, Turkelson CM. Effects of bias on the results of diagnostic studies of carpal tunnel syndrome. J Hand Surg. 2009;34:1006-1013.
16. Witt JC, Hentz JG, Stevens JC. Carpal tunnel syndrome with normal nerve conduction studies. Muscle Nerve. 2004;29:515-522.
17. Graham B. The value added by electrodiagnostic testing in the diagnosis of carpal tunnel syndrome. J Bone Joint Surg. 2008;90:2587-2593.
18. Jablecki CK, Andary MT, Floeter MK, et al. Practice parameter: electrodiagnostic studies in carpal tunnel syndrome. Report of the American Association of Electrodiagnostic Medicine, American Academy of Neurology, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2002;58:1589-1592.
19. Descatha A, Huard L, Aubert F, et al. Meta-analysis on the performance of sonography for the diagnosis of carpal tunnel syndrome. Semin Arthritis Rheum. 2012;41:914-922.
20. Visser LH, Ngo Q, Groeneweg SJ, et al. Long term effect of local corticosteroid injection for carpal tunnel syndrome: a relation with electrodiagnostic severity. Clin Neurophysiol. 2012;123:838-841.
21. Padua L, Padua R, Aprile I, et al. Multiperspective follow-up of untreated carpal tunnel syndrome: a multicenter study. Neurology. 2001;56:1459-1466.
22. Shi Q, MacDermid JC. Is surgical intervention more effective than non-surgical treatment for carpal tunnel syndrome? A systematic review. J Orthop Surg Res. 2011;6:17.-
23. Walker WC, Metzler M, Cifu DX, et al. Neutral wrist splinting in carpal tunnel syndrome: a comparison of night-only versus full-time wear instructions. Arch Phys Med Rehabil. 2000;81:424-429.
24. Brininger TL, Rogers JC, Holm MB, et al. Efficacy of a fabricated customized splint and tendon and nerve gliding exercises for the treatment of carpal tunnel syndrome: a randomized controlled trial. Arch Phys Med Rehabil. 2007;88:1429-1435.
25. Schmid AB, Elliott JM, Strudwick MW, et al. Effect of splinting and exercise on intraneural edema of the median nerve in carpal tunnel syndrome-an MRI study to reveal therapeutic mechanisms. J Orthop Res. 2012;30:1343-1350.
26. Marshall S, Tardif G, Ashworth N. Local corticosteroid injection for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(2):CD001554.-
27. Kamanli A, Bezgincan M, Kaya A. Comparison of local steroid injection into carpal tunnel via proximal and distal approach in patients with carpal tunnel syndrome. Bratislavske Lek Listy. 2011;112:337-341.
28. Huisstede BM, Hoogvliet P, Randsdorp MS, et al. Carpal tunnel syndrome. Part I: effectiveness of nonsurgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:981-1004.
29. Chang MH, Ger LP, Hsieh PF, et al. A randomised clinical trial of oral steroids in the treatment of carpal tunnel syndrome: a long term follow up. J Neurol Neurosurg Psychiatry. 2002;73:710-714.
30. O’Connor D, Marshall S, Massy-Westropp N. Non-surgical treatment (other than steroid injection) for carpal tunnel syndrome. Cochrane Database Syst Rev. 2003;(1):CD003219.-
31. Garfinkel MS, Singhal A, Katz WA, et al. Yoga-based intervention for carpal tunnel syndrome: a randomized trial. JAMA. 1998;280:1601-1603.
32. Tal-Akabi A, Rushton A. An investigation to compare the effectiveness of carpal bone mobilisation and neurodynamic mobilisation as methods of treatment for carpal tunnel syndrome. Man Ther. 2000;5:214-222.
33. O’Connor D, Page MJ, Marshall SC, et al. Ergonomic positioning or equipment for treating carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009600.-
34. Page MJ, O’Connor D, Pitt V, et al. Therapeutic ultrasound for carpal tunnel syndrome. Cochrane Database Syst Rev. 2012;(1):CD009601.-
35. Yao E, Gerritz PK, Henricson E, et al. Randomized controlled trial comparing acupuncture with placebo acupuncture for the treatment of carpal tunnel syndrome. PMR. 2012;4:367-373.
36. Pomerance J, Zurakowski D, Fine I. The cost-effectiveness of nonsurgical versus surgical treatment for carpal tunnel syndrome. J Hand Surg. 2009;34:1193-1200.
37. Turner A, Kimble F, Gulyas K, et al. Can the outcome of open carpal tunnel release be predicted? A review of the literature. ANZ J Surg. 2010;80:50-54.
38. Keith MW, Masear V, Chung KC, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on the treatment of carpal tunnel syndrome. J Bone Joint Surg. 2010;92:218-219.
39. Scholten RJ, Mink van der Molen A, Uitdehaag BM, et al. Surgical treatment options for carpal tunnel syndrome. Cochrane Database Syst Rev. 2007;(4):CD003905.-
40. Nakamichi K, Tachibana S, Yamamoto S, et al. Percutaneous carpal tunnel release compared with mini-open release using ultrasonographic guidance for both techniques. J Hand Surg Am. 2010;35:437-445.
41. Huisstede BM, Randsdorp MS, Coert JH, et al. Carpal tunnel syndrome. Part II: effectiveness of surgical treatments—a systematic review. Arch Phys Med Rehabil. 2010;91:1005-1024.
42. Ludlow KS, Merla JL, Cox JA, et al. Pillar pain as a postoperative complication of carpal tunnel release: a review of the literature. J Hand Ther. 1997;10:277-282.
43. Pomerance J, Fine I. Outcomes of carpal tunnel surgery with and without supervised postoperative therapy. J Hand Surg. 2007;32:1159-1163.
44. Acharya AD, Auchincloss JM. Return to functional hand use and work following open carpal tunnel surgery. J Hand Surg Br. 2005;30:607-610.
45. Dahlin LB, Salo M, Thomsen N, et al. Carpal tunnel syndrome and treatment of recurrent symptoms. Scand J Plast Reconstr Surg Hand Surg. 2010;44:4-11.
Gynecomastia: When is treatment indicated?
• Examine enlarged male breasts to differentiate between true gynecomastia and pseudogynecomastia (seen with obesity) or a mass suggestive of tumor activity. C
• Ask patients about the use of medications associated with gynecomastia, such as some antihypertensives, antibiotics, psychotropic agents, or hormones. C
• Order renal function tests and measure levels of liver enzymes, testosterone, and other hormones when initial history and examination findings are insufficient for a diagnosis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Harry J is a 57-year-old man who came to us for evaluation and management of hypertension. He also complained of chronic headaches. Our initial examination revealed a body mass index (BMI) of 29 kg/m2 and blood pressure (BP) of 150/100 mm Hg. The hypertension responded well to a combination of valsartan and hydrochlorothiazide. A few months later, he developed left breast soreness, as well as decreased libido. Examination revealed a round movable subareolar nodule 2 cm in diameter, with no associated skin changes or lymphadenopathy. Laboratory results were: total testosterone, 106 ng/dL (normal, 241-827); free testosterone, 23 pg/mL (47-244); thyroid-stimulating hormone (TSH), 2.222 mIU/mL (0.350-5.500); and prolactin, 102.7 ng/mL (2.1-17.7). Magnetic resonance imaging (MRI) of the brain revealed a nodular density <10 mm in the pituitary gland with minimal displacement of the stalk, consistent with a microadenoma.
Enlargement of the male breasts—gynecomastia—is caused by a benign proliferation of the ductal epithelium, due to a relative increase in the ratio of free estrogen to androgen locally in the breast. Gynecomastia of recent onset is often associated with pain and tenderness, as was the case with our patient.
Often self-limiting, age-related influences. Gynecomastia is common in newborns, during adolescence, and in old age.1 In both male and female newborns, maternal and placental estrogens induce bilateral proliferation of breast tissue. This resolves within a few weeks after birth. During the early stages of male puberty, there is a relative increase in estrogens derived mostly from peripheral aromatization of testicular and adrenal androgens. If gynecomastia results, it usually regresses spontaneously as testicular testosterone production increases in late puberty.2 Gynecomastia is also common in elderly men due to a decrease in testosterone production and an increase in sex hormone binding globulin (SHBG) that lowers free testosterone levels.
Deleterious contributing factors. Several other potential causes of gynecomastia exist (TABLE 1),3,4 and these can usually be identified with a systematic approach using a careful history, physical examination, and selected laboratory studies. Many medications are associated with gynecomastia (TABLE 2),5 one of the most common being spironolactone due to its antiandrogenic activity at the receptor level.5 Some drugs, although associated with gynecomastia, cannot be linked to a direct cause-and-effect mechanism. These factors are compounded in elderly, obese men who take medications such as spironolactone, known to cause gynecomastia.
TABLE 1
Causes of gynecomastia3,4
| Physiologic |
| Neonatal Adolescent Aging-related |
| Drug induced |
| Antiandrogens Antibiotics Antihypertensive agents GI agents Hormones Illicit drugs Psychiatric drugs |
| Decreased androgen production |
| Primary (testicular) hypogonadism Secondary (central) hypogonadism |
| Decreased androgen effect or synthesis |
| Androgen insensitivity syndrome 5α-Reductase deficiency 17-β-Hydroxysteroid dehydrogenase deficiency |
| Increased estrogen production |
| Adrenal tumor Testicular tumor hCG-secreting tumor Familial aromatase excess syndrome |
| Other |
| Liver disease Thyrotoxicosis Obesity Renal disease Malnutrition |
| GI, gastrointestinal; hCG, human chorionic gonadotropin. |
TABLE 2
Drugs associated with gynecomastia5
| Antiandrogens | Bicalutamide, flutamide, finasteride, spironolactone |
| Antibiotics | Isoniazid, ketoconazole, metronidazole |
| Antihypertensive agents | Amlodipine, diltiazem, nifedipine, verapamil, captopril, enalapril |
| GI agents | Cimetidine, ranitidine, omeprazole |
| Hormones | Anabolic steroids, estrogens, hCG, growth hormone, GnRH agonists |
| Illicit drugs, alcohol | Marijuana, methadone |
| Psychiatric drugs | Psychotropic agents, tricyclic antidepressants |
| Other | Antiretroviral agents, digitalis, fibrates, methotrexate, statins |
| GI, gastrointestinal; GnRH, gonadotropin-releasing hormone; hCG, human chorionic gonadotropin. | |
A patient’s medical history may reveal chronic conditions associated with gynecomastia. Such disorders include cirrhosis, hyperthyroidism, malnutrition, and chronic kidney disease. Rarely, gynecomastia can be a manifestation of a testicular, adrenal, or other neoplasm.
Despite a thorough evaluation, no detectable abnormality is found initially in 25% of gynecomastia cases.6 Close observation and monitoring is necessary in such instances, to ensure the earliest possible identification of the underlying cause and initiation of appropriate medical or surgical therapy.
First steps in the clinical evaluation
In cases of male breast enlargement, first determine whether you are dealing with true gynecomastia or “pseudogynecomastia,” which involves increased fat deposits typically seen in obese individuals.3 In cases of pseudogynecomastia, the tissue is uniformly enlarged and soft, with the same consistency as adipose tissue.
In about half of the cases of gynecomastia, the condition is bilateral.3 It is characteristically a rubbery or firm mass concentric with the nipple-areolar complex.
Clues to look for in the history. When examination suggests true gynecomastia, conduct a focused history to determine if medications or other substances might be causing the problem. (See “A case where drug therapy was to blame”) Some plant-derived oils used as skin care products have also been associated with gynecomastia due to weak estrogenic or anti-androgenic activity.7
Jed G is a 61-year-old man who reported decreased libido and erectile dysfunction. Examination revealed normal male external genitalia and prostate. Gynecomastia was not present. Laboratory results were: total testosterone, 159 ng/dL (normal, 241-827); free testosterone, 40 pg/mL (47-244); follicle-stimulating hormone (FSH), 9.1 mIU/mL (1.4-18.1); luteinizing hormone (LH), 3.4 mIU/mL (1.5-9.3); prolactin, 2.8 ng/mL (2.1-17.7); and normal values for ferritin and iron. His prostate-specific antigen (PSA) level was 0.8 ng/mL (normal, 0.00-4.00 ng/mL).
Mr. G was started on testosterone 1% gel at 5 g/d. The repeat total testosterone measurement was 215 ng/dL, and free testosterone was 82 pg/mL. The patient discontinued the testosterone gel a few months later due to the medication’s high cost.
Several years later, his total testosterone level had fallen to 110 ng/dL, and he continued to complain of fatigue, decreased libido, and erectile dysfunction. We initiated testosterone enanthate 100 mg IM every 3 weeks, which increased his testosterone level to 285 mg/dL. However, hemoglobin increased to 18.3 g/dL, and he noted bilateral nipple tenderness since the start of the injections. Small bilateral gynecomastia about 1 cm in diameter was noted. Testosterone injections were discontinued due to the erythrocytosis. The breast tenderness and gynecomastia resolved 4 months later.
