User login
Pandemic puts patients with psoriatic disease off seeking medical help
More than half of respondents to a recent survey looking at how the COVID-19 pandemic has affected people with psoriasis or psoriatic arthritis (PsA) said that they had avoided seeking medical care in person with a doctor or at a hospital.
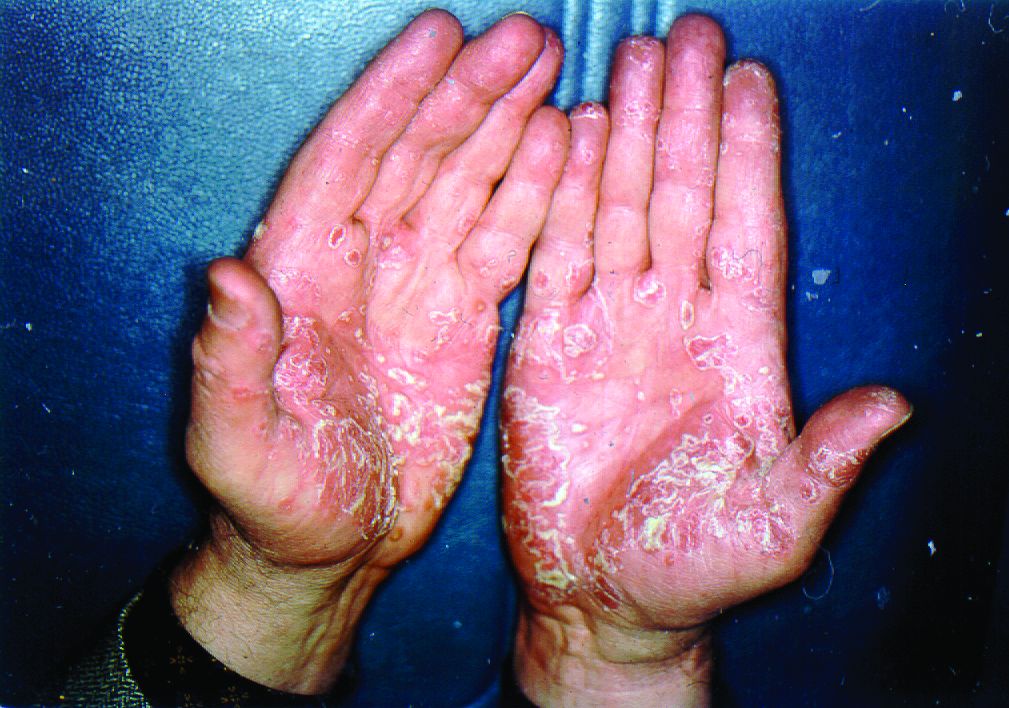
Moreover, around a quarter had their appointment with a rheumatologist canceled, rescheduled, or conducted virtually. Another 1 in 10 had their treatment plan disrupted, and 6% had to change or stop treatment entirely.
The mental health impact of living with these conditions during the pandemic was also notable, said Rachael Manion, the executive director of the Canadian Association of Psoriasis Patients (CAPP), which conducted the survey in collaboration with the Canadian Psoriasis Network (CPN) and Unmasking Psoriasis.
“It’s important to know that there have been a lot of different impacts of the pandemic on people living with psoriatic arthritis and psoriasis. Mental health in particular has had a really big hit as a result,” she said at the Canadian Arthritis Research Conference: Research with Impact.
“About half of the people who responded to our survey noted that their mental health was ‘worse’ or ‘much worse’ during the pandemic,” she said at the meeting, which was sponsored by the Arthritis Society, the Canadian Rheumatology Association, and Canada’s Institute of Musculoskeletal Health and Arthritis. Anxiety and feelings of isolation were reported by a respective 57% and 58% of respondents, and 40% reported depression.
“We can compare that to our earlier information around depression,” Ms. Manion said, which showed that, prior to the pandemic, 24% of people with psoriasis and 23% of those with PsA had said they experienced depression.
“What I found alarming looking at these results was that about a third of people were experiencing despair. Now that’s a really big, scary, overwhelming emotion that has a lot of burden on your mental health,” Ms. Manion said.
Despite the substantial effects on mental health, only 29% of respondents said they had been able to access mental health services during the pandemic.
To look at the impact of the COVID-19 pandemic on the psoriasis and PsA community in Canada, three patient advocacy groups – CAPP, CPN, and Unmasking Psoriasis – codeveloped a survey to look at the disease experience before and after the start of the COVID-19 pandemic. The survey was performed once, with 830 respondents providing information on their lives with psoriasis or PsA in the months before the start of the pandemic and at the time they were surveyed in September and October 2020.
Most of the survey respondents lived in Ontario, Quebec, British Columbia, or Alberta, although other provinces or territories were represented. Almost all respondents (96%) had psoriasis, and 60% also had PsA.
Pre-COVID, nearly half (49%) of patients said that they had not been seen by a rheumatologist, and 39% had not seen a dermatologist for treatment. Asked why, 56% and 27%, respectively, had not been referred, 9% and 15% said they had no specialist located nearby, and 7% and 10% stated that the wait list was too long.
“This tells us that there’s a lot more work that can be done and a lot more education of general practitioners and family medicine professionals about the benefits and the value of specialized care for psoriatic arthritis,” Ms. Manion suggested.
Before the pandemic, joint pain was occurring in 88% of patients, stiffness in 71%, and joint swelling in 67%. Disease flares or sudden periods of worsening occurred on a daily basis for 17%, and around one in five (21%) experienced multiple flares every month.
Prepandemic data also highlighted the negative impact that living with psoriasis or PsA has on people’s ability to sleep, interactions and intimacy with others, and on their school or work lives.
During the pandemic, around a quarter (26%) of respondents said they had worse or much worse access to employment, as well as its benefits such as a stable income (24%). A minority of respondent also described worse access to prescription medication (15%) and over-the-counter medication (13%).
“There are all kinds of things going on for patients in our community: changes to their work, changes to their drug coverage, their ability to sleep and sleep well, their mental health, and their ability to access care and treatments as part of their disease management,” Ms. Manion said.
Her final message to health care professionals was: “I just want to encourage you to continue to check in with your patients about what their experiences have been during the pandemic, and to really consider those impacts as you’re working with them to manage their disease.”
The survey received funding support from AbbVie, Bausch Health, Boehringer Ingelheim, Janssen, LEO Pharma, and Novartis.
More than half of respondents to a recent survey looking at how the COVID-19 pandemic has affected people with psoriasis or psoriatic arthritis (PsA) said that they had avoided seeking medical care in person with a doctor or at a hospital.
Moreover, around a quarter had their appointment with a rheumatologist canceled, rescheduled, or conducted virtually. Another 1 in 10 had their treatment plan disrupted, and 6% had to change or stop treatment entirely.
The mental health impact of living with these conditions during the pandemic was also notable, said Rachael Manion, the executive director of the Canadian Association of Psoriasis Patients (CAPP), which conducted the survey in collaboration with the Canadian Psoriasis Network (CPN) and Unmasking Psoriasis.
“It’s important to know that there have been a lot of different impacts of the pandemic on people living with psoriatic arthritis and psoriasis. Mental health in particular has had a really big hit as a result,” she said at the Canadian Arthritis Research Conference: Research with Impact.
“About half of the people who responded to our survey noted that their mental health was ‘worse’ or ‘much worse’ during the pandemic,” she said at the meeting, which was sponsored by the Arthritis Society, the Canadian Rheumatology Association, and Canada’s Institute of Musculoskeletal Health and Arthritis. Anxiety and feelings of isolation were reported by a respective 57% and 58% of respondents, and 40% reported depression.
“We can compare that to our earlier information around depression,” Ms. Manion said, which showed that, prior to the pandemic, 24% of people with psoriasis and 23% of those with PsA had said they experienced depression.
“What I found alarming looking at these results was that about a third of people were experiencing despair. Now that’s a really big, scary, overwhelming emotion that has a lot of burden on your mental health,” Ms. Manion said.
Despite the substantial effects on mental health, only 29% of respondents said they had been able to access mental health services during the pandemic.
To look at the impact of the COVID-19 pandemic on the psoriasis and PsA community in Canada, three patient advocacy groups – CAPP, CPN, and Unmasking Psoriasis – codeveloped a survey to look at the disease experience before and after the start of the COVID-19 pandemic. The survey was performed once, with 830 respondents providing information on their lives with psoriasis or PsA in the months before the start of the pandemic and at the time they were surveyed in September and October 2020.
Most of the survey respondents lived in Ontario, Quebec, British Columbia, or Alberta, although other provinces or territories were represented. Almost all respondents (96%) had psoriasis, and 60% also had PsA.
Pre-COVID, nearly half (49%) of patients said that they had not been seen by a rheumatologist, and 39% had not seen a dermatologist for treatment. Asked why, 56% and 27%, respectively, had not been referred, 9% and 15% said they had no specialist located nearby, and 7% and 10% stated that the wait list was too long.
“This tells us that there’s a lot more work that can be done and a lot more education of general practitioners and family medicine professionals about the benefits and the value of specialized care for psoriatic arthritis,” Ms. Manion suggested.
Before the pandemic, joint pain was occurring in 88% of patients, stiffness in 71%, and joint swelling in 67%. Disease flares or sudden periods of worsening occurred on a daily basis for 17%, and around one in five (21%) experienced multiple flares every month.
Prepandemic data also highlighted the negative impact that living with psoriasis or PsA has on people’s ability to sleep, interactions and intimacy with others, and on their school or work lives.
During the pandemic, around a quarter (26%) of respondents said they had worse or much worse access to employment, as well as its benefits such as a stable income (24%). A minority of respondent also described worse access to prescription medication (15%) and over-the-counter medication (13%).
“There are all kinds of things going on for patients in our community: changes to their work, changes to their drug coverage, their ability to sleep and sleep well, their mental health, and their ability to access care and treatments as part of their disease management,” Ms. Manion said.
Her final message to health care professionals was: “I just want to encourage you to continue to check in with your patients about what their experiences have been during the pandemic, and to really consider those impacts as you’re working with them to manage their disease.”
The survey received funding support from AbbVie, Bausch Health, Boehringer Ingelheim, Janssen, LEO Pharma, and Novartis.
More than half of respondents to a recent survey looking at how the COVID-19 pandemic has affected people with psoriasis or psoriatic arthritis (PsA) said that they had avoided seeking medical care in person with a doctor or at a hospital.
Moreover, around a quarter had their appointment with a rheumatologist canceled, rescheduled, or conducted virtually. Another 1 in 10 had their treatment plan disrupted, and 6% had to change or stop treatment entirely.
The mental health impact of living with these conditions during the pandemic was also notable, said Rachael Manion, the executive director of the Canadian Association of Psoriasis Patients (CAPP), which conducted the survey in collaboration with the Canadian Psoriasis Network (CPN) and Unmasking Psoriasis.
“It’s important to know that there have been a lot of different impacts of the pandemic on people living with psoriatic arthritis and psoriasis. Mental health in particular has had a really big hit as a result,” she said at the Canadian Arthritis Research Conference: Research with Impact.
“About half of the people who responded to our survey noted that their mental health was ‘worse’ or ‘much worse’ during the pandemic,” she said at the meeting, which was sponsored by the Arthritis Society, the Canadian Rheumatology Association, and Canada’s Institute of Musculoskeletal Health and Arthritis. Anxiety and feelings of isolation were reported by a respective 57% and 58% of respondents, and 40% reported depression.
“We can compare that to our earlier information around depression,” Ms. Manion said, which showed that, prior to the pandemic, 24% of people with psoriasis and 23% of those with PsA had said they experienced depression.
“What I found alarming looking at these results was that about a third of people were experiencing despair. Now that’s a really big, scary, overwhelming emotion that has a lot of burden on your mental health,” Ms. Manion said.
Despite the substantial effects on mental health, only 29% of respondents said they had been able to access mental health services during the pandemic.
To look at the impact of the COVID-19 pandemic on the psoriasis and PsA community in Canada, three patient advocacy groups – CAPP, CPN, and Unmasking Psoriasis – codeveloped a survey to look at the disease experience before and after the start of the COVID-19 pandemic. The survey was performed once, with 830 respondents providing information on their lives with psoriasis or PsA in the months before the start of the pandemic and at the time they were surveyed in September and October 2020.
Most of the survey respondents lived in Ontario, Quebec, British Columbia, or Alberta, although other provinces or territories were represented. Almost all respondents (96%) had psoriasis, and 60% also had PsA.
Pre-COVID, nearly half (49%) of patients said that they had not been seen by a rheumatologist, and 39% had not seen a dermatologist for treatment. Asked why, 56% and 27%, respectively, had not been referred, 9% and 15% said they had no specialist located nearby, and 7% and 10% stated that the wait list was too long.
“This tells us that there’s a lot more work that can be done and a lot more education of general practitioners and family medicine professionals about the benefits and the value of specialized care for psoriatic arthritis,” Ms. Manion suggested.
Before the pandemic, joint pain was occurring in 88% of patients, stiffness in 71%, and joint swelling in 67%. Disease flares or sudden periods of worsening occurred on a daily basis for 17%, and around one in five (21%) experienced multiple flares every month.
Prepandemic data also highlighted the negative impact that living with psoriasis or PsA has on people’s ability to sleep, interactions and intimacy with others, and on their school or work lives.
During the pandemic, around a quarter (26%) of respondents said they had worse or much worse access to employment, as well as its benefits such as a stable income (24%). A minority of respondent also described worse access to prescription medication (15%) and over-the-counter medication (13%).
“There are all kinds of things going on for patients in our community: changes to their work, changes to their drug coverage, their ability to sleep and sleep well, their mental health, and their ability to access care and treatments as part of their disease management,” Ms. Manion said.
Her final message to health care professionals was: “I just want to encourage you to continue to check in with your patients about what their experiences have been during the pandemic, and to really consider those impacts as you’re working with them to manage their disease.”
The survey received funding support from AbbVie, Bausch Health, Boehringer Ingelheim, Janssen, LEO Pharma, and Novartis.
FROM CARC 2021
Psoriasis registry study finds normal pregnancy outcomes
according to one of the largest studies to examine the issue to date.

However, “pregnancy-specific registries that include a larger number of pregnant women with psoriasis ... are needed to more fully characterize the association between psoriasis and treatment and birth outcomes,” acknowledged first author Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, Boston, and colleagues.
The cohort study, published in JAMA Dermatology, used data from the Psoriasis Longitudinal Assessment and Registry (PSOLAR), which “is not a pregnancy specific registry, and medical history is captured only at baseline,” they noted.
