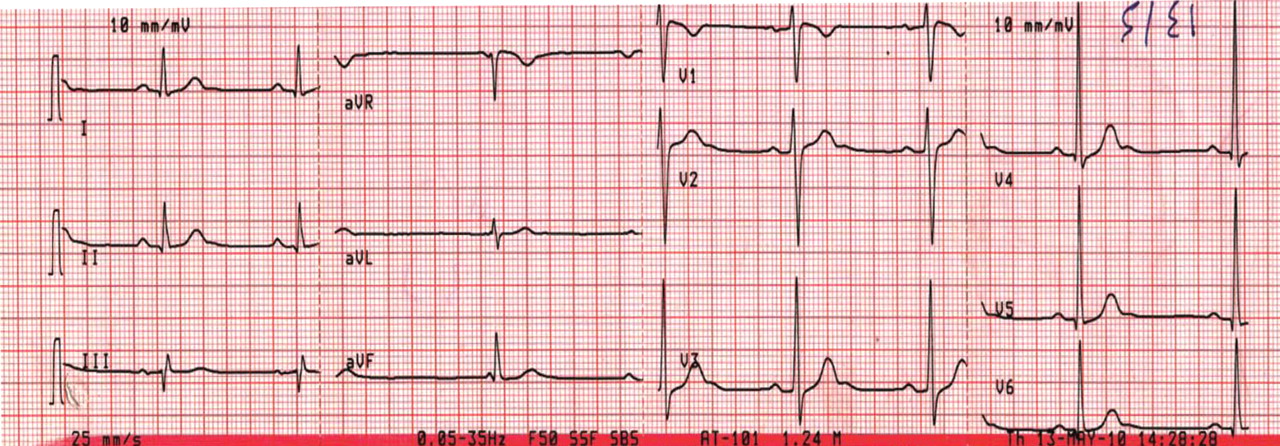
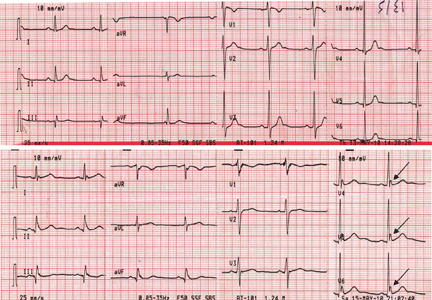
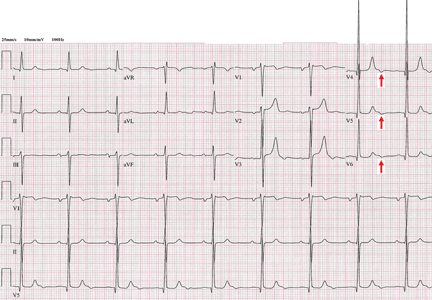
Figure 1. Sinus bradycardia, heart rate 55 beats per minute. The patient’s core body temperature was 36°C (96.8°F). There are no evident J waves.
A 22-year-old man was brought to the emergency room after a motor vehicle accident. He was in a deep coma, with a Glasgow coma score of 4 out of 15 (3 being the worst score) and a core body temperature of 36°C (96.8°F). The next day, clinical evidence of brain death was noted, and his core body temperature dropped as low as 29.6°C (85.3°F). At that time, his electrocardiogram revealed sinus bradycardia, with a rate of 48 beats per minute, PR interval 0.24 second, QRS interval 0.16 second, corrected QT duration 0.5 second, and classic high-amplitude Osborn waves (J waves) that were evident in all leads. Figures 1, 2, and 3 show the effect of various degrees of hypothermia on the electrocardiogram.
