User login
A Painful Coincidence?
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
An 81-year-old woman with a remote history of left proximal femoral fracture (status post–open reduction and internal fixation) acutely developed severe pain in her left lateral thigh while at her home. A few days prior to her left thigh pain, the patient had routine blood work done. Her lab results (prior to the onset of her symptoms) revealed that her hemoglobin decreased from 10 g/dL, noted 9 months earlier, to 6.6 g/dL. Her primary care physician, who was planning to see the patient for her next regularly scheduled follow-up, was made aware of the patient’s decline in hemoglobin prior to the planned visit. The primary care physician called the patient to inform her about her concerning lab findings and coincidentally became aware of the acute, new-onset left thigh pain. The primary care physician requested that the patient be taken by her daughter to the emergency department (ED) for further evaluation.
The acute decrease in hemoglobin carries a broad differential and may or may not be related to the subsequent development of thigh pain. The presentation of an acute onset of pain in the thigh within the context of this patient’s age and gender suggests a femur fracture; this can be osteoporosis-related or a pathologic fracture associated with malignancy. Several malignancies are plausible, including multiple myeloma (given the anemia) or breast cancer. The proximal part of long bones is the most common site of pathologic fractures, and the femur accounts for half of these cases.
In the ED, she denied any recent trauma, hemoptysis, recent dark or bloody stools, vaginal bleeding, abdominal pain, or history of gastric ulcers. She had not experienced any similar episodes of thigh pain in the past. She had a history of atrial fibrillation, hypertension, diabetes mellitus type 2 with diabetic retinopathy and peripheral neuropathy, osteoporosis, nonalcoholic fatty liver disease (NAFLD), and internal hemorrhoids. Her medications included apixaban, metoprolol succinate, metformin, losartan, sitagliptin, calcium, vitamin D, alendronate, and fish oil. She had mild tenderness to palpation of her thigh, but her exam was otherwise normal. Radiography of the left hip and pelvis showed no acute fracture (Figure 1). An upper and lower endoscopy 3 years prior to her presentation revealed internal hemorrhoids.

The patient is taking apixaban, a direct factor Xa inhibitor. The absence of other obvious sources of bleeding suggests that the cause of anemia and pain is most likely bleeding into the anterior thigh compartment, exacerbated by the underlying anticoagulation. Since there was no trauma preceding this episode, the differential diagnosis must be expanded to include other, less common sources of bleeding, including a vascular anomaly such as a pseudoaneurysm or arteriovenous malformation. While the radiographs were normal, a CT scan or MRI may allow for identification of a fracture, other bone lesion, and/or hematoma.
A complete blood count revealed a hemoglobin of 6.6 g/dL (normal, 11.5-14.1 g/dL) with a mean corpuscular volume of 62 fL (normal, 79-96 fL). A CT scan of the abdomen and pelvis with intravenous contrast (Figure 2) was obtained to evaluate for intra-abdominal hemorrhage and retroperitoneal hematoma; it showed mild abdominal and pelvic ascites, a small right pleural effusion with compressive atelectasis, and generalized anasarca, but no evidence of bleeding. She was administered 2 units of packed red blood cells. Apixaban was held and 40 mg intravenous pantoprazole twice daily was started. Her iron level was 12 µg/dL (normal, 50-170

The studies reveal microcytic anemia associated with iron deficiency, as demonstrated by an elevated TIBC and very low ferritin. She also has a low-normal vitamin B12 level, which can contribute to poor red blood cell production; assessing methylmalonic acid levels would help to confirm whether true vitamin B12 deficiency is present. Anasarca can be secondary to severe hypoalbuminemia due to either protein-losing processes (eg, nephrotic syndrome, protein-losing enteropathy) or cirrhosis with poor synthetic function (given her history of NAFLD); it can also be secondary to severe heart failure or end-stage renal disease. The CT scan with contrast ruled out inferior vena cava thrombosis as a cause of ascites and did not reveal an obvious intra-abdominal malignancy as the cause of her anemia. Intestinal edema associated with anasarca can contribute to malabsorption (eg, iron, vitamin B12). The lack of abnormalities with respect to the liver and kidneys makes anasarca secondary to hepatic and renal dysfunction less likely.
The iron deficiency anemia prompted further evaluation for a gastrointestinal source of bleeding. Esophagogastroduodenoscopy showed a single, clean, 3-cm healing ulcer in the antrum, mild gastritis, and a superficial erosion in the duodenal bulb, all of which were biopsied. Because of inadequate bowel preparation, most of the colon was not optimally visualized and evaluation revealed only internal and external hemorrhoids in the rectum. On hospital day 4, the patient’s hemoglobin decreased from 9.6 g/dL to 7.3 g/dL. She had dark stools and also complained of left hip pain and swelling of the left knee and thigh. Another unit of packed red blood cells was given. A push enteroscopy and repeat colonoscopy showed no bleeding from the antral ulcer or from the internal and external hemorrhoids.
The patient has an antral ulcer, which most likely was a source of chronic blood loss and the underlying iron deficiency. However, the presence of healing and lack of signs of bleeding as demonstrated by negative repeat endoscopic studies suggests that the ulcer has little active contribution to the current anemia episode. A capsule enteroscopy could be performed, but most likely would be low yield. The presence of left thigh and knee swelling associated with worsening thigh pain raises the suspicion of a hemorrhagic process within the anterior thigh compartment, perhaps associated with an occult femoral fracture. A CT scan of the thigh would be valuable to identify a fracture or bone lesion as well as the presence of a hematoma. There are no widely available tests to evaluate apixaban anticoagulant activity; the anticoagulant effect would be expected to dissipate completely 36 to 48 hours after discontinuation in the context of normal renal function.
On hospital day 5, the patient’s left leg pain worsened. A physical exam showed edema of her entire left lower extremity with ecchymoses in several areas, including the left knee and lower thigh. A duplex ultrasound was negative for deep venous thrombosis, and X-ray of her left knee was normal. Her repeat hemoglobin was 8.8 g/dL. A repeat CT scan of the abdomen and pelvis again revealed no retroperitoneal bleeding. Orthopedic surgery was consulted on hospital day 7 and had low suspicion for compartment syndrome. Physical exam at that time showed mild swelling of the left thigh, moderate swelling of the left knee joint and pretibial area, two areas of ecchymosis on the left thigh, and diffuse ecchymosis of the left knee; all compartments were soft, and motor and nervous system functions were normal. A CT scan of the left lower extremity (Figure 3) revealed findings suspicious for hemorrhagic myositis with diffuse left thigh swelling with skin thickening and edema. There was no evidence of abscess, gas collection, foreign body, acute osteomyelitis, fracture, or dislocation. The patient’s hemoglobin remained stable.
 that raised suspicion for hemorrhagic myositis and diffuse cellulitis/edema")
Myopathies can be hereditary or acquired. Hereditary myopathies include congenital myopathies, muscular dystrophies, channelopathies, primary metabolic myopathies, and mitochondrial myopathies. Acquired myopathies include infectious myopathies, inflammatory myopathies, endocrine myopathies, secondary metabolic myopathies, and drug-induced and toxic myopathies. The findings of hemorrhagic myositis and skin edema are very intriguing, especially given their localized features. An overt femur fracture was previously ruled out, and an anterior thigh compartment syndrome was considered less likely after orthopedic surgery consultation. There is no description of the patient taking medications that could cause myopathy (such as statins), and there are also no clinical features suggestive of primary inflammatory myopathy, such as dermatomyositis. Increased suspicion of a focal inflammatory process such as localized scleroderma with regional inflammatory myopathy or another focal myopathy must be considered. The next diagnostic steps would include measuring the creatine kinase level, as well as obtaining an MRI of the leg to assess the nature and extent of the myopathy.
Multidisciplinary involvement, including hematology, rheumatology, and surgery, aided in narrowing the differential diagnosis. On hospital day 10, an MRI of the left thigh was performed for suspicion of diabetic myonecrosis (Figure 4). The MRI revealed a 10 cm × 3.6 cm × 22 cm intramuscular hematoma in the belly of the vastus lateralis muscle with associated soft tissue swelling, overlying subcutaneous edema, and skin thickening that was suggestive of hemorrhagic diabetic myonecrosis with some atypical features. A rheumatology consult was requested to evaluate for possible vasculitis in the left lower extremity, and vasculitis was not considered likely. The diagnosis of diabetic myonecrosis with associated intramuscular hemorrhage secondary to apixaban was made after careful reconsideration of the clinical presentation, imaging and laboratory data, and overall picture.
 encapsulated in the muscle belly of the vastus lateralis muscle")
DISCUSSION
A clear schema for approaching the patient with acute, nontraumatic myopathies is important in avoiding diagnostic error. One effective schema is to divide myopathy into infectious and noninfectious categories. Causes of infectious myopathy include bacterial infections (eg, pyomyositis), inflammatory damage to muscles associated with viruses (eg, influenza), as well as rarer causes. Bacterial processes tend to be relatively focal and affect a specific muscle group or anatomic compartment, while viral causes are often more diffuse and occur in the context of a systemic viral syndrome. Bacterial causes range in severity, and life-threatening conditions, such as necrotizing soft tissue infection, must be considered. In this case, bacterial causes were less likely given the patient’s lack of fever, leukocytosis, and systemic signs of infection.1,2 However, these findings are not uniformly sensitive, and clinicians should not exclude potentially life- or limb-threatening infections without thorough evaluation. For example, pyomyositis may present without fever in the subacute stage, without leukocytosis if the patient is immunocompromised, and without overt pus if the infection is not in the suppurative stage.3 Viral causes were made less likely in this patient given the lack of a current or recent systemic viral syndrome.
Once infectious etiologies are deemed unlikely, noninfectious etiologies for nontraumatic myopathies should be considered. Some causes of noninfectious myopathy present with the muscle symptoms as a predominant feature, while others present in the context of another illness such as cancer, metabolic disorders, or other systemic disorders. Many noninfectious causes of myopathy associated with systemic illnesses have diffuse or relatively diffuse symptoms, with pain and/or weakness in multiple muscle groups, often in a bilateral distribution. Such examples include dermatomyositis and polymyositis as well as myositis associated with other rheumatologic conditions. Nontraumatic rhabdomyolysis is diffuse and can occur in association with medications and/or genetic conditions.
Angervall and Stener4 first described diabetic myonecrosis in 1965 as
The mainstay of the diagnosis of diabetic myonecrosis is a thorough history and physical examination and imaging. Routine laboratory evaluation is relatively unhelpful in diagnosing diabetic myonecrosis, but appropriate imaging can provide valuable supportive information. A CT scan and MRI are both helpful in excluding other etiologies as well as identifying features consistent with diabetic myonecrosis. A CT scan can help exclude a localized abscess, tumor, or bone destruction and, in affected patients, may show increased subcutaneous attenuation and increased muscle size with decreased attenuation secondary to edema.2 However, a CT scan may not give optimal assessment of muscle tissue, and therefore MRI may need to be considered. MRI T2 images have a sensitivity nearing 90% for detecting myonecrosis.1 The diagnostic value of MRI often obviates the need for muscle biopsy.
Spontaneous infarction with hemorrhagic features seen on imaging can be explained by a combination of damage from atherosclerotic or microvascular disease, an activated coagulation cascade, and an impaired fibrinolytic pathway.8 Hemorrhagic conversion in diabetic myonecrosis appears to be uncommon.9 In our case, we suspect that it developed because of the combination of bleeding risk from apixaban and the underlying mechanisms of diabetic myonecrosis.
The treatment of diabetic myonecrosis is mainly supportive, with an emphasis on rest, nonsteroidal anti-inflammatory agents, antiplatelet agents, and strict glycemic control.10 There is conflicting information about the value of limb immobilization versus active physical therapy as appropriate treatment modalities.11 Patients who present with clinical concern for sepsis or compartment syndrome require consultation for consideration of acute surgical intervention.10 The short-term prognosis is promising with supportive therapy, but the condition may recur.12 The recurrence rate may be as high as 40%, with a 2-year mortality of 10%.13 Ultimately, patients need to be followed closely in the outpatient setting to reduce the risk of recurrence.
In this patient, the simultaneous occurrence of focal pain and acute blood loss anemia led to a diagnosis of diabetic myonecrosis that was complicated by hemorrhagic conversion, a truly painful coincidence. The patient underwent a thorough evaluation for acute blood loss before the diagnosis was ultimately made. Clinicians should consider diabetic myonecrosis in patients with diabetes who present with acute muscle pain but no evidence of infection.
Key Teaching Points
- Diabetic myonecrosis is an underrecognized entity and should be included in the differential diagnosis for patients with diabetes who present with acute muscle pain and no history of trauma.
- Imaging with CT and/or MRI of the affected region is the mainstay of diagnosis; treatment is predicated on severity and risk factors and can range from conservative therapy to operative intervention.
- Although the prognosis is good in these patients, careful outpatient follow-up is necessary to oversee their recovery to help reduce the risk of recurrence.
Acknowledgment
The authors thank Dr Vijay Singh for his radiology input on image selection for this manuscript.
1. Ivanov M, Asif B, Jaffe R. Don’t move a muscle: a case of diabetic myonecrosis. Am J Med. 2018;131(11):e445-e448. https://doi.org/10.1016/j.amjmed.2018.07.002
2. Morcuende JA, Dobbs MB, Crawford H, Buckwalter JA. Diabetic muscle infarction. Iowa Orthop J. 2000;20:65-74.
3. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21(3):473-494. https://doi.org/10.1128/CMR.00001-08
4. Angervall L, Stener B. Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1(1):39-42. https://doi.org/10.1007/BF01338714
5. Lawrence L, Tovar-Camargo O, Lansang MC, Makin V. Diabetic myonecrosis: a diagnostic and treatment challenge in longstanding diabetes. Case Rep Endocrinol. 2018;2018:1723695. https://doi.org/10.1155/2018/1723695
6. Horton WB, Taylor JS, Ragland TJ, Subauste AR. Diabetic muscle infarction: a systematic review. BMJ Open Diabetes Res Care. 2015;3(1):e000082. https://doi.org/10.1136/bmjdrc-2015-000082
7. Bhasin R, Ghobrial I. Diabetic myonecrosis: a diagnostic challenge in patients with long-standing diabetes. J Community Hosp Intern Med Perspect. 2013;3(1). https://doi.org/10.3402/jchimp.v3i1.20494
8. Bjornskov EK, Carry MR, Katz FH, Lefkowitz J, Ringel SP. Diabetic muscle infarction: a new perspective on pathogenesis and management. Neuromuscul Disord. 1995;5(1):39-45.
9. Cunningham J, Sharma R, Kirzner A, et al. Acute myonecrosis on MRI: etiologies in an oncological cohort and assessment of interobserver variability. Skeletal Radiol. 2016;45(8):1069-1078. https://doi.org/10.1007/s00256-016-2389-4
10. Khanna HK, Stevens AC. Diabetic myonecrosis: a rare complication of diabetes mellitus mimicking deep vein thrombosis. Am J Case Rep. 2017;18:38-41. https://doi.org/10.12659/ajcr.900903
11. Bunch TJ, Birskovich LM, Eiken PW. Diabetic myonecrosis in a previously healthy woman and review of a 25-year Mayo Clinic experience. Endocr Pract. 2002;8(5):343-346. https://doi.org/10.4158/EP.8.5.343
12. Mukherjee S, Aggarwal A, Rastogi A, et al. Spontaneous diabetic myonecrosis: report of four cases from a tertiary care institute. Endocrinol Diabetes Metab Case Rep. 2015;2015:150003. https://doi.org/10.1530/EDM-15-0003
13. Kapur S, McKendry RJ. Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11(1):8-12. https://doi.org/10.1097/01.rhu.0000152142.33358.f1
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
An 81-year-old woman with a remote history of left proximal femoral fracture (status post–open reduction and internal fixation) acutely developed severe pain in her left lateral thigh while at her home. A few days prior to her left thigh pain, the patient had routine blood work done. Her lab results (prior to the onset of her symptoms) revealed that her hemoglobin decreased from 10 g/dL, noted 9 months earlier, to 6.6 g/dL. Her primary care physician, who was planning to see the patient for her next regularly scheduled follow-up, was made aware of the patient’s decline in hemoglobin prior to the planned visit. The primary care physician called the patient to inform her about her concerning lab findings and coincidentally became aware of the acute, new-onset left thigh pain. The primary care physician requested that the patient be taken by her daughter to the emergency department (ED) for further evaluation.
The acute decrease in hemoglobin carries a broad differential and may or may not be related to the subsequent development of thigh pain. The presentation of an acute onset of pain in the thigh within the context of this patient’s age and gender suggests a femur fracture; this can be osteoporosis-related or a pathologic fracture associated with malignancy. Several malignancies are plausible, including multiple myeloma (given the anemia) or breast cancer. The proximal part of long bones is the most common site of pathologic fractures, and the femur accounts for half of these cases.
In the ED, she denied any recent trauma, hemoptysis, recent dark or bloody stools, vaginal bleeding, abdominal pain, or history of gastric ulcers. She had not experienced any similar episodes of thigh pain in the past. She had a history of atrial fibrillation, hypertension, diabetes mellitus type 2 with diabetic retinopathy and peripheral neuropathy, osteoporosis, nonalcoholic fatty liver disease (NAFLD), and internal hemorrhoids. Her medications included apixaban, metoprolol succinate, metformin, losartan, sitagliptin, calcium, vitamin D, alendronate, and fish oil. She had mild tenderness to palpation of her thigh, but her exam was otherwise normal. Radiography of the left hip and pelvis showed no acute fracture (Figure 1). An upper and lower endoscopy 3 years prior to her presentation revealed internal hemorrhoids.
The patient is taking apixaban, a direct factor Xa inhibitor. The absence of other obvious sources of bleeding suggests that the cause of anemia and pain is most likely bleeding into the anterior thigh compartment, exacerbated by the underlying anticoagulation. Since there was no trauma preceding this episode, the differential diagnosis must be expanded to include other, less common sources of bleeding, including a vascular anomaly such as a pseudoaneurysm or arteriovenous malformation. While the radiographs were normal, a CT scan or MRI may allow for identification of a fracture, other bone lesion, and/or hematoma.
A complete blood count revealed a hemoglobin of 6.6 g/dL (normal, 11.5-14.1 g/dL) with a mean corpuscular volume of 62 fL (normal, 79-96 fL). A CT scan of the abdomen and pelvis with intravenous contrast (Figure 2) was obtained to evaluate for intra-abdominal hemorrhage and retroperitoneal hematoma; it showed mild abdominal and pelvic ascites, a small right pleural effusion with compressive atelectasis, and generalized anasarca, but no evidence of bleeding. She was administered 2 units of packed red blood cells. Apixaban was held and 40 mg intravenous pantoprazole twice daily was started. Her iron level was 12 µg/dL (normal, 50-170
The studies reveal microcytic anemia associated with iron deficiency, as demonstrated by an elevated TIBC and very low ferritin. She also has a low-normal vitamin B12 level, which can contribute to poor red blood cell production; assessing methylmalonic acid levels would help to confirm whether true vitamin B12 deficiency is present. Anasarca can be secondary to severe hypoalbuminemia due to either protein-losing processes (eg, nephrotic syndrome, protein-losing enteropathy) or cirrhosis with poor synthetic function (given her history of NAFLD); it can also be secondary to severe heart failure or end-stage renal disease. The CT scan with contrast ruled out inferior vena cava thrombosis as a cause of ascites and did not reveal an obvious intra-abdominal malignancy as the cause of her anemia. Intestinal edema associated with anasarca can contribute to malabsorption (eg, iron, vitamin B12). The lack of abnormalities with respect to the liver and kidneys makes anasarca secondary to hepatic and renal dysfunction less likely.
The iron deficiency anemia prompted further evaluation for a gastrointestinal source of bleeding. Esophagogastroduodenoscopy showed a single, clean, 3-cm healing ulcer in the antrum, mild gastritis, and a superficial erosion in the duodenal bulb, all of which were biopsied. Because of inadequate bowel preparation, most of the colon was not optimally visualized and evaluation revealed only internal and external hemorrhoids in the rectum. On hospital day 4, the patient’s hemoglobin decreased from 9.6 g/dL to 7.3 g/dL. She had dark stools and also complained of left hip pain and swelling of the left knee and thigh. Another unit of packed red blood cells was given. A push enteroscopy and repeat colonoscopy showed no bleeding from the antral ulcer or from the internal and external hemorrhoids.
The patient has an antral ulcer, which most likely was a source of chronic blood loss and the underlying iron deficiency. However, the presence of healing and lack of signs of bleeding as demonstrated by negative repeat endoscopic studies suggests that the ulcer has little active contribution to the current anemia episode. A capsule enteroscopy could be performed, but most likely would be low yield. The presence of left thigh and knee swelling associated with worsening thigh pain raises the suspicion of a hemorrhagic process within the anterior thigh compartment, perhaps associated with an occult femoral fracture. A CT scan of the thigh would be valuable to identify a fracture or bone lesion as well as the presence of a hematoma. There are no widely available tests to evaluate apixaban anticoagulant activity; the anticoagulant effect would be expected to dissipate completely 36 to 48 hours after discontinuation in the context of normal renal function.
On hospital day 5, the patient’s left leg pain worsened. A physical exam showed edema of her entire left lower extremity with ecchymoses in several areas, including the left knee and lower thigh. A duplex ultrasound was negative for deep venous thrombosis, and X-ray of her left knee was normal. Her repeat hemoglobin was 8.8 g/dL. A repeat CT scan of the abdomen and pelvis again revealed no retroperitoneal bleeding. Orthopedic surgery was consulted on hospital day 7 and had low suspicion for compartment syndrome. Physical exam at that time showed mild swelling of the left thigh, moderate swelling of the left knee joint and pretibial area, two areas of ecchymosis on the left thigh, and diffuse ecchymosis of the left knee; all compartments were soft, and motor and nervous system functions were normal. A CT scan of the left lower extremity (Figure 3) revealed findings suspicious for hemorrhagic myositis with diffuse left thigh swelling with skin thickening and edema. There was no evidence of abscess, gas collection, foreign body, acute osteomyelitis, fracture, or dislocation. The patient’s hemoglobin remained stable.
Myopathies can be hereditary or acquired. Hereditary myopathies include congenital myopathies, muscular dystrophies, channelopathies, primary metabolic myopathies, and mitochondrial myopathies. Acquired myopathies include infectious myopathies, inflammatory myopathies, endocrine myopathies, secondary metabolic myopathies, and drug-induced and toxic myopathies. The findings of hemorrhagic myositis and skin edema are very intriguing, especially given their localized features. An overt femur fracture was previously ruled out, and an anterior thigh compartment syndrome was considered less likely after orthopedic surgery consultation. There is no description of the patient taking medications that could cause myopathy (such as statins), and there are also no clinical features suggestive of primary inflammatory myopathy, such as dermatomyositis. Increased suspicion of a focal inflammatory process such as localized scleroderma with regional inflammatory myopathy or another focal myopathy must be considered. The next diagnostic steps would include measuring the creatine kinase level, as well as obtaining an MRI of the leg to assess the nature and extent of the myopathy.
Multidisciplinary involvement, including hematology, rheumatology, and surgery, aided in narrowing the differential diagnosis. On hospital day 10, an MRI of the left thigh was performed for suspicion of diabetic myonecrosis (Figure 4). The MRI revealed a 10 cm × 3.6 cm × 22 cm intramuscular hematoma in the belly of the vastus lateralis muscle with associated soft tissue swelling, overlying subcutaneous edema, and skin thickening that was suggestive of hemorrhagic diabetic myonecrosis with some atypical features. A rheumatology consult was requested to evaluate for possible vasculitis in the left lower extremity, and vasculitis was not considered likely. The diagnosis of diabetic myonecrosis with associated intramuscular hemorrhage secondary to apixaban was made after careful reconsideration of the clinical presentation, imaging and laboratory data, and overall picture.
DISCUSSION
A clear schema for approaching the patient with acute, nontraumatic myopathies is important in avoiding diagnostic error. One effective schema is to divide myopathy into infectious and noninfectious categories. Causes of infectious myopathy include bacterial infections (eg, pyomyositis), inflammatory damage to muscles associated with viruses (eg, influenza), as well as rarer causes. Bacterial processes tend to be relatively focal and affect a specific muscle group or anatomic compartment, while viral causes are often more diffuse and occur in the context of a systemic viral syndrome. Bacterial causes range in severity, and life-threatening conditions, such as necrotizing soft tissue infection, must be considered. In this case, bacterial causes were less likely given the patient’s lack of fever, leukocytosis, and systemic signs of infection.1,2 However, these findings are not uniformly sensitive, and clinicians should not exclude potentially life- or limb-threatening infections without thorough evaluation. For example, pyomyositis may present without fever in the subacute stage, without leukocytosis if the patient is immunocompromised, and without overt pus if the infection is not in the suppurative stage.3 Viral causes were made less likely in this patient given the lack of a current or recent systemic viral syndrome.
Once infectious etiologies are deemed unlikely, noninfectious etiologies for nontraumatic myopathies should be considered. Some causes of noninfectious myopathy present with the muscle symptoms as a predominant feature, while others present in the context of another illness such as cancer, metabolic disorders, or other systemic disorders. Many noninfectious causes of myopathy associated with systemic illnesses have diffuse or relatively diffuse symptoms, with pain and/or weakness in multiple muscle groups, often in a bilateral distribution. Such examples include dermatomyositis and polymyositis as well as myositis associated with other rheumatologic conditions. Nontraumatic rhabdomyolysis is diffuse and can occur in association with medications and/or genetic conditions.
Angervall and Stener4 first described diabetic myonecrosis in 1965 as
The mainstay of the diagnosis of diabetic myonecrosis is a thorough history and physical examination and imaging. Routine laboratory evaluation is relatively unhelpful in diagnosing diabetic myonecrosis, but appropriate imaging can provide valuable supportive information. A CT scan and MRI are both helpful in excluding other etiologies as well as identifying features consistent with diabetic myonecrosis. A CT scan can help exclude a localized abscess, tumor, or bone destruction and, in affected patients, may show increased subcutaneous attenuation and increased muscle size with decreased attenuation secondary to edema.2 However, a CT scan may not give optimal assessment of muscle tissue, and therefore MRI may need to be considered. MRI T2 images have a sensitivity nearing 90% for detecting myonecrosis.1 The diagnostic value of MRI often obviates the need for muscle biopsy.
Spontaneous infarction with hemorrhagic features seen on imaging can be explained by a combination of damage from atherosclerotic or microvascular disease, an activated coagulation cascade, and an impaired fibrinolytic pathway.8 Hemorrhagic conversion in diabetic myonecrosis appears to be uncommon.9 In our case, we suspect that it developed because of the combination of bleeding risk from apixaban and the underlying mechanisms of diabetic myonecrosis.
The treatment of diabetic myonecrosis is mainly supportive, with an emphasis on rest, nonsteroidal anti-inflammatory agents, antiplatelet agents, and strict glycemic control.10 There is conflicting information about the value of limb immobilization versus active physical therapy as appropriate treatment modalities.11 Patients who present with clinical concern for sepsis or compartment syndrome require consultation for consideration of acute surgical intervention.10 The short-term prognosis is promising with supportive therapy, but the condition may recur.12 The recurrence rate may be as high as 40%, with a 2-year mortality of 10%.13 Ultimately, patients need to be followed closely in the outpatient setting to reduce the risk of recurrence.
