User login
Is this adolescent suicidal? Challenges in pediatric inpatient consultation-liaison
CASE Attempted suicide?
Ms. S, a 16-year-old Yemeni-American girl, is brought to the emergency department (ED) by her mother and brother after ingesting an overdose of painkillers and fainting. During the initial evaluation, Ms. S says she had in the past attempted suicide by knife. The medical team suspects that the current overdose is a suicide attempt, and they call the consultation-liaison (C-L) psychiatry/psychology team. Ms. S’s brother strongly denies that his sister had previously attempted suicide, stating, “She’s from a good family, and she is smart. She cannot feel that way.” He also requests the name of the clinician who documented this information in the medical record.
During the consultation, Ms. S reports that the previous morning, she developed strong abdominal pain and discovered that she was menstruating for the first time. She explains that she did not understand what was happening to her and that no one had discussed menstruation with her before. Ms. S took her mother’s opioid pain medication. Ms. S reports she took one pill, but when it did not immediately alleviate her pain, she ingested several more. After this, Ms. S says she went to play with her siblings, but gradually became dizzy and confused, and informed her sister and mother of this. The family was fasting in observance of Ramadan, and as they walked toward the mosque, Ms. S fainted, which prompted her family to bring her to the ED.
During the C-L consultation, Ms. S’s brother, who speaks English, is present, as is her mother, who speaks only Arabic and thus needs a phone interpreter. As the C-L team asks Ms. S a question, it is translated to her mother, and then Ms. S’s response is also translated, and then finally, the mother shares her own response. At times, her brother provides translation. Ms. S speaks in English, but often asks for the translation of words or questions.
Ms. S reports that she and her family emigrated from Yemen to the United States 9 months ago. Ms. S says that she enjoys school and is doing well academically. She denies experiencing any anxiety, worry, or stress related to her life in Yemen, her move to a new country, her parents’ health, school, or other domains. Ms. S also denies any history of depressive episodes or previous suicidal ideation, intention, or attempt, which contradicts her endorsement of a previous suicide attempt to one clinician when she was initially evaluated.
[polldaddy:10040204]
Continue to: The authors' observation
The authors’ observation

The C-L team determined that Ms. S did not meet criteria for major depressive disorder. She did not endorse current feelings of depression and denied anhedonia and other associated symptoms included in DSM-5 criteria for major depressive disorder or adjustment disorder with depressed mood (Table 11).

Although Ms. S and her family recently emigrated from Yemen, she did not report any symptoms consistent with an adjustment disorder with depression. Further,
Accurate case conceptualization and diagnosis is particularly crucial in C-L services, where there is an urgency for clinical decision-making after an initial evaluation without the luxury of amending conceptualization in follow-up sessions. Providing a diagnosis for which a patient does not fully or accurately meet the criteria can have deleterious effects. An inaccurate diagnosis for Ms. S would have unnecessarily added the perceived stigma of a mental disorder to her medical record. Additionally, misdiagnosing or pathologizing a natural process of acculturation could have led to inappropriate or even harmful treatment.
The C-L team evaluated alternative explanations for Ms. S’s statements that suggested she was suicidal. First, they considered her mental status at the time she presented to the ED. An overdose of opioids alters mental status. Complicating reversal of opioid overdose is that some opioids have longer half-lives than naloxone, an opioid antagonist, so the individual can become reintoxicated. Similarly, some opioids are more potent and difficult to reverse.2 An altered mental status may have limited Ms. S’s ability to comprehend and answer questions accurately when she first presented to the ED.
Continue to: Cultural factors and the clinical evaluation
Cultural factors and the clinical evaluation
Next, the C-L team considered Ms. S’s clinical picture as it related to her cultural background. Cultural factors interact with the clinical evaluation in a complex manner, influencing the way patients approach the encounter, the symptoms they report, and the language they use to describe their experiences. While these variables are thoroughly evaluated during comprehensive psychological assessments, within the inpatient consultation service, the goal for pediatric C-L clinicians is to conduct a focused assessment to answer specific and critically important questions about a youth’s psychological functioning. Thus, the fundamental challenge of inpatient consultation is to answer the referral question in a brief period and in a culturally informed manner, to appraise the referring medical team about the relevant clinical and cultural issues, with the goal of ethical and clinically sound decision-making.

The C-L team considered key cultural factors in its assessment of Ms. S (Table 31). Several issues were of concern. First, language is often cited as the top barrier to health care access by Arab Americans, even by those with competency in English.3
Experts in culture argue that even with access to interpreters, many words and phrases lack direct translation, and their implicit meaning may be difficult to reveal. Additionally, at times more significance is placed on nonverbal cues and unspoken expectations.4 This can create barriers to communication with clinicians, especially in the context of an inpatient psychiatric consultation, when thorough understanding of an adolescent and family often needs to occur in a single encounter, and clinicians may not appreciate the subtle nuances of nonverbal communication.
The language barrier also may have influenced Ms. S’s initial endorsement of a previous suicide attempt by knife because the medical staff first interviewed Ms. S without an interpreter. For instance, many medical and psychosocial providers probe patients regarding suicidality with questions such as “Have you ever hurt yourself?” or “Have you ever tried to hurt yourself?” It is possible that in another language, an individual might interpret that question as, “Have you ever gotten hurt?” This interpretation completely alters the meaning of the question and eliminates intention or motivation to harm oneself. Language ambiguity and lack of shared cultural understanding may have influenced Ms. S’s interpretation of and response to such questions. Ms. S and her family were perplexed by the C-L team’s reference to the knife and continued to deny the incident.
Continue to: Cultural attitudes to puberty
Cultural attitudes to puberty
Cultures vary with respect to education of sensitive topics such as puberty. The medical providers assumed that Ms. S was informed about the onset of menses. Therefore, they could not consider the strong impact of such an event on an unsuspecting adolescent. Many adolescent girls in Yemen have poor health and lack menstruation-related knowledge, and many are “prescribed” medications by their mothers without contacting a physician.5 Ms. S reported to the C-L team that no one from her family had discussed menstruation with her. She reported that since arriving at the hospital, nurses had educated her about menstruation, and that she was no longer afraid. She also noted that if she experienced such pain again, she would go to the hospital or “just deal with it.”
Family identification and attitudes toward mental health
Ms. S’s strong identification with her family and attitudes toward mental health may have limited what she chose to disclose regarding her experiences of loss related to leaving her country of origin, adjustment, and acculturation to the new environment, as well as feelings of sadness. Family has a central and critical role in Arab cultures. Commitment to a family’s well-being and enhancement of honor and status is highly valued and encouraged.4 Conversely, being concerned with individual needs may be a source of guilt and feelings of betraying the family.6 Arab Americans tend not to discuss personal problems with people outside their extended family, including counselors and therapists, partly because of cultural stigma against mental illness7,8 and partly because revealing family problems to strangers (ie, clinicians) may be considered a cultural taboo9 and a threat to family honor.10 Although Ms. S was interviewed privately when she first came to the ED and also during the psychiatric consultation, the stigma of psychiatric problems11 and possible concerns about protecting her family’s name may have influenced her readiness to reveal intimate information to “strangers.”
Additionally, family statements that appeared to imply negative beliefs about mental health would have strongly deterred Ms. S from expressing any psychological concerns. For example, Ms. S’s brother took offense when the C-L team said it was evaluating his sister because she had said she had previously attempted suicide.
The tenets of Islam may have provided a framework through which Ms. S interprets emotional concerns and may have defined her explanatory models of psychological stress. For instance, it is not uncommon among American Muslims to view mental health problems as rising from “loss of faith in God,”9 and suicidal ideation may not be disclosed because suicide is forbidden in Islam.12 Therefore, it might be particularly difficult to assess suicidal ideation in a patient who is Muslim, especially those who are less acculturated to Western culture.13
Continue to: Directly asking Ms. S...
Directly asking Ms. S if she had thoughts of harming herself may have been too frightening or guilt-provoking for an adolescent with her background. Asking about passive expression of suicidal ideation would have been more culturally appropriate. For example, asking, “Do you wish that God would let you die?”12 may have elicited more meaningful clinical information about Ms. S’s emotional state and possibly suicide risk.
Furthermore, Ms. S’s identification of coping strategies (ie, “just deal with it”) may have sounded limited to a Western clinician, but this may have been consistent with cultural norms of emotional expression of limiting complaints.4 Also, among Arab Americans, psychiatric symptoms often are expressed through somatization.7,14 Expressing psychological pain through physical symptoms appears protective against public stigma. Public image and opinion is important, and behaviors that would reflect well to others are dictated by the family. These attitudes, beliefs, and values likely impact how Ms. S presented her psychological concerns.
[polldaddy:10040206]
The authors’ observations
Although inpatient hospitalization was initially considered, it was not pursued due to denial of past and current suicidal ideation or suicide attempts, the lack of comorbidity, age-appropriate functioning, and a supportive family environment. Similarly, due to the absence of acute psychiatric symptoms, partial hospitalization was not pursued. The C-L team evaluated treatment options with extreme caution and sensitivity because recommending the wrong treatment option could have deleterious effects on Ms. S and her family’s life. If inpatient hospitalization had been pursued, it could have likely caused the family unnecessary suffering and could have negatively affected familial relationships. Strong feelings of shame, betrayal, and guilt would be intensified, impairing the family’s cohesion, removing environmental and family supports, and putting Ms. S at further risk of developing more severe symptoms of low mood.
Although there were significant concerns about making the wrong recommendation to the family, the C-L team’s highest priority was Ms. S’s safety. Despite cultural concerns, the team would have recommended hospitalization if Ms. S’s clinical picture had warranted this decision.
Continue to: OUTCOME Culturally-appropriate outpatient therapy
OUTCOME Culturally-appropriate outpatient therapy
Due to the lack of substantial evidence of apparent risk for self-harm, the presence of a supportive family, and Ms. S’s high academic performance and future orientation, the C-L team concludes that Ms. S’s concerns were most likely the result of the challenges of acculturation related to the language barrier and a lack of health knowledge. However, the C-L team remains cautious that Ms. S may have minimized or denied her mental health concerns due to various cultural factors. The team recommends that Ms. S seek outpatient psychotherapy from a clinician who specializes in working with Arab American individuals and families in their native language. The C-L team communicates these conclusions to the medical team verbally and in writing.
The authors’ observations
Cultural issues experienced during this consultation may not generalize to other Arab American adolescents and their families because there is diversity even within groups that share common cultural characteristics. Nevertheless, this case underscores the challenge of accurately assessing suicide risk, and making a differential diagnosis in the presence of complex cultural data and the dilemmas clinicians may encounter when attempting to answer important referral questions such as, “Is this adolescent suicidal and in need of psychiatric hospitalization?”
Bottom Line
Cultural factors and attitudes toward mental health and language barriers may play a large role in how patients answer clinical questions. Cultural issues may add a level of intricacy not easily resolved within the restrictions of an inpatient setting, and this complexity may influence clinical judgment, recommendations, and possibly health outcomes. Culturally appropriate psychotherapy is key for patients experiencing difficulty with acculturation.
Related Resources
- Adam B. Caring for Muslim patients: Understanding cultural and religious factors. Current Psychiatry. 2017;16(12):56-57.
- Nassar-McMillan SC, Hakim-Larson J. Counseling considerations among Arab Americans. Journal of Counseling & Development. 2003;81(2):150-159.
- Sue DW. Multidimensional facets of cultural competence. The Counseling Psychologist. 2001;29(6):790-821.
Drug Brand Name
Naloxone • Narcan
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Meehan TJ, Bryant SM, Aks SE. Drugs of abuse: the highs and lows of altered mental states in the emergency department. Emerg Med Clin North Am. 2010;28(3):663-682.
3. Shah SM, Ayash C, Pharaon NA, et al. Arab American immigrants in New York: health care and cancer knowledge, attitudes, and beliefs. J Immigr Minor Health. 2008;10(5):429-436.
4. Budman CL, Lipson JG, Meleis AI. The cultural consultant in mental health care: the case of an Arab adolescent. Am J Orthopsychiatry. 1992; 62(3):359-370.
5. Mohamed EM, Mohamed AG, Al-Ajeal Ly. Knowledge, beliefs and practices regarding menstruation among adolescent schoolgirls in Seiyun City, Yemen. Al-Azhar Assiut Medical Journal. 2011;9(3):67-86.
6. Gorkin M, Masalha S, Yatziv G. Psychotherapy of Israeli-Arab patients: some cultural considerations. Journal of Psychoanalytic Anthropology. 1985;8(4);215-230.
7. Gearing RE, MacKenzie MJ, Ibrahim RW, et al. Stigma and mental health treatment of adolescents with depression in Jordan. Community Ment Health J. 2015;51(1):111-117.
8. Timimi SB. Adolescence in immigrant Arab families. Psychotherapy: theory, research, practice, training. 1995;32(1):141-149.
9. Ahmed S and Reddy LA. Understanding the mental health needs of American Muslims: recommendations and considerations for practice. Journal of Multicultural Counseling and Development. 2007;35(4):207-218.
10. Abudabbeh N, Nydell MK. Transcultural counseling and Arab Americans. In: McFadden J, ed. Transcultural counseling: bilateral and international perspectives. Alexandria, VA. American Counseling Association. 1993:261-284.
11. Erickson CD, al-Timimi NR. Providing mental health services to Arab Americans: recommendations and considerations. Cultur Divers Ethnic Minor Psychol. 2001;7(4):308-327.
12. Ali SR, Liu WM, Humedian M. Islam 101: understanding the religion and therapy implications. Prof Psychol Res Pr. 2004;35(6):635-642.
13. Hedayat-Diba Z. Psychotherapy with Muslims. In: Richards PS, Bergin AE, eds. Handbook of psychotherapy and religious diversity, 2nd ed. Washington, DC: Amercian Psychological Association. 2000:289-314.
14. Al-Krenawi A. Mental health practice in Arab countries. Curr Opin in Psychiatry. 2005;18(5):560-564.
CASE Attempted suicide?
Ms. S, a 16-year-old Yemeni-American girl, is brought to the emergency department (ED) by her mother and brother after ingesting an overdose of painkillers and fainting. During the initial evaluation, Ms. S says she had in the past attempted suicide by knife. The medical team suspects that the current overdose is a suicide attempt, and they call the consultation-liaison (C-L) psychiatry/psychology team. Ms. S’s brother strongly denies that his sister had previously attempted suicide, stating, “She’s from a good family, and she is smart. She cannot feel that way.” He also requests the name of the clinician who documented this information in the medical record.
During the consultation, Ms. S reports that the previous morning, she developed strong abdominal pain and discovered that she was menstruating for the first time. She explains that she did not understand what was happening to her and that no one had discussed menstruation with her before. Ms. S took her mother’s opioid pain medication. Ms. S reports she took one pill, but when it did not immediately alleviate her pain, she ingested several more. After this, Ms. S says she went to play with her siblings, but gradually became dizzy and confused, and informed her sister and mother of this. The family was fasting in observance of Ramadan, and as they walked toward the mosque, Ms. S fainted, which prompted her family to bring her to the ED.
During the C-L consultation, Ms. S’s brother, who speaks English, is present, as is her mother, who speaks only Arabic and thus needs a phone interpreter. As the C-L team asks Ms. S a question, it is translated to her mother, and then Ms. S’s response is also translated, and then finally, the mother shares her own response. At times, her brother provides translation. Ms. S speaks in English, but often asks for the translation of words or questions.
Ms. S reports that she and her family emigrated from Yemen to the United States 9 months ago. Ms. S says that she enjoys school and is doing well academically. She denies experiencing any anxiety, worry, or stress related to her life in Yemen, her move to a new country, her parents’ health, school, or other domains. Ms. S also denies any history of depressive episodes or previous suicidal ideation, intention, or attempt, which contradicts her endorsement of a previous suicide attempt to one clinician when she was initially evaluated.
[polldaddy:10040204]
Continue to: The authors' observation
The authors’ observation
The C-L team determined that Ms. S did not meet criteria for major depressive disorder. She did not endorse current feelings of depression and denied anhedonia and other associated symptoms included in DSM-5 criteria for major depressive disorder or adjustment disorder with depressed mood (Table 11).
Although Ms. S and her family recently emigrated from Yemen, she did not report any symptoms consistent with an adjustment disorder with depression. Further,
Accurate case conceptualization and diagnosis is particularly crucial in C-L services, where there is an urgency for clinical decision-making after an initial evaluation without the luxury of amending conceptualization in follow-up sessions. Providing a diagnosis for which a patient does not fully or accurately meet the criteria can have deleterious effects. An inaccurate diagnosis for Ms. S would have unnecessarily added the perceived stigma of a mental disorder to her medical record. Additionally, misdiagnosing or pathologizing a natural process of acculturation could have led to inappropriate or even harmful treatment.
The C-L team evaluated alternative explanations for Ms. S’s statements that suggested she was suicidal. First, they considered her mental status at the time she presented to the ED. An overdose of opioids alters mental status. Complicating reversal of opioid overdose is that some opioids have longer half-lives than naloxone, an opioid antagonist, so the individual can become reintoxicated. Similarly, some opioids are more potent and difficult to reverse.2 An altered mental status may have limited Ms. S’s ability to comprehend and answer questions accurately when she first presented to the ED.
Continue to: Cultural factors and the clinical evaluation
Cultural factors and the clinical evaluation
Next, the C-L team considered Ms. S’s clinical picture as it related to her cultural background. Cultural factors interact with the clinical evaluation in a complex manner, influencing the way patients approach the encounter, the symptoms they report, and the language they use to describe their experiences. While these variables are thoroughly evaluated during comprehensive psychological assessments, within the inpatient consultation service, the goal for pediatric C-L clinicians is to conduct a focused assessment to answer specific and critically important questions about a youth’s psychological functioning. Thus, the fundamental challenge of inpatient consultation is to answer the referral question in a brief period and in a culturally informed manner, to appraise the referring medical team about the relevant clinical and cultural issues, with the goal of ethical and clinically sound decision-making.
The C-L team considered key cultural factors in its assessment of Ms. S (Table 31). Several issues were of concern. First, language is often cited as the top barrier to health care access by Arab Americans, even by those with competency in English.3
Experts in culture argue that even with access to interpreters, many words and phrases lack direct translation, and their implicit meaning may be difficult to reveal. Additionally, at times more significance is placed on nonverbal cues and unspoken expectations.4 This can create barriers to communication with clinicians, especially in the context of an inpatient psychiatric consultation, when thorough understanding of an adolescent and family often needs to occur in a single encounter, and clinicians may not appreciate the subtle nuances of nonverbal communication.
The language barrier also may have influenced Ms. S’s initial endorsement of a previous suicide attempt by knife because the medical staff first interviewed Ms. S without an interpreter. For instance, many medical and psychosocial providers probe patients regarding suicidality with questions such as “Have you ever hurt yourself?” or “Have you ever tried to hurt yourself?” It is possible that in another language, an individual might interpret that question as, “Have you ever gotten hurt?” This interpretation completely alters the meaning of the question and eliminates intention or motivation to harm oneself. Language ambiguity and lack of shared cultural understanding may have influenced Ms. S’s interpretation of and response to such questions. Ms. S and her family were perplexed by the C-L team’s reference to the knife and continued to deny the incident.
Continue to: Cultural attitudes to puberty
Cultural attitudes to puberty
Cultures vary with respect to education of sensitive topics such as puberty. The medical providers assumed that Ms. S was informed about the onset of menses. Therefore, they could not consider the strong impact of such an event on an unsuspecting adolescent. Many adolescent girls in Yemen have poor health and lack menstruation-related knowledge, and many are “prescribed” medications by their mothers without contacting a physician.5 Ms. S reported to the C-L team that no one from her family had discussed menstruation with her. She reported that since arriving at the hospital, nurses had educated her about menstruation, and that she was no longer afraid. She also noted that if she experienced such pain again, she would go to the hospital or “just deal with it.”
Family identification and attitudes toward mental health
Ms. S’s strong identification with her family and attitudes toward mental health may have limited what she chose to disclose regarding her experiences of loss related to leaving her country of origin, adjustment, and acculturation to the new environment, as well as feelings of sadness. Family has a central and critical role in Arab cultures. Commitment to a family’s well-being and enhancement of honor and status is highly valued and encouraged.4 Conversely, being concerned with individual needs may be a source of guilt and feelings of betraying the family.6 Arab Americans tend not to discuss personal problems with people outside their extended family, including counselors and therapists, partly because of cultural stigma against mental illness7,8 and partly because revealing family problems to strangers (ie, clinicians) may be considered a cultural taboo9 and a threat to family honor.10 Although Ms. S was interviewed privately when she first came to the ED and also during the psychiatric consultation, the stigma of psychiatric problems11 and possible concerns about protecting her family’s name may have influenced her readiness to reveal intimate information to “strangers.”
Additionally, family statements that appeared to imply negative beliefs about mental health would have strongly deterred Ms. S from expressing any psychological concerns. For example, Ms. S’s brother took offense when the C-L team said it was evaluating his sister because she had said she had previously attempted suicide.
The tenets of Islam may have provided a framework through which Ms. S interprets emotional concerns and may have defined her explanatory models of psychological stress. For instance, it is not uncommon among American Muslims to view mental health problems as rising from “loss of faith in God,”9 and suicidal ideation may not be disclosed because suicide is forbidden in Islam.12 Therefore, it might be particularly difficult to assess suicidal ideation in a patient who is Muslim, especially those who are less acculturated to Western culture.13
Continue to: Directly asking Ms. S...
Directly asking Ms. S if she had thoughts of harming herself may have been too frightening or guilt-provoking for an adolescent with her background. Asking about passive expression of suicidal ideation would have been more culturally appropriate. For example, asking, “Do you wish that God would let you die?”12 may have elicited more meaningful clinical information about Ms. S’s emotional state and possibly suicide risk.
Furthermore, Ms. S’s identification of coping strategies (ie, “just deal with it”) may have sounded limited to a Western clinician, but this may have been consistent with cultural norms of emotional expression of limiting complaints.4 Also, among Arab Americans, psychiatric symptoms often are expressed through somatization.7,14 Expressing psychological pain through physical symptoms appears protective against public stigma. Public image and opinion is important, and behaviors that would reflect well to others are dictated by the family. These attitudes, beliefs, and values likely impact how Ms. S presented her psychological concerns.
[polldaddy:10040206]
The authors’ observations
Although inpatient hospitalization was initially considered, it was not pursued due to denial of past and current suicidal ideation or suicide attempts, the lack of comorbidity, age-appropriate functioning, and a supportive family environment. Similarly, due to the absence of acute psychiatric symptoms, partial hospitalization was not pursued. The C-L team evaluated treatment options with extreme caution and sensitivity because recommending the wrong treatment option could have deleterious effects on Ms. S and her family’s life. If inpatient hospitalization had been pursued, it could have likely caused the family unnecessary suffering and could have negatively affected familial relationships. Strong feelings of shame, betrayal, and guilt would be intensified, impairing the family’s cohesion, removing environmental and family supports, and putting Ms. S at further risk of developing more severe symptoms of low mood.
Although there were significant concerns about making the wrong recommendation to the family, the C-L team’s highest priority was Ms. S’s safety. Despite cultural concerns, the team would have recommended hospitalization if Ms. S’s clinical picture had warranted this decision.
Continue to: OUTCOME Culturally-appropriate outpatient therapy
OUTCOME Culturally-appropriate outpatient therapy
Due to the lack of substantial evidence of apparent risk for self-harm, the presence of a supportive family, and Ms. S’s high academic performance and future orientation, the C-L team concludes that Ms. S’s concerns were most likely the result of the challenges of acculturation related to the language barrier and a lack of health knowledge. However, the C-L team remains cautious that Ms. S may have minimized or denied her mental health concerns due to various cultural factors. The team recommends that Ms. S seek outpatient psychotherapy from a clinician who specializes in working with Arab American individuals and families in their native language. The C-L team communicates these conclusions to the medical team verbally and in writing.
The authors’ observations
Cultural issues experienced during this consultation may not generalize to other Arab American adolescents and their families because there is diversity even within groups that share common cultural characteristics. Nevertheless, this case underscores the challenge of accurately assessing suicide risk, and making a differential diagnosis in the presence of complex cultural data and the dilemmas clinicians may encounter when attempting to answer important referral questions such as, “Is this adolescent suicidal and in need of psychiatric hospitalization?”
Bottom Line
Cultural factors and attitudes toward mental health and language barriers may play a large role in how patients answer clinical questions. Cultural issues may add a level of intricacy not easily resolved within the restrictions of an inpatient setting, and this complexity may influence clinical judgment, recommendations, and possibly health outcomes. Culturally appropriate psychotherapy is key for patients experiencing difficulty with acculturation.
Related Resources
- Adam B. Caring for Muslim patients: Understanding cultural and religious factors. Current Psychiatry. 2017;16(12):56-57.
- Nassar-McMillan SC, Hakim-Larson J. Counseling considerations among Arab Americans. Journal of Counseling & Development. 2003;81(2):150-159.
- Sue DW. Multidimensional facets of cultural competence. The Counseling Psychologist. 2001;29(6):790-821.
Drug Brand Name
Naloxone • Narcan
CASE Attempted suicide?
Ms. S, a 16-year-old Yemeni-American girl, is brought to the emergency department (ED) by her mother and brother after ingesting an overdose of painkillers and fainting. During the initial evaluation, Ms. S says she had in the past attempted suicide by knife. The medical team suspects that the current overdose is a suicide attempt, and they call the consultation-liaison (C-L) psychiatry/psychology team. Ms. S’s brother strongly denies that his sister had previously attempted suicide, stating, “She’s from a good family, and she is smart. She cannot feel that way.” He also requests the name of the clinician who documented this information in the medical record.
During the consultation, Ms. S reports that the previous morning, she developed strong abdominal pain and discovered that she was menstruating for the first time. She explains that she did not understand what was happening to her and that no one had discussed menstruation with her before. Ms. S took her mother’s opioid pain medication. Ms. S reports she took one pill, but when it did not immediately alleviate her pain, she ingested several more. After this, Ms. S says she went to play with her siblings, but gradually became dizzy and confused, and informed her sister and mother of this. The family was fasting in observance of Ramadan, and as they walked toward the mosque, Ms. S fainted, which prompted her family to bring her to the ED.
During the C-L consultation, Ms. S’s brother, who speaks English, is present, as is her mother, who speaks only Arabic and thus needs a phone interpreter. As the C-L team asks Ms. S a question, it is translated to her mother, and then Ms. S’s response is also translated, and then finally, the mother shares her own response. At times, her brother provides translation. Ms. S speaks in English, but often asks for the translation of words or questions.
Ms. S reports that she and her family emigrated from Yemen to the United States 9 months ago. Ms. S says that she enjoys school and is doing well academically. She denies experiencing any anxiety, worry, or stress related to her life in Yemen, her move to a new country, her parents’ health, school, or other domains. Ms. S also denies any history of depressive episodes or previous suicidal ideation, intention, or attempt, which contradicts her endorsement of a previous suicide attempt to one clinician when she was initially evaluated.
[polldaddy:10040204]
Continue to: The authors' observation
The authors’ observation
The C-L team determined that Ms. S did not meet criteria for major depressive disorder. She did not endorse current feelings of depression and denied anhedonia and other associated symptoms included in DSM-5 criteria for major depressive disorder or adjustment disorder with depressed mood (Table 11).
Although Ms. S and her family recently emigrated from Yemen, she did not report any symptoms consistent with an adjustment disorder with depression. Further,
Accurate case conceptualization and diagnosis is particularly crucial in C-L services, where there is an urgency for clinical decision-making after an initial evaluation without the luxury of amending conceptualization in follow-up sessions. Providing a diagnosis for which a patient does not fully or accurately meet the criteria can have deleterious effects. An inaccurate diagnosis for Ms. S would have unnecessarily added the perceived stigma of a mental disorder to her medical record. Additionally, misdiagnosing or pathologizing a natural process of acculturation could have led to inappropriate or even harmful treatment.
The C-L team evaluated alternative explanations for Ms. S’s statements that suggested she was suicidal. First, they considered her mental status at the time she presented to the ED. An overdose of opioids alters mental status. Complicating reversal of opioid overdose is that some opioids have longer half-lives than naloxone, an opioid antagonist, so the individual can become reintoxicated. Similarly, some opioids are more potent and difficult to reverse.2 An altered mental status may have limited Ms. S’s ability to comprehend and answer questions accurately when she first presented to the ED.
Continue to: Cultural factors and the clinical evaluation
Cultural factors and the clinical evaluation
Next, the C-L team considered Ms. S’s clinical picture as it related to her cultural background. Cultural factors interact with the clinical evaluation in a complex manner, influencing the way patients approach the encounter, the symptoms they report, and the language they use to describe their experiences. While these variables are thoroughly evaluated during comprehensive psychological assessments, within the inpatient consultation service, the goal for pediatric C-L clinicians is to conduct a focused assessment to answer specific and critically important questions about a youth’s psychological functioning. Thus, the fundamental challenge of inpatient consultation is to answer the referral question in a brief period and in a culturally informed manner, to appraise the referring medical team about the relevant clinical and cultural issues, with the goal of ethical and clinically sound decision-making.
The C-L team considered key cultural factors in its assessment of Ms. S (Table 31). Several issues were of concern. First, language is often cited as the top barrier to health care access by Arab Americans, even by those with competency in English.3
Experts in culture argue that even with access to interpreters, many words and phrases lack direct translation, and their implicit meaning may be difficult to reveal. Additionally, at times more significance is placed on nonverbal cues and unspoken expectations.4 This can create barriers to communication with clinicians, especially in the context of an inpatient psychiatric consultation, when thorough understanding of an adolescent and family often needs to occur in a single encounter, and clinicians may not appreciate the subtle nuances of nonverbal communication.
The language barrier also may have influenced Ms. S’s initial endorsement of a previous suicide attempt by knife because the medical staff first interviewed Ms. S without an interpreter. For instance, many medical and psychosocial providers probe patients regarding suicidality with questions such as “Have you ever hurt yourself?” or “Have you ever tried to hurt yourself?” It is possible that in another language, an individual might interpret that question as, “Have you ever gotten hurt?” This interpretation completely alters the meaning of the question and eliminates intention or motivation to harm oneself. Language ambiguity and lack of shared cultural understanding may have influenced Ms. S’s interpretation of and response to such questions. Ms. S and her family were perplexed by the C-L team’s reference to the knife and continued to deny the incident.
Continue to: Cultural attitudes to puberty
Cultural attitudes to puberty
Cultures vary with respect to education of sensitive topics such as puberty. The medical providers assumed that Ms. S was informed about the onset of menses. Therefore, they could not consider the strong impact of such an event on an unsuspecting adolescent. Many adolescent girls in Yemen have poor health and lack menstruation-related knowledge, and many are “prescribed” medications by their mothers without contacting a physician.5 Ms. S reported to the C-L team that no one from her family had discussed menstruation with her. She reported that since arriving at the hospital, nurses had educated her about menstruation, and that she was no longer afraid. She also noted that if she experienced such pain again, she would go to the hospital or “just deal with it.”
Family identification and attitudes toward mental health
Ms. S’s strong identification with her family and attitudes toward mental health may have limited what she chose to disclose regarding her experiences of loss related to leaving her country of origin, adjustment, and acculturation to the new environment, as well as feelings of sadness. Family has a central and critical role in Arab cultures. Commitment to a family’s well-being and enhancement of honor and status is highly valued and encouraged.4 Conversely, being concerned with individual needs may be a source of guilt and feelings of betraying the family.6 Arab Americans tend not to discuss personal problems with people outside their extended family, including counselors and therapists, partly because of cultural stigma against mental illness7,8 and partly because revealing family problems to strangers (ie, clinicians) may be considered a cultural taboo9 and a threat to family honor.10 Although Ms. S was interviewed privately when she first came to the ED and also during the psychiatric consultation, the stigma of psychiatric problems11 and possible concerns about protecting her family’s name may have influenced her readiness to reveal intimate information to “strangers.”
Additionally, family statements that appeared to imply negative beliefs about mental health would have strongly deterred Ms. S from expressing any psychological concerns. For example, Ms. S’s brother took offense when the C-L team said it was evaluating his sister because she had said she had previously attempted suicide.
The tenets of Islam may have provided a framework through which Ms. S interprets emotional concerns and may have defined her explanatory models of psychological stress. For instance, it is not uncommon among American Muslims to view mental health problems as rising from “loss of faith in God,”9 and suicidal ideation may not be disclosed because suicide is forbidden in Islam.12 Therefore, it might be particularly difficult to assess suicidal ideation in a patient who is Muslim, especially those who are less acculturated to Western culture.13
Continue to: Directly asking Ms. S...
Directly asking Ms. S if she had thoughts of harming herself may have been too frightening or guilt-provoking for an adolescent with her background. Asking about passive expression of suicidal ideation would have been more culturally appropriate. For example, asking, “Do you wish that God would let you die?”12 may have elicited more meaningful clinical information about Ms. S’s emotional state and possibly suicide risk.
Furthermore, Ms. S’s identification of coping strategies (ie, “just deal with it”) may have sounded limited to a Western clinician, but this may have been consistent with cultural norms of emotional expression of limiting complaints.4 Also, among Arab Americans, psychiatric symptoms often are expressed through somatization.7,14 Expressing psychological pain through physical symptoms appears protective against public stigma. Public image and opinion is important, and behaviors that would reflect well to others are dictated by the family. These attitudes, beliefs, and values likely impact how Ms. S presented her psychological concerns.
[polldaddy:10040206]
The authors’ observations
Although inpatient hospitalization was initially considered, it was not pursued due to denial of past and current suicidal ideation or suicide attempts, the lack of comorbidity, age-appropriate functioning, and a supportive family environment. Similarly, due to the absence of acute psychiatric symptoms, partial hospitalization was not pursued. The C-L team evaluated treatment options with extreme caution and sensitivity because recommending the wrong treatment option could have deleterious effects on Ms. S and her family’s life. If inpatient hospitalization had been pursued, it could have likely caused the family unnecessary suffering and could have negatively affected familial relationships. Strong feelings of shame, betrayal, and guilt would be intensified, impairing the family’s cohesion, removing environmental and family supports, and putting Ms. S at further risk of developing more severe symptoms of low mood.
Although there were significant concerns about making the wrong recommendation to the family, the C-L team’s highest priority was Ms. S’s safety. Despite cultural concerns, the team would have recommended hospitalization if Ms. S’s clinical picture had warranted this decision.
Continue to: OUTCOME Culturally-appropriate outpatient therapy
OUTCOME Culturally-appropriate outpatient therapy
Due to the lack of substantial evidence of apparent risk for self-harm, the presence of a supportive family, and Ms. S’s high academic performance and future orientation, the C-L team concludes that Ms. S’s concerns were most likely the result of the challenges of acculturation related to the language barrier and a lack of health knowledge. However, the C-L team remains cautious that Ms. S may have minimized or denied her mental health concerns due to various cultural factors. The team recommends that Ms. S seek outpatient psychotherapy from a clinician who specializes in working with Arab American individuals and families in their native language. The C-L team communicates these conclusions to the medical team verbally and in writing.
The authors’ observations
Cultural issues experienced during this consultation may not generalize to other Arab American adolescents and their families because there is diversity even within groups that share common cultural characteristics. Nevertheless, this case underscores the challenge of accurately assessing suicide risk, and making a differential diagnosis in the presence of complex cultural data and the dilemmas clinicians may encounter when attempting to answer important referral questions such as, “Is this adolescent suicidal and in need of psychiatric hospitalization?”
Bottom Line
Cultural factors and attitudes toward mental health and language barriers may play a large role in how patients answer clinical questions. Cultural issues may add a level of intricacy not easily resolved within the restrictions of an inpatient setting, and this complexity may influence clinical judgment, recommendations, and possibly health outcomes. Culturally appropriate psychotherapy is key for patients experiencing difficulty with acculturation.
Related Resources
- Adam B. Caring for Muslim patients: Understanding cultural and religious factors. Current Psychiatry. 2017;16(12):56-57.
- Nassar-McMillan SC, Hakim-Larson J. Counseling considerations among Arab Americans. Journal of Counseling & Development. 2003;81(2):150-159.
- Sue DW. Multidimensional facets of cultural competence. The Counseling Psychologist. 2001;29(6):790-821.
Drug Brand Name
Naloxone • Narcan
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Meehan TJ, Bryant SM, Aks SE. Drugs of abuse: the highs and lows of altered mental states in the emergency department. Emerg Med Clin North Am. 2010;28(3):663-682.
3. Shah SM, Ayash C, Pharaon NA, et al. Arab American immigrants in New York: health care and cancer knowledge, attitudes, and beliefs. J Immigr Minor Health. 2008;10(5):429-436.
4. Budman CL, Lipson JG, Meleis AI. The cultural consultant in mental health care: the case of an Arab adolescent. Am J Orthopsychiatry. 1992; 62(3):359-370.
5. Mohamed EM, Mohamed AG, Al-Ajeal Ly. Knowledge, beliefs and practices regarding menstruation among adolescent schoolgirls in Seiyun City, Yemen. Al-Azhar Assiut Medical Journal. 2011;9(3):67-86.
6. Gorkin M, Masalha S, Yatziv G. Psychotherapy of Israeli-Arab patients: some cultural considerations. Journal of Psychoanalytic Anthropology. 1985;8(4);215-230.
7. Gearing RE, MacKenzie MJ, Ibrahim RW, et al. Stigma and mental health treatment of adolescents with depression in Jordan. Community Ment Health J. 2015;51(1):111-117.
8. Timimi SB. Adolescence in immigrant Arab families. Psychotherapy: theory, research, practice, training. 1995;32(1):141-149.
9. Ahmed S and Reddy LA. Understanding the mental health needs of American Muslims: recommendations and considerations for practice. Journal of Multicultural Counseling and Development. 2007;35(4):207-218.
10. Abudabbeh N, Nydell MK. Transcultural counseling and Arab Americans. In: McFadden J, ed. Transcultural counseling: bilateral and international perspectives. Alexandria, VA. American Counseling Association. 1993:261-284.
11. Erickson CD, al-Timimi NR. Providing mental health services to Arab Americans: recommendations and considerations. Cultur Divers Ethnic Minor Psychol. 2001;7(4):308-327.
12. Ali SR, Liu WM, Humedian M. Islam 101: understanding the religion and therapy implications. Prof Psychol Res Pr. 2004;35(6):635-642.
13. Hedayat-Diba Z. Psychotherapy with Muslims. In: Richards PS, Bergin AE, eds. Handbook of psychotherapy and religious diversity, 2nd ed. Washington, DC: Amercian Psychological Association. 2000:289-314.
14. Al-Krenawi A. Mental health practice in Arab countries. Curr Opin in Psychiatry. 2005;18(5):560-564.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Meehan TJ, Bryant SM, Aks SE. Drugs of abuse: the highs and lows of altered mental states in the emergency department. Emerg Med Clin North Am. 2010;28(3):663-682.
3. Shah SM, Ayash C, Pharaon NA, et al. Arab American immigrants in New York: health care and cancer knowledge, attitudes, and beliefs. J Immigr Minor Health. 2008;10(5):429-436.
4. Budman CL, Lipson JG, Meleis AI. The cultural consultant in mental health care: the case of an Arab adolescent. Am J Orthopsychiatry. 1992; 62(3):359-370.
5. Mohamed EM, Mohamed AG, Al-Ajeal Ly. Knowledge, beliefs and practices regarding menstruation among adolescent schoolgirls in Seiyun City, Yemen. Al-Azhar Assiut Medical Journal. 2011;9(3):67-86.
6. Gorkin M, Masalha S, Yatziv G. Psychotherapy of Israeli-Arab patients: some cultural considerations. Journal of Psychoanalytic Anthropology. 1985;8(4);215-230.
7. Gearing RE, MacKenzie MJ, Ibrahim RW, et al. Stigma and mental health treatment of adolescents with depression in Jordan. Community Ment Health J. 2015;51(1):111-117.
8. Timimi SB. Adolescence in immigrant Arab families. Psychotherapy: theory, research, practice, training. 1995;32(1):141-149.
9. Ahmed S and Reddy LA. Understanding the mental health needs of American Muslims: recommendations and considerations for practice. Journal of Multicultural Counseling and Development. 2007;35(4):207-218.
10. Abudabbeh N, Nydell MK. Transcultural counseling and Arab Americans. In: McFadden J, ed. Transcultural counseling: bilateral and international perspectives. Alexandria, VA. American Counseling Association. 1993:261-284.
11. Erickson CD, al-Timimi NR. Providing mental health services to Arab Americans: recommendations and considerations. Cultur Divers Ethnic Minor Psychol. 2001;7(4):308-327.
12. Ali SR, Liu WM, Humedian M. Islam 101: understanding the religion and therapy implications. Prof Psychol Res Pr. 2004;35(6):635-642.
13. Hedayat-Diba Z. Psychotherapy with Muslims. In: Richards PS, Bergin AE, eds. Handbook of psychotherapy and religious diversity, 2nd ed. Washington, DC: Amercian Psychological Association. 2000:289-314.
14. Al-Krenawi A. Mental health practice in Arab countries. Curr Opin in Psychiatry. 2005;18(5):560-564.
Management of Short Bowel Syndrome, High-Output Enterostomy, and High-Output Entero-Cutaneous Fistulas in the Inpatient Setting
From the University of Texas Southwestern, Department of Internal Medicine, Dallas, TX.
Abstract
- Objective: To define intestinal failure and associated diseases that often lead to diarrhea and high-output states, and to provide a literature review on the current evidence and practice guidelines for the management of these conditions in the context of a clinical case.
- Methods: Database search on dietary and medical interventions as well as major societal guidelines for the management of intestinal failure and associated conditions.
- Results: Although major societal guidelines exist, the guidelines vary greatly amongst various specialties and are not supported by strong evidence from large randomized controlled trials. The majority of the guidelines recommend consideration of several drug classes, but do not specify medications within the drug class, optimal dose, frequency, mode of administration, and how long to trial a regimen before considering it a failure and adding additional medical therapies.
- Conclusions: Intestinal failure and high-output states affect a very heterogenous population with high morbidity and mortality. This subset of patients should be managed using a multidisciplinary approach involving surgery, gastroenterology, dietetics, internal medicine and ancillary services that include but are not limited to ostomy nurses and home health care. Implementation of a standardized protocol in the electronic medical record including both medical and nutritional therapies may be useful to help optimize efficacy of medications, aid in nutrient absorption, decrease cost, reduce hospital length of stay, and decrease hospital readmissions.
Key words: short bowel syndrome; high-output ostomy; entero-cutaneous fistula; diarrhea; malnutrition.
Intestinal failure includes but is not limited to short bowel syndrome (SBS), high-output enterostomy, and high-output related to entero-cutaneous fistulas (ECF). These conditions are unfortunate complications after major abdominal surgery requiring extensive intestinal resection leading to structural SBS. Absorption of macronutrients and micronutrients is most dependent on the length and specific segment of remaining intact small intestine [1]. The normal small intestine length varies greatly but ranges from 300 to 800 cm, while in those with structural SBS the typical length is 200 cm or less [2,3]. Certain malabsorptive enteropathies and severe intestinal dysmotility conditions may manifest as functional SBS as well. Factors that influence whether an individual will develop functional SBS despite having sufficient small intestinal absorptive area include the degree of jejunal absorptive efficacy and the ability to overcompensate with enough oral caloric intake despite high fecal energy losses, also known as hyperphagia [4].
Pathophysiology
Maintenance of normal bodily functions and homeostasis is dependent on sufficient intestinal absorption of essential macronutrients, micronutrients, and fluids. The hallmark of intestinal failure is based on the presence of decreased small bowel absorptive surface area and subsequent increased losses of key solutes and fluids [1]. Intestinal failure is a broad term that is comprised of 3 distinct phenotypes. The 3 functional classifications of intestinal failure include the following:
- Type 1. Acute intestinal failure is generally self-limiting, occurs after abdominal surgery, and typically lasts less than 28 days.
- Type 2. Subacute intestinal failure frequently occurs in septic, stressed, or metabolically unstable patients and may last up to several months.
- Type 3. Chronic intestinal failure occurs due to a chronic condition that generally requires indefinite parenteral nutrition (PN) [1,3,4].
SBS and enterostomy formation are often associated with excessive diarrhea, such that it is the most common etiology for postoperative readmissions. The definition of “high-output” varies amongst studies, but output is generally considered to be abnormally high if it is greater than 500 mL per 24 hours in ECFs and greater than 1500 mL per 24 hours for enterostomies. There is significant variability from patient to patient, as output largely depends on length of remaining bowel [2,4].
Epidemiology
SBS, high-output enterostomy, and high-output from ECFs comprise a wide spectrum of underlying disease states, including but not limited to inflammatory bowel disease, post-surgical fistula formation, intestinal ischemia, intestinal atresia, radiation enteritis, abdominal trauma, and intussusception [5]. Due to the absence of a United States registry of patients with intestinal failure, the prevalence of these conditions is difficult to ascertain. Most estimations are made using registries for patients on total parenteral nutrition (TPN). The Crohns and Colitis Foundation of America estimates 10,000 to 20,000 people suffer from SBS in the United States. This heterogenous patient population has significant morbidity and mortality for dehydration related to these high-output states. While these conditions are considered rare, they are relatively costly to the health care system. These patients are commonly managed by numerous medical and surgical services, including internal medicine, gastroenterology, surgery, dietitians, wound care nurses, and home health agencies. Management strategies differ amongst these specialties and between professional societies, which makes treatment strategies highly variable and perplexing to providers taking care of this patient population. Furthermore, most of the published guidelines are based on expert opinion and lack high-quality clinical evidence from randomized controlled trials (RCTs). Effectively treating SBS and reducing excess enterostomy output leads to reduced rates of dehydration, electrolyte imbalances, initiation of PN, weight loss and ultimately a reduction in malnutrition. Developing hospital-wide management protocols in the electronic medical record for this heterogenous condition may lead to less complications, fewer hospitalizations, and an improved quality of life for these patients.
