User login
What Is the Best E&M of Heparin-Induced Thrombocytopenia?
Case
A 52-year-old white woman presents to the ED after a motor vehicle accident with a fractured left femur. After surgical repair of the fracture, she is treated with enoxaparin 40 mg daily for VTE prophylaxis. Upon admission to the hospital, her platelet count is 180x109/L. On postoperative day three, it is 140x109/L; on postoperative day six, it is 78x109/L. Because of persistent swelling of the left leg, a venous ultrasound is obtained; results are negative for DVT. Is the decrease in the platelet count concerning for heparin-induced thrombocytopenia?
Overview
Approximately one-third of hospitalized patients are exposed to heparin each year.1 A well-described, life-threatening adverse effect of heparin use is thrombocytopenia, also called heparin-induced thrombocytopenia (HIT). Studies suggest that the frequency of HIT in the U.S. is as high as 1% to 5% in patients exposed to unfractionated heparin.1,2
There are two types of HIT. Type 2 HIT is more serious, with risk for life- or limb-threatening complications. Type 1 HIT is a nonimmune disorder caused by the direct effect of heparin on platelet activation, which is characterized by a drop in thrombocyte count within the first 48 hours of heparin exposure. The platelet count is expected to normalize with continued heparin exposure in Type 1 HIT. Type 2 HIT is an immune-mediated disorder in which heparin-dependent IgG recognizes complexes of heparin and platelet factor 4 (PF4), which subsequently induce platelet activation via the platelet Fc gammaRIIa receptor. A positive feedback loop occurs, causing further release of PF4 and platelet activation, which can lead to devastating prothrombotic complications.
Individuals affected by Type 2 HIT have a 20% to 50% risk of developing new thrombotic events, and also have a 10% rate of major morbidity, including limb ischemia requiring amputation, cerebrovascular events, myocardial infarction, DVT, or pulmonary embolus.1,2
Until recently, the mortality rate in HIT has been reported as high as 20%; however, earlier diagnosis and treatment have resulted in a better prognosis, with mortality and major morbidity of 6% to 10%.2 Low-molecular-weight heparin (LMWH) carries a lower risk for development of HIT; as such, one measure to reduce the risk of HIT is to use LMWH in place of unfractionated heparin.3
Review of the Data
When to suspect HIT. HIT should be considered as a potential diagnosis anytime there is a drop in platelet count, either during or shortly following heparin exposure. The differential diagnosis for thrombocytopenia during heparin exposure is broad and includes:
- Disseminated intravascular coagulation;
- Drug-induced thrombocytopenia;
- Hemolytic-uremic syndrome;
- Immune thrombocytopenic purpura;
- Post-transfusion thrombocytopenia;
- Systemic lupus erythematosus; and
- Thrombotic thrombocytopenic purpura.
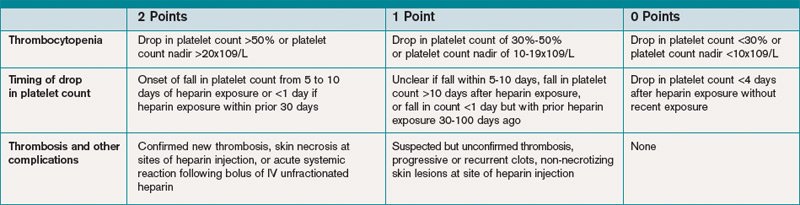
The 2009 Clinical Practice Guideline on Evaluation and Management of HIT provided by the American Society of Hematology recommends the use of Warkentin’s 4Ts clinical probability scoring system as a guide in determining the probability of HIT in patients with thrombocytopenia who are exposed to heparin.4 The 4Ts scoring system is detailed in Table 1.
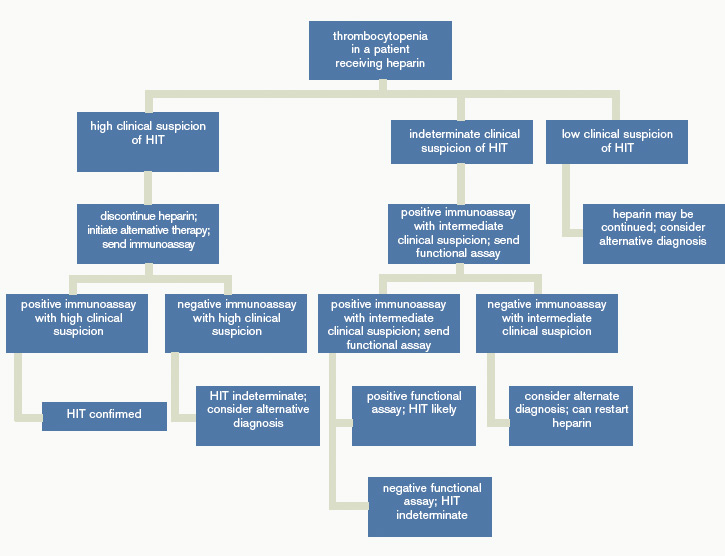
In patients with intermediate to high clinical probability of HIT (4-5 points and 6-8 points, respectively, on the 4Ts scoring system), immunologic and functional assays could further guide management. In patients with a low probability of HIT (4Ts score <3), the diagnosis is unlikely and an alternative diagnoses should be considered. Immunologic and functional assays are not recommended for these patients, and heparin can be continued.
Laboratory and diagnostic workups. Immunologic assays (polyspecific ELISA, IgG-specific ELISA, and particle gel immunoassay) detect antibodies against the PF4 heparin complexes regardless of their capacity to activate platelets. These tests are highly sensitive but less specific for HIT because they also detect PF4-heparin antibodies in patients who do not have HIT; therefore, immunoassays have a lower positive predictive value but a high negative predictive value (>95%).5
Functional assays (serotonin release assay, heparin-induced platelet activation assay, and platelet aggregation test) detect antibodies that induce heparin-dependent platelet activation. These assays are highly sensitive and specific but are not available at many medical centers. The positive predictive value of these assays is higher (89% to 100%).5
Figure 1 provides a diagnostic and initial treatment algorithm for suspected HIT. Immunoassays to detect PF4-heparin antibodies are recommended when clinical probability of HIT is intermediate to high. In these patients, a negative result on serologic testing has a high negative predictive value and suggests that an alternative diagnosis is more likely. In patients with a positive serologic test and intermediate probability of HIT, a functional assay might be beneficial, as a positive result increases the probability of HIT. For patients with high probability of HIT and a positive immunologic assay, functional assays might not be indicated as the diagnosis is likely.
Treatment. If the probability of HIT is intermediate to high based on the 4Ts scoring system, all heparin products, including heparin flushes, should be immediately discontinued and a laboratory investigation for HIT antibodies should be undertaken. An investigation for lower-limb DVT also should be pursued in patients with high probability of HIT, as the risk of thrombosis is more than 30-fold higher than controls, and studies show that approximately 25% of patients with HIT present with both thrombocytopenia and thrombosis.5 In addition, the presence of thrombosis might influence duration of anticoagulation.
Avoid platelet transfusions, as this might propagate thrombosis.
Anticoagulation. With a significant risk of thrombosis associated with this disorder, treatment with an alternative anticoagulant should be started. Vitamin K antagonists, such as warfarin, cannot be given in acute HIT because of the high risk of inducing skin necrosis and venous limb gangrene. Such anticoagulation should not be used until the platelet count increases to greater than 150x109/L. If warfarin already has been given, reversal with vitamin K is indicated.
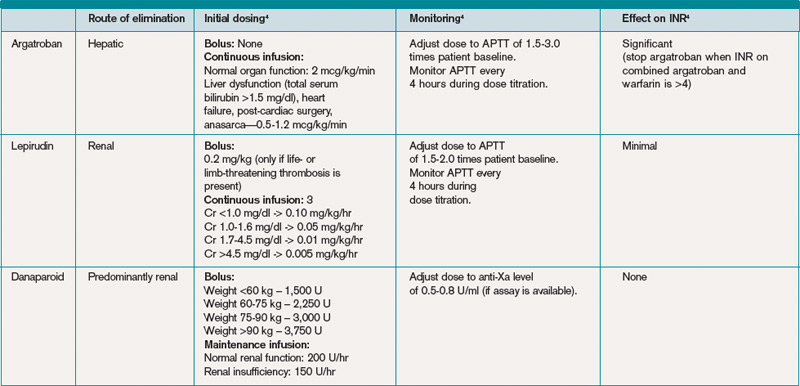
Consequently, an alternative anticoagulant bridge to warfarin therapy must be used. Usually, the bridging agent will be one of two intravenous direct thrombin inhibitors (argatroban and lepirudin) approved for this purpose.6 Both are associated with a higher risk of bleeding. Argatroban is hepatically cleared; lepirudin is renally cleared. Table 2 summarizes dosing information for these agents. A third direct thrombin inhibitor, bivalirudin, is approved for treatment of HIT, but only during percutaneous coronary intervention.6
Finally, the recently FDA-approved oral direct thrombin inhibitor dabigatrin has not been studied in or approved for HIT.
Other rational therapies include the factor Xa inhibitors danaparoid and fondaparinux. However, only danaparoid is FDA-approved for use in the treatment of HIT. It can, in cases of low or moderate suspicion of HIT, be given in prophylactic doses, lowering the risk of major bleeding.
Duration of treatment. Whichever bridging anticoagulant is chosen, it should be continued until the platelet count has fully recovered. Further, prior to discontinuation, warfarin therapy should be administered for at least five days and the international normalized ratio (INR) should be therapeutic for approximately 48 hours.
The subsequent length of warfarin therapy is dependent upon the presence or absence of an associated thrombosis. With the presence of a thrombus, the duration should be as defined for other provoked thromboses (three to six months). With no thrombus, the duration should be at least 30 days.
Future anticoagulation in patients with a prior diagnosis of HIT. A history of HIT does not appear to be a risk factor for a higher frequency of forming heparin antibodies upon re-exposure to heparin.7 Therefore, in patients with an important indication for heparin (i.e. cardiac or vascular surgery) and a remote history of HIT (>100 days), heparin can be used. In patients with a subacute history of HIT in whom surgery cannot be delayed, heparin products should be avoided and laboratory investigation should be pursued.
If the immunoassay is positive but the functional assay is negative, it is reasonable to use heparin. If both the immunologic and the functional assays are positive, the patient should be considered as having acute HIT, and bivalirudin is recommended.4
Back to the Case
Our patient has acute thrombocytopenia with a fall in platelets greater than 50% from baseline. The decrease is within the appropriate time frame for HIT. No thrombosis is found, but no alternate explanation for the thrombocytopenia is apparent. The 4Ts score of 6 indicates high risk for HIT. Heparin was discontinued, and argatroban at a rate of 2 mcg/kg/min was initiated. The immunoassay was positive.
Argatroban was continued until the platelet count reached 150x109/L, at which point warfarin therapy, 5 mg daily, was started. After four days, the INR was 2.2. After another 24 hours, argatroban was discontinued. She was instructed to continue warfarin for another 30 days.
Bottom Line
Evaluation for HIT combines clinical judgment, summarized in the 4Ts, with laboratory evaluation including an immunoassay and possibly a functional assay. Treatment requires immediate discontinuation of heparin, early initiation of a direct thrombin inhibitor, and bridging to warfarin to continue treatment for at least 30 days. TH
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Heparin-Induced Thrombocytopenia. MedScape Reference website. Available at: http://emedicine.medscape.com/article/1357846. Accessed Aug. 31, 2010.
- Heparin-Induced Thrombocytopenia. Orpha.net website. Available at: http://www.orpha.net/data/patho/GB/uk-HIT.pdf. Accessed Aug. 31, 2010.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332(20):1330-1335.
- American Society of Hematology Guidelines: Immune Thrombocytopenia (HIT). American Society of Hematology website. Available at: www.hematology.org/Practice/Guidelines/2934.aspx. Accessed Jan. 28, 2011.
- Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77-90.
- Warkentin TE, Greinacher A, Koster A, Lincoff AM. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:340S-380S.
- Warkentin TE. Agents for the treatment of heparin-induced thrombocytopenia. Hematol Oncol Clin N Am. 2010;24:755-775.
Case
A 52-year-old white woman presents to the ED after a motor vehicle accident with a fractured left femur. After surgical repair of the fracture, she is treated with enoxaparin 40 mg daily for VTE prophylaxis. Upon admission to the hospital, her platelet count is 180x109/L. On postoperative day three, it is 140x109/L; on postoperative day six, it is 78x109/L. Because of persistent swelling of the left leg, a venous ultrasound is obtained; results are negative for DVT. Is the decrease in the platelet count concerning for heparin-induced thrombocytopenia?
Overview
Approximately one-third of hospitalized patients are exposed to heparin each year.1 A well-described, life-threatening adverse effect of heparin use is thrombocytopenia, also called heparin-induced thrombocytopenia (HIT). Studies suggest that the frequency of HIT in the U.S. is as high as 1% to 5% in patients exposed to unfractionated heparin.1,2
There are two types of HIT. Type 2 HIT is more serious, with risk for life- or limb-threatening complications. Type 1 HIT is a nonimmune disorder caused by the direct effect of heparin on platelet activation, which is characterized by a drop in thrombocyte count within the first 48 hours of heparin exposure. The platelet count is expected to normalize with continued heparin exposure in Type 1 HIT. Type 2 HIT is an immune-mediated disorder in which heparin-dependent IgG recognizes complexes of heparin and platelet factor 4 (PF4), which subsequently induce platelet activation via the platelet Fc gammaRIIa receptor. A positive feedback loop occurs, causing further release of PF4 and platelet activation, which can lead to devastating prothrombotic complications.
Individuals affected by Type 2 HIT have a 20% to 50% risk of developing new thrombotic events, and also have a 10% rate of major morbidity, including limb ischemia requiring amputation, cerebrovascular events, myocardial infarction, DVT, or pulmonary embolus.1,2
Until recently, the mortality rate in HIT has been reported as high as 20%; however, earlier diagnosis and treatment have resulted in a better prognosis, with mortality and major morbidity of 6% to 10%.2 Low-molecular-weight heparin (LMWH) carries a lower risk for development of HIT; as such, one measure to reduce the risk of HIT is to use LMWH in place of unfractionated heparin.3
Review of the Data
When to suspect HIT. HIT should be considered as a potential diagnosis anytime there is a drop in platelet count, either during or shortly following heparin exposure. The differential diagnosis for thrombocytopenia during heparin exposure is broad and includes:
- Disseminated intravascular coagulation;
- Drug-induced thrombocytopenia;
- Hemolytic-uremic syndrome;
- Immune thrombocytopenic purpura;
- Post-transfusion thrombocytopenia;
- Systemic lupus erythematosus; and
- Thrombotic thrombocytopenic purpura.
The 2009 Clinical Practice Guideline on Evaluation and Management of HIT provided by the American Society of Hematology recommends the use of Warkentin’s 4Ts clinical probability scoring system as a guide in determining the probability of HIT in patients with thrombocytopenia who are exposed to heparin.4 The 4Ts scoring system is detailed in Table 1.
In patients with intermediate to high clinical probability of HIT (4-5 points and 6-8 points, respectively, on the 4Ts scoring system), immunologic and functional assays could further guide management. In patients with a low probability of HIT (4Ts score <3), the diagnosis is unlikely and an alternative diagnoses should be considered. Immunologic and functional assays are not recommended for these patients, and heparin can be continued.
Laboratory and diagnostic workups. Immunologic assays (polyspecific ELISA, IgG-specific ELISA, and particle gel immunoassay) detect antibodies against the PF4 heparin complexes regardless of their capacity to activate platelets. These tests are highly sensitive but less specific for HIT because they also detect PF4-heparin antibodies in patients who do not have HIT; therefore, immunoassays have a lower positive predictive value but a high negative predictive value (>95%).5
Functional assays (serotonin release assay, heparin-induced platelet activation assay, and platelet aggregation test) detect antibodies that induce heparin-dependent platelet activation. These assays are highly sensitive and specific but are not available at many medical centers. The positive predictive value of these assays is higher (89% to 100%).5
Figure 1 provides a diagnostic and initial treatment algorithm for suspected HIT. Immunoassays to detect PF4-heparin antibodies are recommended when clinical probability of HIT is intermediate to high. In these patients, a negative result on serologic testing has a high negative predictive value and suggests that an alternative diagnosis is more likely. In patients with a positive serologic test and intermediate probability of HIT, a functional assay might be beneficial, as a positive result increases the probability of HIT. For patients with high probability of HIT and a positive immunologic assay, functional assays might not be indicated as the diagnosis is likely.
Treatment. If the probability of HIT is intermediate to high based on the 4Ts scoring system, all heparin products, including heparin flushes, should be immediately discontinued and a laboratory investigation for HIT antibodies should be undertaken. An investigation for lower-limb DVT also should be pursued in patients with high probability of HIT, as the risk of thrombosis is more than 30-fold higher than controls, and studies show that approximately 25% of patients with HIT present with both thrombocytopenia and thrombosis.5 In addition, the presence of thrombosis might influence duration of anticoagulation.
Avoid platelet transfusions, as this might propagate thrombosis.
Anticoagulation. With a significant risk of thrombosis associated with this disorder, treatment with an alternative anticoagulant should be started. Vitamin K antagonists, such as warfarin, cannot be given in acute HIT because of the high risk of inducing skin necrosis and venous limb gangrene. Such anticoagulation should not be used until the platelet count increases to greater than 150x109/L. If warfarin already has been given, reversal with vitamin K is indicated.
Consequently, an alternative anticoagulant bridge to warfarin therapy must be used. Usually, the bridging agent will be one of two intravenous direct thrombin inhibitors (argatroban and lepirudin) approved for this purpose.6 Both are associated with a higher risk of bleeding. Argatroban is hepatically cleared; lepirudin is renally cleared. Table 2 summarizes dosing information for these agents. A third direct thrombin inhibitor, bivalirudin, is approved for treatment of HIT, but only during percutaneous coronary intervention.6
Finally, the recently FDA-approved oral direct thrombin inhibitor dabigatrin has not been studied in or approved for HIT.
Other rational therapies include the factor Xa inhibitors danaparoid and fondaparinux. However, only danaparoid is FDA-approved for use in the treatment of HIT. It can, in cases of low or moderate suspicion of HIT, be given in prophylactic doses, lowering the risk of major bleeding.
Duration of treatment. Whichever bridging anticoagulant is chosen, it should be continued until the platelet count has fully recovered. Further, prior to discontinuation, warfarin therapy should be administered for at least five days and the international normalized ratio (INR) should be therapeutic for approximately 48 hours.
The subsequent length of warfarin therapy is dependent upon the presence or absence of an associated thrombosis. With the presence of a thrombus, the duration should be as defined for other provoked thromboses (three to six months). With no thrombus, the duration should be at least 30 days.
Future anticoagulation in patients with a prior diagnosis of HIT. A history of HIT does not appear to be a risk factor for a higher frequency of forming heparin antibodies upon re-exposure to heparin.7 Therefore, in patients with an important indication for heparin (i.e. cardiac or vascular surgery) and a remote history of HIT (>100 days), heparin can be used. In patients with a subacute history of HIT in whom surgery cannot be delayed, heparin products should be avoided and laboratory investigation should be pursued.
If the immunoassay is positive but the functional assay is negative, it is reasonable to use heparin. If both the immunologic and the functional assays are positive, the patient should be considered as having acute HIT, and bivalirudin is recommended.4
Back to the Case
Our patient has acute thrombocytopenia with a fall in platelets greater than 50% from baseline. The decrease is within the appropriate time frame for HIT. No thrombosis is found, but no alternate explanation for the thrombocytopenia is apparent. The 4Ts score of 6 indicates high risk for HIT. Heparin was discontinued, and argatroban at a rate of 2 mcg/kg/min was initiated. The immunoassay was positive.
Argatroban was continued until the platelet count reached 150x109/L, at which point warfarin therapy, 5 mg daily, was started. After four days, the INR was 2.2. After another 24 hours, argatroban was discontinued. She was instructed to continue warfarin for another 30 days.
Bottom Line
Evaluation for HIT combines clinical judgment, summarized in the 4Ts, with laboratory evaluation including an immunoassay and possibly a functional assay. Treatment requires immediate discontinuation of heparin, early initiation of a direct thrombin inhibitor, and bridging to warfarin to continue treatment for at least 30 days. TH
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Heparin-Induced Thrombocytopenia. MedScape Reference website. Available at: http://emedicine.medscape.com/article/1357846. Accessed Aug. 31, 2010.
- Heparin-Induced Thrombocytopenia. Orpha.net website. Available at: http://www.orpha.net/data/patho/GB/uk-HIT.pdf. Accessed Aug. 31, 2010.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332(20):1330-1335.
- American Society of Hematology Guidelines: Immune Thrombocytopenia (HIT). American Society of Hematology website. Available at: www.hematology.org/Practice/Guidelines/2934.aspx. Accessed Jan. 28, 2011.
- Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77-90.
- Warkentin TE, Greinacher A, Koster A, Lincoff AM. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:340S-380S.
- Warkentin TE. Agents for the treatment of heparin-induced thrombocytopenia. Hematol Oncol Clin N Am. 2010;24:755-775.
Case
A 52-year-old white woman presents to the ED after a motor vehicle accident with a fractured left femur. After surgical repair of the fracture, she is treated with enoxaparin 40 mg daily for VTE prophylaxis. Upon admission to the hospital, her platelet count is 180x109/L. On postoperative day three, it is 140x109/L; on postoperative day six, it is 78x109/L. Because of persistent swelling of the left leg, a venous ultrasound is obtained; results are negative for DVT. Is the decrease in the platelet count concerning for heparin-induced thrombocytopenia?
Overview
Approximately one-third of hospitalized patients are exposed to heparin each year.1 A well-described, life-threatening adverse effect of heparin use is thrombocytopenia, also called heparin-induced thrombocytopenia (HIT). Studies suggest that the frequency of HIT in the U.S. is as high as 1% to 5% in patients exposed to unfractionated heparin.1,2
There are two types of HIT. Type 2 HIT is more serious, with risk for life- or limb-threatening complications. Type 1 HIT is a nonimmune disorder caused by the direct effect of heparin on platelet activation, which is characterized by a drop in thrombocyte count within the first 48 hours of heparin exposure. The platelet count is expected to normalize with continued heparin exposure in Type 1 HIT. Type 2 HIT is an immune-mediated disorder in which heparin-dependent IgG recognizes complexes of heparin and platelet factor 4 (PF4), which subsequently induce platelet activation via the platelet Fc gammaRIIa receptor. A positive feedback loop occurs, causing further release of PF4 and platelet activation, which can lead to devastating prothrombotic complications.
Individuals affected by Type 2 HIT have a 20% to 50% risk of developing new thrombotic events, and also have a 10% rate of major morbidity, including limb ischemia requiring amputation, cerebrovascular events, myocardial infarction, DVT, or pulmonary embolus.1,2
Until recently, the mortality rate in HIT has been reported as high as 20%; however, earlier diagnosis and treatment have resulted in a better prognosis, with mortality and major morbidity of 6% to 10%.2 Low-molecular-weight heparin (LMWH) carries a lower risk for development of HIT; as such, one measure to reduce the risk of HIT is to use LMWH in place of unfractionated heparin.3
Review of the Data
When to suspect HIT. HIT should be considered as a potential diagnosis anytime there is a drop in platelet count, either during or shortly following heparin exposure. The differential diagnosis for thrombocytopenia during heparin exposure is broad and includes:
- Disseminated intravascular coagulation;
- Drug-induced thrombocytopenia;
- Hemolytic-uremic syndrome;
- Immune thrombocytopenic purpura;
- Post-transfusion thrombocytopenia;
- Systemic lupus erythematosus; and
- Thrombotic thrombocytopenic purpura.
The 2009 Clinical Practice Guideline on Evaluation and Management of HIT provided by the American Society of Hematology recommends the use of Warkentin’s 4Ts clinical probability scoring system as a guide in determining the probability of HIT in patients with thrombocytopenia who are exposed to heparin.4 The 4Ts scoring system is detailed in Table 1.
In patients with intermediate to high clinical probability of HIT (4-5 points and 6-8 points, respectively, on the 4Ts scoring system), immunologic and functional assays could further guide management. In patients with a low probability of HIT (4Ts score <3), the diagnosis is unlikely and an alternative diagnoses should be considered. Immunologic and functional assays are not recommended for these patients, and heparin can be continued.
Laboratory and diagnostic workups. Immunologic assays (polyspecific ELISA, IgG-specific ELISA, and particle gel immunoassay) detect antibodies against the PF4 heparin complexes regardless of their capacity to activate platelets. These tests are highly sensitive but less specific for HIT because they also detect PF4-heparin antibodies in patients who do not have HIT; therefore, immunoassays have a lower positive predictive value but a high negative predictive value (>95%).5
Functional assays (serotonin release assay, heparin-induced platelet activation assay, and platelet aggregation test) detect antibodies that induce heparin-dependent platelet activation. These assays are highly sensitive and specific but are not available at many medical centers. The positive predictive value of these assays is higher (89% to 100%).5
Figure 1 provides a diagnostic and initial treatment algorithm for suspected HIT. Immunoassays to detect PF4-heparin antibodies are recommended when clinical probability of HIT is intermediate to high. In these patients, a negative result on serologic testing has a high negative predictive value and suggests that an alternative diagnosis is more likely. In patients with a positive serologic test and intermediate probability of HIT, a functional assay might be beneficial, as a positive result increases the probability of HIT. For patients with high probability of HIT and a positive immunologic assay, functional assays might not be indicated as the diagnosis is likely.
Treatment. If the probability of HIT is intermediate to high based on the 4Ts scoring system, all heparin products, including heparin flushes, should be immediately discontinued and a laboratory investigation for HIT antibodies should be undertaken. An investigation for lower-limb DVT also should be pursued in patients with high probability of HIT, as the risk of thrombosis is more than 30-fold higher than controls, and studies show that approximately 25% of patients with HIT present with both thrombocytopenia and thrombosis.5 In addition, the presence of thrombosis might influence duration of anticoagulation.
Avoid platelet transfusions, as this might propagate thrombosis.
Anticoagulation. With a significant risk of thrombosis associated with this disorder, treatment with an alternative anticoagulant should be started. Vitamin K antagonists, such as warfarin, cannot be given in acute HIT because of the high risk of inducing skin necrosis and venous limb gangrene. Such anticoagulation should not be used until the platelet count increases to greater than 150x109/L. If warfarin already has been given, reversal with vitamin K is indicated.
Consequently, an alternative anticoagulant bridge to warfarin therapy must be used. Usually, the bridging agent will be one of two intravenous direct thrombin inhibitors (argatroban and lepirudin) approved for this purpose.6 Both are associated with a higher risk of bleeding. Argatroban is hepatically cleared; lepirudin is renally cleared. Table 2 summarizes dosing information for these agents. A third direct thrombin inhibitor, bivalirudin, is approved for treatment of HIT, but only during percutaneous coronary intervention.6
Finally, the recently FDA-approved oral direct thrombin inhibitor dabigatrin has not been studied in or approved for HIT.
Other rational therapies include the factor Xa inhibitors danaparoid and fondaparinux. However, only danaparoid is FDA-approved for use in the treatment of HIT. It can, in cases of low or moderate suspicion of HIT, be given in prophylactic doses, lowering the risk of major bleeding.
Duration of treatment. Whichever bridging anticoagulant is chosen, it should be continued until the platelet count has fully recovered. Further, prior to discontinuation, warfarin therapy should be administered for at least five days and the international normalized ratio (INR) should be therapeutic for approximately 48 hours.
The subsequent length of warfarin therapy is dependent upon the presence or absence of an associated thrombosis. With the presence of a thrombus, the duration should be as defined for other provoked thromboses (three to six months). With no thrombus, the duration should be at least 30 days.
Future anticoagulation in patients with a prior diagnosis of HIT. A history of HIT does not appear to be a risk factor for a higher frequency of forming heparin antibodies upon re-exposure to heparin.7 Therefore, in patients with an important indication for heparin (i.e. cardiac or vascular surgery) and a remote history of HIT (>100 days), heparin can be used. In patients with a subacute history of HIT in whom surgery cannot be delayed, heparin products should be avoided and laboratory investigation should be pursued.
If the immunoassay is positive but the functional assay is negative, it is reasonable to use heparin. If both the immunologic and the functional assays are positive, the patient should be considered as having acute HIT, and bivalirudin is recommended.4
Back to the Case
Our patient has acute thrombocytopenia with a fall in platelets greater than 50% from baseline. The decrease is within the appropriate time frame for HIT. No thrombosis is found, but no alternate explanation for the thrombocytopenia is apparent. The 4Ts score of 6 indicates high risk for HIT. Heparin was discontinued, and argatroban at a rate of 2 mcg/kg/min was initiated. The immunoassay was positive.
Argatroban was continued until the platelet count reached 150x109/L, at which point warfarin therapy, 5 mg daily, was started. After four days, the INR was 2.2. After another 24 hours, argatroban was discontinued. She was instructed to continue warfarin for another 30 days.
Bottom Line
Evaluation for HIT combines clinical judgment, summarized in the 4Ts, with laboratory evaluation including an immunoassay and possibly a functional assay. Treatment requires immediate discontinuation of heparin, early initiation of a direct thrombin inhibitor, and bridging to warfarin to continue treatment for at least 30 days. TH
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Heparin-Induced Thrombocytopenia. MedScape Reference website. Available at: http://emedicine.medscape.com/article/1357846. Accessed Aug. 31, 2010.
- Heparin-Induced Thrombocytopenia. Orpha.net website. Available at: http://www.orpha.net/data/patho/GB/uk-HIT.pdf. Accessed Aug. 31, 2010.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332(20):1330-1335.
- American Society of Hematology Guidelines: Immune Thrombocytopenia (HIT). American Society of Hematology website. Available at: www.hematology.org/Practice/Guidelines/2934.aspx. Accessed Jan. 28, 2011.
- Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77-90.
- Warkentin TE, Greinacher A, Koster A, Lincoff AM. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:340S-380S.
- Warkentin TE. Agents for the treatment of heparin-induced thrombocytopenia. Hematol Oncol Clin N Am. 2010;24:755-775.
“Better Late than Never”
A 59‐year‐old man presented to the emergency department with the acute onset of right‐sided abdominal and flank pain. The pain had begun the previous night, was constant and progressively worsening, and radiated to his right groin. He denied fever, nausea, emesis, or change in his bowel habits, but he did notice mild right lower quadrant discomfort with micturition. Upon further questioning, he also complained of mild dyspnea on climbing stairs and an unspecified recent weight loss.
The most common cause of acute severe right‐sided flank and abdominal pain radiating to the groin and associated with dysuria in a middle‐aged man is ureteral colic. Other etiologies important to consider include retrocecal appendicitis, pyelonephritis, and, rarely, a dissecting abdominal aortic aneurysm. This patient's seemingly recent onset exertional dyspnea and weight loss do not neatly fit any of the above, however.
His past medical history was significant for diabetes mellitus and pemphigus vulgaris diagnosed 7 months previously. He had been treated with prednisone, and the dose decreased from 100 to 60 mg daily, 1 month previously, due to poor glycemic control as well as steroid‐induced neuropathy and myopathy. His other medications included naproxen sodium and ibuprofen for back pain, azathioprine, insulin, pioglitazone, and glimiperide. He had no past surgical history. He had lived in the United States since his emigration from Thailand in 1971. His last trip to Thailand was 5 years previously. He was a taxi cab driver. He had a ten‐pack year history of tobacco use, but had quit 20 years prior. He denied history of alcohol or intravenous drug use.
Pemphigus vulgaris is unlikely to be directly related to this patient's presentation, but in light of his poorly controlled diabetes, his azathioprine use, and particularly his high‐dose corticosteroids, he is certainly immunocompromised. Accordingly, a disseminated infection, either newly acquired or reactivated, merits consideration. His history of residence in, and subsequent travel to, Southeast Asia raises the possibility of several diseases, each of which may be protean in their manifestations; these include tuberculosis, melioidosis, and penicilliosis (infection with Penicillium marneffei). The first two may reactivate long after initial exposure, particularly with insults to the immune system. The same is probably true of penicilliosis, although I am not certain of this. On a slightly less exotic note, domestically acquired infection with histoplasmosis or other endemic fungi is possible.
On examination he was afebrile, had a pulse of 130 beats per minute and a blood pressure of 65/46 mmHg. His oxygen saturation was 92%. He appeared markedly cushingoid, and had mild pallor and generalized weakness. Cardiopulmonary examination was unremarkable. His abdominal exam was notable for distention and hypoactive bowel sounds, with tenderness and firmness to palpation on the right side. Peripheral pulses were normal. Examination of the skin demonstrated ecchymoses over the bilateral forearms, and several healed pemphigus lesions on the abdomen and upper extremities.
The patient's severely deranged hemodynamic parameters indicate either current or impending shock, and resuscitative measures should proceed in tandem with diagnostic efforts. The cause of his shock seems most likely to be either hypovolemic (abdominal wall or intra‐abdominal hemorrhage, or conceivably massive third spacing from an intra‐abdominal catastrophe), or distributive (sepsis, or acute adrenal insufficiency if he has missed recent steroid doses). His ecchymoses may simply reflect chronic glucocorticoid use, but also raise suspicion for a coagulopathy. Provided the patient can be stabilized to allow this, I would urgently obtain a computed tomography (CT) scan of the abdomen and pelvis.
Initial laboratory studies demonstrated a hemoglobin of 9.1 g/dL, white blood cell count 8000/L with 33% bands, 48% segmented neutrophils, 18% lymphocytes, and 0.7% eosinophils, platelet count 356,000/L, sodium 128 mmol/L, BUN 52 mg/dL, creatinine 2.3 mg/dL, and glucose of 232 mg/dL. Coagulation studies were normal, and lactic acid was 1.8 mmol/L (normal range, 0.7‐2.1). Fibrinogen was normal at 591 and LDH was mildly elevated at 654 (normal range, 313‐618 U/L). Total protein and albumin were 3.6 and 1.9 g/dL, respectively. Total bilirubin was 0.6 mg/dL. Random serum cortisol was 20.2 g/dL. Liver enzymes, amylase, lipase, iron stores, B12, folate, and stool for occult blood were normal. Initial cardiac biomarkers were negative, but subsequent troponin‐I was 3.81 ng/mL (elevated, >1.00). Urinalysis showed 0‐4 white blood cells per high powered field.
The laboratory studies provide a variety of useful, albeit nonspecific, information. The high percentage of band forms on white blood cell differential further raises concern for an infectious process, although severe noninfectious stress can also cause this. While we do not know whether the patient's renal failure is acute, I suspect that it is, and may result from a variety of insults including sepsis, hypotension, and volume depletion. His moderately elevated troponin‐I likely reflects supplydemand mismatch or sepsis. I would like to see an electrocardiogram, and I remain very interested in obtaining abdominal imaging.
Chest radiography showed pulmonary vascular congestion without evidence of pneumothorax. Computed tomography scan of the abdomen and pelvis showed retroperitoneal fluid bilaterally (Figure 1). This was described as suspicious for ascites versus hemorrhage, but no obvious source of bleeding was identified. There was also a small amount of right perinephric fluid, but no evidence of a renal mass. The abdominal aorta was normal; there was no lymphadenopathy.
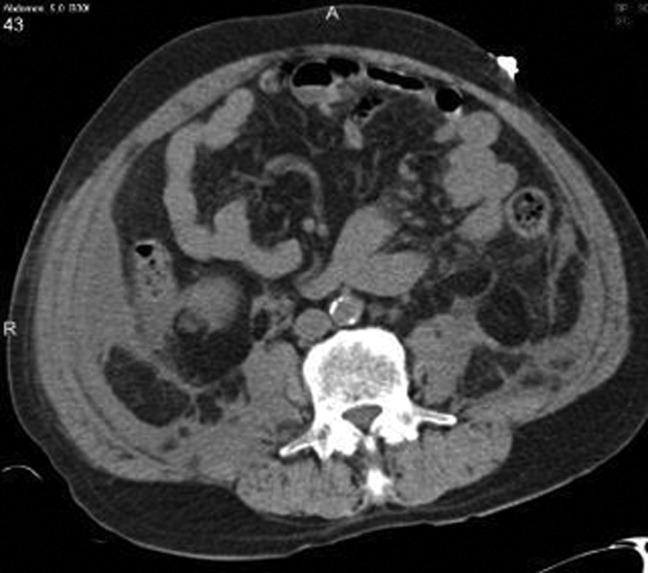
The CT image appears to speak against simple ascites, and seems most consistent with either blood or an infectious process. Consequently, the loculated right retroperitoneal collection should be aspirated, and fluid sent for fungal, acid‐fast, and modified acid‐fast (i.e., for Nocardia) stains and culture, in addition to Gram stain and routine aerobic and anaerobic cultures.
The patient was admitted to the intensive care unit. Stress‐dose steroids were administered, and he improved after resuscitation with fluid and blood. His renal function normalized. Urine and blood cultures returned negative. His hematocrit and multiple repeat CT scans of the abdomen remained stable. A retroperitoneal hemorrhage was diagnosed, and surgical intervention was deemed unnecessary. Both adenosine thallium stress test and echocardiogram were normal. He was continued on 60 mg prednisone daily and discharged home with outpatient follow‐up.
This degree of improvement with volume expansion (and steroids) suggests the patient was markedly volume depleted upon presentation. Although a formal adrenocorticotropic hormone (ACTH) stimulation test was apparently not performed, the random cortisol level suggests adrenal insufficiency was unlikely to have been primarily responsible. While retroperitoneal hemorrhage is possible, the loculated appearance of the collection suggests infection is more likely.
Three weeks later, he was readmitted with recurrent right‐sided abdominal and flank pain. His temperature was 101.3F, and he was tachycardic and hypotensive. His examination was similar to that at the time of his previous presentation. Laboratory data revealed white blood cell count of 13,100/L with 43% bands, hemoglobin of 9.2 g/dL, glucose of 343 mg/dL, bicarbonate 25 mmol/L, normal anion gap and renal function, and lactic acid of 4.5 mmol/L. Liver function tests were normal except for an albumin of 3.0 g/dL. CT scan of the abdomen revealed loculated retroperitoneal fluid collections, increased in size since the prior scan.
The patient is once again evidencing at least early shock, manifested in his deranged hemodynamics and elevated lactate level. I remain puzzled by the fact that he appeared to respond to fluids alone at the time of his initial hospital stay, unless adrenal insufficiency played a greater role than I suspected. Of note, acute adrenal insufficiency could explain much of the current picture, including fever, and bland (uninfected) hematomas are an underappreciated cause of both fever and leukocytosis. Having said this, I remain concerned that his retroperitoneal fluid collections represent abscesses. The most accessible of these should be sampled.
Aspiration of the retroperitoneal fluid yielded purulent material which grew Klebsiella pneumoniae. The cultures were negative for mycobacteria and fungus. Blood and urine cultures were negative. Drains were placed, and he was followed as an outpatient. His fever and leukocytosis subsided, and he completed a 6‐week course of trimethoprim‐sulfamethoxazole. CT imaging confirmed complete evacuation of the fluid.
Retroperitoneal abscesses frequently present in smoldering fashion, although patients may be quite ill by the time of presentation. Most of these are secondary, i.e., they arise from another abnormality in the retroperitoneum. Most commonly this is in the large bowel, kidney, pancreas, or spine. I would carefully scour his follow‐up imaging for additional clues and, if unrevealing, proceed to colonoscopy.
He returned 1 month after drain removal, with 2‐3 days of nausea and abdominal pain. His abdomen was moderately distended but nontender, and multiple persistent petechial and purpuric lesions were present on the upper back, chest, torso, and arms. Abdominal CT scan revealed small bowel obstruction and a collection of fluid in the left paracolic gutter extending into the left retrorenal space.
The patient does not appear to have obvious risk factors for developing a small bowel obstruction. No mention is made of the presence or absence of a transition point on the CT scan, and this should be ascertained. His left‐sided abdominal fluid collection is probably infectious in nature, and I continue to be suspicious of a large bowel (or distal small bowel) source, via either gut perforation or bacterial translocation. The collection needs to be percutaneously drained for both diagnostic and therapeutic reasons, and broadly cultured. Finally, we need to account for the described dermatologic manifestations. The purpuric/petechial lesions sound vasculitic rather than thrombocytopenic in origin based on location; conversely, they may simply reflect a corticosteroid‐related adverse effect. I would like to know whether the purpura was palpable, and to repeat a complete blood count with peripheral smear.
Laboratory data showed hemoglobin of 9.3 g/dL, a platelet count of 444,000/L, and normal coagulation studies. The purpura was nonpalpable (Figure 2). The patient had a nasogastric tube placed for decompression, with bilious drainage. His left retroperitoneal fluid was drained, with cultures yielding Enterococcus faecalis and Enterobacter cloacae. The patient was treated with a course of broad‐spectrum antibiotics. His obstruction improved and the retroperitoneal collection resolved on follow‐up imaging. However, 2 days later, he had recurrent pain; abdominal CT showed a recurrence of small bowel obstruction with an unequivocal transition point in the distal jejunum. A small fluid collection was noted in the left retroperitoneum with a trace of gas in it. He improved with nasogastric suction, his prednisone was tapered to 30 mg daily, and he was discharged home.
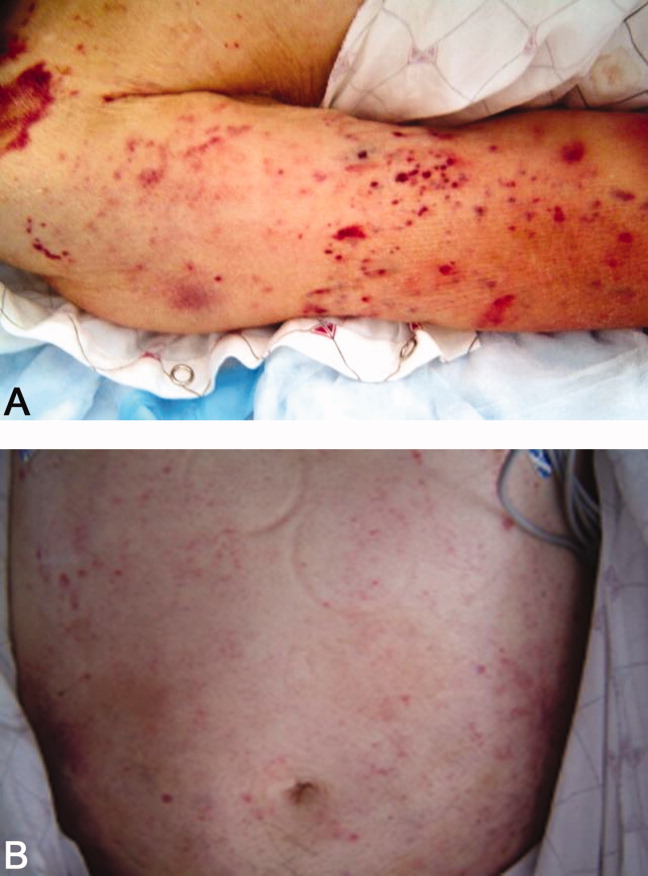
The isolation of both Enterococcus and Enterobacter species from his fluid collection, along with the previous isolation of Klebsiella, strongly suggest a bowel source for his recurrent abscesses. Based on this CT report, the patient has clear evidence of at least partial small bowel obstruction. He lacks a history of prior abdominal surgery or other more typical reasons for obstruction caused by extrinsic compression, such as hernia, although it is possible his recurrent abdominal infections may have led to obstruction due to scarring and adhesions. An intraluminal cause of obstruction also needs to be considered, with causes including malignancy (lymphoma, carcinoid, and adenocarcinoma), Crohn's disease, and infections including tuberculosis as well as parasites such as Taenia and Strongyloides. While the purpura is concerning, given the nonpalpable character along with a normal platelet count and coagulation studies, it may be reasonable to provisionally attribute it to high‐dose corticosteroid use.