In this patient, the simultaneous occurrence of focal pain and acute blood loss anemia led to a diagnosis of diabetic myonecrosis that was complicated by hemorrhagic conversion, a truly painful coincidence. The patient underwent a thorough evaluation for acute blood loss before the diagnosis was ultimately made. Clinicians should consider diabetic myonecrosis in patients with diabetes who present with acute muscle pain but no evidence of infection.
Key Teaching Points
- Diabetic myonecrosis is an underrecognized entity and should be included in the differential diagnosis for patients with diabetes who present with acute muscle pain and no history of trauma.
- Imaging with CT and/or MRI of the affected region is the mainstay of diagnosis; treatment is predicated on severity and risk factors and can range from conservative therapy to operative intervention.
- Although the prognosis is good in these patients, careful outpatient follow-up is necessary to oversee their recovery to help reduce the risk of recurrence.
Acknowledgment
The authors thank Dr Vijay Singh for his radiology input on image selection for this manuscript.
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
An 81-year-old woman with a remote history of left proximal femoral fracture (status post–open reduction and internal fixation) acutely developed severe pain in her left lateral thigh while at her home. A few days prior to her left thigh pain, the patient had routine blood work done. Her lab results (prior to the onset of her symptoms) revealed that her hemoglobin decreased from 10 g/dL, noted 9 months earlier, to 6.6 g/dL. Her primary care physician, who was planning to see the patient for her next regularly scheduled follow-up, was made aware of the patient’s decline in hemoglobin prior to the planned visit. The primary care physician called the patient to inform her about her concerning lab findings and coincidentally became aware of the acute, new-onset left thigh pain. The primary care physician requested that the patient be taken by her daughter to the emergency department (ED) for further evaluation.
The acute decrease in hemoglobin carries a broad differential and may or may not be related to the subsequent development of thigh pain. The presentation of an acute onset of pain in the thigh within the context of this patient’s age and gender suggests a femur fracture; this can be osteoporosis-related or a pathologic fracture associated with malignancy. Several malignancies are plausible, including multiple myeloma (given the anemia) or breast cancer. The proximal part of long bones is the most common site of pathologic fractures, and the femur accounts for half of these cases.
In the ED, she denied any recent trauma, hemoptysis, recent dark or bloody stools, vaginal bleeding, abdominal pain, or history of gastric ulcers. She had not experienced any similar episodes of thigh pain in the past. She had a history of atrial fibrillation, hypertension, diabetes mellitus type 2 with diabetic retinopathy and peripheral neuropathy, osteoporosis, nonalcoholic fatty liver disease (NAFLD), and internal hemorrhoids. Her medications included apixaban, metoprolol succinate, metformin, losartan, sitagliptin, calcium, vitamin D, alendronate, and fish oil. She had mild tenderness to palpation of her thigh, but her exam was otherwise normal. Radiography of the left hip and pelvis showed no acute fracture (Figure 1). An upper and lower endoscopy 3 years prior to her presentation revealed internal hemorrhoids.
The patient is taking apixaban, a direct factor Xa inhibitor. The absence of other obvious sources of bleeding suggests that the cause of anemia and pain is most likely bleeding into the anterior thigh compartment, exacerbated by the underlying anticoagulation. Since there was no trauma preceding this episode, the differential diagnosis must be expanded to include other, less common sources of bleeding, including a vascular anomaly such as a pseudoaneurysm or arteriovenous malformation. While the radiographs were normal, a CT scan or MRI may allow for identification of a fracture, other bone lesion, and/or hematoma.
A complete blood count revealed a hemoglobin of 6.6 g/dL (normal, 11.5-14.1 g/dL) with a mean corpuscular volume of 62 fL (normal, 79-96 fL). A CT scan of the abdomen and pelvis with intravenous contrast (Figure 2) was obtained to evaluate for intra-abdominal hemorrhage and retroperitoneal hematoma; it showed mild abdominal and pelvic ascites, a small right pleural effusion with compressive atelectasis, and generalized anasarca, but no evidence of bleeding. She was administered 2 units of packed red blood cells. Apixaban was held and 40 mg intravenous pantoprazole twice daily was started. Her iron level was 12 µg/dL (normal, 50-170
The studies reveal microcytic anemia associated with iron deficiency, as demonstrated by an elevated TIBC and very low ferritin. She also has a low-normal vitamin B12 level, which can contribute to poor red blood cell production; assessing methylmalonic acid levels would help to confirm whether true vitamin B12 deficiency is present. Anasarca can be secondary to severe hypoalbuminemia due to either protein-losing processes (eg, nephrotic syndrome, protein-losing enteropathy) or cirrhosis with poor synthetic function (given her history of NAFLD); it can also be secondary to severe heart failure or end-stage renal disease. The CT scan with contrast ruled out inferior vena cava thrombosis as a cause of ascites and did not reveal an obvious intra-abdominal malignancy as the cause of her anemia. Intestinal edema associated with anasarca can contribute to malabsorption (eg, iron, vitamin B12). The lack of abnormalities with respect to the liver and kidneys makes anasarca secondary to hepatic and renal dysfunction less likely.
The iron deficiency anemia prompted further evaluation for a gastrointestinal source of bleeding. Esophagogastroduodenoscopy showed a single, clean, 3-cm healing ulcer in the antrum, mild gastritis, and a superficial erosion in the duodenal bulb, all of which were biopsied. Because of inadequate bowel preparation, most of the colon was not optimally visualized and evaluation revealed only internal and external hemorrhoids in the rectum. On hospital day 4, the patient’s hemoglobin decreased from 9.6 g/dL to 7.3 g/dL. She had dark stools and also complained of left hip pain and swelling of the left knee and thigh. Another unit of packed red blood cells was given. A push enteroscopy and repeat colonoscopy showed no bleeding from the antral ulcer or from the internal and external hemorrhoids.
The patient has an antral ulcer, which most likely was a source of chronic blood loss and the underlying iron deficiency. However, the presence of healing and lack of signs of bleeding as demonstrated by negative repeat endoscopic studies suggests that the ulcer has little active contribution to the current anemia episode. A capsule enteroscopy could be performed, but most likely would be low yield. The presence of left thigh and knee swelling associated with worsening thigh pain raises the suspicion of a hemorrhagic process within the anterior thigh compartment, perhaps associated with an occult femoral fracture. A CT scan of the thigh would be valuable to identify a fracture or bone lesion as well as the presence of a hematoma. There are no widely available tests to evaluate apixaban anticoagulant activity; the anticoagulant effect would be expected to dissipate completely 36 to 48 hours after discontinuation in the context of normal renal function.
On hospital day 5, the patient’s left leg pain worsened. A physical exam showed edema of her entire left lower extremity with ecchymoses in several areas, including the left knee and lower thigh. A duplex ultrasound was negative for deep venous thrombosis, and X-ray of her left knee was normal. Her repeat hemoglobin was 8.8 g/dL. A repeat CT scan of the abdomen and pelvis again revealed no retroperitoneal bleeding. Orthopedic surgery was consulted on hospital day 7 and had low suspicion for compartment syndrome. Physical exam at that time showed mild swelling of the left thigh, moderate swelling of the left knee joint and pretibial area, two areas of ecchymosis on the left thigh, and diffuse ecchymosis of the left knee; all compartments were soft, and motor and nervous system functions were normal. A CT scan of the left lower extremity (Figure 3) revealed findings suspicious for hemorrhagic myositis with diffuse left thigh swelling with skin thickening and edema. There was no evidence of abscess, gas collection, foreign body, acute osteomyelitis, fracture, or dislocation. The patient’s hemoglobin remained stable.
Myopathies can be hereditary or acquired. Hereditary myopathies include congenital myopathies, muscular dystrophies, channelopathies, primary metabolic myopathies, and mitochondrial myopathies. Acquired myopathies include infectious myopathies, inflammatory myopathies, endocrine myopathies, secondary metabolic myopathies, and drug-induced and toxic myopathies. The findings of hemorrhagic myositis and skin edema are very intriguing, especially given their localized features. An overt femur fracture was previously ruled out, and an anterior thigh compartment syndrome was considered less likely after orthopedic surgery consultation. There is no description of the patient taking medications that could cause myopathy (such as statins), and there are also no clinical features suggestive of primary inflammatory myopathy, such as dermatomyositis. Increased suspicion of a focal inflammatory process such as localized scleroderma with regional inflammatory myopathy or another focal myopathy must be considered. The next diagnostic steps would include measuring the creatine kinase level, as well as obtaining an MRI of the leg to assess the nature and extent of the myopathy.
Multidisciplinary involvement, including hematology, rheumatology, and surgery, aided in narrowing the differential diagnosis. On hospital day 10, an MRI of the left thigh was performed for suspicion of diabetic myonecrosis (Figure 4). The MRI revealed a 10 cm × 3.6 cm × 22 cm intramuscular hematoma in the belly of the vastus lateralis muscle with associated soft tissue swelling, overlying subcutaneous edema, and skin thickening that was suggestive of hemorrhagic diabetic myonecrosis with some atypical features. A rheumatology consult was requested to evaluate for possible vasculitis in the left lower extremity, and vasculitis was not considered likely. The diagnosis of diabetic myonecrosis with associated intramuscular hemorrhage secondary to apixaban was made after careful reconsideration of the clinical presentation, imaging and laboratory data, and overall picture.
DISCUSSION
A clear schema for approaching the patient with acute, nontraumatic myopathies is important in avoiding diagnostic error. One effective schema is to divide myopathy into infectious and noninfectious categories. Causes of infectious myopathy include bacterial infections (eg, pyomyositis), inflammatory damage to muscles associated with viruses (eg, influenza), as well as rarer causes. Bacterial processes tend to be relatively focal and affect a specific muscle group or anatomic compartment, while viral causes are often more diffuse and occur in the context of a systemic viral syndrome. Bacterial causes range in severity, and life-threatening conditions, such as necrotizing soft tissue infection, must be considered. In this case, bacterial causes were less likely given the patient’s lack of fever, leukocytosis, and systemic signs of infection.1,2 However, these findings are not uniformly sensitive, and clinicians should not exclude potentially life- or limb-threatening infections without thorough evaluation. For example, pyomyositis may present without fever in the subacute stage, without leukocytosis if the patient is immunocompromised, and without overt pus if the infection is not in the suppurative stage.3 Viral causes were made less likely in this patient given the lack of a current or recent systemic viral syndrome.
Once infectious etiologies are deemed unlikely, noninfectious etiologies for nontraumatic myopathies should be considered. Some causes of noninfectious myopathy present with the muscle symptoms as a predominant feature, while others present in the context of another illness such as cancer, metabolic disorders, or other systemic disorders. Many noninfectious causes of myopathy associated with systemic illnesses have diffuse or relatively diffuse symptoms, with pain and/or weakness in multiple muscle groups, often in a bilateral distribution. Such examples include dermatomyositis and polymyositis as well as myositis associated with other rheumatologic conditions. Nontraumatic rhabdomyolysis is diffuse and can occur in association with medications and/or genetic conditions.
Angervall and Stener4 first described diabetic myonecrosis in 1965 as
The mainstay of the diagnosis of diabetic myonecrosis is a thorough history and physical examination and imaging. Routine laboratory evaluation is relatively unhelpful in diagnosing diabetic myonecrosis, but appropriate imaging can provide valuable supportive information. A CT scan and MRI are both helpful in excluding other etiologies as well as identifying features consistent with diabetic myonecrosis. A CT scan can help exclude a localized abscess, tumor, or bone destruction and, in affected patients, may show increased subcutaneous attenuation and increased muscle size with decreased attenuation secondary to edema.2 However, a CT scan may not give optimal assessment of muscle tissue, and therefore MRI may need to be considered. MRI T2 images have a sensitivity nearing 90% for detecting myonecrosis.1 The diagnostic value of MRI often obviates the need for muscle biopsy.
Spontaneous infarction with hemorrhagic features seen on imaging can be explained by a combination of damage from atherosclerotic or microvascular disease, an activated coagulation cascade, and an impaired fibrinolytic pathway.8 Hemorrhagic conversion in diabetic myonecrosis appears to be uncommon.9 In our case, we suspect that it developed because of the combination of bleeding risk from apixaban and the underlying mechanisms of diabetic myonecrosis.
The treatment of diabetic myonecrosis is mainly supportive, with an emphasis on rest, nonsteroidal anti-inflammatory agents, antiplatelet agents, and strict glycemic control.10 There is conflicting information about the value of limb immobilization versus active physical therapy as appropriate treatment modalities.11 Patients who present with clinical concern for sepsis or compartment syndrome require consultation for consideration of acute surgical intervention.10 The short-term prognosis is promising with supportive therapy, but the condition may recur.12 The recurrence rate may be as high as 40%, with a 2-year mortality of 10%.13 Ultimately, patients need to be followed closely in the outpatient setting to reduce the risk of recurrence.
In this patient, the simultaneous occurrence of focal pain and acute blood loss anemia led to a diagnosis of diabetic myonecrosis that was complicated by hemorrhagic conversion, a truly painful coincidence. The patient underwent a thorough evaluation for acute blood loss before the diagnosis was ultimately made. Clinicians should consider diabetic myonecrosis in patients with diabetes who present with acute muscle pain but no evidence of infection.
Key Teaching Points
- Diabetic myonecrosis is an underrecognized entity and should be included in the differential diagnosis for patients with diabetes who present with acute muscle pain and no history of trauma.
- Imaging with CT and/or MRI of the affected region is the mainstay of diagnosis; treatment is predicated on severity and risk factors and can range from conservative therapy to operative intervention.
- Although the prognosis is good in these patients, careful outpatient follow-up is necessary to oversee their recovery to help reduce the risk of recurrence.
Acknowledgment
The authors thank Dr Vijay Singh for his radiology input on image selection for this manuscript.
1. Ivanov M, Asif B, Jaffe R. Don’t move a muscle: a case of diabetic myonecrosis. Am J Med. 2018;131(11):e445-e448. https://doi.org/10.1016/j.amjmed.2018.07.002
2. Morcuende JA, Dobbs MB, Crawford H, Buckwalter JA. Diabetic muscle infarction. Iowa Orthop J. 2000;20:65-74.
3. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21(3):473-494. https://doi.org/10.1128/CMR.00001-08
4. Angervall L, Stener B. Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1(1):39-42. https://doi.org/10.1007/BF01338714
5. Lawrence L, Tovar-Camargo O, Lansang MC, Makin V. Diabetic myonecrosis: a diagnostic and treatment challenge in longstanding diabetes. Case Rep Endocrinol. 2018;2018:1723695. https://doi.org/10.1155/2018/1723695
6. Horton WB, Taylor JS, Ragland TJ, Subauste AR. Diabetic muscle infarction: a systematic review. BMJ Open Diabetes Res Care. 2015;3(1):e000082. https://doi.org/10.1136/bmjdrc-2015-000082
7. Bhasin R, Ghobrial I. Diabetic myonecrosis: a diagnostic challenge in patients with long-standing diabetes. J Community Hosp Intern Med Perspect. 2013;3(1). https://doi.org/10.3402/jchimp.v3i1.20494
8. Bjornskov EK, Carry MR, Katz FH, Lefkowitz J, Ringel SP. Diabetic muscle infarction: a new perspective on pathogenesis and management. Neuromuscul Disord. 1995;5(1):39-45.
9. Cunningham J, Sharma R, Kirzner A, et al. Acute myonecrosis on MRI: etiologies in an oncological cohort and assessment of interobserver variability. Skeletal Radiol. 2016;45(8):1069-1078. https://doi.org/10.1007/s00256-016-2389-4
10. Khanna HK, Stevens AC. Diabetic myonecrosis: a rare complication of diabetes mellitus mimicking deep vein thrombosis. Am J Case Rep. 2017;18:38-41. https://doi.org/10.12659/ajcr.900903
11. Bunch TJ, Birskovich LM, Eiken PW. Diabetic myonecrosis in a previously healthy woman and review of a 25-year Mayo Clinic experience. Endocr Pract. 2002;8(5):343-346. https://doi.org/10.4158/EP.8.5.343
12. Mukherjee S, Aggarwal A, Rastogi A, et al. Spontaneous diabetic myonecrosis: report of four cases from a tertiary care institute. Endocrinol Diabetes Metab Case Rep. 2015;2015:150003. https://doi.org/10.1530/EDM-15-0003
13. Kapur S, McKendry RJ. Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11(1):8-12. https://doi.org/10.1097/01.rhu.0000152142.33358.f1
1. Ivanov M, Asif B, Jaffe R. Don’t move a muscle: a case of diabetic myonecrosis. Am J Med. 2018;131(11):e445-e448. https://doi.org/10.1016/j.amjmed.2018.07.002
2. Morcuende JA, Dobbs MB, Crawford H, Buckwalter JA. Diabetic muscle infarction. Iowa Orthop J. 2000;20:65-74.
3. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21(3):473-494. https://doi.org/10.1128/CMR.00001-08
4. Angervall L, Stener B. Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1(1):39-42. https://doi.org/10.1007/BF01338714
5. Lawrence L, Tovar-Camargo O, Lansang MC, Makin V. Diabetic myonecrosis: a diagnostic and treatment challenge in longstanding diabetes. Case Rep Endocrinol. 2018;2018:1723695. https://doi.org/10.1155/2018/1723695
6. Horton WB, Taylor JS, Ragland TJ, Subauste AR. Diabetic muscle infarction: a systematic review. BMJ Open Diabetes Res Care. 2015;3(1):e000082. https://doi.org/10.1136/bmjdrc-2015-000082
7. Bhasin R, Ghobrial I. Diabetic myonecrosis: a diagnostic challenge in patients with long-standing diabetes. J Community Hosp Intern Med Perspect. 2013;3(1). https://doi.org/10.3402/jchimp.v3i1.20494
8. Bjornskov EK, Carry MR, Katz FH, Lefkowitz J, Ringel SP. Diabetic muscle infarction: a new perspective on pathogenesis and management. Neuromuscul Disord. 1995;5(1):39-45.
9. Cunningham J, Sharma R, Kirzner A, et al. Acute myonecrosis on MRI: etiologies in an oncological cohort and assessment of interobserver variability. Skeletal Radiol. 2016;45(8):1069-1078. https://doi.org/10.1007/s00256-016-2389-4
10. Khanna HK, Stevens AC. Diabetic myonecrosis: a rare complication of diabetes mellitus mimicking deep vein thrombosis. Am J Case Rep. 2017;18:38-41. https://doi.org/10.12659/ajcr.900903
11. Bunch TJ, Birskovich LM, Eiken PW. Diabetic myonecrosis in a previously healthy woman and review of a 25-year Mayo Clinic experience. Endocr Pract. 2002;8(5):343-346. https://doi.org/10.4158/EP.8.5.343
12. Mukherjee S, Aggarwal A, Rastogi A, et al. Spontaneous diabetic myonecrosis: report of four cases from a tertiary care institute. Endocrinol Diabetes Metab Case Rep. 2015;2015:150003. https://doi.org/10.1530/EDM-15-0003
13. Kapur S, McKendry RJ. Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11(1):8-12. https://doi.org/10.1097/01.rhu.0000152142.33358.f1
© 2021 Society of Hospital Medicine
When the dissociation curve shifts to the left
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.

Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
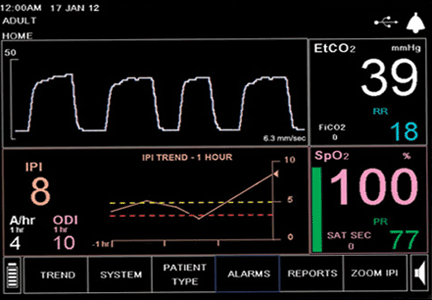
Recognizing, managing medical consequences of eating disorders in primary care
Eating disorders are debilitating biopsychosocial illnesses associated with serious medical illness and a high risk of death.1
Primary care physicians are often the first to see young women who have these problems, diagnose them, and start their evaluation and treatment.2–4 Many patients require acute medical interventions as well as long-term care for chronic medical issues. Therefore, primary care physicians play essential front-line and long-term roles in the multidisciplinary treatment team.
DEFINITIONS OF EATING DISORDERS HAVE CHANGED
Several problems existed in the category of eating disorders in the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-4) and in the DSM-4 Text Revision (DSM-4-TR). These problems have been addressed in the fifth edition (DSM-5), released in 2013.5
One problem in the earlier editions was that many patients referred for treatment of eating disorders—more than 50% in one study6—did not meet the criteria for anorexia nervosa or bulimia nervosa and thus had to be categorized as having “eating disorder not otherwise specified.” Further, the earlier editions did not recognize that young children and adolescent males can be affected.7
Eating disorders are now recognized as an equal-opportunity disease, with all ethnic and socioeconomic groups affected. Children can run into medical trouble with even a small amount of weight loss or falling off the growth curve. Moreover, children and adolescents do not “experience” their bodies in the same way adults do; they may lack the vocabulary for eating-disorder thoughts.
For these reasons, the definitions of eating disorders have changed in the DSM-5.5
Anorexia nervosa. Older editions of the DSM listed amenorrhea as a criterion. This has been eliminated in DSM-5, since amenorrhea does not necessarily predict medical risk or treatment outcome; also, it is not applicable to males or premenorrheal girls and postmenopausal women.8 In addition, the requirement of low weight is now defined in the context of “age, sex, developmental trajectory, and physical health,” rather than the old threshold of 85% of expected weight.9
What remains unchanged is that anorexia nervosa is still characterized by self-starvation in order to maintain an abnormally low body weight, along with an intense fear of being fat and a disturbed self-image.
Bulimia nervosa. In both the old and the new editions of the DSM, bulimia nervosa is characterized by episodes of binge eating followed by inappropriate compensatory behaviors to avoid weight gain, such as vomiting, laxative abuse, diuretic abuse, and overexercise. In DSM-5, bulimia nervosa no longer has subtypes and requires only one binge per week with compensatory behavior, for at least 3 months. This change was based on the finding that there is no clear difference in psychopathology or treatment outcome between patients with one and two binge-purge episodes a week.10
“Eating disorder not otherwise specified” was a wastebasket category, lumping all those who did not meet the criteria for anorexia nervosa or bulimia nervosa or who did not neatly fit into a specific category.10 In DSM-5, subcategories were designed to help distinguish different treatment needs and outcomes between various subtypes.
Binge-eating disorder, one of the new subcategories, is characterized by binge eating without inappropriate compensatory behaviors.9 Patients with binge-eating disorder are often obese, have greater functional impairment, and are more likely to develop components of metabolic syndrome than obese patients without eating disorders.11
Avoidant/restrictive food intake disorder is another new DSM-5 diagnosis, characterized by failure to meet nutritional needs for reasons other than weight control. Reasons include disinterest in eating, dislike of sensory characteristics of food, or avoidance of consequences of eating. This disorder replaces the category “feeding disorder of infancy or early childhood,” since the condition can also occur in adolescents and adults.12
Other new diagnoses are:
- Atypical anorexia nervosa (if the patient is not underweight)
- Purging disorder
- Subthreshold bulimia nervosa (if the patient has < 1 episode per week or has had them for < 3 months)
- Subthreshold binge eating disorder (< 1 time a week or < 3 months)
- Night eating syndrome
- Pica and rumination disorder.
Regardless of the diagnostic label, the medical evaluation and treatment of anyone with an eating disorder should be tailored to the specific behaviors of the eating disorder. Medical complications can be subdivided into those from starvation, from purging, and from refeeding.
MEDICAL COMPLICATIONS OF STARVATION
Cardiovascular effects of starvation
Malnutrition and starvation have multiple adverse effects on the heart.
Electrophysiologic effects. Sinus bradycardia (< 60 bpm) and hypotension are common cardiac manifestations of starvation.13 Bradycardia has been attributed to an adaptive increase in parasympathetic vagal tone.14 QTc prolongation is also seen in patients with malnutrition.15

Together, these electrocardiographic abnormalities predispose the patient to ventricular arrhythmia and sudden cardiac death.16 The risk of ventricular arrhythmia is particularly relevant when treating psychiatric symptoms, since antipsychotics and tricyclic antidepressants are among several drug classes that can cause further QTc prolongation (Table 1).17,18
In patients with QTc prolongation, bradycardia, or both, the standard of care involves acute hospitalization for refeeding using continuous telemetric monitoring until normal rhythm is restored and the heart rate is above 40 at night and 50 by day.4,19
Structural changes. Starvation also causes structural changes in the heart. Loss of lean body mass can reduce cardiac muscle mass, compromise cardiac output, and lead to mitral valve prolapse.20 These changes are fully reversible with restored nutrition and regaining of heart mass.21,22
Effects of starvation on the brain
Starvation can affect brain structure and cognitive function. Undernourished patients have reduced volumes of white and gray matter, a change that can occur within months. Cortical volumes may increase with weight gain, but a reduction in gray matter volume may not be completely reversible.23
Furthermore, starvation impairs cognitive functions that are needed to stop eating-disorder behaviors; namely, decision-making, emotional control, regulation of appetite, and reward path-ways. Therefore, undernourished patients may not have sufficient insight into the disease to be able to make the best choices for recovery. This finding lends support for using the Maudsley method in adolescents, in which parents take control of their child’s eating until the child can maintain a healthy weight.24
Gastrointestinal consequences of starvation
Patients with malnutrition have prolonged gastric emptying and colonic transit time with solid foods.25 They often complain of early satiety, abdominal pain, bloating, and constipation, all symptoms that complicate the refeeding process. A prokinetic such as metoclopramide (Reglan), given 1 hour before meals and at bedtime, may provide some relief from gastrointestinal symptoms.26
Patients may also experience transient lactose or fructose intolerance after prolonged starvation. Taking a lactase supplement (eg, Lactaid 1–10 tabs) before consuming dairy products and dextrose (contained in candies such as Smarties) before eating fruit or fructose-containing foods can sometimes partially relieve symptoms. In general, gastrointestinal function returns over time as nutritional status improves.
Patients with severe or prolonged starvation can develop steatosis accompanied by elevated levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). In reports of starvation-induced steatosis, liver enzyme levels rapidly normalize with nutritional rehabilitation.27
Endocrine consequences of starvation
Amenorrhea. Dysregulation of the hypothalamic-pituitary-gonadal axis is a major endocrine complication of nutritional in-sufficiency. Weight loss disrupts the normal pulsatile secretion of gonadotropin-releasing hormone, reduces secretion of luteinizing hormone and follicle-stimulating hormone, and decreases estrogen levels.28 Leptin deficiency likely plays a role in suppressing gonadotropin secretion with subsequent development of amenorrhea. With weight gain, levels of leptin and gonadotropins normalize and menstruation eventually returns.29,30
Hypothyroidism. Starvation can also lead to dysregulation of the hypothalamic-pituitary-thyroid axis. Typically, the concentration of triiodothyronine (T3) is reduced, the ratio of thyroxine (T4) to T3 is elevated, and thyroid-stimulating hormone (TSH) is close to or within the normal range, creating a euthyroid sick syndrome. In eating disorders, this thyroid disturbance is a result of starvation and resolves with weight restoration. Therefore, thyroid hormone replacement therapy is not medically indicated.28
Osteoporosis. Amenorrhea resulting from low estrogen levels in undernourished patients can raise the risk of osteoporosis and fractures, particularly in patients with a low body mass index. Osteopenia results from a negative balance between bone deposition and resorption.