Case Study
Initial Presentation
A 72-year-old man with history of rectal adenocarcinoma stage T4bN2 status post low anterior resection (LAR) with diverting loop ileostomy and neoadjuvant chemoradiation presented to the hospital with a 3-day history of nausea, vomiting, fatigue, and productive cough.
Additional History
On further questioning, the patient also reported odynophagia and dysphagia related to thrush. Because of his decreased oral intake, he stopped taking his usual insulin regimen prior to admission. His cancer treatment course was notable for a LAR with diverting loop ileostomy which was performed 5 months prior. He had also completed 3 out of 8 cycles of capecitabine and oxaliplatin-based therapy 2 weeks prior to this presentation.
Physical Examination
Significant physical examination findings included dry mucous membranes, oropharyngeal candidiasis, tachycardia, clear lungs, hypoactive bowel sounds, nontender, non-distended abdomen, and a right lower abdominal ileostomy bag with semi-formed stool.
Laboratory test results were pertinent for diabetic ketoacidosis (DKA) with an anion gap of 33, lactic acidosis, acute kidney injury (creatinine 2.7 mg/dL from a baseline of 1.0) and blood glucose of 1059 mg/dL. Remainder of complete blood count and complete metabolic panel were unremarkable.
Hospital Course
The patient was treated for oropharyngeal candidiasis with fluconazole, started on an insulin drip and given intravenous fluids (IVFs) with subsequent resolution of DKA. Once the DKA resolved, his diet was advanced to a mechanical soft, moderate calorie, consistent carbohydrate diet (2000 calories allowed daily with all foods chopped, pureed or cooked, and all meals containing nearly equal amounts of carbohydrates). He was also given Boost supplementation 3 times per day, and daily weights were recorded while assessing for fluid losses. However, during his hospital course the patient developed increasing ileostomy output ranging from 2.7 to 6.5 L per day that only improved when he stopped eating by mouth (NPO).
What conditions should be evaluated prior to starting therapy for high-output enterostomy/diarrhea from either functional or structural SBS?
Prior to starting anti-diarrheal and anti-secretory therapy, infectious and metabolic etiologies for high-enterostomy output should be ruled out. Depending on the patient’s risk factors (eg, recent sick contacts, travel) and whether they are immunocompetent versus immunosuppressed, infectious studies should be obtained. In this patient, Clostridium difficile, stool culture, Giardia antigen, stool ova and parasites were all negative. Additional metabolic labs including thyroid-stimulating hormone, fecal elastase, and fecal fat were obtained and were all within normal limits. In this particular scenario, fecal fat was obtained while he was NPO. Testing for fat malabsorption and pancreatic insufficiency in a patient that is consuming less than 100 grams of fat per day can result in a false-negative outcome, however, and was not an appropriate test in this patient.
Hospital Course Continued
Once infectious etiologies were ruled out, the patient was started on anti-diarrheal medication consisting of loperamide 2 mg every 6 hours and oral pantoprazole 40 mg once per day. The primary internal medicine team speculated that the Boost supplementation may be contributing to the diarrhea because of its hyperosmolar concentration and wanted to discontinue it, but because the patient had protein-calorie malnutrition the dietician recommended continuing Boost supplementation. The primary internal medicine team also encouraged the patient to drink Gatorade with each meal with the approval from the dietician.
What are key dietary recommendations to help reduce high-output enterostomy/diarrhea?
Dietary recommendations are often quite variable depending on the intestinal anatomy (specifically, whether the colon is intact or absent), comorbidities such as renal disease, and severity of fluid and nutrient loses. This patient has the majority of his colon remaining; however, fluid and nutrients are being diverted away from his colon because he has a loop ileostomy. To reduce enterostomy output, it is generally recommended that liquids be consumed separately from solids, and that oral rehydration solutions (ORS) should replace most hyperosmolar and hypoosmolar liquids. Although these recommendations are commonly used, there is sparse data to suggest separating liquids from solids in a medically stable patient with SBS is indeed necessary [6]. In our patient, however, because he has not yet reached medical stability, it would be reasonable to separate the consumption of liquids from solids. The solid component of a SBS diet should consist mainly of protein and carbohydrates, with limited intake of simple sugars and sugar alcohols. If the colon remains intact, it is particularly important to limit fats to less than 30% of the daily caloric intake, to consume a low-oxalate diet, supplement with oral calcium to reduce the risk of calcium-oxalate nephrolithiasis, and increase dietary fiber intake as tolerated. Soluble fiber is fermented by colonic bacteria into short-chain fatty acids (SCFAs) and serve as an additional energy source [7,8]. Medium-chain triglycerides (MCTs) are good sources of fat because the body is able to absorb them into the bloodstream without the use of intestinal lymphatics, which may be damaged or absent in those with intestinal failure. For this particular patient, he would have benefitted from initiation of ORS and counseled to sip on it throughout the day while limiting liquid consumption during meals. He should have also been advised to limit plain Gatorade and Boost as they are both hyperosmolar liquid formulations and can worsen diarrhea. If the patient was unable to tolerate the taste of standard ORS formulations, or the hospital did not have any ORS on formulary, sugar, salt and water at specific amounts may be added to create a homemade ORS. In summary, this patient would have likely tolerated protein in solid form better than liquid protein supplementation.
Hospital Course Continued
The patient continued to have greater than 5 L of output from the ileostomy per day, so the following day the primary team increased the loperamide from 2 mg every 6 hours to 4 mg every 6 hours, added 2 tabs of diphenoxylate-atropine every 8 hours, and made the patient NPO. He continued to require IVFs and frequent electrolyte repletion because of the significant ongoing gastrointestinal losses.
What is the recommended first-line medical therapy for high-output enterostomy/diarrhea?
Anti-diarrheal medications are commonly used in high-output states because they work by reducing the rate of bowel translocation thereby allowing for longer time for nutrient and fluid absorption in the small and large intestine. Loperamide in particular also improves fecal incontinence because it effects the recto-anal inhibitory reflex and increases internal anal sphincter tone [9]. Four RCTs showed that loperamide lead to a significant reduction in enterostomy output compared to placebo with enterostomy output reductions ranging from 22% to 45%; varying dosages of loperamide were used, and ranged from 6 mg per day to 16 total mg per day [10–12]. King et al compared loperamide and codeine to placebo and found that both medications led to reductions in enterostomy output with a greater reduction and better side effect profile in those that received loperamide or combination therapy with loperamide and codeine [13,14]. The majority of studies used a maximum dose of 16 mg per day of loperamide, and this is the maxium daily dose approved by the US Food and Drug Administration (FDA). Interestingly however, loperamide circulates through the enterohepatic circulation which is severely disrupted in SBS, so titrating up to a maximum dose of 32 mg per day while closely monitoring for side effects is also practiced by experts in intestinal failure [15]. It is also important to note that anti-diarrheal medications are most effective when administered 20 to 30 minutes prior to meals and not scheduled every 4 to 6 hours if the patient is eating by mouth. If intestinal transit is so rapid such that undigested anti-diarrheal tablets or capsules are visualized in the stool or stoma, medications can be crushed or opened and mixed with liquids or solids to enhance digestion and absorption.
Hospital Course Continued
The patient continued to have greater than 3 L of ileostomy output per day despite being on scheduled loperamide, diphenoxylate-atropine, and a proton pump inhibitory (PPI), although improved from greater than 5 L per day. He was subsequently started on opium tincture 6 mg every 6 hours, psyllium 3 times per day, the dose of diphenoxylate-atropine was increased from 2 tablets every 8 hours to 2 tablets every 6 hours, and he was encouraged to drink water in between meals. As mentioned previously, the introduction of dietary fiber should be carefully monitored, as this patient population is commonly intolerant of high dietary fiber intake, and hypoosmolar liquids like water should actually be minimized. Within a 48-hour time period, the surgical team recommended increasing the loperamide from 4 mg every 6 hours (16 mg total daily dose) to 12 mg every 6 hours (48 mg total daily dose), increased opium tincture from 6 mg every 6 hours (24 mg total daily dose) to 10 mg every 6 hours (40 mg total daily dose), and increased oral pantoprazole from 40 mg once per day to twice per day.
What are important considerations with regard to dose changes?
Evidence is lacking to suggest an adequate time period to monitor for response to therapy in regards to improvement in diarrheal output. In this scenario, it may have been prudent to wait 24 to 48 hours after each medication change instead of making drastic dose changes in several medications simultaneously. PPIs irreversibly inhibit gastrointestinal acid secretion as do histamine-2 receptor antagonists (H2RAs) but to a lesser degree, and thus reduce high-output enterostomy [16]. Reduction in pH related to elevated gastrin levels after intestinal resection is associated with pancreatic enzyme denaturation and downstream bile salt dysfunction, which can further lead to malabsorption [17]. Gastrin hypersecretion is most prominent within the first 6 months after intestinal resection such that the use of high- dose PPIs for reduction in gastric acid secretion are most efficacious within that time period [18,19]. Jeppesen et al demonstrated that both omeprazole 40 mg oral twice per day and ranitidine 150 mg IV once per day were effective in reducing enterostomy output, although greater reductions were seen with omeprazole [20]. Three studies using cimetidine (both oral and IV formulations) with dosages varying from 200 mg to 800 mg per day showed significant reductions in enterostomy output as well [21–23].
Hospital Course Continued
Despite the previously mentioned interventions, the patient’s ileostomy output remained greater than 3 L per day. Loperamide was increased from 12 mg every 6 hours to 16 mg every 6 hours (64 mg total daily dose) hours and opium tincture was increased from 10 mg to 16 mg every 6 hours (64 mg total daily dose). Despite these changes, no significant reduction in output was noted, so the following day, 4 grams of cholestyramine light was added twice per day.
If the patient continues to have high-output enterostomy/diarrhea, what are additional treatment options?
Bile acid binding resins like cholestyramine, colestipol, and colesevelam are occasionally used if there is a high suspicion for bile acid diarrhea. Bile salt diarrhea typically occurs because of alterations in the enterohepatic circulation of bile salts, which leads to an increased level of bile salts in the colon and stimulation of electrolyte and water secretion and watery diarrhea [24]. Optimal candidates for bile acid binding therapy are those with an intact colon and less than 100 cm of resected ileum. Patients with little to no remaining or functional ileum have a depleted bile salt pool, therefore the addition of bile acid resin binders may actually lead to worsening diarrhea secondary to bile acid deficiency and fat malabsorption. Bile-acid resin binders can also decrease oxalate absorption and precipitate oxalate stone formation in the kidneys. Caution should also be taken to ensure that these medications are administered separately from the remainder of the patient’s medications to limit medication binding.
If the patient exhibits hemodynamic stability, alpha-2 receptor agonists are occasionally used as adjunctive therapy in reducing enterostomy output, although strong evidence to support its use is lacking. The mechanism of action involves stimulation of alpha-2 adrenergic receptors on enteric neurons, which theoretically causes a reduction in gastric and colonic motility and decreases fluid secretion. Buchman et al showed that the effects of a clonidine patch versus placebo did not in fact lead to a significant reduction in enterostomy output; however, a single case report suggested that the combination of 1200 mcg of clonidine per day and somatostatin resulted in decreased enterostomy output via alpha 2-receptor inhibition of adenylate cyclase [25,26].
Hospital Course Continued
The patient’s ileostomy output remained greater than 3 L per day, so loperamide was increased from 14 mg every 6 hours to 20 mg every 6 hours (80 mg total daily dose), cholestyramine was discontinued because of metabolic derangements, and the patient was initiated on 100 mcg of subcutaneous octreotide 3 times per day. Colorectal surgery was consulted for ileostomy takedown given persistently high-output, but surgery was deferred. After a 16-day hospitalization, the patient was eventually discharged home. At the time of discharge, he was having 2–3 L of ileostomy output per day and plans for future chemotherapy were discontinued because of this.
Does hormonal therapy have a role in the management of high-output enterostomy or entero-cutaneous fistulas?
Somatostatin analogues are growth-hormone inhibiting factors that have been used in the treatment of SBS and gastrointestinal fistulas. These medications reduce intestinal and pancreatic fluid secretion, slow intestinal motility, and inhibit the secretion of several hormones including gastrin, vasoactive intestinal peptide, cholecystokinin, and other key intestinal hormones. There is conflicting evidence for the role of these medications in reducing enterostomy output when first-line treatments have failed. Several previous studies using octreotide or somatostatin showed significant reductions in enterostomy output using variable dosages [27–30]. One study using the long-acting release depot octreotide preparation in 8 TPN-dependent patients with SBS showed a significant increase in small bowel transit time, however there was no significant improvement in the following parameters: body weight, stool weight, fecal fat excretion, stool electrolyte excretion, or gastric emptying [31]. Other studies evaluating enterostomy output from gastrointestinal and pancreatic fistulas comparing combined therapy with octreotide and TPN to placebo and TPN failed to show a significant difference in output and spontaneous fistula closure within 20 days of treatment initiation [32]. Because these studies use highly variable somatostatin analogue dosages and routes of administration, the most optimal dosing and route of administration (SQ versus IV) are unknown. In patients with difficult to control blood sugars, initiation of somatostatin analogues should be cautioned since these medications can lead to blood sugar alterations [33]. Additional unintended effects include impairment in intestinal adaptation and an increased risk in gallstone formation [8].
The most recent medical advances in SBS management include gut hormones. Glucagon-like peptide 2 (GLP-2) analogues improve structural and functional intestinal adaptation following intestinal resection by decreasing gastric emptying, decreasing gastric acid secretion, increasing intestinal blood flow, and enhancing nutrient and fluid absorption. Teduglutide, a GLP-2 analog, was successful in reducing fecal energy losses and increasing intestinal wet weight absorption, and reducing the need for PN support in SBS patients [1].
Whose problem is it anyway?
Not only is there variation in management strategies among subspecialties, but recommendations amongst societies within the same subspecialty differ, and thus make management perplexing.
Gastroenterology Guidelines
Several major gastroenterology societies have published guidelines on the management of diarrhea in patients with intestinal failure. The British Society of Gastroenterology (BSG) published guidelines on the management of SBS in 2006 and recommended the following first-line therapy for diarrhea-related complications: start loperamide at 2–8 mg thirty minutes prior to meals, taken up to 4 times per day, and the addition of codeine phosphate 30–60 mg thirty minutes before meals if output remains above goal on loperamide monotherapy. Cholestyramine may be added for those with 100 cm or less of resected terminal ileum to assist with bile-salt-induced diarrhea, though no specific dosage recommendations were reported. In regards to anti-secretory medications, the BSG recommends cimetidine (400 mg oral or IV 4 times per day), ranitidine (300 mg oral twice per day), or omeprazole (40 mg oral once per day or IV twice per day) to reduce jejunostomy output particularly in patients with greater than 2 L of output per day [15,34]. If diarrhea or enterostomy output continues to remain above goal, the guidelines suggest initiating octreotide and/or growth factors (although dosing and duration of therapy is not discussed in detail), and considering evaluation for intestinal transplant once the patient develops complications related to long-term TPN.
The American Gastroenterology Association (AGA) published guidelines and a position statement in 2003 for the management of high-gastric output and fluid losses. For postoperative patients, the AGA recommends the use of PPIs and H2RAs for the first 6 months following bowel resection when hyper-gastrinemia most commonly occurs. The guidelines do not specify which PPI or H2RA is preferred or recommended dosages. For long-term management of diarrhea or excess fluid losses, the guidelines suggest using loperamide or diphenoxylate (4-16 mg per day) first, followed by codeine sulfate 15–60 mg two to three times per day or opium tincture (dosages not specified). The use of octreotide (100 mcg SQ 3 times per day, 30 minutes prior to meals) is recommended only as a last resort if IVF requirements are greater than 3 L per day [8].
Surgical Guidelines
The Cleveland Clinic published institutional guidelines for the management of intestinal failure in 2010 with updated recommendations in 2016. Dietary recommendations include the liberal use of salt, sipping on 1–2 L of ORS between meals, and a slow reintroduction of soluble fiber from foods and/or supplements as tolerated. The guidelines also suggest considering placement of a nasogastric feeding tube or percutaneous gastrostomy tube (PEG) for continuous enteral feeding in addition to oral intake to enhance nutrient absorption [35]. If dietary manipulation is inadequate and medical therapy is required, the following medications are recommended in no particular order: loperamide 4 times per day (maximum dosage of 16 mg), diphenoxylate-atropine 4 times per day (maximum dosage of 20 mg per day), codeine 4 times per day (maximum dosage 240 mg per day), paregoric 5 mL (containing 2 mg of anhydrous morphine) 4 times per day, and opium tincture 0.5 mL (10 mg/mL) 4 times per day. H2RAs and PPIs are recommended for postoperative high-output states, although no dosage recommendations or routes of administration were discussed. 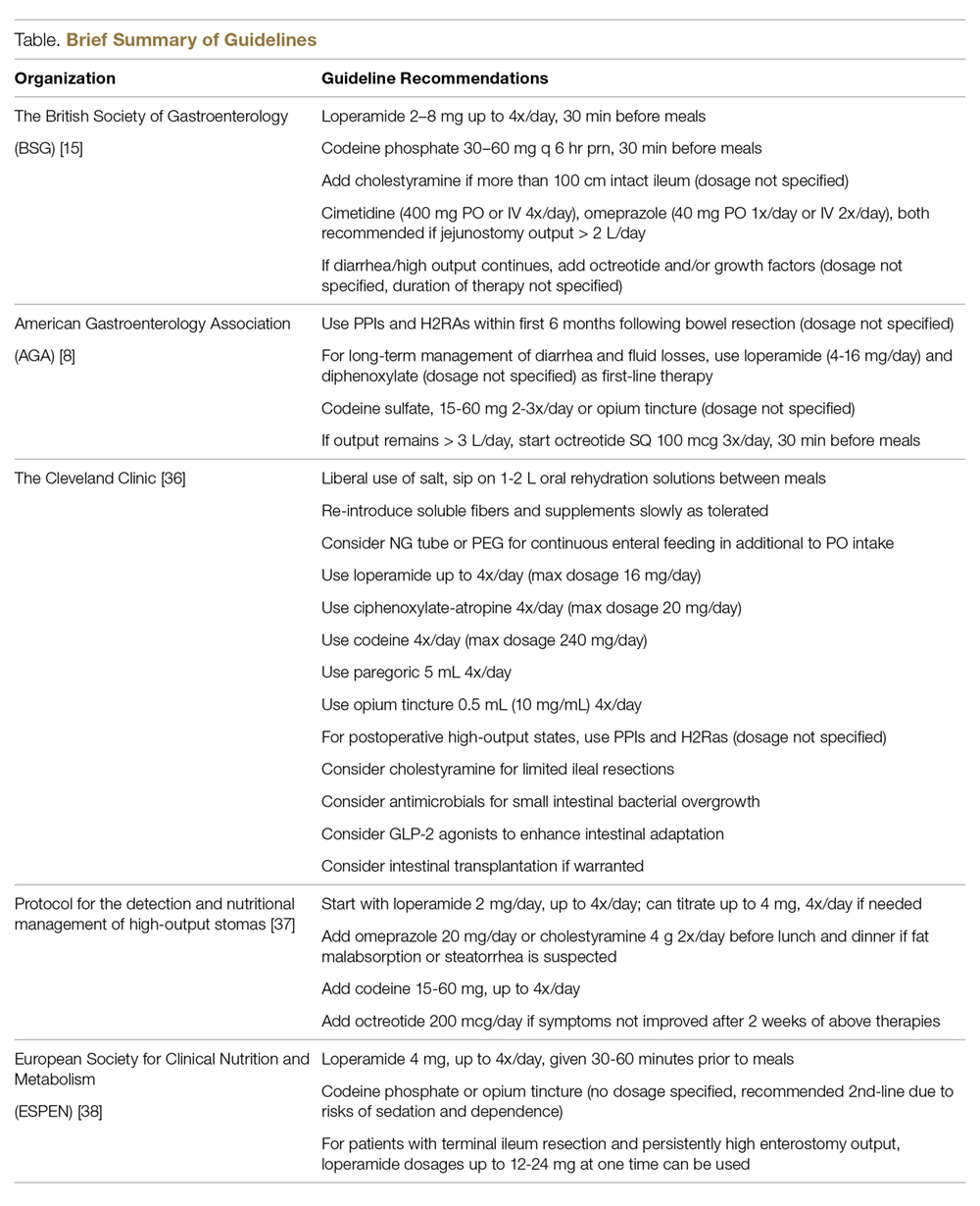
Nutrition Guidelines
Villafranca et al published a protocol for the management of high-output stomas in 2015 that was shown to be effective in reducing high-enterostomy output. The protocol recommended initial treatment with loperamide 2 mg orally up to 4 times per day. If enterostomy output did not improve, the protocol recommended increasing loperamide to 4 mg four times per day, adding omeprazole 20 mg orally or cholestyramine 4 g twice per day before lunch and dinner if fat malabsorption or steatorrhea is suspected, and lastly the addition of codeine 15–60 mg up to 4 times per day and octreotide 200 mcg per day only if symptoms had not improved after 2 weeks [37].
The American Society for Parenteral and Enteral Nutrition (ASPEN) does not have published guidelines for the management of SBS. In 2016 however, the European Society for Clinical Nutrition and Metabolism (ESPEN) published guidelines on the management of chronic intestinal failure in adults. In patients with an intact colon, ESPEN strongly recommends a diet rich in complex carbohydrates and low in fat and using H2RAs or PPIs to treat hyper-gastrinemia within the first 6 months after intestinal resection particularly in those with greater than 2 L per day of fecal output. The ESPEN guidelines do not include whether to start a PPI or H2RA first, which particular drug in each class to try, or dosage recommendations but state that IV soluble formulations should be considered in those that do not seem to respond to tablets. ESPEN does not recommend the addition of soluble fiber to enhance intestinal absorption or probiotics and glutamine to aid in intestinal rehabilitation. For diarrhea and excessive fecal fluid, the guidelines recommend 4 mg of oral loperamide 30–60 minutes prior to meals, 3 to 4 times per day, as first-line treatment in comparison to codeine phosphate or opium tincture given the risks of dependence and sedation with the latter agents. They report, however, that dosages up to 12–24 mg at one time of loperamide are used in patients with terminal ileum resection and persistently high-output enterostomy [38].
Case Conclusion
The services that were closely involved in this patient’s care were general internal medicine, general surgery, colorectal surgery, and ancillary services, including dietary and wound care. Interestingly, despite persistent high ileostomy output during the patient’s 16-day hospital admission, the gastroenterology service was never consulted. This case illustrates the importance of having a multidisciplinary approach to the care of these complicated patients to ensure that the appropriate medications are ordered based on the individual’s anatomy and that medications are ordered at appropriate dosages and timing intervals to maximize drug efficacy. It is also critical to ensure that nursing staff accurately documents all intake and output so that necessary changes can be made after adequate time is given to assess for a true response. There should be close communication between the primary medical or surgical service with the dietician to ensure the patient is counseled on appropriate dietary intake to help minimize diarrhea and fluid losses.
Conclusion
In conclusion, intestinal failure is a heterogenous group of disease states that often occurs after major intestinal resection and is commonly associated with malabsorption and high output states. High-output enterostomy and diarrhea are the most common etiologies leading to hospital re-admission following enterostomy creation or intestinal resection. These patients have high morbidity and mortality rates, and their conditions are costly to the health care system. Lack of high-quality evidence from RCTs and numerous societal guidelines without clear medication and dietary algorithms and low prevalence of these conditions makes management of these patients by general medical and surgical teams challenging. The proper management of intestinal failure and related complications requires a multidisciplinary approach with involvement from medical, surgical, and ancillary services. We propose a multidisciplinary approach with involvement from medical, surgical, and ancillary services in designed and implementing a protocol using electronic medical record based order sets to simplify and improve the management of these patients in the inpatient setting.
Corresponding author: Jake Hutto, 5323 Harry Hines Blvd, Dallas, TX 75390-9030, [email protected].
Financial disclosures: None.
1. Jeppesen PB. Gut hormones in the treatment of short-bowel syndrome and intestinal failure. Current opinion in endocrinology, diabetes, and obesity. Curr Opin Endocrinol Diabetes Obes 2015;22:14–20.
2. Berry SM, Fischer JE. Classification and pathophysiology of enterocutaneous fistulas. Surg Clin North Am 1996;76:1009–18.
3. Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1111–34.
4. de Vries FEE, Reeskamp LF, van Ruler O et al. Systematic review: pharmacotherapy for high-output enterostomies or enteral fistulas. Aliment Pharmacol Ther 2017;46:266–73.
5. Holzheimer RG, Mannick JA. Surgical Treatment: Evidence-Based and Problem-Oriented. Munich: Zuckschwerdt; 2001.
6. Woolf GM, Miller C, Kurian R, Jeejeebhoy KN. Nutritional absorption in short bowel syndrome. Evaluation of fluid, calorie, and divalent cation requirements. Dig Dis Sci 1987;32:8–15.
7. Parrish CR, DiBaise JK. Managing the adult patient with short bowel syndrome. Gastroenterol Hepatol (N Y) 2017;13:600–8.
8. American Gastroenterological Association. American Gastroenterological Association medical position statement: short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1105–10.
9. Musial F, Enck P, Kalveram KT, Erckenbrecht JF. The effect of loperamide on anorectal function in normal healthy men. J Clin Gastroenterol. 1992;15:321–4.
10. Tijtgat GN, Meuwissen SG, Huibregtse K. Loperamide in the symptomatic control of chronic diarrhoea. Double-blind placebo-controlled study. Ann Clin Res 1975;7:325–30.
11. Tytgat GN, Huibregtse K, Dagevos J, van den Ende A. Effect of loperamide on fecal output and composition in well-established ileostomy and ileorectal anastomosis. Am J Dig Dis 1977;22:669–76.
12. Stevens PJ, Dunbar F, Briscoe P. Potential of loperamide oxide in the reduction of ileostomy and colostomy output. Clin Drug Investig 1995;10:158–64.
13. King RF, Norton T, Hill GL. A double-blind crossover study of the effect of loperamide hydrochloride and codeine phosphate on ileostomy output. Aust N Z J Surg 1982;52:121–4.
14. Nightingale JM, Lennard-Jones JE, Walker ER. A patient with jejunostomy liberated from home intravenous therapy after 14 years; contribution of balance studies. Clin Nutr 1992;11:101–5.
15. Nightingale J, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006;55:iv1–12.
16. Nightingale JM, Lennard-Jones JE, Walker ER, Farthing MJ. Jejunal efflux in short bowel syndrome. Lancet 1990;336:765–8.
17. Go VL, Poley JR, Hofmann AF, Summerskill WH. Disturbances in fat digestion induced by acidic jejunal pH due to gastric hypersecretion in man. Gastroenterology 1970;58:638–46.
18. Windsor CW, Fejfar J, Woodward DA. Gastric secretion after massive small bowel resection. Gut 1969;10:779–86.
19. Williams NS, Evans P, King RF. Gastric acid secretion and gastrin production in the short bowel syndrome. Gut 1985;26:914–9.
20. Jeppesen PB, Staun M, Tjellesen L, Mortensen PB. Effect of intravenous ranitidine and omeprazole on intestinal absorption of water, sodium, and macronutrients in patients with intestinal resection. Gut 1998;43:763–9.
21. Aly A, Bárány F, Kollberg B, et al. Effect of an H2-receptor blocking agent on diarrhoeas after extensive small bowel resection in Crohn’s disease. Acta Med Scand 1980;207:119–22.
22. Kato J, Sakamoto J, Teramukai S, et al. A prospective within-patient comparison clinical trial on the effect of parenteral cimetidine for improvement of fluid secretion and electrolyte balance in patients with short bowel syndrome. Hepatogastroenterology. 2004;51:1742–6.
23. Jacobsen O, Ladefoged K, Stage JG, Jarnum S. Effects of cimetidine on jejunostomy effluents in patients with severe short-bowel syndrome. Scand J Gastroenterol 1986;21:824–8.
24. Hofmann AF. The syndrome of ileal disease and the broken enterohepatic circulation: cholerheic enteropathy. Gastroenterology 1967;52:752–7.
25. Buchman AL, Fryer J, Wallin A et al. Clonidine reduces diarrhea and sodium loss in patients with proximal jejunostomy: a controlled study. JPEN J Parenter Enteral Nutr. 2006;30:487–91.
26. Scholz J, Bause H, Reymann A, Dürig M. Treatment with clonidine in a case of the short bowel syndrome with therapy-refractory diarrhea [ in German]. Anasthesiol Intensivmed Notfallmed Schmerzther 1991;26:265–9.
27. Torres AJ, Landa JI, Moreno-Azcoita M, et al. Somatostatin in the management of gastrointestinal fistulas. A multicenter trial. Arch Surg 1992;127:97–9; discussion 100.
28. Nubiola-Calonge P, Badia JM, Sancho J, et al. Blind evaluation of the effect of octreotide (SMS 201-995), a somatostatin analogue, on small-bowel fistula output. Lancet 1987;2:672–4.
29. Kusuhara K, Kusunoki M, Okamoto T, et al. Reduction of the effluent volume in high-output ileostomy patients by a somatostatin analogue, SMS 201-995. Int J Colorectal Dis 1992;7:202–5.
30. O’Keefe SJ, Peterson ME, Fleming CR. Octreotide as an adjunct to home parenteral nutrition in the management of permanent end-jejunostomy syndrome. JPEN J Parenter Enteral Nutr 1994;18:26–34.
31. Nehra V, Camilleri M, Burton D, et al. An open trial of octreotide long-acting release in the management of short bowel syndrome. Am J Gastroenterol 2001;96:1494–8.
32. Sancho JJ, di Costanzo J, Nubiola P, et al. Randomized double-blind placebo-controlled trial of early octreotide in patients with postoperative enterocutaneous fistula. Br J Surg 1995;82:638–41.
33. Alberti KG, Christensen NJ, Christensen SE, et al. Inhibition of insulin secretion by somatostatin. Lancet 1973;2:1299–301.
34. Hofmann AF, Poley JR. Role of bile acid malabsorption in pathogenesis of diarrhea and steatorrhea in patients with ileal resection. I. Response to cholestyramine or replacement of dietary long chain triglyceride by medium chain triglyceride. Gastroenterology 1972;62:918–34.
35. Joly F, Dray X, Corcos O, et al. Tube feeding improves intestinal absorption in short bowel syndrome patients. Gastroenterology 2009;136:824–31.
36. Bharadwaj S, Tandon P, Rivas JM, et al. Update on the management of intestinal failure. Cleveland Cleve Clin J Med 2016;83:841–8.
37. Arenas Villafranca JJ, López-Rodríguez C, Abilés J, et al. Protocol for the detection and nutritional management of high-output stomas. Nutr J 2015;14:45.
38. Pironi L, Arends J, Bozzetti F, et al. ESPEN guidelines on chronic intestinal failure in adults. Clin Nutr 2016;35:247–307.
From the University of Texas Southwestern, Department of Internal Medicine, Dallas, TX.
Abstract
- Objective: To define intestinal failure and associated diseases that often lead to diarrhea and high-output states, and to provide a literature review on the current evidence and practice guidelines for the management of these conditions in the context of a clinical case.
- Methods: Database search on dietary and medical interventions as well as major societal guidelines for the management of intestinal failure and associated conditions.
- Results: Although major societal guidelines exist, the guidelines vary greatly amongst various specialties and are not supported by strong evidence from large randomized controlled trials. The majority of the guidelines recommend consideration of several drug classes, but do not specify medications within the drug class, optimal dose, frequency, mode of administration, and how long to trial a regimen before considering it a failure and adding additional medical therapies.
- Conclusions: Intestinal failure and high-output states affect a very heterogenous population with high morbidity and mortality. This subset of patients should be managed using a multidisciplinary approach involving surgery, gastroenterology, dietetics, internal medicine and ancillary services that include but are not limited to ostomy nurses and home health care. Implementation of a standardized protocol in the electronic medical record including both medical and nutritional therapies may be useful to help optimize efficacy of medications, aid in nutrient absorption, decrease cost, reduce hospital length of stay, and decrease hospital readmissions.
Key words: short bowel syndrome; high-output ostomy; entero-cutaneous fistula; diarrhea; malnutrition.
Intestinal failure includes but is not limited to short bowel syndrome (SBS), high-output enterostomy, and high-output related to entero-cutaneous fistulas (ECF). These conditions are unfortunate complications after major abdominal surgery requiring extensive intestinal resection leading to structural SBS. Absorption of macronutrients and micronutrients is most dependent on the length and specific segment of remaining intact small intestine [1]. The normal small intestine length varies greatly but ranges from 300 to 800 cm, while in those with structural SBS the typical length is 200 cm or less [2,3]. Certain malabsorptive enteropathies and severe intestinal dysmotility conditions may manifest as functional SBS as well. Factors that influence whether an individual will develop functional SBS despite having sufficient small intestinal absorptive area include the degree of jejunal absorptive efficacy and the ability to overcompensate with enough oral caloric intake despite high fecal energy losses, also known as hyperphagia [4].
Pathophysiology
Maintenance of normal bodily functions and homeostasis is dependent on sufficient intestinal absorption of essential macronutrients, micronutrients, and fluids. The hallmark of intestinal failure is based on the presence of decreased small bowel absorptive surface area and subsequent increased losses of key solutes and fluids [1]. Intestinal failure is a broad term that is comprised of 3 distinct phenotypes. The 3 functional classifications of intestinal failure include the following:
- Type 1. Acute intestinal failure is generally self-limiting, occurs after abdominal surgery, and typically lasts less than 28 days.
- Type 2. Subacute intestinal failure frequently occurs in septic, stressed, or metabolically unstable patients and may last up to several months.
- Type 3. Chronic intestinal failure occurs due to a chronic condition that generally requires indefinite parenteral nutrition (PN) [1,3,4].
SBS and enterostomy formation are often associated with excessive diarrhea, such that it is the most common etiology for postoperative readmissions. The definition of “high-output” varies amongst studies, but output is generally considered to be abnormally high if it is greater than 500 mL per 24 hours in ECFs and greater than 1500 mL per 24 hours for enterostomies. There is significant variability from patient to patient, as output largely depends on length of remaining bowel [2,4].
Epidemiology
SBS, high-output enterostomy, and high-output from ECFs comprise a wide spectrum of underlying disease states, including but not limited to inflammatory bowel disease, post-surgical fistula formation, intestinal ischemia, intestinal atresia, radiation enteritis, abdominal trauma, and intussusception [5]. Due to the absence of a United States registry of patients with intestinal failure, the prevalence of these conditions is difficult to ascertain. Most estimations are made using registries for patients on total parenteral nutrition (TPN). The Crohns and Colitis Foundation of America estimates 10,000 to 20,000 people suffer from SBS in the United States. This heterogenous patient population has significant morbidity and mortality for dehydration related to these high-output states. While these conditions are considered rare, they are relatively costly to the health care system. These patients are commonly managed by numerous medical and surgical services, including internal medicine, gastroenterology, surgery, dietitians, wound care nurses, and home health agencies. Management strategies differ amongst these specialties and between professional societies, which makes treatment strategies highly variable and perplexing to providers taking care of this patient population. Furthermore, most of the published guidelines are based on expert opinion and lack high-quality clinical evidence from randomized controlled trials (RCTs). Effectively treating SBS and reducing excess enterostomy output leads to reduced rates of dehydration, electrolyte imbalances, initiation of PN, weight loss and ultimately a reduction in malnutrition. Developing hospital-wide management protocols in the electronic medical record for this heterogenous condition may lead to less complications, fewer hospitalizations, and an improved quality of life for these patients.
Case Study
Initial Presentation
A 72-year-old man with history of rectal adenocarcinoma stage T4bN2 status post low anterior resection (LAR) with diverting loop ileostomy and neoadjuvant chemoradiation presented to the hospital with a 3-day history of nausea, vomiting, fatigue, and productive cough.
Additional History
On further questioning, the patient also reported odynophagia and dysphagia related to thrush. Because of his decreased oral intake, he stopped taking his usual insulin regimen prior to admission. His cancer treatment course was notable for a LAR with diverting loop ileostomy which was performed 5 months prior. He had also completed 3 out of 8 cycles of capecitabine and oxaliplatin-based therapy 2 weeks prior to this presentation.
Physical Examination
Significant physical examination findings included dry mucous membranes, oropharyngeal candidiasis, tachycardia, clear lungs, hypoactive bowel sounds, nontender, non-distended abdomen, and a right lower abdominal ileostomy bag with semi-formed stool.
Laboratory test results were pertinent for diabetic ketoacidosis (DKA) with an anion gap of 33, lactic acidosis, acute kidney injury (creatinine 2.7 mg/dL from a baseline of 1.0) and blood glucose of 1059 mg/dL. Remainder of complete blood count and complete metabolic panel were unremarkable.
Hospital Course
The patient was treated for oropharyngeal candidiasis with fluconazole, started on an insulin drip and given intravenous fluids (IVFs) with subsequent resolution of DKA. Once the DKA resolved, his diet was advanced to a mechanical soft, moderate calorie, consistent carbohydrate diet (2000 calories allowed daily with all foods chopped, pureed or cooked, and all meals containing nearly equal amounts of carbohydrates). He was also given Boost supplementation 3 times per day, and daily weights were recorded while assessing for fluid losses. However, during his hospital course the patient developed increasing ileostomy output ranging from 2.7 to 6.5 L per day that only improved when he stopped eating by mouth (NPO).
What conditions should be evaluated prior to starting therapy for high-output enterostomy/diarrhea from either functional or structural SBS?
Prior to starting anti-diarrheal and anti-secretory therapy, infectious and metabolic etiologies for high-enterostomy output should be ruled out. Depending on the patient’s risk factors (eg, recent sick contacts, travel) and whether they are immunocompetent versus immunosuppressed, infectious studies should be obtained. In this patient, Clostridium difficile, stool culture, Giardia antigen, stool ova and parasites were all negative. Additional metabolic labs including thyroid-stimulating hormone, fecal elastase, and fecal fat were obtained and were all within normal limits. In this particular scenario, fecal fat was obtained while he was NPO. Testing for fat malabsorption and pancreatic insufficiency in a patient that is consuming less than 100 grams of fat per day can result in a false-negative outcome, however, and was not an appropriate test in this patient.
Hospital Course Continued
Once infectious etiologies were ruled out, the patient was started on anti-diarrheal medication consisting of loperamide 2 mg every 6 hours and oral pantoprazole 40 mg once per day. The primary internal medicine team speculated that the Boost supplementation may be contributing to the diarrhea because of its hyperosmolar concentration and wanted to discontinue it, but because the patient had protein-calorie malnutrition the dietician recommended continuing Boost supplementation. The primary internal medicine team also encouraged the patient to drink Gatorade with each meal with the approval from the dietician.
What are key dietary recommendations to help reduce high-output enterostomy/diarrhea?
Dietary recommendations are often quite variable depending on the intestinal anatomy (specifically, whether the colon is intact or absent), comorbidities such as renal disease, and severity of fluid and nutrient loses. This patient has the majority of his colon remaining; however, fluid and nutrients are being diverted away from his colon because he has a loop ileostomy. To reduce enterostomy output, it is generally recommended that liquids be consumed separately from solids, and that oral rehydration solutions (ORS) should replace most hyperosmolar and hypoosmolar liquids. Although these recommendations are commonly used, there is sparse data to suggest separating liquids from solids in a medically stable patient with SBS is indeed necessary [6]. In our patient, however, because he has not yet reached medical stability, it would be reasonable to separate the consumption of liquids from solids. The solid component of a SBS diet should consist mainly of protein and carbohydrates, with limited intake of simple sugars and sugar alcohols. If the colon remains intact, it is particularly important to limit fats to less than 30% of the daily caloric intake, to consume a low-oxalate diet, supplement with oral calcium to reduce the risk of calcium-oxalate nephrolithiasis, and increase dietary fiber intake as tolerated. Soluble fiber is fermented by colonic bacteria into short-chain fatty acids (SCFAs) and serve as an additional energy source [7,8]. Medium-chain triglycerides (MCTs) are good sources of fat because the body is able to absorb them into the bloodstream without the use of intestinal lymphatics, which may be damaged or absent in those with intestinal failure. For this particular patient, he would have benefitted from initiation of ORS and counseled to sip on it throughout the day while limiting liquid consumption during meals. He should have also been advised to limit plain Gatorade and Boost as they are both hyperosmolar liquid formulations and can worsen diarrhea. If the patient was unable to tolerate the taste of standard ORS formulations, or the hospital did not have any ORS on formulary, sugar, salt and water at specific amounts may be added to create a homemade ORS. In summary, this patient would have likely tolerated protein in solid form better than liquid protein supplementation.
Hospital Course Continued
The patient continued to have greater than 5 L of output from the ileostomy per day, so the following day the primary team increased the loperamide from 2 mg every 6 hours to 4 mg every 6 hours, added 2 tabs of diphenoxylate-atropine every 8 hours, and made the patient NPO. He continued to require IVFs and frequent electrolyte repletion because of the significant ongoing gastrointestinal losses.
What is the recommended first-line medical therapy for high-output enterostomy/diarrhea?
Anti-diarrheal medications are commonly used in high-output states because they work by reducing the rate of bowel translocation thereby allowing for longer time for nutrient and fluid absorption in the small and large intestine. Loperamide in particular also improves fecal incontinence because it effects the recto-anal inhibitory reflex and increases internal anal sphincter tone [9]. Four RCTs showed that loperamide lead to a significant reduction in enterostomy output compared to placebo with enterostomy output reductions ranging from 22% to 45%; varying dosages of loperamide were used, and ranged from 6 mg per day to 16 total mg per day [10–12]. King et al compared loperamide and codeine to placebo and found that both medications led to reductions in enterostomy output with a greater reduction and better side effect profile in those that received loperamide or combination therapy with loperamide and codeine [13,14]. The majority of studies used a maximum dose of 16 mg per day of loperamide, and this is the maxium daily dose approved by the US Food and Drug Administration (FDA). Interestingly however, loperamide circulates through the enterohepatic circulation which is severely disrupted in SBS, so titrating up to a maximum dose of 32 mg per day while closely monitoring for side effects is also practiced by experts in intestinal failure [15]. It is also important to note that anti-diarrheal medications are most effective when administered 20 to 30 minutes prior to meals and not scheduled every 4 to 6 hours if the patient is eating by mouth. If intestinal transit is so rapid such that undigested anti-diarrheal tablets or capsules are visualized in the stool or stoma, medications can be crushed or opened and mixed with liquids or solids to enhance digestion and absorption.
Hospital Course Continued
The patient continued to have greater than 3 L of ileostomy output per day despite being on scheduled loperamide, diphenoxylate-atropine, and a proton pump inhibitory (PPI), although improved from greater than 5 L per day. He was subsequently started on opium tincture 6 mg every 6 hours, psyllium 3 times per day, the dose of diphenoxylate-atropine was increased from 2 tablets every 8 hours to 2 tablets every 6 hours, and he was encouraged to drink water in between meals. As mentioned previously, the introduction of dietary fiber should be carefully monitored, as this patient population is commonly intolerant of high dietary fiber intake, and hypoosmolar liquids like water should actually be minimized. Within a 48-hour time period, the surgical team recommended increasing the loperamide from 4 mg every 6 hours (16 mg total daily dose) to 12 mg every 6 hours (48 mg total daily dose), increased opium tincture from 6 mg every 6 hours (24 mg total daily dose) to 10 mg every 6 hours (40 mg total daily dose), and increased oral pantoprazole from 40 mg once per day to twice per day.
What are important considerations with regard to dose changes?
Evidence is lacking to suggest an adequate time period to monitor for response to therapy in regards to improvement in diarrheal output. In this scenario, it may have been prudent to wait 24 to 48 hours after each medication change instead of making drastic dose changes in several medications simultaneously. PPIs irreversibly inhibit gastrointestinal acid secretion as do histamine-2 receptor antagonists (H2RAs) but to a lesser degree, and thus reduce high-output enterostomy [16]. Reduction in pH related to elevated gastrin levels after intestinal resection is associated with pancreatic enzyme denaturation and downstream bile salt dysfunction, which can further lead to malabsorption [17]. Gastrin hypersecretion is most prominent within the first 6 months after intestinal resection such that the use of high- dose PPIs for reduction in gastric acid secretion are most efficacious within that time period [18,19]. Jeppesen et al demonstrated that both omeprazole 40 mg oral twice per day and ranitidine 150 mg IV once per day were effective in reducing enterostomy output, although greater reductions were seen with omeprazole [20]. Three studies using cimetidine (both oral and IV formulations) with dosages varying from 200 mg to 800 mg per day showed significant reductions in enterostomy output as well [21–23].