Their findings showed pregnancy outcomes such as spontaneous abortion, neonatal problems, and congenital anomalies among women with moderate to severe psoriasis were similar to rates in the general U.S. population, and are “consistent with previously reported data,” they reported. “And pregnancy outcomes for women exposed to biologics were similar to those for women with exposure to nonbiologics.”

The study “provides further reassurance that the biologics appear safe at least related to pregnancy outcomes,” commented Jenny Murase, MD, associate professor of dermatology at the University of California, San Francisco, who was not involved in the study. In an interview, she noted that the study “did not examine any potential immunosuppression of the fetus in the first 6 months of life,” which she described as “the heart of the concern, more than whether or not the psoriasis or the biologic affects the pregnancy itself.”
The study used data from the PSOLAR registry collected from June 20, 2007, to Aug.23, 2019, which included 2,224 women of childbearing age (18-45 years) who were collectively followed up for 12,929 patient-years. Among these women, 220 had 298 pregnancies, with 244 live births (81.9%).
“Birth outcomes among all 244 births included 231 healthy newborns (94.7%), 10 infants with a neonatal problem (4.1%), 1 stillbirth (0.4%), and 2 congenital anomalies (0.8%),” the authors reported.
There were also 41 spontaneous abortions (13.8%), and 13 elective terminations (4.4%). “No elective terminations were known to derive from a congenital anomaly or other medical issue,” they added.
Among the documented pregnancies, 252 occurred in women with exposure to biologic therapy either before or during pregnancy, including 168 (56.4%) during the prenatal period, while 46 pregnancies occurred in women with no exposure to biologic therapy.
Dr. Murase, director of medical consultative dermatology for the Palo Alto Foundation Medical Group in Mountain View, Calif., said that a more detailed comparison of the different psoriasis treatments, as well as the offspring outcomes during the first 6 months of life, might offer some further important insight,.
Infants born after exposure to infliximab “and potentially other anti–tumor necrosis factor–alpha agents during the third trimester may be unable to develop an appropriate immune response to live vaccines,” she and her coauthors cautioned in a letter published in 2011, which referred to a case of an infant with disseminated bacillus Calmette-Guérin infection, whose mother had received infliximab for Crohn’s disease throughout pregnancy.
Dr. Murase pointed out that, in the registry study, exposures to certolizumab, which is pegylated and does not cross the placental barrier, were not separated from other cases. It is important to consider “the cross over late in the second trimester and especially third trimester as the infant is getting the ‘antibody boost’ from the mother as it gets ready to set foot in this world and needs the maternal antibodies to prepare its immune system. If the IgG biologics cross third trimester and immunosuppress the infant ... then I think a medication that does not cross the placental barrier is important to consider.”
The study was sponsored by Janssen Scientific Affairs. Dr. Kimball’s disclosures included serving as a consultant and investigator for companies that included AbbVie, Bristol-Myers Squibb, and Janssen; several other authors also had disclosures related to multiple pharmaceutical companies. Dr. Murase’s disclosures included serving as a consultant for Dermira, UCB Pharma, Sanofi, Ferndale, and Regeneron.
according to one of the largest studies to examine the issue to date.
However, “pregnancy-specific registries that include a larger number of pregnant women with psoriasis ... are needed to more fully characterize the association between psoriasis and treatment and birth outcomes,” acknowledged first author Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, Boston, and colleagues.
The cohort study, published in JAMA Dermatology, used data from the Psoriasis Longitudinal Assessment and Registry (PSOLAR), which “is not a pregnancy specific registry, and medical history is captured only at baseline,” they noted.
Their findings showed pregnancy outcomes such as spontaneous abortion, neonatal problems, and congenital anomalies among women with moderate to severe psoriasis were similar to rates in the general U.S. population, and are “consistent with previously reported data,” they reported. “And pregnancy outcomes for women exposed to biologics were similar to those for women with exposure to nonbiologics.”
The study “provides further reassurance that the biologics appear safe at least related to pregnancy outcomes,” commented Jenny Murase, MD, associate professor of dermatology at the University of California, San Francisco, who was not involved in the study. In an interview, she noted that the study “did not examine any potential immunosuppression of the fetus in the first 6 months of life,” which she described as “the heart of the concern, more than whether or not the psoriasis or the biologic affects the pregnancy itself.”
The study used data from the PSOLAR registry collected from June 20, 2007, to Aug.23, 2019, which included 2,224 women of childbearing age (18-45 years) who were collectively followed up for 12,929 patient-years. Among these women, 220 had 298 pregnancies, with 244 live births (81.9%).
“Birth outcomes among all 244 births included 231 healthy newborns (94.7%), 10 infants with a neonatal problem (4.1%), 1 stillbirth (0.4%), and 2 congenital anomalies (0.8%),” the authors reported.
There were also 41 spontaneous abortions (13.8%), and 13 elective terminations (4.4%). “No elective terminations were known to derive from a congenital anomaly or other medical issue,” they added.
Among the documented pregnancies, 252 occurred in women with exposure to biologic therapy either before or during pregnancy, including 168 (56.4%) during the prenatal period, while 46 pregnancies occurred in women with no exposure to biologic therapy.
Dr. Murase, director of medical consultative dermatology for the Palo Alto Foundation Medical Group in Mountain View, Calif., said that a more detailed comparison of the different psoriasis treatments, as well as the offspring outcomes during the first 6 months of life, might offer some further important insight,.
Infants born after exposure to infliximab “and potentially other anti–tumor necrosis factor–alpha agents during the third trimester may be unable to develop an appropriate immune response to live vaccines,” she and her coauthors cautioned in a letter published in 2011, which referred to a case of an infant with disseminated bacillus Calmette-Guérin infection, whose mother had received infliximab for Crohn’s disease throughout pregnancy.
Dr. Murase pointed out that, in the registry study, exposures to certolizumab, which is pegylated and does not cross the placental barrier, were not separated from other cases. It is important to consider “the cross over late in the second trimester and especially third trimester as the infant is getting the ‘antibody boost’ from the mother as it gets ready to set foot in this world and needs the maternal antibodies to prepare its immune system. If the IgG biologics cross third trimester and immunosuppress the infant ... then I think a medication that does not cross the placental barrier is important to consider.”
The study was sponsored by Janssen Scientific Affairs. Dr. Kimball’s disclosures included serving as a consultant and investigator for companies that included AbbVie, Bristol-Myers Squibb, and Janssen; several other authors also had disclosures related to multiple pharmaceutical companies. Dr. Murase’s disclosures included serving as a consultant for Dermira, UCB Pharma, Sanofi, Ferndale, and Regeneron.
according to one of the largest studies to examine the issue to date.
However, “pregnancy-specific registries that include a larger number of pregnant women with psoriasis ... are needed to more fully characterize the association between psoriasis and treatment and birth outcomes,” acknowledged first author Alexa B. Kimball, MD, MPH, professor of dermatology, Harvard Medical School, Boston, and colleagues.
The cohort study, published in JAMA Dermatology, used data from the Psoriasis Longitudinal Assessment and Registry (PSOLAR), which “is not a pregnancy specific registry, and medical history is captured only at baseline,” they noted.
Their findings showed pregnancy outcomes such as spontaneous abortion, neonatal problems, and congenital anomalies among women with moderate to severe psoriasis were similar to rates in the general U.S. population, and are “consistent with previously reported data,” they reported. “And pregnancy outcomes for women exposed to biologics were similar to those for women with exposure to nonbiologics.”
The study “provides further reassurance that the biologics appear safe at least related to pregnancy outcomes,” commented Jenny Murase, MD, associate professor of dermatology at the University of California, San Francisco, who was not involved in the study. In an interview, she noted that the study “did not examine any potential immunosuppression of the fetus in the first 6 months of life,” which she described as “the heart of the concern, more than whether or not the psoriasis or the biologic affects the pregnancy itself.”
The study used data from the PSOLAR registry collected from June 20, 2007, to Aug.23, 2019, which included 2,224 women of childbearing age (18-45 years) who were collectively followed up for 12,929 patient-years. Among these women, 220 had 298 pregnancies, with 244 live births (81.9%).
“Birth outcomes among all 244 births included 231 healthy newborns (94.7%), 10 infants with a neonatal problem (4.1%), 1 stillbirth (0.4%), and 2 congenital anomalies (0.8%),” the authors reported.
There were also 41 spontaneous abortions (13.8%), and 13 elective terminations (4.4%). “No elective terminations were known to derive from a congenital anomaly or other medical issue,” they added.
Among the documented pregnancies, 252 occurred in women with exposure to biologic therapy either before or during pregnancy, including 168 (56.4%) during the prenatal period, while 46 pregnancies occurred in women with no exposure to biologic therapy.
Dr. Murase, director of medical consultative dermatology for the Palo Alto Foundation Medical Group in Mountain View, Calif., said that a more detailed comparison of the different psoriasis treatments, as well as the offspring outcomes during the first 6 months of life, might offer some further important insight,.
Infants born after exposure to infliximab “and potentially other anti–tumor necrosis factor–alpha agents during the third trimester may be unable to develop an appropriate immune response to live vaccines,” she and her coauthors cautioned in a letter published in 2011, which referred to a case of an infant with disseminated bacillus Calmette-Guérin infection, whose mother had received infliximab for Crohn’s disease throughout pregnancy.
Dr. Murase pointed out that, in the registry study, exposures to certolizumab, which is pegylated and does not cross the placental barrier, were not separated from other cases. It is important to consider “the cross over late in the second trimester and especially third trimester as the infant is getting the ‘antibody boost’ from the mother as it gets ready to set foot in this world and needs the maternal antibodies to prepare its immune system. If the IgG biologics cross third trimester and immunosuppress the infant ... then I think a medication that does not cross the placental barrier is important to consider.”
The study was sponsored by Janssen Scientific Affairs. Dr. Kimball’s disclosures included serving as a consultant and investigator for companies that included AbbVie, Bristol-Myers Squibb, and Janssen; several other authors also had disclosures related to multiple pharmaceutical companies. Dr. Murase’s disclosures included serving as a consultant for Dermira, UCB Pharma, Sanofi, Ferndale, and Regeneron.
FROM JAMA DERMATOLOGY
Anybody for a nanobody? Novel psoriasis therapy impresses in phase 2b
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
in a phase 2b randomized trial, Kim A. Papp, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
A nanobody is a tiny antibody fragment with a much smaller molecular weight than the monoclonal antibodies utilized today in treating psoriasis or atopic dermatitis. The sonelokinab nanobody, derived from animals in the camel family, is a recombinant sequence-optimized nanobody specific for human IL-17F, IL-17A, the heterodimer IL-17A/F, and serum albumin. The binding to serum albumin give sonelokinab a lengthy half-life of 10-12 hours, which may be therapeutically relevant, explained Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
He presented the 24-week results of a multicenter, double-blind, double-dummy randomized trial including 313 North American and European adults with an average 18-year history of psoriasis and a baseline Psoriasis Area and Severity Index (PASI) score of about 21. They were randomized to one of six treatment arms for the first 12 weeks: subcutaneous injection of sonelokinab at 30, 60, or 120 mg at weeks 0, 2, 4, and 8; enhanced–loading-dose sonelokinab at 120 mg every 2 weeks through week 10; the IL-17A inhibitor secukinumab (Cosentyx) at its standard dosing as an active comparator; or placebo. Data analysis was by rigorous nonresponder imputation, meaning anyone who didn’t complete the study was scored as a nonresponder.
“This yields a conservative data analysis somewhat biased against sonelokinab,” the dermatologist pointed out.
The primary outcome in the trial was the week-12 rate of an Investigator’s Global Assessment score of 0 or 1, indicative of clear or almost clear skin. This was achieved in 88.2% of patients in the highest-dose arm of sonelokinab. That group also had a week-12 PASI 90 response rate of 76.5% and a PASI 100 response rate of 33.3%. By comparison, patients on standard-dose secukinumab had a less robust week-12 IGA 0/1 rate of 77.4%, a PASI 90 of 64.2%, and a PASI 100 of 28.3%. Of note, however, this secukinumab performance was better than seen in the 30-mg sonelokinab group, and comparable to outcomes with 60 mg of sonelokinab.
Dose escalation was performed from weeks 12-24. Patients with a week-12 IGA score greater than 1 after being on sonelokinab at 30 or 60 mg were upgraded to 120 mg at week 12 and again every 4 weeks thereafter. Placebo-treated controls were switched to 120 mg at weeks 12, 14, 16, and every 4 weeks thereafter. The group on the enhanced–loading-dose sonelokinab moved to 120 mg every 4 weeks, while those who had gotten four doses of sonelokinab at 120 mg during the first 12 weeks were switched to 120 mg every 8 weeks. The secukinumab group remained on the approved dosing through week 24.
At week 24, superior outcomes were seen in the enhanced–loading-dose sonelokinab group, with an IGA 0/1 response rate of 94.2%, a PASI 90 of 90.4%, and a PASI 100 of 56.9%. The corresponding week-24 rates in patients on 120 mg of sonelokinab every 8 weeks from week 12 on were 80.4%, 79.2%, and 40.4%, outcomes similar to those seen with secukinumab.
The rapidity of response to sonelokinab at 120 mg was striking, with approximately one-third of treated patients achieving a PASI 90 response by week 4.
“This could reflect the smaller molecular profile. There is possibly rapid increased absorption or bioavailability, quicker time to achieving serum half-life, better penetration into target tissue, and perhaps more effective engagement at the target. All of those things are possibilities. These are things that are yet to be explored, but it’s very enticing to see that uncharacteristically rapid initial response. It’s all very gratifying – and tantalizing,” Dr. Papp said in response to an audience question.
The safety profile of sonelokinab was reassuring. The most common adverse events were nasopharyngitis in 13.5% of patients and pruritus in 6.7%, with most cases being mild or moderate. As with other IL-17 blockers, there was an increase in oral candidiasis. This side effect appeared to occur in dose-dependent fashion: The incidence was zero in the 30-mg group, 1.9% with 60 mg, 3.8% with sonelokinab at 120 mg without an enhanced loading dose, and 5.9% with the enhanced loading dose.
The study was conducted by Avillion in partnership with Merck. Dr. Papp reported receiving research funding from and serving as a consultant to those and numerous other pharmaceutical companies.
FROM the eadv congress
Rheumatologic disease activity an important influencer of COVID-19 death risk
People with rheumatic and musculoskeletal diseases (RMDs) who contract the SARS-CoV-2 virus appear more likely to die from COVID-19 if their rheumatologic condition is not being well controlled at the time of their infection.

New data from the COVID-19 Global Rheumatology Alliance (GRA) physician registry reported in Annals of the Rheumatic Diseases have found that the odds of dying from COVID-19 were 87% higher in individuals recorded as having moderate to high disease activity versus those reported to be in remission or having low disease activity.