Lack of bone deposition can be especially problematic when disordered eating occurs during peak bone mass development, ie, ages 11 to 14 for girls, and ages 15 to 17 for boys.31,32 Even a 5% to 10% decrease in bone deposition can result in significant risk of osteopenia.33 However, after age 30, bone resorption is a greater contributor.34
Does hormone therapy correct bone loss? Given the association between estrogen deficiency and bone loss, estrogen supplementation was expected to be an effective treatment for bone loss in patients with eating disorders.35 Also, the restoration of menses through hormone replacement may give underweight patients a false sense of achieving a “healthy” weight.36
Golden et al37 prospectively studied 50 adolescents and found no significant difference in bone mineral density at 1 year of follow-up between patients treated with estrogen and those who received only standard nutritional therapy. However, increased bone mineral density was achieved in adolescents with anorexia nervosa treated with transdermally administered estrogen dosed to mimic physiologic pubertal levels.38
Klibanski et al39 found that hormone therapy resulted in a 4% gain in bone density in an extremely low-weight subset of women with anorexia nervosa (< 70% of ideal body weight), whereas similar patients in the control group lost 20%. However, in all groups, only weight gain correlated with bone gain in women who were within 70% of their ideal body weight.
Divasta et al40 evaluated 60 girls and women ages 13 to 27 with anorexia nervosa, randomized to receive either placebo or dehydroepiandrosterone combined with an estrogen-progestin oral contraceptive, and followed for 18 months. As in the study by Klibanski et al,39 bone loss was prevented in the treatment group, but significant bone gain occurred only in the context of weight gain.
The bottom line is that only weight gain has resulted in significant increases in bone density in patients with anorexia nervosa, and hormone therapy without weight gain has not been shown to increase bone density effectively in this population. Although calcium and vitamin D in oral therapeutic doses through foods or through supplementation are required for bone gain, the combination is not enough to augment bone density in the absence of weight gain.37 Although not curative, weight gain is currently the best option for treating bone loss, and no single pharmacologic treatment is effective.
COMPLICATIONS OF PURGING
Oral complications of purging
Patients who purge by vomiting are at risk of complications from exposure of the esophagus, pharynx, and mouth to acidic gastric contents.
Dental problems. Over time, contact with gastric acid wears down enamel on the lingual and occlusal surfaces of teeth, resulting in dental caries and periodontal disease. Until they can give up purging, patients should be instructed to rinse with mouthwash or water immediately after vomiting to reduce the acidity in the mouth.41,42 We recommend that patients not brush their teeth after vomiting, because brushing can deliver acid to otherwise unreachable surfaces and thus worsen tooth erosion. For patients who are determined to brush after vomiting, a bicarbonate toothpaste might mitigate harm.42
Sialadenosis (hypertrophy of the salivary glands) is another consequence of repeated vomiting, with elevated salivary amylase. Both the size of the glands and the salivary amylase level generally normalize on their own after vomiting is stopped, but parotitis can take up to a year to resolve. Similar to smoker’s cough, parotitis may acutely worsen when the patient abruptly stops vomiting and may worsen before it improves.
To reduce discomfort, patients can use hot compresses or sugarless hard candies.44 However, the latter should not be substituted as a chronic habit in a patient with disordered eating. Patients need to be reassured that the swelling is not permanent, since they often interpret it as having fat cheeks (the “chipmunk sign”).
Hypokalemia, metabolic alkalosis, renal dysfunction
Chronic vomiting can cause electrolyte and acid-base imbalances, the most worrisome of which is hypokalemia. With repeated vomiting, loss of potassium and gastric acid causes metabolic alkalosis with hypokalemia, hypochloremia, and hypomagnesemia. Loss of water and the resultant volume contraction activates the renin-angiotensin-aldosterone system, and elevated aldosterone further decreases serum potassium.
In patients with eating disorders, who often have other factors contributing to electrolyte imbalance, vomiting-induced hypokalemia heightens the risk of cardiac arrhythmias.43
Hypokalemia can also cause rhabdomyolysis and kidney damage.41,43 Prolonged hypokalemia and reduced kidney perfusion in the setting of volume depletion causes acute kidney injury and impaired concentrating ability of the renal tubules. Hypovolemia can cause prerenal azotemia and increases the risk for nephrolithiasis and nephrocalcinosis.44,45
When a patient stops vomiting, elevated aldosterone from prior hypovolemia results in water retention and can manifest in significant edema associated with hypochloremic alkalosis. This condition, known as pseudo-Bartter syndrome, usually resolves without treatment. In the meantime, salt restriction and leg elevation can help reduce edema.26
Laxative abuse: A mode of purging
Many patients with eating disorders abuse laxatives to lose weight or to prevent weight gain. Believing that laxatives will prevent calorie absorption, patients commonly take them to compensate for caloric intake (eg, during a binge episode). The immediate weight loss, albeit artificial, is highly reinforcing for an eating-disorder patient. In some cases, patients with eating disorders also abuse laxatives to self-treat the constipation that results from chronic starvation.46
Over time, tolerance to laxatives develops, and patients use increasingly larger doses. This can lead to activation of the renin-angiotensin-aldosterone system.47 Patients interpret the resultant edema as true weight gain and again take laxatives to get rid of it. If laxatives are stopped abruptly, the patient may need inpatient and outpatient support for the resultant fluid shifts.
Gastrointestinal complications of laxative abuse include reflex hypofunction of the bowel, malabsorption, steatorrhea, and gastrointestinal bleeding.47 Reflex hypofunction during laxative withdrawal is a consequence of the bowel becoming tolerant of laxatives.48 Cathartic colon syndrome is a rare complication characterized by loss of the normal haustral markings and slowed or absent peristalsis in segments of the colon.49
Systemically, the major risk of laxative abuse relates to electrolyte and acid-base imbalance. Loss of potassium and water in the stool can cause hypokalemia and metabolic alkalosis.48 The disturbances caused by laxative abuse are similar to those caused by vomiting and diuretic use and have the same treatment.
The most important component of treating laxative abuse is giving patients realistic expectations to help them tolerate temporary discomfort and to help manage the edema and fluid shifts that can happen acutely with shifting of fluid into the intracellular space. In extreme cases, this may need to be managed in the hospital. To help relieve the initial anxiety, doctors should emphasize that any bloating the patient experiences is not true weight gain and will go away within a few days to weeks. In addition, explaining that laxatives reduce nutrient absorption only minimally may lessen the temptation to resume taking them.48
Diuretic abuse: Another form of purging
Diuretic abuse is yet another mode of purging, with its own set of medical complications. Like laxatives, diuretics are not effective weight-loss agents, and the weight reduction they cause is only temporary.
As with vomiting, there is a compensatory activation of the renin-angiotensin-aldosterone system, and therefore subsequent fluid intake will lead to water retention, which encourages further diuretic use.41 Diuretics can also contribute to hypokalemia, hypomagnesemia, hypochloremia, and metabolic alkalosis.
Ipecac abuse can lead to heart failure
Ipecac syrup has long been used to induce vomiting, but this practice has become much less common since ipecac has become harder to obtain in the United States.50 The emetine base contained in ipecac binds irreversibly to cardiac and skeletal muscle. With continued use, irreversible cardiomyopathy develops and can lead to heart failure. Treatment should include supportive care and immediate cessation of ipecac use.
Diabetic patients may skip insulin to lose weight
Patients with diabetes, especially those with type 1 that begins in childhood, are at greater risk of eating disorders over time.51 They may withhold insulin to lose weight, a practice referred to in the nonmedical literature as “diabulimia,” and they seem particularly more likely to develop bulimia nervosa than those without diabetes.52
The medical prognosis is poor for patients with diabetes who develop eating disorders and do not receive intensive treatment.51 In addition, if a diabetic patient on an insulin pump becomes depressed in addition to having an eating disorder, careful monitoring for suicidal thoughts and a rapid follow-up with mental health services are in order.
REFEEDING SYNDROME
When refeeding is started, a high glucose load stimulates insulin secretion, resulting in cellular uptake of phosphorus along with potassium, magnesium, and glucose. In addition, total body phosphorus is depleted by the increased demand for adenosine triphosphate and 2,3-diphosphoglycerate for cellular metabolism.
When liver enzyme levels increase, the astute clinician will closely monitor the patient for evidence of refeeding syndrome. In a child, adolescent, or young adult, the standard of care is inpatient monitoring for acute stabilization.4,19
Hypophosphatemia is the hallmark of refeeding syndrome, although hypomagnesemia, hypokalemia, and hypoglycemia can also occur.53 In addition, sodium and water retention can lead to fluid overload, with shifting of fluid into the intracellular space, resulting in dependent edema.
Cardiovascular complications are the most worrisome manifestations of refeeding syndrome. Electrolyte shifts and increased fluid volume can cause arrhythmias and heart failure. Furthermore, severely undernourished patients may have reduced myocardial mass as well as electrocardiographic abnormalities associated with starvation, which further increase their vulnerability to electrolyte shifts and fluid retention during refeeding.15
Other manifestations of refeeding syndrome include delirium, seizures, rhabdomyolysis, and respiratory failure. In the most extreme cases, refeeding syndrome causes sudden death.53

Fortunately, refeeding syndrome is easily preventable and treatable when recognized early. Electrolytes and cardiovascular and renal function must be carefully monitored, especially during the first week of nutritional restoration.53 In patients with extremely low body mass (< 70% of ideal body weight) or with precipitous weight loss, close monitoring of the complete metabolic panel including electrolytes, AST, ALT, calcium, magnesium, and phosphorus may be required to detect changes that can affect cardiac status. Specific suggestions for refeeding are discussed below and in Table 2.45
ACUTE CARE OF PATIENTS WITH EATING DISORDERS
Refeeding in the inpatient setting

The decision to hospitalize an eating-disorder patient is based on the current or potential risk of serious medical complications and the likelihood of success at home. Medical criteria for hospital admission are outlined in Table 3.4,54
In refeeding undernourished patients, the challenge is to maximize weight gain while preventing refeeding syndrome. Undernourished patients are generally hypometabolic at baseline but become hypermetabolic once refeeding begins.
How many calories should refeeding start with? The traditional principle of “start low and go slow” has been recently challenged.55 Starting at 1,200 kcal/day or less in the typical patient can result in failure to gain weight or even in weight loss in the first week of refeeding.56 The goal is to achieve a weight gain of 0.2 kg/day while the patient is in the hospital. Thus, we start higher, and to date we have seen no cases of life-threatening refeeding syndrome. In all patients who need hospitalization or who are beginning the refeeding process as outpatients, caloric intake should be started at 1,500 to 2,000 kcal/day.45,57 However, for exceptionally low-weight patients, intake may be started lower.
In Australia, patients are started at 1,900 kcal/day.56 All patients in one program there receive nasogastric feeding initially in an intensive care unit and then are moved to a regular nursing floor where they graduate to full oral feeding as they improve cardiovascularly and behaviorally. In the United States, some programs use nasogastric feeding at night for caloric restoration; our program and others use nasogastric feeding as a behavioral modification strategy for patients who refuse food or supplements by mouth.
Phosphorus supplementation. Many centers give phosphorus supplements preventively. In our center, we give potassium phosphate (Neutra-Phos) 500 mg orally twice daily for 5 days, and we have seen no life-threatening cases of refeeding syndrome with that regimen. Other centers give phosphorus supplements in a dose of 250 mg orally twice a day for 5 days, while still others only supplement phosphorus reactively once a deficit has been identified. The latter method requires daily blood draws for monitoring and is reactive rather than proactive. Further studies can help clarify the optimal dosing and timing of phosphorus supplementation.
Managing fluid balance. Fluid-loading these patients may tip them over the edge into refeeding syndrome. Except in cases of shock, patients with eating disorders should not be given intravenous fluids, as it is safer to rehydrate and feed them orally. Electrolyte imbalances can be corrected orally with no need for intravenous supplementation. To avoid fluid overload, fluids can be started at 1,500 mL to 2,000 mL per day, with strict monitoring of intake and output. Fluids are liberalized if ALT and AST levels remain normal and to gradually correct orthostatic hypotension; caloric fluids are ideal to help address energy needs and improve bradycardia.
Laboratory monitoring. On admission, a urinalysis, complete blood cell count, complete metabolic panel, TSH, erythrocyte sedimentation rate, serum magnesium, and phosphorus should be obtained.26 In addition, continuous electrocardiographic recording should begin on admission.45 Inpatient use of a telemetry bed helps identify extreme tachycardia with arrhythmia, as well as profound bradycardia.45,56
Some protocols call for daily laboratory monitoring, although that degree of testing is less cost-effective. If initial results are normal, clinical judgment can be used on when to repeat laboratory evaluation. For instance, patients with edema require repeat complete metabolic panels to assess for elevated ALT and AST, electrolyte imbalances, and other abnormalities.
Signs of refeeding syndrome include tachycardia, hepatosplenomegaly, peripheral edema, altered mental status, and electrolyte disturbances, specifically, acute or severe hypophosphatemia or hypokalemia.26,45 If refeeding syndrome is suspected, the rate of caloric intake should be reduced or not advanced, fluid intake should be urgently reassessed for volume overload, and supportive care with close monitoring should be provided.
KNOWLEDGE SAVES LIVES
Eating disorders can lead to potentially life-threatening medical complications that require attentive care by the primary care clinician and subspecialist. Without thoughtful consideration, it is easy for even a caring medical team to unintentionally enable patients with these illnesses or to cause active harm in the case of underrecognized pathology.58
Acute medical stabilization on an inpatient unit trained to recognize pathology and treat sequelae can be lifesaving. Arming patients and families with medical knowledge, as provided in the Academy for Eating Disorders’ brochure, “Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders”59 can help save patients’ lives.
- Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry 2011; 68:724–731.
- Walsh JM, Wheat ME, Freund K. Detection, evaluation, and treatment of eating disorders the role of the primary care physician. J Gen Intern Med 2000; 15:577–590.
- American Academy of Pediatrics; Committee on Adolescence. Identifying and treating eating disorders. Pediatrics 2003; 111:204–211.
- Rosen DS; American Academy of Pediatrics Committee on Adolescence. Identification and management of eating disorders in children and adolescents. Pediatrics 2010; 126:1240–1253.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington, VA: American Psychiatric Publishing, Incorporated; 2013.
- Eddy KT, Celio Doyle A, Hoste RR, Herzog DB, le Grange D. Eating disorder not otherwise specified in adolescents. J Am Acad Child Adolesc Psychiatry 2008; 47:156–164.
- Muise AM, Stein DG, Arbess G. Eating disorders in adolescent boys: a review of the adolescent and young adult literature. J Adolesc Health 2003; 33:427–435.
- Attia E, Roberto CA. Should amenorrhea be a diagnostic criterion for anorexia nervosa? Int J Eat Disord 2009; 42:581–589.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, fifth edition. http://dsm.psychiatryonline.org/content.aspx?bookid=556§ionid=41101776#103439089. Accessed January 31, 2014.
- Wilfley DE, Bishop ME, Wilson GT, Agras WS. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:S123–S129.
- Wonderlich SA, Gordon KH, Mitchell JE, Crosby RD, Engel SG. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009; 42:687–705.
- Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health 2013: 53:303–305.
- Winston AP, Stafford PJ. Cardiovascular effects of anorexia nervosa. Eur Eat Disord Rev 2000; 8:117–125.
- Galetta F, Franzoni F, Prattichizzo F, Rolla M, Santoro G, Pentimone F. Heart rate variability and left ventricular diastolic function in anorexia nervosa. J Adolesc Health 2003; 32:416–421.
- McCallum K, Bermudez O, Ohlemeyer C, Tyson E, Portilla M, Ferdman B. How should the clinician evaluate and manage the cardiovascular complications of anorexia nervosa? Eat Disord 2006; 14:73–80.
- Akhtar M. Clinical spectrum of ventricular tachycardia. Circulation 1990; 82:1561–1573.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54:1–13.
- The University of Arizona Center for Education and Research on Therapeutics. QT Drug Lists. http://crediblemeds.org/everyone/compos-ite-list-all-qtdrugs/?rf=US. Accessed January 31, 2014.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Romano C, Chinali M, Pasanisi F, et al. Reduced hemodynamic load and cardiac hypotrophy in patients with anorexia nervosa. Am J Clin Nutr 2003; 77:308–312.
- Shamim T, Golden NH, Arden M, Filiberto L, Shenker IR. Resolution of vital sign instability: an objective measure of medical stability in anorexia nervosa. J Adolesc Health 2003; 32:73–77.
- Mont L, Castro J, Herreros B, et al. Reversibility of cardiac abnormalities in adolescents with anorexia nervosa after weight recovery. J Am Acad Child Adolesc Psychiatry 2003; 42:808–813.
- Roberto CA, Mayer LE, Brickman AM, et al. Brain tissue volume changes following weight gain in adults with anorexia nervosa. Int J Eat Disord 2011; 44:406–411.
- Treasure J, Russell G. The case for early intervention in anorexia nervosa: theoretical exploration of maintaining factors. Br J Psychiatry 2011; 199:5–7.
- Hadley SJ, Walsh BT. Gastrointestinal disturbances in anorexia nervosa and bulimia nervosa. Curr Drug Targets CNS Neurol Disord 2003; 2:1–9.
- Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med 2005; 353:1481–1488.
- De Caprio C, Alfano A, Senatore I, Zarrella L, Pasanisi F, Contaldo F. Severe acute liver damage in anorexia nervosa: two case reports. Nutrition 2006; 22:572–575.
- Lawson EA, Klibanski A. Endocrine abnormalities in anorexia nervosa. Nat Clin Pract Endocrinol Metab 2008; 4:407–414.
- Holtkamp K, Mika C, Grzella I, et al. Reproductive function during weight gain in anorexia nervosa. Leptin represents a metabolic gate to gonadotropin secretion. J Neural Transm 2003; 110:427–435.
- Golden NH, Jacobson MS, Schebendach J, Solanto MV, Hertz SM, Shenker IR. Resumption of menses in anorexia nervosa. Arch Pediatr Adolesc Med 1997; 151:16–21.
- Soyka LA, Misra M, Frenchman A, et al. Abnormal bone mineral accrual in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2002; 87:4177–4185.
- Misra M, Klibanski A. Bone metabolism in adolescents with anorexia nervosa. J Endocrinol Invest 2011; 34:324–332.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Biller BM, Saxe V, Herzog DB, Rosenthal DI, Holzman S, Klibanski A. Mechanisms of osteoporosis in adult and adolescent women with anorexia nervosa. J Clin Endocrinol Metab 1989; 68:548–554.
- Hergenroeder AC, Smith EO, Shypailo R, Jones LA, Klish WJ, Ellis K. Bone mineral changes in young women with hypothalamic amenorrhea treated with oral contraceptives, medroxyprogesterone, or placebo over 12 months. Am J Obstet Gynecol 1997; 176:1017–1025.
- Sim LA, McGovern L, Elamin MB, Swiglo BA, Erwin PJ, Montori VM. Effect on bone health of estrogen preparations in premenopausal women with anorexia nervosa: a systematic review and meta-analyses. Int J Eat Disord 2010; 43:218–225.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Misra M, Katzman D, Miller KK, et al. Physiologic estrogen replacement increases bone density in adolescent girls with anorexia nervosa. J Bone Miner Res 2011; 26:2430–2438.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Divasta AD, Feldman HA, Giancaterino C, Rosen CJ, Leboff MS, Gordon CM. The effect of gonadal and adrenal steroid therapy on skeletal health in adolescents and young women with anorexia nervosa. Metabolism 2012; 61:1010–1020.
- Mehler PS. Medical complications of bulimia nervosa and their treatments. Int J Eat Disord 2011; 44:95–104.
- Milosevic A. Eating disorders and the dentist. Br Dent J 1999; 186:109–113.
- Greenfeld D, Mickley D, Quinlan DM, Roloff P. Hypokalemia in outpatients with eating disorders. Am J Psychiatry 1995; 152:60–63.
- Bouquegneau A, Dubois BE, Krzesinski JM, Delanaye P. Anorexia nervosa and the kidney. Am J Kidney Dis 2012; 60:299–307.
- Auron M, Rome E. Anorexia nervosa and bulimia nervosa: what the hospitalist needs to know about CPT 269.9, or nutritional insufficiency. ACP Hospitalist 2011 Sept:28–45.
- Steffen KJ, Mitchell JE, Roerig JL, Lancaster KL. The eating disorders medicine cabinet revisited: a clinician’s guide to ipecac and laxatives. Int J Eat Disord 2007; 40:360–368.
- Roerig JL, Steffen KJ, Mitchell JE, Zunker C. Laxative abuse: epidemiology, diagnosis and management. Drugs 2010; 70:1487–1503.
- Mitchell JE, Boutacoff LI. Laxative abuse complicating bulimia: medical and treatment implications. Int J Eat Disord 1986; 5:325–334.
- Joo JS, Ehrenpreis ED, Gonzalez L, et al. Alterations in colonic anatomy induced by chronic stimulant laxatives: the cathartic colon revisited. J Clin Gastroenterol 1998; 26:283–286.
- Drugs.com. Ipecac syrup. www.drugs.com/monograph/ipecac-syrup.html. Accessed January 31, 2014.
- Peveler RC, Bryden KS, Neil HA, et al. The relationship of disordered eating habits and attitudes to clinical outcomes in young adult females with type 1 diabetes. Diabetes Care 2005; 28:84–88.
- Mannucci E, Rotella F, Ricca V, Moretti S, Placidi GF, Rotella CM. Eating disorders in patients with type 1 diabetes: a meta-analysis. J Endocrinol Invest 2005; 28:417–419.
- Crook MA, Hally V, Panteli JV. The importance of the refeeding syndrome. Nutrition 2001; 17:632–637.
- Fisher M, Golden NH, Katzman DK, et al. Eating disorders in adolescents: a background paper. J Adolesc Health 1995; 16:420–437.
- Kohn MR, Madden S, Clarke SD. Refeeding in anorexia nervosa: increased safety and efficiency through understanding the pathophysiology of protein calorie malnutrition. Curr Opin Pediatr 2011; 23:390–394.
- Garber AK, Michihata N, Hetnal K, Shafer MA, Moscicki AB. A prospective examination of weight gain in hospitalized adolescents with anorexia nervosa on a recommended refeeding protocol. J Adolesc Health 2012; 50:24–29.
- Whitelaw M, Gilbertson H, Lam PY, Sawyer SM. Does aggressive refeeding in hospitalized adolescents with anorexia nervosa result in increased hypophosphatemia? J Adolesc Health 2010; 46:577–582.
- Treasure J, Crane A, McKnight R, Buchanan E, Wolfe M. First do no harm: iatrogenic maintaining factors in anorexia nervosa. Eur Eat Disord Rev 2011; 19:296–302.
- Academy for Eating Disorders (AED). Critical points for early recognition and medical risk management in the care of individuals with eating disorders. http://www.aedweb.org/AM/Template.cfm?Section=Medical_Care_Standards&Template=/CM/ContentDisplay.cfm&ContentID=2413. Accessed January 31, 2014.
Eating disorders are debilitating biopsychosocial illnesses associated with serious medical illness and a high risk of death.1
Primary care physicians are often the first to see young women who have these problems, diagnose them, and start their evaluation and treatment.2–4 Many patients require acute medical interventions as well as long-term care for chronic medical issues. Therefore, primary care physicians play essential front-line and long-term roles in the multidisciplinary treatment team.
DEFINITIONS OF EATING DISORDERS HAVE CHANGED
Several problems existed in the category of eating disorders in the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-4) and in the DSM-4 Text Revision (DSM-4-TR). These problems have been addressed in the fifth edition (DSM-5), released in 2013.5
One problem in the earlier editions was that many patients referred for treatment of eating disorders—more than 50% in one study6—did not meet the criteria for anorexia nervosa or bulimia nervosa and thus had to be categorized as having “eating disorder not otherwise specified.” Further, the earlier editions did not recognize that young children and adolescent males can be affected.7
Eating disorders are now recognized as an equal-opportunity disease, with all ethnic and socioeconomic groups affected. Children can run into medical trouble with even a small amount of weight loss or falling off the growth curve. Moreover, children and adolescents do not “experience” their bodies in the same way adults do; they may lack the vocabulary for eating-disorder thoughts.
For these reasons, the definitions of eating disorders have changed in the DSM-5.5
Anorexia nervosa. Older editions of the DSM listed amenorrhea as a criterion. This has been eliminated in DSM-5, since amenorrhea does not necessarily predict medical risk or treatment outcome; also, it is not applicable to males or premenorrheal girls and postmenopausal women.8 In addition, the requirement of low weight is now defined in the context of “age, sex, developmental trajectory, and physical health,” rather than the old threshold of 85% of expected weight.9
What remains unchanged is that anorexia nervosa is still characterized by self-starvation in order to maintain an abnormally low body weight, along with an intense fear of being fat and a disturbed self-image.
Bulimia nervosa. In both the old and the new editions of the DSM, bulimia nervosa is characterized by episodes of binge eating followed by inappropriate compensatory behaviors to avoid weight gain, such as vomiting, laxative abuse, diuretic abuse, and overexercise. In DSM-5, bulimia nervosa no longer has subtypes and requires only one binge per week with compensatory behavior, for at least 3 months. This change was based on the finding that there is no clear difference in psychopathology or treatment outcome between patients with one and two binge-purge episodes a week.10
“Eating disorder not otherwise specified” was a wastebasket category, lumping all those who did not meet the criteria for anorexia nervosa or bulimia nervosa or who did not neatly fit into a specific category.10 In DSM-5, subcategories were designed to help distinguish different treatment needs and outcomes between various subtypes.
Binge-eating disorder, one of the new subcategories, is characterized by binge eating without inappropriate compensatory behaviors.9 Patients with binge-eating disorder are often obese, have greater functional impairment, and are more likely to develop components of metabolic syndrome than obese patients without eating disorders.11
Avoidant/restrictive food intake disorder is another new DSM-5 diagnosis, characterized by failure to meet nutritional needs for reasons other than weight control. Reasons include disinterest in eating, dislike of sensory characteristics of food, or avoidance of consequences of eating. This disorder replaces the category “feeding disorder of infancy or early childhood,” since the condition can also occur in adolescents and adults.12
Other new diagnoses are:
- Atypical anorexia nervosa (if the patient is not underweight)
- Purging disorder
- Subthreshold bulimia nervosa (if the patient has < 1 episode per week or has had them for < 3 months)
- Subthreshold binge eating disorder (< 1 time a week or < 3 months)
- Night eating syndrome
- Pica and rumination disorder.
Regardless of the diagnostic label, the medical evaluation and treatment of anyone with an eating disorder should be tailored to the specific behaviors of the eating disorder. Medical complications can be subdivided into those from starvation, from purging, and from refeeding.
MEDICAL COMPLICATIONS OF STARVATION
Cardiovascular effects of starvation
Malnutrition and starvation have multiple adverse effects on the heart.