Mr. G had idiopathic hypogonadism. The breast tenderness and gynecomastia he developed were most likely a result of peripheral aromatization of testosterone. This is similar to gynecomastia commonly observed during early puberty and would likely have regressed with continued therapy. However, as noted above, the testosterone injections had to be stopped due to significant erythrocytosis.
The history may also uncover significant weight gain, because obesity is associated with increased aromatase activity resulting in a relative increase in estrogens systemically and locally in the breast. When obesity is the cause of gynecomastia, the breast examination reveals firm, rubbery tissue (unlike the findings in pseudogynecomastia, where there is a soft enlargement of the breast). Alternatively, a history of weight loss is important because it can lead to hypothalamic dysfunction and a decrease in gonadotropin (follicle-stimulating hormone [FSH], luteinizing hormone [LH]) secretion, resulting in decreased testosterone levels.8
Also inquire about prior diagnoses of liver cirrhosis or thyrotoxicosis or the presence of symptoms suggestive of these disorders, such as fatigue, jaundice, bloating, heat intolerance, or heart palpitations. These conditions can alter the metabolism of sex steroids and their binding proteins. A history of decreased libido and erectile dysfunction is suggestive of low testosterone levels, also known as hypogonadism. Headaches, visual disturbances, and behavioral abnormalities suggest a hypothalamic or pituitary disorder resulting in decreased FSH and LH levels and secondary hypogonadism. A family history of gynecomastia is elicited in half the patients with persistent pubertal gynecomastia.9
Physical examination. For all patients (except newborns), calculate the BMI and measure arm span and upper and lower body segments. A eunuchoid proportion—arm span 2 cm or greater than height—is associated with early-onset hypogonadism that precedes fusion of the epiphyses.3 Thus, you’ll need to consider congenital disorders of the testes, such as Klinefelter syndrome, as well as hypothalamic or pituitary disease, such as Kallmann syndrome, resulting in deficient FSH and LH production.
As noted earlier, you’ll need to examine the breasts to determine if true gynecomastia exists, as opposed to increased adipose tissue or the presence of a suspicious mass. A hard or irregular mass outside the areola, especially if associated with skin changes such as dimpling or retraction, should raise the possibility of breast carcinoma. Promptly arrange for diagnostic mammography and possible biopsy in this setting.
Carefully examine the secondary sexual characteristics, including body hair distribution and muscle mass. Inspect the external genitalia, penile development, and position of the urethral meatus. Note testicular size and consistency. Small, firm testes are suggestive of dysgenetic gonads found in patients with Klinefelter syndrome (47 XXY), whereas small, soft testes suggest secondary hypogonadism. A unilateral testicular mass raises suspicion of a neoplasm. Palpate the prostate in older men, especially if contemplating androgen therapy, which could exacerbate a preexisting focal prostate cancer.
Look for signs of hyperthyroidism, such as goiter, exophthalmos, tachycardia, and hyper-reflexia. Examine the abdomen for masses, hepato- or splenomegaly, and signs of cirrhosis, such as ascites and venous congestion. The examination should also include visual fields, cranial nerves, and fundoscopy for possible pituitary (or other) central nervous system lesions. Look for spider angiomas and palmar erythema (as occur in cirrhosis); warm, moist skin and myxedema (as in Graves’ disease); and mucocutaneous lentigines (as in Peutz-Jeghers syndrome).10
When laboratory and radiologic testing may help
Most adolescents with gynecomastia are best managed by reassurance and observation11 (ALGORITHM),3 and no laboratory or radiographic studies are recommended in most cases. Exceptions would be gynecomastia that develops before the onset of puberty; evidence of undervirilization on physical examination; a testicular mass; or persistence of gynecomastia beyond the usual observation period of 12 to 18 months.11
ALGORITHM
Evaluating gynecomastia3
CT, computed tomography; hCG, human chorionic gonadotropin; LH, luteinizing hormone; MRI, magnetic resonance imaging; TSH, thyroid-stimulating hormone.
↑ = elevated; ↓ = lowered; ↔ = normal.
If findings on physical examination are consistent with a breast neoplasm, arrange for mammography immediately. The sensitivity and specificity of mammography for benign and malignant conditions exceed 90%.12 A biopsy may be necessary if uncertainty remains after imaging.
No specific tests are necessary when gynecomastia is clearly associated with intake of a medication known to be associated with the condition, especially if the history and examination are otherwise negative. A prompt regression of gynecomastia after discontinuation of the offending drug will confirm the diagnosis.
If the condition persists in an adolescent or adult and the cause is still unclear, perform renal function tests and measure levels of liver enzymes, early-morning serum human chorionic gonadotropin (hCG), LH, total testosterone, estradiol, TSH, and prolactin.
What lab results may mean. If the total testosterone level is borderline or low-normal (200-350 ng/dL), repeat the test and measure the free testosterone level.
If an elevated hCG level is found, repeat the testicular examination carefully and order ultrasonography. In the absence of a testicular tumor, consider an MRI of the brain and computed tomography (CT) of the abdomen and chest to help identify an extragonadal hCG-secreting tumor.
An elevated LH level and low testosterone level are diagnostic of primary testicular hypogonadism. A karyotype may be necessary in some individuals to diagnose Klinefelter syndrome. Elevated LH and testosterone levels are seen in patients with androgen insensitivity syndromes. These conditions are caused by abnormalities in the androgen receptor with a wide range of possible phenotypes, including ambiguous genitalia.
A low testosterone level with a low or normal LH level indicates secondary hypogonadism of hypothalamic or pituitary origin. An elevated prolactin level in such cases (as was seen in Mr. J.’s case) is usually due to a prolactin secreting pituitary adenoma.
Hereditary hemochromatosis is an important and often overlooked cause of hypogonadism. Obtain iron studies and ferritin levels in this setting.13 Unrecognized hemochromatosis may result in fibrosis and multiple organ failure.
Patients with secondary hypogonadism are best managed by a referral to an endocrinologist, as the potential list of causes is extensive.4,14,15
A low TSH level is consistent with thyrotoxicosis, which may result in increased levels of SHBG and altered metabolism of estrogens and androgens.16 Thus, about 10% of men with thyrotoxicosis present with gynecomastia and erectile dysfunction.16 If the estradiol level is elevated, a testicular ultrasound as well as an adrenal CT scan will help identify a neoplasm.
In a significant number of patients, the diagnostic tests are normal, leading to a diagnosis of idiopathic gynecomastia. In these cases, the alteration in androgen and estrogen levels can be subtle and intermittent.17 Continue surveillance and periodically reevaluate the patient.
Management of gynecomastia
Gynecomastia often results from transient hormonal imbalance and regresses spontaneously. Therefore, no specific treatment is necessary for neonatal, pubertal, or drug-induced gynecomastia. In other situations, prompt diagnosis and treatment are important to maximize the likelihood of successful medical therapy. It has been shown that fibrosis develops 6 to 12 months after the onset of gynecomastia, making it unlikely that medical treatments beyond that stage will result in significant regression of the breast enlargement.18 In such long-standing cases, surgical intervention with subcutaneous mastectomy or liposuction can be considered for patients who have significant psychological problems or esthetic issues. Indications for surgery also include continued growth and tenderness of breast tissue or malignancy.
Available medications include those aimed at decreasing estrogen production or estrogen effect on target breast tissue. Aromatase inhibitors such as testolactone, anastrozole, and letrozole can decrease the synthesis of estrogen by inhibiting aromatization of androgens. Although theoretically promising, results of the few controlled trials with aromatase inhibitors have been generally disappointing.19
Selective estrogen receptor modulators that alter the effect of estrogen on breast tissue are tamoxifen and raloxifene. Tamoxifen is not yet approved for treatment of gynecomastia, but has proven effective in randomized trials.20 At a dose of 20 mg/d for 3 or more months, tamoxifen resulted in complete regression of gynecomastia in 60% of patients and partial regression in 20% of patients.20 Tamoxifen also prevents gynecomastia after medial prostatectomy and treatment with the antiandrogen, bicalutamide.
CASE Mr. J had a pituitary prolactin-secreting microadenoma causing secondary hypogonadism and gynecomastia. He was started on cabergoline (a dopamine agonist) 0.5 mg orally once a week. Four months later, his total testosterone level was 291 ng/dL, and prolactin was 9.3 ng/mL. His headaches and gynecomastia had significantly decreased. He continued to do well on the same regimen and, 6 years later, his prolactin level was 1.4 ng/mL, indicating that treatment had been effective.
CORRESPONDENCE
Roy N. Morcos, MD, Department of Family Medicine, St. Elizabeth Health Center, 1044 Belmont Avenue, Youngstown, OH 44501; [email protected]
1. Haynes B, Mookadem F. Male gynecomastia. Mayo Clin Proc. 2009;84:672.-
2. Nordt C, Divanta A. Gynecomastia in adolescents. Curr Opin Pediatr. 2008;20:375-382.
3. Braunstein G. Gynecomastia. N Engl J Med. 2007;357:1229-1237.
4. Bhasin SI. Testicular disorders. In: Kronenberg HM, Melmed S, Polonsky KS, et al, eds. Williams Textbook of Endocrinology. 11th ed. Philadelphia, Pa: Saunders-Elsevier; 2008:569–671.
5. Eckman A, Dobs A. Drug-induced gynecomastia. Expert Opin Drug Saf. 2008;7:691-702.
6. Derkacz M, Chmiel-Perzyriska I, Nowakowski A. Gynecomastia – a difficult diagnostic problem. Endokrynol Pol. 2011;62:190-202.
7. Henley D, Lipson N, Kovach K, et al. Pubertal gynecomastia linked to lavender and tea tree oils. N Engl J Med. 2007;356:479-485.
8. Ma N, Geffnes M. Gynecomastia in prepubertal and pubertal boys. Curr Opin Pediatr. 2008;20:465-470.
9. Eberle AJ, Sparrow JT, Keenan BS. Treatment of persistent pubertal gynecomastia with dihydrotestosterone heptanoate. J Pediatr. 1986;109:144-149.
10. Kapoor S. Cutaneous manifestations of systemic condition associated with gynecomastia. Skinmed. 2010;8:87-92.
11. Johnson RE, Murad MH. Gynecomastia: pathophysiology, evaluation, and management. Mayo Clin Proc. 2009;84:1010-1015.
12. Evans GF, Anthony T, Turnage RH, et al. The diagnostic accuracy of mammography in the evaluation of male breast disease. Am J Surg. 2001;181:96-102.
13. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-230.
14. Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab. 2002;87:1613-1620.
15. Bhasin SI, Jameson JL. Disorders of the testes and male reproductive system. In: Longo D, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:3019–3020.
16. Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid. 2004;14(suppl 1):S17-S25.
17. Wu FCW, Tajar A, Beynon JM, et al. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med. 2010;363:123-135.
18. Di Lorenzo G, Autorino R, Perdona S, et al. Management of gynaecomastia in patients with prostate cancer: a systematic review. Lancet Oncol. 2005;6:972-979.
19. Mauras N, Bishop K, Merinbaum D, et al. Pharmacokinetics and pharmacodynamics of anastrozole in pubertal boys with recent onset gynecomastia. J Clin Endocrinol Metab. 2009;94:2975-2978.
20. Derman O, Kanbur N, Kilic I, et al. Long-term follow-up of tamoxifen treatment in adolescents with gynecomastia. J Pediatr Endocrinol Metab. 2008;21:449-453.
• Examine enlarged male breasts to differentiate between true gynecomastia and pseudogynecomastia (seen with obesity) or a mass suggestive of tumor activity. C
• Ask patients about the use of medications associated with gynecomastia, such as some antihypertensives, antibiotics, psychotropic agents, or hormones. C
• Order renal function tests and measure levels of liver enzymes, testosterone, and other hormones when initial history and examination findings are insufficient for a diagnosis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Harry J is a 57-year-old man who came to us for evaluation and management of hypertension. He also complained of chronic headaches. Our initial examination revealed a body mass index (BMI) of 29 kg/m2 and blood pressure (BP) of 150/100 mm Hg. The hypertension responded well to a combination of valsartan and hydrochlorothiazide. A few months later, he developed left breast soreness, as well as decreased libido. Examination revealed a round movable subareolar nodule 2 cm in diameter, with no associated skin changes or lymphadenopathy. Laboratory results were: total testosterone, 106 ng/dL (normal, 241-827); free testosterone, 23 pg/mL (47-244); thyroid-stimulating hormone (TSH), 2.222 mIU/mL (0.350-5.500); and prolactin, 102.7 ng/mL (2.1-17.7). Magnetic resonance imaging (MRI) of the brain revealed a nodular density <10 mm in the pituitary gland with minimal displacement of the stalk, consistent with a microadenoma.
Enlargement of the male breasts—gynecomastia—is caused by a benign proliferation of the ductal epithelium, due to a relative increase in the ratio of free estrogen to androgen locally in the breast. Gynecomastia of recent onset is often associated with pain and tenderness, as was the case with our patient.