“I think this really highlights the importance of continuing to appropriately, and actively, treat our patients, and the importance of controlling their disease,” Pedro Machado, MD, PhD, said in an interview. Dr. Machado, an associate professor in rheumatology and muscle diseases at University College London and a consultant rheumatologist at several U.K. hospitals, has been involved in the GRA physician registry from the start, and sits on the GRA steering committee.
Alongside higher disease activity, several other important factors were found to be associated with increased odds of dying from COVID-19 – older age, male gender, and the presence of one or more comorbidities, such as hypertension combined with cardiovascular disease or chronic lung disease.
These demographic and disease-based factors have been linked to an increased risk for COVID-19–related hospitalization before, both in people with RMDs and in the general population, but the latest GRA physician registry data now take that a step further, and link them also to an increased risk for death, together with several other factors more specific to RMDs.
Logging COVID-19 rheumatologic cases
Since the start of the global pandemic, the potential effects that SARS-CoV-2 infection might have on people with RMDs in particular has concerned the rheumatology community. The main worries being that, either because of the underlying RMD itself or to its treatment, there may be immunoregulatory deficits or other risk factors that would make individuals more susceptible to not only infection but also to developing more severe COVID-19 than the general population.
These concerns led to the rapid formation of the GRA and the COVID-19 GRA physician registry in March 2020 to collect and analyze data on adults with rheumatic disease and confirmed or presumptive COVID-19. Entries into the registry are made by or under the direction of rheumatologists, and this is a voluntary process.
“This population cannot ever be entirely representative of the population of patients with rheumatic diseases,” Dr. Machado acknowledged. There will be selection and other biases that affect the reported data. That said, it’s the largest database of reported COVID-19 cases in adult rheumatology patients across the world, with more than 9,000 cases so far included from multiple registries, including those based in Europe and North and South America. Data from one of these – the French RMD cohort – have also recently been published in Annals of the Rheumatic Diseases, showing much the same findings but on a national level.
Hospitalization was the focus of a previous report because “you need large sample sizes” to look at endpoints that occur less frequently. When the first analysis was done, there were around 600 cases from 40 countries in the registry with sufficient data that could be used. Now, with a greater number of recorded cases, factors influencing the risk for death could be examined.
Death rate and risk factors found
Data on 3,729 COVID-19 cases in people with RMDs were included in the current analysis, all recorded in the first few months of the registry being open and up until July 1, 2020. In all, 390 (10.5%) of people died. While this is “clearly higher” than reported in the general population in most countries, the analysis was not designed to calculate a precise estimate.
“It should not be taken as an estimate of the overall death rate among patients with rheumatic diseases and COVID-19,” Dr. Machado and coauthors have been keen to point out.
“Age is always the biggest risk factor,” Dr. Machado explained. “There’s always a gradient: the older the patient, the worse the outcome.”
Indeed, there was a threefold increased risk for death among those aged 66-75 years versus those who were 65 years or younger (odds ratio, 3.00), and a sixfold increased risk for patients older than 75, compared with the younger age group (OR, 6.18).
Having both hypertension and cardiovascular disease was associated with an OR of 1.89, and coexisting chronic lung disease also significantly increased the chances of dying from COVID-19 (OR, 1.68).
Being of male sex was associated with a 46% increased risk for death from COVID-19 versus being of female sex.
The risk for COVID-19 death also rose with the use of corticosteroids. Compared with no steroid use, there was a 69% increased risk for with death at doses of 10 mg or more prednisolone equivalent per day.
“The finding about moderate to high doses of steroids being associated with a worse outcome is consistent with the first report; it was the same for hospitalization,” Dr. Machado observed.
The general consensus on steroid use in the COVID-19 setting is that they should be continued as needed, but at the lowest possible dose, as outlined in provisional recommendations set out by the recently renamed European Alliance of Associations for Rheumatology.
The GRA physician registry findings provide further support for this, suggesting that disease control should be optimized with disease-modifying antirheumatic drugs, ideally without increasing the dose of steroids.
Surprise over sulfasalazine risk
“Taking all medications into account – such as methotrexate, leflunomide, hydroxychloroquine, [tumor necrosis factor] blockers, interleukin-6 blockers, and [Janus kinase] inhibitors – it is quite reassuring because we did not see an association with worse outcome with those drugs overall,” Dr. Machado said.
However, treatment with rituximab (OR, 4.0), sulfasalazine (OR, 3.6), and immunosuppressive agents such as azathioprine, cyclophosphamide, cyclosporine, mycophenolate, or tacrolimus (OR, 2.2), were associated with higher odds of dying from COVID-19 when compared with treatment with methotrexate alone.
The findings for rituximab and immunosuppressant use were perhaps not unexpected, but the possible association between sulfasalazine and COVID-19 death was “a bit intriguing,” Dr. Machado observed. “Sulfasalazine is believed to have low immunosuppressive effect.”
This warrants further investigation, but there are likely a range of confounding factors at play. One could be that people considered to be at higher risk may have been more often prescribed sulfasalazine because it was thought to be less immunosuppressive. Another might be because people taking sulfasalazine were more likely to be smokers, and they were also not advised to protect themselves from exposure to the virus (shielding) during the first wave of the pandemic, at least not in the United Kingdom.
Rituximab caution and vaccination
“Rituximab is a concern,” Dr. Machado acknowledged. “It is a concern that rheumatologists are now aware of and they are addressing, but then it’s a concern for a very specific subgroup of patients.”
While rheumatologists are, and will continue to prescribe it, there will be even more careful consideration over when, in whom, and how to use it during, and possibly even after, the pandemic.
“COVID is here to stay, it will become endemic, and it’s going to be part of our lives like the flu virus is,” Dr. Machado predicted.
Then there is the issue on vaccinating people against COVID-19, should those on rituximab still receive it? The answer is a yes, but, as with other vaccinations it’s all about the timing of when the vaccination is given.
Societies such as the British Society for Rheumatology have already begun to include guidance on this, recommending one of the available COVID-19 vaccines is given at least a month before the next or first dose of rituximab is due. As rituximab is given every few months, with doses sometimes spaced as much as 9 months or even a year apart, this should not be too much of a problem, but it is “better to have the vaccine first,” Dr. Machado said.
Has COVID-19 care improved in RMDs?
In separate research published in The Lancet Rheumatology, April Jorge, MD, of Massachusetts General Hospital and Harvard Medical School, both in Boston, and associates found that the risks of severe COVID-19 outcomes have improved over time, although they still “remain substantial.”
Dr. Jorge and colleagues looked at temporal trends in COVID-19 outcomes in patients with RMDs over the course of the first 6 months of the pandemic in 2020, using data from a large, multicenter, electronic health record network (TriNetX).
They formed two patient cohorts – a late (diagnosed from April 20 to July 20) and an early (diagnosed from January 20 to April 20) cohort – to see if outcomes had improved and discovered lower relative risks among patients in the late cohort for hospitalization (0.67), admission to the ICU (0.56), mechanical ventilation (0.39), acute kidney injury (0.66), renal replacement (0.53), and death (0.39).
“These results are encouraging,” but it’s difficult to match these different populations of patients, Dr. Machado said. “There are always factors that you cannot match for” and were not included in the U.S. analysis.
While there are important caveats in how the analysis was performed and thus in interpreting these data, they do “suggest that one of the reasons why outcomes have improved is because we have become better at treating these patients,” Dr. Machado added.
“Our treatment has improved, and our capacity to treat the complications has improved. We understand better how the disease behaves – we know that they can have thromboembolic complications that we can manage, and we are now able to manage ventilation issues better.”
Moreover, Dr. Machado said that, not only were clinicians more aware of what they should or should not do, there were treatments that were being used routinely or in some cases based on recent clinical trial results. “I think we are indeed treating these patients better.”
The COVID-19 GRA physician registry is financially supported by the American College of Rheumatology and EULAR. Dr. Machado had no relevant conflicts of interest.
People with rheumatic and musculoskeletal diseases (RMDs) who contract the SARS-CoV-2 virus appear more likely to die from COVID-19 if their rheumatologic condition is not being well controlled at the time of their infection.
New data from the COVID-19 Global Rheumatology Alliance (GRA) physician registry reported in Annals of the Rheumatic Diseases have found that the odds of dying from COVID-19 were 87% higher in individuals recorded as having moderate to high disease activity versus those reported to be in remission or having low disease activity.
“I think this really highlights the importance of continuing to appropriately, and actively, treat our patients, and the importance of controlling their disease,” Pedro Machado, MD, PhD, said in an interview. Dr. Machado, an associate professor in rheumatology and muscle diseases at University College London and a consultant rheumatologist at several U.K. hospitals, has been involved in the GRA physician registry from the start, and sits on the GRA steering committee.
Alongside higher disease activity, several other important factors were found to be associated with increased odds of dying from COVID-19 – older age, male gender, and the presence of one or more comorbidities, such as hypertension combined with cardiovascular disease or chronic lung disease.
These demographic and disease-based factors have been linked to an increased risk for COVID-19–related hospitalization before, both in people with RMDs and in the general population, but the latest GRA physician registry data now take that a step further, and link them also to an increased risk for death, together with several other factors more specific to RMDs.
Logging COVID-19 rheumatologic cases
Since the start of the global pandemic, the potential effects that SARS-CoV-2 infection might have on people with RMDs in particular has concerned the rheumatology community. The main worries being that, either because of the underlying RMD itself or to its treatment, there may be immunoregulatory deficits or other risk factors that would make individuals more susceptible to not only infection but also to developing more severe COVID-19 than the general population.
These concerns led to the rapid formation of the GRA and the COVID-19 GRA physician registry in March 2020 to collect and analyze data on adults with rheumatic disease and confirmed or presumptive COVID-19. Entries into the registry are made by or under the direction of rheumatologists, and this is a voluntary process.
“This population cannot ever be entirely representative of the population of patients with rheumatic diseases,” Dr. Machado acknowledged. There will be selection and other biases that affect the reported data. That said, it’s the largest database of reported COVID-19 cases in adult rheumatology patients across the world, with more than 9,000 cases so far included from multiple registries, including those based in Europe and North and South America. Data from one of these – the French RMD cohort – have also recently been published in Annals of the Rheumatic Diseases, showing much the same findings but on a national level.
Hospitalization was the focus of a previous report because “you need large sample sizes” to look at endpoints that occur less frequently. When the first analysis was done, there were around 600 cases from 40 countries in the registry with sufficient data that could be used. Now, with a greater number of recorded cases, factors influencing the risk for death could be examined.
Death rate and risk factors found
Data on 3,729 COVID-19 cases in people with RMDs were included in the current analysis, all recorded in the first few months of the registry being open and up until July 1, 2020. In all, 390 (10.5%) of people died. While this is “clearly higher” than reported in the general population in most countries, the analysis was not designed to calculate a precise estimate.
“It should not be taken as an estimate of the overall death rate among patients with rheumatic diseases and COVID-19,” Dr. Machado and coauthors have been keen to point out.
“Age is always the biggest risk factor,” Dr. Machado explained. “There’s always a gradient: the older the patient, the worse the outcome.”
Indeed, there was a threefold increased risk for death among those aged 66-75 years versus those who were 65 years or younger (odds ratio, 3.00), and a sixfold increased risk for patients older than 75, compared with the younger age group (OR, 6.18).
Having both hypertension and cardiovascular disease was associated with an OR of 1.89, and coexisting chronic lung disease also significantly increased the chances of dying from COVID-19 (OR, 1.68).
Being of male sex was associated with a 46% increased risk for death from COVID-19 versus being of female sex.
The risk for COVID-19 death also rose with the use of corticosteroids. Compared with no steroid use, there was a 69% increased risk for with death at doses of 10 mg or more prednisolone equivalent per day.
“The finding about moderate to high doses of steroids being associated with a worse outcome is consistent with the first report; it was the same for hospitalization,” Dr. Machado observed.
The general consensus on steroid use in the COVID-19 setting is that they should be continued as needed, but at the lowest possible dose, as outlined in provisional recommendations set out by the recently renamed European Alliance of Associations for Rheumatology.
The GRA physician registry findings provide further support for this, suggesting that disease control should be optimized with disease-modifying antirheumatic drugs, ideally without increasing the dose of steroids.
Surprise over sulfasalazine risk
“Taking all medications into account – such as methotrexate, leflunomide, hydroxychloroquine, [tumor necrosis factor] blockers, interleukin-6 blockers, and [Janus kinase] inhibitors – it is quite reassuring because we did not see an association with worse outcome with those drugs overall,” Dr. Machado said.
However, treatment with rituximab (OR, 4.0), sulfasalazine (OR, 3.6), and immunosuppressive agents such as azathioprine, cyclophosphamide, cyclosporine, mycophenolate, or tacrolimus (OR, 2.2), were associated with higher odds of dying from COVID-19 when compared with treatment with methotrexate alone.
The findings for rituximab and immunosuppressant use were perhaps not unexpected, but the possible association between sulfasalazine and COVID-19 death was “a bit intriguing,” Dr. Machado observed. “Sulfasalazine is believed to have low immunosuppressive effect.”
This warrants further investigation, but there are likely a range of confounding factors at play. One could be that people considered to be at higher risk may have been more often prescribed sulfasalazine because it was thought to be less immunosuppressive. Another might be because people taking sulfasalazine were more likely to be smokers, and they were also not advised to protect themselves from exposure to the virus (shielding) during the first wave of the pandemic, at least not in the United Kingdom.
Rituximab caution and vaccination
“Rituximab is a concern,” Dr. Machado acknowledged. “It is a concern that rheumatologists are now aware of and they are addressing, but then it’s a concern for a very specific subgroup of patients.”
While rheumatologists are, and will continue to prescribe it, there will be even more careful consideration over when, in whom, and how to use it during, and possibly even after, the pandemic.
“COVID is here to stay, it will become endemic, and it’s going to be part of our lives like the flu virus is,” Dr. Machado predicted.
Then there is the issue on vaccinating people against COVID-19, should those on rituximab still receive it? The answer is a yes, but, as with other vaccinations it’s all about the timing of when the vaccination is given.
Societies such as the British Society for Rheumatology have already begun to include guidance on this, recommending one of the available COVID-19 vaccines is given at least a month before the next or first dose of rituximab is due. As rituximab is given every few months, with doses sometimes spaced as much as 9 months or even a year apart, this should not be too much of a problem, but it is “better to have the vaccine first,” Dr. Machado said.
Has COVID-19 care improved in RMDs?
In separate research published in The Lancet Rheumatology, April Jorge, MD, of Massachusetts General Hospital and Harvard Medical School, both in Boston, and associates found that the risks of severe COVID-19 outcomes have improved over time, although they still “remain substantial.”