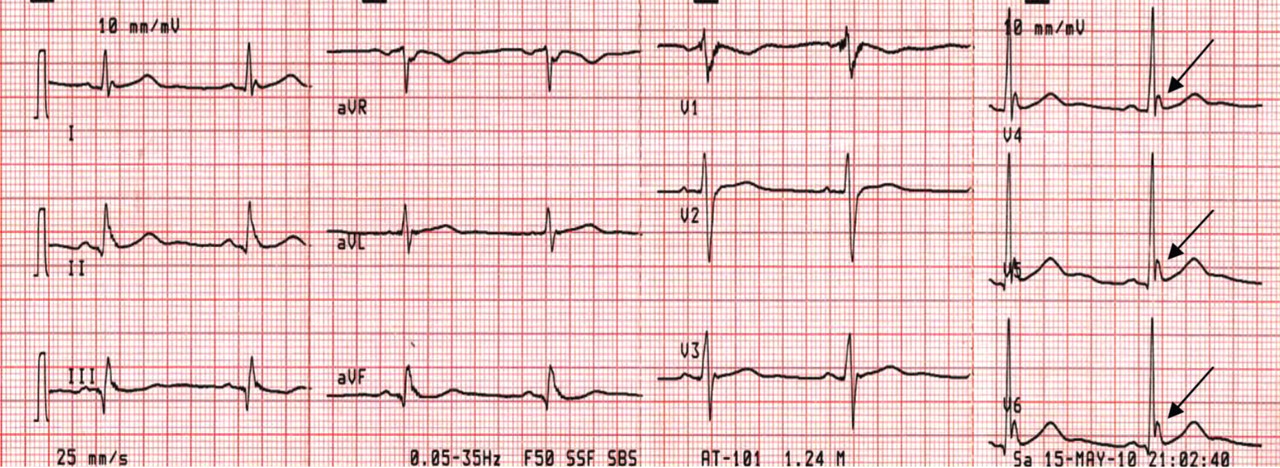
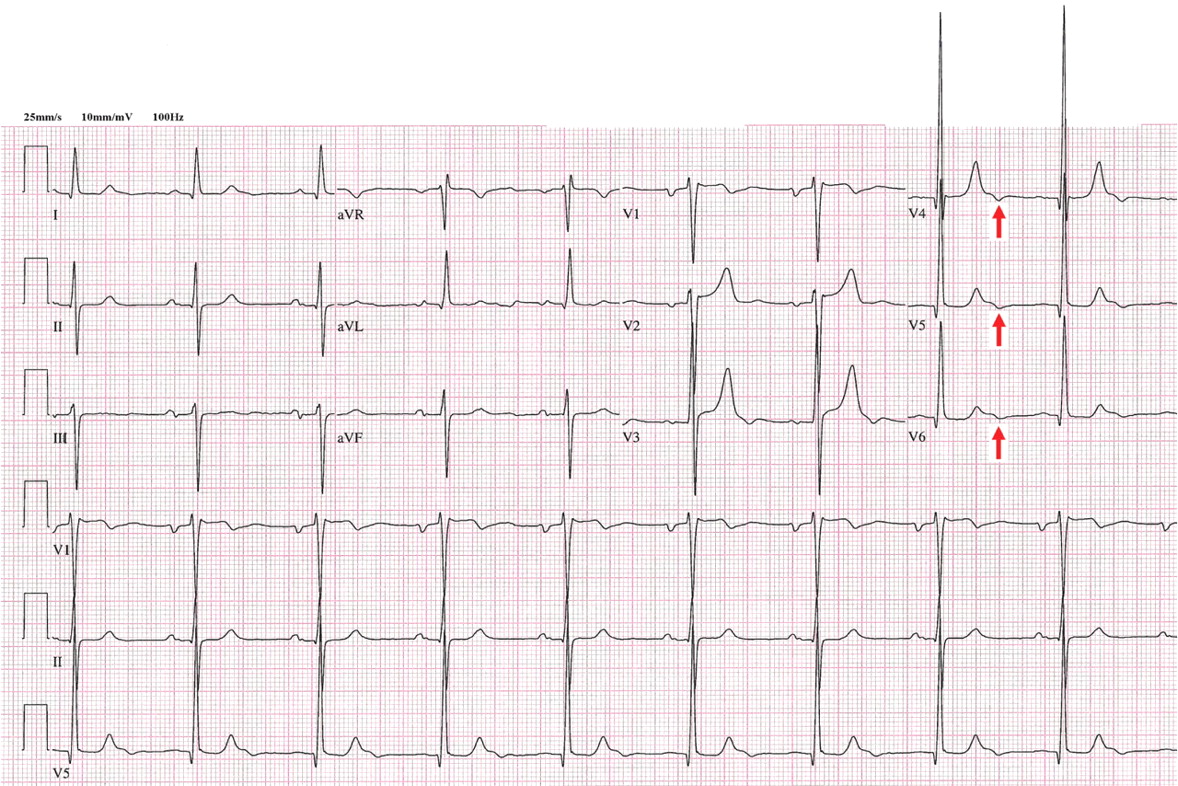
Figure 2. Sinus bradycardia, heart rate 50 beats per minute; low-amplitude J waves are visible in leads V4, V5, and V6 (arrows). The patient’s core body temperature was 31°C (87.8°F).
