User login
IgA Vasculitis in the Setting of Biologic Therapy for Psoriasis and Recurrent Cutaneous Methicillin-Resistant Staphylococcus aureus Colonization
Case Report
A 47-year-old man presented with a sudden-onset rash consisting of red bumps on the abdomen and legs that had been ongoing for several days. He had known psoriasis and psoriatic arthritis that had been well controlled with adalimumab for the last 18 months. He reported concurrent onset of nausea but denied fevers, chills, night sweats, unintentional weight loss, abdominal pain, and pruritus. He endorsed prior cutaneous infections of methicillin-resistant Staphylococcus aureus (MRSA). His medical history also included diabetes mellitus, hypertension, and obesity. His other medications included oral losartan-hydrochlorothiazide, amlodipine, naproxen, and atorvastatin.
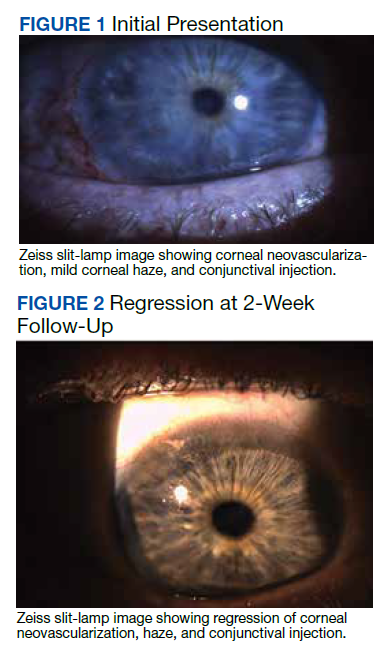
Physical examination revealed numerous thin purpuric papules—some with adherent scale—distributed on the lower legs, extensor forearms, and abdomen. Abdominal lesions were confined to weight-related striae (Figure 1). The palms, soles, oral mucosa, and face were spared. Three punch biopsies were performed, including 1 for direct immunofluorescence (DIF), and the patient was instructed to apply clobetasol to the affected areas twice daily until further notice.

Pathology showed perivascular extravasation of erythrocytes, neutrophils, eosinophils, and leukocytoclasis surrounding blood vessels associated with fibrin (Figure 2). Direct immunofluorescence showed granular deposition of IgA, complement component 3, and fibrinogen in a superficial dermal vascular pattern (Figure 3). These results were consistent with IgA small-vessel vasculitis. One specimen was consistent with the patient’s known psoriasis.
.")
Urinalysis revealed moderate hemoglobinuria, and urine microscopy showed 174 red blood cells per high-power field. Creatinine was high at 1.87 mg/dL (reference range, <1.34 mg/dL; patient’s baseline, 0.81 mg/dL) and glomerular filtration rate was low (42 mL/min, patient’s baseline, >60 mL/min [reference range, 90–120 mL/min]). Erythrocyte sedimentation rate (21 mm/h [reference range, 0–22 mm/h]) and C-reactive protein were elevated (2.2 mg/dL [reference range, 0.3–1.0 mg/dL]). Given his history of cutaneous MRSA infections, a bacterial culture swab was collected from the skin surface to check for colonization, which showed moderate growth of MRSA. Naproxen was discontinued over concern of worsening the patient’s renal status. The patient was instructed to rest at home with his legs elevated, wear compression socks when ambulatory, use chlorhexidine antiseptic daily as a body wash when showering, and apply mupirocin three times daily to the biopsy sites. He was referred to urology for his microhematuria, where cystoscopy revealed no abnormalities.A month passed with no improvement of the patient’s cutaneous vasculitis, and his psoriatic arthritis worsened without his usual use of naproxen. He developed abdominal pain and loss of appetite. A prednisone taper was ordered starting at 40 mg/d (28.8 mg/kg), which provided relief of the skin and joint symptoms only until the course was completed 12 days later.
.")
Five weeks after the initial presentation, the patient returned with a more severe eruption consisting of innumerable purpuric papules that coalesced in plaques on the abdomen, arms, and legs. He also had erythematous facial pustules and mild palmar petechiae (Figure 4). Three biopsies were performed, including 1 for DIF and 1 from a pustule on the forehead. Histology and DIF were again consistent with IgA small-vessel vasculitis. The forehead biopsy was compatible with steroid acne (attributed to recent prednisone use) and psoriasis.

Rheumatology was consulted, and adalimumab was discontinued 6 weeks after the initial presentation out of concern for drug-induced cutaneous vasculitis. Vasculitis work-up was unremarkable, including antineutrophil cytoplasmic antibodies, rheumatoid factor, cyclic citrullinated peptide, and serum protein electrophoresis. Oral dapsone was started at 100 mg/d, with the tentative plan of starting secukinumab if cutaneous symptoms improved. For 3 weeks, the patient’s cutaneous symptoms steadily improved.
Nine weeks after initial presentation to dermatology (3 weeks after discontinuing adalimumab) the patient self-administered his first dose of secukinumab at home. Several hours later, he reported sudden reappearance of vasculitis. He denied diarrhea, abdominal pain, bowel movement urgency, fevers, fatigue, and unintentional weight loss. Antistreptolysin O and hepatitis A antibodies were negative. He was instructed to hold secukinumab indefinitely.
Four weeks after his only secukinumab injection, the patient reported another episode of acute worsening cutaneous symptoms. A 4-week prednisone taper starting at 40 mg/d was ordered. Computed tomography of the chest, abdomen, and pelvis to rule out internal malignancy was unremarkable. Around this time, the patient reported major emotional distress related to an unexpected death in his family, which added to a gradual increase in his stress level related to the COVID-19 pandemic.
Three weeks later, dapsone was increased to 100 mg twice daily on account of the patient’s adiposity and lack of cutaneous improvement on the lower dose. Subsequently, the vasculitis rapidly improved for 2 weeks. The patient then reported symptoms of headache, dizziness, and chills. He was tested for COVID-19 and was negative. Six weeks after increasing the dapsone dose (5 months after initial presentation), the skin was normalizing, showing only faintly hyperpigmented macules confined to areas of resolved vasculitis (forearms, abdomen, legs).
The patient had been on dapsone 100 mg twice daily for 3 months when he was started on ustekinumab (90 mg at weeks 0 and 4, with planned doses every 12 weeks) for psoriatic arthritis in hopes of withdrawing dapsone. His cutaneous symptoms have remained well controlled on this regimen for 18 months. Lowering of dapsone below 100 mg daily has resulted in recurrent mild vasculitis symptoms; he now maintains the once-daily dosing without negative side effects.
Comment
IgA vasculitis is a form of cutaneous small-vessel leukocytoclastic vasculitis (LCV) characterized by episodes of palpable purpura on the extensor surfaces of the arms and legs that may be associated with arthritis, abdominal pain, and/or hematuria. Although vasculitis is a known potential adverse effect of anti–tumor necrosis factor (TNF) α therapy, cases of adalimumab-induced IgA vasculitis are uncommon. As use of more targeted therapies for psoriasis and psoriatic arthritis, such as the IL-17 inhibitor secukinumab, increases so do reports of associated adverse events. Of 6 previously reported cases of secukinumab-associated vasculitis, at least 4 were IgA vasculitis (Table).1-6 Another case described one patient with rheumatoid arthritis undergoing secukinumab treatment who experienced necrotizing glomerulonephritis; however, the authors concluded secukinumab likely was not causative in that case, as serologies and urinalyses suggested gradual onset of the process prior to initiating the medication.7

The exact pathogenesis of IgA vasculitis is unclear, but a prevailing theory involves the dysregulation of IgA synthesis and metabolism. Other than increased serum levels of transforming growth factor β, which is a major stimulating factor for IgA production, it also has been hypothesized that the presence of aberrantly hypoglycosylated IgA exposes an autoepitope for recognition by other pathogenic IgG and IgA, leading to the formation of large immune complexes that can readily deposit in postcapillary venules. The deposition of IgA immune complexes in postcapillary venules and the subsequent activation of the complement system causes direct damage to the endothelial cells of vessel walls. This complement activation is evidenced by vascular complement component 3 deposition on DIF (a nonspecific feature of LCV). Chemotaxis of neutrophils ensues, followed by their firm adherence and transendothelial migration (mediated by monocyte chemoattractant protein 1 [MCP-1]). Neutrophil degranulation releases reactive oxygen species and cytokines, which in turn recruit additional leukocytes to the area of inflammation, subsequently undergoing degeneration (leukocytoclasis). Microvascular permeability also is enhanced by MCP-1, allowing exudation of serum, erythrocytes, and fibrin. In the setting of elevated circulating TNF and IL-1, endothelium is stimulated to activate the intrinsic and extrinsic coagulation pathways. This decreases endothelial fibrinolytic activity, leading to thrombosis. The high venous pressure and low fibrinolytic activity in the lower legs explains why vasculitic lesions often are confined to or begin in this distribution.1,8-10

There also are noteworthy roles for cytokines in LCV. Circulating transforming growth factor β and IL-6—which are necessary for development of T helper 17 (TH17) cells and production of IL-17—are higher in patients with LCV compared to controls. Peripheral blood monocytes in patients with LCV demonstrate higher production of IL-17. Once TH17 cells develop, their survival and phenotype are maintained by IL-23 (considered the master regulator of TH17 differentiation). IL-17 is a potent chemoattractant of IL-8 (CXCL8) and MCP-1, both of which promote neutrophil-mediated perivascular inflammation. The IL-23 and IL-17 pathways implicated in the pathogenesis of psoriasis also cause neutrophil activation and upregulate transcription of proinflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α), which overlap with those implicated in LCV. Autoimmune disease generally entails some positive feedback loop of progressively severe self-recognition and tissue destruction by the immune system. These shared cytokinetic processes may explain how the internal environment of psoriasis could perpetuate IgA vasculitis.1,2,8,10-12
The mechanisms underlying vasculitis associated with adalimumab are unclear, but hypotheses involve direct toxicity on vessels, capillary deposition of anti-TNF/TNF immune complexes, or an inflammatory process resulting in autoantibodies. Similar hypotheses are posited for secukinumab-associated vasculitis, including deposition of secukinumab–IL-17 complexes. Anti–TNF-α medications may increase TH17 cell numbers, leading to increased production of IL-22 and a resultant immunologic microenvironment conducive to vasculitis. All 6 published cases of secukinumab-associated vasculitis that we found had received prior treatment with a TNF-α blocker, but only 1 had occurrence of vasculitis during that treatment.1-6,10
In the 6 cases we reviewed, the time from starting secukinumab to onset of vasculitis ranged from 1 to 18 months. Our patient’s same-day re-emergence of vasculitis after his first secukinumab dose was so acute that we were skeptical of secukinumab as a potential trigger; this may simply have been coincident to the natural waxing and waning of the vasculitis (although onset of IgA vasculitis within 1 day of starting anti–TNF-α therapy has been reported).1-6,13
Specific associations of IgA vasculitis are many and can include bacterial organisms such as Helicobacter pylori, streptococci, and staphylococci. Although internal mucous membrane infections are considered more linked because of the surveillance role of IgA predominantly in mucosal tissues, it is possible that our patient with cutaneous MRSA harbored the same within the nasal mucosa. Our patient also received multiple vaccinations outside our department throughout his clinical course (2 hepatitis B and 1 pneumococcal conjugate), which are known potential triggers for vasculitis. Psychological stress is a known trigger for psoriasis, and given the cytokinetic relationship of psoriasis to vasculitis described previously, it may have indirectly contributed to vasculitis in our case. The anxiety associated with being immunosuppressed during the COVID-19 pandemic and bereavement of losing a family member may have contributed to the refractory nature of our patient’s condition. Renal involvement is relatively common in adults with IgA vasculitis and so should be ruled out, as should occult internal malignancy.8,10,14
It is unclear which of the above factors was causative in our case, but a multifactorial process is likely. Treatment of monoclonal antibody–associated vasculitis entails investigating for triggers and systemic involvement, removing the most likely culprit, quelling the vasculitis acutely, avoiding known potential exacerbators, and introducing an alternative long-term immunomodulant. In all 6 reported similar cases, discontinuation of secukinumab and initiation of prednisone or colchicine led to resolution.1-6 Dapsone also is acceptable for acute control of IgA vasculitis, although this medication is highly lipid soluble and penetrates well into various tissues.15 Thus, lower doses may prove ineffective for obese patients, as was demonstrated in our case. Given the known potential of vaccinations, infections, and other factors (eg, alcohol, penicillin) to trigger IgA vasculitis, these should be avoided.10
Blockade of IL-23 with ustekinumab has been suggested by other authors encountering secukinumab-associated vasculitis, as IL-23 is the main driver and sustainer of TH17 cell differentiation.8 Although 6 previously reported cases of secukinumab-associated vasculitis achieved resolution without long-term recurrence, none did so using an IL-23 inhibitor (nor had any of the described patients received IL-23 inhibitors previously).1-6 Given the established safety of IL-23 inhibitors and that they theoretically are well suited for this unique circumstance (by ceasing the main causative cytokine cascades “upstream”) and were efficacious in quickly resolving our patient’s vasculitis, we suggest that ustekinumab may represent
- Reverte M, Etienne M, Fouchard M, et al. Occurrence of Henoch-Schönlein purpura in a patient treated with secukinumab. J Eur Acad Dermatol Venereol. 2019;33:E455-E457.
- Chelli C, Loget J, Vanhaecke C, et al. Cutaneous vasculitis with gut involvement during secukinumab treatment for psoriatic arthritis. Acta Derm Venereol. 2020;100:adv00077.
- da Silva Cendon Duran C, Santiago MB. Cutaneous vasculitis during secukinumab treatment. Eur J Case Rep Intern Med. 2020;7:001815.
- Bostan E, Gulseren D, Yalici-Armagan B, et al. Vasculitis during certolizumab pegol and secukinumab treatment: report of two cases. Dermatol Ther. 2021;34:E15007.
- Perkovic D, Simac P, Katic J. IgA vasculitis during secukinumab therapy. Clin Rheumatol. 2021;40:2071-2073.
- Villani A, DE Fata Salvatores G, Nappa P, et al. Cutaneous leucocytoclastic vasculitis during secukinumab treatment. Ital J Dermatol Venerol. 2021;156(suppl 1 to no. 6):9-10.
- Góis M, Messias A, Carvalho D, et al. MPO-ANCA-associated necrotizing glomerulonephritis in rheumatoid arthritis; a case report and review of literature. J Nephropathol. 2017;6:58-62.
- Jen HY, Chuang YH, Lin SC, et al. Increased serum interleukin-17 and peripheral Th17 cells in children with acute Henoch-Schönlein purpura. Pediatr Allergy Immunol. 2011;22:862-868.
- Hetland LE, Susrud KS, Lindahl KH, et al. Henoch-Schönlein purpura: a literature review. Acta Derm Venereol 2017;97:1160-1166.
- Weedon D. The vasculopathic reaction pattern. In: Houston M, Davie B, eds. Weedon’s Skin Pathology. 3rd ed. Elsevier Limited; 2010:207-211.
- Puig L. Paradoxical reactions: anti-TNFα ants, ustekinumab, secukinumab, ixekizumab, and others. Curr Probl Dermatol. 2018;53:49-63.
- Nestle F, Kaplan D, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Pinheiro RR, Lencastre A. Henoch-Schönlein purpura during anti-TNFα therapy: a fortuitous event or an indication to stop therapy? Eur J Dermatol. 2017;27:304-305.
- Hello CL, Cohen P, Bousser MG, et al. Suspected hepatitis B vaccination related vasculitis. J Rheumatol. 1999;26:191-194.
- Wolverton SE. Dapsone. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier, Inc; 2021:222-231.
Case Report
A 47-year-old man presented with a sudden-onset rash consisting of red bumps on the abdomen and legs that had been ongoing for several days. He had known psoriasis and psoriatic arthritis that had been well controlled with adalimumab for the last 18 months. He reported concurrent onset of nausea but denied fevers, chills, night sweats, unintentional weight loss, abdominal pain, and pruritus. He endorsed prior cutaneous infections of methicillin-resistant Staphylococcus aureus (MRSA). His medical history also included diabetes mellitus, hypertension, and obesity. His other medications included oral losartan-hydrochlorothiazide, amlodipine, naproxen, and atorvastatin.
Physical examination revealed numerous thin purpuric papules—some with adherent scale—distributed on the lower legs, extensor forearms, and abdomen. Abdominal lesions were confined to weight-related striae (Figure 1). The palms, soles, oral mucosa, and face were spared. Three punch biopsies were performed, including 1 for direct immunofluorescence (DIF), and the patient was instructed to apply clobetasol to the affected areas twice daily until further notice.
Pathology showed perivascular extravasation of erythrocytes, neutrophils, eosinophils, and leukocytoclasis surrounding blood vessels associated with fibrin (Figure 2). Direct immunofluorescence showed granular deposition of IgA, complement component 3, and fibrinogen in a superficial dermal vascular pattern (Figure 3). These results were consistent with IgA small-vessel vasculitis. One specimen was consistent with the patient’s known psoriasis.
Urinalysis revealed moderate hemoglobinuria, and urine microscopy showed 174 red blood cells per high-power field. Creatinine was high at 1.87 mg/dL (reference range, <1.34 mg/dL; patient’s baseline, 0.81 mg/dL) and glomerular filtration rate was low (42 mL/min, patient’s baseline, >60 mL/min [reference range, 90–120 mL/min]). Erythrocyte sedimentation rate (21 mm/h [reference range, 0–22 mm/h]) and C-reactive protein were elevated (2.2 mg/dL [reference range, 0.3–1.0 mg/dL]). Given his history of cutaneous MRSA infections, a bacterial culture swab was collected from the skin surface to check for colonization, which showed moderate growth of MRSA. Naproxen was discontinued over concern of worsening the patient’s renal status. The patient was instructed to rest at home with his legs elevated, wear compression socks when ambulatory, use chlorhexidine antiseptic daily as a body wash when showering, and apply mupirocin three times daily to the biopsy sites. He was referred to urology for his microhematuria, where cystoscopy revealed no abnormalities.A month passed with no improvement of the patient’s cutaneous vasculitis, and his psoriatic arthritis worsened without his usual use of naproxen. He developed abdominal pain and loss of appetite. A prednisone taper was ordered starting at 40 mg/d (28.8 mg/kg), which provided relief of the skin and joint symptoms only until the course was completed 12 days later.
Five weeks after the initial presentation, the patient returned with a more severe eruption consisting of innumerable purpuric papules that coalesced in plaques on the abdomen, arms, and legs. He also had erythematous facial pustules and mild palmar petechiae (Figure 4). Three biopsies were performed, including 1 for DIF and 1 from a pustule on the forehead. Histology and DIF were again consistent with IgA small-vessel vasculitis. The forehead biopsy was compatible with steroid acne (attributed to recent prednisone use) and psoriasis.
Rheumatology was consulted, and adalimumab was discontinued 6 weeks after the initial presentation out of concern for drug-induced cutaneous vasculitis. Vasculitis work-up was unremarkable, including antineutrophil cytoplasmic antibodies, rheumatoid factor, cyclic citrullinated peptide, and serum protein electrophoresis. Oral dapsone was started at 100 mg/d, with the tentative plan of starting secukinumab if cutaneous symptoms improved. For 3 weeks, the patient’s cutaneous symptoms steadily improved.
Nine weeks after initial presentation to dermatology (3 weeks after discontinuing adalimumab) the patient self-administered his first dose of secukinumab at home. Several hours later, he reported sudden reappearance of vasculitis. He denied diarrhea, abdominal pain, bowel movement urgency, fevers, fatigue, and unintentional weight loss. Antistreptolysin O and hepatitis A antibodies were negative. He was instructed to hold secukinumab indefinitely.
Four weeks after his only secukinumab injection, the patient reported another episode of acute worsening cutaneous symptoms. A 4-week prednisone taper starting at 40 mg/d was ordered. Computed tomography of the chest, abdomen, and pelvis to rule out internal malignancy was unremarkable. Around this time, the patient reported major emotional distress related to an unexpected death in his family, which added to a gradual increase in his stress level related to the COVID-19 pandemic.
Three weeks later, dapsone was increased to 100 mg twice daily on account of the patient’s adiposity and lack of cutaneous improvement on the lower dose. Subsequently, the vasculitis rapidly improved for 2 weeks. The patient then reported symptoms of headache, dizziness, and chills. He was tested for COVID-19 and was negative. Six weeks after increasing the dapsone dose (5 months after initial presentation), the skin was normalizing, showing only faintly hyperpigmented macules confined to areas of resolved vasculitis (forearms, abdomen, legs).
The patient had been on dapsone 100 mg twice daily for 3 months when he was started on ustekinumab (90 mg at weeks 0 and 4, with planned doses every 12 weeks) for psoriatic arthritis in hopes of withdrawing dapsone. His cutaneous symptoms have remained well controlled on this regimen for 18 months. Lowering of dapsone below 100 mg daily has resulted in recurrent mild vasculitis symptoms; he now maintains the once-daily dosing without negative side effects.
Comment
IgA vasculitis is a form of cutaneous small-vessel leukocytoclastic vasculitis (LCV) characterized by episodes of palpable purpura on the extensor surfaces of the arms and legs that may be associated with arthritis, abdominal pain, and/or hematuria. Although vasculitis is a known potential adverse effect of anti–tumor necrosis factor (TNF) α therapy, cases of adalimumab-induced IgA vasculitis are uncommon. As use of more targeted therapies for psoriasis and psoriatic arthritis, such as the IL-17 inhibitor secukinumab, increases so do reports of associated adverse events. Of 6 previously reported cases of secukinumab-associated vasculitis, at least 4 were IgA vasculitis (Table).1-6 Another case described one patient with rheumatoid arthritis undergoing secukinumab treatment who experienced necrotizing glomerulonephritis; however, the authors concluded secukinumab likely was not causative in that case, as serologies and urinalyses suggested gradual onset of the process prior to initiating the medication.7
The exact pathogenesis of IgA vasculitis is unclear, but a prevailing theory involves the dysregulation of IgA synthesis and metabolism. Other than increased serum levels of transforming growth factor β, which is a major stimulating factor for IgA production, it also has been hypothesized that the presence of aberrantly hypoglycosylated IgA exposes an autoepitope for recognition by other pathogenic IgG and IgA, leading to the formation of large immune complexes that can readily deposit in postcapillary venules. The deposition of IgA immune complexes in postcapillary venules and the subsequent activation of the complement system causes direct damage to the endothelial cells of vessel walls. This complement activation is evidenced by vascular complement component 3 deposition on DIF (a nonspecific feature of LCV). Chemotaxis of neutrophils ensues, followed by their firm adherence and transendothelial migration (mediated by monocyte chemoattractant protein 1 [MCP-1]). Neutrophil degranulation releases reactive oxygen species and cytokines, which in turn recruit additional leukocytes to the area of inflammation, subsequently undergoing degeneration (leukocytoclasis). Microvascular permeability also is enhanced by MCP-1, allowing exudation of serum, erythrocytes, and fibrin. In the setting of elevated circulating TNF and IL-1, endothelium is stimulated to activate the intrinsic and extrinsic coagulation pathways. This decreases endothelial fibrinolytic activity, leading to thrombosis. The high venous pressure and low fibrinolytic activity in the lower legs explains why vasculitic lesions often are confined to or begin in this distribution.1,8-10
There also are noteworthy roles for cytokines in LCV. Circulating transforming growth factor β and IL-6—which are necessary for development of T helper 17 (TH17) cells and production of IL-17—are higher in patients with LCV compared to controls. Peripheral blood monocytes in patients with LCV demonstrate higher production of IL-17. Once TH17 cells develop, their survival and phenotype are maintained by IL-23 (considered the master regulator of TH17 differentiation). IL-17 is a potent chemoattractant of IL-8 (CXCL8) and MCP-1, both of which promote neutrophil-mediated perivascular inflammation. The IL-23 and IL-17 pathways implicated in the pathogenesis of psoriasis also cause neutrophil activation and upregulate transcription of proinflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α), which overlap with those implicated in LCV. Autoimmune disease generally entails some positive feedback loop of progressively severe self-recognition and tissue destruction by the immune system. These shared cytokinetic processes may explain how the internal environment of psoriasis could perpetuate IgA vasculitis.1,2,8,10-12
The mechanisms underlying vasculitis associated with adalimumab are unclear, but hypotheses involve direct toxicity on vessels, capillary deposition of anti-TNF/TNF immune complexes, or an inflammatory process resulting in autoantibodies. Similar hypotheses are posited for secukinumab-associated vasculitis, including deposition of secukinumab–IL-17 complexes. Anti–TNF-α medications may increase TH17 cell numbers, leading to increased production of IL-22 and a resultant immunologic microenvironment conducive to vasculitis. All 6 published cases of secukinumab-associated vasculitis that we found had received prior treatment with a TNF-α blocker, but only 1 had occurrence of vasculitis during that treatment.1-6,10
In the 6 cases we reviewed, the time from starting secukinumab to onset of vasculitis ranged from 1 to 18 months. Our patient’s same-day re-emergence of vasculitis after his first secukinumab dose was so acute that we were skeptical of secukinumab as a potential trigger; this may simply have been coincident to the natural waxing and waning of the vasculitis (although onset of IgA vasculitis within 1 day of starting anti–TNF-α therapy has been reported).1-6,13
Specific associations of IgA vasculitis are many and can include bacterial organisms such as Helicobacter pylori, streptococci, and staphylococci. Although internal mucous membrane infections are considered more linked because of the surveillance role of IgA predominantly in mucosal tissues, it is possible that our patient with cutaneous MRSA harbored the same within the nasal mucosa. Our patient also received multiple vaccinations outside our department throughout his clinical course (2 hepatitis B and 1 pneumococcal conjugate), which are known potential triggers for vasculitis. Psychological stress is a known trigger for psoriasis, and given the cytokinetic relationship of psoriasis to vasculitis described previously, it may have indirectly contributed to vasculitis in our case. The anxiety associated with being immunosuppressed during the COVID-19 pandemic and bereavement of losing a family member may have contributed to the refractory nature of our patient’s condition. Renal involvement is relatively common in adults with IgA vasculitis and so should be ruled out, as should occult internal malignancy.8,10,14
It is unclear which of the above factors was causative in our case, but a multifactorial process is likely. Treatment of monoclonal antibody–associated vasculitis entails investigating for triggers and systemic involvement, removing the most likely culprit, quelling the vasculitis acutely, avoiding known potential exacerbators, and introducing an alternative long-term immunomodulant. In all 6 reported similar cases, discontinuation of secukinumab and initiation of prednisone or colchicine led to resolution.1-6 Dapsone also is acceptable for acute control of IgA vasculitis, although this medication is highly lipid soluble and penetrates well into various tissues.15 Thus, lower doses may prove ineffective for obese patients, as was demonstrated in our case. Given the known potential of vaccinations, infections, and other factors (eg, alcohol, penicillin) to trigger IgA vasculitis, these should be avoided.10
Blockade of IL-23 with ustekinumab has been suggested by other authors encountering secukinumab-associated vasculitis, as IL-23 is the main driver and sustainer of TH17 cell differentiation.8 Although 6 previously reported cases of secukinumab-associated vasculitis achieved resolution without long-term recurrence, none did so using an IL-23 inhibitor (nor had any of the described patients received IL-23 inhibitors previously).1-6 Given the established safety of IL-23 inhibitors and that they theoretically are well suited for this unique circumstance (by ceasing the main causative cytokine cascades “upstream”) and were efficacious in quickly resolving our patient’s vasculitis, we suggest that ustekinumab may represent
Case Report
A 47-year-old man presented with a sudden-onset rash consisting of red bumps on the abdomen and legs that had been ongoing for several days. He had known psoriasis and psoriatic arthritis that had been well controlled with adalimumab for the last 18 months. He reported concurrent onset of nausea but denied fevers, chills, night sweats, unintentional weight loss, abdominal pain, and pruritus. He endorsed prior cutaneous infections of methicillin-resistant Staphylococcus aureus (MRSA). His medical history also included diabetes mellitus, hypertension, and obesity. His other medications included oral losartan-hydrochlorothiazide, amlodipine, naproxen, and atorvastatin.
Physical examination revealed numerous thin purpuric papules—some with adherent scale—distributed on the lower legs, extensor forearms, and abdomen. Abdominal lesions were confined to weight-related striae (Figure 1). The palms, soles, oral mucosa, and face were spared. Three punch biopsies were performed, including 1 for direct immunofluorescence (DIF), and the patient was instructed to apply clobetasol to the affected areas twice daily until further notice.
Pathology showed perivascular extravasation of erythrocytes, neutrophils, eosinophils, and leukocytoclasis surrounding blood vessels associated with fibrin (Figure 2). Direct immunofluorescence showed granular deposition of IgA, complement component 3, and fibrinogen in a superficial dermal vascular pattern (Figure 3). These results were consistent with IgA small-vessel vasculitis. One specimen was consistent with the patient’s known psoriasis.
Urinalysis revealed moderate hemoglobinuria, and urine microscopy showed 174 red blood cells per high-power field. Creatinine was high at 1.87 mg/dL (reference range, <1.34 mg/dL; patient’s baseline, 0.81 mg/dL) and glomerular filtration rate was low (42 mL/min, patient’s baseline, >60 mL/min [reference range, 90–120 mL/min]). Erythrocyte sedimentation rate (21 mm/h [reference range, 0–22 mm/h]) and C-reactive protein were elevated (2.2 mg/dL [reference range, 0.3–1.0 mg/dL]). Given his history of cutaneous MRSA infections, a bacterial culture swab was collected from the skin surface to check for colonization, which showed moderate growth of MRSA. Naproxen was discontinued over concern of worsening the patient’s renal status. The patient was instructed to rest at home with his legs elevated, wear compression socks when ambulatory, use chlorhexidine antiseptic daily as a body wash when showering, and apply mupirocin three times daily to the biopsy sites. He was referred to urology for his microhematuria, where cystoscopy revealed no abnormalities.A month passed with no improvement of the patient’s cutaneous vasculitis, and his psoriatic arthritis worsened without his usual use of naproxen. He developed abdominal pain and loss of appetite. A prednisone taper was ordered starting at 40 mg/d (28.8 mg/kg), which provided relief of the skin and joint symptoms only until the course was completed 12 days later.
Five weeks after the initial presentation, the patient returned with a more severe eruption consisting of innumerable purpuric papules that coalesced in plaques on the abdomen, arms, and legs. He also had erythematous facial pustules and mild palmar petechiae (Figure 4). Three biopsies were performed, including 1 for DIF and 1 from a pustule on the forehead. Histology and DIF were again consistent with IgA small-vessel vasculitis. The forehead biopsy was compatible with steroid acne (attributed to recent prednisone use) and psoriasis.
Rheumatology was consulted, and adalimumab was discontinued 6 weeks after the initial presentation out of concern for drug-induced cutaneous vasculitis. Vasculitis work-up was unremarkable, including antineutrophil cytoplasmic antibodies, rheumatoid factor, cyclic citrullinated peptide, and serum protein electrophoresis. Oral dapsone was started at 100 mg/d, with the tentative plan of starting secukinumab if cutaneous symptoms improved. For 3 weeks, the patient’s cutaneous symptoms steadily improved.
Nine weeks after initial presentation to dermatology (3 weeks after discontinuing adalimumab) the patient self-administered his first dose of secukinumab at home. Several hours later, he reported sudden reappearance of vasculitis. He denied diarrhea, abdominal pain, bowel movement urgency, fevers, fatigue, and unintentional weight loss. Antistreptolysin O and hepatitis A antibodies were negative. He was instructed to hold secukinumab indefinitely.
Four weeks after his only secukinumab injection, the patient reported another episode of acute worsening cutaneous symptoms. A 4-week prednisone taper starting at 40 mg/d was ordered. Computed tomography of the chest, abdomen, and pelvis to rule out internal malignancy was unremarkable. Around this time, the patient reported major emotional distress related to an unexpected death in his family, which added to a gradual increase in his stress level related to the COVID-19 pandemic.
Three weeks later, dapsone was increased to 100 mg twice daily on account of the patient’s adiposity and lack of cutaneous improvement on the lower dose. Subsequently, the vasculitis rapidly improved for 2 weeks. The patient then reported symptoms of headache, dizziness, and chills. He was tested for COVID-19 and was negative. Six weeks after increasing the dapsone dose (5 months after initial presentation), the skin was normalizing, showing only faintly hyperpigmented macules confined to areas of resolved vasculitis (forearms, abdomen, legs).
The patient had been on dapsone 100 mg twice daily for 3 months when he was started on ustekinumab (90 mg at weeks 0 and 4, with planned doses every 12 weeks) for psoriatic arthritis in hopes of withdrawing dapsone. His cutaneous symptoms have remained well controlled on this regimen for 18 months. Lowering of dapsone below 100 mg daily has resulted in recurrent mild vasculitis symptoms; he now maintains the once-daily dosing without negative side effects.
Comment
IgA vasculitis is a form of cutaneous small-vessel leukocytoclastic vasculitis (LCV) characterized by episodes of palpable purpura on the extensor surfaces of the arms and legs that may be associated with arthritis, abdominal pain, and/or hematuria. Although vasculitis is a known potential adverse effect of anti–tumor necrosis factor (TNF) α therapy, cases of adalimumab-induced IgA vasculitis are uncommon. As use of more targeted therapies for psoriasis and psoriatic arthritis, such as the IL-17 inhibitor secukinumab, increases so do reports of associated adverse events. Of 6 previously reported cases of secukinumab-associated vasculitis, at least 4 were IgA vasculitis (Table).1-6 Another case described one patient with rheumatoid arthritis undergoing secukinumab treatment who experienced necrotizing glomerulonephritis; however, the authors concluded secukinumab likely was not causative in that case, as serologies and urinalyses suggested gradual onset of the process prior to initiating the medication.7
The exact pathogenesis of IgA vasculitis is unclear, but a prevailing theory involves the dysregulation of IgA synthesis and metabolism. Other than increased serum levels of transforming growth factor β, which is a major stimulating factor for IgA production, it also has been hypothesized that the presence of aberrantly hypoglycosylated IgA exposes an autoepitope for recognition by other pathogenic IgG and IgA, leading to the formation of large immune complexes that can readily deposit in postcapillary venules. The deposition of IgA immune complexes in postcapillary venules and the subsequent activation of the complement system causes direct damage to the endothelial cells of vessel walls. This complement activation is evidenced by vascular complement component 3 deposition on DIF (a nonspecific feature of LCV). Chemotaxis of neutrophils ensues, followed by their firm adherence and transendothelial migration (mediated by monocyte chemoattractant protein 1 [MCP-1]). Neutrophil degranulation releases reactive oxygen species and cytokines, which in turn recruit additional leukocytes to the area of inflammation, subsequently undergoing degeneration (leukocytoclasis). Microvascular permeability also is enhanced by MCP-1, allowing exudation of serum, erythrocytes, and fibrin. In the setting of elevated circulating TNF and IL-1, endothelium is stimulated to activate the intrinsic and extrinsic coagulation pathways. This decreases endothelial fibrinolytic activity, leading to thrombosis. The high venous pressure and low fibrinolytic activity in the lower legs explains why vasculitic lesions often are confined to or begin in this distribution.1,8-10
There also are noteworthy roles for cytokines in LCV. Circulating transforming growth factor β and IL-6—which are necessary for development of T helper 17 (TH17) cells and production of IL-17—are higher in patients with LCV compared to controls. Peripheral blood monocytes in patients with LCV demonstrate higher production of IL-17. Once TH17 cells develop, their survival and phenotype are maintained by IL-23 (considered the master regulator of TH17 differentiation). IL-17 is a potent chemoattractant of IL-8 (CXCL8) and MCP-1, both of which promote neutrophil-mediated perivascular inflammation. The IL-23 and IL-17 pathways implicated in the pathogenesis of psoriasis also cause neutrophil activation and upregulate transcription of proinflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α), which overlap with those implicated in LCV. Autoimmune disease generally entails some positive feedback loop of progressively severe self-recognition and tissue destruction by the immune system. These shared cytokinetic processes may explain how the internal environment of psoriasis could perpetuate IgA vasculitis.1,2,8,10-12
The mechanisms underlying vasculitis associated with adalimumab are unclear, but hypotheses involve direct toxicity on vessels, capillary deposition of anti-TNF/TNF immune complexes, or an inflammatory process resulting in autoantibodies. Similar hypotheses are posited for secukinumab-associated vasculitis, including deposition of secukinumab–IL-17 complexes. Anti–TNF-α medications may increase TH17 cell numbers, leading to increased production of IL-22 and a resultant immunologic microenvironment conducive to vasculitis. All 6 published cases of secukinumab-associated vasculitis that we found had received prior treatment with a TNF-α blocker, but only 1 had occurrence of vasculitis during that treatment.1-6,10
In the 6 cases we reviewed, the time from starting secukinumab to onset of vasculitis ranged from 1 to 18 months. Our patient’s same-day re-emergence of vasculitis after his first secukinumab dose was so acute that we were skeptical of secukinumab as a potential trigger; this may simply have been coincident to the natural waxing and waning of the vasculitis (although onset of IgA vasculitis within 1 day of starting anti–TNF-α therapy has been reported).1-6,13
Specific associations of IgA vasculitis are many and can include bacterial organisms such as Helicobacter pylori, streptococci, and staphylococci. Although internal mucous membrane infections are considered more linked because of the surveillance role of IgA predominantly in mucosal tissues, it is possible that our patient with cutaneous MRSA harbored the same within the nasal mucosa. Our patient also received multiple vaccinations outside our department throughout his clinical course (2 hepatitis B and 1 pneumococcal conjugate), which are known potential triggers for vasculitis. Psychological stress is a known trigger for psoriasis, and given the cytokinetic relationship of psoriasis to vasculitis described previously, it may have indirectly contributed to vasculitis in our case. The anxiety associated with being immunosuppressed during the COVID-19 pandemic and bereavement of losing a family member may have contributed to the refractory nature of our patient’s condition. Renal involvement is relatively common in adults with IgA vasculitis and so should be ruled out, as should occult internal malignancy.8,10,14
It is unclear which of the above factors was causative in our case, but a multifactorial process is likely. Treatment of monoclonal antibody–associated vasculitis entails investigating for triggers and systemic involvement, removing the most likely culprit, quelling the vasculitis acutely, avoiding known potential exacerbators, and introducing an alternative long-term immunomodulant. In all 6 reported similar cases, discontinuation of secukinumab and initiation of prednisone or colchicine led to resolution.1-6 Dapsone also is acceptable for acute control of IgA vasculitis, although this medication is highly lipid soluble and penetrates well into various tissues.15 Thus, lower doses may prove ineffective for obese patients, as was demonstrated in our case. Given the known potential of vaccinations, infections, and other factors (eg, alcohol, penicillin) to trigger IgA vasculitis, these should be avoided.10
Blockade of IL-23 with ustekinumab has been suggested by other authors encountering secukinumab-associated vasculitis, as IL-23 is the main driver and sustainer of TH17 cell differentiation.8 Although 6 previously reported cases of secukinumab-associated vasculitis achieved resolution without long-term recurrence, none did so using an IL-23 inhibitor (nor had any of the described patients received IL-23 inhibitors previously).1-6 Given the established safety of IL-23 inhibitors and that they theoretically are well suited for this unique circumstance (by ceasing the main causative cytokine cascades “upstream”) and were efficacious in quickly resolving our patient’s vasculitis, we suggest that ustekinumab may represent
- Reverte M, Etienne M, Fouchard M, et al. Occurrence of Henoch-Schönlein purpura in a patient treated with secukinumab. J Eur Acad Dermatol Venereol. 2019;33:E455-E457.
- Chelli C, Loget J, Vanhaecke C, et al. Cutaneous vasculitis with gut involvement during secukinumab treatment for psoriatic arthritis. Acta Derm Venereol. 2020;100:adv00077.
- da Silva Cendon Duran C, Santiago MB. Cutaneous vasculitis during secukinumab treatment. Eur J Case Rep Intern Med. 2020;7:001815.
- Bostan E, Gulseren D, Yalici-Armagan B, et al. Vasculitis during certolizumab pegol and secukinumab treatment: report of two cases. Dermatol Ther. 2021;34:E15007.
- Perkovic D, Simac P, Katic J. IgA vasculitis during secukinumab therapy. Clin Rheumatol. 2021;40:2071-2073.
- Villani A, DE Fata Salvatores G, Nappa P, et al. Cutaneous leucocytoclastic vasculitis during secukinumab treatment. Ital J Dermatol Venerol. 2021;156(suppl 1 to no. 6):9-10.
- Góis M, Messias A, Carvalho D, et al. MPO-ANCA-associated necrotizing glomerulonephritis in rheumatoid arthritis; a case report and review of literature. J Nephropathol. 2017;6:58-62.
- Jen HY, Chuang YH, Lin SC, et al. Increased serum interleukin-17 and peripheral Th17 cells in children with acute Henoch-Schönlein purpura. Pediatr Allergy Immunol. 2011;22:862-868.
- Hetland LE, Susrud KS, Lindahl KH, et al. Henoch-Schönlein purpura: a literature review. Acta Derm Venereol 2017;97:1160-1166.
- Weedon D. The vasculopathic reaction pattern. In: Houston M, Davie B, eds. Weedon’s Skin Pathology. 3rd ed. Elsevier Limited; 2010:207-211.
- Puig L. Paradoxical reactions: anti-TNFα ants, ustekinumab, secukinumab, ixekizumab, and others. Curr Probl Dermatol. 2018;53:49-63.
- Nestle F, Kaplan D, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Pinheiro RR, Lencastre A. Henoch-Schönlein purpura during anti-TNFα therapy: a fortuitous event or an indication to stop therapy? Eur J Dermatol. 2017;27:304-305.
- Hello CL, Cohen P, Bousser MG, et al. Suspected hepatitis B vaccination related vasculitis. J Rheumatol. 1999;26:191-194.
- Wolverton SE. Dapsone. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier, Inc; 2021:222-231.
- Reverte M, Etienne M, Fouchard M, et al. Occurrence of Henoch-Schönlein purpura in a patient treated with secukinumab. J Eur Acad Dermatol Venereol. 2019;33:E455-E457.
- Chelli C, Loget J, Vanhaecke C, et al. Cutaneous vasculitis with gut involvement during secukinumab treatment for psoriatic arthritis. Acta Derm Venereol. 2020;100:adv00077.
- da Silva Cendon Duran C, Santiago MB. Cutaneous vasculitis during secukinumab treatment. Eur J Case Rep Intern Med. 2020;7:001815.
- Bostan E, Gulseren D, Yalici-Armagan B, et al. Vasculitis during certolizumab pegol and secukinumab treatment: report of two cases. Dermatol Ther. 2021;34:E15007.
- Perkovic D, Simac P, Katic J. IgA vasculitis during secukinumab therapy. Clin Rheumatol. 2021;40:2071-2073.
- Villani A, DE Fata Salvatores G, Nappa P, et al. Cutaneous leucocytoclastic vasculitis during secukinumab treatment. Ital J Dermatol Venerol. 2021;156(suppl 1 to no. 6):9-10.
- Góis M, Messias A, Carvalho D, et al. MPO-ANCA-associated necrotizing glomerulonephritis in rheumatoid arthritis; a case report and review of literature. J Nephropathol. 2017;6:58-62.
- Jen HY, Chuang YH, Lin SC, et al. Increased serum interleukin-17 and peripheral Th17 cells in children with acute Henoch-Schönlein purpura. Pediatr Allergy Immunol. 2011;22:862-868.
- Hetland LE, Susrud KS, Lindahl KH, et al. Henoch-Schönlein purpura: a literature review. Acta Derm Venereol 2017;97:1160-1166.
- Weedon D. The vasculopathic reaction pattern. In: Houston M, Davie B, eds. Weedon’s Skin Pathology. 3rd ed. Elsevier Limited; 2010:207-211.
- Puig L. Paradoxical reactions: anti-TNFα ants, ustekinumab, secukinumab, ixekizumab, and others. Curr Probl Dermatol. 2018;53:49-63.
- Nestle F, Kaplan D, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Pinheiro RR, Lencastre A. Henoch-Schönlein purpura during anti-TNFα therapy: a fortuitous event or an indication to stop therapy? Eur J Dermatol. 2017;27:304-305.
- Hello CL, Cohen P, Bousser MG, et al. Suspected hepatitis B vaccination related vasculitis. J Rheumatol. 1999;26:191-194.
- Wolverton SE. Dapsone. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier, Inc; 2021:222-231.
Practice Points
- Biologic medications including adalimumab and more rarely secukinumab may be associated with leukocytoclastic vasculitis; a smaller subset of patients may experience IgA vasculitis.
- The IL-23 blocker ustekinumab may represent an ideal therapeutic agent when secukinumabassociated vasculitis is suspected. Because IL-23 is the main driver and sustainer of TH17 cell differentiation, it may cease the main causative cytokine cascades “upstream.”