He was admitted a fourth time a week after being discharged, with nausea, generalized weakness, and weight loss. At presentation, he had a blood pressure of 95/65 mmHg. His white blood cell count was 5,900/L, with 79% neutrophils and 20% bands. An AM cortisol was 18.8 /dL. He was thought to have adrenal insufficiency from steroid withdrawal, was treated with intravenous fluids and steroids, and discharged on a higher dose of prednisone at 60 mg daily. One week later, he again returned to the hospital with watery diarrhea, emesis, and generalized weakness. His blood pressure was 82/50 mmHg, and his abdomen appeared benign. He also had an erythematous rash over his mid‐abdomen. Laboratory data was significant for a sodium of 127 mmol/L, potassium of 3.0 mmol/L, chloride of 98 mmol/L, bicarbonate of 26 mmol/L, glucose of 40 mg/dL, lactate of 14 mmol/L, and albumin of 1.0 g/dL. Stool assay for Clostridium difficile was negative. A CT scan of the abdomen and pelvis showed small bilateral pleural effusions and small bowel fluid consistent with gastroenteritis, but without signs of obstruction. Esophagogastroduodenoscopy (EGD) showed bile backwash into the stomach, as well as inflammatory changes in the proximal and mid‐stomach, and inflammatory reaction and edema in the proximal duodenum. Colonoscopy showed normal appearing ileum and colon.
The patient's latest laboratory values appear to reflect his chronic illness and superimposed diarrhea. I am perplexed by his markedly elevated serum lactate value in association with a normal bicarbonate and low anion gap, and would repeat the lactate level to ensure this is not spurious. His hypoglycemia probably reflects a failure to adjust or discontinue his diabetic medications, although both hypoglycemia and type B lactic acidosis are occasionally manifestations of a paraneoplastic syndrome. The normal colonoscopy findings are helpful in exonerating the colon, provided the preparation was adequate. Presumably, the abnormal areas of the stomach and duodenum were biopsied; I remain suspicious that the answer may lie in the jejunum.
The patient was treated with intravenous fluids and stress‐dose steroids, and electrolyte abnormalities were corrected. Biopsies from the EGD and colonoscopy demonstrated numerous larvae within the mucosa of the body and antrum of the stomach, as well as duodenum. There were also rare detached larvae seen in the esophagus, and a few larvae within the ileal mucosa.
The patient appears to have Strongyloides hyperinfection, something he is at clear risk for, given his country of origin and his high‐dose corticosteroids. In retrospect, I was dissuaded from seriously considering a diagnosis of parasitic infection in large part because of the absence of peripheral eosinophilia, but this may not be seen in cases of hyperinfection. Additional clues, again in retrospect, were the repeated abscesses with bowel flora and the seemingly nonspecific abdominal rash. I would treat with a course of ivermectin, and carefully monitor his response.
The characteristics of the larvae were suggestive of Strongyloides species (Figure 3). A subsequent stool test for ova and parasites was positive for Strongyloides larvae. The patient was given a single dose of ivermectin. An endocrinology consultant felt that he did not have adrenal insufficiency, and it was recommended that his steroids be tapered off. He was discharged home once he clinically improved.
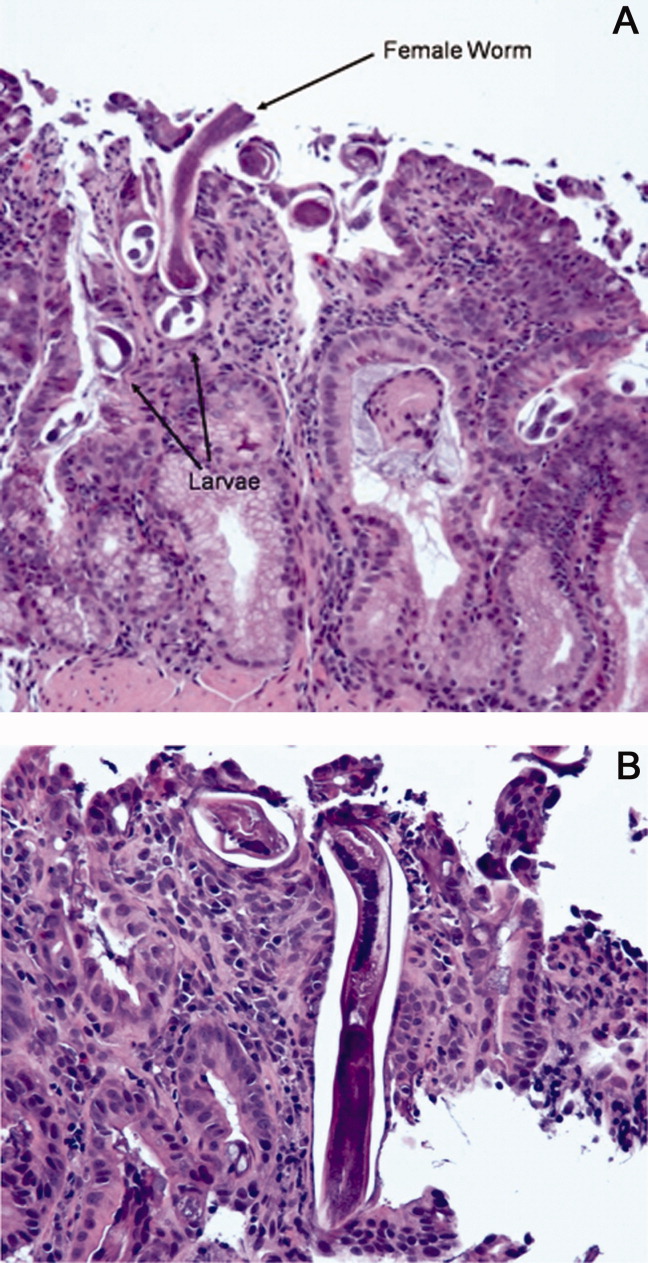
Although one or two doses of ivermectin typically suffices for uncomplicated strongyloidiasis, the risk of failure in hyperinfection mandates a longer treatment course. I don't believe this patient has been adequately treated, although the removal of his steroids will be helpful.
He was readmitted 3 days later with recrudescent symptoms, and his stool remained positive for Strongyloides. He received 2 weeks of ivermectin and albendazole, and was ultimately discharged to a rehabilitation facility after a complicated hospital stay. Nine months later, the patient was reported to be doing well.
COMMENTARY
This patient's immigration status from the developing world, high‐dose corticosteroid use, and complex clinical course all suggested the possibility of an underlying chronic infectious process. Although the discussant recognized this early on and later briefly mentioned strongyloidiasis as a potential cause of intestinal obstruction, the diagnosis of Strongyloides hyperinfection was not suspected until incontrovertible evidence for it was obtained on EGD. Failure to make the diagnosis earlier by both the involved clinicians and the discussant probably stemmed largely from two factors: the absence of eosinophilia; and lack of recognition that purpura may be seen in cases of hyperinfection, presumably reflecting larval infiltration of the dermis.1 Although eosinophilia accompanies most cases of stronglyloidiasis and may be very pronounced, patients with hyperinfection syndrome frequently fail to mount an eosinophilic response due to underlying immunosuppression, with eosinophilia absent in 70% of such patients in a study from Taiwan.2
Strongyloides stercoralis is an intestinal nematode that causes strongyloidiasis. It affects as many as 100 million people globally,3 mainly in tropical and subtropical areas, but is also endemic in the Southeastern United States, Europe, and Japan. Risk factors include male sex, White race, alcoholism, working in contact with soil (farmers, coal mine workers, etc.), chronic care institutionalization, and low socioeconomic status. In nonendemic regions, it more commonly affects travelers, immigrants, or military personnel.4, 5
The life cycle of S. stercoralis is complex. Infective larvae penetrate the skin through contact with contaminated soil, enter the venous system via lymphatics, and travel to the lung.4, 6 Here, they ascend the tracheobronchial tree and migrate to the gut. In the intestine, larvae develop into adult female worms that burrow into the intestinal mucosa. These worms lay eggs that develop into noninfective rhabditiform larvae, which are then expelled in the stool. Some of the rhabditiform larvae, however, develop into infective filariform larvae, which may penetrate colonic mucosa or perianal skin, enter the bloodstream, and lead to the cycle of autoinfection and chronic strongyloidiasis (carrier state). Autoinfection typically involves a low parasite burden, and is controlled by both host immune factors as well as parasitic factors.7 The mechanism of autoinfection can lead to the persistence of strongyloidiasis for decades after the initial infection, as has been documented in former World War II prisoners of war.8
Factors leading to the impairment of cell‐mediated immunity predispose chronically infected individuals to hyperinfection, as occurred in this patient. The most important of these are corticosteroid administration and Human T‐lymphotropic virus Type‐1 (HTLV‐1) infection, both of which cause significant derangement in TH1/TH2 immune system balance.5, 9 In the hyperinfection syndrome, the burden of parasites increases dramatically, leading to a variety of clinical manifestations. Gastrointestinal phenomena frequently predominate, including watery diarrhea, anorexia, weight loss, nausea/vomiting, gastrointestinal bleeding, and occasionally small bowel obstruction. Pulmonary manifestations are likewise common, and include cough, dyspnea, and wheezing. Cutaneous findings are not uncommon, classically pruritic linear lesions of the abdomen, buttocks, and lower extremities which may be rapidly migratory (larva currens), although purpura and petechiae as displayed by our patient appear to be under‐recognized findings in hyperinfection.2, 5 Gram‐negative bacillary meningitis has been well reported as a complication of migrating larvae, and a wide variety of other organs may rarely be involved.5, 10
The presence of chronic strongyloidiasis should be suspected in patients with ongoing gastrointestinal and/or pulmonary symptoms, or unexplained eosinophilia with a potential exposure history, such as immigrants from Southeast Asia. Diagnosis in these individuals is currently most often made serologically, although stool exam provides a somewhat higher specificity for active infection, at the expense of lower sensitivity.3, 11 In the setting of hyperinfection, stool studies are almost uniformly positive for S. stercoralis, and sputum may be diagnostic as well. Consequently, failure to reach the diagnosis usually reflects a lack of clinical suspicion.5
The therapy of choice for strongyloidiasis is currently ivermectin, with a single dose repeated once, 2 weeks later, highly efficacious in eradicating chronic infection. Treatment of hyperinfection is more challenging and less well studied, but clearly necessitates a more prolonged course of treatment. Many experts advocate treating until worms are no longer present in the stool; some have suggested the combination of ivermectin and albendazole as this patient received, although this has not been examined in controlled fashion.
The diagnosis of Strongyloides hyperinfection is typically delayed or missed because of the failure to consider it, with reported mortality rates as high as 50% in hyperinfection and 87% in disseminated disease.3, 12, 13 This patient fortunately was diagnosed, albeit in delayed fashion, proving the maxim better late than never. His case highlights the need for increased clinical awareness of strongyloidiasis, and specifically the need to consider the possibility of chronic Strongyloides infection prior to administering immunosuppressive medications. In particular, serologic screening of individuals from highly endemic areas for strongyloidiasis, when initiating extended courses of corticosteroids, seems prudent.13
Teaching Points
-
Chronic strongyloidiasis is common in the developing world (particularly Southeast Asia), and places infected individuals at significant risk of life‐threatening hyperinfection if not recognized and treated prior to the initiation of immunosuppressive medication, especially corticosteroids.
-
Strongyloides hyperinfection syndrome may be protean in its manifestations, but most commonly includes gastrointestinal, pulmonary, and cutaneous signs and symptoms.
- ,,, et al.Disseminated strongyloidiasis in immunocompromised patients—report of three cases.Int J Dermatol.2009;48(9):975–978.
- ,,, et al.Clinical manifestations of strongyloidiasis in southern Taiwan.J Microbiol Immunol Infect.2002;35(1):29–36.
- ,.Diagnosis of Strongyloides stercoralis infection.Clin Infect Dis.2001;33(7):1040–1047.
- ,,.Intestinal strongyloidiasis and hyperinfection syndrome.Clin Mol Allergy.2006;4:8.
- ,.Strongyloides stercoralis in the immunocompromised population.Clin Microbiol Rev.2004;17(1):208–217.
- ,,.Intestinal strongyloidiasis: recognition, management and determinants of outcome.J Clin Gastroenterol2005;39(3):203–211.
- .Dysregulation of strongyloidiasis: a new hypothesis.Clin Microbiol Rev.1992;5(4):345–355.
- ,,,.Consequences of captivity: health effects of Far East imprisonment in World War II.Q J Med.2009;102:87–96.
- ,,,.Strongyloides hyperinfection syndrome: an emerging global infectious disease.Trans R Soc Trop Med Hyg.2008;102(4):314–318.
- ,,,,,.Strongyloides hyperinfection presenting as acute respiratory failure and Gram‐negative sepsis.Chest.2005;128(5):3681–3684.
- ,,, et al.Use of enzyme‐linked immunosorbent assay and dipstick assay for detection of Strongyloides stercoralis infection in humans.J Clin Microbiol.2007;45:438–442.
- ,,,,,.Complicated and fatal Strongyloides infection in Canadians: risk factors, diagnosis and management.Can Med Assoc J.2004;171:479–484.
- ,,, et al.Maltreatment of Strongyloides infection: case series and worldwide physicians‐in‐training survey.Am J Med.2007;120(6):545.e1–545.e8.
A 59‐year‐old man presented to the emergency department with the acute onset of right‐sided abdominal and flank pain. The pain had begun the previous night, was constant and progressively worsening, and radiated to his right groin. He denied fever, nausea, emesis, or change in his bowel habits, but he did notice mild right lower quadrant discomfort with micturition. Upon further questioning, he also complained of mild dyspnea on climbing stairs and an unspecified recent weight loss.
The most common cause of acute severe right‐sided flank and abdominal pain radiating to the groin and associated with dysuria in a middle‐aged man is ureteral colic. Other etiologies important to consider include retrocecal appendicitis, pyelonephritis, and, rarely, a dissecting abdominal aortic aneurysm. This patient's seemingly recent onset exertional dyspnea and weight loss do not neatly fit any of the above, however.
His past medical history was significant for diabetes mellitus and pemphigus vulgaris diagnosed 7 months previously. He had been treated with prednisone, and the dose decreased from 100 to 60 mg daily, 1 month previously, due to poor glycemic control as well as steroid‐induced neuropathy and myopathy. His other medications included naproxen sodium and ibuprofen for back pain, azathioprine, insulin, pioglitazone, and glimiperide. He had no past surgical history. He had lived in the United States since his emigration from Thailand in 1971. His last trip to Thailand was 5 years previously. He was a taxi cab driver. He had a ten‐pack year history of tobacco use, but had quit 20 years prior. He denied history of alcohol or intravenous drug use.
Pemphigus vulgaris is unlikely to be directly related to this patient's presentation, but in light of his poorly controlled diabetes, his azathioprine use, and particularly his high‐dose corticosteroids, he is certainly immunocompromised. Accordingly, a disseminated infection, either newly acquired or reactivated, merits consideration. His history of residence in, and subsequent travel to, Southeast Asia raises the possibility of several diseases, each of which may be protean in their manifestations; these include tuberculosis, melioidosis, and penicilliosis (infection with Penicillium marneffei). The first two may reactivate long after initial exposure, particularly with insults to the immune system. The same is probably true of penicilliosis, although I am not certain of this. On a slightly less exotic note, domestically acquired infection with histoplasmosis or other endemic fungi is possible.
On examination he was afebrile, had a pulse of 130 beats per minute and a blood pressure of 65/46 mmHg. His oxygen saturation was 92%. He appeared markedly cushingoid, and had mild pallor and generalized weakness. Cardiopulmonary examination was unremarkable. His abdominal exam was notable for distention and hypoactive bowel sounds, with tenderness and firmness to palpation on the right side. Peripheral pulses were normal. Examination of the skin demonstrated ecchymoses over the bilateral forearms, and several healed pemphigus lesions on the abdomen and upper extremities.
The patient's severely deranged hemodynamic parameters indicate either current or impending shock, and resuscitative measures should proceed in tandem with diagnostic efforts. The cause of his shock seems most likely to be either hypovolemic (abdominal wall or intra‐abdominal hemorrhage, or conceivably massive third spacing from an intra‐abdominal catastrophe), or distributive (sepsis, or acute adrenal insufficiency if he has missed recent steroid doses). His ecchymoses may simply reflect chronic glucocorticoid use, but also raise suspicion for a coagulopathy. Provided the patient can be stabilized to allow this, I would urgently obtain a computed tomography (CT) scan of the abdomen and pelvis.
Initial laboratory studies demonstrated a hemoglobin of 9.1 g/dL, white blood cell count 8000/L with 33% bands, 48% segmented neutrophils, 18% lymphocytes, and 0.7% eosinophils, platelet count 356,000/L, sodium 128 mmol/L, BUN 52 mg/dL, creatinine 2.3 mg/dL, and glucose of 232 mg/dL. Coagulation studies were normal, and lactic acid was 1.8 mmol/L (normal range, 0.7‐2.1). Fibrinogen was normal at 591 and LDH was mildly elevated at 654 (normal range, 313‐618 U/L). Total protein and albumin were 3.6 and 1.9 g/dL, respectively. Total bilirubin was 0.6 mg/dL. Random serum cortisol was 20.2 g/dL. Liver enzymes, amylase, lipase, iron stores, B12, folate, and stool for occult blood were normal. Initial cardiac biomarkers were negative, but subsequent troponin‐I was 3.81 ng/mL (elevated, >1.00). Urinalysis showed 0‐4 white blood cells per high powered field.
The laboratory studies provide a variety of useful, albeit nonspecific, information. The high percentage of band forms on white blood cell differential further raises concern for an infectious process, although severe noninfectious stress can also cause this. While we do not know whether the patient's renal failure is acute, I suspect that it is, and may result from a variety of insults including sepsis, hypotension, and volume depletion. His moderately elevated troponin‐I likely reflects supplydemand mismatch or sepsis. I would like to see an electrocardiogram, and I remain very interested in obtaining abdominal imaging.
Chest radiography showed pulmonary vascular congestion without evidence of pneumothorax. Computed tomography scan of the abdomen and pelvis showed retroperitoneal fluid bilaterally (Figure 1). This was described as suspicious for ascites versus hemorrhage, but no obvious source of bleeding was identified. There was also a small amount of right perinephric fluid, but no evidence of a renal mass. The abdominal aorta was normal; there was no lymphadenopathy.
The CT image appears to speak against simple ascites, and seems most consistent with either blood or an infectious process. Consequently, the loculated right retroperitoneal collection should be aspirated, and fluid sent for fungal, acid‐fast, and modified acid‐fast (i.e., for Nocardia) stains and culture, in addition to Gram stain and routine aerobic and anaerobic cultures.
The patient was admitted to the intensive care unit. Stress‐dose steroids were administered, and he improved after resuscitation with fluid and blood. His renal function normalized. Urine and blood cultures returned negative. His hematocrit and multiple repeat CT scans of the abdomen remained stable. A retroperitoneal hemorrhage was diagnosed, and surgical intervention was deemed unnecessary. Both adenosine thallium stress test and echocardiogram were normal. He was continued on 60 mg prednisone daily and discharged home with outpatient follow‐up.
This degree of improvement with volume expansion (and steroids) suggests the patient was markedly volume depleted upon presentation. Although a formal adrenocorticotropic hormone (ACTH) stimulation test was apparently not performed, the random cortisol level suggests adrenal insufficiency was unlikely to have been primarily responsible. While retroperitoneal hemorrhage is possible, the loculated appearance of the collection suggests infection is more likely.
Three weeks later, he was readmitted with recurrent right‐sided abdominal and flank pain. His temperature was 101.3F, and he was tachycardic and hypotensive. His examination was similar to that at the time of his previous presentation. Laboratory data revealed white blood cell count of 13,100/L with 43% bands, hemoglobin of 9.2 g/dL, glucose of 343 mg/dL, bicarbonate 25 mmol/L, normal anion gap and renal function, and lactic acid of 4.5 mmol/L. Liver function tests were normal except for an albumin of 3.0 g/dL. CT scan of the abdomen revealed loculated retroperitoneal fluid collections, increased in size since the prior scan.
The patient is once again evidencing at least early shock, manifested in his deranged hemodynamics and elevated lactate level. I remain puzzled by the fact that he appeared to respond to fluids alone at the time of his initial hospital stay, unless adrenal insufficiency played a greater role than I suspected. Of note, acute adrenal insufficiency could explain much of the current picture, including fever, and bland (uninfected) hematomas are an underappreciated cause of both fever and leukocytosis. Having said this, I remain concerned that his retroperitoneal fluid collections represent abscesses. The most accessible of these should be sampled.
Aspiration of the retroperitoneal fluid yielded purulent material which grew Klebsiella pneumoniae. The cultures were negative for mycobacteria and fungus. Blood and urine cultures were negative. Drains were placed, and he was followed as an outpatient. His fever and leukocytosis subsided, and he completed a 6‐week course of trimethoprim‐sulfamethoxazole. CT imaging confirmed complete evacuation of the fluid.
Retroperitoneal abscesses frequently present in smoldering fashion, although patients may be quite ill by the time of presentation. Most of these are secondary, i.e., they arise from another abnormality in the retroperitoneum. Most commonly this is in the large bowel, kidney, pancreas, or spine. I would carefully scour his follow‐up imaging for additional clues and, if unrevealing, proceed to colonoscopy.
He returned 1 month after drain removal, with 2‐3 days of nausea and abdominal pain. His abdomen was moderately distended but nontender, and multiple persistent petechial and purpuric lesions were present on the upper back, chest, torso, and arms. Abdominal CT scan revealed small bowel obstruction and a collection of fluid in the left paracolic gutter extending into the left retrorenal space.
The patient does not appear to have obvious risk factors for developing a small bowel obstruction. No mention is made of the presence or absence of a transition point on the CT scan, and this should be ascertained. His left‐sided abdominal fluid collection is probably infectious in nature, and I continue to be suspicious of a large bowel (or distal small bowel) source, via either gut perforation or bacterial translocation. The collection needs to be percutaneously drained for both diagnostic and therapeutic reasons, and broadly cultured. Finally, we need to account for the described dermatologic manifestations. The purpuric/petechial lesions sound vasculitic rather than thrombocytopenic in origin based on location; conversely, they may simply reflect a corticosteroid‐related adverse effect. I would like to know whether the purpura was palpable, and to repeat a complete blood count with peripheral smear.
Laboratory data showed hemoglobin of 9.3 g/dL, a platelet count of 444,000/L, and normal coagulation studies. The purpura was nonpalpable (Figure 2). The patient had a nasogastric tube placed for decompression, with bilious drainage. His left retroperitoneal fluid was drained, with cultures yielding Enterococcus faecalis and Enterobacter cloacae. The patient was treated with a course of broad‐spectrum antibiotics. His obstruction improved and the retroperitoneal collection resolved on follow‐up imaging. However, 2 days later, he had recurrent pain; abdominal CT showed a recurrence of small bowel obstruction with an unequivocal transition point in the distal jejunum. A small fluid collection was noted in the left retroperitoneum with a trace of gas in it. He improved with nasogastric suction, his prednisone was tapered to 30 mg daily, and he was discharged home.
The isolation of both Enterococcus and Enterobacter species from his fluid collection, along with the previous isolation of Klebsiella, strongly suggest a bowel source for his recurrent abscesses. Based on this CT report, the patient has clear evidence of at least partial small bowel obstruction. He lacks a history of prior abdominal surgery or other more typical reasons for obstruction caused by extrinsic compression, such as hernia, although it is possible his recurrent abdominal infections may have led to obstruction due to scarring and adhesions. An intraluminal cause of obstruction also needs to be considered, with causes including malignancy (lymphoma, carcinoid, and adenocarcinoma), Crohn's disease, and infections including tuberculosis as well as parasites such as Taenia and Strongyloides. While the purpura is concerning, given the nonpalpable character along with a normal platelet count and coagulation studies, it may be reasonable to provisionally attribute it to high‐dose corticosteroid use.
He was admitted a fourth time a week after being discharged, with nausea, generalized weakness, and weight loss. At presentation, he had a blood pressure of 95/65 mmHg. His white blood cell count was 5,900/L, with 79% neutrophils and 20% bands. An AM cortisol was 18.8 /dL. He was thought to have adrenal insufficiency from steroid withdrawal, was treated with intravenous fluids and steroids, and discharged on a higher dose of prednisone at 60 mg daily. One week later, he again returned to the hospital with watery diarrhea, emesis, and generalized weakness. His blood pressure was 82/50 mmHg, and his abdomen appeared benign. He also had an erythematous rash over his mid‐abdomen. Laboratory data was significant for a sodium of 127 mmol/L, potassium of 3.0 mmol/L, chloride of 98 mmol/L, bicarbonate of 26 mmol/L, glucose of 40 mg/dL, lactate of 14 mmol/L, and albumin of 1.0 g/dL. Stool assay for Clostridium difficile was negative. A CT scan of the abdomen and pelvis showed small bilateral pleural effusions and small bowel fluid consistent with gastroenteritis, but without signs of obstruction. Esophagogastroduodenoscopy (EGD) showed bile backwash into the stomach, as well as inflammatory changes in the proximal and mid‐stomach, and inflammatory reaction and edema in the proximal duodenum. Colonoscopy showed normal appearing ileum and colon.
The patient's latest laboratory values appear to reflect his chronic illness and superimposed diarrhea. I am perplexed by his markedly elevated serum lactate value in association with a normal bicarbonate and low anion gap, and would repeat the lactate level to ensure this is not spurious. His hypoglycemia probably reflects a failure to adjust or discontinue his diabetic medications, although both hypoglycemia and type B lactic acidosis are occasionally manifestations of a paraneoplastic syndrome. The normal colonoscopy findings are helpful in exonerating the colon, provided the preparation was adequate. Presumably, the abnormal areas of the stomach and duodenum were biopsied; I remain suspicious that the answer may lie in the jejunum.
The patient was treated with intravenous fluids and stress‐dose steroids, and electrolyte abnormalities were corrected. Biopsies from the EGD and colonoscopy demonstrated numerous larvae within the mucosa of the body and antrum of the stomach, as well as duodenum. There were also rare detached larvae seen in the esophagus, and a few larvae within the ileal mucosa.
The patient appears to have Strongyloides hyperinfection, something he is at clear risk for, given his country of origin and his high‐dose corticosteroids. In retrospect, I was dissuaded from seriously considering a diagnosis of parasitic infection in large part because of the absence of peripheral eosinophilia, but this may not be seen in cases of hyperinfection. Additional clues, again in retrospect, were the repeated abscesses with bowel flora and the seemingly nonspecific abdominal rash. I would treat with a course of ivermectin, and carefully monitor his response.
The characteristics of the larvae were suggestive of Strongyloides species (Figure 3). A subsequent stool test for ova and parasites was positive for Strongyloides larvae. The patient was given a single dose of ivermectin. An endocrinology consultant felt that he did not have adrenal insufficiency, and it was recommended that his steroids be tapered off. He was discharged home once he clinically improved.
Although one or two doses of ivermectin typically suffices for uncomplicated strongyloidiasis, the risk of failure in hyperinfection mandates a longer treatment course. I don't believe this patient has been adequately treated, although the removal of his steroids will be helpful.
He was readmitted 3 days later with recrudescent symptoms, and his stool remained positive for Strongyloides. He received 2 weeks of ivermectin and albendazole, and was ultimately discharged to a rehabilitation facility after a complicated hospital stay. Nine months later, the patient was reported to be doing well.
COMMENTARY
This patient's immigration status from the developing world, high‐dose corticosteroid use, and complex clinical course all suggested the possibility of an underlying chronic infectious process. Although the discussant recognized this early on and later briefly mentioned strongyloidiasis as a potential cause of intestinal obstruction, the diagnosis of Strongyloides hyperinfection was not suspected until incontrovertible evidence for it was obtained on EGD. Failure to make the diagnosis earlier by both the involved clinicians and the discussant probably stemmed largely from two factors: the absence of eosinophilia; and lack of recognition that purpura may be seen in cases of hyperinfection, presumably reflecting larval infiltration of the dermis.1 Although eosinophilia accompanies most cases of stronglyloidiasis and may be very pronounced, patients with hyperinfection syndrome frequently fail to mount an eosinophilic response due to underlying immunosuppression, with eosinophilia absent in 70% of such patients in a study from Taiwan.2
Strongyloides stercoralis is an intestinal nematode that causes strongyloidiasis. It affects as many as 100 million people globally,3 mainly in tropical and subtropical areas, but is also endemic in the Southeastern United States, Europe, and Japan. Risk factors include male sex, White race, alcoholism, working in contact with soil (farmers, coal mine workers, etc.), chronic care institutionalization, and low socioeconomic status. In nonendemic regions, it more commonly affects travelers, immigrants, or military personnel.4, 5
The life cycle of S. stercoralis is complex. Infective larvae penetrate the skin through contact with contaminated soil, enter the venous system via lymphatics, and travel to the lung.4, 6 Here, they ascend the tracheobronchial tree and migrate to the gut. In the intestine, larvae develop into adult female worms that burrow into the intestinal mucosa. These worms lay eggs that develop into noninfective rhabditiform larvae, which are then expelled in the stool. Some of the rhabditiform larvae, however, develop into infective filariform larvae, which may penetrate colonic mucosa or perianal skin, enter the bloodstream, and lead to the cycle of autoinfection and chronic strongyloidiasis (carrier state). Autoinfection typically involves a low parasite burden, and is controlled by both host immune factors as well as parasitic factors.7 The mechanism of autoinfection can lead to the persistence of strongyloidiasis for decades after the initial infection, as has been documented in former World War II prisoners of war.8
Factors leading to the impairment of cell‐mediated immunity predispose chronically infected individuals to hyperinfection, as occurred in this patient. The most important of these are corticosteroid administration and Human T‐lymphotropic virus Type‐1 (HTLV‐1) infection, both of which cause significant derangement in TH1/TH2 immune system balance.5, 9 In the hyperinfection syndrome, the burden of parasites increases dramatically, leading to a variety of clinical manifestations. Gastrointestinal phenomena frequently predominate, including watery diarrhea, anorexia, weight loss, nausea/vomiting, gastrointestinal bleeding, and occasionally small bowel obstruction. Pulmonary manifestations are likewise common, and include cough, dyspnea, and wheezing. Cutaneous findings are not uncommon, classically pruritic linear lesions of the abdomen, buttocks, and lower extremities which may be rapidly migratory (larva currens), although purpura and petechiae as displayed by our patient appear to be under‐recognized findings in hyperinfection.2, 5 Gram‐negative bacillary meningitis has been well reported as a complication of migrating larvae, and a wide variety of other organs may rarely be involved.5, 10
The presence of chronic strongyloidiasis should be suspected in patients with ongoing gastrointestinal and/or pulmonary symptoms, or unexplained eosinophilia with a potential exposure history, such as immigrants from Southeast Asia. Diagnosis in these individuals is currently most often made serologically, although stool exam provides a somewhat higher specificity for active infection, at the expense of lower sensitivity.3, 11 In the setting of hyperinfection, stool studies are almost uniformly positive for S. stercoralis, and sputum may be diagnostic as well. Consequently, failure to reach the diagnosis usually reflects a lack of clinical suspicion.5
The therapy of choice for strongyloidiasis is currently ivermectin, with a single dose repeated once, 2 weeks later, highly efficacious in eradicating chronic infection. Treatment of hyperinfection is more challenging and less well studied, but clearly necessitates a more prolonged course of treatment. Many experts advocate treating until worms are no longer present in the stool; some have suggested the combination of ivermectin and albendazole as this patient received, although this has not been examined in controlled fashion.
The diagnosis of Strongyloides hyperinfection is typically delayed or missed because of the failure to consider it, with reported mortality rates as high as 50% in hyperinfection and 87% in disseminated disease.3, 12, 13 This patient fortunately was diagnosed, albeit in delayed fashion, proving the maxim better late than never. His case highlights the need for increased clinical awareness of strongyloidiasis, and specifically the need to consider the possibility of chronic Strongyloides infection prior to administering immunosuppressive medications. In particular, serologic screening of individuals from highly endemic areas for strongyloidiasis, when initiating extended courses of corticosteroids, seems prudent.13
Teaching Points
-
Chronic strongyloidiasis is common in the developing world (particularly Southeast Asia), and places infected individuals at significant risk of life‐threatening hyperinfection if not recognized and treated prior to the initiation of immunosuppressive medication, especially corticosteroids.
-
Strongyloides hyperinfection syndrome may be protean in its manifestations, but most commonly includes gastrointestinal, pulmonary, and cutaneous signs and symptoms.
A 59‐year‐old man presented to the emergency department with the acute onset of right‐sided abdominal and flank pain. The pain had begun the previous night, was constant and progressively worsening, and radiated to his right groin. He denied fever, nausea, emesis, or change in his bowel habits, but he did notice mild right lower quadrant discomfort with micturition. Upon further questioning, he also complained of mild dyspnea on climbing stairs and an unspecified recent weight loss.
The most common cause of acute severe right‐sided flank and abdominal pain radiating to the groin and associated with dysuria in a middle‐aged man is ureteral colic. Other etiologies important to consider include retrocecal appendicitis, pyelonephritis, and, rarely, a dissecting abdominal aortic aneurysm. This patient's seemingly recent onset exertional dyspnea and weight loss do not neatly fit any of the above, however.
His past medical history was significant for diabetes mellitus and pemphigus vulgaris diagnosed 7 months previously. He had been treated with prednisone, and the dose decreased from 100 to 60 mg daily, 1 month previously, due to poor glycemic control as well as steroid‐induced neuropathy and myopathy. His other medications included naproxen sodium and ibuprofen for back pain, azathioprine, insulin, pioglitazone, and glimiperide. He had no past surgical history. He had lived in the United States since his emigration from Thailand in 1971. His last trip to Thailand was 5 years previously. He was a taxi cab driver. He had a ten‐pack year history of tobacco use, but had quit 20 years prior. He denied history of alcohol or intravenous drug use.
Pemphigus vulgaris is unlikely to be directly related to this patient's presentation, but in light of his poorly controlled diabetes, his azathioprine use, and particularly his high‐dose corticosteroids, he is certainly immunocompromised. Accordingly, a disseminated infection, either newly acquired or reactivated, merits consideration. His history of residence in, and subsequent travel to, Southeast Asia raises the possibility of several diseases, each of which may be protean in their manifestations; these include tuberculosis, melioidosis, and penicilliosis (infection with Penicillium marneffei). The first two may reactivate long after initial exposure, particularly with insults to the immune system. The same is probably true of penicilliosis, although I am not certain of this. On a slightly less exotic note, domestically acquired infection with histoplasmosis or other endemic fungi is possible.
On examination he was afebrile, had a pulse of 130 beats per minute and a blood pressure of 65/46 mmHg. His oxygen saturation was 92%. He appeared markedly cushingoid, and had mild pallor and generalized weakness. Cardiopulmonary examination was unremarkable. His abdominal exam was notable for distention and hypoactive bowel sounds, with tenderness and firmness to palpation on the right side. Peripheral pulses were normal. Examination of the skin demonstrated ecchymoses over the bilateral forearms, and several healed pemphigus lesions on the abdomen and upper extremities.
The patient's severely deranged hemodynamic parameters indicate either current or impending shock, and resuscitative measures should proceed in tandem with diagnostic efforts. The cause of his shock seems most likely to be either hypovolemic (abdominal wall or intra‐abdominal hemorrhage, or conceivably massive third spacing from an intra‐abdominal catastrophe), or distributive (sepsis, or acute adrenal insufficiency if he has missed recent steroid doses). His ecchymoses may simply reflect chronic glucocorticoid use, but also raise suspicion for a coagulopathy. Provided the patient can be stabilized to allow this, I would urgently obtain a computed tomography (CT) scan of the abdomen and pelvis.
Initial laboratory studies demonstrated a hemoglobin of 9.1 g/dL, white blood cell count 8000/L with 33% bands, 48% segmented neutrophils, 18% lymphocytes, and 0.7% eosinophils, platelet count 356,000/L, sodium 128 mmol/L, BUN 52 mg/dL, creatinine 2.3 mg/dL, and glucose of 232 mg/dL. Coagulation studies were normal, and lactic acid was 1.8 mmol/L (normal range, 0.7‐2.1). Fibrinogen was normal at 591 and LDH was mildly elevated at 654 (normal range, 313‐618 U/L). Total protein and albumin were 3.6 and 1.9 g/dL, respectively. Total bilirubin was 0.6 mg/dL. Random serum cortisol was 20.2 g/dL. Liver enzymes, amylase, lipase, iron stores, B12, folate, and stool for occult blood were normal. Initial cardiac biomarkers were negative, but subsequent troponin‐I was 3.81 ng/mL (elevated, >1.00). Urinalysis showed 0‐4 white blood cells per high powered field.
The laboratory studies provide a variety of useful, albeit nonspecific, information. The high percentage of band forms on white blood cell differential further raises concern for an infectious process, although severe noninfectious stress can also cause this. While we do not know whether the patient's renal failure is acute, I suspect that it is, and may result from a variety of insults including sepsis, hypotension, and volume depletion. His moderately elevated troponin‐I likely reflects supplydemand mismatch or sepsis. I would like to see an electrocardiogram, and I remain very interested in obtaining abdominal imaging.
Chest radiography showed pulmonary vascular congestion without evidence of pneumothorax. Computed tomography scan of the abdomen and pelvis showed retroperitoneal fluid bilaterally (Figure 1). This was described as suspicious for ascites versus hemorrhage, but no obvious source of bleeding was identified. There was also a small amount of right perinephric fluid, but no evidence of a renal mass. The abdominal aorta was normal; there was no lymphadenopathy.
The CT image appears to speak against simple ascites, and seems most consistent with either blood or an infectious process. Consequently, the loculated right retroperitoneal collection should be aspirated, and fluid sent for fungal, acid‐fast, and modified acid‐fast (i.e., for Nocardia) stains and culture, in addition to Gram stain and routine aerobic and anaerobic cultures.
The patient was admitted to the intensive care unit. Stress‐dose steroids were administered, and he improved after resuscitation with fluid and blood. His renal function normalized. Urine and blood cultures returned negative. His hematocrit and multiple repeat CT scans of the abdomen remained stable. A retroperitoneal hemorrhage was diagnosed, and surgical intervention was deemed unnecessary. Both adenosine thallium stress test and echocardiogram were normal. He was continued on 60 mg prednisone daily and discharged home with outpatient follow‐up.
This degree of improvement with volume expansion (and steroids) suggests the patient was markedly volume depleted upon presentation. Although a formal adrenocorticotropic hormone (ACTH) stimulation test was apparently not performed, the random cortisol level suggests adrenal insufficiency was unlikely to have been primarily responsible. While retroperitoneal hemorrhage is possible, the loculated appearance of the collection suggests infection is more likely.
Three weeks later, he was readmitted with recurrent right‐sided abdominal and flank pain. His temperature was 101.3F, and he was tachycardic and hypotensive. His examination was similar to that at the time of his previous presentation. Laboratory data revealed white blood cell count of 13,100/L with 43% bands, hemoglobin of 9.2 g/dL, glucose of 343 mg/dL, bicarbonate 25 mmol/L, normal anion gap and renal function, and lactic acid of 4.5 mmol/L. Liver function tests were normal except for an albumin of 3.0 g/dL. CT scan of the abdomen revealed loculated retroperitoneal fluid collections, increased in size since the prior scan.
The patient is once again evidencing at least early shock, manifested in his deranged hemodynamics and elevated lactate level. I remain puzzled by the fact that he appeared to respond to fluids alone at the time of his initial hospital stay, unless adrenal insufficiency played a greater role than I suspected. Of note, acute adrenal insufficiency could explain much of the current picture, including fever, and bland (uninfected) hematomas are an underappreciated cause of both fever and leukocytosis. Having said this, I remain concerned that his retroperitoneal fluid collections represent abscesses. The most accessible of these should be sampled.
Aspiration of the retroperitoneal fluid yielded purulent material which grew Klebsiella pneumoniae. The cultures were negative for mycobacteria and fungus. Blood and urine cultures were negative. Drains were placed, and he was followed as an outpatient. His fever and leukocytosis subsided, and he completed a 6‐week course of trimethoprim‐sulfamethoxazole. CT imaging confirmed complete evacuation of the fluid.
Retroperitoneal abscesses frequently present in smoldering fashion, although patients may be quite ill by the time of presentation. Most of these are secondary, i.e., they arise from another abnormality in the retroperitoneum. Most commonly this is in the large bowel, kidney, pancreas, or spine. I would carefully scour his follow‐up imaging for additional clues and, if unrevealing, proceed to colonoscopy.
He returned 1 month after drain removal, with 2‐3 days of nausea and abdominal pain. His abdomen was moderately distended but nontender, and multiple persistent petechial and purpuric lesions were present on the upper back, chest, torso, and arms. Abdominal CT scan revealed small bowel obstruction and a collection of fluid in the left paracolic gutter extending into the left retrorenal space.
The patient does not appear to have obvious risk factors for developing a small bowel obstruction. No mention is made of the presence or absence of a transition point on the CT scan, and this should be ascertained. His left‐sided abdominal fluid collection is probably infectious in nature, and I continue to be suspicious of a large bowel (or distal small bowel) source, via either gut perforation or bacterial translocation. The collection needs to be percutaneously drained for both diagnostic and therapeutic reasons, and broadly cultured. Finally, we need to account for the described dermatologic manifestations. The purpuric/petechial lesions sound vasculitic rather than thrombocytopenic in origin based on location; conversely, they may simply reflect a corticosteroid‐related adverse effect. I would like to know whether the purpura was palpable, and to repeat a complete blood count with peripheral smear.
Laboratory data showed hemoglobin of 9.3 g/dL, a platelet count of 444,000/L, and normal coagulation studies. The purpura was nonpalpable (Figure 2). The patient had a nasogastric tube placed for decompression, with bilious drainage. His left retroperitoneal fluid was drained, with cultures yielding Enterococcus faecalis and Enterobacter cloacae. The patient was treated with a course of broad‐spectrum antibiotics. His obstruction improved and the retroperitoneal collection resolved on follow‐up imaging. However, 2 days later, he had recurrent pain; abdominal CT showed a recurrence of small bowel obstruction with an unequivocal transition point in the distal jejunum. A small fluid collection was noted in the left retroperitoneum with a trace of gas in it. He improved with nasogastric suction, his prednisone was tapered to 30 mg daily, and he was discharged home.
The isolation of both Enterococcus and Enterobacter species from his fluid collection, along with the previous isolation of Klebsiella, strongly suggest a bowel source for his recurrent abscesses. Based on this CT report, the patient has clear evidence of at least partial small bowel obstruction. He lacks a history of prior abdominal surgery or other more typical reasons for obstruction caused by extrinsic compression, such as hernia, although it is possible his recurrent abdominal infections may have led to obstruction due to scarring and adhesions. An intraluminal cause of obstruction also needs to be considered, with causes including malignancy (lymphoma, carcinoid, and adenocarcinoma), Crohn's disease, and infections including tuberculosis as well as parasites such as Taenia and Strongyloides. While the purpura is concerning, given the nonpalpable character along with a normal platelet count and coagulation studies, it may be reasonable to provisionally attribute it to high‐dose corticosteroid use.
He was admitted a fourth time a week after being discharged, with nausea, generalized weakness, and weight loss. At presentation, he had a blood pressure of 95/65 mmHg. His white blood cell count was 5,900/L, with 79% neutrophils and 20% bands. An AM cortisol was 18.8 /dL. He was thought to have adrenal insufficiency from steroid withdrawal, was treated with intravenous fluids and steroids, and discharged on a higher dose of prednisone at 60 mg daily. One week later, he again returned to the hospital with watery diarrhea, emesis, and generalized weakness. His blood pressure was 82/50 mmHg, and his abdomen appeared benign. He also had an erythematous rash over his mid‐abdomen. Laboratory data was significant for a sodium of 127 mmol/L, potassium of 3.0 mmol/L, chloride of 98 mmol/L, bicarbonate of 26 mmol/L, glucose of 40 mg/dL, lactate of 14 mmol/L, and albumin of 1.0 g/dL. Stool assay for Clostridium difficile was negative. A CT scan of the abdomen and pelvis showed small bilateral pleural effusions and small bowel fluid consistent with gastroenteritis, but without signs of obstruction. Esophagogastroduodenoscopy (EGD) showed bile backwash into the stomach, as well as inflammatory changes in the proximal and mid‐stomach, and inflammatory reaction and edema in the proximal duodenum. Colonoscopy showed normal appearing ileum and colon.
The patient's latest laboratory values appear to reflect his chronic illness and superimposed diarrhea. I am perplexed by his markedly elevated serum lactate value in association with a normal bicarbonate and low anion gap, and would repeat the lactate level to ensure this is not spurious. His hypoglycemia probably reflects a failure to adjust or discontinue his diabetic medications, although both hypoglycemia and type B lactic acidosis are occasionally manifestations of a paraneoplastic syndrome. The normal colonoscopy findings are helpful in exonerating the colon, provided the preparation was adequate. Presumably, the abnormal areas of the stomach and duodenum were biopsied; I remain suspicious that the answer may lie in the jejunum.