The Osborn wave1 (J wave) is the result of a transient, outward, potassium-mediated current in the ventricular epicardium but not the endocardium, corresponding to a notch in the action potential. This gives rise to a transmural voltage gradient during early repolarization, which appears as the J wave on electrocardiography. It is more pronounced in hypothermia, disappears after normalization of the body temperature, and is usually evident in the inferolateral leads.
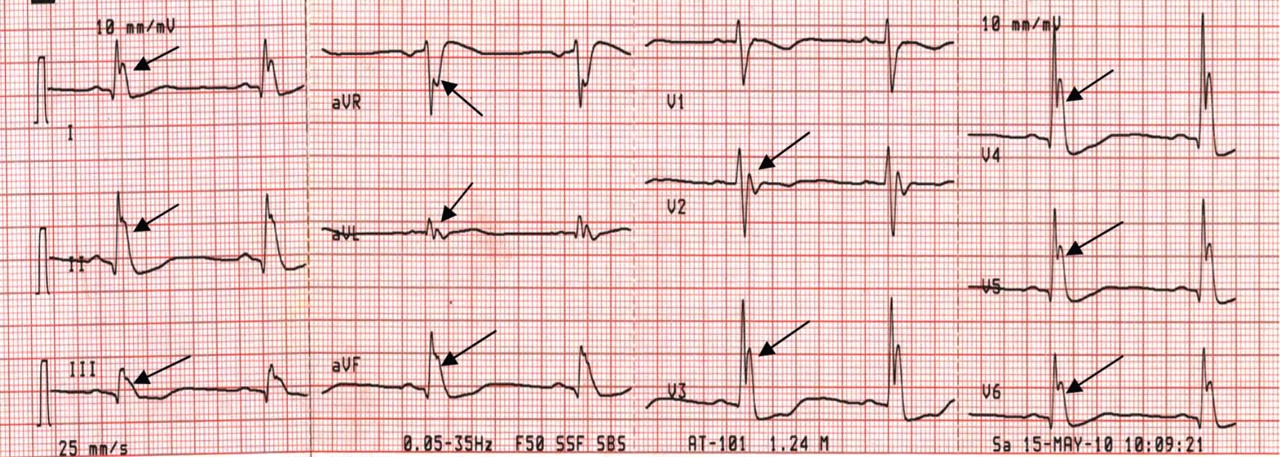
Figure 3. Sinus bradycardia, 48 beats per minute; the PR interval is prolonged at 0.24 second, the QRS interval is prolonged at 0.16 second, the corrected QT interval is 0.5 second, and classic high-amplitude J waves are visible in all leads (arrows). Core body temperature was 29.6°C (85.3°F).
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
The lower the core body temperature, the higher the amplitude of the J wave.
The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
References
Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol1953; 175:389–398.