Dr. Jorge and colleagues looked at temporal trends in COVID-19 outcomes in patients with RMDs over the course of the first 6 months of the pandemic in 2020, using data from a large, multicenter, electronic health record network (TriNetX).
They formed two patient cohorts – a late (diagnosed from April 20 to July 20) and an early (diagnosed from January 20 to April 20) cohort – to see if outcomes had improved and discovered lower relative risks among patients in the late cohort for hospitalization (0.67), admission to the ICU (0.56), mechanical ventilation (0.39), acute kidney injury (0.66), renal replacement (0.53), and death (0.39).
“These results are encouraging,” but it’s difficult to match these different populations of patients, Dr. Machado said. “There are always factors that you cannot match for” and were not included in the U.S. analysis.
While there are important caveats in how the analysis was performed and thus in interpreting these data, they do “suggest that one of the reasons why outcomes have improved is because we have become better at treating these patients,” Dr. Machado added.
“Our treatment has improved, and our capacity to treat the complications has improved. We understand better how the disease behaves – we know that they can have thromboembolic complications that we can manage, and we are now able to manage ventilation issues better.”
Moreover, Dr. Machado said that, not only were clinicians more aware of what they should or should not do, there were treatments that were being used routinely or in some cases based on recent clinical trial results. “I think we are indeed treating these patients better.”
The COVID-19 GRA physician registry is financially supported by the American College of Rheumatology and EULAR. Dr. Machado had no relevant conflicts of interest.
People with rheumatic and musculoskeletal diseases (RMDs) who contract the SARS-CoV-2 virus appear more likely to die from COVID-19 if their rheumatologic condition is not being well controlled at the time of their infection.
New data from the COVID-19 Global Rheumatology Alliance (GRA) physician registry reported in Annals of the Rheumatic Diseases have found that the odds of dying from COVID-19 were 87% higher in individuals recorded as having moderate to high disease activity versus those reported to be in remission or having low disease activity.
“I think this really highlights the importance of continuing to appropriately, and actively, treat our patients, and the importance of controlling their disease,” Pedro Machado, MD, PhD, said in an interview. Dr. Machado, an associate professor in rheumatology and muscle diseases at University College London and a consultant rheumatologist at several U.K. hospitals, has been involved in the GRA physician registry from the start, and sits on the GRA steering committee.
Alongside higher disease activity, several other important factors were found to be associated with increased odds of dying from COVID-19 – older age, male gender, and the presence of one or more comorbidities, such as hypertension combined with cardiovascular disease or chronic lung disease.
These demographic and disease-based factors have been linked to an increased risk for COVID-19–related hospitalization before, both in people with RMDs and in the general population, but the latest GRA physician registry data now take that a step further, and link them also to an increased risk for death, together with several other factors more specific to RMDs.
Logging COVID-19 rheumatologic cases
Since the start of the global pandemic, the potential effects that SARS-CoV-2 infection might have on people with RMDs in particular has concerned the rheumatology community. The main worries being that, either because of the underlying RMD itself or to its treatment, there may be immunoregulatory deficits or other risk factors that would make individuals more susceptible to not only infection but also to developing more severe COVID-19 than the general population.
These concerns led to the rapid formation of the GRA and the COVID-19 GRA physician registry in March 2020 to collect and analyze data on adults with rheumatic disease and confirmed or presumptive COVID-19. Entries into the registry are made by or under the direction of rheumatologists, and this is a voluntary process.
“This population cannot ever be entirely representative of the population of patients with rheumatic diseases,” Dr. Machado acknowledged. There will be selection and other biases that affect the reported data. That said, it’s the largest database of reported COVID-19 cases in adult rheumatology patients across the world, with more than 9,000 cases so far included from multiple registries, including those based in Europe and North and South America. Data from one of these – the French RMD cohort – have also recently been published in Annals of the Rheumatic Diseases, showing much the same findings but on a national level.
Hospitalization was the focus of a previous report because “you need large sample sizes” to look at endpoints that occur less frequently. When the first analysis was done, there were around 600 cases from 40 countries in the registry with sufficient data that could be used. Now, with a greater number of recorded cases, factors influencing the risk for death could be examined.
Death rate and risk factors found
Data on 3,729 COVID-19 cases in people with RMDs were included in the current analysis, all recorded in the first few months of the registry being open and up until July 1, 2020. In all, 390 (10.5%) of people died. While this is “clearly higher” than reported in the general population in most countries, the analysis was not designed to calculate a precise estimate.
“It should not be taken as an estimate of the overall death rate among patients with rheumatic diseases and COVID-19,” Dr. Machado and coauthors have been keen to point out.
“Age is always the biggest risk factor,” Dr. Machado explained. “There’s always a gradient: the older the patient, the worse the outcome.”
Indeed, there was a threefold increased risk for death among those aged 66-75 years versus those who were 65 years or younger (odds ratio, 3.00), and a sixfold increased risk for patients older than 75, compared with the younger age group (OR, 6.18).
Having both hypertension and cardiovascular disease was associated with an OR of 1.89, and coexisting chronic lung disease also significantly increased the chances of dying from COVID-19 (OR, 1.68).
Being of male sex was associated with a 46% increased risk for death from COVID-19 versus being of female sex.
The risk for COVID-19 death also rose with the use of corticosteroids. Compared with no steroid use, there was a 69% increased risk for with death at doses of 10 mg or more prednisolone equivalent per day.
“The finding about moderate to high doses of steroids being associated with a worse outcome is consistent with the first report; it was the same for hospitalization,” Dr. Machado observed.
The general consensus on steroid use in the COVID-19 setting is that they should be continued as needed, but at the lowest possible dose, as outlined in provisional recommendations set out by the recently renamed European Alliance of Associations for Rheumatology.
The GRA physician registry findings provide further support for this, suggesting that disease control should be optimized with disease-modifying antirheumatic drugs, ideally without increasing the dose of steroids.
Surprise over sulfasalazine risk
“Taking all medications into account – such as methotrexate, leflunomide, hydroxychloroquine, [tumor necrosis factor] blockers, interleukin-6 blockers, and [Janus kinase] inhibitors – it is quite reassuring because we did not see an association with worse outcome with those drugs overall,” Dr. Machado said.
However, treatment with rituximab (OR, 4.0), sulfasalazine (OR, 3.6), and immunosuppressive agents such as azathioprine, cyclophosphamide, cyclosporine, mycophenolate, or tacrolimus (OR, 2.2), were associated with higher odds of dying from COVID-19 when compared with treatment with methotrexate alone.
The findings for rituximab and immunosuppressant use were perhaps not unexpected, but the possible association between sulfasalazine and COVID-19 death was “a bit intriguing,” Dr. Machado observed. “Sulfasalazine is believed to have low immunosuppressive effect.”
This warrants further investigation, but there are likely a range of confounding factors at play. One could be that people considered to be at higher risk may have been more often prescribed sulfasalazine because it was thought to be less immunosuppressive. Another might be because people taking sulfasalazine were more likely to be smokers, and they were also not advised to protect themselves from exposure to the virus (shielding) during the first wave of the pandemic, at least not in the United Kingdom.
Rituximab caution and vaccination
“Rituximab is a concern,” Dr. Machado acknowledged. “It is a concern that rheumatologists are now aware of and they are addressing, but then it’s a concern for a very specific subgroup of patients.”
While rheumatologists are, and will continue to prescribe it, there will be even more careful consideration over when, in whom, and how to use it during, and possibly even after, the pandemic.
“COVID is here to stay, it will become endemic, and it’s going to be part of our lives like the flu virus is,” Dr. Machado predicted.
Then there is the issue on vaccinating people against COVID-19, should those on rituximab still receive it? The answer is a yes, but, as with other vaccinations it’s all about the timing of when the vaccination is given.
Societies such as the British Society for Rheumatology have already begun to include guidance on this, recommending one of the available COVID-19 vaccines is given at least a month before the next or first dose of rituximab is due. As rituximab is given every few months, with doses sometimes spaced as much as 9 months or even a year apart, this should not be too much of a problem, but it is “better to have the vaccine first,” Dr. Machado said.
Has COVID-19 care improved in RMDs?
In separate research published in The Lancet Rheumatology, April Jorge, MD, of Massachusetts General Hospital and Harvard Medical School, both in Boston, and associates found that the risks of severe COVID-19 outcomes have improved over time, although they still “remain substantial.”
Dr. Jorge and colleagues looked at temporal trends in COVID-19 outcomes in patients with RMDs over the course of the first 6 months of the pandemic in 2020, using data from a large, multicenter, electronic health record network (TriNetX).
They formed two patient cohorts – a late (diagnosed from April 20 to July 20) and an early (diagnosed from January 20 to April 20) cohort – to see if outcomes had improved and discovered lower relative risks among patients in the late cohort for hospitalization (0.67), admission to the ICU (0.56), mechanical ventilation (0.39), acute kidney injury (0.66), renal replacement (0.53), and death (0.39).
“These results are encouraging,” but it’s difficult to match these different populations of patients, Dr. Machado said. “There are always factors that you cannot match for” and were not included in the U.S. analysis.
While there are important caveats in how the analysis was performed and thus in interpreting these data, they do “suggest that one of the reasons why outcomes have improved is because we have become better at treating these patients,” Dr. Machado added.
“Our treatment has improved, and our capacity to treat the complications has improved. We understand better how the disease behaves – we know that they can have thromboembolic complications that we can manage, and we are now able to manage ventilation issues better.”
Moreover, Dr. Machado said that, not only were clinicians more aware of what they should or should not do, there were treatments that were being used routinely or in some cases based on recent clinical trial results. “I think we are indeed treating these patients better.”
The COVID-19 GRA physician registry is financially supported by the American College of Rheumatology and EULAR. Dr. Machado had no relevant conflicts of interest.
FROM ANNALS OF THE RHEUMATIC DISEASES
FDA alert confirms heart and cancer risks with tofacitinib (Xeljanz)
The Food and Drug Administration has alerted the public to an increased risk of serious heart-related problems and cancer risk associated with the Janus kinase inhibitor tofacitinib (Xeljanz, Xeljanz XR), based on early results from a safety clinical trial comparing tofacitinib and tumor necrosis factor inhibitors in patients with rheumatoid arthritis (RA).

The FDA is awaiting further results from the trial, but in a safety communication issued on Feb. 4, the agency advised patients not to discontinue tofacitinib without consulting their health care providers and advised health care professionals to weigh the risks and benefits when prescribing the drug and continue to follow the current prescribing information.
Tofacitinib was approved for treatment of RA in 2012 at a 5-mg dose. After this approval, the FDA required drug manufacturer Pfizer to conduct a safety clinical trial that included the 5-mg twice-daily dose and a 10-mg twice-daily dose that is currently approved only for ulcerative colitis. In addition to RA and ulcerative colitis, tofacitinib is approved for adults with active psoriatic arthritis and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis.
Pfizer announced partial results of the study, known as the ORAL Surveillance trial, in a press release on Jan. 27. The randomized trial included 4,362 RA patients aged 50 years and older who received either 5-mg or 10-mg doses of tofacitinib or a TNF inhibitor (adalimumab or etanercept).
The full results have yet to be released, but based on data from approximately 10,000 person-years for the combined tofacitinib groups and approximately 5,000 person-years for the TNF inhibitor group, the rate of major cardiovascular adverse events was significantly higher in the combined tofacitinib group, compared with the TNF inhibitor group (0.98 vs. 0.73 per 100 person-years; hazard ratio, 1.33). In addition, the rate of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNF inhibitor group (1.13 vs. 0.77 per 100 person-years; HR, 1.48).
In February 2019, the FDA issued a warning stating an increased risk of pulmonary embolism and death associated with the 10-mg twice-daily dose of tofacitinib, following interims results from the safety study.
In July 2019, the FDA added a boxed warning to tofacitinib advising of the increased risk for pulmonary embolism and death associated with the 10-mg twice-daily dose.
The FDA encouraged health care professionals and patients to report any side effects from tofacitinib or other medications through the FDA MedWatch program online or by phone at 1-800-332-1088.
Until nuances revealed, no change in practice
The preliminary study findings contain some nuances that are a bit complicated from a statistical standpoint, according to Daniel Furst, MD, professor emeritus of medicine at the University of California, Los Angeles; adjunct professor at the University of Washington, Seattle; and research professor at the University of Florence (Italy).

This is supposed to be a noninferiority study, so something might not be noninferior, “but that doesn’t mean it is inferior,” explained Dr. Furst, who is also a member of the MDedge Rheumatology Editorial Advisory Board.
Dr. Furst said he was surprised by the study findings, because “I didn’t expect there to be any differences, and in fact it is not clear how great the differences are” among the groups in the study, he said.
When the complete findings are released, in one of the instances, “the statistics may show a very small statistical difference that indicates we may have to be more careful in this particularly high-risk group,” Dr. Furst noted.
“When we understand the data more closely, we may find that there are some nuances we need to be careful about,” he said. However, “until those data are out, I would not make any changes in my practice.”
Whether the current study findings represent a class effect is “impossible to say,” since tofacitinib affects three enzymes, while other JAK inhibitors affect only one or two, he noted.
Dr. Furst disclosed receiving grant/research support from and/or consulting for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, Pfizer, and Roche/Genentech.
Updated on 2/8/2021.
The Food and Drug Administration has alerted the public to an increased risk of serious heart-related problems and cancer risk associated with the Janus kinase inhibitor tofacitinib (Xeljanz, Xeljanz XR), based on early results from a safety clinical trial comparing tofacitinib and tumor necrosis factor inhibitors in patients with rheumatoid arthritis (RA).
The FDA is awaiting further results from the trial, but in a safety communication issued on Feb. 4, the agency advised patients not to discontinue tofacitinib without consulting their health care providers and advised health care professionals to weigh the risks and benefits when prescribing the drug and continue to follow the current prescribing information.
Tofacitinib was approved for treatment of RA in 2012 at a 5-mg dose. After this approval, the FDA required drug manufacturer Pfizer to conduct a safety clinical trial that included the 5-mg twice-daily dose and a 10-mg twice-daily dose that is currently approved only for ulcerative colitis. In addition to RA and ulcerative colitis, tofacitinib is approved for adults with active psoriatic arthritis and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis.
Pfizer announced partial results of the study, known as the ORAL Surveillance trial, in a press release on Jan. 27. The randomized trial included 4,362 RA patients aged 50 years and older who received either 5-mg or 10-mg doses of tofacitinib or a TNF inhibitor (adalimumab or etanercept).
The full results have yet to be released, but based on data from approximately 10,000 person-years for the combined tofacitinib groups and approximately 5,000 person-years for the TNF inhibitor group, the rate of major cardiovascular adverse events was significantly higher in the combined tofacitinib group, compared with the TNF inhibitor group (0.98 vs. 0.73 per 100 person-years; hazard ratio, 1.33). In addition, the rate of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNF inhibitor group (1.13 vs. 0.77 per 100 person-years; HR, 1.48).