Electrophysiologic effects. Sinus bradycardia (< 60 bpm) and hypotension are common cardiac manifestations of starvation.13 Bradycardia has been attributed to an adaptive increase in parasympathetic vagal tone.14 QTc prolongation is also seen in patients with malnutrition.15
Together, these electrocardiographic abnormalities predispose the patient to ventricular arrhythmia and sudden cardiac death.16 The risk of ventricular arrhythmia is particularly relevant when treating psychiatric symptoms, since antipsychotics and tricyclic antidepressants are among several drug classes that can cause further QTc prolongation (Table 1).17,18
In patients with QTc prolongation, bradycardia, or both, the standard of care involves acute hospitalization for refeeding using continuous telemetric monitoring until normal rhythm is restored and the heart rate is above 40 at night and 50 by day.4,19
Structural changes. Starvation also causes structural changes in the heart. Loss of lean body mass can reduce cardiac muscle mass, compromise cardiac output, and lead to mitral valve prolapse.20 These changes are fully reversible with restored nutrition and regaining of heart mass.21,22
Effects of starvation on the brain
Starvation can affect brain structure and cognitive function. Undernourished patients have reduced volumes of white and gray matter, a change that can occur within months. Cortical volumes may increase with weight gain, but a reduction in gray matter volume may not be completely reversible.23
Furthermore, starvation impairs cognitive functions that are needed to stop eating-disorder behaviors; namely, decision-making, emotional control, regulation of appetite, and reward path-ways. Therefore, undernourished patients may not have sufficient insight into the disease to be able to make the best choices for recovery. This finding lends support for using the Maudsley method in adolescents, in which parents take control of their child’s eating until the child can maintain a healthy weight.24
Gastrointestinal consequences of starvation
Patients with malnutrition have prolonged gastric emptying and colonic transit time with solid foods.25 They often complain of early satiety, abdominal pain, bloating, and constipation, all symptoms that complicate the refeeding process. A prokinetic such as metoclopramide (Reglan), given 1 hour before meals and at bedtime, may provide some relief from gastrointestinal symptoms.26
Patients may also experience transient lactose or fructose intolerance after prolonged starvation. Taking a lactase supplement (eg, Lactaid 1–10 tabs) before consuming dairy products and dextrose (contained in candies such as Smarties) before eating fruit or fructose-containing foods can sometimes partially relieve symptoms. In general, gastrointestinal function returns over time as nutritional status improves.
Patients with severe or prolonged starvation can develop steatosis accompanied by elevated levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). In reports of starvation-induced steatosis, liver enzyme levels rapidly normalize with nutritional rehabilitation.27
Endocrine consequences of starvation
Amenorrhea. Dysregulation of the hypothalamic-pituitary-gonadal axis is a major endocrine complication of nutritional in-sufficiency. Weight loss disrupts the normal pulsatile secretion of gonadotropin-releasing hormone, reduces secretion of luteinizing hormone and follicle-stimulating hormone, and decreases estrogen levels.28 Leptin deficiency likely plays a role in suppressing gonadotropin secretion with subsequent development of amenorrhea. With weight gain, levels of leptin and gonadotropins normalize and menstruation eventually returns.29,30
Hypothyroidism. Starvation can also lead to dysregulation of the hypothalamic-pituitary-thyroid axis. Typically, the concentration of triiodothyronine (T3) is reduced, the ratio of thyroxine (T4) to T3 is elevated, and thyroid-stimulating hormone (TSH) is close to or within the normal range, creating a euthyroid sick syndrome. In eating disorders, this thyroid disturbance is a result of starvation and resolves with weight restoration. Therefore, thyroid hormone replacement therapy is not medically indicated.28
Osteoporosis. Amenorrhea resulting from low estrogen levels in undernourished patients can raise the risk of osteoporosis and fractures, particularly in patients with a low body mass index. Osteopenia results from a negative balance between bone deposition and resorption.
Lack of bone deposition can be especially problematic when disordered eating occurs during peak bone mass development, ie, ages 11 to 14 for girls, and ages 15 to 17 for boys.31,32 Even a 5% to 10% decrease in bone deposition can result in significant risk of osteopenia.33 However, after age 30, bone resorption is a greater contributor.34
Does hormone therapy correct bone loss? Given the association between estrogen deficiency and bone loss, estrogen supplementation was expected to be an effective treatment for bone loss in patients with eating disorders.35 Also, the restoration of menses through hormone replacement may give underweight patients a false sense of achieving a “healthy” weight.36
Golden et al37 prospectively studied 50 adolescents and found no significant difference in bone mineral density at 1 year of follow-up between patients treated with estrogen and those who received only standard nutritional therapy. However, increased bone mineral density was achieved in adolescents with anorexia nervosa treated with transdermally administered estrogen dosed to mimic physiologic pubertal levels.38
Klibanski et al39 found that hormone therapy resulted in a 4% gain in bone density in an extremely low-weight subset of women with anorexia nervosa (< 70% of ideal body weight), whereas similar patients in the control group lost 20%. However, in all groups, only weight gain correlated with bone gain in women who were within 70% of their ideal body weight.
Divasta et al40 evaluated 60 girls and women ages 13 to 27 with anorexia nervosa, randomized to receive either placebo or dehydroepiandrosterone combined with an estrogen-progestin oral contraceptive, and followed for 18 months. As in the study by Klibanski et al,39 bone loss was prevented in the treatment group, but significant bone gain occurred only in the context of weight gain.
The bottom line is that only weight gain has resulted in significant increases in bone density in patients with anorexia nervosa, and hormone therapy without weight gain has not been shown to increase bone density effectively in this population. Although calcium and vitamin D in oral therapeutic doses through foods or through supplementation are required for bone gain, the combination is not enough to augment bone density in the absence of weight gain.37 Although not curative, weight gain is currently the best option for treating bone loss, and no single pharmacologic treatment is effective.
COMPLICATIONS OF PURGING
Oral complications of purging
Patients who purge by vomiting are at risk of complications from exposure of the esophagus, pharynx, and mouth to acidic gastric contents.
Dental problems. Over time, contact with gastric acid wears down enamel on the lingual and occlusal surfaces of teeth, resulting in dental caries and periodontal disease. Until they can give up purging, patients should be instructed to rinse with mouthwash or water immediately after vomiting to reduce the acidity in the mouth.41,42 We recommend that patients not brush their teeth after vomiting, because brushing can deliver acid to otherwise unreachable surfaces and thus worsen tooth erosion. For patients who are determined to brush after vomiting, a bicarbonate toothpaste might mitigate harm.42
Sialadenosis (hypertrophy of the salivary glands) is another consequence of repeated vomiting, with elevated salivary amylase. Both the size of the glands and the salivary amylase level generally normalize on their own after vomiting is stopped, but parotitis can take up to a year to resolve. Similar to smoker’s cough, parotitis may acutely worsen when the patient abruptly stops vomiting and may worsen before it improves.
To reduce discomfort, patients can use hot compresses or sugarless hard candies.44 However, the latter should not be substituted as a chronic habit in a patient with disordered eating. Patients need to be reassured that the swelling is not permanent, since they often interpret it as having fat cheeks (the “chipmunk sign”).
Hypokalemia, metabolic alkalosis, renal dysfunction
Chronic vomiting can cause electrolyte and acid-base imbalances, the most worrisome of which is hypokalemia. With repeated vomiting, loss of potassium and gastric acid causes metabolic alkalosis with hypokalemia, hypochloremia, and hypomagnesemia. Loss of water and the resultant volume contraction activates the renin-angiotensin-aldosterone system, and elevated aldosterone further decreases serum potassium.
In patients with eating disorders, who often have other factors contributing to electrolyte imbalance, vomiting-induced hypokalemia heightens the risk of cardiac arrhythmias.43
Hypokalemia can also cause rhabdomyolysis and kidney damage.41,43 Prolonged hypokalemia and reduced kidney perfusion in the setting of volume depletion causes acute kidney injury and impaired concentrating ability of the renal tubules. Hypovolemia can cause prerenal azotemia and increases the risk for nephrolithiasis and nephrocalcinosis.44,45
When a patient stops vomiting, elevated aldosterone from prior hypovolemia results in water retention and can manifest in significant edema associated with hypochloremic alkalosis. This condition, known as pseudo-Bartter syndrome, usually resolves without treatment. In the meantime, salt restriction and leg elevation can help reduce edema.26
Laxative abuse: A mode of purging
Many patients with eating disorders abuse laxatives to lose weight or to prevent weight gain. Believing that laxatives will prevent calorie absorption, patients commonly take them to compensate for caloric intake (eg, during a binge episode). The immediate weight loss, albeit artificial, is highly reinforcing for an eating-disorder patient. In some cases, patients with eating disorders also abuse laxatives to self-treat the constipation that results from chronic starvation.46
Over time, tolerance to laxatives develops, and patients use increasingly larger doses. This can lead to activation of the renin-angiotensin-aldosterone system.47 Patients interpret the resultant edema as true weight gain and again take laxatives to get rid of it. If laxatives are stopped abruptly, the patient may need inpatient and outpatient support for the resultant fluid shifts.
Gastrointestinal complications of laxative abuse include reflex hypofunction of the bowel, malabsorption, steatorrhea, and gastrointestinal bleeding.47 Reflex hypofunction during laxative withdrawal is a consequence of the bowel becoming tolerant of laxatives.48 Cathartic colon syndrome is a rare complication characterized by loss of the normal haustral markings and slowed or absent peristalsis in segments of the colon.49
Systemically, the major risk of laxative abuse relates to electrolyte and acid-base imbalance. Loss of potassium and water in the stool can cause hypokalemia and metabolic alkalosis.48 The disturbances caused by laxative abuse are similar to those caused by vomiting and diuretic use and have the same treatment.
The most important component of treating laxative abuse is giving patients realistic expectations to help them tolerate temporary discomfort and to help manage the edema and fluid shifts that can happen acutely with shifting of fluid into the intracellular space. In extreme cases, this may need to be managed in the hospital. To help relieve the initial anxiety, doctors should emphasize that any bloating the patient experiences is not true weight gain and will go away within a few days to weeks. In addition, explaining that laxatives reduce nutrient absorption only minimally may lessen the temptation to resume taking them.48
Diuretic abuse: Another form of purging
Diuretic abuse is yet another mode of purging, with its own set of medical complications. Like laxatives, diuretics are not effective weight-loss agents, and the weight reduction they cause is only temporary.
As with vomiting, there is a compensatory activation of the renin-angiotensin-aldosterone system, and therefore subsequent fluid intake will lead to water retention, which encourages further diuretic use.41 Diuretics can also contribute to hypokalemia, hypomagnesemia, hypochloremia, and metabolic alkalosis.
Ipecac abuse can lead to heart failure
Ipecac syrup has long been used to induce vomiting, but this practice has become much less common since ipecac has become harder to obtain in the United States.50 The emetine base contained in ipecac binds irreversibly to cardiac and skeletal muscle. With continued use, irreversible cardiomyopathy develops and can lead to heart failure. Treatment should include supportive care and immediate cessation of ipecac use.
Diabetic patients may skip insulin to lose weight
Patients with diabetes, especially those with type 1 that begins in childhood, are at greater risk of eating disorders over time.51 They may withhold insulin to lose weight, a practice referred to in the nonmedical literature as “diabulimia,” and they seem particularly more likely to develop bulimia nervosa than those without diabetes.52
The medical prognosis is poor for patients with diabetes who develop eating disorders and do not receive intensive treatment.51 In addition, if a diabetic patient on an insulin pump becomes depressed in addition to having an eating disorder, careful monitoring for suicidal thoughts and a rapid follow-up with mental health services are in order.
REFEEDING SYNDROME
When refeeding is started, a high glucose load stimulates insulin secretion, resulting in cellular uptake of phosphorus along with potassium, magnesium, and glucose. In addition, total body phosphorus is depleted by the increased demand for adenosine triphosphate and 2,3-diphosphoglycerate for cellular metabolism.
When liver enzyme levels increase, the astute clinician will closely monitor the patient for evidence of refeeding syndrome. In a child, adolescent, or young adult, the standard of care is inpatient monitoring for acute stabilization.4,19
Hypophosphatemia is the hallmark of refeeding syndrome, although hypomagnesemia, hypokalemia, and hypoglycemia can also occur.53 In addition, sodium and water retention can lead to fluid overload, with shifting of fluid into the intracellular space, resulting in dependent edema.
Cardiovascular complications are the most worrisome manifestations of refeeding syndrome. Electrolyte shifts and increased fluid volume can cause arrhythmias and heart failure. Furthermore, severely undernourished patients may have reduced myocardial mass as well as electrocardiographic abnormalities associated with starvation, which further increase their vulnerability to electrolyte shifts and fluid retention during refeeding.15
Other manifestations of refeeding syndrome include delirium, seizures, rhabdomyolysis, and respiratory failure. In the most extreme cases, refeeding syndrome causes sudden death.53
Fortunately, refeeding syndrome is easily preventable and treatable when recognized early. Electrolytes and cardiovascular and renal function must be carefully monitored, especially during the first week of nutritional restoration.53 In patients with extremely low body mass (< 70% of ideal body weight) or with precipitous weight loss, close monitoring of the complete metabolic panel including electrolytes, AST, ALT, calcium, magnesium, and phosphorus may be required to detect changes that can affect cardiac status. Specific suggestions for refeeding are discussed below and in Table 2.45
ACUTE CARE OF PATIENTS WITH EATING DISORDERS
Refeeding in the inpatient setting
The decision to hospitalize an eating-disorder patient is based on the current or potential risk of serious medical complications and the likelihood of success at home. Medical criteria for hospital admission are outlined in Table 3.4,54
In refeeding undernourished patients, the challenge is to maximize weight gain while preventing refeeding syndrome. Undernourished patients are generally hypometabolic at baseline but become hypermetabolic once refeeding begins.
How many calories should refeeding start with? The traditional principle of “start low and go slow” has been recently challenged.55 Starting at 1,200 kcal/day or less in the typical patient can result in failure to gain weight or even in weight loss in the first week of refeeding.56 The goal is to achieve a weight gain of 0.2 kg/day while the patient is in the hospital. Thus, we start higher, and to date we have seen no cases of life-threatening refeeding syndrome. In all patients who need hospitalization or who are beginning the refeeding process as outpatients, caloric intake should be started at 1,500 to 2,000 kcal/day.45,57 However, for exceptionally low-weight patients, intake may be started lower.
In Australia, patients are started at 1,900 kcal/day.56 All patients in one program there receive nasogastric feeding initially in an intensive care unit and then are moved to a regular nursing floor where they graduate to full oral feeding as they improve cardiovascularly and behaviorally. In the United States, some programs use nasogastric feeding at night for caloric restoration; our program and others use nasogastric feeding as a behavioral modification strategy for patients who refuse food or supplements by mouth.
Phosphorus supplementation. Many centers give phosphorus supplements preventively. In our center, we give potassium phosphate (Neutra-Phos) 500 mg orally twice daily for 5 days, and we have seen no life-threatening cases of refeeding syndrome with that regimen. Other centers give phosphorus supplements in a dose of 250 mg orally twice a day for 5 days, while still others only supplement phosphorus reactively once a deficit has been identified. The latter method requires daily blood draws for monitoring and is reactive rather than proactive. Further studies can help clarify the optimal dosing and timing of phosphorus supplementation.
Managing fluid balance. Fluid-loading these patients may tip them over the edge into refeeding syndrome. Except in cases of shock, patients with eating disorders should not be given intravenous fluids, as it is safer to rehydrate and feed them orally. Electrolyte imbalances can be corrected orally with no need for intravenous supplementation. To avoid fluid overload, fluids can be started at 1,500 mL to 2,000 mL per day, with strict monitoring of intake and output. Fluids are liberalized if ALT and AST levels remain normal and to gradually correct orthostatic hypotension; caloric fluids are ideal to help address energy needs and improve bradycardia.
Laboratory monitoring. On admission, a urinalysis, complete blood cell count, complete metabolic panel, TSH, erythrocyte sedimentation rate, serum magnesium, and phosphorus should be obtained.26 In addition, continuous electrocardiographic recording should begin on admission.45 Inpatient use of a telemetry bed helps identify extreme tachycardia with arrhythmia, as well as profound bradycardia.45,56
Some protocols call for daily laboratory monitoring, although that degree of testing is less cost-effective. If initial results are normal, clinical judgment can be used on when to repeat laboratory evaluation. For instance, patients with edema require repeat complete metabolic panels to assess for elevated ALT and AST, electrolyte imbalances, and other abnormalities.
Signs of refeeding syndrome include tachycardia, hepatosplenomegaly, peripheral edema, altered mental status, and electrolyte disturbances, specifically, acute or severe hypophosphatemia or hypokalemia.26,45 If refeeding syndrome is suspected, the rate of caloric intake should be reduced or not advanced, fluid intake should be urgently reassessed for volume overload, and supportive care with close monitoring should be provided.
KNOWLEDGE SAVES LIVES
Eating disorders can lead to potentially life-threatening medical complications that require attentive care by the primary care clinician and subspecialist. Without thoughtful consideration, it is easy for even a caring medical team to unintentionally enable patients with these illnesses or to cause active harm in the case of underrecognized pathology.58
Acute medical stabilization on an inpatient unit trained to recognize pathology and treat sequelae can be lifesaving. Arming patients and families with medical knowledge, as provided in the Academy for Eating Disorders’ brochure, “Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders”59 can help save patients’ lives.
Eating disorders are debilitating biopsychosocial illnesses associated with serious medical illness and a high risk of death.1
Primary care physicians are often the first to see young women who have these problems, diagnose them, and start their evaluation and treatment.2–4 Many patients require acute medical interventions as well as long-term care for chronic medical issues. Therefore, primary care physicians play essential front-line and long-term roles in the multidisciplinary treatment team.
DEFINITIONS OF EATING DISORDERS HAVE CHANGED
Several problems existed in the category of eating disorders in the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-4) and in the DSM-4 Text Revision (DSM-4-TR). These problems have been addressed in the fifth edition (DSM-5), released in 2013.5
One problem in the earlier editions was that many patients referred for treatment of eating disorders—more than 50% in one study6—did not meet the criteria for anorexia nervosa or bulimia nervosa and thus had to be categorized as having “eating disorder not otherwise specified.” Further, the earlier editions did not recognize that young children and adolescent males can be affected.7
Eating disorders are now recognized as an equal-opportunity disease, with all ethnic and socioeconomic groups affected. Children can run into medical trouble with even a small amount of weight loss or falling off the growth curve. Moreover, children and adolescents do not “experience” their bodies in the same way adults do; they may lack the vocabulary for eating-disorder thoughts.
For these reasons, the definitions of eating disorders have changed in the DSM-5.5
Anorexia nervosa. Older editions of the DSM listed amenorrhea as a criterion. This has been eliminated in DSM-5, since amenorrhea does not necessarily predict medical risk or treatment outcome; also, it is not applicable to males or premenorrheal girls and postmenopausal women.8 In addition, the requirement of low weight is now defined in the context of “age, sex, developmental trajectory, and physical health,” rather than the old threshold of 85% of expected weight.9
What remains unchanged is that anorexia nervosa is still characterized by self-starvation in order to maintain an abnormally low body weight, along with an intense fear of being fat and a disturbed self-image.
Bulimia nervosa. In both the old and the new editions of the DSM, bulimia nervosa is characterized by episodes of binge eating followed by inappropriate compensatory behaviors to avoid weight gain, such as vomiting, laxative abuse, diuretic abuse, and overexercise. In DSM-5, bulimia nervosa no longer has subtypes and requires only one binge per week with compensatory behavior, for at least 3 months. This change was based on the finding that there is no clear difference in psychopathology or treatment outcome between patients with one and two binge-purge episodes a week.10
“Eating disorder not otherwise specified” was a wastebasket category, lumping all those who did not meet the criteria for anorexia nervosa or bulimia nervosa or who did not neatly fit into a specific category.10 In DSM-5, subcategories were designed to help distinguish different treatment needs and outcomes between various subtypes.
Binge-eating disorder, one of the new subcategories, is characterized by binge eating without inappropriate compensatory behaviors.9 Patients with binge-eating disorder are often obese, have greater functional impairment, and are more likely to develop components of metabolic syndrome than obese patients without eating disorders.11
Avoidant/restrictive food intake disorder is another new DSM-5 diagnosis, characterized by failure to meet nutritional needs for reasons other than weight control. Reasons include disinterest in eating, dislike of sensory characteristics of food, or avoidance of consequences of eating. This disorder replaces the category “feeding disorder of infancy or early childhood,” since the condition can also occur in adolescents and adults.12
Other new diagnoses are:
- Atypical anorexia nervosa (if the patient is not underweight)
- Purging disorder
- Subthreshold bulimia nervosa (if the patient has < 1 episode per week or has had them for < 3 months)
- Subthreshold binge eating disorder (< 1 time a week or < 3 months)
- Night eating syndrome
- Pica and rumination disorder.
Regardless of the diagnostic label, the medical evaluation and treatment of anyone with an eating disorder should be tailored to the specific behaviors of the eating disorder. Medical complications can be subdivided into those from starvation, from purging, and from refeeding.
MEDICAL COMPLICATIONS OF STARVATION
Cardiovascular effects of starvation
Malnutrition and starvation have multiple adverse effects on the heart.
Electrophysiologic effects. Sinus bradycardia (< 60 bpm) and hypotension are common cardiac manifestations of starvation.13 Bradycardia has been attributed to an adaptive increase in parasympathetic vagal tone.14 QTc prolongation is also seen in patients with malnutrition.15
Together, these electrocardiographic abnormalities predispose the patient to ventricular arrhythmia and sudden cardiac death.16 The risk of ventricular arrhythmia is particularly relevant when treating psychiatric symptoms, since antipsychotics and tricyclic antidepressants are among several drug classes that can cause further QTc prolongation (Table 1).17,18
In patients with QTc prolongation, bradycardia, or both, the standard of care involves acute hospitalization for refeeding using continuous telemetric monitoring until normal rhythm is restored and the heart rate is above 40 at night and 50 by day.4,19
Structural changes. Starvation also causes structural changes in the heart. Loss of lean body mass can reduce cardiac muscle mass, compromise cardiac output, and lead to mitral valve prolapse.20 These changes are fully reversible with restored nutrition and regaining of heart mass.21,22
Effects of starvation on the brain
Starvation can affect brain structure and cognitive function. Undernourished patients have reduced volumes of white and gray matter, a change that can occur within months. Cortical volumes may increase with weight gain, but a reduction in gray matter volume may not be completely reversible.23
Furthermore, starvation impairs cognitive functions that are needed to stop eating-disorder behaviors; namely, decision-making, emotional control, regulation of appetite, and reward path-ways. Therefore, undernourished patients may not have sufficient insight into the disease to be able to make the best choices for recovery. This finding lends support for using the Maudsley method in adolescents, in which parents take control of their child’s eating until the child can maintain a healthy weight.24
Gastrointestinal consequences of starvation
Patients with malnutrition have prolonged gastric emptying and colonic transit time with solid foods.25 They often complain of early satiety, abdominal pain, bloating, and constipation, all symptoms that complicate the refeeding process. A prokinetic such as metoclopramide (Reglan), given 1 hour before meals and at bedtime, may provide some relief from gastrointestinal symptoms.26
Patients may also experience transient lactose or fructose intolerance after prolonged starvation. Taking a lactase supplement (eg, Lactaid 1–10 tabs) before consuming dairy products and dextrose (contained in candies such as Smarties) before eating fruit or fructose-containing foods can sometimes partially relieve symptoms. In general, gastrointestinal function returns over time as nutritional status improves.
Patients with severe or prolonged starvation can develop steatosis accompanied by elevated levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). In reports of starvation-induced steatosis, liver enzyme levels rapidly normalize with nutritional rehabilitation.27
Endocrine consequences of starvation
Amenorrhea. Dysregulation of the hypothalamic-pituitary-gonadal axis is a major endocrine complication of nutritional in-sufficiency. Weight loss disrupts the normal pulsatile secretion of gonadotropin-releasing hormone, reduces secretion of luteinizing hormone and follicle-stimulating hormone, and decreases estrogen levels.28 Leptin deficiency likely plays a role in suppressing gonadotropin secretion with subsequent development of amenorrhea. With weight gain, levels of leptin and gonadotropins normalize and menstruation eventually returns.29,30
Hypothyroidism. Starvation can also lead to dysregulation of the hypothalamic-pituitary-thyroid axis. Typically, the concentration of triiodothyronine (T3) is reduced, the ratio of thyroxine (T4) to T3 is elevated, and thyroid-stimulating hormone (TSH) is close to or within the normal range, creating a euthyroid sick syndrome. In eating disorders, this thyroid disturbance is a result of starvation and resolves with weight restoration. Therefore, thyroid hormone replacement therapy is not medically indicated.28
Osteoporosis. Amenorrhea resulting from low estrogen levels in undernourished patients can raise the risk of osteoporosis and fractures, particularly in patients with a low body mass index. Osteopenia results from a negative balance between bone deposition and resorption.
Lack of bone deposition can be especially problematic when disordered eating occurs during peak bone mass development, ie, ages 11 to 14 for girls, and ages 15 to 17 for boys.31,32 Even a 5% to 10% decrease in bone deposition can result in significant risk of osteopenia.33 However, after age 30, bone resorption is a greater contributor.34
Does hormone therapy correct bone loss? Given the association between estrogen deficiency and bone loss, estrogen supplementation was expected to be an effective treatment for bone loss in patients with eating disorders.35 Also, the restoration of menses through hormone replacement may give underweight patients a false sense of achieving a “healthy” weight.36
Golden et al37 prospectively studied 50 adolescents and found no significant difference in bone mineral density at 1 year of follow-up between patients treated with estrogen and those who received only standard nutritional therapy. However, increased bone mineral density was achieved in adolescents with anorexia nervosa treated with transdermally administered estrogen dosed to mimic physiologic pubertal levels.38
Klibanski et al39 found that hormone therapy resulted in a 4% gain in bone density in an extremely low-weight subset of women with anorexia nervosa (< 70% of ideal body weight), whereas similar patients in the control group lost 20%. However, in all groups, only weight gain correlated with bone gain in women who were within 70% of their ideal body weight.
Divasta et al40 evaluated 60 girls and women ages 13 to 27 with anorexia nervosa, randomized to receive either placebo or dehydroepiandrosterone combined with an estrogen-progestin oral contraceptive, and followed for 18 months. As in the study by Klibanski et al,39 bone loss was prevented in the treatment group, but significant bone gain occurred only in the context of weight gain.
The bottom line is that only weight gain has resulted in significant increases in bone density in patients with anorexia nervosa, and hormone therapy without weight gain has not been shown to increase bone density effectively in this population. Although calcium and vitamin D in oral therapeutic doses through foods or through supplementation are required for bone gain, the combination is not enough to augment bone density in the absence of weight gain.37 Although not curative, weight gain is currently the best option for treating bone loss, and no single pharmacologic treatment is effective.
COMPLICATIONS OF PURGING
Oral complications of purging
Patients who purge by vomiting are at risk of complications from exposure of the esophagus, pharynx, and mouth to acidic gastric contents.
Dental problems. Over time, contact with gastric acid wears down enamel on the lingual and occlusal surfaces of teeth, resulting in dental caries and periodontal disease. Until they can give up purging, patients should be instructed to rinse with mouthwash or water immediately after vomiting to reduce the acidity in the mouth.41,42 We recommend that patients not brush their teeth after vomiting, because brushing can deliver acid to otherwise unreachable surfaces and thus worsen tooth erosion. For patients who are determined to brush after vomiting, a bicarbonate toothpaste might mitigate harm.42
Sialadenosis (hypertrophy of the salivary glands) is another consequence of repeated vomiting, with elevated salivary amylase. Both the size of the glands and the salivary amylase level generally normalize on their own after vomiting is stopped, but parotitis can take up to a year to resolve. Similar to smoker’s cough, parotitis may acutely worsen when the patient abruptly stops vomiting and may worsen before it improves.
To reduce discomfort, patients can use hot compresses or sugarless hard candies.44 However, the latter should not be substituted as a chronic habit in a patient with disordered eating. Patients need to be reassured that the swelling is not permanent, since they often interpret it as having fat cheeks (the “chipmunk sign”).
