User login
A 46-year-old man with fever, ST-segment elevation
An otherwise healthy 46-year-old man presents with fever, chills, and rigors that began 1 day ago. He reports shortness of breath, nausea, neck pain, sore throat, and right-sided jaw pain, but no chest pain, headache, or photophobia.
His vital signs are within normal limits, except for a temperature of 38.5°C (101.3°F). His jugular venous pressure and heart sounds are normal, and no focal deficit or nuchal rigidity is elicited. Laboratory tests (hemography, biochemistry panel, and cardiac biomarkers) are normal. An enzyme immunoassay for influenza A and B is negative, as is a rapid antigen detection test for streptococcal infection.
Q: What is the most likely diagnosis?
- Acute myocardial infarction
- Coronary vasospasm
- Brugada syndrome
- Acute pericarditis
- Acute meningitis
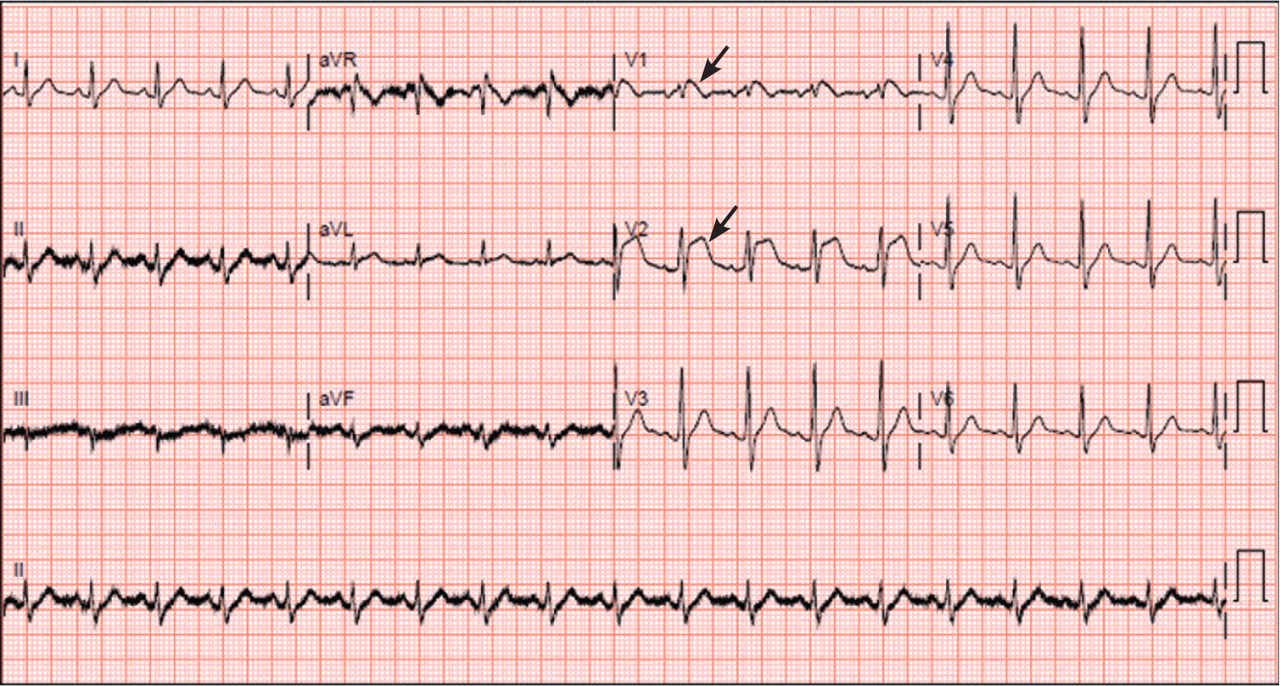
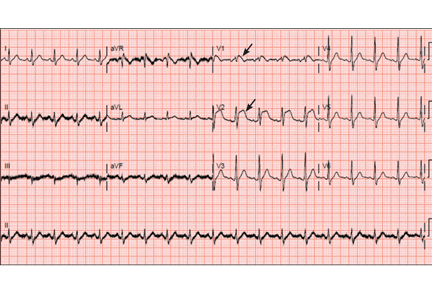
A: ST elevation commonly represents acute myocardial infarction, but it is associated with other conditions, including Prinzmetal angina, hyperkalemia, hypercalcemia, early repolarization, Brugada syndrome, and acute pericarditis.1 These conditions should be considered before an invasive intervention. The ECG findings (ST elevation in the right precordial leads) in this patient were consistent with those of Brugada syndrome.
WHAT IS BRUGADA SYNDROME?
Brugada syndrome is an arrhythmogenic disease characterized by ST-segment elevation in the right precordial leads, right bundle branch block, and a high incidence of sudden cardiac death in younger people.2 It accounts for 4% of all sudden deaths.3
Three different types of changes on ECG have been associated with Brugada syndrome. Type 1 is a coved ST-segment elevation of at least 2 mm, followed by a negative T wave, with little or no isoelectric separation, and present in more than one right precordial lead (from V1 to V3). Type 2 and type 3 patterns on ECG show the same 2-mm or greater J-point elevation, but a positive T wave gives the “saddleback” appearance to the ST-T portion.
Brugada syndrome is confirmed when a type 1 pattern is observed in conjunction with one of the following:
- Documented ventricular fibrillation
- Polymorphic ventricular tachycardia
- A family history of sudden cardiac death at 45 years of age or younger
- Type 1 pattern on ECG in family members
- Ventricular tachycardia that can be induced with programmed electrical stimulation
- Syncope
- Nocturnal agonal respiration.3
This patient had type 1 changes on ECG but none of the above findings.
Brugada syndrome is inherited as an autosomal dominant trait, and mutations in gene SCN5A account for 18% to 30% of cases.3 These mutations impair the function of the sodium channel current, leading to an unopposed outward shift of net transmembrane current at the end of phase 1 of the right ventricular epicardial action potential. Interestingly, changes on ECG that are associated with Brugada syndrome are often dynamic or concealed and are unmasked by sodium channel blockers, fever, vagotonic agents, adrenergic agonists or antagonists, and various electrolyte abnormalities.3
At temperatures above the physiologic range, the inward sodium current is reduced, either because of failure of expression of sodium channels or because of premature closing of the sodium channels in genetically susceptible individuals.4 Therefore, fever can unmask Brugada syndrome, as it did in our patient.
For patients with symptomatic Brugada syndrome, the only current treatment is implantation of a cardioverter-defibrillator.5 Patients without symptoms may benefit from an electrophysiologic study for risk stratification, and an implantable cardioverter-defibrillator is recommended for those in whom ventricular fibrillation can be induced.3,5
CASE CONTINUED
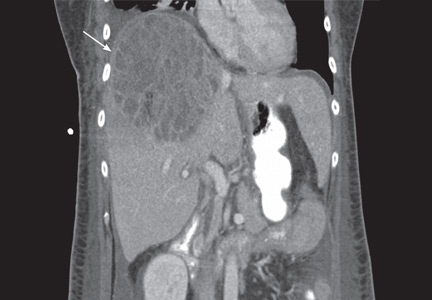
The patient remained febrile after catheterization and received vancomycin (Vancocin) and ceftriaxone (Rocephin) empirically for presumed meningitis. Multiple peripheral blood cultures grew gram-positive cocci in pairs and chains, which were identified as Streptococcus pneumoniae. His fever abated soon after the antibiotic therapy was started.
Lumbar puncture was not done. Transesophageal echocardiography revealed no vegetations, with preserved ejection fraction. The patient has no family history of sudden death and no personal history of syncope or presyncope.
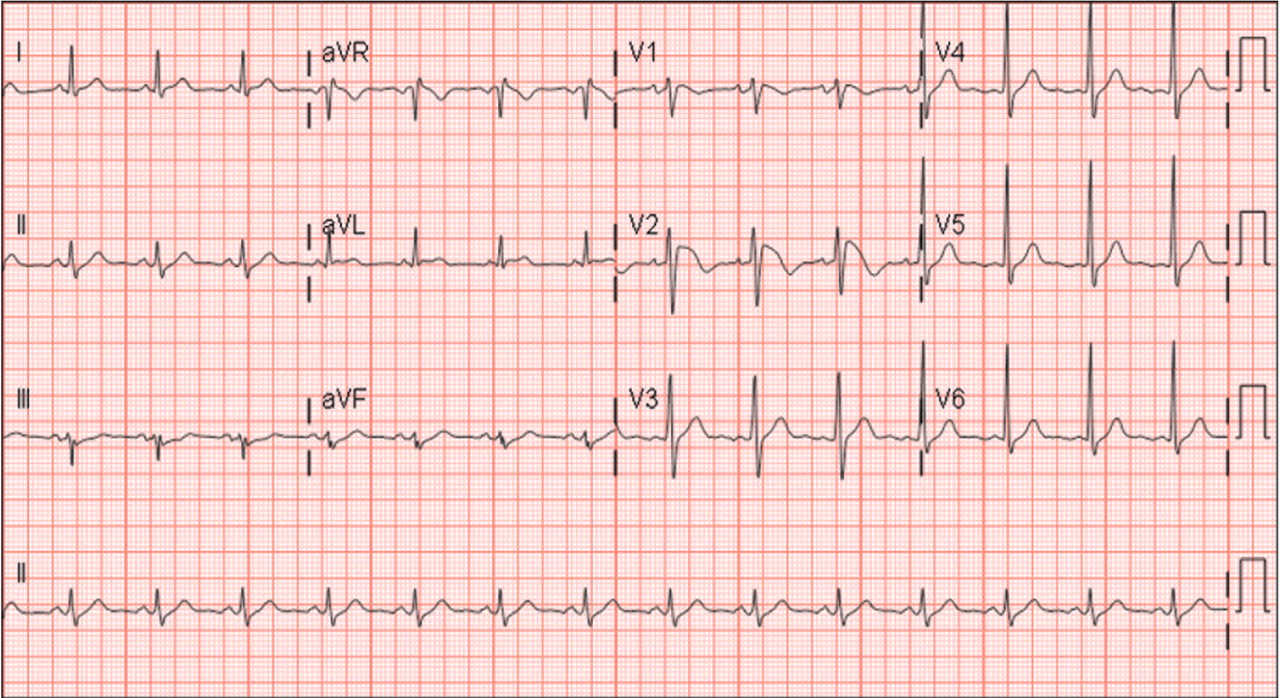
On hospital day 3, although his fever was gone, ECG still showed a Brugada pattern. He was discharged home on a 3-week regimen of intravenous penicillin, with plans for appropriate follow-up, and he was counseled that his family should be screened. An electrophysiologic study was not done, and he had no symptoms 1 year later.
As seen in this patient, Brugada syndrome is important to consider in the differential diagnosis in young patients who present with fever and ST elevations.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20:1391–1396.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85:803–809.
- Antzelevitch C, Nof E. Brugada syndrome: recent advances and controversies. Curr Cardiol Rep 2008; 10:376–383.
An otherwise healthy 46-year-old man presents with fever, chills, and rigors that began 1 day ago. He reports shortness of breath, nausea, neck pain, sore throat, and right-sided jaw pain, but no chest pain, headache, or photophobia.
His vital signs are within normal limits, except for a temperature of 38.5°C (101.3°F). His jugular venous pressure and heart sounds are normal, and no focal deficit or nuchal rigidity is elicited. Laboratory tests (hemography, biochemistry panel, and cardiac biomarkers) are normal. An enzyme immunoassay for influenza A and B is negative, as is a rapid antigen detection test for streptococcal infection.
Q: What is the most likely diagnosis?
- Acute myocardial infarction
- Coronary vasospasm
- Brugada syndrome
- Acute pericarditis
- Acute meningitis
A: ST elevation commonly represents acute myocardial infarction, but it is associated with other conditions, including Prinzmetal angina, hyperkalemia, hypercalcemia, early repolarization, Brugada syndrome, and acute pericarditis.1 These conditions should be considered before an invasive intervention. The ECG findings (ST elevation in the right precordial leads) in this patient were consistent with those of Brugada syndrome.
WHAT IS BRUGADA SYNDROME?
Brugada syndrome is an arrhythmogenic disease characterized by ST-segment elevation in the right precordial leads, right bundle branch block, and a high incidence of sudden cardiac death in younger people.2 It accounts for 4% of all sudden deaths.3
Three different types of changes on ECG have been associated with Brugada syndrome. Type 1 is a coved ST-segment elevation of at least 2 mm, followed by a negative T wave, with little or no isoelectric separation, and present in more than one right precordial lead (from V1 to V3). Type 2 and type 3 patterns on ECG show the same 2-mm or greater J-point elevation, but a positive T wave gives the “saddleback” appearance to the ST-T portion.
Brugada syndrome is confirmed when a type 1 pattern is observed in conjunction with one of the following:
- Documented ventricular fibrillation
- Polymorphic ventricular tachycardia
- A family history of sudden cardiac death at 45 years of age or younger
- Type 1 pattern on ECG in family members
- Ventricular tachycardia that can be induced with programmed electrical stimulation
- Syncope
- Nocturnal agonal respiration.3
This patient had type 1 changes on ECG but none of the above findings.
Brugada syndrome is inherited as an autosomal dominant trait, and mutations in gene SCN5A account for 18% to 30% of cases.3 These mutations impair the function of the sodium channel current, leading to an unopposed outward shift of net transmembrane current at the end of phase 1 of the right ventricular epicardial action potential. Interestingly, changes on ECG that are associated with Brugada syndrome are often dynamic or concealed and are unmasked by sodium channel blockers, fever, vagotonic agents, adrenergic agonists or antagonists, and various electrolyte abnormalities.3
At temperatures above the physiologic range, the inward sodium current is reduced, either because of failure of expression of sodium channels or because of premature closing of the sodium channels in genetically susceptible individuals.4 Therefore, fever can unmask Brugada syndrome, as it did in our patient.
For patients with symptomatic Brugada syndrome, the only current treatment is implantation of a cardioverter-defibrillator.5 Patients without symptoms may benefit from an electrophysiologic study for risk stratification, and an implantable cardioverter-defibrillator is recommended for those in whom ventricular fibrillation can be induced.3,5
CASE CONTINUED
The patient remained febrile after catheterization and received vancomycin (Vancocin) and ceftriaxone (Rocephin) empirically for presumed meningitis. Multiple peripheral blood cultures grew gram-positive cocci in pairs and chains, which were identified as Streptococcus pneumoniae. His fever abated soon after the antibiotic therapy was started.
Lumbar puncture was not done. Transesophageal echocardiography revealed no vegetations, with preserved ejection fraction. The patient has no family history of sudden death and no personal history of syncope or presyncope.
On hospital day 3, although his fever was gone, ECG still showed a Brugada pattern. He was discharged home on a 3-week regimen of intravenous penicillin, with plans for appropriate follow-up, and he was counseled that his family should be screened. An electrophysiologic study was not done, and he had no symptoms 1 year later.
As seen in this patient, Brugada syndrome is important to consider in the differential diagnosis in young patients who present with fever and ST elevations.
An otherwise healthy 46-year-old man presents with fever, chills, and rigors that began 1 day ago. He reports shortness of breath, nausea, neck pain, sore throat, and right-sided jaw pain, but no chest pain, headache, or photophobia.
His vital signs are within normal limits, except for a temperature of 38.5°C (101.3°F). His jugular venous pressure and heart sounds are normal, and no focal deficit or nuchal rigidity is elicited. Laboratory tests (hemography, biochemistry panel, and cardiac biomarkers) are normal. An enzyme immunoassay for influenza A and B is negative, as is a rapid antigen detection test for streptococcal infection.
Q: What is the most likely diagnosis?
- Acute myocardial infarction
- Coronary vasospasm
- Brugada syndrome
- Acute pericarditis
- Acute meningitis
A: ST elevation commonly represents acute myocardial infarction, but it is associated with other conditions, including Prinzmetal angina, hyperkalemia, hypercalcemia, early repolarization, Brugada syndrome, and acute pericarditis.1 These conditions should be considered before an invasive intervention. The ECG findings (ST elevation in the right precordial leads) in this patient were consistent with those of Brugada syndrome.
WHAT IS BRUGADA SYNDROME?
Brugada syndrome is an arrhythmogenic disease characterized by ST-segment elevation in the right precordial leads, right bundle branch block, and a high incidence of sudden cardiac death in younger people.2 It accounts for 4% of all sudden deaths.3
Three different types of changes on ECG have been associated with Brugada syndrome. Type 1 is a coved ST-segment elevation of at least 2 mm, followed by a negative T wave, with little or no isoelectric separation, and present in more than one right precordial lead (from V1 to V3). Type 2 and type 3 patterns on ECG show the same 2-mm or greater J-point elevation, but a positive T wave gives the “saddleback” appearance to the ST-T portion.
Brugada syndrome is confirmed when a type 1 pattern is observed in conjunction with one of the following:
- Documented ventricular fibrillation
- Polymorphic ventricular tachycardia
- A family history of sudden cardiac death at 45 years of age or younger
- Type 1 pattern on ECG in family members
- Ventricular tachycardia that can be induced with programmed electrical stimulation
- Syncope
- Nocturnal agonal respiration.3
This patient had type 1 changes on ECG but none of the above findings.
Brugada syndrome is inherited as an autosomal dominant trait, and mutations in gene SCN5A account for 18% to 30% of cases.3 These mutations impair the function of the sodium channel current, leading to an unopposed outward shift of net transmembrane current at the end of phase 1 of the right ventricular epicardial action potential. Interestingly, changes on ECG that are associated with Brugada syndrome are often dynamic or concealed and are unmasked by sodium channel blockers, fever, vagotonic agents, adrenergic agonists or antagonists, and various electrolyte abnormalities.3
At temperatures above the physiologic range, the inward sodium current is reduced, either because of failure of expression of sodium channels or because of premature closing of the sodium channels in genetically susceptible individuals.4 Therefore, fever can unmask Brugada syndrome, as it did in our patient.