The patient was treated with intravenous fluids and stress‐dose steroids, and electrolyte abnormalities were corrected. Biopsies from the EGD and colonoscopy demonstrated numerous larvae within the mucosa of the body and antrum of the stomach, as well as duodenum. There were also rare detached larvae seen in the esophagus, and a few larvae within the ileal mucosa.
The patient appears to have Strongyloides hyperinfection, something he is at clear risk for, given his country of origin and his high‐dose corticosteroids. In retrospect, I was dissuaded from seriously considering a diagnosis of parasitic infection in large part because of the absence of peripheral eosinophilia, but this may not be seen in cases of hyperinfection. Additional clues, again in retrospect, were the repeated abscesses with bowel flora and the seemingly nonspecific abdominal rash. I would treat with a course of ivermectin, and carefully monitor his response.
The characteristics of the larvae were suggestive of Strongyloides species (Figure 3). A subsequent stool test for ova and parasites was positive for Strongyloides larvae. The patient was given a single dose of ivermectin. An endocrinology consultant felt that he did not have adrenal insufficiency, and it was recommended that his steroids be tapered off. He was discharged home once he clinically improved.
Although one or two doses of ivermectin typically suffices for uncomplicated strongyloidiasis, the risk of failure in hyperinfection mandates a longer treatment course. I don't believe this patient has been adequately treated, although the removal of his steroids will be helpful.
He was readmitted 3 days later with recrudescent symptoms, and his stool remained positive for Strongyloides. He received 2 weeks of ivermectin and albendazole, and was ultimately discharged to a rehabilitation facility after a complicated hospital stay. Nine months later, the patient was reported to be doing well.
COMMENTARY
This patient's immigration status from the developing world, high‐dose corticosteroid use, and complex clinical course all suggested the possibility of an underlying chronic infectious process. Although the discussant recognized this early on and later briefly mentioned strongyloidiasis as a potential cause of intestinal obstruction, the diagnosis of Strongyloides hyperinfection was not suspected until incontrovertible evidence for it was obtained on EGD. Failure to make the diagnosis earlier by both the involved clinicians and the discussant probably stemmed largely from two factors: the absence of eosinophilia; and lack of recognition that purpura may be seen in cases of hyperinfection, presumably reflecting larval infiltration of the dermis.1 Although eosinophilia accompanies most cases of stronglyloidiasis and may be very pronounced, patients with hyperinfection syndrome frequently fail to mount an eosinophilic response due to underlying immunosuppression, with eosinophilia absent in 70% of such patients in a study from Taiwan.2
Strongyloides stercoralis is an intestinal nematode that causes strongyloidiasis. It affects as many as 100 million people globally,3 mainly in tropical and subtropical areas, but is also endemic in the Southeastern United States, Europe, and Japan. Risk factors include male sex, White race, alcoholism, working in contact with soil (farmers, coal mine workers, etc.), chronic care institutionalization, and low socioeconomic status. In nonendemic regions, it more commonly affects travelers, immigrants, or military personnel.4, 5
The life cycle of S. stercoralis is complex. Infective larvae penetrate the skin through contact with contaminated soil, enter the venous system via lymphatics, and travel to the lung.4, 6 Here, they ascend the tracheobronchial tree and migrate to the gut. In the intestine, larvae develop into adult female worms that burrow into the intestinal mucosa. These worms lay eggs that develop into noninfective rhabditiform larvae, which are then expelled in the stool. Some of the rhabditiform larvae, however, develop into infective filariform larvae, which may penetrate colonic mucosa or perianal skin, enter the bloodstream, and lead to the cycle of autoinfection and chronic strongyloidiasis (carrier state). Autoinfection typically involves a low parasite burden, and is controlled by both host immune factors as well as parasitic factors.7 The mechanism of autoinfection can lead to the persistence of strongyloidiasis for decades after the initial infection, as has been documented in former World War II prisoners of war.8
Factors leading to the impairment of cell‐mediated immunity predispose chronically infected individuals to hyperinfection, as occurred in this patient. The most important of these are corticosteroid administration and Human T‐lymphotropic virus Type‐1 (HTLV‐1) infection, both of which cause significant derangement in TH1/TH2 immune system balance.5, 9 In the hyperinfection syndrome, the burden of parasites increases dramatically, leading to a variety of clinical manifestations. Gastrointestinal phenomena frequently predominate, including watery diarrhea, anorexia, weight loss, nausea/vomiting, gastrointestinal bleeding, and occasionally small bowel obstruction. Pulmonary manifestations are likewise common, and include cough, dyspnea, and wheezing. Cutaneous findings are not uncommon, classically pruritic linear lesions of the abdomen, buttocks, and lower extremities which may be rapidly migratory (larva currens), although purpura and petechiae as displayed by our patient appear to be under‐recognized findings in hyperinfection.2, 5 Gram‐negative bacillary meningitis has been well reported as a complication of migrating larvae, and a wide variety of other organs may rarely be involved.5, 10
The presence of chronic strongyloidiasis should be suspected in patients with ongoing gastrointestinal and/or pulmonary symptoms, or unexplained eosinophilia with a potential exposure history, such as immigrants from Southeast Asia. Diagnosis in these individuals is currently most often made serologically, although stool exam provides a somewhat higher specificity for active infection, at the expense of lower sensitivity.3, 11 In the setting of hyperinfection, stool studies are almost uniformly positive for S. stercoralis, and sputum may be diagnostic as well. Consequently, failure to reach the diagnosis usually reflects a lack of clinical suspicion.5
The therapy of choice for strongyloidiasis is currently ivermectin, with a single dose repeated once, 2 weeks later, highly efficacious in eradicating chronic infection. Treatment of hyperinfection is more challenging and less well studied, but clearly necessitates a more prolonged course of treatment. Many experts advocate treating until worms are no longer present in the stool; some have suggested the combination of ivermectin and albendazole as this patient received, although this has not been examined in controlled fashion.
The diagnosis of Strongyloides hyperinfection is typically delayed or missed because of the failure to consider it, with reported mortality rates as high as 50% in hyperinfection and 87% in disseminated disease.3, 12, 13 This patient fortunately was diagnosed, albeit in delayed fashion, proving the maxim better late than never. His case highlights the need for increased clinical awareness of strongyloidiasis, and specifically the need to consider the possibility of chronic Strongyloides infection prior to administering immunosuppressive medications. In particular, serologic screening of individuals from highly endemic areas for strongyloidiasis, when initiating extended courses of corticosteroids, seems prudent.13
Teaching Points
-
Chronic strongyloidiasis is common in the developing world (particularly Southeast Asia), and places infected individuals at significant risk of life‐threatening hyperinfection if not recognized and treated prior to the initiation of immunosuppressive medication, especially corticosteroids.
-
Strongyloides hyperinfection syndrome may be protean in its manifestations, but most commonly includes gastrointestinal, pulmonary, and cutaneous signs and symptoms.
- ,,, et al.Disseminated strongyloidiasis in immunocompromised patients—report of three cases.Int J Dermatol.2009;48(9):975–978.
- ,,, et al.Clinical manifestations of strongyloidiasis in southern Taiwan.J Microbiol Immunol Infect.2002;35(1):29–36.
- ,.Diagnosis of Strongyloides stercoralis infection.Clin Infect Dis.2001;33(7):1040–1047.
- ,,.Intestinal strongyloidiasis and hyperinfection syndrome.Clin Mol Allergy.2006;4:8.
- ,.Strongyloides stercoralis in the immunocompromised population.Clin Microbiol Rev.2004;17(1):208–217.
- ,,.Intestinal strongyloidiasis: recognition, management and determinants of outcome.J Clin Gastroenterol2005;39(3):203–211.
- .Dysregulation of strongyloidiasis: a new hypothesis.Clin Microbiol Rev.1992;5(4):345–355.
- ,,,.Consequences of captivity: health effects of Far East imprisonment in World War II.Q J Med.2009;102:87–96.
- ,,,.Strongyloides hyperinfection syndrome: an emerging global infectious disease.Trans R Soc Trop Med Hyg.2008;102(4):314–318.
- ,,,,,.Strongyloides hyperinfection presenting as acute respiratory failure and Gram‐negative sepsis.Chest.2005;128(5):3681–3684.
- ,,, et al.Use of enzyme‐linked immunosorbent assay and dipstick assay for detection of Strongyloides stercoralis infection in humans.J Clin Microbiol.2007;45:438–442.
- ,,,,,.Complicated and fatal Strongyloides infection in Canadians: risk factors, diagnosis and management.Can Med Assoc J.2004;171:479–484.
- ,,, et al.Maltreatment of Strongyloides infection: case series and worldwide physicians‐in‐training survey.Am J Med.2007;120(6):545.e1–545.e8.
- ,,, et al.Disseminated strongyloidiasis in immunocompromised patients—report of three cases.Int J Dermatol.2009;48(9):975–978.
- ,,, et al.Clinical manifestations of strongyloidiasis in southern Taiwan.J Microbiol Immunol Infect.2002;35(1):29–36.
- ,.Diagnosis of Strongyloides stercoralis infection.Clin Infect Dis.2001;33(7):1040–1047.
- ,,.Intestinal strongyloidiasis and hyperinfection syndrome.Clin Mol Allergy.2006;4:8.
- ,.Strongyloides stercoralis in the immunocompromised population.Clin Microbiol Rev.2004;17(1):208–217.
- ,,.Intestinal strongyloidiasis: recognition, management and determinants of outcome.J Clin Gastroenterol2005;39(3):203–211.
- .Dysregulation of strongyloidiasis: a new hypothesis.Clin Microbiol Rev.1992;5(4):345–355.
- ,,,.Consequences of captivity: health effects of Far East imprisonment in World War II.Q J Med.2009;102:87–96.
- ,,,.Strongyloides hyperinfection syndrome: an emerging global infectious disease.Trans R Soc Trop Med Hyg.2008;102(4):314–318.
- ,,,,,.Strongyloides hyperinfection presenting as acute respiratory failure and Gram‐negative sepsis.Chest.2005;128(5):3681–3684.
- ,,, et al.Use of enzyme‐linked immunosorbent assay and dipstick assay for detection of Strongyloides stercoralis infection in humans.J Clin Microbiol.2007;45:438–442.
- ,,,,,.Complicated and fatal Strongyloides infection in Canadians: risk factors, diagnosis and management.Can Med Assoc J.2004;171:479–484.
- ,,, et al.Maltreatment of Strongyloides infection: case series and worldwide physicians‐in‐training survey.Am J Med.2007;120(6):545.e1–545.e8.
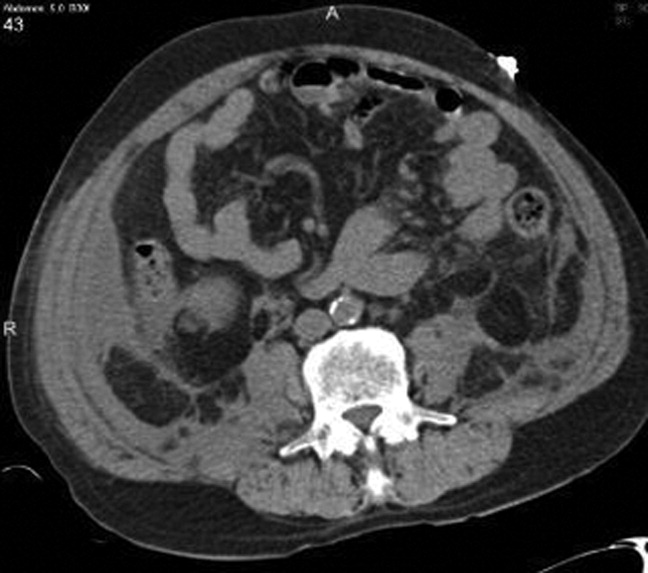
How Do I Determine if My Patient has Decision-Making Capacity?
Case
A 79-year-old male with coronary artery disease, hypertension, non-insulin-dependent mellitus, moderate dementia, and chronic renal insufficiency is admitted after a fall evaluation. He is widowed and lives in an assisted living facility. He’s accompanied by his niece, is alert, and oriented to person. He thinks he is in a clinic and is unable to state the year, but the remainder of the examination is unremarkable. His labs are notable for potassium of 6.3 mmol/L, BUN of 78 mg/dL, and Cr of 3.7 mg/dL. The niece reports that the patient is not fond of medical care, thus the most recent labs are from two years ago (and indicate a BUN of 39 and Cr of 2.8, with an upward trend over the past decade). You discuss possible long-term need for dialysis with the patient and niece, and the patient clearly states "no." However, he also states that it is 1988. How do you determine if he has the capacity to make decisions?
Overview
Hospitalists are familiar with the doctrine of informed consent—describing a disease, treatment options, associated risks and benefits, potential for complications, and alternatives, including no treatment. Not only must the patient be informed, and the decision free from any coercion, but the patient also must have capacity to make the decision.
Hospitalists often care for patients in whom decision-making capacity comes into question. This includes populations with depression, psychosis, dementia, stroke, severe personality disorders, developmental delay, comatose patients, as well as those with impaired attentional capacity (e.g. acute pain) or general debility (e.g. metastatic cancer).1,2
ave for the comatose patient, whether the patient has capacity might not be obvious. However, addressing the components of capacity (communication, understanding, appreciation, and rationalization) by using a validated clinical tool, such as the MacCAT-T, or more simply by systematically applying those four components to the clinical scenario under consideration, hospitalists can make this determination.
Review of the Literature
It is important to differentiate capacity from competency. Competency is a global assessment and a legal determination made by a judge in court. Capacity, on the other hand, is a functional assessment regarding a particular decision. Capacity is not static, and it can be performed by any clinician familiar with the patient. A hospitalist often is well positioned to make a capacity determination given established rapport with the patient and familiarity with the details of the case.
To make this determination, a hospitalist needs to know how to assess capacity. Although capacity usually is defined by state law and varies by jurisdiction, clinicians generally can assume it includes one or more of the four key components:
- Communication. The patient needs to be able to express a treatment choice, and this decision needs to be stable enough for the treatment to be implemented. Changing one’s decision in itself would not bring a patient’s capacity into question, so long as the patient was able to explain the rationale behind the switch. Frequent changes back and forth in the decision-making, however, could be indicative of an underlying psychiatric disorder or extreme indecision, which could bring capacity into question.
- Understanding. The patient needs to recall conversations about treatment, to make the link between causal relationships, and to process probabilities for outcomes. Problems with memory, attention span, and intelligence can affect one’s understanding.
- Appreciation. The patient should be able to identify the illness, treatment options, and likely outcomes as things that will affect him or her directly. A lack of appreciation usually stems from a denial based on intelligence (lack of a capability to understand) or emotion, or a delusion that the patient is not affected by this situation the same way and will have a different outcome.
- Rationalization or reasoning. The patient needs to be able to weigh the risks and benefits of the treatment options presented to come to a conclusion in keeping with their goals and best interests, as defined by their personal set of values. This often is affected in psychosis, depression, anxiety, phobias, delirium, and dementia.3
Several clinical capacity tools have been developed to assess these components:
Clinical tools.
The Mini-Mental Status Examination (MMSE) is a bedside test of a patient’s cognitive function, with scores ranging from 0 to 30.4 Although it wasn’t developed for assessing decision-making capacity, it has been compared with expert evaluation for assessment of capacity; the test performs reasonably well, particularly with high and low scores. Specifically, a MMSE >24 has a negative likelihood ratio (LR) of 0.05 for lack of capacity, while a MMSE <16 has a positive LR of 15.5 Scores from 17 to 23 do not correlate well with capacity, and further testing would be necessary. It is easy to administer, requires no formal training, and is familiar to most hospitalists. However, it does not address any specific aspects of informed consent, such as understanding or choice, and has not been validated in patients with mental illness.
The MacArthur Competence Assessment Tools for Treatment (MacCAT-T) is regarded as the gold standard for capacity assessment aids. It utilizes hospital chart review followed by a semi-structured interview to address clinical issues relevant to the patient being assessed; it takes 15 to 20 minutes to complete.6 The test provides scores in each of the four domains (choice, understanding, appreciation, and reasoning) of capacity. It has been validated in patients with dementia, schizophrenia, and depression. Limiting its clinical applicability is the fact that the MacCAT-T requires training to administer and interpret the results, though this is a relatively brief process.
The Capacity to Consent to Treatment Instrument (CCTI) uses hypothetical clinical vignettes in a structured interview to assess capacity across all four domains. The tool was developed and validated in patients with dementia and Parkinson’s disease, and takes 20 to 25 minutes to complete.7 A potential limitation is the CCTI’s use of vignettes as opposed to a patient-specific discussion, which could lead to different patient answers and a false assessment of the patient’s capacity.
The Hopemont Capacity Assessment Interview (HCAI) utilizes hypothetical vignettes in a semi-structured interview format to assess understanding, appreciation, choice, and likely reasoning.8,9 Similar to CCTI, HCAI is not modified for individual patients. Rather, it uses clinical vignettes to gauge a patient’s ability to make decisions. The test takes 30 to 60 minutes to administer and performs less well in assessing appreciation and reasoning than the MacCAT-T and CCTI.10
It is not necessary to perform a formal assessment of capacity on every inpatient. For most, there is no reasonable concern for impaired capacity, obviating the need for formal testing. Likewise, in patients who clearly lack capacity, such as those with end-stage dementia or established guardians, formal reassessment usually is not required. Formal testing is most useful in situations in which capacity is unclear, disagreement amongst surrogate decision-makers exists, or judicial involvement is anticipated.
The MacCAT-T has been validated in the broadest population and is probably the most clinically useful tool currently available. The MMSE is an attractive alternative because of its widespread use and familiarity; however, it is imprecise with scores from 17 to 23, limiting its applicability.
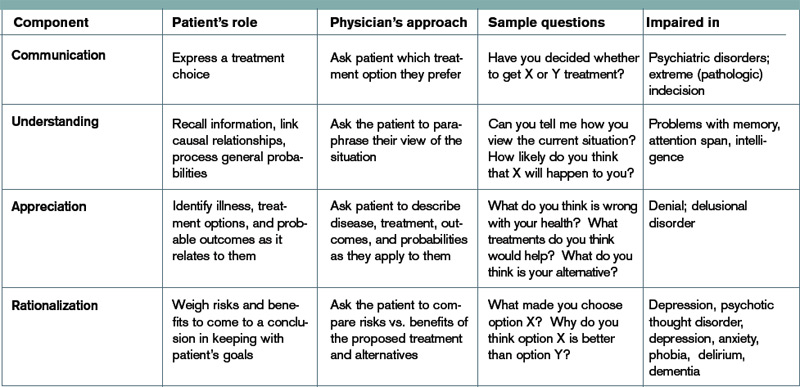
At a minimum, familiarity with the core legal standards of capacity (communication of choice, understanding, appreciation, and reasoning) will improve a hospitalist’s ability to identify patients who lack capacity. Understanding and applying the defined markers most often provides a sufficient capacity evaluation in itself. As capacity is not static, the decision usually requires more than one assessment.
Equally, deciding that a patient lacks capacity is not an end in itself, and the underlying cause should be addressed. Certain factors, such as infection, medication, time of day, and relationship with the clinician doing the assessment, can affect a patient’s capacity. These should be addressed through treatment, education, and social support whenever possible in order to optimize a patient’s performance during the capacity evaluation. If the decision can be delayed until a time when the patient can regain capacity, this should be done in order to maximize the patient’s autonomy.11
Risk-related standards of capacity.
Although some question the notion, given our desire to facilitate management beneficial to the patient, the general consensus is that we have a lower threshold for capacity for consent to treatments that are low-risk and high-benefit.12,13 We would then have a somewhat higher threshold for capacity to refuse that same treatment. Stemming from a desire to protect patients from harm, we have a relatively higher threshold for capacity to make decisions regarding high-risk, low-benefit treatments. For the remainder of cases (low risk/low benefit; high risk/high benefit), as well as treatments that significantly impact a patient’s lifestyle (e.g. dialysis, amputation), we have a low capacity to let patients decide for themselves.11,14
Other considerations.
Clinicians should be thorough in documenting details in coming to a capacity determination, both as a means to formalize the thought process running through the four determinants of capacity, and in order to document for future reference. Cases in which it could be reasonable to call a consultant for those familiar with the assessment basics include:
- Cases in which a determination of lack of capacity could adversely affect the hospitalist’s relationship with the patient;
- Cases in which the hospitalist lacks the time to properly perform the evaluation;
- Particularly difficult or high-stakes cases (e.g. cases that might involve legal proceedings); and
- Cases in which significant mental illness affects a patient’s capacity.11
Early involvement of potential surrogate decision-makers is wise for patients in whom capacity is questioned, both for obtaining collateral history as well as initiating dialogue as to the patient’s wishes. When a patient is found to lack capacity, resources to utilize to help make a treatment decision include existing advance directives and substitute decision-makers, such as durable power of attorneys (DPOAs) and family members. In those rare cases in which clinicians are unable to reach a consensus about a patient’s capacity, an ethics consult should be considered.
Back to the Case.
Following the patient’s declaration that dialysis is not something he is interested in, his niece reports that he is a minimalist when it comes to interventions, and that he had similarly refused a cardiac catheterization in the 1990s. You review with the patient and niece that dialysis would be a procedure to replace his failing kidney function, and that failure to pursue this would ultimately be life-threatening and likely result in death, especially in regard to electrolyte abnormalities and his lack of any other terminal illness.
The consulting nephrologist reviews their recommendations with the patient and niece as well, and the patient consistently refuses. Having clearly communicated his choice, you ask the patient if he understands the situation. He says, "My kidneys are failing. That’s how I got the high potassium." You ask him what that means. "They aren’t going to function on their own much longer," he says. "I could die from it."
You confirm his ideas, and ask him why he doesn’t want dialysis. "I don’t want dialysis because I don’t want to spend my life hooked up to machines three times a week," the patient explains. "I just want to let things run their natural course." The niece says her uncle wouldn’t have wanted dialysis even if it were 10 years ago, so she’s not surprised he is refusing now.
Following this discussion, you feel comfortable that the patient has capacity to make this decision. Having documented this discussion, you discharge him to a subacute rehabilitation facility.
Bottom Line.
In cases in which capacity is in question, a hospitalist’s case-by-case review of the four components of capacity—communicating a choice, understanding, appreciation, and rationalization and reasoning—is warranted to help determine whether a patient has capacity. In cases in which a second opinion is warranted, psychiatry, geriatrics, or ethics consults could be utilized.
Drs. Dastidar and Odden are hospitalists at the University of Michigan in Ann Arbor.
References
- Buchanan A, Brock DW. Deciding for others. Milbank Q. 1986;64(Suppl. 2):17-94.
- Guidelines for assessing the decision-making capacities of potential research subjects with cognitive impairment. American Psychiatric Association. Am J Psychiatry. 1998;155(11):1649-50.
- Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
- Etchells E, Darzins P, Silberfeld M, et al. Assessment of patient capacity to consent to treatment. J Gen Intern Med. 1999;14:27-34.
- Grisso T, Appelbaum PS, Hill-Fotouhi C. The MacCAT-T: a clinical tool to assess patients’ capacities to make treatment decisions. Psychiatr Serv. 1997;48:1415- 1419.
- Marson DC, Ingram KK, Cody HA, Harrell LE. Assessing the competency of patients with Alzheimer’s disease under different legal standards. A prototype instrument. Arch Neurol. 1995;52:949-954.
- Edelstein B. Hopemont Capacity Assessment Interview Manual and Scoring Guide. 1999: Morgantown, W.V.: West Virginia University.
- Pruchno RA, Smyer MA, Rose MS, Hartman-Stein PE, Henderson-Laribee DL. Competence of long-term care residents to participate in decisions about their medical care: a brief, objective assessment. Gerontologist. 1995;35:622-629.
- Moye J, Karel M, Azar AR, Gurrera R. Capacity to consent to treatment: empirical comparison of three instruments in older adults with and without dementia. Gerontologist. 2004;44:166-175.
- Grisso T, Appelbaum PS. Assessing competence to consent to treatment: a guide for physicians and other health professionals. 1998; New York: Oxford University Press, 211.
- Cale GS. Risk-related standards of competence: continuing the debate over risk-related standards of competence. Bioethics. 1999;13(2):131-148.
- Checkland D. On risk and decisional capacity. J Med Philos. 2001;26(1):35-59.
- Wilks I. The debate over risk-related standards of competence. Bioethics. 1997;11(5):413-426.
- Ganzini L, Volicer L, Nelson WA, Fox E, Derse AR. Ten myths about decision-making capacity. J Am Med Dir Assoc. 2004;5(4):263-267.
Acknowledgements:The authors would like to thank Dr. Jeff Rohde for reviewing a copy of the manuscript, and Dr. Amy Rosinski for providing direction from the psychiatry standpoint
Case
A 79-year-old male with coronary artery disease, hypertension, non-insulin-dependent mellitus, moderate dementia, and chronic renal insufficiency is admitted after a fall evaluation. He is widowed and lives in an assisted living facility. He’s accompanied by his niece, is alert, and oriented to person. He thinks he is in a clinic and is unable to state the year, but the remainder of the examination is unremarkable. His labs are notable for potassium of 6.3 mmol/L, BUN of 78 mg/dL, and Cr of 3.7 mg/dL. The niece reports that the patient is not fond of medical care, thus the most recent labs are from two years ago (and indicate a BUN of 39 and Cr of 2.8, with an upward trend over the past decade). You discuss possible long-term need for dialysis with the patient and niece, and the patient clearly states "no." However, he also states that it is 1988. How do you determine if he has the capacity to make decisions?
Overview
Hospitalists are familiar with the doctrine of informed consent—describing a disease, treatment options, associated risks and benefits, potential for complications, and alternatives, including no treatment. Not only must the patient be informed, and the decision free from any coercion, but the patient also must have capacity to make the decision.
Hospitalists often care for patients in whom decision-making capacity comes into question. This includes populations with depression, psychosis, dementia, stroke, severe personality disorders, developmental delay, comatose patients, as well as those with impaired attentional capacity (e.g. acute pain) or general debility (e.g. metastatic cancer).1,2
ave for the comatose patient, whether the patient has capacity might not be obvious. However, addressing the components of capacity (communication, understanding, appreciation, and rationalization) by using a validated clinical tool, such as the MacCAT-T, or more simply by systematically applying those four components to the clinical scenario under consideration, hospitalists can make this determination.
Review of the Literature
It is important to differentiate capacity from competency. Competency is a global assessment and a legal determination made by a judge in court. Capacity, on the other hand, is a functional assessment regarding a particular decision. Capacity is not static, and it can be performed by any clinician familiar with the patient. A hospitalist often is well positioned to make a capacity determination given established rapport with the patient and familiarity with the details of the case.
To make this determination, a hospitalist needs to know how to assess capacity. Although capacity usually is defined by state law and varies by jurisdiction, clinicians generally can assume it includes one or more of the four key components:
- Communication. The patient needs to be able to express a treatment choice, and this decision needs to be stable enough for the treatment to be implemented. Changing one’s decision in itself would not bring a patient’s capacity into question, so long as the patient was able to explain the rationale behind the switch. Frequent changes back and forth in the decision-making, however, could be indicative of an underlying psychiatric disorder or extreme indecision, which could bring capacity into question.
- Understanding. The patient needs to recall conversations about treatment, to make the link between causal relationships, and to process probabilities for outcomes. Problems with memory, attention span, and intelligence can affect one’s understanding.
- Appreciation. The patient should be able to identify the illness, treatment options, and likely outcomes as things that will affect him or her directly. A lack of appreciation usually stems from a denial based on intelligence (lack of a capability to understand) or emotion, or a delusion that the patient is not affected by this situation the same way and will have a different outcome.
- Rationalization or reasoning. The patient needs to be able to weigh the risks and benefits of the treatment options presented to come to a conclusion in keeping with their goals and best interests, as defined by their personal set of values. This often is affected in psychosis, depression, anxiety, phobias, delirium, and dementia.3
Several clinical capacity tools have been developed to assess these components:
Clinical tools.
The Mini-Mental Status Examination (MMSE) is a bedside test of a patient’s cognitive function, with scores ranging from 0 to 30.4 Although it wasn’t developed for assessing decision-making capacity, it has been compared with expert evaluation for assessment of capacity; the test performs reasonably well, particularly with high and low scores. Specifically, a MMSE >24 has a negative likelihood ratio (LR) of 0.05 for lack of capacity, while a MMSE <16 has a positive LR of 15.5 Scores from 17 to 23 do not correlate well with capacity, and further testing would be necessary. It is easy to administer, requires no formal training, and is familiar to most hospitalists. However, it does not address any specific aspects of informed consent, such as understanding or choice, and has not been validated in patients with mental illness.
The MacArthur Competence Assessment Tools for Treatment (MacCAT-T) is regarded as the gold standard for capacity assessment aids. It utilizes hospital chart review followed by a semi-structured interview to address clinical issues relevant to the patient being assessed; it takes 15 to 20 minutes to complete.6 The test provides scores in each of the four domains (choice, understanding, appreciation, and reasoning) of capacity. It has been validated in patients with dementia, schizophrenia, and depression. Limiting its clinical applicability is the fact that the MacCAT-T requires training to administer and interpret the results, though this is a relatively brief process.
The Capacity to Consent to Treatment Instrument (CCTI) uses hypothetical clinical vignettes in a structured interview to assess capacity across all four domains. The tool was developed and validated in patients with dementia and Parkinson’s disease, and takes 20 to 25 minutes to complete.7 A potential limitation is the CCTI’s use of vignettes as opposed to a patient-specific discussion, which could lead to different patient answers and a false assessment of the patient’s capacity.
The Hopemont Capacity Assessment Interview (HCAI) utilizes hypothetical vignettes in a semi-structured interview format to assess understanding, appreciation, choice, and likely reasoning.8,9 Similar to CCTI, HCAI is not modified for individual patients. Rather, it uses clinical vignettes to gauge a patient’s ability to make decisions. The test takes 30 to 60 minutes to administer and performs less well in assessing appreciation and reasoning than the MacCAT-T and CCTI.10
It is not necessary to perform a formal assessment of capacity on every inpatient. For most, there is no reasonable concern for impaired capacity, obviating the need for formal testing. Likewise, in patients who clearly lack capacity, such as those with end-stage dementia or established guardians, formal reassessment usually is not required. Formal testing is most useful in situations in which capacity is unclear, disagreement amongst surrogate decision-makers exists, or judicial involvement is anticipated.
The MacCAT-T has been validated in the broadest population and is probably the most clinically useful tool currently available. The MMSE is an attractive alternative because of its widespread use and familiarity; however, it is imprecise with scores from 17 to 23, limiting its applicability.
At a minimum, familiarity with the core legal standards of capacity (communication of choice, understanding, appreciation, and reasoning) will improve a hospitalist’s ability to identify patients who lack capacity. Understanding and applying the defined markers most often provides a sufficient capacity evaluation in itself. As capacity is not static, the decision usually requires more than one assessment.
Equally, deciding that a patient lacks capacity is not an end in itself, and the underlying cause should be addressed. Certain factors, such as infection, medication, time of day, and relationship with the clinician doing the assessment, can affect a patient’s capacity. These should be addressed through treatment, education, and social support whenever possible in order to optimize a patient’s performance during the capacity evaluation. If the decision can be delayed until a time when the patient can regain capacity, this should be done in order to maximize the patient’s autonomy.11
Risk-related standards of capacity.
Although some question the notion, given our desire to facilitate management beneficial to the patient, the general consensus is that we have a lower threshold for capacity for consent to treatments that are low-risk and high-benefit.12,13 We would then have a somewhat higher threshold for capacity to refuse that same treatment. Stemming from a desire to protect patients from harm, we have a relatively higher threshold for capacity to make decisions regarding high-risk, low-benefit treatments. For the remainder of cases (low risk/low benefit; high risk/high benefit), as well as treatments that significantly impact a patient’s lifestyle (e.g. dialysis, amputation), we have a low capacity to let patients decide for themselves.11,14
Other considerations.
Clinicians should be thorough in documenting details in coming to a capacity determination, both as a means to formalize the thought process running through the four determinants of capacity, and in order to document for future reference. Cases in which it could be reasonable to call a consultant for those familiar with the assessment basics include:
- Cases in which a determination of lack of capacity could adversely affect the hospitalist’s relationship with the patient;
- Cases in which the hospitalist lacks the time to properly perform the evaluation;
- Particularly difficult or high-stakes cases (e.g. cases that might involve legal proceedings); and
- Cases in which significant mental illness affects a patient’s capacity.11
Early involvement of potential surrogate decision-makers is wise for patients in whom capacity is questioned, both for obtaining collateral history as well as initiating dialogue as to the patient’s wishes. When a patient is found to lack capacity, resources to utilize to help make a treatment decision include existing advance directives and substitute decision-makers, such as durable power of attorneys (DPOAs) and family members. In those rare cases in which clinicians are unable to reach a consensus about a patient’s capacity, an ethics consult should be considered.
Back to the Case.
Following the patient’s declaration that dialysis is not something he is interested in, his niece reports that he is a minimalist when it comes to interventions, and that he had similarly refused a cardiac catheterization in the 1990s. You review with the patient and niece that dialysis would be a procedure to replace his failing kidney function, and that failure to pursue this would ultimately be life-threatening and likely result in death, especially in regard to electrolyte abnormalities and his lack of any other terminal illness.
The consulting nephrologist reviews their recommendations with the patient and niece as well, and the patient consistently refuses. Having clearly communicated his choice, you ask the patient if he understands the situation. He says, "My kidneys are failing. That’s how I got the high potassium." You ask him what that means. "They aren’t going to function on their own much longer," he says. "I could die from it."
You confirm his ideas, and ask him why he doesn’t want dialysis. "I don’t want dialysis because I don’t want to spend my life hooked up to machines three times a week," the patient explains. "I just want to let things run their natural course." The niece says her uncle wouldn’t have wanted dialysis even if it were 10 years ago, so she’s not surprised he is refusing now.
Following this discussion, you feel comfortable that the patient has capacity to make this decision. Having documented this discussion, you discharge him to a subacute rehabilitation facility.
Bottom Line.
In cases in which capacity is in question, a hospitalist’s case-by-case review of the four components of capacity—communicating a choice, understanding, appreciation, and rationalization and reasoning—is warranted to help determine whether a patient has capacity. In cases in which a second opinion is warranted, psychiatry, geriatrics, or ethics consults could be utilized.
Drs. Dastidar and Odden are hospitalists at the University of Michigan in Ann Arbor.
References
- Buchanan A, Brock DW. Deciding for others. Milbank Q. 1986;64(Suppl. 2):17-94.
- Guidelines for assessing the decision-making capacities of potential research subjects with cognitive impairment. American Psychiatric Association. Am J Psychiatry. 1998;155(11):1649-50.
- Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
- Etchells E, Darzins P, Silberfeld M, et al. Assessment of patient capacity to consent to treatment. J Gen Intern Med. 1999;14:27-34.
- Grisso T, Appelbaum PS, Hill-Fotouhi C. The MacCAT-T: a clinical tool to assess patients’ capacities to make treatment decisions. Psychiatr Serv. 1997;48:1415- 1419.
- Marson DC, Ingram KK, Cody HA, Harrell LE. Assessing the competency of patients with Alzheimer’s disease under different legal standards. A prototype instrument. Arch Neurol. 1995;52:949-954.
- Edelstein B. Hopemont Capacity Assessment Interview Manual and Scoring Guide. 1999: Morgantown, W.V.: West Virginia University.
- Pruchno RA, Smyer MA, Rose MS, Hartman-Stein PE, Henderson-Laribee DL. Competence of long-term care residents to participate in decisions about their medical care: a brief, objective assessment. Gerontologist. 1995;35:622-629.
- Moye J, Karel M, Azar AR, Gurrera R. Capacity to consent to treatment: empirical comparison of three instruments in older adults with and without dementia. Gerontologist. 2004;44:166-175.
- Grisso T, Appelbaum PS. Assessing competence to consent to treatment: a guide for physicians and other health professionals. 1998; New York: Oxford University Press, 211.
- Cale GS. Risk-related standards of competence: continuing the debate over risk-related standards of competence. Bioethics. 1999;13(2):131-148.
- Checkland D. On risk and decisional capacity. J Med Philos. 2001;26(1):35-59.
- Wilks I. The debate over risk-related standards of competence. Bioethics. 1997;11(5):413-426.
- Ganzini L, Volicer L, Nelson WA, Fox E, Derse AR. Ten myths about decision-making capacity. J Am Med Dir Assoc. 2004;5(4):263-267.
Acknowledgements:The authors would like to thank Dr. Jeff Rohde for reviewing a copy of the manuscript, and Dr. Amy Rosinski for providing direction from the psychiatry standpoint
Case
A 79-year-old male with coronary artery disease, hypertension, non-insulin-dependent mellitus, moderate dementia, and chronic renal insufficiency is admitted after a fall evaluation. He is widowed and lives in an assisted living facility. He’s accompanied by his niece, is alert, and oriented to person. He thinks he is in a clinic and is unable to state the year, but the remainder of the examination is unremarkable. His labs are notable for potassium of 6.3 mmol/L, BUN of 78 mg/dL, and Cr of 3.7 mg/dL. The niece reports that the patient is not fond of medical care, thus the most recent labs are from two years ago (and indicate a BUN of 39 and Cr of 2.8, with an upward trend over the past decade). You discuss possible long-term need for dialysis with the patient and niece, and the patient clearly states "no." However, he also states that it is 1988. How do you determine if he has the capacity to make decisions?
Overview
Hospitalists are familiar with the doctrine of informed consent—describing a disease, treatment options, associated risks and benefits, potential for complications, and alternatives, including no treatment. Not only must the patient be informed, and the decision free from any coercion, but the patient also must have capacity to make the decision.
Hospitalists often care for patients in whom decision-making capacity comes into question. This includes populations with depression, psychosis, dementia, stroke, severe personality disorders, developmental delay, comatose patients, as well as those with impaired attentional capacity (e.g. acute pain) or general debility (e.g. metastatic cancer).1,2
ave for the comatose patient, whether the patient has capacity might not be obvious. However, addressing the components of capacity (communication, understanding, appreciation, and rationalization) by using a validated clinical tool, such as the MacCAT-T, or more simply by systematically applying those four components to the clinical scenario under consideration, hospitalists can make this determination.
Review of the Literature
It is important to differentiate capacity from competency. Competency is a global assessment and a legal determination made by a judge in court. Capacity, on the other hand, is a functional assessment regarding a particular decision. Capacity is not static, and it can be performed by any clinician familiar with the patient. A hospitalist often is well positioned to make a capacity determination given established rapport with the patient and familiarity with the details of the case.
To make this determination, a hospitalist needs to know how to assess capacity. Although capacity usually is defined by state law and varies by jurisdiction, clinicians generally can assume it includes one or more of the four key components:
- Communication. The patient needs to be able to express a treatment choice, and this decision needs to be stable enough for the treatment to be implemented. Changing one’s decision in itself would not bring a patient’s capacity into question, so long as the patient was able to explain the rationale behind the switch. Frequent changes back and forth in the decision-making, however, could be indicative of an underlying psychiatric disorder or extreme indecision, which could bring capacity into question.
- Understanding. The patient needs to recall conversations about treatment, to make the link between causal relationships, and to process probabilities for outcomes. Problems with memory, attention span, and intelligence can affect one’s understanding.
- Appreciation. The patient should be able to identify the illness, treatment options, and likely outcomes as things that will affect him or her directly. A lack of appreciation usually stems from a denial based on intelligence (lack of a capability to understand) or emotion, or a delusion that the patient is not affected by this situation the same way and will have a different outcome.
- Rationalization or reasoning. The patient needs to be able to weigh the risks and benefits of the treatment options presented to come to a conclusion in keeping with their goals and best interests, as defined by their personal set of values. This often is affected in psychosis, depression, anxiety, phobias, delirium, and dementia.3
Several clinical capacity tools have been developed to assess these components:
Clinical tools.
The Mini-Mental Status Examination (MMSE) is a bedside test of a patient’s cognitive function, with scores ranging from 0 to 30.4 Although it wasn’t developed for assessing decision-making capacity, it has been compared with expert evaluation for assessment of capacity; the test performs reasonably well, particularly with high and low scores. Specifically, a MMSE >24 has a negative likelihood ratio (LR) of 0.05 for lack of capacity, while a MMSE <16 has a positive LR of 15.5 Scores from 17 to 23 do not correlate well with capacity, and further testing would be necessary. It is easy to administer, requires no formal training, and is familiar to most hospitalists. However, it does not address any specific aspects of informed consent, such as understanding or choice, and has not been validated in patients with mental illness.
The MacArthur Competence Assessment Tools for Treatment (MacCAT-T) is regarded as the gold standard for capacity assessment aids. It utilizes hospital chart review followed by a semi-structured interview to address clinical issues relevant to the patient being assessed; it takes 15 to 20 minutes to complete.6 The test provides scores in each of the four domains (choice, understanding, appreciation, and reasoning) of capacity. It has been validated in patients with dementia, schizophrenia, and depression. Limiting its clinical applicability is the fact that the MacCAT-T requires training to administer and interpret the results, though this is a relatively brief process.
The Capacity to Consent to Treatment Instrument (CCTI) uses hypothetical clinical vignettes in a structured interview to assess capacity across all four domains. The tool was developed and validated in patients with dementia and Parkinson’s disease, and takes 20 to 25 minutes to complete.7 A potential limitation is the CCTI’s use of vignettes as opposed to a patient-specific discussion, which could lead to different patient answers and a false assessment of the patient’s capacity.
The Hopemont Capacity Assessment Interview (HCAI) utilizes hypothetical vignettes in a semi-structured interview format to assess understanding, appreciation, choice, and likely reasoning.8,9 Similar to CCTI, HCAI is not modified for individual patients. Rather, it uses clinical vignettes to gauge a patient’s ability to make decisions. The test takes 30 to 60 minutes to administer and performs less well in assessing appreciation and reasoning than the MacCAT-T and CCTI.10
It is not necessary to perform a formal assessment of capacity on every inpatient. For most, there is no reasonable concern for impaired capacity, obviating the need for formal testing. Likewise, in patients who clearly lack capacity, such as those with end-stage dementia or established guardians, formal reassessment usually is not required. Formal testing is most useful in situations in which capacity is unclear, disagreement amongst surrogate decision-makers exists, or judicial involvement is anticipated.
The MacCAT-T has been validated in the broadest population and is probably the most clinically useful tool currently available. The MMSE is an attractive alternative because of its widespread use and familiarity; however, it is imprecise with scores from 17 to 23, limiting its applicability.
At a minimum, familiarity with the core legal standards of capacity (communication of choice, understanding, appreciation, and reasoning) will improve a hospitalist’s ability to identify patients who lack capacity. Understanding and applying the defined markers most often provides a sufficient capacity evaluation in itself. As capacity is not static, the decision usually requires more than one assessment.
Equally, deciding that a patient lacks capacity is not an end in itself, and the underlying cause should be addressed. Certain factors, such as infection, medication, time of day, and relationship with the clinician doing the assessment, can affect a patient’s capacity. These should be addressed through treatment, education, and social support whenever possible in order to optimize a patient’s performance during the capacity evaluation. If the decision can be delayed until a time when the patient can regain capacity, this should be done in order to maximize the patient’s autonomy.11
Risk-related standards of capacity.
Although some question the notion, given our desire to facilitate management beneficial to the patient, the general consensus is that we have a lower threshold for capacity for consent to treatments that are low-risk and high-benefit.12,13 We would then have a somewhat higher threshold for capacity to refuse that same treatment. Stemming from a desire to protect patients from harm, we have a relatively higher threshold for capacity to make decisions regarding high-risk, low-benefit treatments. For the remainder of cases (low risk/low benefit; high risk/high benefit), as well as treatments that significantly impact a patient’s lifestyle (e.g. dialysis, amputation), we have a low capacity to let patients decide for themselves.11,14
Other considerations.