Hospital Course Continued
Despite the previously mentioned interventions, the patient’s ileostomy output remained greater than 3 L per day. Loperamide was increased from 12 mg every 6 hours to 16 mg every 6 hours (64 mg total daily dose) hours and opium tincture was increased from 10 mg to 16 mg every 6 hours (64 mg total daily dose). Despite these changes, no significant reduction in output was noted, so the following day, 4 grams of cholestyramine light was added twice per day.
If the patient continues to have high-output enterostomy/diarrhea, what are additional treatment options?
Bile acid binding resins like cholestyramine, colestipol, and colesevelam are occasionally used if there is a high suspicion for bile acid diarrhea. Bile salt diarrhea typically occurs because of alterations in the enterohepatic circulation of bile salts, which leads to an increased level of bile salts in the colon and stimulation of electrolyte and water secretion and watery diarrhea [24]. Optimal candidates for bile acid binding therapy are those with an intact colon and less than 100 cm of resected ileum. Patients with little to no remaining or functional ileum have a depleted bile salt pool, therefore the addition of bile acid resin binders may actually lead to worsening diarrhea secondary to bile acid deficiency and fat malabsorption. Bile-acid resin binders can also decrease oxalate absorption and precipitate oxalate stone formation in the kidneys. Caution should also be taken to ensure that these medications are administered separately from the remainder of the patient’s medications to limit medication binding.
If the patient exhibits hemodynamic stability, alpha-2 receptor agonists are occasionally used as adjunctive therapy in reducing enterostomy output, although strong evidence to support its use is lacking. The mechanism of action involves stimulation of alpha-2 adrenergic receptors on enteric neurons, which theoretically causes a reduction in gastric and colonic motility and decreases fluid secretion. Buchman et al showed that the effects of a clonidine patch versus placebo did not in fact lead to a significant reduction in enterostomy output; however, a single case report suggested that the combination of 1200 mcg of clonidine per day and somatostatin resulted in decreased enterostomy output via alpha 2-receptor inhibition of adenylate cyclase [25,26].
Hospital Course Continued
The patient’s ileostomy output remained greater than 3 L per day, so loperamide was increased from 14 mg every 6 hours to 20 mg every 6 hours (80 mg total daily dose), cholestyramine was discontinued because of metabolic derangements, and the patient was initiated on 100 mcg of subcutaneous octreotide 3 times per day. Colorectal surgery was consulted for ileostomy takedown given persistently high-output, but surgery was deferred. After a 16-day hospitalization, the patient was eventually discharged home. At the time of discharge, he was having 2–3 L of ileostomy output per day and plans for future chemotherapy were discontinued because of this.
Does hormonal therapy have a role in the management of high-output enterostomy or entero-cutaneous fistulas?
Somatostatin analogues are growth-hormone inhibiting factors that have been used in the treatment of SBS and gastrointestinal fistulas. These medications reduce intestinal and pancreatic fluid secretion, slow intestinal motility, and inhibit the secretion of several hormones including gastrin, vasoactive intestinal peptide, cholecystokinin, and other key intestinal hormones. There is conflicting evidence for the role of these medications in reducing enterostomy output when first-line treatments have failed. Several previous studies using octreotide or somatostatin showed significant reductions in enterostomy output using variable dosages [27–30]. One study using the long-acting release depot octreotide preparation in 8 TPN-dependent patients with SBS showed a significant increase in small bowel transit time, however there was no significant improvement in the following parameters: body weight, stool weight, fecal fat excretion, stool electrolyte excretion, or gastric emptying [31]. Other studies evaluating enterostomy output from gastrointestinal and pancreatic fistulas comparing combined therapy with octreotide and TPN to placebo and TPN failed to show a significant difference in output and spontaneous fistula closure within 20 days of treatment initiation [32]. Because these studies use highly variable somatostatin analogue dosages and routes of administration, the most optimal dosing and route of administration (SQ versus IV) are unknown. In patients with difficult to control blood sugars, initiation of somatostatin analogues should be cautioned since these medications can lead to blood sugar alterations [33]. Additional unintended effects include impairment in intestinal adaptation and an increased risk in gallstone formation [8].
The most recent medical advances in SBS management include gut hormones. Glucagon-like peptide 2 (GLP-2) analogues improve structural and functional intestinal adaptation following intestinal resection by decreasing gastric emptying, decreasing gastric acid secretion, increasing intestinal blood flow, and enhancing nutrient and fluid absorption. Teduglutide, a GLP-2 analog, was successful in reducing fecal energy losses and increasing intestinal wet weight absorption, and reducing the need for PN support in SBS patients [1].
Whose problem is it anyway?
Not only is there variation in management strategies among subspecialties, but recommendations amongst societies within the same subspecialty differ, and thus make management perplexing.
Gastroenterology Guidelines
Several major gastroenterology societies have published guidelines on the management of diarrhea in patients with intestinal failure. The British Society of Gastroenterology (BSG) published guidelines on the management of SBS in 2006 and recommended the following first-line therapy for diarrhea-related complications: start loperamide at 2–8 mg thirty minutes prior to meals, taken up to 4 times per day, and the addition of codeine phosphate 30–60 mg thirty minutes before meals if output remains above goal on loperamide monotherapy. Cholestyramine may be added for those with 100 cm or less of resected terminal ileum to assist with bile-salt-induced diarrhea, though no specific dosage recommendations were reported. In regards to anti-secretory medications, the BSG recommends cimetidine (400 mg oral or IV 4 times per day), ranitidine (300 mg oral twice per day), or omeprazole (40 mg oral once per day or IV twice per day) to reduce jejunostomy output particularly in patients with greater than 2 L of output per day [15,34]. If diarrhea or enterostomy output continues to remain above goal, the guidelines suggest initiating octreotide and/or growth factors (although dosing and duration of therapy is not discussed in detail), and considering evaluation for intestinal transplant once the patient develops complications related to long-term TPN.
The American Gastroenterology Association (AGA) published guidelines and a position statement in 2003 for the management of high-gastric output and fluid losses. For postoperative patients, the AGA recommends the use of PPIs and H2RAs for the first 6 months following bowel resection when hyper-gastrinemia most commonly occurs. The guidelines do not specify which PPI or H2RA is preferred or recommended dosages. For long-term management of diarrhea or excess fluid losses, the guidelines suggest using loperamide or diphenoxylate (4-16 mg per day) first, followed by codeine sulfate 15–60 mg two to three times per day or opium tincture (dosages not specified). The use of octreotide (100 mcg SQ 3 times per day, 30 minutes prior to meals) is recommended only as a last resort if IVF requirements are greater than 3 L per day [8].
Surgical Guidelines
The Cleveland Clinic published institutional guidelines for the management of intestinal failure in 2010 with updated recommendations in 2016. Dietary recommendations include the liberal use of salt, sipping on 1–2 L of ORS between meals, and a slow reintroduction of soluble fiber from foods and/or supplements as tolerated. The guidelines also suggest considering placement of a nasogastric feeding tube or percutaneous gastrostomy tube (PEG) for continuous enteral feeding in addition to oral intake to enhance nutrient absorption [35]. If dietary manipulation is inadequate and medical therapy is required, the following medications are recommended in no particular order: loperamide 4 times per day (maximum dosage of 16 mg), diphenoxylate-atropine 4 times per day (maximum dosage of 20 mg per day), codeine 4 times per day (maximum dosage 240 mg per day), paregoric 5 mL (containing 2 mg of anhydrous morphine) 4 times per day, and opium tincture 0.5 mL (10 mg/mL) 4 times per day. H2RAs and PPIs are recommended for postoperative high-output states, although no dosage recommendations or routes of administration were discussed.
Nutrition Guidelines
Villafranca et al published a protocol for the management of high-output stomas in 2015 that was shown to be effective in reducing high-enterostomy output. The protocol recommended initial treatment with loperamide 2 mg orally up to 4 times per day. If enterostomy output did not improve, the protocol recommended increasing loperamide to 4 mg four times per day, adding omeprazole 20 mg orally or cholestyramine 4 g twice per day before lunch and dinner if fat malabsorption or steatorrhea is suspected, and lastly the addition of codeine 15–60 mg up to 4 times per day and octreotide 200 mcg per day only if symptoms had not improved after 2 weeks [37].
The American Society for Parenteral and Enteral Nutrition (ASPEN) does not have published guidelines for the management of SBS. In 2016 however, the European Society for Clinical Nutrition and Metabolism (ESPEN) published guidelines on the management of chronic intestinal failure in adults. In patients with an intact colon, ESPEN strongly recommends a diet rich in complex carbohydrates and low in fat and using H2RAs or PPIs to treat hyper-gastrinemia within the first 6 months after intestinal resection particularly in those with greater than 2 L per day of fecal output. The ESPEN guidelines do not include whether to start a PPI or H2RA first, which particular drug in each class to try, or dosage recommendations but state that IV soluble formulations should be considered in those that do not seem to respond to tablets. ESPEN does not recommend the addition of soluble fiber to enhance intestinal absorption or probiotics and glutamine to aid in intestinal rehabilitation. For diarrhea and excessive fecal fluid, the guidelines recommend 4 mg of oral loperamide 30–60 minutes prior to meals, 3 to 4 times per day, as first-line treatment in comparison to codeine phosphate or opium tincture given the risks of dependence and sedation with the latter agents. They report, however, that dosages up to 12–24 mg at one time of loperamide are used in patients with terminal ileum resection and persistently high-output enterostomy [38].
Case Conclusion
The services that were closely involved in this patient’s care were general internal medicine, general surgery, colorectal surgery, and ancillary services, including dietary and wound care. Interestingly, despite persistent high ileostomy output during the patient’s 16-day hospital admission, the gastroenterology service was never consulted. This case illustrates the importance of having a multidisciplinary approach to the care of these complicated patients to ensure that the appropriate medications are ordered based on the individual’s anatomy and that medications are ordered at appropriate dosages and timing intervals to maximize drug efficacy. It is also critical to ensure that nursing staff accurately documents all intake and output so that necessary changes can be made after adequate time is given to assess for a true response. There should be close communication between the primary medical or surgical service with the dietician to ensure the patient is counseled on appropriate dietary intake to help minimize diarrhea and fluid losses.
Conclusion
In conclusion, intestinal failure is a heterogenous group of disease states that often occurs after major intestinal resection and is commonly associated with malabsorption and high output states. High-output enterostomy and diarrhea are the most common etiologies leading to hospital re-admission following enterostomy creation or intestinal resection. These patients have high morbidity and mortality rates, and their conditions are costly to the health care system. Lack of high-quality evidence from RCTs and numerous societal guidelines without clear medication and dietary algorithms and low prevalence of these conditions makes management of these patients by general medical and surgical teams challenging. The proper management of intestinal failure and related complications requires a multidisciplinary approach with involvement from medical, surgical, and ancillary services. We propose a multidisciplinary approach with involvement from medical, surgical, and ancillary services in designed and implementing a protocol using electronic medical record based order sets to simplify and improve the management of these patients in the inpatient setting.
Corresponding author: Jake Hutto, 5323 Harry Hines Blvd, Dallas, TX 75390-9030, [email protected].
Financial disclosures: None.
From the University of Texas Southwestern, Department of Internal Medicine, Dallas, TX.
Abstract
- Objective: To define intestinal failure and associated diseases that often lead to diarrhea and high-output states, and to provide a literature review on the current evidence and practice guidelines for the management of these conditions in the context of a clinical case.
- Methods: Database search on dietary and medical interventions as well as major societal guidelines for the management of intestinal failure and associated conditions.
- Results: Although major societal guidelines exist, the guidelines vary greatly amongst various specialties and are not supported by strong evidence from large randomized controlled trials. The majority of the guidelines recommend consideration of several drug classes, but do not specify medications within the drug class, optimal dose, frequency, mode of administration, and how long to trial a regimen before considering it a failure and adding additional medical therapies.
- Conclusions: Intestinal failure and high-output states affect a very heterogenous population with high morbidity and mortality. This subset of patients should be managed using a multidisciplinary approach involving surgery, gastroenterology, dietetics, internal medicine and ancillary services that include but are not limited to ostomy nurses and home health care. Implementation of a standardized protocol in the electronic medical record including both medical and nutritional therapies may be useful to help optimize efficacy of medications, aid in nutrient absorption, decrease cost, reduce hospital length of stay, and decrease hospital readmissions.
Key words: short bowel syndrome; high-output ostomy; entero-cutaneous fistula; diarrhea; malnutrition.
Intestinal failure includes but is not limited to short bowel syndrome (SBS), high-output enterostomy, and high-output related to entero-cutaneous fistulas (ECF). These conditions are unfortunate complications after major abdominal surgery requiring extensive intestinal resection leading to structural SBS. Absorption of macronutrients and micronutrients is most dependent on the length and specific segment of remaining intact small intestine [1]. The normal small intestine length varies greatly but ranges from 300 to 800 cm, while in those with structural SBS the typical length is 200 cm or less [2,3]. Certain malabsorptive enteropathies and severe intestinal dysmotility conditions may manifest as functional SBS as well. Factors that influence whether an individual will develop functional SBS despite having sufficient small intestinal absorptive area include the degree of jejunal absorptive efficacy and the ability to overcompensate with enough oral caloric intake despite high fecal energy losses, also known as hyperphagia [4].
Pathophysiology
Maintenance of normal bodily functions and homeostasis is dependent on sufficient intestinal absorption of essential macronutrients, micronutrients, and fluids. The hallmark of intestinal failure is based on the presence of decreased small bowel absorptive surface area and subsequent increased losses of key solutes and fluids [1]. Intestinal failure is a broad term that is comprised of 3 distinct phenotypes. The 3 functional classifications of intestinal failure include the following:
- Type 1. Acute intestinal failure is generally self-limiting, occurs after abdominal surgery, and typically lasts less than 28 days.
- Type 2. Subacute intestinal failure frequently occurs in septic, stressed, or metabolically unstable patients and may last up to several months.
- Type 3. Chronic intestinal failure occurs due to a chronic condition that generally requires indefinite parenteral nutrition (PN) [1,3,4].
SBS and enterostomy formation are often associated with excessive diarrhea, such that it is the most common etiology for postoperative readmissions. The definition of “high-output” varies amongst studies, but output is generally considered to be abnormally high if it is greater than 500 mL per 24 hours in ECFs and greater than 1500 mL per 24 hours for enterostomies. There is significant variability from patient to patient, as output largely depends on length of remaining bowel [2,4].
Epidemiology
SBS, high-output enterostomy, and high-output from ECFs comprise a wide spectrum of underlying disease states, including but not limited to inflammatory bowel disease, post-surgical fistula formation, intestinal ischemia, intestinal atresia, radiation enteritis, abdominal trauma, and intussusception [5]. Due to the absence of a United States registry of patients with intestinal failure, the prevalence of these conditions is difficult to ascertain. Most estimations are made using registries for patients on total parenteral nutrition (TPN). The Crohns and Colitis Foundation of America estimates 10,000 to 20,000 people suffer from SBS in the United States. This heterogenous patient population has significant morbidity and mortality for dehydration related to these high-output states. While these conditions are considered rare, they are relatively costly to the health care system. These patients are commonly managed by numerous medical and surgical services, including internal medicine, gastroenterology, surgery, dietitians, wound care nurses, and home health agencies. Management strategies differ amongst these specialties and between professional societies, which makes treatment strategies highly variable and perplexing to providers taking care of this patient population. Furthermore, most of the published guidelines are based on expert opinion and lack high-quality clinical evidence from randomized controlled trials (RCTs). Effectively treating SBS and reducing excess enterostomy output leads to reduced rates of dehydration, electrolyte imbalances, initiation of PN, weight loss and ultimately a reduction in malnutrition. Developing hospital-wide management protocols in the electronic medical record for this heterogenous condition may lead to less complications, fewer hospitalizations, and an improved quality of life for these patients.
Case Study
Initial Presentation
A 72-year-old man with history of rectal adenocarcinoma stage T4bN2 status post low anterior resection (LAR) with diverting loop ileostomy and neoadjuvant chemoradiation presented to the hospital with a 3-day history of nausea, vomiting, fatigue, and productive cough.
Additional History
On further questioning, the patient also reported odynophagia and dysphagia related to thrush. Because of his decreased oral intake, he stopped taking his usual insulin regimen prior to admission. His cancer treatment course was notable for a LAR with diverting loop ileostomy which was performed 5 months prior. He had also completed 3 out of 8 cycles of capecitabine and oxaliplatin-based therapy 2 weeks prior to this presentation.
Physical Examination
Significant physical examination findings included dry mucous membranes, oropharyngeal candidiasis, tachycardia, clear lungs, hypoactive bowel sounds, nontender, non-distended abdomen, and a right lower abdominal ileostomy bag with semi-formed stool.
Laboratory test results were pertinent for diabetic ketoacidosis (DKA) with an anion gap of 33, lactic acidosis, acute kidney injury (creatinine 2.7 mg/dL from a baseline of 1.0) and blood glucose of 1059 mg/dL. Remainder of complete blood count and complete metabolic panel were unremarkable.
Hospital Course
The patient was treated for oropharyngeal candidiasis with fluconazole, started on an insulin drip and given intravenous fluids (IVFs) with subsequent resolution of DKA. Once the DKA resolved, his diet was advanced to a mechanical soft, moderate calorie, consistent carbohydrate diet (2000 calories allowed daily with all foods chopped, pureed or cooked, and all meals containing nearly equal amounts of carbohydrates). He was also given Boost supplementation 3 times per day, and daily weights were recorded while assessing for fluid losses. However, during his hospital course the patient developed increasing ileostomy output ranging from 2.7 to 6.5 L per day that only improved when he stopped eating by mouth (NPO).
What conditions should be evaluated prior to starting therapy for high-output enterostomy/diarrhea from either functional or structural SBS?
Prior to starting anti-diarrheal and anti-secretory therapy, infectious and metabolic etiologies for high-enterostomy output should be ruled out. Depending on the patient’s risk factors (eg, recent sick contacts, travel) and whether they are immunocompetent versus immunosuppressed, infectious studies should be obtained. In this patient, Clostridium difficile, stool culture, Giardia antigen, stool ova and parasites were all negative. Additional metabolic labs including thyroid-stimulating hormone, fecal elastase, and fecal fat were obtained and were all within normal limits. In this particular scenario, fecal fat was obtained while he was NPO. Testing for fat malabsorption and pancreatic insufficiency in a patient that is consuming less than 100 grams of fat per day can result in a false-negative outcome, however, and was not an appropriate test in this patient.
Hospital Course Continued
Once infectious etiologies were ruled out, the patient was started on anti-diarrheal medication consisting of loperamide 2 mg every 6 hours and oral pantoprazole 40 mg once per day. The primary internal medicine team speculated that the Boost supplementation may be contributing to the diarrhea because of its hyperosmolar concentration and wanted to discontinue it, but because the patient had protein-calorie malnutrition the dietician recommended continuing Boost supplementation. The primary internal medicine team also encouraged the patient to drink Gatorade with each meal with the approval from the dietician.
What are key dietary recommendations to help reduce high-output enterostomy/diarrhea?
Dietary recommendations are often quite variable depending on the intestinal anatomy (specifically, whether the colon is intact or absent), comorbidities such as renal disease, and severity of fluid and nutrient loses. This patient has the majority of his colon remaining; however, fluid and nutrients are being diverted away from his colon because he has a loop ileostomy. To reduce enterostomy output, it is generally recommended that liquids be consumed separately from solids, and that oral rehydration solutions (ORS) should replace most hyperosmolar and hypoosmolar liquids. Although these recommendations are commonly used, there is sparse data to suggest separating liquids from solids in a medically stable patient with SBS is indeed necessary [6]. In our patient, however, because he has not yet reached medical stability, it would be reasonable to separate the consumption of liquids from solids. The solid component of a SBS diet should consist mainly of protein and carbohydrates, with limited intake of simple sugars and sugar alcohols. If the colon remains intact, it is particularly important to limit fats to less than 30% of the daily caloric intake, to consume a low-oxalate diet, supplement with oral calcium to reduce the risk of calcium-oxalate nephrolithiasis, and increase dietary fiber intake as tolerated. Soluble fiber is fermented by colonic bacteria into short-chain fatty acids (SCFAs) and serve as an additional energy source [7,8]. Medium-chain triglycerides (MCTs) are good sources of fat because the body is able to absorb them into the bloodstream without the use of intestinal lymphatics, which may be damaged or absent in those with intestinal failure. For this particular patient, he would have benefitted from initiation of ORS and counseled to sip on it throughout the day while limiting liquid consumption during meals. He should have also been advised to limit plain Gatorade and Boost as they are both hyperosmolar liquid formulations and can worsen diarrhea. If the patient was unable to tolerate the taste of standard ORS formulations, or the hospital did not have any ORS on formulary, sugar, salt and water at specific amounts may be added to create a homemade ORS. In summary, this patient would have likely tolerated protein in solid form better than liquid protein supplementation.
Hospital Course Continued
The patient continued to have greater than 5 L of output from the ileostomy per day, so the following day the primary team increased the loperamide from 2 mg every 6 hours to 4 mg every 6 hours, added 2 tabs of diphenoxylate-atropine every 8 hours, and made the patient NPO. He continued to require IVFs and frequent electrolyte repletion because of the significant ongoing gastrointestinal losses.
What is the recommended first-line medical therapy for high-output enterostomy/diarrhea?
Anti-diarrheal medications are commonly used in high-output states because they work by reducing the rate of bowel translocation thereby allowing for longer time for nutrient and fluid absorption in the small and large intestine. Loperamide in particular also improves fecal incontinence because it effects the recto-anal inhibitory reflex and increases internal anal sphincter tone [9]. Four RCTs showed that loperamide lead to a significant reduction in enterostomy output compared to placebo with enterostomy output reductions ranging from 22% to 45%; varying dosages of loperamide were used, and ranged from 6 mg per day to 16 total mg per day [10–12]. King et al compared loperamide and codeine to placebo and found that both medications led to reductions in enterostomy output with a greater reduction and better side effect profile in those that received loperamide or combination therapy with loperamide and codeine [13,14]. The majority of studies used a maximum dose of 16 mg per day of loperamide, and this is the maxium daily dose approved by the US Food and Drug Administration (FDA). Interestingly however, loperamide circulates through the enterohepatic circulation which is severely disrupted in SBS, so titrating up to a maximum dose of 32 mg per day while closely monitoring for side effects is also practiced by experts in intestinal failure [15]. It is also important to note that anti-diarrheal medications are most effective when administered 20 to 30 minutes prior to meals and not scheduled every 4 to 6 hours if the patient is eating by mouth. If intestinal transit is so rapid such that undigested anti-diarrheal tablets or capsules are visualized in the stool or stoma, medications can be crushed or opened and mixed with liquids or solids to enhance digestion and absorption.
Hospital Course Continued
The patient continued to have greater than 3 L of ileostomy output per day despite being on scheduled loperamide, diphenoxylate-atropine, and a proton pump inhibitory (PPI), although improved from greater than 5 L per day. He was subsequently started on opium tincture 6 mg every 6 hours, psyllium 3 times per day, the dose of diphenoxylate-atropine was increased from 2 tablets every 8 hours to 2 tablets every 6 hours, and he was encouraged to drink water in between meals. As mentioned previously, the introduction of dietary fiber should be carefully monitored, as this patient population is commonly intolerant of high dietary fiber intake, and hypoosmolar liquids like water should actually be minimized. Within a 48-hour time period, the surgical team recommended increasing the loperamide from 4 mg every 6 hours (16 mg total daily dose) to 12 mg every 6 hours (48 mg total daily dose), increased opium tincture from 6 mg every 6 hours (24 mg total daily dose) to 10 mg every 6 hours (40 mg total daily dose), and increased oral pantoprazole from 40 mg once per day to twice per day.
What are important considerations with regard to dose changes?
Evidence is lacking to suggest an adequate time period to monitor for response to therapy in regards to improvement in diarrheal output. In this scenario, it may have been prudent to wait 24 to 48 hours after each medication change instead of making drastic dose changes in several medications simultaneously. PPIs irreversibly inhibit gastrointestinal acid secretion as do histamine-2 receptor antagonists (H2RAs) but to a lesser degree, and thus reduce high-output enterostomy [16]. Reduction in pH related to elevated gastrin levels after intestinal resection is associated with pancreatic enzyme denaturation and downstream bile salt dysfunction, which can further lead to malabsorption [17]. Gastrin hypersecretion is most prominent within the first 6 months after intestinal resection such that the use of high- dose PPIs for reduction in gastric acid secretion are most efficacious within that time period [18,19]. Jeppesen et al demonstrated that both omeprazole 40 mg oral twice per day and ranitidine 150 mg IV once per day were effective in reducing enterostomy output, although greater reductions were seen with omeprazole [20]. Three studies using cimetidine (both oral and IV formulations) with dosages varying from 200 mg to 800 mg per day showed significant reductions in enterostomy output as well [21–23].
Hospital Course Continued
Despite the previously mentioned interventions, the patient’s ileostomy output remained greater than 3 L per day. Loperamide was increased from 12 mg every 6 hours to 16 mg every 6 hours (64 mg total daily dose) hours and opium tincture was increased from 10 mg to 16 mg every 6 hours (64 mg total daily dose). Despite these changes, no significant reduction in output was noted, so the following day, 4 grams of cholestyramine light was added twice per day.
If the patient continues to have high-output enterostomy/diarrhea, what are additional treatment options?
Bile acid binding resins like cholestyramine, colestipol, and colesevelam are occasionally used if there is a high suspicion for bile acid diarrhea. Bile salt diarrhea typically occurs because of alterations in the enterohepatic circulation of bile salts, which leads to an increased level of bile salts in the colon and stimulation of electrolyte and water secretion and watery diarrhea [24]. Optimal candidates for bile acid binding therapy are those with an intact colon and less than 100 cm of resected ileum. Patients with little to no remaining or functional ileum have a depleted bile salt pool, therefore the addition of bile acid resin binders may actually lead to worsening diarrhea secondary to bile acid deficiency and fat malabsorption. Bile-acid resin binders can also decrease oxalate absorption and precipitate oxalate stone formation in the kidneys. Caution should also be taken to ensure that these medications are administered separately from the remainder of the patient’s medications to limit medication binding.
If the patient exhibits hemodynamic stability, alpha-2 receptor agonists are occasionally used as adjunctive therapy in reducing enterostomy output, although strong evidence to support its use is lacking. The mechanism of action involves stimulation of alpha-2 adrenergic receptors on enteric neurons, which theoretically causes a reduction in gastric and colonic motility and decreases fluid secretion. Buchman et al showed that the effects of a clonidine patch versus placebo did not in fact lead to a significant reduction in enterostomy output; however, a single case report suggested that the combination of 1200 mcg of clonidine per day and somatostatin resulted in decreased enterostomy output via alpha 2-receptor inhibition of adenylate cyclase [25,26].
Hospital Course Continued
The patient’s ileostomy output remained greater than 3 L per day, so loperamide was increased from 14 mg every 6 hours to 20 mg every 6 hours (80 mg total daily dose), cholestyramine was discontinued because of metabolic derangements, and the patient was initiated on 100 mcg of subcutaneous octreotide 3 times per day. Colorectal surgery was consulted for ileostomy takedown given persistently high-output, but surgery was deferred. After a 16-day hospitalization, the patient was eventually discharged home. At the time of discharge, he was having 2–3 L of ileostomy output per day and plans for future chemotherapy were discontinued because of this.
Does hormonal therapy have a role in the management of high-output enterostomy or entero-cutaneous fistulas?
Somatostatin analogues are growth-hormone inhibiting factors that have been used in the treatment of SBS and gastrointestinal fistulas. These medications reduce intestinal and pancreatic fluid secretion, slow intestinal motility, and inhibit the secretion of several hormones including gastrin, vasoactive intestinal peptide, cholecystokinin, and other key intestinal hormones. There is conflicting evidence for the role of these medications in reducing enterostomy output when first-line treatments have failed. Several previous studies using octreotide or somatostatin showed significant reductions in enterostomy output using variable dosages [27–30]. One study using the long-acting release depot octreotide preparation in 8 TPN-dependent patients with SBS showed a significant increase in small bowel transit time, however there was no significant improvement in the following parameters: body weight, stool weight, fecal fat excretion, stool electrolyte excretion, or gastric emptying [31]. Other studies evaluating enterostomy output from gastrointestinal and pancreatic fistulas comparing combined therapy with octreotide and TPN to placebo and TPN failed to show a significant difference in output and spontaneous fistula closure within 20 days of treatment initiation [32]. Because these studies use highly variable somatostatin analogue dosages and routes of administration, the most optimal dosing and route of administration (SQ versus IV) are unknown. In patients with difficult to control blood sugars, initiation of somatostatin analogues should be cautioned since these medications can lead to blood sugar alterations [33]. Additional unintended effects include impairment in intestinal adaptation and an increased risk in gallstone formation [8].
The most recent medical advances in SBS management include gut hormones. Glucagon-like peptide 2 (GLP-2) analogues improve structural and functional intestinal adaptation following intestinal resection by decreasing gastric emptying, decreasing gastric acid secretion, increasing intestinal blood flow, and enhancing nutrient and fluid absorption. Teduglutide, a GLP-2 analog, was successful in reducing fecal energy losses and increasing intestinal wet weight absorption, and reducing the need for PN support in SBS patients [1].
Whose problem is it anyway?
Not only is there variation in management strategies among subspecialties, but recommendations amongst societies within the same subspecialty differ, and thus make management perplexing.
Gastroenterology Guidelines
Several major gastroenterology societies have published guidelines on the management of diarrhea in patients with intestinal failure. The British Society of Gastroenterology (BSG) published guidelines on the management of SBS in 2006 and recommended the following first-line therapy for diarrhea-related complications: start loperamide at 2–8 mg thirty minutes prior to meals, taken up to 4 times per day, and the addition of codeine phosphate 30–60 mg thirty minutes before meals if output remains above goal on loperamide monotherapy. Cholestyramine may be added for those with 100 cm or less of resected terminal ileum to assist with bile-salt-induced diarrhea, though no specific dosage recommendations were reported. In regards to anti-secretory medications, the BSG recommends cimetidine (400 mg oral or IV 4 times per day), ranitidine (300 mg oral twice per day), or omeprazole (40 mg oral once per day or IV twice per day) to reduce jejunostomy output particularly in patients with greater than 2 L of output per day [15,34]. If diarrhea or enterostomy output continues to remain above goal, the guidelines suggest initiating octreotide and/or growth factors (although dosing and duration of therapy is not discussed in detail), and considering evaluation for intestinal transplant once the patient develops complications related to long-term TPN.
The American Gastroenterology Association (AGA) published guidelines and a position statement in 2003 for the management of high-gastric output and fluid losses. For postoperative patients, the AGA recommends the use of PPIs and H2RAs for the first 6 months following bowel resection when hyper-gastrinemia most commonly occurs. The guidelines do not specify which PPI or H2RA is preferred or recommended dosages. For long-term management of diarrhea or excess fluid losses, the guidelines suggest using loperamide or diphenoxylate (4-16 mg per day) first, followed by codeine sulfate 15–60 mg two to three times per day or opium tincture (dosages not specified). The use of octreotide (100 mcg SQ 3 times per day, 30 minutes prior to meals) is recommended only as a last resort if IVF requirements are greater than 3 L per day [8].
Surgical Guidelines
The Cleveland Clinic published institutional guidelines for the management of intestinal failure in 2010 with updated recommendations in 2016. Dietary recommendations include the liberal use of salt, sipping on 1–2 L of ORS between meals, and a slow reintroduction of soluble fiber from foods and/or supplements as tolerated. The guidelines also suggest considering placement of a nasogastric feeding tube or percutaneous gastrostomy tube (PEG) for continuous enteral feeding in addition to oral intake to enhance nutrient absorption [35]. If dietary manipulation is inadequate and medical therapy is required, the following medications are recommended in no particular order: loperamide 4 times per day (maximum dosage of 16 mg), diphenoxylate-atropine 4 times per day (maximum dosage of 20 mg per day), codeine 4 times per day (maximum dosage 240 mg per day), paregoric 5 mL (containing 2 mg of anhydrous morphine) 4 times per day, and opium tincture 0.5 mL (10 mg/mL) 4 times per day. H2RAs and PPIs are recommended for postoperative high-output states, although no dosage recommendations or routes of administration were discussed.
Nutrition Guidelines
Villafranca et al published a protocol for the management of high-output stomas in 2015 that was shown to be effective in reducing high-enterostomy output. The protocol recommended initial treatment with loperamide 2 mg orally up to 4 times per day. If enterostomy output did not improve, the protocol recommended increasing loperamide to 4 mg four times per day, adding omeprazole 20 mg orally or cholestyramine 4 g twice per day before lunch and dinner if fat malabsorption or steatorrhea is suspected, and lastly the addition of codeine 15–60 mg up to 4 times per day and octreotide 200 mcg per day only if symptoms had not improved after 2 weeks [37].
The American Society for Parenteral and Enteral Nutrition (ASPEN) does not have published guidelines for the management of SBS. In 2016 however, the European Society for Clinical Nutrition and Metabolism (ESPEN) published guidelines on the management of chronic intestinal failure in adults. In patients with an intact colon, ESPEN strongly recommends a diet rich in complex carbohydrates and low in fat and using H2RAs or PPIs to treat hyper-gastrinemia within the first 6 months after intestinal resection particularly in those with greater than 2 L per day of fecal output. The ESPEN guidelines do not include whether to start a PPI or H2RA first, which particular drug in each class to try, or dosage recommendations but state that IV soluble formulations should be considered in those that do not seem to respond to tablets. ESPEN does not recommend the addition of soluble fiber to enhance intestinal absorption or probiotics and glutamine to aid in intestinal rehabilitation. For diarrhea and excessive fecal fluid, the guidelines recommend 4 mg of oral loperamide 30–60 minutes prior to meals, 3 to 4 times per day, as first-line treatment in comparison to codeine phosphate or opium tincture given the risks of dependence and sedation with the latter agents. They report, however, that dosages up to 12–24 mg at one time of loperamide are used in patients with terminal ileum resection and persistently high-output enterostomy [38].
Case Conclusion
The services that were closely involved in this patient’s care were general internal medicine, general surgery, colorectal surgery, and ancillary services, including dietary and wound care. Interestingly, despite persistent high ileostomy output during the patient’s 16-day hospital admission, the gastroenterology service was never consulted. This case illustrates the importance of having a multidisciplinary approach to the care of these complicated patients to ensure that the appropriate medications are ordered based on the individual’s anatomy and that medications are ordered at appropriate dosages and timing intervals to maximize drug efficacy. It is also critical to ensure that nursing staff accurately documents all intake and output so that necessary changes can be made after adequate time is given to assess for a true response. There should be close communication between the primary medical or surgical service with the dietician to ensure the patient is counseled on appropriate dietary intake to help minimize diarrhea and fluid losses.
Conclusion
In conclusion, intestinal failure is a heterogenous group of disease states that often occurs after major intestinal resection and is commonly associated with malabsorption and high output states. High-output enterostomy and diarrhea are the most common etiologies leading to hospital re-admission following enterostomy creation or intestinal resection. These patients have high morbidity and mortality rates, and their conditions are costly to the health care system. Lack of high-quality evidence from RCTs and numerous societal guidelines without clear medication and dietary algorithms and low prevalence of these conditions makes management of these patients by general medical and surgical teams challenging. The proper management of intestinal failure and related complications requires a multidisciplinary approach with involvement from medical, surgical, and ancillary services. We propose a multidisciplinary approach with involvement from medical, surgical, and ancillary services in designed and implementing a protocol using electronic medical record based order sets to simplify and improve the management of these patients in the inpatient setting.
Corresponding author: Jake Hutto, 5323 Harry Hines Blvd, Dallas, TX 75390-9030, [email protected].
Financial disclosures: None.
1. Jeppesen PB. Gut hormones in the treatment of short-bowel syndrome and intestinal failure. Current opinion in endocrinology, diabetes, and obesity. Curr Opin Endocrinol Diabetes Obes 2015;22:14–20.
2. Berry SM, Fischer JE. Classification and pathophysiology of enterocutaneous fistulas. Surg Clin North Am 1996;76:1009–18.
3. Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1111–34.
4. de Vries FEE, Reeskamp LF, van Ruler O et al. Systematic review: pharmacotherapy for high-output enterostomies or enteral fistulas. Aliment Pharmacol Ther 2017;46:266–73.
5. Holzheimer RG, Mannick JA. Surgical Treatment: Evidence-Based and Problem-Oriented. Munich: Zuckschwerdt; 2001.
6. Woolf GM, Miller C, Kurian R, Jeejeebhoy KN. Nutritional absorption in short bowel syndrome. Evaluation of fluid, calorie, and divalent cation requirements. Dig Dis Sci 1987;32:8–15.
7. Parrish CR, DiBaise JK. Managing the adult patient with short bowel syndrome. Gastroenterol Hepatol (N Y) 2017;13:600–8.
8. American Gastroenterological Association. American Gastroenterological Association medical position statement: short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1105–10.
9. Musial F, Enck P, Kalveram KT, Erckenbrecht JF. The effect of loperamide on anorectal function in normal healthy men. J Clin Gastroenterol. 1992;15:321–4.
10. Tijtgat GN, Meuwissen SG, Huibregtse K. Loperamide in the symptomatic control of chronic diarrhoea. Double-blind placebo-controlled study. Ann Clin Res 1975;7:325–30.
11. Tytgat GN, Huibregtse K, Dagevos J, van den Ende A. Effect of loperamide on fecal output and composition in well-established ileostomy and ileorectal anastomosis. Am J Dig Dis 1977;22:669–76.
12. Stevens PJ, Dunbar F, Briscoe P. Potential of loperamide oxide in the reduction of ileostomy and colostomy output. Clin Drug Investig 1995;10:158–64.
13. King RF, Norton T, Hill GL. A double-blind crossover study of the effect of loperamide hydrochloride and codeine phosphate on ileostomy output. Aust N Z J Surg 1982;52:121–4.
14. Nightingale JM, Lennard-Jones JE, Walker ER. A patient with jejunostomy liberated from home intravenous therapy after 14 years; contribution of balance studies. Clin Nutr 1992;11:101–5.
15. Nightingale J, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006;55:iv1–12.
16. Nightingale JM, Lennard-Jones JE, Walker ER, Farthing MJ. Jejunal efflux in short bowel syndrome. Lancet 1990;336:765–8.
17. Go VL, Poley JR, Hofmann AF, Summerskill WH. Disturbances in fat digestion induced by acidic jejunal pH due to gastric hypersecretion in man. Gastroenterology 1970;58:638–46.
18. Windsor CW, Fejfar J, Woodward DA. Gastric secretion after massive small bowel resection. Gut 1969;10:779–86.
19. Williams NS, Evans P, King RF. Gastric acid secretion and gastrin production in the short bowel syndrome. Gut 1985;26:914–9.
20. Jeppesen PB, Staun M, Tjellesen L, Mortensen PB. Effect of intravenous ranitidine and omeprazole on intestinal absorption of water, sodium, and macronutrients in patients with intestinal resection. Gut 1998;43:763–9.
21. Aly A, Bárány F, Kollberg B, et al. Effect of an H2-receptor blocking agent on diarrhoeas after extensive small bowel resection in Crohn’s disease. Acta Med Scand 1980;207:119–22.
22. Kato J, Sakamoto J, Teramukai S, et al. A prospective within-patient comparison clinical trial on the effect of parenteral cimetidine for improvement of fluid secretion and electrolyte balance in patients with short bowel syndrome. Hepatogastroenterology. 2004;51:1742–6.
23. Jacobsen O, Ladefoged K, Stage JG, Jarnum S. Effects of cimetidine on jejunostomy effluents in patients with severe short-bowel syndrome. Scand J Gastroenterol 1986;21:824–8.
24. Hofmann AF. The syndrome of ileal disease and the broken enterohepatic circulation: cholerheic enteropathy. Gastroenterology 1967;52:752–7.
25. Buchman AL, Fryer J, Wallin A et al. Clonidine reduces diarrhea and sodium loss in patients with proximal jejunostomy: a controlled study. JPEN J Parenter Enteral Nutr. 2006;30:487–91.
26. Scholz J, Bause H, Reymann A, Dürig M. Treatment with clonidine in a case of the short bowel syndrome with therapy-refractory diarrhea [ in German]. Anasthesiol Intensivmed Notfallmed Schmerzther 1991;26:265–9.
27. Torres AJ, Landa JI, Moreno-Azcoita M, et al. Somatostatin in the management of gastrointestinal fistulas. A multicenter trial. Arch Surg 1992;127:97–9; discussion 100.
28. Nubiola-Calonge P, Badia JM, Sancho J, et al. Blind evaluation of the effect of octreotide (SMS 201-995), a somatostatin analogue, on small-bowel fistula output. Lancet 1987;2:672–4.
29. Kusuhara K, Kusunoki M, Okamoto T, et al. Reduction of the effluent volume in high-output ileostomy patients by a somatostatin analogue, SMS 201-995. Int J Colorectal Dis 1992;7:202–5.
30. O’Keefe SJ, Peterson ME, Fleming CR. Octreotide as an adjunct to home parenteral nutrition in the management of permanent end-jejunostomy syndrome. JPEN J Parenter Enteral Nutr 1994;18:26–34.
31. Nehra V, Camilleri M, Burton D, et al. An open trial of octreotide long-acting release in the management of short bowel syndrome. Am J Gastroenterol 2001;96:1494–8.
32. Sancho JJ, di Costanzo J, Nubiola P, et al. Randomized double-blind placebo-controlled trial of early octreotide in patients with postoperative enterocutaneous fistula. Br J Surg 1995;82:638–41.
33. Alberti KG, Christensen NJ, Christensen SE, et al. Inhibition of insulin secretion by somatostatin. Lancet 1973;2:1299–301.
34. Hofmann AF, Poley JR. Role of bile acid malabsorption in pathogenesis of diarrhea and steatorrhea in patients with ileal resection. I. Response to cholestyramine or replacement of dietary long chain triglyceride by medium chain triglyceride. Gastroenterology 1972;62:918–34.
35. Joly F, Dray X, Corcos O, et al. Tube feeding improves intestinal absorption in short bowel syndrome patients. Gastroenterology 2009;136:824–31.
36. Bharadwaj S, Tandon P, Rivas JM, et al. Update on the management of intestinal failure. Cleveland Cleve Clin J Med 2016;83:841–8.
37. Arenas Villafranca JJ, López-Rodríguez C, Abilés J, et al. Protocol for the detection and nutritional management of high-output stomas. Nutr J 2015;14:45.
38. Pironi L, Arends J, Bozzetti F, et al. ESPEN guidelines on chronic intestinal failure in adults. Clin Nutr 2016;35:247–307.
1. Jeppesen PB. Gut hormones in the treatment of short-bowel syndrome and intestinal failure. Current opinion in endocrinology, diabetes, and obesity. Curr Opin Endocrinol Diabetes Obes 2015;22:14–20.
2. Berry SM, Fischer JE. Classification and pathophysiology of enterocutaneous fistulas. Surg Clin North Am 1996;76:1009–18.
3. Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1111–34.
4. de Vries FEE, Reeskamp LF, van Ruler O et al. Systematic review: pharmacotherapy for high-output enterostomies or enteral fistulas. Aliment Pharmacol Ther 2017;46:266–73.
5. Holzheimer RG, Mannick JA. Surgical Treatment: Evidence-Based and Problem-Oriented. Munich: Zuckschwerdt; 2001.
6. Woolf GM, Miller C, Kurian R, Jeejeebhoy KN. Nutritional absorption in short bowel syndrome. Evaluation of fluid, calorie, and divalent cation requirements. Dig Dis Sci 1987;32:8–15.
7. Parrish CR, DiBaise JK. Managing the adult patient with short bowel syndrome. Gastroenterol Hepatol (N Y) 2017;13:600–8.
8. American Gastroenterological Association. American Gastroenterological Association medical position statement: short bowel syndrome and intestinal transplantation. Gastroenterology 2003;124:1105–10.
9. Musial F, Enck P, Kalveram KT, Erckenbrecht JF. The effect of loperamide on anorectal function in normal healthy men. J Clin Gastroenterol. 1992;15:321–4.
10. Tijtgat GN, Meuwissen SG, Huibregtse K. Loperamide in the symptomatic control of chronic diarrhoea. Double-blind placebo-controlled study. Ann Clin Res 1975;7:325–30.
11. Tytgat GN, Huibregtse K, Dagevos J, van den Ende A. Effect of loperamide on fecal output and composition in well-established ileostomy and ileorectal anastomosis. Am J Dig Dis 1977;22:669–76.
12. Stevens PJ, Dunbar F, Briscoe P. Potential of loperamide oxide in the reduction of ileostomy and colostomy output. Clin Drug Investig 1995;10:158–64.
13. King RF, Norton T, Hill GL. A double-blind crossover study of the effect of loperamide hydrochloride and codeine phosphate on ileostomy output. Aust N Z J Surg 1982;52:121–4.
14. Nightingale JM, Lennard-Jones JE, Walker ER. A patient with jejunostomy liberated from home intravenous therapy after 14 years; contribution of balance studies. Clin Nutr 1992;11:101–5.
15. Nightingale J, Woodward JM. Guidelines for management of patients with a short bowel. Gut 2006;55:iv1–12.
16. Nightingale JM, Lennard-Jones JE, Walker ER, Farthing MJ. Jejunal efflux in short bowel syndrome. Lancet 1990;336:765–8.