For patients with symptomatic Brugada syndrome, the only current treatment is implantation of a cardioverter-defibrillator.5 Patients without symptoms may benefit from an electrophysiologic study for risk stratification, and an implantable cardioverter-defibrillator is recommended for those in whom ventricular fibrillation can be induced.3,5
CASE CONTINUED
The patient remained febrile after catheterization and received vancomycin (Vancocin) and ceftriaxone (Rocephin) empirically for presumed meningitis. Multiple peripheral blood cultures grew gram-positive cocci in pairs and chains, which were identified as Streptococcus pneumoniae. His fever abated soon after the antibiotic therapy was started.
Lumbar puncture was not done. Transesophageal echocardiography revealed no vegetations, with preserved ejection fraction. The patient has no family history of sudden death and no personal history of syncope or presyncope.
On hospital day 3, although his fever was gone, ECG still showed a Brugada pattern. He was discharged home on a 3-week regimen of intravenous penicillin, with plans for appropriate follow-up, and he was counseled that his family should be screened. An electrophysiologic study was not done, and he had no symptoms 1 year later.
As seen in this patient, Brugada syndrome is important to consider in the differential diagnosis in young patients who present with fever and ST elevations.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20:1391–1396.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85:803–809.
- Antzelevitch C, Nof E. Brugada syndrome: recent advances and controversies. Curr Cardiol Rep 2008; 10:376–383.
- Wang K, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 1992; 20:1391–1396.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85:803–809.
- Antzelevitch C, Nof E. Brugada syndrome: recent advances and controversies. Curr Cardiol Rep 2008; 10:376–383.
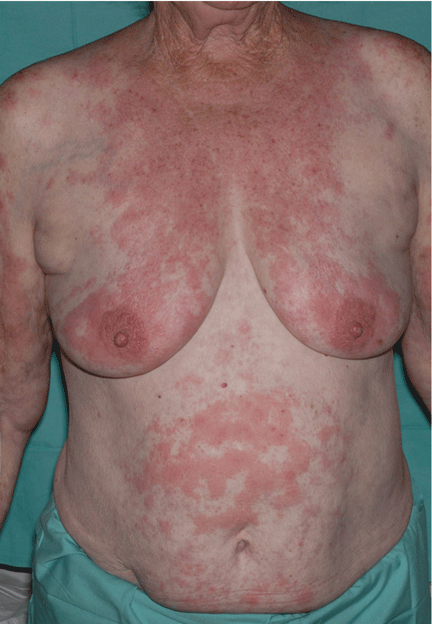
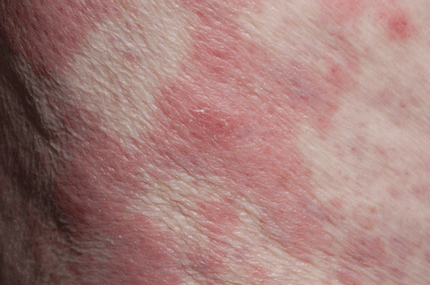
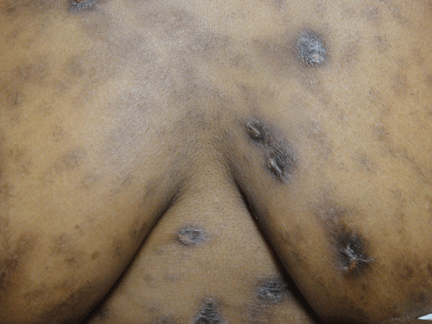
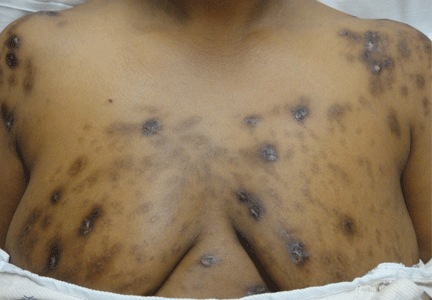
Leukemia cutis
Q: What is the most likely diagnosis?
- Leukemia cutis
- Drug reaction
- Sweet syndrome
- Erythema multiforme
- Urticaria
A: The correct answer is leukemia cutis, defined as a cutaneous infiltration by neoplastic leukocytes.1 When the leukocytes are primarily granulocytic precursors, the terms myeloid sarcoma, granulocytic sarcoma, chloroma, and primary extramedullary leukemia have been used.2 The term monoblastic sarcoma has been used when the cutaneous infiltrate is composed of neoplastic monocytic precursors.2
Leukemia cutis most commonly manifests as erythematous papules and nodules, single or multiple, of varying sizes, and afflicting one or more body sites; it typically is asymptomatic.3 It occurs in 10% to 15% of patients with acute myeloid leukemia4 and is itself a poor prognostic sign.5 The cutaneous changes may pre-date the hematologic manifestations and may even herald a relapse.6
In patients presenting with extramedullary leukemia and no bone marrow or blood involvement, the importance of preemptive chemotherapy for acute myelogenous leukemia has recently been emphasized.7
CASE CONTINUED
The patient undergoes further testing with bone marrow aspiration and trephination, which are diagnostic of acute myeloid leukemia with myelodysplastic changes: the studies reveal a clear excess of myeloblasts (accounting for 40% to 50% of nucleated cells) and clearly dysplastic erythropoiesis and myelopoiesis. Bone marrow cytogenetic analysis reveals a complex abnormal female karyotype with multiple numerical and structural abnormalities, in particular deletion of the long arm of chromosome 5, suggestive of a poor prognosis.
Skin biopsy reveals a normal epidermis but dermal perivascular involvement with a reactive T-lymphocyte infiltrate associated with immature myeloid elements, characterized by positive staining to myeloperoxidase, in keeping with leukemia cutis. It should be noted that histopathologic confirmation of leukemia cutis can be challenging, as the condition can adopt a variety of patterns, and clinicopathologic correlation is often warranted.
The patient is treated with a cycle of cytarabine-based chemotherapy, and her skin eruption transiently improves. However, her clinical condition subsequently deteriorates; she has a relapse of leukemia, with the rash returning more florid and angry-looking than previously. She is subsequently managed palliatively and passes away 3 weeks later.
THE OTHER DIAGNOSTIC CHOICES
Sweet syndrome or acute febrile neutrophilic dermatosis is often seen in association with hematologic malignancies, but the lesions are typically tender, and histopathology reveals an intense dermal neutrophilic infiltrate.6
Erythema multiforme is associated with malignancy, but its characteristic concentric “target” lesions are typically acral and symmetrical in their distribution; their histopathology is inflammatory.8
The patient had not been on any regular medications and her rash could not have been medication-induced.
Urticaria presents with pruritic evanescent wheals, which rarely last more than 12 hours.9 Our patient had a fixed and entirely asymptomatic rash, which in addition did not have the histopathologic features of urticaria—namely, dermal edema involved with an infiltrate made of lymphocytes and eosinophils.9
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In:Weedon D, editor. Skin Pathology, 2nd ed. New York, NY: Churchill Livingstone, 2002:1118–1120.
- Brunning RD, Matutes E, Flandria F, et al. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2001:104–105.
- Watson KM, Mufti G, Salisbury JR, du Vivier AW, Creamer D. Spectrum of clinical presentation, treatment and prognosis in a series of eight patients with leukaemia cutis. Clin Exp Dermatol 2006; 31:218–221.
- Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol 2002; 81:90–95.
- Kaddu S, Zenahlik P, Beham-Schmid C, Kerl H, Cerroni L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol 1999; 40:966–978.
- Cutaneous aspects of leukemia. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC, editors. Dermatology, 2nd ed. Berlin: Springer, 2000:1640–1648.
- Tsimberidou AM, Kantarjian HM, Wen S, et al. Myeloid sarcoma is associated with superior event-free survival and overall survival compared with acute myeloid leukemia. Cancer 2008; 113:1370–1378.
- Erythemato-papulo-squamous diseases. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
- Urticaria, angioedema and anaphylaxis. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
Q: What is the most likely diagnosis?
- Leukemia cutis
- Drug reaction
- Sweet syndrome
- Erythema multiforme
- Urticaria
A: The correct answer is leukemia cutis, defined as a cutaneous infiltration by neoplastic leukocytes.1 When the leukocytes are primarily granulocytic precursors, the terms myeloid sarcoma, granulocytic sarcoma, chloroma, and primary extramedullary leukemia have been used.2 The term monoblastic sarcoma has been used when the cutaneous infiltrate is composed of neoplastic monocytic precursors.2
Leukemia cutis most commonly manifests as erythematous papules and nodules, single or multiple, of varying sizes, and afflicting one or more body sites; it typically is asymptomatic.3 It occurs in 10% to 15% of patients with acute myeloid leukemia4 and is itself a poor prognostic sign.5 The cutaneous changes may pre-date the hematologic manifestations and may even herald a relapse.6
In patients presenting with extramedullary leukemia and no bone marrow or blood involvement, the importance of preemptive chemotherapy for acute myelogenous leukemia has recently been emphasized.7
CASE CONTINUED
The patient undergoes further testing with bone marrow aspiration and trephination, which are diagnostic of acute myeloid leukemia with myelodysplastic changes: the studies reveal a clear excess of myeloblasts (accounting for 40% to 50% of nucleated cells) and clearly dysplastic erythropoiesis and myelopoiesis. Bone marrow cytogenetic analysis reveals a complex abnormal female karyotype with multiple numerical and structural abnormalities, in particular deletion of the long arm of chromosome 5, suggestive of a poor prognosis.
Skin biopsy reveals a normal epidermis but dermal perivascular involvement with a reactive T-lymphocyte infiltrate associated with immature myeloid elements, characterized by positive staining to myeloperoxidase, in keeping with leukemia cutis. It should be noted that histopathologic confirmation of leukemia cutis can be challenging, as the condition can adopt a variety of patterns, and clinicopathologic correlation is often warranted.
The patient is treated with a cycle of cytarabine-based chemotherapy, and her skin eruption transiently improves. However, her clinical condition subsequently deteriorates; she has a relapse of leukemia, with the rash returning more florid and angry-looking than previously. She is subsequently managed palliatively and passes away 3 weeks later.
THE OTHER DIAGNOSTIC CHOICES
Sweet syndrome or acute febrile neutrophilic dermatosis is often seen in association with hematologic malignancies, but the lesions are typically tender, and histopathology reveals an intense dermal neutrophilic infiltrate.6
Erythema multiforme is associated with malignancy, but its characteristic concentric “target” lesions are typically acral and symmetrical in their distribution; their histopathology is inflammatory.8
The patient had not been on any regular medications and her rash could not have been medication-induced.
Urticaria presents with pruritic evanescent wheals, which rarely last more than 12 hours.9 Our patient had a fixed and entirely asymptomatic rash, which in addition did not have the histopathologic features of urticaria—namely, dermal edema involved with an infiltrate made of lymphocytes and eosinophils.9
Q: What is the most likely diagnosis?
- Leukemia cutis
- Drug reaction
- Sweet syndrome
- Erythema multiforme
- Urticaria
A: The correct answer is leukemia cutis, defined as a cutaneous infiltration by neoplastic leukocytes.1 When the leukocytes are primarily granulocytic precursors, the terms myeloid sarcoma, granulocytic sarcoma, chloroma, and primary extramedullary leukemia have been used.2 The term monoblastic sarcoma has been used when the cutaneous infiltrate is composed of neoplastic monocytic precursors.2
Leukemia cutis most commonly manifests as erythematous papules and nodules, single or multiple, of varying sizes, and afflicting one or more body sites; it typically is asymptomatic.3 It occurs in 10% to 15% of patients with acute myeloid leukemia4 and is itself a poor prognostic sign.5 The cutaneous changes may pre-date the hematologic manifestations and may even herald a relapse.6
In patients presenting with extramedullary leukemia and no bone marrow or blood involvement, the importance of preemptive chemotherapy for acute myelogenous leukemia has recently been emphasized.7
CASE CONTINUED
The patient undergoes further testing with bone marrow aspiration and trephination, which are diagnostic of acute myeloid leukemia with myelodysplastic changes: the studies reveal a clear excess of myeloblasts (accounting for 40% to 50% of nucleated cells) and clearly dysplastic erythropoiesis and myelopoiesis. Bone marrow cytogenetic analysis reveals a complex abnormal female karyotype with multiple numerical and structural abnormalities, in particular deletion of the long arm of chromosome 5, suggestive of a poor prognosis.
Skin biopsy reveals a normal epidermis but dermal perivascular involvement with a reactive T-lymphocyte infiltrate associated with immature myeloid elements, characterized by positive staining to myeloperoxidase, in keeping with leukemia cutis. It should be noted that histopathologic confirmation of leukemia cutis can be challenging, as the condition can adopt a variety of patterns, and clinicopathologic correlation is often warranted.
The patient is treated with a cycle of cytarabine-based chemotherapy, and her skin eruption transiently improves. However, her clinical condition subsequently deteriorates; she has a relapse of leukemia, with the rash returning more florid and angry-looking than previously. She is subsequently managed palliatively and passes away 3 weeks later.
THE OTHER DIAGNOSTIC CHOICES
Sweet syndrome or acute febrile neutrophilic dermatosis is often seen in association with hematologic malignancies, but the lesions are typically tender, and histopathology reveals an intense dermal neutrophilic infiltrate.6
Erythema multiforme is associated with malignancy, but its characteristic concentric “target” lesions are typically acral and symmetrical in their distribution; their histopathology is inflammatory.8
The patient had not been on any regular medications and her rash could not have been medication-induced.
Urticaria presents with pruritic evanescent wheals, which rarely last more than 12 hours.9 Our patient had a fixed and entirely asymptomatic rash, which in addition did not have the histopathologic features of urticaria—namely, dermal edema involved with an infiltrate made of lymphocytes and eosinophils.9
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In:Weedon D, editor. Skin Pathology, 2nd ed. New York, NY: Churchill Livingstone, 2002:1118–1120.
- Brunning RD, Matutes E, Flandria F, et al. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2001:104–105.
- Watson KM, Mufti G, Salisbury JR, du Vivier AW, Creamer D. Spectrum of clinical presentation, treatment and prognosis in a series of eight patients with leukaemia cutis. Clin Exp Dermatol 2006; 31:218–221.
- Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol 2002; 81:90–95.
- Kaddu S, Zenahlik P, Beham-Schmid C, Kerl H, Cerroni L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol 1999; 40:966–978.
- Cutaneous aspects of leukemia. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC, editors. Dermatology, 2nd ed. Berlin: Springer, 2000:1640–1648.
- Tsimberidou AM, Kantarjian HM, Wen S, et al. Myeloid sarcoma is associated with superior event-free survival and overall survival compared with acute myeloid leukemia. Cancer 2008; 113:1370–1378.
- Erythemato-papulo-squamous diseases. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
- Urticaria, angioedema and anaphylaxis. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
- Strutton G. Cutaneous infiltrates: lymphomatous and leukemic. In:Weedon D, editor. Skin Pathology, 2nd ed. New York, NY: Churchill Livingstone, 2002:1118–1120.
- Brunning RD, Matutes E, Flandria F, et al. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2001:104–105.
- Watson KM, Mufti G, Salisbury JR, du Vivier AW, Creamer D. Spectrum of clinical presentation, treatment and prognosis in a series of eight patients with leukaemia cutis. Clin Exp Dermatol 2006; 31:218–221.
- Agis H, Weltermann A, Fonatsch C, et al. A comparative study on demographic, hematological, and cytogenetic findings and prognosis in acute myeloid leukemia with and without leukemia cutis. Ann Hematol 2002; 81:90–95.
- Kaddu S, Zenahlik P, Beham-Schmid C, Kerl H, Cerroni L. Specific cutaneous infiltrates in patients with myelogenous leukemia: a clinicopathologic study of 26 patients with assessment of diagnostic criteria. J Am Acad Dermatol 1999; 40:966–978.
- Cutaneous aspects of leukemia. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC, editors. Dermatology, 2nd ed. Berlin: Springer, 2000:1640–1648.
- Tsimberidou AM, Kantarjian HM, Wen S, et al. Myeloid sarcoma is associated with superior event-free survival and overall survival compared with acute myeloid leukemia. Cancer 2008; 113:1370–1378.
- Erythemato-papulo-squamous diseases. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
- Urticaria, angioedema and anaphylaxis. In: Braun-Falco O, Plewig G, Wolff HH, Burgdorf WHC. Dermatology, 2nd ed. Berlin: Springer, 2000.
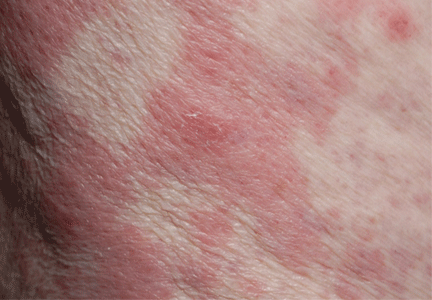
A 40-year-old woman with excoriated skin lesions
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
She denies any history of itching, insect bites, exposure to new medications or jewelry, allergies, recent change in medications, travel, or intravenous drug abuse.
Q: Which is the most likely diagnosis?
- Allergic contact dermatitis
- Xerosis
- Dermatotillomania
- Folliculitis
- Infestation (scabies)
A: Dermatotillomania, ie, pathologic skin picking, is the correct diagnosis. On further questioning, the patient reveals that the wounds have been self-inflicted over many years, starting in her adolescence. The wounds are located only in areas she can reach. She admits that social and emotional stressors had made the condition significantly worse and that lately she had lost control of her skin-picking. She denies nail-biting, trichotillomania, or obsessive-compulsive behavior.