Clinicians should be thorough in documenting details in coming to a capacity determination, both as a means to formalize the thought process running through the four determinants of capacity, and in order to document for future reference. Cases in which it could be reasonable to call a consultant for those familiar with the assessment basics include:
- Cases in which a determination of lack of capacity could adversely affect the hospitalist’s relationship with the patient;
- Cases in which the hospitalist lacks the time to properly perform the evaluation;
- Particularly difficult or high-stakes cases (e.g. cases that might involve legal proceedings); and
- Cases in which significant mental illness affects a patient’s capacity.11
Early involvement of potential surrogate decision-makers is wise for patients in whom capacity is questioned, both for obtaining collateral history as well as initiating dialogue as to the patient’s wishes. When a patient is found to lack capacity, resources to utilize to help make a treatment decision include existing advance directives and substitute decision-makers, such as durable power of attorneys (DPOAs) and family members. In those rare cases in which clinicians are unable to reach a consensus about a patient’s capacity, an ethics consult should be considered.
Back to the Case.
Following the patient’s declaration that dialysis is not something he is interested in, his niece reports that he is a minimalist when it comes to interventions, and that he had similarly refused a cardiac catheterization in the 1990s. You review with the patient and niece that dialysis would be a procedure to replace his failing kidney function, and that failure to pursue this would ultimately be life-threatening and likely result in death, especially in regard to electrolyte abnormalities and his lack of any other terminal illness.
The consulting nephrologist reviews their recommendations with the patient and niece as well, and the patient consistently refuses. Having clearly communicated his choice, you ask the patient if he understands the situation. He says, "My kidneys are failing. That’s how I got the high potassium." You ask him what that means. "They aren’t going to function on their own much longer," he says. "I could die from it."
You confirm his ideas, and ask him why he doesn’t want dialysis. "I don’t want dialysis because I don’t want to spend my life hooked up to machines three times a week," the patient explains. "I just want to let things run their natural course." The niece says her uncle wouldn’t have wanted dialysis even if it were 10 years ago, so she’s not surprised he is refusing now.
Following this discussion, you feel comfortable that the patient has capacity to make this decision. Having documented this discussion, you discharge him to a subacute rehabilitation facility.
Bottom Line.
In cases in which capacity is in question, a hospitalist’s case-by-case review of the four components of capacity—communicating a choice, understanding, appreciation, and rationalization and reasoning—is warranted to help determine whether a patient has capacity. In cases in which a second opinion is warranted, psychiatry, geriatrics, or ethics consults could be utilized.
Drs. Dastidar and Odden are hospitalists at the University of Michigan in Ann Arbor.
References
- Buchanan A, Brock DW. Deciding for others. Milbank Q. 1986;64(Suppl. 2):17-94.
- Guidelines for assessing the decision-making capacities of potential research subjects with cognitive impairment. American Psychiatric Association. Am J Psychiatry. 1998;155(11):1649-50.
- Appelbaum PS, Grisso T. Assessing patients’ capacities to consent to treatment. N Engl J Med. 1988;319(25):1635-1638.
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189-198.
- Etchells E, Darzins P, Silberfeld M, et al. Assessment of patient capacity to consent to treatment. J Gen Intern Med. 1999;14:27-34.
- Grisso T, Appelbaum PS, Hill-Fotouhi C. The MacCAT-T: a clinical tool to assess patients’ capacities to make treatment decisions. Psychiatr Serv. 1997;48:1415- 1419.
- Marson DC, Ingram KK, Cody HA, Harrell LE. Assessing the competency of patients with Alzheimer’s disease under different legal standards. A prototype instrument. Arch Neurol. 1995;52:949-954.
- Edelstein B. Hopemont Capacity Assessment Interview Manual and Scoring Guide. 1999: Morgantown, W.V.: West Virginia University.
- Pruchno RA, Smyer MA, Rose MS, Hartman-Stein PE, Henderson-Laribee DL. Competence of long-term care residents to participate in decisions about their medical care: a brief, objective assessment. Gerontologist. 1995;35:622-629.
- Moye J, Karel M, Azar AR, Gurrera R. Capacity to consent to treatment: empirical comparison of three instruments in older adults with and without dementia. Gerontologist. 2004;44:166-175.
- Grisso T, Appelbaum PS. Assessing competence to consent to treatment: a guide for physicians and other health professionals. 1998; New York: Oxford University Press, 211.
- Cale GS. Risk-related standards of competence: continuing the debate over risk-related standards of competence. Bioethics. 1999;13(2):131-148.
- Checkland D. On risk and decisional capacity. J Med Philos. 2001;26(1):35-59.
- Wilks I. The debate over risk-related standards of competence. Bioethics. 1997;11(5):413-426.
- Ganzini L, Volicer L, Nelson WA, Fox E, Derse AR. Ten myths about decision-making capacity. J Am Med Dir Assoc. 2004;5(4):263-267.
Acknowledgements:The authors would like to thank Dr. Jeff Rohde for reviewing a copy of the manuscript, and Dr. Amy Rosinski for providing direction from the psychiatry standpoint
How Is SIADH Diagnosed and Managed?
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4

Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.

Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
A Lifetime in the Making
A 66‐year‐old man presented to the emergency department with 3 weeks of progressive exertional dyspnea. He also reported a single episode of chest pain 1 day prior to admission.
Cardiac and pulmonary causes of dyspnea are the most common. Other causes include anemia or a neuromuscular process. Given the recent episode of chest pain, coronary ischemia, congestive heart failure, chronic obstructive pulmonary disease (COPD), pulmonary embolism, and pericardial effusion must be considered.
Up until 3 weeks ago, he had no exercise intolerance, and had been relatively active. He began noticing progressive dyspnea to the point where he had considerable difficulty walking up stairs, and performing minor household chores. He also complained of orthopnea and paroxysmal nocturnal dyspnea for the last 3 weeks.He denied chest pain at presentation, but 24 hours prior, he experienced one episode of sharp, left‐sided, nonradiating, nonpositional chest pain that occurred at rest. It lasted approximately 20 minutes and was not associated with diaphoresis, nausea, vomiting, or palpitations. He had never experienced chest discomfort prior to this episode. He denied fever, chills, cough, or wheezing.
Progressive dyspnea on exertion with associated orthopnea and paroxysmal nocturnal dyspnea is classically seen in patients with heart failure and is typically associated with left ventricular failure. However, paroxysmal nocturnal dyspnea and orthopnea are only moderately specific for heart failure. Orthopnea can also be seen in pericardial disease, and in numerous pulmonary diseases, including asthma, COPD, pulmonary hypertension, diaphragmatic weakness, pleural effusion, pulmonary embolism, and any apical lung process including lung cancer or pneumonia. Paroxysmal nocturnal dyspnea can be seen in many of the same disorders and can also be reported in obstructive sleep apnea.
His past medical history was remarkable for two episodes of syncope, occurring 5 and 3 years ago, both while working outside in warm weather. Neither was associated with chest pain, diaphoresis, palpitations, or post‐ictal symptoms. He was diagnosed with prostate cancer 8 years ago, and underwent 2 years of androgen‐deprivation therapy with goserelin along with local radiation therapy. Medications included subcutaneous goserelin every 3 months and daily omeprazole. He denied any other prescription, over‐the‐counter, or herbal medications. He reported a 50‐pack‐year history of smoking, but denied alcohol or illicit drug abuse. He denied any travel history or recent immobilization. He had no children, and there was no known history of heart disease in his family.
The past medical history of two episodes of likely exertional syncope is interesting, but the episodes were sporadic and in the distant past, arguing against a serious and ongoing process. Nonetheless, this history still raises the possibility of cardiac causes of syncope, especially causes such as hypertrophic obstructive cardiomyopathy or aortic stenosis which are classically associated with exertional syncope. Either of these two conditions can result in heart failure if untreated. The history of goserelin therapy does make the possibility of heart failure higher, as there has been an association reported between use of this drug and heart failure. His history of tobacco use is a risk factor for coronary artery disease (CAD) and COPD. An active cancer history is also a risk factor for thromboembolic disease, which remains a consideration.
On admission, his temperature was 36.9C, heart rate 94 bpm, respiratory rate 22 breaths per minute, blood pressure 200/108 mmHg, and oxygen saturation 93% breathing ambient air. He was a thin man in no acute distress. Cardiovascular examination was significant for normal first and second heart sounds, with a soft left‐sided S3; the point of maximal impulse was diffuse, but displaced laterally. His jugular venous pressure was estimated at 9 cm of H2O while positioned at a 45‐degree angle. Rales were heard at the lung bases bilaterally. Abdominal exam was normal. His lower extremities were without edema. There were no focal neurological deficits appreciated. Skin examination was unremarkable.
His combination of physical exam findings strongly suggests heart failure, most likely related to a dilated cardiomyopathy and left ventricular dysfunction. The presence of a left‐sided S3 and rales, and the lack of markedly elevated central venous pressure and peripheral edema, suggest heart failure predominantly due to left ventricular dysfunction. Of note, he is very hypertensive. This would not be the typical finding with severely decompensated heart failure. It would be important to determine whether his elevated blood pressure is due to an acute, reversible cause (e.g., pain, dyspnea, anxiety) or whether cocaine use, psychotropic agents, rare causes such as catecholamine‐producing tumors, other neuroendocrine tumors or thyroid toxic states are at play. In addition, one might see hypertension early in the course of heart failure, from a left ventricular outflow obstructive etiology such as severe aortic stenosis or hypertrophic obstructive cardiomyopathy.
Laboratory evaluation revealed a white blood cell count of 8900/mm3, with a normal differential; hemoglobin was 13.9 g/dL; platelet count was 264,000/mm3. Serum electrolytes and liver enzymes were unremarkable, with serum creatinine 1.1 mg/dL and blood urea nitrogen 7 mg/dL. Serial cardiac troponin‐I levels drawn 8 hours apart were 0.04, 0.07, 0.08, and 0.04 ng/mL (normal 0.04). Brain natriuretic peptide was 1420 pg/mL (normal 100). Thyroid stimulating hormone was 1.19 uIU/mL (normal 0.34‐5.60). Chest radiography revealed mild cardiomegaly, with peripheral interstitial opacities in the mid and lower lobes bilaterally, with fluid within the minor fissure. A 12‐lead electrocardiogram (ECG) revealed normal sinus rhythm at 95 bpm with left anterior fascicular block; intraventricular conduction delay was present (QRS width 106 ms) and QS complexes were present in V1‐V3. In addition, there was a left atrial abnormality and voltage criteria for left ventricular hypertrophy with secondary T‐wave inversions laterally (Figure 1). No previous ECGs were available for comparison. A chest computed tomography scan with contrast showed no evidence of pulmonary embolus. It did show interlobular septal thickening and small bilateral pleural effusions, consistent with left ventricular dysfunction.
The patient's initial lab, imaging, and diagnostic work‐up continues to be consistent with the diagnosis of heart failure. The patient appears to have cardiomegaly and mild pulmonary edema by imaging. The etiology of heart failure remains unknown, but ischemia remains in the differential, given the mildly elevated troponins initially and the ECG findings of left anterior fascicular block and T‐wave inversions in the lateral leads. Left anterior fascicular block can be seen with ischemic heart disease (especially involving the left anterior descending coronary artery), hypertensive heart disease, valvular disease, and some infiltrative cardiac processes. The lateral T‐wave inversions are likely secondary to left ventricular hypertrophy (a so‐called strain pattern), rather than ischemia. Left ventricular hypertrophy is consistent with his hypertension, suggesting that it is chronic; his presentation may be due to hypertensive heart disease with new onset heart failure.
He was admitted to the hospital, and metoprolol, lisinopril, and intravenous furosemide were given. Transthoracic echocardiography demonstrated severe global hypokinesis with a left ventricular ejection fraction of 10%. There was no evidence of ventricular thrombus or valvular disease; however, prominent left ventricular trabeculation with deep recesses was noted (see Figure 2).
The echocardiographic findings of deep recesses and prominent left ventricular trabeculation are seen in only a few disorders. Sometimes these findings are thought to be due to hypertrophic obstructive cardiomyopathy. The deep trabeculations can be seen in patients with some forms of congenital heart disease associated with ventricular pressure overload during fetal development. The other cause is left ventricular noncompaction, a genetic cardiomyopathy which is becoming increasingly recognized. The disorder, along with causing heart failure, is associated with a high risk of ventricular thrombus and thromboembolic events, and a high risk of arrhythmias and sudden death. The overall prognosis appears to be poor, compared to some other cardiomyopathies. The imaging findings of left ventricular noncompaction are nearly pathognomonic, and experienced echocardiographers can usually make the diagnosis. Finally, left heart catheterization or noninvasive stress testing should be part of the workup to definitively exclude an ischemic cardiomyopathy, even in the setting of noncompaction, and especially given his recent history of chest pain.
A left heart catheterization with coronary arteriography demonstrated no angiographic evidence of obstructive coronary disease. Left ventriculography revealed severe global hypokinesis. The patient was diagnosed with left ventricular noncompaction.
The initial medical management centers upon the treatment of heart failure with a beta‐blocker, ACE‐inhibitor, and diuretics for fluid management. Patients with left ventricular noncompaction are at particularly high risk of both embolic events (thought due to propensity to develop left ventricular clots within the deep recesses of the endocardium) and sudden death from arrhythmias. Thus, anticoagulation with warfarin is often indicated and would be reasonable in this patient, given the extremely low ejection fraction. The patient does meet established criteria for primary prophylaxis of sudden death with an implantable cardioverter‐defibrillator in nonischemic cardiomyopathy (left ventricular ejection fraction 35% and New York Heart Association class II failure), and this would also be appropriate therapy as well, given the high‐risk profile of this patient population.
He was discharged in stable condition with a medical regimen consisting of diuretics, metoprolol, and lisinopril. Given the risk for thromboembolism, he was started on warfarin. On subsequent follow‐up, repeat echocardiogram revealed a persistently low left ventricular ejection fraction at 10%. Despite his marked improvement in exercise tolerance and overall well‐being after 4 months of treatment, his ejection fraction did not improve. As a result, he was evaluated and counseled for placement of an implantable cardioverter‐defibrillator, and received a dual‐chamber device shortly afterward.
COMMENTARY
Left ventricular noncompaction is a form of cardiomyopathy increasingly recognized in both pediatric and adult populations. The hallmark features are a pattern of prominent trabeculations and deep recesses in the left ventricular wall. During normal gestation, the myocardium compacts and matures while deep recesses evolve into capillary precursors of the coronary circulation. Left ventricular noncompaction may result from an arrest in this process, with cardiac myofibers failing to compact from their initial spongiform architecture into a developed endocardium.1 Restrictive relaxation from persistent trabeculae predisposes to diastolic dysfunction, while systolic dysfunction may be related to subendocardial hypoperfusion and mechanical dyssynchrony between compacted and noncompacted myocardium.2
Differentiation of left ventricular noncompaction from other cardiomyopathies, based on history and physical examination alone, is essentially impossible. There is high variability and lack of specificity in both clinical profile and onset of symptoms. Electrocardiographic findings are also nonspecific, and the diagnosis typically becomes evident only with transthoracic echocardiography. Current diagnostic criteria include: 1) absence of coexisting cardiac abnormalities; 2) a two‐layer structure with >2:1 ratio of noncompacted to compacted myocardium; 3) predominant involvement of the apical segment of myocardium; and 4) deep intertrabecular recesses demonstrated on Doppler imaging.2, 3 Although echocardiography remains the standard in clinical practice, cardiac magnetic resonance imaging is being increasingly employed as well.4
With more awareness of the disease and the development of higher resolution imaging, the reported incidence has risen. In one single‐center study performed at a heart failure/transplant clinic, 3% of 960 patients referred to heart failure clinic were diagnosed with left ventricular noncompaction, a prevalence similar to hypertensive disease and hypertrophic cardiomyopathy.5 In another community‐hospitalbased study of 4929 adult patients referred for echocardiography, 3.7% of those with systolic dysfunction were diagnosed with noncompaction.6
Left ventricular noncompaction is considered a genetic cardiomyopathy; a family history of heart failure is often present.7 Despite its congenital origin and genetic involvement,2 it is unclear why symptoms may first present at an advanced age. Chest pain and shortness of breath are common complaints, and approximately 62% of patients will have congestive heart failure at presentation.8
Tachyarrhythmia and ventricular tachycardia are commonly seen, as are systemic embolic events and pulmonary embolism. Significant predictors of death include New York Heart Association class III‐IV, sustained ventricular arrhythmias, and increased left atrial size.9
Management is focused on the treatment of arrhythmias, heart failure, and thromboembolic events. The use of standard medical therapy for heart failure (including ACE‐inhibitors and beta‐blockers) is not based on large‐scale studies, yet remains the cornerstone of therapy. An implantable cardioverter‐defibrillator is indicated after hemodynamically compromising sustained ventricular tachycardia or aborted sudden cardiac death, but there are no guidelines for primary prophylaxis outside of patients with heart failure and a depressed ejection fraction.10 Cardiac resynchronization therapy has been successful in some patients with isolated left ventricular noncompaction. Long‐term oral anticoagulation is recommended, especially when impaired left ventricular function, thrombi, or atrial fibrillation have been documented. Patients with left ventricular dysfunction in concert with left ventricular noncompaction are at 10% higher risk for embolic complications when compared to those without noncompaction.11 Familial screening with echocardiography is indicated once the diagnosis has been made.2
In this Clinical Care Conundrum, we describe a rare but increasingly recognized condition, and highlight the importance of delineating the underlying cause of cardiomyopathy when possible. Treatment of heart failure in the hospital setting is sometimes more focused on initiation of diuresis and further stabilization of the patient, and less focused on elucidation of the etiology. While recognition of left ventricular failure led to early treatment with standard therapy in this case, identification of the underlying cause allowed for targeted interventions directed at cardiac arrhythmias, embolic events, and familial screening. Of note, the discussant was careful not to let the prior history of syncopal events distract him from the central issues in this case.
This case also serves as a reminder that congenital anomalies should remain on the differential diagnosis when evaluating new complaints in adult patients. The discussant approached the presentation of new‐onset left ventricular dysfunction in a thorough manner, weighing the likelihood of ischemic and nonischemic causes in the context of the history and physical examination. Careful consideration of the patient's new clinical manifestationscoupled with characteristic echocardiographic findings and normal coronary anatomysolidified the diagnosis. By developing a broad differential, the discussant and clinical team arrived at a diagnosis that for this 66‐year‐old gentleman was a lifetime in the making.
Teaching Points
-
Left ventricular noncompaction is characterized by a pattern of prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular cavity. Heightened awareness among clinicians and echocardiographers has led to increased detection of this condition.
-
This disease needs to be considered in patients of all ages presenting with heart failure, especially in cases characterized by ventricular arrhythmias, thromboembolism, and a family history of similar events.
-
Left ventricular noncompaction management is mainly focused on the treatment of arrhythmias, heart failure, and thromboembolic events.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,,.Isolated ventricular non‐compaction of the myocardium in adults.Heart.2006;93:11–15.
- .Left ventricular noncompaction.Circ J.2009;73:19–26.
- ,,,,.Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy.Heart.2001;86:666–671.
- ,,, et al.Left ventricular non‐compaction: insights from cardiovascular magnetic resonance imaging.J Am Coll Cardiol.2005;46:101–105.
- ,,,,,.Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience.Cardiology.2009;112:158–164.
- ,,,.Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction.Echocardiography.2008;25(1):8–12.
- ,,, et al.Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention.Circulation.2006;113:1801–1816.
- ,,,,.Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis.J Am Coll Cardiol.2000;36:493–500.
- ,,, et al.Wide spectrum of presentation and variable outcomes of isolated left ventricular non‐compaction.Heart.2007;93(1):65–71.
- ,,, et al.Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy.N Engl J Med.2004;350:2151–2159.
- ,.Left ventricular hypertrabeculation/noncompaction and stroke or embolism.Cardiology.2005;103:68–72.
A 66‐year‐old man presented to the emergency department with 3 weeks of progressive exertional dyspnea. He also reported a single episode of chest pain 1 day prior to admission.
Cardiac and pulmonary causes of dyspnea are the most common. Other causes include anemia or a neuromuscular process. Given the recent episode of chest pain, coronary ischemia, congestive heart failure, chronic obstructive pulmonary disease (COPD), pulmonary embolism, and pericardial effusion must be considered.
Up until 3 weeks ago, he had no exercise intolerance, and had been relatively active. He began noticing progressive dyspnea to the point where he had considerable difficulty walking up stairs, and performing minor household chores. He also complained of orthopnea and paroxysmal nocturnal dyspnea for the last 3 weeks.He denied chest pain at presentation, but 24 hours prior, he experienced one episode of sharp, left‐sided, nonradiating, nonpositional chest pain that occurred at rest. It lasted approximately 20 minutes and was not associated with diaphoresis, nausea, vomiting, or palpitations. He had never experienced chest discomfort prior to this episode. He denied fever, chills, cough, or wheezing.
Progressive dyspnea on exertion with associated orthopnea and paroxysmal nocturnal dyspnea is classically seen in patients with heart failure and is typically associated with left ventricular failure. However, paroxysmal nocturnal dyspnea and orthopnea are only moderately specific for heart failure. Orthopnea can also be seen in pericardial disease, and in numerous pulmonary diseases, including asthma, COPD, pulmonary hypertension, diaphragmatic weakness, pleural effusion, pulmonary embolism, and any apical lung process including lung cancer or pneumonia. Paroxysmal nocturnal dyspnea can be seen in many of the same disorders and can also be reported in obstructive sleep apnea.
His past medical history was remarkable for two episodes of syncope, occurring 5 and 3 years ago, both while working outside in warm weather. Neither was associated with chest pain, diaphoresis, palpitations, or post‐ictal symptoms. He was diagnosed with prostate cancer 8 years ago, and underwent 2 years of androgen‐deprivation therapy with goserelin along with local radiation therapy. Medications included subcutaneous goserelin every 3 months and daily omeprazole. He denied any other prescription, over‐the‐counter, or herbal medications. He reported a 50‐pack‐year history of smoking, but denied alcohol or illicit drug abuse. He denied any travel history or recent immobilization. He had no children, and there was no known history of heart disease in his family.
The past medical history of two episodes of likely exertional syncope is interesting, but the episodes were sporadic and in the distant past, arguing against a serious and ongoing process. Nonetheless, this history still raises the possibility of cardiac causes of syncope, especially causes such as hypertrophic obstructive cardiomyopathy or aortic stenosis which are classically associated with exertional syncope. Either of these two conditions can result in heart failure if untreated. The history of goserelin therapy does make the possibility of heart failure higher, as there has been an association reported between use of this drug and heart failure. His history of tobacco use is a risk factor for coronary artery disease (CAD) and COPD. An active cancer history is also a risk factor for thromboembolic disease, which remains a consideration.
On admission, his temperature was 36.9C, heart rate 94 bpm, respiratory rate 22 breaths per minute, blood pressure 200/108 mmHg, and oxygen saturation 93% breathing ambient air. He was a thin man in no acute distress. Cardiovascular examination was significant for normal first and second heart sounds, with a soft left‐sided S3; the point of maximal impulse was diffuse, but displaced laterally. His jugular venous pressure was estimated at 9 cm of H2O while positioned at a 45‐degree angle. Rales were heard at the lung bases bilaterally. Abdominal exam was normal. His lower extremities were without edema. There were no focal neurological deficits appreciated. Skin examination was unremarkable.
His combination of physical exam findings strongly suggests heart failure, most likely related to a dilated cardiomyopathy and left ventricular dysfunction. The presence of a left‐sided S3 and rales, and the lack of markedly elevated central venous pressure and peripheral edema, suggest heart failure predominantly due to left ventricular dysfunction. Of note, he is very hypertensive. This would not be the typical finding with severely decompensated heart failure. It would be important to determine whether his elevated blood pressure is due to an acute, reversible cause (e.g., pain, dyspnea, anxiety) or whether cocaine use, psychotropic agents, rare causes such as catecholamine‐producing tumors, other neuroendocrine tumors or thyroid toxic states are at play. In addition, one might see hypertension early in the course of heart failure, from a left ventricular outflow obstructive etiology such as severe aortic stenosis or hypertrophic obstructive cardiomyopathy.
Laboratory evaluation revealed a white blood cell count of 8900/mm3, with a normal differential; hemoglobin was 13.9 g/dL; platelet count was 264,000/mm3. Serum electrolytes and liver enzymes were unremarkable, with serum creatinine 1.1 mg/dL and blood urea nitrogen 7 mg/dL. Serial cardiac troponin‐I levels drawn 8 hours apart were 0.04, 0.07, 0.08, and 0.04 ng/mL (normal 0.04). Brain natriuretic peptide was 1420 pg/mL (normal 100). Thyroid stimulating hormone was 1.19 uIU/mL (normal 0.34‐5.60). Chest radiography revealed mild cardiomegaly, with peripheral interstitial opacities in the mid and lower lobes bilaterally, with fluid within the minor fissure. A 12‐lead electrocardiogram (ECG) revealed normal sinus rhythm at 95 bpm with left anterior fascicular block; intraventricular conduction delay was present (QRS width 106 ms) and QS complexes were present in V1‐V3. In addition, there was a left atrial abnormality and voltage criteria for left ventricular hypertrophy with secondary T‐wave inversions laterally (Figure 1). No previous ECGs were available for comparison. A chest computed tomography scan with contrast showed no evidence of pulmonary embolus. It did show interlobular septal thickening and small bilateral pleural effusions, consistent with left ventricular dysfunction.
The patient's initial lab, imaging, and diagnostic work‐up continues to be consistent with the diagnosis of heart failure. The patient appears to have cardiomegaly and mild pulmonary edema by imaging. The etiology of heart failure remains unknown, but ischemia remains in the differential, given the mildly elevated troponins initially and the ECG findings of left anterior fascicular block and T‐wave inversions in the lateral leads. Left anterior fascicular block can be seen with ischemic heart disease (especially involving the left anterior descending coronary artery), hypertensive heart disease, valvular disease, and some infiltrative cardiac processes. The lateral T‐wave inversions are likely secondary to left ventricular hypertrophy (a so‐called strain pattern), rather than ischemia. Left ventricular hypertrophy is consistent with his hypertension, suggesting that it is chronic; his presentation may be due to hypertensive heart disease with new onset heart failure.
He was admitted to the hospital, and metoprolol, lisinopril, and intravenous furosemide were given. Transthoracic echocardiography demonstrated severe global hypokinesis with a left ventricular ejection fraction of 10%. There was no evidence of ventricular thrombus or valvular disease; however, prominent left ventricular trabeculation with deep recesses was noted (see Figure 2).
The echocardiographic findings of deep recesses and prominent left ventricular trabeculation are seen in only a few disorders. Sometimes these findings are thought to be due to hypertrophic obstructive cardiomyopathy. The deep trabeculations can be seen in patients with some forms of congenital heart disease associated with ventricular pressure overload during fetal development. The other cause is left ventricular noncompaction, a genetic cardiomyopathy which is becoming increasingly recognized. The disorder, along with causing heart failure, is associated with a high risk of ventricular thrombus and thromboembolic events, and a high risk of arrhythmias and sudden death. The overall prognosis appears to be poor, compared to some other cardiomyopathies. The imaging findings of left ventricular noncompaction are nearly pathognomonic, and experienced echocardiographers can usually make the diagnosis. Finally, left heart catheterization or noninvasive stress testing should be part of the workup to definitively exclude an ischemic cardiomyopathy, even in the setting of noncompaction, and especially given his recent history of chest pain.
A left heart catheterization with coronary arteriography demonstrated no angiographic evidence of obstructive coronary disease. Left ventriculography revealed severe global hypokinesis. The patient was diagnosed with left ventricular noncompaction.
The initial medical management centers upon the treatment of heart failure with a beta‐blocker, ACE‐inhibitor, and diuretics for fluid management. Patients with left ventricular noncompaction are at particularly high risk of both embolic events (thought due to propensity to develop left ventricular clots within the deep recesses of the endocardium) and sudden death from arrhythmias. Thus, anticoagulation with warfarin is often indicated and would be reasonable in this patient, given the extremely low ejection fraction. The patient does meet established criteria for primary prophylaxis of sudden death with an implantable cardioverter‐defibrillator in nonischemic cardiomyopathy (left ventricular ejection fraction 35% and New York Heart Association class II failure), and this would also be appropriate therapy as well, given the high‐risk profile of this patient population.
He was discharged in stable condition with a medical regimen consisting of diuretics, metoprolol, and lisinopril. Given the risk for thromboembolism, he was started on warfarin. On subsequent follow‐up, repeat echocardiogram revealed a persistently low left ventricular ejection fraction at 10%. Despite his marked improvement in exercise tolerance and overall well‐being after 4 months of treatment, his ejection fraction did not improve. As a result, he was evaluated and counseled for placement of an implantable cardioverter‐defibrillator, and received a dual‐chamber device shortly afterward.
COMMENTARY
Left ventricular noncompaction is a form of cardiomyopathy increasingly recognized in both pediatric and adult populations. The hallmark features are a pattern of prominent trabeculations and deep recesses in the left ventricular wall. During normal gestation, the myocardium compacts and matures while deep recesses evolve into capillary precursors of the coronary circulation. Left ventricular noncompaction may result from an arrest in this process, with cardiac myofibers failing to compact from their initial spongiform architecture into a developed endocardium.1 Restrictive relaxation from persistent trabeculae predisposes to diastolic dysfunction, while systolic dysfunction may be related to subendocardial hypoperfusion and mechanical dyssynchrony between compacted and noncompacted myocardium.2
Differentiation of left ventricular noncompaction from other cardiomyopathies, based on history and physical examination alone, is essentially impossible. There is high variability and lack of specificity in both clinical profile and onset of symptoms. Electrocardiographic findings are also nonspecific, and the diagnosis typically becomes evident only with transthoracic echocardiography. Current diagnostic criteria include: 1) absence of coexisting cardiac abnormalities; 2) a two‐layer structure with >2:1 ratio of noncompacted to compacted myocardium; 3) predominant involvement of the apical segment of myocardium; and 4) deep intertrabecular recesses demonstrated on Doppler imaging.2, 3 Although echocardiography remains the standard in clinical practice, cardiac magnetic resonance imaging is being increasingly employed as well.4
With more awareness of the disease and the development of higher resolution imaging, the reported incidence has risen. In one single‐center study performed at a heart failure/transplant clinic, 3% of 960 patients referred to heart failure clinic were diagnosed with left ventricular noncompaction, a prevalence similar to hypertensive disease and hypertrophic cardiomyopathy.5 In another community‐hospitalbased study of 4929 adult patients referred for echocardiography, 3.7% of those with systolic dysfunction were diagnosed with noncompaction.6
Left ventricular noncompaction is considered a genetic cardiomyopathy; a family history of heart failure is often present.7 Despite its congenital origin and genetic involvement,2 it is unclear why symptoms may first present at an advanced age. Chest pain and shortness of breath are common complaints, and approximately 62% of patients will have congestive heart failure at presentation.8
Tachyarrhythmia and ventricular tachycardia are commonly seen, as are systemic embolic events and pulmonary embolism. Significant predictors of death include New York Heart Association class III‐IV, sustained ventricular arrhythmias, and increased left atrial size.9
Management is focused on the treatment of arrhythmias, heart failure, and thromboembolic events. The use of standard medical therapy for heart failure (including ACE‐inhibitors and beta‐blockers) is not based on large‐scale studies, yet remains the cornerstone of therapy. An implantable cardioverter‐defibrillator is indicated after hemodynamically compromising sustained ventricular tachycardia or aborted sudden cardiac death, but there are no guidelines for primary prophylaxis outside of patients with heart failure and a depressed ejection fraction.10 Cardiac resynchronization therapy has been successful in some patients with isolated left ventricular noncompaction. Long‐term oral anticoagulation is recommended, especially when impaired left ventricular function, thrombi, or atrial fibrillation have been documented. Patients with left ventricular dysfunction in concert with left ventricular noncompaction are at 10% higher risk for embolic complications when compared to those without noncompaction.11 Familial screening with echocardiography is indicated once the diagnosis has been made.2
In this Clinical Care Conundrum, we describe a rare but increasingly recognized condition, and highlight the importance of delineating the underlying cause of cardiomyopathy when possible. Treatment of heart failure in the hospital setting is sometimes more focused on initiation of diuresis and further stabilization of the patient, and less focused on elucidation of the etiology. While recognition of left ventricular failure led to early treatment with standard therapy in this case, identification of the underlying cause allowed for targeted interventions directed at cardiac arrhythmias, embolic events, and familial screening. Of note, the discussant was careful not to let the prior history of syncopal events distract him from the central issues in this case.
This case also serves as a reminder that congenital anomalies should remain on the differential diagnosis when evaluating new complaints in adult patients. The discussant approached the presentation of new‐onset left ventricular dysfunction in a thorough manner, weighing the likelihood of ischemic and nonischemic causes in the context of the history and physical examination. Careful consideration of the patient's new clinical manifestationscoupled with characteristic echocardiographic findings and normal coronary anatomysolidified the diagnosis. By developing a broad differential, the discussant and clinical team arrived at a diagnosis that for this 66‐year‐old gentleman was a lifetime in the making.
Teaching Points
-
Left ventricular noncompaction is characterized by a pattern of prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular cavity. Heightened awareness among clinicians and echocardiographers has led to increased detection of this condition.
-
This disease needs to be considered in patients of all ages presenting with heart failure, especially in cases characterized by ventricular arrhythmias, thromboembolism, and a family history of similar events.
-
Left ventricular noncompaction management is mainly focused on the treatment of arrhythmias, heart failure, and thromboembolic events.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 66‐year‐old man presented to the emergency department with 3 weeks of progressive exertional dyspnea. He also reported a single episode of chest pain 1 day prior to admission.
Cardiac and pulmonary causes of dyspnea are the most common. Other causes include anemia or a neuromuscular process. Given the recent episode of chest pain, coronary ischemia, congestive heart failure, chronic obstructive pulmonary disease (COPD), pulmonary embolism, and pericardial effusion must be considered.
Up until 3 weeks ago, he had no exercise intolerance, and had been relatively active. He began noticing progressive dyspnea to the point where he had considerable difficulty walking up stairs, and performing minor household chores. He also complained of orthopnea and paroxysmal nocturnal dyspnea for the last 3 weeks.He denied chest pain at presentation, but 24 hours prior, he experienced one episode of sharp, left‐sided, nonradiating, nonpositional chest pain that occurred at rest. It lasted approximately 20 minutes and was not associated with diaphoresis, nausea, vomiting, or palpitations. He had never experienced chest discomfort prior to this episode. He denied fever, chills, cough, or wheezing.
Progressive dyspnea on exertion with associated orthopnea and paroxysmal nocturnal dyspnea is classically seen in patients with heart failure and is typically associated with left ventricular failure. However, paroxysmal nocturnal dyspnea and orthopnea are only moderately specific for heart failure. Orthopnea can also be seen in pericardial disease, and in numerous pulmonary diseases, including asthma, COPD, pulmonary hypertension, diaphragmatic weakness, pleural effusion, pulmonary embolism, and any apical lung process including lung cancer or pneumonia. Paroxysmal nocturnal dyspnea can be seen in many of the same disorders and can also be reported in obstructive sleep apnea.
His past medical history was remarkable for two episodes of syncope, occurring 5 and 3 years ago, both while working outside in warm weather. Neither was associated with chest pain, diaphoresis, palpitations, or post‐ictal symptoms. He was diagnosed with prostate cancer 8 years ago, and underwent 2 years of androgen‐deprivation therapy with goserelin along with local radiation therapy. Medications included subcutaneous goserelin every 3 months and daily omeprazole. He denied any other prescription, over‐the‐counter, or herbal medications. He reported a 50‐pack‐year history of smoking, but denied alcohol or illicit drug abuse. He denied any travel history or recent immobilization. He had no children, and there was no known history of heart disease in his family.
The past medical history of two episodes of likely exertional syncope is interesting, but the episodes were sporadic and in the distant past, arguing against a serious and ongoing process. Nonetheless, this history still raises the possibility of cardiac causes of syncope, especially causes such as hypertrophic obstructive cardiomyopathy or aortic stenosis which are classically associated with exertional syncope. Either of these two conditions can result in heart failure if untreated. The history of goserelin therapy does make the possibility of heart failure higher, as there has been an association reported between use of this drug and heart failure. His history of tobacco use is a risk factor for coronary artery disease (CAD) and COPD. An active cancer history is also a risk factor for thromboembolic disease, which remains a consideration.
On admission, his temperature was 36.9C, heart rate 94 bpm, respiratory rate 22 breaths per minute, blood pressure 200/108 mmHg, and oxygen saturation 93% breathing ambient air. He was a thin man in no acute distress. Cardiovascular examination was significant for normal first and second heart sounds, with a soft left‐sided S3; the point of maximal impulse was diffuse, but displaced laterally. His jugular venous pressure was estimated at 9 cm of H2O while positioned at a 45‐degree angle. Rales were heard at the lung bases bilaterally. Abdominal exam was normal. His lower extremities were without edema. There were no focal neurological deficits appreciated. Skin examination was unremarkable.
His combination of physical exam findings strongly suggests heart failure, most likely related to a dilated cardiomyopathy and left ventricular dysfunction. The presence of a left‐sided S3 and rales, and the lack of markedly elevated central venous pressure and peripheral edema, suggest heart failure predominantly due to left ventricular dysfunction. Of note, he is very hypertensive. This would not be the typical finding with severely decompensated heart failure. It would be important to determine whether his elevated blood pressure is due to an acute, reversible cause (e.g., pain, dyspnea, anxiety) or whether cocaine use, psychotropic agents, rare causes such as catecholamine‐producing tumors, other neuroendocrine tumors or thyroid toxic states are at play. In addition, one might see hypertension early in the course of heart failure, from a left ventricular outflow obstructive etiology such as severe aortic stenosis or hypertrophic obstructive cardiomyopathy.
Laboratory evaluation revealed a white blood cell count of 8900/mm3, with a normal differential; hemoglobin was 13.9 g/dL; platelet count was 264,000/mm3. Serum electrolytes and liver enzymes were unremarkable, with serum creatinine 1.1 mg/dL and blood urea nitrogen 7 mg/dL. Serial cardiac troponin‐I levels drawn 8 hours apart were 0.04, 0.07, 0.08, and 0.04 ng/mL (normal 0.04). Brain natriuretic peptide was 1420 pg/mL (normal 100). Thyroid stimulating hormone was 1.19 uIU/mL (normal 0.34‐5.60). Chest radiography revealed mild cardiomegaly, with peripheral interstitial opacities in the mid and lower lobes bilaterally, with fluid within the minor fissure. A 12‐lead electrocardiogram (ECG) revealed normal sinus rhythm at 95 bpm with left anterior fascicular block; intraventricular conduction delay was present (QRS width 106 ms) and QS complexes were present in V1‐V3. In addition, there was a left atrial abnormality and voltage criteria for left ventricular hypertrophy with secondary T‐wave inversions laterally (Figure 1). No previous ECGs were available for comparison. A chest computed tomography scan with contrast showed no evidence of pulmonary embolus. It did show interlobular septal thickening and small bilateral pleural effusions, consistent with left ventricular dysfunction.
The patient's initial lab, imaging, and diagnostic work‐up continues to be consistent with the diagnosis of heart failure. The patient appears to have cardiomegaly and mild pulmonary edema by imaging. The etiology of heart failure remains unknown, but ischemia remains in the differential, given the mildly elevated troponins initially and the ECG findings of left anterior fascicular block and T‐wave inversions in the lateral leads. Left anterior fascicular block can be seen with ischemic heart disease (especially involving the left anterior descending coronary artery), hypertensive heart disease, valvular disease, and some infiltrative cardiac processes. The lateral T‐wave inversions are likely secondary to left ventricular hypertrophy (a so‐called strain pattern), rather than ischemia. Left ventricular hypertrophy is consistent with his hypertension, suggesting that it is chronic; his presentation may be due to hypertensive heart disease with new onset heart failure.
He was admitted to the hospital, and metoprolol, lisinopril, and intravenous furosemide were given. Transthoracic echocardiography demonstrated severe global hypokinesis with a left ventricular ejection fraction of 10%. There was no evidence of ventricular thrombus or valvular disease; however, prominent left ventricular trabeculation with deep recesses was noted (see Figure 2).
The echocardiographic findings of deep recesses and prominent left ventricular trabeculation are seen in only a few disorders. Sometimes these findings are thought to be due to hypertrophic obstructive cardiomyopathy. The deep trabeculations can be seen in patients with some forms of congenital heart disease associated with ventricular pressure overload during fetal development. The other cause is left ventricular noncompaction, a genetic cardiomyopathy which is becoming increasingly recognized. The disorder, along with causing heart failure, is associated with a high risk of ventricular thrombus and thromboembolic events, and a high risk of arrhythmias and sudden death. The overall prognosis appears to be poor, compared to some other cardiomyopathies. The imaging findings of left ventricular noncompaction are nearly pathognomonic, and experienced echocardiographers can usually make the diagnosis. Finally, left heart catheterization or noninvasive stress testing should be part of the workup to definitively exclude an ischemic cardiomyopathy, even in the setting of noncompaction, and especially given his recent history of chest pain.
A left heart catheterization with coronary arteriography demonstrated no angiographic evidence of obstructive coronary disease. Left ventriculography revealed severe global hypokinesis. The patient was diagnosed with left ventricular noncompaction.
The initial medical management centers upon the treatment of heart failure with a beta‐blocker, ACE‐inhibitor, and diuretics for fluid management. Patients with left ventricular noncompaction are at particularly high risk of both embolic events (thought due to propensity to develop left ventricular clots within the deep recesses of the endocardium) and sudden death from arrhythmias. Thus, anticoagulation with warfarin is often indicated and would be reasonable in this patient, given the extremely low ejection fraction. The patient does meet established criteria for primary prophylaxis of sudden death with an implantable cardioverter‐defibrillator in nonischemic cardiomyopathy (left ventricular ejection fraction 35% and New York Heart Association class II failure), and this would also be appropriate therapy as well, given the high‐risk profile of this patient population.
He was discharged in stable condition with a medical regimen consisting of diuretics, metoprolol, and lisinopril. Given the risk for thromboembolism, he was started on warfarin. On subsequent follow‐up, repeat echocardiogram revealed a persistently low left ventricular ejection fraction at 10%. Despite his marked improvement in exercise tolerance and overall well‐being after 4 months of treatment, his ejection fraction did not improve. As a result, he was evaluated and counseled for placement of an implantable cardioverter‐defibrillator, and received a dual‐chamber device shortly afterward.
COMMENTARY
Left ventricular noncompaction is a form of cardiomyopathy increasingly recognized in both pediatric and adult populations. The hallmark features are a pattern of prominent trabeculations and deep recesses in the left ventricular wall. During normal gestation, the myocardium compacts and matures while deep recesses evolve into capillary precursors of the coronary circulation. Left ventricular noncompaction may result from an arrest in this process, with cardiac myofibers failing to compact from their initial spongiform architecture into a developed endocardium.1 Restrictive relaxation from persistent trabeculae predisposes to diastolic dysfunction, while systolic dysfunction may be related to subendocardial hypoperfusion and mechanical dyssynchrony between compacted and noncompacted myocardium.2
Differentiation of left ventricular noncompaction from other cardiomyopathies, based on history and physical examination alone, is essentially impossible. There is high variability and lack of specificity in both clinical profile and onset of symptoms. Electrocardiographic findings are also nonspecific, and the diagnosis typically becomes evident only with transthoracic echocardiography. Current diagnostic criteria include: 1) absence of coexisting cardiac abnormalities; 2) a two‐layer structure with >2:1 ratio of noncompacted to compacted myocardium; 3) predominant involvement of the apical segment of myocardium; and 4) deep intertrabecular recesses demonstrated on Doppler imaging.2, 3 Although echocardiography remains the standard in clinical practice, cardiac magnetic resonance imaging is being increasingly employed as well.4
With more awareness of the disease and the development of higher resolution imaging, the reported incidence has risen. In one single‐center study performed at a heart failure/transplant clinic, 3% of 960 patients referred to heart failure clinic were diagnosed with left ventricular noncompaction, a prevalence similar to hypertensive disease and hypertrophic cardiomyopathy.5 In another community‐hospitalbased study of 4929 adult patients referred for echocardiography, 3.7% of those with systolic dysfunction were diagnosed with noncompaction.6
Left ventricular noncompaction is considered a genetic cardiomyopathy; a family history of heart failure is often present.7 Despite its congenital origin and genetic involvement,2 it is unclear why symptoms may first present at an advanced age. Chest pain and shortness of breath are common complaints, and approximately 62% of patients will have congestive heart failure at presentation.8
Tachyarrhythmia and ventricular tachycardia are commonly seen, as are systemic embolic events and pulmonary embolism. Significant predictors of death include New York Heart Association class III‐IV, sustained ventricular arrhythmias, and increased left atrial size.9
Management is focused on the treatment of arrhythmias, heart failure, and thromboembolic events. The use of standard medical therapy for heart failure (including ACE‐inhibitors and beta‐blockers) is not based on large‐scale studies, yet remains the cornerstone of therapy. An implantable cardioverter‐defibrillator is indicated after hemodynamically compromising sustained ventricular tachycardia or aborted sudden cardiac death, but there are no guidelines for primary prophylaxis outside of patients with heart failure and a depressed ejection fraction.10 Cardiac resynchronization therapy has been successful in some patients with isolated left ventricular noncompaction. Long‐term oral anticoagulation is recommended, especially when impaired left ventricular function, thrombi, or atrial fibrillation have been documented. Patients with left ventricular dysfunction in concert with left ventricular noncompaction are at 10% higher risk for embolic complications when compared to those without noncompaction.11 Familial screening with echocardiography is indicated once the diagnosis has been made.2
In this Clinical Care Conundrum, we describe a rare but increasingly recognized condition, and highlight the importance of delineating the underlying cause of cardiomyopathy when possible. Treatment of heart failure in the hospital setting is sometimes more focused on initiation of diuresis and further stabilization of the patient, and less focused on elucidation of the etiology. While recognition of left ventricular failure led to early treatment with standard therapy in this case, identification of the underlying cause allowed for targeted interventions directed at cardiac arrhythmias, embolic events, and familial screening. Of note, the discussant was careful not to let the prior history of syncopal events distract him from the central issues in this case.