In February 2019, the FDA issued a warning stating an increased risk of pulmonary embolism and death associated with the 10-mg twice-daily dose of tofacitinib, following interims results from the safety study.
In July 2019, the FDA added a boxed warning to tofacitinib advising of the increased risk for pulmonary embolism and death associated with the 10-mg twice-daily dose.
The FDA encouraged health care professionals and patients to report any side effects from tofacitinib or other medications through the FDA MedWatch program online or by phone at 1-800-332-1088.
Until nuances revealed, no change in practice
The preliminary study findings contain some nuances that are a bit complicated from a statistical standpoint, according to Daniel Furst, MD, professor emeritus of medicine at the University of California, Los Angeles; adjunct professor at the University of Washington, Seattle; and research professor at the University of Florence (Italy).
This is supposed to be a noninferiority study, so something might not be noninferior, “but that doesn’t mean it is inferior,” explained Dr. Furst, who is also a member of the MDedge Rheumatology Editorial Advisory Board.
Dr. Furst said he was surprised by the study findings, because “I didn’t expect there to be any differences, and in fact it is not clear how great the differences are” among the groups in the study, he said.
When the complete findings are released, in one of the instances, “the statistics may show a very small statistical difference that indicates we may have to be more careful in this particularly high-risk group,” Dr. Furst noted.
“When we understand the data more closely, we may find that there are some nuances we need to be careful about,” he said. However, “until those data are out, I would not make any changes in my practice.”
Whether the current study findings represent a class effect is “impossible to say,” since tofacitinib affects three enzymes, while other JAK inhibitors affect only one or two, he noted.
Dr. Furst disclosed receiving grant/research support from and/or consulting for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, Pfizer, and Roche/Genentech.
Updated on 2/8/2021.
The Food and Drug Administration has alerted the public to an increased risk of serious heart-related problems and cancer risk associated with the Janus kinase inhibitor tofacitinib (Xeljanz, Xeljanz XR), based on early results from a safety clinical trial comparing tofacitinib and tumor necrosis factor inhibitors in patients with rheumatoid arthritis (RA).
The FDA is awaiting further results from the trial, but in a safety communication issued on Feb. 4, the agency advised patients not to discontinue tofacitinib without consulting their health care providers and advised health care professionals to weigh the risks and benefits when prescribing the drug and continue to follow the current prescribing information.
Tofacitinib was approved for treatment of RA in 2012 at a 5-mg dose. After this approval, the FDA required drug manufacturer Pfizer to conduct a safety clinical trial that included the 5-mg twice-daily dose and a 10-mg twice-daily dose that is currently approved only for ulcerative colitis. In addition to RA and ulcerative colitis, tofacitinib is approved for adults with active psoriatic arthritis and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis.
Pfizer announced partial results of the study, known as the ORAL Surveillance trial, in a press release on Jan. 27. The randomized trial included 4,362 RA patients aged 50 years and older who received either 5-mg or 10-mg doses of tofacitinib or a TNF inhibitor (adalimumab or etanercept).
The full results have yet to be released, but based on data from approximately 10,000 person-years for the combined tofacitinib groups and approximately 5,000 person-years for the TNF inhibitor group, the rate of major cardiovascular adverse events was significantly higher in the combined tofacitinib group, compared with the TNF inhibitor group (0.98 vs. 0.73 per 100 person-years; hazard ratio, 1.33). In addition, the rate of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNF inhibitor group (1.13 vs. 0.77 per 100 person-years; HR, 1.48).
In February 2019, the FDA issued a warning stating an increased risk of pulmonary embolism and death associated with the 10-mg twice-daily dose of tofacitinib, following interims results from the safety study.
In July 2019, the FDA added a boxed warning to tofacitinib advising of the increased risk for pulmonary embolism and death associated with the 10-mg twice-daily dose.
The FDA encouraged health care professionals and patients to report any side effects from tofacitinib or other medications through the FDA MedWatch program online or by phone at 1-800-332-1088.
Until nuances revealed, no change in practice
The preliminary study findings contain some nuances that are a bit complicated from a statistical standpoint, according to Daniel Furst, MD, professor emeritus of medicine at the University of California, Los Angeles; adjunct professor at the University of Washington, Seattle; and research professor at the University of Florence (Italy).
This is supposed to be a noninferiority study, so something might not be noninferior, “but that doesn’t mean it is inferior,” explained Dr. Furst, who is also a member of the MDedge Rheumatology Editorial Advisory Board.
Dr. Furst said he was surprised by the study findings, because “I didn’t expect there to be any differences, and in fact it is not clear how great the differences are” among the groups in the study, he said.
When the complete findings are released, in one of the instances, “the statistics may show a very small statistical difference that indicates we may have to be more careful in this particularly high-risk group,” Dr. Furst noted.
“When we understand the data more closely, we may find that there are some nuances we need to be careful about,” he said. However, “until those data are out, I would not make any changes in my practice.”
Whether the current study findings represent a class effect is “impossible to say,” since tofacitinib affects three enzymes, while other JAK inhibitors affect only one or two, he noted.
Dr. Furst disclosed receiving grant/research support from and/or consulting for AbbVie, Actelion, Amgen, Bristol-Myers Squibb, Corbus, the National Institutes of Health, Novartis, Pfizer, and Roche/Genentech.
Updated on 2/8/2021.
In head-to-head trial, two biologics differ markedly for control of psoriasis
with other biologics, according to data from two simultaneously published trials, one of which was a head-to-head comparison with ustekinumab.
In the head-to-head trial called BE VIVID, which included a placebo arm, there was a large advantage of bimekizumab over ustekinumab, a biologic that targets IL-12 and IL-23 and is approved for treating psoriasis, for both coprimary endpoints, according to a multinational group of investigators led by Kristian Reich, MD, PhD, professor of dermatology at the University Medical Center, Hamburg-Eppendorf, Germany.
The proportion of patients with skin clearance was not only greater but faster, “with responses observed after one dose,” Dr. Reich and coinvestigators reported.
The data from the BE VIVID trial was published simultaneously with the BE READY trial, which was placebo-controlled but did not include an active comparator.
Evaluated at week 16, the coprimary endpoints in both studies were skin clearance as measured by a Psoriasis Area Severity Index greater than 90% (PASI 90) and Investigators Global Assessment (IGA) score of 0 (clear) or 1 (almost clear).
In BE VIVID, 567 patients were randomized in 11 countries, including the United States. The dose of bimekizumab was 320 mg administered subcutaneously every 4 weeks. In a randomization scheme of 4:2:1, half as many patients (163) were randomized to ustekinumab (Stelara), which was administered in weight-based dosing of 45 mg or 90 mg at enrollment, at 4 weeks, and then every 12 weeks. The placebo arm had 83 patients. All were switched to bimekizumab at 16 weeks.
At week 16, PASI 90 was achieved in 85% of patients randomized to bimekizumab, compared with 50% of patients randomized to ustekinumab (P < .0001). The rate in the placebo group was 5%.
The bimekizumab advantage for an IGA response of 0 or 1 was of similar magnitude, relative to ustekinumab (84% vs. 53%; P < .0001) and placebo (5%). All secondary efficacy endpoints, such as PASI 90 at week 12 (85% vs. 44%) and PASI 100 at week 16 (59% vs. 21%), favored bimekizumab over ustekinumab.
In the BE READY trial, which evaluated the same dose and schedule of bimekizumab, the rates of PASI 90 at week 16 were 91% and 1% (P < .0001) for the experimental arm and placebo, respectively. The proportion of patients with an IGA score of 0 or 1 were 93% and 1% (P < .0001), respectively.
In BE READY, patients who achieved PASI 90 at week 16 were reallocated to receive bimekizumab every 4 weeks, bimekizumab every 8 weeks (also 320 mg), or placebo. Both schedules of bimekizumab maintained responses through week 56, according to the authors, led by Kenneth B. Gordon, MD, professor and chair of dermatology, Medical College of Wisconsin, Milwaukee.
In both trials, safety was evaluated over the first 16 weeks as well as over a subsequent maintenance period, which extended to 52 weeks in BE VIVID and 56 weeks in BE READY. For bimekizumab, oral candidiasis was the most common treatment-related adverse event. In BE VIVID, this adverse event was reported in 9% of bimekizumab patients, compared with 0% of either the ustekinumab or placebo groups, up to week 16. Out to week 52, the rates were 15% in the bimekizumab group and 1% in the ustekinumab group.
In the BE READY trial, the rates of oral candidiasis were 6% and 0% for bimekizumab and placebo, respectively, through week 16. Over the maintenance periods, the rates were 9% and 11% for the every-8-week and every-4-week doses, respectively.
Discontinuation for adverse events was not higher on bimekizumab than placebo in either trial, nor was the proportion of serious treatment-emergent adverse events.
Nevertheless, the potential for adverse events was a key part of the discussion regarding the future role of bimekizumab, if approved, in an editorial that accompanied the publication of these studies.

“Bimekizumab might be our most effective biologic for psoriasis yet,” coauthors, William W. Huang, MD, PhD, associate professor of dermatology, and Steven R. Feldman, MD, PhD, professor of dermatology, both at Wake Forest University, Winston-Salem, NC, wrote in the editorial. “If the goal of psoriasis treatment is complete clearance, bimekizumab seems like a good option from an efficacy perspective.”
However, they noted that other IL-17 blockers, like secukinumab (Cosentyx) and brodalumab (Siliq), have been associated with risks, including the development of inflammatory bowel disease. In addition to the oral candidiasis seen in the BE VIVID and BE READY trials, they cautioned that other issues might arise with longer follow-up and greater numbers of patients exposed to this therapy.
In an interview, Dr. Feldman said adequately informed patients might be willing to accept these risks for the potential of greater efficacy, but he emphasized the need for appropriate warnings and education.
“We have a lot of very good treatments that offer patients an excellent chance of an excellent outcome – treatments that have been around and in use in large numbers of people for years,” Dr. Feldman said. “Unless the doctor and patient felt strongly about the need to use this new, perhaps more potent option, I would be personally inclined to use treatment with well-established safety profiles first.”

The senior author of the BE VIVID trial, Mark Lebwohl, MD, dean for clinical therapeutics and professor of dermatology, at the Icahn School of Medicine at Mount Sinai, New York, disagreed. He acknowledged that other agents targeting IL-17 have been associated with IBD, but risk of IBD is already elevated in patients with psoriasis and the risk appears to be lower with bimekizumab relative to prior agents in this class.
“Bimekizumab has now been studied in thousands of patients over several years. We can say with support from a sizable amount of data that IBD is very uncommon,” he said. While oral candidiasis is associated with bimekizumab, it is “easy to treat.”
Asked specifically if he will consider using bimekizumab as a first-line agent in psoriasis patients who are candidates for a biologic, Dr. Lebwohl said he would. Based on the evidence that this agent is more effective than other options and has manageable side effects, he believes it will be an important new treatment option.
Dr. Reich, Dr. Lebwohl, Dr. Gordon, and Dr. Feldman have financial relationships with multiple companies that produce therapies for psoriasis, including UCB Pharma, the sponsor of these studies.
with other biologics, according to data from two simultaneously published trials, one of which was a head-to-head comparison with ustekinumab.
In the head-to-head trial called BE VIVID, which included a placebo arm, there was a large advantage of bimekizumab over ustekinumab, a biologic that targets IL-12 and IL-23 and is approved for treating psoriasis, for both coprimary endpoints, according to a multinational group of investigators led by Kristian Reich, MD, PhD, professor of dermatology at the University Medical Center, Hamburg-Eppendorf, Germany.
The proportion of patients with skin clearance was not only greater but faster, “with responses observed after one dose,” Dr. Reich and coinvestigators reported.
The data from the BE VIVID trial was published simultaneously with the BE READY trial, which was placebo-controlled but did not include an active comparator.
Evaluated at week 16, the coprimary endpoints in both studies were skin clearance as measured by a Psoriasis Area Severity Index greater than 90% (PASI 90) and Investigators Global Assessment (IGA) score of 0 (clear) or 1 (almost clear).
In BE VIVID, 567 patients were randomized in 11 countries, including the United States. The dose of bimekizumab was 320 mg administered subcutaneously every 4 weeks. In a randomization scheme of 4:2:1, half as many patients (163) were randomized to ustekinumab (Stelara), which was administered in weight-based dosing of 45 mg or 90 mg at enrollment, at 4 weeks, and then every 12 weeks. The placebo arm had 83 patients. All were switched to bimekizumab at 16 weeks.
At week 16, PASI 90 was achieved in 85% of patients randomized to bimekizumab, compared with 50% of patients randomized to ustekinumab (P < .0001). The rate in the placebo group was 5%.
The bimekizumab advantage for an IGA response of 0 or 1 was of similar magnitude, relative to ustekinumab (84% vs. 53%; P < .0001) and placebo (5%). All secondary efficacy endpoints, such as PASI 90 at week 12 (85% vs. 44%) and PASI 100 at week 16 (59% vs. 21%), favored bimekizumab over ustekinumab.
In the BE READY trial, which evaluated the same dose and schedule of bimekizumab, the rates of PASI 90 at week 16 were 91% and 1% (P < .0001) for the experimental arm and placebo, respectively. The proportion of patients with an IGA score of 0 or 1 were 93% and 1% (P < .0001), respectively.
In BE READY, patients who achieved PASI 90 at week 16 were reallocated to receive bimekizumab every 4 weeks, bimekizumab every 8 weeks (also 320 mg), or placebo. Both schedules of bimekizumab maintained responses through week 56, according to the authors, led by Kenneth B. Gordon, MD, professor and chair of dermatology, Medical College of Wisconsin, Milwaukee.
In both trials, safety was evaluated over the first 16 weeks as well as over a subsequent maintenance period, which extended to 52 weeks in BE VIVID and 56 weeks in BE READY. For bimekizumab, oral candidiasis was the most common treatment-related adverse event. In BE VIVID, this adverse event was reported in 9% of bimekizumab patients, compared with 0% of either the ustekinumab or placebo groups, up to week 16. Out to week 52, the rates were 15% in the bimekizumab group and 1% in the ustekinumab group.
In the BE READY trial, the rates of oral candidiasis were 6% and 0% for bimekizumab and placebo, respectively, through week 16. Over the maintenance periods, the rates were 9% and 11% for the every-8-week and every-4-week doses, respectively.
Discontinuation for adverse events was not higher on bimekizumab than placebo in either trial, nor was the proportion of serious treatment-emergent adverse events.
Nevertheless, the potential for adverse events was a key part of the discussion regarding the future role of bimekizumab, if approved, in an editorial that accompanied the publication of these studies.