As for the other possible diagnoses:
Allergic contact dermatitis occurs when contact with a particular substance elicits a hypersensitivity reaction. This reaction is of the delayed type (type IV). The affected individual can develop skin erythema and swelling with vesicles that are intensely pruritic at the contact site. The erythema may become evident hours after exposure, or not until weeks later, which can make the diagnosis difficult at times.
Our patient’s lesions were not pruritic, and she denied recent exposure to allergens.
Xerosis. Xerotic (dry) skin is usually rough, with fine scales and fissures. Xerosis can affect people of all ages and is often more intense during the winter. It affects mainly the arms, legs, and hands. Patients note pruritus, which can be treated with liberal use of emollients and tepid water baths.
Our patient’s lesions were scarred, hyperpigmented, and nonpruritic.
Folliculitis is a superficial infection of the hair follicle that presents as an erythematous pustule on the extremities, buttocks, or scalp. The pustule can be tender to palpation and can progress to an abscess that requires incision and drainage and intravenous antibiotics. A moist environment and poor hygiene are predisposing factors. Staphylococcus aureus is the culprit in most cases.
Our patient’s lesions were on the chest and upper back, where hair follicles were sparse or absent, and there was no erythema or tenderness.
Scabies is a skin infestation with Sarcoptes scabiei mites, which burrow in the skin and cause intense pruritus, especially at night. Scabies usually affects the sides and webs of the fingers and skin folds. Sexual contact is a common way of transmission; however, transmission can also occur by sharing beds and towels.
Patients with dermatotillomania lack intense pruritus, and skin-picking occurs during the day, while the patient is awake.
SELF-INFLICTED WOUNDS
Pathologic skin-picking, neurotic excoriation, excoriated acne, or dermatotillomania results from scratching, picking, gouging, or squeezing of one’s skin via teeth, fingernails, tweezers, or other objects.1–3 Lesions are usually found on skin that the patient can easily reach, such as the face, chest, upper and lower extremities, and upper back.4
The prevalence of pathologic skin-picking is estimated at 2% in dermatology patients.5 The overall prevalence of psychiatric disorders in all dermatology outpatients is estimated at 30% to 40%. Women outnumber men with this disorder.6
Dermatotillomania is thought to be on the spectrum of obsessive-compulsive disorder, in which patients exhibit impulses and compulsions.5 It starts in childhood or early adulthood, with an average lifetime duration of 21 years.7 It is usually associated with anxiety, depression, obsessive-compulsive traits, eating disorders, body dysmorphic disorders, or hypochondriasis. Psychosocial stress is the main trigger. Patients report feelings of tension and stress before picking and relief while picking; there is no suicidal ideation.8
Treatments are both pharmacologic and behavioral.9 Cognitive behavioral therapy and habit reversal therapy have each been successful when used alone.8 In addition, several case reports10 and double-blind studies11,12 have shown that treatment with a selective serotonin-reuptake inhibitor (SSRI) can reduce pathologic skin-picking.13,14 However, SSRIs have also been reported to induce or aggravate this behavior in patients with underlying mild skin-picking and a family history of skin-picking.15 Thus, it is pertinent to extract a detailed history from the patient before prescribing an SSRI.
We referred our patient for behavioral therapy and prescribed fluoxetine (Prozac) 20 mg daily. She showed improvement in symptoms in 4 weeks and has since stopped skin-picking completely.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.
- Arnold LM. Phenomenology and therapeutic options for dermatotillomania. Expert Rev Neurother 2002; 2:725–730.
- Bohne A, Keuthen N, Wilhelm S. Pathologic hairpulling, skin picking, and nail biting. Ann Clin Psychiatry 2005; 17:227–232.
- Gattu S, Rashid RM, Khachemoune A. Self-induced skin lesions: a review of dermatitis artefacta. Cutis 2009; 84:247–251.
- Keuthen NJ, Deckersbach T, Wilhelm S, et al. Repetitive skin-picking in a student population and comparison with a sample of self-injurious skin-pickers. Psychosomatics 2000; 41:210–215.
- Arnold LM, Auchenbach MB, McElroy SL. Psychogenic excoriation. Clinical features, proposed diagnostic criteria, epidemiology and approaches to treatment. CNS Drugs 2001; 15:351–359.
- Wilhelm S, Keuthen NJ, Deckersbach T, et al. Self-injurious skin picking: clinical characteristics and comorbidity. J Clin Psychiatry 1999; 60:454–459.
- Gupta MA, Gupta AK, Haberman HF. Neurotic excoriations: a review and some new perspectives. Compr Psychiatry 1986; 27:381–386.
- Rosenbaum MS, Ayllon T. The behavioral treatment of neurodermatitis through habit-reversal. Behav Res Ther 1981; 19:313–318.
- Deckersbach T, Wilhelm S, Keuthen N. Self-injurious skin picking: clinical characteristics, assessment methods, and treatment modalities. Brief Treatment and Crisis Intervention 2003; 3:249–260.
- Sharma H. Psychogenic excoriation responding to fluoxetine: a case report. J Indian Med Assoc 2008; 106:245,262.
- Bloch MR, Elliott M, Thompson H, Koran LM. Fluoxetine in pathologic skin-picking: open-label and double-blind results. Psychosomatics 2001; 42:314–319.
- Simeon D, Stein DJ, Gross S, Islam N, Schmeidler J, Hollander E. A double-blind trial of fluoxetine in pathologic skin picking. J Clin Psychiatry 1997; 58:341–347.
- Gupta MA, Gupta AK. The use of antidepressant drugs in dermatology. J Eur Acad Dermatol Venereol 2001; 15:512–518.
- Keuthen NJ, Jameson M, Loh R, Deckersbach T, Wilhelm S, Dougherty DD. Open-label escitalopram treatment for pathological skin picking. Int Clin Psychopharmacol 2007; 22:268–274.
- Denys D, van Megen HJ, Westenberg HG. Emerging skin-picking behaviour after serotonin reuptake inhibitor-treatment in patients with obsessive-compulsive disorder: possible mechanisms and implications for clinical care. J Psychopharmacol 2003; 17:127–129.
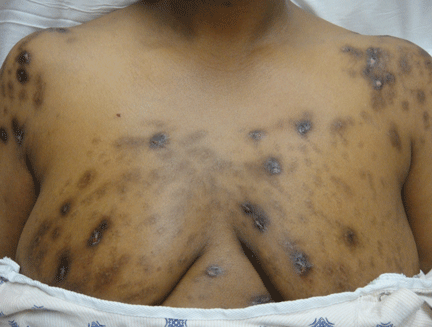
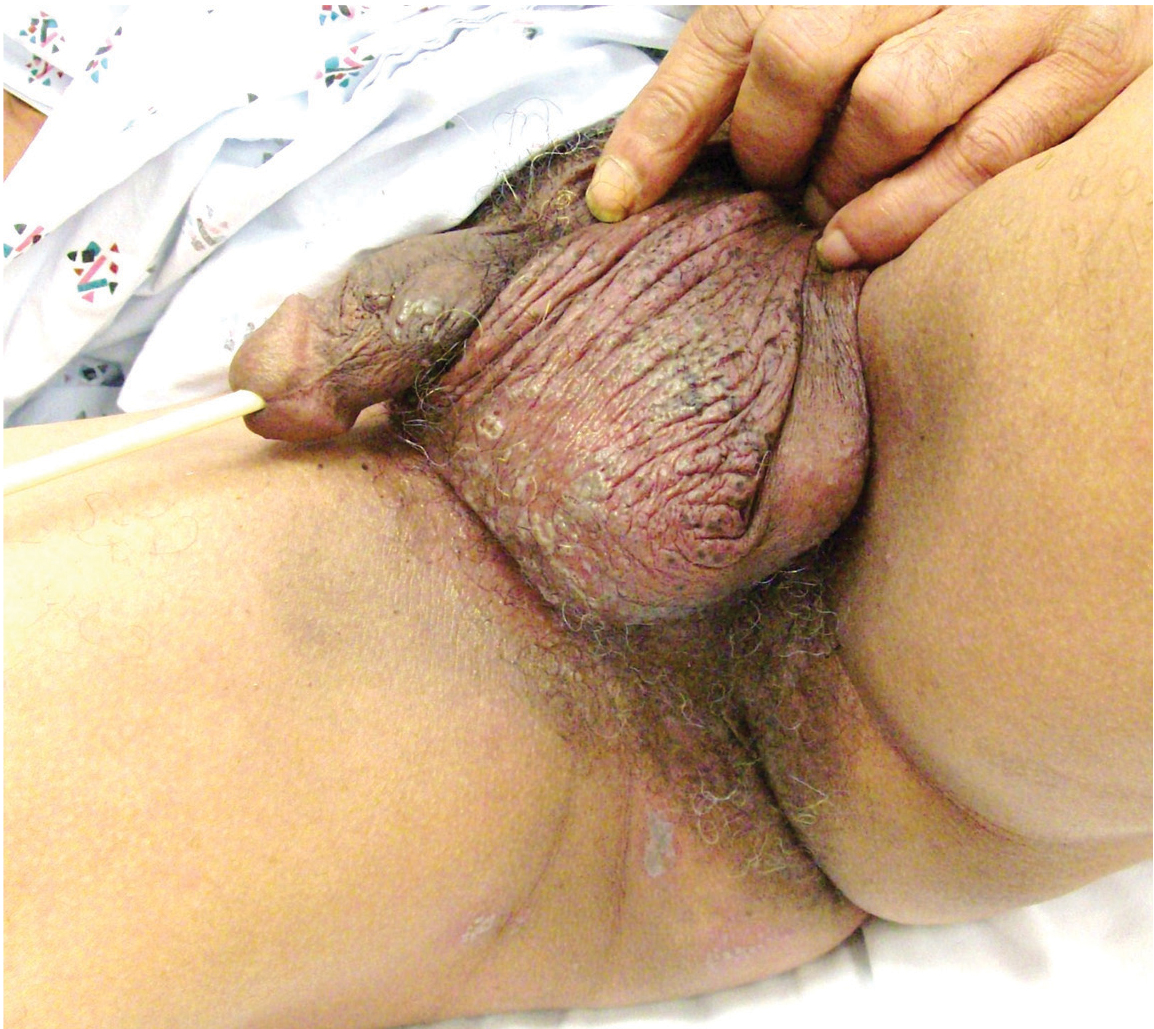
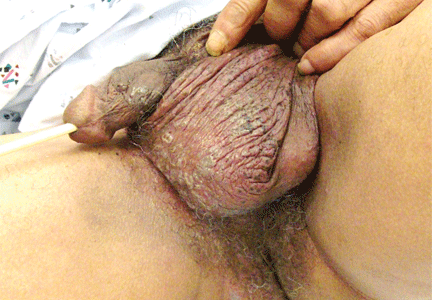
Genital lesions and acute urinary retention
He is sexually active. Nothing in his medical and surgical history would appear to contribute to his symptoms. He is not taking any medications.
His temperature is 38.5°C (101.3°F). His bladder is distended on palpation and percussion, and he has patches of vesicular and ulcerative lesions on an erythematous base over the scrotum and penis and the proximal inner thighs, lacking a dermatomal pattern. He also has decreased sensation to touch and pain perianally. Anal sphincter tone, deep tendon reflexes, and motor examination of the lower extremities are normal. The patient also lacks any positive meningeal findings. Bladder catheterization yields 1,250 mL of urine.
Q: What is the next step?
- Urine culture
- Immunofluorescence for chlamydial infection
- Cerebrospinal fluid (CSF) analysis
- Spinal myelography
- Urine toxicologic screen
A: CSF analysis is a vital diagnostic step in the evaluation of patients who present with skin rash around the genital area and acute urinary retention, to rule out acute sacral myeloradiculitis. In this disease, CSF analysis typically shows an increase in lymphocytes and a mild increase in protein. Urinary tract infection, gonorrhea, central spinal cord compression, and drug intoxication are unlikely to have this presentation. Hence, CSF analysis is the correct answer.
The patient undergoes a spinal tap. CSF analysis reveals an elevated white cell count of 85 cells/μL (reference range 0–5), with 79% lymphocytes (reference range 60%–70%), a mildly elevated protein concentration at 80 mg/dL (reference range 15–50), and a normal glucose level. Polymerase chain reaction testing of the CSF is negative for herpes simplex virus (HSV).
Magnetic resonance images of the brain and spinal cord are normal.
Polymerase chain reaction testing of specimens collected from the genital lesions detects HSV type 2 (HSV-2). Serum Venereal Disease Research Laboratory and serum human immunodeficiency virus tests are negative.
Diagnosis: HSV-2 sacral radiculitis (Elsberg syndrome).
THE PATIENT’S COURSE AND TREATMENT
The patient was initially treated symptomatically by draining the urinary bladder via a Foley catheter, followed by intermittent self-catheterization and topical application of lidocaine cream. He was also given intravenous acyclovir (Zovirax) 10 mg/kg every 8 hours for 2 days, followed by 400 mg orally every 8 hours for 10 days.
By 2 weeks, the skin lesions had resolved, and the patient reported regaining sensation in his anal and sacral areas. He also said he was able to void urine without any difficulty.
ELSBERG SYNDROME
Elsberg syndrome describes multiple disorders characterized by sacral myeloradiculitis, which can be secondary to viral infection, most commonly HSV type 2.1,2 CSF analysis and cultures should be performed. However, cultures and HSV studies of the CSF are often negative, as in this patient.3 CSF lymphocytosis along with a slightly elevated protein level is typical of this disease. It is the latent HSV reactivation in the sacral sensory ganglia that results in sensory neuronal dysfunction and subsequent loss of bladder sensation along with areflexia.
Elsberg syndrome is self-limiting and has a generally good prognosis, but complications such as necrotizing myelitis have been described.4 The combination of acute urinary retention associated with vesicular skin lesions and sensory nerve dysfunction in the sacral nerve root distribution in a young, sexually active patient is a strong clue to the diagnosis of Elsberg syndrome, a serious but treatable disease.
- Caplan LR, Kleeman FJ, Berg S. Urinary retention probably secondary to herpes genitalis. N Engl J Med 1977; 297:920–921.
- Eberhardt O, Küker W, Dichgans J, Weller M. HSV-2 sacral radiculitis (Elsberg syndrome). Neurology 2004; 63:758–759.
- Sakakibara R, Uchiyama T, Liu Z, et al. Meningitis-retention syndrome. An unrecognized clinical condition. J Neurol 2005; 252:1495–1499.
- Koskiniemi ML, Vaheri A, Manninen V, Nikki P. Ascending myelitis with high antibody titer to herpes simplex virus in the cerebrospinal fluid. J Neurol 1982; 227:187–191.
He is sexually active. Nothing in his medical and surgical history would appear to contribute to his symptoms. He is not taking any medications.
His temperature is 38.5°C (101.3°F). His bladder is distended on palpation and percussion, and he has patches of vesicular and ulcerative lesions on an erythematous base over the scrotum and penis and the proximal inner thighs, lacking a dermatomal pattern. He also has decreased sensation to touch and pain perianally. Anal sphincter tone, deep tendon reflexes, and motor examination of the lower extremities are normal. The patient also lacks any positive meningeal findings. Bladder catheterization yields 1,250 mL of urine.
Q: What is the next step?
- Urine culture
- Immunofluorescence for chlamydial infection
- Cerebrospinal fluid (CSF) analysis
- Spinal myelography
- Urine toxicologic screen
A: CSF analysis is a vital diagnostic step in the evaluation of patients who present with skin rash around the genital area and acute urinary retention, to rule out acute sacral myeloradiculitis. In this disease, CSF analysis typically shows an increase in lymphocytes and a mild increase in protein. Urinary tract infection, gonorrhea, central spinal cord compression, and drug intoxication are unlikely to have this presentation. Hence, CSF analysis is the correct answer.
The patient undergoes a spinal tap. CSF analysis reveals an elevated white cell count of 85 cells/μL (reference range 0–5), with 79% lymphocytes (reference range 60%–70%), a mildly elevated protein concentration at 80 mg/dL (reference range 15–50), and a normal glucose level. Polymerase chain reaction testing of the CSF is negative for herpes simplex virus (HSV).
Magnetic resonance images of the brain and spinal cord are normal.
Polymerase chain reaction testing of specimens collected from the genital lesions detects HSV type 2 (HSV-2). Serum Venereal Disease Research Laboratory and serum human immunodeficiency virus tests are negative.
Diagnosis: HSV-2 sacral radiculitis (Elsberg syndrome).
THE PATIENT’S COURSE AND TREATMENT
The patient was initially treated symptomatically by draining the urinary bladder via a Foley catheter, followed by intermittent self-catheterization and topical application of lidocaine cream. He was also given intravenous acyclovir (Zovirax) 10 mg/kg every 8 hours for 2 days, followed by 400 mg orally every 8 hours for 10 days.
By 2 weeks, the skin lesions had resolved, and the patient reported regaining sensation in his anal and sacral areas. He also said he was able to void urine without any difficulty.
ELSBERG SYNDROME
Elsberg syndrome describes multiple disorders characterized by sacral myeloradiculitis, which can be secondary to viral infection, most commonly HSV type 2.1,2 CSF analysis and cultures should be performed. However, cultures and HSV studies of the CSF are often negative, as in this patient.3 CSF lymphocytosis along with a slightly elevated protein level is typical of this disease. It is the latent HSV reactivation in the sacral sensory ganglia that results in sensory neuronal dysfunction and subsequent loss of bladder sensation along with areflexia.
Elsberg syndrome is self-limiting and has a generally good prognosis, but complications such as necrotizing myelitis have been described.4 The combination of acute urinary retention associated with vesicular skin lesions and sensory nerve dysfunction in the sacral nerve root distribution in a young, sexually active patient is a strong clue to the diagnosis of Elsberg syndrome, a serious but treatable disease.