Hypokalemia, metabolic alkalosis, renal dysfunction
Chronic vomiting can cause electrolyte and acid-base imbalances, the most worrisome of which is hypokalemia. With repeated vomiting, loss of potassium and gastric acid causes metabolic alkalosis with hypokalemia, hypochloremia, and hypomagnesemia. Loss of water and the resultant volume contraction activates the renin-angiotensin-aldosterone system, and elevated aldosterone further decreases serum potassium.
In patients with eating disorders, who often have other factors contributing to electrolyte imbalance, vomiting-induced hypokalemia heightens the risk of cardiac arrhythmias.43
Hypokalemia can also cause rhabdomyolysis and kidney damage.41,43 Prolonged hypokalemia and reduced kidney perfusion in the setting of volume depletion causes acute kidney injury and impaired concentrating ability of the renal tubules. Hypovolemia can cause prerenal azotemia and increases the risk for nephrolithiasis and nephrocalcinosis.44,45
When a patient stops vomiting, elevated aldosterone from prior hypovolemia results in water retention and can manifest in significant edema associated with hypochloremic alkalosis. This condition, known as pseudo-Bartter syndrome, usually resolves without treatment. In the meantime, salt restriction and leg elevation can help reduce edema.26
Laxative abuse: A mode of purging
Many patients with eating disorders abuse laxatives to lose weight or to prevent weight gain. Believing that laxatives will prevent calorie absorption, patients commonly take them to compensate for caloric intake (eg, during a binge episode). The immediate weight loss, albeit artificial, is highly reinforcing for an eating-disorder patient. In some cases, patients with eating disorders also abuse laxatives to self-treat the constipation that results from chronic starvation.46
Over time, tolerance to laxatives develops, and patients use increasingly larger doses. This can lead to activation of the renin-angiotensin-aldosterone system.47 Patients interpret the resultant edema as true weight gain and again take laxatives to get rid of it. If laxatives are stopped abruptly, the patient may need inpatient and outpatient support for the resultant fluid shifts.
Gastrointestinal complications of laxative abuse include reflex hypofunction of the bowel, malabsorption, steatorrhea, and gastrointestinal bleeding.47 Reflex hypofunction during laxative withdrawal is a consequence of the bowel becoming tolerant of laxatives.48 Cathartic colon syndrome is a rare complication characterized by loss of the normal haustral markings and slowed or absent peristalsis in segments of the colon.49
Systemically, the major risk of laxative abuse relates to electrolyte and acid-base imbalance. Loss of potassium and water in the stool can cause hypokalemia and metabolic alkalosis.48 The disturbances caused by laxative abuse are similar to those caused by vomiting and diuretic use and have the same treatment.
The most important component of treating laxative abuse is giving patients realistic expectations to help them tolerate temporary discomfort and to help manage the edema and fluid shifts that can happen acutely with shifting of fluid into the intracellular space. In extreme cases, this may need to be managed in the hospital. To help relieve the initial anxiety, doctors should emphasize that any bloating the patient experiences is not true weight gain and will go away within a few days to weeks. In addition, explaining that laxatives reduce nutrient absorption only minimally may lessen the temptation to resume taking them.48
Diuretic abuse: Another form of purging
Diuretic abuse is yet another mode of purging, with its own set of medical complications. Like laxatives, diuretics are not effective weight-loss agents, and the weight reduction they cause is only temporary.
As with vomiting, there is a compensatory activation of the renin-angiotensin-aldosterone system, and therefore subsequent fluid intake will lead to water retention, which encourages further diuretic use.41 Diuretics can also contribute to hypokalemia, hypomagnesemia, hypochloremia, and metabolic alkalosis.
Ipecac abuse can lead to heart failure
Ipecac syrup has long been used to induce vomiting, but this practice has become much less common since ipecac has become harder to obtain in the United States.50 The emetine base contained in ipecac binds irreversibly to cardiac and skeletal muscle. With continued use, irreversible cardiomyopathy develops and can lead to heart failure. Treatment should include supportive care and immediate cessation of ipecac use.
Diabetic patients may skip insulin to lose weight
Patients with diabetes, especially those with type 1 that begins in childhood, are at greater risk of eating disorders over time.51 They may withhold insulin to lose weight, a practice referred to in the nonmedical literature as “diabulimia,” and they seem particularly more likely to develop bulimia nervosa than those without diabetes.52
The medical prognosis is poor for patients with diabetes who develop eating disorders and do not receive intensive treatment.51 In addition, if a diabetic patient on an insulin pump becomes depressed in addition to having an eating disorder, careful monitoring for suicidal thoughts and a rapid follow-up with mental health services are in order.
REFEEDING SYNDROME
When refeeding is started, a high glucose load stimulates insulin secretion, resulting in cellular uptake of phosphorus along with potassium, magnesium, and glucose. In addition, total body phosphorus is depleted by the increased demand for adenosine triphosphate and 2,3-diphosphoglycerate for cellular metabolism.
When liver enzyme levels increase, the astute clinician will closely monitor the patient for evidence of refeeding syndrome. In a child, adolescent, or young adult, the standard of care is inpatient monitoring for acute stabilization.4,19
Hypophosphatemia is the hallmark of refeeding syndrome, although hypomagnesemia, hypokalemia, and hypoglycemia can also occur.53 In addition, sodium and water retention can lead to fluid overload, with shifting of fluid into the intracellular space, resulting in dependent edema.
Cardiovascular complications are the most worrisome manifestations of refeeding syndrome. Electrolyte shifts and increased fluid volume can cause arrhythmias and heart failure. Furthermore, severely undernourished patients may have reduced myocardial mass as well as electrocardiographic abnormalities associated with starvation, which further increase their vulnerability to electrolyte shifts and fluid retention during refeeding.15
Other manifestations of refeeding syndrome include delirium, seizures, rhabdomyolysis, and respiratory failure. In the most extreme cases, refeeding syndrome causes sudden death.53
Fortunately, refeeding syndrome is easily preventable and treatable when recognized early. Electrolytes and cardiovascular and renal function must be carefully monitored, especially during the first week of nutritional restoration.53 In patients with extremely low body mass (< 70% of ideal body weight) or with precipitous weight loss, close monitoring of the complete metabolic panel including electrolytes, AST, ALT, calcium, magnesium, and phosphorus may be required to detect changes that can affect cardiac status. Specific suggestions for refeeding are discussed below and in Table 2.45
ACUTE CARE OF PATIENTS WITH EATING DISORDERS
Refeeding in the inpatient setting
The decision to hospitalize an eating-disorder patient is based on the current or potential risk of serious medical complications and the likelihood of success at home. Medical criteria for hospital admission are outlined in Table 3.4,54
In refeeding undernourished patients, the challenge is to maximize weight gain while preventing refeeding syndrome. Undernourished patients are generally hypometabolic at baseline but become hypermetabolic once refeeding begins.
How many calories should refeeding start with? The traditional principle of “start low and go slow” has been recently challenged.55 Starting at 1,200 kcal/day or less in the typical patient can result in failure to gain weight or even in weight loss in the first week of refeeding.56 The goal is to achieve a weight gain of 0.2 kg/day while the patient is in the hospital. Thus, we start higher, and to date we have seen no cases of life-threatening refeeding syndrome. In all patients who need hospitalization or who are beginning the refeeding process as outpatients, caloric intake should be started at 1,500 to 2,000 kcal/day.45,57 However, for exceptionally low-weight patients, intake may be started lower.
In Australia, patients are started at 1,900 kcal/day.56 All patients in one program there receive nasogastric feeding initially in an intensive care unit and then are moved to a regular nursing floor where they graduate to full oral feeding as they improve cardiovascularly and behaviorally. In the United States, some programs use nasogastric feeding at night for caloric restoration; our program and others use nasogastric feeding as a behavioral modification strategy for patients who refuse food or supplements by mouth.
Phosphorus supplementation. Many centers give phosphorus supplements preventively. In our center, we give potassium phosphate (Neutra-Phos) 500 mg orally twice daily for 5 days, and we have seen no life-threatening cases of refeeding syndrome with that regimen. Other centers give phosphorus supplements in a dose of 250 mg orally twice a day for 5 days, while still others only supplement phosphorus reactively once a deficit has been identified. The latter method requires daily blood draws for monitoring and is reactive rather than proactive. Further studies can help clarify the optimal dosing and timing of phosphorus supplementation.
Managing fluid balance. Fluid-loading these patients may tip them over the edge into refeeding syndrome. Except in cases of shock, patients with eating disorders should not be given intravenous fluids, as it is safer to rehydrate and feed them orally. Electrolyte imbalances can be corrected orally with no need for intravenous supplementation. To avoid fluid overload, fluids can be started at 1,500 mL to 2,000 mL per day, with strict monitoring of intake and output. Fluids are liberalized if ALT and AST levels remain normal and to gradually correct orthostatic hypotension; caloric fluids are ideal to help address energy needs and improve bradycardia.
Laboratory monitoring. On admission, a urinalysis, complete blood cell count, complete metabolic panel, TSH, erythrocyte sedimentation rate, serum magnesium, and phosphorus should be obtained.26 In addition, continuous electrocardiographic recording should begin on admission.45 Inpatient use of a telemetry bed helps identify extreme tachycardia with arrhythmia, as well as profound bradycardia.45,56
Some protocols call for daily laboratory monitoring, although that degree of testing is less cost-effective. If initial results are normal, clinical judgment can be used on when to repeat laboratory evaluation. For instance, patients with edema require repeat complete metabolic panels to assess for elevated ALT and AST, electrolyte imbalances, and other abnormalities.
Signs of refeeding syndrome include tachycardia, hepatosplenomegaly, peripheral edema, altered mental status, and electrolyte disturbances, specifically, acute or severe hypophosphatemia or hypokalemia.26,45 If refeeding syndrome is suspected, the rate of caloric intake should be reduced or not advanced, fluid intake should be urgently reassessed for volume overload, and supportive care with close monitoring should be provided.
KNOWLEDGE SAVES LIVES
Eating disorders can lead to potentially life-threatening medical complications that require attentive care by the primary care clinician and subspecialist. Without thoughtful consideration, it is easy for even a caring medical team to unintentionally enable patients with these illnesses or to cause active harm in the case of underrecognized pathology.58
Acute medical stabilization on an inpatient unit trained to recognize pathology and treat sequelae can be lifesaving. Arming patients and families with medical knowledge, as provided in the Academy for Eating Disorders’ brochure, “Critical Points for Early Recognition and Medical Risk Management in the Care of Individuals with Eating Disorders”59 can help save patients’ lives.
- Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry 2011; 68:724–731.
- Walsh JM, Wheat ME, Freund K. Detection, evaluation, and treatment of eating disorders the role of the primary care physician. J Gen Intern Med 2000; 15:577–590.
- American Academy of Pediatrics; Committee on Adolescence. Identifying and treating eating disorders. Pediatrics 2003; 111:204–211.
- Rosen DS; American Academy of Pediatrics Committee on Adolescence. Identification and management of eating disorders in children and adolescents. Pediatrics 2010; 126:1240–1253.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington, VA: American Psychiatric Publishing, Incorporated; 2013.
- Eddy KT, Celio Doyle A, Hoste RR, Herzog DB, le Grange D. Eating disorder not otherwise specified in adolescents. J Am Acad Child Adolesc Psychiatry 2008; 47:156–164.
- Muise AM, Stein DG, Arbess G. Eating disorders in adolescent boys: a review of the adolescent and young adult literature. J Adolesc Health 2003; 33:427–435.
- Attia E, Roberto CA. Should amenorrhea be a diagnostic criterion for anorexia nervosa? Int J Eat Disord 2009; 42:581–589.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, fifth edition. http://dsm.psychiatryonline.org/content.aspx?bookid=556§ionid=41101776#103439089. Accessed January 31, 2014.
- Wilfley DE, Bishop ME, Wilson GT, Agras WS. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:S123–S129.
- Wonderlich SA, Gordon KH, Mitchell JE, Crosby RD, Engel SG. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009; 42:687–705.
- Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health 2013: 53:303–305.
- Winston AP, Stafford PJ. Cardiovascular effects of anorexia nervosa. Eur Eat Disord Rev 2000; 8:117–125.
- Galetta F, Franzoni F, Prattichizzo F, Rolla M, Santoro G, Pentimone F. Heart rate variability and left ventricular diastolic function in anorexia nervosa. J Adolesc Health 2003; 32:416–421.
- McCallum K, Bermudez O, Ohlemeyer C, Tyson E, Portilla M, Ferdman B. How should the clinician evaluate and manage the cardiovascular complications of anorexia nervosa? Eat Disord 2006; 14:73–80.
- Akhtar M. Clinical spectrum of ventricular tachycardia. Circulation 1990; 82:1561–1573.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54:1–13.
- The University of Arizona Center for Education and Research on Therapeutics. QT Drug Lists. http://crediblemeds.org/everyone/compos-ite-list-all-qtdrugs/?rf=US. Accessed January 31, 2014.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Romano C, Chinali M, Pasanisi F, et al. Reduced hemodynamic load and cardiac hypotrophy in patients with anorexia nervosa. Am J Clin Nutr 2003; 77:308–312.
- Shamim T, Golden NH, Arden M, Filiberto L, Shenker IR. Resolution of vital sign instability: an objective measure of medical stability in anorexia nervosa. J Adolesc Health 2003; 32:73–77.
- Mont L, Castro J, Herreros B, et al. Reversibility of cardiac abnormalities in adolescents with anorexia nervosa after weight recovery. J Am Acad Child Adolesc Psychiatry 2003; 42:808–813.
- Roberto CA, Mayer LE, Brickman AM, et al. Brain tissue volume changes following weight gain in adults with anorexia nervosa. Int J Eat Disord 2011; 44:406–411.
- Treasure J, Russell G. The case for early intervention in anorexia nervosa: theoretical exploration of maintaining factors. Br J Psychiatry 2011; 199:5–7.
- Hadley SJ, Walsh BT. Gastrointestinal disturbances in anorexia nervosa and bulimia nervosa. Curr Drug Targets CNS Neurol Disord 2003; 2:1–9.
- Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med 2005; 353:1481–1488.
- De Caprio C, Alfano A, Senatore I, Zarrella L, Pasanisi F, Contaldo F. Severe acute liver damage in anorexia nervosa: two case reports. Nutrition 2006; 22:572–575.
- Lawson EA, Klibanski A. Endocrine abnormalities in anorexia nervosa. Nat Clin Pract Endocrinol Metab 2008; 4:407–414.
- Holtkamp K, Mika C, Grzella I, et al. Reproductive function during weight gain in anorexia nervosa. Leptin represents a metabolic gate to gonadotropin secretion. J Neural Transm 2003; 110:427–435.
- Golden NH, Jacobson MS, Schebendach J, Solanto MV, Hertz SM, Shenker IR. Resumption of menses in anorexia nervosa. Arch Pediatr Adolesc Med 1997; 151:16–21.
- Soyka LA, Misra M, Frenchman A, et al. Abnormal bone mineral accrual in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2002; 87:4177–4185.
- Misra M, Klibanski A. Bone metabolism in adolescents with anorexia nervosa. J Endocrinol Invest 2011; 34:324–332.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Biller BM, Saxe V, Herzog DB, Rosenthal DI, Holzman S, Klibanski A. Mechanisms of osteoporosis in adult and adolescent women with anorexia nervosa. J Clin Endocrinol Metab 1989; 68:548–554.
- Hergenroeder AC, Smith EO, Shypailo R, Jones LA, Klish WJ, Ellis K. Bone mineral changes in young women with hypothalamic amenorrhea treated with oral contraceptives, medroxyprogesterone, or placebo over 12 months. Am J Obstet Gynecol 1997; 176:1017–1025.
- Sim LA, McGovern L, Elamin MB, Swiglo BA, Erwin PJ, Montori VM. Effect on bone health of estrogen preparations in premenopausal women with anorexia nervosa: a systematic review and meta-analyses. Int J Eat Disord 2010; 43:218–225.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Misra M, Katzman D, Miller KK, et al. Physiologic estrogen replacement increases bone density in adolescent girls with anorexia nervosa. J Bone Miner Res 2011; 26:2430–2438.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Divasta AD, Feldman HA, Giancaterino C, Rosen CJ, Leboff MS, Gordon CM. The effect of gonadal and adrenal steroid therapy on skeletal health in adolescents and young women with anorexia nervosa. Metabolism 2012; 61:1010–1020.
- Mehler PS. Medical complications of bulimia nervosa and their treatments. Int J Eat Disord 2011; 44:95–104.
- Milosevic A. Eating disorders and the dentist. Br Dent J 1999; 186:109–113.
- Greenfeld D, Mickley D, Quinlan DM, Roloff P. Hypokalemia in outpatients with eating disorders. Am J Psychiatry 1995; 152:60–63.
- Bouquegneau A, Dubois BE, Krzesinski JM, Delanaye P. Anorexia nervosa and the kidney. Am J Kidney Dis 2012; 60:299–307.
- Auron M, Rome E. Anorexia nervosa and bulimia nervosa: what the hospitalist needs to know about CPT 269.9, or nutritional insufficiency. ACP Hospitalist 2011 Sept:28–45.
- Steffen KJ, Mitchell JE, Roerig JL, Lancaster KL. The eating disorders medicine cabinet revisited: a clinician’s guide to ipecac and laxatives. Int J Eat Disord 2007; 40:360–368.
- Roerig JL, Steffen KJ, Mitchell JE, Zunker C. Laxative abuse: epidemiology, diagnosis and management. Drugs 2010; 70:1487–1503.
- Mitchell JE, Boutacoff LI. Laxative abuse complicating bulimia: medical and treatment implications. Int J Eat Disord 1986; 5:325–334.
- Joo JS, Ehrenpreis ED, Gonzalez L, et al. Alterations in colonic anatomy induced by chronic stimulant laxatives: the cathartic colon revisited. J Clin Gastroenterol 1998; 26:283–286.
- Drugs.com. Ipecac syrup. www.drugs.com/monograph/ipecac-syrup.html. Accessed January 31, 2014.
- Peveler RC, Bryden KS, Neil HA, et al. The relationship of disordered eating habits and attitudes to clinical outcomes in young adult females with type 1 diabetes. Diabetes Care 2005; 28:84–88.
- Mannucci E, Rotella F, Ricca V, Moretti S, Placidi GF, Rotella CM. Eating disorders in patients with type 1 diabetes: a meta-analysis. J Endocrinol Invest 2005; 28:417–419.
- Crook MA, Hally V, Panteli JV. The importance of the refeeding syndrome. Nutrition 2001; 17:632–637.
- Fisher M, Golden NH, Katzman DK, et al. Eating disorders in adolescents: a background paper. J Adolesc Health 1995; 16:420–437.
- Kohn MR, Madden S, Clarke SD. Refeeding in anorexia nervosa: increased safety and efficiency through understanding the pathophysiology of protein calorie malnutrition. Curr Opin Pediatr 2011; 23:390–394.
- Garber AK, Michihata N, Hetnal K, Shafer MA, Moscicki AB. A prospective examination of weight gain in hospitalized adolescents with anorexia nervosa on a recommended refeeding protocol. J Adolesc Health 2012; 50:24–29.
- Whitelaw M, Gilbertson H, Lam PY, Sawyer SM. Does aggressive refeeding in hospitalized adolescents with anorexia nervosa result in increased hypophosphatemia? J Adolesc Health 2010; 46:577–582.
- Treasure J, Crane A, McKnight R, Buchanan E, Wolfe M. First do no harm: iatrogenic maintaining factors in anorexia nervosa. Eur Eat Disord Rev 2011; 19:296–302.
- Academy for Eating Disorders (AED). Critical points for early recognition and medical risk management in the care of individuals with eating disorders. http://www.aedweb.org/AM/Template.cfm?Section=Medical_Care_Standards&Template=/CM/ContentDisplay.cfm&ContentID=2413. Accessed January 31, 2014.
- Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Arch Gen Psychiatry 2011; 68:724–731.
- Walsh JM, Wheat ME, Freund K. Detection, evaluation, and treatment of eating disorders the role of the primary care physician. J Gen Intern Med 2000; 15:577–590.
- American Academy of Pediatrics; Committee on Adolescence. Identifying and treating eating disorders. Pediatrics 2003; 111:204–211.
- Rosen DS; American Academy of Pediatrics Committee on Adolescence. Identification and management of eating disorders in children and adolescents. Pediatrics 2010; 126:1240–1253.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. Arlington, VA: American Psychiatric Publishing, Incorporated; 2013.
- Eddy KT, Celio Doyle A, Hoste RR, Herzog DB, le Grange D. Eating disorder not otherwise specified in adolescents. J Am Acad Child Adolesc Psychiatry 2008; 47:156–164.
- Muise AM, Stein DG, Arbess G. Eating disorders in adolescent boys: a review of the adolescent and young adult literature. J Adolesc Health 2003; 33:427–435.
- Attia E, Roberto CA. Should amenorrhea be a diagnostic criterion for anorexia nervosa? Int J Eat Disord 2009; 42:581–589.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, fifth edition. http://dsm.psychiatryonline.org/content.aspx?bookid=556§ionid=41101776#103439089. Accessed January 31, 2014.
- Wilfley DE, Bishop ME, Wilson GT, Agras WS. Classification of eating disorders: toward DSM-V. Int J Eat Disord 2007; 40:S123–S129.
- Wonderlich SA, Gordon KH, Mitchell JE, Crosby RD, Engel SG. The validity and clinical utility of binge eating disorder. Int J Eat Disord 2009; 42:687–705.
- Ornstein RM, Rosen DS, Mammel KA, et al. Distribution of eating disorders in children and adolescents using the proposed DSM-5 criteria for feeding and eating disorders. J Adolesc Health 2013: 53:303–305.
- Winston AP, Stafford PJ. Cardiovascular effects of anorexia nervosa. Eur Eat Disord Rev 2000; 8:117–125.
- Galetta F, Franzoni F, Prattichizzo F, Rolla M, Santoro G, Pentimone F. Heart rate variability and left ventricular diastolic function in anorexia nervosa. J Adolesc Health 2003; 32:416–421.
- McCallum K, Bermudez O, Ohlemeyer C, Tyson E, Portilla M, Ferdman B. How should the clinician evaluate and manage the cardiovascular complications of anorexia nervosa? Eat Disord 2006; 14:73–80.
- Akhtar M. Clinical spectrum of ventricular tachycardia. Circulation 1990; 82:1561–1573.
- Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013; 54:1–13.
- The University of Arizona Center for Education and Research on Therapeutics. QT Drug Lists. http://crediblemeds.org/everyone/compos-ite-list-all-qtdrugs/?rf=US. Accessed January 31, 2014.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Romano C, Chinali M, Pasanisi F, et al. Reduced hemodynamic load and cardiac hypotrophy in patients with anorexia nervosa. Am J Clin Nutr 2003; 77:308–312.
- Shamim T, Golden NH, Arden M, Filiberto L, Shenker IR. Resolution of vital sign instability: an objective measure of medical stability in anorexia nervosa. J Adolesc Health 2003; 32:73–77.
- Mont L, Castro J, Herreros B, et al. Reversibility of cardiac abnormalities in adolescents with anorexia nervosa after weight recovery. J Am Acad Child Adolesc Psychiatry 2003; 42:808–813.
- Roberto CA, Mayer LE, Brickman AM, et al. Brain tissue volume changes following weight gain in adults with anorexia nervosa. Int J Eat Disord 2011; 44:406–411.
- Treasure J, Russell G. The case for early intervention in anorexia nervosa: theoretical exploration of maintaining factors. Br J Psychiatry 2011; 199:5–7.
- Hadley SJ, Walsh BT. Gastrointestinal disturbances in anorexia nervosa and bulimia nervosa. Curr Drug Targets CNS Neurol Disord 2003; 2:1–9.
- Yager J, Andersen AE. Clinical practice. Anorexia nervosa. N Engl J Med 2005; 353:1481–1488.
- De Caprio C, Alfano A, Senatore I, Zarrella L, Pasanisi F, Contaldo F. Severe acute liver damage in anorexia nervosa: two case reports. Nutrition 2006; 22:572–575.
- Lawson EA, Klibanski A. Endocrine abnormalities in anorexia nervosa. Nat Clin Pract Endocrinol Metab 2008; 4:407–414.
- Holtkamp K, Mika C, Grzella I, et al. Reproductive function during weight gain in anorexia nervosa. Leptin represents a metabolic gate to gonadotropin secretion. J Neural Transm 2003; 110:427–435.
- Golden NH, Jacobson MS, Schebendach J, Solanto MV, Hertz SM, Shenker IR. Resumption of menses in anorexia nervosa. Arch Pediatr Adolesc Med 1997; 151:16–21.
- Soyka LA, Misra M, Frenchman A, et al. Abnormal bone mineral accrual in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2002; 87:4177–4185.
- Misra M, Klibanski A. Bone metabolism in adolescents with anorexia nervosa. J Endocrinol Invest 2011; 34:324–332.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Biller BM, Saxe V, Herzog DB, Rosenthal DI, Holzman S, Klibanski A. Mechanisms of osteoporosis in adult and adolescent women with anorexia nervosa. J Clin Endocrinol Metab 1989; 68:548–554.
- Hergenroeder AC, Smith EO, Shypailo R, Jones LA, Klish WJ, Ellis K. Bone mineral changes in young women with hypothalamic amenorrhea treated with oral contraceptives, medroxyprogesterone, or placebo over 12 months. Am J Obstet Gynecol 1997; 176:1017–1025.
- Sim LA, McGovern L, Elamin MB, Swiglo BA, Erwin PJ, Montori VM. Effect on bone health of estrogen preparations in premenopausal women with anorexia nervosa: a systematic review and meta-analyses. Int J Eat Disord 2010; 43:218–225.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Misra M, Katzman D, Miller KK, et al. Physiologic estrogen replacement increases bone density in adolescent girls with anorexia nervosa. J Bone Miner Res 2011; 26:2430–2438.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Divasta AD, Feldman HA, Giancaterino C, Rosen CJ, Leboff MS, Gordon CM. The effect of gonadal and adrenal steroid therapy on skeletal health in adolescents and young women with anorexia nervosa. Metabolism 2012; 61:1010–1020.
- Mehler PS. Medical complications of bulimia nervosa and their treatments. Int J Eat Disord 2011; 44:95–104.
- Milosevic A. Eating disorders and the dentist. Br Dent J 1999; 186:109–113.
- Greenfeld D, Mickley D, Quinlan DM, Roloff P. Hypokalemia in outpatients with eating disorders. Am J Psychiatry 1995; 152:60–63.
- Bouquegneau A, Dubois BE, Krzesinski JM, Delanaye P. Anorexia nervosa and the kidney. Am J Kidney Dis 2012; 60:299–307.
- Auron M, Rome E. Anorexia nervosa and bulimia nervosa: what the hospitalist needs to know about CPT 269.9, or nutritional insufficiency. ACP Hospitalist 2011 Sept:28–45.
- Steffen KJ, Mitchell JE, Roerig JL, Lancaster KL. The eating disorders medicine cabinet revisited: a clinician’s guide to ipecac and laxatives. Int J Eat Disord 2007; 40:360–368.
- Roerig JL, Steffen KJ, Mitchell JE, Zunker C. Laxative abuse: epidemiology, diagnosis and management. Drugs 2010; 70:1487–1503.
- Mitchell JE, Boutacoff LI. Laxative abuse complicating bulimia: medical and treatment implications. Int J Eat Disord 1986; 5:325–334.
- Joo JS, Ehrenpreis ED, Gonzalez L, et al. Alterations in colonic anatomy induced by chronic stimulant laxatives: the cathartic colon revisited. J Clin Gastroenterol 1998; 26:283–286.