Often self-limiting, age-related influences. Gynecomastia is common in newborns, during adolescence, and in old age.1 In both male and female newborns, maternal and placental estrogens induce bilateral proliferation of breast tissue. This resolves within a few weeks after birth. During the early stages of male puberty, there is a relative increase in estrogens derived mostly from peripheral aromatization of testicular and adrenal androgens. If gynecomastia results, it usually regresses spontaneously as testicular testosterone production increases in late puberty.2 Gynecomastia is also common in elderly men due to a decrease in testosterone production and an increase in sex hormone binding globulin (SHBG) that lowers free testosterone levels.
Deleterious contributing factors. Several other potential causes of gynecomastia exist (TABLE 1),3,4 and these can usually be identified with a systematic approach using a careful history, physical examination, and selected laboratory studies. Many medications are associated with gynecomastia (TABLE 2),5 one of the most common being spironolactone due to its antiandrogenic activity at the receptor level.5 Some drugs, although associated with gynecomastia, cannot be linked to a direct cause-and-effect mechanism. These factors are compounded in elderly, obese men who take medications such as spironolactone, known to cause gynecomastia.
TABLE 1
Causes of gynecomastia3,4
| Physiologic |
| Neonatal Adolescent Aging-related |
| Drug induced |
| Antiandrogens Antibiotics Antihypertensive agents GI agents Hormones Illicit drugs Psychiatric drugs |
| Decreased androgen production |
| Primary (testicular) hypogonadism Secondary (central) hypogonadism |
| Decreased androgen effect or synthesis |
| Androgen insensitivity syndrome 5α-Reductase deficiency 17-β-Hydroxysteroid dehydrogenase deficiency |
| Increased estrogen production |
| Adrenal tumor Testicular tumor hCG-secreting tumor Familial aromatase excess syndrome |
| Other |
| Liver disease Thyrotoxicosis Obesity Renal disease Malnutrition |
| GI, gastrointestinal; hCG, human chorionic gonadotropin. |
TABLE 2
Drugs associated with gynecomastia5
| Antiandrogens | Bicalutamide, flutamide, finasteride, spironolactone |
| Antibiotics | Isoniazid, ketoconazole, metronidazole |
| Antihypertensive agents | Amlodipine, diltiazem, nifedipine, verapamil, captopril, enalapril |
| GI agents | Cimetidine, ranitidine, omeprazole |
| Hormones | Anabolic steroids, estrogens, hCG, growth hormone, GnRH agonists |
| Illicit drugs, alcohol | Marijuana, methadone |
| Psychiatric drugs | Psychotropic agents, tricyclic antidepressants |
| Other | Antiretroviral agents, digitalis, fibrates, methotrexate, statins |
| GI, gastrointestinal; GnRH, gonadotropin-releasing hormone; hCG, human chorionic gonadotropin. | |
A patient’s medical history may reveal chronic conditions associated with gynecomastia. Such disorders include cirrhosis, hyperthyroidism, malnutrition, and chronic kidney disease. Rarely, gynecomastia can be a manifestation of a testicular, adrenal, or other neoplasm.
Despite a thorough evaluation, no detectable abnormality is found initially in 25% of gynecomastia cases.6 Close observation and monitoring is necessary in such instances, to ensure the earliest possible identification of the underlying cause and initiation of appropriate medical or surgical therapy.
First steps in the clinical evaluation
In cases of male breast enlargement, first determine whether you are dealing with true gynecomastia or “pseudogynecomastia,” which involves increased fat deposits typically seen in obese individuals.3 In cases of pseudogynecomastia, the tissue is uniformly enlarged and soft, with the same consistency as adipose tissue.
In about half of the cases of gynecomastia, the condition is bilateral.3 It is characteristically a rubbery or firm mass concentric with the nipple-areolar complex.
Clues to look for in the history. When examination suggests true gynecomastia, conduct a focused history to determine if medications or other substances might be causing the problem. (See “A case where drug therapy was to blame”) Some plant-derived oils used as skin care products have also been associated with gynecomastia due to weak estrogenic or anti-androgenic activity.7
Jed G is a 61-year-old man who reported decreased libido and erectile dysfunction. Examination revealed normal male external genitalia and prostate. Gynecomastia was not present. Laboratory results were: total testosterone, 159 ng/dL (normal, 241-827); free testosterone, 40 pg/mL (47-244); follicle-stimulating hormone (FSH), 9.1 mIU/mL (1.4-18.1); luteinizing hormone (LH), 3.4 mIU/mL (1.5-9.3); prolactin, 2.8 ng/mL (2.1-17.7); and normal values for ferritin and iron. His prostate-specific antigen (PSA) level was 0.8 ng/mL (normal, 0.00-4.00 ng/mL).
Mr. G was started on testosterone 1% gel at 5 g/d. The repeat total testosterone measurement was 215 ng/dL, and free testosterone was 82 pg/mL. The patient discontinued the testosterone gel a few months later due to the medication’s high cost.
Several years later, his total testosterone level had fallen to 110 ng/dL, and he continued to complain of fatigue, decreased libido, and erectile dysfunction. We initiated testosterone enanthate 100 mg IM every 3 weeks, which increased his testosterone level to 285 mg/dL. However, hemoglobin increased to 18.3 g/dL, and he noted bilateral nipple tenderness since the start of the injections. Small bilateral gynecomastia about 1 cm in diameter was noted. Testosterone injections were discontinued due to the erythrocytosis. The breast tenderness and gynecomastia resolved 4 months later.
Mr. G had idiopathic hypogonadism. The breast tenderness and gynecomastia he developed were most likely a result of peripheral aromatization of testosterone. This is similar to gynecomastia commonly observed during early puberty and would likely have regressed with continued therapy. However, as noted above, the testosterone injections had to be stopped due to significant erythrocytosis.
The history may also uncover significant weight gain, because obesity is associated with increased aromatase activity resulting in a relative increase in estrogens systemically and locally in the breast. When obesity is the cause of gynecomastia, the breast examination reveals firm, rubbery tissue (unlike the findings in pseudogynecomastia, where there is a soft enlargement of the breast). Alternatively, a history of weight loss is important because it can lead to hypothalamic dysfunction and a decrease in gonadotropin (follicle-stimulating hormone [FSH], luteinizing hormone [LH]) secretion, resulting in decreased testosterone levels.8
Also inquire about prior diagnoses of liver cirrhosis or thyrotoxicosis or the presence of symptoms suggestive of these disorders, such as fatigue, jaundice, bloating, heat intolerance, or heart palpitations. These conditions can alter the metabolism of sex steroids and their binding proteins. A history of decreased libido and erectile dysfunction is suggestive of low testosterone levels, also known as hypogonadism. Headaches, visual disturbances, and behavioral abnormalities suggest a hypothalamic or pituitary disorder resulting in decreased FSH and LH levels and secondary hypogonadism. A family history of gynecomastia is elicited in half the patients with persistent pubertal gynecomastia.9
Physical examination. For all patients (except newborns), calculate the BMI and measure arm span and upper and lower body segments. A eunuchoid proportion—arm span 2 cm or greater than height—is associated with early-onset hypogonadism that precedes fusion of the epiphyses.3 Thus, you’ll need to consider congenital disorders of the testes, such as Klinefelter syndrome, as well as hypothalamic or pituitary disease, such as Kallmann syndrome, resulting in deficient FSH and LH production.
As noted earlier, you’ll need to examine the breasts to determine if true gynecomastia exists, as opposed to increased adipose tissue or the presence of a suspicious mass. A hard or irregular mass outside the areola, especially if associated with skin changes such as dimpling or retraction, should raise the possibility of breast carcinoma. Promptly arrange for diagnostic mammography and possible biopsy in this setting.
Carefully examine the secondary sexual characteristics, including body hair distribution and muscle mass. Inspect the external genitalia, penile development, and position of the urethral meatus. Note testicular size and consistency. Small, firm testes are suggestive of dysgenetic gonads found in patients with Klinefelter syndrome (47 XXY), whereas small, soft testes suggest secondary hypogonadism. A unilateral testicular mass raises suspicion of a neoplasm. Palpate the prostate in older men, especially if contemplating androgen therapy, which could exacerbate a preexisting focal prostate cancer.
Look for signs of hyperthyroidism, such as goiter, exophthalmos, tachycardia, and hyper-reflexia. Examine the abdomen for masses, hepato- or splenomegaly, and signs of cirrhosis, such as ascites and venous congestion. The examination should also include visual fields, cranial nerves, and fundoscopy for possible pituitary (or other) central nervous system lesions. Look for spider angiomas and palmar erythema (as occur in cirrhosis); warm, moist skin and myxedema (as in Graves’ disease); and mucocutaneous lentigines (as in Peutz-Jeghers syndrome).10
When laboratory and radiologic testing may help
Most adolescents with gynecomastia are best managed by reassurance and observation11 (ALGORITHM),3 and no laboratory or radiographic studies are recommended in most cases. Exceptions would be gynecomastia that develops before the onset of puberty; evidence of undervirilization on physical examination; a testicular mass; or persistence of gynecomastia beyond the usual observation period of 12 to 18 months.11
ALGORITHM
Evaluating gynecomastia3
CT, computed tomography; hCG, human chorionic gonadotropin; LH, luteinizing hormone; MRI, magnetic resonance imaging; TSH, thyroid-stimulating hormone.
↑ = elevated; ↓ = lowered; ↔ = normal.
If findings on physical examination are consistent with a breast neoplasm, arrange for mammography immediately. The sensitivity and specificity of mammography for benign and malignant conditions exceed 90%.12 A biopsy may be necessary if uncertainty remains after imaging.
No specific tests are necessary when gynecomastia is clearly associated with intake of a medication known to be associated with the condition, especially if the history and examination are otherwise negative. A prompt regression of gynecomastia after discontinuation of the offending drug will confirm the diagnosis.
If the condition persists in an adolescent or adult and the cause is still unclear, perform renal function tests and measure levels of liver enzymes, early-morning serum human chorionic gonadotropin (hCG), LH, total testosterone, estradiol, TSH, and prolactin.
What lab results may mean. If the total testosterone level is borderline or low-normal (200-350 ng/dL), repeat the test and measure the free testosterone level.
If an elevated hCG level is found, repeat the testicular examination carefully and order ultrasonography. In the absence of a testicular tumor, consider an MRI of the brain and computed tomography (CT) of the abdomen and chest to help identify an extragonadal hCG-secreting tumor.
An elevated LH level and low testosterone level are diagnostic of primary testicular hypogonadism. A karyotype may be necessary in some individuals to diagnose Klinefelter syndrome. Elevated LH and testosterone levels are seen in patients with androgen insensitivity syndromes. These conditions are caused by abnormalities in the androgen receptor with a wide range of possible phenotypes, including ambiguous genitalia.
A low testosterone level with a low or normal LH level indicates secondary hypogonadism of hypothalamic or pituitary origin. An elevated prolactin level in such cases (as was seen in Mr. J.’s case) is usually due to a prolactin secreting pituitary adenoma.
Hereditary hemochromatosis is an important and often overlooked cause of hypogonadism. Obtain iron studies and ferritin levels in this setting.13 Unrecognized hemochromatosis may result in fibrosis and multiple organ failure.
Patients with secondary hypogonadism are best managed by a referral to an endocrinologist, as the potential list of causes is extensive.4,14,15
A low TSH level is consistent with thyrotoxicosis, which may result in increased levels of SHBG and altered metabolism of estrogens and androgens.16 Thus, about 10% of men with thyrotoxicosis present with gynecomastia and erectile dysfunction.16 If the estradiol level is elevated, a testicular ultrasound as well as an adrenal CT scan will help identify a neoplasm.
In a significant number of patients, the diagnostic tests are normal, leading to a diagnosis of idiopathic gynecomastia. In these cases, the alteration in androgen and estrogen levels can be subtle and intermittent.17 Continue surveillance and periodically reevaluate the patient.
Management of gynecomastia
Gynecomastia often results from transient hormonal imbalance and regresses spontaneously. Therefore, no specific treatment is necessary for neonatal, pubertal, or drug-induced gynecomastia. In other situations, prompt diagnosis and treatment are important to maximize the likelihood of successful medical therapy. It has been shown that fibrosis develops 6 to 12 months after the onset of gynecomastia, making it unlikely that medical treatments beyond that stage will result in significant regression of the breast enlargement.18 In such long-standing cases, surgical intervention with subcutaneous mastectomy or liposuction can be considered for patients who have significant psychological problems or esthetic issues. Indications for surgery also include continued growth and tenderness of breast tissue or malignancy.
Available medications include those aimed at decreasing estrogen production or estrogen effect on target breast tissue. Aromatase inhibitors such as testolactone, anastrozole, and letrozole can decrease the synthesis of estrogen by inhibiting aromatization of androgens. Although theoretically promising, results of the few controlled trials with aromatase inhibitors have been generally disappointing.19
Selective estrogen receptor modulators that alter the effect of estrogen on breast tissue are tamoxifen and raloxifene. Tamoxifen is not yet approved for treatment of gynecomastia, but has proven effective in randomized trials.20 At a dose of 20 mg/d for 3 or more months, tamoxifen resulted in complete regression of gynecomastia in 60% of patients and partial regression in 20% of patients.20 Tamoxifen also prevents gynecomastia after medial prostatectomy and treatment with the antiandrogen, bicalutamide.