He is sexually active. Nothing in his medical and surgical history would appear to contribute to his symptoms. He is not taking any medications.
His temperature is 38.5°C (101.3°F). His bladder is distended on palpation and percussion, and he has patches of vesicular and ulcerative lesions on an erythematous base over the scrotum and penis and the proximal inner thighs, lacking a dermatomal pattern. He also has decreased sensation to touch and pain perianally. Anal sphincter tone, deep tendon reflexes, and motor examination of the lower extremities are normal. The patient also lacks any positive meningeal findings. Bladder catheterization yields 1,250 mL of urine.
Q: What is the next step?
- Urine culture
- Immunofluorescence for chlamydial infection
- Cerebrospinal fluid (CSF) analysis
- Spinal myelography
- Urine toxicologic screen
A: CSF analysis is a vital diagnostic step in the evaluation of patients who present with skin rash around the genital area and acute urinary retention, to rule out acute sacral myeloradiculitis. In this disease, CSF analysis typically shows an increase in lymphocytes and a mild increase in protein. Urinary tract infection, gonorrhea, central spinal cord compression, and drug intoxication are unlikely to have this presentation. Hence, CSF analysis is the correct answer.
The patient undergoes a spinal tap. CSF analysis reveals an elevated white cell count of 85 cells/μL (reference range 0–5), with 79% lymphocytes (reference range 60%–70%), a mildly elevated protein concentration at 80 mg/dL (reference range 15–50), and a normal glucose level. Polymerase chain reaction testing of the CSF is negative for herpes simplex virus (HSV).
Magnetic resonance images of the brain and spinal cord are normal.
Polymerase chain reaction testing of specimens collected from the genital lesions detects HSV type 2 (HSV-2). Serum Venereal Disease Research Laboratory and serum human immunodeficiency virus tests are negative.
Diagnosis: HSV-2 sacral radiculitis (Elsberg syndrome).
THE PATIENT’S COURSE AND TREATMENT
The patient was initially treated symptomatically by draining the urinary bladder via a Foley catheter, followed by intermittent self-catheterization and topical application of lidocaine cream. He was also given intravenous acyclovir (Zovirax) 10 mg/kg every 8 hours for 2 days, followed by 400 mg orally every 8 hours for 10 days.
By 2 weeks, the skin lesions had resolved, and the patient reported regaining sensation in his anal and sacral areas. He also said he was able to void urine without any difficulty.
ELSBERG SYNDROME
Elsberg syndrome describes multiple disorders characterized by sacral myeloradiculitis, which can be secondary to viral infection, most commonly HSV type 2.1,2 CSF analysis and cultures should be performed. However, cultures and HSV studies of the CSF are often negative, as in this patient.3 CSF lymphocytosis along with a slightly elevated protein level is typical of this disease. It is the latent HSV reactivation in the sacral sensory ganglia that results in sensory neuronal dysfunction and subsequent loss of bladder sensation along with areflexia.
Elsberg syndrome is self-limiting and has a generally good prognosis, but complications such as necrotizing myelitis have been described.4 The combination of acute urinary retention associated with vesicular skin lesions and sensory nerve dysfunction in the sacral nerve root distribution in a young, sexually active patient is a strong clue to the diagnosis of Elsberg syndrome, a serious but treatable disease.
- Caplan LR, Kleeman FJ, Berg S. Urinary retention probably secondary to herpes genitalis. N Engl J Med 1977; 297:920–921.
- Eberhardt O, Küker W, Dichgans J, Weller M. HSV-2 sacral radiculitis (Elsberg syndrome). Neurology 2004; 63:758–759.
- Sakakibara R, Uchiyama T, Liu Z, et al. Meningitis-retention syndrome. An unrecognized clinical condition. J Neurol 2005; 252:1495–1499.
- Koskiniemi ML, Vaheri A, Manninen V, Nikki P. Ascending myelitis with high antibody titer to herpes simplex virus in the cerebrospinal fluid. J Neurol 1982; 227:187–191.
- Caplan LR, Kleeman FJ, Berg S. Urinary retention probably secondary to herpes genitalis. N Engl J Med 1977; 297:920–921.
- Eberhardt O, Küker W, Dichgans J, Weller M. HSV-2 sacral radiculitis (Elsberg syndrome). Neurology 2004; 63:758–759.
- Sakakibara R, Uchiyama T, Liu Z, et al. Meningitis-retention syndrome. An unrecognized clinical condition. J Neurol 2005; 252:1495–1499.
- Koskiniemi ML, Vaheri A, Manninen V, Nikki P. Ascending myelitis with high antibody titer to herpes simplex virus in the cerebrospinal fluid. J Neurol 1982; 227:187–191.
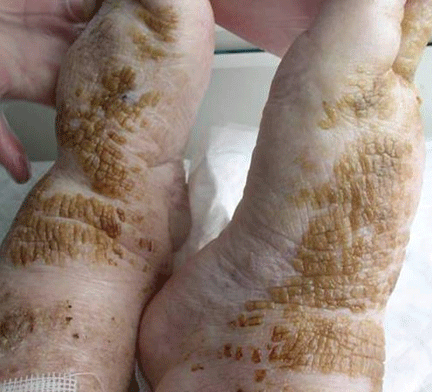
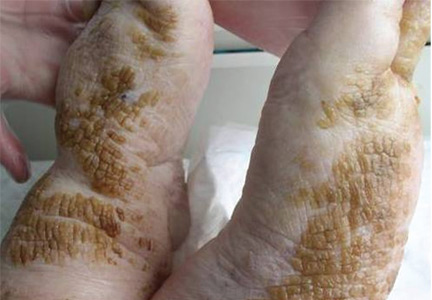
Cornflake-like scales on the ankles and feet
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Biopsy of the lesions
- Hemoglobin A1C
- Fungal culture
- Brush with soap and water or alcohol
- Bacterial culture
A: Patchy brown, waxy scales in a patient who neglects normal cleansing should raise the possibility of dermatosis neglecta. Hence, brushing with soap and water or alcohol is the correct answer.
In this patient, the plaques were easily removed using an alcohol-soaked swab, revealing underlying normal skin with bleeding spots. The patient’s family was instructed to lightly scrub the area daily with a washcloth, soap, and water. The lesions resolved completely over the next 3 weeks.
DERMATOSIS NEGLECTA
Dermatosis neglecta is a condition that results from the accumulation in the epidermal stratum corium of sebum, sweat, corneocytes, and bacterias in areas of unwashed skin. It affects all age groups, and it has a similar prevalence in both sexes. Sometimes, an underlying physical disability or emotional problem is the cause, other times it is due to prior trauma or hyperesthesia of the affected area. Dermatosis neglecta presents with asymptomatic areas of dark brown patches or plaque with cornflake-like scales and a verrucous surface.1–4 The diagnosis is usually easy and is based on the characteristic findings and the simple test described above. For treatment, in some cases a keratolytic lotion containing lactic acid, glycolic acid, urea, or salicylic acid is helpful.4
MISDIAGNOSIS IS EASY
Dermatosis neglecta could easily be misdiagnosed as verrucous epidermal nevus, terra firma-forme dermatosis, acanthosis nigricans, or another less common dermatosis of “dirty skin” lesions, such as confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome).2–4
Verrucous epidermal nevus appears at birth or early childhood. Patients have verrucous papules and plaques that commonly follow a narrow linear distribution.
Acanthosis nigricans is characterized by broad, velvety, hyperpigmented smooth plaques on the neck, axillae, and flexures. It is related to insulin resistance, obesity, and underlying malignancy.
Confluent and reticulate papillomatosis is a rare dermatosis of unknown cause, with a pigmented reticular pattern distributed on the central trunk. Patients tend to practice normal hygiene.
Terra firma-forme dermatosis is characterized by dirt-like patches that are resistant to usual hygiene measures but that can be eliminated by rubbing with isopropyl alcohol. The cause is unknown, and it is not related to a lack of cleansing. It is considered a variant of dermatosis neglecta.
The medical literature contains few reports of dermatosis neglecta, but the true incidence is probably underestimated. Early recognition by a physician avoids the need for aggressive diagnostic and therapeutic procedures.
- Ruiz-Maldonado R, Durán-McKinster C, Tamayo-Sánchez L, Orozco-Covarrubias ML. Dermatosis neglecta: dirt crusts simulating verrucous nevi. Arch Dermatol 1999; 135:728–729.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. Case series and review of the literature. J Dtsch Dermatol Ges 2009; 7:102–107.
- Qadir SN, Ejaz A, Raza N. Dermatosis neglecta in a case of multiple fractures, shoulder dislocation and radial nerve palsy in a 35-year-old man: a case report. J Med Case Reports 2008; 2:347.
- Lucas JL, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J 2006; 12:5.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Biopsy of the lesions
- Hemoglobin A1C
- Fungal culture
- Brush with soap and water or alcohol
- Bacterial culture
A: Patchy brown, waxy scales in a patient who neglects normal cleansing should raise the possibility of dermatosis neglecta. Hence, brushing with soap and water or alcohol is the correct answer.
In this patient, the plaques were easily removed using an alcohol-soaked swab, revealing underlying normal skin with bleeding spots. The patient’s family was instructed to lightly scrub the area daily with a washcloth, soap, and water. The lesions resolved completely over the next 3 weeks.
DERMATOSIS NEGLECTA
Dermatosis neglecta is a condition that results from the accumulation in the epidermal stratum corium of sebum, sweat, corneocytes, and bacterias in areas of unwashed skin. It affects all age groups, and it has a similar prevalence in both sexes. Sometimes, an underlying physical disability or emotional problem is the cause, other times it is due to prior trauma or hyperesthesia of the affected area. Dermatosis neglecta presents with asymptomatic areas of dark brown patches or plaque with cornflake-like scales and a verrucous surface.1–4 The diagnosis is usually easy and is based on the characteristic findings and the simple test described above. For treatment, in some cases a keratolytic lotion containing lactic acid, glycolic acid, urea, or salicylic acid is helpful.4
MISDIAGNOSIS IS EASY
Dermatosis neglecta could easily be misdiagnosed as verrucous epidermal nevus, terra firma-forme dermatosis, acanthosis nigricans, or another less common dermatosis of “dirty skin” lesions, such as confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome).2–4
Verrucous epidermal nevus appears at birth or early childhood. Patients have verrucous papules and plaques that commonly follow a narrow linear distribution.
Acanthosis nigricans is characterized by broad, velvety, hyperpigmented smooth plaques on the neck, axillae, and flexures. It is related to insulin resistance, obesity, and underlying malignancy.
Confluent and reticulate papillomatosis is a rare dermatosis of unknown cause, with a pigmented reticular pattern distributed on the central trunk. Patients tend to practice normal hygiene.
Terra firma-forme dermatosis is characterized by dirt-like patches that are resistant to usual hygiene measures but that can be eliminated by rubbing with isopropyl alcohol. The cause is unknown, and it is not related to a lack of cleansing. It is considered a variant of dermatosis neglecta.
The medical literature contains few reports of dermatosis neglecta, but the true incidence is probably underestimated. Early recognition by a physician avoids the need for aggressive diagnostic and therapeutic procedures.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Biopsy of the lesions
- Hemoglobin A1C
- Fungal culture
- Brush with soap and water or alcohol
- Bacterial culture
A: Patchy brown, waxy scales in a patient who neglects normal cleansing should raise the possibility of dermatosis neglecta. Hence, brushing with soap and water or alcohol is the correct answer.
In this patient, the plaques were easily removed using an alcohol-soaked swab, revealing underlying normal skin with bleeding spots. The patient’s family was instructed to lightly scrub the area daily with a washcloth, soap, and water. The lesions resolved completely over the next 3 weeks.
DERMATOSIS NEGLECTA
Dermatosis neglecta is a condition that results from the accumulation in the epidermal stratum corium of sebum, sweat, corneocytes, and bacterias in areas of unwashed skin. It affects all age groups, and it has a similar prevalence in both sexes. Sometimes, an underlying physical disability or emotional problem is the cause, other times it is due to prior trauma or hyperesthesia of the affected area. Dermatosis neglecta presents with asymptomatic areas of dark brown patches or plaque with cornflake-like scales and a verrucous surface.1–4 The diagnosis is usually easy and is based on the characteristic findings and the simple test described above. For treatment, in some cases a keratolytic lotion containing lactic acid, glycolic acid, urea, or salicylic acid is helpful.4
MISDIAGNOSIS IS EASY
Dermatosis neglecta could easily be misdiagnosed as verrucous epidermal nevus, terra firma-forme dermatosis, acanthosis nigricans, or another less common dermatosis of “dirty skin” lesions, such as confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome).2–4
Verrucous epidermal nevus appears at birth or early childhood. Patients have verrucous papules and plaques that commonly follow a narrow linear distribution.
Acanthosis nigricans is characterized by broad, velvety, hyperpigmented smooth plaques on the neck, axillae, and flexures. It is related to insulin resistance, obesity, and underlying malignancy.
Confluent and reticulate papillomatosis is a rare dermatosis of unknown cause, with a pigmented reticular pattern distributed on the central trunk. Patients tend to practice normal hygiene.
Terra firma-forme dermatosis is characterized by dirt-like patches that are resistant to usual hygiene measures but that can be eliminated by rubbing with isopropyl alcohol. The cause is unknown, and it is not related to a lack of cleansing. It is considered a variant of dermatosis neglecta.
The medical literature contains few reports of dermatosis neglecta, but the true incidence is probably underestimated. Early recognition by a physician avoids the need for aggressive diagnostic and therapeutic procedures.
- Ruiz-Maldonado R, Durán-McKinster C, Tamayo-Sánchez L, Orozco-Covarrubias ML. Dermatosis neglecta: dirt crusts simulating verrucous nevi. Arch Dermatol 1999; 135:728–729.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. Case series and review of the literature. J Dtsch Dermatol Ges 2009; 7:102–107.
- Qadir SN, Ejaz A, Raza N. Dermatosis neglecta in a case of multiple fractures, shoulder dislocation and radial nerve palsy in a 35-year-old man: a case report. J Med Case Reports 2008; 2:347.
- Lucas JL, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J 2006; 12:5.
- Ruiz-Maldonado R, Durán-McKinster C, Tamayo-Sánchez L, Orozco-Covarrubias ML. Dermatosis neglecta: dirt crusts simulating verrucous nevi. Arch Dermatol 1999; 135:728–729.
- Akkash L, Badran D, Al-Omari AQ. Terra firma forme dermatosis. Case series and review of the literature. J Dtsch Dermatol Ges 2009; 7:102–107.
- Qadir SN, Ejaz A, Raza N. Dermatosis neglecta in a case of multiple fractures, shoulder dislocation and radial nerve palsy in a 35-year-old man: a case report. J Med Case Reports 2008; 2:347.
- Lucas JL, Brodell RT, Feldman SR. Dermatosis neglecta: a series of case reports and review of other dirty-appearing dermatoses. Dermatol Online J 2006; 12:5.
Palmoplantar eruption
A 38-year-old woman presents with recurrent asymptomatic lesions on the palms and soles and on the sides of both feet. The lesions have been developing for 2 months, unaccompanied by fever or other systemic symptoms.
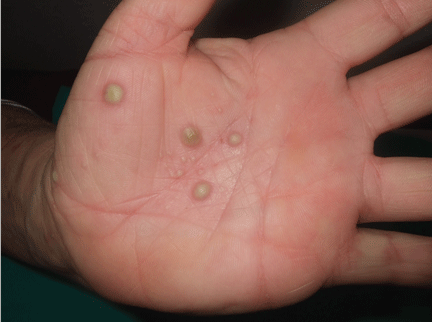
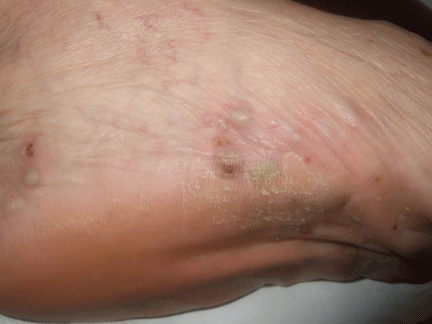
Laboratory tests of C-reactive protein, erythrocyte sedimentation rate, viral serologies, and antinuclear antibodies are normal. A pustule culture is negative, and a cutaneous biopsy shows parakeratosis and elongation of rete ridges, neutrophils migrating from papillary capillaries to the epidermis, and spongiform Kogoj pustule.
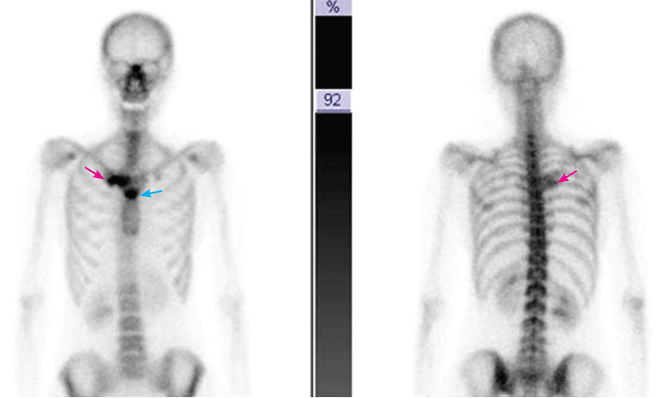
Q: Which is the most likely diagnosis?
- Pustular psoriasis
- Impetigo contagiosa
- Syndrome of synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO)
- Dyshidrotic eczema
- Acute exanthematous pustulosis (drug eruption)
A: SAPHO syndrome is the most likely diagnosis. The presence of pustules and aseptic osteitis of the anterior chest wall is compatible with SAPHO syndrome. Treatment with topical clobetasol propionate (Temovate) for pustules and NSAIDs for osteitis brought a good response.