“Bimekizumab might be our most effective biologic for psoriasis yet,” coauthors, William W. Huang, MD, PhD, associate professor of dermatology, and Steven R. Feldman, MD, PhD, professor of dermatology, both at Wake Forest University, Winston-Salem, NC, wrote in the editorial. “If the goal of psoriasis treatment is complete clearance, bimekizumab seems like a good option from an efficacy perspective.”
However, they noted that other IL-17 blockers, like secukinumab (Cosentyx) and brodalumab (Siliq), have been associated with risks, including the development of inflammatory bowel disease. In addition to the oral candidiasis seen in the BE VIVID and BE READY trials, they cautioned that other issues might arise with longer follow-up and greater numbers of patients exposed to this therapy.
In an interview, Dr. Feldman said adequately informed patients might be willing to accept these risks for the potential of greater efficacy, but he emphasized the need for appropriate warnings and education.
“We have a lot of very good treatments that offer patients an excellent chance of an excellent outcome – treatments that have been around and in use in large numbers of people for years,” Dr. Feldman said. “Unless the doctor and patient felt strongly about the need to use this new, perhaps more potent option, I would be personally inclined to use treatment with well-established safety profiles first.”
The senior author of the BE VIVID trial, Mark Lebwohl, MD, dean for clinical therapeutics and professor of dermatology, at the Icahn School of Medicine at Mount Sinai, New York, disagreed. He acknowledged that other agents targeting IL-17 have been associated with IBD, but risk of IBD is already elevated in patients with psoriasis and the risk appears to be lower with bimekizumab relative to prior agents in this class.
“Bimekizumab has now been studied in thousands of patients over several years. We can say with support from a sizable amount of data that IBD is very uncommon,” he said. While oral candidiasis is associated with bimekizumab, it is “easy to treat.”
Asked specifically if he will consider using bimekizumab as a first-line agent in psoriasis patients who are candidates for a biologic, Dr. Lebwohl said he would. Based on the evidence that this agent is more effective than other options and has manageable side effects, he believes it will be an important new treatment option.
Dr. Reich, Dr. Lebwohl, Dr. Gordon, and Dr. Feldman have financial relationships with multiple companies that produce therapies for psoriasis, including UCB Pharma, the sponsor of these studies.
with other biologics, according to data from two simultaneously published trials, one of which was a head-to-head comparison with ustekinumab.
In the head-to-head trial called BE VIVID, which included a placebo arm, there was a large advantage of bimekizumab over ustekinumab, a biologic that targets IL-12 and IL-23 and is approved for treating psoriasis, for both coprimary endpoints, according to a multinational group of investigators led by Kristian Reich, MD, PhD, professor of dermatology at the University Medical Center, Hamburg-Eppendorf, Germany.
The proportion of patients with skin clearance was not only greater but faster, “with responses observed after one dose,” Dr. Reich and coinvestigators reported.
The data from the BE VIVID trial was published simultaneously with the BE READY trial, which was placebo-controlled but did not include an active comparator.
Evaluated at week 16, the coprimary endpoints in both studies were skin clearance as measured by a Psoriasis Area Severity Index greater than 90% (PASI 90) and Investigators Global Assessment (IGA) score of 0 (clear) or 1 (almost clear).
In BE VIVID, 567 patients were randomized in 11 countries, including the United States. The dose of bimekizumab was 320 mg administered subcutaneously every 4 weeks. In a randomization scheme of 4:2:1, half as many patients (163) were randomized to ustekinumab (Stelara), which was administered in weight-based dosing of 45 mg or 90 mg at enrollment, at 4 weeks, and then every 12 weeks. The placebo arm had 83 patients. All were switched to bimekizumab at 16 weeks.
At week 16, PASI 90 was achieved in 85% of patients randomized to bimekizumab, compared with 50% of patients randomized to ustekinumab (P < .0001). The rate in the placebo group was 5%.
The bimekizumab advantage for an IGA response of 0 or 1 was of similar magnitude, relative to ustekinumab (84% vs. 53%; P < .0001) and placebo (5%). All secondary efficacy endpoints, such as PASI 90 at week 12 (85% vs. 44%) and PASI 100 at week 16 (59% vs. 21%), favored bimekizumab over ustekinumab.
In the BE READY trial, which evaluated the same dose and schedule of bimekizumab, the rates of PASI 90 at week 16 were 91% and 1% (P < .0001) for the experimental arm and placebo, respectively. The proportion of patients with an IGA score of 0 or 1 were 93% and 1% (P < .0001), respectively.
In BE READY, patients who achieved PASI 90 at week 16 were reallocated to receive bimekizumab every 4 weeks, bimekizumab every 8 weeks (also 320 mg), or placebo. Both schedules of bimekizumab maintained responses through week 56, according to the authors, led by Kenneth B. Gordon, MD, professor and chair of dermatology, Medical College of Wisconsin, Milwaukee.
In both trials, safety was evaluated over the first 16 weeks as well as over a subsequent maintenance period, which extended to 52 weeks in BE VIVID and 56 weeks in BE READY. For bimekizumab, oral candidiasis was the most common treatment-related adverse event. In BE VIVID, this adverse event was reported in 9% of bimekizumab patients, compared with 0% of either the ustekinumab or placebo groups, up to week 16. Out to week 52, the rates were 15% in the bimekizumab group and 1% in the ustekinumab group.
In the BE READY trial, the rates of oral candidiasis were 6% and 0% for bimekizumab and placebo, respectively, through week 16. Over the maintenance periods, the rates were 9% and 11% for the every-8-week and every-4-week doses, respectively.
Discontinuation for adverse events was not higher on bimekizumab than placebo in either trial, nor was the proportion of serious treatment-emergent adverse events.
Nevertheless, the potential for adverse events was a key part of the discussion regarding the future role of bimekizumab, if approved, in an editorial that accompanied the publication of these studies.
“Bimekizumab might be our most effective biologic for psoriasis yet,” coauthors, William W. Huang, MD, PhD, associate professor of dermatology, and Steven R. Feldman, MD, PhD, professor of dermatology, both at Wake Forest University, Winston-Salem, NC, wrote in the editorial. “If the goal of psoriasis treatment is complete clearance, bimekizumab seems like a good option from an efficacy perspective.”
However, they noted that other IL-17 blockers, like secukinumab (Cosentyx) and brodalumab (Siliq), have been associated with risks, including the development of inflammatory bowel disease. In addition to the oral candidiasis seen in the BE VIVID and BE READY trials, they cautioned that other issues might arise with longer follow-up and greater numbers of patients exposed to this therapy.
In an interview, Dr. Feldman said adequately informed patients might be willing to accept these risks for the potential of greater efficacy, but he emphasized the need for appropriate warnings and education.
“We have a lot of very good treatments that offer patients an excellent chance of an excellent outcome – treatments that have been around and in use in large numbers of people for years,” Dr. Feldman said. “Unless the doctor and patient felt strongly about the need to use this new, perhaps more potent option, I would be personally inclined to use treatment with well-established safety profiles first.”
The senior author of the BE VIVID trial, Mark Lebwohl, MD, dean for clinical therapeutics and professor of dermatology, at the Icahn School of Medicine at Mount Sinai, New York, disagreed. He acknowledged that other agents targeting IL-17 have been associated with IBD, but risk of IBD is already elevated in patients with psoriasis and the risk appears to be lower with bimekizumab relative to prior agents in this class.
“Bimekizumab has now been studied in thousands of patients over several years. We can say with support from a sizable amount of data that IBD is very uncommon,” he said. While oral candidiasis is associated with bimekizumab, it is “easy to treat.”
Asked specifically if he will consider using bimekizumab as a first-line agent in psoriasis patients who are candidates for a biologic, Dr. Lebwohl said he would. Based on the evidence that this agent is more effective than other options and has manageable side effects, he believes it will be an important new treatment option.
Dr. Reich, Dr. Lebwohl, Dr. Gordon, and Dr. Feldman have financial relationships with multiple companies that produce therapies for psoriasis, including UCB Pharma, the sponsor of these studies.
FROM THE LANCET

Patients with early arthritis may need tailored treatments
Patients with early, undifferentiated arthritis may benefit from milder or stronger treatments, depending on the number of their risk factors for developing rheumatoid arthritis, researchers say.
If the finding is borne out by further research, clinicians could consider treating some of these patients with hydroxychloroquine, steroids, or nonsteroidal anti-inflammatory drugs (NSAIDs) rather than methotrexate, said Pascal de Jong, MD, PhD, a rheumatologist at Erasmus Medical Center in Rotterdam, the Netherlands.
“Maybe those patients with fewer risk factors should get less intensive treatment,” he said in an interview. The study by de Jong and colleagues was published online Jan. 23 in Rheumatology.
The European Alliance of Associations for Rheumatology recommends starting treatment with methotrexate for patients who are at risk for persistent arthritis, which it says is “factually synonymous” with rheumatoid arthritis.
But these recommendations are based on studies involving patients with established rheumatoid arthritis, Dr. de Jong said.
In an earlier study, he and his colleagues found that hydroxychloroquine can be just as effective as methotrexate for patients newly diagnosed with rheumatoid arthritis who don’t have autoantibodies. This led them to wonder whether their findings might apply to some subgroups of patients with early arthritis.
As an initial test of this idea, they identified 130 patients from the Rotterdam Early Arthritis Cohort (tREACH) trial who had at least one swollen joint but who did meet the diagnostic criteria for rheumatoid arthritis.
They sorted the patients into groups on the basis of the number of risk factors for persistent arthritis. The risk factors were autoantibody positivity (rheumatoid factor and/or anticitrullinated protein antibody), polyarthritis (more than four swollen joints), erosive disease, and elevations in levels of acute-phase reactants.
Thirty-one patients had none of these risk factors, 66 patients had one risk factor, and the remaining 33 patients had at least two risk factors.
After 2 years of follow-up, 74% of the patients who had had no risk factors had recovered from their arthritis and had not taken disease-modifying antirheumatic drugs (DMARDs) for at least 6 months (DMARD-free remission). Among the patients who had had one risk factor, 48% achieved DMARD-free remission. Among those who had had two risk factors, 45% achieved DMARD-free remission. The differences between the group that had had no risk factors and the other two groups were statistically significant (P < .05).
The researchers found that those patients who had been experiencing their symptoms for fewer than 6 months were more likely to achieve disease-free remission.
They also sorted patients into different groups on the basis of the treatments they received. One group of 30 comprised all patients who had been initially treated with methotrexate and included patients who had also received other drugs. One group of 40 received hydroxychloroquine initially, and one group of 60 comprised patients who had received no DMARDs initially and included those who had received NSAIDs or glucocorticoids.
There was no statistically significant difference in DMARD-free remission rates among the treatment groups. However, among those patients who were not treated initially with DMARDs, the chance of sustaining DMARD-free remission for more than a year was lower in comparison with the patients who received methotrexate initially (odds ratio, 4.28; 95% confidence interval, 1.34-13.72; P < .05).
Those patients who had fewer baseline risk factors were more likely to have their medication dosages tapered, and they were at lower risk for flares. Patients with more risk factors were more likely to require an intensificiation of treatment, such as with the use of biologicals.
Methotrexate is more likely to cause side effects such as nausea, fatigue, and hair loss than hydroxychloroquine, Dr. de Jong said. “If the medication is better tolerated, it also influences the compliance of the patient,” he said.
The study could help rheumatologists determine which patients need the most aggressive treatment, agreed Kevin Deane, MD, PhD, associate professor of medicine, division of rheumatology, the University of Colorado, Aurora.

“That’s a common clinical problem,” he said. “Somebody comes in with sort of mild arthritis, and you don’t quite know what it is yet.”
But he added that more research is needed to understand what treatment works best for those patients whose arthritis has not yet been differentiated. Primary care physicians who suspect inflammatory arthritis should refer their patients to rheumatologists and should test for rheumatoid factors and anticyclic citrullinated peptide, Dr. Deane said.
The authors and Dr. Deane have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with early, undifferentiated arthritis may benefit from milder or stronger treatments, depending on the number of their risk factors for developing rheumatoid arthritis, researchers say.
If the finding is borne out by further research, clinicians could consider treating some of these patients with hydroxychloroquine, steroids, or nonsteroidal anti-inflammatory drugs (NSAIDs) rather than methotrexate, said Pascal de Jong, MD, PhD, a rheumatologist at Erasmus Medical Center in Rotterdam, the Netherlands.
“Maybe those patients with fewer risk factors should get less intensive treatment,” he said in an interview. The study by de Jong and colleagues was published online Jan. 23 in Rheumatology.
The European Alliance of Associations for Rheumatology recommends starting treatment with methotrexate for patients who are at risk for persistent arthritis, which it says is “factually synonymous” with rheumatoid arthritis.
But these recommendations are based on studies involving patients with established rheumatoid arthritis, Dr. de Jong said.
In an earlier study, he and his colleagues found that hydroxychloroquine can be just as effective as methotrexate for patients newly diagnosed with rheumatoid arthritis who don’t have autoantibodies. This led them to wonder whether their findings might apply to some subgroups of patients with early arthritis.
As an initial test of this idea, they identified 130 patients from the Rotterdam Early Arthritis Cohort (tREACH) trial who had at least one swollen joint but who did meet the diagnostic criteria for rheumatoid arthritis.
They sorted the patients into groups on the basis of the number of risk factors for persistent arthritis. The risk factors were autoantibody positivity (rheumatoid factor and/or anticitrullinated protein antibody), polyarthritis (more than four swollen joints), erosive disease, and elevations in levels of acute-phase reactants.
Thirty-one patients had none of these risk factors, 66 patients had one risk factor, and the remaining 33 patients had at least two risk factors.
After 2 years of follow-up, 74% of the patients who had had no risk factors had recovered from their arthritis and had not taken disease-modifying antirheumatic drugs (DMARDs) for at least 6 months (DMARD-free remission). Among the patients who had had one risk factor, 48% achieved DMARD-free remission. Among those who had had two risk factors, 45% achieved DMARD-free remission. The differences between the group that had had no risk factors and the other two groups were statistically significant (P < .05).
The researchers found that those patients who had been experiencing their symptoms for fewer than 6 months were more likely to achieve disease-free remission.
They also sorted patients into different groups on the basis of the treatments they received. One group of 30 comprised all patients who had been initially treated with methotrexate and included patients who had also received other drugs. One group of 40 received hydroxychloroquine initially, and one group of 60 comprised patients who had received no DMARDs initially and included those who had received NSAIDs or glucocorticoids.