- Drugs.com. Ipecac syrup. www.drugs.com/monograph/ipecac-syrup.html. Accessed January 31, 2014.
- Peveler RC, Bryden KS, Neil HA, et al. The relationship of disordered eating habits and attitudes to clinical outcomes in young adult females with type 1 diabetes. Diabetes Care 2005; 28:84–88.
- Mannucci E, Rotella F, Ricca V, Moretti S, Placidi GF, Rotella CM. Eating disorders in patients with type 1 diabetes: a meta-analysis. J Endocrinol Invest 2005; 28:417–419.
- Crook MA, Hally V, Panteli JV. The importance of the refeeding syndrome. Nutrition 2001; 17:632–637.
- Fisher M, Golden NH, Katzman DK, et al. Eating disorders in adolescents: a background paper. J Adolesc Health 1995; 16:420–437.
- Kohn MR, Madden S, Clarke SD. Refeeding in anorexia nervosa: increased safety and efficiency through understanding the pathophysiology of protein calorie malnutrition. Curr Opin Pediatr 2011; 23:390–394.
- Garber AK, Michihata N, Hetnal K, Shafer MA, Moscicki AB. A prospective examination of weight gain in hospitalized adolescents with anorexia nervosa on a recommended refeeding protocol. J Adolesc Health 2012; 50:24–29.
- Whitelaw M, Gilbertson H, Lam PY, Sawyer SM. Does aggressive refeeding in hospitalized adolescents with anorexia nervosa result in increased hypophosphatemia? J Adolesc Health 2010; 46:577–582.
- Treasure J, Crane A, McKnight R, Buchanan E, Wolfe M. First do no harm: iatrogenic maintaining factors in anorexia nervosa. Eur Eat Disord Rev 2011; 19:296–302.
- Academy for Eating Disorders (AED). Critical points for early recognition and medical risk management in the care of individuals with eating disorders. http://www.aedweb.org/AM/Template.cfm?Section=Medical_Care_Standards&Template=/CM/ContentDisplay.cfm&ContentID=2413. Accessed January 31, 2014.
KEY POINTS
- The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5), released in 2013, has updated the criteria for some eating disorders and has added some new disorders.
- Starvation can cause cardiac, cerebral, gastrointestinal, and endocrine problems.
- Purging can lead to problems with oral health, electrolyte imbalances, and even renal failure.
- Refeeding poses the risk of refeeding syndrome, with fluid overload and electrolyte imbalances. Many patients undergoing refeeding are best managed in the hospital.
Choosing Wisely in Hospital Medicine
The overuse of medical tests and treatments is a growing concern. A recent survey revealed that 2 in 5 primary care physicians perceive that patients in their own practice are receiving too much care.[1] Twenty‐eight percent of the physicians indicated they provide more care than they should. When queried about reasons for the aggressiveness of care, responses included fear of malpractice litigation, adherence to clinical performance measures that require following protocols, and inadequacy of time spent with patients. Overutilization of healthcare resources is a complex issue promulgated not only by the factors cited by the physicians but also a culture in the United States habituated to believe more care is better care.[2, 3, 4] In 2010, $2.6 trillion was spent on healthcare, an increase of $1.3 trillion between 2000 and 2010.5 As much as 30% of healthcare spending may be wasted.[6] Because physicians influence approximately 80% of healthcare expenditures, including ordering tests and treatments, it is imperative that physicians take a leadership role in reversing this trend.[7]
In response to this need, several physician‐led projects have emerged.[8, 9, 10] One such initiative is the American Board of Internal Medicine Foundation's (ABIM‐F's) Choosing Wisely campaign.[11] The ABIM‐F contacted a variety of specialty societies and asked each to identify the 5 top tests or treatments relevant to their specialty that may frequently be overused. Phase 1 of the Choosing Wisely campaign was launched in April 2012 with 9 specialty societies participating. The second phase was unveiled in February 2013 and comprised of 16 additional groups including the Society of Hospital Medicine (SHM). The SHM represents 35,000 hospitalists in the United States whose primary focus is the general medical care of hospitalized patients. This is especially important because almost one‐third of total US healthcare expenditures are on hospital care,[12] and hospitalists care for an increasing number of hospitalized patients.[13] In this article, we describe the used to derive the adult hospital medicine Choosing processes Wisely list, review the tests and treatments that the SHM's Choosing Wisely Subcommittee chose, and discuss potential next steps in implementation of the adult hospital medicine recommendations.
METHODOLOGY
Upon invitation to participate in the Choosing Wisely campaign, SHM's Hospital Quality and Patient Safety (HQPS) Committee formally convened the Choosing Wisely Subcommittee. The subcommittee identified and executed a methodology (see Supporting Figure 1 and Supporting Table 1 in the online version of this article) to create the list of 5 tests and treatments that the SHM submitted to the ABIM‐F. All subcommittee members participated fully in the voting and refinement process. The Choosing Wisely Subcommittee worked closely with the SHM's Pediatrics Choosing Wisely Subcommittee to develop both adult and pediatric lists.
Convening the Choosing Wisely Subcommittee
The HQPS Committee convened a subcommittee consisting of 9 members. The subcommittee represented a diverse group of hospitalists reflecting different institution types, geographic regions, and experience. All Choosing Wisely Subcommittee members signed conflict of interest statements and reported no conflict related to the conclusions, implications, or opinions stated. The subcommittee did not consult other external stakeholders in the development of recommendations.
Identification and Refinement of Potential Wasteful Practices
To generate an initial list of potential recommendations, members of all of the SHM committees were surveyed and asked to submit 5 tests and treatments that are inappropriately used or overused. SHM staff removed duplicates and categorized submissions by topic, highlighting overlapping recommendations. Tests and treatments that are used infrequently and items included in phase 1 society lists were also excluded. Subcommittee members then ranked the resultant list using a 5‐point Likert scale. All SHM members were then given the opportunity to rank their agreement with the tests and treatments on the list, as refined at the time based upon their own experience and consideration of the following criteria: tests and procedures within the control and purview of hospital medicine, the frequency with which the tests or procedures occur, and the significance of associated costs. This was accomplished via electronic survey.
Establishing an Evidence Base
SHM staff conducted a literature review of the list of tests and treatments that was further refined by the SHM membership's ranking using a standard template. Two reviewers (W.N. and J.G.) conducted an independent literature review of the remaining tests and treatments using PubMed, MEDLINE, and Cochrane Library. The reviewers also conducted generic Internet searches. The literature review included all literature published through 2012 as well as nonEnglish language publications. The reviewers included clinical research guidelines and primary and secondary research studies. Studies included in the review were based upon common criteria including whether the article discussed an evaluation of efficacy and/or utility of treatment, reviewed the harm associated with the administration of a test or treatment, and explored the cost associated with the test or treatment as well as the overall strength of evidence. Additionally, the reference lists included in articles were reviewed to identify supplementary literature sources. The reviewers read and analyzed the articles identified in the initial search for relevant subject matter and summarized the findings in a table.
Delphi Panels
A Delphi scoring process was utilized to complete list refinement.[14] Subcommittee members anonymously voted via email for the strength of the test and treatment recommendation based upon specific criteria. To assist with this process, they received a copy of the completed literature review and an evidence summary of the literature. The following categories were used to guide the scoring: validity/evidence base to support, feasibility of implementation, frequency of occurrence, cost of occurrence, yield/emmpact, harm, and potential to improve. Results were aggregated and shared with the Choosing Wisely Subcommittee. The subcommittee conferred a final time, editing the recommendations for clarification and improved wording. A second anonymous vote was then conducted for the remaining tests and treatments through a revised scoring spreadsheet. The penultimate list was presented to the SHM's Board. Upon the Board's approval, the final list was submitted to the ABIM‐F.
RESULTS
The results of each stage of the list development process are shown in the online supporting information (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). The initial survey of SHM committee members garnered in excess of 150 tests and treatments from approximately 40 SHM committee members. The subsequent list refinement by SHM staff narrowed this list to 65 items, which were then further reduced to 15 items after ranking by members of the subcommittee (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). Voting by members of the general SHM membership further reduced the list to 11 tests and treatments.
The final list of 5 tests and treatments submitted to the ABIM‐F were:
- Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
- Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for gastrointestinal (GI) complications.
- Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
- Do not order continuous telemetry monitoring outside of the intensive care unit (ICU) without using a protocol that governs continuation.
- Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability (Table 1).
|
| Test/Treatment Recommendations |
| Do not place, or leave in place, urinary catheters for incontinence or convenience, or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days or urologic procedures; use weights instead to monitor diuresis).[21, 50] |
| Do not prescribe GI prophylaxis to medical inpatients without clear‐cut indication or high risk for GI complication.[24] |
| Avoid transfusing red blood cells just because hemoglobin levels are below arbitrary thresholds such as 10, 9, or even 8 mg/dL in the absence of symptoms.[29, 51] |
| Avoid overuse/unnecessary use of telemetry monitoring in the hospital, particularly for patients at low risk for adverse cardiac outcomes.[35, 43, 52, 53] |
| Do not perform repetitive CBC and chemistry testing in the face of clinical and lab stability.[44, 54, 55] |
RECOMMENDATIONS
Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
Despite guidelines identifying appropriate indications for the placement of urinary catheters, urinary tract infections due to catheter use remain the most frequent type of infection in acute care settings. Nearly 1 in every 5 patients in the hospital receives an indwelling catheter, and up to half are placed inappropriately.[15] Twenty‐six percent of patients who have indwelling catheters for 2 to 10 days will develop bacteriuria; subsequently, 24% of those patients will develop a catheter‐associated urinary tract infection (CAUTI).[15] More than 13,000 deaths due to CAUTI occur annually.[16] In addition to urinary tract infections and their complications, additional adverse outcomes related to indwelling catheters include formation of encrustations and restrictions to flow, prolonged hospital stay, and exposure to multidrug resistant organisms due to increased use of antibiotics. Evidence suggests that infections due to catheters are frequently preventable.[17, 18]
The economic burden associated with indwelling catheter complications is also substantial. Each episode of symptomatic urinary tract infection adds $676 in incremental costs, and catheter‐related bacteremia costs at least $2836.15 According to Scott, nearly 450,000 CAUTIs were estimated to have occurred in 2007, resulting in direct medical costs of between $340 to $370 million.[19]
Several organizations simultaneously released guidelines to provide a roadmap for appropriate catheter use and prevention of CAUTIs.[20, 21] Despite explicit guidelines, the Centers for Disease Control and Prevention recently reported that there was no improvement in CAUTIs between 2010 and 2011.[22] Implementing these strategies for CAUTI reduction include establishing a multidisciplinary team that applies a clear protocol, with daily reminders about catheters and stop orders for catheter discontinuation.
Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for GI complications.
Stress ulcer prophylaxis in the hospital with proton pump inhibitors (PPIs) or histamine‐2 antagonists are common. As many as 71% of patients admitted to the hospital receive some form of prophylaxis without appropriate indication.[23] Guidelines exist for appropriate use; however, therapy is commonly used in the inpatient setting for indications not investigated or supported by the literature.[24]
Inappropriate prescribing practices have been associated with multiple adverse events, including drug interactions, hospital‐acquired infections, and increased costs of care. Although consensus among physicians regarding whether GI prophylaxis causes harm is lacking, studies demonstrate a strong correlation between use of PPIs and common adverse events such as pneumonia and Clostridium difficile infection.[25, 26] For instance, inpatients receiving PPIs were 3.6 times more likely to develop C. difficile‐associated diarrhea than inpatients not exposed to PPIs.[27]
The American Society of Health‐System Pharmacists Therapeutic Guidelines on Stress Ulcer Prophylaxis provide guidance regarding the optimal indication for administration of acid‐suppression medication for patients in the hospital setting. The clinical guidelines specify that stress ulcer prophylaxis is not recommended for adult patients in non‐ICU settings. The recommendations are applicable to general medical and surgical patients with fewer than 2 risk factors for clinically important bleeding. Indications for use of stress ulcer prophylaxis in the ICU include coagulopathy and mechanical ventilation.[24]
Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
Anemia is a frequent comorbid condition in hospitalized patients. Correcting anemia by means of allogeneic blood transfusions with the goal of maximizing oxygen delivery is common practice in many hospitals. Varied threshold levels of hemoglobin and hematocrit are used, which is unsupported by evidence.[28, 29]
Acute anemia with normovolemic hemodilution has been proven safe in patients with coronary artery disease, heart valve disease, and the elderly. A restrictive transfusion approach with hemoglobin cutoff of 7 g/dL, as opposed to higher thresholds, has shown improved outcomes (lower mortality and lower rate of rebleeding) in adult and pediatric critical care as well as surgical patients.[30] Large studies in patients with acute myocardial infarction demonstrated that restrictive transfusional strategies are associated with decreased in‐hospital mortality, rate of reinfarction, and worsening heart failure, as well as 30‐day mortality.[31] A randomized trial in patients with active GI bleeding showed that a restrictive strategy of hemoglobin threshold of 7 g/dL was associated with improved outcomes (less mortality, less rate of rebleeding), compared with a strategy to transfuse patients with hemoglobin less than 9 g/dL.[32] In addition, increased awareness of the high cost of blood ($700$900 per unit) associated with the blood banking process as well as risk of potential infectious and noninfectious adverse reactions (eg, human immunodeficiency virus, hepatitis C virus, transfusion‐related lung injury, transfusion‐related circulatory overload) must be considered in the risk/benefit equation.[28]
Based on current available evidence, the American Association of Blood Banks recommends adhering to a restrictive transfusion strategy (7 g/dL) in hospitalized stable patients, and this threshold is raised to 8 g/dL in patients with preexisting cardiovascular disease or with active symptoms.[28] This should be combined with techniques such as preoperative anemia optimization by hematinics replacement (eg, iron, vitamin B12, folate, erythropoietin), intraoperative strategies (eg, antifibrinolytics, hypotension, normovolemic hemodilution, etc.), and postoperative strategies (eg, intraoperative cell salvage). These strategies have been shown to result in parsimonious red blood cell utilization as well as in substantial healthcare cost savings.[33]
Do not order continuous telemetry monitoring outside of the ICU without using a protocol that governs continuation.
Telemetry use in the hospital is common and clearly has a role for patients with certain cardiac conditions and those at risk for cardiac events. Telemetry is resource intensive, requiring dedicated multidisciplinary staff with specialized training. Many hospitals lack the ability to maintain and staff telemetry beds.[34] Physicians may overestimate the role of telemetry in guiding patient management.[35] One study concluded that only 12.6% of patients on a non‐ICU cardiac telemetry unit required telemetric monitoring, and only 7% received modified management as a result of telemetry findings.[36]
Inappropriate utilization of telemetry can be linked to increased length of stay or boarding in the emergency department, reduced hospital throughput, increased ambulance diversion, and increased operational costs.[37] In addition, the use of telemetry can lead to a false sense of security and alarm fatigue.[38] Telemetry artifacts may result in unnecessary testing and procedures for patients.[39] Furthermore, to accommodate the need for telemetry, frequent room changes may occur that may lead to decreased patient satisfaction. Low‐risk chest pain patients (hemodynamically stable with negative biomarkers, no electrocardiogram changes, and no indication for invasive procedure) do not require telemetry monitoring, because it rarely affects direct care of these patients.[36, 40] A 2009 study concluded that telemetry monitoring does not affect the care or the outcome of low‐risk patients.[41] Patients with other diagnoses, such as chronic obstructive pulmonary disease exacerbation or hemodynamically stable pulmonary embolism, and those requiring blood transfusions, are often placed in monitored beds without evidence that this will impact their care.[37]
The American Heart Association has published guidelines on the use of cardiac telemetry.[35] Patients are risk stratified into 3 categories, with class III patients being those who are low risk and do not require telemetry. Seventy percent of patients with the top 10 diagnoses that were admitted from the emergency department may clinically warrant telemetry.[37] Implementing a systematic evidence‐based approach to telemetry use can decrease unnecessary telemetry days,[42] reduce costs, and avoid unnecessary testing for rhythm artifacts.[39, 43]
Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability.
Although unnecessary laboratory testing is widely perceived as ineffective and wasteful, no national guideline or consensus statement exists regarding the utility or timing of repetitive laboratory testing. Multiple studies showed no difference in readmission rates, transfers to ICUs, lengths of stay, rates of adverse events, or mortality when the frequency of laboratory testing was reduced. Charges for daily laboratory testing were estimated to be $150/patient/day.[44] In a study at a university‐associated teaching hospital, an intervention to reduce the frequency of laboratory testing was associated with a total decrease of nearly 98,000 tests over a 3‐year period.[45] The cost savings in this study was estimated to be almost $2 million over the same time period. A second study at a teaching hospital, involving a computerized physician order entry (CPOE)‐based intervention, showed a reduction of almost 72,000 tests over a 1‐year period, which reduced the total number of inpatient phlebotomies by approximately 21%.[46]
The cost of routine, daily laboratory testing for a given patient or health system is not insignificant. When healthcare providers are made aware of the cost of daily laboratory testing, this might reduce the number of laboratory tests ordered and result in significant savings for a health system, as well as improve the patient experience.[44]
Developing guidelines or strategies to reduce repetitive laboratory testing in the face of clinical or laboratory stability would likely produce significant cost savings for both the individual patient as well as the health system, and could possibly would likely improve the hospital experience for many patients. Widespread adoption of CPOE by the US healthcare system has the potential to facilitate decision support that can change laboratory ordering practices.
DISCUSSION
Eliminating waste in healthcare is a priority for physicians,[6, 7, 8, 9, 10] and the ABIM‐F's Choosing Wisely campaign is a key component of this effort.[11] The SHM chose 5 tests and treatments relevant to the specialty of hospital medicine that occur at a high frequency, have significant cost and affect to patients, and that can feasibly be impacted. Given that a high percentage of healthcare costs occur in the hospital[5] and hospitalists care for an increasing number of these patients,[13] successful implementation of the SHM's adult hospital medicine Choosing Wisely list has great potential to decrease waste in the hospital, reduce harm, and improve patient outcomes.
The methodology chosen to develop the adult Choosing Wisely recommendations was intended to be both pragmatic and evidence based. A broad range of opinions was solicited, including from the SHM's general membership. The final refinement included a literature review and a Delphi process.
Review of cost and utilization data to determine the scope of the problem was used for decision‐making by subcommittee members to formulate the SHM's recommendations. For some recommendations, there were significant data, whereas for others, this information was sparse. As has been noted, we were unable to identify the total number of patients in the United States who receive telemetry on an annual basis, and thus were unable to make an estimate about the total population that would be impacted by improved utilization. However, several studies do indicate inappropriate use in significant patient populations and widespread use of the resource. Similarly, we were able to identify the costs associated with a CBC, but were unable to calculate the total number of CBCs administered annually. In the absence of these data, subcommittee members utilized other criteria, including frequency of test or treatment, patient harm or benefit, and utility for making treatment/management decisions.
In general, the tests and treatments contained in the adult hospital medicine Choosing Wisely list are not requested by patients. As such, physicians' choices play a greater role, potentially magnifying the impact hospitalists could make. Overuse of medical tests is multifactorial, and culture plays a significant role in the United States.[2, 3, 4] Although each of the tests and treatments identified by the SHM is within the purview of hospitalists, ensuring that guidelines are reliably followed will require interdisciplinary process changes. Ample opportunity exists to partner with nurses (urinary catheters and telemetry), pharmacists (stress ulcer prophylaxis), blood banks, and laboratories (transfusions and lab testing), as well as other healthcare providers and physicians in multiple specialties.
Successful implementation of each guideline will require improvement of systems within hospitals to drive reliability.[47] Provider education, training programs, protocols and reminders may prove to be significant catalysts in overcoming misinformation or no information about specific guidelines. More importantly, interdisciplinary teams will need to assess the current practice patterns within their hospitals prior to implementing solutions that standardize and automate the ordering processes for these tests and treatments.[48] Additionally, the culture within individual patient care units will need to be modified.[49] The challenge of changing the behavior of multiple stakeholders and hardwiring systems changes represent significant potential barriers to success.
There are several potential concerns with the recommendations. Concepts such as high risk and clinical stability exist in several of the recommendations. In most cases, specific guidelines exist that explicitly define the appropriate use of the test or treatment. Where they do not, implementers will need to define the operational definitions, such as the number of normal CBCs that define stability. Although the recommendations are based on the best evidence available, consensus still plays a role. As has been noted, the risk of malpractice litigation influences physicians' decisions.[1] Although evidence‐based recommendations such as these help shape the standard of care and mitigate risk, they may not completely eliminate this concern. Providers should always weigh the risks and benefits of any test or treatment. Finally, the approach taken to establish the list was both pragmatic and evidence based. Published evidence was not reviewed until the list was honed to 11. When the evidence was reviewed, the strength of the evidence was judged in a subjective manner by members of the committee as part of the Delphi panel voting.
CONCLUSION
As healthcare providers enter an era of more cost conscious decision‐making about provision of care based upon necessity, hospitalists have an excellent opportunity to impact overutilization. The 5 recommendations comprising the adult hospital medicine Choosing Wisely list offer an explicit starting point. The SHM hopes to lead this process during the coming months and years and to offer additional recommendations, providing a foundation for hospitalists to decrease unnecessary tests and treatments and improve healthcare value.
Acknowledgments
The authors thank the additional members of the Choosing Wisely subcommittee of the SHM's Healthcare Quality and Patient Safety Committee: Krishna Das, MD; Shelley Taylor, MD; Kevin O'Leary, MD; and Nasim Afsarmanesh, MD. The authors also thank SHM staff who were involved in all facets of the recommendation development process, particularly Brendon Shank, who provided significant input into the survey and dissemination process.
Disclosure
Nothing to report.
- , , . Too little? Too much? Primary care physicians' views on US health care: a brief report. Arch Intern Med. 2011;171:1582–1585.
- , , , et al. Evidence that consumers are skeptical about evidence‐based health care. Health Aff (Millwood). 2010;29:1400–1406.
- , . Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801–1802.
- Too much treatment? Aggressive medical care can lead to more pain, with no gain. Consum Rep. 2008;73:40–44.
- Centers for Medicare and Medicaid Services. Historical national health expenditure data. Available at: http://www.cms.gov/Research‐Statistics‐Data‐and‐Systems/Statistics‐Trends‐and‐Reports/NationalHealthExpendData/NationalHealthAccountsHistorical.html. Accessed February 12, 2013.
- , . Eliminating waste in US health care. JAMA. 2012;307:1513–1516.
- . Change the microenvironment: delivery system reform essential to controlling costs. Available at: http://www.commonwealthfund.org/Publications/Commentaries/2009/Apr/Change‐the‐Microenvironment.aspx. Accessed February 12, 2013.
- Costs of care. Available at: http://www.costsofcare.org. Accessed February 12, 2013.
- , . Less is more: how less health care can result in better health. Arch Intern Med. 2010;170:749–750.
- , , , ; Clinical Guidelines Committee of the American College of Physicians. High‐value, cost‐conscious health care: concepts for clinicians to evaluate the benefits, harms, and costs of medical interventions. Ann Intern Med. 2011;154:174–180.
- ABIM Foundation. U.S. physician groups identify commonly used tests or procedures they say are often not necessary. Available at: http://www.abimfoundation.org/News/ABIM‐Foundation‐News/2012/Choosing‐Wisely.aspx. Accessed February 12, 2013.
- , . Addressing requests by patients for nonbeneficial interventions. JAMA. 2012;307:149–150.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360:1102–1112.
- , , , . The appropriateness method has acceptable reliability and validity for assessing overuse and underuse of surgical procedures. J Clin Epidemiol. 2012;65:1133–1143.
- . Clinical and economic consequences of nosocomial catheter‐related bacteriuria. Am J Infect Control. 2000;28:68–75.
- , , , et al. Estimating health care‐associated infections and deaths in U.S. hospitals, 2002. Public Health Rep. 2007;122:160–166.
- , , , et al. A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1): S12–S21.
- , , , et al. Strategies to prevent catheter‐associated urinary tract infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1):S41–S50.
- . The direct medical costs of healthcare‐associated infections in U.S. hospitals and the benefits of prevention. Available at: http://www.cdc.gov/hai/pdfs/hai/scott_costpaper.pdf. Accessed February 12, 2013.
- , , , , . Healthcare Infection Control Practices Advisory Committee, guideline for prevention of catheter‐associated urinary tract infections 2009. Infect Control Hosp Epidemiol. 2010;31:319–326.
- , , , et al. Diagnosis, prevention, and treatment of catheter‐associated urinary tract infection in adults: 2009 International Clinical Practice Guidelines from the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:625–663.
- , , , et al. 2011 National and State Healthcare‐Associated Infection Standardized Infection Ratio Report. Available at: http://www.cdc.gov/hai/pdfs/SIR/SIR‐Report_02_07_2013.pdf. Accessed February 13, 2013.
- , . Stress ulcer prophylaxis in hospitalized patients not in intensive care units. Am J Health Syst Pharm. 2007;64:1396–1400.
- . ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm. 1999;56:347–379.
- , , , , , . Risk of community‐acquired pneumonia and use of gastric acid‐suppressive drugs. JAMA. 2004;292:1955–1960.
- , , , . Acid‐suppressive medication use and the risk for hospital‐acquired pneumonia. JAMA. 2009;301:2120–2128.
- , , , . Gastric acid suppression by proton pump inhibitors as a risk factor for clostridium difficile‐associated diarrhea in hospitalized patients. Am J Gastroenterol. 2008;103:2308–2313.
- , , , et al. Red blood cell transfusion: a clinical practice guideline from the AABB*. Ann Intern Med. 2012;157:49–58.
- , , , et al. Guidelines for the clinical use of red cell transfusions. Br J Haematol. 2001;113:24–31.
- , , , . Mortality and morbidity in patients with very low postoperative Hb levels who decline blood transfusion. Transfusion. 2002;42:812–818.
- , , , , . Association of blood transfusion with increased mortality in myocardial infarction: a meta‐analysis and diversity‐adjusted study sequential analysis. JAMA Intern Med 2013;173:132–139.
- , , , et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368:11–21.
- , , , . Economic impact of inappropriate blood transfusions in coronary artery bypass graft surgery. Am J Med. 1993;94:509–514.
- , , , , , . The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97:1483–1487.
- , , , et al. ACC/AHA guidelines for ambulatory electrocardiography: executive summary and recommendations. A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee to Revise the Guidelines for Ambulatory Electrocardiography). Circulation. 1999;100:886–893.
- , , , et al. Role of telemetry monitoring in the non‐intensive care unit. Am J Cardiol. 1995;76:960–965.
- , . When do patients need admission to a telemetry bed? J Emerg Med. 2007;33:53–60.
- , . Electrocardiographic monitoring in the hospitalized patient: a diagnostic intervention of uncertain clinical impact. Am J Emerg Med. 2008;26:1047–1055.
- , , , , . Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med. 1999;341:1270–1274.
- , , , . Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in‐hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci. 2005;60:605–606.
- , , , , . Telemetry monitoring guidelines for efficient and safe delivery of cardiac rhythm monitoring to noncritical hospital inpatients. Crit Pathw Cardiol. 2009;8:125–126.
- Agency for Healthcare Research and Quality. Winawer N. Redesign of telemetry unit admission and transfer criteria leads to improved patient flow and reduced emergency department waiting times. Available at: http://www.innovations.ahrq.gov/content.aspx?id=2239. Accessed February 12, 2013.
- , , , et al. Is telemetry monitoring necessary in low‐risk suspected acute chest pain syndromes? Chest. 2002;122:517–523.
- , . Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg. 2011;146:524–527.
- , , , et al. A cost‐effective method for reducing the volume of laboratory tests in a university‐associated teaching hospital. Mt Sinai J Med. 2006;73:787–794.
- , , , et al. Reducing unnecessary inpatient laboratory testing in a teaching hospital. Am J Clin Pathol. 2006;126:200–206.
- . Making noncatastrophic health care processes reliable: learning to walk before running in creating high‐reliability organizations. Health Serv Res. 2006;41:1677–1689.
- , , , et al. What have we learned about interventions to reduce medical errors? Annu Rev Public Health. 2010;31:479–497.
- , . Safe Patients, Smart Hospitals: How One Doctor's Checklist Can Help Us Change Health Care From The Inside Out. New York, NY: Hudson Street Press; 2010.
- , , , , . Catheter‐associated urinary tract infection and the Medicare rule changes. Ann Intern Med. 2009;150:877–884.
- Consensus conference. Perioperative red blood cell transfusion. JAMA. 1988;260:2700–2703.
- , , , , , . Is telemetry overused? Is it as helpful as thought? Cleve Clin J Med. 2009;76:368–372.
- , , , et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711.
- , , , et al. Diagnostic blood loss from phlebotomy and hospital‐acquired anemia during acute myocardial infarction. Arch Intern Med. 2011;171:1646–1653.
- , , , , . Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med. 2005;20:520–524.
The overuse of medical tests and treatments is a growing concern. A recent survey revealed that 2 in 5 primary care physicians perceive that patients in their own practice are receiving too much care.[1] Twenty‐eight percent of the physicians indicated they provide more care than they should. When queried about reasons for the aggressiveness of care, responses included fear of malpractice litigation, adherence to clinical performance measures that require following protocols, and inadequacy of time spent with patients. Overutilization of healthcare resources is a complex issue promulgated not only by the factors cited by the physicians but also a culture in the United States habituated to believe more care is better care.[2, 3, 4] In 2010, $2.6 trillion was spent on healthcare, an increase of $1.3 trillion between 2000 and 2010.5 As much as 30% of healthcare spending may be wasted.[6] Because physicians influence approximately 80% of healthcare expenditures, including ordering tests and treatments, it is imperative that physicians take a leadership role in reversing this trend.[7]
In response to this need, several physician‐led projects have emerged.[8, 9, 10] One such initiative is the American Board of Internal Medicine Foundation's (ABIM‐F's) Choosing Wisely campaign.[11] The ABIM‐F contacted a variety of specialty societies and asked each to identify the 5 top tests or treatments relevant to their specialty that may frequently be overused. Phase 1 of the Choosing Wisely campaign was launched in April 2012 with 9 specialty societies participating. The second phase was unveiled in February 2013 and comprised of 16 additional groups including the Society of Hospital Medicine (SHM). The SHM represents 35,000 hospitalists in the United States whose primary focus is the general medical care of hospitalized patients. This is especially important because almost one‐third of total US healthcare expenditures are on hospital care,[12] and hospitalists care for an increasing number of hospitalized patients.[13] In this article, we describe the used to derive the adult hospital medicine Choosing processes Wisely list, review the tests and treatments that the SHM's Choosing Wisely Subcommittee chose, and discuss potential next steps in implementation of the adult hospital medicine recommendations.
METHODOLOGY
Upon invitation to participate in the Choosing Wisely campaign, SHM's Hospital Quality and Patient Safety (HQPS) Committee formally convened the Choosing Wisely Subcommittee. The subcommittee identified and executed a methodology (see Supporting Figure 1 and Supporting Table 1 in the online version of this article) to create the list of 5 tests and treatments that the SHM submitted to the ABIM‐F. All subcommittee members participated fully in the voting and refinement process. The Choosing Wisely Subcommittee worked closely with the SHM's Pediatrics Choosing Wisely Subcommittee to develop both adult and pediatric lists.
Convening the Choosing Wisely Subcommittee
The HQPS Committee convened a subcommittee consisting of 9 members. The subcommittee represented a diverse group of hospitalists reflecting different institution types, geographic regions, and experience. All Choosing Wisely Subcommittee members signed conflict of interest statements and reported no conflict related to the conclusions, implications, or opinions stated. The subcommittee did not consult other external stakeholders in the development of recommendations.
Identification and Refinement of Potential Wasteful Practices
To generate an initial list of potential recommendations, members of all of the SHM committees were surveyed and asked to submit 5 tests and treatments that are inappropriately used or overused. SHM staff removed duplicates and categorized submissions by topic, highlighting overlapping recommendations. Tests and treatments that are used infrequently and items included in phase 1 society lists were also excluded. Subcommittee members then ranked the resultant list using a 5‐point Likert scale. All SHM members were then given the opportunity to rank their agreement with the tests and treatments on the list, as refined at the time based upon their own experience and consideration of the following criteria: tests and procedures within the control and purview of hospital medicine, the frequency with which the tests or procedures occur, and the significance of associated costs. This was accomplished via electronic survey.
Establishing an Evidence Base
SHM staff conducted a literature review of the list of tests and treatments that was further refined by the SHM membership's ranking using a standard template. Two reviewers (W.N. and J.G.) conducted an independent literature review of the remaining tests and treatments using PubMed, MEDLINE, and Cochrane Library. The reviewers also conducted generic Internet searches. The literature review included all literature published through 2012 as well as nonEnglish language publications. The reviewers included clinical research guidelines and primary and secondary research studies. Studies included in the review were based upon common criteria including whether the article discussed an evaluation of efficacy and/or utility of treatment, reviewed the harm associated with the administration of a test or treatment, and explored the cost associated with the test or treatment as well as the overall strength of evidence. Additionally, the reference lists included in articles were reviewed to identify supplementary literature sources. The reviewers read and analyzed the articles identified in the initial search for relevant subject matter and summarized the findings in a table.
Delphi Panels
A Delphi scoring process was utilized to complete list refinement.[14] Subcommittee members anonymously voted via email for the strength of the test and treatment recommendation based upon specific criteria. To assist with this process, they received a copy of the completed literature review and an evidence summary of the literature. The following categories were used to guide the scoring: validity/evidence base to support, feasibility of implementation, frequency of occurrence, cost of occurrence, yield/emmpact, harm, and potential to improve. Results were aggregated and shared with the Choosing Wisely Subcommittee. The subcommittee conferred a final time, editing the recommendations for clarification and improved wording. A second anonymous vote was then conducted for the remaining tests and treatments through a revised scoring spreadsheet. The penultimate list was presented to the SHM's Board. Upon the Board's approval, the final list was submitted to the ABIM‐F.
RESULTS
The results of each stage of the list development process are shown in the online supporting information (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). The initial survey of SHM committee members garnered in excess of 150 tests and treatments from approximately 40 SHM committee members. The subsequent list refinement by SHM staff narrowed this list to 65 items, which were then further reduced to 15 items after ranking by members of the subcommittee (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). Voting by members of the general SHM membership further reduced the list to 11 tests and treatments.
The final list of 5 tests and treatments submitted to the ABIM‐F were:
- Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
- Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for gastrointestinal (GI) complications.
- Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
- Do not order continuous telemetry monitoring outside of the intensive care unit (ICU) without using a protocol that governs continuation.
- Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability (Table 1).
|
| Test/Treatment Recommendations |
| Do not place, or leave in place, urinary catheters for incontinence or convenience, or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days or urologic procedures; use weights instead to monitor diuresis).[21, 50] |
| Do not prescribe GI prophylaxis to medical inpatients without clear‐cut indication or high risk for GI complication.[24] |
| Avoid transfusing red blood cells just because hemoglobin levels are below arbitrary thresholds such as 10, 9, or even 8 mg/dL in the absence of symptoms.[29, 51] |
| Avoid overuse/unnecessary use of telemetry monitoring in the hospital, particularly for patients at low risk for adverse cardiac outcomes.[35, 43, 52, 53] |
| Do not perform repetitive CBC and chemistry testing in the face of clinical and lab stability.[44, 54, 55] |
RECOMMENDATIONS
Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
Despite guidelines identifying appropriate indications for the placement of urinary catheters, urinary tract infections due to catheter use remain the most frequent type of infection in acute care settings. Nearly 1 in every 5 patients in the hospital receives an indwelling catheter, and up to half are placed inappropriately.[15] Twenty‐six percent of patients who have indwelling catheters for 2 to 10 days will develop bacteriuria; subsequently, 24% of those patients will develop a catheter‐associated urinary tract infection (CAUTI).[15] More than 13,000 deaths due to CAUTI occur annually.[16] In addition to urinary tract infections and their complications, additional adverse outcomes related to indwelling catheters include formation of encrustations and restrictions to flow, prolonged hospital stay, and exposure to multidrug resistant organisms due to increased use of antibiotics. Evidence suggests that infections due to catheters are frequently preventable.[17, 18]
The economic burden associated with indwelling catheter complications is also substantial. Each episode of symptomatic urinary tract infection adds $676 in incremental costs, and catheter‐related bacteremia costs at least $2836.15 According to Scott, nearly 450,000 CAUTIs were estimated to have occurred in 2007, resulting in direct medical costs of between $340 to $370 million.[19]
Several organizations simultaneously released guidelines to provide a roadmap for appropriate catheter use and prevention of CAUTIs.[20, 21] Despite explicit guidelines, the Centers for Disease Control and Prevention recently reported that there was no improvement in CAUTIs between 2010 and 2011.[22] Implementing these strategies for CAUTI reduction include establishing a multidisciplinary team that applies a clear protocol, with daily reminders about catheters and stop orders for catheter discontinuation.
Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for GI complications.
Stress ulcer prophylaxis in the hospital with proton pump inhibitors (PPIs) or histamine‐2 antagonists are common. As many as 71% of patients admitted to the hospital receive some form of prophylaxis without appropriate indication.[23] Guidelines exist for appropriate use; however, therapy is commonly used in the inpatient setting for indications not investigated or supported by the literature.[24]
Inappropriate prescribing practices have been associated with multiple adverse events, including drug interactions, hospital‐acquired infections, and increased costs of care. Although consensus among physicians regarding whether GI prophylaxis causes harm is lacking, studies demonstrate a strong correlation between use of PPIs and common adverse events such as pneumonia and Clostridium difficile infection.[25, 26] For instance, inpatients receiving PPIs were 3.6 times more likely to develop C. difficile‐associated diarrhea than inpatients not exposed to PPIs.[27]
The American Society of Health‐System Pharmacists Therapeutic Guidelines on Stress Ulcer Prophylaxis provide guidance regarding the optimal indication for administration of acid‐suppression medication for patients in the hospital setting. The clinical guidelines specify that stress ulcer prophylaxis is not recommended for adult patients in non‐ICU settings. The recommendations are applicable to general medical and surgical patients with fewer than 2 risk factors for clinically important bleeding. Indications for use of stress ulcer prophylaxis in the ICU include coagulopathy and mechanical ventilation.[24]
Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
Anemia is a frequent comorbid condition in hospitalized patients. Correcting anemia by means of allogeneic blood transfusions with the goal of maximizing oxygen delivery is common practice in many hospitals. Varied threshold levels of hemoglobin and hematocrit are used, which is unsupported by evidence.[28, 29]
Acute anemia with normovolemic hemodilution has been proven safe in patients with coronary artery disease, heart valve disease, and the elderly. A restrictive transfusion approach with hemoglobin cutoff of 7 g/dL, as opposed to higher thresholds, has shown improved outcomes (lower mortality and lower rate of rebleeding) in adult and pediatric critical care as well as surgical patients.[30] Large studies in patients with acute myocardial infarction demonstrated that restrictive transfusional strategies are associated with decreased in‐hospital mortality, rate of reinfarction, and worsening heart failure, as well as 30‐day mortality.[31] A randomized trial in patients with active GI bleeding showed that a restrictive strategy of hemoglobin threshold of 7 g/dL was associated with improved outcomes (less mortality, less rate of rebleeding), compared with a strategy to transfuse patients with hemoglobin less than 9 g/dL.[32] In addition, increased awareness of the high cost of blood ($700$900 per unit) associated with the blood banking process as well as risk of potential infectious and noninfectious adverse reactions (eg, human immunodeficiency virus, hepatitis C virus, transfusion‐related lung injury, transfusion‐related circulatory overload) must be considered in the risk/benefit equation.[28]
Based on current available evidence, the American Association of Blood Banks recommends adhering to a restrictive transfusion strategy (7 g/dL) in hospitalized stable patients, and this threshold is raised to 8 g/dL in patients with preexisting cardiovascular disease or with active symptoms.[28] This should be combined with techniques such as preoperative anemia optimization by hematinics replacement (eg, iron, vitamin B12, folate, erythropoietin), intraoperative strategies (eg, antifibrinolytics, hypotension, normovolemic hemodilution, etc.), and postoperative strategies (eg, intraoperative cell salvage). These strategies have been shown to result in parsimonious red blood cell utilization as well as in substantial healthcare cost savings.[33]
Do not order continuous telemetry monitoring outside of the ICU without using a protocol that governs continuation.
Telemetry use in the hospital is common and clearly has a role for patients with certain cardiac conditions and those at risk for cardiac events. Telemetry is resource intensive, requiring dedicated multidisciplinary staff with specialized training. Many hospitals lack the ability to maintain and staff telemetry beds.[34] Physicians may overestimate the role of telemetry in guiding patient management.[35] One study concluded that only 12.6% of patients on a non‐ICU cardiac telemetry unit required telemetric monitoring, and only 7% received modified management as a result of telemetry findings.[36]
Inappropriate utilization of telemetry can be linked to increased length of stay or boarding in the emergency department, reduced hospital throughput, increased ambulance diversion, and increased operational costs.[37] In addition, the use of telemetry can lead to a false sense of security and alarm fatigue.[38] Telemetry artifacts may result in unnecessary testing and procedures for patients.[39] Furthermore, to accommodate the need for telemetry, frequent room changes may occur that may lead to decreased patient satisfaction. Low‐risk chest pain patients (hemodynamically stable with negative biomarkers, no electrocardiogram changes, and no indication for invasive procedure) do not require telemetry monitoring, because it rarely affects direct care of these patients.[36, 40] A 2009 study concluded that telemetry monitoring does not affect the care or the outcome of low‐risk patients.[41] Patients with other diagnoses, such as chronic obstructive pulmonary disease exacerbation or hemodynamically stable pulmonary embolism, and those requiring blood transfusions, are often placed in monitored beds without evidence that this will impact their care.[37]
The American Heart Association has published guidelines on the use of cardiac telemetry.[35] Patients are risk stratified into 3 categories, with class III patients being those who are low risk and do not require telemetry. Seventy percent of patients with the top 10 diagnoses that were admitted from the emergency department may clinically warrant telemetry.[37] Implementing a systematic evidence‐based approach to telemetry use can decrease unnecessary telemetry days,[42] reduce costs, and avoid unnecessary testing for rhythm artifacts.[39, 43]
Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability.
Although unnecessary laboratory testing is widely perceived as ineffective and wasteful, no national guideline or consensus statement exists regarding the utility or timing of repetitive laboratory testing. Multiple studies showed no difference in readmission rates, transfers to ICUs, lengths of stay, rates of adverse events, or mortality when the frequency of laboratory testing was reduced. Charges for daily laboratory testing were estimated to be $150/patient/day.[44] In a study at a university‐associated teaching hospital, an intervention to reduce the frequency of laboratory testing was associated with a total decrease of nearly 98,000 tests over a 3‐year period.[45] The cost savings in this study was estimated to be almost $2 million over the same time period. A second study at a teaching hospital, involving a computerized physician order entry (CPOE)‐based intervention, showed a reduction of almost 72,000 tests over a 1‐year period, which reduced the total number of inpatient phlebotomies by approximately 21%.[46]
The cost of routine, daily laboratory testing for a given patient or health system is not insignificant. When healthcare providers are made aware of the cost of daily laboratory testing, this might reduce the number of laboratory tests ordered and result in significant savings for a health system, as well as improve the patient experience.[44]
Developing guidelines or strategies to reduce repetitive laboratory testing in the face of clinical or laboratory stability would likely produce significant cost savings for both the individual patient as well as the health system, and could possibly would likely improve the hospital experience for many patients. Widespread adoption of CPOE by the US healthcare system has the potential to facilitate decision support that can change laboratory ordering practices.
DISCUSSION
Eliminating waste in healthcare is a priority for physicians,[6, 7, 8, 9, 10] and the ABIM‐F's Choosing Wisely campaign is a key component of this effort.[11] The SHM chose 5 tests and treatments relevant to the specialty of hospital medicine that occur at a high frequency, have significant cost and affect to patients, and that can feasibly be impacted. Given that a high percentage of healthcare costs occur in the hospital[5] and hospitalists care for an increasing number of these patients,[13] successful implementation of the SHM's adult hospital medicine Choosing Wisely list has great potential to decrease waste in the hospital, reduce harm, and improve patient outcomes.
The methodology chosen to develop the adult Choosing Wisely recommendations was intended to be both pragmatic and evidence based. A broad range of opinions was solicited, including from the SHM's general membership. The final refinement included a literature review and a Delphi process.
Review of cost and utilization data to determine the scope of the problem was used for decision‐making by subcommittee members to formulate the SHM's recommendations. For some recommendations, there were significant data, whereas for others, this information was sparse. As has been noted, we were unable to identify the total number of patients in the United States who receive telemetry on an annual basis, and thus were unable to make an estimate about the total population that would be impacted by improved utilization. However, several studies do indicate inappropriate use in significant patient populations and widespread use of the resource. Similarly, we were able to identify the costs associated with a CBC, but were unable to calculate the total number of CBCs administered annually. In the absence of these data, subcommittee members utilized other criteria, including frequency of test or treatment, patient harm or benefit, and utility for making treatment/management decisions.
In general, the tests and treatments contained in the adult hospital medicine Choosing Wisely list are not requested by patients. As such, physicians' choices play a greater role, potentially magnifying the impact hospitalists could make. Overuse of medical tests is multifactorial, and culture plays a significant role in the United States.[2, 3, 4] Although each of the tests and treatments identified by the SHM is within the purview of hospitalists, ensuring that guidelines are reliably followed will require interdisciplinary process changes. Ample opportunity exists to partner with nurses (urinary catheters and telemetry), pharmacists (stress ulcer prophylaxis), blood banks, and laboratories (transfusions and lab testing), as well as other healthcare providers and physicians in multiple specialties.
Successful implementation of each guideline will require improvement of systems within hospitals to drive reliability.[47] Provider education, training programs, protocols and reminders may prove to be significant catalysts in overcoming misinformation or no information about specific guidelines. More importantly, interdisciplinary teams will need to assess the current practice patterns within their hospitals prior to implementing solutions that standardize and automate the ordering processes for these tests and treatments.[48] Additionally, the culture within individual patient care units will need to be modified.[49] The challenge of changing the behavior of multiple stakeholders and hardwiring systems changes represent significant potential barriers to success.
There are several potential concerns with the recommendations. Concepts such as high risk and clinical stability exist in several of the recommendations. In most cases, specific guidelines exist that explicitly define the appropriate use of the test or treatment. Where they do not, implementers will need to define the operational definitions, such as the number of normal CBCs that define stability. Although the recommendations are based on the best evidence available, consensus still plays a role. As has been noted, the risk of malpractice litigation influences physicians' decisions.[1] Although evidence‐based recommendations such as these help shape the standard of care and mitigate risk, they may not completely eliminate this concern. Providers should always weigh the risks and benefits of any test or treatment. Finally, the approach taken to establish the list was both pragmatic and evidence based. Published evidence was not reviewed until the list was honed to 11. When the evidence was reviewed, the strength of the evidence was judged in a subjective manner by members of the committee as part of the Delphi panel voting.
CONCLUSION
As healthcare providers enter an era of more cost conscious decision‐making about provision of care based upon necessity, hospitalists have an excellent opportunity to impact overutilization. The 5 recommendations comprising the adult hospital medicine Choosing Wisely list offer an explicit starting point. The SHM hopes to lead this process during the coming months and years and to offer additional recommendations, providing a foundation for hospitalists to decrease unnecessary tests and treatments and improve healthcare value.
Acknowledgments
The authors thank the additional members of the Choosing Wisely subcommittee of the SHM's Healthcare Quality and Patient Safety Committee: Krishna Das, MD; Shelley Taylor, MD; Kevin O'Leary, MD; and Nasim Afsarmanesh, MD. The authors also thank SHM staff who were involved in all facets of the recommendation development process, particularly Brendon Shank, who provided significant input into the survey and dissemination process.
Disclosure
Nothing to report.
The overuse of medical tests and treatments is a growing concern. A recent survey revealed that 2 in 5 primary care physicians perceive that patients in their own practice are receiving too much care.[1] Twenty‐eight percent of the physicians indicated they provide more care than they should. When queried about reasons for the aggressiveness of care, responses included fear of malpractice litigation, adherence to clinical performance measures that require following protocols, and inadequacy of time spent with patients. Overutilization of healthcare resources is a complex issue promulgated not only by the factors cited by the physicians but also a culture in the United States habituated to believe more care is better care.[2, 3, 4] In 2010, $2.6 trillion was spent on healthcare, an increase of $1.3 trillion between 2000 and 2010.5 As much as 30% of healthcare spending may be wasted.[6] Because physicians influence approximately 80% of healthcare expenditures, including ordering tests and treatments, it is imperative that physicians take a leadership role in reversing this trend.[7]
In response to this need, several physician‐led projects have emerged.[8, 9, 10] One such initiative is the American Board of Internal Medicine Foundation's (ABIM‐F's) Choosing Wisely campaign.[11] The ABIM‐F contacted a variety of specialty societies and asked each to identify the 5 top tests or treatments relevant to their specialty that may frequently be overused. Phase 1 of the Choosing Wisely campaign was launched in April 2012 with 9 specialty societies participating. The second phase was unveiled in February 2013 and comprised of 16 additional groups including the Society of Hospital Medicine (SHM). The SHM represents 35,000 hospitalists in the United States whose primary focus is the general medical care of hospitalized patients. This is especially important because almost one‐third of total US healthcare expenditures are on hospital care,[12] and hospitalists care for an increasing number of hospitalized patients.[13] In this article, we describe the used to derive the adult hospital medicine Choosing processes Wisely list, review the tests and treatments that the SHM's Choosing Wisely Subcommittee chose, and discuss potential next steps in implementation of the adult hospital medicine recommendations.
METHODOLOGY
Upon invitation to participate in the Choosing Wisely campaign, SHM's Hospital Quality and Patient Safety (HQPS) Committee formally convened the Choosing Wisely Subcommittee. The subcommittee identified and executed a methodology (see Supporting Figure 1 and Supporting Table 1 in the online version of this article) to create the list of 5 tests and treatments that the SHM submitted to the ABIM‐F. All subcommittee members participated fully in the voting and refinement process. The Choosing Wisely Subcommittee worked closely with the SHM's Pediatrics Choosing Wisely Subcommittee to develop both adult and pediatric lists.
Convening the Choosing Wisely Subcommittee
The HQPS Committee convened a subcommittee consisting of 9 members. The subcommittee represented a diverse group of hospitalists reflecting different institution types, geographic regions, and experience. All Choosing Wisely Subcommittee members signed conflict of interest statements and reported no conflict related to the conclusions, implications, or opinions stated. The subcommittee did not consult other external stakeholders in the development of recommendations.
Identification and Refinement of Potential Wasteful Practices
To generate an initial list of potential recommendations, members of all of the SHM committees were surveyed and asked to submit 5 tests and treatments that are inappropriately used or overused. SHM staff removed duplicates and categorized submissions by topic, highlighting overlapping recommendations. Tests and treatments that are used infrequently and items included in phase 1 society lists were also excluded. Subcommittee members then ranked the resultant list using a 5‐point Likert scale. All SHM members were then given the opportunity to rank their agreement with the tests and treatments on the list, as refined at the time based upon their own experience and consideration of the following criteria: tests and procedures within the control and purview of hospital medicine, the frequency with which the tests or procedures occur, and the significance of associated costs. This was accomplished via electronic survey.
Establishing an Evidence Base
SHM staff conducted a literature review of the list of tests and treatments that was further refined by the SHM membership's ranking using a standard template. Two reviewers (W.N. and J.G.) conducted an independent literature review of the remaining tests and treatments using PubMed, MEDLINE, and Cochrane Library. The reviewers also conducted generic Internet searches. The literature review included all literature published through 2012 as well as nonEnglish language publications. The reviewers included clinical research guidelines and primary and secondary research studies. Studies included in the review were based upon common criteria including whether the article discussed an evaluation of efficacy and/or utility of treatment, reviewed the harm associated with the administration of a test or treatment, and explored the cost associated with the test or treatment as well as the overall strength of evidence. Additionally, the reference lists included in articles were reviewed to identify supplementary literature sources. The reviewers read and analyzed the articles identified in the initial search for relevant subject matter and summarized the findings in a table.
Delphi Panels
A Delphi scoring process was utilized to complete list refinement.[14] Subcommittee members anonymously voted via email for the strength of the test and treatment recommendation based upon specific criteria. To assist with this process, they received a copy of the completed literature review and an evidence summary of the literature. The following categories were used to guide the scoring: validity/evidence base to support, feasibility of implementation, frequency of occurrence, cost of occurrence, yield/emmpact, harm, and potential to improve. Results were aggregated and shared with the Choosing Wisely Subcommittee. The subcommittee conferred a final time, editing the recommendations for clarification and improved wording. A second anonymous vote was then conducted for the remaining tests and treatments through a revised scoring spreadsheet. The penultimate list was presented to the SHM's Board. Upon the Board's approval, the final list was submitted to the ABIM‐F.
RESULTS
The results of each stage of the list development process are shown in the online supporting information (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). The initial survey of SHM committee members garnered in excess of 150 tests and treatments from approximately 40 SHM committee members. The subsequent list refinement by SHM staff narrowed this list to 65 items, which were then further reduced to 15 items after ranking by members of the subcommittee (see Supporting Figure 1 and Supporting Table 1 in the online version of this article). Voting by members of the general SHM membership further reduced the list to 11 tests and treatments.
The final list of 5 tests and treatments submitted to the ABIM‐F were:
- Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
- Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for gastrointestinal (GI) complications.
- Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
- Do not order continuous telemetry monitoring outside of the intensive care unit (ICU) without using a protocol that governs continuation.
- Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability (Table 1).
|
| Test/Treatment Recommendations |
| Do not place, or leave in place, urinary catheters for incontinence or convenience, or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days or urologic procedures; use weights instead to monitor diuresis).[21, 50] |
| Do not prescribe GI prophylaxis to medical inpatients without clear‐cut indication or high risk for GI complication.[24] |
| Avoid transfusing red blood cells just because hemoglobin levels are below arbitrary thresholds such as 10, 9, or even 8 mg/dL in the absence of symptoms.[29, 51] |
| Avoid overuse/unnecessary use of telemetry monitoring in the hospital, particularly for patients at low risk for adverse cardiac outcomes.[35, 43, 52, 53] |
| Do not perform repetitive CBC and chemistry testing in the face of clinical and lab stability.[44, 54, 55] |
RECOMMENDATIONS
Do not place, or leave in place, urinary catheters for incontinence or convenience or monitoring of output for noncritically ill patients (acceptable indications: critical illness, obstruction, hospice, perioperatively for <2 days for urologic procedures; use weights instead to monitor diuresis).