Pustular psoriasis is an uncommon type of psoriasis characterized by erythema and pustules involving the flexural and anogenital areas. Cutaneous lesions of psoriasis vulgaris may be present before an acute pustular episode. Withdrawal of systemic corticosteroids in a patient with psoriasis has been reported as a precipitating factor.
Impetigo contagiosa is a superficial cutaneous infection characterized by an erythematous macule that evolves into a vesicle or pustule. These lesions are more common in children. Culture of the fluid usually reveals Staphylococcus aureus or S pyogenes.
Dyshidrotic eczema is a pruritic vesicular eruption of unknown cause on the palm and soles (bilateral and symmetric). The typical histologic findings are spongiotic and intraepidermal vesicles.
Acute exanthematous pustulosis is a drug-induced reaction characterized by confluent erythema, blisters, and pustules, mucous membrane erosions with fever, and lymphadenopathy. Cultures of the pustules are negative, and biopsy can help confirm the diagnosis of drug eruption.
SAPHO SYNDROME
SAPHO syndrome is a rare condition of unknown pathogenesis originally described by Chamot et al1 in 1987. The onset is usually in young adulthood, and is similar in men and women. It is characterized by synovitis, acne, pustulosis, hyperostosis, and osteitis. Of paramount importance is the finding of a non-infectious inflammatory osteitis in a patient with skin lesions.
Clinical findings: Pustules plus rheumatic pain
SAPHO syndrome must be suspected when a patient is affected by a pustular skin disease associated with rheumatic pain. If examination shows that the pain is caused by a sterile inflammation of bone or joints, the diagnosis tends to be confirmed.2
Osteoarticular involvement tends to be limited to the anterior chest wall. It may include aseptic osteitis, hyperostosis, and symmetrical arthritis. Peripheral and axial osteitis is one of the main characteristics of the syndrome and is found in around 90% of cases.
Cutaneous manifestations are present in two-thirds of patients and consist chiefly of severe acne (acne fulminans, acne conglobata, and hidradenitis suppurativa), pustular psoriasis, and palmoplantar pustulosis. Neutrophilic dermatoses associated with this syndrome include Sweet syndrome and pyoderma gangrenosum. Acne lesions are usually seen in men, whereas palmoplantar pustulosis is seen in women,3 often accompanying osteoarticular manifestations.
Radiologic findings in the spine are spondylodiskitis, osteosclerosis, sacroiliac joint involvement, and paravertebral ossification. In anterior chest wall hyperostosis, common findings are bone hypertrophy and sclerosis with a soft-tissue component. Laboratory test results are uncharacteristic, with variable signs of inflammation, and the C-reactive protein and sedimentation rate are usually elevated in the absence of leukocytosis.
Pathogenesis remains elusive
The pathogenesis of this syndrome remains elusive. Since SAPHO syndrome usually involves the axial skeleton, some investigators have suggested a possible link between the SAPHO syndrome and the seronegative spondyloarthopathies.3 It has also been related to an infection by Propionibacterium acnes and Corynebacterium species,4 which have been isolated in cultures of bone and skin lesions. However, the fact that these bacteria are contaminant agents makes their involvement in the pathogenesis of this syndrome unlikely. More recently, high concentrations of tumor necrosis factor alpha in bone specimens of patients with SAPHO syndrome have been reported, thus highlighting the central role of this cytokine in maintaining inflammation.
Treatment is to relieve symptoms
Since understanding of the pathogenesis of SAPHO syndrome is limited, a wide range of therapies has been used,5 mostly to relieve symptoms. These include NSAIDs; steroids; antibiotics; bisphosphonates such as pamidronate (Aredia) and zoledronic acid (Reclast); and immunosuppressors and immunomodulators such as methotrexate (Trexall), leflunomide (Arava), sulfasalazine (Azulfidine), cyclosporine (Sandimmune). The results with these therapies have been quite varied. Good response has been reported with tumor necrosis factor alpha blockers—infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira).6
- Chamot AM, Benhamou CL, Kahn MF, Beraneck L, Kaplan G, Prost A. Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases [in French]. Rev Rhum Mal Osteoartic 1987; 54:187–196.
- Benhamou CL, Chamot AM, Kahn MF. Synovitis-acnepustulosis hyperostosis-osteomyelitis syndrome (SAPHO). A new syndrome among the spondyloarthropathies? Clin Exp Rheumatol 1988; 6:109–112.
- Hayem G, Bouchaud-Chabot A, Benali K, et al. SAPHO syndrome: a long-term follow-up study of 120 cases. Semin Arthritis Rheum 1999; 29:159–171.
- Moll C, Hernández MV, Cañete JD, et al. Ilium osteitis as the main manifestation of the SAPHO syndrome: response to infliximab therapy and review of the literature. Semin Arthritis Rheum 2008; 37:299–306.
- Olivieri I, Padula A, Palazzi C. Pharmacological management of SAPHO syndrome. Expert Opin Investig Drugs 2006; 15:1229–1233.
- Arias-Santiago S, Sanchez-Cano D, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Adalimumab treatment for SAPHO syndrome. Acta Derm Venereol 2010; 90:301–302.
A 38-year-old woman presents with recurrent asymptomatic lesions on the palms and soles and on the sides of both feet. The lesions have been developing for 2 months, unaccompanied by fever or other systemic symptoms.
Laboratory tests of C-reactive protein, erythrocyte sedimentation rate, viral serologies, and antinuclear antibodies are normal. A pustule culture is negative, and a cutaneous biopsy shows parakeratosis and elongation of rete ridges, neutrophils migrating from papillary capillaries to the epidermis, and spongiform Kogoj pustule.
Q: Which is the most likely diagnosis?
- Pustular psoriasis
- Impetigo contagiosa
- Syndrome of synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO)
- Dyshidrotic eczema
- Acute exanthematous pustulosis (drug eruption)
A: SAPHO syndrome is the most likely diagnosis. The presence of pustules and aseptic osteitis of the anterior chest wall is compatible with SAPHO syndrome. Treatment with topical clobetasol propionate (Temovate) for pustules and NSAIDs for osteitis brought a good response.
Pustular psoriasis is an uncommon type of psoriasis characterized by erythema and pustules involving the flexural and anogenital areas. Cutaneous lesions of psoriasis vulgaris may be present before an acute pustular episode. Withdrawal of systemic corticosteroids in a patient with psoriasis has been reported as a precipitating factor.
Impetigo contagiosa is a superficial cutaneous infection characterized by an erythematous macule that evolves into a vesicle or pustule. These lesions are more common in children. Culture of the fluid usually reveals Staphylococcus aureus or S pyogenes.
Dyshidrotic eczema is a pruritic vesicular eruption of unknown cause on the palm and soles (bilateral and symmetric). The typical histologic findings are spongiotic and intraepidermal vesicles.
Acute exanthematous pustulosis is a drug-induced reaction characterized by confluent erythema, blisters, and pustules, mucous membrane erosions with fever, and lymphadenopathy. Cultures of the pustules are negative, and biopsy can help confirm the diagnosis of drug eruption.
SAPHO SYNDROME
SAPHO syndrome is a rare condition of unknown pathogenesis originally described by Chamot et al1 in 1987. The onset is usually in young adulthood, and is similar in men and women. It is characterized by synovitis, acne, pustulosis, hyperostosis, and osteitis. Of paramount importance is the finding of a non-infectious inflammatory osteitis in a patient with skin lesions.
Clinical findings: Pustules plus rheumatic pain
SAPHO syndrome must be suspected when a patient is affected by a pustular skin disease associated with rheumatic pain. If examination shows that the pain is caused by a sterile inflammation of bone or joints, the diagnosis tends to be confirmed.2
Osteoarticular involvement tends to be limited to the anterior chest wall. It may include aseptic osteitis, hyperostosis, and symmetrical arthritis. Peripheral and axial osteitis is one of the main characteristics of the syndrome and is found in around 90% of cases.
Cutaneous manifestations are present in two-thirds of patients and consist chiefly of severe acne (acne fulminans, acne conglobata, and hidradenitis suppurativa), pustular psoriasis, and palmoplantar pustulosis. Neutrophilic dermatoses associated with this syndrome include Sweet syndrome and pyoderma gangrenosum. Acne lesions are usually seen in men, whereas palmoplantar pustulosis is seen in women,3 often accompanying osteoarticular manifestations.
Radiologic findings in the spine are spondylodiskitis, osteosclerosis, sacroiliac joint involvement, and paravertebral ossification. In anterior chest wall hyperostosis, common findings are bone hypertrophy and sclerosis with a soft-tissue component. Laboratory test results are uncharacteristic, with variable signs of inflammation, and the C-reactive protein and sedimentation rate are usually elevated in the absence of leukocytosis.
Pathogenesis remains elusive
The pathogenesis of this syndrome remains elusive. Since SAPHO syndrome usually involves the axial skeleton, some investigators have suggested a possible link between the SAPHO syndrome and the seronegative spondyloarthopathies.3 It has also been related to an infection by Propionibacterium acnes and Corynebacterium species,4 which have been isolated in cultures of bone and skin lesions. However, the fact that these bacteria are contaminant agents makes their involvement in the pathogenesis of this syndrome unlikely. More recently, high concentrations of tumor necrosis factor alpha in bone specimens of patients with SAPHO syndrome have been reported, thus highlighting the central role of this cytokine in maintaining inflammation.
Treatment is to relieve symptoms
Since understanding of the pathogenesis of SAPHO syndrome is limited, a wide range of therapies has been used,5 mostly to relieve symptoms. These include NSAIDs; steroids; antibiotics; bisphosphonates such as pamidronate (Aredia) and zoledronic acid (Reclast); and immunosuppressors and immunomodulators such as methotrexate (Trexall), leflunomide (Arava), sulfasalazine (Azulfidine), cyclosporine (Sandimmune). The results with these therapies have been quite varied. Good response has been reported with tumor necrosis factor alpha blockers—infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira).6
A 38-year-old woman presents with recurrent asymptomatic lesions on the palms and soles and on the sides of both feet. The lesions have been developing for 2 months, unaccompanied by fever or other systemic symptoms.
Laboratory tests of C-reactive protein, erythrocyte sedimentation rate, viral serologies, and antinuclear antibodies are normal. A pustule culture is negative, and a cutaneous biopsy shows parakeratosis and elongation of rete ridges, neutrophils migrating from papillary capillaries to the epidermis, and spongiform Kogoj pustule.
Q: Which is the most likely diagnosis?
- Pustular psoriasis
- Impetigo contagiosa
- Syndrome of synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO)
- Dyshidrotic eczema
- Acute exanthematous pustulosis (drug eruption)
A: SAPHO syndrome is the most likely diagnosis. The presence of pustules and aseptic osteitis of the anterior chest wall is compatible with SAPHO syndrome. Treatment with topical clobetasol propionate (Temovate) for pustules and NSAIDs for osteitis brought a good response.
Pustular psoriasis is an uncommon type of psoriasis characterized by erythema and pustules involving the flexural and anogenital areas. Cutaneous lesions of psoriasis vulgaris may be present before an acute pustular episode. Withdrawal of systemic corticosteroids in a patient with psoriasis has been reported as a precipitating factor.
Impetigo contagiosa is a superficial cutaneous infection characterized by an erythematous macule that evolves into a vesicle or pustule. These lesions are more common in children. Culture of the fluid usually reveals Staphylococcus aureus or S pyogenes.
Dyshidrotic eczema is a pruritic vesicular eruption of unknown cause on the palm and soles (bilateral and symmetric). The typical histologic findings are spongiotic and intraepidermal vesicles.
Acute exanthematous pustulosis is a drug-induced reaction characterized by confluent erythema, blisters, and pustules, mucous membrane erosions with fever, and lymphadenopathy. Cultures of the pustules are negative, and biopsy can help confirm the diagnosis of drug eruption.
SAPHO SYNDROME
SAPHO syndrome is a rare condition of unknown pathogenesis originally described by Chamot et al1 in 1987. The onset is usually in young adulthood, and is similar in men and women. It is characterized by synovitis, acne, pustulosis, hyperostosis, and osteitis. Of paramount importance is the finding of a non-infectious inflammatory osteitis in a patient with skin lesions.
Clinical findings: Pustules plus rheumatic pain
SAPHO syndrome must be suspected when a patient is affected by a pustular skin disease associated with rheumatic pain. If examination shows that the pain is caused by a sterile inflammation of bone or joints, the diagnosis tends to be confirmed.2
Osteoarticular involvement tends to be limited to the anterior chest wall. It may include aseptic osteitis, hyperostosis, and symmetrical arthritis. Peripheral and axial osteitis is one of the main characteristics of the syndrome and is found in around 90% of cases.
Cutaneous manifestations are present in two-thirds of patients and consist chiefly of severe acne (acne fulminans, acne conglobata, and hidradenitis suppurativa), pustular psoriasis, and palmoplantar pustulosis. Neutrophilic dermatoses associated with this syndrome include Sweet syndrome and pyoderma gangrenosum. Acne lesions are usually seen in men, whereas palmoplantar pustulosis is seen in women,3 often accompanying osteoarticular manifestations.
Radiologic findings in the spine are spondylodiskitis, osteosclerosis, sacroiliac joint involvement, and paravertebral ossification. In anterior chest wall hyperostosis, common findings are bone hypertrophy and sclerosis with a soft-tissue component. Laboratory test results are uncharacteristic, with variable signs of inflammation, and the C-reactive protein and sedimentation rate are usually elevated in the absence of leukocytosis.
Pathogenesis remains elusive
The pathogenesis of this syndrome remains elusive. Since SAPHO syndrome usually involves the axial skeleton, some investigators have suggested a possible link between the SAPHO syndrome and the seronegative spondyloarthopathies.3 It has also been related to an infection by Propionibacterium acnes and Corynebacterium species,4 which have been isolated in cultures of bone and skin lesions. However, the fact that these bacteria are contaminant agents makes their involvement in the pathogenesis of this syndrome unlikely. More recently, high concentrations of tumor necrosis factor alpha in bone specimens of patients with SAPHO syndrome have been reported, thus highlighting the central role of this cytokine in maintaining inflammation.
Treatment is to relieve symptoms
Since understanding of the pathogenesis of SAPHO syndrome is limited, a wide range of therapies has been used,5 mostly to relieve symptoms. These include NSAIDs; steroids; antibiotics; bisphosphonates such as pamidronate (Aredia) and zoledronic acid (Reclast); and immunosuppressors and immunomodulators such as methotrexate (Trexall), leflunomide (Arava), sulfasalazine (Azulfidine), cyclosporine (Sandimmune). The results with these therapies have been quite varied. Good response has been reported with tumor necrosis factor alpha blockers—infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira).6
- Chamot AM, Benhamou CL, Kahn MF, Beraneck L, Kaplan G, Prost A. Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases [in French]. Rev Rhum Mal Osteoartic 1987; 54:187–196.
- Benhamou CL, Chamot AM, Kahn MF. Synovitis-acnepustulosis hyperostosis-osteomyelitis syndrome (SAPHO). A new syndrome among the spondyloarthropathies? Clin Exp Rheumatol 1988; 6:109–112.
- Hayem G, Bouchaud-Chabot A, Benali K, et al. SAPHO syndrome: a long-term follow-up study of 120 cases. Semin Arthritis Rheum 1999; 29:159–171.
- Moll C, Hernández MV, Cañete JD, et al. Ilium osteitis as the main manifestation of the SAPHO syndrome: response to infliximab therapy and review of the literature. Semin Arthritis Rheum 2008; 37:299–306.
- Olivieri I, Padula A, Palazzi C. Pharmacological management of SAPHO syndrome. Expert Opin Investig Drugs 2006; 15:1229–1233.
- Arias-Santiago S, Sanchez-Cano D, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Adalimumab treatment for SAPHO syndrome. Acta Derm Venereol 2010; 90:301–302.
- Chamot AM, Benhamou CL, Kahn MF, Beraneck L, Kaplan G, Prost A. Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases [in French]. Rev Rhum Mal Osteoartic 1987; 54:187–196.
- Benhamou CL, Chamot AM, Kahn MF. Synovitis-acnepustulosis hyperostosis-osteomyelitis syndrome (SAPHO). A new syndrome among the spondyloarthropathies? Clin Exp Rheumatol 1988; 6:109–112.
- Hayem G, Bouchaud-Chabot A, Benali K, et al. SAPHO syndrome: a long-term follow-up study of 120 cases. Semin Arthritis Rheum 1999; 29:159–171.
- Moll C, Hernández MV, Cañete JD, et al. Ilium osteitis as the main manifestation of the SAPHO syndrome: response to infliximab therapy and review of the literature. Semin Arthritis Rheum 2008; 37:299–306.
- Olivieri I, Padula A, Palazzi C. Pharmacological management of SAPHO syndrome. Expert Opin Investig Drugs 2006; 15:1229–1233.
- Arias-Santiago S, Sanchez-Cano D, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Adalimumab treatment for SAPHO syndrome. Acta Derm Venereol 2010; 90:301–302.
An ulcerated plaque on the hand
Q: Which is the most likely diagnosis?
- Blastomycosis
- Keratoacanthoma
- Basal cell carcinoma
- Squamous cell carcinoma
- Extramammary Paget disease
A: Squamous cell carcinoma is the most likely diagnosis. The lesions may take the form of a patch, plaque, or nodule, sometimes with scaling or an ulcerated center. The borders often are irregular, fleshy, and bleed easily.
Blastomycosis is a rare fungal infection caused by inhaling the spores of the fungus Blastomyces dermatitidis. Skin features begin as papules, pustules, or subcutaneous nodules. Over a period of months, lesions heal to form raised wart-like scars, which are often irreversible.