Hesham R. Omar, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Hany D. Abdelmalak, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Address: Hesham R. Omar, MD, Internal Medicine Department, Mercy Hospital and Medical Center, 2525 South Michigan Avenue, Chicago, IL 60616; e-mail [email protected]
Hesham R. Omar, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Hany D. Abdelmalak, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Address: Hesham R. Omar, MD, Internal Medicine Department, Mercy Hospital and Medical Center, 2525 South Michigan Avenue, Chicago, IL 60616; e-mail [email protected]
Author and Disclosure Information
Hesham R. Omar, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Hany D. Abdelmalak, MD Internal Medicine Program, Department of Medicine, Mercy Hospital and Medical Center, Chicago, IL
Address: Hesham R. Omar, MD, Internal Medicine Department, Mercy Hospital and Medical Center, 2525 South Michigan Avenue, Chicago, IL 60616; e-mail [email protected]
Figure 1. Sinus bradycardia, heart rate 55 beats per minute. The patient’s core body temperature was 36°C (96.8°F). There are no evident J waves.
A 22-year-old man was brought to the emergency room after a motor vehicle accident. He was in a deep coma, with a Glasgow coma score of 4 out of 15 (3 being the worst score) and a core body temperature of 36°C (96.8°F). The next day, clinical evidence of brain death was noted, and his core body temperature dropped as low as 29.6°C (85.3°F). At that time, his electrocardiogram revealed sinus bradycardia, with a rate of 48 beats per minute, PR interval 0.24 second, QRS interval 0.16 second, corrected QT duration 0.5 second, and classic high-amplitude Osborn waves (J waves) that were evident in all leads. Figures 1, 2, and 3 show the effect of various degrees of hypothermia on the electrocardiogram.
Figure 2. Sinus bradycardia, heart rate 50 beats per minute; low-amplitude J waves are visible in leads V4, V5, and V6 (arrows). The patient’s core body temperature was 31°C (87.8°F).
The Osborn wave1 (J wave) is the result of a transient, outward, potassium-mediated current in the ventricular epicardium but not the endocardium, corresponding to a notch in the action potential. This gives rise to a transmural voltage gradient during early repolarization, which appears as the J wave on electrocardiography. It is more pronounced in hypothermia, disappears after normalization of the body temperature, and is usually evident in the inferolateral leads.
Figure 3. Sinus bradycardia, 48 beats per minute; the PR interval is prolonged at 0.24 second, the QRS interval is prolonged at 0.16 second, the corrected QT interval is 0.5 second, and classic high-amplitude J waves are visible in all leads (arrows). Core body temperature was 29.6°C (85.3°F).
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
The lower the core body temperature, the higher the amplitude of the J wave.
The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
Figure 1. Sinus bradycardia, heart rate 55 beats per minute. The patient’s core body temperature was 36°C (96.8°F). There are no evident J waves.
A 22-year-old man was brought to the emergency room after a motor vehicle accident. He was in a deep coma, with a Glasgow coma score of 4 out of 15 (3 being the worst score) and a core body temperature of 36°C (96.8°F). The next day, clinical evidence of brain death was noted, and his core body temperature dropped as low as 29.6°C (85.3°F). At that time, his electrocardiogram revealed sinus bradycardia, with a rate of 48 beats per minute, PR interval 0.24 second, QRS interval 0.16 second, corrected QT duration 0.5 second, and classic high-amplitude Osborn waves (J waves) that were evident in all leads. Figures 1, 2, and 3 show the effect of various degrees of hypothermia on the electrocardiogram.
Figure 2. Sinus bradycardia, heart rate 50 beats per minute; low-amplitude J waves are visible in leads V4, V5, and V6 (arrows). The patient’s core body temperature was 31°C (87.8°F).
The Osborn wave1 (J wave) is the result of a transient, outward, potassium-mediated current in the ventricular epicardium but not the endocardium, corresponding to a notch in the action potential. This gives rise to a transmural voltage gradient during early repolarization, which appears as the J wave on electrocardiography. It is more pronounced in hypothermia, disappears after normalization of the body temperature, and is usually evident in the inferolateral leads.
Figure 3. Sinus bradycardia, 48 beats per minute; the PR interval is prolonged at 0.24 second, the QRS interval is prolonged at 0.16 second, the corrected QT interval is 0.5 second, and classic high-amplitude J waves are visible in all leads (arrows). Core body temperature was 29.6°C (85.3°F).