CASE Mr. J had a pituitary prolactin-secreting microadenoma causing secondary hypogonadism and gynecomastia. He was started on cabergoline (a dopamine agonist) 0.5 mg orally once a week. Four months later, his total testosterone level was 291 ng/dL, and prolactin was 9.3 ng/mL. His headaches and gynecomastia had significantly decreased. He continued to do well on the same regimen and, 6 years later, his prolactin level was 1.4 ng/mL, indicating that treatment had been effective.
CORRESPONDENCE
Roy N. Morcos, MD, Department of Family Medicine, St. Elizabeth Health Center, 1044 Belmont Avenue, Youngstown, OH 44501; [email protected]
• Examine enlarged male breasts to differentiate between true gynecomastia and pseudogynecomastia (seen with obesity) or a mass suggestive of tumor activity. C
• Ask patients about the use of medications associated with gynecomastia, such as some antihypertensives, antibiotics, psychotropic agents, or hormones. C
• Order renal function tests and measure levels of liver enzymes, testosterone, and other hormones when initial history and examination findings are insufficient for a diagnosis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Harry J is a 57-year-old man who came to us for evaluation and management of hypertension. He also complained of chronic headaches. Our initial examination revealed a body mass index (BMI) of 29 kg/m2 and blood pressure (BP) of 150/100 mm Hg. The hypertension responded well to a combination of valsartan and hydrochlorothiazide. A few months later, he developed left breast soreness, as well as decreased libido. Examination revealed a round movable subareolar nodule 2 cm in diameter, with no associated skin changes or lymphadenopathy. Laboratory results were: total testosterone, 106 ng/dL (normal, 241-827); free testosterone, 23 pg/mL (47-244); thyroid-stimulating hormone (TSH), 2.222 mIU/mL (0.350-5.500); and prolactin, 102.7 ng/mL (2.1-17.7). Magnetic resonance imaging (MRI) of the brain revealed a nodular density <10 mm in the pituitary gland with minimal displacement of the stalk, consistent with a microadenoma.
Enlargement of the male breasts—gynecomastia—is caused by a benign proliferation of the ductal epithelium, due to a relative increase in the ratio of free estrogen to androgen locally in the breast. Gynecomastia of recent onset is often associated with pain and tenderness, as was the case with our patient.
Often self-limiting, age-related influences. Gynecomastia is common in newborns, during adolescence, and in old age.1 In both male and female newborns, maternal and placental estrogens induce bilateral proliferation of breast tissue. This resolves within a few weeks after birth. During the early stages of male puberty, there is a relative increase in estrogens derived mostly from peripheral aromatization of testicular and adrenal androgens. If gynecomastia results, it usually regresses spontaneously as testicular testosterone production increases in late puberty.2 Gynecomastia is also common in elderly men due to a decrease in testosterone production and an increase in sex hormone binding globulin (SHBG) that lowers free testosterone levels.
Deleterious contributing factors. Several other potential causes of gynecomastia exist (TABLE 1),3,4 and these can usually be identified with a systematic approach using a careful history, physical examination, and selected laboratory studies. Many medications are associated with gynecomastia (TABLE 2),5 one of the most common being spironolactone due to its antiandrogenic activity at the receptor level.5 Some drugs, although associated with gynecomastia, cannot be linked to a direct cause-and-effect mechanism. These factors are compounded in elderly, obese men who take medications such as spironolactone, known to cause gynecomastia.
TABLE 1
Causes of gynecomastia3,4
| Physiologic |
| Neonatal Adolescent Aging-related |
| Drug induced |
| Antiandrogens Antibiotics Antihypertensive agents GI agents Hormones Illicit drugs Psychiatric drugs |
| Decreased androgen production |
| Primary (testicular) hypogonadism Secondary (central) hypogonadism |
| Decreased androgen effect or synthesis |
| Androgen insensitivity syndrome 5α-Reductase deficiency 17-β-Hydroxysteroid dehydrogenase deficiency |
| Increased estrogen production |
| Adrenal tumor Testicular tumor hCG-secreting tumor Familial aromatase excess syndrome |
| Other |
| Liver disease Thyrotoxicosis Obesity Renal disease Malnutrition |
| GI, gastrointestinal; hCG, human chorionic gonadotropin. |
TABLE 2
Drugs associated with gynecomastia5
| Antiandrogens | Bicalutamide, flutamide, finasteride, spironolactone |
| Antibiotics | Isoniazid, ketoconazole, metronidazole |
| Antihypertensive agents | Amlodipine, diltiazem, nifedipine, verapamil, captopril, enalapril |
| GI agents | Cimetidine, ranitidine, omeprazole |
| Hormones | Anabolic steroids, estrogens, hCG, growth hormone, GnRH agonists |
| Illicit drugs, alcohol | Marijuana, methadone |
| Psychiatric drugs | Psychotropic agents, tricyclic antidepressants |
| Other | Antiretroviral agents, digitalis, fibrates, methotrexate, statins |
| GI, gastrointestinal; GnRH, gonadotropin-releasing hormone; hCG, human chorionic gonadotropin. | |
A patient’s medical history may reveal chronic conditions associated with gynecomastia. Such disorders include cirrhosis, hyperthyroidism, malnutrition, and chronic kidney disease. Rarely, gynecomastia can be a manifestation of a testicular, adrenal, or other neoplasm.
Despite a thorough evaluation, no detectable abnormality is found initially in 25% of gynecomastia cases.6 Close observation and monitoring is necessary in such instances, to ensure the earliest possible identification of the underlying cause and initiation of appropriate medical or surgical therapy.
First steps in the clinical evaluation
In cases of male breast enlargement, first determine whether you are dealing with true gynecomastia or “pseudogynecomastia,” which involves increased fat deposits typically seen in obese individuals.3 In cases of pseudogynecomastia, the tissue is uniformly enlarged and soft, with the same consistency as adipose tissue.
In about half of the cases of gynecomastia, the condition is bilateral.3 It is characteristically a rubbery or firm mass concentric with the nipple-areolar complex.
Clues to look for in the history. When examination suggests true gynecomastia, conduct a focused history to determine if medications or other substances might be causing the problem. (See “A case where drug therapy was to blame”) Some plant-derived oils used as skin care products have also been associated with gynecomastia due to weak estrogenic or anti-androgenic activity.7
Jed G is a 61-year-old man who reported decreased libido and erectile dysfunction. Examination revealed normal male external genitalia and prostate. Gynecomastia was not present. Laboratory results were: total testosterone, 159 ng/dL (normal, 241-827); free testosterone, 40 pg/mL (47-244); follicle-stimulating hormone (FSH), 9.1 mIU/mL (1.4-18.1); luteinizing hormone (LH), 3.4 mIU/mL (1.5-9.3); prolactin, 2.8 ng/mL (2.1-17.7); and normal values for ferritin and iron. His prostate-specific antigen (PSA) level was 0.8 ng/mL (normal, 0.00-4.00 ng/mL).
Mr. G was started on testosterone 1% gel at 5 g/d. The repeat total testosterone measurement was 215 ng/dL, and free testosterone was 82 pg/mL. The patient discontinued the testosterone gel a few months later due to the medication’s high cost.
Several years later, his total testosterone level had fallen to 110 ng/dL, and he continued to complain of fatigue, decreased libido, and erectile dysfunction. We initiated testosterone enanthate 100 mg IM every 3 weeks, which increased his testosterone level to 285 mg/dL. However, hemoglobin increased to 18.3 g/dL, and he noted bilateral nipple tenderness since the start of the injections. Small bilateral gynecomastia about 1 cm in diameter was noted. Testosterone injections were discontinued due to the erythrocytosis. The breast tenderness and gynecomastia resolved 4 months later.
Mr. G had idiopathic hypogonadism. The breast tenderness and gynecomastia he developed were most likely a result of peripheral aromatization of testosterone. This is similar to gynecomastia commonly observed during early puberty and would likely have regressed with continued therapy. However, as noted above, the testosterone injections had to be stopped due to significant erythrocytosis.
The history may also uncover significant weight gain, because obesity is associated with increased aromatase activity resulting in a relative increase in estrogens systemically and locally in the breast. When obesity is the cause of gynecomastia, the breast examination reveals firm, rubbery tissue (unlike the findings in pseudogynecomastia, where there is a soft enlargement of the breast). Alternatively, a history of weight loss is important because it can lead to hypothalamic dysfunction and a decrease in gonadotropin (follicle-stimulating hormone [FSH], luteinizing hormone [LH]) secretion, resulting in decreased testosterone levels.8
Also inquire about prior diagnoses of liver cirrhosis or thyrotoxicosis or the presence of symptoms suggestive of these disorders, such as fatigue, jaundice, bloating, heat intolerance, or heart palpitations. These conditions can alter the metabolism of sex steroids and their binding proteins. A history of decreased libido and erectile dysfunction is suggestive of low testosterone levels, also known as hypogonadism. Headaches, visual disturbances, and behavioral abnormalities suggest a hypothalamic or pituitary disorder resulting in decreased FSH and LH levels and secondary hypogonadism. A family history of gynecomastia is elicited in half the patients with persistent pubertal gynecomastia.9
Physical examination. For all patients (except newborns), calculate the BMI and measure arm span and upper and lower body segments. A eunuchoid proportion—arm span 2 cm or greater than height—is associated with early-onset hypogonadism that precedes fusion of the epiphyses.3 Thus, you’ll need to consider congenital disorders of the testes, such as Klinefelter syndrome, as well as hypothalamic or pituitary disease, such as Kallmann syndrome, resulting in deficient FSH and LH production.
As noted earlier, you’ll need to examine the breasts to determine if true gynecomastia exists, as opposed to increased adipose tissue or the presence of a suspicious mass. A hard or irregular mass outside the areola, especially if associated with skin changes such as dimpling or retraction, should raise the possibility of breast carcinoma. Promptly arrange for diagnostic mammography and possible biopsy in this setting.
Carefully examine the secondary sexual characteristics, including body hair distribution and muscle mass. Inspect the external genitalia, penile development, and position of the urethral meatus. Note testicular size and consistency. Small, firm testes are suggestive of dysgenetic gonads found in patients with Klinefelter syndrome (47 XXY), whereas small, soft testes suggest secondary hypogonadism. A unilateral testicular mass raises suspicion of a neoplasm. Palpate the prostate in older men, especially if contemplating androgen therapy, which could exacerbate a preexisting focal prostate cancer.
Look for signs of hyperthyroidism, such as goiter, exophthalmos, tachycardia, and hyper-reflexia. Examine the abdomen for masses, hepato- or splenomegaly, and signs of cirrhosis, such as ascites and venous congestion. The examination should also include visual fields, cranial nerves, and fundoscopy for possible pituitary (or other) central nervous system lesions. Look for spider angiomas and palmar erythema (as occur in cirrhosis); warm, moist skin and myxedema (as in Graves’ disease); and mucocutaneous lentigines (as in Peutz-Jeghers syndrome).10
When laboratory and radiologic testing may help
Most adolescents with gynecomastia are best managed by reassurance and observation11 (ALGORITHM),3 and no laboratory or radiographic studies are recommended in most cases. Exceptions would be gynecomastia that develops before the onset of puberty; evidence of undervirilization on physical examination; a testicular mass; or persistence of gynecomastia beyond the usual observation period of 12 to 18 months.11
ALGORITHM
Evaluating gynecomastia3
CT, computed tomography; hCG, human chorionic gonadotropin; LH, luteinizing hormone; MRI, magnetic resonance imaging; TSH, thyroid-stimulating hormone.
↑ = elevated; ↓ = lowered; ↔ = normal.
If findings on physical examination are consistent with a breast neoplasm, arrange for mammography immediately. The sensitivity and specificity of mammography for benign and malignant conditions exceed 90%.12 A biopsy may be necessary if uncertainty remains after imaging.
No specific tests are necessary when gynecomastia is clearly associated with intake of a medication known to be associated with the condition, especially if the history and examination are otherwise negative. A prompt regression of gynecomastia after discontinuation of the offending drug will confirm the diagnosis.
If the condition persists in an adolescent or adult and the cause is still unclear, perform renal function tests and measure levels of liver enzymes, early-morning serum human chorionic gonadotropin (hCG), LH, total testosterone, estradiol, TSH, and prolactin.
What lab results may mean. If the total testosterone level is borderline or low-normal (200-350 ng/dL), repeat the test and measure the free testosterone level.
If an elevated hCG level is found, repeat the testicular examination carefully and order ultrasonography. In the absence of a testicular tumor, consider an MRI of the brain and computed tomography (CT) of the abdomen and chest to help identify an extragonadal hCG-secreting tumor.