Keratoacanthoma is a benign nonmelanomatous cancer that grows rapidly and remits spontaneously. This diagnosis is unlikely in our patient, whose lesion evolved over 2 years.
Basal cell carcinoma has a waxy, translucent, or pearly appearance, without a keratotic component. Telangiectasias are common. Unlike squamous cell carcinomas, the edges of basal cell lesions are clear rather than fleshy.
Extramammary Paget disease usually manifests as intense pruritus in affected areas, which appear as chronic intertrigo or nonresolving eczema. The dorsum of the hand is a very rare location for this disease. Given our patient’s presentation, extrammary Paget disease is an unlikely diagnosis.
CHRONIC SUN EXPOSURE IS A KEY RISK FACTOR
Our patient has worked outdoors for many years, and chronic sun exposure is a key risk factor for squamous cell carcinoma.1
Squamous cell carcinoma is a nonmelanomatous skin cancer arising from the more superficial layers of keratinocytes. Unlike basal cell carcinoma, it can metastasize,2 so the diagnosis must be made as soon as possible.
The key to diagnosis is a high degree of suspicion. Patients usually present with chronic sun-damaged skin with lesions as actinic keratoses or keratin horns. Squamous cell carcinoma appears as a thick, adherent scale that does not heal and may intermittently bleed. Because it spreads into the dermis, it can appear like an ulcer, with hard, raised edges, as in our patient.
TREATMENT AND PREVENTION
The standard effective treatment is complete surgical excision.1 Nonsurgical treatments include curettage plus cautery, cryosurgery, radiotherapy, photodynamic therapy, and imiquimod (Aldara) 5% cream. Minimizing sun exposure and regular checkups are important preventive measures.
Our patient underwent total surgical excision of the lesion. Due to the size of the lesion, a skin graft was required and was obtained from the abdomen. No relapse was observed after 1 year of follow-up.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Rowe DE, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis and survival rates in squamous cell carcinoma of the skin, ear, and lip: implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
Q: Which is the most likely diagnosis?
- Blastomycosis
- Keratoacanthoma
- Basal cell carcinoma
- Squamous cell carcinoma
- Extramammary Paget disease
A: Squamous cell carcinoma is the most likely diagnosis. The lesions may take the form of a patch, plaque, or nodule, sometimes with scaling or an ulcerated center. The borders often are irregular, fleshy, and bleed easily.
Blastomycosis is a rare fungal infection caused by inhaling the spores of the fungus Blastomyces dermatitidis. Skin features begin as papules, pustules, or subcutaneous nodules. Over a period of months, lesions heal to form raised wart-like scars, which are often irreversible.
Keratoacanthoma is a benign nonmelanomatous cancer that grows rapidly and remits spontaneously. This diagnosis is unlikely in our patient, whose lesion evolved over 2 years.
Basal cell carcinoma has a waxy, translucent, or pearly appearance, without a keratotic component. Telangiectasias are common. Unlike squamous cell carcinomas, the edges of basal cell lesions are clear rather than fleshy.
Extramammary Paget disease usually manifests as intense pruritus in affected areas, which appear as chronic intertrigo or nonresolving eczema. The dorsum of the hand is a very rare location for this disease. Given our patient’s presentation, extrammary Paget disease is an unlikely diagnosis.
CHRONIC SUN EXPOSURE IS A KEY RISK FACTOR
Our patient has worked outdoors for many years, and chronic sun exposure is a key risk factor for squamous cell carcinoma.1
Squamous cell carcinoma is a nonmelanomatous skin cancer arising from the more superficial layers of keratinocytes. Unlike basal cell carcinoma, it can metastasize,2 so the diagnosis must be made as soon as possible.
The key to diagnosis is a high degree of suspicion. Patients usually present with chronic sun-damaged skin with lesions as actinic keratoses or keratin horns. Squamous cell carcinoma appears as a thick, adherent scale that does not heal and may intermittently bleed. Because it spreads into the dermis, it can appear like an ulcer, with hard, raised edges, as in our patient.
TREATMENT AND PREVENTION
The standard effective treatment is complete surgical excision.1 Nonsurgical treatments include curettage plus cautery, cryosurgery, radiotherapy, photodynamic therapy, and imiquimod (Aldara) 5% cream. Minimizing sun exposure and regular checkups are important preventive measures.
Our patient underwent total surgical excision of the lesion. Due to the size of the lesion, a skin graft was required and was obtained from the abdomen. No relapse was observed after 1 year of follow-up.
Q: Which is the most likely diagnosis?
- Blastomycosis
- Keratoacanthoma
- Basal cell carcinoma
- Squamous cell carcinoma
- Extramammary Paget disease
A: Squamous cell carcinoma is the most likely diagnosis. The lesions may take the form of a patch, plaque, or nodule, sometimes with scaling or an ulcerated center. The borders often are irregular, fleshy, and bleed easily.
Blastomycosis is a rare fungal infection caused by inhaling the spores of the fungus Blastomyces dermatitidis. Skin features begin as papules, pustules, or subcutaneous nodules. Over a period of months, lesions heal to form raised wart-like scars, which are often irreversible.
Keratoacanthoma is a benign nonmelanomatous cancer that grows rapidly and remits spontaneously. This diagnosis is unlikely in our patient, whose lesion evolved over 2 years.
Basal cell carcinoma has a waxy, translucent, or pearly appearance, without a keratotic component. Telangiectasias are common. Unlike squamous cell carcinomas, the edges of basal cell lesions are clear rather than fleshy.
Extramammary Paget disease usually manifests as intense pruritus in affected areas, which appear as chronic intertrigo or nonresolving eczema. The dorsum of the hand is a very rare location for this disease. Given our patient’s presentation, extrammary Paget disease is an unlikely diagnosis.
CHRONIC SUN EXPOSURE IS A KEY RISK FACTOR
Our patient has worked outdoors for many years, and chronic sun exposure is a key risk factor for squamous cell carcinoma.1
Squamous cell carcinoma is a nonmelanomatous skin cancer arising from the more superficial layers of keratinocytes. Unlike basal cell carcinoma, it can metastasize,2 so the diagnosis must be made as soon as possible.
The key to diagnosis is a high degree of suspicion. Patients usually present with chronic sun-damaged skin with lesions as actinic keratoses or keratin horns. Squamous cell carcinoma appears as a thick, adherent scale that does not heal and may intermittently bleed. Because it spreads into the dermis, it can appear like an ulcer, with hard, raised edges, as in our patient.
TREATMENT AND PREVENTION
The standard effective treatment is complete surgical excision.1 Nonsurgical treatments include curettage plus cautery, cryosurgery, radiotherapy, photodynamic therapy, and imiquimod (Aldara) 5% cream. Minimizing sun exposure and regular checkups are important preventive measures.
Our patient underwent total surgical excision of the lesion. Due to the size of the lesion, a skin graft was required and was obtained from the abdomen. No relapse was observed after 1 year of follow-up.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Rowe DE, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis and survival rates in squamous cell carcinoma of the skin, ear, and lip: implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Rowe DE, Carroll RJ, Day CL. Prognostic factors for local recurrence, metastasis and survival rates in squamous cell carcinoma of the skin, ear, and lip: implications for treatment modality selection. J Am Acad Dermatol 1992; 26:976–990.
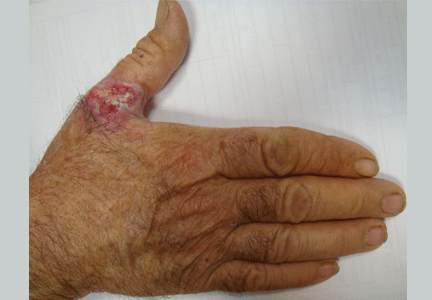
The shrinking woman
A 45-year-old woman who has been undergoing hemodialysis for 20 years presents with diffuse bone pain, pruritic skin, muscle weakness, and disability. About 8 years ago, she was diagnosed with uremic hyperparathyroidism and underwent two parathyroidectomy procedures and eight sessions of percutaneous alcohol ablation of the parathyroid gland.
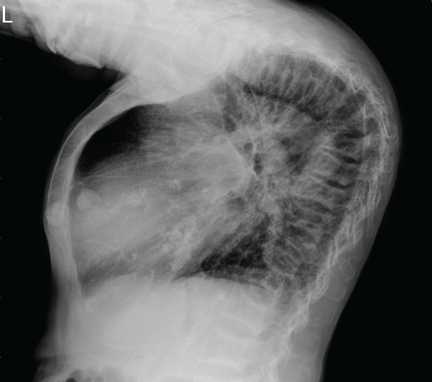
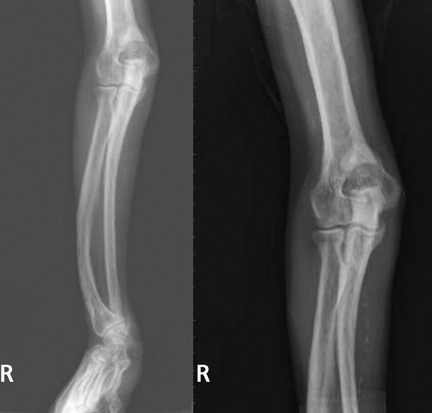
A technetium-99m sestamibi radionuclide scan shows bilateral parathyroid hyperplasia with no ectopic parathyroid adenomas. Although a surgeon she consulted 1 month ago declined to perform another parathyroidectomy for technical reasons, another surgeon agreed to do it at this time. Parathyroidectomy was successfully performed, after which the parathyroid hormone level decreased drastically, to 85 pg/mL.
To our surprise, her total body length increased by 6 to 7 cm after surgery, with partial straightening of the back and legs noted about 4 to 5 weeks after surgery. Furthermore, she was able to walk a short distance just a few weeks after surgery. Unfortunately, she died from sepsis the next year.
SEVERE UREMIC HYPERPARATHYROIDISM: A CLINICAL DILEMMA
The clinical appearance of reduced body length and diffuse bony deformity leading to “shrinking” as a consequence of prolonged severe uremic hyperparathyroidism has only rarely been reported.1 However, it is not uncommon for surgeons to decide against parathyroidectomy because of concerns of extensive subcutaneous fibrosis and recurrent laryngeal nerve damage associated with previous operations. The result is that the patient’s uremic hyperparathyroidism goes untreated, increasing the risk of long-term complications, as in this patient.
TYPICAL RADIOGRAPHIC FEATURES
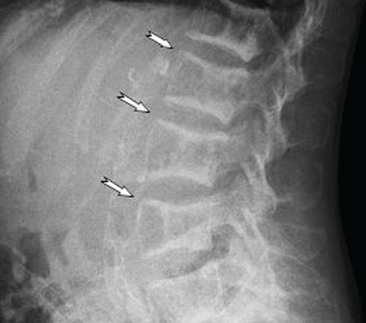
Why the body length increases after parathyroidectomy is not yet known, but plausible mechanisms are the effects of a recovery of muscle strength and the vigorous bony remineralization that strengthens weight-bearing bones after resolution of uremic hyperparathyroidism.
THE DANGERS OF DELAYED TREATMENT
Delaying parathyroidectomy may induce prolonged and severe uremic hyperparathyroidism, as in this patient. Nevertheless, despite the delay, surgery was able to partially ameliorate the symptoms of hyperparathyroidism and improve the extreme bone deformity. However, the patient’s informed consent, a detailed preoperative evaluation, and exclusion of ectopic parathyroid adenomas are imperative before surgical treatment.
- Horensten ML, Boner G, Rosenfeld JB. The shrinking man. A manifestation of severe renal osteodystrophy. JAMA 1980; 244:267–268.
- Jevtic V. Imaging of renal osteodystrophy. Eur J Radiol 2003; 46:85–95.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
A 45-year-old woman who has been undergoing hemodialysis for 20 years presents with diffuse bone pain, pruritic skin, muscle weakness, and disability. About 8 years ago, she was diagnosed with uremic hyperparathyroidism and underwent two parathyroidectomy procedures and eight sessions of percutaneous alcohol ablation of the parathyroid gland.
A technetium-99m sestamibi radionuclide scan shows bilateral parathyroid hyperplasia with no ectopic parathyroid adenomas. Although a surgeon she consulted 1 month ago declined to perform another parathyroidectomy for technical reasons, another surgeon agreed to do it at this time. Parathyroidectomy was successfully performed, after which the parathyroid hormone level decreased drastically, to 85 pg/mL.
To our surprise, her total body length increased by 6 to 7 cm after surgery, with partial straightening of the back and legs noted about 4 to 5 weeks after surgery. Furthermore, she was able to walk a short distance just a few weeks after surgery. Unfortunately, she died from sepsis the next year.
SEVERE UREMIC HYPERPARATHYROIDISM: A CLINICAL DILEMMA
The clinical appearance of reduced body length and diffuse bony deformity leading to “shrinking” as a consequence of prolonged severe uremic hyperparathyroidism has only rarely been reported.1 However, it is not uncommon for surgeons to decide against parathyroidectomy because of concerns of extensive subcutaneous fibrosis and recurrent laryngeal nerve damage associated with previous operations. The result is that the patient’s uremic hyperparathyroidism goes untreated, increasing the risk of long-term complications, as in this patient.
TYPICAL RADIOGRAPHIC FEATURES
Why the body length increases after parathyroidectomy is not yet known, but plausible mechanisms are the effects of a recovery of muscle strength and the vigorous bony remineralization that strengthens weight-bearing bones after resolution of uremic hyperparathyroidism.
THE DANGERS OF DELAYED TREATMENT
Delaying parathyroidectomy may induce prolonged and severe uremic hyperparathyroidism, as in this patient. Nevertheless, despite the delay, surgery was able to partially ameliorate the symptoms of hyperparathyroidism and improve the extreme bone deformity. However, the patient’s informed consent, a detailed preoperative evaluation, and exclusion of ectopic parathyroid adenomas are imperative before surgical treatment.
A 45-year-old woman who has been undergoing hemodialysis for 20 years presents with diffuse bone pain, pruritic skin, muscle weakness, and disability. About 8 years ago, she was diagnosed with uremic hyperparathyroidism and underwent two parathyroidectomy procedures and eight sessions of percutaneous alcohol ablation of the parathyroid gland.
A technetium-99m sestamibi radionuclide scan shows bilateral parathyroid hyperplasia with no ectopic parathyroid adenomas. Although a surgeon she consulted 1 month ago declined to perform another parathyroidectomy for technical reasons, another surgeon agreed to do it at this time. Parathyroidectomy was successfully performed, after which the parathyroid hormone level decreased drastically, to 85 pg/mL.
To our surprise, her total body length increased by 6 to 7 cm after surgery, with partial straightening of the back and legs noted about 4 to 5 weeks after surgery. Furthermore, she was able to walk a short distance just a few weeks after surgery. Unfortunately, she died from sepsis the next year.
SEVERE UREMIC HYPERPARATHYROIDISM: A CLINICAL DILEMMA
The clinical appearance of reduced body length and diffuse bony deformity leading to “shrinking” as a consequence of prolonged severe uremic hyperparathyroidism has only rarely been reported.1 However, it is not uncommon for surgeons to decide against parathyroidectomy because of concerns of extensive subcutaneous fibrosis and recurrent laryngeal nerve damage associated with previous operations. The result is that the patient’s uremic hyperparathyroidism goes untreated, increasing the risk of long-term complications, as in this patient.
TYPICAL RADIOGRAPHIC FEATURES
Why the body length increases after parathyroidectomy is not yet known, but plausible mechanisms are the effects of a recovery of muscle strength and the vigorous bony remineralization that strengthens weight-bearing bones after resolution of uremic hyperparathyroidism.
THE DANGERS OF DELAYED TREATMENT
Delaying parathyroidectomy may induce prolonged and severe uremic hyperparathyroidism, as in this patient. Nevertheless, despite the delay, surgery was able to partially ameliorate the symptoms of hyperparathyroidism and improve the extreme bone deformity. However, the patient’s informed consent, a detailed preoperative evaluation, and exclusion of ectopic parathyroid adenomas are imperative before surgical treatment.
- Horensten ML, Boner G, Rosenfeld JB. The shrinking man. A manifestation of severe renal osteodystrophy. JAMA 1980; 244:267–268.
- Jevtic V. Imaging of renal osteodystrophy. Eur J Radiol 2003; 46:85–95.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Horensten ML, Boner G, Rosenfeld JB. The shrinking man. A manifestation of severe renal osteodystrophy. JAMA 1980; 244:267–268.
- Jevtic V. Imaging of renal osteodystrophy. Eur J Radiol 2003; 46:85–95.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
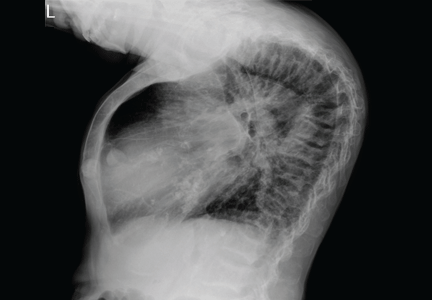
Painful red nodule on the right hand
A 46-year-old healthy man presents with a 15-day history of a tender subcutaneous nodule on the dorsum of the right hand that appeared after cleaning his aquarium. He has no fever or systemic symptoms. For 2 weeks he has been taking amoxicillin-clavulanate (Augmentin) and metronidazole (Flagyl), but without an adequate response.