A Patient With Recurrent Immune Stromal Keratitis and Adherence Challenges
Herpes simplex keratitis (HSK) is a common yet potentially blinding condition caused by a primary or reactivated herpetic infection of the cornea.1 The Herpetic Eye Disease Study established the standard of care in HSK management.2 Treatments range from oral antivirals and artificial tears to topical antibiotics, amniotic membranes, and corneal transplantation.3 Patients with immune stromal keratitis (ISK) may experience low-grade chronic keratitis for years.4 ISK is classified by a cellular and neovascularization infiltration of the cornea.5 We present a case of a patient with recurrent ISK and review its presentation, diagnosis, and management.
Case Presentation
A 52-year-old man presented to the eye clinic with a watery and itchy right eye with mildly blurred vision. His ocular history was unremarkable. His medical history was notable for hepatitis C, hypertension, alcohol and drug dependence, homelessness, and a COVID-19–induced coma. His medications included trazodone, nifedipine, clonidine HCl, and buprenorphine/naloxone.
On clinical examination, the patient’s best-corrected visual acuity was 20/40 in the right eye and 20/20 in the left. Corneal sensitivity was absent in the right eye and intact in the left. Anterior segment findings in the right eye included 360-degree superficial corneal neovascularization, deep neovascularization temporally, scattered patches of corneal haze, epithelial irregularity, and 2+ diffuse bulbar conjunctival injection (Figure 1). The anterior segment of the left eye and the posterior segments of both eyes were unremarkable. The differential diagnosis included HSK, syphilis, Cogan syndrome, varicella-zoster virus keratitis, Epstein-Barr virus keratitis, and Lyme disease. With consultation from a corneal specialist, the patient was given the presumptive diagnosis of ISK in the right eye based on unilateral corneal presentation and lack of corneal sensitivity. He was treated with
The patient returned a week later having only used the prednisolone drops for 2 days before discontinuing. Examination showed no change in his corneal appearance from the previous week. The patient was counseled on the importance of adherence to the regimen of topical prednisolone and oral valacyclovir.
The patient followed up 2 weeks later. He reported good adherence to the ISK medication regimen. His symptoms had resolved, and his visual acuity returned to 20/20 in the right eye. Slit-lamp examination showed improvement in injection, and the superficial corneal neovascularization had cleared. A trace ghost vessel was seen temporally at a site of deep neovascularization (Figure 2). He was instructed to continue valacyclovir once daily and prednisolone drops once daily in the right eye and to follow up in 1 month.
At the 1-month follow-up, the patient’s signs and symptoms had reverted to his original presentation. The patient reported poor adherence to the medication regimen, having missed multiple doses of prednisolone drops as well as valacyclovir. The patient was counseled again on the ISK regimen, and the prednisolone drops and 1-g oral valacyclovir were refilled. A follow-up visit was scheduled for 2 weeks. Additional follow-up revealed a resolved corneal appearance and bimonthly follow-ups were scheduled thereafter.
Discussion
HSK is the most common infectious cause of unilateral blindness and vision impairment in the world.2 This case highlights the diagnosis and management of a patient with ISK, a type of HSK characterized by decreased corneal sensitivity and unilateral stromal opacification or neovascularization.6
ISK is caused by the herpes simplex virus (HSV), a double-stranded enveloped DNA virus that occurs worldwide with little variation, replicates in many types of cells, has rapid growth, and is cytolytic, causing necrosis of nearby cells. Transmission is via direct contact and there is a lifelong latency period in the trigeminal ganglia. Both primary and reactivation infections of HSK can affect a broad array of ocular structures, from the lids to the retina. Infectious epithelial keratitis, also known as dendritic keratitis, is the reactivation of the live virus and is the most common presentation of HSK. ISK is responsible for 20% to 48% of recurrent HSV disease and is the leading cause of vision loss. ISK is the result of an immune-mediated inflammatory response due to a retained viral antigen within the stromal tissue.7 Inflammation in the corneal stroma leads to corneal haze and eventually focal or diffuse scarring, reducing the visual potential.7 This presentation may occur days to years after the initial epithelial episode and may persist for years. Although this patient did not present with infectious epithelial keratitis, it is possible he had a previous episode not mentioned as a history was difficult to obtain, and it can be subtle or innocuous, like pink eye.
Symptoms of ISK include unilateral redness, photophobia, tearing, eye pain, and blurred vision, as described by this patient. On examination, initial manifestations of ISK include corneal haze, edema, scarring, and neovascularization.7 Again, this patient presented with edema and neovascularization. These signs may improve with prompt diagnosis and treatment. More frequent reactivated disease leads to a higher propensity of corneal scarring and irregular astigmatism, reducing the visual outcome.
The standard of care established by the Herpetic Eye Disease Study recommends that a patient with presumed ISK should be started on oral antiviral therapy and, in the absence of epithelial disease, topical steroids. Oral antivirals, such as acyclovir and valacyclovir, have good ocular penetration, a good safety profile, a low susceptibility of resistance, and are well tolerated with long-term treatment.2,8 There were no known interactions between any of the patient’s medications and valacyclovir. Oral antivirals should be used in the initial presentation and for maintenance therapy to help reduce the chance of recurrent disease. Initial treatment for ISK is 1-g valacyclovir 3 times daily. When the eye becomes quiet, that dosage can be tapered to 1 g twice daily, to 1 g once daily, and eventually to a maintenance dose of 500 mg daily. Topical steroids block the inflammatory cascade, therefore reducing the corneal inflammation and potential scarring, further reducing the risk of visual impairment.9 Initial treatment is 1 drop 3 times daily, then can be tapered at the same schedule as the oral acyclovir to help simplify adherence for the patient. After 1 drop once daily, steroids may be discontinued while the oral antiviral maintenance dosage continues. Follow-ups should be performed on a monthly to bimonthly basis to evaluate intraocular pressure, ensuring there is no steroid response.
As seen in this patient, adherence with a treatment regimen and awareness of factors, such as a complex psychosocial history that may impact this adherence, are of utmost importance.7
Conclusions
ISK presents unilaterally with decreased or absent corneal sensitivity and nonspecific symptoms. It should be at the top of the list in the differential diagnosis in any patient with unilateral corneal edema, opacification, or neovascularization, and the patient should be started on oral antiviral therapy.
1. Sibley D, Larkin DFP. Update on Herpes simplex keratitis management. Eye (Lond). 2020;34(12):2219-2226. doi:10.1038/s41433-020-01153-x
2. Chodosh J, Ung L. Adoption of innovation in herpes simplex virus keratitis. Cornea. 2020;39(1)(suppl 1):S7-S18. doi:10.1097/ICO.0000000000002425
3. Pérez-Bartolomé F, Botín DM, de Dompablo P, de Arriba P, Arnalich Montiel F, Muñoz Negrete FJ. Post-herpes neurotrophic keratopathy: pathogenesis, clinical signs and current therapies. Arch Soc Esp Oftalmol. 2019;94(4):171-183. doi:10.1016/j.oftal.2019.01.002
4. Holland EJ, Schwartz GS. Classification of herpes simplex virus keratitis. Cornea. 1999;18(2):144-154.
5. Gauthier AS, Noureddine S, Delbosc B. Interstitial keratitis diagnosis and treatment. J Fr Ophtalmol. 2019;42(6):e229-e237. doi:10.1016/j.jfo.2019.04.001
6. Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol. 2012;5(57):448-462. doi:10.1016/jsurvophthal.2012.01.005
7. Wang L, Wang R, Xu C, Zhou H. Pathogenesis of herpes stromal keratitis: immune inflammatory response mediated by inflammatory regulators. Front Immunol. 2020;11:766. Published 2020 May 13. doi:10.3389/fimmu.2020.00766
8. Tyring SK, Baker D, Snowden W. Valacyclovir for herpes simplex virus infection: long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J Infect Dis. 2002;186(suppl 1):S40-S46. doi:10.1086/342966
9. Dawson CR. The herpetic eye disease study. Arch Ophthalmol. 1990;108(2):191-192. doi:10.1001/archopht.1990.01070040043027
Herpes simplex keratitis (HSK) is a common yet potentially blinding condition caused by a primary or reactivated herpetic infection of the cornea.1 The Herpetic Eye Disease Study established the standard of care in HSK management.2 Treatments range from oral antivirals and artificial tears to topical antibiotics, amniotic membranes, and corneal transplantation.3 Patients with immune stromal keratitis (ISK) may experience low-grade chronic keratitis for years.4 ISK is classified by a cellular and neovascularization infiltration of the cornea.5 We present a case of a patient with recurrent ISK and review its presentation, diagnosis, and management.
Case Presentation
A 52-year-old man presented to the eye clinic with a watery and itchy right eye with mildly blurred vision. His ocular history was unremarkable. His medical history was notable for hepatitis C, hypertension, alcohol and drug dependence, homelessness, and a COVID-19–induced coma. His medications included trazodone, nifedipine, clonidine HCl, and buprenorphine/naloxone.
On clinical examination, the patient’s best-corrected visual acuity was 20/40 in the right eye and 20/20 in the left. Corneal sensitivity was absent in the right eye and intact in the left. Anterior segment findings in the right eye included 360-degree superficial corneal neovascularization, deep neovascularization temporally, scattered patches of corneal haze, epithelial irregularity, and 2+ diffuse bulbar conjunctival injection (Figure 1). The anterior segment of the left eye and the posterior segments of both eyes were unremarkable. The differential diagnosis included HSK, syphilis, Cogan syndrome, varicella-zoster virus keratitis, Epstein-Barr virus keratitis, and Lyme disease. With consultation from a corneal specialist, the patient was given the presumptive diagnosis of ISK in the right eye based on unilateral corneal presentation and lack of corneal sensitivity. He was treated with
The patient returned a week later having only used the prednisolone drops for 2 days before discontinuing. Examination showed no change in his corneal appearance from the previous week. The patient was counseled on the importance of adherence to the regimen of topical prednisolone and oral valacyclovir.
The patient followed up 2 weeks later. He reported good adherence to the ISK medication regimen. His symptoms had resolved, and his visual acuity returned to 20/20 in the right eye. Slit-lamp examination showed improvement in injection, and the superficial corneal neovascularization had cleared. A trace ghost vessel was seen temporally at a site of deep neovascularization (Figure 2). He was instructed to continue valacyclovir once daily and prednisolone drops once daily in the right eye and to follow up in 1 month.
At the 1-month follow-up, the patient’s signs and symptoms had reverted to his original presentation. The patient reported poor adherence to the medication regimen, having missed multiple doses of prednisolone drops as well as valacyclovir. The patient was counseled again on the ISK regimen, and the prednisolone drops and 1-g oral valacyclovir were refilled. A follow-up visit was scheduled for 2 weeks. Additional follow-up revealed a resolved corneal appearance and bimonthly follow-ups were scheduled thereafter.
Discussion
HSK is the most common infectious cause of unilateral blindness and vision impairment in the world.2 This case highlights the diagnosis and management of a patient with ISK, a type of HSK characterized by decreased corneal sensitivity and unilateral stromal opacification or neovascularization.6
ISK is caused by the herpes simplex virus (HSV), a double-stranded enveloped DNA virus that occurs worldwide with little variation, replicates in many types of cells, has rapid growth, and is cytolytic, causing necrosis of nearby cells. Transmission is via direct contact and there is a lifelong latency period in the trigeminal ganglia. Both primary and reactivation infections of HSK can affect a broad array of ocular structures, from the lids to the retina. Infectious epithelial keratitis, also known as dendritic keratitis, is the reactivation of the live virus and is the most common presentation of HSK. ISK is responsible for 20% to 48% of recurrent HSV disease and is the leading cause of vision loss. ISK is the result of an immune-mediated inflammatory response due to a retained viral antigen within the stromal tissue.7 Inflammation in the corneal stroma leads to corneal haze and eventually focal or diffuse scarring, reducing the visual potential.7 This presentation may occur days to years after the initial epithelial episode and may persist for years. Although this patient did not present with infectious epithelial keratitis, it is possible he had a previous episode not mentioned as a history was difficult to obtain, and it can be subtle or innocuous, like pink eye.
Symptoms of ISK include unilateral redness, photophobia, tearing, eye pain, and blurred vision, as described by this patient. On examination, initial manifestations of ISK include corneal haze, edema, scarring, and neovascularization.7 Again, this patient presented with edema and neovascularization. These signs may improve with prompt diagnosis and treatment. More frequent reactivated disease leads to a higher propensity of corneal scarring and irregular astigmatism, reducing the visual outcome.
The standard of care established by the Herpetic Eye Disease Study recommends that a patient with presumed ISK should be started on oral antiviral therapy and, in the absence of epithelial disease, topical steroids. Oral antivirals, such as acyclovir and valacyclovir, have good ocular penetration, a good safety profile, a low susceptibility of resistance, and are well tolerated with long-term treatment.2,8 There were no known interactions between any of the patient’s medications and valacyclovir. Oral antivirals should be used in the initial presentation and for maintenance therapy to help reduce the chance of recurrent disease. Initial treatment for ISK is 1-g valacyclovir 3 times daily. When the eye becomes quiet, that dosage can be tapered to 1 g twice daily, to 1 g once daily, and eventually to a maintenance dose of 500 mg daily. Topical steroids block the inflammatory cascade, therefore reducing the corneal inflammation and potential scarring, further reducing the risk of visual impairment.9 Initial treatment is 1 drop 3 times daily, then can be tapered at the same schedule as the oral acyclovir to help simplify adherence for the patient. After 1 drop once daily, steroids may be discontinued while the oral antiviral maintenance dosage continues. Follow-ups should be performed on a monthly to bimonthly basis to evaluate intraocular pressure, ensuring there is no steroid response.
As seen in this patient, adherence with a treatment regimen and awareness of factors, such as a complex psychosocial history that may impact this adherence, are of utmost importance.7
Conclusions
ISK presents unilaterally with decreased or absent corneal sensitivity and nonspecific symptoms. It should be at the top of the list in the differential diagnosis in any patient with unilateral corneal edema, opacification, or neovascularization, and the patient should be started on oral antiviral therapy.
Herpes simplex keratitis (HSK) is a common yet potentially blinding condition caused by a primary or reactivated herpetic infection of the cornea.1 The Herpetic Eye Disease Study established the standard of care in HSK management.2 Treatments range from oral antivirals and artificial tears to topical antibiotics, amniotic membranes, and corneal transplantation.3 Patients with immune stromal keratitis (ISK) may experience low-grade chronic keratitis for years.4 ISK is classified by a cellular and neovascularization infiltration of the cornea.5 We present a case of a patient with recurrent ISK and review its presentation, diagnosis, and management.
Case Presentation
A 52-year-old man presented to the eye clinic with a watery and itchy right eye with mildly blurred vision. His ocular history was unremarkable. His medical history was notable for hepatitis C, hypertension, alcohol and drug dependence, homelessness, and a COVID-19–induced coma. His medications included trazodone, nifedipine, clonidine HCl, and buprenorphine/naloxone.
On clinical examination, the patient’s best-corrected visual acuity was 20/40 in the right eye and 20/20 in the left. Corneal sensitivity was absent in the right eye and intact in the left. Anterior segment findings in the right eye included 360-degree superficial corneal neovascularization, deep neovascularization temporally, scattered patches of corneal haze, epithelial irregularity, and 2+ diffuse bulbar conjunctival injection (Figure 1). The anterior segment of the left eye and the posterior segments of both eyes were unremarkable. The differential diagnosis included HSK, syphilis, Cogan syndrome, varicella-zoster virus keratitis, Epstein-Barr virus keratitis, and Lyme disease. With consultation from a corneal specialist, the patient was given the presumptive diagnosis of ISK in the right eye based on unilateral corneal presentation and lack of corneal sensitivity. He was treated with
The patient returned a week later having only used the prednisolone drops for 2 days before discontinuing. Examination showed no change in his corneal appearance from the previous week. The patient was counseled on the importance of adherence to the regimen of topical prednisolone and oral valacyclovir.
The patient followed up 2 weeks later. He reported good adherence to the ISK medication regimen. His symptoms had resolved, and his visual acuity returned to 20/20 in the right eye. Slit-lamp examination showed improvement in injection, and the superficial corneal neovascularization had cleared. A trace ghost vessel was seen temporally at a site of deep neovascularization (Figure 2). He was instructed to continue valacyclovir once daily and prednisolone drops once daily in the right eye and to follow up in 1 month.
At the 1-month follow-up, the patient’s signs and symptoms had reverted to his original presentation. The patient reported poor adherence to the medication regimen, having missed multiple doses of prednisolone drops as well as valacyclovir. The patient was counseled again on the ISK regimen, and the prednisolone drops and 1-g oral valacyclovir were refilled. A follow-up visit was scheduled for 2 weeks. Additional follow-up revealed a resolved corneal appearance and bimonthly follow-ups were scheduled thereafter.
Discussion
HSK is the most common infectious cause of unilateral blindness and vision impairment in the world.2 This case highlights the diagnosis and management of a patient with ISK, a type of HSK characterized by decreased corneal sensitivity and unilateral stromal opacification or neovascularization.6
ISK is caused by the herpes simplex virus (HSV), a double-stranded enveloped DNA virus that occurs worldwide with little variation, replicates in many types of cells, has rapid growth, and is cytolytic, causing necrosis of nearby cells. Transmission is via direct contact and there is a lifelong latency period in the trigeminal ganglia. Both primary and reactivation infections of HSK can affect a broad array of ocular structures, from the lids to the retina. Infectious epithelial keratitis, also known as dendritic keratitis, is the reactivation of the live virus and is the most common presentation of HSK. ISK is responsible for 20% to 48% of recurrent HSV disease and is the leading cause of vision loss. ISK is the result of an immune-mediated inflammatory response due to a retained viral antigen within the stromal tissue.7 Inflammation in the corneal stroma leads to corneal haze and eventually focal or diffuse scarring, reducing the visual potential.7 This presentation may occur days to years after the initial epithelial episode and may persist for years. Although this patient did not present with infectious epithelial keratitis, it is possible he had a previous episode not mentioned as a history was difficult to obtain, and it can be subtle or innocuous, like pink eye.
Symptoms of ISK include unilateral redness, photophobia, tearing, eye pain, and blurred vision, as described by this patient. On examination, initial manifestations of ISK include corneal haze, edema, scarring, and neovascularization.7 Again, this patient presented with edema and neovascularization. These signs may improve with prompt diagnosis and treatment. More frequent reactivated disease leads to a higher propensity of corneal scarring and irregular astigmatism, reducing the visual outcome.
The standard of care established by the Herpetic Eye Disease Study recommends that a patient with presumed ISK should be started on oral antiviral therapy and, in the absence of epithelial disease, topical steroids. Oral antivirals, such as acyclovir and valacyclovir, have good ocular penetration, a good safety profile, a low susceptibility of resistance, and are well tolerated with long-term treatment.2,8 There were no known interactions between any of the patient’s medications and valacyclovir. Oral antivirals should be used in the initial presentation and for maintenance therapy to help reduce the chance of recurrent disease. Initial treatment for ISK is 1-g valacyclovir 3 times daily. When the eye becomes quiet, that dosage can be tapered to 1 g twice daily, to 1 g once daily, and eventually to a maintenance dose of 500 mg daily. Topical steroids block the inflammatory cascade, therefore reducing the corneal inflammation and potential scarring, further reducing the risk of visual impairment.9 Initial treatment is 1 drop 3 times daily, then can be tapered at the same schedule as the oral acyclovir to help simplify adherence for the patient. After 1 drop once daily, steroids may be discontinued while the oral antiviral maintenance dosage continues. Follow-ups should be performed on a monthly to bimonthly basis to evaluate intraocular pressure, ensuring there is no steroid response.
As seen in this patient, adherence with a treatment regimen and awareness of factors, such as a complex psychosocial history that may impact this adherence, are of utmost importance.7
Conclusions
ISK presents unilaterally with decreased or absent corneal sensitivity and nonspecific symptoms. It should be at the top of the list in the differential diagnosis in any patient with unilateral corneal edema, opacification, or neovascularization, and the patient should be started on oral antiviral therapy.
1. Sibley D, Larkin DFP. Update on Herpes simplex keratitis management. Eye (Lond). 2020;34(12):2219-2226. doi:10.1038/s41433-020-01153-x
2. Chodosh J, Ung L. Adoption of innovation in herpes simplex virus keratitis. Cornea. 2020;39(1)(suppl 1):S7-S18. doi:10.1097/ICO.0000000000002425
3. Pérez-Bartolomé F, Botín DM, de Dompablo P, de Arriba P, Arnalich Montiel F, Muñoz Negrete FJ. Post-herpes neurotrophic keratopathy: pathogenesis, clinical signs and current therapies. Arch Soc Esp Oftalmol. 2019;94(4):171-183. doi:10.1016/j.oftal.2019.01.002
4. Holland EJ, Schwartz GS. Classification of herpes simplex virus keratitis. Cornea. 1999;18(2):144-154.
5. Gauthier AS, Noureddine S, Delbosc B. Interstitial keratitis diagnosis and treatment. J Fr Ophtalmol. 2019;42(6):e229-e237. doi:10.1016/j.jfo.2019.04.001
6. Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol. 2012;5(57):448-462. doi:10.1016/jsurvophthal.2012.01.005
7. Wang L, Wang R, Xu C, Zhou H. Pathogenesis of herpes stromal keratitis: immune inflammatory response mediated by inflammatory regulators. Front Immunol. 2020;11:766. Published 2020 May 13. doi:10.3389/fimmu.2020.00766
8. Tyring SK, Baker D, Snowden W. Valacyclovir for herpes simplex virus infection: long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J Infect Dis. 2002;186(suppl 1):S40-S46. doi:10.1086/342966
9. Dawson CR. The herpetic eye disease study. Arch Ophthalmol. 1990;108(2):191-192. doi:10.1001/archopht.1990.01070040043027
1. Sibley D, Larkin DFP. Update on Herpes simplex keratitis management. Eye (Lond). 2020;34(12):2219-2226. doi:10.1038/s41433-020-01153-x
2. Chodosh J, Ung L. Adoption of innovation in herpes simplex virus keratitis. Cornea. 2020;39(1)(suppl 1):S7-S18. doi:10.1097/ICO.0000000000002425
3. Pérez-Bartolomé F, Botín DM, de Dompablo P, de Arriba P, Arnalich Montiel F, Muñoz Negrete FJ. Post-herpes neurotrophic keratopathy: pathogenesis, clinical signs and current therapies. Arch Soc Esp Oftalmol. 2019;94(4):171-183. doi:10.1016/j.oftal.2019.01.002
4. Holland EJ, Schwartz GS. Classification of herpes simplex virus keratitis. Cornea. 1999;18(2):144-154.
5. Gauthier AS, Noureddine S, Delbosc B. Interstitial keratitis diagnosis and treatment. J Fr Ophtalmol. 2019;42(6):e229-e237. doi:10.1016/j.jfo.2019.04.001
6. Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol. 2012;5(57):448-462. doi:10.1016/jsurvophthal.2012.01.005
7. Wang L, Wang R, Xu C, Zhou H. Pathogenesis of herpes stromal keratitis: immune inflammatory response mediated by inflammatory regulators. Front Immunol. 2020;11:766. Published 2020 May 13. doi:10.3389/fimmu.2020.00766
8. Tyring SK, Baker D, Snowden W. Valacyclovir for herpes simplex virus infection: long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J Infect Dis. 2002;186(suppl 1):S40-S46. doi:10.1086/342966
9. Dawson CR. The herpetic eye disease study. Arch Ophthalmol. 1990;108(2):191-192. doi:10.1001/archopht.1990.01070040043027
57-year-old man • type 2 diabetes • neuropathy • bilateral foot blisters • Dx?
THE CASE
A 57-year-old man with type 2 diabetes, hyperlipidemia, and obesity presented to the emergency department (ED) for bilateral foot blisters, both of which appeared 1 day prior to evaluation. The patient’s history also included right-side Charcot foot diagnosed 4 years earlier and right foot osteomyelitis diagnosed 2 years prior. He had ongoing neuropathy in both feet but denied any significant pain.
The patient wore orthotics daily and he’d had new orthotics made 6 months prior; however, a recent COVID-19 diagnosis and prolonged hospital stay resulted in a 30-pound weight loss and decreased swelling in his ankles. He acquired new shoes 2 weeks prior to ED presentation.
Physical examination revealed large blisters along the medial aspect of the patient’s feet, with both hemorrhagic and serous fluid-filled bullae. The lesions were flaccid but intact, without drainage or surrounding erythema, warmth, or tenderness. The blister on the left foot measured 8 x 5 cm and extended from the great toe to mid-arch (FIGURE), while the one on the right foot measured 8 x 3 cm and extended from the great toe to the base of the proximal arch. Sensation was decreased in the bilateral first and second digits but unchanged from prior documented exams. Bilateral dorsalis pedis pulses were normal.

Work-up included imaging and lab work. The patient’s complete blood count was normal, as were his erythrocyte sedimentation rate and C-reactive protein level. Radiographs of the right foot were normal, but those of the left foot were concerning, although inconclusive, for osteomyelitis. Further evaluation with magnetic resonance imaging of his left foot revealed a deformity of the first digit with some subchondral signal change that was thought to be posttraumatic or degenerative, but unlikely osteomyelitis.
THE DIAGNOSIS
Podiatry was consulted for blister management. Based on atraumatic history, rapid appearance, location of blisters, unremarkable lab work and imaging, and concurrent diabetes, the patient received a diagnosis of bilateral bullous diabeticorum (BD).
DISCUSSION
Roughly one-third of patients with diabetes will experience some cutaneous adverse effect because of the disease.1 Common iterations include acanthosis nigricans, rash, or even infection.2 BD is a rare bullous skin lesion that occurs in patients with diabetes; it has a reported annual incidence of 0.16% and may be underdiagnosed.1
Cases of BD have been described both in patients with longstanding diabetes and in those newly diagnosed, although the former group is more often affected.1 BD is reported more frequently in males than females, at a ratio of 2:1.1,3 Patients ages 17 to 80 years (average age, 55 years) have received a diagnosis of BD.1 Most affected patients will have a concomitant peripheral neuropathy and sometimes nephropathy or retinopathy.1
Continue to: The etiology of BD...
The etiology of BD is unclear but appears to be multifactorial. Hypotheses suggest that there’s a link to neuropathy/nephropathy, excessive exposure to ultraviolet light, or a vascular cause secondary to hyaline deposition in the capillary walls.4,5
What you’ll see at presentation
The typical manifestation of BD is the rapid appearance of tense blisters, which may occur overnight or even within hours.1 They are usually painless; common locations include the feet, distal legs, hands, and forearms.1,5 The bullae can be serous or hemorrhagic.1
Most notable in the patient’s history will be a lack of trauma or injury to the area.1 Although A1C values do not correlate with blister formation, patients with hypoglycemic episodes and highly varying blood glucose values seem to have higher rates of occurrence.1
Other sources of blistering must be ruled out
The diagnosis of BD is clinical and based on history, exam, and exclusion of other bullous diagnoses.6 A key clue in the history is the spontaneous and rapid onset without associated trauma in a patient with diabetes.6 Direct immunofluorescence, although nonspecific, can be helpful to rule out other disorders (such as porphyria cutanea tarda and bullous pemphigoid) if the history and exam are inconclusive. Direct and indirect immunofluorescence is typically negative in BD.4,6
The differential diagnosis includes other conditions that involve bullae—such as frictional bullae, bullous pemphigoid, and bullous systemic lupus erythematosus—as well as porphyria, erythema multiforme, insect bites, or even fixed drug eruption.2,7
Continue to: Porphyria
Porphyria tends to develop on the hands, whereas BD most commonly occurs on the feet.5
Erythema multiforme typically includes inflammatory skin changes.5
Trauma or fixed drug eruption as a cause of blistering lesions would be revealed during history taking.
Considerations for treatment and follow-up
Without treatment, blisters often self-resolve in 2 to 6 weeks, but there is high likelihood of recurrence.6,8 There is no consensus on treatment, although a typical course of action is to deroof the blister and examine the area to rule out infection.6 The wound is then covered with wet-to-dry gauze that is changed regularly. If there is suspicion for or signs of underlying infection, such as an ulcer or skin necrosis, antibiotics should be included in the treatment plan.7
Additional considerations. Patients will often need therapeutic footwear if the blisters are located on the feet. Given the higher prevalence of microvascular complications in patients with diabetes who develop BD, routine ophthalmologic examination and renal function testing to monitor for microalbuminuria are recommended.5
Our patient underwent bedside incision and drainage and was discharged home with appropriate wound care and follow-up.
THE TAKEAWAY
BD cases may be underdiagnosed in clinical practice, perhaps due to patients not seeking help for a seemingly nonthreatening condition or lack of clinician recognition that bullae are related to a patient’s diabetes status. Prompt recognition and proper wound care are important to prevent poor outcomes, such as ulceration or necrosis.
CORRESPONDENCE
Kathleen S. Kinderwater, MD, 101 Heart Drive, Greenville, NC 27834; [email protected]
1. Larsen K, Jensen T, Karlsmark T, et al. Incidence of bullosis diabeticorum—a controversial cause of chronic foot ulceration. Int Wound J. 2008;5:591-596. doi: 10.1111/j.1742-481X.2008.00476.x
2. Lipsky BA, Baker PD, Ahroni JH. Diabetic bullae: 12 cases of a purportedly rare cutaneous disorder. Int J Dermatol. 2000;39:196-200. doi: 10.1046/j.1365-4362.2000.00947.x
3. Gupta V, Gulati N, Bahl J, et al. Bullosis diabeticorum: rare presentation in a common disease. Case Rep Endocrinol. 2014;2014:862912.
4. Sonani H, Abdul Salim S, Garla VV, et al. Bullosis diabeticorum: a rare presentation with immunoglobulin G (IgG) deposition related vasculopathy. Case report and focused review. Am J Case Rep. 2018;19:52-56. doi: 10.12659/ajcr.905452
5. Chouk C, Litaiem N. Bullosis diabeticorum. StatPearls [Internet]. Updated June 5, 2021. Accessed July 14, 2022. www.ncbi.nlm.nih.gov/books/NBK539872/
6. Chatterjee D, Radotra A, Radotra BD, et al. Bullous diabeticorum: a rare blistering manifestation of diabetes. Indian Dermatol Online J. 2017;8:274-275. doi: 10.4103/idoj.IDOJ_340_16
7. Kansal NK, Anuragi RP. Bullous lesions in diabetes mellitus: bullous diabeticorum (diabetic bulla). BMJ Case Rep. 2020;13:e238617. doi: 10.1136/bcr-2020-238617
8. Bello F, Samaila OM, Lawal Y, et al. 2 cases of bullosis diabeticorum following long-distance journeys by road: a report of 2 cases. Case Rep Endocrinol. 2012;2012:367218. doi: 10.1155/2012/367218
THE CASE
A 57-year-old man with type 2 diabetes, hyperlipidemia, and obesity presented to the emergency department (ED) for bilateral foot blisters, both of which appeared 1 day prior to evaluation. The patient’s history also included right-side Charcot foot diagnosed 4 years earlier and right foot osteomyelitis diagnosed 2 years prior. He had ongoing neuropathy in both feet but denied any significant pain.
The patient wore orthotics daily and he’d had new orthotics made 6 months prior; however, a recent COVID-19 diagnosis and prolonged hospital stay resulted in a 30-pound weight loss and decreased swelling in his ankles. He acquired new shoes 2 weeks prior to ED presentation.
Physical examination revealed large blisters along the medial aspect of the patient’s feet, with both hemorrhagic and serous fluid-filled bullae. The lesions were flaccid but intact, without drainage or surrounding erythema, warmth, or tenderness. The blister on the left foot measured 8 x 5 cm and extended from the great toe to mid-arch (FIGURE), while the one on the right foot measured 8 x 3 cm and extended from the great toe to the base of the proximal arch. Sensation was decreased in the bilateral first and second digits but unchanged from prior documented exams. Bilateral dorsalis pedis pulses were normal.
Work-up included imaging and lab work. The patient’s complete blood count was normal, as were his erythrocyte sedimentation rate and C-reactive protein level. Radiographs of the right foot were normal, but those of the left foot were concerning, although inconclusive, for osteomyelitis. Further evaluation with magnetic resonance imaging of his left foot revealed a deformity of the first digit with some subchondral signal change that was thought to be posttraumatic or degenerative, but unlikely osteomyelitis.
THE DIAGNOSIS
Podiatry was consulted for blister management. Based on atraumatic history, rapid appearance, location of blisters, unremarkable lab work and imaging, and concurrent diabetes, the patient received a diagnosis of bilateral bullous diabeticorum (BD).
DISCUSSION
Roughly one-third of patients with diabetes will experience some cutaneous adverse effect because of the disease.1 Common iterations include acanthosis nigricans, rash, or even infection.2 BD is a rare bullous skin lesion that occurs in patients with diabetes; it has a reported annual incidence of 0.16% and may be underdiagnosed.1
Cases of BD have been described both in patients with longstanding diabetes and in those newly diagnosed, although the former group is more often affected.1 BD is reported more frequently in males than females, at a ratio of 2:1.1,3 Patients ages 17 to 80 years (average age, 55 years) have received a diagnosis of BD.1 Most affected patients will have a concomitant peripheral neuropathy and sometimes nephropathy or retinopathy.1
Continue to: The etiology of BD...
The etiology of BD is unclear but appears to be multifactorial. Hypotheses suggest that there’s a link to neuropathy/nephropathy, excessive exposure to ultraviolet light, or a vascular cause secondary to hyaline deposition in the capillary walls.4,5
What you’ll see at presentation
The typical manifestation of BD is the rapid appearance of tense blisters, which may occur overnight or even within hours.1 They are usually painless; common locations include the feet, distal legs, hands, and forearms.1,5 The bullae can be serous or hemorrhagic.1
Most notable in the patient’s history will be a lack of trauma or injury to the area.1 Although A1C values do not correlate with blister formation, patients with hypoglycemic episodes and highly varying blood glucose values seem to have higher rates of occurrence.1
Other sources of blistering must be ruled out
The diagnosis of BD is clinical and based on history, exam, and exclusion of other bullous diagnoses.6 A key clue in the history is the spontaneous and rapid onset without associated trauma in a patient with diabetes.6 Direct immunofluorescence, although nonspecific, can be helpful to rule out other disorders (such as porphyria cutanea tarda and bullous pemphigoid) if the history and exam are inconclusive. Direct and indirect immunofluorescence is typically negative in BD.4,6
The differential diagnosis includes other conditions that involve bullae—such as frictional bullae, bullous pemphigoid, and bullous systemic lupus erythematosus—as well as porphyria, erythema multiforme, insect bites, or even fixed drug eruption.2,7
Continue to: Porphyria
Porphyria tends to develop on the hands, whereas BD most commonly occurs on the feet.5
Erythema multiforme typically includes inflammatory skin changes.5
Trauma or fixed drug eruption as a cause of blistering lesions would be revealed during history taking.
Considerations for treatment and follow-up
Without treatment, blisters often self-resolve in 2 to 6 weeks, but there is high likelihood of recurrence.6,8 There is no consensus on treatment, although a typical course of action is to deroof the blister and examine the area to rule out infection.6 The wound is then covered with wet-to-dry gauze that is changed regularly. If there is suspicion for or signs of underlying infection, such as an ulcer or skin necrosis, antibiotics should be included in the treatment plan.7
Additional considerations. Patients will often need therapeutic footwear if the blisters are located on the feet. Given the higher prevalence of microvascular complications in patients with diabetes who develop BD, routine ophthalmologic examination and renal function testing to monitor for microalbuminuria are recommended.5
Our patient underwent bedside incision and drainage and was discharged home with appropriate wound care and follow-up.
THE TAKEAWAY
BD cases may be underdiagnosed in clinical practice, perhaps due to patients not seeking help for a seemingly nonthreatening condition or lack of clinician recognition that bullae are related to a patient’s diabetes status. Prompt recognition and proper wound care are important to prevent poor outcomes, such as ulceration or necrosis.
CORRESPONDENCE
Kathleen S. Kinderwater, MD, 101 Heart Drive, Greenville, NC 27834; [email protected]
THE CASE
A 57-year-old man with type 2 diabetes, hyperlipidemia, and obesity presented to the emergency department (ED) for bilateral foot blisters, both of which appeared 1 day prior to evaluation. The patient’s history also included right-side Charcot foot diagnosed 4 years earlier and right foot osteomyelitis diagnosed 2 years prior. He had ongoing neuropathy in both feet but denied any significant pain.
The patient wore orthotics daily and he’d had new orthotics made 6 months prior; however, a recent COVID-19 diagnosis and prolonged hospital stay resulted in a 30-pound weight loss and decreased swelling in his ankles. He acquired new shoes 2 weeks prior to ED presentation.
Physical examination revealed large blisters along the medial aspect of the patient’s feet, with both hemorrhagic and serous fluid-filled bullae. The lesions were flaccid but intact, without drainage or surrounding erythema, warmth, or tenderness. The blister on the left foot measured 8 x 5 cm and extended from the great toe to mid-arch (FIGURE), while the one on the right foot measured 8 x 3 cm and extended from the great toe to the base of the proximal arch. Sensation was decreased in the bilateral first and second digits but unchanged from prior documented exams. Bilateral dorsalis pedis pulses were normal.
Work-up included imaging and lab work. The patient’s complete blood count was normal, as were his erythrocyte sedimentation rate and C-reactive protein level. Radiographs of the right foot were normal, but those of the left foot were concerning, although inconclusive, for osteomyelitis. Further evaluation with magnetic resonance imaging of his left foot revealed a deformity of the first digit with some subchondral signal change that was thought to be posttraumatic or degenerative, but unlikely osteomyelitis.
THE DIAGNOSIS
Podiatry was consulted for blister management. Based on atraumatic history, rapid appearance, location of blisters, unremarkable lab work and imaging, and concurrent diabetes, the patient received a diagnosis of bilateral bullous diabeticorum (BD).
DISCUSSION
Roughly one-third of patients with diabetes will experience some cutaneous adverse effect because of the disease.1 Common iterations include acanthosis nigricans, rash, or even infection.2 BD is a rare bullous skin lesion that occurs in patients with diabetes; it has a reported annual incidence of 0.16% and may be underdiagnosed.1
Cases of BD have been described both in patients with longstanding diabetes and in those newly diagnosed, although the former group is more often affected.1 BD is reported more frequently in males than females, at a ratio of 2:1.1,3 Patients ages 17 to 80 years (average age, 55 years) have received a diagnosis of BD.1 Most affected patients will have a concomitant peripheral neuropathy and sometimes nephropathy or retinopathy.1
Continue to: The etiology of BD...
The etiology of BD is unclear but appears to be multifactorial. Hypotheses suggest that there’s a link to neuropathy/nephropathy, excessive exposure to ultraviolet light, or a vascular cause secondary to hyaline deposition in the capillary walls.4,5
What you’ll see at presentation
The typical manifestation of BD is the rapid appearance of tense blisters, which may occur overnight or even within hours.1 They are usually painless; common locations include the feet, distal legs, hands, and forearms.1,5 The bullae can be serous or hemorrhagic.1
Most notable in the patient’s history will be a lack of trauma or injury to the area.1 Although A1C values do not correlate with blister formation, patients with hypoglycemic episodes and highly varying blood glucose values seem to have higher rates of occurrence.1
Other sources of blistering must be ruled out
The diagnosis of BD is clinical and based on history, exam, and exclusion of other bullous diagnoses.6 A key clue in the history is the spontaneous and rapid onset without associated trauma in a patient with diabetes.6 Direct immunofluorescence, although nonspecific, can be helpful to rule out other disorders (such as porphyria cutanea tarda and bullous pemphigoid) if the history and exam are inconclusive. Direct and indirect immunofluorescence is typically negative in BD.4,6
The differential diagnosis includes other conditions that involve bullae—such as frictional bullae, bullous pemphigoid, and bullous systemic lupus erythematosus—as well as porphyria, erythema multiforme, insect bites, or even fixed drug eruption.2,7
Continue to: Porphyria
Porphyria tends to develop on the hands, whereas BD most commonly occurs on the feet.5
Erythema multiforme typically includes inflammatory skin changes.5
Trauma or fixed drug eruption as a cause of blistering lesions would be revealed during history taking.
Considerations for treatment and follow-up
Without treatment, blisters often self-resolve in 2 to 6 weeks, but there is high likelihood of recurrence.6,8 There is no consensus on treatment, although a typical course of action is to deroof the blister and examine the area to rule out infection.6 The wound is then covered with wet-to-dry gauze that is changed regularly. If there is suspicion for or signs of underlying infection, such as an ulcer or skin necrosis, antibiotics should be included in the treatment plan.7
Additional considerations. Patients will often need therapeutic footwear if the blisters are located on the feet. Given the higher prevalence of microvascular complications in patients with diabetes who develop BD, routine ophthalmologic examination and renal function testing to monitor for microalbuminuria are recommended.5
Our patient underwent bedside incision and drainage and was discharged home with appropriate wound care and follow-up.
THE TAKEAWAY
BD cases may be underdiagnosed in clinical practice, perhaps due to patients not seeking help for a seemingly nonthreatening condition or lack of clinician recognition that bullae are related to a patient’s diabetes status. Prompt recognition and proper wound care are important to prevent poor outcomes, such as ulceration or necrosis.
CORRESPONDENCE
Kathleen S. Kinderwater, MD, 101 Heart Drive, Greenville, NC 27834; [email protected]
1. Larsen K, Jensen T, Karlsmark T, et al. Incidence of bullosis diabeticorum—a controversial cause of chronic foot ulceration. Int Wound J. 2008;5:591-596. doi: 10.1111/j.1742-481X.2008.00476.x
2. Lipsky BA, Baker PD, Ahroni JH. Diabetic bullae: 12 cases of a purportedly rare cutaneous disorder. Int J Dermatol. 2000;39:196-200. doi: 10.1046/j.1365-4362.2000.00947.x
3. Gupta V, Gulati N, Bahl J, et al. Bullosis diabeticorum: rare presentation in a common disease. Case Rep Endocrinol. 2014;2014:862912.
4. Sonani H, Abdul Salim S, Garla VV, et al. Bullosis diabeticorum: a rare presentation with immunoglobulin G (IgG) deposition related vasculopathy. Case report and focused review. Am J Case Rep. 2018;19:52-56. doi: 10.12659/ajcr.905452
5. Chouk C, Litaiem N. Bullosis diabeticorum. StatPearls [Internet]. Updated June 5, 2021. Accessed July 14, 2022. www.ncbi.nlm.nih.gov/books/NBK539872/
6. Chatterjee D, Radotra A, Radotra BD, et al. Bullous diabeticorum: a rare blistering manifestation of diabetes. Indian Dermatol Online J. 2017;8:274-275. doi: 10.4103/idoj.IDOJ_340_16
7. Kansal NK, Anuragi RP. Bullous lesions in diabetes mellitus: bullous diabeticorum (diabetic bulla). BMJ Case Rep. 2020;13:e238617. doi: 10.1136/bcr-2020-238617
8. Bello F, Samaila OM, Lawal Y, et al. 2 cases of bullosis diabeticorum following long-distance journeys by road: a report of 2 cases. Case Rep Endocrinol. 2012;2012:367218. doi: 10.1155/2012/367218
1. Larsen K, Jensen T, Karlsmark T, et al. Incidence of bullosis diabeticorum—a controversial cause of chronic foot ulceration. Int Wound J. 2008;5:591-596. doi: 10.1111/j.1742-481X.2008.00476.x
2. Lipsky BA, Baker PD, Ahroni JH. Diabetic bullae: 12 cases of a purportedly rare cutaneous disorder. Int J Dermatol. 2000;39:196-200. doi: 10.1046/j.1365-4362.2000.00947.x
3. Gupta V, Gulati N, Bahl J, et al. Bullosis diabeticorum: rare presentation in a common disease. Case Rep Endocrinol. 2014;2014:862912.
4. Sonani H, Abdul Salim S, Garla VV, et al. Bullosis diabeticorum: a rare presentation with immunoglobulin G (IgG) deposition related vasculopathy. Case report and focused review. Am J Case Rep. 2018;19:52-56. doi: 10.12659/ajcr.905452
5. Chouk C, Litaiem N. Bullosis diabeticorum. StatPearls [Internet]. Updated June 5, 2021. Accessed July 14, 2022. www.ncbi.nlm.nih.gov/books/NBK539872/
6. Chatterjee D, Radotra A, Radotra BD, et al. Bullous diabeticorum: a rare blistering manifestation of diabetes. Indian Dermatol Online J. 2017;8:274-275. doi: 10.4103/idoj.IDOJ_340_16
7. Kansal NK, Anuragi RP. Bullous lesions in diabetes mellitus: bullous diabeticorum (diabetic bulla). BMJ Case Rep. 2020;13:e238617. doi: 10.1136/bcr-2020-238617
8. Bello F, Samaila OM, Lawal Y, et al. 2 cases of bullosis diabeticorum following long-distance journeys by road: a report of 2 cases. Case Rep Endocrinol. 2012;2012:367218. doi: 10.1155/2012/367218

Catheter-Directed Retrieval of an Infected Fragment in a Vietnam War Veteran
Shrapnel injuries are commonly encountered in war zones.1 Shrapnel injuries can remain asymptomatic or become systemic, with health effects of the retained foreign body ranging from local to systemic toxicities depending on the patient’s reaction to the chemical composition and corrosiveness of the fragments in vivo.2 We present a case of a reactivating shrapnel injury in the form of a retroperitoneal infection and subsequent iliopsoas abscess. A collaborative procedure was performed between surgery and interventional radiology to snare and remove the infected fragment and drain the abscess.
Case Presentation
While serving in Vietnam, a soldier sustained a fragment injury to his left lower abdomen. He underwent a laparotomy, small bowel resection, and a temporary ileostomy at the time of the injury. Nearly 50 years later, the patient presented with chronic left lower quadrant pain and a low-grade fever. He was diagnosed clinically in the emergency department (ED) with diverticulitis and treated with antibiotics. The patient initially responded to treatment but returned 6 months later with similar symptoms, low-grade fever, and mild leukocytosis. A computed tomography (CT) scan during that encounter without IV contrast revealed a few scattered colonic diverticula without definite diverticulitis as well as a metallic fragment embedded in the left iliopsoas with increased soft tissue density.
The patient was diagnosed with a pelvic/abdominal wall hematoma and was discharged with pain medication. The patient reported recurrent attacks of left lower quadrant pain, fever, and changes in bowel habits, prompting gastrointestinal consultation and a colonoscopy that was unremarkable. Ten months later, the patient again presented to the ED, with recurrent symptoms, a fever of 102 °F, and leukocytosis with a white blood cell count of 11.7 × 109/L. CT scan with IV contrast revealed a large left iliopsoas abscess associated with an approximately 1-cm metallic fragment (Figure 1). A drainage catheter was placed under CT guidance and approximately 270 mL of purulent fluid was drained. Culture of the fluid was positive for Escherichia coli (E coli). Two days after drain placement, the fragment was removed as a joint procedure with interventional radiology and surgery. Using the drainage catheter tract as a point of entry, multiple attempts were made to retrieve the fragment with Olympus EndoJaw endoscopic forceps without success.

Ultimately a stiff directional sheath from a Cook Medical transjugular liver biopsy kit was used with a Merit Medical EnSnare to relocate the fragment to the left inguinal region for surgical excision (Figures 2, 3, and 4). The fragment was removed and swabbed for culture and sensitivity and a BLAKE drain was placed in the evacuated abscess cavity. The patient tolerated the procedure well and was discharged the following day. Three days later, culture and sensitivity grew E coli and Acinetobacter, thus confirming infection and a nidus for the surrounding abscess formation. On follow-up with general surgery 7 days later, the patient reported he was doing well, and the drain was removed without difficulty.
Discussion
Foreign body injuries can be benign or debilitating depending on the initial damage, anatomical location of the foreign body, composition of the foreign body, and the patient’s response to it. Retained shrapnel deep within the muscle tissue rarely causes complications. Although many times embedded objects can be asymptomatic and require no further management, migration of the foreign body or the formation of a fistula is possible, causing symptoms and requiring surgical intervention.1 One case involved the formation of a purulent fistula appearing a year after an explosive wound to the lumbosacral spine, which was treated with antimicrobials. Recurrence of the fistula several times after treatment led to surgical removal of the shrapnel along with antibiotic treatment of the osteomyelitis.3 Although uncommon, lead exposure that occurs due to retained foreign body fragments from gunshot or military-related injuries can cause systemic lead toxicity. Symptoms may range from abdominal pain, nausea, and constipation to jaundice and hepatitis.4 The severity has also been stated to correlate with the surface area of the lead exposed for dissolution.5 Migration of foreign bodies and shrapnel to other sites in the body, such as movement from soft tissues into distantly located body cavities, have been reported as well. Such a case involved the spontaneous onset of knee synovitis due to an intra-articular metallic object that was introduced via a blast injury to the upper third of the ipsilateral thigh.1
In this patient’s case, a large intramuscular abscess had formed nearly 50 years after the initial combat injury, requiring drainage of the abscess and removal of the fragment. By snaring the foreign body to a more superficial site, the surgical removal only required a minor incision, decreasing recovery time and the likelihood of postoperative complications that would have been associated with a large retroperitoneal dissection. While loop snare is often the first-line technique for the removal of intravascular foreign bodies, its use in soft tissue retained materials is scarcely reported.6 The more typical uses involve the removal of intraluminal materials, such as partially fractured venous catheters, guide wires, stents, and vena cava filters. The same report mentioned that in all 16 cases of percutaneous foreign body retrieval, no surgical intervention was required.7 In the case of most nonvascular foreign bodies, however, surgical retrieval is usually performed.8
Surgical removal of foreign bodies can be difficult in cases where a foreign body is anatomically located next to vital structures.9 An additional challenge with a sole surgical approach to foreign body retrieval is when it is small in size and lies deep within the soft tissue, as was the case for our patient. In such cases, the surgical procedure can be time consuming and lead to more trauma to the surrounding tissues.10 These factors alone necessitate consideration of postoperative morbidity and mortality.
In our patient, the retained fragment was embedded in the wall of an abscess located retroperitoneally in his iliopsoas muscle. When considering the proximity of the iliopsoas muscle to the digestive tract, urinary tract, and iliac lymph nodes, it is reasonable for infectious material to come in contact with the foreign body from these nearby structures, resulting in secondary infection.11 Surgery was previously considered the first-line treatment for retroperitoneal abscesses until the advent of imaging-guided percutaneous drainage.12
In some instances, surgical drainage may still be attempted, such as if there are different disease processes requiring open surgery or if percutaneous catheter drainage is not technically possible due to the location of the abscess, thick exudate, loculation/septations, or phlegmon. In these cases, laparoscopic drainage as opposed to open surgical drainage can provide the benefits of an open procedure (ie, total drainage and resection of infected tissue) but is less invasive, requires a smaller incision, and heals faster.13 Percutaneous drainage is the current first-line treatment due to the lack of need for general anesthesia, lower cost, and better morbidity and mortality outcomes compared to surgical methods.12 While percutaneous drainage proved to be immediately therapeutic for our patient, the risk of abscess recurrence with the retained infected fragment necessitated coordination of procedures across specialties to provide the best outcome for the patient.
Conclusions
This case demonstrates a multidisciplinary approach to transforming an otherwise large retroperitoneal dissection to a minimally invasive and technically efficient abscess drainage and foreign body retrieval.
1. Schroeder JE, Lowe J, Chaimsky G, Liebergall M, Mosheiff R. Migrating shrapnel: a rare cause of knee synovitis. Mil Med. 2010;175(11):929-930. doi:10.7205/milmed-d-09-00254
2. Centeno JA, Rogers DA, van der Voet GB, et al. Embedded fragments from U.S. military personnel—chemical analysis and potential health implications. Int J Environ Res Public Health. 2014;11(2):1261-1278. Published 2014 Jan 23. doi:10.3390/ijerph110201261
3. Carija R, Busic Z, Bradaric N, Bulovic B, Borzic Z, Pavicic-Perkovic S. Surgical removal of metallic foreign body (shrapnel) from the lumbosacral spine and the treatment of chronic osteomyelitis: a case report. West Indian Med J. 2014;63(4):373-375. doi:10.7727/wimj.2012.290
4. Grasso I, Blattner M, Short T, Downs J. Severe systemic lead toxicity resulting from extra-articular retained shrapnel presenting as jaundice and hepatitis: a case report and review of the literature. Mil Med. 2017;182(3-4):e1843-e1848. doi:10.7205/MILMED-D-16-00231
5. Dillman RO, Crumb CK, Lidsky MJ. Lead poisoning from a gunshot wound: report of a case and review of the literature. Am J Med. 1979;66(3):509-514. doi:10.1016/0002-9343(79)91083-0
6. Woodhouse JB, Uberoi R. Techniques for intravascular foreign body retrieval. Cardiovasc Intervent Radiol. 2013;36(4):888-897. doi:10.1007/s00270-012-0488-8
7. Mallmann CV, Wolf KJ, Wacker FK. Retrieval of vascular foreign bodies using a self-made wire snare. Acta Radiol. 2008;49(10):1124-1128. doi:10.1080/02841850802454741
8. Nosher JL, Siegel R. Percutaneous retrieval of nonvascular foreign bodies. Radiology. 1993;187(3):649-651. doi:10.1148/radiology.187.3.8497610
9. Fu Y, Cui LG, Romagnoli C, Li ZQ, Lei YT. Ultrasound-guided removal of retained soft tissue foreign body with late presentation. Chin Med J (Engl). 2017;130(14):1753-1754. doi:10.4103/0366-6999.209910
10. Liang HD, Li H, Feng H, Zhao ZN, Song WJ, Yuan B. Application of intraoperative navigation and positioning system in the removal of deep foreign bodies in the limbs. Chin Med J (Engl). 2019;132(11):1375-1377. doi:10.1097/CM9.0000000000000253
11. Moriarty CM, Baker RJ. A pain in the psoas. Sports Health. 2016;8(6):568-572. doi:10.1177/1941738116665112
12. Akhan O, Durmaz H, Balcı S, Birgi E, Çiftçi T, Akıncı D. Percutaneous drainage of retroperitoneal abscesses: variables for success, failure, and recurrence. Diagn Interv Radiol. 2020;26(2):124-130. doi:10.5152/dir.2019.19199
13. Hong CH, Hong YC, Bae SH, et al. Laparoscopic drainage as a minimally invasive treatment for a psoas abscess: a single center case series and literature review. Medicine (Baltimore). 2020;99(14):e19640. doi:10.1097/MD.0000000000019640
Shrapnel injuries are commonly encountered in war zones.1 Shrapnel injuries can remain asymptomatic or become systemic, with health effects of the retained foreign body ranging from local to systemic toxicities depending on the patient’s reaction to the chemical composition and corrosiveness of the fragments in vivo.2 We present a case of a reactivating shrapnel injury in the form of a retroperitoneal infection and subsequent iliopsoas abscess. A collaborative procedure was performed between surgery and interventional radiology to snare and remove the infected fragment and drain the abscess.
Case Presentation
While serving in Vietnam, a soldier sustained a fragment injury to his left lower abdomen. He underwent a laparotomy, small bowel resection, and a temporary ileostomy at the time of the injury. Nearly 50 years later, the patient presented with chronic left lower quadrant pain and a low-grade fever. He was diagnosed clinically in the emergency department (ED) with diverticulitis and treated with antibiotics. The patient initially responded to treatment but returned 6 months later with similar symptoms, low-grade fever, and mild leukocytosis. A computed tomography (CT) scan during that encounter without IV contrast revealed a few scattered colonic diverticula without definite diverticulitis as well as a metallic fragment embedded in the left iliopsoas with increased soft tissue density.
The patient was diagnosed with a pelvic/abdominal wall hematoma and was discharged with pain medication. The patient reported recurrent attacks of left lower quadrant pain, fever, and changes in bowel habits, prompting gastrointestinal consultation and a colonoscopy that was unremarkable. Ten months later, the patient again presented to the ED, with recurrent symptoms, a fever of 102 °F, and leukocytosis with a white blood cell count of 11.7 × 109/L. CT scan with IV contrast revealed a large left iliopsoas abscess associated with an approximately 1-cm metallic fragment (Figure 1). A drainage catheter was placed under CT guidance and approximately 270 mL of purulent fluid was drained. Culture of the fluid was positive for Escherichia coli (E coli). Two days after drain placement, the fragment was removed as a joint procedure with interventional radiology and surgery. Using the drainage catheter tract as a point of entry, multiple attempts were made to retrieve the fragment with Olympus EndoJaw endoscopic forceps without success.
Ultimately a stiff directional sheath from a Cook Medical transjugular liver biopsy kit was used with a Merit Medical EnSnare to relocate the fragment to the left inguinal region for surgical excision (Figures 2, 3, and 4). The fragment was removed and swabbed for culture and sensitivity and a BLAKE drain was placed in the evacuated abscess cavity. The patient tolerated the procedure well and was discharged the following day. Three days later, culture and sensitivity grew E coli and Acinetobacter, thus confirming infection and a nidus for the surrounding abscess formation. On follow-up with general surgery 7 days later, the patient reported he was doing well, and the drain was removed without difficulty.
Discussion
Foreign body injuries can be benign or debilitating depending on the initial damage, anatomical location of the foreign body, composition of the foreign body, and the patient’s response to it. Retained shrapnel deep within the muscle tissue rarely causes complications. Although many times embedded objects can be asymptomatic and require no further management, migration of the foreign body or the formation of a fistula is possible, causing symptoms and requiring surgical intervention.1 One case involved the formation of a purulent fistula appearing a year after an explosive wound to the lumbosacral spine, which was treated with antimicrobials. Recurrence of the fistula several times after treatment led to surgical removal of the shrapnel along with antibiotic treatment of the osteomyelitis.3 Although uncommon, lead exposure that occurs due to retained foreign body fragments from gunshot or military-related injuries can cause systemic lead toxicity. Symptoms may range from abdominal pain, nausea, and constipation to jaundice and hepatitis.4 The severity has also been stated to correlate with the surface area of the lead exposed for dissolution.5 Migration of foreign bodies and shrapnel to other sites in the body, such as movement from soft tissues into distantly located body cavities, have been reported as well. Such a case involved the spontaneous onset of knee synovitis due to an intra-articular metallic object that was introduced via a blast injury to the upper third of the ipsilateral thigh.1
In this patient’s case, a large intramuscular abscess had formed nearly 50 years after the initial combat injury, requiring drainage of the abscess and removal of the fragment. By snaring the foreign body to a more superficial site, the surgical removal only required a minor incision, decreasing recovery time and the likelihood of postoperative complications that would have been associated with a large retroperitoneal dissection. While loop snare is often the first-line technique for the removal of intravascular foreign bodies, its use in soft tissue retained materials is scarcely reported.6 The more typical uses involve the removal of intraluminal materials, such as partially fractured venous catheters, guide wires, stents, and vena cava filters. The same report mentioned that in all 16 cases of percutaneous foreign body retrieval, no surgical intervention was required.7 In the case of most nonvascular foreign bodies, however, surgical retrieval is usually performed.8
Surgical removal of foreign bodies can be difficult in cases where a foreign body is anatomically located next to vital structures.9 An additional challenge with a sole surgical approach to foreign body retrieval is when it is small in size and lies deep within the soft tissue, as was the case for our patient. In such cases, the surgical procedure can be time consuming and lead to more trauma to the surrounding tissues.10 These factors alone necessitate consideration of postoperative morbidity and mortality.
In our patient, the retained fragment was embedded in the wall of an abscess located retroperitoneally in his iliopsoas muscle. When considering the proximity of the iliopsoas muscle to the digestive tract, urinary tract, and iliac lymph nodes, it is reasonable for infectious material to come in contact with the foreign body from these nearby structures, resulting in secondary infection.11 Surgery was previously considered the first-line treatment for retroperitoneal abscesses until the advent of imaging-guided percutaneous drainage.12
In some instances, surgical drainage may still be attempted, such as if there are different disease processes requiring open surgery or if percutaneous catheter drainage is not technically possible due to the location of the abscess, thick exudate, loculation/septations, or phlegmon. In these cases, laparoscopic drainage as opposed to open surgical drainage can provide the benefits of an open procedure (ie, total drainage and resection of infected tissue) but is less invasive, requires a smaller incision, and heals faster.13 Percutaneous drainage is the current first-line treatment due to the lack of need for general anesthesia, lower cost, and better morbidity and mortality outcomes compared to surgical methods.12 While percutaneous drainage proved to be immediately therapeutic for our patient, the risk of abscess recurrence with the retained infected fragment necessitated coordination of procedures across specialties to provide the best outcome for the patient.
Conclusions
This case demonstrates a multidisciplinary approach to transforming an otherwise large retroperitoneal dissection to a minimally invasive and technically efficient abscess drainage and foreign body retrieval.
Shrapnel injuries are commonly encountered in war zones.1 Shrapnel injuries can remain asymptomatic or become systemic, with health effects of the retained foreign body ranging from local to systemic toxicities depending on the patient’s reaction to the chemical composition and corrosiveness of the fragments in vivo.2 We present a case of a reactivating shrapnel injury in the form of a retroperitoneal infection and subsequent iliopsoas abscess. A collaborative procedure was performed between surgery and interventional radiology to snare and remove the infected fragment and drain the abscess.
Case Presentation
While serving in Vietnam, a soldier sustained a fragment injury to his left lower abdomen. He underwent a laparotomy, small bowel resection, and a temporary ileostomy at the time of the injury. Nearly 50 years later, the patient presented with chronic left lower quadrant pain and a low-grade fever. He was diagnosed clinically in the emergency department (ED) with diverticulitis and treated with antibiotics. The patient initially responded to treatment but returned 6 months later with similar symptoms, low-grade fever, and mild leukocytosis. A computed tomography (CT) scan during that encounter without IV contrast revealed a few scattered colonic diverticula without definite diverticulitis as well as a metallic fragment embedded in the left iliopsoas with increased soft tissue density.
The patient was diagnosed with a pelvic/abdominal wall hematoma and was discharged with pain medication. The patient reported recurrent attacks of left lower quadrant pain, fever, and changes in bowel habits, prompting gastrointestinal consultation and a colonoscopy that was unremarkable. Ten months later, the patient again presented to the ED, with recurrent symptoms, a fever of 102 °F, and leukocytosis with a white blood cell count of 11.7 × 109/L. CT scan with IV contrast revealed a large left iliopsoas abscess associated with an approximately 1-cm metallic fragment (Figure 1). A drainage catheter was placed under CT guidance and approximately 270 mL of purulent fluid was drained. Culture of the fluid was positive for Escherichia coli (E coli). Two days after drain placement, the fragment was removed as a joint procedure with interventional radiology and surgery. Using the drainage catheter tract as a point of entry, multiple attempts were made to retrieve the fragment with Olympus EndoJaw endoscopic forceps without success.
Ultimately a stiff directional sheath from a Cook Medical transjugular liver biopsy kit was used with a Merit Medical EnSnare to relocate the fragment to the left inguinal region for surgical excision (Figures 2, 3, and 4). The fragment was removed and swabbed for culture and sensitivity and a BLAKE drain was placed in the evacuated abscess cavity. The patient tolerated the procedure well and was discharged the following day. Three days later, culture and sensitivity grew E coli and Acinetobacter, thus confirming infection and a nidus for the surrounding abscess formation. On follow-up with general surgery 7 days later, the patient reported he was doing well, and the drain was removed without difficulty.
Discussion
Foreign body injuries can be benign or debilitating depending on the initial damage, anatomical location of the foreign body, composition of the foreign body, and the patient’s response to it. Retained shrapnel deep within the muscle tissue rarely causes complications. Although many times embedded objects can be asymptomatic and require no further management, migration of the foreign body or the formation of a fistula is possible, causing symptoms and requiring surgical intervention.1 One case involved the formation of a purulent fistula appearing a year after an explosive wound to the lumbosacral spine, which was treated with antimicrobials. Recurrence of the fistula several times after treatment led to surgical removal of the shrapnel along with antibiotic treatment of the osteomyelitis.3 Although uncommon, lead exposure that occurs due to retained foreign body fragments from gunshot or military-related injuries can cause systemic lead toxicity. Symptoms may range from abdominal pain, nausea, and constipation to jaundice and hepatitis.4 The severity has also been stated to correlate with the surface area of the lead exposed for dissolution.5 Migration of foreign bodies and shrapnel to other sites in the body, such as movement from soft tissues into distantly located body cavities, have been reported as well. Such a case involved the spontaneous onset of knee synovitis due to an intra-articular metallic object that was introduced via a blast injury to the upper third of the ipsilateral thigh.1
In this patient’s case, a large intramuscular abscess had formed nearly 50 years after the initial combat injury, requiring drainage of the abscess and removal of the fragment. By snaring the foreign body to a more superficial site, the surgical removal only required a minor incision, decreasing recovery time and the likelihood of postoperative complications that would have been associated with a large retroperitoneal dissection. While loop snare is often the first-line technique for the removal of intravascular foreign bodies, its use in soft tissue retained materials is scarcely reported.6 The more typical uses involve the removal of intraluminal materials, such as partially fractured venous catheters, guide wires, stents, and vena cava filters. The same report mentioned that in all 16 cases of percutaneous foreign body retrieval, no surgical intervention was required.7 In the case of most nonvascular foreign bodies, however, surgical retrieval is usually performed.8
Surgical removal of foreign bodies can be difficult in cases where a foreign body is anatomically located next to vital structures.9 An additional challenge with a sole surgical approach to foreign body retrieval is when it is small in size and lies deep within the soft tissue, as was the case for our patient. In such cases, the surgical procedure can be time consuming and lead to more trauma to the surrounding tissues.10 These factors alone necessitate consideration of postoperative morbidity and mortality.
In our patient, the retained fragment was embedded in the wall of an abscess located retroperitoneally in his iliopsoas muscle. When considering the proximity of the iliopsoas muscle to the digestive tract, urinary tract, and iliac lymph nodes, it is reasonable for infectious material to come in contact with the foreign body from these nearby structures, resulting in secondary infection.11 Surgery was previously considered the first-line treatment for retroperitoneal abscesses until the advent of imaging-guided percutaneous drainage.12
In some instances, surgical drainage may still be attempted, such as if there are different disease processes requiring open surgery or if percutaneous catheter drainage is not technically possible due to the location of the abscess, thick exudate, loculation/septations, or phlegmon. In these cases, laparoscopic drainage as opposed to open surgical drainage can provide the benefits of an open procedure (ie, total drainage and resection of infected tissue) but is less invasive, requires a smaller incision, and heals faster.13 Percutaneous drainage is the current first-line treatment due to the lack of need for general anesthesia, lower cost, and better morbidity and mortality outcomes compared to surgical methods.12 While percutaneous drainage proved to be immediately therapeutic for our patient, the risk of abscess recurrence with the retained infected fragment necessitated coordination of procedures across specialties to provide the best outcome for the patient.
Conclusions
This case demonstrates a multidisciplinary approach to transforming an otherwise large retroperitoneal dissection to a minimally invasive and technically efficient abscess drainage and foreign body retrieval.
1. Schroeder JE, Lowe J, Chaimsky G, Liebergall M, Mosheiff R. Migrating shrapnel: a rare cause of knee synovitis. Mil Med. 2010;175(11):929-930. doi:10.7205/milmed-d-09-00254
2. Centeno JA, Rogers DA, van der Voet GB, et al. Embedded fragments from U.S. military personnel—chemical analysis and potential health implications. Int J Environ Res Public Health. 2014;11(2):1261-1278. Published 2014 Jan 23. doi:10.3390/ijerph110201261
3. Carija R, Busic Z, Bradaric N, Bulovic B, Borzic Z, Pavicic-Perkovic S. Surgical removal of metallic foreign body (shrapnel) from the lumbosacral spine and the treatment of chronic osteomyelitis: a case report. West Indian Med J. 2014;63(4):373-375. doi:10.7727/wimj.2012.290
4. Grasso I, Blattner M, Short T, Downs J. Severe systemic lead toxicity resulting from extra-articular retained shrapnel presenting as jaundice and hepatitis: a case report and review of the literature. Mil Med. 2017;182(3-4):e1843-e1848. doi:10.7205/MILMED-D-16-00231
5. Dillman RO, Crumb CK, Lidsky MJ. Lead poisoning from a gunshot wound: report of a case and review of the literature. Am J Med. 1979;66(3):509-514. doi:10.1016/0002-9343(79)91083-0
6. Woodhouse JB, Uberoi R. Techniques for intravascular foreign body retrieval. Cardiovasc Intervent Radiol. 2013;36(4):888-897. doi:10.1007/s00270-012-0488-8
7. Mallmann CV, Wolf KJ, Wacker FK. Retrieval of vascular foreign bodies using a self-made wire snare. Acta Radiol. 2008;49(10):1124-1128. doi:10.1080/02841850802454741
8. Nosher JL, Siegel R. Percutaneous retrieval of nonvascular foreign bodies. Radiology. 1993;187(3):649-651. doi:10.1148/radiology.187.3.8497610
9. Fu Y, Cui LG, Romagnoli C, Li ZQ, Lei YT. Ultrasound-guided removal of retained soft tissue foreign body with late presentation. Chin Med J (Engl). 2017;130(14):1753-1754. doi:10.4103/0366-6999.209910
10. Liang HD, Li H, Feng H, Zhao ZN, Song WJ, Yuan B. Application of intraoperative navigation and positioning system in the removal of deep foreign bodies in the limbs. Chin Med J (Engl). 2019;132(11):1375-1377. doi:10.1097/CM9.0000000000000253
11. Moriarty CM, Baker RJ. A pain in the psoas. Sports Health. 2016;8(6):568-572. doi:10.1177/1941738116665112
12. Akhan O, Durmaz H, Balcı S, Birgi E, Çiftçi T, Akıncı D. Percutaneous drainage of retroperitoneal abscesses: variables for success, failure, and recurrence. Diagn Interv Radiol. 2020;26(2):124-130. doi:10.5152/dir.2019.19199
13. Hong CH, Hong YC, Bae SH, et al. Laparoscopic drainage as a minimally invasive treatment for a psoas abscess: a single center case series and literature review. Medicine (Baltimore). 2020;99(14):e19640. doi:10.1097/MD.0000000000019640
1. Schroeder JE, Lowe J, Chaimsky G, Liebergall M, Mosheiff R. Migrating shrapnel: a rare cause of knee synovitis. Mil Med. 2010;175(11):929-930. doi:10.7205/milmed-d-09-00254
2. Centeno JA, Rogers DA, van der Voet GB, et al. Embedded fragments from U.S. military personnel—chemical analysis and potential health implications. Int J Environ Res Public Health. 2014;11(2):1261-1278. Published 2014 Jan 23. doi:10.3390/ijerph110201261
3. Carija R, Busic Z, Bradaric N, Bulovic B, Borzic Z, Pavicic-Perkovic S. Surgical removal of metallic foreign body (shrapnel) from the lumbosacral spine and the treatment of chronic osteomyelitis: a case report. West Indian Med J. 2014;63(4):373-375. doi:10.7727/wimj.2012.290
4. Grasso I, Blattner M, Short T, Downs J. Severe systemic lead toxicity resulting from extra-articular retained shrapnel presenting as jaundice and hepatitis: a case report and review of the literature. Mil Med. 2017;182(3-4):e1843-e1848. doi:10.7205/MILMED-D-16-00231
5. Dillman RO, Crumb CK, Lidsky MJ. Lead poisoning from a gunshot wound: report of a case and review of the literature. Am J Med. 1979;66(3):509-514. doi:10.1016/0002-9343(79)91083-0
6. Woodhouse JB, Uberoi R. Techniques for intravascular foreign body retrieval. Cardiovasc Intervent Radiol. 2013;36(4):888-897. doi:10.1007/s00270-012-0488-8
7. Mallmann CV, Wolf KJ, Wacker FK. Retrieval of vascular foreign bodies using a self-made wire snare. Acta Radiol. 2008;49(10):1124-1128. doi:10.1080/02841850802454741
8. Nosher JL, Siegel R. Percutaneous retrieval of nonvascular foreign bodies. Radiology. 1993;187(3):649-651. doi:10.1148/radiology.187.3.8497610
9. Fu Y, Cui LG, Romagnoli C, Li ZQ, Lei YT. Ultrasound-guided removal of retained soft tissue foreign body with late presentation. Chin Med J (Engl). 2017;130(14):1753-1754. doi:10.4103/0366-6999.209910
10. Liang HD, Li H, Feng H, Zhao ZN, Song WJ, Yuan B. Application of intraoperative navigation and positioning system in the removal of deep foreign bodies in the limbs. Chin Med J (Engl). 2019;132(11):1375-1377. doi:10.1097/CM9.0000000000000253
11. Moriarty CM, Baker RJ. A pain in the psoas. Sports Health. 2016;8(6):568-572. doi:10.1177/1941738116665112
12. Akhan O, Durmaz H, Balcı S, Birgi E, Çiftçi T, Akıncı D. Percutaneous drainage of retroperitoneal abscesses: variables for success, failure, and recurrence. Diagn Interv Radiol. 2020;26(2):124-130. doi:10.5152/dir.2019.19199
13. Hong CH, Hong YC, Bae SH, et al. Laparoscopic drainage as a minimally invasive treatment for a psoas abscess: a single center case series and literature review. Medicine (Baltimore). 2020;99(14):e19640. doi:10.1097/MD.0000000000019640
Successful Use of Lanadelumab in an Older Patient With Type II Hereditary Angioedema
Hereditary angioedema (HAE) is a rare genetic disorder affecting about 1 in 67,000 individuals and may lead to increased morbidity and mortality.1,2 HAE is characterized by recurring episodes of subcutaneous and/or submucosal edema without urticaria due to an excess of bradykinin.2,3 Autosomal dominant inheritance is present in 75% of patients with HAE and is classified into 2 main types.2 Type I HAE is caused by deficiency of C1 esterase inhibitor, accounting for 85% of cases.2 Type II HAE is marked by normal to elevated levels of C1 esterase inhibitor but with reduced activity.2
Cutaneous and abdominal angioedema attacks are the most common presentation.1 However, any location may be affected, including the face, oropharynx, and larynx.1 Only 0.9% of all HAE attacks cause laryngeal edema, but 50% of HAE patients have experienced a laryngeal attack, which may be lethal.1 An angioedema attack can range in severity, depending on the location and degree of edema.3 In addition, patients with HAE often are diagnosed with anxiety and depression secondary to their poor quality of life.4 Thus, long-term prophylaxis of attacks is crucial to reduce the physical and psychological implications.
Previously, HAE was treated with antifibrinolytic agents and attenuated androgens for short- and long-term prophylaxis.1 These treatment modalities are now considered second-line since the development of novel medications with improved efficacy and limited adverse effects (AEs).1 For long-term prophylaxis, subcutaneous and IV C1 esterase inhibitor has been proven effective in both types I and II HAE.1 Another option, lanadelumab, a subcutaneously delivered monoclonal antibody inhibitor of plasma kallikrein, has been proven to decrease the frequency of HAE attacks without significant AEs.5 Lanadelumab works by binding to the active site of plasma kallikrein, which reduces its activity and slows the production of bradykinin.6 This results in decreasing vascular permeability and swelling episodes in patients with HAE.7 Data, however, are limited, specifically regarding patients with type II HAE and patients aged ≥ 65 years.5 This article reports on an older male with type II HAE successfully treated with lanadelumab.
Case Presentation
An 81-year-old male patient with hypertension, hypertriglyceridemia, and aortic aneurysm had recurrent, frequent episodes of severe abdominal pain with a remote history of extremity and scrotal swelling since adolescence. He was misdiagnosed for years and was eventually determined to have HAE at age 75 years after his niece was diagnosed, prompting him to be reevaluated for his frequent bouts of abdominal pain. His laboratory findings were consistent with HAE type II with low C4 (7.8 mg/dL), normal C1 esterase inhibitor levels (24 mg/dL), and low levels of C1 esterase inhibitor activity (28% of normal).
Initially, he described having weekly attacks of abdominal pain that could last 1 to several days. At worst, these attacks would last up to a month, causing a decrease in appetite and weight loss. At age 77 years, he began an on-demand treatment, icatibant, a bradykinin receptor blocker. After initiating icatibant during an acute attack, the pain would diminish within 1 to 2 hours, and within several hours, he would be pain free. Previously, pain relief would take several days to weeks. He continued to use icatibant on-demand, typically requiring treatment every 1 to 2 months for only the more severe attacks.
After an increasing frequency of abdominal pain attacks, prophylactic medication was recommended. Therefore, subcutaneous lanadelumab 300 mg every 2 weeks was initiated for long-term prophylaxis. The patient went from requiring on-demand treatment 2 to 3 times per month to once in 6 months after starting lanadelumab. In addition, he tolerated the medication well without any AEs.
Discussion
According to the international WAO/EAACI 2021 guidelines, HAE treatment goals are “to achieve complete control of the disease and to normalize patients’ lives.”8 On-demand treatment options include C1 esterase inhibitor, icatibant, or ecallantide (a kallikrein inhibitor).8 Long-term prophylaxis in HAE should be considered, accounting for disease activity, burden, control, and patient preference. Five medications have been used for long-term prophylaxis: antifibrinolytic agents (not recommended), attenuated androgens (considered second-line), C1 esterase inhibitor, berotralstat, and lanadelumab.8
Antifibrinolytics are no longer recommended for long-term prophylactic treatment as their efficacy is poor and was not considered for our patient. Attenuated androgens, such as danazol, have a history of prophylactic use in patients with HAE due to their good efficacy but are suboptimal due to their significant AE profile and many drug-drug interactions.8 In addition, androgens have many contraindications, including hypertension and hypertriglyceridemia, which were both present in our patient. Consequently, danazol was not an advised treatment for our patient. C1 esterase inhibitor is often used to prevent HAE attacks and can be given intravenously or subcutaneously, typically administered biweekly. A potential AE of C1 esterase inhibitor is thrombosis.Therefore, C1 esterase inhibitor was not a preferred choice in our older patient with a history of hypercoagulability. Berotralstat, a plasma kallikrein inhibitor, is an oral treatment option that also has shown efficacy in long-term prophylaxis. The most common AEs of berotralstat tend to be gastrointestinal symptoms, and the medication requires dose adjustment for patients with hepatic impairment.8 Berotralstat was not considered because it was not an approved treatment option at the time of this patient’s treatment. Lanadelumab is a human monoclonal antibody against plasma kallikrein, which decreases bradykinin production in patients with HAE, thus preventing angioedema attacks.5 Data regarding the use of lanadelumab in patients with type II HAE are limited, but because HAE with normal C1 esterase inhibitor levels involves the production of bradykinin via kallikrein, lanadelumab should still be effective.1 Lanadelumab was chosen for our patient because of its minimal AEs and is not known to increase the risk of thrombosis.
Lanadelumab is a novel medication, recently approved in 2018 by the US Food and Drug Administration for the treatment of type I and type 2 HAE in patients aged ≥ 12 years.7 The phase 3 Hereditary Angioedema Long-term Prophylaxis (HELP) study concluded that treatment with subcutaneous lanadelumab for 26 weeks significantly decreased the frequency of angioedema attacks compared with placebo.5 However, 113 (90.4%) of patients in the phase III HELP study had type I HAE.5 Of the 125 patients that completed this randomized, double-blind study, only 12 had type II HAE.5 In addition, this study only included 5 patients aged ≥ 65 years.5 Also, no patients aged ≥ 65 years were part of the treatment arms that included a lanadelumab dose of 300 mg.5 In a case series of 12 patients in Canada, treatment with lanadelumab decreased angioedema attacks by 72%.9 However, the series only included 1 patient with type II HAE who was aged 36 years.9 Therefore, our case demonstrates the efficacy of lanadelumab in a patient aged ≥ 65 years with type II HAE.
Conclusions
HAE is a rare and potentially fatal disease characterized by recurrent, unpredictable attacks of edema throughout the body. The disease burden adversely affects a patient’s quality of life. Therefore, long-term prophylaxis is critical to managing patients with HAE. Lanadelumab has been proven as an effective long-term prophylactic treatment option for HAE attacks. This case supports the use of lanadelumab in patients with type II HAE and patients aged ≥ 65 years.
Acknowledgments
The patient was initially written up based on his delayed diagnosis as a case report.3 An earlier version of this article was presented by Samuel Weiss, MD, and Derek Smith, MD, as a poster at the American Academy of Allergy, Asthma, and Immunology virtual conference February 26 to March 1, 2021.
1. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136-1148. doi:10.1056/NEJMra1808012
2. Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care. 2018;24(14)(suppl):S292-S298.
3. Berger J, Carroll MP Jr, Champoux E, Coop CA. Extremely delayed diagnosis of type II hereditary angioedema: case report and review of the literature. Mil Med. 2018;183(11-12):e765-e767. doi:10.1093/milmed/usy031
4. Fouche AS, Saunders EF, Craig T. Depression and anxiety in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(4):371-375. doi:10.1016/j.anai.2013.05.028
5. Banerji A, Riedl MA, Bernstein JA, et al; HELP Investigators. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320(20):2108-2121. doi:10.1001/jama.2018.16773
6. Busse PJ, Farkas H, Banerji A, et al. Lanadelumab for the prophylactic treatment of hereditary angioedema with C1 inhibitor deficiency: a review of preclinical and phase I studies. BioDrugs. 2019;33(1):33-43. doi:10.1007/s40259-018-0325-y
7. Riedl MA, Maurer M, Bernstein JA, et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy. 2020;75(11):2879-2887. doi:10.1111/all.14416
8. Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. 2022;77(7):1961-1990. doi:10.1111/all.15214
9. Iaboni A, Kanani A, Lacuesta G, Song C, Kan M, Betschel SD. Impact of lanadelumab in hereditary angioedema: a case series of 12 patients in Canada. Allergy Asthma Clin Immunol. 2021;17(1):78. Published 2021 Jul 23. doi:10.1186/s13223-021-00579-6
Hereditary angioedema (HAE) is a rare genetic disorder affecting about 1 in 67,000 individuals and may lead to increased morbidity and mortality.1,2 HAE is characterized by recurring episodes of subcutaneous and/or submucosal edema without urticaria due to an excess of bradykinin.2,3 Autosomal dominant inheritance is present in 75% of patients with HAE and is classified into 2 main types.2 Type I HAE is caused by deficiency of C1 esterase inhibitor, accounting for 85% of cases.2 Type II HAE is marked by normal to elevated levels of C1 esterase inhibitor but with reduced activity.2
Cutaneous and abdominal angioedema attacks are the most common presentation.1 However, any location may be affected, including the face, oropharynx, and larynx.1 Only 0.9% of all HAE attacks cause laryngeal edema, but 50% of HAE patients have experienced a laryngeal attack, which may be lethal.1 An angioedema attack can range in severity, depending on the location and degree of edema.3 In addition, patients with HAE often are diagnosed with anxiety and depression secondary to their poor quality of life.4 Thus, long-term prophylaxis of attacks is crucial to reduce the physical and psychological implications.
Previously, HAE was treated with antifibrinolytic agents and attenuated androgens for short- and long-term prophylaxis.1 These treatment modalities are now considered second-line since the development of novel medications with improved efficacy and limited adverse effects (AEs).1 For long-term prophylaxis, subcutaneous and IV C1 esterase inhibitor has been proven effective in both types I and II HAE.1 Another option, lanadelumab, a subcutaneously delivered monoclonal antibody inhibitor of plasma kallikrein, has been proven to decrease the frequency of HAE attacks without significant AEs.5 Lanadelumab works by binding to the active site of plasma kallikrein, which reduces its activity and slows the production of bradykinin.6 This results in decreasing vascular permeability and swelling episodes in patients with HAE.7 Data, however, are limited, specifically regarding patients with type II HAE and patients aged ≥ 65 years.5 This article reports on an older male with type II HAE successfully treated with lanadelumab.
Case Presentation
An 81-year-old male patient with hypertension, hypertriglyceridemia, and aortic aneurysm had recurrent, frequent episodes of severe abdominal pain with a remote history of extremity and scrotal swelling since adolescence. He was misdiagnosed for years and was eventually determined to have HAE at age 75 years after his niece was diagnosed, prompting him to be reevaluated for his frequent bouts of abdominal pain. His laboratory findings were consistent with HAE type II with low C4 (7.8 mg/dL), normal C1 esterase inhibitor levels (24 mg/dL), and low levels of C1 esterase inhibitor activity (28% of normal).
Initially, he described having weekly attacks of abdominal pain that could last 1 to several days. At worst, these attacks would last up to a month, causing a decrease in appetite and weight loss. At age 77 years, he began an on-demand treatment, icatibant, a bradykinin receptor blocker. After initiating icatibant during an acute attack, the pain would diminish within 1 to 2 hours, and within several hours, he would be pain free. Previously, pain relief would take several days to weeks. He continued to use icatibant on-demand, typically requiring treatment every 1 to 2 months for only the more severe attacks.
After an increasing frequency of abdominal pain attacks, prophylactic medication was recommended. Therefore, subcutaneous lanadelumab 300 mg every 2 weeks was initiated for long-term prophylaxis. The patient went from requiring on-demand treatment 2 to 3 times per month to once in 6 months after starting lanadelumab. In addition, he tolerated the medication well without any AEs.
Discussion
According to the international WAO/EAACI 2021 guidelines, HAE treatment goals are “to achieve complete control of the disease and to normalize patients’ lives.”8 On-demand treatment options include C1 esterase inhibitor, icatibant, or ecallantide (a kallikrein inhibitor).8 Long-term prophylaxis in HAE should be considered, accounting for disease activity, burden, control, and patient preference. Five medications have been used for long-term prophylaxis: antifibrinolytic agents (not recommended), attenuated androgens (considered second-line), C1 esterase inhibitor, berotralstat, and lanadelumab.8
Antifibrinolytics are no longer recommended for long-term prophylactic treatment as their efficacy is poor and was not considered for our patient. Attenuated androgens, such as danazol, have a history of prophylactic use in patients with HAE due to their good efficacy but are suboptimal due to their significant AE profile and many drug-drug interactions.8 In addition, androgens have many contraindications, including hypertension and hypertriglyceridemia, which were both present in our patient. Consequently, danazol was not an advised treatment for our patient. C1 esterase inhibitor is often used to prevent HAE attacks and can be given intravenously or subcutaneously, typically administered biweekly. A potential AE of C1 esterase inhibitor is thrombosis.Therefore, C1 esterase inhibitor was not a preferred choice in our older patient with a history of hypercoagulability. Berotralstat, a plasma kallikrein inhibitor, is an oral treatment option that also has shown efficacy in long-term prophylaxis. The most common AEs of berotralstat tend to be gastrointestinal symptoms, and the medication requires dose adjustment for patients with hepatic impairment.8 Berotralstat was not considered because it was not an approved treatment option at the time of this patient’s treatment. Lanadelumab is a human monoclonal antibody against plasma kallikrein, which decreases bradykinin production in patients with HAE, thus preventing angioedema attacks.5 Data regarding the use of lanadelumab in patients with type II HAE are limited, but because HAE with normal C1 esterase inhibitor levels involves the production of bradykinin via kallikrein, lanadelumab should still be effective.1 Lanadelumab was chosen for our patient because of its minimal AEs and is not known to increase the risk of thrombosis.
Lanadelumab is a novel medication, recently approved in 2018 by the US Food and Drug Administration for the treatment of type I and type 2 HAE in patients aged ≥ 12 years.7 The phase 3 Hereditary Angioedema Long-term Prophylaxis (HELP) study concluded that treatment with subcutaneous lanadelumab for 26 weeks significantly decreased the frequency of angioedema attacks compared with placebo.5 However, 113 (90.4%) of patients in the phase III HELP study had type I HAE.5 Of the 125 patients that completed this randomized, double-blind study, only 12 had type II HAE.5 In addition, this study only included 5 patients aged ≥ 65 years.5 Also, no patients aged ≥ 65 years were part of the treatment arms that included a lanadelumab dose of 300 mg.5 In a case series of 12 patients in Canada, treatment with lanadelumab decreased angioedema attacks by 72%.9 However, the series only included 1 patient with type II HAE who was aged 36 years.9 Therefore, our case demonstrates the efficacy of lanadelumab in a patient aged ≥ 65 years with type II HAE.
Conclusions
HAE is a rare and potentially fatal disease characterized by recurrent, unpredictable attacks of edema throughout the body. The disease burden adversely affects a patient’s quality of life. Therefore, long-term prophylaxis is critical to managing patients with HAE. Lanadelumab has been proven as an effective long-term prophylactic treatment option for HAE attacks. This case supports the use of lanadelumab in patients with type II HAE and patients aged ≥ 65 years.
Acknowledgments
The patient was initially written up based on his delayed diagnosis as a case report.3 An earlier version of this article was presented by Samuel Weiss, MD, and Derek Smith, MD, as a poster at the American Academy of Allergy, Asthma, and Immunology virtual conference February 26 to March 1, 2021.
Hereditary angioedema (HAE) is a rare genetic disorder affecting about 1 in 67,000 individuals and may lead to increased morbidity and mortality.1,2 HAE is characterized by recurring episodes of subcutaneous and/or submucosal edema without urticaria due to an excess of bradykinin.2,3 Autosomal dominant inheritance is present in 75% of patients with HAE and is classified into 2 main types.2 Type I HAE is caused by deficiency of C1 esterase inhibitor, accounting for 85% of cases.2 Type II HAE is marked by normal to elevated levels of C1 esterase inhibitor but with reduced activity.2
Cutaneous and abdominal angioedema attacks are the most common presentation.1 However, any location may be affected, including the face, oropharynx, and larynx.1 Only 0.9% of all HAE attacks cause laryngeal edema, but 50% of HAE patients have experienced a laryngeal attack, which may be lethal.1 An angioedema attack can range in severity, depending on the location and degree of edema.3 In addition, patients with HAE often are diagnosed with anxiety and depression secondary to their poor quality of life.4 Thus, long-term prophylaxis of attacks is crucial to reduce the physical and psychological implications.
Previously, HAE was treated with antifibrinolytic agents and attenuated androgens for short- and long-term prophylaxis.1 These treatment modalities are now considered second-line since the development of novel medications with improved efficacy and limited adverse effects (AEs).1 For long-term prophylaxis, subcutaneous and IV C1 esterase inhibitor has been proven effective in both types I and II HAE.1 Another option, lanadelumab, a subcutaneously delivered monoclonal antibody inhibitor of plasma kallikrein, has been proven to decrease the frequency of HAE attacks without significant AEs.5 Lanadelumab works by binding to the active site of plasma kallikrein, which reduces its activity and slows the production of bradykinin.6 This results in decreasing vascular permeability and swelling episodes in patients with HAE.7 Data, however, are limited, specifically regarding patients with type II HAE and patients aged ≥ 65 years.5 This article reports on an older male with type II HAE successfully treated with lanadelumab.
Case Presentation
An 81-year-old male patient with hypertension, hypertriglyceridemia, and aortic aneurysm had recurrent, frequent episodes of severe abdominal pain with a remote history of extremity and scrotal swelling since adolescence. He was misdiagnosed for years and was eventually determined to have HAE at age 75 years after his niece was diagnosed, prompting him to be reevaluated for his frequent bouts of abdominal pain. His laboratory findings were consistent with HAE type II with low C4 (7.8 mg/dL), normal C1 esterase inhibitor levels (24 mg/dL), and low levels of C1 esterase inhibitor activity (28% of normal).
Initially, he described having weekly attacks of abdominal pain that could last 1 to several days. At worst, these attacks would last up to a month, causing a decrease in appetite and weight loss. At age 77 years, he began an on-demand treatment, icatibant, a bradykinin receptor blocker. After initiating icatibant during an acute attack, the pain would diminish within 1 to 2 hours, and within several hours, he would be pain free. Previously, pain relief would take several days to weeks. He continued to use icatibant on-demand, typically requiring treatment every 1 to 2 months for only the more severe attacks.
After an increasing frequency of abdominal pain attacks, prophylactic medication was recommended. Therefore, subcutaneous lanadelumab 300 mg every 2 weeks was initiated for long-term prophylaxis. The patient went from requiring on-demand treatment 2 to 3 times per month to once in 6 months after starting lanadelumab. In addition, he tolerated the medication well without any AEs.
Discussion
According to the international WAO/EAACI 2021 guidelines, HAE treatment goals are “to achieve complete control of the disease and to normalize patients’ lives.”8 On-demand treatment options include C1 esterase inhibitor, icatibant, or ecallantide (a kallikrein inhibitor).8 Long-term prophylaxis in HAE should be considered, accounting for disease activity, burden, control, and patient preference. Five medications have been used for long-term prophylaxis: antifibrinolytic agents (not recommended), attenuated androgens (considered second-line), C1 esterase inhibitor, berotralstat, and lanadelumab.8
Antifibrinolytics are no longer recommended for long-term prophylactic treatment as their efficacy is poor and was not considered for our patient. Attenuated androgens, such as danazol, have a history of prophylactic use in patients with HAE due to their good efficacy but are suboptimal due to their significant AE profile and many drug-drug interactions.8 In addition, androgens have many contraindications, including hypertension and hypertriglyceridemia, which were both present in our patient. Consequently, danazol was not an advised treatment for our patient. C1 esterase inhibitor is often used to prevent HAE attacks and can be given intravenously or subcutaneously, typically administered biweekly. A potential AE of C1 esterase inhibitor is thrombosis.Therefore, C1 esterase inhibitor was not a preferred choice in our older patient with a history of hypercoagulability. Berotralstat, a plasma kallikrein inhibitor, is an oral treatment option that also has shown efficacy in long-term prophylaxis. The most common AEs of berotralstat tend to be gastrointestinal symptoms, and the medication requires dose adjustment for patients with hepatic impairment.8 Berotralstat was not considered because it was not an approved treatment option at the time of this patient’s treatment. Lanadelumab is a human monoclonal antibody against plasma kallikrein, which decreases bradykinin production in patients with HAE, thus preventing angioedema attacks.5 Data regarding the use of lanadelumab in patients with type II HAE are limited, but because HAE with normal C1 esterase inhibitor levels involves the production of bradykinin via kallikrein, lanadelumab should still be effective.1 Lanadelumab was chosen for our patient because of its minimal AEs and is not known to increase the risk of thrombosis.
Lanadelumab is a novel medication, recently approved in 2018 by the US Food and Drug Administration for the treatment of type I and type 2 HAE in patients aged ≥ 12 years.7 The phase 3 Hereditary Angioedema Long-term Prophylaxis (HELP) study concluded that treatment with subcutaneous lanadelumab for 26 weeks significantly decreased the frequency of angioedema attacks compared with placebo.5 However, 113 (90.4%) of patients in the phase III HELP study had type I HAE.5 Of the 125 patients that completed this randomized, double-blind study, only 12 had type II HAE.5 In addition, this study only included 5 patients aged ≥ 65 years.5 Also, no patients aged ≥ 65 years were part of the treatment arms that included a lanadelumab dose of 300 mg.5 In a case series of 12 patients in Canada, treatment with lanadelumab decreased angioedema attacks by 72%.9 However, the series only included 1 patient with type II HAE who was aged 36 years.9 Therefore, our case demonstrates the efficacy of lanadelumab in a patient aged ≥ 65 years with type II HAE.
Conclusions
HAE is a rare and potentially fatal disease characterized by recurrent, unpredictable attacks of edema throughout the body. The disease burden adversely affects a patient’s quality of life. Therefore, long-term prophylaxis is critical to managing patients with HAE. Lanadelumab has been proven as an effective long-term prophylactic treatment option for HAE attacks. This case supports the use of lanadelumab in patients with type II HAE and patients aged ≥ 65 years.
Acknowledgments
The patient was initially written up based on his delayed diagnosis as a case report.3 An earlier version of this article was presented by Samuel Weiss, MD, and Derek Smith, MD, as a poster at the American Academy of Allergy, Asthma, and Immunology virtual conference February 26 to March 1, 2021.
1. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136-1148. doi:10.1056/NEJMra1808012
2. Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care. 2018;24(14)(suppl):S292-S298.
3. Berger J, Carroll MP Jr, Champoux E, Coop CA. Extremely delayed diagnosis of type II hereditary angioedema: case report and review of the literature. Mil Med. 2018;183(11-12):e765-e767. doi:10.1093/milmed/usy031
4. Fouche AS, Saunders EF, Craig T. Depression and anxiety in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(4):371-375. doi:10.1016/j.anai.2013.05.028
5. Banerji A, Riedl MA, Bernstein JA, et al; HELP Investigators. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320(20):2108-2121. doi:10.1001/jama.2018.16773
6. Busse PJ, Farkas H, Banerji A, et al. Lanadelumab for the prophylactic treatment of hereditary angioedema with C1 inhibitor deficiency: a review of preclinical and phase I studies. BioDrugs. 2019;33(1):33-43. doi:10.1007/s40259-018-0325-y
7. Riedl MA, Maurer M, Bernstein JA, et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy. 2020;75(11):2879-2887. doi:10.1111/all.14416
8. Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. 2022;77(7):1961-1990. doi:10.1111/all.15214
9. Iaboni A, Kanani A, Lacuesta G, Song C, Kan M, Betschel SD. Impact of lanadelumab in hereditary angioedema: a case series of 12 patients in Canada. Allergy Asthma Clin Immunol. 2021;17(1):78. Published 2021 Jul 23. doi:10.1186/s13223-021-00579-6
1. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136-1148. doi:10.1056/NEJMra1808012
2. Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care. 2018;24(14)(suppl):S292-S298.
3. Berger J, Carroll MP Jr, Champoux E, Coop CA. Extremely delayed diagnosis of type II hereditary angioedema: case report and review of the literature. Mil Med. 2018;183(11-12):e765-e767. doi:10.1093/milmed/usy031
4. Fouche AS, Saunders EF, Craig T. Depression and anxiety in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(4):371-375. doi:10.1016/j.anai.2013.05.028
5. Banerji A, Riedl MA, Bernstein JA, et al; HELP Investigators. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320(20):2108-2121. doi:10.1001/jama.2018.16773
6. Busse PJ, Farkas H, Banerji A, et al. Lanadelumab for the prophylactic treatment of hereditary angioedema with C1 inhibitor deficiency: a review of preclinical and phase I studies. BioDrugs. 2019;33(1):33-43. doi:10.1007/s40259-018-0325-y
7. Riedl MA, Maurer M, Bernstein JA, et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy. 2020;75(11):2879-2887. doi:10.1111/all.14416
8. Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema—the 2021 revision and update. Allergy. 2022;77(7):1961-1990. doi:10.1111/all.15214
9. Iaboni A, Kanani A, Lacuesta G, Song C, Kan M, Betschel SD. Impact of lanadelumab in hereditary angioedema: a case series of 12 patients in Canada. Allergy Asthma Clin Immunol. 2021;17(1):78. Published 2021 Jul 23. doi:10.1186/s13223-021-00579-6
56-year-old man • increased heart rate • weakness • intense sweating • horseradish consumption • Dx?
THE CASE
A 56-year-old physician (CUL) visited a local seafood restaurant, after having fasted since the prior evening. He had a history of hypertension that was well controlled with lisinopril/hydrochlorothiazide.
The physician and his party were seated outside, where the temperature was in the mid-70s. The group ordered oysters on the half shell accompanied by mignonette sauce, cocktail sauce, and horseradish. The physician ate an olive-size amount of horseradish with an oyster. He immediately complained of a sharp burning sensation in his stomach and remarked that the horseradish was significantly stronger than what he was accustomed to. Within 30 seconds, he noted an increased heart rate, weakness, and intense sweating. There was no increase in nasal secretions. Observers noted that he was very pale.
About 5 minutes after eating the horseradish, the physician leaned his head back and briefly lost consciousness. His wife, while supporting his head and checking his pulse, instructed other diners to call for emergency services, at which point the physician regained consciousness and the dispatcher was told that an ambulance was no longer necessary. Within a matter of minutes, all symptoms had abated, except for some mild weakness.
THE DIAGNOSIS
Ten minutes after the event, the physician identified his symptoms as a horseradish-induced vasovagal syncope (VVS), based on a case report published in JAMA in 1988, which his wife found after he asked her to do an Internet search of his symptoms.1
THE DISCUSSION
Horseradish’s active component is isothiocyanate. Horseradish-induced syncope is also called Seder syncope after the Jewish Passover holiday dinner at which observant Jews are required to eat “bitter herbs.”1,2 This type of syncope is thought to occur when horseradish vapors directly irritate the gastric or respiratory tract mucosa.
VVS commonly manifests for the first time at around age 13 years; however, the timing of that first occurrence can vary significantly among individuals (as in this case)
The loss of consciousness may be caused by an emotional trigger (eg, sight of blood, cast removal,8 blood or platelet donations9,10), a painful event (eg, an injection11), an orthostatic trigger12 (eg, prolonged standing), or visceral reflexes such as swallowing.13 In approximately 30% of cases, loss of consciousness is associated with memory loss.14 Loss of consciousness with VVS may be associated with injury in 33% of cases.15
Continue to: The recovery with awareness
The recovery with awareness of time, place, and person may be a feature of VVS, which would differentiate it from seizures and brainstem vascular events. Autonomic prodromal symptoms—including abdominal discomfort, pallor, sweating, and nausea—may precede the loss of consciousness.8
An evolutionary response?
VVS may have developed as a trait through evolution, although modern medicine treats it as a disease. Many potential explanations for VVS as a body defense mechanism have been proposed. Examples include fainting at the sight of blood, which developed during the Old Stone Age—a period with extreme human-to-human violence—or acting like a “possum playing dead” as a tactic designed to confuse an attacker.16
Another theory involves clot production and suggests that VVS-induced hypotension is a defense against bleeding by improving clot formation.17
A psychological defense theory maintains that the fainting and memory loss are designed to prevent a painful or overwhelming experience from being remembered. None of these theories, however, explain orthostatic VVS.18
The brain defense theory could explain all forms of VVS. It postulates that hypotension causes decreased cerebral perfusion, which leads to syncope resulting in the body returning to a more orthostatic position with increased cerebral profusion.19
Continue to: The patient
The patient in this case was able to leave the restaurant on his own volition 30 minutes after the event and resume normal activities. Ten days later, an electrocardiogram was performed, with negative results. In this case, the use of a potassium-wasting diuretic exacerbated the risk of a fluid-deprived state, hypokalemia, and hypotension, possibly contributing to the syncope. The patient has since “gotten back on the horseradish” without ill effect.
THE TAKEAWAY
Consumers and health care providers should be aware of the risks associated with consumption of fresh horseradish and should allow it to rest prior to ingestion to allow some evaporation of its active ingredient. An old case report saved the patient from an unnecessary (and costly) emergency department visit.
ACKNOWLEDGEMENTS
The authors would like to thank Terry J. Hannan, MBBS, FRACP, FACHI, FACMI for his critical review of the manuscript.
CORRESPONDENCE
Christoph U. Lehmann, MD, Clinical Informatics Center, 5323 Harry Hines Boulevard, Dallas, TX 75390; [email protected]
1. Rubin HR, Wu AW. The bitter herbs of Seder: more on horseradish horrors. JAMA. 1988;259:1943. doi: 10.1001/jama.259.13.1943b
2. Seder syncope. The Free Dictionary. Accessed July 20, 2022. https://medical-dictionary.thefreedictionary.com/Horseradish+Syncope
3. Sheldon RS, Sheldon AG, Connolly SJ, et al. Age of first faint in patients with vasovagal syncope. J Cardiovasc Electrophysiol. 2006;17:49-54. doi: 10.1111/j.1540-8167.2005.00267.x
4. Wallin BG, Sundlöf G. Sympathetic outflow to muscles during vasovagal syncope. J Auton Nerv Syst. 1982;6:287-291. doi: 10.1016/0165-1838(82)90001-7
5. Jardine DL, Melton IC, Crozier IG, et al. Decrease in cardiac output and muscle sympathetic activity during vasovagal syncope. Am J Physiol Heart Circ Physiol. 2002;282:H1804-H1809. doi: 10.1152/ajpheart.00640.2001
6. Waxman MB, Asta JA, Cameron DA. Localization of the reflex pathway responsible for the vasodepressor reaction induced by inferior vena caval occlusion and isoproterenol. Can J Physiol Pharmacol. 1992;70:882-889. doi: 10.1139/y92-118
7. Alboni P, Alboni M. Typical vasovagal syncope as a “defense mechanism” for the heart by contrasting sympathetic overactivity. Clin Auton Res. 2017;27:253-261. doi: 10.1007/s10286-017-0446-2
8. Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30:2631-2671. doi: 10.1093/eurheartj/ehp298
9. Davies J, MacDonald L, Sivakumar B, et al. Prospective analysis of syncope/pre-syncope in a tertiary paediatric orthopaedic fracture outpatient clinic. ANZ J Surg. 2021;91:668-672. doi: 10.1111/ans.16664
10. Almutairi H, Salam M, Batarfi K, et al. Incidence and severity of adverse events among platelet donors: a three-year retrospective study. Medicine (Baltimore). 2020;99:e23648. doi: 10.1097/MD.0000000000023648
11. Coakley A, Bailey A, Tao J, et al. Video education to improve clinical skills in the prevention of and response to vasovagal syncopal episodes. Int J Womens Dermatol. 2020;6:186-190. doi: 10.1016/j.ijwd.2020.02.002
12. Thijs RD, Brignole M, Falup-Pecurariu C, et al. Recommendations for tilt table testing and other provocative cardiovascular autonomic tests in conditions that may cause transient loss of consciousness: consensus statement of the European Federation of Autonomic Societies (EFAS) endorsed by the American Autonomic Society (AAS) and the European Academy of Neurology (EAN). Auton Neurosci. 2021;233:102792. doi: 10.1016/j.autneu.2021.102792
13. Nakagawa S, Hisanaga S, Kondoh H, et al. A case of swallow syncope induced by vagotonic visceral reflex resulting in atrioventricular node suppression. J Electrocardiol. 1987;20:65-69. doi: 10.1016/0022-0736(87)90010-0
14. O’Dwyer C, Bennett K, Langan Y, et al. Amnesia for loss of consciousness is common in vasovagal syncope. Europace. 2011;13:1040-1045. doi: 10.1093/europace/eur069
15. Jorge JG, Raj SR, Teixeira PS, et al. Likelihood of injury due to vasovagal syncope: a systematic review and meta-analysis. Europace. 2021;23:1092-1099. doi: 10.1093/europace/euab041
16. Bracha HS, Bracha AS, Williams AE, et al. The human fear-circuitry and fear-induced fainting in healthy individuals—the paleolithic-threat hypothesis. Clin Auton Res. 2005;15:238-241. doi: 10.1007/s10286-005-0245-z
17. Diehl RR. Vasovagal syncope and Darwinian fitness. Clin Auton Res. 2005;15:126-129. doi: 10.1007/s10286-005-0244-0
18. Engel CL, Romano J. Studies of syncope; biologic interpretation of vasodepressor syncope. Psychosom Med. 1947;9:288-294. doi: 10.1097/00006842-194709000-00002
19. Blanc JJ, Benditt DG. Vasovagal syncope: hypothesis focusing on its being a clinical feature unique to humans. J Cardiovasc Electrophysiol. 2016;27:623-629. doi: 10.1111/jce.12945
THE CASE
A 56-year-old physician (CUL) visited a local seafood restaurant, after having fasted since the prior evening. He had a history of hypertension that was well controlled with lisinopril/hydrochlorothiazide.
The physician and his party were seated outside, where the temperature was in the mid-70s. The group ordered oysters on the half shell accompanied by mignonette sauce, cocktail sauce, and horseradish. The physician ate an olive-size amount of horseradish with an oyster. He immediately complained of a sharp burning sensation in his stomach and remarked that the horseradish was significantly stronger than what he was accustomed to. Within 30 seconds, he noted an increased heart rate, weakness, and intense sweating. There was no increase in nasal secretions. Observers noted that he was very pale.
About 5 minutes after eating the horseradish, the physician leaned his head back and briefly lost consciousness. His wife, while supporting his head and checking his pulse, instructed other diners to call for emergency services, at which point the physician regained consciousness and the dispatcher was told that an ambulance was no longer necessary. Within a matter of minutes, all symptoms had abated, except for some mild weakness.
THE DIAGNOSIS
Ten minutes after the event, the physician identified his symptoms as a horseradish-induced vasovagal syncope (VVS), based on a case report published in JAMA in 1988, which his wife found after he asked her to do an Internet search of his symptoms.1
THE DISCUSSION
Horseradish’s active component is isothiocyanate. Horseradish-induced syncope is also called Seder syncope after the Jewish Passover holiday dinner at which observant Jews are required to eat “bitter herbs.”1,2 This type of syncope is thought to occur when horseradish vapors directly irritate the gastric or respiratory tract mucosa.
VVS commonly manifests for the first time at around age 13 years; however, the timing of that first occurrence can vary significantly among individuals (as in this case)
The loss of consciousness may be caused by an emotional trigger (eg, sight of blood, cast removal,8 blood or platelet donations9,10), a painful event (eg, an injection11), an orthostatic trigger12 (eg, prolonged standing), or visceral reflexes such as swallowing.13 In approximately 30% of cases, loss of consciousness is associated with memory loss.14 Loss of consciousness with VVS may be associated with injury in 33% of cases.15
Continue to: The recovery with awareness
The recovery with awareness of time, place, and person may be a feature of VVS, which would differentiate it from seizures and brainstem vascular events. Autonomic prodromal symptoms—including abdominal discomfort, pallor, sweating, and nausea—may precede the loss of consciousness.8
An evolutionary response?
VVS may have developed as a trait through evolution, although modern medicine treats it as a disease. Many potential explanations for VVS as a body defense mechanism have been proposed. Examples include fainting at the sight of blood, which developed during the Old Stone Age—a period with extreme human-to-human violence—or acting like a “possum playing dead” as a tactic designed to confuse an attacker.16
Another theory involves clot production and suggests that VVS-induced hypotension is a defense against bleeding by improving clot formation.17
A psychological defense theory maintains that the fainting and memory loss are designed to prevent a painful or overwhelming experience from being remembered. None of these theories, however, explain orthostatic VVS.18
The brain defense theory could explain all forms of VVS. It postulates that hypotension causes decreased cerebral perfusion, which leads to syncope resulting in the body returning to a more orthostatic position with increased cerebral profusion.19
Continue to: The patient
The patient in this case was able to leave the restaurant on his own volition 30 minutes after the event and resume normal activities. Ten days later, an electrocardiogram was performed, with negative results. In this case, the use of a potassium-wasting diuretic exacerbated the risk of a fluid-deprived state, hypokalemia, and hypotension, possibly contributing to the syncope. The patient has since “gotten back on the horseradish” without ill effect.
THE TAKEAWAY
Consumers and health care providers should be aware of the risks associated with consumption of fresh horseradish and should allow it to rest prior to ingestion to allow some evaporation of its active ingredient. An old case report saved the patient from an unnecessary (and costly) emergency department visit.
ACKNOWLEDGEMENTS
The authors would like to thank Terry J. Hannan, MBBS, FRACP, FACHI, FACMI for his critical review of the manuscript.
CORRESPONDENCE
Christoph U. Lehmann, MD, Clinical Informatics Center, 5323 Harry Hines Boulevard, Dallas, TX 75390; [email protected]
THE CASE
A 56-year-old physician (CUL) visited a local seafood restaurant, after having fasted since the prior evening. He had a history of hypertension that was well controlled with lisinopril/hydrochlorothiazide.
The physician and his party were seated outside, where the temperature was in the mid-70s. The group ordered oysters on the half shell accompanied by mignonette sauce, cocktail sauce, and horseradish. The physician ate an olive-size amount of horseradish with an oyster. He immediately complained of a sharp burning sensation in his stomach and remarked that the horseradish was significantly stronger than what he was accustomed to. Within 30 seconds, he noted an increased heart rate, weakness, and intense sweating. There was no increase in nasal secretions. Observers noted that he was very pale.
About 5 minutes after eating the horseradish, the physician leaned his head back and briefly lost consciousness. His wife, while supporting his head and checking his pulse, instructed other diners to call for emergency services, at which point the physician regained consciousness and the dispatcher was told that an ambulance was no longer necessary. Within a matter of minutes, all symptoms had abated, except for some mild weakness.
THE DIAGNOSIS
Ten minutes after the event, the physician identified his symptoms as a horseradish-induced vasovagal syncope (VVS), based on a case report published in JAMA in 1988, which his wife found after he asked her to do an Internet search of his symptoms.1
THE DISCUSSION
Horseradish’s active component is isothiocyanate. Horseradish-induced syncope is also called Seder syncope after the Jewish Passover holiday dinner at which observant Jews are required to eat “bitter herbs.”1,2 This type of syncope is thought to occur when horseradish vapors directly irritate the gastric or respiratory tract mucosa.
VVS commonly manifests for the first time at around age 13 years; however, the timing of that first occurrence can vary significantly among individuals (as in this case)
The loss of consciousness may be caused by an emotional trigger (eg, sight of blood, cast removal,8 blood or platelet donations9,10), a painful event (eg, an injection11), an orthostatic trigger12 (eg, prolonged standing), or visceral reflexes such as swallowing.13 In approximately 30% of cases, loss of consciousness is associated with memory loss.14 Loss of consciousness with VVS may be associated with injury in 33% of cases.15
Continue to: The recovery with awareness
The recovery with awareness of time, place, and person may be a feature of VVS, which would differentiate it from seizures and brainstem vascular events. Autonomic prodromal symptoms—including abdominal discomfort, pallor, sweating, and nausea—may precede the loss of consciousness.8
An evolutionary response?
VVS may have developed as a trait through evolution, although modern medicine treats it as a disease. Many potential explanations for VVS as a body defense mechanism have been proposed. Examples include fainting at the sight of blood, which developed during the Old Stone Age—a period with extreme human-to-human violence—or acting like a “possum playing dead” as a tactic designed to confuse an attacker.16
Another theory involves clot production and suggests that VVS-induced hypotension is a defense against bleeding by improving clot formation.17
A psychological defense theory maintains that the fainting and memory loss are designed to prevent a painful or overwhelming experience from being remembered. None of these theories, however, explain orthostatic VVS.18
The brain defense theory could explain all forms of VVS. It postulates that hypotension causes decreased cerebral perfusion, which leads to syncope resulting in the body returning to a more orthostatic position with increased cerebral profusion.19
Continue to: The patient
The patient in this case was able to leave the restaurant on his own volition 30 minutes after the event and resume normal activities. Ten days later, an electrocardiogram was performed, with negative results. In this case, the use of a potassium-wasting diuretic exacerbated the risk of a fluid-deprived state, hypokalemia, and hypotension, possibly contributing to the syncope. The patient has since “gotten back on the horseradish” without ill effect.
THE TAKEAWAY
Consumers and health care providers should be aware of the risks associated with consumption of fresh horseradish and should allow it to rest prior to ingestion to allow some evaporation of its active ingredient. An old case report saved the patient from an unnecessary (and costly) emergency department visit.
ACKNOWLEDGEMENTS
The authors would like to thank Terry J. Hannan, MBBS, FRACP, FACHI, FACMI for his critical review of the manuscript.
CORRESPONDENCE
Christoph U. Lehmann, MD, Clinical Informatics Center, 5323 Harry Hines Boulevard, Dallas, TX 75390; [email protected]
1. Rubin HR, Wu AW. The bitter herbs of Seder: more on horseradish horrors. JAMA. 1988;259:1943. doi: 10.1001/jama.259.13.1943b
2. Seder syncope. The Free Dictionary. Accessed July 20, 2022. https://medical-dictionary.thefreedictionary.com/Horseradish+Syncope
3. Sheldon RS, Sheldon AG, Connolly SJ, et al. Age of first faint in patients with vasovagal syncope. J Cardiovasc Electrophysiol. 2006;17:49-54. doi: 10.1111/j.1540-8167.2005.00267.x
4. Wallin BG, Sundlöf G. Sympathetic outflow to muscles during vasovagal syncope. J Auton Nerv Syst. 1982;6:287-291. doi: 10.1016/0165-1838(82)90001-7
5. Jardine DL, Melton IC, Crozier IG, et al. Decrease in cardiac output and muscle sympathetic activity during vasovagal syncope. Am J Physiol Heart Circ Physiol. 2002;282:H1804-H1809. doi: 10.1152/ajpheart.00640.2001
6. Waxman MB, Asta JA, Cameron DA. Localization of the reflex pathway responsible for the vasodepressor reaction induced by inferior vena caval occlusion and isoproterenol. Can J Physiol Pharmacol. 1992;70:882-889. doi: 10.1139/y92-118
7. Alboni P, Alboni M. Typical vasovagal syncope as a “defense mechanism” for the heart by contrasting sympathetic overactivity. Clin Auton Res. 2017;27:253-261. doi: 10.1007/s10286-017-0446-2
8. Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30:2631-2671. doi: 10.1093/eurheartj/ehp298
9. Davies J, MacDonald L, Sivakumar B, et al. Prospective analysis of syncope/pre-syncope in a tertiary paediatric orthopaedic fracture outpatient clinic. ANZ J Surg. 2021;91:668-672. doi: 10.1111/ans.16664
10. Almutairi H, Salam M, Batarfi K, et al. Incidence and severity of adverse events among platelet donors: a three-year retrospective study. Medicine (Baltimore). 2020;99:e23648. doi: 10.1097/MD.0000000000023648
11. Coakley A, Bailey A, Tao J, et al. Video education to improve clinical skills in the prevention of and response to vasovagal syncopal episodes. Int J Womens Dermatol. 2020;6:186-190. doi: 10.1016/j.ijwd.2020.02.002
12. Thijs RD, Brignole M, Falup-Pecurariu C, et al. Recommendations for tilt table testing and other provocative cardiovascular autonomic tests in conditions that may cause transient loss of consciousness: consensus statement of the European Federation of Autonomic Societies (EFAS) endorsed by the American Autonomic Society (AAS) and the European Academy of Neurology (EAN). Auton Neurosci. 2021;233:102792. doi: 10.1016/j.autneu.2021.102792
13. Nakagawa S, Hisanaga S, Kondoh H, et al. A case of swallow syncope induced by vagotonic visceral reflex resulting in atrioventricular node suppression. J Electrocardiol. 1987;20:65-69. doi: 10.1016/0022-0736(87)90010-0
14. O’Dwyer C, Bennett K, Langan Y, et al. Amnesia for loss of consciousness is common in vasovagal syncope. Europace. 2011;13:1040-1045. doi: 10.1093/europace/eur069
15. Jorge JG, Raj SR, Teixeira PS, et al. Likelihood of injury due to vasovagal syncope: a systematic review and meta-analysis. Europace. 2021;23:1092-1099. doi: 10.1093/europace/euab041
16. Bracha HS, Bracha AS, Williams AE, et al. The human fear-circuitry and fear-induced fainting in healthy individuals—the paleolithic-threat hypothesis. Clin Auton Res. 2005;15:238-241. doi: 10.1007/s10286-005-0245-z
17. Diehl RR. Vasovagal syncope and Darwinian fitness. Clin Auton Res. 2005;15:126-129. doi: 10.1007/s10286-005-0244-0
18. Engel CL, Romano J. Studies of syncope; biologic interpretation of vasodepressor syncope. Psychosom Med. 1947;9:288-294. doi: 10.1097/00006842-194709000-00002
19. Blanc JJ, Benditt DG. Vasovagal syncope: hypothesis focusing on its being a clinical feature unique to humans. J Cardiovasc Electrophysiol. 2016;27:623-629. doi: 10.1111/jce.12945
1. Rubin HR, Wu AW. The bitter herbs of Seder: more on horseradish horrors. JAMA. 1988;259:1943. doi: 10.1001/jama.259.13.1943b
2. Seder syncope. The Free Dictionary. Accessed July 20, 2022. https://medical-dictionary.thefreedictionary.com/Horseradish+Syncope
3. Sheldon RS, Sheldon AG, Connolly SJ, et al. Age of first faint in patients with vasovagal syncope. J Cardiovasc Electrophysiol. 2006;17:49-54. doi: 10.1111/j.1540-8167.2005.00267.x
4. Wallin BG, Sundlöf G. Sympathetic outflow to muscles during vasovagal syncope. J Auton Nerv Syst. 1982;6:287-291. doi: 10.1016/0165-1838(82)90001-7
5. Jardine DL, Melton IC, Crozier IG, et al. Decrease in cardiac output and muscle sympathetic activity during vasovagal syncope. Am J Physiol Heart Circ Physiol. 2002;282:H1804-H1809. doi: 10.1152/ajpheart.00640.2001
6. Waxman MB, Asta JA, Cameron DA. Localization of the reflex pathway responsible for the vasodepressor reaction induced by inferior vena caval occlusion and isoproterenol. Can J Physiol Pharmacol. 1992;70:882-889. doi: 10.1139/y92-118
7. Alboni P, Alboni M. Typical vasovagal syncope as a “defense mechanism” for the heart by contrasting sympathetic overactivity. Clin Auton Res. 2017;27:253-261. doi: 10.1007/s10286-017-0446-2
8. Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30:2631-2671. doi: 10.1093/eurheartj/ehp298
9. Davies J, MacDonald L, Sivakumar B, et al. Prospective analysis of syncope/pre-syncope in a tertiary paediatric orthopaedic fracture outpatient clinic. ANZ J Surg. 2021;91:668-672. doi: 10.1111/ans.16664
10. Almutairi H, Salam M, Batarfi K, et al. Incidence and severity of adverse events among platelet donors: a three-year retrospective study. Medicine (Baltimore). 2020;99:e23648. doi: 10.1097/MD.0000000000023648
11. Coakley A, Bailey A, Tao J, et al. Video education to improve clinical skills in the prevention of and response to vasovagal syncopal episodes. Int J Womens Dermatol. 2020;6:186-190. doi: 10.1016/j.ijwd.2020.02.002
12. Thijs RD, Brignole M, Falup-Pecurariu C, et al. Recommendations for tilt table testing and other provocative cardiovascular autonomic tests in conditions that may cause transient loss of consciousness: consensus statement of the European Federation of Autonomic Societies (EFAS) endorsed by the American Autonomic Society (AAS) and the European Academy of Neurology (EAN). Auton Neurosci. 2021;233:102792. doi: 10.1016/j.autneu.2021.102792
13. Nakagawa S, Hisanaga S, Kondoh H, et al. A case of swallow syncope induced by vagotonic visceral reflex resulting in atrioventricular node suppression. J Electrocardiol. 1987;20:65-69. doi: 10.1016/0022-0736(87)90010-0
14. O’Dwyer C, Bennett K, Langan Y, et al. Amnesia for loss of consciousness is common in vasovagal syncope. Europace. 2011;13:1040-1045. doi: 10.1093/europace/eur069
15. Jorge JG, Raj SR, Teixeira PS, et al. Likelihood of injury due to vasovagal syncope: a systematic review and meta-analysis. Europace. 2021;23:1092-1099. doi: 10.1093/europace/euab041
16. Bracha HS, Bracha AS, Williams AE, et al. The human fear-circuitry and fear-induced fainting in healthy individuals—the paleolithic-threat hypothesis. Clin Auton Res. 2005;15:238-241. doi: 10.1007/s10286-005-0245-z
17. Diehl RR. Vasovagal syncope and Darwinian fitness. Clin Auton Res. 2005;15:126-129. doi: 10.1007/s10286-005-0244-0
18. Engel CL, Romano J. Studies of syncope; biologic interpretation of vasodepressor syncope. Psychosom Med. 1947;9:288-294. doi: 10.1097/00006842-194709000-00002
19. Blanc JJ, Benditt DG. Vasovagal syncope: hypothesis focusing on its being a clinical feature unique to humans. J Cardiovasc Electrophysiol. 2016;27:623-629. doi: 10.1111/jce.12945

Angiolymphoid Hyperplasia with Eosinophilia in a Patient With Coccidioidomycosis
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare nodular unencapsulated mass that is characterized by benign anomalous vascular hyperplasia of epithelioidlike endothelial cells attached to dilated blood vessels. The mass is surrounded by lymphocytes and eosinophils that can present clinically as papules, plaques, or nodules.1 The etiology of ALHE is unknown; it is hypothesized that it is a vascular neoplasm or a lymphoproliferative disorder.
Coccidioidomycosis (CM) is a prevalent deep fungal infection endemic to the southwestern United States caused by Coccidioides immitis and Coccidioides posadasii. Infection can occur from direct inoculation through abrasions or direct trauma but usually occurs through the inhalation of spores and can result in a reactive rash (eg, Sweet syndrome, erythema nodosum, interstitial granulomatous dermatitis).2 Coccidioidomycosis also can result in respiratory pneumonia and dissemination from pulmonary infection of the skin. As such, it is important to distinguish CM and its immunologically mediated eruptions for accurate diagnosis and treatment.
We report a novel case of ALHE as a reactive dermatologic presentation in a patient with CM.
Case Report
A 72-year-old woman presented to the dermatology clinic with itchy papules and plaques on the arms and legs of 17 years’ duration. Her medical history included coronary artery disease and hypercholesterolemia as well as a remote history of cutaneous marginal zone B-cell lymphoma of the nose, which was confirmed by histology and treated more than 10 years prior and has remained in remission for 6 years. Her current medications included aspirin, atorvastatin, lisinopril, and metoprolol succinate.
Our patient first presented to our dermatology clinic for itchy nodules and papules on the legs and arms. The patient previously had been seen by another dermatologist 2 months prior for the same condition. At that time, biopsies of the lesions were reported as prurigo nodules. Physical examination at the current presentation revealed round, pink to flesh-colored, raised papules and plaques scattered on the arms and legs (Figure 1). The differential diagnosis included lymphomatoid papulosis, cutaneous B-cell lymphoma, pseudolymphoma, cutaneous CM, and papular mucinosis.

Four-mm punch biopsies of the right proximal pretibial region and left knee region were taken and sent for histologic analysis, direct immunofluorescence testing, and tissue culture. Testing for atypical mycobacteria and deep fungal infection was negative; bacterial cultures and sensitivity testing were negative. Direct immunofluorescence testing was negative. Microscopic examination of material from the right proximal pretibial region showed widely dilated, variously shaped, large blood vessels in a multinodular pattern; the vessels also were surrounded by an inflammatory cell infiltrate containing eosinophils. Histologic findings were consistent with ALHE.
Subsequent biopsies were completed 2 weeks and 1 month from the initial presentation. Both histology reports—from 2 different histopathology laboratories—were consistent with ALHE (Figure 2). Additional work-up during the patient’s initial visit to our clinic for the rash included CM serologic testing, which demonstrated IgM and IgG antibodies. Subsequently, chest radiography revealed a 2.2×2.3-cm mass in the right lower lobe of the lung. Follow-up computed tomography 1 month later confirmed the nodule in the same area to be 2.3×2.1×1.8 cm.
.")
The patient was referred to pulmonology and was treated for pulmonary CM with oral fluconazole 200 mg twice daily for 4 months. Initial treatment also included clobetasol cream 0.05% applied twice daily, which did not produce marked improvement in pruritus. Narrowband UVB phototherapy was attempted, but the patient could not complete the course because of travel time to the office; however, the patient’s ALHE improved considerably with the fluconazole treatment for pulmonary CM.
Oral doxycycline 100 mg twice daily was added to the fluconazole 2 months after her initial visit to our office, which kept the ALHE at bay and helped with the pruritus (Figure 3). Pulmonology and primary care comanaged the pulmonary CM with oral fluconazole 200 mg twice daily. Repeat serologic testing for CM was negative for IgG and IgM after 14 months since the initial visit to the office.

Comment
Pulmonary CM infection has varying dermatologic manifestations. A PubMed search of articles indexed for MEDLINE using the terms ALHE and coccidioidomycosis yielded no case reports; in fact, there have been few reported cases of ALHE at all. Notable conditions associated with ALHE include membranous nephropathy and arteriovenous malformations treated with corticosteroids and surgery, respectively.3,4 Our case is a rare presentation of CM infection manifesting with ALHE. Following treatment and remission for our patient’s CM infection, the ALHE lesion decreased in size.
Standard treatment of uncomplicated CM involves azole antifungals, typically oral fluconazole or itraconazole 400 to 600 mg/d. In more severe cases (eg, immunocompromised patients) amphotericin B can be used.5 Our patient was treated with oral fluconazole 200 mg twice daily for 4 months.
In the literature, treatment via surgical excision, steroid injection, pulsed-dye laser therapy, and radiotherapy also has been described.6-8 Antibiotics including clindamycin, doxycycline, and amoxicillin-clavulanate also have been shown to be effective.9
In our patient, ALHE improved when oral doxycycline 100 mg twice daily was added to the oral fluconazole. In fact, after 4 months of treatment, the CM infection and ALHE lesions both improved to a point at which the lesions were not visible. When those lesions recurred 15 months later, they responded with another course of doxycycline and fluconazole.
Upon recurrence, the patient was asked to have her care transferred to her pulmonologist, who then managed the fluconazole regimen. During the pulmonologist’s workup, no peripheral eosinophilia was found. This is important because eosinophils can be a marker for CM infection; in this case, however, the ALHE lesion was a reactive process to the infection. Classically known to play a reactive role in fungal infection, these white blood cells demonstrate reactivity to the environmental fungus Alternaria alternata by contact-dependent killing, utilizing β2 integrins and CD11b to recognize and adhere to β-glucan. Eosinophils react through contact-dependent killing, releasing cytotoxic granule proteins and proinflammatory mediators, and have been documented to occur in CM and Paracoccidioides brasiliensis infection, in which they deposit major basic protein on the organism.10 Most pertinent to our case with ALHE and CM is the ability of eosinophils to communicate with other immune cells. Eosinophils play a role in the active inflammation of CM through cytokine signaling, which may propagate formation of ALHE.
The function of eosinophils in ALHE is poorly understood; it is unclear whether they act as a primary driver of pathogenesis or are simply indicators of secondary infiltration or infection. Our review of the current literature suggests that eosinophils are unnecessary for progression of ALHE but might be involved at its onset. As reported, even monoclonal antibody therapy (eg, mepolizumab and benralizumab) that effectively depletes eosinophil levels by negating IL-5 signaling do not slow progression of ALHE.11 Symptomatic changes are modest at best (ie, simply softening the ALHE nodules).
Our patient had no peripheral eosinophilia, suggesting that the onset of ALHE might not be caused by eosinophilia but a different inflammatory process—in this patient, by CM. Because peripheral eosinophilia was not seen in our patient, the presence of eosinophils in the ALHE lesion likely is unnecessary for its onset or progression but is a secondary process that exacerbates the lesion. The pathogenesis is unknown but could be directed toward lymphocytes and plasma cells, with eosinophils as part of the dynamic process.11
Conclusion
Because reports of an association between CM and ALHE are limited, our case is distinguished by a unique clinical presentation of ALHE. When a patient is given a diagnosis of ALHE, it therefore is important to consider exposure to CM as a cause, especially in patients who reside in or travel to a region where CM is endemic.
- Wells GC, Whimster IW. Subcutaneous angiolymphoid hyperplasia with eosinophilia. Br J Dermatol. 1969;81:1-14. doi:10.1111/j.1365-2133.1969.tb15914.x
- DiCaudo D. Coccidioidomycosis. Semin Cutan Med Surg. 2014;33:140-145. doi:10.12788/j.sder.0111
- Onishi Y, Ohara K. Angiolymphoid hyperplasia with eosinophilia associated with arteriovenous malformation: a clinicopathological correlation with angiography and serial estimation of serum levels of renin, eosinophil cationic protein and interleukin 5. Br J Dermatol. 1999;140:1153-1156. doi:10.1046/j.1365-2133.1999.02880.x
- Matsumoto A, Matsui I, Namba T, et al. VEGF-A links angiolymphoid hyperplasia with eosinophilia (ALHE) to THSD7A membranous nephropathy: a report of 2 cases. Am J Kidney Dis. 2019;73:880-885. doi:10.1053/j.ajkd.2018.10.009
- Bercovitch RS, Catanzaro A, Schwartz BS, et al. Coccidioidomycosis during pregnancy: a review and recommendations for management. Clin Infect Dis. 2011;53:363-368. doi:10.1093/cid/cir410
- Youssef A, Hasan AR, Youssef Y, et al. Angiolymphoid hyperplasia with eosinophilia: a case report. J Med Case Rep. 2018;12:89. doi:10.1186/s13256-018-1599-x
- Abrahamson TG, Davis DA. Angiolymphoid hyperplasia witheosinophilia responsive to pulsed dye laser. J Am Acad Dermatol. 2003;49(2 suppl case reports):S195-S196. doi:10.1067/mjd.2003.314
- Lembo S, Balato A, Cirillo T, et al. A long-term follow-up of angiolymphoid hyperplasia with eosinophilia treated by corticosteroids: when a traditional therapy is still up-to-date. Case Rep Dermatol. 2011;3:64-67. doi:10.1159/000323182
- Cleveland E. Atypical presentation of angiolymphomatous hyperplasia with eosinophilia. J Am Acad Dermatol. 2018;79(3 suppl 1):AB53. doi:10.1016/j.jaad.2018.05.249
- Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol. 2015;50:214-227. doi:10.1007/s12016-015-8525-4
- Grünewald M, Stölzl D, Wehkamp U, et al. Role of eosinophils in angiolymphoid hyperplasia with eosinophilia. JAMA Dermatol. 2021;157:1241-1243. doi:10.1001/jamadermatol.2021.2732
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare nodular unencapsulated mass that is characterized by benign anomalous vascular hyperplasia of epithelioidlike endothelial cells attached to dilated blood vessels. The mass is surrounded by lymphocytes and eosinophils that can present clinically as papules, plaques, or nodules.1 The etiology of ALHE is unknown; it is hypothesized that it is a vascular neoplasm or a lymphoproliferative disorder.
Coccidioidomycosis (CM) is a prevalent deep fungal infection endemic to the southwestern United States caused by Coccidioides immitis and Coccidioides posadasii. Infection can occur from direct inoculation through abrasions or direct trauma but usually occurs through the inhalation of spores and can result in a reactive rash (eg, Sweet syndrome, erythema nodosum, interstitial granulomatous dermatitis).2 Coccidioidomycosis also can result in respiratory pneumonia and dissemination from pulmonary infection of the skin. As such, it is important to distinguish CM and its immunologically mediated eruptions for accurate diagnosis and treatment.
We report a novel case of ALHE as a reactive dermatologic presentation in a patient with CM.
Case Report
A 72-year-old woman presented to the dermatology clinic with itchy papules and plaques on the arms and legs of 17 years’ duration. Her medical history included coronary artery disease and hypercholesterolemia as well as a remote history of cutaneous marginal zone B-cell lymphoma of the nose, which was confirmed by histology and treated more than 10 years prior and has remained in remission for 6 years. Her current medications included aspirin, atorvastatin, lisinopril, and metoprolol succinate.
Our patient first presented to our dermatology clinic for itchy nodules and papules on the legs and arms. The patient previously had been seen by another dermatologist 2 months prior for the same condition. At that time, biopsies of the lesions were reported as prurigo nodules. Physical examination at the current presentation revealed round, pink to flesh-colored, raised papules and plaques scattered on the arms and legs (Figure 1). The differential diagnosis included lymphomatoid papulosis, cutaneous B-cell lymphoma, pseudolymphoma, cutaneous CM, and papular mucinosis.
Four-mm punch biopsies of the right proximal pretibial region and left knee region were taken and sent for histologic analysis, direct immunofluorescence testing, and tissue culture. Testing for atypical mycobacteria and deep fungal infection was negative; bacterial cultures and sensitivity testing were negative. Direct immunofluorescence testing was negative. Microscopic examination of material from the right proximal pretibial region showed widely dilated, variously shaped, large blood vessels in a multinodular pattern; the vessels also were surrounded by an inflammatory cell infiltrate containing eosinophils. Histologic findings were consistent with ALHE.
Subsequent biopsies were completed 2 weeks and 1 month from the initial presentation. Both histology reports—from 2 different histopathology laboratories—were consistent with ALHE (Figure 2). Additional work-up during the patient’s initial visit to our clinic for the rash included CM serologic testing, which demonstrated IgM and IgG antibodies. Subsequently, chest radiography revealed a 2.2×2.3-cm mass in the right lower lobe of the lung. Follow-up computed tomography 1 month later confirmed the nodule in the same area to be 2.3×2.1×1.8 cm.
The patient was referred to pulmonology and was treated for pulmonary CM with oral fluconazole 200 mg twice daily for 4 months. Initial treatment also included clobetasol cream 0.05% applied twice daily, which did not produce marked improvement in pruritus. Narrowband UVB phototherapy was attempted, but the patient could not complete the course because of travel time to the office; however, the patient’s ALHE improved considerably with the fluconazole treatment for pulmonary CM.
Oral doxycycline 100 mg twice daily was added to the fluconazole 2 months after her initial visit to our office, which kept the ALHE at bay and helped with the pruritus (Figure 3). Pulmonology and primary care comanaged the pulmonary CM with oral fluconazole 200 mg twice daily. Repeat serologic testing for CM was negative for IgG and IgM after 14 months since the initial visit to the office.
Comment
Pulmonary CM infection has varying dermatologic manifestations. A PubMed search of articles indexed for MEDLINE using the terms ALHE and coccidioidomycosis yielded no case reports; in fact, there have been few reported cases of ALHE at all. Notable conditions associated with ALHE include membranous nephropathy and arteriovenous malformations treated with corticosteroids and surgery, respectively.3,4 Our case is a rare presentation of CM infection manifesting with ALHE. Following treatment and remission for our patient’s CM infection, the ALHE lesion decreased in size.
Standard treatment of uncomplicated CM involves azole antifungals, typically oral fluconazole or itraconazole 400 to 600 mg/d. In more severe cases (eg, immunocompromised patients) amphotericin B can be used.5 Our patient was treated with oral fluconazole 200 mg twice daily for 4 months.
In the literature, treatment via surgical excision, steroid injection, pulsed-dye laser therapy, and radiotherapy also has been described.6-8 Antibiotics including clindamycin, doxycycline, and amoxicillin-clavulanate also have been shown to be effective.9
In our patient, ALHE improved when oral doxycycline 100 mg twice daily was added to the oral fluconazole. In fact, after 4 months of treatment, the CM infection and ALHE lesions both improved to a point at which the lesions were not visible. When those lesions recurred 15 months later, they responded with another course of doxycycline and fluconazole.
Upon recurrence, the patient was asked to have her care transferred to her pulmonologist, who then managed the fluconazole regimen. During the pulmonologist’s workup, no peripheral eosinophilia was found. This is important because eosinophils can be a marker for CM infection; in this case, however, the ALHE lesion was a reactive process to the infection. Classically known to play a reactive role in fungal infection, these white blood cells demonstrate reactivity to the environmental fungus Alternaria alternata by contact-dependent killing, utilizing β2 integrins and CD11b to recognize and adhere to β-glucan. Eosinophils react through contact-dependent killing, releasing cytotoxic granule proteins and proinflammatory mediators, and have been documented to occur in CM and Paracoccidioides brasiliensis infection, in which they deposit major basic protein on the organism.10 Most pertinent to our case with ALHE and CM is the ability of eosinophils to communicate with other immune cells. Eosinophils play a role in the active inflammation of CM through cytokine signaling, which may propagate formation of ALHE.
The function of eosinophils in ALHE is poorly understood; it is unclear whether they act as a primary driver of pathogenesis or are simply indicators of secondary infiltration or infection. Our review of the current literature suggests that eosinophils are unnecessary for progression of ALHE but might be involved at its onset. As reported, even monoclonal antibody therapy (eg, mepolizumab and benralizumab) that effectively depletes eosinophil levels by negating IL-5 signaling do not slow progression of ALHE.11 Symptomatic changes are modest at best (ie, simply softening the ALHE nodules).
Our patient had no peripheral eosinophilia, suggesting that the onset of ALHE might not be caused by eosinophilia but a different inflammatory process—in this patient, by CM. Because peripheral eosinophilia was not seen in our patient, the presence of eosinophils in the ALHE lesion likely is unnecessary for its onset or progression but is a secondary process that exacerbates the lesion. The pathogenesis is unknown but could be directed toward lymphocytes and plasma cells, with eosinophils as part of the dynamic process.11
Conclusion
Because reports of an association between CM and ALHE are limited, our case is distinguished by a unique clinical presentation of ALHE. When a patient is given a diagnosis of ALHE, it therefore is important to consider exposure to CM as a cause, especially in patients who reside in or travel to a region where CM is endemic.
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare nodular unencapsulated mass that is characterized by benign anomalous vascular hyperplasia of epithelioidlike endothelial cells attached to dilated blood vessels. The mass is surrounded by lymphocytes and eosinophils that can present clinically as papules, plaques, or nodules.1 The etiology of ALHE is unknown; it is hypothesized that it is a vascular neoplasm or a lymphoproliferative disorder.
Coccidioidomycosis (CM) is a prevalent deep fungal infection endemic to the southwestern United States caused by Coccidioides immitis and Coccidioides posadasii. Infection can occur from direct inoculation through abrasions or direct trauma but usually occurs through the inhalation of spores and can result in a reactive rash (eg, Sweet syndrome, erythema nodosum, interstitial granulomatous dermatitis).2 Coccidioidomycosis also can result in respiratory pneumonia and dissemination from pulmonary infection of the skin. As such, it is important to distinguish CM and its immunologically mediated eruptions for accurate diagnosis and treatment.
We report a novel case of ALHE as a reactive dermatologic presentation in a patient with CM.
Case Report
A 72-year-old woman presented to the dermatology clinic with itchy papules and plaques on the arms and legs of 17 years’ duration. Her medical history included coronary artery disease and hypercholesterolemia as well as a remote history of cutaneous marginal zone B-cell lymphoma of the nose, which was confirmed by histology and treated more than 10 years prior and has remained in remission for 6 years. Her current medications included aspirin, atorvastatin, lisinopril, and metoprolol succinate.
Our patient first presented to our dermatology clinic for itchy nodules and papules on the legs and arms. The patient previously had been seen by another dermatologist 2 months prior for the same condition. At that time, biopsies of the lesions were reported as prurigo nodules. Physical examination at the current presentation revealed round, pink to flesh-colored, raised papules and plaques scattered on the arms and legs (Figure 1). The differential diagnosis included lymphomatoid papulosis, cutaneous B-cell lymphoma, pseudolymphoma, cutaneous CM, and papular mucinosis.
Four-mm punch biopsies of the right proximal pretibial region and left knee region were taken and sent for histologic analysis, direct immunofluorescence testing, and tissue culture. Testing for atypical mycobacteria and deep fungal infection was negative; bacterial cultures and sensitivity testing were negative. Direct immunofluorescence testing was negative. Microscopic examination of material from the right proximal pretibial region showed widely dilated, variously shaped, large blood vessels in a multinodular pattern; the vessels also were surrounded by an inflammatory cell infiltrate containing eosinophils. Histologic findings were consistent with ALHE.
Subsequent biopsies were completed 2 weeks and 1 month from the initial presentation. Both histology reports—from 2 different histopathology laboratories—were consistent with ALHE (Figure 2). Additional work-up during the patient’s initial visit to our clinic for the rash included CM serologic testing, which demonstrated IgM and IgG antibodies. Subsequently, chest radiography revealed a 2.2×2.3-cm mass in the right lower lobe of the lung. Follow-up computed tomography 1 month later confirmed the nodule in the same area to be 2.3×2.1×1.8 cm.
The patient was referred to pulmonology and was treated for pulmonary CM with oral fluconazole 200 mg twice daily for 4 months. Initial treatment also included clobetasol cream 0.05% applied twice daily, which did not produce marked improvement in pruritus. Narrowband UVB phototherapy was attempted, but the patient could not complete the course because of travel time to the office; however, the patient’s ALHE improved considerably with the fluconazole treatment for pulmonary CM.
Oral doxycycline 100 mg twice daily was added to the fluconazole 2 months after her initial visit to our office, which kept the ALHE at bay and helped with the pruritus (Figure 3). Pulmonology and primary care comanaged the pulmonary CM with oral fluconazole 200 mg twice daily. Repeat serologic testing for CM was negative for IgG and IgM after 14 months since the initial visit to the office.
Comment
Pulmonary CM infection has varying dermatologic manifestations. A PubMed search of articles indexed for MEDLINE using the terms ALHE and coccidioidomycosis yielded no case reports; in fact, there have been few reported cases of ALHE at all. Notable conditions associated with ALHE include membranous nephropathy and arteriovenous malformations treated with corticosteroids and surgery, respectively.3,4 Our case is a rare presentation of CM infection manifesting with ALHE. Following treatment and remission for our patient’s CM infection, the ALHE lesion decreased in size.
Standard treatment of uncomplicated CM involves azole antifungals, typically oral fluconazole or itraconazole 400 to 600 mg/d. In more severe cases (eg, immunocompromised patients) amphotericin B can be used.5 Our patient was treated with oral fluconazole 200 mg twice daily for 4 months.
In the literature, treatment via surgical excision, steroid injection, pulsed-dye laser therapy, and radiotherapy also has been described.6-8 Antibiotics including clindamycin, doxycycline, and amoxicillin-clavulanate also have been shown to be effective.9
In our patient, ALHE improved when oral doxycycline 100 mg twice daily was added to the oral fluconazole. In fact, after 4 months of treatment, the CM infection and ALHE lesions both improved to a point at which the lesions were not visible. When those lesions recurred 15 months later, they responded with another course of doxycycline and fluconazole.
Upon recurrence, the patient was asked to have her care transferred to her pulmonologist, who then managed the fluconazole regimen. During the pulmonologist’s workup, no peripheral eosinophilia was found. This is important because eosinophils can be a marker for CM infection; in this case, however, the ALHE lesion was a reactive process to the infection. Classically known to play a reactive role in fungal infection, these white blood cells demonstrate reactivity to the environmental fungus Alternaria alternata by contact-dependent killing, utilizing β2 integrins and CD11b to recognize and adhere to β-glucan. Eosinophils react through contact-dependent killing, releasing cytotoxic granule proteins and proinflammatory mediators, and have been documented to occur in CM and Paracoccidioides brasiliensis infection, in which they deposit major basic protein on the organism.10 Most pertinent to our case with ALHE and CM is the ability of eosinophils to communicate with other immune cells. Eosinophils play a role in the active inflammation of CM through cytokine signaling, which may propagate formation of ALHE.
The function of eosinophils in ALHE is poorly understood; it is unclear whether they act as a primary driver of pathogenesis or are simply indicators of secondary infiltration or infection. Our review of the current literature suggests that eosinophils are unnecessary for progression of ALHE but might be involved at its onset. As reported, even monoclonal antibody therapy (eg, mepolizumab and benralizumab) that effectively depletes eosinophil levels by negating IL-5 signaling do not slow progression of ALHE.11 Symptomatic changes are modest at best (ie, simply softening the ALHE nodules).
Our patient had no peripheral eosinophilia, suggesting that the onset of ALHE might not be caused by eosinophilia but a different inflammatory process—in this patient, by CM. Because peripheral eosinophilia was not seen in our patient, the presence of eosinophils in the ALHE lesion likely is unnecessary for its onset or progression but is a secondary process that exacerbates the lesion. The pathogenesis is unknown but could be directed toward lymphocytes and plasma cells, with eosinophils as part of the dynamic process.11
Conclusion
Because reports of an association between CM and ALHE are limited, our case is distinguished by a unique clinical presentation of ALHE. When a patient is given a diagnosis of ALHE, it therefore is important to consider exposure to CM as a cause, especially in patients who reside in or travel to a region where CM is endemic.
- Wells GC, Whimster IW. Subcutaneous angiolymphoid hyperplasia with eosinophilia. Br J Dermatol. 1969;81:1-14. doi:10.1111/j.1365-2133.1969.tb15914.x
- DiCaudo D. Coccidioidomycosis. Semin Cutan Med Surg. 2014;33:140-145. doi:10.12788/j.sder.0111
- Onishi Y, Ohara K. Angiolymphoid hyperplasia with eosinophilia associated with arteriovenous malformation: a clinicopathological correlation with angiography and serial estimation of serum levels of renin, eosinophil cationic protein and interleukin 5. Br J Dermatol. 1999;140:1153-1156. doi:10.1046/j.1365-2133.1999.02880.x
- Matsumoto A, Matsui I, Namba T, et al. VEGF-A links angiolymphoid hyperplasia with eosinophilia (ALHE) to THSD7A membranous nephropathy: a report of 2 cases. Am J Kidney Dis. 2019;73:880-885. doi:10.1053/j.ajkd.2018.10.009
- Bercovitch RS, Catanzaro A, Schwartz BS, et al. Coccidioidomycosis during pregnancy: a review and recommendations for management. Clin Infect Dis. 2011;53:363-368. doi:10.1093/cid/cir410
- Youssef A, Hasan AR, Youssef Y, et al. Angiolymphoid hyperplasia with eosinophilia: a case report. J Med Case Rep. 2018;12:89. doi:10.1186/s13256-018-1599-x
- Abrahamson TG, Davis DA. Angiolymphoid hyperplasia witheosinophilia responsive to pulsed dye laser. J Am Acad Dermatol. 2003;49(2 suppl case reports):S195-S196. doi:10.1067/mjd.2003.314
- Lembo S, Balato A, Cirillo T, et al. A long-term follow-up of angiolymphoid hyperplasia with eosinophilia treated by corticosteroids: when a traditional therapy is still up-to-date. Case Rep Dermatol. 2011;3:64-67. doi:10.1159/000323182
- Cleveland E. Atypical presentation of angiolymphomatous hyperplasia with eosinophilia. J Am Acad Dermatol. 2018;79(3 suppl 1):AB53. doi:10.1016/j.jaad.2018.05.249
- Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol. 2015;50:214-227. doi:10.1007/s12016-015-8525-4
- Grünewald M, Stölzl D, Wehkamp U, et al. Role of eosinophils in angiolymphoid hyperplasia with eosinophilia. JAMA Dermatol. 2021;157:1241-1243. doi:10.1001/jamadermatol.2021.2732
- Wells GC, Whimster IW. Subcutaneous angiolymphoid hyperplasia with eosinophilia. Br J Dermatol. 1969;81:1-14. doi:10.1111/j.1365-2133.1969.tb15914.x
- DiCaudo D. Coccidioidomycosis. Semin Cutan Med Surg. 2014;33:140-145. doi:10.12788/j.sder.0111
- Onishi Y, Ohara K. Angiolymphoid hyperplasia with eosinophilia associated with arteriovenous malformation: a clinicopathological correlation with angiography and serial estimation of serum levels of renin, eosinophil cationic protein and interleukin 5. Br J Dermatol. 1999;140:1153-1156. doi:10.1046/j.1365-2133.1999.02880.x
- Matsumoto A, Matsui I, Namba T, et al. VEGF-A links angiolymphoid hyperplasia with eosinophilia (ALHE) to THSD7A membranous nephropathy: a report of 2 cases. Am J Kidney Dis. 2019;73:880-885. doi:10.1053/j.ajkd.2018.10.009
- Bercovitch RS, Catanzaro A, Schwartz BS, et al. Coccidioidomycosis during pregnancy: a review and recommendations for management. Clin Infect Dis. 2011;53:363-368. doi:10.1093/cid/cir410
- Youssef A, Hasan AR, Youssef Y, et al. Angiolymphoid hyperplasia with eosinophilia: a case report. J Med Case Rep. 2018;12:89. doi:10.1186/s13256-018-1599-x
- Abrahamson TG, Davis DA. Angiolymphoid hyperplasia witheosinophilia responsive to pulsed dye laser. J Am Acad Dermatol. 2003;49(2 suppl case reports):S195-S196. doi:10.1067/mjd.2003.314
- Lembo S, Balato A, Cirillo T, et al. A long-term follow-up of angiolymphoid hyperplasia with eosinophilia treated by corticosteroids: when a traditional therapy is still up-to-date. Case Rep Dermatol. 2011;3:64-67. doi:10.1159/000323182
- Cleveland E. Atypical presentation of angiolymphomatous hyperplasia with eosinophilia. J Am Acad Dermatol. 2018;79(3 suppl 1):AB53. doi:10.1016/j.jaad.2018.05.249
- Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol. 2015;50:214-227. doi:10.1007/s12016-015-8525-4
- Grünewald M, Stölzl D, Wehkamp U, et al. Role of eosinophils in angiolymphoid hyperplasia with eosinophilia. JAMA Dermatol. 2021;157:1241-1243. doi:10.1001/jamadermatol.2021.2732
Practice Points
- Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare entity of unknown etiology.
- There is an association between ALHE and coccidioidomycosis (CM). Patients who present with ALHE and reside in a CM-endemic region should be examined for CM.

Prolonged Drug-Induced Hypersensitivity Syndrome/DRESS With Alopecia Areata and Autoimmune Thyroiditis
Drug-induced hypersensitivity syndrome (DIHS), also called drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, is a potentially fatal drug-induced hypersensitivity reaction that is characterized by a cutaneous eruption, multiorgan involvement, viral reactivation, and hematologic abnormalities. As the nomenclature of this disease advances, consensus groups have adopted DIHS/DRESS to underscore that both names refer to the same clinical phenomenon.1 Autoimmune sequelae have been reported after DIHS/DRESS that include vitiligo, thyroid disease, and type 1 diabetes mellitus (T1DM). We present a case of lamotrigine-associated DIHS/DRESS complicated by an unusually prolonged course requiring oral corticosteroids and narrow-band ultraviolet B (UVB) treatment and with development of extensive alopecia areata and autoimmune thyroiditis.
Case Presentation
A 35-year-old female Filipino patient was prescribed lamotrigine 25 mg daily for bipolar II disorder and titrated to 100 mg twice daily after 1 month. One week after the increase, the patient developed a diffuse morbilliform rash covering their entire body along with facial swelling and generalized pruritus. Lamotrigine was discontinued after lamotrigine allergy was diagnosed. The patient improved following a 9-day oral prednisone taper and was placed on oxcarbazepine 300 mg twice daily to manage their bipolar disorder. One day after completing the taper, the patient presented again with worsening rash, swelling, and cervical lymphadenopathy. Oxcarbazepine was discontinued, and oral prednisone 60 mg was reinstituted for an additional 11 days.
Dermatology evaluated the patient 10 days after completion of the second oral steroid taper (1 month after cessation of lamotrigine). The patient had erythroderma along with malaise, fevers, chills, and fatigue and a diffuse burning sensation (Figure 1). The patient was hypotensive and tachycardic with significant eosinophilia (42%; reference range, 0%-8%), transaminitis, and renal insufficiency. The patient was diagnosed with DIHS/DRESS based on their clinical presentation and calculated RegiSCAR score of 7 (score > 5 corresponds with definite DIHS/DRESS and points were given for fever, enlarged lymph nodes, eosinophilia ≥ 20%, skin rash extending > 50% of their body, edema and scaling, and 2 organs involved).2 A punch biopsy was confirmatory (Figure 2A).3 The patient was started on prednisone 80 mg once daily along with topical fluocinonide 0.05% ointment. However, the patient’s clinical status deteriorated, requiring hospital admission for heart failure evaluation. The echocardiogram revealed hyperdynamic circulation but was otherwise unremarkable.
The patient was maintained on prednisone 70 to 80 mg daily for 2 months before improvement of the rash and pruritus. The prednisone was slowly tapered over a 6-week period and then discontinued. Shortly after discontinuation, the patient redeveloped erythroderma. Skin biopsy and complete blood count (17.3% eosinophilia) confirmed the suspected DIHS/DRESS relapse (Figure 2B). In addition, the patient reported upper respiratory tract symptoms and concurrently tested positive for human herpesvirus 6 (HHV-6). The patient was restarted on prednisone and low-dose narrow-band UVB (nbUVB) therapy was added. Over the following 2 months, they responded well to low-dose nbUVB therapy. By the end of nbUVB treatment, about 5 months after initial presentation, the patient’s erythroderma improved, eosinophilia resolved, and they were able to tolerate prednisone taper. Ten months after cessation of lamotrigine, prednisone was finally discontinued. Two weeks later, the patient was screened for adrenal insufficiency (AI) given the prolonged steroid course. Their serum morning cortisol level was within normal limits.
Four months after DIHS/DRESS resolution and cessation of steroids, the patient noted significant patches of smooth alopecia on their posterior scalp and was diagnosed with alopecia areata. Treatment with intralesional triamcinolone over 2 months resulted in regrowth of hair (Figure 3). A month later, the patient reported increasing fatigue and anorexia. The patient was evaluated once more for AI, this time with low morning cortisol and low adrenocorticotrophic hormone (ACTH) levels—consistent with AI secondary to prolonged glucocorticoid therapy. The patient also was concomitantly evaluated for hypothyroidism with significantly elevated thyroperoxidase antibodies—confirming the diagnosis of Hashimoto thyroiditis.
Discussion
DIHS/DRESS syndrome is a rare, but potentially life-threatening hypersensitivity to a medication, often beginning 2 to 6 weeks after exposure to the causative agent. The incidence of DIHS/DRESS in the general population is about 2 per 100,000.3 Our patient presented with DIHS/DRESS 33 days after starting lamotrigine, which corresponds with the published mean onset of anticonvulsant-induced DIHS/DRESS (29.7-33.3 days).4 Recent evidence shows that time from drug exposure to DIHS/DRESS symptoms may vary by drug class, with antibiotics implicated as precipitating DIHS/DRESS in < 15 days.3 The diagnosis of DIHS/DRESS may be complicated for many reasons. The accompanying rash may be morbilliform, erythroderma, or exfoliative dermatitis with multiple anatomic regions affected.5 Systemic involvement with various internal organs occurs in > 90% of cases, with the liver and kidney involved most frequently.5 Overall mortality rate may be as high as 10% most commonly due to acute liver failure.5 Biopsy may be helpful in the diagnosis but is not always specific.5 Diagnostic criteria include RegiSCAR and J-SCAR scores; our patient met criteria for both (Table).5
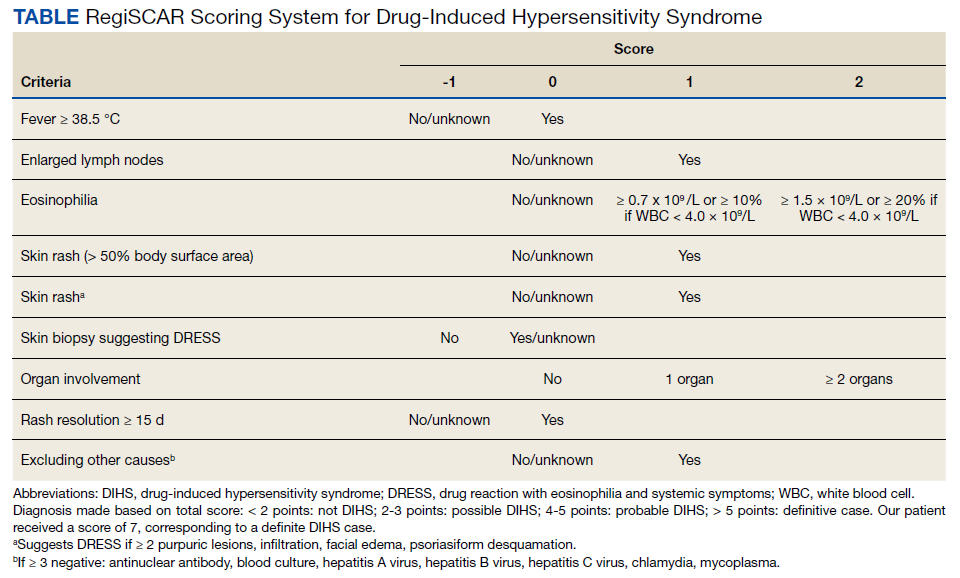
The pathogenesis of DIHS/DRESS remains unclear. Proposed mechanisms include genetic predisposition with human leukocyte antigen (HLA) haplotypes, autoimmune with a delayed cell-mediated immune response associated with herpesviruses, and abnormal enzymatic pathways that metabolize medications.2 Although no HLA has been identified between lamotrigine and DIHS, HLA-A*02:07 and HLA-B*15:02 have been associated with lamotrigine-induced cutaneous drug reactions in patients of Thai ancestry.6 Immunosuppression also is a risk factor, especially when accompanied by a primary or reactivated HHV-6 infection, as seen in our patient.2 Additionally, HHV-6 infection may be a common link between DIHS/DRESS and autoimmune thyroiditis but is believed to involve elevated levels of interferon-γ-induced protein-10 (IP-10) that may lead to excessive recruitment of cytotoxic T cells into target tissues.7 Elevated levels of IP-10 are seen in many autoimmune conditions, such as autoimmune thyroiditis, Sjögren syndrome, and Graves disease.8
DIHS/DRESS syndrome has been associated with development of autoimmune diseases as long-term sequelae. The most commonly affected organs are the thyroid and pancreas; approximately 4.8% of patients develop autoimmune thyroiditis and 3.5% develop fulminant T1DM.9 The time from onset of DIHS/DRESS to development of autoimmune thyroiditis can range from 2 months to 2 years, whereas the range from DIHS/DRESS onset to fulminant T1DM is about 40 days.9 Alopecia had been reported in 1, occurring 4 months after DIHS/DRESS onset. Our patient’s alopecia areata and Hashimoto thyroiditis occurred 14 and 15 months after DIHS/DRESS presentation, respectively.
Treatment
For management, early recognition and discontinuation of the offending agent is paramount. Systemic corticosteroids are the accepted treatment standard. Symptoms of DIHS/DRESS usually resolve between 3 and 18 weeks, with the mean resolution time at 7 weeks.10 Our patient developed a prolonged course with persistent eosinophilia for 20 weeks and cutaneous symptoms for 32 weeks—requiring 40 weeks of oral prednisone. The most significant clinical improvement occurred during the 8-week period low-dose nbUVB was used (Figure 4). There also are reports outlining the successful use of intravenous immunoglobulin, cyclosporine, cyclophosphamide, rituximab, or plasma exchange in cases refractory to oral corticosteroids.11
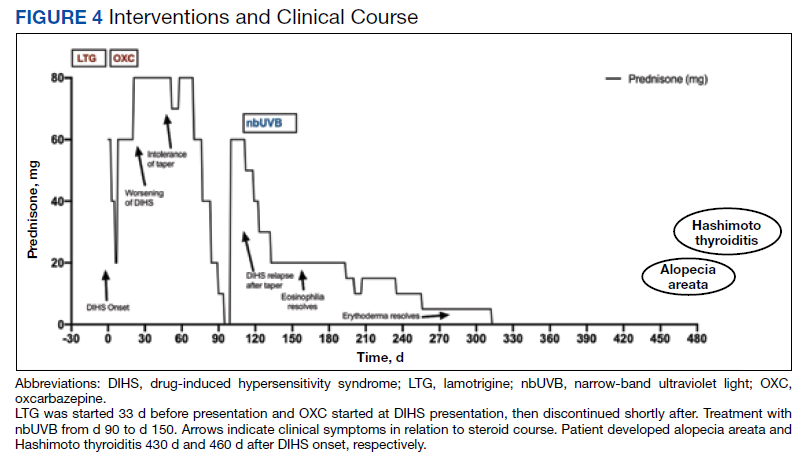
A recent retrospective case control study showed that treatment of DIHS/DRESS with cyclosporine in patients who had a contraindication to steroids resulted in faster resolution of symptoms, shorter treatment durations, and shorter hospitalizations than did those treated with corticosteroids.12 However, the data are limited by a significantly smaller number of patients treated with cyclosporine than steroids and the cyclosporine treatment group having milder cases of DIHS/DRESS.12
The risk of AI is increased for patients who have taken > 20 mg of prednisone daily ≥ 3 weeks, an evening dose ≥ 5 mg for a few weeks, or have a Cushingoid appearance.13 Patients may not regain full adrenal function for 12 to 18 months.14 Our patient had a normal basal serum cortisol level 2 weeks after prednisone cessation and then presented 5 months later with AI. While the reason for this period of normality is unclear, it may partly be due to the variable length of hypothalamic-pituitary-adrenal axis recovery time. Thus, ACTH stimulation tests in addition to serum cortisol may be done in patients with suspected AI for higher diagnostic certainty.10
Conclusions
DIHS/DRESS is a severe cutaneous adverse reaction that may require a prolonged treatment course until symptom resolution (40 weeks of oral prednisone in our patient). Oral corticosteroids are the mainstay of treatment, but long-term use is associated with significant adverse effects, such as AI in our patient. Alternative therapies, such as cyclosporine, look promising, but further studies are needed to determine safety profile and efficacy.12 Additionally, patients with DIHS/DRESS should be educated and followed for potential autoimmune sequelae; in our patient alopecia areata and autoimmune thyroiditis were late sequelae, occurring 14 and 15 months, respectively, after onset of DIHS/DRESS.
1. RegiSCAR. Accessed June 3, 2022. http://www.regiscar.org
2. Shiohara T, Mizukawa Y. Drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS): an update in 2019. Allergol Int. 2019;68(3):301-308. doi:10.1016/j.alit.2019.03.006
3. Wolfson AR, Zhou L, Li Y, Phadke NA, Chow OA, Blumenthal KG. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome identified in the electronic health record allergy module. J Allergy Clin Immunol Pract. 2019;7(2):633-640. doi:10.1016/j.jaip.2018.08.013
4. Sasidharanpillai S, Govindan A, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a histopathology based analysis. Indian J Dermatol Venereol Leprol. 2016;82(1):28. doi:10.4103/0378-6323.168934
5. Kardaun SH, Sekula P, Valeyrie‐Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169(5):1071-1080. doi:10.1111/bjd.12501
6. Koomdee N, Pratoomwun J, Jantararoungtong T, et al. Association of HLA-A and HLA-B alleles with lamotrigine-induced cutaneous adverse drug reactions in the Thai population. Front Pharmacol. 2017;8. doi:10.3389/fphar.2017.00879
7. Yang C-W, Cho Y-T, Hsieh Y-C, Hsu S-H, Chen K-L, Chu C-Y. The interferon-γ-induced protein 10/CXCR3 axis is associated with human herpesvirus-6 reactivation and the development of sequelae in drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2020;183(5):909-919. doi:10.1111/bjd.18942
8. Ruffilli I, Ferrari SM, Colaci M, Ferri C, Fallahi P, Antonelli A. IP-10 in autoimmune thyroiditis. Horm Metab Res. 2014;46(9):597-602. doi:10.1055/s-0034-1382053
9. Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42(3):276-282. doi:10.1111/1346-8138.12770
10. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124(7):588-597. doi:10.1016/j.amjmed.2011.01.017
11. Bommersbach TJ, Lapid MI, Leung JG, Cunningham JL, Rummans TA, Kung S. Management of psychotropic drug-induced dress syndrome: a systematic review. Mayo Clin Proc. 2016;91(6):787-801. doi:10.1016/j.mayocp.2016.03.006
12. Nguyen E, Yanes D, Imadojemu S, Kroshinsky D. Evaluation of cyclosporine for the treatment of DRESS syndrome. JAMA Dermatol. 2020;156(6):704-706. doi:10.1001/jamadermatol.2020.0048
13. Joseph RM, Hunter AL, Ray DW, Dixon WG. Systemic glucocorticoid therapy and adrenal insufficiency in adults: a systematic review. Semin Arthritis Rheum. 2016;46(1):133-141. doi:10.1016/j.semarthrit.2016.03.001
14. Jamilloux Y, Liozon E, Pugnet G, et al. Recovery of adrenal function after long-term glucocorticoid therapy for giant cell arteritis: a cohort study. PLoS ONE. 2013;8(7):e68713. doi:10.1371/journal.pone.0068713
Drug-induced hypersensitivity syndrome (DIHS), also called drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, is a potentially fatal drug-induced hypersensitivity reaction that is characterized by a cutaneous eruption, multiorgan involvement, viral reactivation, and hematologic abnormalities. As the nomenclature of this disease advances, consensus groups have adopted DIHS/DRESS to underscore that both names refer to the same clinical phenomenon.1 Autoimmune sequelae have been reported after DIHS/DRESS that include vitiligo, thyroid disease, and type 1 diabetes mellitus (T1DM). We present a case of lamotrigine-associated DIHS/DRESS complicated by an unusually prolonged course requiring oral corticosteroids and narrow-band ultraviolet B (UVB) treatment and with development of extensive alopecia areata and autoimmune thyroiditis.
Case Presentation
A 35-year-old female Filipino patient was prescribed lamotrigine 25 mg daily for bipolar II disorder and titrated to 100 mg twice daily after 1 month. One week after the increase, the patient developed a diffuse morbilliform rash covering their entire body along with facial swelling and generalized pruritus. Lamotrigine was discontinued after lamotrigine allergy was diagnosed. The patient improved following a 9-day oral prednisone taper and was placed on oxcarbazepine 300 mg twice daily to manage their bipolar disorder. One day after completing the taper, the patient presented again with worsening rash, swelling, and cervical lymphadenopathy. Oxcarbazepine was discontinued, and oral prednisone 60 mg was reinstituted for an additional 11 days.
Dermatology evaluated the patient 10 days after completion of the second oral steroid taper (1 month after cessation of lamotrigine). The patient had erythroderma along with malaise, fevers, chills, and fatigue and a diffuse burning sensation (Figure 1). The patient was hypotensive and tachycardic with significant eosinophilia (42%; reference range, 0%-8%), transaminitis, and renal insufficiency. The patient was diagnosed with DIHS/DRESS based on their clinical presentation and calculated RegiSCAR score of 7 (score > 5 corresponds with definite DIHS/DRESS and points were given for fever, enlarged lymph nodes, eosinophilia ≥ 20%, skin rash extending > 50% of their body, edema and scaling, and 2 organs involved).2 A punch biopsy was confirmatory (Figure 2A).3 The patient was started on prednisone 80 mg once daily along with topical fluocinonide 0.05% ointment. However, the patient’s clinical status deteriorated, requiring hospital admission for heart failure evaluation. The echocardiogram revealed hyperdynamic circulation but was otherwise unremarkable.
The patient was maintained on prednisone 70 to 80 mg daily for 2 months before improvement of the rash and pruritus. The prednisone was slowly tapered over a 6-week period and then discontinued. Shortly after discontinuation, the patient redeveloped erythroderma. Skin biopsy and complete blood count (17.3% eosinophilia) confirmed the suspected DIHS/DRESS relapse (Figure 2B). In addition, the patient reported upper respiratory tract symptoms and concurrently tested positive for human herpesvirus 6 (HHV-6). The patient was restarted on prednisone and low-dose narrow-band UVB (nbUVB) therapy was added. Over the following 2 months, they responded well to low-dose nbUVB therapy. By the end of nbUVB treatment, about 5 months after initial presentation, the patient’s erythroderma improved, eosinophilia resolved, and they were able to tolerate prednisone taper. Ten months after cessation of lamotrigine, prednisone was finally discontinued. Two weeks later, the patient was screened for adrenal insufficiency (AI) given the prolonged steroid course. Their serum morning cortisol level was within normal limits.
Four months after DIHS/DRESS resolution and cessation of steroids, the patient noted significant patches of smooth alopecia on their posterior scalp and was diagnosed with alopecia areata. Treatment with intralesional triamcinolone over 2 months resulted in regrowth of hair (Figure 3). A month later, the patient reported increasing fatigue and anorexia. The patient was evaluated once more for AI, this time with low morning cortisol and low adrenocorticotrophic hormone (ACTH) levels—consistent with AI secondary to prolonged glucocorticoid therapy. The patient also was concomitantly evaluated for hypothyroidism with significantly elevated thyroperoxidase antibodies—confirming the diagnosis of Hashimoto thyroiditis.
Discussion
DIHS/DRESS syndrome is a rare, but potentially life-threatening hypersensitivity to a medication, often beginning 2 to 6 weeks after exposure to the causative agent. The incidence of DIHS/DRESS in the general population is about 2 per 100,000.3 Our patient presented with DIHS/DRESS 33 days after starting lamotrigine, which corresponds with the published mean onset of anticonvulsant-induced DIHS/DRESS (29.7-33.3 days).4 Recent evidence shows that time from drug exposure to DIHS/DRESS symptoms may vary by drug class, with antibiotics implicated as precipitating DIHS/DRESS in < 15 days.3 The diagnosis of DIHS/DRESS may be complicated for many reasons. The accompanying rash may be morbilliform, erythroderma, or exfoliative dermatitis with multiple anatomic regions affected.5 Systemic involvement with various internal organs occurs in > 90% of cases, with the liver and kidney involved most frequently.5 Overall mortality rate may be as high as 10% most commonly due to acute liver failure.5 Biopsy may be helpful in the diagnosis but is not always specific.5 Diagnostic criteria include RegiSCAR and J-SCAR scores; our patient met criteria for both (Table).5
The pathogenesis of DIHS/DRESS remains unclear. Proposed mechanisms include genetic predisposition with human leukocyte antigen (HLA) haplotypes, autoimmune with a delayed cell-mediated immune response associated with herpesviruses, and abnormal enzymatic pathways that metabolize medications.2 Although no HLA has been identified between lamotrigine and DIHS, HLA-A*02:07 and HLA-B*15:02 have been associated with lamotrigine-induced cutaneous drug reactions in patients of Thai ancestry.6 Immunosuppression also is a risk factor, especially when accompanied by a primary or reactivated HHV-6 infection, as seen in our patient.2 Additionally, HHV-6 infection may be a common link between DIHS/DRESS and autoimmune thyroiditis but is believed to involve elevated levels of interferon-γ-induced protein-10 (IP-10) that may lead to excessive recruitment of cytotoxic T cells into target tissues.7 Elevated levels of IP-10 are seen in many autoimmune conditions, such as autoimmune thyroiditis, Sjögren syndrome, and Graves disease.8
DIHS/DRESS syndrome has been associated with development of autoimmune diseases as long-term sequelae. The most commonly affected organs are the thyroid and pancreas; approximately 4.8% of patients develop autoimmune thyroiditis and 3.5% develop fulminant T1DM.9 The time from onset of DIHS/DRESS to development of autoimmune thyroiditis can range from 2 months to 2 years, whereas the range from DIHS/DRESS onset to fulminant T1DM is about 40 days.9 Alopecia had been reported in 1, occurring 4 months after DIHS/DRESS onset. Our patient’s alopecia areata and Hashimoto thyroiditis occurred 14 and 15 months after DIHS/DRESS presentation, respectively.
Treatment
For management, early recognition and discontinuation of the offending agent is paramount. Systemic corticosteroids are the accepted treatment standard. Symptoms of DIHS/DRESS usually resolve between 3 and 18 weeks, with the mean resolution time at 7 weeks.10 Our patient developed a prolonged course with persistent eosinophilia for 20 weeks and cutaneous symptoms for 32 weeks—requiring 40 weeks of oral prednisone. The most significant clinical improvement occurred during the 8-week period low-dose nbUVB was used (Figure 4). There also are reports outlining the successful use of intravenous immunoglobulin, cyclosporine, cyclophosphamide, rituximab, or plasma exchange in cases refractory to oral corticosteroids.11
A recent retrospective case control study showed that treatment of DIHS/DRESS with cyclosporine in patients who had a contraindication to steroids resulted in faster resolution of symptoms, shorter treatment durations, and shorter hospitalizations than did those treated with corticosteroids.12 However, the data are limited by a significantly smaller number of patients treated with cyclosporine than steroids and the cyclosporine treatment group having milder cases of DIHS/DRESS.12
The risk of AI is increased for patients who have taken > 20 mg of prednisone daily ≥ 3 weeks, an evening dose ≥ 5 mg for a few weeks, or have a Cushingoid appearance.13 Patients may not regain full adrenal function for 12 to 18 months.14 Our patient had a normal basal serum cortisol level 2 weeks after prednisone cessation and then presented 5 months later with AI. While the reason for this period of normality is unclear, it may partly be due to the variable length of hypothalamic-pituitary-adrenal axis recovery time. Thus, ACTH stimulation tests in addition to serum cortisol may be done in patients with suspected AI for higher diagnostic certainty.10
Conclusions
DIHS/DRESS is a severe cutaneous adverse reaction that may require a prolonged treatment course until symptom resolution (40 weeks of oral prednisone in our patient). Oral corticosteroids are the mainstay of treatment, but long-term use is associated with significant adverse effects, such as AI in our patient. Alternative therapies, such as cyclosporine, look promising, but further studies are needed to determine safety profile and efficacy.12 Additionally, patients with DIHS/DRESS should be educated and followed for potential autoimmune sequelae; in our patient alopecia areata and autoimmune thyroiditis were late sequelae, occurring 14 and 15 months, respectively, after onset of DIHS/DRESS.
Drug-induced hypersensitivity syndrome (DIHS), also called drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, is a potentially fatal drug-induced hypersensitivity reaction that is characterized by a cutaneous eruption, multiorgan involvement, viral reactivation, and hematologic abnormalities. As the nomenclature of this disease advances, consensus groups have adopted DIHS/DRESS to underscore that both names refer to the same clinical phenomenon.1 Autoimmune sequelae have been reported after DIHS/DRESS that include vitiligo, thyroid disease, and type 1 diabetes mellitus (T1DM). We present a case of lamotrigine-associated DIHS/DRESS complicated by an unusually prolonged course requiring oral corticosteroids and narrow-band ultraviolet B (UVB) treatment and with development of extensive alopecia areata and autoimmune thyroiditis.
Case Presentation
A 35-year-old female Filipino patient was prescribed lamotrigine 25 mg daily for bipolar II disorder and titrated to 100 mg twice daily after 1 month. One week after the increase, the patient developed a diffuse morbilliform rash covering their entire body along with facial swelling and generalized pruritus. Lamotrigine was discontinued after lamotrigine allergy was diagnosed. The patient improved following a 9-day oral prednisone taper and was placed on oxcarbazepine 300 mg twice daily to manage their bipolar disorder. One day after completing the taper, the patient presented again with worsening rash, swelling, and cervical lymphadenopathy. Oxcarbazepine was discontinued, and oral prednisone 60 mg was reinstituted for an additional 11 days.
Dermatology evaluated the patient 10 days after completion of the second oral steroid taper (1 month after cessation of lamotrigine). The patient had erythroderma along with malaise, fevers, chills, and fatigue and a diffuse burning sensation (Figure 1). The patient was hypotensive and tachycardic with significant eosinophilia (42%; reference range, 0%-8%), transaminitis, and renal insufficiency. The patient was diagnosed with DIHS/DRESS based on their clinical presentation and calculated RegiSCAR score of 7 (score > 5 corresponds with definite DIHS/DRESS and points were given for fever, enlarged lymph nodes, eosinophilia ≥ 20%, skin rash extending > 50% of their body, edema and scaling, and 2 organs involved).2 A punch biopsy was confirmatory (Figure 2A).3 The patient was started on prednisone 80 mg once daily along with topical fluocinonide 0.05% ointment. However, the patient’s clinical status deteriorated, requiring hospital admission for heart failure evaluation. The echocardiogram revealed hyperdynamic circulation but was otherwise unremarkable.
The patient was maintained on prednisone 70 to 80 mg daily for 2 months before improvement of the rash and pruritus. The prednisone was slowly tapered over a 6-week period and then discontinued. Shortly after discontinuation, the patient redeveloped erythroderma. Skin biopsy and complete blood count (17.3% eosinophilia) confirmed the suspected DIHS/DRESS relapse (Figure 2B). In addition, the patient reported upper respiratory tract symptoms and concurrently tested positive for human herpesvirus 6 (HHV-6). The patient was restarted on prednisone and low-dose narrow-band UVB (nbUVB) therapy was added. Over the following 2 months, they responded well to low-dose nbUVB therapy. By the end of nbUVB treatment, about 5 months after initial presentation, the patient’s erythroderma improved, eosinophilia resolved, and they were able to tolerate prednisone taper. Ten months after cessation of lamotrigine, prednisone was finally discontinued. Two weeks later, the patient was screened for adrenal insufficiency (AI) given the prolonged steroid course. Their serum morning cortisol level was within normal limits.
Four months after DIHS/DRESS resolution and cessation of steroids, the patient noted significant patches of smooth alopecia on their posterior scalp and was diagnosed with alopecia areata. Treatment with intralesional triamcinolone over 2 months resulted in regrowth of hair (Figure 3). A month later, the patient reported increasing fatigue and anorexia. The patient was evaluated once more for AI, this time with low morning cortisol and low adrenocorticotrophic hormone (ACTH) levels—consistent with AI secondary to prolonged glucocorticoid therapy. The patient also was concomitantly evaluated for hypothyroidism with significantly elevated thyroperoxidase antibodies—confirming the diagnosis of Hashimoto thyroiditis.
Discussion
DIHS/DRESS syndrome is a rare, but potentially life-threatening hypersensitivity to a medication, often beginning 2 to 6 weeks after exposure to the causative agent. The incidence of DIHS/DRESS in the general population is about 2 per 100,000.3 Our patient presented with DIHS/DRESS 33 days after starting lamotrigine, which corresponds with the published mean onset of anticonvulsant-induced DIHS/DRESS (29.7-33.3 days).4 Recent evidence shows that time from drug exposure to DIHS/DRESS symptoms may vary by drug class, with antibiotics implicated as precipitating DIHS/DRESS in < 15 days.3 The diagnosis of DIHS/DRESS may be complicated for many reasons. The accompanying rash may be morbilliform, erythroderma, or exfoliative dermatitis with multiple anatomic regions affected.5 Systemic involvement with various internal organs occurs in > 90% of cases, with the liver and kidney involved most frequently.5 Overall mortality rate may be as high as 10% most commonly due to acute liver failure.5 Biopsy may be helpful in the diagnosis but is not always specific.5 Diagnostic criteria include RegiSCAR and J-SCAR scores; our patient met criteria for both (Table).5
The pathogenesis of DIHS/DRESS remains unclear. Proposed mechanisms include genetic predisposition with human leukocyte antigen (HLA) haplotypes, autoimmune with a delayed cell-mediated immune response associated with herpesviruses, and abnormal enzymatic pathways that metabolize medications.2 Although no HLA has been identified between lamotrigine and DIHS, HLA-A*02:07 and HLA-B*15:02 have been associated with lamotrigine-induced cutaneous drug reactions in patients of Thai ancestry.6 Immunosuppression also is a risk factor, especially when accompanied by a primary or reactivated HHV-6 infection, as seen in our patient.2 Additionally, HHV-6 infection may be a common link between DIHS/DRESS and autoimmune thyroiditis but is believed to involve elevated levels of interferon-γ-induced protein-10 (IP-10) that may lead to excessive recruitment of cytotoxic T cells into target tissues.7 Elevated levels of IP-10 are seen in many autoimmune conditions, such as autoimmune thyroiditis, Sjögren syndrome, and Graves disease.8
DIHS/DRESS syndrome has been associated with development of autoimmune diseases as long-term sequelae. The most commonly affected organs are the thyroid and pancreas; approximately 4.8% of patients develop autoimmune thyroiditis and 3.5% develop fulminant T1DM.9 The time from onset of DIHS/DRESS to development of autoimmune thyroiditis can range from 2 months to 2 years, whereas the range from DIHS/DRESS onset to fulminant T1DM is about 40 days.9 Alopecia had been reported in 1, occurring 4 months after DIHS/DRESS onset. Our patient’s alopecia areata and Hashimoto thyroiditis occurred 14 and 15 months after DIHS/DRESS presentation, respectively.
Treatment
For management, early recognition and discontinuation of the offending agent is paramount. Systemic corticosteroids are the accepted treatment standard. Symptoms of DIHS/DRESS usually resolve between 3 and 18 weeks, with the mean resolution time at 7 weeks.10 Our patient developed a prolonged course with persistent eosinophilia for 20 weeks and cutaneous symptoms for 32 weeks—requiring 40 weeks of oral prednisone. The most significant clinical improvement occurred during the 8-week period low-dose nbUVB was used (Figure 4). There also are reports outlining the successful use of intravenous immunoglobulin, cyclosporine, cyclophosphamide, rituximab, or plasma exchange in cases refractory to oral corticosteroids.11
A recent retrospective case control study showed that treatment of DIHS/DRESS with cyclosporine in patients who had a contraindication to steroids resulted in faster resolution of symptoms, shorter treatment durations, and shorter hospitalizations than did those treated with corticosteroids.12 However, the data are limited by a significantly smaller number of patients treated with cyclosporine than steroids and the cyclosporine treatment group having milder cases of DIHS/DRESS.12
The risk of AI is increased for patients who have taken > 20 mg of prednisone daily ≥ 3 weeks, an evening dose ≥ 5 mg for a few weeks, or have a Cushingoid appearance.13 Patients may not regain full adrenal function for 12 to 18 months.14 Our patient had a normal basal serum cortisol level 2 weeks after prednisone cessation and then presented 5 months later with AI. While the reason for this period of normality is unclear, it may partly be due to the variable length of hypothalamic-pituitary-adrenal axis recovery time. Thus, ACTH stimulation tests in addition to serum cortisol may be done in patients with suspected AI for higher diagnostic certainty.10
Conclusions
DIHS/DRESS is a severe cutaneous adverse reaction that may require a prolonged treatment course until symptom resolution (40 weeks of oral prednisone in our patient). Oral corticosteroids are the mainstay of treatment, but long-term use is associated with significant adverse effects, such as AI in our patient. Alternative therapies, such as cyclosporine, look promising, but further studies are needed to determine safety profile and efficacy.12 Additionally, patients with DIHS/DRESS should be educated and followed for potential autoimmune sequelae; in our patient alopecia areata and autoimmune thyroiditis were late sequelae, occurring 14 and 15 months, respectively, after onset of DIHS/DRESS.
1. RegiSCAR. Accessed June 3, 2022. http://www.regiscar.org
2. Shiohara T, Mizukawa Y. Drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS): an update in 2019. Allergol Int. 2019;68(3):301-308. doi:10.1016/j.alit.2019.03.006
3. Wolfson AR, Zhou L, Li Y, Phadke NA, Chow OA, Blumenthal KG. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome identified in the electronic health record allergy module. J Allergy Clin Immunol Pract. 2019;7(2):633-640. doi:10.1016/j.jaip.2018.08.013
4. Sasidharanpillai S, Govindan A, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a histopathology based analysis. Indian J Dermatol Venereol Leprol. 2016;82(1):28. doi:10.4103/0378-6323.168934
5. Kardaun SH, Sekula P, Valeyrie‐Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169(5):1071-1080. doi:10.1111/bjd.12501
6. Koomdee N, Pratoomwun J, Jantararoungtong T, et al. Association of HLA-A and HLA-B alleles with lamotrigine-induced cutaneous adverse drug reactions in the Thai population. Front Pharmacol. 2017;8. doi:10.3389/fphar.2017.00879
7. Yang C-W, Cho Y-T, Hsieh Y-C, Hsu S-H, Chen K-L, Chu C-Y. The interferon-γ-induced protein 10/CXCR3 axis is associated with human herpesvirus-6 reactivation and the development of sequelae in drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2020;183(5):909-919. doi:10.1111/bjd.18942
8. Ruffilli I, Ferrari SM, Colaci M, Ferri C, Fallahi P, Antonelli A. IP-10 in autoimmune thyroiditis. Horm Metab Res. 2014;46(9):597-602. doi:10.1055/s-0034-1382053
9. Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42(3):276-282. doi:10.1111/1346-8138.12770
10. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124(7):588-597. doi:10.1016/j.amjmed.2011.01.017
11. Bommersbach TJ, Lapid MI, Leung JG, Cunningham JL, Rummans TA, Kung S. Management of psychotropic drug-induced dress syndrome: a systematic review. Mayo Clin Proc. 2016;91(6):787-801. doi:10.1016/j.mayocp.2016.03.006
12. Nguyen E, Yanes D, Imadojemu S, Kroshinsky D. Evaluation of cyclosporine for the treatment of DRESS syndrome. JAMA Dermatol. 2020;156(6):704-706. doi:10.1001/jamadermatol.2020.0048
13. Joseph RM, Hunter AL, Ray DW, Dixon WG. Systemic glucocorticoid therapy and adrenal insufficiency in adults: a systematic review. Semin Arthritis Rheum. 2016;46(1):133-141. doi:10.1016/j.semarthrit.2016.03.001
14. Jamilloux Y, Liozon E, Pugnet G, et al. Recovery of adrenal function after long-term glucocorticoid therapy for giant cell arteritis: a cohort study. PLoS ONE. 2013;8(7):e68713. doi:10.1371/journal.pone.0068713
1. RegiSCAR. Accessed June 3, 2022. http://www.regiscar.org
2. Shiohara T, Mizukawa Y. Drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS): an update in 2019. Allergol Int. 2019;68(3):301-308. doi:10.1016/j.alit.2019.03.006
3. Wolfson AR, Zhou L, Li Y, Phadke NA, Chow OA, Blumenthal KG. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome identified in the electronic health record allergy module. J Allergy Clin Immunol Pract. 2019;7(2):633-640. doi:10.1016/j.jaip.2018.08.013
4. Sasidharanpillai S, Govindan A, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): a histopathology based analysis. Indian J Dermatol Venereol Leprol. 2016;82(1):28. doi:10.4103/0378-6323.168934
5. Kardaun SH, Sekula P, Valeyrie‐Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169(5):1071-1080. doi:10.1111/bjd.12501
6. Koomdee N, Pratoomwun J, Jantararoungtong T, et al. Association of HLA-A and HLA-B alleles with lamotrigine-induced cutaneous adverse drug reactions in the Thai population. Front Pharmacol. 2017;8. doi:10.3389/fphar.2017.00879
7. Yang C-W, Cho Y-T, Hsieh Y-C, Hsu S-H, Chen K-L, Chu C-Y. The interferon-γ-induced protein 10/CXCR3 axis is associated with human herpesvirus-6 reactivation and the development of sequelae in drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2020;183(5):909-919. doi:10.1111/bjd.18942
8. Ruffilli I, Ferrari SM, Colaci M, Ferri C, Fallahi P, Antonelli A. IP-10 in autoimmune thyroiditis. Horm Metab Res. 2014;46(9):597-602. doi:10.1055/s-0034-1382053
9. Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42(3):276-282. doi:10.1111/1346-8138.12770
10. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124(7):588-597. doi:10.1016/j.amjmed.2011.01.017
11. Bommersbach TJ, Lapid MI, Leung JG, Cunningham JL, Rummans TA, Kung S. Management of psychotropic drug-induced dress syndrome: a systematic review. Mayo Clin Proc. 2016;91(6):787-801. doi:10.1016/j.mayocp.2016.03.006
12. Nguyen E, Yanes D, Imadojemu S, Kroshinsky D. Evaluation of cyclosporine for the treatment of DRESS syndrome. JAMA Dermatol. 2020;156(6):704-706. doi:10.1001/jamadermatol.2020.0048
13. Joseph RM, Hunter AL, Ray DW, Dixon WG. Systemic glucocorticoid therapy and adrenal insufficiency in adults: a systematic review. Semin Arthritis Rheum. 2016;46(1):133-141. doi:10.1016/j.semarthrit.2016.03.001
14. Jamilloux Y, Liozon E, Pugnet G, et al. Recovery of adrenal function after long-term glucocorticoid therapy for giant cell arteritis: a cohort study. PLoS ONE. 2013;8(7):e68713. doi:10.1371/journal.pone.0068713
Unique Treatment for Alopecia Areata Combining Epinephrine With an Intralesional Steroid
Alopecia areata (AA) is an autoimmune disorder characterized by transient hair loss with preservation of the hair follicle (HF). The lifetime incidence risk of AA is approximately 2%,1 with a mean age of onset of 25 to 36 years and with no clinically relevant significant differences between sex or ethnicity.2 Most commonly, it presents as round, well-demarcated patches of alopecia on the scalp and spontaneously resolves in nearly 30% of patients. However, severe disease is associated with younger age of presentation and can progress to a total loss of scalp or body hair—referred to as alopecia totalis and alopecia universalis, respectively—thus severely impacting quality of life.3,4
First-line treatment options for AA include potent topical steroids5,6 and intralesional (IL) steroids, most commonly IL triamcinolone acetonide (ILTA). Intralesional steroids have been found to be more effective than topicals in stimulating hair growth at the injection site.7,8 A recent systemic therapy—the Janus kinase inhibitor baricitinib—was approved by the US Food and Drug Administration for AA.9 Other systemic therapies such as oral corticosteroids have been studied in small trials with promising results.10 However, the risks of systemic therapies may outweigh the benefits.9,10
Another less common topical therapy is contact immunotherapy, which involves topical application of an unlicensed non–pharmaceutical-grade agent to areas affected with AA. It is reported to have a wide range of response rates (29%–87%).11
We report 2 cases of extensive AA that were treated with a novel combination regimen— 2.5 mg/mL of ILTA diluted with lidocaine 1% and epinephrine 1:100,000 in place of normal saline (NS)— which is a modification to an already widely used first-line treatment.
Case Reports
Patient 1—An 11-year-old girl presented with nonscarring alopecia of the vertex and occipital scalp. Three years prior she was treated with topical and IL corticosteroids by a different provider. Physical examination revealed almost complete alopecia involving the bottom two-thirds of the occipital scalp as well as the medial eyebrows (Figures 1A and 1B). Over the span of 1 year she was treated with betamethasone dipropionate cream 0.05% and several rounds of ILTA 2.5 mg/mL buffered with NS, with minimal improvement. A year after the initial presentation, the decision was made to initiate monthly injections of ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000. Some hair regrowth of the occipital scalp was noted by 3 months, with near-complete regrowth of the scalp hair and eyebrows by 7 months and 5 months, respectively (Figures 1C and 1D). During this period, the patient continued to develop new areas of alopecia of the scalp and eyebrows, which also were injected with this combination. In total, the patient received 8 rounds of IL injections 4 to 6 weeks apart in the scalp and 6 rounds in the eyebrows. The treated areas showed resolution over a follow-up period of 14 months, though there was recurrence at the right medial eyebrow at 5 months. No localized skin atrophy or other adverse effects were noted.