There was no statistically significant difference in DMARD-free remission rates among the treatment groups. However, among those patients who were not treated initially with DMARDs, the chance of sustaining DMARD-free remission for more than a year was lower in comparison with the patients who received methotrexate initially (odds ratio, 4.28; 95% confidence interval, 1.34-13.72; P < .05).
Those patients who had fewer baseline risk factors were more likely to have their medication dosages tapered, and they were at lower risk for flares. Patients with more risk factors were more likely to require an intensificiation of treatment, such as with the use of biologicals.
Methotrexate is more likely to cause side effects such as nausea, fatigue, and hair loss than hydroxychloroquine, Dr. de Jong said. “If the medication is better tolerated, it also influences the compliance of the patient,” he said.
The study could help rheumatologists determine which patients need the most aggressive treatment, agreed Kevin Deane, MD, PhD, associate professor of medicine, division of rheumatology, the University of Colorado, Aurora.
“That’s a common clinical problem,” he said. “Somebody comes in with sort of mild arthritis, and you don’t quite know what it is yet.”
But he added that more research is needed to understand what treatment works best for those patients whose arthritis has not yet been differentiated. Primary care physicians who suspect inflammatory arthritis should refer their patients to rheumatologists and should test for rheumatoid factors and anticyclic citrullinated peptide, Dr. Deane said.
The authors and Dr. Deane have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Patients with early, undifferentiated arthritis may benefit from milder or stronger treatments, depending on the number of their risk factors for developing rheumatoid arthritis, researchers say.
If the finding is borne out by further research, clinicians could consider treating some of these patients with hydroxychloroquine, steroids, or nonsteroidal anti-inflammatory drugs (NSAIDs) rather than methotrexate, said Pascal de Jong, MD, PhD, a rheumatologist at Erasmus Medical Center in Rotterdam, the Netherlands.
“Maybe those patients with fewer risk factors should get less intensive treatment,” he said in an interview. The study by de Jong and colleagues was published online Jan. 23 in Rheumatology.
The European Alliance of Associations for Rheumatology recommends starting treatment with methotrexate for patients who are at risk for persistent arthritis, which it says is “factually synonymous” with rheumatoid arthritis.
But these recommendations are based on studies involving patients with established rheumatoid arthritis, Dr. de Jong said.
In an earlier study, he and his colleagues found that hydroxychloroquine can be just as effective as methotrexate for patients newly diagnosed with rheumatoid arthritis who don’t have autoantibodies. This led them to wonder whether their findings might apply to some subgroups of patients with early arthritis.
As an initial test of this idea, they identified 130 patients from the Rotterdam Early Arthritis Cohort (tREACH) trial who had at least one swollen joint but who did meet the diagnostic criteria for rheumatoid arthritis.
They sorted the patients into groups on the basis of the number of risk factors for persistent arthritis. The risk factors were autoantibody positivity (rheumatoid factor and/or anticitrullinated protein antibody), polyarthritis (more than four swollen joints), erosive disease, and elevations in levels of acute-phase reactants.
Thirty-one patients had none of these risk factors, 66 patients had one risk factor, and the remaining 33 patients had at least two risk factors.
After 2 years of follow-up, 74% of the patients who had had no risk factors had recovered from their arthritis and had not taken disease-modifying antirheumatic drugs (DMARDs) for at least 6 months (DMARD-free remission). Among the patients who had had one risk factor, 48% achieved DMARD-free remission. Among those who had had two risk factors, 45% achieved DMARD-free remission. The differences between the group that had had no risk factors and the other two groups were statistically significant (P < .05).
The researchers found that those patients who had been experiencing their symptoms for fewer than 6 months were more likely to achieve disease-free remission.
They also sorted patients into different groups on the basis of the treatments they received. One group of 30 comprised all patients who had been initially treated with methotrexate and included patients who had also received other drugs. One group of 40 received hydroxychloroquine initially, and one group of 60 comprised patients who had received no DMARDs initially and included those who had received NSAIDs or glucocorticoids.
There was no statistically significant difference in DMARD-free remission rates among the treatment groups. However, among those patients who were not treated initially with DMARDs, the chance of sustaining DMARD-free remission for more than a year was lower in comparison with the patients who received methotrexate initially (odds ratio, 4.28; 95% confidence interval, 1.34-13.72; P < .05).
Those patients who had fewer baseline risk factors were more likely to have their medication dosages tapered, and they were at lower risk for flares. Patients with more risk factors were more likely to require an intensificiation of treatment, such as with the use of biologicals.
Methotrexate is more likely to cause side effects such as nausea, fatigue, and hair loss than hydroxychloroquine, Dr. de Jong said. “If the medication is better tolerated, it also influences the compliance of the patient,” he said.
The study could help rheumatologists determine which patients need the most aggressive treatment, agreed Kevin Deane, MD, PhD, associate professor of medicine, division of rheumatology, the University of Colorado, Aurora.
“That’s a common clinical problem,” he said. “Somebody comes in with sort of mild arthritis, and you don’t quite know what it is yet.”
But he added that more research is needed to understand what treatment works best for those patients whose arthritis has not yet been differentiated. Primary care physicians who suspect inflammatory arthritis should refer their patients to rheumatologists and should test for rheumatoid factors and anticyclic citrullinated peptide, Dr. Deane said.
The authors and Dr. Deane have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Tofacitinib for RA misses the mark in safety study
Daily treatment with tofacitinib (Xeljanz) led to more malignancies and adverse cardiovascular events in older rheumatoid arthritis patients compared with treatment with a tumor necrosis factor (TNF) inhibitor, according to the partial results of a safety study announced last week by Pfizer.
The postmarketing study known as ORAL Surveillance began in 2014 to evaluate the safety of the Janus kinase (JAK) inhibitor tofacitinib compared to a TNF inhibitor in RA patients 50 years of age or older with at least one additional cardiovascular risk factor. Its 4,362 participants were randomized to either daily doses of 5 mg (n = 1,455) or 10 mg (n = 1,456) of tofacitinib or the TNFi (n = 1,451), which was adalimumab for patients in the United States, Canada, and Puerto Rico, and etanercept elsewhere. During analysis, adverse events were pooled for all patients on tofacitinib.
Overall, 135 patients developed major adverse cardiovascular events (MACE) and 164 developed malignancies – excluding nonmelanoma skin cancer. The incidence of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNFi group (1.13 vs. 0.77 per 100 person-years; hazard ratio, 1.48; 95% confidence interval, 1.04-2.09). The rate of MACE was also higher in the combined tofacitinib group (0.98 vs. 0.73 per 100 person-years; HR, 1.33; 95% CI, 0.91-1.94). Both rates for tofacitinib did not meet the trial’s noninferiority criteria.
Among the patients on tofacitinib, the most reported MACE was myocardial infarction and the most reported malignancy was lung cancer. Study participants with noted risk factors – including older age and smoking – were more likely to experience adverse events.
In February 2019, patients in the 10-mg tofacitinib group were switched to the 5-mg because of a safety signal indicating increased risk of pulmonary embolism and death.
Tofacitinib was approved for RA in November 2012, though concerns about serious side effects had been noted during clinical trials and a boxed warning was ultimately added to the drug’s label. Tofacitinib is also approved for adults with active psoriatic arthritis, adults with moderately to severely active ulcerative colitis, and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis. Other JAK inhibitors such as baricitinib and upadacitinib have been approved for RA in the interim as well, though the higher dose of baricitinib was rejected in committee because of safety concerns and both their boxes also warn against infections, thrombosis, and cancer.
A postmarketing safety study on baricitinib is expected to be completed in 2025.
The full results of the ORAL Surveillance study – which should address safety regarding pulmonary embolism and mortality, as well as efficacy data – have not yet been released. “Pfizer is working with the [FDA] and other regulatory agencies to review the full results and analyses as they become available,” the press release said.
Daily treatment with tofacitinib (Xeljanz) led to more malignancies and adverse cardiovascular events in older rheumatoid arthritis patients compared with treatment with a tumor necrosis factor (TNF) inhibitor, according to the partial results of a safety study announced last week by Pfizer.
The postmarketing study known as ORAL Surveillance began in 2014 to evaluate the safety of the Janus kinase (JAK) inhibitor tofacitinib compared to a TNF inhibitor in RA patients 50 years of age or older with at least one additional cardiovascular risk factor. Its 4,362 participants were randomized to either daily doses of 5 mg (n = 1,455) or 10 mg (n = 1,456) of tofacitinib or the TNFi (n = 1,451), which was adalimumab for patients in the United States, Canada, and Puerto Rico, and etanercept elsewhere. During analysis, adverse events were pooled for all patients on tofacitinib.
Overall, 135 patients developed major adverse cardiovascular events (MACE) and 164 developed malignancies – excluding nonmelanoma skin cancer. The incidence of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNFi group (1.13 vs. 0.77 per 100 person-years; hazard ratio, 1.48; 95% confidence interval, 1.04-2.09). The rate of MACE was also higher in the combined tofacitinib group (0.98 vs. 0.73 per 100 person-years; HR, 1.33; 95% CI, 0.91-1.94). Both rates for tofacitinib did not meet the trial’s noninferiority criteria.
Among the patients on tofacitinib, the most reported MACE was myocardial infarction and the most reported malignancy was lung cancer. Study participants with noted risk factors – including older age and smoking – were more likely to experience adverse events.
In February 2019, patients in the 10-mg tofacitinib group were switched to the 5-mg because of a safety signal indicating increased risk of pulmonary embolism and death.
Tofacitinib was approved for RA in November 2012, though concerns about serious side effects had been noted during clinical trials and a boxed warning was ultimately added to the drug’s label. Tofacitinib is also approved for adults with active psoriatic arthritis, adults with moderately to severely active ulcerative colitis, and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis. Other JAK inhibitors such as baricitinib and upadacitinib have been approved for RA in the interim as well, though the higher dose of baricitinib was rejected in committee because of safety concerns and both their boxes also warn against infections, thrombosis, and cancer.
A postmarketing safety study on baricitinib is expected to be completed in 2025.
The full results of the ORAL Surveillance study – which should address safety regarding pulmonary embolism and mortality, as well as efficacy data – have not yet been released. “Pfizer is working with the [FDA] and other regulatory agencies to review the full results and analyses as they become available,” the press release said.
Daily treatment with tofacitinib (Xeljanz) led to more malignancies and adverse cardiovascular events in older rheumatoid arthritis patients compared with treatment with a tumor necrosis factor (TNF) inhibitor, according to the partial results of a safety study announced last week by Pfizer.
The postmarketing study known as ORAL Surveillance began in 2014 to evaluate the safety of the Janus kinase (JAK) inhibitor tofacitinib compared to a TNF inhibitor in RA patients 50 years of age or older with at least one additional cardiovascular risk factor. Its 4,362 participants were randomized to either daily doses of 5 mg (n = 1,455) or 10 mg (n = 1,456) of tofacitinib or the TNFi (n = 1,451), which was adalimumab for patients in the United States, Canada, and Puerto Rico, and etanercept elsewhere. During analysis, adverse events were pooled for all patients on tofacitinib.
Overall, 135 patients developed major adverse cardiovascular events (MACE) and 164 developed malignancies – excluding nonmelanoma skin cancer. The incidence of adjudicated malignancies was significantly higher in the tofacitinib group, compared with the TNFi group (1.13 vs. 0.77 per 100 person-years; hazard ratio, 1.48; 95% confidence interval, 1.04-2.09). The rate of MACE was also higher in the combined tofacitinib group (0.98 vs. 0.73 per 100 person-years; HR, 1.33; 95% CI, 0.91-1.94). Both rates for tofacitinib did not meet the trial’s noninferiority criteria.
Among the patients on tofacitinib, the most reported MACE was myocardial infarction and the most reported malignancy was lung cancer. Study participants with noted risk factors – including older age and smoking – were more likely to experience adverse events.
In February 2019, patients in the 10-mg tofacitinib group were switched to the 5-mg because of a safety signal indicating increased risk of pulmonary embolism and death.
Tofacitinib was approved for RA in November 2012, though concerns about serious side effects had been noted during clinical trials and a boxed warning was ultimately added to the drug’s label. Tofacitinib is also approved for adults with active psoriatic arthritis, adults with moderately to severely active ulcerative colitis, and patients aged 2 years or older with active polyarticular course juvenile idiopathic arthritis. Other JAK inhibitors such as baricitinib and upadacitinib have been approved for RA in the interim as well, though the higher dose of baricitinib was rejected in committee because of safety concerns and both their boxes also warn against infections, thrombosis, and cancer.
A postmarketing safety study on baricitinib is expected to be completed in 2025.
The full results of the ORAL Surveillance study – which should address safety regarding pulmonary embolism and mortality, as well as efficacy data – have not yet been released. “Pfizer is working with the [FDA] and other regulatory agencies to review the full results and analyses as they become available,” the press release said.
Guselkumab maintains psoriasis efficacy long after discontinuation
Fully half of patients with moderate to severe psoriasis who achieve complete clearance after their first four doses of guselkumab (Tremfya) continue to maintain a PASI 90 response nearly 6 months after withdrawal of the biologic, according to a post hoc analysis of the pivotal phase 3 VOYAGE 2 trial.
“That’s impressive maintenance of efficacy,” said Curdin Conrad, MD, who presented the data at the virtual annual congress of the European Academy of Dermatology and Venereology.
“These findings are reassuring when you have to interrupt guselkumab therapy: For example, due to acute infection, pregnancy, or surgery. But it might also help when considering in the future a flexible dosing interval, particularly for patients who had complete clearance,” added Dr. Conrad, professor of dermatology and head of the polyclinic and the Center of Excellence for Psoriasis at Lausanne (Switzerland) University Hospital.
The intriguing implication from VOYAGE 2 that guselkumab might lend itself to flexible dosing featuring lengthy drug-free intervals is being prospectively examined in the ongoing phase 3b GUIDE trial. This is a double-blind, placebo-controlled trial including 888 French and German patients with moderate to severe psoriasis and a study hypothesis that those who have a Psoriasis Area and Severity Index score of 0 at weeks 20 and 28 in response to on-label dosing – the so-called ‘super responders’ – will maintain disease control until week 68 if their dosing is reduced to 100 mg of guselkumab every 16 weeks instead of the standard 8-week intervals.
Dr. Conrad reported that in VOYAGE 2, 106 patients on standard-dose guselkumab who had a PASI score of 0 at weeks 20 and 28 were randomized to discontinue the interleukin-23 inhibitor after receiving their fourth dose at week 20. It took 25 weeks for 50% of them to lose their PASI 90 response as defined by regression to a PASI score of 1 or greater. Using a less stringent definition of maintenance of efficacy, the super responders’ median time off guselkumab until reaching a PASI score of 3 or more was 30.7 weeks, with a median of 35.4 weeks to a PASI score of 5 or more.