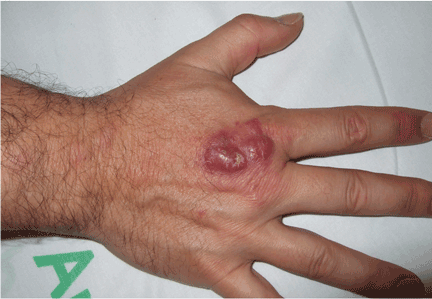
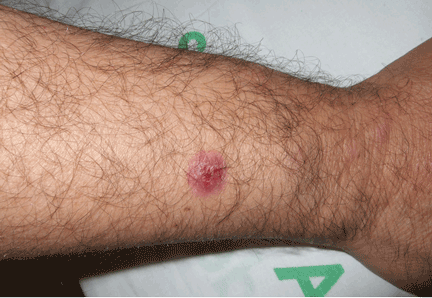
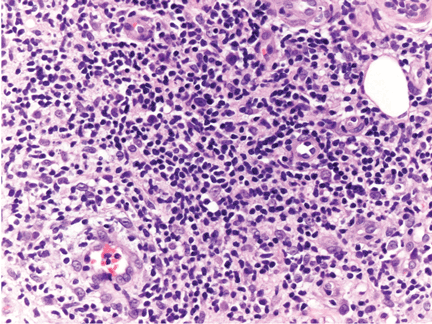
Q: Which is the most likely diagnosis?
- Tuberculosis verrucosa cutis
- Sporotrichosis
- Nontuberculous mycobacterial infection
- Leishmaniasis
- Nocardiosis
A: The correct answer is nontuberculous mycobacterial infection. Mycobacterium marinum was subsequently isolated from culture (Löwenstein-Jensen medium) of wound drainage.
Tuberculosis verrucosa cutis is an indolent, warty plaque that occurs after direct inoculation of M tuberculosis into the skin of someone previously infected with M tuberculosis. It can result from accidental exposure to tuberculous tissue in high-risk groups such as laboratory workers, physicians, and pathologists.
Sporothrix schenckii is the dimorphic fungus that causes sporotrichosis. Activities associated with acquisition of sporotrichosis include gardening (rose-gardener's disease), landscaping, farming, berry-picking, horticulture, and carpentry. The characteristic infection involves suppurating subcutaneous nodules that progress proximally along lymphatic channels (lymphocutaneous sporotrichosis).
Leishmaniasis is a disease caused by the protozoa of the Leishmania species, which is transmitted by the bite of a female sandfly. Initially, the lesion is a small, red papule up to 2 cm in diameter. Over several weeks, the papule becomes darker, develops a crust in the center, and eventually ulcerates to present a typical appearance of an ulcer with raised edges and surrounding dusky red skin.
Nocardia is a genus of filamentous grampositive bacteria that stains acid-fast, just as M marinum does. Nocardia asteroides primarily affects the lungs and can disseminate systemically. However, Nocardia brasiliensis is often associated with sporotrichoid-spreading subcutaneous nodules.
CLINICAL PRESENTATION AND DIAGNOSIS
M marinum is a genus of nontuberculous mycobacteria that usually causes disease in fish but can cause human skin infection by penetrating through a break in the skin. It can spread to deeper structures, resulting in tenosynovitis, arthritis, or osteomyelitis.1 The disease caused by M marinum is sometimes called “swimming pool granuloma” or “fish-tank granuloma.”2
In this case, the patient probably acquired the infection while cleaning his home aquarium,3 so asking about contact with pet fish is very important for the clinical diagnosis.
Lesions are usually localized to the site of the inoculation, with a predilection for areas predisposed to trauma, such as the hands and the fingers, after an incubation period of 2 to 8 weeks. Lesions are self-limited and usually appear as pruriginous bluish-red papules or pustules that may increase in size to form a verrucous or violaceous plaque or nodule. Superficial lesions often undergo central ulceration. Disseminated infection is rare and occurs mainly in patients who are immunocompromised because of renal transplant, systemic lupus erythematosus, chronic steroid therapy, or anti-tumor necrosis factor alpha therapy.4 In such cases, infection can be life-threatening.
On histopathologic study, a nonspecific inflammation consisting of a mixed infiltrate with lymphocytes, neutrophils, and histiocytes is usually observed. Sometimes a granulomatous inflammatory infiltrate mimicking tuberculoid granuloma may be noted. M marinum is an aerobic, nonmotile acid-fast bacillus. It grows at 30°C to 32°C on Löwenstein-Jensen medium in 2 to 5 weeks, but cultures are rarely positive. Polymerase chain reaction and enzyme-linked immunosorbent assays can help identify the organism. Other diagnoses to be considered are leishmaniasis, tuberculosis verrucosa cutis, blastomycosis, histoplasmosis, and sporotrichosis.
TREATMENT
Treatment of M marinum infection is not based on any specific criteria. Spontaneous resolution in 24 to 36 months has been described. 5 Antibiotics are the first-line therapy, and sometimes surgical treatment may be necessary for deeper infection with necrotic tissue. Cryotherapy, electrode therapy, and irradiation have been reported to be effective.
Few studies have been conducted to determine the first-line antibiotic treatment for M marinum infection. In this case, treatment with sulfamethoxazole-trimethoprim6 (Bactrim) resulted in resolution of the lesions in 6 months. Other antibiotics often used as monotherapy are tetracycline, minocycline (Minocin), and doxycycline (Vibramycin) 100 mg twice daily for 3 months.7 However, treatment failure has been described with many antibiotics.
Multidrug therapy is usually prescribed to minimize the risk of resistance. A combination of clarithromycin (Biaxin), streptomycin, and ethionamide (Trecator) has been prescribed successfully.3 Another combined regimen—clarithromycin 500 mg twice a day, rifampin (Rifadin) 600 mg daily, and ethambutol (Myambutol) 25 mg/kg daily—has been shown to be effective.5 Also, rifampin 600 mg daily and ethambutol 15 to 25 mg/kg/day were effective in a patient with a sporotrichoid M marinum infection associated with infliximab (Remicade). 8 The duration of treatment ranged between 2 and 6 months, or up to 2 months after the disappearance of the cutaneous lesions.
Clinical awareness of M marinum infection is important so that the diagnosis can be made and appropriate therapy can be initiated promptly, because some cases with disseminated disease and serious complications have been described.9
- Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol 2006; 33:817–819.
- Lewis FM, Marsh BJ, von Reyn CF. Fish tank exposure and cutaneous infections due to Mycobacterium marinum: tuberculin skin testing, treatment, and prevention. Clin Infect Dis 2003; 37:390–397.
- Dodiuk-Gad R, Dyachenko P, Ziv M, et al. Nontuberculous mycobacterial infections of the skin: a retrospective study of 25 cases. J Am Acad Dermatol 2007; 57:413–420.
- Streit M, Böhlen LM, Hunziker T, et al. Disseminated Mycobacterium marinum infection with extensive cutaneous eruption and bacteremia in an immunocompromised patient. Eur J Dermatol 2006; 16:79–83.
- Jogi R, Tyring SK. Therapy of nontuberculous mycobacterial infections. Dermatol Ther 2004; 17:491–498.
- Alinovi A, Vecchini F, Bassissi P. Sporothricoid mycobacterial infection. A case report. Acta Derm Venereol 1993; 73:146–147.
- Mahaisavariya P, Chaiprasert A, Khemngern S, et al. Nontuberculous mycobacterial skin infections: clinical and bacteriological studies. J Med Assoc Thai 2003; 86:52–60.
- Rallis E, Koumantaki-Mathioudaki E, Frangoulis E, Chatziolou E, Katsambas A. Severe sporotrichoid fish tank granuloma following infliximab therapy. Am J Clin Dermatol 2007; 8:385–388.
- Imakado S, Kojima Y, Hiyoshi T, Morimoto S. Disseminated Mycobacterium marinum infection in a patient with diabetic nephropathy. Diabetes Res Clin Pract 2009; 83:e35–e36.
A 46-year-old healthy man presents with a 15-day history of a tender subcutaneous nodule on the dorsum of the right hand that appeared after cleaning his aquarium. He has no fever or systemic symptoms. For 2 weeks he has been taking amoxicillin-clavulanate (Augmentin) and metronidazole (Flagyl), but without an adequate response.
Q: Which is the most likely diagnosis?
- Tuberculosis verrucosa cutis
- Sporotrichosis
- Nontuberculous mycobacterial infection
- Leishmaniasis
- Nocardiosis
A: The correct answer is nontuberculous mycobacterial infection. Mycobacterium marinum was subsequently isolated from culture (Löwenstein-Jensen medium) of wound drainage.
Tuberculosis verrucosa cutis is an indolent, warty plaque that occurs after direct inoculation of M tuberculosis into the skin of someone previously infected with M tuberculosis. It can result from accidental exposure to tuberculous tissue in high-risk groups such as laboratory workers, physicians, and pathologists.
Sporothrix schenckii is the dimorphic fungus that causes sporotrichosis. Activities associated with acquisition of sporotrichosis include gardening (rose-gardener's disease), landscaping, farming, berry-picking, horticulture, and carpentry. The characteristic infection involves suppurating subcutaneous nodules that progress proximally along lymphatic channels (lymphocutaneous sporotrichosis).
Leishmaniasis is a disease caused by the protozoa of the Leishmania species, which is transmitted by the bite of a female sandfly. Initially, the lesion is a small, red papule up to 2 cm in diameter. Over several weeks, the papule becomes darker, develops a crust in the center, and eventually ulcerates to present a typical appearance of an ulcer with raised edges and surrounding dusky red skin.
Nocardia is a genus of filamentous grampositive bacteria that stains acid-fast, just as M marinum does. Nocardia asteroides primarily affects the lungs and can disseminate systemically. However, Nocardia brasiliensis is often associated with sporotrichoid-spreading subcutaneous nodules.
CLINICAL PRESENTATION AND DIAGNOSIS
M marinum is a genus of nontuberculous mycobacteria that usually causes disease in fish but can cause human skin infection by penetrating through a break in the skin. It can spread to deeper structures, resulting in tenosynovitis, arthritis, or osteomyelitis.1 The disease caused by M marinum is sometimes called “swimming pool granuloma” or “fish-tank granuloma.”2
In this case, the patient probably acquired the infection while cleaning his home aquarium,3 so asking about contact with pet fish is very important for the clinical diagnosis.
Lesions are usually localized to the site of the inoculation, with a predilection for areas predisposed to trauma, such as the hands and the fingers, after an incubation period of 2 to 8 weeks. Lesions are self-limited and usually appear as pruriginous bluish-red papules or pustules that may increase in size to form a verrucous or violaceous plaque or nodule. Superficial lesions often undergo central ulceration. Disseminated infection is rare and occurs mainly in patients who are immunocompromised because of renal transplant, systemic lupus erythematosus, chronic steroid therapy, or anti-tumor necrosis factor alpha therapy.4 In such cases, infection can be life-threatening.
On histopathologic study, a nonspecific inflammation consisting of a mixed infiltrate with lymphocytes, neutrophils, and histiocytes is usually observed. Sometimes a granulomatous inflammatory infiltrate mimicking tuberculoid granuloma may be noted. M marinum is an aerobic, nonmotile acid-fast bacillus. It grows at 30°C to 32°C on Löwenstein-Jensen medium in 2 to 5 weeks, but cultures are rarely positive. Polymerase chain reaction and enzyme-linked immunosorbent assays can help identify the organism. Other diagnoses to be considered are leishmaniasis, tuberculosis verrucosa cutis, blastomycosis, histoplasmosis, and sporotrichosis.
TREATMENT
Treatment of M marinum infection is not based on any specific criteria. Spontaneous resolution in 24 to 36 months has been described. 5 Antibiotics are the first-line therapy, and sometimes surgical treatment may be necessary for deeper infection with necrotic tissue. Cryotherapy, electrode therapy, and irradiation have been reported to be effective.
Few studies have been conducted to determine the first-line antibiotic treatment for M marinum infection. In this case, treatment with sulfamethoxazole-trimethoprim6 (Bactrim) resulted in resolution of the lesions in 6 months. Other antibiotics often used as monotherapy are tetracycline, minocycline (Minocin), and doxycycline (Vibramycin) 100 mg twice daily for 3 months.7 However, treatment failure has been described with many antibiotics.
Multidrug therapy is usually prescribed to minimize the risk of resistance. A combination of clarithromycin (Biaxin), streptomycin, and ethionamide (Trecator) has been prescribed successfully.3 Another combined regimen—clarithromycin 500 mg twice a day, rifampin (Rifadin) 600 mg daily, and ethambutol (Myambutol) 25 mg/kg daily—has been shown to be effective.5 Also, rifampin 600 mg daily and ethambutol 15 to 25 mg/kg/day were effective in a patient with a sporotrichoid M marinum infection associated with infliximab (Remicade). 8 The duration of treatment ranged between 2 and 6 months, or up to 2 months after the disappearance of the cutaneous lesions.
Clinical awareness of M marinum infection is important so that the diagnosis can be made and appropriate therapy can be initiated promptly, because some cases with disseminated disease and serious complications have been described.9
A 46-year-old healthy man presents with a 15-day history of a tender subcutaneous nodule on the dorsum of the right hand that appeared after cleaning his aquarium. He has no fever or systemic symptoms. For 2 weeks he has been taking amoxicillin-clavulanate (Augmentin) and metronidazole (Flagyl), but without an adequate response.
Q: Which is the most likely diagnosis?
- Tuberculosis verrucosa cutis
- Sporotrichosis
- Nontuberculous mycobacterial infection
- Leishmaniasis
- Nocardiosis
A: The correct answer is nontuberculous mycobacterial infection. Mycobacterium marinum was subsequently isolated from culture (Löwenstein-Jensen medium) of wound drainage.
Tuberculosis verrucosa cutis is an indolent, warty plaque that occurs after direct inoculation of M tuberculosis into the skin of someone previously infected with M tuberculosis. It can result from accidental exposure to tuberculous tissue in high-risk groups such as laboratory workers, physicians, and pathologists.
Sporothrix schenckii is the dimorphic fungus that causes sporotrichosis. Activities associated with acquisition of sporotrichosis include gardening (rose-gardener's disease), landscaping, farming, berry-picking, horticulture, and carpentry. The characteristic infection involves suppurating subcutaneous nodules that progress proximally along lymphatic channels (lymphocutaneous sporotrichosis).
Leishmaniasis is a disease caused by the protozoa of the Leishmania species, which is transmitted by the bite of a female sandfly. Initially, the lesion is a small, red papule up to 2 cm in diameter. Over several weeks, the papule becomes darker, develops a crust in the center, and eventually ulcerates to present a typical appearance of an ulcer with raised edges and surrounding dusky red skin.
Nocardia is a genus of filamentous grampositive bacteria that stains acid-fast, just as M marinum does. Nocardia asteroides primarily affects the lungs and can disseminate systemically. However, Nocardia brasiliensis is often associated with sporotrichoid-spreading subcutaneous nodules.
CLINICAL PRESENTATION AND DIAGNOSIS
M marinum is a genus of nontuberculous mycobacteria that usually causes disease in fish but can cause human skin infection by penetrating through a break in the skin. It can spread to deeper structures, resulting in tenosynovitis, arthritis, or osteomyelitis.1 The disease caused by M marinum is sometimes called “swimming pool granuloma” or “fish-tank granuloma.”2
In this case, the patient probably acquired the infection while cleaning his home aquarium,3 so asking about contact with pet fish is very important for the clinical diagnosis.
Lesions are usually localized to the site of the inoculation, with a predilection for areas predisposed to trauma, such as the hands and the fingers, after an incubation period of 2 to 8 weeks. Lesions are self-limited and usually appear as pruriginous bluish-red papules or pustules that may increase in size to form a verrucous or violaceous plaque or nodule. Superficial lesions often undergo central ulceration. Disseminated infection is rare and occurs mainly in patients who are immunocompromised because of renal transplant, systemic lupus erythematosus, chronic steroid therapy, or anti-tumor necrosis factor alpha therapy.4 In such cases, infection can be life-threatening.
On histopathologic study, a nonspecific inflammation consisting of a mixed infiltrate with lymphocytes, neutrophils, and histiocytes is usually observed. Sometimes a granulomatous inflammatory infiltrate mimicking tuberculoid granuloma may be noted. M marinum is an aerobic, nonmotile acid-fast bacillus. It grows at 30°C to 32°C on Löwenstein-Jensen medium in 2 to 5 weeks, but cultures are rarely positive. Polymerase chain reaction and enzyme-linked immunosorbent assays can help identify the organism. Other diagnoses to be considered are leishmaniasis, tuberculosis verrucosa cutis, blastomycosis, histoplasmosis, and sporotrichosis.
TREATMENT
Treatment of M marinum infection is not based on any specific criteria. Spontaneous resolution in 24 to 36 months has been described. 5 Antibiotics are the first-line therapy, and sometimes surgical treatment may be necessary for deeper infection with necrotic tissue. Cryotherapy, electrode therapy, and irradiation have been reported to be effective.
Few studies have been conducted to determine the first-line antibiotic treatment for M marinum infection. In this case, treatment with sulfamethoxazole-trimethoprim6 (Bactrim) resulted in resolution of the lesions in 6 months. Other antibiotics often used as monotherapy are tetracycline, minocycline (Minocin), and doxycycline (Vibramycin) 100 mg twice daily for 3 months.7 However, treatment failure has been described with many antibiotics.
Multidrug therapy is usually prescribed to minimize the risk of resistance. A combination of clarithromycin (Biaxin), streptomycin, and ethionamide (Trecator) has been prescribed successfully.3 Another combined regimen—clarithromycin 500 mg twice a day, rifampin (Rifadin) 600 mg daily, and ethambutol (Myambutol) 25 mg/kg daily—has been shown to be effective.5 Also, rifampin 600 mg daily and ethambutol 15 to 25 mg/kg/day were effective in a patient with a sporotrichoid M marinum infection associated with infliximab (Remicade). 8 The duration of treatment ranged between 2 and 6 months, or up to 2 months after the disappearance of the cutaneous lesions.