Despite guidelines identifying appropriate indications for the placement of urinary catheters, urinary tract infections due to catheter use remain the most frequent type of infection in acute care settings. Nearly 1 in every 5 patients in the hospital receives an indwelling catheter, and up to half are placed inappropriately.[15] Twenty‐six percent of patients who have indwelling catheters for 2 to 10 days will develop bacteriuria; subsequently, 24% of those patients will develop a catheter‐associated urinary tract infection (CAUTI).[15] More than 13,000 deaths due to CAUTI occur annually.[16] In addition to urinary tract infections and their complications, additional adverse outcomes related to indwelling catheters include formation of encrustations and restrictions to flow, prolonged hospital stay, and exposure to multidrug resistant organisms due to increased use of antibiotics. Evidence suggests that infections due to catheters are frequently preventable.[17, 18]
The economic burden associated with indwelling catheter complications is also substantial. Each episode of symptomatic urinary tract infection adds $676 in incremental costs, and catheter‐related bacteremia costs at least $2836.15 According to Scott, nearly 450,000 CAUTIs were estimated to have occurred in 2007, resulting in direct medical costs of between $340 to $370 million.[19]
Several organizations simultaneously released guidelines to provide a roadmap for appropriate catheter use and prevention of CAUTIs.[20, 21] Despite explicit guidelines, the Centers for Disease Control and Prevention recently reported that there was no improvement in CAUTIs between 2010 and 2011.[22] Implementing these strategies for CAUTI reduction include establishing a multidisciplinary team that applies a clear protocol, with daily reminders about catheters and stop orders for catheter discontinuation.
Do not prescribe medications for stress ulcer prophylaxis to medical inpatients unless at high risk for GI complications.
Stress ulcer prophylaxis in the hospital with proton pump inhibitors (PPIs) or histamine‐2 antagonists are common. As many as 71% of patients admitted to the hospital receive some form of prophylaxis without appropriate indication.[23] Guidelines exist for appropriate use; however, therapy is commonly used in the inpatient setting for indications not investigated or supported by the literature.[24]
Inappropriate prescribing practices have been associated with multiple adverse events, including drug interactions, hospital‐acquired infections, and increased costs of care. Although consensus among physicians regarding whether GI prophylaxis causes harm is lacking, studies demonstrate a strong correlation between use of PPIs and common adverse events such as pneumonia and Clostridium difficile infection.[25, 26] For instance, inpatients receiving PPIs were 3.6 times more likely to develop C. difficile‐associated diarrhea than inpatients not exposed to PPIs.[27]
The American Society of Health‐System Pharmacists Therapeutic Guidelines on Stress Ulcer Prophylaxis provide guidance regarding the optimal indication for administration of acid‐suppression medication for patients in the hospital setting. The clinical guidelines specify that stress ulcer prophylaxis is not recommended for adult patients in non‐ICU settings. The recommendations are applicable to general medical and surgical patients with fewer than 2 risk factors for clinically important bleeding. Indications for use of stress ulcer prophylaxis in the ICU include coagulopathy and mechanical ventilation.[24]
Avoid transfusions of red blood cells for arbitrary hemoglobin or hematocrit thresholds and in the absence of symptoms or active coronary disease, heart failure, or stroke.
Anemia is a frequent comorbid condition in hospitalized patients. Correcting anemia by means of allogeneic blood transfusions with the goal of maximizing oxygen delivery is common practice in many hospitals. Varied threshold levels of hemoglobin and hematocrit are used, which is unsupported by evidence.[28, 29]
Acute anemia with normovolemic hemodilution has been proven safe in patients with coronary artery disease, heart valve disease, and the elderly. A restrictive transfusion approach with hemoglobin cutoff of 7 g/dL, as opposed to higher thresholds, has shown improved outcomes (lower mortality and lower rate of rebleeding) in adult and pediatric critical care as well as surgical patients.[30] Large studies in patients with acute myocardial infarction demonstrated that restrictive transfusional strategies are associated with decreased in‐hospital mortality, rate of reinfarction, and worsening heart failure, as well as 30‐day mortality.[31] A randomized trial in patients with active GI bleeding showed that a restrictive strategy of hemoglobin threshold of 7 g/dL was associated with improved outcomes (less mortality, less rate of rebleeding), compared with a strategy to transfuse patients with hemoglobin less than 9 g/dL.[32] In addition, increased awareness of the high cost of blood ($700$900 per unit) associated with the blood banking process as well as risk of potential infectious and noninfectious adverse reactions (eg, human immunodeficiency virus, hepatitis C virus, transfusion‐related lung injury, transfusion‐related circulatory overload) must be considered in the risk/benefit equation.[28]
Based on current available evidence, the American Association of Blood Banks recommends adhering to a restrictive transfusion strategy (7 g/dL) in hospitalized stable patients, and this threshold is raised to 8 g/dL in patients with preexisting cardiovascular disease or with active symptoms.[28] This should be combined with techniques such as preoperative anemia optimization by hematinics replacement (eg, iron, vitamin B12, folate, erythropoietin), intraoperative strategies (eg, antifibrinolytics, hypotension, normovolemic hemodilution, etc.), and postoperative strategies (eg, intraoperative cell salvage). These strategies have been shown to result in parsimonious red blood cell utilization as well as in substantial healthcare cost savings.[33]
Do not order continuous telemetry monitoring outside of the ICU without using a protocol that governs continuation.
Telemetry use in the hospital is common and clearly has a role for patients with certain cardiac conditions and those at risk for cardiac events. Telemetry is resource intensive, requiring dedicated multidisciplinary staff with specialized training. Many hospitals lack the ability to maintain and staff telemetry beds.[34] Physicians may overestimate the role of telemetry in guiding patient management.[35] One study concluded that only 12.6% of patients on a non‐ICU cardiac telemetry unit required telemetric monitoring, and only 7% received modified management as a result of telemetry findings.[36]
Inappropriate utilization of telemetry can be linked to increased length of stay or boarding in the emergency department, reduced hospital throughput, increased ambulance diversion, and increased operational costs.[37] In addition, the use of telemetry can lead to a false sense of security and alarm fatigue.[38] Telemetry artifacts may result in unnecessary testing and procedures for patients.[39] Furthermore, to accommodate the need for telemetry, frequent room changes may occur that may lead to decreased patient satisfaction. Low‐risk chest pain patients (hemodynamically stable with negative biomarkers, no electrocardiogram changes, and no indication for invasive procedure) do not require telemetry monitoring, because it rarely affects direct care of these patients.[36, 40] A 2009 study concluded that telemetry monitoring does not affect the care or the outcome of low‐risk patients.[41] Patients with other diagnoses, such as chronic obstructive pulmonary disease exacerbation or hemodynamically stable pulmonary embolism, and those requiring blood transfusions, are often placed in monitored beds without evidence that this will impact their care.[37]
The American Heart Association has published guidelines on the use of cardiac telemetry.[35] Patients are risk stratified into 3 categories, with class III patients being those who are low risk and do not require telemetry. Seventy percent of patients with the top 10 diagnoses that were admitted from the emergency department may clinically warrant telemetry.[37] Implementing a systematic evidence‐based approach to telemetry use can decrease unnecessary telemetry days,[42] reduce costs, and avoid unnecessary testing for rhythm artifacts.[39, 43]
Do not perform repetitive complete blood count (CBC) and chemistry testing in the face of clinical and lab stability.
Although unnecessary laboratory testing is widely perceived as ineffective and wasteful, no national guideline or consensus statement exists regarding the utility or timing of repetitive laboratory testing. Multiple studies showed no difference in readmission rates, transfers to ICUs, lengths of stay, rates of adverse events, or mortality when the frequency of laboratory testing was reduced. Charges for daily laboratory testing were estimated to be $150/patient/day.[44] In a study at a university‐associated teaching hospital, an intervention to reduce the frequency of laboratory testing was associated with a total decrease of nearly 98,000 tests over a 3‐year period.[45] The cost savings in this study was estimated to be almost $2 million over the same time period. A second study at a teaching hospital, involving a computerized physician order entry (CPOE)‐based intervention, showed a reduction of almost 72,000 tests over a 1‐year period, which reduced the total number of inpatient phlebotomies by approximately 21%.[46]
The cost of routine, daily laboratory testing for a given patient or health system is not insignificant. When healthcare providers are made aware of the cost of daily laboratory testing, this might reduce the number of laboratory tests ordered and result in significant savings for a health system, as well as improve the patient experience.[44]
Developing guidelines or strategies to reduce repetitive laboratory testing in the face of clinical or laboratory stability would likely produce significant cost savings for both the individual patient as well as the health system, and could possibly would likely improve the hospital experience for many patients. Widespread adoption of CPOE by the US healthcare system has the potential to facilitate decision support that can change laboratory ordering practices.
DISCUSSION
Eliminating waste in healthcare is a priority for physicians,[6, 7, 8, 9, 10] and the ABIM‐F's Choosing Wisely campaign is a key component of this effort.[11] The SHM chose 5 tests and treatments relevant to the specialty of hospital medicine that occur at a high frequency, have significant cost and affect to patients, and that can feasibly be impacted. Given that a high percentage of healthcare costs occur in the hospital[5] and hospitalists care for an increasing number of these patients,[13] successful implementation of the SHM's adult hospital medicine Choosing Wisely list has great potential to decrease waste in the hospital, reduce harm, and improve patient outcomes.
The methodology chosen to develop the adult Choosing Wisely recommendations was intended to be both pragmatic and evidence based. A broad range of opinions was solicited, including from the SHM's general membership. The final refinement included a literature review and a Delphi process.
Review of cost and utilization data to determine the scope of the problem was used for decision‐making by subcommittee members to formulate the SHM's recommendations. For some recommendations, there were significant data, whereas for others, this information was sparse. As has been noted, we were unable to identify the total number of patients in the United States who receive telemetry on an annual basis, and thus were unable to make an estimate about the total population that would be impacted by improved utilization. However, several studies do indicate inappropriate use in significant patient populations and widespread use of the resource. Similarly, we were able to identify the costs associated with a CBC, but were unable to calculate the total number of CBCs administered annually. In the absence of these data, subcommittee members utilized other criteria, including frequency of test or treatment, patient harm or benefit, and utility for making treatment/management decisions.
In general, the tests and treatments contained in the adult hospital medicine Choosing Wisely list are not requested by patients. As such, physicians' choices play a greater role, potentially magnifying the impact hospitalists could make. Overuse of medical tests is multifactorial, and culture plays a significant role in the United States.[2, 3, 4] Although each of the tests and treatments identified by the SHM is within the purview of hospitalists, ensuring that guidelines are reliably followed will require interdisciplinary process changes. Ample opportunity exists to partner with nurses (urinary catheters and telemetry), pharmacists (stress ulcer prophylaxis), blood banks, and laboratories (transfusions and lab testing), as well as other healthcare providers and physicians in multiple specialties.
Successful implementation of each guideline will require improvement of systems within hospitals to drive reliability.[47] Provider education, training programs, protocols and reminders may prove to be significant catalysts in overcoming misinformation or no information about specific guidelines. More importantly, interdisciplinary teams will need to assess the current practice patterns within their hospitals prior to implementing solutions that standardize and automate the ordering processes for these tests and treatments.[48] Additionally, the culture within individual patient care units will need to be modified.[49] The challenge of changing the behavior of multiple stakeholders and hardwiring systems changes represent significant potential barriers to success.
There are several potential concerns with the recommendations. Concepts such as high risk and clinical stability exist in several of the recommendations. In most cases, specific guidelines exist that explicitly define the appropriate use of the test or treatment. Where they do not, implementers will need to define the operational definitions, such as the number of normal CBCs that define stability. Although the recommendations are based on the best evidence available, consensus still plays a role. As has been noted, the risk of malpractice litigation influences physicians' decisions.[1] Although evidence‐based recommendations such as these help shape the standard of care and mitigate risk, they may not completely eliminate this concern. Providers should always weigh the risks and benefits of any test or treatment. Finally, the approach taken to establish the list was both pragmatic and evidence based. Published evidence was not reviewed until the list was honed to 11. When the evidence was reviewed, the strength of the evidence was judged in a subjective manner by members of the committee as part of the Delphi panel voting.
CONCLUSION
As healthcare providers enter an era of more cost conscious decision‐making about provision of care based upon necessity, hospitalists have an excellent opportunity to impact overutilization. The 5 recommendations comprising the adult hospital medicine Choosing Wisely list offer an explicit starting point. The SHM hopes to lead this process during the coming months and years and to offer additional recommendations, providing a foundation for hospitalists to decrease unnecessary tests and treatments and improve healthcare value.
Acknowledgments
The authors thank the additional members of the Choosing Wisely subcommittee of the SHM's Healthcare Quality and Patient Safety Committee: Krishna Das, MD; Shelley Taylor, MD; Kevin O'Leary, MD; and Nasim Afsarmanesh, MD. The authors also thank SHM staff who were involved in all facets of the recommendation development process, particularly Brendon Shank, who provided significant input into the survey and dissemination process.
Disclosure
Nothing to report.
- , , . Too little? Too much? Primary care physicians' views on US health care: a brief report. Arch Intern Med. 2011;171:1582–1585.
- , , , et al. Evidence that consumers are skeptical about evidence‐based health care. Health Aff (Millwood). 2010;29:1400–1406.
- , . Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801–1802.
- Too much treatment? Aggressive medical care can lead to more pain, with no gain. Consum Rep. 2008;73:40–44.
- Centers for Medicare and Medicaid Services. Historical national health expenditure data. Available at: http://www.cms.gov/Research‐Statistics‐Data‐and‐Systems/Statistics‐Trends‐and‐Reports/NationalHealthExpendData/NationalHealthAccountsHistorical.html. Accessed February 12, 2013.
- , . Eliminating waste in US health care. JAMA. 2012;307:1513–1516.
- . Change the microenvironment: delivery system reform essential to controlling costs. Available at: http://www.commonwealthfund.org/Publications/Commentaries/2009/Apr/Change‐the‐Microenvironment.aspx. Accessed February 12, 2013.
- Costs of care. Available at: http://www.costsofcare.org. Accessed February 12, 2013.
- , . Less is more: how less health care can result in better health. Arch Intern Med. 2010;170:749–750.
- , , , ; Clinical Guidelines Committee of the American College of Physicians. High‐value, cost‐conscious health care: concepts for clinicians to evaluate the benefits, harms, and costs of medical interventions. Ann Intern Med. 2011;154:174–180.
- ABIM Foundation. U.S. physician groups identify commonly used tests or procedures they say are often not necessary. Available at: http://www.abimfoundation.org/News/ABIM‐Foundation‐News/2012/Choosing‐Wisely.aspx. Accessed February 12, 2013.
- , . Addressing requests by patients for nonbeneficial interventions. JAMA. 2012;307:149–150.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360:1102–1112.
- , , , . The appropriateness method has acceptable reliability and validity for assessing overuse and underuse of surgical procedures. J Clin Epidemiol. 2012;65:1133–1143.
- . Clinical and economic consequences of nosocomial catheter‐related bacteriuria. Am J Infect Control. 2000;28:68–75.
- , , , et al. Estimating health care‐associated infections and deaths in U.S. hospitals, 2002. Public Health Rep. 2007;122:160–166.
- , , , et al. A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1): S12–S21.
- , , , et al. Strategies to prevent catheter‐associated urinary tract infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1):S41–S50.
- . The direct medical costs of healthcare‐associated infections in U.S. hospitals and the benefits of prevention. Available at: http://www.cdc.gov/hai/pdfs/hai/scott_costpaper.pdf. Accessed February 12, 2013.
- , , , , . Healthcare Infection Control Practices Advisory Committee, guideline for prevention of catheter‐associated urinary tract infections 2009. Infect Control Hosp Epidemiol. 2010;31:319–326.
- , , , et al. Diagnosis, prevention, and treatment of catheter‐associated urinary tract infection in adults: 2009 International Clinical Practice Guidelines from the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:625–663.
- , , , et al. 2011 National and State Healthcare‐Associated Infection Standardized Infection Ratio Report. Available at: http://www.cdc.gov/hai/pdfs/SIR/SIR‐Report_02_07_2013.pdf. Accessed February 13, 2013.
- , . Stress ulcer prophylaxis in hospitalized patients not in intensive care units. Am J Health Syst Pharm. 2007;64:1396–1400.
- . ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm. 1999;56:347–379.
- , , , , , . Risk of community‐acquired pneumonia and use of gastric acid‐suppressive drugs. JAMA. 2004;292:1955–1960.
- , , , . Acid‐suppressive medication use and the risk for hospital‐acquired pneumonia. JAMA. 2009;301:2120–2128.
- , , , . Gastric acid suppression by proton pump inhibitors as a risk factor for clostridium difficile‐associated diarrhea in hospitalized patients. Am J Gastroenterol. 2008;103:2308–2313.
- , , , et al. Red blood cell transfusion: a clinical practice guideline from the AABB*. Ann Intern Med. 2012;157:49–58.
- , , , et al. Guidelines for the clinical use of red cell transfusions. Br J Haematol. 2001;113:24–31.
- , , , . Mortality and morbidity in patients with very low postoperative Hb levels who decline blood transfusion. Transfusion. 2002;42:812–818.
- , , , , . Association of blood transfusion with increased mortality in myocardial infarction: a meta‐analysis and diversity‐adjusted study sequential analysis. JAMA Intern Med 2013;173:132–139.
- , , , et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368:11–21.
- , , , . Economic impact of inappropriate blood transfusions in coronary artery bypass graft surgery. Am J Med. 1993;94:509–514.
- , , , , , . The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97:1483–1487.
- , , , et al. ACC/AHA guidelines for ambulatory electrocardiography: executive summary and recommendations. A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee to Revise the Guidelines for Ambulatory Electrocardiography). Circulation. 1999;100:886–893.
- , , , et al. Role of telemetry monitoring in the non‐intensive care unit. Am J Cardiol. 1995;76:960–965.
- , . When do patients need admission to a telemetry bed? J Emerg Med. 2007;33:53–60.
- , . Electrocardiographic monitoring in the hospitalized patient: a diagnostic intervention of uncertain clinical impact. Am J Emerg Med. 2008;26:1047–1055.
- , , , , . Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med. 1999;341:1270–1274.
- , , , . Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in‐hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci. 2005;60:605–606.
- , , , , . Telemetry monitoring guidelines for efficient and safe delivery of cardiac rhythm monitoring to noncritical hospital inpatients. Crit Pathw Cardiol. 2009;8:125–126.
- Agency for Healthcare Research and Quality. Winawer N. Redesign of telemetry unit admission and transfer criteria leads to improved patient flow and reduced emergency department waiting times. Available at: http://www.innovations.ahrq.gov/content.aspx?id=2239. Accessed February 12, 2013.
- , , , et al. Is telemetry monitoring necessary in low‐risk suspected acute chest pain syndromes? Chest. 2002;122:517–523.
- , . Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg. 2011;146:524–527.
- , , , et al. A cost‐effective method for reducing the volume of laboratory tests in a university‐associated teaching hospital. Mt Sinai J Med. 2006;73:787–794.
- , , , et al. Reducing unnecessary inpatient laboratory testing in a teaching hospital. Am J Clin Pathol. 2006;126:200–206.
- . Making noncatastrophic health care processes reliable: learning to walk before running in creating high‐reliability organizations. Health Serv Res. 2006;41:1677–1689.
- , , , et al. What have we learned about interventions to reduce medical errors? Annu Rev Public Health. 2010;31:479–497.
- , . Safe Patients, Smart Hospitals: How One Doctor's Checklist Can Help Us Change Health Care From The Inside Out. New York, NY: Hudson Street Press; 2010.
- , , , , . Catheter‐associated urinary tract infection and the Medicare rule changes. Ann Intern Med. 2009;150:877–884.
- Consensus conference. Perioperative red blood cell transfusion. JAMA. 1988;260:2700–2703.
- , , , , , . Is telemetry overused? Is it as helpful as thought? Cleve Clin J Med. 2009;76:368–372.
- , , , et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711.
- , , , et al. Diagnostic blood loss from phlebotomy and hospital‐acquired anemia during acute myocardial infarction. Arch Intern Med. 2011;171:1646–1653.
- , , , , . Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med. 2005;20:520–524.
- , , . Too little? Too much? Primary care physicians' views on US health care: a brief report. Arch Intern Med. 2011;171:1582–1585.
- , , , et al. Evidence that consumers are skeptical about evidence‐based health care. Health Aff (Millwood). 2010;29:1400–1406.
- , . Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801–1802.
- Too much treatment? Aggressive medical care can lead to more pain, with no gain. Consum Rep. 2008;73:40–44.
- Centers for Medicare and Medicaid Services. Historical national health expenditure data. Available at: http://www.cms.gov/Research‐Statistics‐Data‐and‐Systems/Statistics‐Trends‐and‐Reports/NationalHealthExpendData/NationalHealthAccountsHistorical.html. Accessed February 12, 2013.
- , . Eliminating waste in US health care. JAMA. 2012;307:1513–1516.
- . Change the microenvironment: delivery system reform essential to controlling costs. Available at: http://www.commonwealthfund.org/Publications/Commentaries/2009/Apr/Change‐the‐Microenvironment.aspx. Accessed February 12, 2013.
- Costs of care. Available at: http://www.costsofcare.org. Accessed February 12, 2013.
- , . Less is more: how less health care can result in better health. Arch Intern Med. 2010;170:749–750.
- , , , ; Clinical Guidelines Committee of the American College of Physicians. High‐value, cost‐conscious health care: concepts for clinicians to evaluate the benefits, harms, and costs of medical interventions. Ann Intern Med. 2011;154:174–180.
- ABIM Foundation. U.S. physician groups identify commonly used tests or procedures they say are often not necessary. Available at: http://www.abimfoundation.org/News/ABIM‐Foundation‐News/2012/Choosing‐Wisely.aspx. Accessed February 12, 2013.
- , . Addressing requests by patients for nonbeneficial interventions. JAMA. 2012;307:149–150.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360:1102–1112.
- , , , . The appropriateness method has acceptable reliability and validity for assessing overuse and underuse of surgical procedures. J Clin Epidemiol. 2012;65:1133–1143.
- . Clinical and economic consequences of nosocomial catheter‐related bacteriuria. Am J Infect Control. 2000;28:68–75.
- , , , et al. Estimating health care‐associated infections and deaths in U.S. hospitals, 2002. Public Health Rep. 2007;122:160–166.
- , , , et al. A compendium of strategies to prevent healthcare‐associated infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1): S12–S21.
- , , , et al. Strategies to prevent catheter‐associated urinary tract infections in acute care hospitals. Infect Control Hosp Epidemiol. 2008;29(suppl 1):S41–S50.
- . The direct medical costs of healthcare‐associated infections in U.S. hospitals and the benefits of prevention. Available at: http://www.cdc.gov/hai/pdfs/hai/scott_costpaper.pdf. Accessed February 12, 2013.
- , , , , . Healthcare Infection Control Practices Advisory Committee, guideline for prevention of catheter‐associated urinary tract infections 2009. Infect Control Hosp Epidemiol. 2010;31:319–326.
- , , , et al. Diagnosis, prevention, and treatment of catheter‐associated urinary tract infection in adults: 2009 International Clinical Practice Guidelines from the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:625–663.
- , , , et al. 2011 National and State Healthcare‐Associated Infection Standardized Infection Ratio Report. Available at: http://www.cdc.gov/hai/pdfs/SIR/SIR‐Report_02_07_2013.pdf. Accessed February 13, 2013.
- , . Stress ulcer prophylaxis in hospitalized patients not in intensive care units. Am J Health Syst Pharm. 2007;64:1396–1400.
- . ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm. 1999;56:347–379.
- , , , , , . Risk of community‐acquired pneumonia and use of gastric acid‐suppressive drugs. JAMA. 2004;292:1955–1960.
- , , , . Acid‐suppressive medication use and the risk for hospital‐acquired pneumonia. JAMA. 2009;301:2120–2128.
- , , , . Gastric acid suppression by proton pump inhibitors as a risk factor for clostridium difficile‐associated diarrhea in hospitalized patients. Am J Gastroenterol. 2008;103:2308–2313.
- , , , et al. Red blood cell transfusion: a clinical practice guideline from the AABB*. Ann Intern Med. 2012;157:49–58.
- , , , et al. Guidelines for the clinical use of red cell transfusions. Br J Haematol. 2001;113:24–31.
- , , , . Mortality and morbidity in patients with very low postoperative Hb levels who decline blood transfusion. Transfusion. 2002;42:812–818.
- , , , , . Association of blood transfusion with increased mortality in myocardial infarction: a meta‐analysis and diversity‐adjusted study sequential analysis. JAMA Intern Med 2013;173:132–139.
- , , , et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368:11–21.
- , , , . Economic impact of inappropriate blood transfusions in coronary artery bypass graft surgery. Am J Med. 1993;94:509–514.
- , , , , , . The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97:1483–1487.
- , , , et al. ACC/AHA guidelines for ambulatory electrocardiography: executive summary and recommendations. A report of the American College of Cardiology/American Heart Association task force on practice guidelines (Committee to Revise the Guidelines for Ambulatory Electrocardiography). Circulation. 1999;100:886–893.
- , , , et al. Role of telemetry monitoring in the non‐intensive care unit. Am J Cardiol. 1995;76:960–965.
- , . When do patients need admission to a telemetry bed? J Emerg Med. 2007;33:53–60.
- , . Electrocardiographic monitoring in the hospitalized patient: a diagnostic intervention of uncertain clinical impact. Am J Emerg Med. 2008;26:1047–1055.
- , , , , . Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med. 1999;341:1270–1274.
- , , , . Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in‐hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci. 2005;60:605–606.
- , , , , . Telemetry monitoring guidelines for efficient and safe delivery of cardiac rhythm monitoring to noncritical hospital inpatients. Crit Pathw Cardiol. 2009;8:125–126.
- Agency for Healthcare Research and Quality. Winawer N. Redesign of telemetry unit admission and transfer criteria leads to improved patient flow and reduced emergency department waiting times. Available at: http://www.innovations.ahrq.gov/content.aspx?id=2239. Accessed February 12, 2013.
- , , , et al. Is telemetry monitoring necessary in low‐risk suspected acute chest pain syndromes? Chest. 2002;122:517–523.
- , . Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg. 2011;146:524–527.
- , , , et al. A cost‐effective method for reducing the volume of laboratory tests in a university‐associated teaching hospital. Mt Sinai J Med. 2006;73:787–794.
- , , , et al. Reducing unnecessary inpatient laboratory testing in a teaching hospital. Am J Clin Pathol. 2006;126:200–206.
- . Making noncatastrophic health care processes reliable: learning to walk before running in creating high‐reliability organizations. Health Serv Res. 2006;41:1677–1689.
- , , , et al. What have we learned about interventions to reduce medical errors? Annu Rev Public Health. 2010;31:479–497.
- , . Safe Patients, Smart Hospitals: How One Doctor's Checklist Can Help Us Change Health Care From The Inside Out. New York, NY: Hudson Street Press; 2010.
- , , , , . Catheter‐associated urinary tract infection and the Medicare rule changes. Ann Intern Med. 2009;150:877–884.
- Consensus conference. Perioperative red blood cell transfusion. JAMA. 1988;260:2700–2703.
- , , , , , . Is telemetry overused? Is it as helpful as thought? Cleve Clin J Med. 2009;76:368–372.
- , , , et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711.
- , , , et al. Diagnostic blood loss from phlebotomy and hospital‐acquired anemia during acute myocardial infarction. Arch Intern Med. 2011;171:1646–1653.
- , , , , . Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med. 2005;20:520–524.
Copyright © 2013 Society of Hospital Medicine
Rounding up the usual suspects
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.