17. Go VL, Poley JR, Hofmann AF, Summerskill WH. Disturbances in fat digestion induced by acidic jejunal pH due to gastric hypersecretion in man. Gastroenterology 1970;58:638–46.
18. Windsor CW, Fejfar J, Woodward DA. Gastric secretion after massive small bowel resection. Gut 1969;10:779–86.
19. Williams NS, Evans P, King RF. Gastric acid secretion and gastrin production in the short bowel syndrome. Gut 1985;26:914–9.
20. Jeppesen PB, Staun M, Tjellesen L, Mortensen PB. Effect of intravenous ranitidine and omeprazole on intestinal absorption of water, sodium, and macronutrients in patients with intestinal resection. Gut 1998;43:763–9.
21. Aly A, Bárány F, Kollberg B, et al. Effect of an H2-receptor blocking agent on diarrhoeas after extensive small bowel resection in Crohn’s disease. Acta Med Scand 1980;207:119–22.
22. Kato J, Sakamoto J, Teramukai S, et al. A prospective within-patient comparison clinical trial on the effect of parenteral cimetidine for improvement of fluid secretion and electrolyte balance in patients with short bowel syndrome. Hepatogastroenterology. 2004;51:1742–6.
23. Jacobsen O, Ladefoged K, Stage JG, Jarnum S. Effects of cimetidine on jejunostomy effluents in patients with severe short-bowel syndrome. Scand J Gastroenterol 1986;21:824–8.
24. Hofmann AF. The syndrome of ileal disease and the broken enterohepatic circulation: cholerheic enteropathy. Gastroenterology 1967;52:752–7.
25. Buchman AL, Fryer J, Wallin A et al. Clonidine reduces diarrhea and sodium loss in patients with proximal jejunostomy: a controlled study. JPEN J Parenter Enteral Nutr. 2006;30:487–91.
26. Scholz J, Bause H, Reymann A, Dürig M. Treatment with clonidine in a case of the short bowel syndrome with therapy-refractory diarrhea [ in German]. Anasthesiol Intensivmed Notfallmed Schmerzther 1991;26:265–9.
27. Torres AJ, Landa JI, Moreno-Azcoita M, et al. Somatostatin in the management of gastrointestinal fistulas. A multicenter trial. Arch Surg 1992;127:97–9; discussion 100.
28. Nubiola-Calonge P, Badia JM, Sancho J, et al. Blind evaluation of the effect of octreotide (SMS 201-995), a somatostatin analogue, on small-bowel fistula output. Lancet 1987;2:672–4.
29. Kusuhara K, Kusunoki M, Okamoto T, et al. Reduction of the effluent volume in high-output ileostomy patients by a somatostatin analogue, SMS 201-995. Int J Colorectal Dis 1992;7:202–5.
30. O’Keefe SJ, Peterson ME, Fleming CR. Octreotide as an adjunct to home parenteral nutrition in the management of permanent end-jejunostomy syndrome. JPEN J Parenter Enteral Nutr 1994;18:26–34.
31. Nehra V, Camilleri M, Burton D, et al. An open trial of octreotide long-acting release in the management of short bowel syndrome. Am J Gastroenterol 2001;96:1494–8.
32. Sancho JJ, di Costanzo J, Nubiola P, et al. Randomized double-blind placebo-controlled trial of early octreotide in patients with postoperative enterocutaneous fistula. Br J Surg 1995;82:638–41.
33. Alberti KG, Christensen NJ, Christensen SE, et al. Inhibition of insulin secretion by somatostatin. Lancet 1973;2:1299–301.
34. Hofmann AF, Poley JR. Role of bile acid malabsorption in pathogenesis of diarrhea and steatorrhea in patients with ileal resection. I. Response to cholestyramine or replacement of dietary long chain triglyceride by medium chain triglyceride. Gastroenterology 1972;62:918–34.
35. Joly F, Dray X, Corcos O, et al. Tube feeding improves intestinal absorption in short bowel syndrome patients. Gastroenterology 2009;136:824–31.
36. Bharadwaj S, Tandon P, Rivas JM, et al. Update on the management of intestinal failure. Cleveland Cleve Clin J Med 2016;83:841–8.
37. Arenas Villafranca JJ, López-Rodríguez C, Abilés J, et al. Protocol for the detection and nutritional management of high-output stomas. Nutr J 2015;14:45.
38. Pironi L, Arends J, Bozzetti F, et al. ESPEN guidelines on chronic intestinal failure in adults. Clin Nutr 2016;35:247–307.
Pancreatic Adenocarcinoma: Management of Advanced Unresectable and Metastatic Disease
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
HER2-Positive Breast Cancer: Current Management
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and 
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).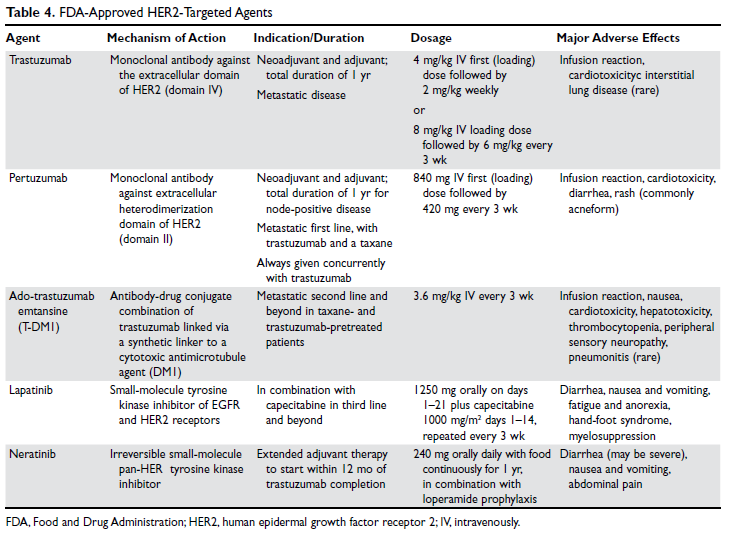
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2). 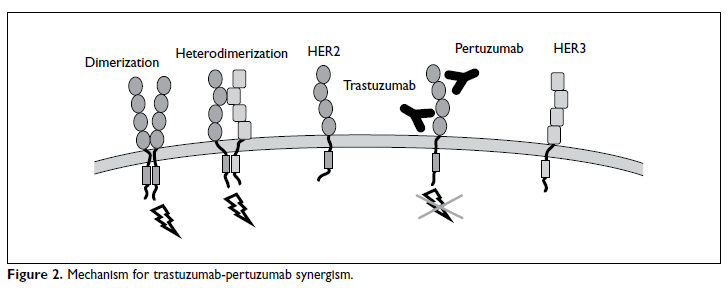
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3). 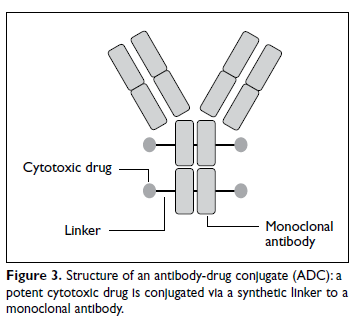
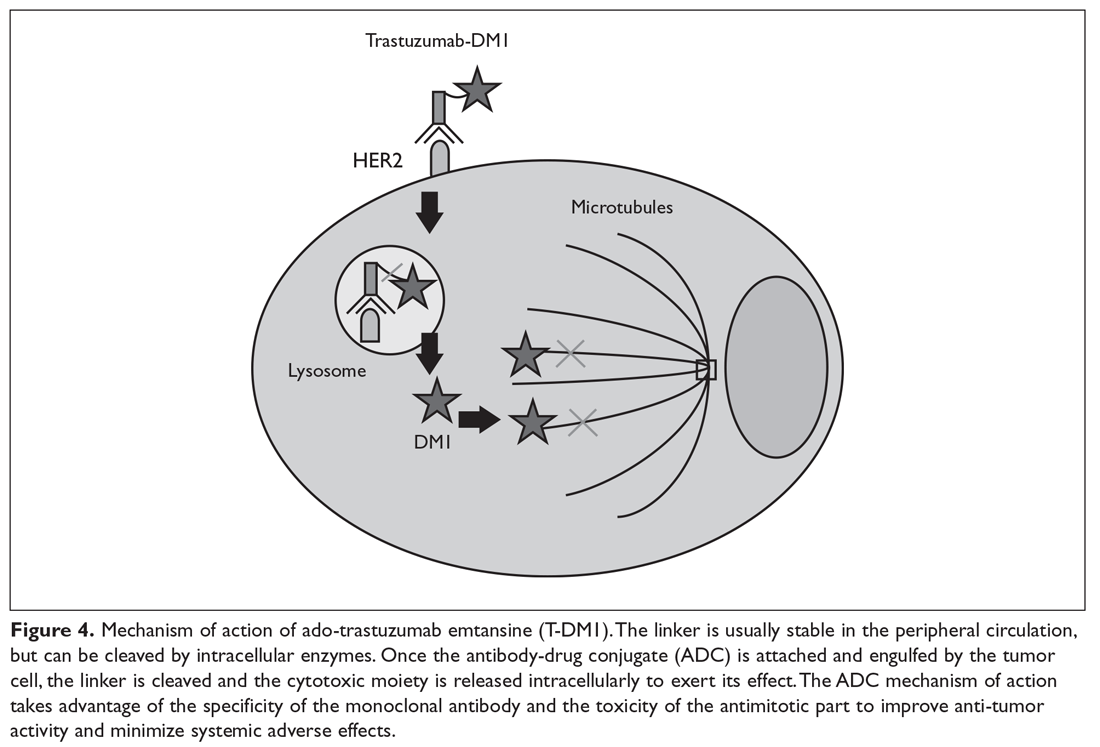
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2).
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3).
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
Introduction
Breast cancer is the second leading cause of cancer deaths among women in the United States, according to the SEER database. It is estimated that 1 in 8 women will be diagnosed with breast cancer at some point during their lifetime (12.4% lifetime risk).1,2 Because breast tumors are clinically and histopathologically heterogeneous, different diagnostic and therapeutic approaches are required for each subtype. Among the subtypes, tumors that are positive for human epidermal growth factor receptor 2 (HER2) account for approximately 15% to 20% of all newly diagnosed localized and metastatic invasive breast tumors.3,4 Historically, this subset of tumors has been considered the most aggressive due to a higher propensity to relapse and metastasize, translating into poorer prognosis compared with other subtypes.5–7 However, with the advent of HER2-targeted therapy in the late 1990s, prognosis has significantly improved for both early- and late-stage HER2-positive tumors.8
Pathogenesis
The HER2 proto-oncogene belongs to a family of human epidermal growth factor receptors that includes 4 transmembrane tyrosine kinase receptors: HER1 (also commonly known as epidermal growth factor receptor, EGFR), HER2, HER3, and HER4. Another commonly used nomenclature for this family of receptors is ERBB1 to ERBB4. Each of the receptors has a similar structure consisting of a growth factor–binding extracellular domain, a single transmembrane segment, an intracellular protein-tyrosine kinase catalytic domain, and a tyrosine-containing cytoplasmic tail. Activation of the extracellular domain leads to conformational changes that initiate a cascade of reactions resulting in protein kinase activation. ERBB tyrosine receptor kinases subsequently activate several intracellular pathways that are critical for cellular function and survival, including the PI3K-AKT, RAS-MAPK, and mTOR pathways. Hyperactivation or overexpression of these receptors leads to uncontrolled cell growth and proliferation, and eventually cancerogenesis.9,10
HER2 gene amplification can cause activation of the receptor’s extramembranous domain by way of either dimerization of two HER2 receptors or heterodimerization with other ERBB family receptors, leading to ligand-independent activation of cell signaling (ie, activation in the absence of external growth factors). Besides breast cancer, HER2 protein is overexpressed in several other tumor types, including esophageal and gastric adenocarcinomas, colon and gynecological malignancies, and to a lesser extent in other malignancies.
Biomarker Testing
All patients with newly diagnosed breast cancer should have their tumor tissue submitted for biomarker testing for estrogen receptors (ER), progesterone receptors (PR), and HER2 overexpression, as the result this testing dictates therapy choices. The purpose of HER2 testing is to investigate whether the HER2 gene, located on chromosome 17, is overexpressed or amplified. HER2 status provides the basis for treatment selection, which impacts long-term outcome measures such as recurrence and survival. Routine testing of carcinoma in situ for HER2 expression/amplification is not recommended and has no implication on choice of therapy at this time.
In 2013, the American Society of Clinical Oncology and the College of American Pathologists (ASCO/CAP) updated their clinical guideline recommendations for HER2 testing in breast cancer to improve its accuracy and its utility as a predictive marker.11 There are currently 2 approved modalities for HER2 testing: detection of HER2 protein overexpression by i
Fluorescence in-situ hybridization (FISH) testing assesses for HER2 amplification by determining the number of HER2 signals and
Neoadjuvant and Adjuvant Therapy for Locoregional Disease
Case Patient 1
A 56-year-old woman undergoes ultrasound-guided biopsy of a self-palpated breast lump. Pathology shows invasive ductal carcinoma that is ER-positive, PR-negative, and HER2 equivocal by IHC (2+ staining). Follow-up FISH testing shows a HER2/CEP17 ratio of 2.5. The tumor is estimated to be 2 cm in diameter by imaging and exam with no clinically palpable axillary lymphadenopathy. The patient exhibits no constitutional or localized symptoms concerning for metastases.
- What is the recommended management approach for this patient?
According to the ASCO/CAP guidelines, this patient’s tumor qualifies as HER2-positive based upon testing results showing amplification of the gene. This result has important implications for management since nearly all patients with macroscopically invasive HER2-positive tumors should be considered for adjuvant chemotherapy in combination with anti-HER2 therapy. The patient should proceed with upfront tumor resection and sentinel lymph node biopsy. Systemic staging imaging (ie, computed tomography [CT] or bone scan) is not indicated in early stage breast cancer.12,13 Systemic staging scans are indicated when (1) any anatomical stage III disease is suspected (eg, with involvement of the skin or chest wall, the presence of enlarged matted or fixed axillary lymph nodes, and involvement of nodal stations other than in the axilla), and (2) when symptoms or abnormal laboratory values raise suspicion for distant metastases (eg, unexplained bone pain, unintentional weight loss, elevated serum alkaline phosphatase, and transaminitis).
Case 1 Continued
The patient presents to discuss treatment options after undergoing a lumpectomy and sentinel node biopsy procedure. The pathology report notes a single focus of carcinoma measuring 2 cm with negative sentinel lymph nodes.
- What agents are used for adjuvant therapy in HER2-postive breast cancer?
Nearly all patients with macroscopically invasive (> 1 mm) breast carcinoma should be considered for adjuvant therapy using a regimen that contains a taxane and trastuzumab. However, the benefit may be small for patients with tumors ≤ 5 mm (T1a, N0), so it is important to carefully weigh the risk against the benefit. Among the agents that targeting HER2, only trastuzumab has been shown to improve overall survival (OS) in the adjuvant setting; long-term follow-up data are awaited for other agents.8 A trastuzumab biosimilar, trastuzumab-dkst, was recently approved by the US Food and Drug Administration (FDA) for the same indications as trastuzumab.14 The regimens most commonly used in the adjuvant and neoadjuvant settings for nonmetastatic breast cancer are summarized in Table 2.
Patients with small (≤ 3 cm), node-negative tumors can generally be considered for a reduced-intensity regimen that includes weekly paclitaxel plus trastuzumab. This combination proved efficacious in a single-group, multicenter study that enrolled 406 patients.15 Paclitaxel and trastuzumab were given once weekly for 12 weeks, followed by trastuzumab, either weekly or every 3 weeks, to complete 1 year of therapy.After a median follow-up of more than 6 years, the rates of distant and locoregional recurrence were 1% and 1.2%, respectively.16
A combination of docetaxel, carboplatin, and trastuzumab is a nonanthracycline regimen that is also appropriate in this setting, based on the results of the Breast Cancer International Research Group 006 (BCIRG-006) trial.17 This phase 3 randomized trial enrolled 3222 women with HER2-positive, invasive, high-risk adenocarcinoma. Eligible patients had a T1–3 tumor and either lymph node–negative or –positive disease and were randomly assigned to receive 1 of 3 regimens: group 1 received doxorubicin and cyclophosphamide every 3 weeks for 4 cycles followed by docetaxel every 3 weeks for 4 cycles (AC-T); group 2 received the AC-T regimen in combination with trastuzumab; and group 3 received docetaxel, carboplatin, and trastuzumab once every 3 weeks for 6 cycles (TCH). Groups 2 and 3 also received trastuzumab for an additional 34 weeks to complete 1 year of therapy. Trastuzumab-containing regimens were found to offer superior disease-free survival (DFS) and OS. When comparing the 2 trastuzumab arms after more than 10 years of follow-up, no statistically significant advantage of an anthracycline regimen over a nonanthracycline regimen was found.18 Furthermore, the anthracycline arm had a fivefold higher incidence of symptomatic congestive heart failure (grades 3 and 4), and the nonanthracycline regimen was associated with a lower incidence of treatment-related leukemia, a clinically significant finding despite not reaching statistical significance due to low overall numbers.
BCIRG-006, NSABP B-31, NCCTG N9831, and HERA are all large randomized trials with consistent results confirming trastuzumab’s role in reducing recurrence and improving survival in HER2-positive breast cancer in the adjuvant settings. The estimated overall benefit from addition of this agent was a 34% to 41% improvement in survival and a 33% to 52% improvement in DFS.8,17–20
Dual anti-HER2 therapy containing both trastuzumab and pertuzumab should be strongly considered for patients with macroscopic lymph node involvement based on the results of the APHINITY trial.21 In this study, the addition of pertuzumab to standard trastuzumab-based therapy led to a significant improvement in invasive-disease-free survival at 3 years. In subgroup analysis, the benefit was restricted to the node-positive group (3-year invasive-disease-free survival rates of 92% in the pertuzumab group versus 90.2% in the placebo group, P = 0.02). Patients with hormone receptor–negative disease derived greater benefit from the addition of pertuzumab. Regimens used in the APHINITY trial included the anti-HER2 agents trastuzumab and pertuzumab in combination with 1 of the following chemotherapy regimens: sequential cyclophosphamide plus either doxorubicin or epirubicin, followed by either 4 cycles of docetaxel or 12 weekly doses of paclitaxel; sequential fluorouracil plus either epirubicin or doxorubicin plus cyclophosphamide (3 or 4 cycles), followed by 3 or 4 cycles of docetaxel or 12 weekly cycles of paclitaxel; or 6 cycles of concurrent docetaxel plus carboplatin.
One-year therapy with neratinib, an oral tyrosine kinase inhibitor of HER2, is now approved by the FDA after completion of trastuzumab in the adjuvant setting, based on the results of the ExteNET trial.22 In this study, patients who had completed trastuzumab within the preceding 12 months, without evidence of recurrence, were randomly assigned to receive either oral neratinib or placebo daily for 1 year. The 2-year DFS rate was 93.9% and 91.6% for the neratinib and placebo groups, respectively. The most common adverse effect of neratinib was diarrhea, with approximately 40% of patients experiencing grade 3 diarrhea. In subgroup analyses, hormone receptor–positive patients derived the most benefit, while hormone receptor–negative patients derived no or marginal benefit.22 OS benefit has not yet been established.23
Trastuzumab therapy (with pertuzumab if indicated) should be offered for an optimal duration of 12 months (17 cycles, including those given with chemotherapy backbone). A shorter duration of therapy, 6 months, has been shown to be inferior,24 while a longer duration, 24 months, has been shown to provide no additional benefit.25
Finally, sequential addition of anti-estrogen endocrine therapy is indicated for hormone-positive tumors. Endocrine therapy is usually added after completion of the chemotherapy backbone of the regimen, but may be given concurrently with anti-HER2 therapy. If radiation is being administered, endocrine therapy can be given concurrently or started after radiation therapy is completed.
Case 1 Conclusion
The patient can be offered 1 of 2 adjuvant treatment regimens, either TH or TCH (Table 2). Since the patient had lumpectomy, she is an appropriate candidate for adjuvant radiation, which would be started after completion of the chemotherapy backbone (taxane/platinum). Endocrine therapy for at least 5 years should be offered sequentially or concurrently with radiation. Her long-term prognosis is very favorable.
Case Patient 2
A 43-year-old woman presents with a 4-cm breast mass, a separate skin nodule, and palpable matted axillary lymphadenopathy. Biopsies of the breast mass and subcutaneous nodule reveal invasive ductal carcinoma that is ER-negative, PR-negative, and HER2-positive by IHC (3+ staining). Based on clinical findings, the patient is staged as T4b (separate tumor nodule), N2 (matted axillary lymph nodes). Systemic staging with CT scan of the chest, abdomen, and pelvis shows no evidence of distant metastases.
- What is the recommended approach to management for this patient?
Recommendations for neoadjuvant therapy, given before definitive surgery, follow the same path as with other subtypes of breast cancer. Patients with suspected anatomical stage III disease are strongly encouraged to undergo upfront (neoadjuvant) chemotherapy in combination with HER2-targeted agents. In addition, all HER2-positive patients with clinically node-positive disease can be offered neoadjuvant therapy using chemotherapy plus dual anti-HER2 therapy (trastuzumab and pertuzumab), with complete pathological response expected in more than 60% of patients.26,27 Because this patient has locally advanced disease, especially skin involvement and matted axillary nodes, she should undergo neoadjuvant therapy. Preferred regimens contain both trastuzumab and pertuzumab in combination with cytotoxic chemotherapy. The latter may be given concurrently (nonanthracycline regimens, such as docetaxel plus carboplatin) or sequentially (anthracycline-based regimens), as outlined in Table 2. Administration of anthracyclines and trastuzumab simultaneously is contraindicated due to increased risk of cardiomyopathy.28
Endocrine therapy is not indicated for this patient per the current standard of care because the tumor was ER- and PR-negative. Had the tumor been hormone receptor–positive, endocrine therapy for a minimum of 5 years would have been indicated. Likewise, in the case of hormone receptor–positive disease, 12 months of neratinib therapy after completion of trastuzumab may add further benefit, as shown in the ExteNET trial.22,23 Neratinib seems to have a propensity to prevent or delay trastuzumab-induced overexpression of estrogen receptors. This is mainly due to hormone receptor/HER2 crosstalk, a potential mechanism of resistance to trastuzumab.29,30
In addition to the medical therapy options discussed here, this patient would be expected to benefit from adjuvant radiation to the breast and regional lymph nodes, given the presence of T4 disease and bulky adenopathy in the axilla.31
Case 2 Conclusion
The patient undergoes neoadjuvant treatment (docetaxel, carboplatin, trastuzumab, and pertuzumab every 21 days for a total of 6 cycles), followed by surgical resection (modified radical mastectomy) that reveals complete pathological response (no residual invasive carcinoma). Subsequently, she receives radiation therapy to the primary tumor site and regional lymph nodes while continuing trastuzumab and pertuzumab for 11 more cycles (17 total). Despite presenting with locally advanced disease, the patient has a favorable overall prognosis due to an excellent pathological response.
- What is the approach to follow-up after completion of primary therapy?
Patients may follow up every 3 to 6 months for clinical evaluation in the first 5 years after completing primary adjuvant therapy. An annual screening mammogram is recommended as well. Body imaging can be done if dictated by symptoms. However, routine CT, positron emission tomography, or bone scans are not recommended as part of follow-up in the absence of symptoms, mainly because of a lack of evidence that such surveillance improves survival.32
Metastatic HER2-Positive Breast Cancer
Metastatic breast cancer most commonly presents as a distant recurrence of previously treated local disease. However, 6% to 18% of patients have no prior history of breast cancer and present with de novo metastatic disease.33,34 The most commonly involved distant organs are the skeletal bones, liver, lung, distant lymph node stations, and brain. Compared to other subtypes, HER2-positive tumors have an increased tendency to involve the central nervous system.35–38 Although metastatic HER2-positive breast cancer is not considered curable, significant improvement in survival has been achieved, and patients with metastatic disease have median survival approaching 5 years.39
Case Presentation 3
A 69-year-old woman with a history of breast cancer 4 years ago presents with new-onset back pain and unintentional weight loss. On exam, she is found to have palpable axillary adenopathy on the same side as her previous cancer. Her initial disease was stage IIB ER-positive and HER2-positive and was treated with chemotherapy, mastectomy, and anastrozole, which the patient is still taking. She undergoes CT scan of the chest, abdomen, and pelvis and radionucleotide bone scan, which show multiple liver and bony lesions suspicious for metastatic disease. Axillary lymph node biopsy confirms recurrent invasive carcinoma that is ER-positive and HER2-positive by IHC (3+).
- What is the approach to management of a patient who presents with symptoms of recurrent HER2-positive disease?
This patient likely has metastatic breast cancer based on the imaging findings. In such cases, a biopsy of the recurrent disease should always be considered, if feasible, to confirm the diagnosis and rule out other etiologies such as different malignances and benign conditions. Hormone-receptor and HER2 testing should also be performed on recurrent disease, since a change in HER2 status can be seen in 15% to 33% of cases.40–42
Based on data from the phase 3 CLEOPATRA trial, first-line systemic regimens for patients with metastatic breast cancer that is positive for HER2 should consist of a combination of docetaxel, trastuzumab, and pertuzumab. Compared to placebo, adding pertuzumab yielded superior progression-free survival of 18.4 months (versus 12.4 months for placebo) and an unprecedented OS of 56.5 months (versus 40.8 for placebo).39 Weekly paclitaxel can replace docetaxel with comparable efficacy (Table 3).43
Patients can develop significant neuropathy as well as skin and nail changes after multiple cycles of taxane-based chemotherapy. Therefore, the taxane backbone may be dropped after 6 to 8 cycles, while patients continue the trastuzumab and pertuzumab combination until disease progression or unacceptable toxicity. Some patients may enjoy remarkable long-term survival on “maintenance” anti-HER2 therapy.44 Despite lack of high-level evidence, such as from large randomized trials, some experts recommend the addition of a hormone blocker after discontinuation of the taxane in ER-positive tumors.45
Premenopausal and perimenopausal women with hormone receptor–positive metastatic disease should be considered for simultaneous ovarian suppression. Ovarian suppression can be accomplished medically using a gonadotropin-releasing hormone agonist (goserelin) or surgically via salpingo-oophorectomy.46–48
Case 3 Conclusion
The patient receives 6 cycles of docetaxel, trastuzumab, and pertuzumab, after which the docetaxel is discontinued due to neuropathy while she continues the other 2 agents. After 26 months of disease control, the patient is found to have new liver metastatic lesions, indicating progression of disease.
- What therapeutic options are available for this patient?
Patients whose disease progresses after receiving taxane- and trastuzumab-containing regimens are candidates to receive the novel antibody-drug conjugate ado-trastuzumab emtansine (T-DM1). Early progressors (ie, patients with early stage disease who have progression of disease while receiving adjuvant trastuzumab or within 6 months of completion of adjuvant trastuzumab) are also candidates for T-DM1. Treatment usually fits in the second line or beyond based on data from the EMILIA trial, which randomly assigned patients to receive either capecitabine plus lapatinib or T-DM1.49,50 Progression-free survival in the T-DM1 group was 9.6 months versus 6.4 months for the comparator. Improvement of 4 months in OS persisted with longer follow-up despite a crossover rate of 27%. Furthermore, a significantly higher objective response rate and fewer adverse effects were reported in the T-DM1 patients. Most patients included in the EMILIA trial were pertuzumab-naive. However, the benefit of T-DM1 appears to persist, albeit to a lesser extent, for pertuzumab-pretreated patients.51,52
Patients in whom treatment fails with 2 or more lines of therapy containing taxane-trastuzumab (with or without pertuzumab) and T-DM1 are candidates to receive a combination of capecitabine and lapatinib, a TKI, in the third line and beyond. Similarly, the combination of capecitabine with trastuzumab in the same settings appears to have equal efficacy.53,54 Trastuzumab may be continued beyond progression while changing the single-agent chemotherapy drug for subsequent lines of therapy, per ASCO guidelines,55 although improvement in OS has not been demonstrated beyond the third line in a large randomized trial (Table 3).
Approved HER2-Targeted Drugs
HER2-directed therapy is implemented in the management of nearly all stages of HER2-positive invasive breast cancer, including early and late stages (Table 4).
Trastuzumab
Trastuzumab was the first anti-HER2 agent to be approved by the FDA in 1998. It is a humanized monoclonal antibody directed against the extracellular domain of the HER2 receptor (domain IV). Trastuzumab functions by interrupting HER2 signal transduction and by flagging tumor cells for immune destruction.56 Cardiotoxicity, usually manifested as left ventricular systolic dysfunction, is the most noteworthy adverse effect of trastuzumab. The most prominent risk factors for cardiomyopathy in patients receiving trastuzumab are low baseline ejection fraction (< 55%), age > 50 years, co-administration and higher cumulative dose of anthracyclines, and increased body mass index and obesity.57–59 Whether patients receive therapy in the neoadjuvant, adjuvant, or metastatic settings, baseline cardiac function assessment with echocardiogram or multiple-gated acquisition scan is required. While well-designed randomized trials validating the value and frequency of monitoring are lacking, repeated cardiac testing every 3 months is generally recommended for patients undergoing adjuvant therapy. Patients with metastatic disease who are receiving treatment with palliative intent may be monitored less frequently.60,61
An asymptomatic drop in ejection fraction is the most common manifestation of cardiac toxicity. Other cardiac manifestations have also been reported with much less frequency, including arrhythmias, severe congestive heart failure, ventricular thrombus formation, and even cardiac death. Until monitoring and dose-adjustment guidelines are issued, the guidance provided in the FDA-approved prescribing information should be followed, which recommends holding trastuzumab when there is ≥ 16% absolute reduction in left ventricular ejection fraction (LVEF) from the baseline value; or if the LVEF value is below the institutional lower limit of normal and the drop is ≥ 10%. After holding the drug, cardiac function can be re-evaluated every 4 weeks. In most patients, trastuzumab-induced cardiotoxicity can be reversed by withholding trastuzumab and initiating cardioprotective therapy, although the latter remains controversial. Re-challenging after recovery of ejection fraction is possible and toxicity does not appear to be proportional to cumulative dose. Cardiomyopathy due to trastuzumab therapy is potentially reversible within 6 months in more than 80% of cases.28,57,60–63
Other notable adverse effects of trastuzumab include pulmonary toxicity (such as interstitial lung disease) and infusion reactions (usually during or within 24 hours of first dose).
Pertuzumab
Pertuzumab is another humanized monoclonal antibody directed to a different extracellular domain of the HER2 receptor, the dimerization domain (domain II), which is responsible for heterodimerization of HER2 with other HER receptors, especially HER3. This agent should always be co-administered with trastuzumab as the 2 drugs produce synergistic anti-tumor effect, without competition for the receptor. Activation of HER3, via dimerization with HER2, produces an alternative mechanism of downstream signaling, even in the presence of trastuzumab and in the absence of growth factors (Figure 2).
Ado-Trastuzumab Emtansine
Ado-trastuzumab emtansine (T-DM1) is an antibody-drug conjugate that combines the monoclonal antibody trastuzumab with the cytotoxic agent DM1 (emtansine), a potent microtubule inhibitor and a derivative of maytansine, in a single structure (Figure 3).
Lapatinib
Lapatinib is an oral small-molecule tyrosine kinase inhibitor of EGFR (HER1) and HER2 receptors. It is approved in combination with capecitabine for patients with HER2-expressing metastatic breast cancer who previously received trastuzumab, an anthracycline, and a taxane chemotherapy or T-DM1. Lapatinib is also approved in combination with letrozole in postmenopausal women with HER2-positive, hormone receptor–positive metastatic disease, although it is unclear where this regimen would fit in the current schema. It may be considered for patients with hormone receptor–positive disease who are not candidates for therapy with taxane-trastuzumab and T-DM1 or who decline this therapy. Diarrhea is seen in most patients treated with lapatinib and may be severe in 20% of cases when lapatinib is combined with capecitabine. Decreases in LVEF have been reported and cardiac function monitoring at baseline and periodically may be considered.69,70 Lapatinib is not approved for use in adjuvant settings.
Neratinib
Neratinib is an oral small-molecule irreversible tyrosine kinase inhibitor of HER1, HER2, and HER4. It is currently approved only for extended adjuvant therapy after completion of 1 year of standard trastuzumab therapy. It is given orally every day for 1 year. The main side effect, expected in nearly all patients, is diarrhea, which can be severe in up to 40% of patients and may lead to dehydration and electrolyte imbalance. Diarrhea usually starts early in the course of therapy and can be most intense during the first cycle. Therefore, prophylactic antidiarrheal therapy is recommended to reduce the intensity of diarrhea. Loperamide prophylaxis may be initiated simultaneously for all patients using a tapering schedule. Drug interruption or dose reduction may be required if diarrhea is severe or refractory.21,71 Neratinib is not FDA-approved in the metastatic settings.
Conclusion
HER2-positive tumors represent a distinct subset(s) of breast tumors with unique pathological and clinical characteristics. Treatment with a combination of cytotoxic chemotherapy and HER2-targeted agents has led to a dramatic improvement in survival for patients with locoregional and advanced disease. Trastuzumab is an integral part of adjuvant therapy for HER2-positive invasive disease. Pertuzumab should be added to trastuzumab in node-positive disease. Neratinib may be considered after completion of trastuzumab therapy in patients with hormone receptor–positive disease. For metastatic HER2-positive breast cancer, a regimen consisting of docetaxel plus trastuzumab and pertuzumab is the standard first-line therapy. Ado-trastuzumab is an ideal next line option for patients whose disease progresses on trastuzumab and taxanes.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
1. Yedjou CG, Tchounwou PB, Payton M, et al. Assessing the racial and ethnic disparities in breast cancer mortality in the United States. Int J Environ Res Public Health 2017;14(5).
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–89.
3. Huang HJ, Neven P, Drijkoningen M, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol 2005;58:611–6.
4. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
5. Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest 2010;28:963–-8.
6. Huang HJ, Neven P, Drijkoningen M, et al. Hormone receptors do not predict the HER2/neu status in all age groups of women with an operable breast cancer. Ann Oncol 2005;16:1755–61.
7. Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–502.
8. Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol 2014;32:3744–52.
9. Brennan PJ, Kumagai T, Berezov A, et al. HER2/neu: mechanisms of dimerization/oligomerization. Oncogene 2000;19:6093–101.
10. Roskoski R Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11.
11. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
12. Ravaioli A, Pasini G, Polselli A, et al. Staging of breast cancer: new recommended standard procedure. Breast Cancer Res Treat 2002;72:53–60.
13. Puglisi F, Follador A, Minisini AM, et al. Baseline staging tests after a new diagnosis of breast cancer: further evidence of their limited indications. Ann Oncol 2005;16:263–6.
14. FDA approves trastuzumab biosimilar. Cancer Discov 2018;8:130.
15. Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med 2015;372:134–41.
16. Tolaney SM, Barry WT, Guo H, Dillon D, et al. Seven-year (yr) follow-up of adjuvant paclitaxel (T) and trastuzumab (H) (APT trial) for node-negative, HER2-positive breast cancer (BC) [ASCO abstract]. J Clin Oncol. 2017;35(suppl):511.
17. Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 2011;365:1273–83.
18. Slamon DJ, Eiermann W, Robert NJ, et al. Ten year follow-up of BCIRG-006 comparing doxorubicin plus cyclophosphamide followed by docetaxel (AC -> T) with doxorubicin plus cyclophosphamide followed by docetaxel and trastuzumab (AC -> TH) with docetaxel, carboplatin and trastuzumab (TCH) in HER2+early breast cancer [SABC abstract]. Cancer Res 2016;76(4 supplement):S5-04.
19. Jahanzeb M. Adjuvant trastuzumab therapy for HER2-positive breast cancer. Clin Breast Cancer 2008;8:324–33.
20. Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017;389:1195–205.
21. von Minckwitz G, Procter M, de Azambuja E, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med 2017;377:122–31.
22. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77.
23. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1688–700.
24. Pivot X, Romieu G, Debled M, et al. 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): a randomised phase 3 trial. Lancet Oncol 2013;14:741–8.
25. Goldhirsch A, Gelber RD, Piccart-Gebhart MJ, et al. 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 2013;382:1021–8.
26. Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol 2013;24:2278–84.
27. Schneeweiss A, Chia S, Hickish T, et al. Long-term efficacy analysis of the randomised, phase II TRYPHAENA cardiac safety study: Evaluating pertuzumab and trastuzumab plus standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer. Eur J Cancer 2018;89:27–35
28. de Azambuja E, Procter MJ, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac events at 8 years of median follow-up in the Herceptin Adjuvant trial (BIG 1-01). J Clin Oncol 2014;32:2159–65.
29. Dowsett M, Harper-Wynne C, Boeddinghaus I, et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001;61:8452–8.
30. Nahta R, O’Regan RM. Therapeutic implications of estrogen receptor signaling in HER2-positive breast cancers. Breast Cancer Res Treat 2012;135:39–48.
31. Recht A, Comen EA, Fine RE, et al. Postmastectomy radiotherapy: An American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology focused guideline update. Pract Radiat Oncol 2016;6:e219-e34.
32. Runowicz CD, Leach CR, Henry NL, et al. American Cancer Society/American Society of Clinical Oncology breast cancer survivorship care guideline. J Clin Oncol 2016;34:611–35.
33. Zeichner SB, Herna S, Mani A, et al. Survival of patients with de-novo metastatic breast cancer: analysis of data from a large breast cancer-specific private practice, a university-based cancer center and review of the literature. Breast Cancer Res Treat 2015;153:617–24.
34. Dawood S, Broglio K, Ensor J, et al. Survival differences among women with de novo stage IV and relapsed breast cancer. Ann Oncol 2010;21:2169–74.
35. Savci-Heijink CD, Halfwerk H, Hooijer GK, et al. Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 2015;150:547–57.
36. Kimbung S, Loman N, Hedenfalk I. Clinical and molecular complexity of breast cancer metastases. Semin Cancer Biol 2015;35:85–95.
37. Bendell JC, Domchek SM, Burstein HJ, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003;97:2972–7.
38. Burstein HJ, Lieberman G, Slamon DJ, et al. Isolated central nervous system metastases in patients with HER2-overexpressing advanced breast cancer treated with first-line trastuzumab-based therapy. Ann Oncol 2005;16:1772–7.
39. Swain SM, Baselga J, Kim SB, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015;372:724–34.
40. Lindstrom LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol 2012;30:2601–8.
41. Guarneri V, Giovannelli S, Ficarra G, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist 2008;13:838–44.
42. Salkeni MA, Hall SJ. Metastatic breast cancer: Endocrine therapy landscape reshaped. Avicenna J Med 2017;7:144–52.
43. Dang C, Iyengar N, Datko F, et al. Phase II study of paclitaxel given once per week along with trastuzumab and pertuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:442–7.
44. Cantini L, Pistelli M, Savini A, et al. Long-responders to anti-HER2 therapies: A case report and review of the literature. Mol Clin Oncol 2018;8:147–52.
45. Sutherland S, Miles D, Makris A. Use of maintenance endocrine therapy after chemotherapy in metastatic breast cancer. Eur J Cancer 2016;69:216–22.
46. Falkson G, Holcroft C, Gelman RS, et al. Ten-year follow-up study of premenopausal women with metastatic breast cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol 1995;13:1453–8.
47. Boccardo F, Rubagotti A, Perrotta A, et al. Ovarian ablation versus goserelin with or without tamoxifen in pre-perimenopausal patients with advanced breast cancer: results of a multicentric Italian study. Ann Oncol 1994;5:337–42.
48 Taylor CW, Green S, Dalton WS, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998;16:994–9.
49. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 2012;367:1783–91.
50. Dieras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:732–42.
51. Dzimitrowicz H, Berger M, Vargo C, et al. T-DM1 Activity in metastatic human epidermal growth factor receptor 2-positive breast cancers that received prior therapy with trastuzumab and pertuzumab. J Clin Oncol 2016;34:3511–7.
52. Fabi A, Giannarelli D, Moscetti L, et al. Ado-trastuzumab emtansine (T-DM1) in HER2+ advanced breast cancer patients: does pretreatment with pertuzumab matter? Future Oncol 2017;13:2791–7.
53. Madden R, Kosari S, Peterson GM, et al. Lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer: A systematic review. Int J Clin Pharmacol Ther 2018;56:72–80.
54. Pivot X, Manikhas A, Zurawski B, et al. CEREBEL (EGF111438): A phase III, randomized, open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J Clin Oncol 2015;33:1564–73.
55. Giordano SH, Temin S, Kirshner JJ, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–99.
56. Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
57. Russell SD, Blackwell KL, Lawrence J, et al. Independent adjudication of symptomatic heart failure with the use of doxorubicin and cyclophosphamide followed by trastuzumab adjuvant therapy: a combined review of cardiac data from the National Surgical Adjuvant breast and Bowel Project B-31 and the North Central Cancer Treatment Group N9831 clinical trials. J Clin Oncol 2010;28:3416–21.
58. Ewer SM, Ewer MS. Cardiotoxicity profile of trastuzumab. Drug Saf 2008;31:459–67.
59. Guenancia C, Lefebvre A, Cardinale D, et al. Obesity as a risk factor for anthracyclines and trastuzumab cardiotoxicity in breast cancer: a systematic review and meta-analysis. J Clin Oncol 2016;34:3157–65.
60. Dang CT, Yu AF, Jones LW, et al. Cardiac surveillance guidelines for trastuzumab-containing therapy in early-stage breast cancer: getting to the heart of the matter. J Clin Oncol 2016;34:1030–3.
61. Brann AM, Cobleigh MA, Okwuosa TM. Cardiovascular monitoring with trastuzumab therapy: how frequent is too frequent? JAMA Oncol 2016;2:1123–4.
62. Suter TM, Procter M, van Veldhuisen DJ, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol 2007;25:3859–65.
63. Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol 2010;28:3422–8.
64. Yamashita-Kashima Y, Shu S, Yorozu K, et al. Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncol Lett 2017;14:4197–205.
65. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117:1736–42.
66. Girish S, Gupta M, Wang B, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol 2012;69:1229–40.
67. Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015;21:123–33.
68. Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther 2016;15:480–90.
69. Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol 2005;23:2502–12.
70. Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009;27:5538–46.
71. Neratinib (Nerlynx) for HER2-positive breast cancer. Med Lett Drugs Ther 2018;60(1539):23.
Sudden-onset memory problems, visual hallucinations, and odd behaviors
CASE A rapid decline
Ms. D, age 62, presents to a psychiatric emergency room (ER) after experiencing visual hallucinations, exhibiting odd behaviors, and having memory problems. On interview, she is disoriented, distractible, tearful, and tangential. She plays with her shirt and glasses, and occasionally shouts. She perseverates on “the aerialists,” acrobatic children she has been seeing in her apartment. She becomes distressed and shouts, “I would love to just get them!”
Ms. D is unable to provide an account of her history. Collateral information is obtained from her daughter, who has brought Ms. D to the ER for evaluation. She reports that her mother has no relevant medical or psychiatric history, and does not take any medications, except a mixture of Chinese herbs that she brews into a tea.
Ms. D’s daughter says that her mother began to deteriorate 5 months ago, after she traveled to California to care for her sister, who was seriously ill and passed away. After Ms. D returned, she would cry frequently. She also appeared “spaced out,” complained of feeling dizzy, and frequently misplaced belongings. Three months before presenting to the ER, she began to experience weakness, fatigue, and difficulty walking. Her daughter became more worried 2 months ago, when Ms. D began sleeping with her purse and hiding her belongings around their house. When asked about these odd behaviors, Ms. D claimed that “the aerialists” were climbing through her windows at night and stealing her things.
A week before seeking treatment at the ER, Ms. D’s daughter had taken her to a neurologist at another facility for clinical evaluation. An MRI of the brain showed minimal dilation in the subarachnoid space and a focal 1 cm lipoma in the anterior falx cerebri, but was otherwise unremarkable. However, Ms. D’s symptoms continued to worsen, and began to interfere with her ability to care for herself.
The team in the psychiatric ER attributes Ms. D’s symptoms to a severe, psychotic depressive episode. They admit her to the psychiatric inpatient unit for further evaluation.
[polldaddy:10012742]
Continue to: The authors' observations
The authors’ observations
Ms. D was plagued by several mood and psychotic symptoms. Such symptoms can arise from many different psychiatric or organic etiologies. In Ms. D’s case, several aspects of her presentation suggest that her illness was psychiatric. The severe illness of a beloved family member is a significant stressor that could cause a great deal of grief and devastation, possibly leading to depression. Indeed, Ms. D’s daughter noticed that her mother was crying frequently, which is consistent with grief or depression.
Memory problems, which might manifest as misplacing belongings, can also indicate a depressive illness, especially in older patients. Moreover, impaired concentration, which can cause one to appear “spaced out” or distractible, is a core symptom of major depressive disorder. Sadness and grief also can be appropriate during bereavement and in response to significant losses. Therefore, in Ms. D’s case, it is possible her frequent crying, “spaced out” appearance, and other mood symptoms she experienced immediately after caring for her sister were an appropriate response to her sister’s illness and death.
However, other aspects of Ms. D’s presentation suggested an organic etiology. Her rapid deterioration and symptom onset relatively late in life were consistent with dementia and malignancy. Her complaint of feeling dizzy suggested a neurologic process was affecting her vestibular system. Finally, while psychiatric disorders can certainly cause visual hallucinations, they occur in only a small percentage of cases.1 Visual hallucinations are commonly associated with delirium, intoxication, and neurologic illness.