An elevated LH level and low testosterone level are diagnostic of primary testicular hypogonadism. A karyotype may be necessary in some individuals to diagnose Klinefelter syndrome. Elevated LH and testosterone levels are seen in patients with androgen insensitivity syndromes. These conditions are caused by abnormalities in the androgen receptor with a wide range of possible phenotypes, including ambiguous genitalia.
A low testosterone level with a low or normal LH level indicates secondary hypogonadism of hypothalamic or pituitary origin. An elevated prolactin level in such cases (as was seen in Mr. J.’s case) is usually due to a prolactin secreting pituitary adenoma.
Hereditary hemochromatosis is an important and often overlooked cause of hypogonadism. Obtain iron studies and ferritin levels in this setting.13 Unrecognized hemochromatosis may result in fibrosis and multiple organ failure.
Patients with secondary hypogonadism are best managed by a referral to an endocrinologist, as the potential list of causes is extensive.4,14,15
A low TSH level is consistent with thyrotoxicosis, which may result in increased levels of SHBG and altered metabolism of estrogens and androgens.16 Thus, about 10% of men with thyrotoxicosis present with gynecomastia and erectile dysfunction.16 If the estradiol level is elevated, a testicular ultrasound as well as an adrenal CT scan will help identify a neoplasm.
In a significant number of patients, the diagnostic tests are normal, leading to a diagnosis of idiopathic gynecomastia. In these cases, the alteration in androgen and estrogen levels can be subtle and intermittent.17 Continue surveillance and periodically reevaluate the patient.
Management of gynecomastia
Gynecomastia often results from transient hormonal imbalance and regresses spontaneously. Therefore, no specific treatment is necessary for neonatal, pubertal, or drug-induced gynecomastia. In other situations, prompt diagnosis and treatment are important to maximize the likelihood of successful medical therapy. It has been shown that fibrosis develops 6 to 12 months after the onset of gynecomastia, making it unlikely that medical treatments beyond that stage will result in significant regression of the breast enlargement.18 In such long-standing cases, surgical intervention with subcutaneous mastectomy or liposuction can be considered for patients who have significant psychological problems or esthetic issues. Indications for surgery also include continued growth and tenderness of breast tissue or malignancy.
Available medications include those aimed at decreasing estrogen production or estrogen effect on target breast tissue. Aromatase inhibitors such as testolactone, anastrozole, and letrozole can decrease the synthesis of estrogen by inhibiting aromatization of androgens. Although theoretically promising, results of the few controlled trials with aromatase inhibitors have been generally disappointing.19
Selective estrogen receptor modulators that alter the effect of estrogen on breast tissue are tamoxifen and raloxifene. Tamoxifen is not yet approved for treatment of gynecomastia, but has proven effective in randomized trials.20 At a dose of 20 mg/d for 3 or more months, tamoxifen resulted in complete regression of gynecomastia in 60% of patients and partial regression in 20% of patients.20 Tamoxifen also prevents gynecomastia after medial prostatectomy and treatment with the antiandrogen, bicalutamide.
CASE Mr. J had a pituitary prolactin-secreting microadenoma causing secondary hypogonadism and gynecomastia. He was started on cabergoline (a dopamine agonist) 0.5 mg orally once a week. Four months later, his total testosterone level was 291 ng/dL, and prolactin was 9.3 ng/mL. His headaches and gynecomastia had significantly decreased. He continued to do well on the same regimen and, 6 years later, his prolactin level was 1.4 ng/mL, indicating that treatment had been effective.
CORRESPONDENCE
Roy N. Morcos, MD, Department of Family Medicine, St. Elizabeth Health Center, 1044 Belmont Avenue, Youngstown, OH 44501; [email protected]
1. Haynes B, Mookadem F. Male gynecomastia. Mayo Clin Proc. 2009;84:672.-
2. Nordt C, Divanta A. Gynecomastia in adolescents. Curr Opin Pediatr. 2008;20:375-382.
3. Braunstein G. Gynecomastia. N Engl J Med. 2007;357:1229-1237.
4. Bhasin SI. Testicular disorders. In: Kronenberg HM, Melmed S, Polonsky KS, et al, eds. Williams Textbook of Endocrinology. 11th ed. Philadelphia, Pa: Saunders-Elsevier; 2008:569–671.
5. Eckman A, Dobs A. Drug-induced gynecomastia. Expert Opin Drug Saf. 2008;7:691-702.
6. Derkacz M, Chmiel-Perzyriska I, Nowakowski A. Gynecomastia – a difficult diagnostic problem. Endokrynol Pol. 2011;62:190-202.
7. Henley D, Lipson N, Kovach K, et al. Pubertal gynecomastia linked to lavender and tea tree oils. N Engl J Med. 2007;356:479-485.
8. Ma N, Geffnes M. Gynecomastia in prepubertal and pubertal boys. Curr Opin Pediatr. 2008;20:465-470.
9. Eberle AJ, Sparrow JT, Keenan BS. Treatment of persistent pubertal gynecomastia with dihydrotestosterone heptanoate. J Pediatr. 1986;109:144-149.
10. Kapoor S. Cutaneous manifestations of systemic condition associated with gynecomastia. Skinmed. 2010;8:87-92.
11. Johnson RE, Murad MH. Gynecomastia: pathophysiology, evaluation, and management. Mayo Clin Proc. 2009;84:1010-1015.
12. Evans GF, Anthony T, Turnage RH, et al. The diagnostic accuracy of mammography in the evaluation of male breast disease. Am J Surg. 2001;181:96-102.
13. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-230.
14. Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab. 2002;87:1613-1620.
15. Bhasin SI, Jameson JL. Disorders of the testes and male reproductive system. In: Longo D, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:3019–3020.
16. Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid. 2004;14(suppl 1):S17-S25.
17. Wu FCW, Tajar A, Beynon JM, et al. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med. 2010;363:123-135.
18. Di Lorenzo G, Autorino R, Perdona S, et al. Management of gynaecomastia in patients with prostate cancer: a systematic review. Lancet Oncol. 2005;6:972-979.
19. Mauras N, Bishop K, Merinbaum D, et al. Pharmacokinetics and pharmacodynamics of anastrozole in pubertal boys with recent onset gynecomastia. J Clin Endocrinol Metab. 2009;94:2975-2978.
20. Derman O, Kanbur N, Kilic I, et al. Long-term follow-up of tamoxifen treatment in adolescents with gynecomastia. J Pediatr Endocrinol Metab. 2008;21:449-453.
1. Haynes B, Mookadem F. Male gynecomastia. Mayo Clin Proc. 2009;84:672.-
2. Nordt C, Divanta A. Gynecomastia in adolescents. Curr Opin Pediatr. 2008;20:375-382.
3. Braunstein G. Gynecomastia. N Engl J Med. 2007;357:1229-1237.
4. Bhasin SI. Testicular disorders. In: Kronenberg HM, Melmed S, Polonsky KS, et al, eds. Williams Textbook of Endocrinology. 11th ed. Philadelphia, Pa: Saunders-Elsevier; 2008:569–671.
5. Eckman A, Dobs A. Drug-induced gynecomastia. Expert Opin Drug Saf. 2008;7:691-702.
6. Derkacz M, Chmiel-Perzyriska I, Nowakowski A. Gynecomastia – a difficult diagnostic problem. Endokrynol Pol. 2011;62:190-202.
7. Henley D, Lipson N, Kovach K, et al. Pubertal gynecomastia linked to lavender and tea tree oils. N Engl J Med. 2007;356:479-485.
8. Ma N, Geffnes M. Gynecomastia in prepubertal and pubertal boys. Curr Opin Pediatr. 2008;20:465-470.
9. Eberle AJ, Sparrow JT, Keenan BS. Treatment of persistent pubertal gynecomastia with dihydrotestosterone heptanoate. J Pediatr. 1986;109:144-149.
10. Kapoor S. Cutaneous manifestations of systemic condition associated with gynecomastia. Skinmed. 2010;8:87-92.
11. Johnson RE, Murad MH. Gynecomastia: pathophysiology, evaluation, and management. Mayo Clin Proc. 2009;84:1010-1015.
12. Evans GF, Anthony T, Turnage RH, et al. The diagnostic accuracy of mammography in the evaluation of male breast disease. Am J Surg. 2001;181:96-102.
13. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-230.
14. Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab. 2002;87:1613-1620.
15. Bhasin SI, Jameson JL. Disorders of the testes and male reproductive system. In: Longo D, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:3019–3020.
16. Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid. 2004;14(suppl 1):S17-S25.
17. Wu FCW, Tajar A, Beynon JM, et al. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med. 2010;363:123-135.
18. Di Lorenzo G, Autorino R, Perdona S, et al. Management of gynaecomastia in patients with prostate cancer: a systematic review. Lancet Oncol. 2005;6:972-979.
19. Mauras N, Bishop K, Merinbaum D, et al. Pharmacokinetics and pharmacodynamics of anastrozole in pubertal boys with recent onset gynecomastia. J Clin Endocrinol Metab. 2009;94:2975-2978.
20. Derman O, Kanbur N, Kilic I, et al. Long-term follow-up of tamoxifen treatment in adolescents with gynecomastia. J Pediatr Endocrinol Metab. 2008;21:449-453.
When your patients disclose ‘insider information’
Discuss this article at www.facebook.com/CurrentPsychiatry
Dear Dr. Mossman:
My patient is an officer in a large corporation. During therapy, he sometimes talks about how the company is doing. Would I risk malpractice liability if I used this information in managing my retirement investments?
Submitted by “Dr. B”
As most physicians find out within a short time of finishing medical school, doctors learn all kinds of useful things from their patients, including information that can help them manage personal matters outside their practices. But are you allowed to use nonpublic business information to make investment decisions?
As this article explains, legal rules and case law suggest that if psychiatrists or therapists act on potentially profitable business information incidentally mentioned by a patient during treatment, they may be subject to serious legal problems. To explain why, we’ll begin with a brief overview of business terms, including “securities” and “insider trading.” Then, to answer Dr. B’s question, we’ll look at what kind of legal consequences may result if mental health professionals are found guilty of “misappropriating” confidential business information.
Securities and security rules
Approximately one-half to two-thirds of Americans have money invested in the stock market—either through their retirement plans, by owning mutual funds, or by holding stocks of individual companies.1 Stocks are a type of financial instrument, or security, that companies issue to raise capital. Companies also raise money by issuing debt, typically in the form of bonds that pay interest to the holder, who in buying the bond has in effect loaned money to the company. Derivatives refer to securities that have prices that move up or down depending on the value of some underlying asset, such as stock prices.2
Stock prices fluctuate in reaction to general economic developments—changes in the unemployment rate, in the cost of basic materials (eg, oil or metals used in manufacturing), or in government policies that influence consumers’ purchasing decisions. But the key factor in determining the price of a company’s stock is investors’ beliefs about the company’s future earnings.3 Because investors usually have to make educated guesses about a company’s future, actually knowing something about a company before the general public finds out would give an investor a huge—but possibly unfair—advantage over other investors.
Making markets fair for all investors is the key purpose of U.S. laws on trading securities. In the 1930s, Congress created the Securities and Exchange Commission (SEC), a federal agency charged with ensuring that companies report the truth about their financial situation and that potential investors receive full, fair disclosure of available public information.4 Among the many ways that the SEC does this is by enforcing regulations concerning “insider trading.”
‘Insider trading’
Corporate “insiders” (eg, directors or employees) often know a lot about how their businesses are doing, and they buy or sell stock in their own companies. Such trading is legal if the insiders follow federal regulations about the timing of their investments and report them publicly.
Insider trading is illegal, however, if an individual acquires material, nonpublic information about a corporation through a relationship that involves trust and confidence and then uses that information when buying or selling a security. The SEC has prosecuted corporate employees who traded securities after learning of confidential developments in their companies, friends and family members of corporate officers who bought or sold securities after getting such information, and employees of law firms who misused information they received while providing services to corporations whose securities they traded.5
To be guilty of insider trading, a person must:
- buy or sell a security based on information that the person realizes is material and nonpublic,6 and
- have received the confidential information under circumstances that create a duty of trust or confidence.7
If both of these conditions are met, the person has wrongfully used confidential information with which he was entrusted, or “misappropriated” that information for personal gain.8
Physicians sometimes gain information that, if used for investment decisions, might lead to accusations of insider trading. Stock prices of pharmaceutical companies rise before public announcements of clinical drug trials, which suggests that information about those results leaks out in advance.9 Recently, physicians have gotten into well-publicized legal trouble by making investment decisions based on information they obtained while participating on an institution’s board10 and from learning early results of clinical drug trials.11
But would it be wrong for a psychiatrist to make a potentially profitable investment based on information obtained incidentally during a treatment encounter? After all, it’s not as though the psychiatrist would be a corporate insider or would have acquired the information improperly. Yet courts have ruled that a psychiatrist’s trading on such information might constitute malpractice and could be grounds for even more serious legal consequences.