This case also serves as a reminder that congenital anomalies should remain on the differential diagnosis when evaluating new complaints in adult patients. The discussant approached the presentation of new‐onset left ventricular dysfunction in a thorough manner, weighing the likelihood of ischemic and nonischemic causes in the context of the history and physical examination. Careful consideration of the patient's new clinical manifestationscoupled with characteristic echocardiographic findings and normal coronary anatomysolidified the diagnosis. By developing a broad differential, the discussant and clinical team arrived at a diagnosis that for this 66‐year‐old gentleman was a lifetime in the making.
Teaching Points
-
Left ventricular noncompaction is characterized by a pattern of prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular cavity. Heightened awareness among clinicians and echocardiographers has led to increased detection of this condition.
-
This disease needs to be considered in patients of all ages presenting with heart failure, especially in cases characterized by ventricular arrhythmias, thromboembolism, and a family history of similar events.
-
Left ventricular noncompaction management is mainly focused on the treatment of arrhythmias, heart failure, and thromboembolic events.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,,.Isolated ventricular non‐compaction of the myocardium in adults.Heart.2006;93:11–15.
- .Left ventricular noncompaction.Circ J.2009;73:19–26.
- ,,,,.Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy.Heart.2001;86:666–671.
- ,,, et al.Left ventricular non‐compaction: insights from cardiovascular magnetic resonance imaging.J Am Coll Cardiol.2005;46:101–105.
- ,,,,,.Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience.Cardiology.2009;112:158–164.
- ,,,.Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction.Echocardiography.2008;25(1):8–12.
- ,,, et al.Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention.Circulation.2006;113:1801–1816.
- ,,,,.Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis.J Am Coll Cardiol.2000;36:493–500.
- ,,, et al.Wide spectrum of presentation and variable outcomes of isolated left ventricular non‐compaction.Heart.2007;93(1):65–71.
- ,,, et al.Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy.N Engl J Med.2004;350:2151–2159.
- ,.Left ventricular hypertrabeculation/noncompaction and stroke or embolism.Cardiology.2005;103:68–72.
- ,,.Isolated ventricular non‐compaction of the myocardium in adults.Heart.2006;93:11–15.
- .Left ventricular noncompaction.Circ J.2009;73:19–26.
- ,,,,.Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: a step towards classification as a distinct cardiomyopathy.Heart.2001;86:666–671.
- ,,, et al.Left ventricular non‐compaction: insights from cardiovascular magnetic resonance imaging.J Am Coll Cardiol.2005;46:101–105.
- ,,,,,.Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience.Cardiology.2009;112:158–164.
- ,,,.Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction.Echocardiography.2008;25(1):8–12.
- ,,, et al.Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention.Circulation.2006;113:1801–1816.
- ,,,,.Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis.J Am Coll Cardiol.2000;36:493–500.
- ,,, et al.Wide spectrum of presentation and variable outcomes of isolated left ventricular non‐compaction.Heart.2007;93(1):65–71.
- ,,, et al.Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy.N Engl J Med.2004;350:2151–2159.
- ,.Left ventricular hypertrabeculation/noncompaction and stroke or embolism.Cardiology.2005;103:68–72.
What Corticosteroid is Most Appropriate for treating Acute Exacerbations of CoPD?
Case
A 66-year-old Caucasian female with moderate chronic obstructive pulmonary disease (COPD) (FEV1 55% predicted), obesity, hypertension, and Type 2 diabetes mellitus on insulin therapy presents to the ED with four days of increased cough productive of yellow sputum and progressive shortness of breath. Her physical exam is notable for an oxygen saturation of 87% on room air, along with diffuse expiratory wheezing with use of accessory muscles; her chest X-ray is unchanged from previous. The patient is given oxygen, nebulized bronchodilators, and one dose of IV methylprednisolone. Her symptoms do not improve significantly, and she is admitted for further management. What regimen of corticosteroids is most appropriate to treat her acute exacerbation of COPD?
Overview
COPD is the fourth-leading cause of death in the United States and continues to increase in prevalence.1 Acute exacerbations of COPD (AECOPD) contribute significantly to this high mortality rate, which approaches 40% at one year in those patients requiring mechanical support.1 An exacerbation of COPD has been defined as an acute change in a patient’s baseline dyspnea, cough, and/or sputum beyond day-to-day variability sufficient to warrant a change in therapy.2 Exacerbations commonly occur in COPD patients and often necessitate hospital admission. In fact, COPD consistently is one of the 10 most common reasons for hospitalization, with billions of dollars in associated healthcare costs.3
The goals for inpatient management of AECOPD are to provide acute symptom relief and to minimize the potential for subsequent exacerbations. These are accomplished via a multifaceted approach, including the use of bronchodilators, antibiotics, supplemental oxygen, noninvasive positive pressure ventilation in certain circumstances, and systemic corticosteroids.
The administration of systemic steroids in AECOPD has been prevalent for several decades, with initial studies showing positive effects on lung function, specifically FEV1.4 Studies have demonstrated the benefit of steroids in prolonging the time to subsequent exacerbation, reducing the rate of treatment failure, and reducing length of stay (LOS).5 Corticosteroids have since become an essential component of the standard of care in AECOPD management.
Despite consensus that systemic steroids should be used in COPD exacerbations, a great deal of controversy still surrounds the optimal steroid regimen.6 Steroid use is not without risk, as steroids can lead to adverse outcomes in medically complex hospitalized patients (see Table 1, below). Current guidelines provide limited guidance as to the optimal route of administration, dosing regimen, or length of therapy; clinical practice varies widely.
Review of the Data
Administration route: intravenous (IV) vs. oral. The use of steroids in AECOPD began with such IV formulations as methylprednisolone, and this became the typical method of treating hospitalized patients. This practice was validated in a multicenter Veterans Affairs trial, which demonstrated decreased risk of treatment failure (defined as all-cause mortality, need for intubation, readmission for COPD, or intensification of pharmacologic therapy) for patients randomized to receive an IV-to-oral steroid regimen compared with those randomized to placebo.5 Patients receiving steroids also had shorter LOS and improvements in FEV1 after the first day of treatment. Subsequent randomized controlled trials in patients with AECOPD demonstrated the benefit of oral regimens compared with placebo with regard to FEV1, LOS, and risk of treatment failure.6,7,8
Similarities in the bioavailability of oral and IV steroids have been known for a long time.9 Comparisons in efficacy initially were completed in the management of acute asthma exacerbations, with increasing evidence, including a meta-analysis, demonstrating no difference in improvement in pulmonary function and in preventing relapse of exacerbations for oral compared with IV steroids.10 However, only recently have oral and IV steroids been compared in the treatment of AECOPD. De Jong et al randomized more than 200 patients hospitalized for AECOPD to 60 mg of either IV or oral prednisolone for five days, followed by a week of an oral taper.11 There were no significant differences in treatment failure between the IV and oral groups (62% vs. 56%, respectively, at 90 days; one-sided lower bound of the 95% confidence interval [CI], −5.8%).
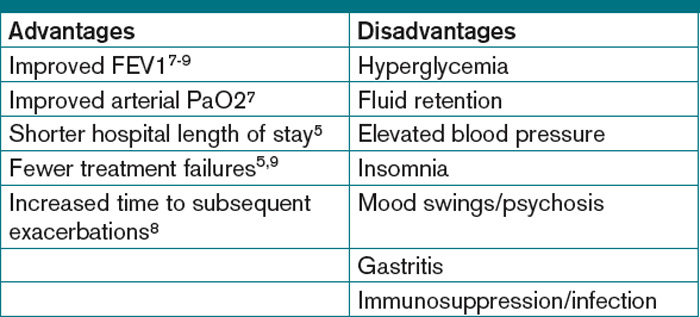
A large observational study by Lindenauer et al, including nearly 80,000 AECOPD patients admitted at more than 400 hospitals, added further support to the idea that oral and IV steroids were comparable in efficacy.12 In this study, multivariate analysis found no difference in treatment failure between oral and IV groups (odds ratio [OR] 0.93; 95% CI, 0.84-1.02). The authors also found, however, that current clinical practice still overwhelmingly favors intravenous steroids, with 92% of study patients initially being administered IV steroids.12
Based on the evidence from de Jong and Lindenauer, it appears that there is no significant benefit to the use of IV over oral steroids. Additionally, there is evidence for oral administration being associated with beneficial effects on cost and hospital LOS.12 Oral steroids, therefore, are the preferred route of administration to treat a hospitalized patient with AECOPD, unless the patient is unable to tolerate oral medications. Current guidelines support the practice of giving oral steroids as first-line treatment for AECOPD (see Table 2, above).
High dose vs. low dose. Another important clinical issue concerns the dosing of steroids. The randomized trials examining the use of corticosteroids in AECOPD vary widely in the dosages studied. Further, the majority of these trials have compared steroids to placebo, rather than comparing different dosage regimens. The agents studied have included prednisone, prednisolone, methylprednisolone, and hydrocortisone, or combinations thereof. In order to compare regimens of these different drugs, steroid doses often are converted into prednisone equivalents (see Table 3, below). Though no guidelines define “high dose” and “low dose,” some studies have designated doses of >80 mg prednisone equivalents daily as high-dose and prednisone equivalents of ≤80 mg daily as low-dose.13,14
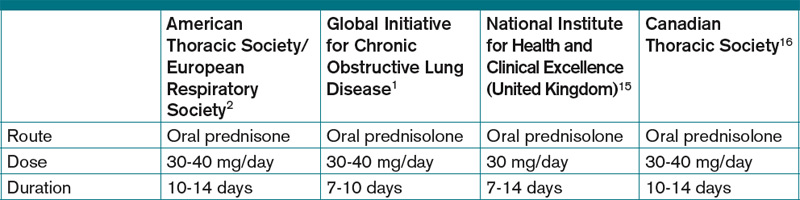
Starting doses of systemic corticosteroids in the treatment of AECOPD in clinical studies range from prednisone equivalents of 30 mg daily to 625 mg on the first day of treatment.5,8 No randomized studies of high- versus low-dose steroid regimens have been conducted. One retrospective chart review of 145 AECOPD admissions evaluated outcomes among patients who were given higher (mean daily dose >80 mg prednisone equivalent) and lower (mean daily dose of ≤80 mg prednisone) doses.14 The authors found that patients who received higher doses of steroids had significantly longer LOS compared with those who received lower doses, especially among the subset of patients who were admitted to the floor rather than the ICU, though this analysis did not adjust for severity of illness. In this study, the most striking finding noted by the authors was the wide variability in the steroid doses prescribed for the inpatient treatment of AECOPD.
More recently, the study by Lindenauer et al examined outcomes between patients treated with high-dose IV steroids (equivalent of 120 mg-800 mg of prednisone on the first or second day of treatment) compared to low-dose oral steroids (prednisone equivalents of 20 mg-80 mg per day).12 The authors found no differences between the two groups regarding the rate of treatment failure, defined by initiation of mechanical ventilation after the second hospital day, in-hospital mortality, or readmission for COPD within 30 days of discharge. After multivariate adjustment, including the propensity for oral treatment, the low-dose oral therapy group was found to have lower risk of treatment failure, shorter LOS, and lower total hospital cost.
Despite the heterogeneity of the published data and the lack of randomized trials, the existing evidence suggests that low-dose prednisone (or equivalent) is similar in efficacy to higher doses and generally is associated with shorter hospital stays. Recognizing these benefits, guidelines do favor initiating treatment with low-dose steroids in patients admitted with AECOPD. The most recent publications from the American Thoracic Society/European Respiratory Society Task Force (ATS/ERS), the Global Initiative for Chronic Obstructive Lung Disease (GOLD), the National Clinical Guidelines Centre in the United Kingdom, and the Canadian Thoracic Society all recommend equivalent dosing of prednisone in patients admitted with AECOPD who are able to tolerate oral intake (see Table 2).1,2,15,16
Duration. As with the dosing of systemic corticosteroids in AECOPD, the optimal duration of treatment is not well-established. National and international consensus panels vary in their recommendations, as outlined in Table 2. This may be related to the variability in length of treatment found in the literature.
Treatment durations ranging from one day to eight weeks have been studied in inpatients with AECOPD. The landmark randomized controlled trial by Niewoehner and colleagues compared two-week and eight-week courses of systemic corticosteroids and found no difference in the rates of treatment failure, which included death, need for mechanical ventilation, readmission for COPD, and intensification of pharmacologic therapy.5 Based on these results, many experts have concluded that there is no benefit to steroid courses lasting beyond two weeks.
Although improvements in outcomes have been demonstrated with corticosteroid regimens as short as three days compared with placebo, most of the randomized controlled trials have included courses of seven to 14 days.4 Given the risks of adverse events (e.g. hyperglycemia) that are associated with systemic administration of steroids, the shortest effective duration should be considered.
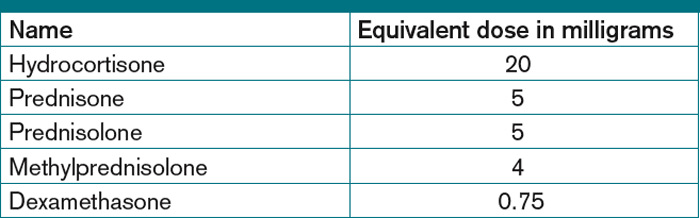
In both clinical practice and clinical studies, steroid regimens often include a taper. A study by Vondracek and Hemstreet found that 79% of hospital discharges for AECOPD included a tapered corticosteroid regimen.14 From a physiologic standpoint, durations of corticosteroid treatment approximately three weeks or less, regardless of dosage, should not lead to adrenal suppression.17 There also is no evidence to suggest that abrupt discontinuation of steroids leads to clinical worsening of disease, and complicated steroid tapers are a potential source of medication errors after hospital discharge.18 Furthermore, the clinical guidelines do not address the tapering of corticosteroids. Therefore, there is a lack of evidence advocating for or against the use of tapered steroid regimens in AECOPD.
Back to the Case
In addition to standard treatment modalities for AECOPD, our patient was administered oral prednisone 40 mg daily. She experienced steroid-induced hyperglycemia, which was corrected with adjustment of her insulin regimen. The patient’s pulmonary symptoms improved within 72 hours, and she was discharged home on hospital day four to complete a seven-day steroid course. At hospital discharge, she was administered influenza and pneumococcal vaccinations, and she was instructed to resume her usual insulin dosing once she finished her prednisone course.
Overview
In the management of AECOPD, there remains a lack of consensus in defining the ideal steroid regimen. Based on current literature, the use of low-dose oral corticosteroids, such as prednisone 40 mg daily, for a seven- to 14-day course is recommended. TH
Dr. Cunningham is an assistant professor of internal medicine and academic hospitalist in the section of hospital medicine at Vanderbilt University School of Medicine in Nashville, Tenn. Dr. LaBrin is an assistant professor of internal medicine and pediatrics and academic hospitalist at Vanderbilt University School of Medicine.
References
- From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2010. Global Initiative for Chronic Obstructive Lung Disease website. Available at: www.goldcopd.org/GuidelineItem.asp?intId=989. Accessed Feb. 21, 2011.
- Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932-946.
- Morbidity and mortality: 2009 chart book on cardiovascular, lung, and blood diseases. National Institutes of Health’s National Heart, Lung, and Blood Institute website. Available at: www.nhlbi.nih.gov/resources/docs/2009_ChartBook.pdf. Accessed Feb. 24, 2011.
- Albert RK, Martin TR, Lewis SW. Controlled trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med. 1980;92(6):753-758.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340(25):1941-1947.
- Thompson WH, Nielson C, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med. 1996;154:407-412.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608-1613.
- Davies L, Angus RM, Calverley PM. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet. 1999;354(9177):456-460.
- Al-Habet S, Rogers HJ. Pharmacokinetics of intravenous and oral prednisolone. Br J Clin Pharmacol. 1980;10(5):503-508.
- Rowe BH, Keller JL, Oxman AD. Effectiveness of steroid therapy in acute exacerbations of asthma: a meta-analysis. Am J Emerg Med. 1992;10:301-310.
- De Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: A randomized, controlled, double-blind study. Chest. 2007;132(6):1741-1747.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA. 2010;303(23):2359-2367.
- Manser R, Reid D, Abramsom MJ. Corticosteroids for acute severe asthma in hospitalized patients. Cochrane Database Syst Rev. 2000;(2):CD001740.
- Vondracek SF, Hemstreet BA. Retrospective evaluation of systemic corticosteroids for the management of acute exacerbations of chronic obstructive pulmonary disease. Am J Health Syst Pharm. 2006;63:645-652.
- Chronic obstructive pulmonary disease: management of chronic obstructive pulmonary disease in adults in primary and secondary care. National Institute for Health and Clinical Excellence website. Available at: guidance.nice.org.uk/CG101/Guidance/pdf/English. Accessed Feb. 21, 2011.
- O’Donnell DE, Aaron S, Bourbeau J, et al. Canadian Thoracic Society recommendations for management of chronic obstructive pulmonary disease—2007 update. Can Respir J. 2007;14 Suppl B:5B-32B.
- Webb J, Clark TJ. Recovery of plasma corticotrophin and cortisol levels after three-week course of prednisolone. Thorax. 1981;36:22-24.
- O’Driscoll BR, Kalra S, Wilson M, Pickering CA, Carroll KB, Woodcock AA. Double-blind trial of steroid tapering in acute asthma. Lancet. 1993; 341:324-7.
Case
A 66-year-old Caucasian female with moderate chronic obstructive pulmonary disease (COPD) (FEV1 55% predicted), obesity, hypertension, and Type 2 diabetes mellitus on insulin therapy presents to the ED with four days of increased cough productive of yellow sputum and progressive shortness of breath. Her physical exam is notable for an oxygen saturation of 87% on room air, along with diffuse expiratory wheezing with use of accessory muscles; her chest X-ray is unchanged from previous. The patient is given oxygen, nebulized bronchodilators, and one dose of IV methylprednisolone. Her symptoms do not improve significantly, and she is admitted for further management. What regimen of corticosteroids is most appropriate to treat her acute exacerbation of COPD?
Overview
COPD is the fourth-leading cause of death in the United States and continues to increase in prevalence.1 Acute exacerbations of COPD (AECOPD) contribute significantly to this high mortality rate, which approaches 40% at one year in those patients requiring mechanical support.1 An exacerbation of COPD has been defined as an acute change in a patient’s baseline dyspnea, cough, and/or sputum beyond day-to-day variability sufficient to warrant a change in therapy.2 Exacerbations commonly occur in COPD patients and often necessitate hospital admission. In fact, COPD consistently is one of the 10 most common reasons for hospitalization, with billions of dollars in associated healthcare costs.3
The goals for inpatient management of AECOPD are to provide acute symptom relief and to minimize the potential for subsequent exacerbations. These are accomplished via a multifaceted approach, including the use of bronchodilators, antibiotics, supplemental oxygen, noninvasive positive pressure ventilation in certain circumstances, and systemic corticosteroids.
The administration of systemic steroids in AECOPD has been prevalent for several decades, with initial studies showing positive effects on lung function, specifically FEV1.4 Studies have demonstrated the benefit of steroids in prolonging the time to subsequent exacerbation, reducing the rate of treatment failure, and reducing length of stay (LOS).5 Corticosteroids have since become an essential component of the standard of care in AECOPD management.
Despite consensus that systemic steroids should be used in COPD exacerbations, a great deal of controversy still surrounds the optimal steroid regimen.6 Steroid use is not without risk, as steroids can lead to adverse outcomes in medically complex hospitalized patients (see Table 1, below). Current guidelines provide limited guidance as to the optimal route of administration, dosing regimen, or length of therapy; clinical practice varies widely.
Review of the Data
Administration route: intravenous (IV) vs. oral. The use of steroids in AECOPD began with such IV formulations as methylprednisolone, and this became the typical method of treating hospitalized patients. This practice was validated in a multicenter Veterans Affairs trial, which demonstrated decreased risk of treatment failure (defined as all-cause mortality, need for intubation, readmission for COPD, or intensification of pharmacologic therapy) for patients randomized to receive an IV-to-oral steroid regimen compared with those randomized to placebo.5 Patients receiving steroids also had shorter LOS and improvements in FEV1 after the first day of treatment. Subsequent randomized controlled trials in patients with AECOPD demonstrated the benefit of oral regimens compared with placebo with regard to FEV1, LOS, and risk of treatment failure.6,7,8
Similarities in the bioavailability of oral and IV steroids have been known for a long time.9 Comparisons in efficacy initially were completed in the management of acute asthma exacerbations, with increasing evidence, including a meta-analysis, demonstrating no difference in improvement in pulmonary function and in preventing relapse of exacerbations for oral compared with IV steroids.10 However, only recently have oral and IV steroids been compared in the treatment of AECOPD. De Jong et al randomized more than 200 patients hospitalized for AECOPD to 60 mg of either IV or oral prednisolone for five days, followed by a week of an oral taper.11 There were no significant differences in treatment failure between the IV and oral groups (62% vs. 56%, respectively, at 90 days; one-sided lower bound of the 95% confidence interval [CI], −5.8%).
A large observational study by Lindenauer et al, including nearly 80,000 AECOPD patients admitted at more than 400 hospitals, added further support to the idea that oral and IV steroids were comparable in efficacy.12 In this study, multivariate analysis found no difference in treatment failure between oral and IV groups (odds ratio [OR] 0.93; 95% CI, 0.84-1.02). The authors also found, however, that current clinical practice still overwhelmingly favors intravenous steroids, with 92% of study patients initially being administered IV steroids.12
Based on the evidence from de Jong and Lindenauer, it appears that there is no significant benefit to the use of IV over oral steroids. Additionally, there is evidence for oral administration being associated with beneficial effects on cost and hospital LOS.12 Oral steroids, therefore, are the preferred route of administration to treat a hospitalized patient with AECOPD, unless the patient is unable to tolerate oral medications. Current guidelines support the practice of giving oral steroids as first-line treatment for AECOPD (see Table 2, above).
High dose vs. low dose. Another important clinical issue concerns the dosing of steroids. The randomized trials examining the use of corticosteroids in AECOPD vary widely in the dosages studied. Further, the majority of these trials have compared steroids to placebo, rather than comparing different dosage regimens. The agents studied have included prednisone, prednisolone, methylprednisolone, and hydrocortisone, or combinations thereof. In order to compare regimens of these different drugs, steroid doses often are converted into prednisone equivalents (see Table 3, below). Though no guidelines define “high dose” and “low dose,” some studies have designated doses of >80 mg prednisone equivalents daily as high-dose and prednisone equivalents of ≤80 mg daily as low-dose.13,14
Starting doses of systemic corticosteroids in the treatment of AECOPD in clinical studies range from prednisone equivalents of 30 mg daily to 625 mg on the first day of treatment.5,8 No randomized studies of high- versus low-dose steroid regimens have been conducted. One retrospective chart review of 145 AECOPD admissions evaluated outcomes among patients who were given higher (mean daily dose >80 mg prednisone equivalent) and lower (mean daily dose of ≤80 mg prednisone) doses.14 The authors found that patients who received higher doses of steroids had significantly longer LOS compared with those who received lower doses, especially among the subset of patients who were admitted to the floor rather than the ICU, though this analysis did not adjust for severity of illness. In this study, the most striking finding noted by the authors was the wide variability in the steroid doses prescribed for the inpatient treatment of AECOPD.
More recently, the study by Lindenauer et al examined outcomes between patients treated with high-dose IV steroids (equivalent of 120 mg-800 mg of prednisone on the first or second day of treatment) compared to low-dose oral steroids (prednisone equivalents of 20 mg-80 mg per day).12 The authors found no differences between the two groups regarding the rate of treatment failure, defined by initiation of mechanical ventilation after the second hospital day, in-hospital mortality, or readmission for COPD within 30 days of discharge. After multivariate adjustment, including the propensity for oral treatment, the low-dose oral therapy group was found to have lower risk of treatment failure, shorter LOS, and lower total hospital cost.
Despite the heterogeneity of the published data and the lack of randomized trials, the existing evidence suggests that low-dose prednisone (or equivalent) is similar in efficacy to higher doses and generally is associated with shorter hospital stays. Recognizing these benefits, guidelines do favor initiating treatment with low-dose steroids in patients admitted with AECOPD. The most recent publications from the American Thoracic Society/European Respiratory Society Task Force (ATS/ERS), the Global Initiative for Chronic Obstructive Lung Disease (GOLD), the National Clinical Guidelines Centre in the United Kingdom, and the Canadian Thoracic Society all recommend equivalent dosing of prednisone in patients admitted with AECOPD who are able to tolerate oral intake (see Table 2).1,2,15,16
Duration. As with the dosing of systemic corticosteroids in AECOPD, the optimal duration of treatment is not well-established. National and international consensus panels vary in their recommendations, as outlined in Table 2. This may be related to the variability in length of treatment found in the literature.
Treatment durations ranging from one day to eight weeks have been studied in inpatients with AECOPD. The landmark randomized controlled trial by Niewoehner and colleagues compared two-week and eight-week courses of systemic corticosteroids and found no difference in the rates of treatment failure, which included death, need for mechanical ventilation, readmission for COPD, and intensification of pharmacologic therapy.5 Based on these results, many experts have concluded that there is no benefit to steroid courses lasting beyond two weeks.
Although improvements in outcomes have been demonstrated with corticosteroid regimens as short as three days compared with placebo, most of the randomized controlled trials have included courses of seven to 14 days.4 Given the risks of adverse events (e.g. hyperglycemia) that are associated with systemic administration of steroids, the shortest effective duration should be considered.
In both clinical practice and clinical studies, steroid regimens often include a taper. A study by Vondracek and Hemstreet found that 79% of hospital discharges for AECOPD included a tapered corticosteroid regimen.14 From a physiologic standpoint, durations of corticosteroid treatment approximately three weeks or less, regardless of dosage, should not lead to adrenal suppression.17 There also is no evidence to suggest that abrupt discontinuation of steroids leads to clinical worsening of disease, and complicated steroid tapers are a potential source of medication errors after hospital discharge.18 Furthermore, the clinical guidelines do not address the tapering of corticosteroids. Therefore, there is a lack of evidence advocating for or against the use of tapered steroid regimens in AECOPD.
Back to the Case
In addition to standard treatment modalities for AECOPD, our patient was administered oral prednisone 40 mg daily. She experienced steroid-induced hyperglycemia, which was corrected with adjustment of her insulin regimen. The patient’s pulmonary symptoms improved within 72 hours, and she was discharged home on hospital day four to complete a seven-day steroid course. At hospital discharge, she was administered influenza and pneumococcal vaccinations, and she was instructed to resume her usual insulin dosing once she finished her prednisone course.
Overview
In the management of AECOPD, there remains a lack of consensus in defining the ideal steroid regimen. Based on current literature, the use of low-dose oral corticosteroids, such as prednisone 40 mg daily, for a seven- to 14-day course is recommended. TH
Dr. Cunningham is an assistant professor of internal medicine and academic hospitalist in the section of hospital medicine at Vanderbilt University School of Medicine in Nashville, Tenn. Dr. LaBrin is an assistant professor of internal medicine and pediatrics and academic hospitalist at Vanderbilt University School of Medicine.
References
- From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2010. Global Initiative for Chronic Obstructive Lung Disease website. Available at: www.goldcopd.org/GuidelineItem.asp?intId=989. Accessed Feb. 21, 2011.
- Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932-946.
- Morbidity and mortality: 2009 chart book on cardiovascular, lung, and blood diseases. National Institutes of Health’s National Heart, Lung, and Blood Institute website. Available at: www.nhlbi.nih.gov/resources/docs/2009_ChartBook.pdf. Accessed Feb. 24, 2011.
- Albert RK, Martin TR, Lewis SW. Controlled trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med. 1980;92(6):753-758.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340(25):1941-1947.
- Thompson WH, Nielson C, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med. 1996;154:407-412.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608-1613.
- Davies L, Angus RM, Calverley PM. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet. 1999;354(9177):456-460.
- Al-Habet S, Rogers HJ. Pharmacokinetics of intravenous and oral prednisolone. Br J Clin Pharmacol. 1980;10(5):503-508.
- Rowe BH, Keller JL, Oxman AD. Effectiveness of steroid therapy in acute exacerbations of asthma: a meta-analysis. Am J Emerg Med. 1992;10:301-310.
- De Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: A randomized, controlled, double-blind study. Chest. 2007;132(6):1741-1747.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA. 2010;303(23):2359-2367.
- Manser R, Reid D, Abramsom MJ. Corticosteroids for acute severe asthma in hospitalized patients. Cochrane Database Syst Rev. 2000;(2):CD001740.
- Vondracek SF, Hemstreet BA. Retrospective evaluation of systemic corticosteroids for the management of acute exacerbations of chronic obstructive pulmonary disease. Am J Health Syst Pharm. 2006;63:645-652.
- Chronic obstructive pulmonary disease: management of chronic obstructive pulmonary disease in adults in primary and secondary care. National Institute for Health and Clinical Excellence website. Available at: guidance.nice.org.uk/CG101/Guidance/pdf/English. Accessed Feb. 21, 2011.
- O’Donnell DE, Aaron S, Bourbeau J, et al. Canadian Thoracic Society recommendations for management of chronic obstructive pulmonary disease—2007 update. Can Respir J. 2007;14 Suppl B:5B-32B.
- Webb J, Clark TJ. Recovery of plasma corticotrophin and cortisol levels after three-week course of prednisolone. Thorax. 1981;36:22-24.
- O’Driscoll BR, Kalra S, Wilson M, Pickering CA, Carroll KB, Woodcock AA. Double-blind trial of steroid tapering in acute asthma. Lancet. 1993; 341:324-7.
Case
A 66-year-old Caucasian female with moderate chronic obstructive pulmonary disease (COPD) (FEV1 55% predicted), obesity, hypertension, and Type 2 diabetes mellitus on insulin therapy presents to the ED with four days of increased cough productive of yellow sputum and progressive shortness of breath. Her physical exam is notable for an oxygen saturation of 87% on room air, along with diffuse expiratory wheezing with use of accessory muscles; her chest X-ray is unchanged from previous. The patient is given oxygen, nebulized bronchodilators, and one dose of IV methylprednisolone. Her symptoms do not improve significantly, and she is admitted for further management. What regimen of corticosteroids is most appropriate to treat her acute exacerbation of COPD?
Overview
COPD is the fourth-leading cause of death in the United States and continues to increase in prevalence.1 Acute exacerbations of COPD (AECOPD) contribute significantly to this high mortality rate, which approaches 40% at one year in those patients requiring mechanical support.1 An exacerbation of COPD has been defined as an acute change in a patient’s baseline dyspnea, cough, and/or sputum beyond day-to-day variability sufficient to warrant a change in therapy.2 Exacerbations commonly occur in COPD patients and often necessitate hospital admission. In fact, COPD consistently is one of the 10 most common reasons for hospitalization, with billions of dollars in associated healthcare costs.3
The goals for inpatient management of AECOPD are to provide acute symptom relief and to minimize the potential for subsequent exacerbations. These are accomplished via a multifaceted approach, including the use of bronchodilators, antibiotics, supplemental oxygen, noninvasive positive pressure ventilation in certain circumstances, and systemic corticosteroids.
The administration of systemic steroids in AECOPD has been prevalent for several decades, with initial studies showing positive effects on lung function, specifically FEV1.4 Studies have demonstrated the benefit of steroids in prolonging the time to subsequent exacerbation, reducing the rate of treatment failure, and reducing length of stay (LOS).5 Corticosteroids have since become an essential component of the standard of care in AECOPD management.
Despite consensus that systemic steroids should be used in COPD exacerbations, a great deal of controversy still surrounds the optimal steroid regimen.6 Steroid use is not without risk, as steroids can lead to adverse outcomes in medically complex hospitalized patients (see Table 1, below). Current guidelines provide limited guidance as to the optimal route of administration, dosing regimen, or length of therapy; clinical practice varies widely.
Review of the Data
Administration route: intravenous (IV) vs. oral. The use of steroids in AECOPD began with such IV formulations as methylprednisolone, and this became the typical method of treating hospitalized patients. This practice was validated in a multicenter Veterans Affairs trial, which demonstrated decreased risk of treatment failure (defined as all-cause mortality, need for intubation, readmission for COPD, or intensification of pharmacologic therapy) for patients randomized to receive an IV-to-oral steroid regimen compared with those randomized to placebo.5 Patients receiving steroids also had shorter LOS and improvements in FEV1 after the first day of treatment. Subsequent randomized controlled trials in patients with AECOPD demonstrated the benefit of oral regimens compared with placebo with regard to FEV1, LOS, and risk of treatment failure.6,7,8
Similarities in the bioavailability of oral and IV steroids have been known for a long time.9 Comparisons in efficacy initially were completed in the management of acute asthma exacerbations, with increasing evidence, including a meta-analysis, demonstrating no difference in improvement in pulmonary function and in preventing relapse of exacerbations for oral compared with IV steroids.10 However, only recently have oral and IV steroids been compared in the treatment of AECOPD. De Jong et al randomized more than 200 patients hospitalized for AECOPD to 60 mg of either IV or oral prednisolone for five days, followed by a week of an oral taper.11 There were no significant differences in treatment failure between the IV and oral groups (62% vs. 56%, respectively, at 90 days; one-sided lower bound of the 95% confidence interval [CI], −5.8%).
A large observational study by Lindenauer et al, including nearly 80,000 AECOPD patients admitted at more than 400 hospitals, added further support to the idea that oral and IV steroids were comparable in efficacy.12 In this study, multivariate analysis found no difference in treatment failure between oral and IV groups (odds ratio [OR] 0.93; 95% CI, 0.84-1.02). The authors also found, however, that current clinical practice still overwhelmingly favors intravenous steroids, with 92% of study patients initially being administered IV steroids.12
Based on the evidence from de Jong and Lindenauer, it appears that there is no significant benefit to the use of IV over oral steroids. Additionally, there is evidence for oral administration being associated with beneficial effects on cost and hospital LOS.12 Oral steroids, therefore, are the preferred route of administration to treat a hospitalized patient with AECOPD, unless the patient is unable to tolerate oral medications. Current guidelines support the practice of giving oral steroids as first-line treatment for AECOPD (see Table 2, above).
High dose vs. low dose. Another important clinical issue concerns the dosing of steroids. The randomized trials examining the use of corticosteroids in AECOPD vary widely in the dosages studied. Further, the majority of these trials have compared steroids to placebo, rather than comparing different dosage regimens. The agents studied have included prednisone, prednisolone, methylprednisolone, and hydrocortisone, or combinations thereof. In order to compare regimens of these different drugs, steroid doses often are converted into prednisone equivalents (see Table 3, below). Though no guidelines define “high dose” and “low dose,” some studies have designated doses of >80 mg prednisone equivalents daily as high-dose and prednisone equivalents of ≤80 mg daily as low-dose.13,14
Starting doses of systemic corticosteroids in the treatment of AECOPD in clinical studies range from prednisone equivalents of 30 mg daily to 625 mg on the first day of treatment.5,8 No randomized studies of high- versus low-dose steroid regimens have been conducted. One retrospective chart review of 145 AECOPD admissions evaluated outcomes among patients who were given higher (mean daily dose >80 mg prednisone equivalent) and lower (mean daily dose of ≤80 mg prednisone) doses.14 The authors found that patients who received higher doses of steroids had significantly longer LOS compared with those who received lower doses, especially among the subset of patients who were admitted to the floor rather than the ICU, though this analysis did not adjust for severity of illness. In this study, the most striking finding noted by the authors was the wide variability in the steroid doses prescribed for the inpatient treatment of AECOPD.
More recently, the study by Lindenauer et al examined outcomes between patients treated with high-dose IV steroids (equivalent of 120 mg-800 mg of prednisone on the first or second day of treatment) compared to low-dose oral steroids (prednisone equivalents of 20 mg-80 mg per day).12 The authors found no differences between the two groups regarding the rate of treatment failure, defined by initiation of mechanical ventilation after the second hospital day, in-hospital mortality, or readmission for COPD within 30 days of discharge. After multivariate adjustment, including the propensity for oral treatment, the low-dose oral therapy group was found to have lower risk of treatment failure, shorter LOS, and lower total hospital cost.
Despite the heterogeneity of the published data and the lack of randomized trials, the existing evidence suggests that low-dose prednisone (or equivalent) is similar in efficacy to higher doses and generally is associated with shorter hospital stays. Recognizing these benefits, guidelines do favor initiating treatment with low-dose steroids in patients admitted with AECOPD. The most recent publications from the American Thoracic Society/European Respiratory Society Task Force (ATS/ERS), the Global Initiative for Chronic Obstructive Lung Disease (GOLD), the National Clinical Guidelines Centre in the United Kingdom, and the Canadian Thoracic Society all recommend equivalent dosing of prednisone in patients admitted with AECOPD who are able to tolerate oral intake (see Table 2).1,2,15,16
Duration. As with the dosing of systemic corticosteroids in AECOPD, the optimal duration of treatment is not well-established. National and international consensus panels vary in their recommendations, as outlined in Table 2. This may be related to the variability in length of treatment found in the literature.
Treatment durations ranging from one day to eight weeks have been studied in inpatients with AECOPD. The landmark randomized controlled trial by Niewoehner and colleagues compared two-week and eight-week courses of systemic corticosteroids and found no difference in the rates of treatment failure, which included death, need for mechanical ventilation, readmission for COPD, and intensification of pharmacologic therapy.5 Based on these results, many experts have concluded that there is no benefit to steroid courses lasting beyond two weeks.
Although improvements in outcomes have been demonstrated with corticosteroid regimens as short as three days compared with placebo, most of the randomized controlled trials have included courses of seven to 14 days.4 Given the risks of adverse events (e.g. hyperglycemia) that are associated with systemic administration of steroids, the shortest effective duration should be considered.
In both clinical practice and clinical studies, steroid regimens often include a taper. A study by Vondracek and Hemstreet found that 79% of hospital discharges for AECOPD included a tapered corticosteroid regimen.14 From a physiologic standpoint, durations of corticosteroid treatment approximately three weeks or less, regardless of dosage, should not lead to adrenal suppression.17 There also is no evidence to suggest that abrupt discontinuation of steroids leads to clinical worsening of disease, and complicated steroid tapers are a potential source of medication errors after hospital discharge.18 Furthermore, the clinical guidelines do not address the tapering of corticosteroids. Therefore, there is a lack of evidence advocating for or against the use of tapered steroid regimens in AECOPD.
Back to the Case
In addition to standard treatment modalities for AECOPD, our patient was administered oral prednisone 40 mg daily. She experienced steroid-induced hyperglycemia, which was corrected with adjustment of her insulin regimen. The patient’s pulmonary symptoms improved within 72 hours, and she was discharged home on hospital day four to complete a seven-day steroid course. At hospital discharge, she was administered influenza and pneumococcal vaccinations, and she was instructed to resume her usual insulin dosing once she finished her prednisone course.
Overview
In the management of AECOPD, there remains a lack of consensus in defining the ideal steroid regimen. Based on current literature, the use of low-dose oral corticosteroids, such as prednisone 40 mg daily, for a seven- to 14-day course is recommended. TH
Dr. Cunningham is an assistant professor of internal medicine and academic hospitalist in the section of hospital medicine at Vanderbilt University School of Medicine in Nashville, Tenn. Dr. LaBrin is an assistant professor of internal medicine and pediatrics and academic hospitalist at Vanderbilt University School of Medicine.
References
- From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2010. Global Initiative for Chronic Obstructive Lung Disease website. Available at: www.goldcopd.org/GuidelineItem.asp?intId=989. Accessed Feb. 21, 2011.
- Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932-946.
- Morbidity and mortality: 2009 chart book on cardiovascular, lung, and blood diseases. National Institutes of Health’s National Heart, Lung, and Blood Institute website. Available at: www.nhlbi.nih.gov/resources/docs/2009_ChartBook.pdf. Accessed Feb. 24, 2011.
- Albert RK, Martin TR, Lewis SW. Controlled trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med. 1980;92(6):753-758.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340(25):1941-1947.
- Thompson WH, Nielson C, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med. 1996;154:407-412.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608-1613.
- Davies L, Angus RM, Calverley PM. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet. 1999;354(9177):456-460.
- Al-Habet S, Rogers HJ. Pharmacokinetics of intravenous and oral prednisolone. Br J Clin Pharmacol. 1980;10(5):503-508.
- Rowe BH, Keller JL, Oxman AD. Effectiveness of steroid therapy in acute exacerbations of asthma: a meta-analysis. Am J Emerg Med. 1992;10:301-310.
- De Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: A randomized, controlled, double-blind study. Chest. 2007;132(6):1741-1747.
- Lindenauer PK, Pekow PS, Lahti MC, Lee Y, Benjamin EM, Rothberg MB. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA. 2010;303(23):2359-2367.
- Manser R, Reid D, Abramsom MJ. Corticosteroids for acute severe asthma in hospitalized patients. Cochrane Database Syst Rev. 2000;(2):CD001740.
- Vondracek SF, Hemstreet BA. Retrospective evaluation of systemic corticosteroids for the management of acute exacerbations of chronic obstructive pulmonary disease. Am J Health Syst Pharm. 2006;63:645-652.
- Chronic obstructive pulmonary disease: management of chronic obstructive pulmonary disease in adults in primary and secondary care. National Institute for Health and Clinical Excellence website. Available at: guidance.nice.org.uk/CG101/Guidance/pdf/English. Accessed Feb. 21, 2011.
- O’Donnell DE, Aaron S, Bourbeau J, et al. Canadian Thoracic Society recommendations for management of chronic obstructive pulmonary disease—2007 update. Can Respir J. 2007;14 Suppl B:5B-32B.
- Webb J, Clark TJ. Recovery of plasma corticotrophin and cortisol levels after three-week course of prednisolone. Thorax. 1981;36:22-24.
- O’Driscoll BR, Kalra S, Wilson M, Pickering CA, Carroll KB, Woodcock AA. Double-blind trial of steroid tapering in acute asthma. Lancet. 1993; 341:324-7.
What Is the Best Approach to Medical Therapy for Patients with Ischemic Stroke?
Case
A 58-year-old woman with diabetes mellitus and hypertension presents with dysarthria and weakness on the right side of her body starting six hours prior to presentation. She is afebrile and has a blood pressure of 162/84 mmHg. Exam reveals the absence of a heart murmur and no lower-extremity swelling or calf tenderness. There is weakness of the right side of the body on exam with diminished proprioception. A noncontrast head CT shows no intracranial hemorrhage. She is admitted to the hospital with the diagnosis of acute ischemic stroke. What anticlotting or antiplatelet medications should she receive?
Overview
Stroke remains a significant cause of morbidity and mortality in the U.S. and around the world. The majority of strokes are ischemic in etiology. Although thrombolytic therapy is the most effective way to salvage ischemic brain tissue that has not yet infarcted, there is a narrow window for the use of thrombolytics in the treatment of acute ischemic stroke. As a result, many patients will not be eligible for thrombolysis. Outside of 4.5 hours from symptom onset, evidence suggests that the risk outweighs the benefit of using the thrombolytic alteplase. For patients ineligible for thrombolytic therapy, antiplatelet therapy remains the best choice for treatment.