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
The lower the core body temperature, the higher the amplitude of the J wave.
The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
References
Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol1953; 175:389–398.
References
Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol1953; 175:389–398.
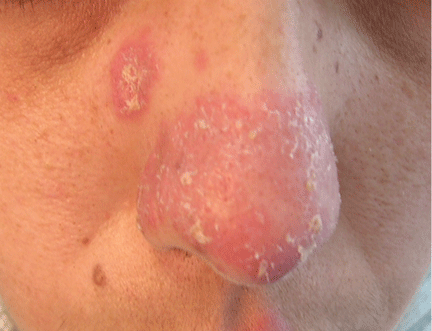
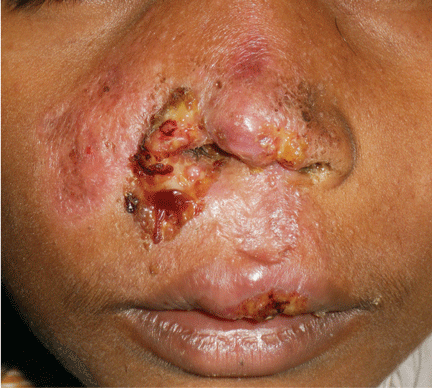
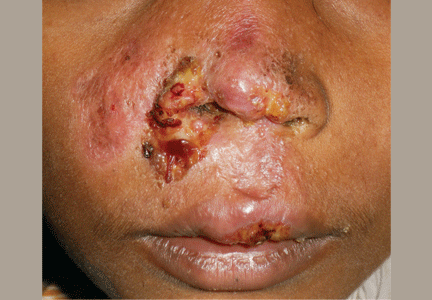
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
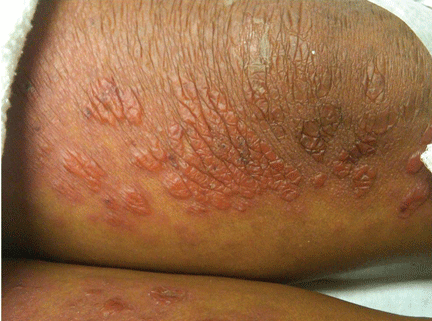
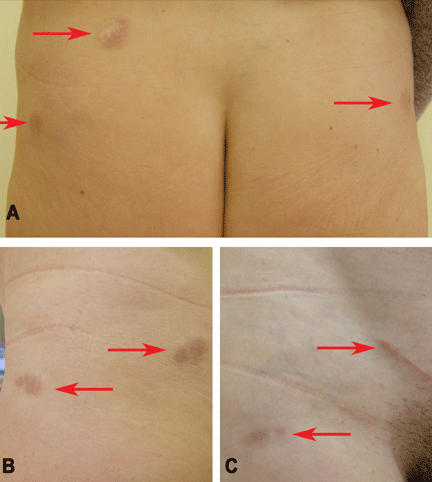
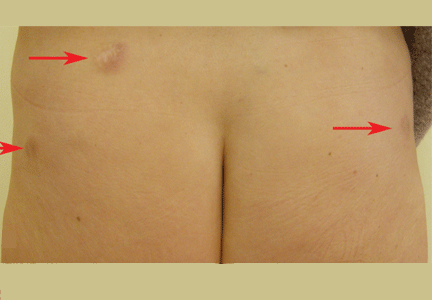
Figure 1. The acrocyanotic lesions were covered with scales.
A physical examination revealed acrocyanotic lesions on the nose that were covered with scales (Figure 1). Laboratory testing showed increased cholesterol levels, a positive antinuclear antibody titer (1:160 or higher is positive), and a positive anti-Ro/SS-A antibody titer (1:80 or higher is positive). Tests for cryoglobulin, cold agglutinins, anti-double-stranded DNA antibody, anti-extractable nuclear antigens, C3 and C4 complement proteins, and anticardiolipin antibody were normal or negative.