Continue to: EVALUATION Severe impairment
EVALUATION Severe impairment
On the psychiatric inpatient unit, Ms. D remains unable to give a coherent account of her illness or recent events. During interviews, she abruptly shifts from laughing to crying for no apparent reason. While answering questions, her responses trail off and she appears to forget what she had been saying. However, she continues to speak at length about “the aerialists,” stating that she sees them living in her wardrobe and jumping from rooftop to rooftop in her neighborhood.
A mental status examination finds evidence of severe cognitive impairment. Ms. D is unable to identify the correct date, time, or place, and appears surprised when told she is in a hospital. She can repeat the names of 3 objects but cannot recall them a few minutes later. Finally, she scores a 14 on the Mini-Mental State Examination (MMSE) and a 5 on the Montreal Cognitive Assessment (MoCA), indicating severe impairment.
On the unit, Ms. D cannot remember the location of her room or bathroom, and even when given directions, she needs to be escorted to her destination. Her gait is unsteady and wide-spaced, and she walks on her toes at times. When food is placed before her, she needs to be shown how to take the lids off containers, pick up utensils, and start eating.
All laboratory results are unremarkable, including a complete blood count, basic metabolic panel, liver function tests, gamma-glutamyl transpeptidase, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, methylmalonic acid, homocysteine, folate, erythrocyte sedimentation rate, C-reactive protein, antinuclear antibodies, rapid plasma reagin, human immunodeficiency virus, and Lyme titers. The team also considers Ms. D’s history of herbal medicine use, because herbal mixtures can contain heavy metals and other contaminants. However, all toxicology results are normal, including arsenic, mercury, lead, copper, and zinc.
To address her symptoms, Ms. D is given risperidone, 0.5 mg twice a day, and donepezil, 5 mg/d.
[polldaddy:10012743]
Continue to: The authors' observations
The authors’ observations
Despite her persistent psychiatric symptoms, Ms. D had several neurologic symptoms that warranted further investigation. Her abrupt shifts from laughter to tears for no apparent reason were consistent with pseudobulbar affect. Her inability to remember how to use utensils during meals was consistent with apraxia. Finally, her abnormal gait raised concern for a process affecting her motor system.
OUTCOME A rare disorder
Given the psychiatry team’s suspicions for a neurologic etiology of Ms. D’s symptoms, an MRI of her brain is repeated. The results are notable for abnormal restricted diffusion in the caudate and putamen bilaterally, which is consistent with Creutzfeldt-Jakob disease (CJD). EEG shows moderate diffuse cerebral dysfunction, frontal intermittent delta activity, and diffuse cortical hyperexcitability, consistent with early- to mid-onset prion disease. Upon evaluation by the neurology team, Ms. D appears fearful, suspicious, and disorganized, but her examination does not reveal additional significant sensorimotor findings.
Ms. D is transferred to the neurology service for further workup and management. A lumbar puncture is positive for real-time quaking-induced conversion (RT-QuIC) and 14-3-3 protein with elevated tau proteins; these findings also are consistent with CJD. She develops transaminitis, with an alanine transaminase (ALT) of 127 and aspartate transaminase (AST) of 355, and a malignancy is suspected. However, CT scans of the chest, abdomen, and pelvis show no evidence of malignancy, and an extensive gastrointestinal workup is unremarkable, including anti-smooth muscle antibodies, anti-liver-kidney microsomal antibody, antimitochondrial antibodies, gliadin antibody, alpha-1 antitrypsin, liver/kidney microsomal antibody, and hepatitis serologies. While on the neurology service, risperidone and donepezil are discontinued because
After discontinuing these medications, she is evaluated by the psychiatry consult team for mood lability. The psychiatry consult team recommends quetiapine, which is later started at 25 mg nightly at bedtime.
Clinically, Ms. D’s mental status continues to deteriorate. She becomes nonverbal and minimally able to follow commands. She is ultimately discharged to an inpatient hospice for end-of-life care and the team recommends that she continue with quetiapine once there.
Continue to: The authors' observations
The authors’ observations
CJD is a rare, rapidly progressive, fatal form of dementia. In the United States, the incidence is approximately 1 to 1.5 cases per 1 million people each year.2 There are various forms of the disease. Sporadic CJD is the most common, representing 85% of cases.3 Sporadic CJD typically occurs in patients in their 60s and quickly leads to death—50% of patients die within 5 months, and 90% of patients die within 1 year.2,3 The illness is hypothesized to arise from the production of misfolded prion proteins, ultimately leading to vacuolation, neuronal loss, and the spongiform appearance characteristic of CJD.3,4
Psychiatric symptoms have long been acknowledged as a feature of CJD. Recent data indicates that psychiatric symptoms occur in 90% to 92% of cases.5,6 Sleep disturbances and depressive symptoms, including vegetative symptoms, anhedonia, and tearfulness, appear to be most common.5 Psychotic symptoms occur in approximately 42% of cases and may include persecutory and paranoid delusions, as well as an array of vivid auditory, visual, and tactile hallucinations.5,7
There is also evidence that psychiatric symptoms may be an early marker of CJD.5,8 A Mayo Clinic study found that psychiatric symptoms occurred within the prodromal phase of CJD in 26% of cases, and psychiatric symptoms occurred within the first 100 days of illness in 86% of cases.5
Case reports have described patients with CJD who initially presented with depression, psychosis, and other psychiatric symptoms.9-11 Interestingly, there have been cases with only psychiatric symptoms, and no neurologic symptoms until relatively late in the illness.10,11 Several patients with CJD have been evaluated in psychiatric ERs, admitted to psychiatric hospitals, and treated with psychiatric medications and ECT.5,9 In one study, 44% of CJD cases were misdiagnosed as “psychiatric patients” due to the prominence of their psychiatric symptomatology.8
Continue to: Making the diagnosis in psychiatric settings
Making the diagnosis in psychiatric settings. Often, the most difficult aspect of CJD is making the diagnosis.3,12 Sporadic CJD in particular can vary widely in its clinical presentation.3 The core clinical feature of CJD is rapidly progressive dementia, so suspect CJD in these patients. However, CJD can be difficult to distinguish from other rapidly progressive dementias, such as autoimmune and paraneoplastic encephalopathies.2,3 The presence of neurologic features, specifically myoclonus, akinetic mutism, and visual, cerebellar, and extrapyramidal symptoms, should also be considered a red flag for the disorder3 (Table).

Finally, positive findings on MRI, EEG, or CSF assay can indicate a probable diagnosis of CJD.13 MRI, particularly diffusion weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR), is recognized as the most studied, sensitive, and overall useful neuroimaging modality for detecting CJD.2,3,12 Although the appearance of CJD on MRI can vary widely, asymmetric hyperintensities in ≥3 cortical gyri, particularly in the frontal and parietal lobes, provide strong evidence of CJD and are observed in 80% to 81% of cases.4,12 Asymmetric hyperintensities in the basal ganglia, particularly the caudate and rostral putamen, are observed in 69% to 70% of cases.4,12,13
EEG and CSF assay also can be useful for making the diagnosis. While diffuse slowing and frontal rhythmic delta activity appear early in the course of CJD, periodic sharp wave complexes emerge later in the illness.4 However, EEG findings are not diagnostic, because periodic sharp wave complexes are seen in only two-thirds of CJD cases and also occur in other neurologic illnesses.3,4 In recent years, lumbar puncture with subsequent CSF testing has become increasingly useful in detecting the illness. The presence of the 14-3-3 protein and tau protein is highly sensitive, although not specific, for CJD.3 A definite diagnosis of CJD requires discovery of the misfolded prion proteins, such as by RT-QuIC or brain biopsy.2,3,13
Management of CJD in psychiatric patients. CJD is an invariably fatal disease for which there is no effective cure or disease modifying treatment.2 Therefore, supportive therapies are the mainstay of care. Psychotropic medications can be used to provide symptom relief. While the sleep disturbances, anxiety, and agitation/hallucinations associated with CJD appear to respond well to hypnotic, anxiolytic, and antipsychotic medications, respectively, antidepressants and mood-stabilizing medications appear to have little benefit for patients with CJD.5 During the final stages of the disease, patients may suffer from akinetic mutism and inability to swallow, which often leads to aspiration pneumonia.14 Patients should also be offered end-of-life counseling, planning, and care, and provided with other comfort measures wherever possible (Figure).
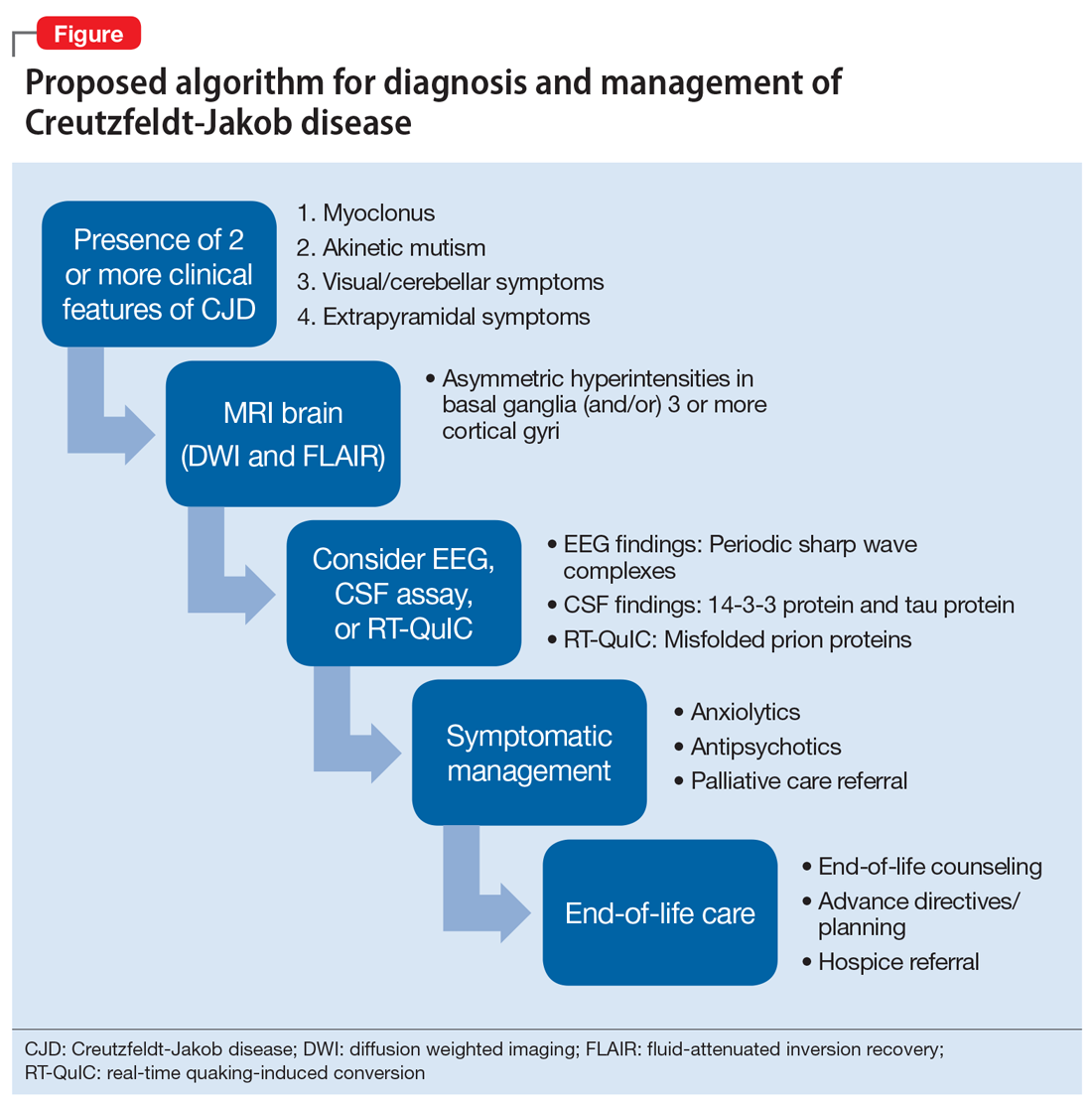
Continue to: Bottom Line
Bottom Line
Patients with Creutzfeldt-Jakob disease (CJD) may present to psychiatric settings, particularly to a psychiatric emergency room. Consider CJD as a possible etiology in patients with rapidly progressive dementia, depression, and psychosis. CJD is invariably fatal and there is no effective disease-modifying treatment. Supportive therapies are the mainstay of care.
Related Resources
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Centers for Disease Control and Prevention. Creutzfeldt-Jakob disease, classic (CJD). http://www.cdc.gov/prions/cjd.
Drug Brand Names
Donepezil • Aricept
Risperidone • Risperdal
Quetiapine • Seroquel
1. Resnick PJ. The detection of malingered psychosis. Psychiatr Clin North Am. 1999;22(1):159-172.
2. Bucelli RC, Ances BM. Diagnosis and evaluation of a patient with rapidly progressive dementia. Mo Med. 2013;110(5):422-428.
3. Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2.
4. Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-628.
5. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17(4):489-495.
6. Krasnianski A, Bohling GT, Harden M, et al. Psychiatric symptoms in patients with sporadic Creutzfeldt-Jakob disease in Germany. J Clin Psychiatry. 2015;76(9):1209-1215.
7. Javed Q, Alam F, Krishna S, et al. An unusual case of sporadic Creutzfeldt-Jakob disease (CJD). BMJ Case Rep. 2010;pii: bcr1220092576. doi:10.1136/bcr.12.2009.2576.
8. Abudy A, Juven-Wetzler A, Zohar J. The different faces of Creutzfeldt-Jacob disease CJD in psychiatry. Gen Hosp Psychiatry. 2014;36(3):245-248.
9. Jiang TT, Moses H, Gordon H, et al. Sporadic Creutzfeldt-Jakob disease presenting as major depression. South Med J. 1999;92(8):807-808.
10. Ali R, Baborie A, Larner AJ et al. Psychiatric presentation of sporadic Creutzfeldt-Jakob disease: a challenge to current diagnostic criteria. J Neuropsychiatry Clin Neurosci. 2013;25(4):335-338.
11. Gençer AG, Pelin Z, Küçükali CI., et al. Creutzfeldt-Jakob disease. Psychogeriatrics. 2011;11(2):119-124.
12. Caobelli F, Cobelli M, Pizzocaro C, et al. The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literature. J Neuroimaging. 2015;25(1):2-13.
13. Centers for Disease Control and Prevention. CDC's diagnostic criteria for Creutzfeldt-Jakob disease, 2010. http://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Updated February 11, 2015. Accessed August 2, 2016.
14. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease. Curr Psychiatry Rep. 2003;5(1):43-46.
CASE A rapid decline
Ms. D, age 62, presents to a psychiatric emergency room (ER) after experiencing visual hallucinations, exhibiting odd behaviors, and having memory problems. On interview, she is disoriented, distractible, tearful, and tangential. She plays with her shirt and glasses, and occasionally shouts. She perseverates on “the aerialists,” acrobatic children she has been seeing in her apartment. She becomes distressed and shouts, “I would love to just get them!”
Ms. D is unable to provide an account of her history. Collateral information is obtained from her daughter, who has brought Ms. D to the ER for evaluation. She reports that her mother has no relevant medical or psychiatric history, and does not take any medications, except a mixture of Chinese herbs that she brews into a tea.
Ms. D’s daughter says that her mother began to deteriorate 5 months ago, after she traveled to California to care for her sister, who was seriously ill and passed away. After Ms. D returned, she would cry frequently. She also appeared “spaced out,” complained of feeling dizzy, and frequently misplaced belongings. Three months before presenting to the ER, she began to experience weakness, fatigue, and difficulty walking. Her daughter became more worried 2 months ago, when Ms. D began sleeping with her purse and hiding her belongings around their house. When asked about these odd behaviors, Ms. D claimed that “the aerialists” were climbing through her windows at night and stealing her things.
A week before seeking treatment at the ER, Ms. D’s daughter had taken her to a neurologist at another facility for clinical evaluation. An MRI of the brain showed minimal dilation in the subarachnoid space and a focal 1 cm lipoma in the anterior falx cerebri, but was otherwise unremarkable. However, Ms. D’s symptoms continued to worsen, and began to interfere with her ability to care for herself.
The team in the psychiatric ER attributes Ms. D’s symptoms to a severe, psychotic depressive episode. They admit her to the psychiatric inpatient unit for further evaluation.
[polldaddy:10012742]
Continue to: The authors' observations
The authors’ observations
Ms. D was plagued by several mood and psychotic symptoms. Such symptoms can arise from many different psychiatric or organic etiologies. In Ms. D’s case, several aspects of her presentation suggest that her illness was psychiatric. The severe illness of a beloved family member is a significant stressor that could cause a great deal of grief and devastation, possibly leading to depression. Indeed, Ms. D’s daughter noticed that her mother was crying frequently, which is consistent with grief or depression.
Memory problems, which might manifest as misplacing belongings, can also indicate a depressive illness, especially in older patients. Moreover, impaired concentration, which can cause one to appear “spaced out” or distractible, is a core symptom of major depressive disorder. Sadness and grief also can be appropriate during bereavement and in response to significant losses. Therefore, in Ms. D’s case, it is possible her frequent crying, “spaced out” appearance, and other mood symptoms she experienced immediately after caring for her sister were an appropriate response to her sister’s illness and death.
However, other aspects of Ms. D’s presentation suggested an organic etiology. Her rapid deterioration and symptom onset relatively late in life were consistent with dementia and malignancy. Her complaint of feeling dizzy suggested a neurologic process was affecting her vestibular system. Finally, while psychiatric disorders can certainly cause visual hallucinations, they occur in only a small percentage of cases.1 Visual hallucinations are commonly associated with delirium, intoxication, and neurologic illness.
Continue to: EVALUATION Severe impairment
EVALUATION Severe impairment
On the psychiatric inpatient unit, Ms. D remains unable to give a coherent account of her illness or recent events. During interviews, she abruptly shifts from laughing to crying for no apparent reason. While answering questions, her responses trail off and she appears to forget what she had been saying. However, she continues to speak at length about “the aerialists,” stating that she sees them living in her wardrobe and jumping from rooftop to rooftop in her neighborhood.
A mental status examination finds evidence of severe cognitive impairment. Ms. D is unable to identify the correct date, time, or place, and appears surprised when told she is in a hospital. She can repeat the names of 3 objects but cannot recall them a few minutes later. Finally, she scores a 14 on the Mini-Mental State Examination (MMSE) and a 5 on the Montreal Cognitive Assessment (MoCA), indicating severe impairment.
On the unit, Ms. D cannot remember the location of her room or bathroom, and even when given directions, she needs to be escorted to her destination. Her gait is unsteady and wide-spaced, and she walks on her toes at times. When food is placed before her, she needs to be shown how to take the lids off containers, pick up utensils, and start eating.
All laboratory results are unremarkable, including a complete blood count, basic metabolic panel, liver function tests, gamma-glutamyl transpeptidase, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, methylmalonic acid, homocysteine, folate, erythrocyte sedimentation rate, C-reactive protein, antinuclear antibodies, rapid plasma reagin, human immunodeficiency virus, and Lyme titers. The team also considers Ms. D’s history of herbal medicine use, because herbal mixtures can contain heavy metals and other contaminants. However, all toxicology results are normal, including arsenic, mercury, lead, copper, and zinc.
To address her symptoms, Ms. D is given risperidone, 0.5 mg twice a day, and donepezil, 5 mg/d.
[polldaddy:10012743]
Continue to: The authors' observations
The authors’ observations
Despite her persistent psychiatric symptoms, Ms. D had several neurologic symptoms that warranted further investigation. Her abrupt shifts from laughter to tears for no apparent reason were consistent with pseudobulbar affect. Her inability to remember how to use utensils during meals was consistent with apraxia. Finally, her abnormal gait raised concern for a process affecting her motor system.
OUTCOME A rare disorder
Given the psychiatry team’s suspicions for a neurologic etiology of Ms. D’s symptoms, an MRI of her brain is repeated. The results are notable for abnormal restricted diffusion in the caudate and putamen bilaterally, which is consistent with Creutzfeldt-Jakob disease (CJD). EEG shows moderate diffuse cerebral dysfunction, frontal intermittent delta activity, and diffuse cortical hyperexcitability, consistent with early- to mid-onset prion disease. Upon evaluation by the neurology team, Ms. D appears fearful, suspicious, and disorganized, but her examination does not reveal additional significant sensorimotor findings.
Ms. D is transferred to the neurology service for further workup and management. A lumbar puncture is positive for real-time quaking-induced conversion (RT-QuIC) and 14-3-3 protein with elevated tau proteins; these findings also are consistent with CJD. She develops transaminitis, with an alanine transaminase (ALT) of 127 and aspartate transaminase (AST) of 355, and a malignancy is suspected. However, CT scans of the chest, abdomen, and pelvis show no evidence of malignancy, and an extensive gastrointestinal workup is unremarkable, including anti-smooth muscle antibodies, anti-liver-kidney microsomal antibody, antimitochondrial antibodies, gliadin antibody, alpha-1 antitrypsin, liver/kidney microsomal antibody, and hepatitis serologies. While on the neurology service, risperidone and donepezil are discontinued because
After discontinuing these medications, she is evaluated by the psychiatry consult team for mood lability. The psychiatry consult team recommends quetiapine, which is later started at 25 mg nightly at bedtime.
Clinically, Ms. D’s mental status continues to deteriorate. She becomes nonverbal and minimally able to follow commands. She is ultimately discharged to an inpatient hospice for end-of-life care and the team recommends that she continue with quetiapine once there.
Continue to: The authors' observations
The authors’ observations
CJD is a rare, rapidly progressive, fatal form of dementia. In the United States, the incidence is approximately 1 to 1.5 cases per 1 million people each year.2 There are various forms of the disease. Sporadic CJD is the most common, representing 85% of cases.3 Sporadic CJD typically occurs in patients in their 60s and quickly leads to death—50% of patients die within 5 months, and 90% of patients die within 1 year.2,3 The illness is hypothesized to arise from the production of misfolded prion proteins, ultimately leading to vacuolation, neuronal loss, and the spongiform appearance characteristic of CJD.3,4
Psychiatric symptoms have long been acknowledged as a feature of CJD. Recent data indicates that psychiatric symptoms occur in 90% to 92% of cases.5,6 Sleep disturbances and depressive symptoms, including vegetative symptoms, anhedonia, and tearfulness, appear to be most common.5 Psychotic symptoms occur in approximately 42% of cases and may include persecutory and paranoid delusions, as well as an array of vivid auditory, visual, and tactile hallucinations.5,7
There is also evidence that psychiatric symptoms may be an early marker of CJD.5,8 A Mayo Clinic study found that psychiatric symptoms occurred within the prodromal phase of CJD in 26% of cases, and psychiatric symptoms occurred within the first 100 days of illness in 86% of cases.5
Case reports have described patients with CJD who initially presented with depression, psychosis, and other psychiatric symptoms.9-11 Interestingly, there have been cases with only psychiatric symptoms, and no neurologic symptoms until relatively late in the illness.10,11 Several patients with CJD have been evaluated in psychiatric ERs, admitted to psychiatric hospitals, and treated with psychiatric medications and ECT.5,9 In one study, 44% of CJD cases were misdiagnosed as “psychiatric patients” due to the prominence of their psychiatric symptomatology.8
Continue to: Making the diagnosis in psychiatric settings
Making the diagnosis in psychiatric settings. Often, the most difficult aspect of CJD is making the diagnosis.3,12 Sporadic CJD in particular can vary widely in its clinical presentation.3 The core clinical feature of CJD is rapidly progressive dementia, so suspect CJD in these patients. However, CJD can be difficult to distinguish from other rapidly progressive dementias, such as autoimmune and paraneoplastic encephalopathies.2,3 The presence of neurologic features, specifically myoclonus, akinetic mutism, and visual, cerebellar, and extrapyramidal symptoms, should also be considered a red flag for the disorder3 (Table).
Finally, positive findings on MRI, EEG, or CSF assay can indicate a probable diagnosis of CJD.13 MRI, particularly diffusion weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR), is recognized as the most studied, sensitive, and overall useful neuroimaging modality for detecting CJD.2,3,12 Although the appearance of CJD on MRI can vary widely, asymmetric hyperintensities in ≥3 cortical gyri, particularly in the frontal and parietal lobes, provide strong evidence of CJD and are observed in 80% to 81% of cases.4,12 Asymmetric hyperintensities in the basal ganglia, particularly the caudate and rostral putamen, are observed in 69% to 70% of cases.4,12,13
EEG and CSF assay also can be useful for making the diagnosis. While diffuse slowing and frontal rhythmic delta activity appear early in the course of CJD, periodic sharp wave complexes emerge later in the illness.4 However, EEG findings are not diagnostic, because periodic sharp wave complexes are seen in only two-thirds of CJD cases and also occur in other neurologic illnesses.3,4 In recent years, lumbar puncture with subsequent CSF testing has become increasingly useful in detecting the illness. The presence of the 14-3-3 protein and tau protein is highly sensitive, although not specific, for CJD.3 A definite diagnosis of CJD requires discovery of the misfolded prion proteins, such as by RT-QuIC or brain biopsy.2,3,13
Management of CJD in psychiatric patients. CJD is an invariably fatal disease for which there is no effective cure or disease modifying treatment.2 Therefore, supportive therapies are the mainstay of care. Psychotropic medications can be used to provide symptom relief. While the sleep disturbances, anxiety, and agitation/hallucinations associated with CJD appear to respond well to hypnotic, anxiolytic, and antipsychotic medications, respectively, antidepressants and mood-stabilizing medications appear to have little benefit for patients with CJD.5 During the final stages of the disease, patients may suffer from akinetic mutism and inability to swallow, which often leads to aspiration pneumonia.14 Patients should also be offered end-of-life counseling, planning, and care, and provided with other comfort measures wherever possible (Figure).
Continue to: Bottom Line
Bottom Line
Patients with Creutzfeldt-Jakob disease (CJD) may present to psychiatric settings, particularly to a psychiatric emergency room. Consider CJD as a possible etiology in patients with rapidly progressive dementia, depression, and psychosis. CJD is invariably fatal and there is no effective disease-modifying treatment. Supportive therapies are the mainstay of care.
Related Resources
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Centers for Disease Control and Prevention. Creutzfeldt-Jakob disease, classic (CJD). http://www.cdc.gov/prions/cjd.
Drug Brand Names
Donepezil • Aricept
Risperidone • Risperdal
Quetiapine • Seroquel
CASE A rapid decline
Ms. D, age 62, presents to a psychiatric emergency room (ER) after experiencing visual hallucinations, exhibiting odd behaviors, and having memory problems. On interview, she is disoriented, distractible, tearful, and tangential. She plays with her shirt and glasses, and occasionally shouts. She perseverates on “the aerialists,” acrobatic children she has been seeing in her apartment. She becomes distressed and shouts, “I would love to just get them!”
Ms. D is unable to provide an account of her history. Collateral information is obtained from her daughter, who has brought Ms. D to the ER for evaluation. She reports that her mother has no relevant medical or psychiatric history, and does not take any medications, except a mixture of Chinese herbs that she brews into a tea.
Ms. D’s daughter says that her mother began to deteriorate 5 months ago, after she traveled to California to care for her sister, who was seriously ill and passed away. After Ms. D returned, she would cry frequently. She also appeared “spaced out,” complained of feeling dizzy, and frequently misplaced belongings. Three months before presenting to the ER, she began to experience weakness, fatigue, and difficulty walking. Her daughter became more worried 2 months ago, when Ms. D began sleeping with her purse and hiding her belongings around their house. When asked about these odd behaviors, Ms. D claimed that “the aerialists” were climbing through her windows at night and stealing her things.
A week before seeking treatment at the ER, Ms. D’s daughter had taken her to a neurologist at another facility for clinical evaluation. An MRI of the brain showed minimal dilation in the subarachnoid space and a focal 1 cm lipoma in the anterior falx cerebri, but was otherwise unremarkable. However, Ms. D’s symptoms continued to worsen, and began to interfere with her ability to care for herself.
The team in the psychiatric ER attributes Ms. D’s symptoms to a severe, psychotic depressive episode. They admit her to the psychiatric inpatient unit for further evaluation.
[polldaddy:10012742]
Continue to: The authors' observations
The authors’ observations
Ms. D was plagued by several mood and psychotic symptoms. Such symptoms can arise from many different psychiatric or organic etiologies. In Ms. D’s case, several aspects of her presentation suggest that her illness was psychiatric. The severe illness of a beloved family member is a significant stressor that could cause a great deal of grief and devastation, possibly leading to depression. Indeed, Ms. D’s daughter noticed that her mother was crying frequently, which is consistent with grief or depression.
Memory problems, which might manifest as misplacing belongings, can also indicate a depressive illness, especially in older patients. Moreover, impaired concentration, which can cause one to appear “spaced out” or distractible, is a core symptom of major depressive disorder. Sadness and grief also can be appropriate during bereavement and in response to significant losses. Therefore, in Ms. D’s case, it is possible her frequent crying, “spaced out” appearance, and other mood symptoms she experienced immediately after caring for her sister were an appropriate response to her sister’s illness and death.
However, other aspects of Ms. D’s presentation suggested an organic etiology. Her rapid deterioration and symptom onset relatively late in life were consistent with dementia and malignancy. Her complaint of feeling dizzy suggested a neurologic process was affecting her vestibular system. Finally, while psychiatric disorders can certainly cause visual hallucinations, they occur in only a small percentage of cases.1 Visual hallucinations are commonly associated with delirium, intoxication, and neurologic illness.
Continue to: EVALUATION Severe impairment
EVALUATION Severe impairment
On the psychiatric inpatient unit, Ms. D remains unable to give a coherent account of her illness or recent events. During interviews, she abruptly shifts from laughing to crying for no apparent reason. While answering questions, her responses trail off and she appears to forget what she had been saying. However, she continues to speak at length about “the aerialists,” stating that she sees them living in her wardrobe and jumping from rooftop to rooftop in her neighborhood.
A mental status examination finds evidence of severe cognitive impairment. Ms. D is unable to identify the correct date, time, or place, and appears surprised when told she is in a hospital. She can repeat the names of 3 objects but cannot recall them a few minutes later. Finally, she scores a 14 on the Mini-Mental State Examination (MMSE) and a 5 on the Montreal Cognitive Assessment (MoCA), indicating severe impairment.
On the unit, Ms. D cannot remember the location of her room or bathroom, and even when given directions, she needs to be escorted to her destination. Her gait is unsteady and wide-spaced, and she walks on her toes at times. When food is placed before her, she needs to be shown how to take the lids off containers, pick up utensils, and start eating.
All laboratory results are unremarkable, including a complete blood count, basic metabolic panel, liver function tests, gamma-glutamyl transpeptidase, magnesium, phosphate, thyroid-stimulating hormone, vitamin B12, methylmalonic acid, homocysteine, folate, erythrocyte sedimentation rate, C-reactive protein, antinuclear antibodies, rapid plasma reagin, human immunodeficiency virus, and Lyme titers. The team also considers Ms. D’s history of herbal medicine use, because herbal mixtures can contain heavy metals and other contaminants. However, all toxicology results are normal, including arsenic, mercury, lead, copper, and zinc.
To address her symptoms, Ms. D is given risperidone, 0.5 mg twice a day, and donepezil, 5 mg/d.
[polldaddy:10012743]
Continue to: The authors' observations
The authors’ observations
Despite her persistent psychiatric symptoms, Ms. D had several neurologic symptoms that warranted further investigation. Her abrupt shifts from laughter to tears for no apparent reason were consistent with pseudobulbar affect. Her inability to remember how to use utensils during meals was consistent with apraxia. Finally, her abnormal gait raised concern for a process affecting her motor system.
OUTCOME A rare disorder
Given the psychiatry team’s suspicions for a neurologic etiology of Ms. D’s symptoms, an MRI of her brain is repeated. The results are notable for abnormal restricted diffusion in the caudate and putamen bilaterally, which is consistent with Creutzfeldt-Jakob disease (CJD). EEG shows moderate diffuse cerebral dysfunction, frontal intermittent delta activity, and diffuse cortical hyperexcitability, consistent with early- to mid-onset prion disease. Upon evaluation by the neurology team, Ms. D appears fearful, suspicious, and disorganized, but her examination does not reveal additional significant sensorimotor findings.
Ms. D is transferred to the neurology service for further workup and management. A lumbar puncture is positive for real-time quaking-induced conversion (RT-QuIC) and 14-3-3 protein with elevated tau proteins; these findings also are consistent with CJD. She develops transaminitis, with an alanine transaminase (ALT) of 127 and aspartate transaminase (AST) of 355, and a malignancy is suspected. However, CT scans of the chest, abdomen, and pelvis show no evidence of malignancy, and an extensive gastrointestinal workup is unremarkable, including anti-smooth muscle antibodies, anti-liver-kidney microsomal antibody, antimitochondrial antibodies, gliadin antibody, alpha-1 antitrypsin, liver/kidney microsomal antibody, and hepatitis serologies. While on the neurology service, risperidone and donepezil are discontinued because
After discontinuing these medications, she is evaluated by the psychiatry consult team for mood lability. The psychiatry consult team recommends quetiapine, which is later started at 25 mg nightly at bedtime.
Clinically, Ms. D’s mental status continues to deteriorate. She becomes nonverbal and minimally able to follow commands. She is ultimately discharged to an inpatient hospice for end-of-life care and the team recommends that she continue with quetiapine once there.
Continue to: The authors' observations
The authors’ observations
CJD is a rare, rapidly progressive, fatal form of dementia. In the United States, the incidence is approximately 1 to 1.5 cases per 1 million people each year.2 There are various forms of the disease. Sporadic CJD is the most common, representing 85% of cases.3 Sporadic CJD typically occurs in patients in their 60s and quickly leads to death—50% of patients die within 5 months, and 90% of patients die within 1 year.2,3 The illness is hypothesized to arise from the production of misfolded prion proteins, ultimately leading to vacuolation, neuronal loss, and the spongiform appearance characteristic of CJD.3,4
Psychiatric symptoms have long been acknowledged as a feature of CJD. Recent data indicates that psychiatric symptoms occur in 90% to 92% of cases.5,6 Sleep disturbances and depressive symptoms, including vegetative symptoms, anhedonia, and tearfulness, appear to be most common.5 Psychotic symptoms occur in approximately 42% of cases and may include persecutory and paranoid delusions, as well as an array of vivid auditory, visual, and tactile hallucinations.5,7
There is also evidence that psychiatric symptoms may be an early marker of CJD.5,8 A Mayo Clinic study found that psychiatric symptoms occurred within the prodromal phase of CJD in 26% of cases, and psychiatric symptoms occurred within the first 100 days of illness in 86% of cases.5
Case reports have described patients with CJD who initially presented with depression, psychosis, and other psychiatric symptoms.9-11 Interestingly, there have been cases with only psychiatric symptoms, and no neurologic symptoms until relatively late in the illness.10,11 Several patients with CJD have been evaluated in psychiatric ERs, admitted to psychiatric hospitals, and treated with psychiatric medications and ECT.5,9 In one study, 44% of CJD cases were misdiagnosed as “psychiatric patients” due to the prominence of their psychiatric symptomatology.8
Continue to: Making the diagnosis in psychiatric settings
Making the diagnosis in psychiatric settings. Often, the most difficult aspect of CJD is making the diagnosis.3,12 Sporadic CJD in particular can vary widely in its clinical presentation.3 The core clinical feature of CJD is rapidly progressive dementia, so suspect CJD in these patients. However, CJD can be difficult to distinguish from other rapidly progressive dementias, such as autoimmune and paraneoplastic encephalopathies.2,3 The presence of neurologic features, specifically myoclonus, akinetic mutism, and visual, cerebellar, and extrapyramidal symptoms, should also be considered a red flag for the disorder3 (Table).
Finally, positive findings on MRI, EEG, or CSF assay can indicate a probable diagnosis of CJD.13 MRI, particularly diffusion weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR), is recognized as the most studied, sensitive, and overall useful neuroimaging modality for detecting CJD.2,3,12 Although the appearance of CJD on MRI can vary widely, asymmetric hyperintensities in ≥3 cortical gyri, particularly in the frontal and parietal lobes, provide strong evidence of CJD and are observed in 80% to 81% of cases.4,12 Asymmetric hyperintensities in the basal ganglia, particularly the caudate and rostral putamen, are observed in 69% to 70% of cases.4,12,13
EEG and CSF assay also can be useful for making the diagnosis. While diffuse slowing and frontal rhythmic delta activity appear early in the course of CJD, periodic sharp wave complexes emerge later in the illness.4 However, EEG findings are not diagnostic, because periodic sharp wave complexes are seen in only two-thirds of CJD cases and also occur in other neurologic illnesses.3,4 In recent years, lumbar puncture with subsequent CSF testing has become increasingly useful in detecting the illness. The presence of the 14-3-3 protein and tau protein is highly sensitive, although not specific, for CJD.3 A definite diagnosis of CJD requires discovery of the misfolded prion proteins, such as by RT-QuIC or brain biopsy.2,3,13
Management of CJD in psychiatric patients. CJD is an invariably fatal disease for which there is no effective cure or disease modifying treatment.2 Therefore, supportive therapies are the mainstay of care. Psychotropic medications can be used to provide symptom relief. While the sleep disturbances, anxiety, and agitation/hallucinations associated with CJD appear to respond well to hypnotic, anxiolytic, and antipsychotic medications, respectively, antidepressants and mood-stabilizing medications appear to have little benefit for patients with CJD.5 During the final stages of the disease, patients may suffer from akinetic mutism and inability to swallow, which often leads to aspiration pneumonia.14 Patients should also be offered end-of-life counseling, planning, and care, and provided with other comfort measures wherever possible (Figure).
Continue to: Bottom Line
Bottom Line
Patients with Creutzfeldt-Jakob disease (CJD) may present to psychiatric settings, particularly to a psychiatric emergency room. Consider CJD as a possible etiology in patients with rapidly progressive dementia, depression, and psychosis. CJD is invariably fatal and there is no effective disease-modifying treatment. Supportive therapies are the mainstay of care.
Related Resources
- National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease fact sheet. http://www.ninds.nih.gov/disorders/cjd/detail_cjd.htm.
- Centers for Disease Control and Prevention. Creutzfeldt-Jakob disease, classic (CJD). http://www.cdc.gov/prions/cjd.
Drug Brand Names
Donepezil • Aricept
Risperidone • Risperdal
Quetiapine • Seroquel
1. Resnick PJ. The detection of malingered psychosis. Psychiatr Clin North Am. 1999;22(1):159-172.
2. Bucelli RC, Ances BM. Diagnosis and evaluation of a patient with rapidly progressive dementia. Mo Med. 2013;110(5):422-428.
3. Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2.
4. Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-628.
5. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17(4):489-495.
6. Krasnianski A, Bohling GT, Harden M, et al. Psychiatric symptoms in patients with sporadic Creutzfeldt-Jakob disease in Germany. J Clin Psychiatry. 2015;76(9):1209-1215.
7. Javed Q, Alam F, Krishna S, et al. An unusual case of sporadic Creutzfeldt-Jakob disease (CJD). BMJ Case Rep. 2010;pii: bcr1220092576. doi:10.1136/bcr.12.2009.2576.
8. Abudy A, Juven-Wetzler A, Zohar J. The different faces of Creutzfeldt-Jacob disease CJD in psychiatry. Gen Hosp Psychiatry. 2014;36(3):245-248.
9. Jiang TT, Moses H, Gordon H, et al. Sporadic Creutzfeldt-Jakob disease presenting as major depression. South Med J. 1999;92(8):807-808.
10. Ali R, Baborie A, Larner AJ et al. Psychiatric presentation of sporadic Creutzfeldt-Jakob disease: a challenge to current diagnostic criteria. J Neuropsychiatry Clin Neurosci. 2013;25(4):335-338.
11. Gençer AG, Pelin Z, Küçükali CI., et al. Creutzfeldt-Jakob disease. Psychogeriatrics. 2011;11(2):119-124.
12. Caobelli F, Cobelli M, Pizzocaro C, et al. The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literature. J Neuroimaging. 2015;25(1):2-13.
13. Centers for Disease Control and Prevention. CDC's diagnostic criteria for Creutzfeldt-Jakob disease, 2010. http://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Updated February 11, 2015. Accessed August 2, 2016.
14. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease. Curr Psychiatry Rep. 2003;5(1):43-46.
1. Resnick PJ. The detection of malingered psychosis. Psychiatr Clin North Am. 1999;22(1):159-172.
2. Bucelli RC, Ances BM. Diagnosis and evaluation of a patient with rapidly progressive dementia. Mo Med. 2013;110(5):422-428.
3. Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2.
4. Puoti G, Bizzi A, Forloni G, et al. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618-628.
5. Wall CA, Rummans TA, Aksamit AJ, et al. Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci. 2005;17(4):489-495.
6. Krasnianski A, Bohling GT, Harden M, et al. Psychiatric symptoms in patients with sporadic Creutzfeldt-Jakob disease in Germany. J Clin Psychiatry. 2015;76(9):1209-1215.
7. Javed Q, Alam F, Krishna S, et al. An unusual case of sporadic Creutzfeldt-Jakob disease (CJD). BMJ Case Rep. 2010;pii: bcr1220092576. doi:10.1136/bcr.12.2009.2576.
8. Abudy A, Juven-Wetzler A, Zohar J. The different faces of Creutzfeldt-Jacob disease CJD in psychiatry. Gen Hosp Psychiatry. 2014;36(3):245-248.
9. Jiang TT, Moses H, Gordon H, et al. Sporadic Creutzfeldt-Jakob disease presenting as major depression. South Med J. 1999;92(8):807-808.
10. Ali R, Baborie A, Larner AJ et al. Psychiatric presentation of sporadic Creutzfeldt-Jakob disease: a challenge to current diagnostic criteria. J Neuropsychiatry Clin Neurosci. 2013;25(4):335-338.
11. Gençer AG, Pelin Z, Küçükali CI., et al. Creutzfeldt-Jakob disease. Psychogeriatrics. 2011;11(2):119-124.
12. Caobelli F, Cobelli M, Pizzocaro C, et al. The role of neuroimaging in evaluating patients affected by Creutzfeldt-Jakob disease: a systematic review of the literature. J Neuroimaging. 2015;25(1):2-13.
13. Centers for Disease Control and Prevention. CDC's diagnostic criteria for Creutzfeldt-Jakob disease, 2010. http://www.cdc.gov/prions/cjd/diagnostic-criteria.html. Updated February 11, 2015. Accessed August 2, 2016.
14. Martindale JL, Geschwind MD, Miller BL. Psychiatric and neuroimaging findings in Creutzfeldt-Jakob disease. Curr Psychiatry Rep. 2003;5(1):43-46.
Small-Cell Lung Cancer
From the Karmanos Cancer Institute, Detroit, MI (Dr. Mamdani) and the Indiana University School of Medicine, Indianapolis, IN (Dr. Jalal).
Abstract
- Objective: To review the clinical aspects and current practices of management of small cell lung cancer (SCLC).
- Methods: Review of the literature.
- Results: SCLC is an aggressive cancer of neuroendocrine origin with a very strong association with smoking. Approximately 25% of patients present with limited-stage disease while the remaining majority of patients have extensive-stage disease, defined as disease extending beyond one hemithorax at the time of diagnosis. SCLC is often associated with endocrine or neurologic paraneoplastic syndromes. The treatment of limited-stage disease consists of platinum-based chemotherapy administered concurrently with radiation. Patients with partial or complete response should be offered prophylactic cranial radiation (PCI). Extensive-stage disease is largely treated with platinum-based chemotherapy and the role of PCI is more controversial. The efficacy of second-line chemotherapy after disease progression on platinum based chemotherapy is limited.
- Conclusion: Despite a number of advances in the treatment of various malignancies over the period of past several years, the prognosis of patients with SCLC remains poor. There have been a number of clinical trials utilizing novel therapeutic agents to improve outcomes of these patients; however, few of them have shown marginal success in a very select subgroup of patients.
Key words: lung cancer; small-cell lung cancer.
Small-cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin, accounting for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually [1]. The incidence of SCLC in the United States has steadily declined over the past 30 years presumably because of decrease in the percentage of smokers and change to low-tar filter cigarettes [2]. Although the incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1 [3]. Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with complex genomic landscape consisting of thousands of mutations [4,5]. Despite a number of advances in the treatment of non-small cell lung cancer over the past decade, the therapeutic landscape of SCLC remains narrow with median overall survival (OS) of 9 months in patients with advanced disease.
Case Study
Initial Presentation
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the period of past 6 weeks. He also reports having had 20-lb weight loss over the same period of time. He is a current smoker and has been smoking one pack of cigarettes per day since the age of 18 years. A chest x-ray performed in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5 cm right hilar mass with presence of enlarged mediastinal lymph nodes bilaterally.