In addition, 34 other VOYAGE 2 participants who were almost clear on guselkumab at weeks 20 and 28, with a PASI score of more than 0 but less than 1, were randomized to guselkumab withdrawal after their week-20 dose. Median time to loss of their PASI 90 response was shorter than that of the super responders – not surprising since their mean PASI score when the biologic was halted was 0.5, rather than 0 as for the super responders. But Dr. Conrad said the maintenance of response was still impressive: A median of 16.2 weeks to reach a PASI score of 1 or more, 27.2 weeks for a PASI 3, and 33.7 weeks for a PASI score of 5.
He reported receiving research funding from and serving as a scientific adviser to Janssen, the study sponsor, as well as to more than a dozen other pharmaceutical companies.
Fully half of patients with moderate to severe psoriasis who achieve complete clearance after their first four doses of guselkumab (Tremfya) continue to maintain a PASI 90 response nearly 6 months after withdrawal of the biologic, according to a post hoc analysis of the pivotal phase 3 VOYAGE 2 trial.
“That’s impressive maintenance of efficacy,” said Curdin Conrad, MD, who presented the data at the virtual annual congress of the European Academy of Dermatology and Venereology.
“These findings are reassuring when you have to interrupt guselkumab therapy: For example, due to acute infection, pregnancy, or surgery. But it might also help when considering in the future a flexible dosing interval, particularly for patients who had complete clearance,” added Dr. Conrad, professor of dermatology and head of the polyclinic and the Center of Excellence for Psoriasis at Lausanne (Switzerland) University Hospital.
The intriguing implication from VOYAGE 2 that guselkumab might lend itself to flexible dosing featuring lengthy drug-free intervals is being prospectively examined in the ongoing phase 3b GUIDE trial. This is a double-blind, placebo-controlled trial including 888 French and German patients with moderate to severe psoriasis and a study hypothesis that those who have a Psoriasis Area and Severity Index score of 0 at weeks 20 and 28 in response to on-label dosing – the so-called ‘super responders’ – will maintain disease control until week 68 if their dosing is reduced to 100 mg of guselkumab every 16 weeks instead of the standard 8-week intervals.
Dr. Conrad reported that in VOYAGE 2, 106 patients on standard-dose guselkumab who had a PASI score of 0 at weeks 20 and 28 were randomized to discontinue the interleukin-23 inhibitor after receiving their fourth dose at week 20. It took 25 weeks for 50% of them to lose their PASI 90 response as defined by regression to a PASI score of 1 or greater. Using a less stringent definition of maintenance of efficacy, the super responders’ median time off guselkumab until reaching a PASI score of 3 or more was 30.7 weeks, with a median of 35.4 weeks to a PASI score of 5 or more.
In addition, 34 other VOYAGE 2 participants who were almost clear on guselkumab at weeks 20 and 28, with a PASI score of more than 0 but less than 1, were randomized to guselkumab withdrawal after their week-20 dose. Median time to loss of their PASI 90 response was shorter than that of the super responders – not surprising since their mean PASI score when the biologic was halted was 0.5, rather than 0 as for the super responders. But Dr. Conrad said the maintenance of response was still impressive: A median of 16.2 weeks to reach a PASI score of 1 or more, 27.2 weeks for a PASI 3, and 33.7 weeks for a PASI score of 5.
He reported receiving research funding from and serving as a scientific adviser to Janssen, the study sponsor, as well as to more than a dozen other pharmaceutical companies.
Fully half of patients with moderate to severe psoriasis who achieve complete clearance after their first four doses of guselkumab (Tremfya) continue to maintain a PASI 90 response nearly 6 months after withdrawal of the biologic, according to a post hoc analysis of the pivotal phase 3 VOYAGE 2 trial.
“That’s impressive maintenance of efficacy,” said Curdin Conrad, MD, who presented the data at the virtual annual congress of the European Academy of Dermatology and Venereology.
“These findings are reassuring when you have to interrupt guselkumab therapy: For example, due to acute infection, pregnancy, or surgery. But it might also help when considering in the future a flexible dosing interval, particularly for patients who had complete clearance,” added Dr. Conrad, professor of dermatology and head of the polyclinic and the Center of Excellence for Psoriasis at Lausanne (Switzerland) University Hospital.
The intriguing implication from VOYAGE 2 that guselkumab might lend itself to flexible dosing featuring lengthy drug-free intervals is being prospectively examined in the ongoing phase 3b GUIDE trial. This is a double-blind, placebo-controlled trial including 888 French and German patients with moderate to severe psoriasis and a study hypothesis that those who have a Psoriasis Area and Severity Index score of 0 at weeks 20 and 28 in response to on-label dosing – the so-called ‘super responders’ – will maintain disease control until week 68 if their dosing is reduced to 100 mg of guselkumab every 16 weeks instead of the standard 8-week intervals.
Dr. Conrad reported that in VOYAGE 2, 106 patients on standard-dose guselkumab who had a PASI score of 0 at weeks 20 and 28 were randomized to discontinue the interleukin-23 inhibitor after receiving their fourth dose at week 20. It took 25 weeks for 50% of them to lose their PASI 90 response as defined by regression to a PASI score of 1 or greater. Using a less stringent definition of maintenance of efficacy, the super responders’ median time off guselkumab until reaching a PASI score of 3 or more was 30.7 weeks, with a median of 35.4 weeks to a PASI score of 5 or more.
In addition, 34 other VOYAGE 2 participants who were almost clear on guselkumab at weeks 20 and 28, with a PASI score of more than 0 but less than 1, were randomized to guselkumab withdrawal after their week-20 dose. Median time to loss of their PASI 90 response was shorter than that of the super responders – not surprising since their mean PASI score when the biologic was halted was 0.5, rather than 0 as for the super responders. But Dr. Conrad said the maintenance of response was still impressive: A median of 16.2 weeks to reach a PASI score of 1 or more, 27.2 weeks for a PASI 3, and 33.7 weeks for a PASI score of 5.
He reported receiving research funding from and serving as a scientific adviser to Janssen, the study sponsor, as well as to more than a dozen other pharmaceutical companies.
FROM THE EADV CONGRESS
Deucravacitinib offers biologic-like psoriasis efficacy in oral form
and a range of other chronic inflammatory diseases, Bruce E. Strober, MD, PhD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.

Deucravacitinib solely blocks tyrosine kinase 2 (TYK2) signaling without touching Janus kinase (JAK) 1, 2, or 3. In so doing, it inhibits several cytokines important for inflammation: interleukin-12, IL-13, and interferon-alpha and -beta. Yet it doesn’t affect the numerous pathways mediated by JAKs 1-3, many of which relate to growth and development of cell lineages, including production of erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, prolactin, growth hormone, and leptin. These deucravacitinib characteristics should translate into fewer off-target side effects than with oral JAK inhibitors.
“The promise of TYK2 inhibition that’s brought to you by deucravacitinib is there will be no laboratory monitoring and the effects will be narrow in blocking inflammation,” said Dr. Strober, a dermatologist at Yale University, New Haven, Conn., and in private practice in Cromwell, Conn.
He highlighted the positive results of a randomized, phase 2, dose-ranging study conducted in 267 patients with moderate or severe plaque psoriasis. Participants had an average baseline Psoriasis Area and Severity Index (PASI) score of 19, with a Dermatology Life Quality Index score of about 12. At the top dose of 12 mg once daily, 75% of patients achieved a PASI 75 response at week 12, and 44% reached a PASI 90, as did 69% and 44%, respectively, who were on deucravacitinib at 3 mg twice daily. Those are collective efficacy numbers similar to adalimumab (Humira) or ustekinumab (Stelara).
Deucravacitinib may provide efficacy “like one of our second-tier biological therapies, yet it will be oral,” Dr. Strober commented.
Importantly, no laboratory abnormalities were detected in this trial. Only mild side effects were documented, most prominently acne, which occurred in dose-dependent fashion in 2% of patients on 3 mg of deucravacitinib twice daily and 4% at 12 mg once daily.
“The treatment of the acne that is elicited by this drug is yet to be fully described, but I’m sure we’ll learn the best approaches, given that acne is in our wheel house,” the dermatologist added.
Bristol-Myers Squibb has announced positive results from the pivotal phase 3 POETYK PSO-1 trial. Deucravacitinib at 6 mg once daily met both of its coprimary efficacy endpoints in the study, which included 666 patients with moderate to severe psoriasis. The TYK 2 inhibitor demonstrated superiority to both placebo and oral apremilast (Otezla) at week 16. The company said the safety profile was consistent with the phase 2 results, and that the full details of the phase 3 trial will be presented next year at a major medical meeting.
In addition, positive phase 2 results were reported for deucravacitinib in the treatment of psoriatic arthritis in a randomized trial presented at the fall 2020 meeting of the American College of Rheumatology. Deucravacitinib is also under study for lupus and inflammatory bowel disease.
Dr. Strober, an active clinical trialist, reported serving as a consultant to more than two dozen pharmaceutical companies, including Bristol-Myers Squibb.
MedscapeLive and this news organization are owned by the same parent company.
and a range of other chronic inflammatory diseases, Bruce E. Strober, MD, PhD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
Deucravacitinib solely blocks tyrosine kinase 2 (TYK2) signaling without touching Janus kinase (JAK) 1, 2, or 3. In so doing, it inhibits several cytokines important for inflammation: interleukin-12, IL-13, and interferon-alpha and -beta. Yet it doesn’t affect the numerous pathways mediated by JAKs 1-3, many of which relate to growth and development of cell lineages, including production of erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, prolactin, growth hormone, and leptin. These deucravacitinib characteristics should translate into fewer off-target side effects than with oral JAK inhibitors.
“The promise of TYK2 inhibition that’s brought to you by deucravacitinib is there will be no laboratory monitoring and the effects will be narrow in blocking inflammation,” said Dr. Strober, a dermatologist at Yale University, New Haven, Conn., and in private practice in Cromwell, Conn.
He highlighted the positive results of a randomized, phase 2, dose-ranging study conducted in 267 patients with moderate or severe plaque psoriasis. Participants had an average baseline Psoriasis Area and Severity Index (PASI) score of 19, with a Dermatology Life Quality Index score of about 12. At the top dose of 12 mg once daily, 75% of patients achieved a PASI 75 response at week 12, and 44% reached a PASI 90, as did 69% and 44%, respectively, who were on deucravacitinib at 3 mg twice daily. Those are collective efficacy numbers similar to adalimumab (Humira) or ustekinumab (Stelara).
Deucravacitinib may provide efficacy “like one of our second-tier biological therapies, yet it will be oral,” Dr. Strober commented.
Importantly, no laboratory abnormalities were detected in this trial. Only mild side effects were documented, most prominently acne, which occurred in dose-dependent fashion in 2% of patients on 3 mg of deucravacitinib twice daily and 4% at 12 mg once daily.
“The treatment of the acne that is elicited by this drug is yet to be fully described, but I’m sure we’ll learn the best approaches, given that acne is in our wheel house,” the dermatologist added.
Bristol-Myers Squibb has announced positive results from the pivotal phase 3 POETYK PSO-1 trial. Deucravacitinib at 6 mg once daily met both of its coprimary efficacy endpoints in the study, which included 666 patients with moderate to severe psoriasis. The TYK 2 inhibitor demonstrated superiority to both placebo and oral apremilast (Otezla) at week 16. The company said the safety profile was consistent with the phase 2 results, and that the full details of the phase 3 trial will be presented next year at a major medical meeting.
In addition, positive phase 2 results were reported for deucravacitinib in the treatment of psoriatic arthritis in a randomized trial presented at the fall 2020 meeting of the American College of Rheumatology. Deucravacitinib is also under study for lupus and inflammatory bowel disease.
Dr. Strober, an active clinical trialist, reported serving as a consultant to more than two dozen pharmaceutical companies, including Bristol-Myers Squibb.
MedscapeLive and this news organization are owned by the same parent company.
and a range of other chronic inflammatory diseases, Bruce E. Strober, MD, PhD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
Deucravacitinib solely blocks tyrosine kinase 2 (TYK2) signaling without touching Janus kinase (JAK) 1, 2, or 3. In so doing, it inhibits several cytokines important for inflammation: interleukin-12, IL-13, and interferon-alpha and -beta. Yet it doesn’t affect the numerous pathways mediated by JAKs 1-3, many of which relate to growth and development of cell lineages, including production of erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, prolactin, growth hormone, and leptin. These deucravacitinib characteristics should translate into fewer off-target side effects than with oral JAK inhibitors.
“The promise of TYK2 inhibition that’s brought to you by deucravacitinib is there will be no laboratory monitoring and the effects will be narrow in blocking inflammation,” said Dr. Strober, a dermatologist at Yale University, New Haven, Conn., and in private practice in Cromwell, Conn.
He highlighted the positive results of a randomized, phase 2, dose-ranging study conducted in 267 patients with moderate or severe plaque psoriasis. Participants had an average baseline Psoriasis Area and Severity Index (PASI) score of 19, with a Dermatology Life Quality Index score of about 12. At the top dose of 12 mg once daily, 75% of patients achieved a PASI 75 response at week 12, and 44% reached a PASI 90, as did 69% and 44%, respectively, who were on deucravacitinib at 3 mg twice daily. Those are collective efficacy numbers similar to adalimumab (Humira) or ustekinumab (Stelara).
Deucravacitinib may provide efficacy “like one of our second-tier biological therapies, yet it will be oral,” Dr. Strober commented.
Importantly, no laboratory abnormalities were detected in this trial. Only mild side effects were documented, most prominently acne, which occurred in dose-dependent fashion in 2% of patients on 3 mg of deucravacitinib twice daily and 4% at 12 mg once daily.
“The treatment of the acne that is elicited by this drug is yet to be fully described, but I’m sure we’ll learn the best approaches, given that acne is in our wheel house,” the dermatologist added.
Bristol-Myers Squibb has announced positive results from the pivotal phase 3 POETYK PSO-1 trial. Deucravacitinib at 6 mg once daily met both of its coprimary efficacy endpoints in the study, which included 666 patients with moderate to severe psoriasis. The TYK 2 inhibitor demonstrated superiority to both placebo and oral apremilast (Otezla) at week 16. The company said the safety profile was consistent with the phase 2 results, and that the full details of the phase 3 trial will be presented next year at a major medical meeting.
In addition, positive phase 2 results were reported for deucravacitinib in the treatment of psoriatic arthritis in a randomized trial presented at the fall 2020 meeting of the American College of Rheumatology. Deucravacitinib is also under study for lupus and inflammatory bowel disease.
Dr. Strober, an active clinical trialist, reported serving as a consultant to more than two dozen pharmaceutical companies, including Bristol-Myers Squibb.
MedscapeLive and this news organization are owned by the same parent company.
FROM MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR