User login
Fever, tachycardia, and tachypnea during a psychotic exacerbation
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.

Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.

EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
Delirious after undergoing workup for stroke
CASE Altered mental status after stroke workup
Ms. L, age 91, is admitted to the hospital for a neurologic evaluation of a recent episode of left-sided weakness that occurred 1 week ago. This left-sided weakness resolved without intervention within 2 hours while at home. This presentation is typical of a transient ischemic attack (TIA). She has a history of hypertension, bradycardia, and pacemaker implantation. On initial evaluation, her memory is intact, and she is able to walk normally. Her score on the St. Louis University Mental Status (SLUMS) exam is 25, which suggests normal cognitive functioning for her academic background. A CT scan of the head reveals a subacute stroke of the right posterior limb of the internal capsule consistent with recent TIA.
Ms. L is admitted for a routine stroke workup and prepares to undergo a CT angiogram (CTA) with the use of the iodinated agent iopamidol (100 mL, 76%) to evaluate patency of cerebral vessels. Her baseline blood urea nitrogen (BUN) and creatinine levels are within normal limits.
A day after undergoing CTA, Ms. L starts mumbling to herself, has unpredictable mood outbursts, and is not oriented to time, place, or person.
[polldaddy:10199351]
The authors’ observations
Due to her acute altered mental status (AMS), Ms. L underwent an emergent CT scan of the head to rule out any acute intracranial hemorrhages or thromboembolic events. The results of this test were negative. Urinalysis, BUN, creatinine, basic chemistry, and complete blood count panels were unrevealing. On a repeat SLUMS exam, Ms. L scored 9, indicating cognitive impairment.
Ms. L also underwent a comprehensive metabolic profile, which excluded any electrolyte abnormalities, or any hepatic or renal causes of AMS. There was no sign of dehydration, acidosis, hypoglycemia, hypoxemia, hypotension, or bradycardia/tachycardia. A urinalysis, chest X-ray, complete blood count, and 2 blood cultures conducted 24 hours apart did not reveal any signs of infection. There were no recent changes in her medications and she was not taking any sleep medications or other psychiatric medications that might precipitate a withdrawal syndrome.
There have been multiple reports of contrast-induced nephropathy (CIN), which may be evidenced by high BUN-to-creatinine ratios and could cause AMS in geriatric patients. However, CIN was ruled out as a potential cause in our patient because her BUN-to-creatinine was unremarkable.
Continue to: Routine EEG was clinically...
Routine EEG was clinically inconclusive. Diffusion-weighted MRI may have been helpful to identify ischemic strokes that a CT scan of the head might miss,1 but we were unable to conduct this test because Ms. L had a pacemaker. Barber et al2 suggested that in the setting of acute stroke, the use of MRI may not have an added advantage over the CT scan of the head.
[polldaddy:10199352]
TREATMENT Rapid improvement with supportive therapy
Intravenous fluids are administered as supportive therapy to Ms. L for suspected contrast-induced encephalopathy (CIE). The next day, Ms. L experiences a notable improvement in cognition, beyond that attributed to IV hydration. By 3 days post-contrast injection, her SLUMS score increases to 15. By 72 hours after contrast administration, Ms. L’s cognition returns to baseline. She is monitored for 24 hours after returning to baseline cognitive functioning. After observing her to be in no physical or medical distress and at baseline functioning, she is discharged home under the care of her son with outpatient follow-up and rehab services.
The authors’ observations
For Ms. L, the differential diagnosis included post-ictal phenomenon, new-onset ischemic or hemorrhagic changes, hyperperfusion syndrome, and CIE.
Seizures were ruled out because EEG was inconclusive, and Ms. L did not have the clinical features one would expect in an ictal episode. Transient ischemic attack is, by definition, an ischemic event with clinical return to baseline within 24 hours. Although a CT scan of the head may not be the most sensitive way to detect early ischemic changes and small ischemic zones, the self-limiting course and complete resolution of Ms. L’s symptoms with return to baseline is indicative of a more benign pathology, such as CIE. New hemorrhagic conversions have a dramatic presentation on radiologic studies. Historically, CIE presentations on imaging have been closely associated with the hyperattentuation seen in subarachnoid hemorrhage (SAH). The absence of typical radiologic and clinical findings in our case ruled out SAH.
Continue to: Typical CT scan findings in CIE include...
Typical CT scan findings in CIE include abnormal cortical contrast enhancement and edema, subarachnoid contrast enhancement, and striatal contrast enhancement (Figure 1, Figure 2, and Figure 3). Since the first clinical description, reports of 39 CT-/MRI-confirmed cases of CIE have been published in English language medical literature, with documented clinical follow-up3 and a median recovery time of 2.5 days. In a case report by Ito et al,4 there were no supportive radiographic findings. Ours is the second documented case that showed no radiologic signs of CIE. With a paucity of other etiologic evidence, negative lab tests for other causes of delirium, and the rapid resolution of Ms. L’s AMS after providing IV fluids as supportive treatment, a temporal correlation can be deduced, which implicates iodine-based contrast as the inciting factor.

Iodine-based contrast agents have been used since the 1920s. Today, >75 million procedures requiring iodine dyes are performed annually worldwide.5 This level of routine iodine contrast usage compels a mention of risk factors and complications from using such dyes. As a general rule, contrast agent reactions can be categorized as immediate (<1 day) or delayed (1 to 7 days after contrast administration). Immediate reactions are immunoglobulin E (IgE)-mediated anaphylactic reactions. Delayed reactions involve a T-cell mediated response that ranges from pruritus and urticaria (approximately 70%) to cardiac complications such as cardiovascular shock, arrhythmia, arrest, and Kounis syndrome. Other less prevalent complications include hypotension, bronchospasm, and CIN. Patients with the following factors may be at higher risk for contrast-induced reactions:
- asthma
- cardiac arrhythmias
- central myasthenia gravis
- >70 years of age
- pheochromocytoma
- sickle cell anemia
- hyperthyroidism
- dehydration
- hypotension.
Although some older literature reported correlations between seafood and shellfish allergies and iodine contrast reactions, more recent reports suggest there may not be a direct correlation, or any correlation at all.5,6

Iodinated CIE is a rare complication of contrast angiography. It was first reported in 1970 as transient cortical blindness after coronary angiography.7 Clinical manifestations include encephalopathy evidenced by AMS, affected orientation, and acute psychotic changes, including paranoia and hallucinations, seizures, cortical blindness, and focal neurologic deficits. Neuroimaging has been pivotal in confirming the diagnosis and in excluding thromboembolic and hemorrhagic complications of angiography.8
Encephalopathy has been documented after administration of

Continue to: Regardless of the mechanism...
Regardless of the mechanism, all the above-mentioned studies note a reversal of radiologic and neurologic findings without any deficits within 48 to 72 hours (median recovery time of 2.5 days).3 All reported cases of CIE, including ours, were found to be completely reversible without any neurologic or radiologic deficits after resolution (48 to 72 hours post-contrast administration).
Clinicians should have a high index of suspicion for CIE in patients with recent iodine-based contrast exposure. From a practical standpoint, such a mechanism could be easily missed because while use of a single-administration contrast agent may appear in procedure notes or medication administration records, it might not necessarily appear in documentation of currently administered medications. Also, such cases might not always present with unique radiologic findings, as illustrated by Ms. L’s case.
Bottom Line
Have a high index of suspicion for contrast-induced encephalopathy, especially in geriatric patients, even in the absence of radiologic findings. A full delirium/dementia workup is warranted to rule out other life-threatening causes of altered mental status. Timely recognition could enable implementation of medicationsparing approaches to the disorder, such as IV fluids and frequent reorientation.
Related Resources
- Donepudi B, Trottier S. A seizure and hemiplegia following contrast exposure: Understanding contrast-induced encephalopathy. Case Rep Med. 2018;2018:9278526. doi:10.1155/2018/9278526.
- Hamra M, Bakhit Y, Khan M, et al. Case report and literature review on contrast-induced encephalopathy. Future Cardiol. 2017;13(4):331-335.
Drug Brand Names
Iohexol • Omnipaque
Iopamidol • Isovue-370
Iopromide • Ultravist
Ioxilan • Oxilan
1. Moreau F, Asdaghi N, Modi J, et al. Magnetic resonance imaging versus computed tomography in transient ischemic attack and minor stroke: the more you see the more you know. Cerebrovasc Dis Extra. 2013;3(1):130-136.
2. Barber PA, Hill MD, Eliasziw M, et al. Imaging of the brain in acute ischaemic stroke: comparison of computed tomography and magnetic resonance diffusion-weighted imaging. J Neurol Neurosurg Psychiatry. 2005;76(11):1528-1533.
3. Leong S, Fanning NF. Persistent neurological deficit from iodinated contrast encephalopathy following intracranial aneurysm coiling: a case report and review of the literature. Interv Neuroradiol. 2012;18(1):33-41.
4. Ito N, Nishio R, Ozuki T, et al. A state of delirium (confusion) following cerebral angiography with ioxilan: a case report. Nihon Igaku Hoshasen Gakkai Zasshi. 2002; 62(7):370-371.
5. Bottinor W, Polkampally P, Jovin I. Adverse reactions to iodinated contrast media. Int J Angiol. 2013;22:149-154.
6. Cohan R. AHRQ Patient Safety Network Reaction to Dye. US Department of Health and Human Services Agency for Healthcare Research and Quality. https://psnet.ahrq.gov/webmm/case/75/reaction-to-dye. Published September 2004. Accessed March 5, 2017.
7. Fischer-Williams M, Gottschalk PG, Browell JN. Transient cortical blindness: an unusual complication of coronary angiography. Neurology. 1970;20(4):353-355.
8. Lantos G. Cortical blindness due to osmotic disruption of the blood-brain barrier by angiographic contrast material: CT and MRI studies. Neurology. 1989;39(4):567-571.
9. Kocabay G, Karabay CY. Iopromide-induced encephalopathy following coronary angioplasty. Perfusion. 2011;26:67-70.
10. Dangas G, Monsein LH, Laureno R, et al. Transient contrast encephalopathy after carotid artery stenting. Journal of Endovascular Therapy. 2001;8:111-113.
11. Sawaya RA, Hammoud R, Arnaout SJ, et al. Contrast induced encephalopathy following coronary angioplasty with iohexol. Southern Medical Journal. 2007;100(10):1054-1055.
CASE Altered mental status after stroke workup
Ms. L, age 91, is admitted to the hospital for a neurologic evaluation of a recent episode of left-sided weakness that occurred 1 week ago. This left-sided weakness resolved without intervention within 2 hours while at home. This presentation is typical of a transient ischemic attack (TIA). She has a history of hypertension, bradycardia, and pacemaker implantation. On initial evaluation, her memory is intact, and she is able to walk normally. Her score on the St. Louis University Mental Status (SLUMS) exam is 25, which suggests normal cognitive functioning for her academic background. A CT scan of the head reveals a subacute stroke of the right posterior limb of the internal capsule consistent with recent TIA.
Ms. L is admitted for a routine stroke workup and prepares to undergo a CT angiogram (CTA) with the use of the iodinated agent iopamidol (100 mL, 76%) to evaluate patency of cerebral vessels. Her baseline blood urea nitrogen (BUN) and creatinine levels are within normal limits.
A day after undergoing CTA, Ms. L starts mumbling to herself, has unpredictable mood outbursts, and is not oriented to time, place, or person.
[polldaddy:10199351]
The authors’ observations
Due to her acute altered mental status (AMS), Ms. L underwent an emergent CT scan of the head to rule out any acute intracranial hemorrhages or thromboembolic events. The results of this test were negative. Urinalysis, BUN, creatinine, basic chemistry, and complete blood count panels were unrevealing. On a repeat SLUMS exam, Ms. L scored 9, indicating cognitive impairment.
Ms. L also underwent a comprehensive metabolic profile, which excluded any electrolyte abnormalities, or any hepatic or renal causes of AMS. There was no sign of dehydration, acidosis, hypoglycemia, hypoxemia, hypotension, or bradycardia/tachycardia. A urinalysis, chest X-ray, complete blood count, and 2 blood cultures conducted 24 hours apart did not reveal any signs of infection. There were no recent changes in her medications and she was not taking any sleep medications or other psychiatric medications that might precipitate a withdrawal syndrome.
There have been multiple reports of contrast-induced nephropathy (CIN), which may be evidenced by high BUN-to-creatinine ratios and could cause AMS in geriatric patients. However, CIN was ruled out as a potential cause in our patient because her BUN-to-creatinine was unremarkable.
Continue to: Routine EEG was clinically...
Routine EEG was clinically inconclusive. Diffusion-weighted MRI may have been helpful to identify ischemic strokes that a CT scan of the head might miss,1 but we were unable to conduct this test because Ms. L had a pacemaker. Barber et al2 suggested that in the setting of acute stroke, the use of MRI may not have an added advantage over the CT scan of the head.
[polldaddy:10199352]
TREATMENT Rapid improvement with supportive therapy
Intravenous fluids are administered as supportive therapy to Ms. L for suspected contrast-induced encephalopathy (CIE). The next day, Ms. L experiences a notable improvement in cognition, beyond that attributed to IV hydration. By 3 days post-contrast injection, her SLUMS score increases to 15. By 72 hours after contrast administration, Ms. L’s cognition returns to baseline. She is monitored for 24 hours after returning to baseline cognitive functioning. After observing her to be in no physical or medical distress and at baseline functioning, she is discharged home under the care of her son with outpatient follow-up and rehab services.
The authors’ observations
For Ms. L, the differential diagnosis included post-ictal phenomenon, new-onset ischemic or hemorrhagic changes, hyperperfusion syndrome, and CIE.
Seizures were ruled out because EEG was inconclusive, and Ms. L did not have the clinical features one would expect in an ictal episode. Transient ischemic attack is, by definition, an ischemic event with clinical return to baseline within 24 hours. Although a CT scan of the head may not be the most sensitive way to detect early ischemic changes and small ischemic zones, the self-limiting course and complete resolution of Ms. L’s symptoms with return to baseline is indicative of a more benign pathology, such as CIE. New hemorrhagic conversions have a dramatic presentation on radiologic studies. Historically, CIE presentations on imaging have been closely associated with the hyperattentuation seen in subarachnoid hemorrhage (SAH). The absence of typical radiologic and clinical findings in our case ruled out SAH.
Continue to: Typical CT scan findings in CIE include...
Typical CT scan findings in CIE include abnormal cortical contrast enhancement and edema, subarachnoid contrast enhancement, and striatal contrast enhancement (Figure 1, Figure 2, and Figure 3). Since the first clinical description, reports of 39 CT-/MRI-confirmed cases of CIE have been published in English language medical literature, with documented clinical follow-up3 and a median recovery time of 2.5 days. In a case report by Ito et al,4 there were no supportive radiographic findings. Ours is the second documented case that showed no radiologic signs of CIE. With a paucity of other etiologic evidence, negative lab tests for other causes of delirium, and the rapid resolution of Ms. L’s AMS after providing IV fluids as supportive treatment, a temporal correlation can be deduced, which implicates iodine-based contrast as the inciting factor.
Iodine-based contrast agents have been used since the 1920s. Today, >75 million procedures requiring iodine dyes are performed annually worldwide.5 This level of routine iodine contrast usage compels a mention of risk factors and complications from using such dyes. As a general rule, contrast agent reactions can be categorized as immediate (<1 day) or delayed (1 to 7 days after contrast administration). Immediate reactions are immunoglobulin E (IgE)-mediated anaphylactic reactions. Delayed reactions involve a T-cell mediated response that ranges from pruritus and urticaria (approximately 70%) to cardiac complications such as cardiovascular shock, arrhythmia, arrest, and Kounis syndrome. Other less prevalent complications include hypotension, bronchospasm, and CIN. Patients with the following factors may be at higher risk for contrast-induced reactions:
- asthma
- cardiac arrhythmias
- central myasthenia gravis
- >70 years of age
- pheochromocytoma
- sickle cell anemia
- hyperthyroidism
- dehydration
- hypotension.
Although some older literature reported correlations between seafood and shellfish allergies and iodine contrast reactions, more recent reports suggest there may not be a direct correlation, or any correlation at all.5,6
Iodinated CIE is a rare complication of contrast angiography. It was first reported in 1970 as transient cortical blindness after coronary angiography.7 Clinical manifestations include encephalopathy evidenced by AMS, affected orientation, and acute psychotic changes, including paranoia and hallucinations, seizures, cortical blindness, and focal neurologic deficits. Neuroimaging has been pivotal in confirming the diagnosis and in excluding thromboembolic and hemorrhagic complications of angiography.8
Encephalopathy has been documented after administration of
Continue to: Regardless of the mechanism...
Regardless of the mechanism, all the above-mentioned studies note a reversal of radiologic and neurologic findings without any deficits within 48 to 72 hours (median recovery time of 2.5 days).3 All reported cases of CIE, including ours, were found to be completely reversible without any neurologic or radiologic deficits after resolution (48 to 72 hours post-contrast administration).
Clinicians should have a high index of suspicion for CIE in patients with recent iodine-based contrast exposure. From a practical standpoint, such a mechanism could be easily missed because while use of a single-administration contrast agent may appear in procedure notes or medication administration records, it might not necessarily appear in documentation of currently administered medications. Also, such cases might not always present with unique radiologic findings, as illustrated by Ms. L’s case.
Bottom Line
Have a high index of suspicion for contrast-induced encephalopathy, especially in geriatric patients, even in the absence of radiologic findings. A full delirium/dementia workup is warranted to rule out other life-threatening causes of altered mental status. Timely recognition could enable implementation of medicationsparing approaches to the disorder, such as IV fluids and frequent reorientation.
Related Resources
- Donepudi B, Trottier S. A seizure and hemiplegia following contrast exposure: Understanding contrast-induced encephalopathy. Case Rep Med. 2018;2018:9278526. doi:10.1155/2018/9278526.
- Hamra M, Bakhit Y, Khan M, et al. Case report and literature review on contrast-induced encephalopathy. Future Cardiol. 2017;13(4):331-335.
Drug Brand Names
Iohexol • Omnipaque
Iopamidol • Isovue-370
Iopromide • Ultravist
Ioxilan • Oxilan
CASE Altered mental status after stroke workup
Ms. L, age 91, is admitted to the hospital for a neurologic evaluation of a recent episode of left-sided weakness that occurred 1 week ago. This left-sided weakness resolved without intervention within 2 hours while at home. This presentation is typical of a transient ischemic attack (TIA). She has a history of hypertension, bradycardia, and pacemaker implantation. On initial evaluation, her memory is intact, and she is able to walk normally. Her score on the St. Louis University Mental Status (SLUMS) exam is 25, which suggests normal cognitive functioning for her academic background. A CT scan of the head reveals a subacute stroke of the right posterior limb of the internal capsule consistent with recent TIA.
Ms. L is admitted for a routine stroke workup and prepares to undergo a CT angiogram (CTA) with the use of the iodinated agent iopamidol (100 mL, 76%) to evaluate patency of cerebral vessels. Her baseline blood urea nitrogen (BUN) and creatinine levels are within normal limits.
A day after undergoing CTA, Ms. L starts mumbling to herself, has unpredictable mood outbursts, and is not oriented to time, place, or person.
[polldaddy:10199351]
The authors’ observations
Due to her acute altered mental status (AMS), Ms. L underwent an emergent CT scan of the head to rule out any acute intracranial hemorrhages or thromboembolic events. The results of this test were negative. Urinalysis, BUN, creatinine, basic chemistry, and complete blood count panels were unrevealing. On a repeat SLUMS exam, Ms. L scored 9, indicating cognitive impairment.
Ms. L also underwent a comprehensive metabolic profile, which excluded any electrolyte abnormalities, or any hepatic or renal causes of AMS. There was no sign of dehydration, acidosis, hypoglycemia, hypoxemia, hypotension, or bradycardia/tachycardia. A urinalysis, chest X-ray, complete blood count, and 2 blood cultures conducted 24 hours apart did not reveal any signs of infection. There were no recent changes in her medications and she was not taking any sleep medications or other psychiatric medications that might precipitate a withdrawal syndrome.
There have been multiple reports of contrast-induced nephropathy (CIN), which may be evidenced by high BUN-to-creatinine ratios and could cause AMS in geriatric patients. However, CIN was ruled out as a potential cause in our patient because her BUN-to-creatinine was unremarkable.
Continue to: Routine EEG was clinically...
Routine EEG was clinically inconclusive. Diffusion-weighted MRI may have been helpful to identify ischemic strokes that a CT scan of the head might miss,1 but we were unable to conduct this test because Ms. L had a pacemaker. Barber et al2 suggested that in the setting of acute stroke, the use of MRI may not have an added advantage over the CT scan of the head.
[polldaddy:10199352]
TREATMENT Rapid improvement with supportive therapy
Intravenous fluids are administered as supportive therapy to Ms. L for suspected contrast-induced encephalopathy (CIE). The next day, Ms. L experiences a notable improvement in cognition, beyond that attributed to IV hydration. By 3 days post-contrast injection, her SLUMS score increases to 15. By 72 hours after contrast administration, Ms. L’s cognition returns to baseline. She is monitored for 24 hours after returning to baseline cognitive functioning. After observing her to be in no physical or medical distress and at baseline functioning, she is discharged home under the care of her son with outpatient follow-up and rehab services.
The authors’ observations
For Ms. L, the differential diagnosis included post-ictal phenomenon, new-onset ischemic or hemorrhagic changes, hyperperfusion syndrome, and CIE.
Seizures were ruled out because EEG was inconclusive, and Ms. L did not have the clinical features one would expect in an ictal episode. Transient ischemic attack is, by definition, an ischemic event with clinical return to baseline within 24 hours. Although a CT scan of the head may not be the most sensitive way to detect early ischemic changes and small ischemic zones, the self-limiting course and complete resolution of Ms. L’s symptoms with return to baseline is indicative of a more benign pathology, such as CIE. New hemorrhagic conversions have a dramatic presentation on radiologic studies. Historically, CIE presentations on imaging have been closely associated with the hyperattentuation seen in subarachnoid hemorrhage (SAH). The absence of typical radiologic and clinical findings in our case ruled out SAH.
Continue to: Typical CT scan findings in CIE include...
Typical CT scan findings in CIE include abnormal cortical contrast enhancement and edema, subarachnoid contrast enhancement, and striatal contrast enhancement (Figure 1, Figure 2, and Figure 3). Since the first clinical description, reports of 39 CT-/MRI-confirmed cases of CIE have been published in English language medical literature, with documented clinical follow-up3 and a median recovery time of 2.5 days. In a case report by Ito et al,4 there were no supportive radiographic findings. Ours is the second documented case that showed no radiologic signs of CIE. With a paucity of other etiologic evidence, negative lab tests for other causes of delirium, and the rapid resolution of Ms. L’s AMS after providing IV fluids as supportive treatment, a temporal correlation can be deduced, which implicates iodine-based contrast as the inciting factor.
Iodine-based contrast agents have been used since the 1920s. Today, >75 million procedures requiring iodine dyes are performed annually worldwide.5 This level of routine iodine contrast usage compels a mention of risk factors and complications from using such dyes. As a general rule, contrast agent reactions can be categorized as immediate (<1 day) or delayed (1 to 7 days after contrast administration). Immediate reactions are immunoglobulin E (IgE)-mediated anaphylactic reactions. Delayed reactions involve a T-cell mediated response that ranges from pruritus and urticaria (approximately 70%) to cardiac complications such as cardiovascular shock, arrhythmia, arrest, and Kounis syndrome. Other less prevalent complications include hypotension, bronchospasm, and CIN. Patients with the following factors may be at higher risk for contrast-induced reactions:
- asthma
- cardiac arrhythmias
- central myasthenia gravis
- >70 years of age
- pheochromocytoma
- sickle cell anemia
- hyperthyroidism
- dehydration
- hypotension.
Although some older literature reported correlations between seafood and shellfish allergies and iodine contrast reactions, more recent reports suggest there may not be a direct correlation, or any correlation at all.5,6
Iodinated CIE is a rare complication of contrast angiography. It was first reported in 1970 as transient cortical blindness after coronary angiography.7 Clinical manifestations include encephalopathy evidenced by AMS, affected orientation, and acute psychotic changes, including paranoia and hallucinations, seizures, cortical blindness, and focal neurologic deficits. Neuroimaging has been pivotal in confirming the diagnosis and in excluding thromboembolic and hemorrhagic complications of angiography.8
Encephalopathy has been documented after administration of
Continue to: Regardless of the mechanism...
Regardless of the mechanism, all the above-mentioned studies note a reversal of radiologic and neurologic findings without any deficits within 48 to 72 hours (median recovery time of 2.5 days).3 All reported cases of CIE, including ours, were found to be completely reversible without any neurologic or radiologic deficits after resolution (48 to 72 hours post-contrast administration).
Clinicians should have a high index of suspicion for CIE in patients with recent iodine-based contrast exposure. From a practical standpoint, such a mechanism could be easily missed because while use of a single-administration contrast agent may appear in procedure notes or medication administration records, it might not necessarily appear in documentation of currently administered medications. Also, such cases might not always present with unique radiologic findings, as illustrated by Ms. L’s case.
Bottom Line
Have a high index of suspicion for contrast-induced encephalopathy, especially in geriatric patients, even in the absence of radiologic findings. A full delirium/dementia workup is warranted to rule out other life-threatening causes of altered mental status. Timely recognition could enable implementation of medicationsparing approaches to the disorder, such as IV fluids and frequent reorientation.
Related Resources
- Donepudi B, Trottier S. A seizure and hemiplegia following contrast exposure: Understanding contrast-induced encephalopathy. Case Rep Med. 2018;2018:9278526. doi:10.1155/2018/9278526.
- Hamra M, Bakhit Y, Khan M, et al. Case report and literature review on contrast-induced encephalopathy. Future Cardiol. 2017;13(4):331-335.
Drug Brand Names
Iohexol • Omnipaque
Iopamidol • Isovue-370
Iopromide • Ultravist
Ioxilan • Oxilan
1. Moreau F, Asdaghi N, Modi J, et al. Magnetic resonance imaging versus computed tomography in transient ischemic attack and minor stroke: the more you see the more you know. Cerebrovasc Dis Extra. 2013;3(1):130-136.
2. Barber PA, Hill MD, Eliasziw M, et al. Imaging of the brain in acute ischaemic stroke: comparison of computed tomography and magnetic resonance diffusion-weighted imaging. J Neurol Neurosurg Psychiatry. 2005;76(11):1528-1533.
3. Leong S, Fanning NF. Persistent neurological deficit from iodinated contrast encephalopathy following intracranial aneurysm coiling: a case report and review of the literature. Interv Neuroradiol. 2012;18(1):33-41.
4. Ito N, Nishio R, Ozuki T, et al. A state of delirium (confusion) following cerebral angiography with ioxilan: a case report. Nihon Igaku Hoshasen Gakkai Zasshi. 2002; 62(7):370-371.
5. Bottinor W, Polkampally P, Jovin I. Adverse reactions to iodinated contrast media. Int J Angiol. 2013;22:149-154.
6. Cohan R. AHRQ Patient Safety Network Reaction to Dye. US Department of Health and Human Services Agency for Healthcare Research and Quality. https://psnet.ahrq.gov/webmm/case/75/reaction-to-dye. Published September 2004. Accessed March 5, 2017.
7. Fischer-Williams M, Gottschalk PG, Browell JN. Transient cortical blindness: an unusual complication of coronary angiography. Neurology. 1970;20(4):353-355.
8. Lantos G. Cortical blindness due to osmotic disruption of the blood-brain barrier by angiographic contrast material: CT and MRI studies. Neurology. 1989;39(4):567-571.
9. Kocabay G, Karabay CY. Iopromide-induced encephalopathy following coronary angioplasty. Perfusion. 2011;26:67-70.
10. Dangas G, Monsein LH, Laureno R, et al. Transient contrast encephalopathy after carotid artery stenting. Journal of Endovascular Therapy. 2001;8:111-113.
11. Sawaya RA, Hammoud R, Arnaout SJ, et al. Contrast induced encephalopathy following coronary angioplasty with iohexol. Southern Medical Journal. 2007;100(10):1054-1055.
1. Moreau F, Asdaghi N, Modi J, et al. Magnetic resonance imaging versus computed tomography in transient ischemic attack and minor stroke: the more you see the more you know. Cerebrovasc Dis Extra. 2013;3(1):130-136.
2. Barber PA, Hill MD, Eliasziw M, et al. Imaging of the brain in acute ischaemic stroke: comparison of computed tomography and magnetic resonance diffusion-weighted imaging. J Neurol Neurosurg Psychiatry. 2005;76(11):1528-1533.
3. Leong S, Fanning NF. Persistent neurological deficit from iodinated contrast encephalopathy following intracranial aneurysm coiling: a case report and review of the literature. Interv Neuroradiol. 2012;18(1):33-41.
4. Ito N, Nishio R, Ozuki T, et al. A state of delirium (confusion) following cerebral angiography with ioxilan: a case report. Nihon Igaku Hoshasen Gakkai Zasshi. 2002; 62(7):370-371.
5. Bottinor W, Polkampally P, Jovin I. Adverse reactions to iodinated contrast media. Int J Angiol. 2013;22:149-154.
6. Cohan R. AHRQ Patient Safety Network Reaction to Dye. US Department of Health and Human Services Agency for Healthcare Research and Quality. https://psnet.ahrq.gov/webmm/case/75/reaction-to-dye. Published September 2004. Accessed March 5, 2017.
7. Fischer-Williams M, Gottschalk PG, Browell JN. Transient cortical blindness: an unusual complication of coronary angiography. Neurology. 1970;20(4):353-355.
8. Lantos G. Cortical blindness due to osmotic disruption of the blood-brain barrier by angiographic contrast material: CT and MRI studies. Neurology. 1989;39(4):567-571.
9. Kocabay G, Karabay CY. Iopromide-induced encephalopathy following coronary angioplasty. Perfusion. 2011;26:67-70.
10. Dangas G, Monsein LH, Laureno R, et al. Transient contrast encephalopathy after carotid artery stenting. Journal of Endovascular Therapy. 2001;8:111-113.
11. Sawaya RA, Hammoud R, Arnaout SJ, et al. Contrast induced encephalopathy following coronary angioplasty with iohexol. Southern Medical Journal. 2007;100(10):1054-1055.
A mood disorder complicated by multiple sclerosis
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.

Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
The teenager who couldn’t stay awake
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
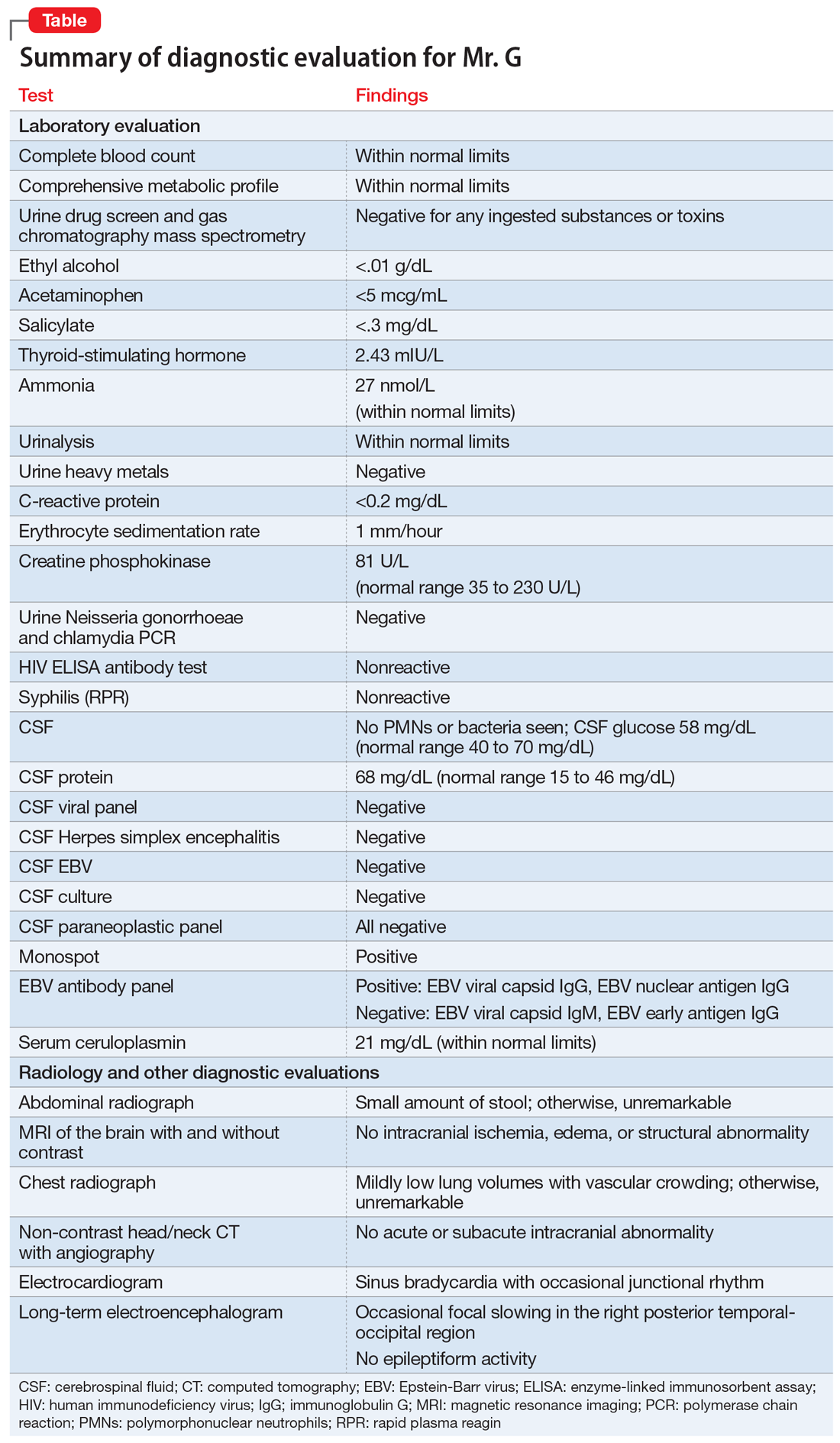
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3

[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
Unrelenting depression: ‘I would rather be dead than feel this way’
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
Manic after having found a ‘cure’ for Alzheimer’s disease
CASE Reckless driving, impulse buying
Mr. A, age 73, is admitted to the inpatient psychiatric unit at a community hospital for evaluation of a psychotic episode. His admission to the unit was initiated by his primary care physician, who noted that Mr. A was “not making sense” during a routine visit. Mr. A was speaking rapidly about how he had discovered that high-dose omega-3 fatty acid supplements were a “cure” for Alzheimer’s disease. He also believes that he was recently appointed as CEO of Microsoft and Apple for his discoveries.
Three months earlier, Mr. A had started taking high doses of omega-3 fatty acid supplements (10 to 15 g/d) because he believed they were the cure for memory problems, pain, and depression. At that time, he discontinued taking nortriptyline, 25 mg/d, and citalopram, 40 mg/d, which his outpatient psychiatrist had prescribed for major depressive disorder (MDD). Mr. A also had stopped taking buprenorphine, 2 mg, sublingual, 4 times a day, which he had been prescribed for chronic pain.
Mr. A’s wife reports that during the last 2 months, her husband had become irritable, impulsive, grandiose, and was sleeping very little. She added that although her husband’s ophthalmologist had advised him to not drive due to impaired vision, he had been driving recklessly across the metropolitan area. He had also spent nearly $15,000 buying furniture and other items for their home.
In addition to MDD, Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain. He has been taking vitamin D3, 2,000 U/d, as a nutritional supplement.
[polldaddy:10091672]
The authors’ observations
Mr. A met the DSM-5 criteria for a manic episode (Table 11). His manic and delusional symptoms are new. He has a long-standing diagnosis of MDD, which for many years had been successfully treated with antidepressants without a manic switch. The absence of a manic switch when treated with antidepressants without a mood stabilizer suggested that Mr. A did not have bipolarity in terms of a mood disorder diathesis.2 In addition, it would be unusual for an individual to develop a new-onset or primary bipolar disorder after age 60. Individuals in this age group who present with manic symptoms for the first time are preponderantly found to have a general medical or iatrogenic cause for the emergence of these symptoms.3 Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain.

Typically a sedentary man, Mr. A had been exhibiting disinhibited behavior, grandiosity, insomnia, and psychosis. These symptoms began 3 months before he was admitted to the psychiatric unit, when he had started taking high doses of omega-3 fatty acid supplements.
Continue to: EVALUATION Persistent mania
EVALUATION Persistent mania
On initial examination, Mr. A is upset and irritable. He is casually dressed and well-groomed. He lacks insight and says he was brought to the hospital against his will, and it is his wife “who is the one who is crazy.” He is oriented to person, place, and time. At times he is found roaming the hallways, being intrusive, hyperverbal, and tangential with pressured speech. He is very difficult to redirect, and regularly interrupts the interview. His vital signs are stable. He walks well, with slow and steady gait, and displays no tremor or bradykinesia.
[polldaddy:10091674]
The authors’ observations
In order to rule out organic causes, a complete blood count, comprehensive metabolic panel, thyroid profile, urine drug screen, and brain MRI were ordered. No abnormalities were found. DHA and EPA levels were not measured because such testing was not available at the laboratory at the hospital.
Mania emerging after the sixth decade of life is a rare occurrence. Therefore, we made a substantial effort to try to find another cause that might explain Mr. A’s unusual presentation (Table 2).

Omega-3 fatty acid–induced mania. The major types of omega-3 polyunsaturated fatty acids are EPA and DHA and their precursor, alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish, such as salmon, and in fish oil supplements. Omega-3 fatty acids have beneficial anti-inflammatory, antioxidative, and neuroplastic effects.4 Having properties similar to selective serotonin reuptake inhibitors, omega-3 fatty acids are thought to help prevent depression, have few interactions with other medications, and have a lower adverse-effect burden than antidepressants. They have been found to be beneficial as a maintenance treatment and for prevention of depressive episodes in bipolar depression, but no positive association has been found for bipolar mania.5
Continue to: However, very limited evidence suggests...
However, very limited evidence suggests that omega-3 fatty acid supplements, particularly those with flaxseed oil, can induce hypomania or mania. This association was first reported by Rudin6 in 1981, and later reported in other studies.7 How omega-3 fatty acids might induce mania is unclear.
Mr. A was reportedly taking high doses of an omega-3 fatty acid supplement. We hypothesized that the antidepressant effect of this supplement may have precipitated a manic episode. Mr. A had no history of manic episodes in the past and was stable during the treatment with the outpatient psychiatrist. A first episode mania in the seventh decade of life would be highly unusual without an organic etiology. After laboratory tests found no abnormalities that would point to an organic etiology, iatrogenic causes were considered. After a review of the literature, there was anecdotal evidence for the induction of mania in clinical trials studying the effects of omega-3 supplements on affective disorders.
This led us to ask: How much omega-3 fatty acid supplements, if any, can a patient with a depressive or bipolar disorder safely take? Currently, omega-3 fatty acid supplements are not FDA-approved for the treatment of depression or bipolar disorder. However, patients may take 1.5 to 2 g/d for MDD. Further research is needed to determine the optimal dose. It is unclear at this time if omega-3 fatty acid supplementation has any benefit in the acute or maintenance treatment of bipolar disorder.

Alternative nutritional supplements for mood disorders. Traditionally, mood disorders, such as MDD and bipolar disorder, have been treated with psychotropic medications. However, through the years, sporadic studies have examined the efficacy of nutritional interventions as a cost-effective approach to preventing and treating these conditions.5 Proponents of this approach believe such supplements can increase efficacy, as well as decrease the required dose of psychotropic medications, thus potentially minimizing adverse effects. However, their overuse can pose a potential threat of toxicity or unexpected adverse effects, such as precipitation of mania. Table 38 lists over-the-counter nutritional and/or herbal agents that may cause mania.
Continue to: TREATMENT Nonadherence leads to a court order
TREATMENT Nonadherence leads to a court order
On admission, Mr. A receives a dose of
[polldaddy:10091676]
The authors’ observations
During an acute manic episode, the goal of treatment is urgent mood stabilization. Monotherapy can be used; however, in emergent settings, a combination is often used for a rapid response. The most commonly used agents are lithium, anticonvulsants such as valproic acid, and antipsychotics.9 In addition, benzodiazepines can be used for insomnia, agitation, or anxiety. The decision to use lithium, an anticonvulsant, or an antipsychotic depends upon the specific medication’s adverse effects, the patient’s medical history, previous medication trials, drug–drug interactions, patient preference, and cost.
Because Mr. A has a history of chronic kidney disease, lithium was contraindicated.
[polldaddy:10091678]
Continue to: The authors' observations
The authors’ observations
After the acute episode of mania resolves, maintenance pharmacotherapy typically involves continuing the same regimen that achieved mood stabilization. Monotherapy is typically preferred to combination therapy, but it is not always possible after a manic episode.10 A reasonable approach is to slowly taper the antipsychotic after several months of dual therapy if symptoms continue to be well-controlled. Further adjustments may be necessary, depending on the medications’ adverse effects. Moreover, further acute episodes of mania or depression will also determine future treatment.
OUTCOME Resolution of delusions
Mr. A is discharged 30 days after admission. At this point, his acute manic episode has resolved with non-tangential, non-pressured speech, improved sleep, and decreased impulsivity. His grandiose delusions also have resolved. He is prescribed valproic acid, 1,000 mg/d, and risperidone, 6 mg/d at bedtime, under the care of his outpatient psychiatrist.
Bottom Line
Initial presentation of a manic episode in an older patient is rare. It is important to rule out organic causes. Weak evidence suggests omega-3 fatty acid supplements may have the potential to induce mania in certain patients.
Related Resource
- Ramaswamy S, Driscoll D, Rodriguez A, et al. Nutraceuticals for traumatic brain injury: Should you recommend their use? Current Psychiatry. 2017;16(7):34-38,40,41-45.
Drug Brand Names
Buprenorphine • Suboxone, Subutex
Citalopram • Celexa
Hydrocodone/acetaminophen • Vicodin
Lithium • Eskalith, Lithobid
Lorazepam• Ativan
Nortriptyline • Pamelor
Risperidone • Risperdal
Valproic acid • Depakote
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Goodwin GM, Haddad PM, Ferrier IN, et al. Evidence-based guidelines for treating bipolar disorder: revised third edition recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2016;30(6):495-553.
3. Sami M, Khan H, Nilforooshan R. Late onset mania as an organic syndrome: a review of case reports in the literature. J Affect Disord. 2015:188:226-231.
4. Su KP, Matsuoka Y, Pae CU. Omega-3 polyunsaturated fatty acids in prevention of mood and anxiety disorders. Clin Psychopharmacol Neurosci. 2015;13(2):129-137.
5. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
6. Rudin DO. The major psychoses and neuroses as omega-3 essential fatty acid deficiency syndrome: substrate pellagra. Biol Psychiatry. 1981;16(9):837-850.
7. Su KP, Shen WW, Huang SY. Are omega3 fatty acids beneficial in depression but not mania? Arch Gen Psychiatry. 2000;57(7):716-717.
8. Joshi K, Faubion M. Mania and psychosis associated with St. John’s wort and ginseng. Psychiatry (Edgmont). 2005;2(9):56-61.
9. Grunze H, Vieta E, Goodwin GM, et al. The world federation of societies of biological psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry. 2009;10(2):85-116.
10. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166(4):476-488.
CASE Reckless driving, impulse buying
Mr. A, age 73, is admitted to the inpatient psychiatric unit at a community hospital for evaluation of a psychotic episode. His admission to the unit was initiated by his primary care physician, who noted that Mr. A was “not making sense” during a routine visit. Mr. A was speaking rapidly about how he had discovered that high-dose omega-3 fatty acid supplements were a “cure” for Alzheimer’s disease. He also believes that he was recently appointed as CEO of Microsoft and Apple for his discoveries.
Three months earlier, Mr. A had started taking high doses of omega-3 fatty acid supplements (10 to 15 g/d) because he believed they were the cure for memory problems, pain, and depression. At that time, he discontinued taking nortriptyline, 25 mg/d, and citalopram, 40 mg/d, which his outpatient psychiatrist had prescribed for major depressive disorder (MDD). Mr. A also had stopped taking buprenorphine, 2 mg, sublingual, 4 times a day, which he had been prescribed for chronic pain.
Mr. A’s wife reports that during the last 2 months, her husband had become irritable, impulsive, grandiose, and was sleeping very little. She added that although her husband’s ophthalmologist had advised him to not drive due to impaired vision, he had been driving recklessly across the metropolitan area. He had also spent nearly $15,000 buying furniture and other items for their home.
In addition to MDD, Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain. He has been taking vitamin D3, 2,000 U/d, as a nutritional supplement.
[polldaddy:10091672]
The authors’ observations
Mr. A met the DSM-5 criteria for a manic episode (Table 11). His manic and delusional symptoms are new. He has a long-standing diagnosis of MDD, which for many years had been successfully treated with antidepressants without a manic switch. The absence of a manic switch when treated with antidepressants without a mood stabilizer suggested that Mr. A did not have bipolarity in terms of a mood disorder diathesis.2 In addition, it would be unusual for an individual to develop a new-onset or primary bipolar disorder after age 60. Individuals in this age group who present with manic symptoms for the first time are preponderantly found to have a general medical or iatrogenic cause for the emergence of these symptoms.3 Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain.
Typically a sedentary man, Mr. A had been exhibiting disinhibited behavior, grandiosity, insomnia, and psychosis. These symptoms began 3 months before he was admitted to the psychiatric unit, when he had started taking high doses of omega-3 fatty acid supplements.
Continue to: EVALUATION Persistent mania
EVALUATION Persistent mania
On initial examination, Mr. A is upset and irritable. He is casually dressed and well-groomed. He lacks insight and says he was brought to the hospital against his will, and it is his wife “who is the one who is crazy.” He is oriented to person, place, and time. At times he is found roaming the hallways, being intrusive, hyperverbal, and tangential with pressured speech. He is very difficult to redirect, and regularly interrupts the interview. His vital signs are stable. He walks well, with slow and steady gait, and displays no tremor or bradykinesia.
[polldaddy:10091674]
The authors’ observations
In order to rule out organic causes, a complete blood count, comprehensive metabolic panel, thyroid profile, urine drug screen, and brain MRI were ordered. No abnormalities were found. DHA and EPA levels were not measured because such testing was not available at the laboratory at the hospital.
Mania emerging after the sixth decade of life is a rare occurrence. Therefore, we made a substantial effort to try to find another cause that might explain Mr. A’s unusual presentation (Table 2).
Omega-3 fatty acid–induced mania. The major types of omega-3 polyunsaturated fatty acids are EPA and DHA and their precursor, alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish, such as salmon, and in fish oil supplements. Omega-3 fatty acids have beneficial anti-inflammatory, antioxidative, and neuroplastic effects.4 Having properties similar to selective serotonin reuptake inhibitors, omega-3 fatty acids are thought to help prevent depression, have few interactions with other medications, and have a lower adverse-effect burden than antidepressants. They have been found to be beneficial as a maintenance treatment and for prevention of depressive episodes in bipolar depression, but no positive association has been found for bipolar mania.5
Continue to: However, very limited evidence suggests...
However, very limited evidence suggests that omega-3 fatty acid supplements, particularly those with flaxseed oil, can induce hypomania or mania. This association was first reported by Rudin6 in 1981, and later reported in other studies.7 How omega-3 fatty acids might induce mania is unclear.
Mr. A was reportedly taking high doses of an omega-3 fatty acid supplement. We hypothesized that the antidepressant effect of this supplement may have precipitated a manic episode. Mr. A had no history of manic episodes in the past and was stable during the treatment with the outpatient psychiatrist. A first episode mania in the seventh decade of life would be highly unusual without an organic etiology. After laboratory tests found no abnormalities that would point to an organic etiology, iatrogenic causes were considered. After a review of the literature, there was anecdotal evidence for the induction of mania in clinical trials studying the effects of omega-3 supplements on affective disorders.
This led us to ask: How much omega-3 fatty acid supplements, if any, can a patient with a depressive or bipolar disorder safely take? Currently, omega-3 fatty acid supplements are not FDA-approved for the treatment of depression or bipolar disorder. However, patients may take 1.5 to 2 g/d for MDD. Further research is needed to determine the optimal dose. It is unclear at this time if omega-3 fatty acid supplementation has any benefit in the acute or maintenance treatment of bipolar disorder.
Alternative nutritional supplements for mood disorders. Traditionally, mood disorders, such as MDD and bipolar disorder, have been treated with psychotropic medications. However, through the years, sporadic studies have examined the efficacy of nutritional interventions as a cost-effective approach to preventing and treating these conditions.5 Proponents of this approach believe such supplements can increase efficacy, as well as decrease the required dose of psychotropic medications, thus potentially minimizing adverse effects. However, their overuse can pose a potential threat of toxicity or unexpected adverse effects, such as precipitation of mania. Table 38 lists over-the-counter nutritional and/or herbal agents that may cause mania.
Continue to: TREATMENT Nonadherence leads to a court order
TREATMENT Nonadherence leads to a court order
On admission, Mr. A receives a dose of
[polldaddy:10091676]
The authors’ observations
During an acute manic episode, the goal of treatment is urgent mood stabilization. Monotherapy can be used; however, in emergent settings, a combination is often used for a rapid response. The most commonly used agents are lithium, anticonvulsants such as valproic acid, and antipsychotics.9 In addition, benzodiazepines can be used for insomnia, agitation, or anxiety. The decision to use lithium, an anticonvulsant, or an antipsychotic depends upon the specific medication’s adverse effects, the patient’s medical history, previous medication trials, drug–drug interactions, patient preference, and cost.
Because Mr. A has a history of chronic kidney disease, lithium was contraindicated.
[polldaddy:10091678]
Continue to: The authors' observations
The authors’ observations
After the acute episode of mania resolves, maintenance pharmacotherapy typically involves continuing the same regimen that achieved mood stabilization. Monotherapy is typically preferred to combination therapy, but it is not always possible after a manic episode.10 A reasonable approach is to slowly taper the antipsychotic after several months of dual therapy if symptoms continue to be well-controlled. Further adjustments may be necessary, depending on the medications’ adverse effects. Moreover, further acute episodes of mania or depression will also determine future treatment.
OUTCOME Resolution of delusions
Mr. A is discharged 30 days after admission. At this point, his acute manic episode has resolved with non-tangential, non-pressured speech, improved sleep, and decreased impulsivity. His grandiose delusions also have resolved. He is prescribed valproic acid, 1,000 mg/d, and risperidone, 6 mg/d at bedtime, under the care of his outpatient psychiatrist.
Bottom Line
Initial presentation of a manic episode in an older patient is rare. It is important to rule out organic causes. Weak evidence suggests omega-3 fatty acid supplements may have the potential to induce mania in certain patients.
Related Resource
- Ramaswamy S, Driscoll D, Rodriguez A, et al. Nutraceuticals for traumatic brain injury: Should you recommend their use? Current Psychiatry. 2017;16(7):34-38,40,41-45.
Drug Brand Names
Buprenorphine • Suboxone, Subutex
Citalopram • Celexa
Hydrocodone/acetaminophen • Vicodin
Lithium • Eskalith, Lithobid
Lorazepam• Ativan
Nortriptyline • Pamelor
Risperidone • Risperdal
Valproic acid • Depakote
CASE Reckless driving, impulse buying
Mr. A, age 73, is admitted to the inpatient psychiatric unit at a community hospital for evaluation of a psychotic episode. His admission to the unit was initiated by his primary care physician, who noted that Mr. A was “not making sense” during a routine visit. Mr. A was speaking rapidly about how he had discovered that high-dose omega-3 fatty acid supplements were a “cure” for Alzheimer’s disease. He also believes that he was recently appointed as CEO of Microsoft and Apple for his discoveries.
Three months earlier, Mr. A had started taking high doses of omega-3 fatty acid supplements (10 to 15 g/d) because he believed they were the cure for memory problems, pain, and depression. At that time, he discontinued taking nortriptyline, 25 mg/d, and citalopram, 40 mg/d, which his outpatient psychiatrist had prescribed for major depressive disorder (MDD). Mr. A also had stopped taking buprenorphine, 2 mg, sublingual, 4 times a day, which he had been prescribed for chronic pain.
Mr. A’s wife reports that during the last 2 months, her husband had become irritable, impulsive, grandiose, and was sleeping very little. She added that although her husband’s ophthalmologist had advised him to not drive due to impaired vision, he had been driving recklessly across the metropolitan area. He had also spent nearly $15,000 buying furniture and other items for their home.
In addition to MDD, Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain. He has been taking vitamin D3, 2,000 U/d, as a nutritional supplement.
[polldaddy:10091672]
The authors’ observations
Mr. A met the DSM-5 criteria for a manic episode (Table 11). His manic and delusional symptoms are new. He has a long-standing diagnosis of MDD, which for many years had been successfully treated with antidepressants without a manic switch. The absence of a manic switch when treated with antidepressants without a mood stabilizer suggested that Mr. A did not have bipolarity in terms of a mood disorder diathesis.2 In addition, it would be unusual for an individual to develop a new-onset or primary bipolar disorder after age 60. Individuals in this age group who present with manic symptoms for the first time are preponderantly found to have a general medical or iatrogenic cause for the emergence of these symptoms.3 Mr. A has a history of chronic kidney disease, Leber’s hereditary optic neuropathy, and chronic pain.
Typically a sedentary man, Mr. A had been exhibiting disinhibited behavior, grandiosity, insomnia, and psychosis. These symptoms began 3 months before he was admitted to the psychiatric unit, when he had started taking high doses of omega-3 fatty acid supplements.
Continue to: EVALUATION Persistent mania
EVALUATION Persistent mania
On initial examination, Mr. A is upset and irritable. He is casually dressed and well-groomed. He lacks insight and says he was brought to the hospital against his will, and it is his wife “who is the one who is crazy.” He is oriented to person, place, and time. At times he is found roaming the hallways, being intrusive, hyperverbal, and tangential with pressured speech. He is very difficult to redirect, and regularly interrupts the interview. His vital signs are stable. He walks well, with slow and steady gait, and displays no tremor or bradykinesia.
[polldaddy:10091674]
The authors’ observations
In order to rule out organic causes, a complete blood count, comprehensive metabolic panel, thyroid profile, urine drug screen, and brain MRI were ordered. No abnormalities were found. DHA and EPA levels were not measured because such testing was not available at the laboratory at the hospital.
Mania emerging after the sixth decade of life is a rare occurrence. Therefore, we made a substantial effort to try to find another cause that might explain Mr. A’s unusual presentation (Table 2).
Omega-3 fatty acid–induced mania. The major types of omega-3 polyunsaturated fatty acids are EPA and DHA and their precursor, alpha-linolenic acid (ALA). EPA and DHA are found primarily in fatty fish, such as salmon, and in fish oil supplements. Omega-3 fatty acids have beneficial anti-inflammatory, antioxidative, and neuroplastic effects.4 Having properties similar to selective serotonin reuptake inhibitors, omega-3 fatty acids are thought to help prevent depression, have few interactions with other medications, and have a lower adverse-effect burden than antidepressants. They have been found to be beneficial as a maintenance treatment and for prevention of depressive episodes in bipolar depression, but no positive association has been found for bipolar mania.5
Continue to: However, very limited evidence suggests...
However, very limited evidence suggests that omega-3 fatty acid supplements, particularly those with flaxseed oil, can induce hypomania or mania. This association was first reported by Rudin6 in 1981, and later reported in other studies.7 How omega-3 fatty acids might induce mania is unclear.
Mr. A was reportedly taking high doses of an omega-3 fatty acid supplement. We hypothesized that the antidepressant effect of this supplement may have precipitated a manic episode. Mr. A had no history of manic episodes in the past and was stable during the treatment with the outpatient psychiatrist. A first episode mania in the seventh decade of life would be highly unusual without an organic etiology. After laboratory tests found no abnormalities that would point to an organic etiology, iatrogenic causes were considered. After a review of the literature, there was anecdotal evidence for the induction of mania in clinical trials studying the effects of omega-3 supplements on affective disorders.
This led us to ask: How much omega-3 fatty acid supplements, if any, can a patient with a depressive or bipolar disorder safely take? Currently, omega-3 fatty acid supplements are not FDA-approved for the treatment of depression or bipolar disorder. However, patients may take 1.5 to 2 g/d for MDD. Further research is needed to determine the optimal dose. It is unclear at this time if omega-3 fatty acid supplementation has any benefit in the acute or maintenance treatment of bipolar disorder.
Alternative nutritional supplements for mood disorders. Traditionally, mood disorders, such as MDD and bipolar disorder, have been treated with psychotropic medications. However, through the years, sporadic studies have examined the efficacy of nutritional interventions as a cost-effective approach to preventing and treating these conditions.5 Proponents of this approach believe such supplements can increase efficacy, as well as decrease the required dose of psychotropic medications, thus potentially minimizing adverse effects. However, their overuse can pose a potential threat of toxicity or unexpected adverse effects, such as precipitation of mania. Table 38 lists over-the-counter nutritional and/or herbal agents that may cause mania.
Continue to: TREATMENT Nonadherence leads to a court order
TREATMENT Nonadherence leads to a court order
On admission, Mr. A receives a dose of
[polldaddy:10091676]
The authors’ observations
During an acute manic episode, the goal of treatment is urgent mood stabilization. Monotherapy can be used; however, in emergent settings, a combination is often used for a rapid response. The most commonly used agents are lithium, anticonvulsants such as valproic acid, and antipsychotics.9 In addition, benzodiazepines can be used for insomnia, agitation, or anxiety. The decision to use lithium, an anticonvulsant, or an antipsychotic depends upon the specific medication’s adverse effects, the patient’s medical history, previous medication trials, drug–drug interactions, patient preference, and cost.
Because Mr. A has a history of chronic kidney disease, lithium was contraindicated.
[polldaddy:10091678]
Continue to: The authors' observations
The authors’ observations
After the acute episode of mania resolves, maintenance pharmacotherapy typically involves continuing the same regimen that achieved mood stabilization. Monotherapy is typically preferred to combination therapy, but it is not always possible after a manic episode.10 A reasonable approach is to slowly taper the antipsychotic after several months of dual therapy if symptoms continue to be well-controlled. Further adjustments may be necessary, depending on the medications’ adverse effects. Moreover, further acute episodes of mania or depression will also determine future treatment.
OUTCOME Resolution of delusions
Mr. A is discharged 30 days after admission. At this point, his acute manic episode has resolved with non-tangential, non-pressured speech, improved sleep, and decreased impulsivity. His grandiose delusions also have resolved. He is prescribed valproic acid, 1,000 mg/d, and risperidone, 6 mg/d at bedtime, under the care of his outpatient psychiatrist.
Bottom Line
Initial presentation of a manic episode in an older patient is rare. It is important to rule out organic causes. Weak evidence suggests omega-3 fatty acid supplements may have the potential to induce mania in certain patients.
Related Resource
- Ramaswamy S, Driscoll D, Rodriguez A, et al. Nutraceuticals for traumatic brain injury: Should you recommend their use? Current Psychiatry. 2017;16(7):34-38,40,41-45.
Drug Brand Names
Buprenorphine • Suboxone, Subutex
Citalopram • Celexa
Hydrocodone/acetaminophen • Vicodin
Lithium • Eskalith, Lithobid
Lorazepam• Ativan
Nortriptyline • Pamelor
Risperidone • Risperdal
Valproic acid • Depakote
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Goodwin GM, Haddad PM, Ferrier IN, et al. Evidence-based guidelines for treating bipolar disorder: revised third edition recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2016;30(6):495-553.
3. Sami M, Khan H, Nilforooshan R. Late onset mania as an organic syndrome: a review of case reports in the literature. J Affect Disord. 2015:188:226-231.
4. Su KP, Matsuoka Y, Pae CU. Omega-3 polyunsaturated fatty acids in prevention of mood and anxiety disorders. Clin Psychopharmacol Neurosci. 2015;13(2):129-137.
5. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
6. Rudin DO. The major psychoses and neuroses as omega-3 essential fatty acid deficiency syndrome: substrate pellagra. Biol Psychiatry. 1981;16(9):837-850.
7. Su KP, Shen WW, Huang SY. Are omega3 fatty acids beneficial in depression but not mania? Arch Gen Psychiatry. 2000;57(7):716-717.
8. Joshi K, Faubion M. Mania and psychosis associated with St. John’s wort and ginseng. Psychiatry (Edgmont). 2005;2(9):56-61.
9. Grunze H, Vieta E, Goodwin GM, et al. The world federation of societies of biological psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry. 2009;10(2):85-116.
10. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166(4):476-488.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Goodwin GM, Haddad PM, Ferrier IN, et al. Evidence-based guidelines for treating bipolar disorder: revised third edition recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2016;30(6):495-553.
3. Sami M, Khan H, Nilforooshan R. Late onset mania as an organic syndrome: a review of case reports in the literature. J Affect Disord. 2015:188:226-231.
4. Su KP, Matsuoka Y, Pae CU. Omega-3 polyunsaturated fatty acids in prevention of mood and anxiety disorders. Clin Psychopharmacol Neurosci. 2015;13(2):129-137.
5. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
6. Rudin DO. The major psychoses and neuroses as omega-3 essential fatty acid deficiency syndrome: substrate pellagra. Biol Psychiatry. 1981;16(9):837-850.
7. Su KP, Shen WW, Huang SY. Are omega3 fatty acids beneficial in depression but not mania? Arch Gen Psychiatry. 2000;57(7):716-717.
8. Joshi K, Faubion M. Mania and psychosis associated with St. John’s wort and ginseng. Psychiatry (Edgmont). 2005;2(9):56-61.
9. Grunze H, Vieta E, Goodwin GM, et al. The world federation of societies of biological psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry. 2009;10(2):85-116.
10. Suppes T, Vieta E, Liu S, et al; Trial 127 Investigators. Maintenance treatment for patients with bipolar I disorder: results from a North American study of quetiapine in combination with lithium or divalproex (trial 127). Am J Psychiatry. 2009;166(4):476-488.