What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastases. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the VA staging system, limited-stage disease is defined as tumor that is confined to one hemithorax and can be encompassed within one radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Extensive-stage disease is the presentation in 75% of the patients where the disease extends beyond one hemithorax. Extensive-stage disease includes presence of malignant pleural effusion and/or distant metastasis [6]. The Veterans Administration Lung Study Group (VALG) classification and staging system is more commonly used compared to the AJCC TNM staging system since it is less complex, directs treatment decisions, and correlates closely with prognosis. Given its propensity to metastasize quickly, none of the currently available screening methods have proven to be successful in early detection of SCLC. Eighty-six percent of the 125 patients that were diagnosed with SCLC while undergoing annual low-dose chest CT scans on National Lung Cancer Screening Trial had advanced disease at diagnosis [7,8]. These results highlight the fact that he majority of the SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. In addition, SCLC usually presents with centrally located tumors and bulky mediastinal adenopathy. Common symptoms include shortness of breath and cough. SCLC is commonly located submucosally in the bronchus and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava (SVC) syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes (PNS) owing to the ectopic secretion of hormones or antibodies by the tumor cells. The PNS can be broadly categorized into endocrine and neurologic; and are summarized in Table 1. 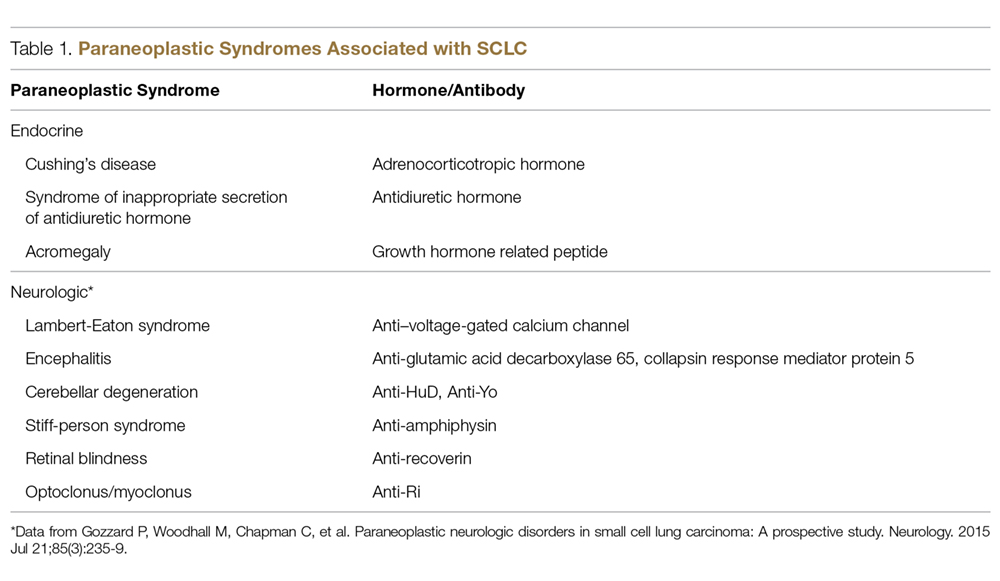
The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using the PET-CT. Therefore, brain imaging with contrast enhanced CT or MRI is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy is warranted in case of unexplained cytopenias, especially when associated with teardrop red cells or nucleated red cells on peripheral blood smear indicative of marrow infiltrative process. The tissue diagnosis is established by obtaining a biopsy of the primary tumor or one of the metastatic sites. In case of localized disease, bronchoscopy (if necessary, with endobronchial ultrasound) with biopsy of centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nucleus:cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry. Very frequently the cells are also positive for TTF1. Although serum tumor markers, including neuron-specific enolase (NSE) and progastrin-releasing peptide (prGRP), are frequently elevated in patients with SCLC, they are of limited value in clinical practice owing to their lack of sensitivity and specificity.
Case Continued
The patient underwent FDG-PET scan that showed the presence of hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There were no other areas of FDG avidity. His brain MRI did not show any evidence of brain metastasis. Thus, he was confirmed to have limited-stage SCLC.
What is the standard of care for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care for the treatment of limited stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1). 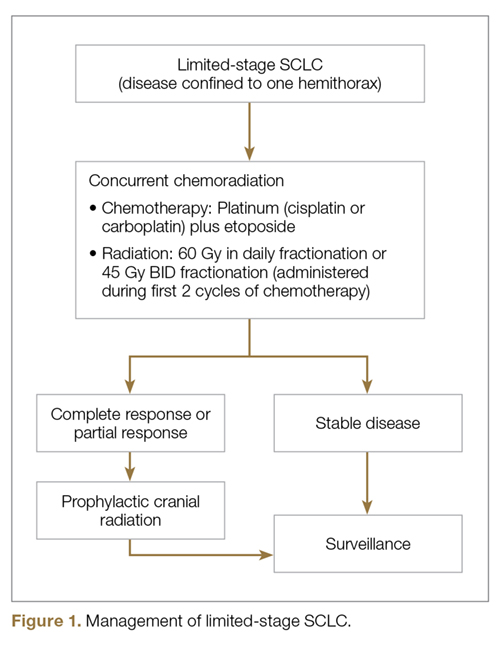
Choice of Chemotherapy
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen [9]. This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile [10–12]. Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive stage SCLC [13–15]. A meta-analysis of 4 randomized trials, including 663 patients with SCLC, comparing cisplatin-based versus carboplatin-based regimens where 32% of patients had limited stage disease and 68% had extensive stage disease showed no statistically significant difference in the response rate, progression free survival (PFS), or OS between the two regimens [16]. Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the two regimens is different. Cisplatin based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy induced nausea/vomiting [13], while carboplatin-based regimens are more myelosuppressive [17]. In addition, the combination of thoracic radiation with either of these regiments is associated with higher risk of esophagitis, pneumonitis, and myelosuppression [18]. The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation [19]. Of note, intravenous (IV) etoposide is always preferred over oral etoposide, especially in curative setting given unreliable absorption and bioavailability of oral formulations.
Thoracic Radiation
The addition of thoracic radiation to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown 25% to 30% decrease in local failure and 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited stage SCLC [20,21]. Early (with the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS [18,22,23]. The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The ECOG/RTOG randomized trial compared 45 Gy radiation delivered twice daily over a period of 3 weeks with once a day over 5 weeks, concurrently with chemotherapy. The twice a day regimen led to 10% improvement in 5-year OS (26% vs 16%), but higher incidence of grade 3 and 4 adverse events [24]. Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase III trial (CONVERT) compared 45 Gy twice daily with 66 Gy once daily radiation in this setting. This trial did not show any difference in OS. The patients in twice daily arm had higher incidence of grade 4 neutropenia [25]. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often hyperfractionated regimen is not feasible for the patients and many radiation oncology centers. Hopefully the CALBG 30610 study, which is ongoing, will clarify the optimal radiation schedule for limited-stage disease.
Prophylactic Cranial Irradiation
Approximately 75% of patients with limited-stage disease experience disease recurrence and brain is the site of recurrence in approximately half of these patients. Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiation delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases [26–28]. Although individual small studies have not shown survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS [27]. The majority of patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
Role of Surgery
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging [29,30]. Most of the data in this setting are derived from retrospective studies [31,32]. A 5-year OS of 40% to 60% has been has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if no nodal involvement) or chemoradiation (if nodal involvement) is recommended [33,34]. Wedge or segmental resections are not considered to be optimum surgical options.
Case Continued
The patient received 4 cycles of cisplatin and etoposide along with 70 Gy radiation concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans showed partial response (PR). The patient underwent PCI 6 weeks after completion of treatment. Eighteen months later, the patient comes to the clinic for routine follow-up. He is doing generally well except for mildly decreased appetite and unintentional loss of 5 lb weight. His CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2 cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirmed the adrenal and liver lesions to be hypermetabolic. In addition, the PET showed multiple FDG avid bone lesions throughout the spine. Brain MRI was negative for any brain metastasis.
What is the standard of care for extensive-stage SCLC?
For extensive-stage SCLC, chemotherapy is the mainstay of treatment, with the goals of treatment being prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option (Figure 2). 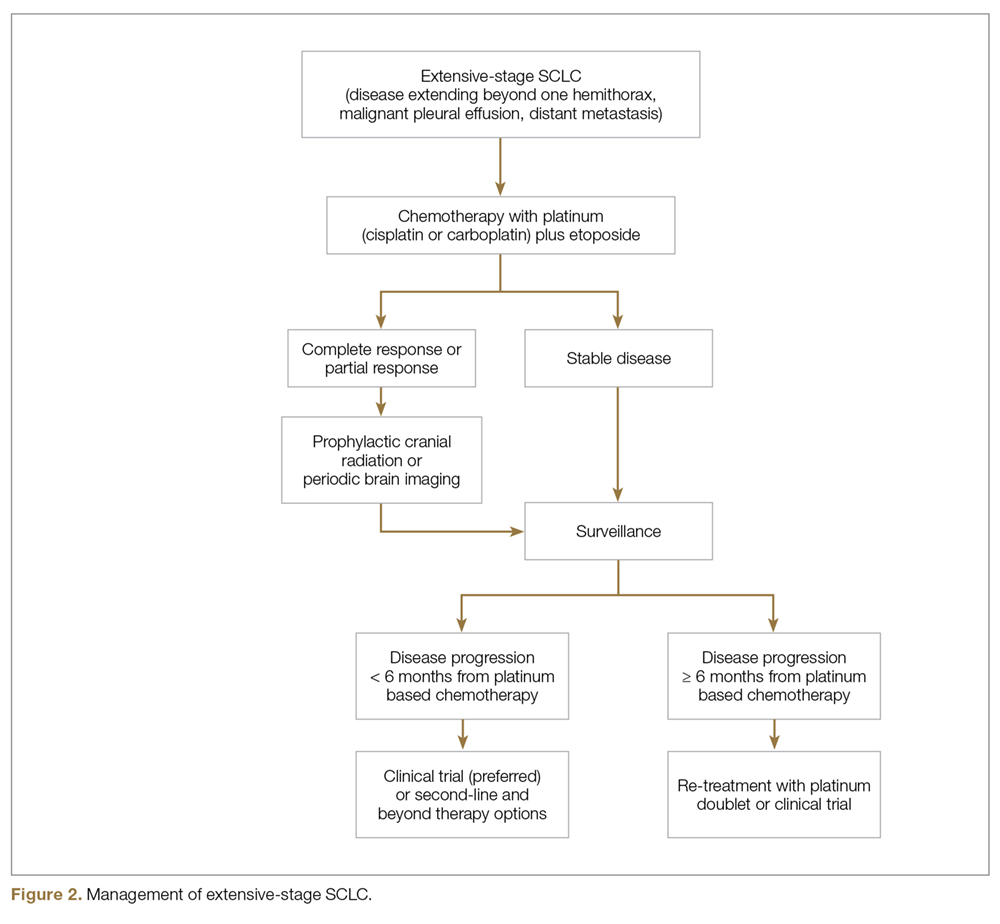
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity [39–42]. Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit [43–45]. The addition of antiangiogenic agent bevacizumab to standard platinum-based doublet has not yielded prolongation of OS in SCLC and led to unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease [46–51]. Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase III trial and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS [52,53].
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
Second-Line Therapy
Despite being exquisitely chemo-sensitive, SCLC is associated with very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse since first-line platinum based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If the disease progression occurs between 3 to 6 months since platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25% [54,55]. A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (cyclophosphamide, adriamycin, vincristine) (Table 2). 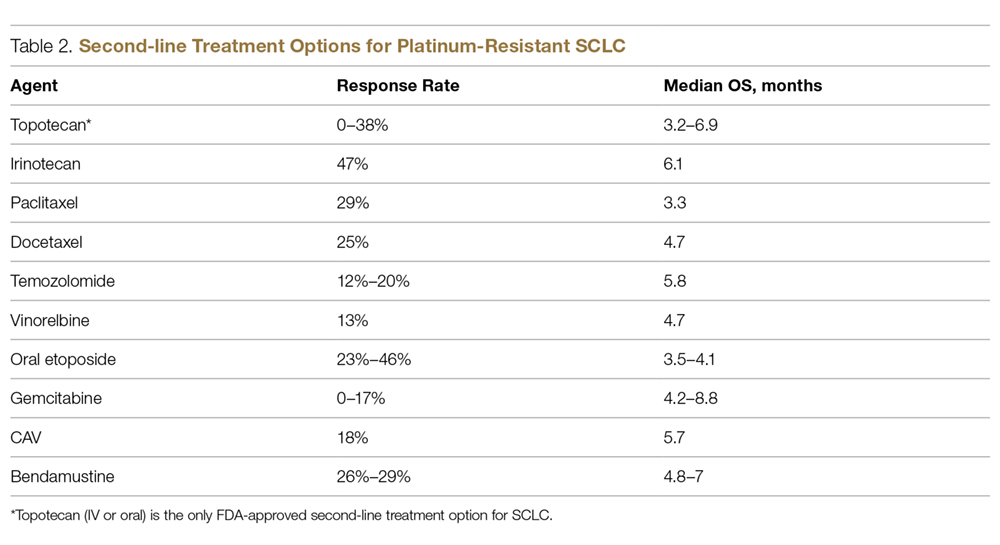
Immunotherapy
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving and currently there are no FDA-approved immunotherapy agents in SCLC. A recently conducted phase I/II trial (CheckMate 032) of anti-PD-1 antibody nivolumab with or without anti-CTLA-1 antibody ipilimumab in patients with relapsed SCLC reported a response rate of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg + ipilimumab 3 mg/kg. The 2-year OS was 26% with the combination and 14% with single agent nivolumab [56,57]. Only 18% of patients had PD-L1 expression of ≥ 1% and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20% and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the NCCN guidelines as one of the options for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy; however, it is questionable whether routine use of this combination is justified based on currently available data. However the evidence for the combination of nivolumab and ipilimumab remains limited. This efficacy and toxicity data of both randomized and nonrandomized cohorts were presented together making it hard to interpret the results.
Another phase Ib study (KEYNOTE-028) utilizing anti-PD-1 antibody pembrolizumab 10 mg/kg IV every 2 weeks in patients with relapsed SCLC after receiving one or more prior lines of therapy and PD-L1 expression of ≥ 1% showed a response rate of 33% with median duration of response of 19 months and 1-year OS of 38% [58]. Although only 28% of screened patients had PD-L1 expression of ≥ 1% , these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
Role of Prophylactic Cranial Irradiation
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase III trial conducted by EORTC comparing PCI with no PCI in patients with extensive-stage SCLC who had attained partial or complete response to initial platinum-based chemotherapy showed decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI. However, this trial did not require mandatory brain imaging prior to PCI and therefore it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups. A more recent phase III study conducted in Japan that compared PCI (25Gy in 10 fractions) with no PCI reported no difference in survival between the two groups. As opposed to EORTC study, the Japanese study did require baseline brain imaging to confirm absence of brain metastasis prior to enrollment. In addition, the patients in the control arm underwent periodic brain MRI to allow early detection of brain metastasis [59]. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven and PCI likely has a role in a highly select small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG PS 3–4) or underlying neurocognitive disorders [33,60]. NMDA receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction [61]. Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiation [61].
Role of Radiation
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiation after completion of platinum-based chemotherapy. A randomized trial including patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiation in combination with continued chemotherapy versus chemotherapy alone [62]. The median OS was longer with the addition of thoracic radiation compared to chemotherapy alone. Another phase III trial did not show improvement in 1-year OS with consolidative thoracic radiation, but 2-year OS and 6-month PFS were longer [63]. In general, consolidative thoracic radiation benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy [64]. In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiation should be utilized for palliative purposes in patients with painful bone metastasis, spine cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy with extensive stage disease. Whole brain radiotherapy is preferred over SRS because of frequent presence of micrometastasis even in the setting of one or two radiographically evident brain metastasis.
Novel Therapies
A very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and a high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major limitations to the development of targeted therapies in SCLC is limited availability of tissue owing to small tissue samples and frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been under investigation for their potential role in SCLC. Several of them, including EGFR TKIs, BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have been unsuccessful in showing a survival advantage in this disease. Several others including PARP inhibitors, cellular developmental pathway inhibitors and antibody drug conjugates are being tested. A phase I study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated complete response in 14.3%, partial response in 57.1%, and stable disease in 28.6% of patients with acceptable safety profile [65]. So far, none of these agents are approved for use in SCLC and the majority are in early phase clinical trials [66].
One of the emerging targets in the treatment of SCLC is DLL3. DLL3 is expressed on > 80% SCLCL tumor cells and cancer stem cells. Rovalpituzumab tesirine (ROVA-T) is an antibody drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase I trial of ROVA-T in patients with relapsed SCLC after 1 or 2 prior lines of therapies reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and mPFS were 4.6 months [67]. ROVA-T is currently in later phases of clinical trials and has a potential to serve as one of the options for patients with extensive-stage disease after disease progression on platinum-based therapy.
Response Assessment/Surveillance
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation. The surveillance guidelines consist of history, physical exam, and imaging every 3 months during 1st 2 years, every 6 months during the 3rdyear, and annually thereafter. If PCI is not performed, brain MRI or contrast enhanced CT scan should be performed every 3 to 4 months during the first 2 years of follow-up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the 1st year, every 3 to 4 months during year 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
Corresponding author: Hirva Mamdani, MD, Karmanos Cancer Institute, 4100 John R, Detroit, MI 48201, [email protected].
Financial disclosures: None.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed April 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed April 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
8. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
9. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
10. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
11. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
12. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
13. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12(7):666–73.
14. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
15. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
16. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
17. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
18. Takada M, Fukuoka M, Kawahara M, Sugiura T, Yokoyama A, Yokota S, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
19. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
20. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
21. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10(6):890–5.
22. Murray N, Coy P, Pater JL, Hodson I, Arnold A, Zee BC, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
23. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27(10):1818–28.
24. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340(4):265–71.
25. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18(8):1116–25.
26. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87(3):183–90.
27. Aupérin A, Arriagada R, Pignon JP, Le Pechoux C, Gregor A, Stephens RJ, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
28. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10(5):467–74.
29. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9(10):1132–9.
30. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
31. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
32. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
33. Yang CF, Chan DY, Speicher PJ, Gulack BC, Wang X, Hartwig MG, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
34. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
35. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
36. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
37. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
38. Zhou H, Zeng C, Wei Y, Zhou J, Yao W. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
39. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol 1995 Oct;13:2594–9.
40. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
41. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
42. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
43. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
44. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10(2):282–91.
45. Miles DW, Earl HM, Souhami RL, Harper PG, Rudd R, Ash CM, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
46. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
47. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4(12):1555–60.
48. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29(16):2215–22.
49. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27(35):6006–11.
50. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
51. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trialdagger. Ann Oncol 2015;26:908–14.
52. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts). J Clin Oncol 2017;35(15_suppl):8504-. [Can’t find the rest of the reference]
53. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
54. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
55. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
56. Hellmann MD, Ott PA, Zugazagoitia J, Ready NE, Hann CL, Braud FGD, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032. J Clin Oncol 2017;35(15_suppl):8503-. [Can’t find the rest of the reference]
57. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
58. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
59. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
60. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27(1):78–84.
61. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
62. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17(7):2092–9.
63. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
64. Slotman BJ, van Tinteren H, Praag JO, Knegjens JL, El Sharouni SY, Hatton M, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
65. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer. 2015 ;89:66–70.
66. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Translational lung cancer research. 2015;4:533–44.
67. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.
From the Karmanos Cancer Institute, Detroit, MI (Dr. Mamdani) and the Indiana University School of Medicine, Indianapolis, IN (Dr. Jalal).
Abstract
- Objective: To review the clinical aspects and current practices of management of small cell lung cancer (SCLC).
- Methods: Review of the literature.
- Results: SCLC is an aggressive cancer of neuroendocrine origin with a very strong association with smoking. Approximately 25% of patients present with limited-stage disease while the remaining majority of patients have extensive-stage disease, defined as disease extending beyond one hemithorax at the time of diagnosis. SCLC is often associated with endocrine or neurologic paraneoplastic syndromes. The treatment of limited-stage disease consists of platinum-based chemotherapy administered concurrently with radiation. Patients with partial or complete response should be offered prophylactic cranial radiation (PCI). Extensive-stage disease is largely treated with platinum-based chemotherapy and the role of PCI is more controversial. The efficacy of second-line chemotherapy after disease progression on platinum based chemotherapy is limited.
- Conclusion: Despite a number of advances in the treatment of various malignancies over the period of past several years, the prognosis of patients with SCLC remains poor. There have been a number of clinical trials utilizing novel therapeutic agents to improve outcomes of these patients; however, few of them have shown marginal success in a very select subgroup of patients.
Key words: lung cancer; small-cell lung cancer.
Small-cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin, accounting for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually [1]. The incidence of SCLC in the United States has steadily declined over the past 30 years presumably because of decrease in the percentage of smokers and change to low-tar filter cigarettes [2]. Although the incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1 [3]. Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with complex genomic landscape consisting of thousands of mutations [4,5]. Despite a number of advances in the treatment of non-small cell lung cancer over the past decade, the therapeutic landscape of SCLC remains narrow with median overall survival (OS) of 9 months in patients with advanced disease.
Case Study
Initial Presentation
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the period of past 6 weeks. He also reports having had 20-lb weight loss over the same period of time. He is a current smoker and has been smoking one pack of cigarettes per day since the age of 18 years. A chest x-ray performed in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5 cm right hilar mass with presence of enlarged mediastinal lymph nodes bilaterally.
What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastases. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the VA staging system, limited-stage disease is defined as tumor that is confined to one hemithorax and can be encompassed within one radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Extensive-stage disease is the presentation in 75% of the patients where the disease extends beyond one hemithorax. Extensive-stage disease includes presence of malignant pleural effusion and/or distant metastasis [6]. The Veterans Administration Lung Study Group (VALG) classification and staging system is more commonly used compared to the AJCC TNM staging system since it is less complex, directs treatment decisions, and correlates closely with prognosis. Given its propensity to metastasize quickly, none of the currently available screening methods have proven to be successful in early detection of SCLC. Eighty-six percent of the 125 patients that were diagnosed with SCLC while undergoing annual low-dose chest CT scans on National Lung Cancer Screening Trial had advanced disease at diagnosis [7,8]. These results highlight the fact that he majority of the SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. In addition, SCLC usually presents with centrally located tumors and bulky mediastinal adenopathy. Common symptoms include shortness of breath and cough. SCLC is commonly located submucosally in the bronchus and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava (SVC) syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes (PNS) owing to the ectopic secretion of hormones or antibodies by the tumor cells. The PNS can be broadly categorized into endocrine and neurologic; and are summarized in Table 1.
The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using the PET-CT. Therefore, brain imaging with contrast enhanced CT or MRI is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy is warranted in case of unexplained cytopenias, especially when associated with teardrop red cells or nucleated red cells on peripheral blood smear indicative of marrow infiltrative process. The tissue diagnosis is established by obtaining a biopsy of the primary tumor or one of the metastatic sites. In case of localized disease, bronchoscopy (if necessary, with endobronchial ultrasound) with biopsy of centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nucleus:cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry. Very frequently the cells are also positive for TTF1. Although serum tumor markers, including neuron-specific enolase (NSE) and progastrin-releasing peptide (prGRP), are frequently elevated in patients with SCLC, they are of limited value in clinical practice owing to their lack of sensitivity and specificity.
Case Continued
The patient underwent FDG-PET scan that showed the presence of hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There were no other areas of FDG avidity. His brain MRI did not show any evidence of brain metastasis. Thus, he was confirmed to have limited-stage SCLC.
What is the standard of care for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care for the treatment of limited stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1).
Choice of Chemotherapy
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen [9]. This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile [10–12]. Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive stage SCLC [13–15]. A meta-analysis of 4 randomized trials, including 663 patients with SCLC, comparing cisplatin-based versus carboplatin-based regimens where 32% of patients had limited stage disease and 68% had extensive stage disease showed no statistically significant difference in the response rate, progression free survival (PFS), or OS between the two regimens [16]. Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the two regimens is different. Cisplatin based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy induced nausea/vomiting [13], while carboplatin-based regimens are more myelosuppressive [17]. In addition, the combination of thoracic radiation with either of these regiments is associated with higher risk of esophagitis, pneumonitis, and myelosuppression [18]. The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation [19]. Of note, intravenous (IV) etoposide is always preferred over oral etoposide, especially in curative setting given unreliable absorption and bioavailability of oral formulations.
Thoracic Radiation
The addition of thoracic radiation to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown 25% to 30% decrease in local failure and 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited stage SCLC [20,21]. Early (with the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS [18,22,23]. The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The ECOG/RTOG randomized trial compared 45 Gy radiation delivered twice daily over a period of 3 weeks with once a day over 5 weeks, concurrently with chemotherapy. The twice a day regimen led to 10% improvement in 5-year OS (26% vs 16%), but higher incidence of grade 3 and 4 adverse events [24]. Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase III trial (CONVERT) compared 45 Gy twice daily with 66 Gy once daily radiation in this setting. This trial did not show any difference in OS. The patients in twice daily arm had higher incidence of grade 4 neutropenia [25]. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often hyperfractionated regimen is not feasible for the patients and many radiation oncology centers. Hopefully the CALBG 30610 study, which is ongoing, will clarify the optimal radiation schedule for limited-stage disease.
Prophylactic Cranial Irradiation
Approximately 75% of patients with limited-stage disease experience disease recurrence and brain is the site of recurrence in approximately half of these patients. Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiation delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases [26–28]. Although individual small studies have not shown survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS [27]. The majority of patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
Role of Surgery
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging [29,30]. Most of the data in this setting are derived from retrospective studies [31,32]. A 5-year OS of 40% to 60% has been has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if no nodal involvement) or chemoradiation (if nodal involvement) is recommended [33,34]. Wedge or segmental resections are not considered to be optimum surgical options.
Case Continued
The patient received 4 cycles of cisplatin and etoposide along with 70 Gy radiation concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans showed partial response (PR). The patient underwent PCI 6 weeks after completion of treatment. Eighteen months later, the patient comes to the clinic for routine follow-up. He is doing generally well except for mildly decreased appetite and unintentional loss of 5 lb weight. His CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2 cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirmed the adrenal and liver lesions to be hypermetabolic. In addition, the PET showed multiple FDG avid bone lesions throughout the spine. Brain MRI was negative for any brain metastasis.
What is the standard of care for extensive-stage SCLC?
For extensive-stage SCLC, chemotherapy is the mainstay of treatment, with the goals of treatment being prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option (Figure 2).
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity [39–42]. Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit [43–45]. The addition of antiangiogenic agent bevacizumab to standard platinum-based doublet has not yielded prolongation of OS in SCLC and led to unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease [46–51]. Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase III trial and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS [52,53].
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
Second-Line Therapy
Despite being exquisitely chemo-sensitive, SCLC is associated with very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse since first-line platinum based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If the disease progression occurs between 3 to 6 months since platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25% [54,55]. A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (cyclophosphamide, adriamycin, vincristine) (Table 2).
Immunotherapy
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving and currently there are no FDA-approved immunotherapy agents in SCLC. A recently conducted phase I/II trial (CheckMate 032) of anti-PD-1 antibody nivolumab with or without anti-CTLA-1 antibody ipilimumab in patients with relapsed SCLC reported a response rate of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg + ipilimumab 3 mg/kg. The 2-year OS was 26% with the combination and 14% with single agent nivolumab [56,57]. Only 18% of patients had PD-L1 expression of ≥ 1% and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20% and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the NCCN guidelines as one of the options for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy; however, it is questionable whether routine use of this combination is justified based on currently available data. However the evidence for the combination of nivolumab and ipilimumab remains limited. This efficacy and toxicity data of both randomized and nonrandomized cohorts were presented together making it hard to interpret the results.
Another phase Ib study (KEYNOTE-028) utilizing anti-PD-1 antibody pembrolizumab 10 mg/kg IV every 2 weeks in patients with relapsed SCLC after receiving one or more prior lines of therapy and PD-L1 expression of ≥ 1% showed a response rate of 33% with median duration of response of 19 months and 1-year OS of 38% [58]. Although only 28% of screened patients had PD-L1 expression of ≥ 1% , these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
Role of Prophylactic Cranial Irradiation
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase III trial conducted by EORTC comparing PCI with no PCI in patients with extensive-stage SCLC who had attained partial or complete response to initial platinum-based chemotherapy showed decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI. However, this trial did not require mandatory brain imaging prior to PCI and therefore it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups. A more recent phase III study conducted in Japan that compared PCI (25Gy in 10 fractions) with no PCI reported no difference in survival between the two groups. As opposed to EORTC study, the Japanese study did require baseline brain imaging to confirm absence of brain metastasis prior to enrollment. In addition, the patients in the control arm underwent periodic brain MRI to allow early detection of brain metastasis [59]. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven and PCI likely has a role in a highly select small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG PS 3–4) or underlying neurocognitive disorders [33,60]. NMDA receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction [61]. Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiation [61].
Role of Radiation
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiation after completion of platinum-based chemotherapy. A randomized trial including patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiation in combination with continued chemotherapy versus chemotherapy alone [62]. The median OS was longer with the addition of thoracic radiation compared to chemotherapy alone. Another phase III trial did not show improvement in 1-year OS with consolidative thoracic radiation, but 2-year OS and 6-month PFS were longer [63]. In general, consolidative thoracic radiation benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy [64]. In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiation should be utilized for palliative purposes in patients with painful bone metastasis, spine cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy with extensive stage disease. Whole brain radiotherapy is preferred over SRS because of frequent presence of micrometastasis even in the setting of one or two radiographically evident brain metastasis.
Novel Therapies
A very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and a high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major limitations to the development of targeted therapies in SCLC is limited availability of tissue owing to small tissue samples and frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been under investigation for their potential role in SCLC. Several of them, including EGFR TKIs, BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have been unsuccessful in showing a survival advantage in this disease. Several others including PARP inhibitors, cellular developmental pathway inhibitors and antibody drug conjugates are being tested. A phase I study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated complete response in 14.3%, partial response in 57.1%, and stable disease in 28.6% of patients with acceptable safety profile [65]. So far, none of these agents are approved for use in SCLC and the majority are in early phase clinical trials [66].
One of the emerging targets in the treatment of SCLC is DLL3. DLL3 is expressed on > 80% SCLCL tumor cells and cancer stem cells. Rovalpituzumab tesirine (ROVA-T) is an antibody drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase I trial of ROVA-T in patients with relapsed SCLC after 1 or 2 prior lines of therapies reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and mPFS were 4.6 months [67]. ROVA-T is currently in later phases of clinical trials and has a potential to serve as one of the options for patients with extensive-stage disease after disease progression on platinum-based therapy.
Response Assessment/Surveillance
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation. The surveillance guidelines consist of history, physical exam, and imaging every 3 months during 1st 2 years, every 6 months during the 3rdyear, and annually thereafter. If PCI is not performed, brain MRI or contrast enhanced CT scan should be performed every 3 to 4 months during the first 2 years of follow-up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the 1st year, every 3 to 4 months during year 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
Corresponding author: Hirva Mamdani, MD, Karmanos Cancer Institute, 4100 John R, Detroit, MI 48201, [email protected].
Financial disclosures: None.
From the Karmanos Cancer Institute, Detroit, MI (Dr. Mamdani) and the Indiana University School of Medicine, Indianapolis, IN (Dr. Jalal).
Abstract
- Objective: To review the clinical aspects and current practices of management of small cell lung cancer (SCLC).
- Methods: Review of the literature.
- Results: SCLC is an aggressive cancer of neuroendocrine origin with a very strong association with smoking. Approximately 25% of patients present with limited-stage disease while the remaining majority of patients have extensive-stage disease, defined as disease extending beyond one hemithorax at the time of diagnosis. SCLC is often associated with endocrine or neurologic paraneoplastic syndromes. The treatment of limited-stage disease consists of platinum-based chemotherapy administered concurrently with radiation. Patients with partial or complete response should be offered prophylactic cranial radiation (PCI). Extensive-stage disease is largely treated with platinum-based chemotherapy and the role of PCI is more controversial. The efficacy of second-line chemotherapy after disease progression on platinum based chemotherapy is limited.
- Conclusion: Despite a number of advances in the treatment of various malignancies over the period of past several years, the prognosis of patients with SCLC remains poor. There have been a number of clinical trials utilizing novel therapeutic agents to improve outcomes of these patients; however, few of them have shown marginal success in a very select subgroup of patients.
Key words: lung cancer; small-cell lung cancer.
Small-cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin, accounting for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually [1]. The incidence of SCLC in the United States has steadily declined over the past 30 years presumably because of decrease in the percentage of smokers and change to low-tar filter cigarettes [2]. Although the incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1 [3]. Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with complex genomic landscape consisting of thousands of mutations [4,5]. Despite a number of advances in the treatment of non-small cell lung cancer over the past decade, the therapeutic landscape of SCLC remains narrow with median overall survival (OS) of 9 months in patients with advanced disease.
Case Study
Initial Presentation
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the period of past 6 weeks. He also reports having had 20-lb weight loss over the same period of time. He is a current smoker and has been smoking one pack of cigarettes per day since the age of 18 years. A chest x-ray performed in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5 cm right hilar mass with presence of enlarged mediastinal lymph nodes bilaterally.
What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastases. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the VA staging system, limited-stage disease is defined as tumor that is confined to one hemithorax and can be encompassed within one radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Extensive-stage disease is the presentation in 75% of the patients where the disease extends beyond one hemithorax. Extensive-stage disease includes presence of malignant pleural effusion and/or distant metastasis [6]. The Veterans Administration Lung Study Group (VALG) classification and staging system is more commonly used compared to the AJCC TNM staging system since it is less complex, directs treatment decisions, and correlates closely with prognosis. Given its propensity to metastasize quickly, none of the currently available screening methods have proven to be successful in early detection of SCLC. Eighty-six percent of the 125 patients that were diagnosed with SCLC while undergoing annual low-dose chest CT scans on National Lung Cancer Screening Trial had advanced disease at diagnosis [7,8]. These results highlight the fact that he majority of the SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. In addition, SCLC usually presents with centrally located tumors and bulky mediastinal adenopathy. Common symptoms include shortness of breath and cough. SCLC is commonly located submucosally in the bronchus and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava (SVC) syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes (PNS) owing to the ectopic secretion of hormones or antibodies by the tumor cells. The PNS can be broadly categorized into endocrine and neurologic; and are summarized in Table 1.
The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using the PET-CT. Therefore, brain imaging with contrast enhanced CT or MRI is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy is warranted in case of unexplained cytopenias, especially when associated with teardrop red cells or nucleated red cells on peripheral blood smear indicative of marrow infiltrative process. The tissue diagnosis is established by obtaining a biopsy of the primary tumor or one of the metastatic sites. In case of localized disease, bronchoscopy (if necessary, with endobronchial ultrasound) with biopsy of centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nucleus:cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry. Very frequently the cells are also positive for TTF1. Although serum tumor markers, including neuron-specific enolase (NSE) and progastrin-releasing peptide (prGRP), are frequently elevated in patients with SCLC, they are of limited value in clinical practice owing to their lack of sensitivity and specificity.
Case Continued
The patient underwent FDG-PET scan that showed the presence of hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There were no other areas of FDG avidity. His brain MRI did not show any evidence of brain metastasis. Thus, he was confirmed to have limited-stage SCLC.
What is the standard of care for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care for the treatment of limited stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1).
Choice of Chemotherapy
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen [9]. This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile [10–12]. Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive stage SCLC [13–15]. A meta-analysis of 4 randomized trials, including 663 patients with SCLC, comparing cisplatin-based versus carboplatin-based regimens where 32% of patients had limited stage disease and 68% had extensive stage disease showed no statistically significant difference in the response rate, progression free survival (PFS), or OS between the two regimens [16]. Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the two regimens is different. Cisplatin based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy induced nausea/vomiting [13], while carboplatin-based regimens are more myelosuppressive [17]. In addition, the combination of thoracic radiation with either of these regiments is associated with higher risk of esophagitis, pneumonitis, and myelosuppression [18]. The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation [19]. Of note, intravenous (IV) etoposide is always preferred over oral etoposide, especially in curative setting given unreliable absorption and bioavailability of oral formulations.
Thoracic Radiation
The addition of thoracic radiation to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown 25% to 30% decrease in local failure and 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited stage SCLC [20,21]. Early (with the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS [18,22,23]. The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The ECOG/RTOG randomized trial compared 45 Gy radiation delivered twice daily over a period of 3 weeks with once a day over 5 weeks, concurrently with chemotherapy. The twice a day regimen led to 10% improvement in 5-year OS (26% vs 16%), but higher incidence of grade 3 and 4 adverse events [24]. Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase III trial (CONVERT) compared 45 Gy twice daily with 66 Gy once daily radiation in this setting. This trial did not show any difference in OS. The patients in twice daily arm had higher incidence of grade 4 neutropenia [25]. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often hyperfractionated regimen is not feasible for the patients and many radiation oncology centers. Hopefully the CALBG 30610 study, which is ongoing, will clarify the optimal radiation schedule for limited-stage disease.
Prophylactic Cranial Irradiation
Approximately 75% of patients with limited-stage disease experience disease recurrence and brain is the site of recurrence in approximately half of these patients. Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiation delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases [26–28]. Although individual small studies have not shown survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS [27]. The majority of patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
Role of Surgery
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging [29,30]. Most of the data in this setting are derived from retrospective studies [31,32]. A 5-year OS of 40% to 60% has been has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if no nodal involvement) or chemoradiation (if nodal involvement) is recommended [33,34]. Wedge or segmental resections are not considered to be optimum surgical options.
Case Continued
The patient received 4 cycles of cisplatin and etoposide along with 70 Gy radiation concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans showed partial response (PR). The patient underwent PCI 6 weeks after completion of treatment. Eighteen months later, the patient comes to the clinic for routine follow-up. He is doing generally well except for mildly decreased appetite and unintentional loss of 5 lb weight. His CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2 cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirmed the adrenal and liver lesions to be hypermetabolic. In addition, the PET showed multiple FDG avid bone lesions throughout the spine. Brain MRI was negative for any brain metastasis.
What is the standard of care for extensive-stage SCLC?
For extensive-stage SCLC, chemotherapy is the mainstay of treatment, with the goals of treatment being prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option (Figure 2).
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity [39–42]. Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit [43–45]. The addition of antiangiogenic agent bevacizumab to standard platinum-based doublet has not yielded prolongation of OS in SCLC and led to unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease [46–51]. Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase III trial and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS [52,53].
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
Second-Line Therapy
Despite being exquisitely chemo-sensitive, SCLC is associated with very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse since first-line platinum based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If the disease progression occurs between 3 to 6 months since platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25% [54,55]. A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (cyclophosphamide, adriamycin, vincristine) (Table 2).
Immunotherapy
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving and currently there are no FDA-approved immunotherapy agents in SCLC. A recently conducted phase I/II trial (CheckMate 032) of anti-PD-1 antibody nivolumab with or without anti-CTLA-1 antibody ipilimumab in patients with relapsed SCLC reported a response rate of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg + ipilimumab 3 mg/kg. The 2-year OS was 26% with the combination and 14% with single agent nivolumab [56,57]. Only 18% of patients had PD-L1 expression of ≥ 1% and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20% and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the NCCN guidelines as one of the options for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy; however, it is questionable whether routine use of this combination is justified based on currently available data. However the evidence for the combination of nivolumab and ipilimumab remains limited. This efficacy and toxicity data of both randomized and nonrandomized cohorts were presented together making it hard to interpret the results.
Another phase Ib study (KEYNOTE-028) utilizing anti-PD-1 antibody pembrolizumab 10 mg/kg IV every 2 weeks in patients with relapsed SCLC after receiving one or more prior lines of therapy and PD-L1 expression of ≥ 1% showed a response rate of 33% with median duration of response of 19 months and 1-year OS of 38% [58]. Although only 28% of screened patients had PD-L1 expression of ≥ 1% , these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
Role of Prophylactic Cranial Irradiation
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase III trial conducted by EORTC comparing PCI with no PCI in patients with extensive-stage SCLC who had attained partial or complete response to initial platinum-based chemotherapy showed decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI. However, this trial did not require mandatory brain imaging prior to PCI and therefore it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups. A more recent phase III study conducted in Japan that compared PCI (25Gy in 10 fractions) with no PCI reported no difference in survival between the two groups. As opposed to EORTC study, the Japanese study did require baseline brain imaging to confirm absence of brain metastasis prior to enrollment. In addition, the patients in the control arm underwent periodic brain MRI to allow early detection of brain metastasis [59]. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven and PCI likely has a role in a highly select small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG PS 3–4) or underlying neurocognitive disorders [33,60]. NMDA receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction [61]. Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiation [61].
Role of Radiation
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiation after completion of platinum-based chemotherapy. A randomized trial including patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiation in combination with continued chemotherapy versus chemotherapy alone [62]. The median OS was longer with the addition of thoracic radiation compared to chemotherapy alone. Another phase III trial did not show improvement in 1-year OS with consolidative thoracic radiation, but 2-year OS and 6-month PFS were longer [63]. In general, consolidative thoracic radiation benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy [64]. In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiation should be utilized for palliative purposes in patients with painful bone metastasis, spine cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy with extensive stage disease. Whole brain radiotherapy is preferred over SRS because of frequent presence of micrometastasis even in the setting of one or two radiographically evident brain metastasis.
Novel Therapies
A very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and a high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major limitations to the development of targeted therapies in SCLC is limited availability of tissue owing to small tissue samples and frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been under investigation for their potential role in SCLC. Several of them, including EGFR TKIs, BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have been unsuccessful in showing a survival advantage in this disease. Several others including PARP inhibitors, cellular developmental pathway inhibitors and antibody drug conjugates are being tested. A phase I study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated complete response in 14.3%, partial response in 57.1%, and stable disease in 28.6% of patients with acceptable safety profile [65]. So far, none of these agents are approved for use in SCLC and the majority are in early phase clinical trials [66].
One of the emerging targets in the treatment of SCLC is DLL3. DLL3 is expressed on > 80% SCLCL tumor cells and cancer stem cells. Rovalpituzumab tesirine (ROVA-T) is an antibody drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase I trial of ROVA-T in patients with relapsed SCLC after 1 or 2 prior lines of therapies reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and mPFS were 4.6 months [67]. ROVA-T is currently in later phases of clinical trials and has a potential to serve as one of the options for patients with extensive-stage disease after disease progression on platinum-based therapy.
Response Assessment/Surveillance
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation. The surveillance guidelines consist of history, physical exam, and imaging every 3 months during 1st 2 years, every 6 months during the 3rdyear, and annually thereafter. If PCI is not performed, brain MRI or contrast enhanced CT scan should be performed every 3 to 4 months during the first 2 years of follow-up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the 1st year, every 3 to 4 months during year 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
Corresponding author: Hirva Mamdani, MD, Karmanos Cancer Institute, 4100 John R, Detroit, MI 48201, [email protected].
Financial disclosures: None.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed April 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed April 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
8. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
9. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
10. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
11. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
12. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
13. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12(7):666–73.
14. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
15. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
16. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
17. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
18. Takada M, Fukuoka M, Kawahara M, Sugiura T, Yokoyama A, Yokota S, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
19. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
20. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
21. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10(6):890–5.
22. Murray N, Coy P, Pater JL, Hodson I, Arnold A, Zee BC, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
23. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27(10):1818–28.
24. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340(4):265–71.
25. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18(8):1116–25.
26. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87(3):183–90.
27. Aupérin A, Arriagada R, Pignon JP, Le Pechoux C, Gregor A, Stephens RJ, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
28. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10(5):467–74.
29. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9(10):1132–9.
30. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
31. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
32. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
33. Yang CF, Chan DY, Speicher PJ, Gulack BC, Wang X, Hartwig MG, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
34. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
35. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
36. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
37. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
38. Zhou H, Zeng C, Wei Y, Zhou J, Yao W. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
39. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol 1995 Oct;13:2594–9.
40. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
41. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
42. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
43. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
44. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10(2):282–91.
45. Miles DW, Earl HM, Souhami RL, Harper PG, Rudd R, Ash CM, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
46. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
47. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4(12):1555–60.
48. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29(16):2215–22.
49. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27(35):6006–11.
50. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
51. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trialdagger. Ann Oncol 2015;26:908–14.
52. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts). J Clin Oncol 2017;35(15_suppl):8504-. [Can’t find the rest of the reference]
53. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
54. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
55. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
56. Hellmann MD, Ott PA, Zugazagoitia J, Ready NE, Hann CL, Braud FGD, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032. J Clin Oncol 2017;35(15_suppl):8503-. [Can’t find the rest of the reference]
57. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
58. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
59. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
60. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27(1):78–84.
61. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
62. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17(7):2092–9.
63. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
64. Slotman BJ, van Tinteren H, Praag JO, Knegjens JL, El Sharouni SY, Hatton M, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
65. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer. 2015 ;89:66–70.
66. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Translational lung cancer research. 2015;4:533–44.
67. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed April 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed April 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
8. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
9. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
10. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
11. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
12. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
13. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12(7):666–73.
14. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
15. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
16. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
17. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
18. Takada M, Fukuoka M, Kawahara M, Sugiura T, Yokoyama A, Yokota S, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
19. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
20. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
21. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10(6):890–5.
22. Murray N, Coy P, Pater JL, Hodson I, Arnold A, Zee BC, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
23. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27(10):1818–28.
24. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340(4):265–71.
25. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18(8):1116–25.
26. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87(3):183–90.
27. Aupérin A, Arriagada R, Pignon JP, Le Pechoux C, Gregor A, Stephens RJ, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
28. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10(5):467–74.
29. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9(10):1132–9.
30. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
31. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
32. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
33. Yang CF, Chan DY, Speicher PJ, Gulack BC, Wang X, Hartwig MG, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
34. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
35. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
36. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
37. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
38. Zhou H, Zeng C, Wei Y, Zhou J, Yao W. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
39. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol 1995 Oct;13:2594–9.
40. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
41. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
42. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
43. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
44. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10(2):282–91.
45. Miles DW, Earl HM, Souhami RL, Harper PG, Rudd R, Ash CM, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
46. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
47. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4(12):1555–60.
48. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29(16):2215–22.
49. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27(35):6006–11.
50. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
51. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trialdagger. Ann Oncol 2015;26:908–14.
52. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts). J Clin Oncol 2017;35(15_suppl):8504-. [Can’t find the rest of the reference]
53. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
54. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
55. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
56. Hellmann MD, Ott PA, Zugazagoitia J, Ready NE, Hann CL, Braud FGD, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032. J Clin Oncol 2017;35(15_suppl):8503-. [Can’t find the rest of the reference]
57. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
58. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
59. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
60. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27(1):78–84.
61. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
62. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17(7):2092–9.
63. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
64. Slotman BJ, van Tinteren H, Praag JO, Knegjens JL, El Sharouni SY, Hatton M, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
65. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer. 2015 ;89:66–70.
66. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Translational lung cancer research. 2015;4:533–44.
67. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.
Treating psychosis in patients with HIV/AIDS

Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
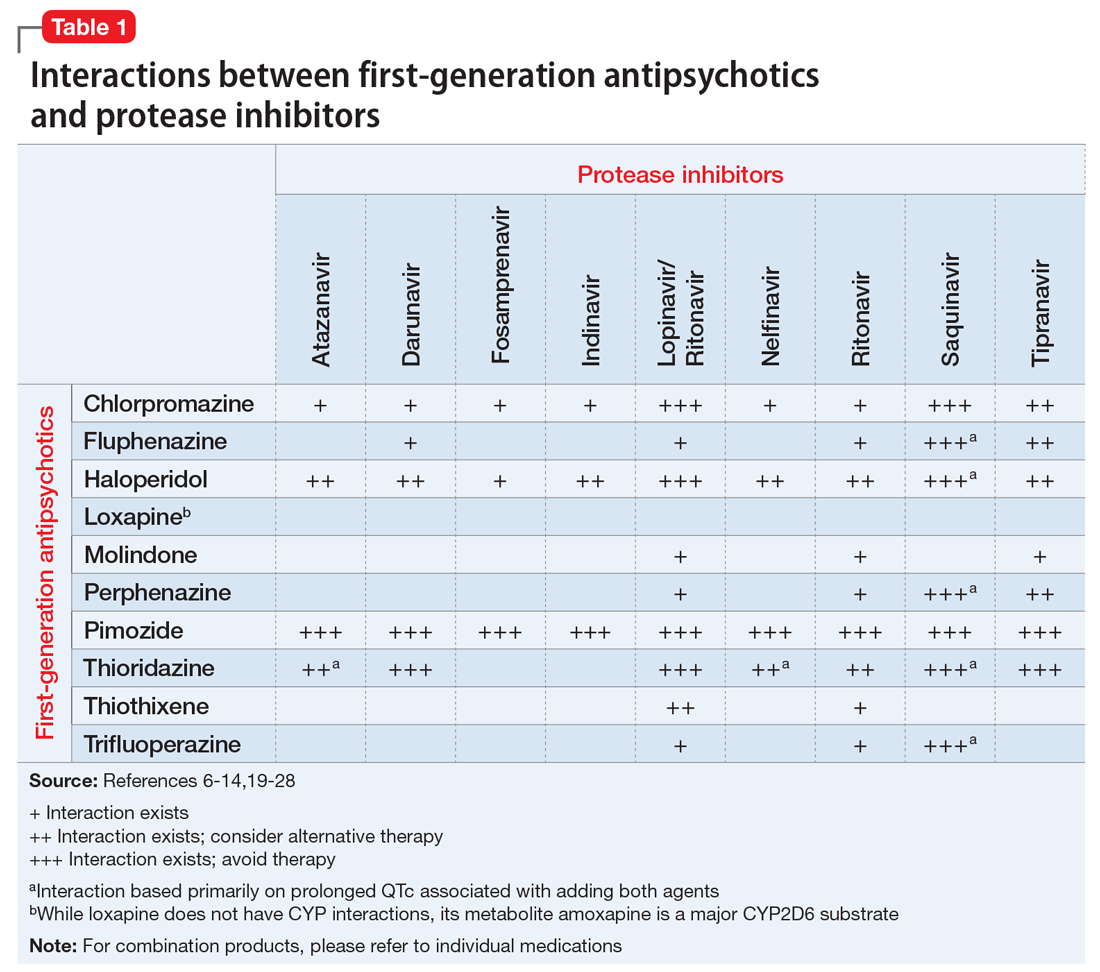
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
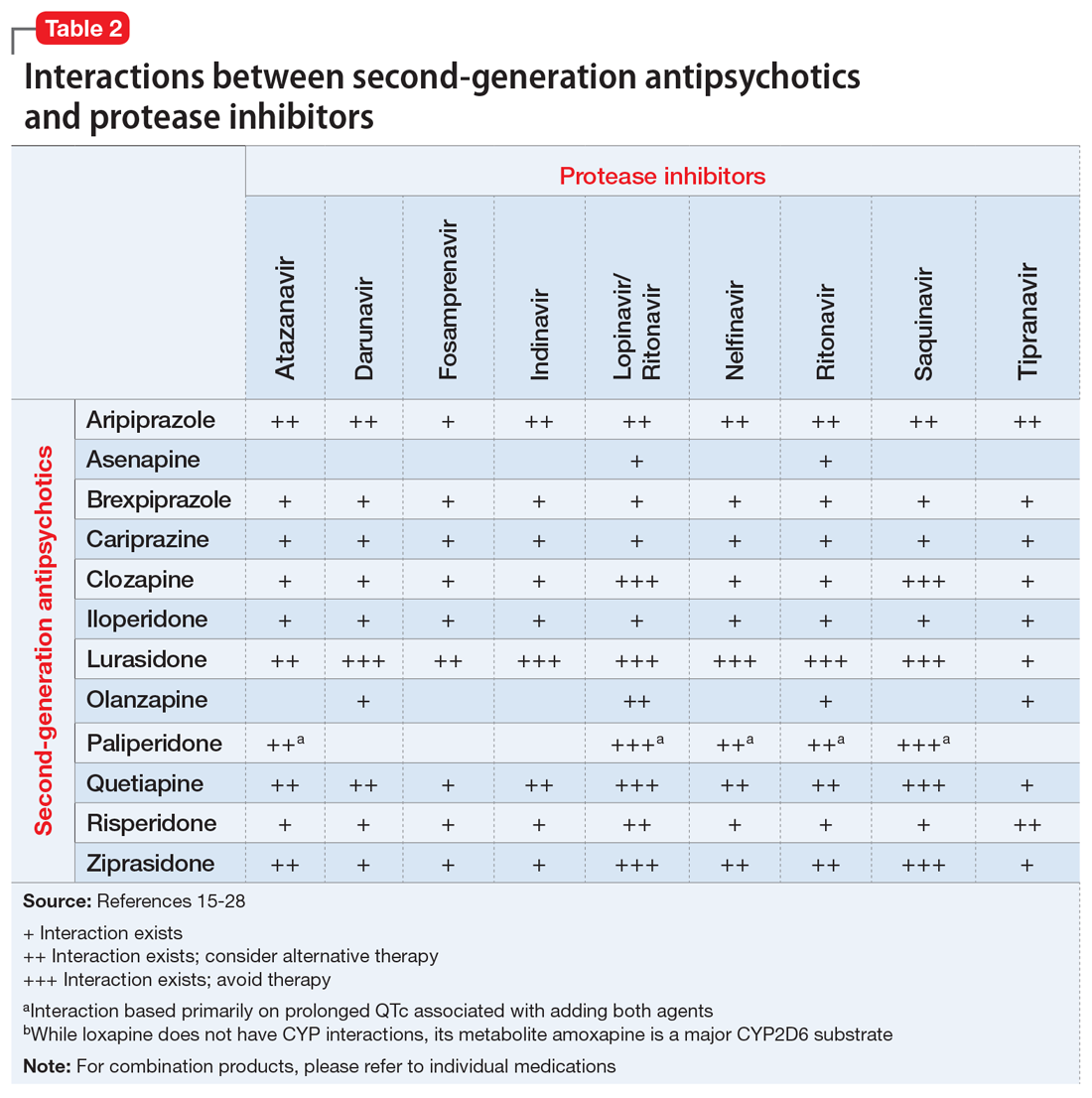
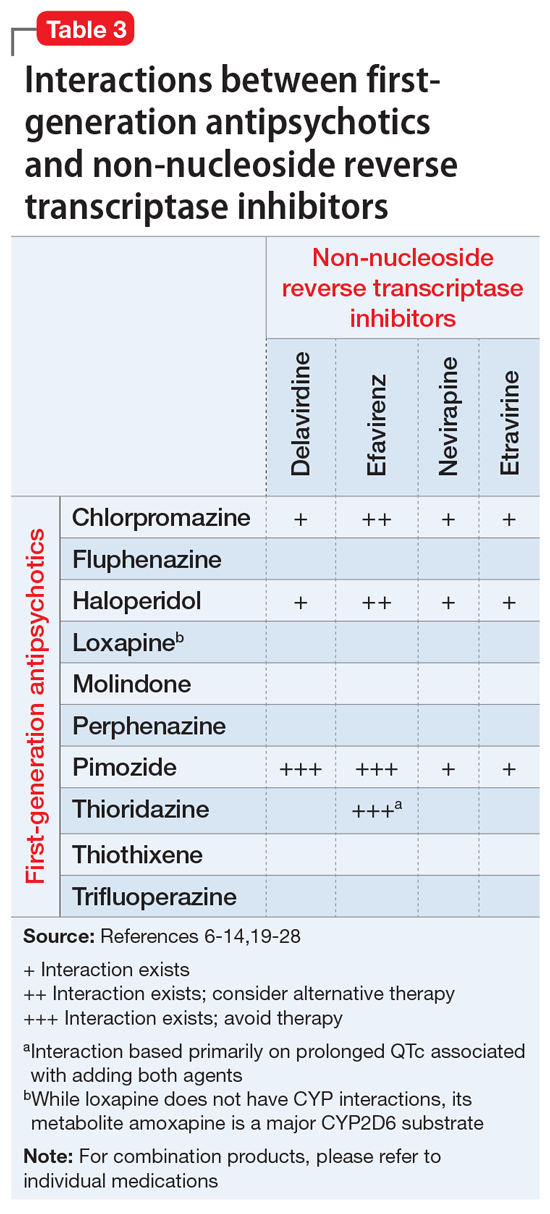
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.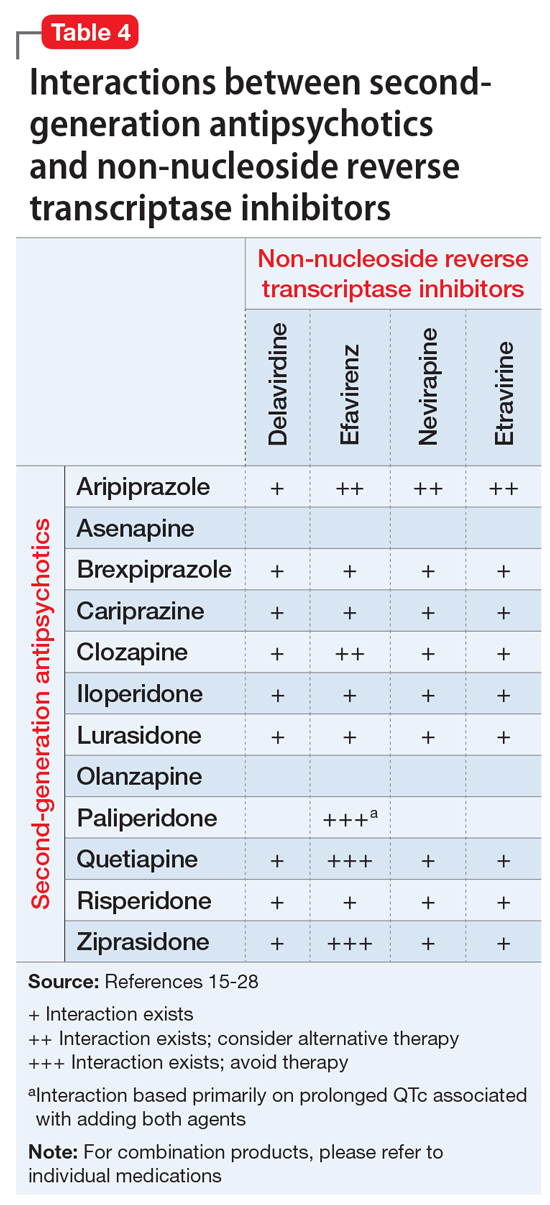
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.
Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.
Aggressive outbursts and emotional lability in a 16-year-old boy
Mr. X, age 16, has cerebral palsy (CP), idiopathic normal pressure hydrocephalus (iNPH), and a history of impulse control disorder and behavioral instability, including episodes of aggression or combativeness. Mr. X’s mother reports that these episodes are almost always preceded by inappropriate laughing or crying. His outbursts and emotional lability have gotten worse during the last 6 months. Due to his disruptive behaviors, Mr. X has been unable to attend school, and his parents are considering group home placement. Although they were previously able to control their son’s aggressive behaviors, they fear for his safety, and after one such episode, they call 911. Mr. X is transported by police in handcuffs to the comprehensive psychiatric emergency room (CPEP) for evaluation.
While in CPEP, Mr. X remains uncooperative and disruptive; subsequently, he is placed in 4-point restraints and given
[polldaddy:9991896]
The authors’ observations
Pseudobulbar affect (PBA) is a disorder characterized by sporadic episodes of inappropriate laughing and/or crying that are incongruent with situational context and are frequently exaggerated in comparison with the actual feelings of the patient. The duration of PBA episodes can last seconds to minutes and arise unpredictably.
PBA typically develops secondary to a neurologic disorder, most commonly Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Parkinson’s disease (PD), stroke, or traumatic brain injury (TBI).1 PBA symptoms are present in an estimated 29.3% of patients with AD, 44.8% of patients with ALS, 45.8% of patients with MS, 26% of patients with PD, 37.8% of patients with stroke, and 52.4% of patients with TBI.2 Although PBA appears far more frequently in patients with MS or ALS compared with those with PD, PD represents an under-recognized and larger patient population. A small fraction of patients also develops PBA secondary to hyperthyroidism, hypothyroidism, Graves’ disease, Wilson’s disease, brain tumors, and a multitude of encephalopathies.3 These neurologic disorders cause dysregulation of the corticopontine-cerebellar circuitry, resulting in functional impediment to the normal affect modulator action of the cerebellum.4
The neurologic insults that can result in PBA may include CP or iNPH. Cerebellar injury is a frequent pathological finding in CP.5 In patients with iNPH, in addition to altered CSF flow, enlarged ventricles compress the corticospinal tracts in the lateral ventricles,6 which is theorized to induce PBA symptoms.
PBA is diagnosed by subjective clinical evaluation and by using the Center for Neurologic Study–Lability Scale (CNS-LS). The CNS-LS is a 7-question survey that addresses the severity of affect lability (Table 17). It may be completed by the patient or caregiver. Each question ranges in score from 1 to 5, with the total score ranging from 7 to 35. The minimum score required for the diagnosis of PBA is 13.7
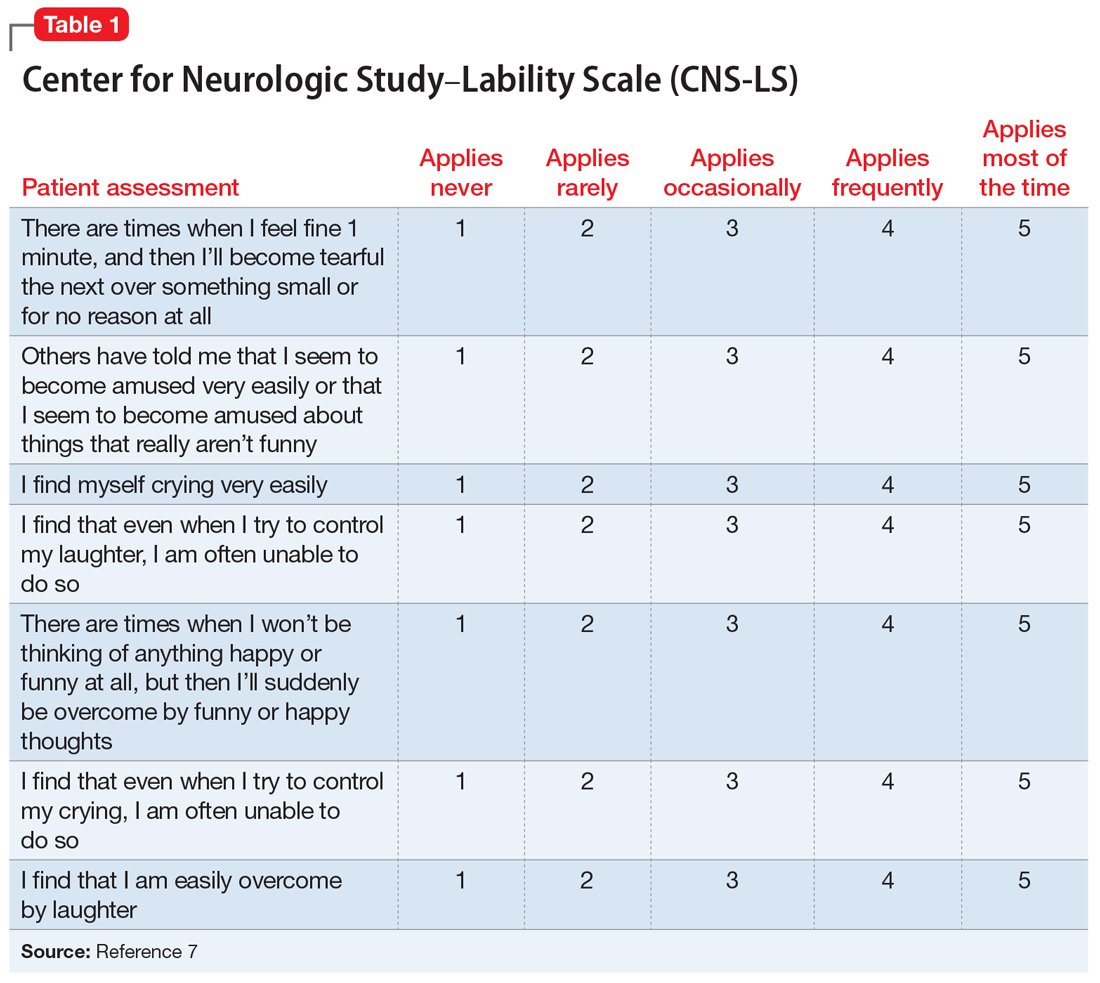
PBA is frequently misdiagnosed as depression, although the 2 disorders can occur simultaneously (Table 21,8). A crucial distinguishing factor between depression and PBA is the extent of symptoms. Depression presents as feelings of sadness associated with crying and disinterest that occur for weeks to months. In contrast, PBA presents as brief, uncontrollable episodes of laughing and/or crying that last seconds to minutes. Unlike depression, the behaviors associated with PBA are exaggerated or do not match the patient’s feelings. Furthermore, a neurologic disease or brain injury is always present in a patient with PBA, but is not imperative for the diagnosis of depression.
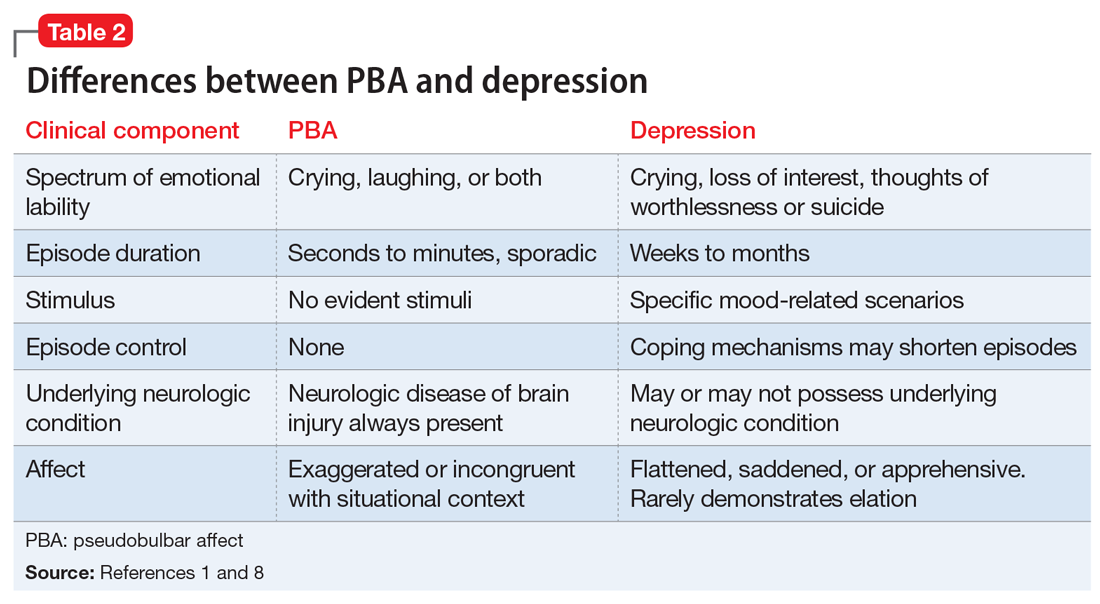
Continue to: Compared with individuals without PBA...
Compared with individuals without PBA, patients with PBA also experience more distress, embarrassment, and social disability, and are consequently more likely to suffer from other psychiatric conditions, including depression, anxiety/panic attacks, bipolar disorder, posttraumatic stress disorder, psychotic disorder, and schizophrenia.1 The Patient Health Questionnaire (PHQ-9), a tool for measuring depression severity, can be used in addition to the CNS-LS to determine if the patient has both depression and PBA.
HISTORY Poor response to anxiolytics and antipsychotics
Mr. X previously received a ventriculoperitoneal shunt for treating iNPH. He was not taking any medications for CP. To address his impulse control disorder, he was prescribed olanzapine, 20 mg/d, risperidone, 2 mg/d, and diazepam, 5 mg three times a day. Mr. X is uncontrolled on these medications, experiencing frequent behavioral outbursts at home. His mother completes a CNS-LS for him. He receives a score of 20, which suggests a diagnosis of PBA. His PHQ-9 score is 8, indicating mild depression.
[polldaddy:9991899]
TREATMENT Introducing a new medication
Mr. X is started on
Continue to: The authors' observations
The authors’ observations
Decreasing the severity and frequency of episodes constitutes the mainstay of treating PBA. In the past, off-label treatments, including selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants, were prescribed to reduce PBA symptoms.5 Currently, dextromethorphan/quinidine is the only FDA-approved medication for treating PBA; however, its use in patients younger than age 18 is considered investigational.
Atypical antipsychotics, such as olanzapine and risperidone, have more warnings and precautions than dextromethorphan/quinidine. Risperidone has a “black-box” warning for QT prolongation, in addition to death and stroke in elderly patients.10 Although dextromethorphan/quinidine does not have a black-box warning, it does increase the risk of QT prolongation, and patients with cardiac risk factors should undergo an electrocardiogram before starting this medication. Additionally, risperidone and olanzapine are known to cause significant weight gain, which can increase the risk of developing hyperlipidemia, metabolic syndrome, and type 2 diabetes mellitus.10,11 Neuroleptic malignant syndrome (NMS) is a potentially life-threatening adverse effect of all antipsychotics. NMS is characterized by fever, rigidity, altered consciousness, and increased heart and respiratory rates.12
Quinidine increases the bioavailability of dextromethorphan by inhibiting CYP2D6. When dextromethorphan/quinidine is simultaneously used with an SSRI that also inhibits CYP2D6, such as paroxetine or fluoxetine, the patient may be at increased risk for developing adverse effects such as respiratory depression and serotonin syndrome.13
[polldaddy:9991902]
Continue to: The authors' observations
The authors’ observations
Although the exact pathophysiology of PBA is unknown, multiple theories may explain the principle elements of the condition. In the absence of a neurologic insult, the cerebellum acts as an affect regulator, inhibiting laughter and crying at times in which they are considered inappropriate. Parvizi et al4 have theorized that the lesions involved in PBA disrupt the corticopontine-cerebellar circuitry, which impedes the ability of the cerebellum to function as an affect modulator.3 In addition to the dysregulation of cerebellar circuitry, altered serotonin and glutamate levels are believed to contribute to the deficient affect regulation observed in PBA; therefore, adding dextromethorphan/quinidine potentiates serotonin and glutamate levels in the synaptic cleft, resulting in a reduction in PBA episodes.4
OUTCOME Affect stability
Seven months after beginning dextromethorphan/quinidine, Mr. X has experienced resolution of his PBA episodes. His PHQ-9 score was reduced to 0 (no clinical signs of depression) within 1 month of starting this medication and his PHQ-9 scores remain below 5, representing minimal depressive severity. The CNS-LS scale is not conducted at further visits because the patient’s mother reported no further PBA episodes. Mr. X no longer exhibits episodes of aggression. These episodes seemed to have been a manifestation of his frustration and difficulty in controlling his PBA episodes. Furthermore, his dosage of diazepam was reduced, and he was weaned off risperidone. Mr. X’s parents report that he has a drastically improved affect. He continues to tolerate his medication well and no longer demonstrates any exacerbations of his psychiatric symptoms.
Bottom Line
Pseudobulbar affect (PBA) may occur secondary to various neurologic insults, including cerebral palsy and idiopathic normal pressure hydrocephalus. The condition is diagnosed by a subjective clinical evaluation and use of the Center for Neurologic Study–Lability Scale. Dextromethorphan/quinidine can significantly reduce PBA symptoms.
Acknowledgements
The authors thank Anthony S. Graziano and Rachel M. Watt, both Physician Assistant students, Daemen College, Amherst, New York.
Related Resources
- Frock B, Williams A, Caplan JP. Pseudobulbar affect: when patients laugh or cry, but don’t know why. Current Psychiatry. 2016;15(9):56-60,63.
- Crumpacker DW. Enhancing approaches to the identification and management of pseudobulbar affect. J Clin Psychiatry. 2016;77(9):e1155.
Drug Brand Names
Dextromethorphan/quinidine • Nuedexta
Diazepam • Valium
Fluoxetine • Prozac
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Paroxetine • Paxil
Risperidone • Risperdal
1. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
2. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
3. Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17(4):447-454.
4. Parvizi J, Anderson SW, Martin CO, et al. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124(pt 9):1708-1719.
5. Johnsen SD, Bodensteiner JB, Lotze TE. Frequency and nature of cerebellar injury in the extremely premature survivor with cerebral palsy. J Child Neurol. 2005;20(1):60-64.
6. Kamiya K, Hori M, Miyajima M, et al. Axon diameter and intra-axonal volume fraction of the corticospinal tract in idiopathic normal pressure hydrocephalus measured by Q-Space imaging. PLoS One. 2014;9(8):e103842. doi: 10.1371/journal.pone.0103842.
7. Moore SR, Gresham LS, Bromberg MB, et al. A self report measuredextromethorphan of affective lability. J Neurol Neurosurg Psychiatry. 1997;63(1):89-93.
8. Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Ther Clinical Risk Manag. 2013;9:483-489.
9. Cruz MP. Nuedexta for the treatment of pseudobulbar affect. A condition of involuntary crying or laughing. P T. 2013;38(6):325-328.
10. Goëb JL, Marco S, Duhamel A, et al. Metabolic side effects of risperidone in children and adolescents with early onset schizophrenia. Prim Care Companion J Clin Psychiatry. 2008;10(6):486-487.
11. Nemeroff CB. Dosing the antipsychotic medication olanzapine. J Clin Psychiatry. 1997;58(suppl 10):45-49.
12. Troller JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23(6):477-492.
13. Schoedel KA, Pope LE, Sellers EM. Randomized open-label drug-drug interaction trial of dextromethorphan/quinidine and paroxetine in healthy volunteers. Clin Drug Investig. 2012;32(3):157-169.
Mr. X, age 16, has cerebral palsy (CP), idiopathic normal pressure hydrocephalus (iNPH), and a history of impulse control disorder and behavioral instability, including episodes of aggression or combativeness. Mr. X’s mother reports that these episodes are almost always preceded by inappropriate laughing or crying. His outbursts and emotional lability have gotten worse during the last 6 months. Due to his disruptive behaviors, Mr. X has been unable to attend school, and his parents are considering group home placement. Although they were previously able to control their son’s aggressive behaviors, they fear for his safety, and after one such episode, they call 911. Mr. X is transported by police in handcuffs to the comprehensive psychiatric emergency room (CPEP) for evaluation.
While in CPEP, Mr. X remains uncooperative and disruptive; subsequently, he is placed in 4-point restraints and given
[polldaddy:9991896]
The authors’ observations
Pseudobulbar affect (PBA) is a disorder characterized by sporadic episodes of inappropriate laughing and/or crying that are incongruent with situational context and are frequently exaggerated in comparison with the actual feelings of the patient. The duration of PBA episodes can last seconds to minutes and arise unpredictably.
PBA typically develops secondary to a neurologic disorder, most commonly Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Parkinson’s disease (PD), stroke, or traumatic brain injury (TBI).1 PBA symptoms are present in an estimated 29.3% of patients with AD, 44.8% of patients with ALS, 45.8% of patients with MS, 26% of patients with PD, 37.8% of patients with stroke, and 52.4% of patients with TBI.2 Although PBA appears far more frequently in patients with MS or ALS compared with those with PD, PD represents an under-recognized and larger patient population. A small fraction of patients also develops PBA secondary to hyperthyroidism, hypothyroidism, Graves’ disease, Wilson’s disease, brain tumors, and a multitude of encephalopathies.3 These neurologic disorders cause dysregulation of the corticopontine-cerebellar circuitry, resulting in functional impediment to the normal affect modulator action of the cerebellum.4
The neurologic insults that can result in PBA may include CP or iNPH. Cerebellar injury is a frequent pathological finding in CP.5 In patients with iNPH, in addition to altered CSF flow, enlarged ventricles compress the corticospinal tracts in the lateral ventricles,6 which is theorized to induce PBA symptoms.
PBA is diagnosed by subjective clinical evaluation and by using the Center for Neurologic Study–Lability Scale (CNS-LS). The CNS-LS is a 7-question survey that addresses the severity of affect lability (Table 17). It may be completed by the patient or caregiver. Each question ranges in score from 1 to 5, with the total score ranging from 7 to 35. The minimum score required for the diagnosis of PBA is 13.7
PBA is frequently misdiagnosed as depression, although the 2 disorders can occur simultaneously (Table 21,8). A crucial distinguishing factor between depression and PBA is the extent of symptoms. Depression presents as feelings of sadness associated with crying and disinterest that occur for weeks to months. In contrast, PBA presents as brief, uncontrollable episodes of laughing and/or crying that last seconds to minutes. Unlike depression, the behaviors associated with PBA are exaggerated or do not match the patient’s feelings. Furthermore, a neurologic disease or brain injury is always present in a patient with PBA, but is not imperative for the diagnosis of depression.
Continue to: Compared with individuals without PBA...
Compared with individuals without PBA, patients with PBA also experience more distress, embarrassment, and social disability, and are consequently more likely to suffer from other psychiatric conditions, including depression, anxiety/panic attacks, bipolar disorder, posttraumatic stress disorder, psychotic disorder, and schizophrenia.1 The Patient Health Questionnaire (PHQ-9), a tool for measuring depression severity, can be used in addition to the CNS-LS to determine if the patient has both depression and PBA.
HISTORY Poor response to anxiolytics and antipsychotics
Mr. X previously received a ventriculoperitoneal shunt for treating iNPH. He was not taking any medications for CP. To address his impulse control disorder, he was prescribed olanzapine, 20 mg/d, risperidone, 2 mg/d, and diazepam, 5 mg three times a day. Mr. X is uncontrolled on these medications, experiencing frequent behavioral outbursts at home. His mother completes a CNS-LS for him. He receives a score of 20, which suggests a diagnosis of PBA. His PHQ-9 score is 8, indicating mild depression.
[polldaddy:9991899]
TREATMENT Introducing a new medication
Mr. X is started on
Continue to: The authors' observations
The authors’ observations
Decreasing the severity and frequency of episodes constitutes the mainstay of treating PBA. In the past, off-label treatments, including selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants, were prescribed to reduce PBA symptoms.5 Currently, dextromethorphan/quinidine is the only FDA-approved medication for treating PBA; however, its use in patients younger than age 18 is considered investigational.
Atypical antipsychotics, such as olanzapine and risperidone, have more warnings and precautions than dextromethorphan/quinidine. Risperidone has a “black-box” warning for QT prolongation, in addition to death and stroke in elderly patients.10 Although dextromethorphan/quinidine does not have a black-box warning, it does increase the risk of QT prolongation, and patients with cardiac risk factors should undergo an electrocardiogram before starting this medication. Additionally, risperidone and olanzapine are known to cause significant weight gain, which can increase the risk of developing hyperlipidemia, metabolic syndrome, and type 2 diabetes mellitus.10,11 Neuroleptic malignant syndrome (NMS) is a potentially life-threatening adverse effect of all antipsychotics. NMS is characterized by fever, rigidity, altered consciousness, and increased heart and respiratory rates.12
Quinidine increases the bioavailability of dextromethorphan by inhibiting CYP2D6. When dextromethorphan/quinidine is simultaneously used with an SSRI that also inhibits CYP2D6, such as paroxetine or fluoxetine, the patient may be at increased risk for developing adverse effects such as respiratory depression and serotonin syndrome.13
[polldaddy:9991902]
Continue to: The authors' observations
The authors’ observations
Although the exact pathophysiology of PBA is unknown, multiple theories may explain the principle elements of the condition. In the absence of a neurologic insult, the cerebellum acts as an affect regulator, inhibiting laughter and crying at times in which they are considered inappropriate. Parvizi et al4 have theorized that the lesions involved in PBA disrupt the corticopontine-cerebellar circuitry, which impedes the ability of the cerebellum to function as an affect modulator.3 In addition to the dysregulation of cerebellar circuitry, altered serotonin and glutamate levels are believed to contribute to the deficient affect regulation observed in PBA; therefore, adding dextromethorphan/quinidine potentiates serotonin and glutamate levels in the synaptic cleft, resulting in a reduction in PBA episodes.4
OUTCOME Affect stability
Seven months after beginning dextromethorphan/quinidine, Mr. X has experienced resolution of his PBA episodes. His PHQ-9 score was reduced to 0 (no clinical signs of depression) within 1 month of starting this medication and his PHQ-9 scores remain below 5, representing minimal depressive severity. The CNS-LS scale is not conducted at further visits because the patient’s mother reported no further PBA episodes. Mr. X no longer exhibits episodes of aggression. These episodes seemed to have been a manifestation of his frustration and difficulty in controlling his PBA episodes. Furthermore, his dosage of diazepam was reduced, and he was weaned off risperidone. Mr. X’s parents report that he has a drastically improved affect. He continues to tolerate his medication well and no longer demonstrates any exacerbations of his psychiatric symptoms.
Bottom Line
Pseudobulbar affect (PBA) may occur secondary to various neurologic insults, including cerebral palsy and idiopathic normal pressure hydrocephalus. The condition is diagnosed by a subjective clinical evaluation and use of the Center for Neurologic Study–Lability Scale. Dextromethorphan/quinidine can significantly reduce PBA symptoms.
Acknowledgements
The authors thank Anthony S. Graziano and Rachel M. Watt, both Physician Assistant students, Daemen College, Amherst, New York.
Related Resources
- Frock B, Williams A, Caplan JP. Pseudobulbar affect: when patients laugh or cry, but don’t know why. Current Psychiatry. 2016;15(9):56-60,63.
- Crumpacker DW. Enhancing approaches to the identification and management of pseudobulbar affect. J Clin Psychiatry. 2016;77(9):e1155.
Drug Brand Names
Dextromethorphan/quinidine • Nuedexta
Diazepam • Valium
Fluoxetine • Prozac
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Paroxetine • Paxil
Risperidone • Risperdal
Mr. X, age 16, has cerebral palsy (CP), idiopathic normal pressure hydrocephalus (iNPH), and a history of impulse control disorder and behavioral instability, including episodes of aggression or combativeness. Mr. X’s mother reports that these episodes are almost always preceded by inappropriate laughing or crying. His outbursts and emotional lability have gotten worse during the last 6 months. Due to his disruptive behaviors, Mr. X has been unable to attend school, and his parents are considering group home placement. Although they were previously able to control their son’s aggressive behaviors, they fear for his safety, and after one such episode, they call 911. Mr. X is transported by police in handcuffs to the comprehensive psychiatric emergency room (CPEP) for evaluation.
While in CPEP, Mr. X remains uncooperative and disruptive; subsequently, he is placed in 4-point restraints and given
[polldaddy:9991896]
The authors’ observations
Pseudobulbar affect (PBA) is a disorder characterized by sporadic episodes of inappropriate laughing and/or crying that are incongruent with situational context and are frequently exaggerated in comparison with the actual feelings of the patient. The duration of PBA episodes can last seconds to minutes and arise unpredictably.
PBA typically develops secondary to a neurologic disorder, most commonly Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Parkinson’s disease (PD), stroke, or traumatic brain injury (TBI).1 PBA symptoms are present in an estimated 29.3% of patients with AD, 44.8% of patients with ALS, 45.8% of patients with MS, 26% of patients with PD, 37.8% of patients with stroke, and 52.4% of patients with TBI.2 Although PBA appears far more frequently in patients with MS or ALS compared with those with PD, PD represents an under-recognized and larger patient population. A small fraction of patients also develops PBA secondary to hyperthyroidism, hypothyroidism, Graves’ disease, Wilson’s disease, brain tumors, and a multitude of encephalopathies.3 These neurologic disorders cause dysregulation of the corticopontine-cerebellar circuitry, resulting in functional impediment to the normal affect modulator action of the cerebellum.4
The neurologic insults that can result in PBA may include CP or iNPH. Cerebellar injury is a frequent pathological finding in CP.5 In patients with iNPH, in addition to altered CSF flow, enlarged ventricles compress the corticospinal tracts in the lateral ventricles,6 which is theorized to induce PBA symptoms.
PBA is diagnosed by subjective clinical evaluation and by using the Center for Neurologic Study–Lability Scale (CNS-LS). The CNS-LS is a 7-question survey that addresses the severity of affect lability (Table 17). It may be completed by the patient or caregiver. Each question ranges in score from 1 to 5, with the total score ranging from 7 to 35. The minimum score required for the diagnosis of PBA is 13.7
PBA is frequently misdiagnosed as depression, although the 2 disorders can occur simultaneously (Table 21,8). A crucial distinguishing factor between depression and PBA is the extent of symptoms. Depression presents as feelings of sadness associated with crying and disinterest that occur for weeks to months. In contrast, PBA presents as brief, uncontrollable episodes of laughing and/or crying that last seconds to minutes. Unlike depression, the behaviors associated with PBA are exaggerated or do not match the patient’s feelings. Furthermore, a neurologic disease or brain injury is always present in a patient with PBA, but is not imperative for the diagnosis of depression.
Continue to: Compared with individuals without PBA...
Compared with individuals without PBA, patients with PBA also experience more distress, embarrassment, and social disability, and are consequently more likely to suffer from other psychiatric conditions, including depression, anxiety/panic attacks, bipolar disorder, posttraumatic stress disorder, psychotic disorder, and schizophrenia.1 The Patient Health Questionnaire (PHQ-9), a tool for measuring depression severity, can be used in addition to the CNS-LS to determine if the patient has both depression and PBA.
HISTORY Poor response to anxiolytics and antipsychotics
Mr. X previously received a ventriculoperitoneal shunt for treating iNPH. He was not taking any medications for CP. To address his impulse control disorder, he was prescribed olanzapine, 20 mg/d, risperidone, 2 mg/d, and diazepam, 5 mg three times a day. Mr. X is uncontrolled on these medications, experiencing frequent behavioral outbursts at home. His mother completes a CNS-LS for him. He receives a score of 20, which suggests a diagnosis of PBA. His PHQ-9 score is 8, indicating mild depression.
[polldaddy:9991899]
TREATMENT Introducing a new medication
Mr. X is started on
Continue to: The authors' observations
The authors’ observations
Decreasing the severity and frequency of episodes constitutes the mainstay of treating PBA. In the past, off-label treatments, including selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants, were prescribed to reduce PBA symptoms.5 Currently, dextromethorphan/quinidine is the only FDA-approved medication for treating PBA; however, its use in patients younger than age 18 is considered investigational.
Atypical antipsychotics, such as olanzapine and risperidone, have more warnings and precautions than dextromethorphan/quinidine. Risperidone has a “black-box” warning for QT prolongation, in addition to death and stroke in elderly patients.10 Although dextromethorphan/quinidine does not have a black-box warning, it does increase the risk of QT prolongation, and patients with cardiac risk factors should undergo an electrocardiogram before starting this medication. Additionally, risperidone and olanzapine are known to cause significant weight gain, which can increase the risk of developing hyperlipidemia, metabolic syndrome, and type 2 diabetes mellitus.10,11 Neuroleptic malignant syndrome (NMS) is a potentially life-threatening adverse effect of all antipsychotics. NMS is characterized by fever, rigidity, altered consciousness, and increased heart and respiratory rates.12
Quinidine increases the bioavailability of dextromethorphan by inhibiting CYP2D6. When dextromethorphan/quinidine is simultaneously used with an SSRI that also inhibits CYP2D6, such as paroxetine or fluoxetine, the patient may be at increased risk for developing adverse effects such as respiratory depression and serotonin syndrome.13
[polldaddy:9991902]
Continue to: The authors' observations
The authors’ observations
Although the exact pathophysiology of PBA is unknown, multiple theories may explain the principle elements of the condition. In the absence of a neurologic insult, the cerebellum acts as an affect regulator, inhibiting laughter and crying at times in which they are considered inappropriate. Parvizi et al4 have theorized that the lesions involved in PBA disrupt the corticopontine-cerebellar circuitry, which impedes the ability of the cerebellum to function as an affect modulator.3 In addition to the dysregulation of cerebellar circuitry, altered serotonin and glutamate levels are believed to contribute to the deficient affect regulation observed in PBA; therefore, adding dextromethorphan/quinidine potentiates serotonin and glutamate levels in the synaptic cleft, resulting in a reduction in PBA episodes.4
OUTCOME Affect stability
Seven months after beginning dextromethorphan/quinidine, Mr. X has experienced resolution of his PBA episodes. His PHQ-9 score was reduced to 0 (no clinical signs of depression) within 1 month of starting this medication and his PHQ-9 scores remain below 5, representing minimal depressive severity. The CNS-LS scale is not conducted at further visits because the patient’s mother reported no further PBA episodes. Mr. X no longer exhibits episodes of aggression. These episodes seemed to have been a manifestation of his frustration and difficulty in controlling his PBA episodes. Furthermore, his dosage of diazepam was reduced, and he was weaned off risperidone. Mr. X’s parents report that he has a drastically improved affect. He continues to tolerate his medication well and no longer demonstrates any exacerbations of his psychiatric symptoms.
Bottom Line
Pseudobulbar affect (PBA) may occur secondary to various neurologic insults, including cerebral palsy and idiopathic normal pressure hydrocephalus. The condition is diagnosed by a subjective clinical evaluation and use of the Center for Neurologic Study–Lability Scale. Dextromethorphan/quinidine can significantly reduce PBA symptoms.
Acknowledgements
The authors thank Anthony S. Graziano and Rachel M. Watt, both Physician Assistant students, Daemen College, Amherst, New York.
Related Resources
- Frock B, Williams A, Caplan JP. Pseudobulbar affect: when patients laugh or cry, but don’t know why. Current Psychiatry. 2016;15(9):56-60,63.
- Crumpacker DW. Enhancing approaches to the identification and management of pseudobulbar affect. J Clin Psychiatry. 2016;77(9):e1155.
Drug Brand Names
Dextromethorphan/quinidine • Nuedexta
Diazepam • Valium
Fluoxetine • Prozac
Haloperidol • Haldol
Lorazepam • Ativan
Olanzapine • Zyprexa
Paroxetine • Paxil
Risperidone • Risperdal
1. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
2. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
3. Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17(4):447-454.
4. Parvizi J, Anderson SW, Martin CO, et al. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124(pt 9):1708-1719.
5. Johnsen SD, Bodensteiner JB, Lotze TE. Frequency and nature of cerebellar injury in the extremely premature survivor with cerebral palsy. J Child Neurol. 2005;20(1):60-64.
6. Kamiya K, Hori M, Miyajima M, et al. Axon diameter and intra-axonal volume fraction of the corticospinal tract in idiopathic normal pressure hydrocephalus measured by Q-Space imaging. PLoS One. 2014;9(8):e103842. doi: 10.1371/journal.pone.0103842.