Medications that prevent blood from coagulating or clotting are used to treat and prevent a recurring or second stroke. Typically, an antiplatelet agent (most often aspirin) is initiated within 48 hours of an ischemic stroke and continued in low doses as maintenance. Multiple studies suggest that antiplatelet therapy can reduce the risk for a second stroke by 25%. Specific anticlotting agents might be warranted in some patients with high-risk conditions for a stroke.
Review of Data
Early initiation of aspirin has shown benefit in the treatment of an acute ischemic stroke. Two major trials—the International Stroke Trial (IST) and the Chinese Acute Stroke Trial (CAST)— evaluated the role of aspirin (see Table 1, p. 15).1,2 The IST and CAST trials showed that roughly nine nonfatal strokes were avoided per every 1,000 early treatments. Taking the endpoint of death, as well as focal deficits, the two trials confirmed a rate of reduction of 13 per 1,000 patients.
Overall, the consensus was that initiating aspirin within 48 hours of a presumed ischemic cerebrovascular accident posed no major risk of hemorrhagic complication and improved the long-term outcomes.
Along with aspirin, other antiplatelet agents have been studied, most commonly dipyridamole and clopidrogel. The EARLY trial demonstrated no significant differences in the aspirin and dipyridamole groups at 90 days.3
Another large trial, which focused on clopidrogel and aspirin, looked at aspirin plus clopidrogel or aspirin alone. The FASTER trial enrolled mostly patients with mild cerebrovasular accidents (CVA) or transient ischemic attacks (TIA), and there was no difference in outcome measures between the groups.4 However, the MATCH trial found that aspirin and clopidrogel did not provide improved stroke preventions versus clopidogrel alone but had a larger risk of hemorrhagic/bleeding complications.5
Aspirin dosage is somewhat controversial. Fewer side effects occur with lower doses. Combining the trials, consensus treatment includes early aspirin dosing (325 mg initially, then 150 mg-325 mg daily) given to patients with ischemic stroke. Early aspirin should be avoided in those patients who qualify for and are receiving alteplase, heparin, or oral warfarin therapy.
There are other antiplatelet agents for long-term management of ischemic stroke. Whereas aspirin alone is used in the early management of acute ischemic stroke in those ineligible for thrombolytic therapy, many patients are transitioned to other antiplatelet strategies for secondary prevention long-term. The number needed to treat for aspirin to reduce one future stroke, myocardial infarction (MI), or vascular death when compared to placebo is quite high at 33. However, the combination of aspirin and dipyradimole does not prevent MI, vascular death, or the combined endpoint of either stroke or death.
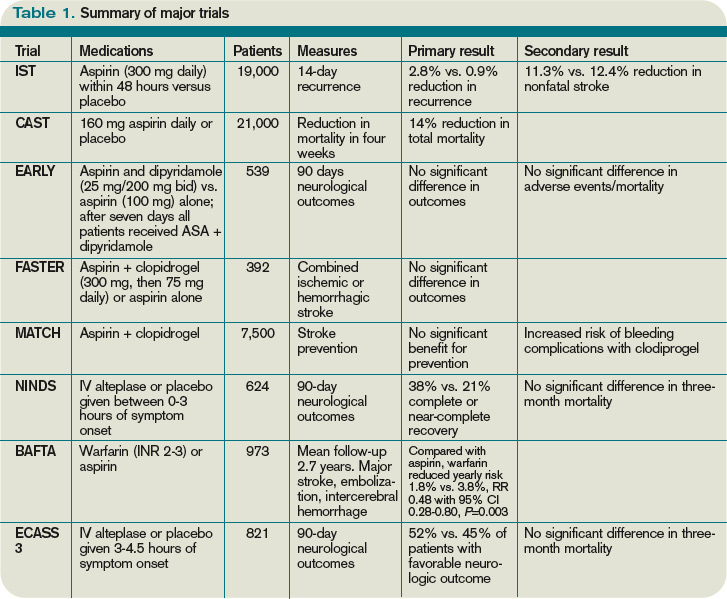
Clopidogrel is more effective than aspirin in preventing a combined endpoint of ischemic stroke, MI, or vascular death, but it is not superior to aspirin in preventing recurrent stroke in TIA or stroke patients. The effects of clopidrogel are greater in patients with peripheral arterial disease, previous coronary artery bypass grafting, insulin-dependent diabetes, or recurrent vascular events.
There is a substantially high cost of treatment and long-term disability associated with stroke. Costs can vary from 3% to 5% of the annual healthcare budget. The newer antiplatelet agents are more expensive than aspirin, and overall cost-effectiveness is difficult to estimate. Yet, from an economic standpoint, the combination of aspirin and dipyradimole can be recommended as an alternative for secondary stroke prevention in patients without major comorbidities. In those patients with higher risk factors and/or comorbidities, clopidogrel might be more cost-effective than aspirin alone. Furthermore, in patients with aspirin intolerance, clopidogrel is a useful, but expensive, alternative.
Thrombolytic therapy. Restora-tion of blood flow with thrombolytic therapy is the most effective way of salvaging ischemic brain tissue that has not already infarcted. The window for use of the thrombolytic alteplase is narrow; studies suggest that its benefit diminishes with increasing time to treatment. Indeed, after 4.5 hours from the onset of symptoms, evidence suggests that the harm might outweigh the benefit, so the determination of who is eligible for its use has to be made quickly.
Guidelines published by the American Heart Association/American Stoke Association stroke council outline strict inclusion and exclusion criteria for the use of alteplase in the management of acute ischemic stroke.6 Obtaining informed consent and emergent neuroimaging are vital in preventing delays in alteplase administration.
Two major trials that illustrate the benefit of alteplase in the treatment of acute ischemic stroke are the NINDS trial and the ECASS 3 trial. NINDS showed that when intravenous alteplase was used within three hours of symptom onset, patients had improved functional outcome at three months.7 The ECASS 3 trial showed that intravenous alteplase has benefit when given up to 4.5 hours after symptom onset.8 Treatment with intravenous alteplase from three-4.5 hours in the ECASS 3 trial showed a modest improvement in patient outcomes at three months, with a number needed to treat of 14 for a favorable outcome.

A 2010 meta-analysis looked specifically at outcomes in stroke based on time to treat with alteplase using pooled data from the NINDS, ATLANTIS, ECASS (1, 2, and 3), and EPITHET trials.9 It showed that the number needed to treat for a favorable outcome at three months increased steadily when time to treatment was delayed. It also showed that the risk of death after alteplase administration increased significantly after 4.5 hours. Thus, after 4.5 hours, it suggests that harm might exceed the benefits of treatment.
Anticoagulant use in ischemic stroke. Clinical trials have not been effective in demonstrating the use of heparin and low-molecular-weight heparins (LMWHs). A 2008 systematic review of 24 trials (approximately 24,000 patients) demonstrated:
- Anticoagulant therapy did not reduce odds of death;
- Therapy was associated with nine fewer recurrent ischemic strokes per 1,000 patients, but also showed a similar increase in symptomatic intracranial hemorrhages; and
- Overall, researchers could not specify a particular anticoagulant mode or regimen that had an overall net patient benefit.
The use of heparin in atrial fibrillation and stroke has generated controversy in recent years. Review of the data, however, indicates that early treatment with heparin might cause more harm than benefit. A 2007 meta-analysis did not support the use of early anticoagulant therapy. Seven trials (4,200 patients) compared heparin or LMWH started within 48 hours to other treatments (aspirin, placebo). The study authors found:
- Nonsignificant reduction in recurrent ischemic stroke within seven to 14 days;
- Statistically significant increase in symptomatic intracranial hemorrhages; and
- Similar rates of death/disability at final follow-up of studies.
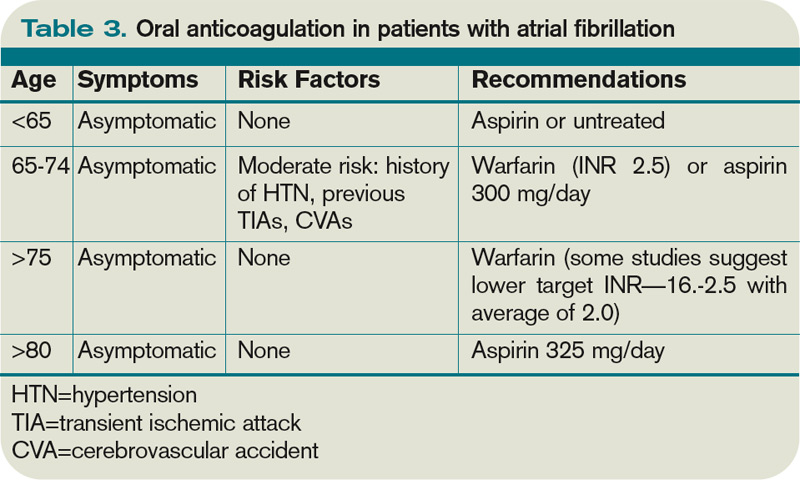
For those patients who continue to demonstrate neurological deterioration, heparin and LMWH use did not appear to improve outcomes. Therefore, based on a consensus of national guidelines, the use of full-dose anticoagulation with heparin or LMWH is not recommended.
The data suggest that in patients with stroke secondary to:
- Dissection of cervical or intracranial arteries;
- Intracardiac thrombus and valvular disease; and
- Mechanical heart valves, full-dose anticoagulation can be initiated. However, the benefit is unproven.
Back to the Case
Our patient with acute ischemic stroke with right-sided weakness on exam presented outside of the window within which alteplase could be administered safely. She was started on aspirin 325 mg daily. There was no indication for full anticoagulation with intravenous heparin or warfarin. Her weakness showed slight improvement on exam during the hospitalization. As an insulin-dependent diabetic, she was thought to be at high risk for recurrent stroke. As such, she was transitioned to a combination of aspirin and clopidogrel prior to her discharge to an acute inpatient rehabilitation hospital.
Bottom Line
Early aspirin therapy (within 48 hours) is recommended (initial dose 325 mg, then 150 mg-325 mg daily) for patients with ischemic stroke who are not candidates for alteplase, IV heparin, or oral anticoagulants.10 Aspirin is the only antiplatelet agent that has been shown to be effective for the early treatment of acute ischemic stroke. In patients without contraindications, aspirin, the combination of aspirin-dipyradimole, or clopidogrel is appropriate for secondary prevention.
The subset of patients at high risk of recurrent stroke should be transitioned to clopidogrel or aspirin/clopidogrel, unless otherwise contraindicated. TH
Dr. Chaturvedi is an instructor in the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and medical director of HM at Northwestern Lake Forest Hospital. Dr. Abraham is an instructor in the Division of Hospital Medicine at Northwestern University Feinberg School of Medicine.
References
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19,435 patients with acute ischemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569-1581.
- CAST: randomised placebo-controlled trial of early aspirin use in 20,000 patients with acute ischaemic stroke. CAST (Chinese Acute Stroke Trial) Collaborative Group. Lancet. 1997;349:1641-1649.
- Dengler R, Diener HC, Schwartz A, et al. Early treatment with aspirin plus extended-release dipyridamole for transient ischaemic attack or ischaemic stroke within 24 h of symptom onset (EARLY trial): a randomised, open-label, blinded-endpoint trial. Lancet Neurol. 2010;9:159-166.
- Kennedy J, Hill MD, Ryckborst KJ, et al. Fast assessment of stroke and transient ischaemic attack to prevent early recurrence (FASTER): a randomised controlled pilot trial. Lancet Neurol. 2007;6:961-969.
- Diener HC, Bogousslavsky J, Brass LM, et al. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:331-337.
- Adams HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655-1711.
- Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695-1703.
- Hacke W, Kaste M, Bluhmki E, et al. Thombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317-1329.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581-1587.
- Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:630S-669S.
Case
A 58-year-old woman with diabetes mellitus and hypertension presents with dysarthria and weakness on the right side of her body starting six hours prior to presentation. She is afebrile and has a blood pressure of 162/84 mmHg. Exam reveals the absence of a heart murmur and no lower-extremity swelling or calf tenderness. There is weakness of the right side of the body on exam with diminished proprioception. A noncontrast head CT shows no intracranial hemorrhage. She is admitted to the hospital with the diagnosis of acute ischemic stroke. What anticlotting or antiplatelet medications should she receive?
Overview
Stroke remains a significant cause of morbidity and mortality in the U.S. and around the world. The majority of strokes are ischemic in etiology. Although thrombolytic therapy is the most effective way to salvage ischemic brain tissue that has not yet infarcted, there is a narrow window for the use of thrombolytics in the treatment of acute ischemic stroke. As a result, many patients will not be eligible for thrombolysis. Outside of 4.5 hours from symptom onset, evidence suggests that the risk outweighs the benefit of using the thrombolytic alteplase. For patients ineligible for thrombolytic therapy, antiplatelet therapy remains the best choice for treatment.
Medications that prevent blood from coagulating or clotting are used to treat and prevent a recurring or second stroke. Typically, an antiplatelet agent (most often aspirin) is initiated within 48 hours of an ischemic stroke and continued in low doses as maintenance. Multiple studies suggest that antiplatelet therapy can reduce the risk for a second stroke by 25%. Specific anticlotting agents might be warranted in some patients with high-risk conditions for a stroke.
Review of Data
Early initiation of aspirin has shown benefit in the treatment of an acute ischemic stroke. Two major trials—the International Stroke Trial (IST) and the Chinese Acute Stroke Trial (CAST)— evaluated the role of aspirin (see Table 1, p. 15).1,2 The IST and CAST trials showed that roughly nine nonfatal strokes were avoided per every 1,000 early treatments. Taking the endpoint of death, as well as focal deficits, the two trials confirmed a rate of reduction of 13 per 1,000 patients.
Overall, the consensus was that initiating aspirin within 48 hours of a presumed ischemic cerebrovascular accident posed no major risk of hemorrhagic complication and improved the long-term outcomes.
Along with aspirin, other antiplatelet agents have been studied, most commonly dipyridamole and clopidrogel. The EARLY trial demonstrated no significant differences in the aspirin and dipyridamole groups at 90 days.3
Another large trial, which focused on clopidrogel and aspirin, looked at aspirin plus clopidrogel or aspirin alone. The FASTER trial enrolled mostly patients with mild cerebrovasular accidents (CVA) or transient ischemic attacks (TIA), and there was no difference in outcome measures between the groups.4 However, the MATCH trial found that aspirin and clopidrogel did not provide improved stroke preventions versus clopidogrel alone but had a larger risk of hemorrhagic/bleeding complications.5
Aspirin dosage is somewhat controversial. Fewer side effects occur with lower doses. Combining the trials, consensus treatment includes early aspirin dosing (325 mg initially, then 150 mg-325 mg daily) given to patients with ischemic stroke. Early aspirin should be avoided in those patients who qualify for and are receiving alteplase, heparin, or oral warfarin therapy.
There are other antiplatelet agents for long-term management of ischemic stroke. Whereas aspirin alone is used in the early management of acute ischemic stroke in those ineligible for thrombolytic therapy, many patients are transitioned to other antiplatelet strategies for secondary prevention long-term. The number needed to treat for aspirin to reduce one future stroke, myocardial infarction (MI), or vascular death when compared to placebo is quite high at 33. However, the combination of aspirin and dipyradimole does not prevent MI, vascular death, or the combined endpoint of either stroke or death.
Clopidogrel is more effective than aspirin in preventing a combined endpoint of ischemic stroke, MI, or vascular death, but it is not superior to aspirin in preventing recurrent stroke in TIA or stroke patients. The effects of clopidrogel are greater in patients with peripheral arterial disease, previous coronary artery bypass grafting, insulin-dependent diabetes, or recurrent vascular events.
There is a substantially high cost of treatment and long-term disability associated with stroke. Costs can vary from 3% to 5% of the annual healthcare budget. The newer antiplatelet agents are more expensive than aspirin, and overall cost-effectiveness is difficult to estimate. Yet, from an economic standpoint, the combination of aspirin and dipyradimole can be recommended as an alternative for secondary stroke prevention in patients without major comorbidities. In those patients with higher risk factors and/or comorbidities, clopidogrel might be more cost-effective than aspirin alone. Furthermore, in patients with aspirin intolerance, clopidogrel is a useful, but expensive, alternative.
Thrombolytic therapy. Restora-tion of blood flow with thrombolytic therapy is the most effective way of salvaging ischemic brain tissue that has not already infarcted. The window for use of the thrombolytic alteplase is narrow; studies suggest that its benefit diminishes with increasing time to treatment. Indeed, after 4.5 hours from the onset of symptoms, evidence suggests that the harm might outweigh the benefit, so the determination of who is eligible for its use has to be made quickly.
Guidelines published by the American Heart Association/American Stoke Association stroke council outline strict inclusion and exclusion criteria for the use of alteplase in the management of acute ischemic stroke.6 Obtaining informed consent and emergent neuroimaging are vital in preventing delays in alteplase administration.
Two major trials that illustrate the benefit of alteplase in the treatment of acute ischemic stroke are the NINDS trial and the ECASS 3 trial. NINDS showed that when intravenous alteplase was used within three hours of symptom onset, patients had improved functional outcome at three months.7 The ECASS 3 trial showed that intravenous alteplase has benefit when given up to 4.5 hours after symptom onset.8 Treatment with intravenous alteplase from three-4.5 hours in the ECASS 3 trial showed a modest improvement in patient outcomes at three months, with a number needed to treat of 14 for a favorable outcome.
A 2010 meta-analysis looked specifically at outcomes in stroke based on time to treat with alteplase using pooled data from the NINDS, ATLANTIS, ECASS (1, 2, and 3), and EPITHET trials.9 It showed that the number needed to treat for a favorable outcome at three months increased steadily when time to treatment was delayed. It also showed that the risk of death after alteplase administration increased significantly after 4.5 hours. Thus, after 4.5 hours, it suggests that harm might exceed the benefits of treatment.
Anticoagulant use in ischemic stroke. Clinical trials have not been effective in demonstrating the use of heparin and low-molecular-weight heparins (LMWHs). A 2008 systematic review of 24 trials (approximately 24,000 patients) demonstrated:
- Anticoagulant therapy did not reduce odds of death;
- Therapy was associated with nine fewer recurrent ischemic strokes per 1,000 patients, but also showed a similar increase in symptomatic intracranial hemorrhages; and
- Overall, researchers could not specify a particular anticoagulant mode or regimen that had an overall net patient benefit.
The use of heparin in atrial fibrillation and stroke has generated controversy in recent years. Review of the data, however, indicates that early treatment with heparin might cause more harm than benefit. A 2007 meta-analysis did not support the use of early anticoagulant therapy. Seven trials (4,200 patients) compared heparin or LMWH started within 48 hours to other treatments (aspirin, placebo). The study authors found:
- Nonsignificant reduction in recurrent ischemic stroke within seven to 14 days;
- Statistically significant increase in symptomatic intracranial hemorrhages; and
- Similar rates of death/disability at final follow-up of studies.
For those patients who continue to demonstrate neurological deterioration, heparin and LMWH use did not appear to improve outcomes. Therefore, based on a consensus of national guidelines, the use of full-dose anticoagulation with heparin or LMWH is not recommended.
The data suggest that in patients with stroke secondary to:
- Dissection of cervical or intracranial arteries;
- Intracardiac thrombus and valvular disease; and
- Mechanical heart valves, full-dose anticoagulation can be initiated. However, the benefit is unproven.
Back to the Case
Our patient with acute ischemic stroke with right-sided weakness on exam presented outside of the window within which alteplase could be administered safely. She was started on aspirin 325 mg daily. There was no indication for full anticoagulation with intravenous heparin or warfarin. Her weakness showed slight improvement on exam during the hospitalization. As an insulin-dependent diabetic, she was thought to be at high risk for recurrent stroke. As such, she was transitioned to a combination of aspirin and clopidogrel prior to her discharge to an acute inpatient rehabilitation hospital.
Bottom Line
Early aspirin therapy (within 48 hours) is recommended (initial dose 325 mg, then 150 mg-325 mg daily) for patients with ischemic stroke who are not candidates for alteplase, IV heparin, or oral anticoagulants.10 Aspirin is the only antiplatelet agent that has been shown to be effective for the early treatment of acute ischemic stroke. In patients without contraindications, aspirin, the combination of aspirin-dipyradimole, or clopidogrel is appropriate for secondary prevention.
The subset of patients at high risk of recurrent stroke should be transitioned to clopidogrel or aspirin/clopidogrel, unless otherwise contraindicated. TH
Dr. Chaturvedi is an instructor in the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and medical director of HM at Northwestern Lake Forest Hospital. Dr. Abraham is an instructor in the Division of Hospital Medicine at Northwestern University Feinberg School of Medicine.
References
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19,435 patients with acute ischemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569-1581.
- CAST: randomised placebo-controlled trial of early aspirin use in 20,000 patients with acute ischaemic stroke. CAST (Chinese Acute Stroke Trial) Collaborative Group. Lancet. 1997;349:1641-1649.
- Dengler R, Diener HC, Schwartz A, et al. Early treatment with aspirin plus extended-release dipyridamole for transient ischaemic attack or ischaemic stroke within 24 h of symptom onset (EARLY trial): a randomised, open-label, blinded-endpoint trial. Lancet Neurol. 2010;9:159-166.
- Kennedy J, Hill MD, Ryckborst KJ, et al. Fast assessment of stroke and transient ischaemic attack to prevent early recurrence (FASTER): a randomised controlled pilot trial. Lancet Neurol. 2007;6:961-969.
- Diener HC, Bogousslavsky J, Brass LM, et al. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:331-337.
- Adams HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655-1711.
- Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695-1703.
- Hacke W, Kaste M, Bluhmki E, et al. Thombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317-1329.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581-1587.
- Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:630S-669S.
Case
A 58-year-old woman with diabetes mellitus and hypertension presents with dysarthria and weakness on the right side of her body starting six hours prior to presentation. She is afebrile and has a blood pressure of 162/84 mmHg. Exam reveals the absence of a heart murmur and no lower-extremity swelling or calf tenderness. There is weakness of the right side of the body on exam with diminished proprioception. A noncontrast head CT shows no intracranial hemorrhage. She is admitted to the hospital with the diagnosis of acute ischemic stroke. What anticlotting or antiplatelet medications should she receive?
Overview
Stroke remains a significant cause of morbidity and mortality in the U.S. and around the world. The majority of strokes are ischemic in etiology. Although thrombolytic therapy is the most effective way to salvage ischemic brain tissue that has not yet infarcted, there is a narrow window for the use of thrombolytics in the treatment of acute ischemic stroke. As a result, many patients will not be eligible for thrombolysis. Outside of 4.5 hours from symptom onset, evidence suggests that the risk outweighs the benefit of using the thrombolytic alteplase. For patients ineligible for thrombolytic therapy, antiplatelet therapy remains the best choice for treatment.
Medications that prevent blood from coagulating or clotting are used to treat and prevent a recurring or second stroke. Typically, an antiplatelet agent (most often aspirin) is initiated within 48 hours of an ischemic stroke and continued in low doses as maintenance. Multiple studies suggest that antiplatelet therapy can reduce the risk for a second stroke by 25%. Specific anticlotting agents might be warranted in some patients with high-risk conditions for a stroke.
Review of Data
Early initiation of aspirin has shown benefit in the treatment of an acute ischemic stroke. Two major trials—the International Stroke Trial (IST) and the Chinese Acute Stroke Trial (CAST)— evaluated the role of aspirin (see Table 1, p. 15).1,2 The IST and CAST trials showed that roughly nine nonfatal strokes were avoided per every 1,000 early treatments. Taking the endpoint of death, as well as focal deficits, the two trials confirmed a rate of reduction of 13 per 1,000 patients.
Overall, the consensus was that initiating aspirin within 48 hours of a presumed ischemic cerebrovascular accident posed no major risk of hemorrhagic complication and improved the long-term outcomes.
Along with aspirin, other antiplatelet agents have been studied, most commonly dipyridamole and clopidrogel. The EARLY trial demonstrated no significant differences in the aspirin and dipyridamole groups at 90 days.3
Another large trial, which focused on clopidrogel and aspirin, looked at aspirin plus clopidrogel or aspirin alone. The FASTER trial enrolled mostly patients with mild cerebrovasular accidents (CVA) or transient ischemic attacks (TIA), and there was no difference in outcome measures between the groups.4 However, the MATCH trial found that aspirin and clopidrogel did not provide improved stroke preventions versus clopidogrel alone but had a larger risk of hemorrhagic/bleeding complications.5
Aspirin dosage is somewhat controversial. Fewer side effects occur with lower doses. Combining the trials, consensus treatment includes early aspirin dosing (325 mg initially, then 150 mg-325 mg daily) given to patients with ischemic stroke. Early aspirin should be avoided in those patients who qualify for and are receiving alteplase, heparin, or oral warfarin therapy.
There are other antiplatelet agents for long-term management of ischemic stroke. Whereas aspirin alone is used in the early management of acute ischemic stroke in those ineligible for thrombolytic therapy, many patients are transitioned to other antiplatelet strategies for secondary prevention long-term. The number needed to treat for aspirin to reduce one future stroke, myocardial infarction (MI), or vascular death when compared to placebo is quite high at 33. However, the combination of aspirin and dipyradimole does not prevent MI, vascular death, or the combined endpoint of either stroke or death.
Clopidogrel is more effective than aspirin in preventing a combined endpoint of ischemic stroke, MI, or vascular death, but it is not superior to aspirin in preventing recurrent stroke in TIA or stroke patients. The effects of clopidrogel are greater in patients with peripheral arterial disease, previous coronary artery bypass grafting, insulin-dependent diabetes, or recurrent vascular events.
There is a substantially high cost of treatment and long-term disability associated with stroke. Costs can vary from 3% to 5% of the annual healthcare budget. The newer antiplatelet agents are more expensive than aspirin, and overall cost-effectiveness is difficult to estimate. Yet, from an economic standpoint, the combination of aspirin and dipyradimole can be recommended as an alternative for secondary stroke prevention in patients without major comorbidities. In those patients with higher risk factors and/or comorbidities, clopidogrel might be more cost-effective than aspirin alone. Furthermore, in patients with aspirin intolerance, clopidogrel is a useful, but expensive, alternative.
Thrombolytic therapy. Restora-tion of blood flow with thrombolytic therapy is the most effective way of salvaging ischemic brain tissue that has not already infarcted. The window for use of the thrombolytic alteplase is narrow; studies suggest that its benefit diminishes with increasing time to treatment. Indeed, after 4.5 hours from the onset of symptoms, evidence suggests that the harm might outweigh the benefit, so the determination of who is eligible for its use has to be made quickly.
Guidelines published by the American Heart Association/American Stoke Association stroke council outline strict inclusion and exclusion criteria for the use of alteplase in the management of acute ischemic stroke.6 Obtaining informed consent and emergent neuroimaging are vital in preventing delays in alteplase administration.
Two major trials that illustrate the benefit of alteplase in the treatment of acute ischemic stroke are the NINDS trial and the ECASS 3 trial. NINDS showed that when intravenous alteplase was used within three hours of symptom onset, patients had improved functional outcome at three months.7 The ECASS 3 trial showed that intravenous alteplase has benefit when given up to 4.5 hours after symptom onset.8 Treatment with intravenous alteplase from three-4.5 hours in the ECASS 3 trial showed a modest improvement in patient outcomes at three months, with a number needed to treat of 14 for a favorable outcome.
A 2010 meta-analysis looked specifically at outcomes in stroke based on time to treat with alteplase using pooled data from the NINDS, ATLANTIS, ECASS (1, 2, and 3), and EPITHET trials.9 It showed that the number needed to treat for a favorable outcome at three months increased steadily when time to treatment was delayed. It also showed that the risk of death after alteplase administration increased significantly after 4.5 hours. Thus, after 4.5 hours, it suggests that harm might exceed the benefits of treatment.
Anticoagulant use in ischemic stroke. Clinical trials have not been effective in demonstrating the use of heparin and low-molecular-weight heparins (LMWHs). A 2008 systematic review of 24 trials (approximately 24,000 patients) demonstrated:
- Anticoagulant therapy did not reduce odds of death;
- Therapy was associated with nine fewer recurrent ischemic strokes per 1,000 patients, but also showed a similar increase in symptomatic intracranial hemorrhages; and
- Overall, researchers could not specify a particular anticoagulant mode or regimen that had an overall net patient benefit.
The use of heparin in atrial fibrillation and stroke has generated controversy in recent years. Review of the data, however, indicates that early treatment with heparin might cause more harm than benefit. A 2007 meta-analysis did not support the use of early anticoagulant therapy. Seven trials (4,200 patients) compared heparin or LMWH started within 48 hours to other treatments (aspirin, placebo). The study authors found:
- Nonsignificant reduction in recurrent ischemic stroke within seven to 14 days;
- Statistically significant increase in symptomatic intracranial hemorrhages; and
- Similar rates of death/disability at final follow-up of studies.
For those patients who continue to demonstrate neurological deterioration, heparin and LMWH use did not appear to improve outcomes. Therefore, based on a consensus of national guidelines, the use of full-dose anticoagulation with heparin or LMWH is not recommended.
The data suggest that in patients with stroke secondary to:
- Dissection of cervical or intracranial arteries;
- Intracardiac thrombus and valvular disease; and
- Mechanical heart valves, full-dose anticoagulation can be initiated. However, the benefit is unproven.
Back to the Case
Our patient with acute ischemic stroke with right-sided weakness on exam presented outside of the window within which alteplase could be administered safely. She was started on aspirin 325 mg daily. There was no indication for full anticoagulation with intravenous heparin or warfarin. Her weakness showed slight improvement on exam during the hospitalization. As an insulin-dependent diabetic, she was thought to be at high risk for recurrent stroke. As such, she was transitioned to a combination of aspirin and clopidogrel prior to her discharge to an acute inpatient rehabilitation hospital.
Bottom Line
Early aspirin therapy (within 48 hours) is recommended (initial dose 325 mg, then 150 mg-325 mg daily) for patients with ischemic stroke who are not candidates for alteplase, IV heparin, or oral anticoagulants.10 Aspirin is the only antiplatelet agent that has been shown to be effective for the early treatment of acute ischemic stroke. In patients without contraindications, aspirin, the combination of aspirin-dipyradimole, or clopidogrel is appropriate for secondary prevention.
The subset of patients at high risk of recurrent stroke should be transitioned to clopidogrel or aspirin/clopidogrel, unless otherwise contraindicated. TH
Dr. Chaturvedi is an instructor in the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and medical director of HM at Northwestern Lake Forest Hospital. Dr. Abraham is an instructor in the Division of Hospital Medicine at Northwestern University Feinberg School of Medicine.
References
- The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19,435 patients with acute ischemic stroke. International Stroke Trial Collaborative Group. Lancet. 1997;349:1569-1581.
- CAST: randomised placebo-controlled trial of early aspirin use in 20,000 patients with acute ischaemic stroke. CAST (Chinese Acute Stroke Trial) Collaborative Group. Lancet. 1997;349:1641-1649.
- Dengler R, Diener HC, Schwartz A, et al. Early treatment with aspirin plus extended-release dipyridamole for transient ischaemic attack or ischaemic stroke within 24 h of symptom onset (EARLY trial): a randomised, open-label, blinded-endpoint trial. Lancet Neurol. 2010;9:159-166.
- Kennedy J, Hill MD, Ryckborst KJ, et al. Fast assessment of stroke and transient ischaemic attack to prevent early recurrence (FASTER): a randomised controlled pilot trial. Lancet Neurol. 2007;6:961-969.
- Diener HC, Bogousslavsky J, Brass LM, et al. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:331-337.
- Adams HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655-1711.
- Lees KR, Bluhmki E, von Kummer R, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695-1703.
- Hacke W, Kaste M, Bluhmki E, et al. Thombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317-1329.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581-1587.
- Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:630S-669S.
Making a List, Check It Twice
In October, a 36‐year‐old woman with no significant past medical history presented to the Emergency Department (ED) with a 3‐day history of headache and fever. The headache was severe, throbbing, and frontal in location. She also complained of daily fevers measured up to 103F, generalized malaise, and fatigue. She did not report neck stiffness or photophobia. She felt better after receiving intravenous fluids and was discharged home with a diagnosis of a nonspecific viral illness. Two days later, she returned to the ED with worsening headache, fever, mild photophobia, and poor oral intake. She also complained of a dry cough that made her headache worse, as did bending over. She did not report confusion, neck stiffness, shortness of breath, sore throat, runny nose, abdominal symptoms, or rash.
This patient presents a second time to the ED with worsening headache and fever raising concerns about meningitis. At the time of her first ED visit, it can be assumed that she had a nontoxic appearance because she was discharged shortly thereafter. Thus, acute bacterial meningitis seems less likely, but occasionally patients with meningococcal meningitis may not appear significantly ill until later in the process. Nonetheless, acute meningitis, possibly viral, is the initial concern. The time of the year is an important variable because many viral infections are seasonal. Enteroviruses are the most common cause of viral meningitis in the United States, particularly in the summer and fall. In contrast, mumps, measles, and varicella zoster viruses occur more commonly in winter and spring. Herpetic meningoencephalitis is a life‐threatening condition with a guarded prognosis. Therefore, early recognition and treatment is necessary to decrease morbidity and mortality. Drugs such as nonsteroidal anti‐inflammatory agents, trimethoprim‐sulfamethoxazole, amoxicillin, and rarely vaccines can also cause aseptic meningitis. Infections from fungi, spirochetes, mycobacteria, and rarely parasites also cause meningitis, but would be of greater concern in a patient with risk factors such as recent travel or an immunocompromised state.
Increased headache with bending and cough might indicate elevated intracranial pressure. However, this is a nonspecific complaint, and headache is often worse with the Valsalva maneuver. Because she reports a cough, a chest x‐ray would be useful. In addition to routine initial tests, cerebrospinal fluid (CSF) analysis and human immunodeficiency virus (HIV) testing is recommended.
Her past medical history was notable for depression. Her medications included bupropion, multivitamins, and fish oil. She was also taking milk thistle pills daily to protect her liver because she had been drinking alcohol heavily for the past 2 weeks since her husband left her. She smoked 1 pack of cigarettes daily. She had not traveled recently. She reported no recent animal or wildlife exposure but did recall falling into a midwestern river while canoeing 2 weeks prior to presentation. She worked as a hairstylist and described no sick contacts or risk factors for HIV disease.
An important new historical element is that the patient fell into a river. If she swallowed a significant amount of water during her fall overboard, meningitis from waterborne infections such as Aeromonas, Acanthamoeba, and Naegleria need to be considered. Fortunately, these are rare in the Midwest. Her canoeing history may suggest exposure to wooded areas. Certainly, tickborne infections such as ehrlichiosis, babesiosis, Lyme disease, and Rocky Mountain spotted fever can also cause meningitis. Histoplasmosis and blastomycosis are also endemic to the midwestern United States and can disseminate and cause central nervous system disease.
At this time, viral and bacterial infections are highest on the differential diagnosis. However, the microbiology laboratory needs to be alerted to the possibility of fungal or parasitic organisms depending on the initial CSF analysis results.
The patient was a Caucasian woman who appeared comfortable. Her blood pressure was 130/62 mm Hg, heart rate was 83 beats per minute, respiratory rate was 18 per minute, temperature was 100.8F, and oxygen saturation was 98% on room air. She was fully alert and oriented. Her pupils were bilaterally equal, reactive to light and accommodation with intact extraocular movement and no nystagmus. There was conjunctival injection bilaterally without noticeable pallor or icterus. Fundoscopic examination, which the patient tolerated without difficulty, was normal. Inspection of the oral cavity showed mild tonsillar enlargement. The neck was supple with no stiffness. No cervical, axillary, or inguinal lymph nodes were palpable. Faint bilateral basilar crackles were audible over the posterior chest. There was very mild right upper quadrant abdominal tenderness without guarding. The liver and spleen were normal size and bowel sounds were present. No rash, peripheral edema, or spinal tenderness was noted. A complete neurological examination was normal.
Her general appearance and vital signs seem reassuring. Conjunctival injection and mild tonsillar enlargement are nonspecific findings and may occur in systemic inflammatory states especially viral infections. Atelectasis may account for faint bilateral basilar crackles especially if associated with post‐tussive change. Her alcohol use puts her at risk of aspiration. A right lower lobe process (pneumonia) can sometimes present with right upper quadrant tenderness. However, this tenderness may also represent muscle soreness from repeated coughing, liver, or gallbladder disease. The same infectious process affecting the central nervous system and possibly her lungs, may also be affecting the liver.
A complete blood count revealed a white blood cell count of 3000/mm3 (79% neutrophils, 15% lymphocytes, 5% monocytes), hemoglobin of 11.7 g/dL, and platelets of 110,000/mm3. The serum sodium was 133 mmol/L, potassium was 3.7 mmol/L, bicarbonate was 22 mmol/L, and blood urea nitrogen was 20 mg/dL. The serum creatinine was 1.5 compared to 1.0 mg/dL on testing 2 days prior. A liver function panel showed protein of 5.1 g/dL, albumin of 3 g/dL, aspartate aminotransferase (AST) of 576 IU/L, alanine aminotransferase (ALT) of 584 IU/L, alkaline phosphatase of 282 IU/L, and total bilirubin of 1 mg/dL. The coagulation profile, creatinine phosphokinase, acetaminophen level, urine pregnancy test, urine drug screen, and urinalysis (including urine microscopy) were normal.
The CSF opening pressure was 13 cm H2O. CSF analysis showed 4 mononuclear leukocytes per high‐power field, CSF protein was 27 mg/dL, and glucose was 76 mg/dL. No organisms were noted on gram stain. A chest x‐ray showed focal airspace opacity in the left lower lobe (Figure 1) and the patient was hospitalized for further management.
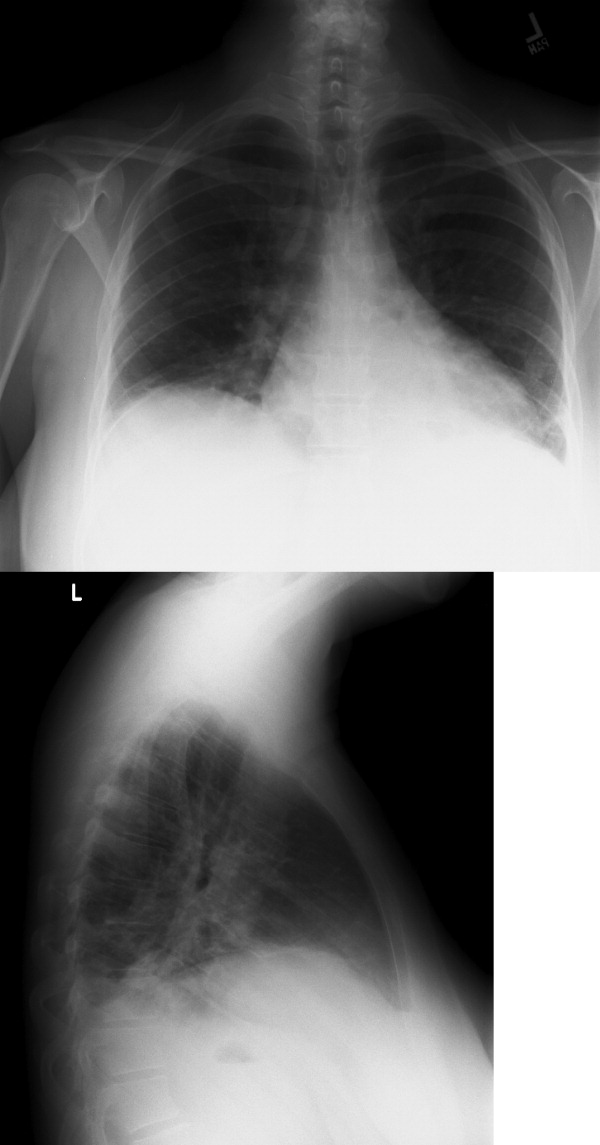
The normal CSF analysis makes acute meningitis much less likely. It is interesting to note that the aminotransferase levels are nearly equal. Usually, in viral and many other causes of hepatitis, the ALT is higher than the AST, whereas the contrary is true in alcoholic hepatitis. Because the patient has been consuming significant amounts of alcohol recently, these levels may become equal in the setting of another primary liver process. The elevation in liver enzymes also raises the possibility of autoimmune hepatitis secondary to a systemic vasculitis such as systemic lupus erythematosus. Nonetheless, the focus should be on infectious causes of hepatitis such as hepatitis C, adenovirus, parvovirus, Epstein‐Barr virus (EBV), cytomegalovirus, and herpes simplex virus that can cause pneumonia either as a primary or secondary infection. Acute HIV infection can also present in this fashion, and anti‐HIV antibody testing may be negative early in the disease. In the setting of a normal urinalysis and bland urine sediment, prerenal azotemia is the most likely cause of her acute renal injury and can be confirmed by testing the urinary sodium and creatinine. A peripheral smear should be reviewed to evaluate the pancytopenia.
Severe headache, fever, conjunctival injection, pancytopenia, acute kidney injury, hepatitis, and pneumonia may occur in leptospirosis, particularly in a patient with recent freshwater exposure. Alternatively, ehrlichiosis can also account for fever, headache, pancytopenia, renal failure, hepatitis, and pneumonia, but conjunctival suffusion is not often present. At this time, treatment for community‐acquired pneumonia that includes coverage for leptospirosis should be started.
The patient was hydrated with intravenous fluids and treated with intravenous ceftriaxone and azithromycin for community‐acquired pneumonia. An abdominal ultrasound was normal. The serologic assays for acute hepatitis A, B, and C infection were negative. The following morning, she reported worsening headache, increased cough now productive of whitish‐yellow sputum, and diffuse body aches. She appeared more lethargic and toxic. Her blood pressure was 100/83 mm Hg, heart rate was 84 beats per minute, respiratory rate was 24 per minute, and temperature was 101.3F. She had increased crackles on chest auscultation bilaterally and required supplemental oxygen at 4 L/minute by nasal cannula. Examination of both legs now revealed multiple scattered, faintly erythematous, 2‐cm‐sized patches overlying tender subtle subcutaneous nodules. Additionally, a mildly pruritic, V‐shaped area of blanchable erythema was also seen on her chest. The white blood cell count was 2500/mm3 (77% neutrophils, 15% lymphocytes), serum creatinine was 1.8 mg/dL, AST was 351 IU/L, and ALT was 485 IU/L. Blood cultures showed no growth and a peripheral smear examination was unrevealing. A noncontrast chest computed tomographic scan showed findings consistent with multifocal pneumonia (Figure 2).
It would be prudent at this time to expand her antimicrobial coverage (such as with vancomycin and piperacillin‐tazobactam) for activity against methicillin‐resistant Staphylococcus aureus and Pseudomonas because of her clinical worsening. Although ceftriaxone or piperacillin would cover leptospirosis, given the possibility of ehrlichiosis, the addition of doxycycline should be strongly considered.
The description of the rash on her legs seems consistent with erythema nodosum, which is associated with a number of infections (streptococcal, fungal, syphilis, EBV, cat‐scratch disease, tuberculosis), inflammatory conditions (inflammatory bowel disease, autoimmune disease, malignancy), and pregnancy. The blanchable rash on the chest is also a cause of concern for a possible drug reaction (ceftriaxone). A Jarisch‐Herxheimer reaction is possible given her acute worsening of symptoms with initiation of antibiotic therapy.
An antineutrophil cytoplasmic antibodyassociated vasculitis or another autoimmune condition such as systemic lupus erythematosus can account for erythema nodosum, rash, pancytopenia, and hepatitis. This diagnosis might also fit if she had a vasculitic pulmonary hemorrhage that caused her lung infiltrates and worsening hypoxia. A complete antinuclear antibody panel, antineutrophil cytoplasmic antibody, and antismooth muscle antibody testing is recommended. A skin and bronchoscopic biopsy should be considered.
Her dose of ceftriaxone was increased for possible severe pneumococcal pneumonia. The dermatology consultant felt that her leg lesions were consistent with erythema nodosum and the chest rash consistent with cutaneous photodamage. Bronchoscopic examination was normal and a bronchoalveolar lavage sample showed 2905 red blood cells/mm3 and 605 white blood cells/mm3 (70% neutrophils, 7% lymphocytes, 16% histiocytes), normal cytology, and negative cultures. There was no significant clinical improvement by the fourth hospital day and oral doxycycline was started. The next day, her skin lesions had resolved and she felt better. The serologic tests for Legionella, Mycoplasma, cytomegalovirus, EBV, Toxoplasma, Chlamydophila, Ehrlichia, Leptospira, Q‐fever, parvovirus, and adenovirus were negative. A fungal serology panel, HIV polymerase chain reaction, cryoglobulin level, and several rheumatologic tests (antinuclear antibody, extractable nuclear antigen panel, rheumatoid factor, antineutrophil cytoplasmic antibody, antiproteinase 3, and antiglomerular basement membrane antibodies) were normal. Blood cultures continued to show no growth.
The apparent response to doxycycline suggests she might have ehrlichiosis. A buffy coat review for morulae should be done. It is also possible that she may have improved on her initial therapy alone before starting doxycycline and her clinical worsening (including the chest rash) was due to a Jarisch‐Herxheimer reaction. Serologic tests for leptospirosis and ehrlichiosis should be repeated in 12 weeks because such infections may not cause detectable antibody levels early in the illness.
Ceftriaxone and doxycycline were continued and she showed rapid and significant clinical improvement. She was discharged 4 days later with instructions to complete a 10‐day course of antibiotics. At her 3‐month follow‐up, she was doing well and a repeat Leptospira antibody test by the Indirect Hemagglutination Assay (MRL Diagnostics, Cypress, California; normal titer 1:50) was positive at a titer of 1:100, which is highly suggestive of leptospirosis.
Commentary
Leptospirosis is a zoonotic infection caused by spirochetes of the genus Leptospira. The infection is usually transmitted indirectly to humans through contact with water, food, or soil contaminated with the urine of infected mammals.1 Risk factors for infection include participation in recreational activities (such as freshwater swimming, canoeing, and camping), occupational exposure, and exposure to infected pets or domesticated livestock. Approximately 100200 cases are identified annually in the United States, and approximately half occur in the state of Hawaii.2 Outbreaks of leptospirosis have been reported previously in the Midwest.3 These organisms inoculate humans through contact with mucous membranes or broken skin, or enter by swallowing infected food or water. A large number of these infections remain subclinical or result in a very mild illness with spontaneous clearance by the host's immune mechanism. Following an incubation period of 230 days, infected individuals may develop clinically significant disease (Table 1). Clinical presentations may overlap as the disease progresses. Although much remains to be learned about the exact pathogenic mechanism, disruption of the cell membranes of small vessel endothelia (a toxin‐like effect), and cytokine‐mediated tissue injury are believed to cause organ hemorrhage and ischemia.4
|
| 1. Mild influenza‐like self‐remitting disease (90% of cases) |
| Undifferentiated fever (usually 100F105F), severe headache, and myalgia (especially lower limbs). |
| 2. Moderately severe disease usually requiring hospitalization (5%9% of cases) |
| Marked prostration, anorexia, nausea, and vomiting, conjunctival suffusion, transient rash, frequently abdominal pain, constipation or diarrhea, and occasionally epistaxis. |
| 3. Severe disease involving multiple organ systems (1%5% of cases) |
| Hepatorenal Syndrome (Weil's syndrome) |
| Constellation of jaundice, hemorrhagic diathesis, and acute renal failure. Hepatic failure is rarely fatal. Renal involvement is usually more severe and the common cause of death. Cardiac (myocarditis with arrhythmias) and pulmonary complications are frequent. Confusion and restlessness may occur. |
| Hemorrhagic pneumonitis |
| Usually presents as a dry cough initially but becomes blood‐streaked after 23 days. Often characterized by a rapid progression to involve extensive areas of lungs, massive intra‐alveolar hemorrhage, acute respiratory failure, and death. |
| Central nervous system involvement |
| Meningismus, meningitis, or meningoencephalitis. |
The clinical diagnosis of leptospirosis is difficult because of its protean manifestations. Although nonspecific, 2 clinical features may provide a clue to the clinical diagnosis. First, the presence of conjunctival suffusion occurs in the early stage of the disease and is often associated with subconjunctival hemorrhage. Second, severe myalgia, commonly involving the lower limbs, is also characteristically present.1, 5 In 1 series of 58 patients with acute leptospirosis, conjunctival suffusion was observed in 50% of cases, and subconjunctival hemorrhage in 29%. Body ache and muscle tenderness was described in almost all cases.6
As seen in this case, the presence of a rash may pose a clinical challenge. A transient macular, maculopapular, purpuric, or urticarial rash may be seen in acute leptospirosis, but rashes may also be representative of a complication of treatment.1 First described in 1895 in patients with syphilis treated with mercury, the Jarisch‐Herxheimer reaction typically occurs within a few hours of antimicrobial treatment of spirochete infections and often presents with a rash, headache, fever, rigors, hypotension, sweating, and worsening symptoms of the underlying illness.7 Other skin findings such as the occurrence of erythema nodosum have been previously reported in cases of leptospirosis.8
Human ehrlichiosis (HE) is caused by tickborne, obligatory intracellular bacteria that infect leukocytes. There are 3 distinct clinical conditions: human monocytic ehrlichiosis (HME, caused by Ehrlichia chaffeensis), human granulocytic anaplasmosis (HGA, caused by Anaplasma phagocytophilum), and human ewingii ehrlichiosis (HEE, caused by E. ewingii). Although most cases of HME and HEE are seen in the southeastern and south‐central United States and California, the highest incidence of HGA is reported in the northeastern and upper Midwest regions.9 As with leptospirosis, the clinical range of HE spans from asymptomatic infection to life‐threatening illness. Following an incubation period of 12 weeks, symptomatic cases usually present with nonspecific complaints such as high fevers, chills, headache, nausea, arthralgia, myalgia, and malaise.10 The majority of cases will report a tick bite or an exposure to ticks. Laboratory tests often reveal leukopenia (white blood cell count 4000/mm3), thrombocytopenia, hyponatremia, and elevated AST and ALT. Patients with severe disease may develop renal, respiratory, and hepatic failure. Thus, differentiating ehrlichiosis from leptospirosis is often challenging for the clinician.
However, there are a few clinical clues that help distinguish between these illnesses in this case. HGA as a cause of HE would be more likely in the Midwest. Although a rash is present in one‐third of patients with HME, it is seldom present in HGA unless coinfected with Borrelia burgdorferi, the causative agent for Lyme disease. Additionally, her history of freshwater exposure and the absence of a history of a tick bite also favor leptospirosis. As noted previously, conjunctival suffusion, a characteristic clinical feature of leptospirosis, has only been described in case reports of HE.11, 12
Serologic tests are often used to establish the diagnosis of leptospirosis and ehrlichiosis. Leptospires are fastidious organisms that are difficult to isolate on inoculated growth media. The microscopic agglutination test for leptospirosis is considered the diagnostic gold standard due to its high specificity, but its use is limited by its technical complexity, lack of availability (other than in reference laboratories), and low sensitivity early in the disease (antibody levels detected by this method usually do not appear until 7 days after symptom onset).13 A variety of rapid serologic assays are also available. Although these tests have good overall sensitivity (ranging between 79% and 93%), they perform relatively poorly for acute‐phase sera (sensitivity of 38.5%52.7%).13 The high early false negative rate is believed to be a result of inadequate Leptospira antibody titers in the acute phase of the illness. Seroconversion or a 4‐fold rise between acute and convalescent‐phase antibody titers is the most definitive criterion for the diagnosis of leptospirosis. However, without paired sera samples, a single high microscopic agglutination test titer can be taken as diagnostic for leptospirosis depending on the degree of regional endemicity.14
Similarly, currently available serologic assays for ehrlichiosis produce negative results in most patients in the first week of illness, and it is important to obtain a convalescent phase serum specimen for confirmatory diagnosis of HME and HGA. Seroconversion or a 4‐fold increase in titer between acute and convalescent phase sera is considered diagnostic. The sensitivity of finding morulae (intracytoplasmic vacuolar microcolonies of Ehrlichia) on a peripheral smear is unknown, and data suggest that this finding is more common in cases of HGA compared to HME.15
Although doxycycline is the drug of choice for the treatment of ehrlichiosis, Leptospira is susceptible to a wide variety of antibiotics because it exhibits a double membrane surface architecture with components common to both gram‐negative and gram‐positive bacteria.1 Recommended treatment regimens for severe leptospirosis include the use of high‐dose intravenous penicillin or a third‐generation cephalosporin. Less severe cases can be treated with oral amoxicillin or doxycycline.16 The fact that this patient's clinical improvement appeared to lag after initiation of ceftriaxone does not necessarily indicate a lack of efficacy but perhaps a Jarisch‐Herxheimer reaction in response to appropriate antibiotic therapy.
Teaching Points
-
Establishing a diagnosis of leptospirosis is challenging and requires a high index of suspicion. Clinicians should be aware of the limitations of the diagnostic accuracy of the serologic assays for leptospirosis because they are frequently negative in the first week after symptom onset.
-
The classic finding of conjunctival suffusion is helpful in differentiating leptospirosis from human ehrlichiosis.
-
This case also highlights the importance of the clinical practice of making a list of suspected diagnoses, remaining open to these possibilities, and checking serologic tests again in convalescence to confirm the diagnosis.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Acknowledgements
The authors thank Dr. Brian Harte for his valuable guidance in the preparation of this manuscript.
- ,,.Leptospirosis: an emerging global public health problem.J Biosci.2008;33:557–569.
- Centers for Disease Control and Prevention. Leptospirosis.2005. http://www.cdc.gov/ncidod/dbmd/diseaseinfo/leptospirosis_t.htm. Accessed November 15,year="2010"2010.
- Morbidity and Mortality Weekly Report.From the Centers for Disease Control and Prevention. Update: leptospirosis and unexplained acute febrile illness among athletes participating in triathlons—Illinois and Wisconsin, 1998.JAMA.1998;280:1474–1475.
- ,.Optimal treatment of leptospirosis: queries and projections.Int J Antimicrob Agents.2006;28:491–496.
- ,.Leptospirosis in the tropics and in travelers.Curr Infect Dis Rep.2006;8:51–58.
- ,,,,,.Clinico‐epidemiological study of hospitalized cases of severe leptospirosis.Indian J Med Res.1999;109:94–99.
- ,.Proposed mechanisms and preventative options of Jarisch‐Herxheimer reactions.J Clin Pharm Ther.2005;30:291–295.
- .Leptospirosis presenting with erythema nodosum.Arch Dis Child.1977;52:418–419.
- ,,.Emerging and re‐emerging tick‐transmitted rickettsial and ehrlichial infections.Med Clin N Am.2008;92:1345–1361.
- ,.Tick‐borne ehrlichiosis infection in human beings.J Vector Borne Dis.2008;45:273–280.
- ,.Ehrlichia in Tennessee.South Med J.1989;82:669.
- ,,,,,.Ehrlichial meningitis with cerebrospinal fluid morulae.Pediatr Infect Dis J.1999;18:552–555.
- ,,, et al.Evaluation of four commercially available rapid serologic tests for diagnosis of leptospirosis.J Clin Microbiol.2003;41:803–809.
- ,.Diagnosis of leptospirosis utilizing modified Faine's criteria.J Assoc Physicians India.2004;52:678–679.
- ,.Ehrlichiosis in children.J Pediatr.1997;131:184–192.
- ,World Health Organization, International Leptospirosis Society. Human leptospirosis: guidance for diagnosis, surveillance and control.Geneva, Switzerland:World Health Organization;2003.
In October, a 36‐year‐old woman with no significant past medical history presented to the Emergency Department (ED) with a 3‐day history of headache and fever. The headache was severe, throbbing, and frontal in location. She also complained of daily fevers measured up to 103F, generalized malaise, and fatigue. She did not report neck stiffness or photophobia. She felt better after receiving intravenous fluids and was discharged home with a diagnosis of a nonspecific viral illness. Two days later, she returned to the ED with worsening headache, fever, mild photophobia, and poor oral intake. She also complained of a dry cough that made her headache worse, as did bending over. She did not report confusion, neck stiffness, shortness of breath, sore throat, runny nose, abdominal symptoms, or rash.
This patient presents a second time to the ED with worsening headache and fever raising concerns about meningitis. At the time of her first ED visit, it can be assumed that she had a nontoxic appearance because she was discharged shortly thereafter. Thus, acute bacterial meningitis seems less likely, but occasionally patients with meningococcal meningitis may not appear significantly ill until later in the process. Nonetheless, acute meningitis, possibly viral, is the initial concern. The time of the year is an important variable because many viral infections are seasonal. Enteroviruses are the most common cause of viral meningitis in the United States, particularly in the summer and fall. In contrast, mumps, measles, and varicella zoster viruses occur more commonly in winter and spring. Herpetic meningoencephalitis is a life‐threatening condition with a guarded prognosis. Therefore, early recognition and treatment is necessary to decrease morbidity and mortality. Drugs such as nonsteroidal anti‐inflammatory agents, trimethoprim‐sulfamethoxazole, amoxicillin, and rarely vaccines can also cause aseptic meningitis. Infections from fungi, spirochetes, mycobacteria, and rarely parasites also cause meningitis, but would be of greater concern in a patient with risk factors such as recent travel or an immunocompromised state.
Increased headache with bending and cough might indicate elevated intracranial pressure. However, this is a nonspecific complaint, and headache is often worse with the Valsalva maneuver. Because she reports a cough, a chest x‐ray would be useful. In addition to routine initial tests, cerebrospinal fluid (CSF) analysis and human immunodeficiency virus (HIV) testing is recommended.
Her past medical history was notable for depression. Her medications included bupropion, multivitamins, and fish oil. She was also taking milk thistle pills daily to protect her liver because she had been drinking alcohol heavily for the past 2 weeks since her husband left her. She smoked 1 pack of cigarettes daily. She had not traveled recently. She reported no recent animal or wildlife exposure but did recall falling into a midwestern river while canoeing 2 weeks prior to presentation. She worked as a hairstylist and described no sick contacts or risk factors for HIV disease.
An important new historical element is that the patient fell into a river. If she swallowed a significant amount of water during her fall overboard, meningitis from waterborne infections such as Aeromonas, Acanthamoeba, and Naegleria need to be considered. Fortunately, these are rare in the Midwest. Her canoeing history may suggest exposure to wooded areas. Certainly, tickborne infections such as ehrlichiosis, babesiosis, Lyme disease, and Rocky Mountain spotted fever can also cause meningitis. Histoplasmosis and blastomycosis are also endemic to the midwestern United States and can disseminate and cause central nervous system disease.
At this time, viral and bacterial infections are highest on the differential diagnosis. However, the microbiology laboratory needs to be alerted to the possibility of fungal or parasitic organisms depending on the initial CSF analysis results.
The patient was a Caucasian woman who appeared comfortable. Her blood pressure was 130/62 mm Hg, heart rate was 83 beats per minute, respiratory rate was 18 per minute, temperature was 100.8F, and oxygen saturation was 98% on room air. She was fully alert and oriented. Her pupils were bilaterally equal, reactive to light and accommodation with intact extraocular movement and no nystagmus. There was conjunctival injection bilaterally without noticeable pallor or icterus. Fundoscopic examination, which the patient tolerated without difficulty, was normal. Inspection of the oral cavity showed mild tonsillar enlargement. The neck was supple with no stiffness. No cervical, axillary, or inguinal lymph nodes were palpable. Faint bilateral basilar crackles were audible over the posterior chest. There was very mild right upper quadrant abdominal tenderness without guarding. The liver and spleen were normal size and bowel sounds were present. No rash, peripheral edema, or spinal tenderness was noted. A complete neurological examination was normal.
Her general appearance and vital signs seem reassuring. Conjunctival injection and mild tonsillar enlargement are nonspecific findings and may occur in systemic inflammatory states especially viral infections. Atelectasis may account for faint bilateral basilar crackles especially if associated with post‐tussive change. Her alcohol use puts her at risk of aspiration. A right lower lobe process (pneumonia) can sometimes present with right upper quadrant tenderness. However, this tenderness may also represent muscle soreness from repeated coughing, liver, or gallbladder disease. The same infectious process affecting the central nervous system and possibly her lungs, may also be affecting the liver.
A complete blood count revealed a white blood cell count of 3000/mm3 (79% neutrophils, 15% lymphocytes, 5% monocytes), hemoglobin of 11.7 g/dL, and platelets of 110,000/mm3. The serum sodium was 133 mmol/L, potassium was 3.7 mmol/L, bicarbonate was 22 mmol/L, and blood urea nitrogen was 20 mg/dL. The serum creatinine was 1.5 compared to 1.0 mg/dL on testing 2 days prior. A liver function panel showed protein of 5.1 g/dL, albumin of 3 g/dL, aspartate aminotransferase (AST) of 576 IU/L, alanine aminotransferase (ALT) of 584 IU/L, alkaline phosphatase of 282 IU/L, and total bilirubin of 1 mg/dL. The coagulation profile, creatinine phosphokinase, acetaminophen level, urine pregnancy test, urine drug screen, and urinalysis (including urine microscopy) were normal.
The CSF opening pressure was 13 cm H2O. CSF analysis showed 4 mononuclear leukocytes per high‐power field, CSF protein was 27 mg/dL, and glucose was 76 mg/dL. No organisms were noted on gram stain. A chest x‐ray showed focal airspace opacity in the left lower lobe (Figure 1) and the patient was hospitalized for further management.
The normal CSF analysis makes acute meningitis much less likely. It is interesting to note that the aminotransferase levels are nearly equal. Usually, in viral and many other causes of hepatitis, the ALT is higher than the AST, whereas the contrary is true in alcoholic hepatitis. Because the patient has been consuming significant amounts of alcohol recently, these levels may become equal in the setting of another primary liver process. The elevation in liver enzymes also raises the possibility of autoimmune hepatitis secondary to a systemic vasculitis such as systemic lupus erythematosus. Nonetheless, the focus should be on infectious causes of hepatitis such as hepatitis C, adenovirus, parvovirus, Epstein‐Barr virus (EBV), cytomegalovirus, and herpes simplex virus that can cause pneumonia either as a primary or secondary infection. Acute HIV infection can also present in this fashion, and anti‐HIV antibody testing may be negative early in the disease. In the setting of a normal urinalysis and bland urine sediment, prerenal azotemia is the most likely cause of her acute renal injury and can be confirmed by testing the urinary sodium and creatinine. A peripheral smear should be reviewed to evaluate the pancytopenia.
Severe headache, fever, conjunctival injection, pancytopenia, acute kidney injury, hepatitis, and pneumonia may occur in leptospirosis, particularly in a patient with recent freshwater exposure. Alternatively, ehrlichiosis can also account for fever, headache, pancytopenia, renal failure, hepatitis, and pneumonia, but conjunctival suffusion is not often present. At this time, treatment for community‐acquired pneumonia that includes coverage for leptospirosis should be started.
The patient was hydrated with intravenous fluids and treated with intravenous ceftriaxone and azithromycin for community‐acquired pneumonia. An abdominal ultrasound was normal. The serologic assays for acute hepatitis A, B, and C infection were negative. The following morning, she reported worsening headache, increased cough now productive of whitish‐yellow sputum, and diffuse body aches. She appeared more lethargic and toxic. Her blood pressure was 100/83 mm Hg, heart rate was 84 beats per minute, respiratory rate was 24 per minute, and temperature was 101.3F. She had increased crackles on chest auscultation bilaterally and required supplemental oxygen at 4 L/minute by nasal cannula. Examination of both legs now revealed multiple scattered, faintly erythematous, 2‐cm‐sized patches overlying tender subtle subcutaneous nodules. Additionally, a mildly pruritic, V‐shaped area of blanchable erythema was also seen on her chest. The white blood cell count was 2500/mm3 (77% neutrophils, 15% lymphocytes), serum creatinine was 1.8 mg/dL, AST was 351 IU/L, and ALT was 485 IU/L. Blood cultures showed no growth and a peripheral smear examination was unrevealing. A noncontrast chest computed tomographic scan showed findings consistent with multifocal pneumonia (Figure 2).
It would be prudent at this time to expand her antimicrobial coverage (such as with vancomycin and piperacillin‐tazobactam) for activity against methicillin‐resistant Staphylococcus aureus and Pseudomonas because of her clinical worsening. Although ceftriaxone or piperacillin would cover leptospirosis, given the possibility of ehrlichiosis, the addition of doxycycline should be strongly considered.
The description of the rash on her legs seems consistent with erythema nodosum, which is associated with a number of infections (streptococcal, fungal, syphilis, EBV, cat‐scratch disease, tuberculosis), inflammatory conditions (inflammatory bowel disease, autoimmune disease, malignancy), and pregnancy. The blanchable rash on the chest is also a cause of concern for a possible drug reaction (ceftriaxone). A Jarisch‐Herxheimer reaction is possible given her acute worsening of symptoms with initiation of antibiotic therapy.
An antineutrophil cytoplasmic antibodyassociated vasculitis or another autoimmune condition such as systemic lupus erythematosus can account for erythema nodosum, rash, pancytopenia, and hepatitis. This diagnosis might also fit if she had a vasculitic pulmonary hemorrhage that caused her lung infiltrates and worsening hypoxia. A complete antinuclear antibody panel, antineutrophil cytoplasmic antibody, and antismooth muscle antibody testing is recommended. A skin and bronchoscopic biopsy should be considered.
Her dose of ceftriaxone was increased for possible severe pneumococcal pneumonia. The dermatology consultant felt that her leg lesions were consistent with erythema nodosum and the chest rash consistent with cutaneous photodamage. Bronchoscopic examination was normal and a bronchoalveolar lavage sample showed 2905 red blood cells/mm3 and 605 white blood cells/mm3 (70% neutrophils, 7% lymphocytes, 16% histiocytes), normal cytology, and negative cultures. There was no significant clinical improvement by the fourth hospital day and oral doxycycline was started. The next day, her skin lesions had resolved and she felt better. The serologic tests for Legionella, Mycoplasma, cytomegalovirus, EBV, Toxoplasma, Chlamydophila, Ehrlichia, Leptospira, Q‐fever, parvovirus, and adenovirus were negative. A fungal serology panel, HIV polymerase chain reaction, cryoglobulin level, and several rheumatologic tests (antinuclear antibody, extractable nuclear antigen panel, rheumatoid factor, antineutrophil cytoplasmic antibody, antiproteinase 3, and antiglomerular basement membrane antibodies) were normal. Blood cultures continued to show no growth.
The apparent response to doxycycline suggests she might have ehrlichiosis. A buffy coat review for morulae should be done. It is also possible that she may have improved on her initial therapy alone before starting doxycycline and her clinical worsening (including the chest rash) was due to a Jarisch‐Herxheimer reaction. Serologic tests for leptospirosis and ehrlichiosis should be repeated in 12 weeks because such infections may not cause detectable antibody levels early in the illness.
Ceftriaxone and doxycycline were continued and she showed rapid and significant clinical improvement. She was discharged 4 days later with instructions to complete a 10‐day course of antibiotics. At her 3‐month follow‐up, she was doing well and a repeat Leptospira antibody test by the Indirect Hemagglutination Assay (MRL Diagnostics, Cypress, California; normal titer 1:50) was positive at a titer of 1:100, which is highly suggestive of leptospirosis.
Commentary
Leptospirosis is a zoonotic infection caused by spirochetes of the genus Leptospira. The infection is usually transmitted indirectly to humans through contact with water, food, or soil contaminated with the urine of infected mammals.1 Risk factors for infection include participation in recreational activities (such as freshwater swimming, canoeing, and camping), occupational exposure, and exposure to infected pets or domesticated livestock. Approximately 100200 cases are identified annually in the United States, and approximately half occur in the state of Hawaii.2 Outbreaks of leptospirosis have been reported previously in the Midwest.3 These organisms inoculate humans through contact with mucous membranes or broken skin, or enter by swallowing infected food or water. A large number of these infections remain subclinical or result in a very mild illness with spontaneous clearance by the host's immune mechanism. Following an incubation period of 230 days, infected individuals may develop clinically significant disease (Table 1). Clinical presentations may overlap as the disease progresses. Although much remains to be learned about the exact pathogenic mechanism, disruption of the cell membranes of small vessel endothelia (a toxin‐like effect), and cytokine‐mediated tissue injury are believed to cause organ hemorrhage and ischemia.4
|
| 1. Mild influenza‐like self‐remitting disease (90% of cases) |
| Undifferentiated fever (usually 100F105F), severe headache, and myalgia (especially lower limbs). |
| 2. Moderately severe disease usually requiring hospitalization (5%9% of cases) |
| Marked prostration, anorexia, nausea, and vomiting, conjunctival suffusion, transient rash, frequently abdominal pain, constipation or diarrhea, and occasionally epistaxis. |
| 3. Severe disease involving multiple organ systems (1%5% of cases) |
| Hepatorenal Syndrome (Weil's syndrome) |
| Constellation of jaundice, hemorrhagic diathesis, and acute renal failure. Hepatic failure is rarely fatal. Renal involvement is usually more severe and the common cause of death. Cardiac (myocarditis with arrhythmias) and pulmonary complications are frequent. Confusion and restlessness may occur. |
| Hemorrhagic pneumonitis |
| Usually presents as a dry cough initially but becomes blood‐streaked after 23 days. Often characterized by a rapid progression to involve extensive areas of lungs, massive intra‐alveolar hemorrhage, acute respiratory failure, and death. |
| Central nervous system involvement |
| Meningismus, meningitis, or meningoencephalitis. |
The clinical diagnosis of leptospirosis is difficult because of its protean manifestations. Although nonspecific, 2 clinical features may provide a clue to the clinical diagnosis. First, the presence of conjunctival suffusion occurs in the early stage of the disease and is often associated with subconjunctival hemorrhage. Second, severe myalgia, commonly involving the lower limbs, is also characteristically present.1, 5 In 1 series of 58 patients with acute leptospirosis, conjunctival suffusion was observed in 50% of cases, and subconjunctival hemorrhage in 29%. Body ache and muscle tenderness was described in almost all cases.6
As seen in this case, the presence of a rash may pose a clinical challenge. A transient macular, maculopapular, purpuric, or urticarial rash may be seen in acute leptospirosis, but rashes may also be representative of a complication of treatment.1 First described in 1895 in patients with syphilis treated with mercury, the Jarisch‐Herxheimer reaction typically occurs within a few hours of antimicrobial treatment of spirochete infections and often presents with a rash, headache, fever, rigors, hypotension, sweating, and worsening symptoms of the underlying illness.7 Other skin findings such as the occurrence of erythema nodosum have been previously reported in cases of leptospirosis.8
Human ehrlichiosis (HE) is caused by tickborne, obligatory intracellular bacteria that infect leukocytes. There are 3 distinct clinical conditions: human monocytic ehrlichiosis (HME, caused by Ehrlichia chaffeensis), human granulocytic anaplasmosis (HGA, caused by Anaplasma phagocytophilum), and human ewingii ehrlichiosis (HEE, caused by E. ewingii). Although most cases of HME and HEE are seen in the southeastern and south‐central United States and California, the highest incidence of HGA is reported in the northeastern and upper Midwest regions.9 As with leptospirosis, the clinical range of HE spans from asymptomatic infection to life‐threatening illness. Following an incubation period of 12 weeks, symptomatic cases usually present with nonspecific complaints such as high fevers, chills, headache, nausea, arthralgia, myalgia, and malaise.10 The majority of cases will report a tick bite or an exposure to ticks. Laboratory tests often reveal leukopenia (white blood cell count 4000/mm3), thrombocytopenia, hyponatremia, and elevated AST and ALT. Patients with severe disease may develop renal, respiratory, and hepatic failure. Thus, differentiating ehrlichiosis from leptospirosis is often challenging for the clinician.
However, there are a few clinical clues that help distinguish between these illnesses in this case. HGA as a cause of HE would be more likely in the Midwest. Although a rash is present in one‐third of patients with HME, it is seldom present in HGA unless coinfected with Borrelia burgdorferi, the causative agent for Lyme disease. Additionally, her history of freshwater exposure and the absence of a history of a tick bite also favor leptospirosis. As noted previously, conjunctival suffusion, a characteristic clinical feature of leptospirosis, has only been described in case reports of HE.11, 12
Serologic tests are often used to establish the diagnosis of leptospirosis and ehrlichiosis. Leptospires are fastidious organisms that are difficult to isolate on inoculated growth media. The microscopic agglutination test for leptospirosis is considered the diagnostic gold standard due to its high specificity, but its use is limited by its technical complexity, lack of availability (other than in reference laboratories), and low sensitivity early in the disease (antibody levels detected by this method usually do not appear until 7 days after symptom onset).13 A variety of rapid serologic assays are also available. Although these tests have good overall sensitivity (ranging between 79% and 93%), they perform relatively poorly for acute‐phase sera (sensitivity of 38.5%52.7%).13 The high early false negative rate is believed to be a result of inadequate Leptospira antibody titers in the acute phase of the illness. Seroconversion or a 4‐fold rise between acute and convalescent‐phase antibody titers is the most definitive criterion for the diagnosis of leptospirosis. However, without paired sera samples, a single high microscopic agglutination test titer can be taken as diagnostic for leptospirosis depending on the degree of regional endemicity.14
Similarly, currently available serologic assays for ehrlichiosis produce negative results in most patients in the first week of illness, and it is important to obtain a convalescent phase serum specimen for confirmatory diagnosis of HME and HGA. Seroconversion or a 4‐fold increase in titer between acute and convalescent phase sera is considered diagnostic. The sensitivity of finding morulae (intracytoplasmic vacuolar microcolonies of Ehrlichia) on a peripheral smear is unknown, and data suggest that this finding is more common in cases of HGA compared to HME.15
Although doxycycline is the drug of choice for the treatment of ehrlichiosis, Leptospira is susceptible to a wide variety of antibiotics because it exhibits a double membrane surface architecture with components common to both gram‐negative and gram‐positive bacteria.1 Recommended treatment regimens for severe leptospirosis include the use of high‐dose intravenous penicillin or a third‐generation cephalosporin. Less severe cases can be treated with oral amoxicillin or doxycycline.16 The fact that this patient's clinical improvement appeared to lag after initiation of ceftriaxone does not necessarily indicate a lack of efficacy but perhaps a Jarisch‐Herxheimer reaction in response to appropriate antibiotic therapy.
Teaching Points
-
Establishing a diagnosis of leptospirosis is challenging and requires a high index of suspicion. Clinicians should be aware of the limitations of the diagnostic accuracy of the serologic assays for leptospirosis because they are frequently negative in the first week after symptom onset.
-
The classic finding of conjunctival suffusion is helpful in differentiating leptospirosis from human ehrlichiosis.
-
This case also highlights the importance of the clinical practice of making a list of suspected diagnoses, remaining open to these possibilities, and checking serologic tests again in convalescence to confirm the diagnosis.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Acknowledgements
The authors thank Dr. Brian Harte for his valuable guidance in the preparation of this manuscript.
In October, a 36‐year‐old woman with no significant past medical history presented to the Emergency Department (ED) with a 3‐day history of headache and fever. The headache was severe, throbbing, and frontal in location. She also complained of daily fevers measured up to 103F, generalized malaise, and fatigue. She did not report neck stiffness or photophobia. She felt better after receiving intravenous fluids and was discharged home with a diagnosis of a nonspecific viral illness. Two days later, she returned to the ED with worsening headache, fever, mild photophobia, and poor oral intake. She also complained of a dry cough that made her headache worse, as did bending over. She did not report confusion, neck stiffness, shortness of breath, sore throat, runny nose, abdominal symptoms, or rash.
This patient presents a second time to the ED with worsening headache and fever raising concerns about meningitis. At the time of her first ED visit, it can be assumed that she had a nontoxic appearance because she was discharged shortly thereafter. Thus, acute bacterial meningitis seems less likely, but occasionally patients with meningococcal meningitis may not appear significantly ill until later in the process. Nonetheless, acute meningitis, possibly viral, is the initial concern. The time of the year is an important variable because many viral infections are seasonal. Enteroviruses are the most common cause of viral meningitis in the United States, particularly in the summer and fall. In contrast, mumps, measles, and varicella zoster viruses occur more commonly in winter and spring. Herpetic meningoencephalitis is a life‐threatening condition with a guarded prognosis. Therefore, early recognition and treatment is necessary to decrease morbidity and mortality. Drugs such as nonsteroidal anti‐inflammatory agents, trimethoprim‐sulfamethoxazole, amoxicillin, and rarely vaccines can also cause aseptic meningitis. Infections from fungi, spirochetes, mycobacteria, and rarely parasites also cause meningitis, but would be of greater concern in a patient with risk factors such as recent travel or an immunocompromised state.
Increased headache with bending and cough might indicate elevated intracranial pressure. However, this is a nonspecific complaint, and headache is often worse with the Valsalva maneuver. Because she reports a cough, a chest x‐ray would be useful. In addition to routine initial tests, cerebrospinal fluid (CSF) analysis and human immunodeficiency virus (HIV) testing is recommended.
Her past medical history was notable for depression. Her medications included bupropion, multivitamins, and fish oil. She was also taking milk thistle pills daily to protect her liver because she had been drinking alcohol heavily for the past 2 weeks since her husband left her. She smoked 1 pack of cigarettes daily. She had not traveled recently. She reported no recent animal or wildlife exposure but did recall falling into a midwestern river while canoeing 2 weeks prior to presentation. She worked as a hairstylist and described no sick contacts or risk factors for HIV disease.
An important new historical element is that the patient fell into a river. If she swallowed a significant amount of water during her fall overboard, meningitis from waterborne infections such as Aeromonas, Acanthamoeba, and Naegleria need to be considered. Fortunately, these are rare in the Midwest. Her canoeing history may suggest exposure to wooded areas. Certainly, tickborne infections such as ehrlichiosis, babesiosis, Lyme disease, and Rocky Mountain spotted fever can also cause meningitis. Histoplasmosis and blastomycosis are also endemic to the midwestern United States and can disseminate and cause central nervous system disease.
At this time, viral and bacterial infections are highest on the differential diagnosis. However, the microbiology laboratory needs to be alerted to the possibility of fungal or parasitic organisms depending on the initial CSF analysis results.
The patient was a Caucasian woman who appeared comfortable. Her blood pressure was 130/62 mm Hg, heart rate was 83 beats per minute, respiratory rate was 18 per minute, temperature was 100.8F, and oxygen saturation was 98% on room air. She was fully alert and oriented. Her pupils were bilaterally equal, reactive to light and accommodation with intact extraocular movement and no nystagmus. There was conjunctival injection bilaterally without noticeable pallor or icterus. Fundoscopic examination, which the patient tolerated without difficulty, was normal. Inspection of the oral cavity showed mild tonsillar enlargement. The neck was supple with no stiffness. No cervical, axillary, or inguinal lymph nodes were palpable. Faint bilateral basilar crackles were audible over the posterior chest. There was very mild right upper quadrant abdominal tenderness without guarding. The liver and spleen were normal size and bowel sounds were present. No rash, peripheral edema, or spinal tenderness was noted. A complete neurological examination was normal.
Her general appearance and vital signs seem reassuring. Conjunctival injection and mild tonsillar enlargement are nonspecific findings and may occur in systemic inflammatory states especially viral infections. Atelectasis may account for faint bilateral basilar crackles especially if associated with post‐tussive change. Her alcohol use puts her at risk of aspiration. A right lower lobe process (pneumonia) can sometimes present with right upper quadrant tenderness. However, this tenderness may also represent muscle soreness from repeated coughing, liver, or gallbladder disease. The same infectious process affecting the central nervous system and possibly her lungs, may also be affecting the liver.
A complete blood count revealed a white blood cell count of 3000/mm3 (79% neutrophils, 15% lymphocytes, 5% monocytes), hemoglobin of 11.7 g/dL, and platelets of 110,000/mm3. The serum sodium was 133 mmol/L, potassium was 3.7 mmol/L, bicarbonate was 22 mmol/L, and blood urea nitrogen was 20 mg/dL. The serum creatinine was 1.5 compared to 1.0 mg/dL on testing 2 days prior. A liver function panel showed protein of 5.1 g/dL, albumin of 3 g/dL, aspartate aminotransferase (AST) of 576 IU/L, alanine aminotransferase (ALT) of 584 IU/L, alkaline phosphatase of 282 IU/L, and total bilirubin of 1 mg/dL. The coagulation profile, creatinine phosphokinase, acetaminophen level, urine pregnancy test, urine drug screen, and urinalysis (including urine microscopy) were normal.
The CSF opening pressure was 13 cm H2O. CSF analysis showed 4 mononuclear leukocytes per high‐power field, CSF protein was 27 mg/dL, and glucose was 76 mg/dL. No organisms were noted on gram stain. A chest x‐ray showed focal airspace opacity in the left lower lobe (Figure 1) and the patient was hospitalized for further management.
The normal CSF analysis makes acute meningitis much less likely. It is interesting to note that the aminotransferase levels are nearly equal. Usually, in viral and many other causes of hepatitis, the ALT is higher than the AST, whereas the contrary is true in alcoholic hepatitis. Because the patient has been consuming significant amounts of alcohol recently, these levels may become equal in the setting of another primary liver process. The elevation in liver enzymes also raises the possibility of autoimmune hepatitis secondary to a systemic vasculitis such as systemic lupus erythematosus. Nonetheless, the focus should be on infectious causes of hepatitis such as hepatitis C, adenovirus, parvovirus, Epstein‐Barr virus (EBV), cytomegalovirus, and herpes simplex virus that can cause pneumonia either as a primary or secondary infection. Acute HIV infection can also present in this fashion, and anti‐HIV antibody testing may be negative early in the disease. In the setting of a normal urinalysis and bland urine sediment, prerenal azotemia is the most likely cause of her acute renal injury and can be confirmed by testing the urinary sodium and creatinine. A peripheral smear should be reviewed to evaluate the pancytopenia.
Severe headache, fever, conjunctival injection, pancytopenia, acute kidney injury, hepatitis, and pneumonia may occur in leptospirosis, particularly in a patient with recent freshwater exposure. Alternatively, ehrlichiosis can also account for fever, headache, pancytopenia, renal failure, hepatitis, and pneumonia, but conjunctival suffusion is not often present. At this time, treatment for community‐acquired pneumonia that includes coverage for leptospirosis should be started.
The patient was hydrated with intravenous fluids and treated with intravenous ceftriaxone and azithromycin for community‐acquired pneumonia. An abdominal ultrasound was normal. The serologic assays for acute hepatitis A, B, and C infection were negative. The following morning, she reported worsening headache, increased cough now productive of whitish‐yellow sputum, and diffuse body aches. She appeared more lethargic and toxic. Her blood pressure was 100/83 mm Hg, heart rate was 84 beats per minute, respiratory rate was 24 per minute, and temperature was 101.3F. She had increased crackles on chest auscultation bilaterally and required supplemental oxygen at 4 L/minute by nasal cannula. Examination of both legs now revealed multiple scattered, faintly erythematous, 2‐cm‐sized patches overlying tender subtle subcutaneous nodules. Additionally, a mildly pruritic, V‐shaped area of blanchable erythema was also seen on her chest. The white blood cell count was 2500/mm3 (77% neutrophils, 15% lymphocytes), serum creatinine was 1.8 mg/dL, AST was 351 IU/L, and ALT was 485 IU/L. Blood cultures showed no growth and a peripheral smear examination was unrevealing. A noncontrast chest computed tomographic scan showed findings consistent with multifocal pneumonia (Figure 2).
It would be prudent at this time to expand her antimicrobial coverage (such as with vancomycin and piperacillin‐tazobactam) for activity against methicillin‐resistant Staphylococcus aureus and Pseudomonas because of her clinical worsening. Although ceftriaxone or piperacillin would cover leptospirosis, given the possibility of ehrlichiosis, the addition of doxycycline should be strongly considered.
The description of the rash on her legs seems consistent with erythema nodosum, which is associated with a number of infections (streptococcal, fungal, syphilis, EBV, cat‐scratch disease, tuberculosis), inflammatory conditions (inflammatory bowel disease, autoimmune disease, malignancy), and pregnancy. The blanchable rash on the chest is also a cause of concern for a possible drug reaction (ceftriaxone). A Jarisch‐Herxheimer reaction is possible given her acute worsening of symptoms with initiation of antibiotic therapy.
An antineutrophil cytoplasmic antibodyassociated vasculitis or another autoimmune condition such as systemic lupus erythematosus can account for erythema nodosum, rash, pancytopenia, and hepatitis. This diagnosis might also fit if she had a vasculitic pulmonary hemorrhage that caused her lung infiltrates and worsening hypoxia. A complete antinuclear antibody panel, antineutrophil cytoplasmic antibody, and antismooth muscle antibody testing is recommended. A skin and bronchoscopic biopsy should be considered.
Her dose of ceftriaxone was increased for possible severe pneumococcal pneumonia. The dermatology consultant felt that her leg lesions were consistent with erythema nodosum and the chest rash consistent with cutaneous photodamage. Bronchoscopic examination was normal and a bronchoalveolar lavage sample showed 2905 red blood cells/mm3 and 605 white blood cells/mm3 (70% neutrophils, 7% lymphocytes, 16% histiocytes), normal cytology, and negative cultures. There was no significant clinical improvement by the fourth hospital day and oral doxycycline was started. The next day, her skin lesions had resolved and she felt better. The serologic tests for Legionella, Mycoplasma, cytomegalovirus, EBV, Toxoplasma, Chlamydophila, Ehrlichia, Leptospira, Q‐fever, parvovirus, and adenovirus were negative. A fungal serology panel, HIV polymerase chain reaction, cryoglobulin level, and several rheumatologic tests (antinuclear antibody, extractable nuclear antigen panel, rheumatoid factor, antineutrophil cytoplasmic antibody, antiproteinase 3, and antiglomerular basement membrane antibodies) were normal. Blood cultures continued to show no growth.
The apparent response to doxycycline suggests she might have ehrlichiosis. A buffy coat review for morulae should be done. It is also possible that she may have improved on her initial therapy alone before starting doxycycline and her clinical worsening (including the chest rash) was due to a Jarisch‐Herxheimer reaction. Serologic tests for leptospirosis and ehrlichiosis should be repeated in 12 weeks because such infections may not cause detectable antibody levels early in the illness.
Ceftriaxone and doxycycline were continued and she showed rapid and significant clinical improvement. She was discharged 4 days later with instructions to complete a 10‐day course of antibiotics. At her 3‐month follow‐up, she was doing well and a repeat Leptospira antibody test by the Indirect Hemagglutination Assay (MRL Diagnostics, Cypress, California; normal titer 1:50) was positive at a titer of 1:100, which is highly suggestive of leptospirosis.
Commentary
Leptospirosis is a zoonotic infection caused by spirochetes of the genus Leptospira. The infection is usually transmitted indirectly to humans through contact with water, food, or soil contaminated with the urine of infected mammals.1 Risk factors for infection include participation in recreational activities (such as freshwater swimming, canoeing, and camping), occupational exposure, and exposure to infected pets or domesticated livestock. Approximately 100200 cases are identified annually in the United States, and approximately half occur in the state of Hawaii.2 Outbreaks of leptospirosis have been reported previously in the Midwest.3 These organisms inoculate humans through contact with mucous membranes or broken skin, or enter by swallowing infected food or water. A large number of these infections remain subclinical or result in a very mild illness with spontaneous clearance by the host's immune mechanism. Following an incubation period of 230 days, infected individuals may develop clinically significant disease (Table 1). Clinical presentations may overlap as the disease progresses. Although much remains to be learned about the exact pathogenic mechanism, disruption of the cell membranes of small vessel endothelia (a toxin‐like effect), and cytokine‐mediated tissue injury are believed to cause organ hemorrhage and ischemia.4
|
| 1. Mild influenza‐like self‐remitting disease (90% of cases) |
| Undifferentiated fever (usually 100F105F), severe headache, and myalgia (especially lower limbs). |
| 2. Moderately severe disease usually requiring hospitalization (5%9% of cases) |
| Marked prostration, anorexia, nausea, and vomiting, conjunctival suffusion, transient rash, frequently abdominal pain, constipation or diarrhea, and occasionally epistaxis. |
| 3. Severe disease involving multiple organ systems (1%5% of cases) |
| Hepatorenal Syndrome (Weil's syndrome) |
| Constellation of jaundice, hemorrhagic diathesis, and acute renal failure. Hepatic failure is rarely fatal. Renal involvement is usually more severe and the common cause of death. Cardiac (myocarditis with arrhythmias) and pulmonary complications are frequent. Confusion and restlessness may occur. |
| Hemorrhagic pneumonitis |
| Usually presents as a dry cough initially but becomes blood‐streaked after 23 days. Often characterized by a rapid progression to involve extensive areas of lungs, massive intra‐alveolar hemorrhage, acute respiratory failure, and death. |
| Central nervous system involvement |
| Meningismus, meningitis, or meningoencephalitis. |
The clinical diagnosis of leptospirosis is difficult because of its protean manifestations. Although nonspecific, 2 clinical features may provide a clue to the clinical diagnosis. First, the presence of conjunctival suffusion occurs in the early stage of the disease and is often associated with subconjunctival hemorrhage. Second, severe myalgia, commonly involving the lower limbs, is also characteristically present.1, 5 In 1 series of 58 patients with acute leptospirosis, conjunctival suffusion was observed in 50% of cases, and subconjunctival hemorrhage in 29%. Body ache and muscle tenderness was described in almost all cases.6
As seen in this case, the presence of a rash may pose a clinical challenge. A transient macular, maculopapular, purpuric, or urticarial rash may be seen in acute leptospirosis, but rashes may also be representative of a complication of treatment.1 First described in 1895 in patients with syphilis treated with mercury, the Jarisch‐Herxheimer reaction typically occurs within a few hours of antimicrobial treatment of spirochete infections and often presents with a rash, headache, fever, rigors, hypotension, sweating, and worsening symptoms of the underlying illness.7 Other skin findings such as the occurrence of erythema nodosum have been previously reported in cases of leptospirosis.8
Human ehrlichiosis (HE) is caused by tickborne, obligatory intracellular bacteria that infect leukocytes. There are 3 distinct clinical conditions: human monocytic ehrlichiosis (HME, caused by Ehrlichia chaffeensis), human granulocytic anaplasmosis (HGA, caused by Anaplasma phagocytophilum), and human ewingii ehrlichiosis (HEE, caused by E. ewingii). Although most cases of HME and HEE are seen in the southeastern and south‐central United States and California, the highest incidence of HGA is reported in the northeastern and upper Midwest regions.9 As with leptospirosis, the clinical range of HE spans from asymptomatic infection to life‐threatening illness. Following an incubation period of 12 weeks, symptomatic cases usually present with nonspecific complaints such as high fevers, chills, headache, nausea, arthralgia, myalgia, and malaise.10 The majority of cases will report a tick bite or an exposure to ticks. Laboratory tests often reveal leukopenia (white blood cell count 4000/mm3), thrombocytopenia, hyponatremia, and elevated AST and ALT. Patients with severe disease may develop renal, respiratory, and hepatic failure. Thus, differentiating ehrlichiosis from leptospirosis is often challenging for the clinician.
However, there are a few clinical clues that help distinguish between these illnesses in this case. HGA as a cause of HE would be more likely in the Midwest. Although a rash is present in one‐third of patients with HME, it is seldom present in HGA unless coinfected with Borrelia burgdorferi, the causative agent for Lyme disease. Additionally, her history of freshwater exposure and the absence of a history of a tick bite also favor leptospirosis. As noted previously, conjunctival suffusion, a characteristic clinical feature of leptospirosis, has only been described in case reports of HE.11, 12
Serologic tests are often used to establish the diagnosis of leptospirosis and ehrlichiosis. Leptospires are fastidious organisms that are difficult to isolate on inoculated growth media. The microscopic agglutination test for leptospirosis is considered the diagnostic gold standard due to its high specificity, but its use is limited by its technical complexity, lack of availability (other than in reference laboratories), and low sensitivity early in the disease (antibody levels detected by this method usually do not appear until 7 days after symptom onset).13 A variety of rapid serologic assays are also available. Although these tests have good overall sensitivity (ranging between 79% and 93%), they perform relatively poorly for acute‐phase sera (sensitivity of 38.5%52.7%).13 The high early false negative rate is believed to be a result of inadequate Leptospira antibody titers in the acute phase of the illness. Seroconversion or a 4‐fold rise between acute and convalescent‐phase antibody titers is the most definitive criterion for the diagnosis of leptospirosis. However, without paired sera samples, a single high microscopic agglutination test titer can be taken as diagnostic for leptospirosis depending on the degree of regional endemicity.14
Similarly, currently available serologic assays for ehrlichiosis produce negative results in most patients in the first week of illness, and it is important to obtain a convalescent phase serum specimen for confirmatory diagnosis of HME and HGA. Seroconversion or a 4‐fold increase in titer between acute and convalescent phase sera is considered diagnostic. The sensitivity of finding morulae (intracytoplasmic vacuolar microcolonies of Ehrlichia) on a peripheral smear is unknown, and data suggest that this finding is more common in cases of HGA compared to HME.15
Although doxycycline is the drug of choice for the treatment of ehrlichiosis, Leptospira is susceptible to a wide variety of antibiotics because it exhibits a double membrane surface architecture with components common to both gram‐negative and gram‐positive bacteria.1 Recommended treatment regimens for severe leptospirosis include the use of high‐dose intravenous penicillin or a third‐generation cephalosporin. Less severe cases can be treated with oral amoxicillin or doxycycline.16 The fact that this patient's clinical improvement appeared to lag after initiation of ceftriaxone does not necessarily indicate a lack of efficacy but perhaps a Jarisch‐Herxheimer reaction in response to appropriate antibiotic therapy.
Teaching Points
-
Establishing a diagnosis of leptospirosis is challenging and requires a high index of suspicion. Clinicians should be aware of the limitations of the diagnostic accuracy of the serologic assays for leptospirosis because they are frequently negative in the first week after symptom onset.
-
The classic finding of conjunctival suffusion is helpful in differentiating leptospirosis from human ehrlichiosis.
-
This case also highlights the importance of the clinical practice of making a list of suspected diagnoses, remaining open to these possibilities, and checking serologic tests again in convalescence to confirm the diagnosis.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Acknowledgements
The authors thank Dr. Brian Harte for his valuable guidance in the preparation of this manuscript.
- ,,.Leptospirosis: an emerging global public health problem.J Biosci.2008;33:557–569.
- Centers for Disease Control and Prevention. Leptospirosis.2005. http://www.cdc.gov/ncidod/dbmd/diseaseinfo/leptospirosis_t.htm. Accessed November 15,year="2010"2010.
- Morbidity and Mortality Weekly Report.From the Centers for Disease Control and Prevention. Update: leptospirosis and unexplained acute febrile illness among athletes participating in triathlons—Illinois and Wisconsin, 1998.JAMA.1998;280:1474–1475.
- ,.Optimal treatment of leptospirosis: queries and projections.Int J Antimicrob Agents.2006;28:491–496.
- ,.Leptospirosis in the tropics and in travelers.Curr Infect Dis Rep.2006;8:51–58.
- ,,,,,.Clinico‐epidemiological study of hospitalized cases of severe leptospirosis.Indian J Med Res.1999;109:94–99.
- ,.Proposed mechanisms and preventative options of Jarisch‐Herxheimer reactions.J Clin Pharm Ther.2005;30:291–295.
- .Leptospirosis presenting with erythema nodosum.Arch Dis Child.1977;52:418–419.
- ,,.Emerging and re‐emerging tick‐transmitted rickettsial and ehrlichial infections.Med Clin N Am.2008;92:1345–1361.
- ,.Tick‐borne ehrlichiosis infection in human beings.J Vector Borne Dis.2008;45:273–280.
- ,.Ehrlichia in Tennessee.South Med J.1989;82:669.
- ,,,,,.Ehrlichial meningitis with cerebrospinal fluid morulae.Pediatr Infect Dis J.1999;18:552–555.
- ,,, et al.Evaluation of four commercially available rapid serologic tests for diagnosis of leptospirosis.J Clin Microbiol.2003;41:803–809.
- ,.Diagnosis of leptospirosis utilizing modified Faine's criteria.J Assoc Physicians India.2004;52:678–679.
- ,.Ehrlichiosis in children.J Pediatr.1997;131:184–192.
- ,World Health Organization, International Leptospirosis Society. Human leptospirosis: guidance for diagnosis, surveillance and control.Geneva, Switzerland:World Health Organization;2003.
- ,,.Leptospirosis: an emerging global public health problem.J Biosci.2008;33:557–569.
- Centers for Disease Control and Prevention. Leptospirosis.2005. http://www.cdc.gov/ncidod/dbmd/diseaseinfo/leptospirosis_t.htm. Accessed November 15,year="2010"2010.
- Morbidity and Mortality Weekly Report.From the Centers for Disease Control and Prevention. Update: leptospirosis and unexplained acute febrile illness among athletes participating in triathlons—Illinois and Wisconsin, 1998.JAMA.1998;280:1474–1475.
- ,.Optimal treatment of leptospirosis: queries and projections.Int J Antimicrob Agents.2006;28:491–496.
- ,.Leptospirosis in the tropics and in travelers.Curr Infect Dis Rep.2006;8:51–58.
- ,,,,,.Clinico‐epidemiological study of hospitalized cases of severe leptospirosis.Indian J Med Res.1999;109:94–99.
- ,.Proposed mechanisms and preventative options of Jarisch‐Herxheimer reactions.J Clin Pharm Ther.2005;30:291–295.
- .Leptospirosis presenting with erythema nodosum.Arch Dis Child.1977;52:418–419.
- ,,.Emerging and re‐emerging tick‐transmitted rickettsial and ehrlichial infections.Med Clin N Am.2008;92:1345–1361.
- ,.Tick‐borne ehrlichiosis infection in human beings.J Vector Borne Dis.2008;45:273–280.
- ,.Ehrlichia in Tennessee.South Med J.1989;82:669.
- ,,,,,.Ehrlichial meningitis with cerebrospinal fluid morulae.Pediatr Infect Dis J.1999;18:552–555.
- ,,, et al.Evaluation of four commercially available rapid serologic tests for diagnosis of leptospirosis.J Clin Microbiol.2003;41:803–809.
- ,.Diagnosis of leptospirosis utilizing modified Faine's criteria.J Assoc Physicians India.2004;52:678–679.
- ,.Ehrlichiosis in children.J Pediatr.1997;131:184–192.
- ,World Health Organization, International Leptospirosis Society. Human leptospirosis: guidance for diagnosis, surveillance and control.Geneva, Switzerland:World Health Organization;2003.
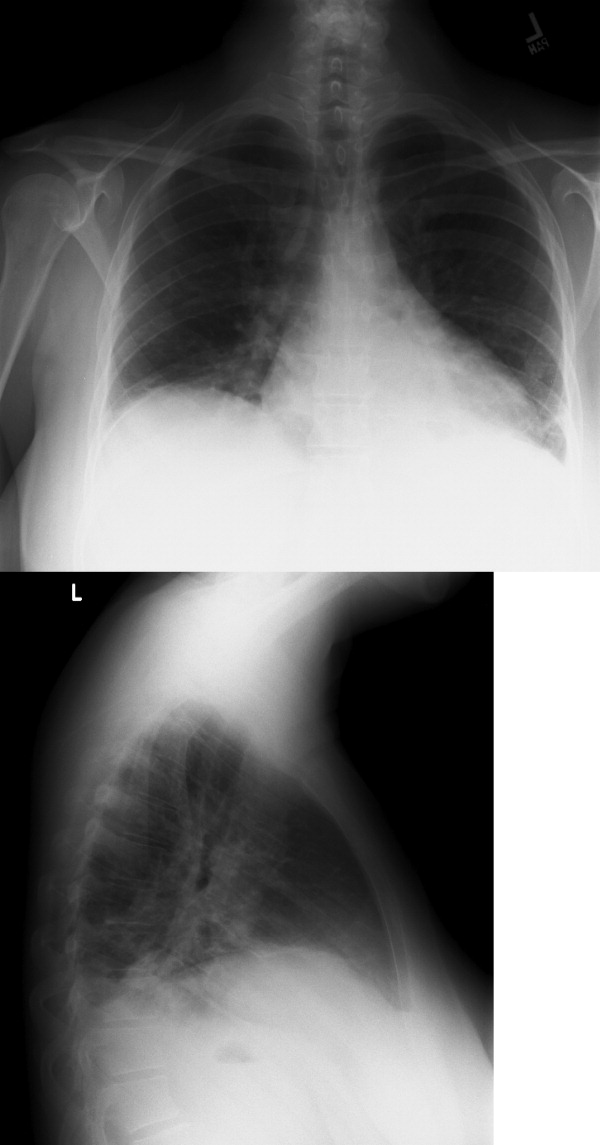
When Should a Patient with Ascites Receive Spontaneous Bacterial Peritonitis (SBP) Prophylaxis?
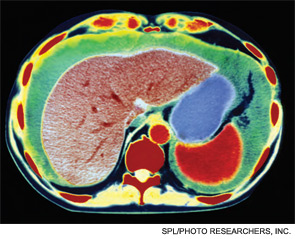
Case
A 54-year-old man with end-stage liver disease (ESLD) and no prior history of spontaneous bacterial peritonitis (SBP) presents with increasing shortness of breath and abdominal distention. He is admitted for worsening volume overload. The patient reveals that he has not been compliant with his diuretics. On the day of admission, a large-volume paracentesis is performed. Results are significant for a white blood cell count of 150 cells/mm3 and a total protein of 0.9 g/ul. The patient is started on furosemide and spironolactone, and his symptoms significantly improve throughout his hospitalization. His medications are reconciled on the day of discharge. He is not on any antibiotics for SBP prophylaxis; should he be? In general, which patients with ascites should receive SBP prophylaxis?
Overview
Spontaneous bacterial peritonitis is an infection of ascitic fluid that occurs in the absence of an indentified intra-abdominal source of infection or inflammation, i.e., perforation or abscess.1 It is diagnosed when the polymorphonuclear cell (PMN) count in the ascitic fluid is equal to or greater than 250 cells/mm3, with or without positive cultures.
SBP is a significant cause of morbidity and mortality in patients with cirrhosis, with the mortality rate approaching 20% to 40%.2 Of the 32% to 34% of cirrhotic patients who present with, or develop, a bacterial infection during their hospitalization, 25% are due to SBP.1 Changes in gut motility, mucosal defense, and microflora allow for translocation of bacteria into enteric lymph nodes and the bloodstream, resulting in seeding of the peritoneal fluid and SBP.1 Alterations in both systemic and localized immune defenses, both of which are reduced in patients with liver disease, also play a role in SBP pathogenesis (see Table 1, p. 41).
Current evidence supports the use of a third-generation cephalosporin or amoxicillin/clavulanate for initial treatment of SBP, as most infections are caused by gram-negative bacilli, in particular E. coli (see Table 2 on p. 41 and Table 3 on p. 42).1 Alternatively, an oral or intravenous fluoroquinolone could be used if the prevalence of fluoroquinolone-resistant organisms is low.1
Due to the frequency and morbidity associated with SBP, there is great interest in preventing it. However, the use of prophylactic antibiotics needs to be restricted to patients who are at highest risk of developing SBP. According to numerous studies, patients at high risk for SBP include:
- Patients with a prior SBP history;
- Patients admitted with a gastrointestinal bleed; and
- Patients with low total protein content in their ascitic fluid (defined as <1.5 g/ul).1
SBP History
Spontaneous bacterial peritonitis portends bad outcomes. The one-year mortality rate after an episode of SBP is 30% to 50%.1 Furthermore, patients who have recovered from a previous episode of SBP have a 70% chance of developing another episode of SBP in that year.1,2 In one study, norfloxacin was shown to decrease the one-year risk of SBP to 20% from 68% in patients with a history of SBP.3 Additionally, the likelihood of developing SBP from gram-negative bacilli was reduced to 3% from 60%. In order to be efficacious, norfloxacin must be given daily. When fluoroquinolones are prescribed less than once daily, there is a higher rate of fluoroquinolone resistant organisms in the stool.1
Though once-daily dosing of norfloxacin is recommended to decrease the promotion of resistant organisms in prophylaxis against SBP, ciprofloxacin once weekly is acceptable. In a group of patients with low ascitic protein content, with or without a history of SBP, weekly ciprofloxacin has been shown to decrease SBP incidence to 4% from 22% at six months.4 In regard to length of treatment, recommendations are to continue prophylactic antibiotics until resolution of ascites, the patient receives a transplant, or the patient passes away.1
Saab et al studied the impact of oral antibiotic prophylaxis in patients with advanced liver disease on morbidity and mortality.5 The authors examined prospective, randomized, controlled trials that compared high-risk cirrhotic patients receiving oral antibiotic prophylaxis for SBP with groups receiving placebo or no intervention. Eight studies totaling 647 patients were included in the analysis.
The overall mortality rate for patients treated with SBP prophylaxis was 16%, compared with 25% for the control group. Groups treated with prophylactic antibiotics also had a lower incidence of all infections (6.2% vs. 22.2% in the control groups). Additionally, a survival benefit was seen at three months in the group that received prophylactic antibiotics.
The absolute risk reduction with prophylactic antibiotics for primary prevention of SBP was 8% with a number needed to treat of 13. The incidence of gastrointestinal (GI) bleeding, renal failure, and hepatic failure did not significantly differ between treatment and control groups. Thus, survival benefit is thought to be related to the reduced incidence of infections in the group receiving prophylactic antibiotics.5
History of GI Bleeding
The incidence of developing SBP in cirrhotics with an active GI bleed is anywhere from 20% to 45%.1,2 For those with ascites of any etiology and a GI bleed, the incidence can be as high as 60%.5 In general, bacterial infections are frequently diagnosed in patients with cirrhosis and GI bleeding, and have been documented in 22% of these patients within the first 48 hours after admission. According to several studies, that percentage can reach as high as 35% to 66% within seven to 14 days of admission.6 A seven-day course of antibiotics, or antibiotics until discharge, is generally acceptable for SBP prophylaxis in the setting of ascites and GI bleeding (see Table 2, right).1
Bernard et al performed a meta-analysis of five trials to assess the efficacy of antibiotic prophylaxis in the prevention of infections and effect on survival in patients with cirrhosis and GI bleeding. Out of 534 patients, 264 were treated with antibiotics between four and 10 days, and 270 did not receive any antibiotics.
The endpoints of the study were infection, bacteremia and/or SBP; incidence of SBP; and death. Antibiotic prophylaxis not only increased the mean survival rate by 9.1%, but also increased the mean percentage of patients free of infection (32% improvement); bacteremia and/or SBP (19% improvement); and SBP (7% improvement).7
Low Ascitic Fluid Protein
Of the three major risk factors for SBP, ascitic fluid protein content is the most debated. Guarner et al studied the risk of first community-acquired SBP in cirrhotics with low ascitic fluid protein.2 Patients were seen immediately after discharge from the hospital and at two- to three-month intervals. Of the 109 hospitalized patients, 23 (21%) developed SBP, nine of which developed SBP during their hospitalization. The one-year cumulative probability of SBP in these patients with low ascitic fluid protein levels was 35%.
During this study, the authors also looked at 20 different patient variables on admission and found that two parameters—high bilirubin (>3.2mg/dL) and low platelet count (<98,000 cells/ul)—were associated with an increased risk of SBP. This is consistent with studies showing that patients with higher Model for End-Stage Liver Disease (MELD) or Child-Pugh scores, indicating more severe liver disease, are at increased risk for SBP. This likely is the reason SBP prophylaxis is recommended for patients with an elevated bilirubin, and higher Child-Pugh scores, by the American Association for the Study of Liver Disease (see Table 2, p. 41).
Runyon et al showed that 15% of patients with low ascitic fluid protein developed SBP during their hospitalization, as compared with 2% of patients with ascitic fluid levels greater than 1 g/dl.8 A randomized, non-placebo-controlled trial by Navasa et al evaluating 70 cirrhotic patients with low ascitic ascitic protein levels showed a lower probability of developing SBP in the group placed on SBP prophylaxis with norfloxacin (5% vs. 31%).9 Six-month mortality rate was also lower (19% vs. 36%).
In contrast to the previous studies, Grothe et al found that the presence of SBP was not related to ascitic protein content.10 Given conflicting studies, controversy still remains on whether patients with low ascitic protein should receive long-term prophylactic antibiotics.
Antibiotic Drawbacks
The consensus in the literature is that patients with ascites who are admitted with a GI bleed, or those with a history of SBP, should be placed on SBP prophylaxis. However, patients placed on long-term antibiotics are at risk for developing bacterial resistance. Bacterial resistance in cultures taken from cirrhotic patients with SBP has increased over the last decade, particularly in gram-negative bacteria.5 Patients who receive antibiotics in the pre-transplant setting also are at risk for post-transplant fungal infections.
Additionally, the antibiotic of choice for SBP prophylaxis is typically a fluoroquinolone, which can be expensive. However, numerous studies have shown that the cost of initiating prophylactic therapy for SBP in patients with a prior episode of SBP can be cheaper than treating SBP after diagnosis.2
Back to the Case
Our patient’s paracentesis was negative for SBP. Additionally, he does not have a history of SBP, nor does he have an active GI bleed. His only possible indication for SBP prophylaxis is low ascitic protein concentration. His electrolytes were all within normal limits. Additionally, total bilirubin was only slightly elevated at 2.3 mg/dL.
Based on the American Association for the Study of Liver Diseases guidelines, the patient was not started on SBP prophylaxis. Additionally, given his history of medication noncompliance, there is concern that he might not take the antibiotics as prescribed, thus leading to the development of bacterial resistance and more serious infections in the future.
Bottom Line
Patients with ascites and a prior episode of SBP, and those admitted to the hospital for GI bleeding, should be placed on SBP prophylaxis. SBP prophylaxis for low protein ascitic fluid remains controversial but is recommended by the American Association for the Study of Liver Diseases. TH
Dr. del Pino Jones is a hospitalist at the University of Colorado Denver.
References
- Ghassemi S, Garcia-Tsao G. Prevention and treatment of infections in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21(1):77-93.
- Guarner C, Solà R, Soriono G, et al. Risk of a first community-acquired spontaneous bacterial peritonitis in cirrhotics with low ascitic fluid protein levels. Gastroenterology. 1999;117(2):414-419.
- Ginés P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology. 1990;12(4 Pt 1):716-724.
- Rolachon A, Cordier L, Bacq Y, et al. Ciprofloxacin and long-term prevention of spontaneous bacterial peritonitis: results of a prospective controlled trial. Hepatology. 1995;22(4 Pt 1):1171-1174.
- Saab S, Hernandez J, Chi AC, Tong MJ. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am J Gastroenterol. 2009;104(4):993-1001.
- Deschênes M, Villeneuve J. Risk factors for the development of bacterial infections in hospitalized patients with cirrhosis. Am J Gastroenterol. 1999;94(8):2193-2197.
- Bernard B, Grangé J, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29(6):1655-1661.
- Runyon B. Low-protein-concentration ascitic fluid is predisposed to spontaneous bacterial peritonitis. Gastroenterology. 1986;91(6):1343-1346.
- Navasa M, Fernandez J, Montoliu S, et al. Randomized, double-blind, placebo-controlled trial evaluating norfloxacin in the primary prophylaxis of spontaneous bacterial peritonitis in cirrhotics with renal impairment, hyponatremia or severe liver failure. J Hepatol. 2006;44(Supp2):S51.
- Grothe W, Lottere E, Fleig W. Factors predictive for spontaneous bacterial peritonitis (SBP) under routine inpatient conditions in patients with cirrhosis: a prospective multicenter trial. J Hepatol. 1990;34(4):547.
Case
A 54-year-old man with end-stage liver disease (ESLD) and no prior history of spontaneous bacterial peritonitis (SBP) presents with increasing shortness of breath and abdominal distention. He is admitted for worsening volume overload. The patient reveals that he has not been compliant with his diuretics. On the day of admission, a large-volume paracentesis is performed. Results are significant for a white blood cell count of 150 cells/mm3 and a total protein of 0.9 g/ul. The patient is started on furosemide and spironolactone, and his symptoms significantly improve throughout his hospitalization. His medications are reconciled on the day of discharge. He is not on any antibiotics for SBP prophylaxis; should he be? In general, which patients with ascites should receive SBP prophylaxis?
Overview
Spontaneous bacterial peritonitis is an infection of ascitic fluid that occurs in the absence of an indentified intra-abdominal source of infection or inflammation, i.e., perforation or abscess.1 It is diagnosed when the polymorphonuclear cell (PMN) count in the ascitic fluid is equal to or greater than 250 cells/mm3, with or without positive cultures.
SBP is a significant cause of morbidity and mortality in patients with cirrhosis, with the mortality rate approaching 20% to 40%.2 Of the 32% to 34% of cirrhotic patients who present with, or develop, a bacterial infection during their hospitalization, 25% are due to SBP.1 Changes in gut motility, mucosal defense, and microflora allow for translocation of bacteria into enteric lymph nodes and the bloodstream, resulting in seeding of the peritoneal fluid and SBP.1 Alterations in both systemic and localized immune defenses, both of which are reduced in patients with liver disease, also play a role in SBP pathogenesis (see Table 1, p. 41).
Current evidence supports the use of a third-generation cephalosporin or amoxicillin/clavulanate for initial treatment of SBP, as most infections are caused by gram-negative bacilli, in particular E. coli (see Table 2 on p. 41 and Table 3 on p. 42).1 Alternatively, an oral or intravenous fluoroquinolone could be used if the prevalence of fluoroquinolone-resistant organisms is low.1
Due to the frequency and morbidity associated with SBP, there is great interest in preventing it. However, the use of prophylactic antibiotics needs to be restricted to patients who are at highest risk of developing SBP. According to numerous studies, patients at high risk for SBP include:
- Patients with a prior SBP history;
- Patients admitted with a gastrointestinal bleed; and
- Patients with low total protein content in their ascitic fluid (defined as <1.5 g/ul).1
SBP History
Spontaneous bacterial peritonitis portends bad outcomes. The one-year mortality rate after an episode of SBP is 30% to 50%.1 Furthermore, patients who have recovered from a previous episode of SBP have a 70% chance of developing another episode of SBP in that year.1,2 In one study, norfloxacin was shown to decrease the one-year risk of SBP to 20% from 68% in patients with a history of SBP.3 Additionally, the likelihood of developing SBP from gram-negative bacilli was reduced to 3% from 60%. In order to be efficacious, norfloxacin must be given daily. When fluoroquinolones are prescribed less than once daily, there is a higher rate of fluoroquinolone resistant organisms in the stool.1
Though once-daily dosing of norfloxacin is recommended to decrease the promotion of resistant organisms in prophylaxis against SBP, ciprofloxacin once weekly is acceptable. In a group of patients with low ascitic protein content, with or without a history of SBP, weekly ciprofloxacin has been shown to decrease SBP incidence to 4% from 22% at six months.4 In regard to length of treatment, recommendations are to continue prophylactic antibiotics until resolution of ascites, the patient receives a transplant, or the patient passes away.1
Saab et al studied the impact of oral antibiotic prophylaxis in patients with advanced liver disease on morbidity and mortality.5 The authors examined prospective, randomized, controlled trials that compared high-risk cirrhotic patients receiving oral antibiotic prophylaxis for SBP with groups receiving placebo or no intervention. Eight studies totaling 647 patients were included in the analysis.
The overall mortality rate for patients treated with SBP prophylaxis was 16%, compared with 25% for the control group. Groups treated with prophylactic antibiotics also had a lower incidence of all infections (6.2% vs. 22.2% in the control groups). Additionally, a survival benefit was seen at three months in the group that received prophylactic antibiotics.
The absolute risk reduction with prophylactic antibiotics for primary prevention of SBP was 8% with a number needed to treat of 13. The incidence of gastrointestinal (GI) bleeding, renal failure, and hepatic failure did not significantly differ between treatment and control groups. Thus, survival benefit is thought to be related to the reduced incidence of infections in the group receiving prophylactic antibiotics.5
History of GI Bleeding
The incidence of developing SBP in cirrhotics with an active GI bleed is anywhere from 20% to 45%.1,2 For those with ascites of any etiology and a GI bleed, the incidence can be as high as 60%.5 In general, bacterial infections are frequently diagnosed in patients with cirrhosis and GI bleeding, and have been documented in 22% of these patients within the first 48 hours after admission. According to several studies, that percentage can reach as high as 35% to 66% within seven to 14 days of admission.6 A seven-day course of antibiotics, or antibiotics until discharge, is generally acceptable for SBP prophylaxis in the setting of ascites and GI bleeding (see Table 2, right).1
Bernard et al performed a meta-analysis of five trials to assess the efficacy of antibiotic prophylaxis in the prevention of infections and effect on survival in patients with cirrhosis and GI bleeding. Out of 534 patients, 264 were treated with antibiotics between four and 10 days, and 270 did not receive any antibiotics.
The endpoints of the study were infection, bacteremia and/or SBP; incidence of SBP; and death. Antibiotic prophylaxis not only increased the mean survival rate by 9.1%, but also increased the mean percentage of patients free of infection (32% improvement); bacteremia and/or SBP (19% improvement); and SBP (7% improvement).7
Low Ascitic Fluid Protein
Of the three major risk factors for SBP, ascitic fluid protein content is the most debated. Guarner et al studied the risk of first community-acquired SBP in cirrhotics with low ascitic fluid protein.2 Patients were seen immediately after discharge from the hospital and at two- to three-month intervals. Of the 109 hospitalized patients, 23 (21%) developed SBP, nine of which developed SBP during their hospitalization. The one-year cumulative probability of SBP in these patients with low ascitic fluid protein levels was 35%.
During this study, the authors also looked at 20 different patient variables on admission and found that two parameters—high bilirubin (>3.2mg/dL) and low platelet count (<98,000 cells/ul)—were associated with an increased risk of SBP. This is consistent with studies showing that patients with higher Model for End-Stage Liver Disease (MELD) or Child-Pugh scores, indicating more severe liver disease, are at increased risk for SBP. This likely is the reason SBP prophylaxis is recommended for patients with an elevated bilirubin, and higher Child-Pugh scores, by the American Association for the Study of Liver Disease (see Table 2, p. 41).
Runyon et al showed that 15% of patients with low ascitic fluid protein developed SBP during their hospitalization, as compared with 2% of patients with ascitic fluid levels greater than 1 g/dl.8 A randomized, non-placebo-controlled trial by Navasa et al evaluating 70 cirrhotic patients with low ascitic ascitic protein levels showed a lower probability of developing SBP in the group placed on SBP prophylaxis with norfloxacin (5% vs. 31%).9 Six-month mortality rate was also lower (19% vs. 36%).
In contrast to the previous studies, Grothe et al found that the presence of SBP was not related to ascitic protein content.10 Given conflicting studies, controversy still remains on whether patients with low ascitic protein should receive long-term prophylactic antibiotics.
Antibiotic Drawbacks
The consensus in the literature is that patients with ascites who are admitted with a GI bleed, or those with a history of SBP, should be placed on SBP prophylaxis. However, patients placed on long-term antibiotics are at risk for developing bacterial resistance. Bacterial resistance in cultures taken from cirrhotic patients with SBP has increased over the last decade, particularly in gram-negative bacteria.5 Patients who receive antibiotics in the pre-transplant setting also are at risk for post-transplant fungal infections.
Additionally, the antibiotic of choice for SBP prophylaxis is typically a fluoroquinolone, which can be expensive. However, numerous studies have shown that the cost of initiating prophylactic therapy for SBP in patients with a prior episode of SBP can be cheaper than treating SBP after diagnosis.2
Back to the Case
Our patient’s paracentesis was negative for SBP. Additionally, he does not have a history of SBP, nor does he have an active GI bleed. His only possible indication for SBP prophylaxis is low ascitic protein concentration. His electrolytes were all within normal limits. Additionally, total bilirubin was only slightly elevated at 2.3 mg/dL.
Based on the American Association for the Study of Liver Diseases guidelines, the patient was not started on SBP prophylaxis. Additionally, given his history of medication noncompliance, there is concern that he might not take the antibiotics as prescribed, thus leading to the development of bacterial resistance and more serious infections in the future.
Bottom Line
Patients with ascites and a prior episode of SBP, and those admitted to the hospital for GI bleeding, should be placed on SBP prophylaxis. SBP prophylaxis for low protein ascitic fluid remains controversial but is recommended by the American Association for the Study of Liver Diseases. TH
Dr. del Pino Jones is a hospitalist at the University of Colorado Denver.
References
- Ghassemi S, Garcia-Tsao G. Prevention and treatment of infections in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21(1):77-93.
- Guarner C, Solà R, Soriono G, et al. Risk of a first community-acquired spontaneous bacterial peritonitis in cirrhotics with low ascitic fluid protein levels. Gastroenterology. 1999;117(2):414-419.
- Ginés P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology. 1990;12(4 Pt 1):716-724.
- Rolachon A, Cordier L, Bacq Y, et al. Ciprofloxacin and long-term prevention of spontaneous bacterial peritonitis: results of a prospective controlled trial. Hepatology. 1995;22(4 Pt 1):1171-1174.
- Saab S, Hernandez J, Chi AC, Tong MJ. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am J Gastroenterol. 2009;104(4):993-1001.
- Deschênes M, Villeneuve J. Risk factors for the development of bacterial infections in hospitalized patients with cirrhosis. Am J Gastroenterol. 1999;94(8):2193-2197.
- Bernard B, Grangé J, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29(6):1655-1661.
- Runyon B. Low-protein-concentration ascitic fluid is predisposed to spontaneous bacterial peritonitis. Gastroenterology. 1986;91(6):1343-1346.
- Navasa M, Fernandez J, Montoliu S, et al. Randomized, double-blind, placebo-controlled trial evaluating norfloxacin in the primary prophylaxis of spontaneous bacterial peritonitis in cirrhotics with renal impairment, hyponatremia or severe liver failure. J Hepatol. 2006;44(Supp2):S51.
- Grothe W, Lottere E, Fleig W. Factors predictive for spontaneous bacterial peritonitis (SBP) under routine inpatient conditions in patients with cirrhosis: a prospective multicenter trial. J Hepatol. 1990;34(4):547.
Case
A 54-year-old man with end-stage liver disease (ESLD) and no prior history of spontaneous bacterial peritonitis (SBP) presents with increasing shortness of breath and abdominal distention. He is admitted for worsening volume overload. The patient reveals that he has not been compliant with his diuretics. On the day of admission, a large-volume paracentesis is performed. Results are significant for a white blood cell count of 150 cells/mm3 and a total protein of 0.9 g/ul. The patient is started on furosemide and spironolactone, and his symptoms significantly improve throughout his hospitalization. His medications are reconciled on the day of discharge. He is not on any antibiotics for SBP prophylaxis; should he be? In general, which patients with ascites should receive SBP prophylaxis?
Overview
Spontaneous bacterial peritonitis is an infection of ascitic fluid that occurs in the absence of an indentified intra-abdominal source of infection or inflammation, i.e., perforation or abscess.1 It is diagnosed when the polymorphonuclear cell (PMN) count in the ascitic fluid is equal to or greater than 250 cells/mm3, with or without positive cultures.
SBP is a significant cause of morbidity and mortality in patients with cirrhosis, with the mortality rate approaching 20% to 40%.2 Of the 32% to 34% of cirrhotic patients who present with, or develop, a bacterial infection during their hospitalization, 25% are due to SBP.1 Changes in gut motility, mucosal defense, and microflora allow for translocation of bacteria into enteric lymph nodes and the bloodstream, resulting in seeding of the peritoneal fluid and SBP.1 Alterations in both systemic and localized immune defenses, both of which are reduced in patients with liver disease, also play a role in SBP pathogenesis (see Table 1, p. 41).
Current evidence supports the use of a third-generation cephalosporin or amoxicillin/clavulanate for initial treatment of SBP, as most infections are caused by gram-negative bacilli, in particular E. coli (see Table 2 on p. 41 and Table 3 on p. 42).1 Alternatively, an oral or intravenous fluoroquinolone could be used if the prevalence of fluoroquinolone-resistant organisms is low.1
Due to the frequency and morbidity associated with SBP, there is great interest in preventing it. However, the use of prophylactic antibiotics needs to be restricted to patients who are at highest risk of developing SBP. According to numerous studies, patients at high risk for SBP include:
- Patients with a prior SBP history;
- Patients admitted with a gastrointestinal bleed; and
- Patients with low total protein content in their ascitic fluid (defined as <1.5 g/ul).1
SBP History
Spontaneous bacterial peritonitis portends bad outcomes. The one-year mortality rate after an episode of SBP is 30% to 50%.1 Furthermore, patients who have recovered from a previous episode of SBP have a 70% chance of developing another episode of SBP in that year.1,2 In one study, norfloxacin was shown to decrease the one-year risk of SBP to 20% from 68% in patients with a history of SBP.3 Additionally, the likelihood of developing SBP from gram-negative bacilli was reduced to 3% from 60%. In order to be efficacious, norfloxacin must be given daily. When fluoroquinolones are prescribed less than once daily, there is a higher rate of fluoroquinolone resistant organisms in the stool.1
Though once-daily dosing of norfloxacin is recommended to decrease the promotion of resistant organisms in prophylaxis against SBP, ciprofloxacin once weekly is acceptable. In a group of patients with low ascitic protein content, with or without a history of SBP, weekly ciprofloxacin has been shown to decrease SBP incidence to 4% from 22% at six months.4 In regard to length of treatment, recommendations are to continue prophylactic antibiotics until resolution of ascites, the patient receives a transplant, or the patient passes away.1
Saab et al studied the impact of oral antibiotic prophylaxis in patients with advanced liver disease on morbidity and mortality.5 The authors examined prospective, randomized, controlled trials that compared high-risk cirrhotic patients receiving oral antibiotic prophylaxis for SBP with groups receiving placebo or no intervention. Eight studies totaling 647 patients were included in the analysis.
The overall mortality rate for patients treated with SBP prophylaxis was 16%, compared with 25% for the control group. Groups treated with prophylactic antibiotics also had a lower incidence of all infections (6.2% vs. 22.2% in the control groups). Additionally, a survival benefit was seen at three months in the group that received prophylactic antibiotics.
The absolute risk reduction with prophylactic antibiotics for primary prevention of SBP was 8% with a number needed to treat of 13. The incidence of gastrointestinal (GI) bleeding, renal failure, and hepatic failure did not significantly differ between treatment and control groups. Thus, survival benefit is thought to be related to the reduced incidence of infections in the group receiving prophylactic antibiotics.5
History of GI Bleeding
The incidence of developing SBP in cirrhotics with an active GI bleed is anywhere from 20% to 45%.1,2 For those with ascites of any etiology and a GI bleed, the incidence can be as high as 60%.5 In general, bacterial infections are frequently diagnosed in patients with cirrhosis and GI bleeding, and have been documented in 22% of these patients within the first 48 hours after admission. According to several studies, that percentage can reach as high as 35% to 66% within seven to 14 days of admission.6 A seven-day course of antibiotics, or antibiotics until discharge, is generally acceptable for SBP prophylaxis in the setting of ascites and GI bleeding (see Table 2, right).1
Bernard et al performed a meta-analysis of five trials to assess the efficacy of antibiotic prophylaxis in the prevention of infections and effect on survival in patients with cirrhosis and GI bleeding. Out of 534 patients, 264 were treated with antibiotics between four and 10 days, and 270 did not receive any antibiotics.
The endpoints of the study were infection, bacteremia and/or SBP; incidence of SBP; and death. Antibiotic prophylaxis not only increased the mean survival rate by 9.1%, but also increased the mean percentage of patients free of infection (32% improvement); bacteremia and/or SBP (19% improvement); and SBP (7% improvement).7
Low Ascitic Fluid Protein
Of the three major risk factors for SBP, ascitic fluid protein content is the most debated. Guarner et al studied the risk of first community-acquired SBP in cirrhotics with low ascitic fluid protein.2 Patients were seen immediately after discharge from the hospital and at two- to three-month intervals. Of the 109 hospitalized patients, 23 (21%) developed SBP, nine of which developed SBP during their hospitalization. The one-year cumulative probability of SBP in these patients with low ascitic fluid protein levels was 35%.
During this study, the authors also looked at 20 different patient variables on admission and found that two parameters—high bilirubin (>3.2mg/dL) and low platelet count (<98,000 cells/ul)—were associated with an increased risk of SBP. This is consistent with studies showing that patients with higher Model for End-Stage Liver Disease (MELD) or Child-Pugh scores, indicating more severe liver disease, are at increased risk for SBP. This likely is the reason SBP prophylaxis is recommended for patients with an elevated bilirubin, and higher Child-Pugh scores, by the American Association for the Study of Liver Disease (see Table 2, p. 41).
Runyon et al showed that 15% of patients with low ascitic fluid protein developed SBP during their hospitalization, as compared with 2% of patients with ascitic fluid levels greater than 1 g/dl.8 A randomized, non-placebo-controlled trial by Navasa et al evaluating 70 cirrhotic patients with low ascitic ascitic protein levels showed a lower probability of developing SBP in the group placed on SBP prophylaxis with norfloxacin (5% vs. 31%).9 Six-month mortality rate was also lower (19% vs. 36%).
In contrast to the previous studies, Grothe et al found that the presence of SBP was not related to ascitic protein content.10 Given conflicting studies, controversy still remains on whether patients with low ascitic protein should receive long-term prophylactic antibiotics.
Antibiotic Drawbacks
The consensus in the literature is that patients with ascites who are admitted with a GI bleed, or those with a history of SBP, should be placed on SBP prophylaxis. However, patients placed on long-term antibiotics are at risk for developing bacterial resistance. Bacterial resistance in cultures taken from cirrhotic patients with SBP has increased over the last decade, particularly in gram-negative bacteria.5 Patients who receive antibiotics in the pre-transplant setting also are at risk for post-transplant fungal infections.
Additionally, the antibiotic of choice for SBP prophylaxis is typically a fluoroquinolone, which can be expensive. However, numerous studies have shown that the cost of initiating prophylactic therapy for SBP in patients with a prior episode of SBP can be cheaper than treating SBP after diagnosis.2
Back to the Case
Our patient’s paracentesis was negative for SBP. Additionally, he does not have a history of SBP, nor does he have an active GI bleed. His only possible indication for SBP prophylaxis is low ascitic protein concentration. His electrolytes were all within normal limits. Additionally, total bilirubin was only slightly elevated at 2.3 mg/dL.
Based on the American Association for the Study of Liver Diseases guidelines, the patient was not started on SBP prophylaxis. Additionally, given his history of medication noncompliance, there is concern that he might not take the antibiotics as prescribed, thus leading to the development of bacterial resistance and more serious infections in the future.
Bottom Line
Patients with ascites and a prior episode of SBP, and those admitted to the hospital for GI bleeding, should be placed on SBP prophylaxis. SBP prophylaxis for low protein ascitic fluid remains controversial but is recommended by the American Association for the Study of Liver Diseases. TH
Dr. del Pino Jones is a hospitalist at the University of Colorado Denver.
References
- Ghassemi S, Garcia-Tsao G. Prevention and treatment of infections in patients with cirrhosis. Best Pract Res Clin Gastroenterol. 2007;21(1):77-93.
- Guarner C, Solà R, Soriono G, et al. Risk of a first community-acquired spontaneous bacterial peritonitis in cirrhotics with low ascitic fluid protein levels. Gastroenterology. 1999;117(2):414-419.
- Ginés P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology. 1990;12(4 Pt 1):716-724.
- Rolachon A, Cordier L, Bacq Y, et al. Ciprofloxacin and long-term prevention of spontaneous bacterial peritonitis: results of a prospective controlled trial. Hepatology. 1995;22(4 Pt 1):1171-1174.
- Saab S, Hernandez J, Chi AC, Tong MJ. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am J Gastroenterol. 2009;104(4):993-1001.
- Deschênes M, Villeneuve J. Risk factors for the development of bacterial infections in hospitalized patients with cirrhosis. Am J Gastroenterol. 1999;94(8):2193-2197.
- Bernard B, Grangé J, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29(6):1655-1661.
- Runyon B. Low-protein-concentration ascitic fluid is predisposed to spontaneous bacterial peritonitis. Gastroenterology. 1986;91(6):1343-1346.
- Navasa M, Fernandez J, Montoliu S, et al. Randomized, double-blind, placebo-controlled trial evaluating norfloxacin in the primary prophylaxis of spontaneous bacterial peritonitis in cirrhotics with renal impairment, hyponatremia or severe liver failure. J Hepatol. 2006;44(Supp2):S51.
- Grothe W, Lottere E, Fleig W. Factors predictive for spontaneous bacterial peritonitis (SBP) under routine inpatient conditions in patients with cirrhosis: a prospective multicenter trial. J Hepatol. 1990;34(4):547.
Mapping Out Diagnosis
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.