User login
When should a hospitalized patient be transfused?
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
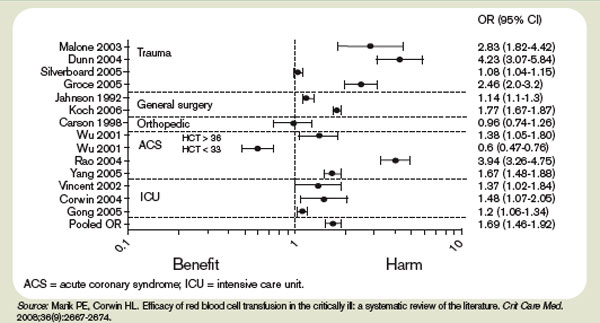
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
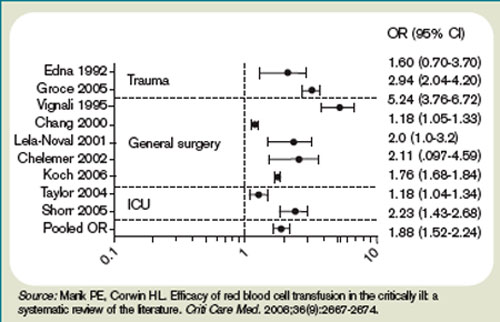
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.

In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.
In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.
In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Face-to-Face Improvement
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
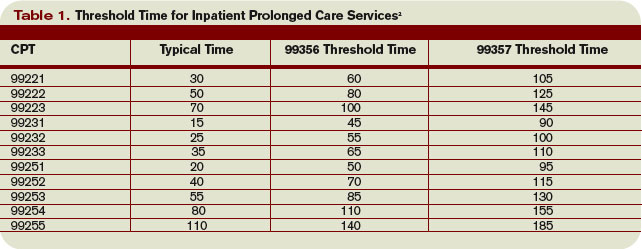
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.

Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.
Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.
Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
Medicare Modifications
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Beware Office Politics
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
The latest research you need to know
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.
Inhaled Insulin: Troubled Drug Rises from the Ashes
Remember all the hype leading up to the approval of the dry-powder formulation of human insulin, produced by means of recombinant DNA technology, a.k.a. inhaled insulin (Exubera)? That was three years ago (January 2006). Remember all the press releases regarding the removal of inhaled insulin from the market? That was October 2007.1,2
Almost immediately after Pfizer “pulled the plug” on inhaled insulin, cases of lung cancer started being reported—albeit it had occurred in Exubera-treated patients that had a history of smoking cigarettes—a contraindication within the drug’s approved label. Some clinicians questioned whether it was due to insulin being a weak growth factor when binding to the type 1 insulin-like growth factor receptor; others wondered if it was related to smoking history.3,4 Three other collaboration efforts for inhaled insulin—NovoNordisk/Aradigm, MannKind, and Alkermes/Eli Lilly AIR insulin— were in the pipeline when Pfizer bowed out of the market. MannKind’s Technosphere insulin and Alkermes/Eli Lilly AIR insulin are still being investigated. Both are in phase 3 clinical trials.5
So, contrary to popular belief, inhaled insulin is not dead, yet. These other companies are looking to improve upon what Pfizer lost out on. The AIR system uses a smaller, breathable inhaler, which would fit into a patient’s hand. The inhaled powder has a smaller particle size and a larger surface area, which provides deeper lung penetration of drug.6
More Drugs Via the Pulmonary Route
Aside from asthma, chronic obstructive pulmonary disease, pulmonary hypertension, and cystic fibrosis, where a hospitalist could expect to use pulmonary delivered drugs, other medicines are being investigated for administration via this route. The pulmonary route may be used for tuberculosis (TB), where lower doses can be given since high doses of systemic therapy lead to significant drug toxicity.7
Inhaled vaccines are being developed, including Bacillus Calmette-Guérin (BCG) TB and respiratory syncytial virus (RSV). Parathyroid hormone for osteoporosis, human factor IX for hemophilia, and interferon α-2b for hepatitis B virus, are potential and current inhaled treatments.
New Delivery Systems: Will They Pan Out?
Knowing the lung absorbs biologic drugs with a wide range of molecular weights, solubility, and charges, is a plus for pulmonary delivery. However, pulmonary drug delivery also presents challenges. These include local toxic effects, such as cell injury, edema, and altered tissue defenses. Drug carriers, preservatives, and propellants, such as sulfites, might harm pulmonary tissue or the body.
Safety is one of the biggest concerns when companies develop new drug delivery systems. These inhaled products and methods of delivering inhaled insulin are quickly moving through clinical trials.
Only time will tell when approvals will take place, but it looks as though there will be some innovative insulin products in the near future. TH
Michele B Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City.
References
1. Alvey L. U.S. Food and Drug Administration. FDA ap-proves first ever inhaled insulin combination product for treatment of diabetes. www.fda.gov/bbs/topics/ news/2006/NEW01304.html. Published Jan. 27, 2006. Accessed Dec. 1, 2008.
2. U.S. Food and Drug Administration. Drug discontinuations. www.fda.gov/cder/drug/shortages/#disc. Published Oct. 19, 2007. Accessed Dec. 1, 2008.
3. von Kriegstein E, von Kriegstein K. Inhaled insulin for diabetes. N Engl J Med. 2007;356:2106-2108.
4. McMahon GT, Arky RA. Inhaled insulin for diabetes. N Engl J Med. 2007;356:497-502.
5. Opar A. Another blow for inhaled protein therapeutics. Nat Rev Drug Discov. 2008;7:189-190.
6. Dubin CH. The state of systemic pulmonary delivery: one year after Exubera’s approval. Nat Rev Drug Discov. 2007;7(4):61-67.
7. Greb E. Inhalable drugs in the launch pad: will they take off? Pharm Tech. 2008;4:48-55.
8. Riley K, Long P. FDA approves treatment for rare neurologic disease. www.fda.gov/bbs/topics/ NEWS/2008/NEW01884.html. Published Sept. 12, 2008. Accessed Dec. 1, 2008.
9. Keppra XR approved in the U.S. hugin.info/133973/ R/1251192/271964.pdf. Published Sept. 15, 2008. Accessed Sept. 15, 2008.
10. U.S. Food and Drug Administration. 2008 safety alerts for human medical products (drugs, biologics, medical devices, special nutritionals, and cosmetics). www.fda.gov/medwatch/safety/2008/rituxan_DHCP_Final%209411700.pdf Published Sept. 2008. Accessed Sept. 15, 2008.
11. U.S. Food and Drug Administration. Pregnacy and lactation labeling. www.fda.gov/cder/regulatory/ pregnancy_labeling/default.htm Published June 11, 2008. Accessed Sept. 15, 2008.
12. Cruzan S. U.S. Food and Drug Administration. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breast-feeding. www. fda.gov/bbs/topics/NEWS/2008/NEW018 41.html. Published May 28, 2008. Accessed Sept. 15, 2008.
13. Peggy P. FDA to take A, B, and C out of pregnancy labeling. www.medpagetoday.com/OBGYN/Pregnancy/ tb/9626. Published May 28, 2008. Accessed Sept. 15, 2008.
Remember all the hype leading up to the approval of the dry-powder formulation of human insulin, produced by means of recombinant DNA technology, a.k.a. inhaled insulin (Exubera)? That was three years ago (January 2006). Remember all the press releases regarding the removal of inhaled insulin from the market? That was October 2007.1,2
Almost immediately after Pfizer “pulled the plug” on inhaled insulin, cases of lung cancer started being reported—albeit it had occurred in Exubera-treated patients that had a history of smoking cigarettes—a contraindication within the drug’s approved label. Some clinicians questioned whether it was due to insulin being a weak growth factor when binding to the type 1 insulin-like growth factor receptor; others wondered if it was related to smoking history.3,4 Three other collaboration efforts for inhaled insulin—NovoNordisk/Aradigm, MannKind, and Alkermes/Eli Lilly AIR insulin— were in the pipeline when Pfizer bowed out of the market. MannKind’s Technosphere insulin and Alkermes/Eli Lilly AIR insulin are still being investigated. Both are in phase 3 clinical trials.5
So, contrary to popular belief, inhaled insulin is not dead, yet. These other companies are looking to improve upon what Pfizer lost out on. The AIR system uses a smaller, breathable inhaler, which would fit into a patient’s hand. The inhaled powder has a smaller particle size and a larger surface area, which provides deeper lung penetration of drug.6
More Drugs Via the Pulmonary Route
Aside from asthma, chronic obstructive pulmonary disease, pulmonary hypertension, and cystic fibrosis, where a hospitalist could expect to use pulmonary delivered drugs, other medicines are being investigated for administration via this route. The pulmonary route may be used for tuberculosis (TB), where lower doses can be given since high doses of systemic therapy lead to significant drug toxicity.7
Inhaled vaccines are being developed, including Bacillus Calmette-Guérin (BCG) TB and respiratory syncytial virus (RSV). Parathyroid hormone for osteoporosis, human factor IX for hemophilia, and interferon α-2b for hepatitis B virus, are potential and current inhaled treatments.
New Delivery Systems: Will They Pan Out?
Knowing the lung absorbs biologic drugs with a wide range of molecular weights, solubility, and charges, is a plus for pulmonary delivery. However, pulmonary drug delivery also presents challenges. These include local toxic effects, such as cell injury, edema, and altered tissue defenses. Drug carriers, preservatives, and propellants, such as sulfites, might harm pulmonary tissue or the body.
Safety is one of the biggest concerns when companies develop new drug delivery systems. These inhaled products and methods of delivering inhaled insulin are quickly moving through clinical trials.
Only time will tell when approvals will take place, but it looks as though there will be some innovative insulin products in the near future. TH
Michele B Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City.
References
1. Alvey L. U.S. Food and Drug Administration. FDA ap-proves first ever inhaled insulin combination product for treatment of diabetes. www.fda.gov/bbs/topics/ news/2006/NEW01304.html. Published Jan. 27, 2006. Accessed Dec. 1, 2008.
2. U.S. Food and Drug Administration. Drug discontinuations. www.fda.gov/cder/drug/shortages/#disc. Published Oct. 19, 2007. Accessed Dec. 1, 2008.
3. von Kriegstein E, von Kriegstein K. Inhaled insulin for diabetes. N Engl J Med. 2007;356:2106-2108.
4. McMahon GT, Arky RA. Inhaled insulin for diabetes. N Engl J Med. 2007;356:497-502.
5. Opar A. Another blow for inhaled protein therapeutics. Nat Rev Drug Discov. 2008;7:189-190.
6. Dubin CH. The state of systemic pulmonary delivery: one year after Exubera’s approval. Nat Rev Drug Discov. 2007;7(4):61-67.
7. Greb E. Inhalable drugs in the launch pad: will they take off? Pharm Tech. 2008;4:48-55.
8. Riley K, Long P. FDA approves treatment for rare neurologic disease. www.fda.gov/bbs/topics/ NEWS/2008/NEW01884.html. Published Sept. 12, 2008. Accessed Dec. 1, 2008.
9. Keppra XR approved in the U.S. hugin.info/133973/ R/1251192/271964.pdf. Published Sept. 15, 2008. Accessed Sept. 15, 2008.
10. U.S. Food and Drug Administration. 2008 safety alerts for human medical products (drugs, biologics, medical devices, special nutritionals, and cosmetics). www.fda.gov/medwatch/safety/2008/rituxan_DHCP_Final%209411700.pdf Published Sept. 2008. Accessed Sept. 15, 2008.
11. U.S. Food and Drug Administration. Pregnacy and lactation labeling. www.fda.gov/cder/regulatory/ pregnancy_labeling/default.htm Published June 11, 2008. Accessed Sept. 15, 2008.
12. Cruzan S. U.S. Food and Drug Administration. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breast-feeding. www. fda.gov/bbs/topics/NEWS/2008/NEW018 41.html. Published May 28, 2008. Accessed Sept. 15, 2008.
13. Peggy P. FDA to take A, B, and C out of pregnancy labeling. www.medpagetoday.com/OBGYN/Pregnancy/ tb/9626. Published May 28, 2008. Accessed Sept. 15, 2008.
Remember all the hype leading up to the approval of the dry-powder formulation of human insulin, produced by means of recombinant DNA technology, a.k.a. inhaled insulin (Exubera)? That was three years ago (January 2006). Remember all the press releases regarding the removal of inhaled insulin from the market? That was October 2007.1,2
Almost immediately after Pfizer “pulled the plug” on inhaled insulin, cases of lung cancer started being reported—albeit it had occurred in Exubera-treated patients that had a history of smoking cigarettes—a contraindication within the drug’s approved label. Some clinicians questioned whether it was due to insulin being a weak growth factor when binding to the type 1 insulin-like growth factor receptor; others wondered if it was related to smoking history.3,4 Three other collaboration efforts for inhaled insulin—NovoNordisk/Aradigm, MannKind, and Alkermes/Eli Lilly AIR insulin— were in the pipeline when Pfizer bowed out of the market. MannKind’s Technosphere insulin and Alkermes/Eli Lilly AIR insulin are still being investigated. Both are in phase 3 clinical trials.5
So, contrary to popular belief, inhaled insulin is not dead, yet. These other companies are looking to improve upon what Pfizer lost out on. The AIR system uses a smaller, breathable inhaler, which would fit into a patient’s hand. The inhaled powder has a smaller particle size and a larger surface area, which provides deeper lung penetration of drug.6
More Drugs Via the Pulmonary Route
Aside from asthma, chronic obstructive pulmonary disease, pulmonary hypertension, and cystic fibrosis, where a hospitalist could expect to use pulmonary delivered drugs, other medicines are being investigated for administration via this route. The pulmonary route may be used for tuberculosis (TB), where lower doses can be given since high doses of systemic therapy lead to significant drug toxicity.7
Inhaled vaccines are being developed, including Bacillus Calmette-Guérin (BCG) TB and respiratory syncytial virus (RSV). Parathyroid hormone for osteoporosis, human factor IX for hemophilia, and interferon α-2b for hepatitis B virus, are potential and current inhaled treatments.
New Delivery Systems: Will They Pan Out?
Knowing the lung absorbs biologic drugs with a wide range of molecular weights, solubility, and charges, is a plus for pulmonary delivery. However, pulmonary drug delivery also presents challenges. These include local toxic effects, such as cell injury, edema, and altered tissue defenses. Drug carriers, preservatives, and propellants, such as sulfites, might harm pulmonary tissue or the body.
Safety is one of the biggest concerns when companies develop new drug delivery systems. These inhaled products and methods of delivering inhaled insulin are quickly moving through clinical trials.
Only time will tell when approvals will take place, but it looks as though there will be some innovative insulin products in the near future. TH
Michele B Kaufman, PharmD, BSc, RPh, is a freelance medical writer based in New York City.
References
1. Alvey L. U.S. Food and Drug Administration. FDA ap-proves first ever inhaled insulin combination product for treatment of diabetes. www.fda.gov/bbs/topics/ news/2006/NEW01304.html. Published Jan. 27, 2006. Accessed Dec. 1, 2008.
2. U.S. Food and Drug Administration. Drug discontinuations. www.fda.gov/cder/drug/shortages/#disc. Published Oct. 19, 2007. Accessed Dec. 1, 2008.
3. von Kriegstein E, von Kriegstein K. Inhaled insulin for diabetes. N Engl J Med. 2007;356:2106-2108.
4. McMahon GT, Arky RA. Inhaled insulin for diabetes. N Engl J Med. 2007;356:497-502.
5. Opar A. Another blow for inhaled protein therapeutics. Nat Rev Drug Discov. 2008;7:189-190.
6. Dubin CH. The state of systemic pulmonary delivery: one year after Exubera’s approval. Nat Rev Drug Discov. 2007;7(4):61-67.
7. Greb E. Inhalable drugs in the launch pad: will they take off? Pharm Tech. 2008;4:48-55.
8. Riley K, Long P. FDA approves treatment for rare neurologic disease. www.fda.gov/bbs/topics/ NEWS/2008/NEW01884.html. Published Sept. 12, 2008. Accessed Dec. 1, 2008.
9. Keppra XR approved in the U.S. hugin.info/133973/ R/1251192/271964.pdf. Published Sept. 15, 2008. Accessed Sept. 15, 2008.
10. U.S. Food and Drug Administration. 2008 safety alerts for human medical products (drugs, biologics, medical devices, special nutritionals, and cosmetics). www.fda.gov/medwatch/safety/2008/rituxan_DHCP_Final%209411700.pdf Published Sept. 2008. Accessed Sept. 15, 2008.
11. U.S. Food and Drug Administration. Pregnacy and lactation labeling. www.fda.gov/cder/regulatory/ pregnancy_labeling/default.htm Published June 11, 2008. Accessed Sept. 15, 2008.
12. Cruzan S. U.S. Food and Drug Administration. FDA proposes new rule to provide updated information on the use of prescription drugs and biological products during pregnancy and breast-feeding. www. fda.gov/bbs/topics/NEWS/2008/NEW018 41.html. Published May 28, 2008. Accessed Sept. 15, 2008.
13. Peggy P. FDA to take A, B, and C out of pregnancy labeling. www.medpagetoday.com/OBGYN/Pregnancy/ tb/9626. Published May 28, 2008. Accessed Sept. 15, 2008.
JUPITER to Earth: A statin helps people with normal LDL-C and high hs-CRP, but what does it mean?
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
KEY POINTS
- LDL-C is the current gold standard diagnostic marker of risk, and elevated values should be aggressively treated in both primary and secondary prevention.
- The optional LDL-C goal of 70 mg/dL for patients at high risk may need to be extended to others at higher global risk, such as those with elevated hs-CRP.
- Although elevated hs-CRP may identify some people with low LDL-C who are nevertheless at higher global risk, more sensitive and specific markers of risk are needed.