Clinical awareness of M marinum infection is important so that the diagnosis can be made and appropriate therapy can be initiated promptly, because some cases with disseminated disease and serious complications have been described.9
- Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol 2006; 33:817–819.
- Lewis FM, Marsh BJ, von Reyn CF. Fish tank exposure and cutaneous infections due to Mycobacterium marinum: tuberculin skin testing, treatment, and prevention. Clin Infect Dis 2003; 37:390–397.
- Dodiuk-Gad R, Dyachenko P, Ziv M, et al. Nontuberculous mycobacterial infections of the skin: a retrospective study of 25 cases. J Am Acad Dermatol 2007; 57:413–420.
- Streit M, Böhlen LM, Hunziker T, et al. Disseminated Mycobacterium marinum infection with extensive cutaneous eruption and bacteremia in an immunocompromised patient. Eur J Dermatol 2006; 16:79–83.
- Jogi R, Tyring SK. Therapy of nontuberculous mycobacterial infections. Dermatol Ther 2004; 17:491–498.
- Alinovi A, Vecchini F, Bassissi P. Sporothricoid mycobacterial infection. A case report. Acta Derm Venereol 1993; 73:146–147.
- Mahaisavariya P, Chaiprasert A, Khemngern S, et al. Nontuberculous mycobacterial skin infections: clinical and bacteriological studies. J Med Assoc Thai 2003; 86:52–60.
- Rallis E, Koumantaki-Mathioudaki E, Frangoulis E, Chatziolou E, Katsambas A. Severe sporotrichoid fish tank granuloma following infliximab therapy. Am J Clin Dermatol 2007; 8:385–388.
- Imakado S, Kojima Y, Hiyoshi T, Morimoto S. Disseminated Mycobacterium marinum infection in a patient with diabetic nephropathy. Diabetes Res Clin Pract 2009; 83:e35–e36.
- Lam A, Toma W, Schlesinger N. Mycobacterium marinum arthritis mimicking rheumatoid arthritis. J Rheumatol 2006; 33:817–819.
- Lewis FM, Marsh BJ, von Reyn CF. Fish tank exposure and cutaneous infections due to Mycobacterium marinum: tuberculin skin testing, treatment, and prevention. Clin Infect Dis 2003; 37:390–397.
- Dodiuk-Gad R, Dyachenko P, Ziv M, et al. Nontuberculous mycobacterial infections of the skin: a retrospective study of 25 cases. J Am Acad Dermatol 2007; 57:413–420.
- Streit M, Böhlen LM, Hunziker T, et al. Disseminated Mycobacterium marinum infection with extensive cutaneous eruption and bacteremia in an immunocompromised patient. Eur J Dermatol 2006; 16:79–83.
- Jogi R, Tyring SK. Therapy of nontuberculous mycobacterial infections. Dermatol Ther 2004; 17:491–498.
- Alinovi A, Vecchini F, Bassissi P. Sporothricoid mycobacterial infection. A case report. Acta Derm Venereol 1993; 73:146–147.
- Mahaisavariya P, Chaiprasert A, Khemngern S, et al. Nontuberculous mycobacterial skin infections: clinical and bacteriological studies. J Med Assoc Thai 2003; 86:52–60.
- Rallis E, Koumantaki-Mathioudaki E, Frangoulis E, Chatziolou E, Katsambas A. Severe sporotrichoid fish tank granuloma following infliximab therapy. Am J Clin Dermatol 2007; 8:385–388.
- Imakado S, Kojima Y, Hiyoshi T, Morimoto S. Disseminated Mycobacterium marinum infection in a patient with diabetic nephropathy. Diabetes Res Clin Pract 2009; 83:e35–e36.
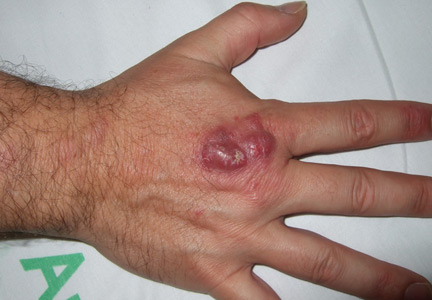
Pyogenic liver abscess
A 58-year-old woman presents with fever, chills, vomiting, and right-upperquadrant abdominal pain. She has had diarrhea for several days and has lost 15 lb over the last 6 weeks. Six months ago, she took a cruise through the Panama Canal to Nicaragua, Costa Rica, Mexico, and El Salvador.
The patient undergoes CT-guided liver biopsy with drain placement. Cultures grow Klebsiella pneumoniae, and she is started on intravenous piperacillin-tazobactam for 4 weeks, followed by 4 weeks of oral ciprofloxacin.
Follow-up evaluation at 4 weeks and at 8 weeks shows improvement in the patient’s condition, with CT of the abdomen and pelvis showing a gradual decrease in the size of the abscess.
INCREASING PREVALENCE
K pneumoniae has emerged as the most common organism seen in pyogenic liver abscess.1 Initially seen in Taiwan in the 1980s, K pneumoniae liver abscess is becoming more common in the United States.2 Diabetes and impaired fasting glucose have both been implicated as potential risk factors, but the condition is also seen in nondiabetic patients.3 Although pyogenic liver abscess is commonly a sequela of biliary disease, K pneumoniae liver abscess is more often cryptogenic. 1 Clinical manifestations usually include fever, abdominal pain in the right upper quadrant, nausea, and vomiting.
In this patient, no clear relationship was established between her travel and her illness.
GREATER RISK OF SPREAD
K pneumoniae liver abscess is more likely to spread than polymicrobial liver abscess.3 It is associated with endophthalmitis, meningitis, brain abscess, and septic pulmonary embolism.3 Diabetic patients are particularly susceptible to metastatic foci.3 The reason is not well understood, but poor glycemic control leading to impaired neutrophil phagocytosis is thought to play a role.4
DIAGNOSIS AND TREATMENT
Liver abscesses are associated with elevated alkaline phosphatase levels, hyperbilirubinemia, leukocytosis, hypoalbuminemia, and anemia. As bacteremia is often seen with K pneumoniae liver abscess, blood cultures should be obtained.1 Imaging studies should include right-upper-quadrant ultrasonography if suspicion is high for concomitant biliary disease, and also CT with intravenous contrast to better quantify the dimensions of the abscess.5
Treatment includes empiric parenteral antibiotics and percutaneous drainage. In addition, culture of purulent material for aerobic and anaerobic organisms helps guide antibiotic treatment.
The antibiotic regimen should consist of a first- or third-generation beta-lactamase inhibitor, with or without an aminoglycoside.3 Patients unable to tolerate beta-lactam antibiotics can be given a fluoroquinolone.
The results of cultures and determination of antibiotic sensitivities help to further modifiy antibiotic therapy. Antibiotic therapy may be needed for 4 to 6 weeks. Parenteral antibiotics are recommended initially, and if a patient responds to therapy, treatment can be switched to oral antibiotics to complete the course of treatment.
Although antibiotics with percutaneous drainage are the recommended course of therapy, surgical drainage is sometimes necessary and is best done with the input of a hepatobiliary surgeon. Patients with abscesses larger than 5 cm who had surgical drainage had better clinical outcomes than those who had percutaneous drainage,6 but monitoring the response to antibiotics and the patient’s clinical course is very important when determining the need for emergency surgical intervention vs percutaneous drainage.6
Follow-up imaging is necessary to evaluate the response to therapy, to determine the continued need for antibiotics, and to assess for any further need for drainage.
- Pope JV, Teich DL, Clardy P, McGillicuddy DC. Klebsiella pneumoniae liver abscess: an emerging problem in North America. J Emerg Med 2008; Epub ahead of print.
- Frazee BW, Hansen S, Lambert L. Invasive infection with hypermucoviscous Klebsiella pneumoniae: multiple cases presenting to a single emergency department in the United States. Ann Emerg Med 2009; 53:639–642.
- Lee SS, Chen YS, Tsai HC, et al. Predictors of septic metastatic infection and mortality among patients with Klebsiella pneumoniae liver abscess. Clin Infect Dis 2008; 47:642–650.
- Lin JC, Siu LK, Fung CP, et al. Impaired phagocytosis of capsular serotypes K1 or K2 Klebsiella pneumoniae in type 2 diabetes mellitus patients with poor glycemic control. J Clin Endocrinol Metab 2006; 91:3084–3087.
- Golia P, Sadler M. Pyogenic liver abscess: Klebsiella as an emerging pathogen. Emerg Radiol 2006; 13:87–88.
- Tan YM, Chung AY, Chow PK, et al. An appraisal of surgical and percutaneous drainage for pyogenic liver abscesses larger than 5 cm. Ann Surg 2005; 241:485–490.
A 58-year-old woman presents with fever, chills, vomiting, and right-upperquadrant abdominal pain. She has had diarrhea for several days and has lost 15 lb over the last 6 weeks. Six months ago, she took a cruise through the Panama Canal to Nicaragua, Costa Rica, Mexico, and El Salvador.
The patient undergoes CT-guided liver biopsy with drain placement. Cultures grow Klebsiella pneumoniae, and she is started on intravenous piperacillin-tazobactam for 4 weeks, followed by 4 weeks of oral ciprofloxacin.
Follow-up evaluation at 4 weeks and at 8 weeks shows improvement in the patient’s condition, with CT of the abdomen and pelvis showing a gradual decrease in the size of the abscess.
INCREASING PREVALENCE
K pneumoniae has emerged as the most common organism seen in pyogenic liver abscess.1 Initially seen in Taiwan in the 1980s, K pneumoniae liver abscess is becoming more common in the United States.2 Diabetes and impaired fasting glucose have both been implicated as potential risk factors, but the condition is also seen in nondiabetic patients.3 Although pyogenic liver abscess is commonly a sequela of biliary disease, K pneumoniae liver abscess is more often cryptogenic. 1 Clinical manifestations usually include fever, abdominal pain in the right upper quadrant, nausea, and vomiting.
In this patient, no clear relationship was established between her travel and her illness.
GREATER RISK OF SPREAD
K pneumoniae liver abscess is more likely to spread than polymicrobial liver abscess.3 It is associated with endophthalmitis, meningitis, brain abscess, and septic pulmonary embolism.3 Diabetic patients are particularly susceptible to metastatic foci.3 The reason is not well understood, but poor glycemic control leading to impaired neutrophil phagocytosis is thought to play a role.4
DIAGNOSIS AND TREATMENT
Liver abscesses are associated with elevated alkaline phosphatase levels, hyperbilirubinemia, leukocytosis, hypoalbuminemia, and anemia. As bacteremia is often seen with K pneumoniae liver abscess, blood cultures should be obtained.1 Imaging studies should include right-upper-quadrant ultrasonography if suspicion is high for concomitant biliary disease, and also CT with intravenous contrast to better quantify the dimensions of the abscess.5
Treatment includes empiric parenteral antibiotics and percutaneous drainage. In addition, culture of purulent material for aerobic and anaerobic organisms helps guide antibiotic treatment.
The antibiotic regimen should consist of a first- or third-generation beta-lactamase inhibitor, with or without an aminoglycoside.3 Patients unable to tolerate beta-lactam antibiotics can be given a fluoroquinolone.
The results of cultures and determination of antibiotic sensitivities help to further modifiy antibiotic therapy. Antibiotic therapy may be needed for 4 to 6 weeks. Parenteral antibiotics are recommended initially, and if a patient responds to therapy, treatment can be switched to oral antibiotics to complete the course of treatment.
Although antibiotics with percutaneous drainage are the recommended course of therapy, surgical drainage is sometimes necessary and is best done with the input of a hepatobiliary surgeon. Patients with abscesses larger than 5 cm who had surgical drainage had better clinical outcomes than those who had percutaneous drainage,6 but monitoring the response to antibiotics and the patient’s clinical course is very important when determining the need for emergency surgical intervention vs percutaneous drainage.6
Follow-up imaging is necessary to evaluate the response to therapy, to determine the continued need for antibiotics, and to assess for any further need for drainage.
A 58-year-old woman presents with fever, chills, vomiting, and right-upperquadrant abdominal pain. She has had diarrhea for several days and has lost 15 lb over the last 6 weeks. Six months ago, she took a cruise through the Panama Canal to Nicaragua, Costa Rica, Mexico, and El Salvador.
The patient undergoes CT-guided liver biopsy with drain placement. Cultures grow Klebsiella pneumoniae, and she is started on intravenous piperacillin-tazobactam for 4 weeks, followed by 4 weeks of oral ciprofloxacin.
Follow-up evaluation at 4 weeks and at 8 weeks shows improvement in the patient’s condition, with CT of the abdomen and pelvis showing a gradual decrease in the size of the abscess.
INCREASING PREVALENCE
K pneumoniae has emerged as the most common organism seen in pyogenic liver abscess.1 Initially seen in Taiwan in the 1980s, K pneumoniae liver abscess is becoming more common in the United States.2 Diabetes and impaired fasting glucose have both been implicated as potential risk factors, but the condition is also seen in nondiabetic patients.3 Although pyogenic liver abscess is commonly a sequela of biliary disease, K pneumoniae liver abscess is more often cryptogenic. 1 Clinical manifestations usually include fever, abdominal pain in the right upper quadrant, nausea, and vomiting.
In this patient, no clear relationship was established between her travel and her illness.
GREATER RISK OF SPREAD
K pneumoniae liver abscess is more likely to spread than polymicrobial liver abscess.3 It is associated with endophthalmitis, meningitis, brain abscess, and septic pulmonary embolism.3 Diabetic patients are particularly susceptible to metastatic foci.3 The reason is not well understood, but poor glycemic control leading to impaired neutrophil phagocytosis is thought to play a role.4
DIAGNOSIS AND TREATMENT
Liver abscesses are associated with elevated alkaline phosphatase levels, hyperbilirubinemia, leukocytosis, hypoalbuminemia, and anemia. As bacteremia is often seen with K pneumoniae liver abscess, blood cultures should be obtained.1 Imaging studies should include right-upper-quadrant ultrasonography if suspicion is high for concomitant biliary disease, and also CT with intravenous contrast to better quantify the dimensions of the abscess.5
Treatment includes empiric parenteral antibiotics and percutaneous drainage. In addition, culture of purulent material for aerobic and anaerobic organisms helps guide antibiotic treatment.
The antibiotic regimen should consist of a first- or third-generation beta-lactamase inhibitor, with or without an aminoglycoside.3 Patients unable to tolerate beta-lactam antibiotics can be given a fluoroquinolone.
The results of cultures and determination of antibiotic sensitivities help to further modifiy antibiotic therapy. Antibiotic therapy may be needed for 4 to 6 weeks. Parenteral antibiotics are recommended initially, and if a patient responds to therapy, treatment can be switched to oral antibiotics to complete the course of treatment.
Although antibiotics with percutaneous drainage are the recommended course of therapy, surgical drainage is sometimes necessary and is best done with the input of a hepatobiliary surgeon. Patients with abscesses larger than 5 cm who had surgical drainage had better clinical outcomes than those who had percutaneous drainage,6 but monitoring the response to antibiotics and the patient’s clinical course is very important when determining the need for emergency surgical intervention vs percutaneous drainage.6
Follow-up imaging is necessary to evaluate the response to therapy, to determine the continued need for antibiotics, and to assess for any further need for drainage.
- Pope JV, Teich DL, Clardy P, McGillicuddy DC. Klebsiella pneumoniae liver abscess: an emerging problem in North America. J Emerg Med 2008; Epub ahead of print.
- Frazee BW, Hansen S, Lambert L. Invasive infection with hypermucoviscous Klebsiella pneumoniae: multiple cases presenting to a single emergency department in the United States. Ann Emerg Med 2009; 53:639–642.
- Lee SS, Chen YS, Tsai HC, et al. Predictors of septic metastatic infection and mortality among patients with Klebsiella pneumoniae liver abscess. Clin Infect Dis 2008; 47:642–650.
- Lin JC, Siu LK, Fung CP, et al. Impaired phagocytosis of capsular serotypes K1 or K2 Klebsiella pneumoniae in type 2 diabetes mellitus patients with poor glycemic control. J Clin Endocrinol Metab 2006; 91:3084–3087.
- Golia P, Sadler M. Pyogenic liver abscess: Klebsiella as an emerging pathogen. Emerg Radiol 2006; 13:87–88.
- Tan YM, Chung AY, Chow PK, et al. An appraisal of surgical and percutaneous drainage for pyogenic liver abscesses larger than 5 cm. Ann Surg 2005; 241:485–490.
- Pope JV, Teich DL, Clardy P, McGillicuddy DC. Klebsiella pneumoniae liver abscess: an emerging problem in North America. J Emerg Med 2008; Epub ahead of print.
- Frazee BW, Hansen S, Lambert L. Invasive infection with hypermucoviscous Klebsiella pneumoniae: multiple cases presenting to a single emergency department in the United States. Ann Emerg Med 2009; 53:639–642.
- Lee SS, Chen YS, Tsai HC, et al. Predictors of septic metastatic infection and mortality among patients with Klebsiella pneumoniae liver abscess. Clin Infect Dis 2008; 47:642–650.
- Lin JC, Siu LK, Fung CP, et al. Impaired phagocytosis of capsular serotypes K1 or K2 Klebsiella pneumoniae in type 2 diabetes mellitus patients with poor glycemic control. J Clin Endocrinol Metab 2006; 91:3084–3087.
- Golia P, Sadler M. Pyogenic liver abscess: Klebsiella as an emerging pathogen. Emerg Radiol 2006; 13:87–88.
- Tan YM, Chung AY, Chow PK, et al. An appraisal of surgical and percutaneous drainage for pyogenic liver abscesses larger than 5 cm. Ann Surg 2005; 241:485–490.