Patient 2—A 34-year-old woman who was otherwise healthy presented with previously untreated AA involving the scalp of 2 months’ duration. Physical examination revealed the following areas of nonscarring alopecia: a 10×10-cm area of the right occipital scalp with some regrowth; a 10×14-cm area of the left parieto-occipital scalp; and a 1-cm area posterior to the vertex (Figure 2A). Given the extensive involvement, the decision was made to initiate ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000 once monthly. Appreciable hair regrowth was noted within 1 month, mostly on the parietal scalp. Substantial improvement was noted after 3 months in all affected areas of the hair-bearing scalp, with near-complete regrowth on the left occipital scalp and greater than 50% regrowth on the right occipital scalp (Figure 2B). No adverse effects were noted. She currently has no alopecia.

Comment
Alopecia Pathogenesis—The most widely adopted theory of AA etiology implicates an aberrant immune response. The HF, which is a dynamic “mini-organ” with its own immune and hormonal microenvironment, is considered an “immune-privileged site”—meaning it is less exposed to immune responses than most other body areas. It is hypothesized that AA results from a breakdown in this immune privilege, with the subsequent attack on the peribulbar part of the follicle by CD8+ T lymphocytes. This lymphocytic infiltrate induces apoptosis in the HF keratinocytes, resulting in inhibition of hair shaft production.12 Other theories suggest a link to the sympathetic-adrenal-medullary system and hypothalamic-pituitary-adrenal axis.13
Therapies for Alopecia—Topical and IL corticosteroids are the first-line therapies for localized AA in patients with less than 50% scalp involvement. Triamcinolone acetonide generally is the IL steroid of choice because it is widely available and less atrophogenic than other steroids. Unlike topicals, ILTA bypasses the epidermis when injected, achieving direct access to the HF.14
High-quality controlled studies regarding the use of ILTA in AA are scarce. A meta-analysis concluded that 5 mg/mL and 10 mg/mL of ILTA diluted in NS were equally effective (80.9% [P<.05] vs 76.4% [P<.005], respectively). Concentrations of less than 5 mg/mL of ILTA resulted in lower rates of hair regrowth (62.3%; P=.04).15 The role of diluents other than NS has not been studied.
Benefits of Epinephrine in ILTA Therapy—The role of epinephrine 1:100,000 is to decrease the rate of clearance of triamcinolone acetonide from the HF, allowing for a better therapeutic effect. Laser Doppler blood flowmeter studies have shown that epinephrine 1:100,000 injected in the scalp causes vasoconstriction, thereby decreasing the blood flow rate of clearance of other substances in the same solution.16 Also, a more gradual systemic absorption is achieved, decreasing systemic side effects such as osteoporosis.17
Another potential benefit of epinephrine has been suggested in animal studies that demonstrate the important role of the sympathetic nervous system in HF growth. In a mouse study, chemical sympathectomy led to diminished norepinephrine levels in the skin, accompanied by a decreased keratinocyte proliferation and hair growth. Conversely, norepinephrine was found to promote HF growth in an organotypic skin culture model.18 Topically applied isoproterenol, a panadrenergic receptor agonist, accelerated HF growth in an organotypic skin culture. It also has been shown that external light and temperature changes stimulate hair growth via the sympathetic nervous system, promoting anagen HF growth in cultured skin explants, further linking HF activity with sympathetic nerve activity.19
In our experience, cases of AA that at first failed ILTA 5 mg/mL in NS have been successfully treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000. One such case was alopecia totalis, though we do not have high-quality photographs to present for this report. The 2 cases presented here are the ones with the best photographs to demonstrate our outcomes. Both were treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000 administered using a 0.5-in long 30-gauge needle, with 0.05 to 0.1 mL per injection approximately 0.51-cm apart. The treatment intervals were 4 weeks, with a maximal dose of 20 mg per session. In addition to the 2 cases reported here, the Table includes 2 other patients in our practice who were successfully treated with this novel regimen.

Prior to adopting this combination regimen, our standard therapy for AA was 5 mg/mL ILTA buffered with NS. Instead of NS, we now use the widely available 1% lidocaine with epinephrine 1:100,000 and dilute the ILTA to 2.5 mg/mL. We postulate that epinephrine 1:100,000 enhances therapeutic efficacy via local vasoconstriction, thus keeping the ILTA in situ longer than NS. This effect allows for a lower concentration of ILTA (2.5 mg/mL) to be effective. Furthermore, epinephrine 1:100,000 may have an independent effect, as suggested in mouse studies.18
Our first case demonstrated the ophiasis subtype of AA (symmetric bandlike hair loss), which has a poorer prognosis and is less responsive to therapy.20 In this patient, prior treatment with topical corticosteroids and ILTA in NS failed to induce a response. After a series of injections with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000, she entered remission. Our second case is one of alopecia subtotalis, which responded quickly, and the patient entered remission after just 3 months of treatment. These 2 cases are illustrative of the results that we regularly get and have come to expect with this treatment.
Conclusion
Our novel modified regimen of 2.5 mg/mL ILTA diluted with 1% lidocaine and epinephrine 1:100,000 has yielded a series of excellent outcomes in many of our most challenging AA cases without any untoward effects. Two cases are presented here. Higher-powered studies are needed to validate this new yet simple approach. A split-scalp or split-lesion study comparing ILTA with and without epinephrine 1:100,000 would be warranted for further investigation.
- Mirzoyev SA, Schrum AG, Davis MDP, et al. Lifetime incidence risk of alopecia areata estimated at 2.1 percent by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397-403.
- Tosti A, Bellavista S, Iorizzo M. Alopecia areata: a long term follow-up study of 191 patients. J Am Acad Dermatol. 2006;55:438-441.
- Walker SA, Rothman S. A statistical study and consideration of endocrine influences. J Invest Dermatol. 1950;14:403-413.
- Charuwichitratana S, Wattanakrai P, Tanrattanakorn S. Randomized double-blind placebo-controlled trial in the treatment of alopecia areata with 0.25% desoximetasone cream. Arch Dermatol. 2000;136:1276-1277.
- Tosti A, Iorizzo M, Botta GL, et al. Efficacy and safety of a new clobetasol propionate 0.05% foam in alopecia areata: a randomized, double-blind placebo-controlled trial. J Eur Acad Dermatol Venereol. 2006;20:1243-1247.
- Kubeyinje EP. Intralesional triamcinolone acetonide in alopecia areata amongst 62 Saudi Arabs. East Afr Med J. 1994;71:674-675.
- Porter D, Burton JL. A comparison of intra-lesional triamcinolonehexacetonide and triamcinolone acetonide in alopecia areata. Br J Dermatol. 1971;85:272-273.
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056/NEJMoa2110343
- Lai VWY, Chen G, Gin D, et al. Systemic treatments for alopeciaareata: a systematic review. Australas J Dermatol. 2019;60:E1-E13. doi:10.1111/ajd.12913
- Rokhsar CK, Shupack JL, Vafai JJ, et al. Efficacy of topical sensitizers in the treatment of alopecia areata. J Am Acad Dermatol. 1998;39:751-761.
- Dainichi T, Kabashima K. Alopecia areata: what’s new in epidemiology, pathogenesis, diagnosis, and therapeutic options? J Dermatol Sci. 2017;86:3-12.
- Ito T. Recent advances in the pathogenesis of autoimmune hair loss disease alopecia areata. Clin Dev Immunol. 2013;2013:348546.
- Ramos PM, Anzai A, Duque-Estrada B, et al. Consensus on the treatment of alopecia areata—Brazilian Society of Dermatology. An Bras Dermatol. 2020;95(suppl 1):39-52.
- Yee BE, Tong Y, Goldenberg A, et al. Efficacy of different concentrations of intralesional triamcinolone acetonide for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1018-1021.
- Na YC, Park R, Jeong HS, et al. Epinephrine vasoconstriction effect time in the scalp differs according to injection site and concentration. Dermatol Surg. 2016;42:1054-1060.
- Samrao A, Fu JM, Harris ST, et al. Bone mineral density in patients with alopecia areata treated with long-term intralesional corticosteroids. J Drugs Dermatol. 2013;12:E36-E40.
- Kong Y, Liu Y, Pan L, et al. Norepinephrine regulates keratinocyte proliferation to promote the growth of hair follicles. Cells Tissues Organs. 2015-2016;201:423-435.
- Fan SM, Chang YT, Chen CL, et al. External light activates hair follicle stem cells through eyes via an ipRGC-SCN-sympathetic neural pathway. Proc Natl Acad Sci U S A. 2018;115:E6880-E6889. Erratum appears in Proc Natl Acad Sci U S A. 2018;115:E12121.
- Spano F, Donovan JC. Alopecia areata: part 1: pathogenesis, diagnosis, and prognosis. Can Fam Physician. 2015;61:751-755.
Alopecia areata (AA) is an autoimmune disorder characterized by transient hair loss with preservation of the hair follicle (HF). The lifetime incidence risk of AA is approximately 2%,1 with a mean age of onset of 25 to 36 years and with no clinically relevant significant differences between sex or ethnicity.2 Most commonly, it presents as round, well-demarcated patches of alopecia on the scalp and spontaneously resolves in nearly 30% of patients. However, severe disease is associated with younger age of presentation and can progress to a total loss of scalp or body hair—referred to as alopecia totalis and alopecia universalis, respectively—thus severely impacting quality of life.3,4
First-line treatment options for AA include potent topical steroids5,6 and intralesional (IL) steroids, most commonly IL triamcinolone acetonide (ILTA). Intralesional steroids have been found to be more effective than topicals in stimulating hair growth at the injection site.7,8 A recent systemic therapy—the Janus kinase inhibitor baricitinib—was approved by the US Food and Drug Administration for AA.9 Other systemic therapies such as oral corticosteroids have been studied in small trials with promising results.10 However, the risks of systemic therapies may outweigh the benefits.9,10
Another less common topical therapy is contact immunotherapy, which involves topical application of an unlicensed non–pharmaceutical-grade agent to areas affected with AA. It is reported to have a wide range of response rates (29%–87%).11
We report 2 cases of extensive AA that were treated with a novel combination regimen— 2.5 mg/mL of ILTA diluted with lidocaine 1% and epinephrine 1:100,000 in place of normal saline (NS)— which is a modification to an already widely used first-line treatment.
Case Reports
Patient 1—An 11-year-old girl presented with nonscarring alopecia of the vertex and occipital scalp. Three years prior she was treated with topical and IL corticosteroids by a different provider. Physical examination revealed almost complete alopecia involving the bottom two-thirds of the occipital scalp as well as the medial eyebrows (Figures 1A and 1B). Over the span of 1 year she was treated with betamethasone dipropionate cream 0.05% and several rounds of ILTA 2.5 mg/mL buffered with NS, with minimal improvement. A year after the initial presentation, the decision was made to initiate monthly injections of ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000. Some hair regrowth of the occipital scalp was noted by 3 months, with near-complete regrowth of the scalp hair and eyebrows by 7 months and 5 months, respectively (Figures 1C and 1D). During this period, the patient continued to develop new areas of alopecia of the scalp and eyebrows, which also were injected with this combination. In total, the patient received 8 rounds of IL injections 4 to 6 weeks apart in the scalp and 6 rounds in the eyebrows. The treated areas showed resolution over a follow-up period of 14 months, though there was recurrence at the right medial eyebrow at 5 months. No localized skin atrophy or other adverse effects were noted.
Patient 2—A 34-year-old woman who was otherwise healthy presented with previously untreated AA involving the scalp of 2 months’ duration. Physical examination revealed the following areas of nonscarring alopecia: a 10×10-cm area of the right occipital scalp with some regrowth; a 10×14-cm area of the left parieto-occipital scalp; and a 1-cm area posterior to the vertex (Figure 2A). Given the extensive involvement, the decision was made to initiate ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000 once monthly. Appreciable hair regrowth was noted within 1 month, mostly on the parietal scalp. Substantial improvement was noted after 3 months in all affected areas of the hair-bearing scalp, with near-complete regrowth on the left occipital scalp and greater than 50% regrowth on the right occipital scalp (Figure 2B). No adverse effects were noted. She currently has no alopecia.
Comment
Alopecia Pathogenesis—The most widely adopted theory of AA etiology implicates an aberrant immune response. The HF, which is a dynamic “mini-organ” with its own immune and hormonal microenvironment, is considered an “immune-privileged site”—meaning it is less exposed to immune responses than most other body areas. It is hypothesized that AA results from a breakdown in this immune privilege, with the subsequent attack on the peribulbar part of the follicle by CD8+ T lymphocytes. This lymphocytic infiltrate induces apoptosis in the HF keratinocytes, resulting in inhibition of hair shaft production.12 Other theories suggest a link to the sympathetic-adrenal-medullary system and hypothalamic-pituitary-adrenal axis.13
Therapies for Alopecia—Topical and IL corticosteroids are the first-line therapies for localized AA in patients with less than 50% scalp involvement. Triamcinolone acetonide generally is the IL steroid of choice because it is widely available and less atrophogenic than other steroids. Unlike topicals, ILTA bypasses the epidermis when injected, achieving direct access to the HF.14
High-quality controlled studies regarding the use of ILTA in AA are scarce. A meta-analysis concluded that 5 mg/mL and 10 mg/mL of ILTA diluted in NS were equally effective (80.9% [P<.05] vs 76.4% [P<.005], respectively). Concentrations of less than 5 mg/mL of ILTA resulted in lower rates of hair regrowth (62.3%; P=.04).15 The role of diluents other than NS has not been studied.
Benefits of Epinephrine in ILTA Therapy—The role of epinephrine 1:100,000 is to decrease the rate of clearance of triamcinolone acetonide from the HF, allowing for a better therapeutic effect. Laser Doppler blood flowmeter studies have shown that epinephrine 1:100,000 injected in the scalp causes vasoconstriction, thereby decreasing the blood flow rate of clearance of other substances in the same solution.16 Also, a more gradual systemic absorption is achieved, decreasing systemic side effects such as osteoporosis.17
Another potential benefit of epinephrine has been suggested in animal studies that demonstrate the important role of the sympathetic nervous system in HF growth. In a mouse study, chemical sympathectomy led to diminished norepinephrine levels in the skin, accompanied by a decreased keratinocyte proliferation and hair growth. Conversely, norepinephrine was found to promote HF growth in an organotypic skin culture model.18 Topically applied isoproterenol, a panadrenergic receptor agonist, accelerated HF growth in an organotypic skin culture. It also has been shown that external light and temperature changes stimulate hair growth via the sympathetic nervous system, promoting anagen HF growth in cultured skin explants, further linking HF activity with sympathetic nerve activity.19
In our experience, cases of AA that at first failed ILTA 5 mg/mL in NS have been successfully treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000. One such case was alopecia totalis, though we do not have high-quality photographs to present for this report. The 2 cases presented here are the ones with the best photographs to demonstrate our outcomes. Both were treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000 administered using a 0.5-in long 30-gauge needle, with 0.05 to 0.1 mL per injection approximately 0.51-cm apart. The treatment intervals were 4 weeks, with a maximal dose of 20 mg per session. In addition to the 2 cases reported here, the Table includes 2 other patients in our practice who were successfully treated with this novel regimen.
Prior to adopting this combination regimen, our standard therapy for AA was 5 mg/mL ILTA buffered with NS. Instead of NS, we now use the widely available 1% lidocaine with epinephrine 1:100,000 and dilute the ILTA to 2.5 mg/mL. We postulate that epinephrine 1:100,000 enhances therapeutic efficacy via local vasoconstriction, thus keeping the ILTA in situ longer than NS. This effect allows for a lower concentration of ILTA (2.5 mg/mL) to be effective. Furthermore, epinephrine 1:100,000 may have an independent effect, as suggested in mouse studies.18
Our first case demonstrated the ophiasis subtype of AA (symmetric bandlike hair loss), which has a poorer prognosis and is less responsive to therapy.20 In this patient, prior treatment with topical corticosteroids and ILTA in NS failed to induce a response. After a series of injections with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000, she entered remission. Our second case is one of alopecia subtotalis, which responded quickly, and the patient entered remission after just 3 months of treatment. These 2 cases are illustrative of the results that we regularly get and have come to expect with this treatment.
Conclusion
Our novel modified regimen of 2.5 mg/mL ILTA diluted with 1% lidocaine and epinephrine 1:100,000 has yielded a series of excellent outcomes in many of our most challenging AA cases without any untoward effects. Two cases are presented here. Higher-powered studies are needed to validate this new yet simple approach. A split-scalp or split-lesion study comparing ILTA with and without epinephrine 1:100,000 would be warranted for further investigation.
Alopecia areata (AA) is an autoimmune disorder characterized by transient hair loss with preservation of the hair follicle (HF). The lifetime incidence risk of AA is approximately 2%,1 with a mean age of onset of 25 to 36 years and with no clinically relevant significant differences between sex or ethnicity.2 Most commonly, it presents as round, well-demarcated patches of alopecia on the scalp and spontaneously resolves in nearly 30% of patients. However, severe disease is associated with younger age of presentation and can progress to a total loss of scalp or body hair—referred to as alopecia totalis and alopecia universalis, respectively—thus severely impacting quality of life.3,4
First-line treatment options for AA include potent topical steroids5,6 and intralesional (IL) steroids, most commonly IL triamcinolone acetonide (ILTA). Intralesional steroids have been found to be more effective than topicals in stimulating hair growth at the injection site.7,8 A recent systemic therapy—the Janus kinase inhibitor baricitinib—was approved by the US Food and Drug Administration for AA.9 Other systemic therapies such as oral corticosteroids have been studied in small trials with promising results.10 However, the risks of systemic therapies may outweigh the benefits.9,10
Another less common topical therapy is contact immunotherapy, which involves topical application of an unlicensed non–pharmaceutical-grade agent to areas affected with AA. It is reported to have a wide range of response rates (29%–87%).11
We report 2 cases of extensive AA that were treated with a novel combination regimen— 2.5 mg/mL of ILTA diluted with lidocaine 1% and epinephrine 1:100,000 in place of normal saline (NS)— which is a modification to an already widely used first-line treatment.
Case Reports
Patient 1—An 11-year-old girl presented with nonscarring alopecia of the vertex and occipital scalp. Three years prior she was treated with topical and IL corticosteroids by a different provider. Physical examination revealed almost complete alopecia involving the bottom two-thirds of the occipital scalp as well as the medial eyebrows (Figures 1A and 1B). Over the span of 1 year she was treated with betamethasone dipropionate cream 0.05% and several rounds of ILTA 2.5 mg/mL buffered with NS, with minimal improvement. A year after the initial presentation, the decision was made to initiate monthly injections of ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000. Some hair regrowth of the occipital scalp was noted by 3 months, with near-complete regrowth of the scalp hair and eyebrows by 7 months and 5 months, respectively (Figures 1C and 1D). During this period, the patient continued to develop new areas of alopecia of the scalp and eyebrows, which also were injected with this combination. In total, the patient received 8 rounds of IL injections 4 to 6 weeks apart in the scalp and 6 rounds in the eyebrows. The treated areas showed resolution over a follow-up period of 14 months, though there was recurrence at the right medial eyebrow at 5 months. No localized skin atrophy or other adverse effects were noted.
Patient 2—A 34-year-old woman who was otherwise healthy presented with previously untreated AA involving the scalp of 2 months’ duration. Physical examination revealed the following areas of nonscarring alopecia: a 10×10-cm area of the right occipital scalp with some regrowth; a 10×14-cm area of the left parieto-occipital scalp; and a 1-cm area posterior to the vertex (Figure 2A). Given the extensive involvement, the decision was made to initiate ILTA 2.5 mg/mL buffered with 1% lidocaine and epinephrine 1:100,000 once monthly. Appreciable hair regrowth was noted within 1 month, mostly on the parietal scalp. Substantial improvement was noted after 3 months in all affected areas of the hair-bearing scalp, with near-complete regrowth on the left occipital scalp and greater than 50% regrowth on the right occipital scalp (Figure 2B). No adverse effects were noted. She currently has no alopecia.
Comment
Alopecia Pathogenesis—The most widely adopted theory of AA etiology implicates an aberrant immune response. The HF, which is a dynamic “mini-organ” with its own immune and hormonal microenvironment, is considered an “immune-privileged site”—meaning it is less exposed to immune responses than most other body areas. It is hypothesized that AA results from a breakdown in this immune privilege, with the subsequent attack on the peribulbar part of the follicle by CD8+ T lymphocytes. This lymphocytic infiltrate induces apoptosis in the HF keratinocytes, resulting in inhibition of hair shaft production.12 Other theories suggest a link to the sympathetic-adrenal-medullary system and hypothalamic-pituitary-adrenal axis.13
Therapies for Alopecia—Topical and IL corticosteroids are the first-line therapies for localized AA in patients with less than 50% scalp involvement. Triamcinolone acetonide generally is the IL steroid of choice because it is widely available and less atrophogenic than other steroids. Unlike topicals, ILTA bypasses the epidermis when injected, achieving direct access to the HF.14
High-quality controlled studies regarding the use of ILTA in AA are scarce. A meta-analysis concluded that 5 mg/mL and 10 mg/mL of ILTA diluted in NS were equally effective (80.9% [P<.05] vs 76.4% [P<.005], respectively). Concentrations of less than 5 mg/mL of ILTA resulted in lower rates of hair regrowth (62.3%; P=.04).15 The role of diluents other than NS has not been studied.
Benefits of Epinephrine in ILTA Therapy—The role of epinephrine 1:100,000 is to decrease the rate of clearance of triamcinolone acetonide from the HF, allowing for a better therapeutic effect. Laser Doppler blood flowmeter studies have shown that epinephrine 1:100,000 injected in the scalp causes vasoconstriction, thereby decreasing the blood flow rate of clearance of other substances in the same solution.16 Also, a more gradual systemic absorption is achieved, decreasing systemic side effects such as osteoporosis.17
Another potential benefit of epinephrine has been suggested in animal studies that demonstrate the important role of the sympathetic nervous system in HF growth. In a mouse study, chemical sympathectomy led to diminished norepinephrine levels in the skin, accompanied by a decreased keratinocyte proliferation and hair growth. Conversely, norepinephrine was found to promote HF growth in an organotypic skin culture model.18 Topically applied isoproterenol, a panadrenergic receptor agonist, accelerated HF growth in an organotypic skin culture. It also has been shown that external light and temperature changes stimulate hair growth via the sympathetic nervous system, promoting anagen HF growth in cultured skin explants, further linking HF activity with sympathetic nerve activity.19
In our experience, cases of AA that at first failed ILTA 5 mg/mL in NS have been successfully treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000. One such case was alopecia totalis, though we do not have high-quality photographs to present for this report. The 2 cases presented here are the ones with the best photographs to demonstrate our outcomes. Both were treated with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000 administered using a 0.5-in long 30-gauge needle, with 0.05 to 0.1 mL per injection approximately 0.51-cm apart. The treatment intervals were 4 weeks, with a maximal dose of 20 mg per session. In addition to the 2 cases reported here, the Table includes 2 other patients in our practice who were successfully treated with this novel regimen.
Prior to adopting this combination regimen, our standard therapy for AA was 5 mg/mL ILTA buffered with NS. Instead of NS, we now use the widely available 1% lidocaine with epinephrine 1:100,000 and dilute the ILTA to 2.5 mg/mL. We postulate that epinephrine 1:100,000 enhances therapeutic efficacy via local vasoconstriction, thus keeping the ILTA in situ longer than NS. This effect allows for a lower concentration of ILTA (2.5 mg/mL) to be effective. Furthermore, epinephrine 1:100,000 may have an independent effect, as suggested in mouse studies.18
Our first case demonstrated the ophiasis subtype of AA (symmetric bandlike hair loss), which has a poorer prognosis and is less responsive to therapy.20 In this patient, prior treatment with topical corticosteroids and ILTA in NS failed to induce a response. After a series of injections with 2.5 mg/mL ILTA in 1% lidocaine and epinephrine 1:100,000, she entered remission. Our second case is one of alopecia subtotalis, which responded quickly, and the patient entered remission after just 3 months of treatment. These 2 cases are illustrative of the results that we regularly get and have come to expect with this treatment.
Conclusion
Our novel modified regimen of 2.5 mg/mL ILTA diluted with 1% lidocaine and epinephrine 1:100,000 has yielded a series of excellent outcomes in many of our most challenging AA cases without any untoward effects. Two cases are presented here. Higher-powered studies are needed to validate this new yet simple approach. A split-scalp or split-lesion study comparing ILTA with and without epinephrine 1:100,000 would be warranted for further investigation.
- Mirzoyev SA, Schrum AG, Davis MDP, et al. Lifetime incidence risk of alopecia areata estimated at 2.1 percent by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397-403.
- Tosti A, Bellavista S, Iorizzo M. Alopecia areata: a long term follow-up study of 191 patients. J Am Acad Dermatol. 2006;55:438-441.
- Walker SA, Rothman S. A statistical study and consideration of endocrine influences. J Invest Dermatol. 1950;14:403-413.
- Charuwichitratana S, Wattanakrai P, Tanrattanakorn S. Randomized double-blind placebo-controlled trial in the treatment of alopecia areata with 0.25% desoximetasone cream. Arch Dermatol. 2000;136:1276-1277.
- Tosti A, Iorizzo M, Botta GL, et al. Efficacy and safety of a new clobetasol propionate 0.05% foam in alopecia areata: a randomized, double-blind placebo-controlled trial. J Eur Acad Dermatol Venereol. 2006;20:1243-1247.
- Kubeyinje EP. Intralesional triamcinolone acetonide in alopecia areata amongst 62 Saudi Arabs. East Afr Med J. 1994;71:674-675.
- Porter D, Burton JL. A comparison of intra-lesional triamcinolonehexacetonide and triamcinolone acetonide in alopecia areata. Br J Dermatol. 1971;85:272-273.
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056/NEJMoa2110343
- Lai VWY, Chen G, Gin D, et al. Systemic treatments for alopeciaareata: a systematic review. Australas J Dermatol. 2019;60:E1-E13. doi:10.1111/ajd.12913
- Rokhsar CK, Shupack JL, Vafai JJ, et al. Efficacy of topical sensitizers in the treatment of alopecia areata. J Am Acad Dermatol. 1998;39:751-761.
- Dainichi T, Kabashima K. Alopecia areata: what’s new in epidemiology, pathogenesis, diagnosis, and therapeutic options? J Dermatol Sci. 2017;86:3-12.
- Ito T. Recent advances in the pathogenesis of autoimmune hair loss disease alopecia areata. Clin Dev Immunol. 2013;2013:348546.
- Ramos PM, Anzai A, Duque-Estrada B, et al. Consensus on the treatment of alopecia areata—Brazilian Society of Dermatology. An Bras Dermatol. 2020;95(suppl 1):39-52.
- Yee BE, Tong Y, Goldenberg A, et al. Efficacy of different concentrations of intralesional triamcinolone acetonide for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1018-1021.
- Na YC, Park R, Jeong HS, et al. Epinephrine vasoconstriction effect time in the scalp differs according to injection site and concentration. Dermatol Surg. 2016;42:1054-1060.
- Samrao A, Fu JM, Harris ST, et al. Bone mineral density in patients with alopecia areata treated with long-term intralesional corticosteroids. J Drugs Dermatol. 2013;12:E36-E40.
- Kong Y, Liu Y, Pan L, et al. Norepinephrine regulates keratinocyte proliferation to promote the growth of hair follicles. Cells Tissues Organs. 2015-2016;201:423-435.
- Fan SM, Chang YT, Chen CL, et al. External light activates hair follicle stem cells through eyes via an ipRGC-SCN-sympathetic neural pathway. Proc Natl Acad Sci U S A. 2018;115:E6880-E6889. Erratum appears in Proc Natl Acad Sci U S A. 2018;115:E12121.
- Spano F, Donovan JC. Alopecia areata: part 1: pathogenesis, diagnosis, and prognosis. Can Fam Physician. 2015;61:751-755.
- Mirzoyev SA, Schrum AG, Davis MDP, et al. Lifetime incidence risk of alopecia areata estimated at 2.1 percent by Rochester Epidemiology Project, 1990-2009. J Invest Dermatol. 2014;134:1141-1142.
- Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397-403.
- Tosti A, Bellavista S, Iorizzo M. Alopecia areata: a long term follow-up study of 191 patients. J Am Acad Dermatol. 2006;55:438-441.
- Walker SA, Rothman S. A statistical study and consideration of endocrine influences. J Invest Dermatol. 1950;14:403-413.
- Charuwichitratana S, Wattanakrai P, Tanrattanakorn S. Randomized double-blind placebo-controlled trial in the treatment of alopecia areata with 0.25% desoximetasone cream. Arch Dermatol. 2000;136:1276-1277.
- Tosti A, Iorizzo M, Botta GL, et al. Efficacy and safety of a new clobetasol propionate 0.05% foam in alopecia areata: a randomized, double-blind placebo-controlled trial. J Eur Acad Dermatol Venereol. 2006;20:1243-1247.
- Kubeyinje EP. Intralesional triamcinolone acetonide in alopecia areata amongst 62 Saudi Arabs. East Afr Med J. 1994;71:674-675.
- Porter D, Burton JL. A comparison of intra-lesional triamcinolonehexacetonide and triamcinolone acetonide in alopecia areata. Br J Dermatol. 1971;85:272-273.
- King B, Ohyama M, Kwon O, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687-1699. doi:10.1056/NEJMoa2110343
- Lai VWY, Chen G, Gin D, et al. Systemic treatments for alopeciaareata: a systematic review. Australas J Dermatol. 2019;60:E1-E13. doi:10.1111/ajd.12913
- Rokhsar CK, Shupack JL, Vafai JJ, et al. Efficacy of topical sensitizers in the treatment of alopecia areata. J Am Acad Dermatol. 1998;39:751-761.
- Dainichi T, Kabashima K. Alopecia areata: what’s new in epidemiology, pathogenesis, diagnosis, and therapeutic options? J Dermatol Sci. 2017;86:3-12.
- Ito T. Recent advances in the pathogenesis of autoimmune hair loss disease alopecia areata. Clin Dev Immunol. 2013;2013:348546.
- Ramos PM, Anzai A, Duque-Estrada B, et al. Consensus on the treatment of alopecia areata—Brazilian Society of Dermatology. An Bras Dermatol. 2020;95(suppl 1):39-52.
- Yee BE, Tong Y, Goldenberg A, et al. Efficacy of different concentrations of intralesional triamcinolone acetonide for alopecia areata: a systematic review and meta-analysis. J Am Acad Dermatol. 2020;82:1018-1021.
- Na YC, Park R, Jeong HS, et al. Epinephrine vasoconstriction effect time in the scalp differs according to injection site and concentration. Dermatol Surg. 2016;42:1054-1060.
- Samrao A, Fu JM, Harris ST, et al. Bone mineral density in patients with alopecia areata treated with long-term intralesional corticosteroids. J Drugs Dermatol. 2013;12:E36-E40.
- Kong Y, Liu Y, Pan L, et al. Norepinephrine regulates keratinocyte proliferation to promote the growth of hair follicles. Cells Tissues Organs. 2015-2016;201:423-435.
- Fan SM, Chang YT, Chen CL, et al. External light activates hair follicle stem cells through eyes via an ipRGC-SCN-sympathetic neural pathway. Proc Natl Acad Sci U S A. 2018;115:E6880-E6889. Erratum appears in Proc Natl Acad Sci U S A. 2018;115:E12121.
- Spano F, Donovan JC. Alopecia areata: part 1: pathogenesis, diagnosis, and prognosis. Can Fam Physician. 2015;61:751-755.
Practice Points
- Patients with alopecia areata that is refractory to first-line treatments may benefit from intralesional triamcinolone acetonide (ILTA) diluted to 2.5 mg/mL in 1% lidocaine and epinephrine 1:100,000 in place of normal saline.
- Local vasoconstriction due to epinephrine may potentiate ILTA effects and play an independent role.