7. Moore SR, Gresham LS, Bromberg MB, et al. A self report measuredextromethorphan of affective lability. J Neurol Neurosurg Psychiatry. 1997;63(1):89-93.
8. Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Ther Clinical Risk Manag. 2013;9:483-489.
9. Cruz MP. Nuedexta for the treatment of pseudobulbar affect. A condition of involuntary crying or laughing. P T. 2013;38(6):325-328.
10. Goëb JL, Marco S, Duhamel A, et al. Metabolic side effects of risperidone in children and adolescents with early onset schizophrenia. Prim Care Companion J Clin Psychiatry. 2008;10(6):486-487.
11. Nemeroff CB. Dosing the antipsychotic medication olanzapine. J Clin Psychiatry. 1997;58(suppl 10):45-49.
12. Troller JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23(6):477-492.
13. Schoedel KA, Pope LE, Sellers EM. Randomized open-label drug-drug interaction trial of dextromethorphan/quinidine and paroxetine in healthy volunteers. Clin Drug Investig. 2012;32(3):157-169.
1. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
2. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
3. Schiffer R, Pope LE. Review of pseudobulbar affect including a novel and potential therapy. J Neuropsychiatry Clin Neurosci. 2005;17(4):447-454.
4. Parvizi J, Anderson SW, Martin CO, et al. Pathological laughter and crying: a link to the cerebellum. Brain. 2001;124(pt 9):1708-1719.
5. Johnsen SD, Bodensteiner JB, Lotze TE. Frequency and nature of cerebellar injury in the extremely premature survivor with cerebral palsy. J Child Neurol. 2005;20(1):60-64.
6. Kamiya K, Hori M, Miyajima M, et al. Axon diameter and intra-axonal volume fraction of the corticospinal tract in idiopathic normal pressure hydrocephalus measured by Q-Space imaging. PLoS One. 2014;9(8):e103842. doi: 10.1371/journal.pone.0103842.
7. Moore SR, Gresham LS, Bromberg MB, et al. A self report measuredextromethorphan of affective lability. J Neurol Neurosurg Psychiatry. 1997;63(1):89-93.
8. Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Ther Clinical Risk Manag. 2013;9:483-489.
9. Cruz MP. Nuedexta for the treatment of pseudobulbar affect. A condition of involuntary crying or laughing. P T. 2013;38(6):325-328.
10. Goëb JL, Marco S, Duhamel A, et al. Metabolic side effects of risperidone in children and adolescents with early onset schizophrenia. Prim Care Companion J Clin Psychiatry. 2008;10(6):486-487.
11. Nemeroff CB. Dosing the antipsychotic medication olanzapine. J Clin Psychiatry. 1997;58(suppl 10):45-49.
12. Troller JN, Chen X, Sachdev PS. Neuroleptic malignant syndrome associated with atypical antipsychotic drugs. CNS Drugs. 2009;23(6):477-492.
13. Schoedel KA, Pope LE, Sellers EM. Randomized open-label drug-drug interaction trial of dextromethorphan/quinidine and paroxetine in healthy volunteers. Clin Drug Investig. 2012;32(3):157-169.

Visual hallucinations and severe anxiety in the ICU after surgery
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.

Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).

Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.
Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).
Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.
Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).
Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
A 10-year-old boy with ‘voices in my head’: Is it a psychotic disorder?
CASE Auditory hallucinations?
M, age 10, has had multiple visits to the pediatric emergency department (PED) with the chief concern of excessive urinary frequency. At each visit, the medical workup has been negative and he was discharged home. After a few months, M’s parents bring their son back to the PED because he reports hearing “voices in my head” and “feeling tense and scared.” When these feelings become too overwhelming, M stops eating and experiences substantial fear and anxiety that require his mother’s repeated reassurances. M’s mother reports that 2 weeks before his most recent PED visit, he became increasingly anxious and disturbed, and said he was afraid most of the time, and worried about the safety of his family for no apparent reason.
The psychiatrist evaluates M in the PED and diagnoses him with unspecified schizophrenia spectrum and other psychotic disorder based on his persistent report of auditory and tactile hallucinations, including hearing a voice of a man telling him he was going to choke on his food and feeling someone touch his arm to soothe him during his anxious moments. M does not meet criteria for acute inpatient hospitalization, and is discharged home with referral to follow-up at our child and adolescent psychiatry outpatient clinic.
On subsequent evaluation in our clinic, M reports most of the same about his experience hearing “voices in my head” that repeatedly suggest “I might choke on my food and end up seriously ill in the hospital.” He started to hear the “voices” after he witnessed his sister choke while eating a few days earlier. He also mentions that the “voices” tell him “you have to use the restroom.” As a result, he uses the restroom several times before leaving for home and is frequently late for school. His parents accommodate his behavior—his mother allows him to use the bathroom multiple times, and his father overlooks the behavior as part of school anxiety.
At school, his teacher reports a concern for attention-deficit/hyperactivity disorder (ADHD) based on M’s continuous inattentiveness in class and dropping grades. He asks for bathroom breaks up to 15 times a day, which disrupts his class work.
These behaviors have led to a gradual 1-year decline in his overall functioning, including difficulty at school for requesting too many bathroom breaks; having to repeat the 3rd grade; and incurring multiple hospital visits for evaluation of his various complaints. M has become socially isolated and withdrawn from friends and family.
M’s developmental history is normal and his family history is negative for any psychiatric disorder. Medical history and physical examination are unremarkable. CT scan of his head is unremarkable, and all hematologic and biochemistry laboratory test values are within normal range.
[polldaddy:9971376]
Continue to: The authors' observations
The authors’ observations
Several factors may contribute to an increased chance of misdiagnosis of a psychiatric illness
On closer sequential evaluations with M and his family, we determined that the “voices” he was hearing were actually intrusive thoughts, and not hallucinations. M clarified this by saying that first he feels a “pressure”-like sensation in his head, followed by repeated intrusive thoughts of voiding his bladder that compel him to go to the restroom to try to urinate. He feels temporary relief after complying with the urge, even when he passes only a small amount of urine or just washes his hands. After a brief period of relief, this process repeats itself. Further, he was able to clarify his experience while eating food, where he first felt a “pressure”-like sensation in his head, followed by intrusive thoughts of choking that result in him not eating.

This led us to a more appropriate diagnosis of OCD (Table 11). The incidence of OCD has 2 peaks, with different gender distributions. The first peak occurs in childhood, with symptoms mostly arising between 7 and 12 years of age and affecting boys more often than girls. The second peak occurs in early adulthood, at a mean age of 21 years, with a slight female majority.2 However, OCD is often under recognized and undertreated, perhaps due to its extensive heterogeneity; symptom presentations and comorbidity patterns can vary noticeably between individual patients as well as age groups.
OCD is characterized by the presence of obsessions or compulsions that wax and wane in severity, are time-consuming (at least 1 hour per day), and cause subjective distress or interfere with life of the patient or the family. Adults with OCD recognize at some level that the obsessions and/or compulsions are excessive and unreasonable, although children are not required to have this insight to meet criteria for the diagnosis.1 Rating scales, such as the Children’s Yale-Brown Obsessive-Compulsive Scale, Dimensional Yale-Brown Obsessive-Compulsive Scale, and Family Accommodation Scale, are useful to obtain detailed information regarding OCD symptoms, tics, and other factors relevant to the diagnosis.
Continue to: M's symptomatology...
M’s symptomatology did not appear to be psychotic. He was screened for positive or negative symptoms of psychosis, which he and his family clearly denied. Moreover, M’s compulsions (going to the restroom) were typically performed in response to his obsessions (urge to void his bladder) to reduce his distress, which is different from schizophrenia, in which repetitive behaviors are performed in response to psychotic ideation, and not obsessions (Table 23-5).
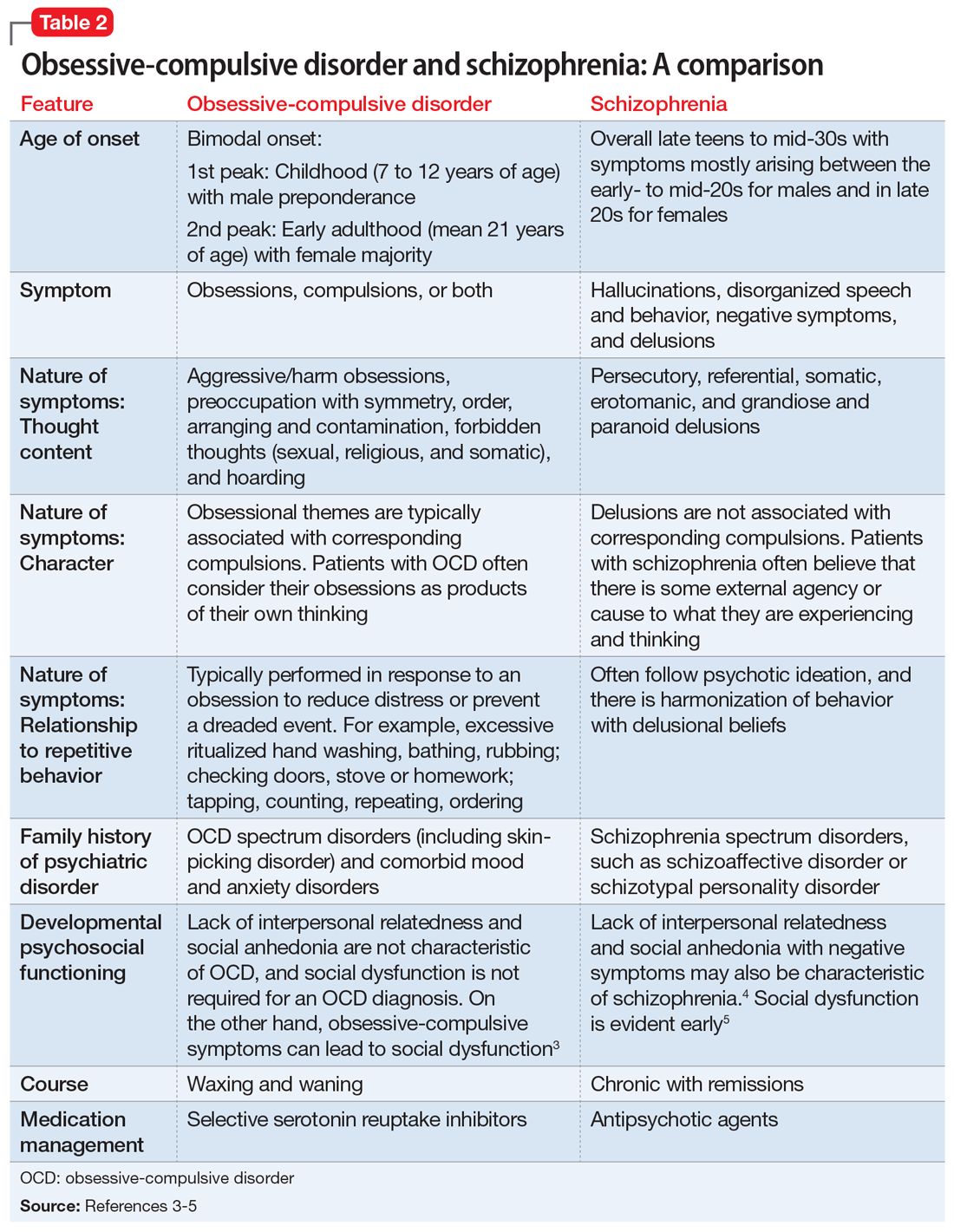
M’s inattentiveness in the classroom was found to be related to his obsessions and compulsions, and not part of a symptom cluster characterizing ADHD. Teachers often interpret inattention and poor classroom performance as ADHD, but having detailed conversations with teachers often is helpful in understanding the nature of a child’s symptomology and making the appropriate diagnosis.
Establishing the correct clinical diagnosis is critical because it is the starting point in treatment. First-line medication for one condition may exacerbate the symptoms of others. For example, in addition to having a large adverse-effect burden, antipsychotics can induce de novo obsessive–compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors (SSRIs) may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.6 Similarly, stimulant medications for ADHD may exacerbate OCS and may even induce them on their own.7,8
[polldaddy:9971377]
Continue to: The authors' observations
The authors’ observations
Studies have reported an average of 2.5 years from the onset of OCD symptoms to diagnosis in the United States.9 A key reason for this delay, which is more frequently encountered in pediatric patients, is secrecy. Children often feel embarrassed about their symptoms and conceal them until the interference with their functioning becomes extremely disabling. In some cases, symptoms may closely resemble normal childhood routines. In fact, some repetitive behaviors may be normal in some developmental stages, and OCD could be conceptualized as a pathological condition with continuity of normal behaviors during different developmental periods.10
Also, symptoms may go unnoticed for quite some time as unsuspecting and well-intentioned parents and family members become overly involved in the child’s rituals (eg, allowing for increasing frequent prolonged bathroom breaks or frequent change of clothing, etc.). This well-established phenomenon, termed accommodation, is defined as participation of family members in a child’s OCD–related rituals.11 Especially when symptoms are mild or the child is functioning well, accommodation can make it difficult for parents to realize the presence or nature of a problem, as they might tend to minimize their child’s symptoms as representing a unique personality trait or a special “quirk.” Parents generally will seek treatment when their child’s symptoms become more impairing and begin to interfere with social functioning, school performance, or family functioning.
The clinical picture is further complicated by comorbidity. Approximately 60% to 80% of children and adolescents with OCD have ≥1 comorbid psychiatric disorders. Some of the most common include tic disorders, ADHD, anxiety disorders, and mood or eating disorders.9
[polldaddy:9971379]
Continue to: TREATMENT Combination therapy
TREATMENT Combination therapy
In keeping with American Academy of Child and Adolescent Psychiatry guidelines on treating OCD (Table 312), we start M on fluoxetine 10 mg/d. He also begins CBT. Fluoxetine is slowly titrated to 40 mg/d while M engages in learning and utilizing CBT techniques to manage his OCD.
The authors’ observations
The combination of CBT and medication has been suggested as the treatment of choice for moderate and severe OCD.12 The Pediatric OCD Treatment Study, a 5-year, 3-site outcome study designed to compare placebo, sertraline, CBT, and combined CBT and sertraline, concluded that the combined treatment (CBT plus sertraline) was more effective than CBT alone or sertraline alone.13 The effect sizes for the combined treatment, CBT alone, and sertraline alone were 1.4, 0.97, and 0.67, respectively. Remission rates for SSRIs alone are <33%.13,14
SSRIs are the first-line medication for OCD in children, adolescents, and adults (Table 312). Well-designed clinical trials have demonstrated the efficacy and safety of the SSRIs fluoxetine, sertraline, and fluvoxamine (alone or combined with CBT) in children and adolescents with OCD.13 Other SSRIs, such as citalopram, paroxetine, and escitalopram, also have demonstrated efficacy in children and adolescents with OCD, even though the FDA has not yet approved their use in pediatric patients.12 Despite a positive trial of paroxetine in pediatric OCD,12 there have been concerns related to its higher rates of treatment-emergent suicidality,15 lower likelihood of treatment response,16 and its particularly short half-life in pediatric patients.17
Clomipramine is a tricyclic antidepressant with serotonergic properties that is used alone or to boost the effect of an SSRI when there is a partial response. It should be introduced at a low dose in pediatric patients (before age 12) and closely monitored for anticholinergic and cardiac adverse effects. A systemic review and meta-analysis of early treatment responses of SSRIs and clomipramine in pediatric OCD indicated that the greatest benefits occurred early in treatment.18 Clomipramine was associated with a greater measured benefit compared with placebo than SSRIs; there was no evidence of a relationship between SSRI dosing and treatment effect, although data were limited. Adults and children with OCD demonstrated a similar degree and time course of response to SSRIs in OCD.18
Treatment should start with a low dose to reduce the risk of adverse effects with an adequate trial for 10 to 16 weeks at adequate doses. Most experts suggest that treatment should continue for at least 12 months after symptom resolution or stabilization, followed by a very gradual cessation.19
Continue to: OUTCOME Improvement in functioning
OUTCOME Improvement in functioning
After 12 months of combined CBT and fluoxetine, M’s global assessment of functioning (GAF) scale score improves from 35 to 80, indicating major improvement in overall functional level.
Acknowledgement
The authors thank Uzoma Osuchukwu, MD, ex-fellow, Department of Child and Adolescent Psychiatry, Columbia University College of Physicians and Surgeons, Harlem Hospital Center, New York, New York, for his assistance with this article.
Bottom Line
Obsessive-compulsive disorder may masquerade as a schizophrenia spectrum disorder, particularly in younger patients. Accurate differentiation is crucial because antipsychotics can induce de novo obsessive-compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.
Related Resource
- Raveendranathan D, Shiva L, Sharma E, et al. Obsessive compulsive disorder masquerading as psychosis. Indian J Psychol Med. 2012;34(2):179-180.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Paroxetine • Paxil
Sertraline • Zoloft
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Geller D, Biederman J, Jones J, et al. Is juvenile obsessive-compulsive disorder a developmental subtype of the disorder? A review of the pediatric literature. J Am Acad Child Adolesc Psychiatry.1998;37(4):420-427.
3. Huppert JD, Simpson HB, Nissenson KJ, et al. Quality of life and functional impairment in obsessive-compulsive disorder: A comparison of patients with and without comorbidity, patients in remission, and healthy controls. Depress Anxiety. 2009;26(1):39-45.
4. Sobel W, Wolski R, Cancro R, et al. Interpersonal relatedness and paranoid schizophrenia. Am J Psychiatry.1996;153(8):1084-1087.
5. Meares A. The diagnosis of prepsychotic schizophrenia. Lancet. 1959;1(7063):55-58.
6. Poyurovsky M, Weizman A, Weizman R. Obsessive-compulsive disorder in schizophrenia: Clinical characteristics and treatment. CNS Drugs. 2004;18(14):989-1010.
7. Kouris S. Methylphenidate-induced obsessive-compulsiveness. J Am Acad Child Adolesc Psychiatry. 1998;37(2):135.
8. Woolley JB, Heyman I. Dexamphetamine for obsessive-compulsive disorder. Am J Psychiatry. 2003;160(1):183.
9. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin N Am. 2006;29(2):352-370.
10. Evans DW, Milanak ME, Medeiros B, et al. Magical beliefs and rituals in young children. Child Psychiatry Hum Dev. 2002;33(1):43-58.
11. Amir N, Freshman M, Foa E. Family distress and involvement in relatives of obsessive-compulsive disorder patients. J Anxiety Disord. 2000;14(3):209-217.
12. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
13. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: The Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
14. Franklin ME, Sapyta J, Freeman JB, et al. Cognitive behavior therapy augmentation of pharmacotherapy in pediatric obsessive-compulsive disorder: The Pediatric OCD Treatment Study II (POTS II) randomized controlled trial. JAMA. 2011;306(11):1224-1232.
15. Wagner KD, Asarnow JR, Vitiello B, et al. Out of the black box: treatment of resistant depression in adolescents and the antidepressant controversy. J Child Adolesc Psychopharmacol. 2012;22(1):5-10.
16. Sakolsky DJ, Perel JM, Emslie GJ, et al. Antidepressant exposure as a predictor of clinical outcomes in the treatment of resistant depression in adolescents (TORDIA) study. J Clin Psychopharmacol. 2011;31(1):92-97.
17. Findling RL. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
18. Varigonda AL, Jakubovski E, Bloch MH. Systematic review and meta-analysis: early treatment responses of selective serotonin reuptake inhibitors and clomipramine in pediatric obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2016 Oct;55(10):851-859.e2.
19. Mancuso E, Faro A, Joshi G, et al. Treatment of pediatric obsessive-compulsive disorder: a review. J Child Adolesc Psychopharmacol. 2010;20(4):299-308.
CASE Auditory hallucinations?
M, age 10, has had multiple visits to the pediatric emergency department (PED) with the chief concern of excessive urinary frequency. At each visit, the medical workup has been negative and he was discharged home. After a few months, M’s parents bring their son back to the PED because he reports hearing “voices in my head” and “feeling tense and scared.” When these feelings become too overwhelming, M stops eating and experiences substantial fear and anxiety that require his mother’s repeated reassurances. M’s mother reports that 2 weeks before his most recent PED visit, he became increasingly anxious and disturbed, and said he was afraid most of the time, and worried about the safety of his family for no apparent reason.
The psychiatrist evaluates M in the PED and diagnoses him with unspecified schizophrenia spectrum and other psychotic disorder based on his persistent report of auditory and tactile hallucinations, including hearing a voice of a man telling him he was going to choke on his food and feeling someone touch his arm to soothe him during his anxious moments. M does not meet criteria for acute inpatient hospitalization, and is discharged home with referral to follow-up at our child and adolescent psychiatry outpatient clinic.
On subsequent evaluation in our clinic, M reports most of the same about his experience hearing “voices in my head” that repeatedly suggest “I might choke on my food and end up seriously ill in the hospital.” He started to hear the “voices” after he witnessed his sister choke while eating a few days earlier. He also mentions that the “voices” tell him “you have to use the restroom.” As a result, he uses the restroom several times before leaving for home and is frequently late for school. His parents accommodate his behavior—his mother allows him to use the bathroom multiple times, and his father overlooks the behavior as part of school anxiety.
At school, his teacher reports a concern for attention-deficit/hyperactivity disorder (ADHD) based on M’s continuous inattentiveness in class and dropping grades. He asks for bathroom breaks up to 15 times a day, which disrupts his class work.
These behaviors have led to a gradual 1-year decline in his overall functioning, including difficulty at school for requesting too many bathroom breaks; having to repeat the 3rd grade; and incurring multiple hospital visits for evaluation of his various complaints. M has become socially isolated and withdrawn from friends and family.
M’s developmental history is normal and his family history is negative for any psychiatric disorder. Medical history and physical examination are unremarkable. CT scan of his head is unremarkable, and all hematologic and biochemistry laboratory test values are within normal range.
[polldaddy:9971376]
Continue to: The authors' observations
The authors’ observations
Several factors may contribute to an increased chance of misdiagnosis of a psychiatric illness
On closer sequential evaluations with M and his family, we determined that the “voices” he was hearing were actually intrusive thoughts, and not hallucinations. M clarified this by saying that first he feels a “pressure”-like sensation in his head, followed by repeated intrusive thoughts of voiding his bladder that compel him to go to the restroom to try to urinate. He feels temporary relief after complying with the urge, even when he passes only a small amount of urine or just washes his hands. After a brief period of relief, this process repeats itself. Further, he was able to clarify his experience while eating food, where he first felt a “pressure”-like sensation in his head, followed by intrusive thoughts of choking that result in him not eating.
This led us to a more appropriate diagnosis of OCD (Table 11). The incidence of OCD has 2 peaks, with different gender distributions. The first peak occurs in childhood, with symptoms mostly arising between 7 and 12 years of age and affecting boys more often than girls. The second peak occurs in early adulthood, at a mean age of 21 years, with a slight female majority.2 However, OCD is often under recognized and undertreated, perhaps due to its extensive heterogeneity; symptom presentations and comorbidity patterns can vary noticeably between individual patients as well as age groups.
OCD is characterized by the presence of obsessions or compulsions that wax and wane in severity, are time-consuming (at least 1 hour per day), and cause subjective distress or interfere with life of the patient or the family. Adults with OCD recognize at some level that the obsessions and/or compulsions are excessive and unreasonable, although children are not required to have this insight to meet criteria for the diagnosis.1 Rating scales, such as the Children’s Yale-Brown Obsessive-Compulsive Scale, Dimensional Yale-Brown Obsessive-Compulsive Scale, and Family Accommodation Scale, are useful to obtain detailed information regarding OCD symptoms, tics, and other factors relevant to the diagnosis.
Continue to: M's symptomatology...
M’s symptomatology did not appear to be psychotic. He was screened for positive or negative symptoms of psychosis, which he and his family clearly denied. Moreover, M’s compulsions (going to the restroom) were typically performed in response to his obsessions (urge to void his bladder) to reduce his distress, which is different from schizophrenia, in which repetitive behaviors are performed in response to psychotic ideation, and not obsessions (Table 23-5).
M’s inattentiveness in the classroom was found to be related to his obsessions and compulsions, and not part of a symptom cluster characterizing ADHD. Teachers often interpret inattention and poor classroom performance as ADHD, but having detailed conversations with teachers often is helpful in understanding the nature of a child’s symptomology and making the appropriate diagnosis.
Establishing the correct clinical diagnosis is critical because it is the starting point in treatment. First-line medication for one condition may exacerbate the symptoms of others. For example, in addition to having a large adverse-effect burden, antipsychotics can induce de novo obsessive–compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors (SSRIs) may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.6 Similarly, stimulant medications for ADHD may exacerbate OCS and may even induce them on their own.7,8
[polldaddy:9971377]
Continue to: The authors' observations
The authors’ observations
Studies have reported an average of 2.5 years from the onset of OCD symptoms to diagnosis in the United States.9 A key reason for this delay, which is more frequently encountered in pediatric patients, is secrecy. Children often feel embarrassed about their symptoms and conceal them until the interference with their functioning becomes extremely disabling. In some cases, symptoms may closely resemble normal childhood routines. In fact, some repetitive behaviors may be normal in some developmental stages, and OCD could be conceptualized as a pathological condition with continuity of normal behaviors during different developmental periods.10
Also, symptoms may go unnoticed for quite some time as unsuspecting and well-intentioned parents and family members become overly involved in the child’s rituals (eg, allowing for increasing frequent prolonged bathroom breaks or frequent change of clothing, etc.). This well-established phenomenon, termed accommodation, is defined as participation of family members in a child’s OCD–related rituals.11 Especially when symptoms are mild or the child is functioning well, accommodation can make it difficult for parents to realize the presence or nature of a problem, as they might tend to minimize their child’s symptoms as representing a unique personality trait or a special “quirk.” Parents generally will seek treatment when their child’s symptoms become more impairing and begin to interfere with social functioning, school performance, or family functioning.
The clinical picture is further complicated by comorbidity. Approximately 60% to 80% of children and adolescents with OCD have ≥1 comorbid psychiatric disorders. Some of the most common include tic disorders, ADHD, anxiety disorders, and mood or eating disorders.9
[polldaddy:9971379]
Continue to: TREATMENT Combination therapy
TREATMENT Combination therapy
In keeping with American Academy of Child and Adolescent Psychiatry guidelines on treating OCD (Table 312), we start M on fluoxetine 10 mg/d. He also begins CBT. Fluoxetine is slowly titrated to 40 mg/d while M engages in learning and utilizing CBT techniques to manage his OCD.
The authors’ observations
The combination of CBT and medication has been suggested as the treatment of choice for moderate and severe OCD.12 The Pediatric OCD Treatment Study, a 5-year, 3-site outcome study designed to compare placebo, sertraline, CBT, and combined CBT and sertraline, concluded that the combined treatment (CBT plus sertraline) was more effective than CBT alone or sertraline alone.13 The effect sizes for the combined treatment, CBT alone, and sertraline alone were 1.4, 0.97, and 0.67, respectively. Remission rates for SSRIs alone are <33%.13,14
SSRIs are the first-line medication for OCD in children, adolescents, and adults (Table 312). Well-designed clinical trials have demonstrated the efficacy and safety of the SSRIs fluoxetine, sertraline, and fluvoxamine (alone or combined with CBT) in children and adolescents with OCD.13 Other SSRIs, such as citalopram, paroxetine, and escitalopram, also have demonstrated efficacy in children and adolescents with OCD, even though the FDA has not yet approved their use in pediatric patients.12 Despite a positive trial of paroxetine in pediatric OCD,12 there have been concerns related to its higher rates of treatment-emergent suicidality,15 lower likelihood of treatment response,16 and its particularly short half-life in pediatric patients.17
Clomipramine is a tricyclic antidepressant with serotonergic properties that is used alone or to boost the effect of an SSRI when there is a partial response. It should be introduced at a low dose in pediatric patients (before age 12) and closely monitored for anticholinergic and cardiac adverse effects. A systemic review and meta-analysis of early treatment responses of SSRIs and clomipramine in pediatric OCD indicated that the greatest benefits occurred early in treatment.18 Clomipramine was associated with a greater measured benefit compared with placebo than SSRIs; there was no evidence of a relationship between SSRI dosing and treatment effect, although data were limited. Adults and children with OCD demonstrated a similar degree and time course of response to SSRIs in OCD.18
Treatment should start with a low dose to reduce the risk of adverse effects with an adequate trial for 10 to 16 weeks at adequate doses. Most experts suggest that treatment should continue for at least 12 months after symptom resolution or stabilization, followed by a very gradual cessation.19
Continue to: OUTCOME Improvement in functioning
OUTCOME Improvement in functioning
After 12 months of combined CBT and fluoxetine, M’s global assessment of functioning (GAF) scale score improves from 35 to 80, indicating major improvement in overall functional level.
Acknowledgement
The authors thank Uzoma Osuchukwu, MD, ex-fellow, Department of Child and Adolescent Psychiatry, Columbia University College of Physicians and Surgeons, Harlem Hospital Center, New York, New York, for his assistance with this article.
Bottom Line
Obsessive-compulsive disorder may masquerade as a schizophrenia spectrum disorder, particularly in younger patients. Accurate differentiation is crucial because antipsychotics can induce de novo obsessive-compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.
Related Resource
- Raveendranathan D, Shiva L, Sharma E, et al. Obsessive compulsive disorder masquerading as psychosis. Indian J Psychol Med. 2012;34(2):179-180.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Paroxetine • Paxil
Sertraline • Zoloft
CASE Auditory hallucinations?
M, age 10, has had multiple visits to the pediatric emergency department (PED) with the chief concern of excessive urinary frequency. At each visit, the medical workup has been negative and he was discharged home. After a few months, M’s parents bring their son back to the PED because he reports hearing “voices in my head” and “feeling tense and scared.” When these feelings become too overwhelming, M stops eating and experiences substantial fear and anxiety that require his mother’s repeated reassurances. M’s mother reports that 2 weeks before his most recent PED visit, he became increasingly anxious and disturbed, and said he was afraid most of the time, and worried about the safety of his family for no apparent reason.
The psychiatrist evaluates M in the PED and diagnoses him with unspecified schizophrenia spectrum and other psychotic disorder based on his persistent report of auditory and tactile hallucinations, including hearing a voice of a man telling him he was going to choke on his food and feeling someone touch his arm to soothe him during his anxious moments. M does not meet criteria for acute inpatient hospitalization, and is discharged home with referral to follow-up at our child and adolescent psychiatry outpatient clinic.
On subsequent evaluation in our clinic, M reports most of the same about his experience hearing “voices in my head” that repeatedly suggest “I might choke on my food and end up seriously ill in the hospital.” He started to hear the “voices” after he witnessed his sister choke while eating a few days earlier. He also mentions that the “voices” tell him “you have to use the restroom.” As a result, he uses the restroom several times before leaving for home and is frequently late for school. His parents accommodate his behavior—his mother allows him to use the bathroom multiple times, and his father overlooks the behavior as part of school anxiety.
At school, his teacher reports a concern for attention-deficit/hyperactivity disorder (ADHD) based on M’s continuous inattentiveness in class and dropping grades. He asks for bathroom breaks up to 15 times a day, which disrupts his class work.
These behaviors have led to a gradual 1-year decline in his overall functioning, including difficulty at school for requesting too many bathroom breaks; having to repeat the 3rd grade; and incurring multiple hospital visits for evaluation of his various complaints. M has become socially isolated and withdrawn from friends and family.
M’s developmental history is normal and his family history is negative for any psychiatric disorder. Medical history and physical examination are unremarkable. CT scan of his head is unremarkable, and all hematologic and biochemistry laboratory test values are within normal range.
[polldaddy:9971376]
Continue to: The authors' observations
The authors’ observations
Several factors may contribute to an increased chance of misdiagnosis of a psychiatric illness
On closer sequential evaluations with M and his family, we determined that the “voices” he was hearing were actually intrusive thoughts, and not hallucinations. M clarified this by saying that first he feels a “pressure”-like sensation in his head, followed by repeated intrusive thoughts of voiding his bladder that compel him to go to the restroom to try to urinate. He feels temporary relief after complying with the urge, even when he passes only a small amount of urine or just washes his hands. After a brief period of relief, this process repeats itself. Further, he was able to clarify his experience while eating food, where he first felt a “pressure”-like sensation in his head, followed by intrusive thoughts of choking that result in him not eating.
This led us to a more appropriate diagnosis of OCD (Table 11). The incidence of OCD has 2 peaks, with different gender distributions. The first peak occurs in childhood, with symptoms mostly arising between 7 and 12 years of age and affecting boys more often than girls. The second peak occurs in early adulthood, at a mean age of 21 years, with a slight female majority.2 However, OCD is often under recognized and undertreated, perhaps due to its extensive heterogeneity; symptom presentations and comorbidity patterns can vary noticeably between individual patients as well as age groups.
OCD is characterized by the presence of obsessions or compulsions that wax and wane in severity, are time-consuming (at least 1 hour per day), and cause subjective distress or interfere with life of the patient or the family. Adults with OCD recognize at some level that the obsessions and/or compulsions are excessive and unreasonable, although children are not required to have this insight to meet criteria for the diagnosis.1 Rating scales, such as the Children’s Yale-Brown Obsessive-Compulsive Scale, Dimensional Yale-Brown Obsessive-Compulsive Scale, and Family Accommodation Scale, are useful to obtain detailed information regarding OCD symptoms, tics, and other factors relevant to the diagnosis.
Continue to: M's symptomatology...
M’s symptomatology did not appear to be psychotic. He was screened for positive or negative symptoms of psychosis, which he and his family clearly denied. Moreover, M’s compulsions (going to the restroom) were typically performed in response to his obsessions (urge to void his bladder) to reduce his distress, which is different from schizophrenia, in which repetitive behaviors are performed in response to psychotic ideation, and not obsessions (Table 23-5).
M’s inattentiveness in the classroom was found to be related to his obsessions and compulsions, and not part of a symptom cluster characterizing ADHD. Teachers often interpret inattention and poor classroom performance as ADHD, but having detailed conversations with teachers often is helpful in understanding the nature of a child’s symptomology and making the appropriate diagnosis.
Establishing the correct clinical diagnosis is critical because it is the starting point in treatment. First-line medication for one condition may exacerbate the symptoms of others. For example, in addition to having a large adverse-effect burden, antipsychotics can induce de novo obsessive–compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors (SSRIs) may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.6 Similarly, stimulant medications for ADHD may exacerbate OCS and may even induce them on their own.7,8
[polldaddy:9971377]
Continue to: The authors' observations
The authors’ observations
Studies have reported an average of 2.5 years from the onset of OCD symptoms to diagnosis in the United States.9 A key reason for this delay, which is more frequently encountered in pediatric patients, is secrecy. Children often feel embarrassed about their symptoms and conceal them until the interference with their functioning becomes extremely disabling. In some cases, symptoms may closely resemble normal childhood routines. In fact, some repetitive behaviors may be normal in some developmental stages, and OCD could be conceptualized as a pathological condition with continuity of normal behaviors during different developmental periods.10
Also, symptoms may go unnoticed for quite some time as unsuspecting and well-intentioned parents and family members become overly involved in the child’s rituals (eg, allowing for increasing frequent prolonged bathroom breaks or frequent change of clothing, etc.). This well-established phenomenon, termed accommodation, is defined as participation of family members in a child’s OCD–related rituals.11 Especially when symptoms are mild or the child is functioning well, accommodation can make it difficult for parents to realize the presence or nature of a problem, as they might tend to minimize their child’s symptoms as representing a unique personality trait or a special “quirk.” Parents generally will seek treatment when their child’s symptoms become more impairing and begin to interfere with social functioning, school performance, or family functioning.
The clinical picture is further complicated by comorbidity. Approximately 60% to 80% of children and adolescents with OCD have ≥1 comorbid psychiatric disorders. Some of the most common include tic disorders, ADHD, anxiety disorders, and mood or eating disorders.9
[polldaddy:9971379]
Continue to: TREATMENT Combination therapy
TREATMENT Combination therapy
In keeping with American Academy of Child and Adolescent Psychiatry guidelines on treating OCD (Table 312), we start M on fluoxetine 10 mg/d. He also begins CBT. Fluoxetine is slowly titrated to 40 mg/d while M engages in learning and utilizing CBT techniques to manage his OCD.
The authors’ observations
The combination of CBT and medication has been suggested as the treatment of choice for moderate and severe OCD.12 The Pediatric OCD Treatment Study, a 5-year, 3-site outcome study designed to compare placebo, sertraline, CBT, and combined CBT and sertraline, concluded that the combined treatment (CBT plus sertraline) was more effective than CBT alone or sertraline alone.13 The effect sizes for the combined treatment, CBT alone, and sertraline alone were 1.4, 0.97, and 0.67, respectively. Remission rates for SSRIs alone are <33%.13,14
SSRIs are the first-line medication for OCD in children, adolescents, and adults (Table 312). Well-designed clinical trials have demonstrated the efficacy and safety of the SSRIs fluoxetine, sertraline, and fluvoxamine (alone or combined with CBT) in children and adolescents with OCD.13 Other SSRIs, such as citalopram, paroxetine, and escitalopram, also have demonstrated efficacy in children and adolescents with OCD, even though the FDA has not yet approved their use in pediatric patients.12 Despite a positive trial of paroxetine in pediatric OCD,12 there have been concerns related to its higher rates of treatment-emergent suicidality,15 lower likelihood of treatment response,16 and its particularly short half-life in pediatric patients.17
Clomipramine is a tricyclic antidepressant with serotonergic properties that is used alone or to boost the effect of an SSRI when there is a partial response. It should be introduced at a low dose in pediatric patients (before age 12) and closely monitored for anticholinergic and cardiac adverse effects. A systemic review and meta-analysis of early treatment responses of SSRIs and clomipramine in pediatric OCD indicated that the greatest benefits occurred early in treatment.18 Clomipramine was associated with a greater measured benefit compared with placebo than SSRIs; there was no evidence of a relationship between SSRI dosing and treatment effect, although data were limited. Adults and children with OCD demonstrated a similar degree and time course of response to SSRIs in OCD.18
Treatment should start with a low dose to reduce the risk of adverse effects with an adequate trial for 10 to 16 weeks at adequate doses. Most experts suggest that treatment should continue for at least 12 months after symptom resolution or stabilization, followed by a very gradual cessation.19
Continue to: OUTCOME Improvement in functioning
OUTCOME Improvement in functioning
After 12 months of combined CBT and fluoxetine, M’s global assessment of functioning (GAF) scale score improves from 35 to 80, indicating major improvement in overall functional level.
Acknowledgement
The authors thank Uzoma Osuchukwu, MD, ex-fellow, Department of Child and Adolescent Psychiatry, Columbia University College of Physicians and Surgeons, Harlem Hospital Center, New York, New York, for his assistance with this article.
Bottom Line
Obsessive-compulsive disorder may masquerade as a schizophrenia spectrum disorder, particularly in younger patients. Accurate differentiation is crucial because antipsychotics can induce de novo obsessive-compulsive symptoms (OCS) or exacerbate preexisting OCS, and selective serotonin reuptake inhibitors may exacerbate psychosis in schizo-obsessive patients with a history of impulsivity and aggressiveness.
Related Resource
- Raveendranathan D, Shiva L, Sharma E, et al. Obsessive compulsive disorder masquerading as psychosis. Indian J Psychol Med. 2012;34(2):179-180.
Drug Brand Names
Citalopram • Celexa
Clomipramine • Anafranil
Escitalopram • Lexapro
Fluoxetine • Prozac
Fluvoxamine • Luvox
Paroxetine • Paxil
Sertraline • Zoloft
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Geller D, Biederman J, Jones J, et al. Is juvenile obsessive-compulsive disorder a developmental subtype of the disorder? A review of the pediatric literature. J Am Acad Child Adolesc Psychiatry.1998;37(4):420-427.
3. Huppert JD, Simpson HB, Nissenson KJ, et al. Quality of life and functional impairment in obsessive-compulsive disorder: A comparison of patients with and without comorbidity, patients in remission, and healthy controls. Depress Anxiety. 2009;26(1):39-45.
4. Sobel W, Wolski R, Cancro R, et al. Interpersonal relatedness and paranoid schizophrenia. Am J Psychiatry.1996;153(8):1084-1087.
5. Meares A. The diagnosis of prepsychotic schizophrenia. Lancet. 1959;1(7063):55-58.
6. Poyurovsky M, Weizman A, Weizman R. Obsessive-compulsive disorder in schizophrenia: Clinical characteristics and treatment. CNS Drugs. 2004;18(14):989-1010.
7. Kouris S. Methylphenidate-induced obsessive-compulsiveness. J Am Acad Child Adolesc Psychiatry. 1998;37(2):135.
8. Woolley JB, Heyman I. Dexamphetamine for obsessive-compulsive disorder. Am J Psychiatry. 2003;160(1):183.
9. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin N Am. 2006;29(2):352-370.
10. Evans DW, Milanak ME, Medeiros B, et al. Magical beliefs and rituals in young children. Child Psychiatry Hum Dev. 2002;33(1):43-58.
11. Amir N, Freshman M, Foa E. Family distress and involvement in relatives of obsessive-compulsive disorder patients. J Anxiety Disord. 2000;14(3):209-217.
12. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
13. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: The Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
14. Franklin ME, Sapyta J, Freeman JB, et al. Cognitive behavior therapy augmentation of pharmacotherapy in pediatric obsessive-compulsive disorder: The Pediatric OCD Treatment Study II (POTS II) randomized controlled trial. JAMA. 2011;306(11):1224-1232.
15. Wagner KD, Asarnow JR, Vitiello B, et al. Out of the black box: treatment of resistant depression in adolescents and the antidepressant controversy. J Child Adolesc Psychopharmacol. 2012;22(1):5-10.
16. Sakolsky DJ, Perel JM, Emslie GJ, et al. Antidepressant exposure as a predictor of clinical outcomes in the treatment of resistant depression in adolescents (TORDIA) study. J Clin Psychopharmacol. 2011;31(1):92-97.
17. Findling RL. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
18. Varigonda AL, Jakubovski E, Bloch MH. Systematic review and meta-analysis: early treatment responses of selective serotonin reuptake inhibitors and clomipramine in pediatric obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2016 Oct;55(10):851-859.e2.
19. Mancuso E, Faro A, Joshi G, et al. Treatment of pediatric obsessive-compulsive disorder: a review. J Child Adolesc Psychopharmacol. 2010;20(4):299-308.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Geller D, Biederman J, Jones J, et al. Is juvenile obsessive-compulsive disorder a developmental subtype of the disorder? A review of the pediatric literature. J Am Acad Child Adolesc Psychiatry.1998;37(4):420-427.
3. Huppert JD, Simpson HB, Nissenson KJ, et al. Quality of life and functional impairment in obsessive-compulsive disorder: A comparison of patients with and without comorbidity, patients in remission, and healthy controls. Depress Anxiety. 2009;26(1):39-45.
4. Sobel W, Wolski R, Cancro R, et al. Interpersonal relatedness and paranoid schizophrenia. Am J Psychiatry.1996;153(8):1084-1087.
5. Meares A. The diagnosis of prepsychotic schizophrenia. Lancet. 1959;1(7063):55-58.
6. Poyurovsky M, Weizman A, Weizman R. Obsessive-compulsive disorder in schizophrenia: Clinical characteristics and treatment. CNS Drugs. 2004;18(14):989-1010.
7. Kouris S. Methylphenidate-induced obsessive-compulsiveness. J Am Acad Child Adolesc Psychiatry. 1998;37(2):135.
8. Woolley JB, Heyman I. Dexamphetamine for obsessive-compulsive disorder. Am J Psychiatry. 2003;160(1):183.
9. Geller DA. Obsessive-compulsive and spectrum disorders in children and adolescents. Psychiatr Clin N Am. 2006;29(2):352-370.
10. Evans DW, Milanak ME, Medeiros B, et al. Magical beliefs and rituals in young children. Child Psychiatry Hum Dev. 2002;33(1):43-58.
11. Amir N, Freshman M, Foa E. Family distress and involvement in relatives of obsessive-compulsive disorder patients. J Anxiety Disord. 2000;14(3):209-217.
12. Practice parameter for the assessment and treatment of children and adolescents with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2012;51(1):98-113.
13. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: The Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969-1976.
14. Franklin ME, Sapyta J, Freeman JB, et al. Cognitive behavior therapy augmentation of pharmacotherapy in pediatric obsessive-compulsive disorder: The Pediatric OCD Treatment Study II (POTS II) randomized controlled trial. JAMA. 2011;306(11):1224-1232.
15. Wagner KD, Asarnow JR, Vitiello B, et al. Out of the black box: treatment of resistant depression in adolescents and the antidepressant controversy. J Child Adolesc Psychopharmacol. 2012;22(1):5-10.
16. Sakolsky DJ, Perel JM, Emslie GJ, et al. Antidepressant exposure as a predictor of clinical outcomes in the treatment of resistant depression in adolescents (TORDIA) study. J Clin Psychopharmacol. 2011;31(1):92-97.
17. Findling RL. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
18. Varigonda AL, Jakubovski E, Bloch MH. Systematic review and meta-analysis: early treatment responses of selective serotonin reuptake inhibitors and clomipramine in pediatric obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2016 Oct;55(10):851-859.e2.
19. Mancuso E, Faro A, Joshi G, et al. Treatment of pediatric obsessive-compulsive disorder: a review. J Child Adolesc Psychopharmacol. 2010;20(4):299-308.