Potential malpractice issues
The federal court ruling in United States v Willis12 describes how a psychiatrist learned during treatment that a patient’s husband was seeking to become CEO of a large bank. Realizing that this development might make the bank more valuable, the psychiatrist told his broker what he had learned and purchased 13,000 shares of the bank’s stock for himself and his children. When the husband’s efforts were announced publicly a few weeks later, the psychiatrist sold the shares at a big profit.
Quoting the vow of confidentiality contained in the Hippocratic Oath (Box),13 the court held that the psychiatrist had an obligation to the patient not to disclose information learned during her treatment without her permission. The court said the patient “had an economic interest in preserving the confidentiality of the information disclosed,” and the psychiatrist’s actions “might have jeopardized her husband’s advancement” and financial benefits the wife would have gained. Also, the psychiatrist’s “disclosures jeopardized the psychiatrist-patient relationship,” which might negate the wife’s financial investment in her care, require her to find a new psychiatrist, or require additional treatment to deal with how the psychiatrist’s behavior had affected her.12
And about whatever I may see or hear in treatment, or even without treatment, in the life of human beings—things that should not ever be blurted out outside—I will remain silent, holding such things to be unutterable.
Source: Reference 13
More legal consequences
Dr. Willis had legal problems more serious than just a malpractice lawsuit. He faced criminal prosecution for insider trading and mail fraud, and the court refused to dismiss these charges. The court reasoned that the psychiatrist received the information while in a position of trust and confidence, and breached that trust when he used that confidential information for his personal benefit—behavior that meets the legal definition of “misappropriation.” Because the psychiatrist received stock trade confirmations through the U.S. mail, he also could face federal charges of mail fraud. Ultimately, Dr. Willis pled guilty and paid $137,000 in fines and penalties. Although Dr. Willis retained his New Jersey medical license and avoided a prison sentence, the district court sentenced him to 5 years of probation and required that he perform 3,000 hours of community service.14,15
In a second case,16 a licensed clinical social worker made investments through a broker based on information learned during a therapy session about upcoming business developments (the 1994 Lockheed-Martin Marietta merger). The social worker pled guilty to insider trading, forfeited the illegal gains, and paid a large fine.
Related Resources
- Insider trading versus medical professionalism. Lancet. 2005;366(9488):781.
- Nijm LM. The online message board controversy. Physicians hit with claims of libel and insider trading by their employers. J Leg Med. 2000;21(2):223-239.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jacobe D. In U.S., 54% have stock market investments, lowest since 1999. Gallup Economy. http://www.gallup.com/poll/147206/stock-market-investments-lowest-1999.aspx. Published April 20, 2011. Accessed October 9, 2012.
2. Roman S. Introduction to the mathematics of finance: from risk management to options pricing. New York NY: Springer-Verlag; 2004.
3. Elton EJ, Gruber MJ, Brown SJ, et al. Modern portfolio theory and investment analysis. Hoboken, NJ: John Wiley & Sons; 2010.
4. Keller E, Gehlmann GA. Introductory comment: a historical introduction to the Securities Act of 1933 and the Securities Exchange Act of 1934. Ohio State Law Journal. 1988;49:329-352.
5. U.S. Securities and Exchange Commission. Insider trading. http://www.sec.gov/answers/insider.htm. Published April 19, 2001. Accessed October 9, 2012.
6. 17 CFR 240. 10b5-1.
7. 17 CFR 240. 10b5-2.
8. United States v O’Hagan, 521 U.S. 642 (1997).
9. Rothenstein JM, Tomlinson G, Tannock IF, et al. Company stock prices before and after public announcements related to oncology drugs. J Natl Cancer Inst. 2011;103(20):1507-1512.
10. U.S. Securities and Exchange Commission. SEC charges five physicians with insider trading in stock of medical professional liability insurer. http://www.sec.gov/news/press/2012/2012-132.htm. Published July 10, 2012. Accessed October 9, 2012.
11. Two more are sentenced in insider trading cases. New York Times. December 21 2011:B9. http://www.nytimes.com/2011/12/22/business/in-crackdown-on-insider-trading-two-more-are-sentenced.html?_r=0. Accessed October 9, 2012.
12. United States v Willis, 737 F Supp 269 (SD NY 1990).
13. von Staden H. “In a pure and holy way”: personal and professional conduct in the Hippocratic Oath? J Hist Med Allied Sci. 1996;51(4):404-437.
14. 24 Sec Reg & L Rep (BNA) 7 (1992).
15. Psychiatrist is sentenced. New York Times. January 8 1992. http://www.nytimes.com/1992/01/08/business/credit-markets-psychiatrist-is-sentenced.html. Accessed November 5, 2012.
16. SEC v Cooper, Litigation Rel. No. 14754, 60 S.E.C. Docket 2430 (1995).

Douglas Mossman, MD
Dr. Mossman is Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Dr. Mossman is Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Douglas Mossman, MD
Dr. Mossman is Professor and Program Director, University of Cincinnati Forensic Psychiatry Fellowship, Cincinnati, OH
Discuss this article at www.facebook.com/CurrentPsychiatry
Dear Dr. Mossman:
My patient is an officer in a large corporation. During therapy, he sometimes talks about how the company is doing. Would I risk malpractice liability if I used this information in managing my retirement investments?
Submitted by “Dr. B”
As most physicians find out within a short time of finishing medical school, doctors learn all kinds of useful things from their patients, including information that can help them manage personal matters outside their practices. But are you allowed to use nonpublic business information to make investment decisions?
As this article explains, legal rules and case law suggest that if psychiatrists or therapists act on potentially profitable business information incidentally mentioned by a patient during treatment, they may be subject to serious legal problems. To explain why, we’ll begin with a brief overview of business terms, including “securities” and “insider trading.” Then, to answer Dr. B’s question, we’ll look at what kind of legal consequences may result if mental health professionals are found guilty of “misappropriating” confidential business information.
Securities and security rules
Approximately one-half to two-thirds of Americans have money invested in the stock market—either through their retirement plans, by owning mutual funds, or by holding stocks of individual companies.1 Stocks are a type of financial instrument, or security, that companies issue to raise capital. Companies also raise money by issuing debt, typically in the form of bonds that pay interest to the holder, who in buying the bond has in effect loaned money to the company. Derivatives refer to securities that have prices that move up or down depending on the value of some underlying asset, such as stock prices.2
Stock prices fluctuate in reaction to general economic developments—changes in the unemployment rate, in the cost of basic materials (eg, oil or metals used in manufacturing), or in government policies that influence consumers’ purchasing decisions. But the key factor in determining the price of a company’s stock is investors’ beliefs about the company’s future earnings.3 Because investors usually have to make educated guesses about a company’s future, actually knowing something about a company before the general public finds out would give an investor a huge—but possibly unfair—advantage over other investors.
Making markets fair for all investors is the key purpose of U.S. laws on trading securities. In the 1930s, Congress created the Securities and Exchange Commission (SEC), a federal agency charged with ensuring that companies report the truth about their financial situation and that potential investors receive full, fair disclosure of available public information.4 Among the many ways that the SEC does this is by enforcing regulations concerning “insider trading.”
‘Insider trading’
Corporate “insiders” (eg, directors or employees) often know a lot about how their businesses are doing, and they buy or sell stock in their own companies. Such trading is legal if the insiders follow federal regulations about the timing of their investments and report them publicly.
Insider trading is illegal, however, if an individual acquires material, nonpublic information about a corporation through a relationship that involves trust and confidence and then uses that information when buying or selling a security. The SEC has prosecuted corporate employees who traded securities after learning of confidential developments in their companies, friends and family members of corporate officers who bought or sold securities after getting such information, and employees of law firms who misused information they received while providing services to corporations whose securities they traded.5
To be guilty of insider trading, a person must:
- buy or sell a security based on information that the person realizes is material and nonpublic,6 and
- have received the confidential information under circumstances that create a duty of trust or confidence.7
If both of these conditions are met, the person has wrongfully used confidential information with which he was entrusted, or “misappropriated” that information for personal gain.8
Physicians sometimes gain information that, if used for investment decisions, might lead to accusations of insider trading. Stock prices of pharmaceutical companies rise before public announcements of clinical drug trials, which suggests that information about those results leaks out in advance.9 Recently, physicians have gotten into well-publicized legal trouble by making investment decisions based on information they obtained while participating on an institution’s board10 and from learning early results of clinical drug trials.11
But would it be wrong for a psychiatrist to make a potentially profitable investment based on information obtained incidentally during a treatment encounter? After all, it’s not as though the psychiatrist would be a corporate insider or would have acquired the information improperly. Yet courts have ruled that a psychiatrist’s trading on such information might constitute malpractice and could be grounds for even more serious legal consequences.
Potential malpractice issues
The federal court ruling in United States v Willis12 describes how a psychiatrist learned during treatment that a patient’s husband was seeking to become CEO of a large bank. Realizing that this development might make the bank more valuable, the psychiatrist told his broker what he had learned and purchased 13,000 shares of the bank’s stock for himself and his children. When the husband’s efforts were announced publicly a few weeks later, the psychiatrist sold the shares at a big profit.
Quoting the vow of confidentiality contained in the Hippocratic Oath (Box),13 the court held that the psychiatrist had an obligation to the patient not to disclose information learned during her treatment without her permission. The court said the patient “had an economic interest in preserving the confidentiality of the information disclosed,” and the psychiatrist’s actions “might have jeopardized her husband’s advancement” and financial benefits the wife would have gained. Also, the psychiatrist’s “disclosures jeopardized the psychiatrist-patient relationship,” which might negate the wife’s financial investment in her care, require her to find a new psychiatrist, or require additional treatment to deal with how the psychiatrist’s behavior had affected her.12
And about whatever I may see or hear in treatment, or even without treatment, in the life of human beings—things that should not ever be blurted out outside—I will remain silent, holding such things to be unutterable.
Source: Reference 13
More legal consequences
Dr. Willis had legal problems more serious than just a malpractice lawsuit. He faced criminal prosecution for insider trading and mail fraud, and the court refused to dismiss these charges. The court reasoned that the psychiatrist received the information while in a position of trust and confidence, and breached that trust when he used that confidential information for his personal benefit—behavior that meets the legal definition of “misappropriation.” Because the psychiatrist received stock trade confirmations through the U.S. mail, he also could face federal charges of mail fraud. Ultimately, Dr. Willis pled guilty and paid $137,000 in fines and penalties. Although Dr. Willis retained his New Jersey medical license and avoided a prison sentence, the district court sentenced him to 5 years of probation and required that he perform 3,000 hours of community service.14,15
In a second case,16 a licensed clinical social worker made investments through a broker based on information learned during a therapy session about upcoming business developments (the 1994 Lockheed-Martin Marietta merger). The social worker pled guilty to insider trading, forfeited the illegal gains, and paid a large fine.
Related Resources
- Insider trading versus medical professionalism. Lancet. 2005;366(9488):781.
- Nijm LM. The online message board controversy. Physicians hit with claims of libel and insider trading by their employers. J Leg Med. 2000;21(2):223-239.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Dear Dr. Mossman:
My patient is an officer in a large corporation. During therapy, he sometimes talks about how the company is doing. Would I risk malpractice liability if I used this information in managing my retirement investments?
Submitted by “Dr. B”
As most physicians find out within a short time of finishing medical school, doctors learn all kinds of useful things from their patients, including information that can help them manage personal matters outside their practices. But are you allowed to use nonpublic business information to make investment decisions?
As this article explains, legal rules and case law suggest that if psychiatrists or therapists act on potentially profitable business information incidentally mentioned by a patient during treatment, they may be subject to serious legal problems. To explain why, we’ll begin with a brief overview of business terms, including “securities” and “insider trading.” Then, to answer Dr. B’s question, we’ll look at what kind of legal consequences may result if mental health professionals are found guilty of “misappropriating” confidential business information.
Securities and security rules
Approximately one-half to two-thirds of Americans have money invested in the stock market—either through their retirement plans, by owning mutual funds, or by holding stocks of individual companies.1 Stocks are a type of financial instrument, or security, that companies issue to raise capital. Companies also raise money by issuing debt, typically in the form of bonds that pay interest to the holder, who in buying the bond has in effect loaned money to the company. Derivatives refer to securities that have prices that move up or down depending on the value of some underlying asset, such as stock prices.2
Stock prices fluctuate in reaction to general economic developments—changes in the unemployment rate, in the cost of basic materials (eg, oil or metals used in manufacturing), or in government policies that influence consumers’ purchasing decisions. But the key factor in determining the price of a company’s stock is investors’ beliefs about the company’s future earnings.3 Because investors usually have to make educated guesses about a company’s future, actually knowing something about a company before the general public finds out would give an investor a huge—but possibly unfair—advantage over other investors.
Making markets fair for all investors is the key purpose of U.S. laws on trading securities. In the 1930s, Congress created the Securities and Exchange Commission (SEC), a federal agency charged with ensuring that companies report the truth about their financial situation and that potential investors receive full, fair disclosure of available public information.4 Among the many ways that the SEC does this is by enforcing regulations concerning “insider trading.”
‘Insider trading’
Corporate “insiders” (eg, directors or employees) often know a lot about how their businesses are doing, and they buy or sell stock in their own companies. Such trading is legal if the insiders follow federal regulations about the timing of their investments and report them publicly.
Insider trading is illegal, however, if an individual acquires material, nonpublic information about a corporation through a relationship that involves trust and confidence and then uses that information when buying or selling a security. The SEC has prosecuted corporate employees who traded securities after learning of confidential developments in their companies, friends and family members of corporate officers who bought or sold securities after getting such information, and employees of law firms who misused information they received while providing services to corporations whose securities they traded.5
To be guilty of insider trading, a person must:
- buy or sell a security based on information that the person realizes is material and nonpublic,6 and
- have received the confidential information under circumstances that create a duty of trust or confidence.7
If both of these conditions are met, the person has wrongfully used confidential information with which he was entrusted, or “misappropriated” that information for personal gain.8
Physicians sometimes gain information that, if used for investment decisions, might lead to accusations of insider trading. Stock prices of pharmaceutical companies rise before public announcements of clinical drug trials, which suggests that information about those results leaks out in advance.9 Recently, physicians have gotten into well-publicized legal trouble by making investment decisions based on information they obtained while participating on an institution’s board10 and from learning early results of clinical drug trials.11
But would it be wrong for a psychiatrist to make a potentially profitable investment based on information obtained incidentally during a treatment encounter? After all, it’s not as though the psychiatrist would be a corporate insider or would have acquired the information improperly. Yet courts have ruled that a psychiatrist’s trading on such information might constitute malpractice and could be grounds for even more serious legal consequences.
Potential malpractice issues
The federal court ruling in United States v Willis12 describes how a psychiatrist learned during treatment that a patient’s husband was seeking to become CEO of a large bank. Realizing that this development might make the bank more valuable, the psychiatrist told his broker what he had learned and purchased 13,000 shares of the bank’s stock for himself and his children. When the husband’s efforts were announced publicly a few weeks later, the psychiatrist sold the shares at a big profit.
Quoting the vow of confidentiality contained in the Hippocratic Oath (Box),13 the court held that the psychiatrist had an obligation to the patient not to disclose information learned during her treatment without her permission. The court said the patient “had an economic interest in preserving the confidentiality of the information disclosed,” and the psychiatrist’s actions “might have jeopardized her husband’s advancement” and financial benefits the wife would have gained. Also, the psychiatrist’s “disclosures jeopardized the psychiatrist-patient relationship,” which might negate the wife’s financial investment in her care, require her to find a new psychiatrist, or require additional treatment to deal with how the psychiatrist’s behavior had affected her.12
And about whatever I may see or hear in treatment, or even without treatment, in the life of human beings—things that should not ever be blurted out outside—I will remain silent, holding such things to be unutterable.
Source: Reference 13
More legal consequences
Dr. Willis had legal problems more serious than just a malpractice lawsuit. He faced criminal prosecution for insider trading and mail fraud, and the court refused to dismiss these charges. The court reasoned that the psychiatrist received the information while in a position of trust and confidence, and breached that trust when he used that confidential information for his personal benefit—behavior that meets the legal definition of “misappropriation.” Because the psychiatrist received stock trade confirmations through the U.S. mail, he also could face federal charges of mail fraud. Ultimately, Dr. Willis pled guilty and paid $137,000 in fines and penalties. Although Dr. Willis retained his New Jersey medical license and avoided a prison sentence, the district court sentenced him to 5 years of probation and required that he perform 3,000 hours of community service.14,15
In a second case,16 a licensed clinical social worker made investments through a broker based on information learned during a therapy session about upcoming business developments (the 1994 Lockheed-Martin Marietta merger). The social worker pled guilty to insider trading, forfeited the illegal gains, and paid a large fine.
Related Resources
- Insider trading versus medical professionalism. Lancet. 2005;366(9488):781.
- Nijm LM. The online message board controversy. Physicians hit with claims of libel and insider trading by their employers. J Leg Med. 2000;21(2):223-239.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jacobe D. In U.S., 54% have stock market investments, lowest since 1999. Gallup Economy. http://www.gallup.com/poll/147206/stock-market-investments-lowest-1999.aspx. Published April 20, 2011. Accessed October 9, 2012.
2. Roman S. Introduction to the mathematics of finance: from risk management to options pricing. New York NY: Springer-Verlag; 2004.
3. Elton EJ, Gruber MJ, Brown SJ, et al. Modern portfolio theory and investment analysis. Hoboken, NJ: John Wiley & Sons; 2010.
4. Keller E, Gehlmann GA. Introductory comment: a historical introduction to the Securities Act of 1933 and the Securities Exchange Act of 1934. Ohio State Law Journal. 1988;49:329-352.
5. U.S. Securities and Exchange Commission. Insider trading. http://www.sec.gov/answers/insider.htm. Published April 19, 2001. Accessed October 9, 2012.
6. 17 CFR 240. 10b5-1.
7. 17 CFR 240. 10b5-2.
8. United States v O’Hagan, 521 U.S. 642 (1997).
9. Rothenstein JM, Tomlinson G, Tannock IF, et al. Company stock prices before and after public announcements related to oncology drugs. J Natl Cancer Inst. 2011;103(20):1507-1512.
10. U.S. Securities and Exchange Commission. SEC charges five physicians with insider trading in stock of medical professional liability insurer. http://www.sec.gov/news/press/2012/2012-132.htm. Published July 10, 2012. Accessed October 9, 2012.
11. Two more are sentenced in insider trading cases. New York Times. December 21 2011:B9. http://www.nytimes.com/2011/12/22/business/in-crackdown-on-insider-trading-two-more-are-sentenced.html?_r=0. Accessed October 9, 2012.
12. United States v Willis, 737 F Supp 269 (SD NY 1990).
13. von Staden H. “In a pure and holy way”: personal and professional conduct in the Hippocratic Oath? J Hist Med Allied Sci. 1996;51(4):404-437.
14. 24 Sec Reg & L Rep (BNA) 7 (1992).
15. Psychiatrist is sentenced. New York Times. January 8 1992. http://www.nytimes.com/1992/01/08/business/credit-markets-psychiatrist-is-sentenced.html. Accessed November 5, 2012.
16. SEC v Cooper, Litigation Rel. No. 14754, 60 S.E.C. Docket 2430 (1995).
1. Jacobe D. In U.S., 54% have stock market investments, lowest since 1999. Gallup Economy. http://www.gallup.com/poll/147206/stock-market-investments-lowest-1999.aspx. Published April 20, 2011. Accessed October 9, 2012.
2. Roman S. Introduction to the mathematics of finance: from risk management to options pricing. New York NY: Springer-Verlag; 2004.
3. Elton EJ, Gruber MJ, Brown SJ, et al. Modern portfolio theory and investment analysis. Hoboken, NJ: John Wiley & Sons; 2010.
4. Keller E, Gehlmann GA. Introductory comment: a historical introduction to the Securities Act of 1933 and the Securities Exchange Act of 1934. Ohio State Law Journal. 1988;49:329-352.
5. U.S. Securities and Exchange Commission. Insider trading. http://www.sec.gov/answers/insider.htm. Published April 19, 2001. Accessed October 9, 2012.
6. 17 CFR 240. 10b5-1.
7. 17 CFR 240. 10b5-2.
8. United States v O’Hagan, 521 U.S. 642 (1997).
9. Rothenstein JM, Tomlinson G, Tannock IF, et al. Company stock prices before and after public announcements related to oncology drugs. J Natl Cancer Inst. 2011;103(20):1507-1512.
10. U.S. Securities and Exchange Commission. SEC charges five physicians with insider trading in stock of medical professional liability insurer. http://www.sec.gov/news/press/2012/2012-132.htm. Published July 10, 2012. Accessed October 9, 2012.
11. Two more are sentenced in insider trading cases. New York Times. December 21 2011:B9. http://www.nytimes.com/2011/12/22/business/in-crackdown-on-insider-trading-two-more-are-sentenced.html?_r=0. Accessed October 9, 2012.
12. United States v Willis, 737 F Supp 269 (SD NY 1990).
13. von Staden H. “In a pure and holy way”: personal and professional conduct in the Hippocratic Oath? J Hist Med Allied Sci. 1996;51(4):404-437.
14. 24 Sec Reg & L Rep (BNA) 7 (1992).
15. Psychiatrist is sentenced. New York Times. January 8 1992. http://www.nytimes.com/1992/01/08/business/credit-markets-psychiatrist-is-sentenced.html. Accessed November 5, 2012.
16. SEC v Cooper, Litigation Rel. No. 14754, 60 S.E.C. Docket 2430 (1995).
BRCA1/2 testing and cancer risk management in underserved women at a public hospital
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Background and objective Genetic test uptake and cancer risk management have been understudied in medically underserved populations. Study aims were to quantify rates of BRCA1/2 genetic testing and evidence-based cancer risk management (ie, prophylactic surgeries and surveillance practices) in women who were seen for breast and ovarian cancer genetic counseling in a public, safety net health system.
Methods We conducted a retrospective medical record abstraction of 195 women who presented for breast or ovarian genetic counseling within a 2-year period (2008-2009) at Parkland Health & Hospital System in Dallas, Texas.
Results The identified women represented a racially and ethnically diverse population: 48% Hispanic, 37% non-Hispanic black, 12% non-Hispanic white, and 3% Asian. Among the 158 women who were medically eligible for genetic testing, 134 (84.8%) received BRCA1/2 results, with most tests funded through a financial assistance program. In all, 29 women (22%) tested positive for BRCA1/2 mutations. Financial and funding barriers were identified for 20 of the untested women. Among the identified high-risk women (mutation carriers, selected variants, and noncarriers with pretest BRCAPRO scores 30 or more), 26% had prophylactic breast surgeries and 33% had prophylactic ovarian surgeries within the follow-up period averaging 35 months. Of those who opted for surveillance, 71% had at least 1 mammogram or MRI and 38% had CA-125 tests. Trends indicated lower rates of all risk management behaviors, except for mammogram or MRI, among non-Hispanic black women.
Conclusions Within this racially and ethnically diverse sample, BRCA1/2 test uptake was high, but financial barriers were identified for nontested women. The rates of breast cancer risk management were generally comparable with other studies, but risk management for ovarian cancer was limited, especially among non-Hispanic black women. The reasons for these apparen disparities should be further explored.
Click on the PDF icon at the top of this introduction to read the full article.
Community Oncology Podcast - Pazopanib in soft tissue sarcoma
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Dr. David Henry's podcast covers highlights of the November issue including pazopanib in soft tissue sarcoma and dasatinib in first-line treatment of chronic myeloid leukemia.
Federal Grant Supports "eHospitalist" Pilot Program in Wisconsin
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
John Almquist, MD, FHM, director of hospitalist services for Ministry Health Care, a 15-hospital system serving rural Wisconsin, believes that an "e-hospitalist" pilot project now being tested at Ministry St. Mary's Hospital in Rhinelander, Wis., could be a boon for rural communities that have difficulty recruiting primary-care physicians (PCPs).
When the hospitals in those communities are unable to offer hospitalist coverage, it makes the setting less attractive to PCPs because they might have to follow their patients in the hospital day and night, he explains.
Ministry recruited and trained two nurse practitioners who will soon be deployed at a critical-access hospital in Eagle River, population 1,443, supported remotely by the eight-member HM group in Rhinelander for consultations, supervision, and multidisciplinary rounds. The training is bolstered by written order sets focused on 30 common medical conditions that lead to admissions to rural hospitals.
"The hospitalist in Rhinelander is also able to talk directly to the patient at the remote site," Dr. Almquist says.
The e-hospitalist program uses a telehealth network developed by Marshfield Clinic, a multispecialty physician group practice based in Marshfield, Wis. The clinic recently received a $1 million grant from the federal government to expand its 15-year-old telemedicine program. Part of the grant money is being used to expand the ehospitalist approach to new sites.
Visit our website for more information about hospitalists and telemedicine.
ITL: Physician Reviews of HM-Relevant Research
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Clinical question: Does the addition of clopidogrel to aspirin reduce the risk of any type of recurrent stroke, or affect the risk of bleeding or death, in patients who recently suffered a lacunar stroke?
Background: There are no prior randomized, multicenter trials on secondary prevention of lacunar stroke; aspirin is the standard antiplatelet therapy in this setting.
Study design: Double-blind, randomized, multicenter trial.
Setting: Eighty-two clinical centers in North America, Latin America, and Spain.
Synopsis: Researchers enrolled 3,020 patients from 2003 to 2011; criteria included age >30 years old and symptomatic lacunar stroke (proven by MRI) in the preceding 180 days.
Results showed no significant difference between recurrent strokes (any type) in the aspirin-only group (2.7% per year) versus the aspirin-plus-clopidogrel group (2.5% per year). Major hemorrhage risk was much higher in the aspirin-plus-clopidogrel group (2.1% per year) versus aspirin-only group (1.1% per year). All-cause mortality also was much higher in the aspirin-plus-clopidogrel group (N=113) versus the aspirin-only group (N=77).
Bottom line: The addition of clopidogrel to aspirin for secondary prevention does not significantly reduce the risk of recurrent stroke, but it does significantly increase the risk of bleeding and death.
Citation: Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med. 2012;367:817-825.
For more physician reviews of recent HM-relevant literature, visit our website.
Woman with “Dull, Achy” Back Pain and Shortness of Breath
ANSWER
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
ANSWER
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
ANSWER
This ECG demonstrates normal sinus rhythm, right-axis deviation, evidence of a lateral MI, and inferolateral ST- and T-wave abnormalities.
Right-axis deviation is indicated by an R-wave axis between 90° and 180° and QS or QR complexes in lead I and/or aVL. While the most common cause of a right-axis deviation is right ventricular hypertrophy, it is also evident in a lateral MI. Evidence for the latter includes the presence of significant Q waves in leads I and aVL. Finally, inferolateral ST- and T-wave changes are evidenced by inverted T waves in leads II, III, aVF, and precordial leads V4 to V6.
ECG evidence of a lateral MI not present on a previous scan (eight months ago), in the presence of a normal troponin level, suggests a recent MI.
A 70-year-old woman has a 10-year history of a dilated nonischemic cardiomyopathy and New York Heart Association Class II heart failure. She presents with a one-week history of back pain and shortness of breath. She describes the pain as a “dull, achy” pressure, exacerbated by exertion and relieved with rest. She says the pain is localized in the back between her scapulas and does not radiate. She denies substernal chest pain, nausea, vomiting, or diaphoresis; the only associated symptom is dyspnea. Her most recent echocardiogram showed a dilated left ventricle, with a left ventricular ejection fraction of 29%, and a normal right ventricle, with mild hypertrophy and mildly reduced systolic function. She was also noted to have atherosclerotic changes in her ascending and descending thoracic aorta. Medical history is remarkable for diabetes, hypertension, chronic renal insufficiency, hyperlipidemia, and cataracts. Her current medications include aspirin, fer-rous sulfate, furosemide, hydralazine, glargine insulin, isosorbide dinitrate, lisinopril, metoprolol, and raloxifene. She is allergic to codeine, amiodarone, and radi-ographic contrast. Family history is positive for coronary artery disease, diabetes, and stroke. The patient is widowed, does not smoke, and does not consume alcohol. She is very active in her local quilting club. The review of systems is positive for increased weakness and diarrhea. She states that approximately two weeks ago, she experienced vague epigastric pain and diaphoresis; she did not seek medical attention, as it resolved. The physical exam reveals a thin, elderly woman in mild distress. Blood pressure is 139/82 mm Hg; pulse, 66 beats/min; respiratory rate, 21 breaths/¬min-1; and temperature, 35.9°C. Her weight is 108 lb. Pertinent physical findings include a grade II/VI diastolic murmur at the left lower sternal border, 2+ peripheral pulses with a bruit present in the right femoral artery, occasional late expiratory wheezes in both lung bases, vertebral tenderness at the T6-T7 level with no evidence of scoliosis or kyphosis, and no evidence of peripheral edema. She is intact from a neurologic standpoint. Significant laboratory data include a serum glucose level of 294 mg/dL; blood urea nitrogen (BUN), 68 mg/dL; creatinine, 1.75 mg/dL; glomerular filtration rate, 30 mL/min; B-type natriuretic peptide, 984 pg/mL; and serum troponin, 0.11 ng/mL. An ECG is obtained that reveals the following: a ventricular rate of 62 beats/min; PR interval, 160 ms; QRS duration, 94 ms; QT/QTc interval, 404/410 ms; P ax-is, 84°; R axis, 151°; and T axis, 253°. What is your interpretation of this ECG?
Topical Steroids: the Solution or the Cause?
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
ANSWER
The correct answer is all of the above (choice “d”). Prolonged injudicious use of topical steroids can cause a number of problems, including these; they are collectively termed iatrogenic since they are ultimately caused by prescribed medication. One of the more difficult aspects of this problem to deal with is the “addictive” state, in which withdrawal symptoms compel the patient to continue applying the offending steroid cream.
DISCUSSION
This is a relatively common scenario in dermatology offices. The misuse of topical steroids is well known, and something we strive to prevent—but with mixed results. It’s one of the reasons we’re stingy with refills of such medications, requiring the patient to be seen at least once a year. Unfortunately, this patient had been getting “refills” from friends in Mexico; patients often “borrow” steroid creams from household members or friends, or use products prescribed for one condition to treat others for which they were not intended.
The primary mode of action of topical steroids is vasoconstriction, a positive thing in terms of reduction of inflammation. The bad news is that continuous use of class 1 (the most powerful) steroids, such as clobetasol, can cause such profound and prolonged vasoconstriction that the skin effectively loses its blood supply and withers, sometimes down to adipose tissue. As one might suspect, this is more likely in already thin-skinned areas, including the antecubital area, face, neck, eyelids, and genitals, where the creation of striae is especially common.
Fairly early on in this process, before frank atrophy occurs, the condition being treated usually resolves. However, when the steroid is stopped, stinging and itching immediately return—which, of course, causes the patient to reapply the medication, perpetuating the vicious cycle.
The cycle is ultimately broken by gradual reduction in the frequency of application of successively weaker steroids. Usually, the skin gradually regenerates and returns to normal. In this case, the process will be lengthy and will almost certainly result in significant scarring.
Even injudicious application of weaker classes of steroids (eg, hydrocortisone 2.5% cream) to areas such as the face can result in a range of deleterious effects, including localized rosacea-like eruption or erythema. It has been reported that approximately 75% of cases of perioral dermatitis are either caused by or exacerbated by the application of topical steroids.
Topical application of even mid-strength steroids can also have systemic effects (eg, adrenal suppression, hyperglycemia) if applied over large areas. This is especially true when pediatric patients are involved.
Prevention of these iatrogenic effects lies in selecting the lowest strength steroid for the condition and area in question, then using them sparingly: no more than twice a day, and for no more than five days in a row, stopping for two consecutive days to allow the skin to regenerate. Even more caution should be exercised in treating children and when applying the product to intertriginous areas (skin-on-skin areas, such as the groin, in axillae, or under the breasts). Covering steroid-treated areas with anything—bandages, socks, even skin—effectively potentiates the positive and negative effects of steroids.
A 59-year-old man presents with skin changes on both antecubital areas. For more than a year, he has applied clobetasol 0.05% cream at least twice daily to the area ostensibly for treatment of long-standing eczema, which has affected not only the antecubital areas but also the patient’s legs. In addition to the eczema, he has a history of atopy, marked by seasonal allergies and asthma. He notes that his stress level has increased in the past several months, which he suspects has contributed to his itching. On examination, marked epidermal atrophy is seen in both antecubital areas, along with extensive purpura. Surface adnexal structures, such as hair, follicles, and skin lines, are sparse at best, but dermal and subdermal vasculature are readily visible. In the midst of the affected area on the right arm, a nickel-sized, full-thickness defect is noted. Beneath it, adipose tissue can be seen. Clearly, these changes are attributable to the effects of the clobetasol, which the patient is advised to stop. But he replies that when he does, the treated areas burn and itch even more, until he obtains relief by applying more clobetasol.

Cold weather and diarrhea: Don't forget yersiniosis
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.

Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.

Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).

The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.
Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.
Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).
The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].
The genus Yersinia includes 11 species. Three species are generally associated with human disease; Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis. Yersinia pestis is the causative agent of plague. Yersinia pseudotuberculosis can manifest with fever, abdominal pain, and scarlatiniform rash. Additional symptoms include diarrhea, sterile joint effusions, erythema nodosum, and septicemia; these symptoms can be indistinguishable from Kawasaki Disease. By report, almost 10 % of cases of Kawasaki Disease in Japan have serologic or bacteriologic evidence of Y. pseudotuberculosis infection [Redbook: 2012 Report of the Committee on Infectious Diseases, 795-7]. Y. enterocolitica is most often associated with yersiniosis.
Although Y. enterocolitica is not the most common cause of diarrheal illness in the United States, it is one of the nine pathogens that have been monitored by the Foodborne Diseases Active Surveillance Network (FoodNet) since 1996. In the United States, it is estimated that Y. enterocolitica causes slightly over 115,000 infections annually (Emerg. Infect. Dis. 2011;17:7-15). The disease is more common in cooler months. It is transmitted by consumption of contaminated food, especially raw or undercooked pork products.
Only a few outbreaks have been reported in the United States, and these were usually associated with consumption of pork, specifically chitterlings (pig intestines), a winter holiday dish prepared most frequently in black households in the South (MMWR 1990;39:819-20). Transmission to infants and young children is thought to occur from caretakers preparing chitterlings who have not adequately cleaned their hands prior to touching objects subsequently handled by the child.
The incubation period is usually 4-6 days (range, 1-14 days). The duration of diarrhea is variable and can persist up to 3 weeks. Organisms can be excreted an average of 6 weeks. Clinical manifestations vary by age. Younger children usually present with fever and diarrhea. Stools frequently contain blood and leucocytes. Vomiting is also reported in most series. In contrast, older children and adults often present with a pseudoappendicitis syndrome with right-sided abdominal pain and fever. Leukocytosis is often present. At surgery, mesenteric adenitis is observed, and the appendix generally is normal.
Bacteremia can occur and is usually associated with infection in children less than 1 year of age and in those with iron-overloaded states, including persons with sickle cell disease, beta-thalassemia, and those receiving deferoxamine therapy. While uncommon, focal manifestations including pharyngitis, osteomyelitis, pyomyositis, pneumonia, empyema, and meningitis may occur.
Diagnosis is confirmed by isolation of the organism from stool, blood, peritoneal fluid, lymph nodes, and throat cultures. Most laboratories do not routinely test for Yersinia in stool cultures. If Y. enterocolitica is suspected, you should notify the laboratory so the stool can be plated on appropriate media (CIN agar). Serologic tests to detect a rise in serum antibody titers to confirm infection are available in reference and research laboratories, but are not generally used for diagnosis. Cross reactivity with Brucella, Salmonella, Vibrio, and Rickettsia may lead to false positive titer results. Y. enterocolitica antibodies also have antigenic similarity with thyroid tissue. You may see persistent elevation of titers in patients with thyroid disease.
Benefit of antimicrobial therapy for isolated Y. enterocolitica gastrointestinal disease and Y. pseudotuberculosis has not been established. Therapy may decrease the duration of fecal shedding. Treatment is indicated for immunocompromised hosts and persons with septicemia and focal infections. Y. enterocolitica and Y. pseudotuberculosis are usually sensitive to trimethoprim-sulfamethoxazole, aminoglycosides, cefotaxime, fluoroquinolones (persons greater than 18 years of age or older), and tetracycline or doxycycline (for children at least 8 years of age and older).
So what is the actual incidence and when should the practitioner be concerned? Initial population based surveillance data for Y. enterocolitica infections in FoodNet sites between 1996 and 1999 reported an overall incidence of 0.9 cases per 100,000 population. The highest incidence was among black and Asian individuals and was 3.2 cases and 1.5 cases per 100,000 population, respectively. The incidence in Hispanics and whites was 0.6 and 0.4 cases per 100,000 respectively. Incidence increased with decreasing age in all racial/ethnic groups. Blacks infants had the highest incidence, 141.9 cases/100,000 population, and the highest incidence in infants was reported from Georgia (207 cases/100,000). Seasonal variation in incidence was noted only in black individuals with peak activity occurring in December (Clin. Infect. Dis. 2004;38[Suppl 3]:S181-9).
The most recent data from FoodNet (1996-2009) reveals an overall incidence of 0.5/100,000. There was a decline in incidence in all racial and ethnic groups. The highest incidence is still observed in black and Asians (0.9 and 0.7 per 100,000). The most dramatic decline occurred in black individuals (3.2 vs. 0.9 per 100,000). In 1998, an educational campaign was initiated in Georgia that targeted high-risk individuals and provided information on the safe handling and preparation of chitterlings. The state of Georgia reported the greatest decline to 0.4/100,000, which has almost eliminated the racial disparity reported in 2009. It is unclear if this campaign was the only reason for the decline in Georgia. The incidence in whites is 0.2/100,000. Since 2007, the incidence in Asian children less than 5 years of ages has been the highest amongst all racial and ethnic groups. Pork consumption is still assumed to be the major source. Seasonal variability persists amongst Black children less 5 years of age, implying that chitterlings may still be the source of infection for individuals in this group (Clin. Infect. Dis. 2012:54 [Suppl 5]:S385-S90).
In general, yersiniosis should be included in the differential of a febrile diarrheal illness, particularly during the cooler months and holiday season. It is prudent to determine if consumption and/or preparation of chitterlings or other pork products by the patient or caretakers has occurred. This will enable you to alert the laboratory so stool specimens can be cultured on the appropriate medium (CIN agar). Consumption of chitterlings is not limited to any specific racial or ethnic group. Individuals from rural and farming areas may also consume this product.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Write to Dr. Word at [email protected].