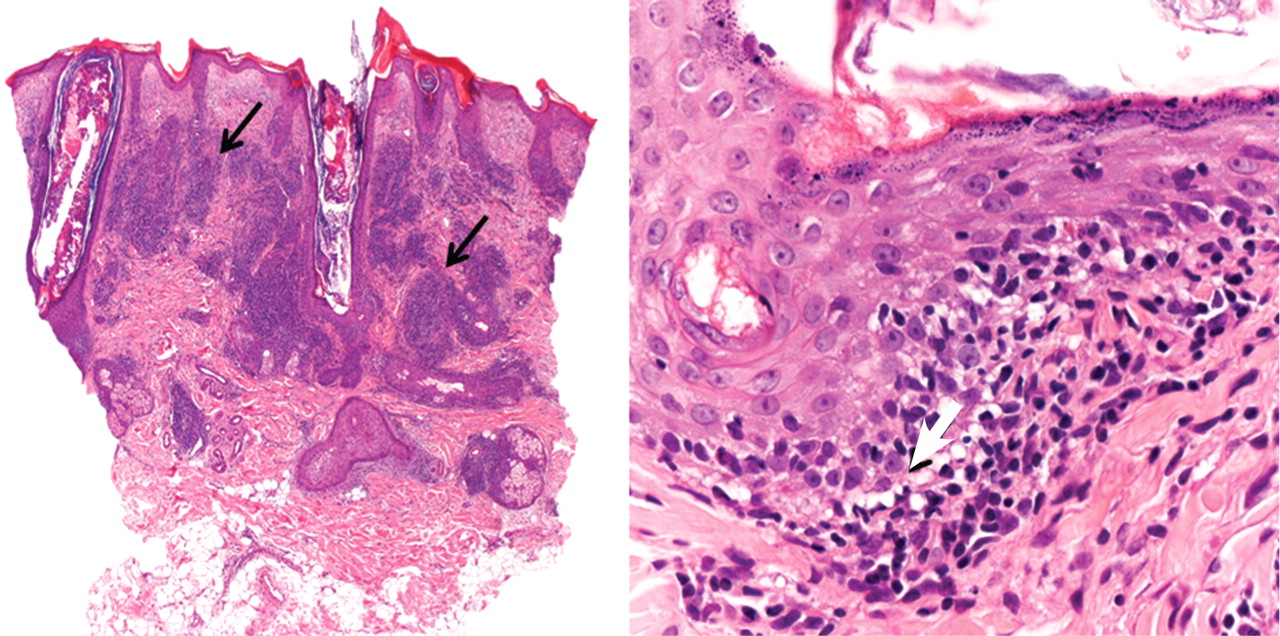
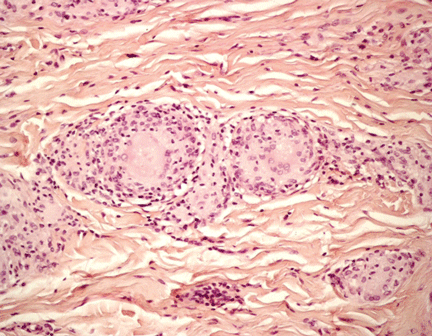
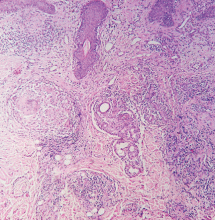
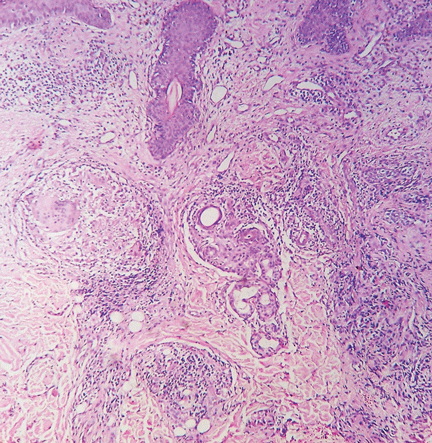
Figure 2. On the left, superficial, interstitial, and deep perivascular and perifollicular dense infiltrate of lymphocytes is seen (arrows) (hematoxylin-eosin, × 4). On the right, hydropic degeneration of the basal cell layer is seen (arrow) (hematoxylin-eosin, × 40).
Histologic examination revealed degeneration of the basal layer of the dermis, with periadnexal and perivascular inflammatory infiltrates (Figure 2). On immunofluorescence testing, linear deposits of immunoglobulin M were noted at the dermoepidermal junction.
Q: What is the most likely diagnosis?
Lupus pernio
Rosacea
Seborrheic dermatitis
Chilblain lupus erythematosus
Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
References
Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics2005; 116:e472–e475.
Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol2001; 44:925–931.
Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol2005; 32:760–761.
Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest2009; 136:946–947.
Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis1994; 54:395–399.
Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol1996; 135:355–362.
Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus1999; 8:215–219.
Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus2007; 16:163–167.
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
María Isabel Soriano-Hernández, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Pilar Burkhardt-Pérez, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Alaminos-Mingorance, PhD Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
María Isabel Soriano-Hernández, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Pilar Burkhardt-Pérez, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Alaminos-Mingorance, PhD Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
Author and Disclosure Information
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
María Isabel Soriano-Hernández, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Pilar Burkhardt-Pérez, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Alaminos-Mingorance, PhD Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
Figure 1. The acrocyanotic lesions were covered with scales.
A physical examination revealed acrocyanotic lesions on the nose that were covered with scales (Figure 1). Laboratory testing showed increased cholesterol levels, a positive antinuclear antibody titer (1:160 or higher is positive), and a positive anti-Ro/SS-A antibody titer (1:80 or higher is positive). Tests for cryoglobulin, cold agglutinins, anti-double-stranded DNA antibody, anti-extractable nuclear antigens, C3 and C4 complement proteins, and anticardiolipin antibody were normal or negative.
Figure 2. On the left, superficial, interstitial, and deep perivascular and perifollicular dense infiltrate of lymphocytes is seen (arrows) (hematoxylin-eosin, × 4). On the right, hydropic degeneration of the basal cell layer is seen (arrow) (hematoxylin-eosin, × 40).
Histologic examination revealed degeneration of the basal layer of the dermis, with periadnexal and perivascular inflammatory infiltrates (Figure 2). On immunofluorescence testing, linear deposits of immunoglobulin M were noted at the dermoepidermal junction.
Q: What is the most likely diagnosis?
Lupus pernio
Rosacea
Seborrheic dermatitis
Chilblain lupus erythematosus
Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
A 38-year-old woman presented with a pruriginous and erythematous lesion on her nose that appeared during periods of cold weather. She said she is completely asymptomatic during the summer months.
Figure 1. The acrocyanotic lesions were covered with scales.
A physical examination revealed acrocyanotic lesions on the nose that were covered with scales (Figure 1). Laboratory testing showed increased cholesterol levels, a positive antinuclear antibody titer (1:160 or higher is positive), and a positive anti-Ro/SS-A antibody titer (1:80 or higher is positive). Tests for cryoglobulin, cold agglutinins, anti-double-stranded DNA antibody, anti-extractable nuclear antigens, C3 and C4 complement proteins, and anticardiolipin antibody were normal or negative.
Figure 2. On the left, superficial, interstitial, and deep perivascular and perifollicular dense infiltrate of lymphocytes is seen (arrows) (hematoxylin-eosin, × 4). On the right, hydropic degeneration of the basal cell layer is seen (arrow) (hematoxylin-eosin, × 40).
Histologic examination revealed degeneration of the basal layer of the dermis, with periadnexal and perivascular inflammatory infiltrates (Figure 2). On immunofluorescence testing, linear deposits of immunoglobulin M were noted at the dermoepidermal junction.
Q: What is the most likely diagnosis?
Lupus pernio
Rosacea
Seborrheic dermatitis
Chilblain lupus erythematosus
Lupus vulgaris
A: The diagnosis is chilblain lupus erythematosus.
The differential diagnosis of an erythematous lesion on the nose of a middle-aged woman also includes rosacea, lupus pernio, lupus vulgaris, and seborrheic dermatitis. Some of these lesions are exacerbated by cold. Usually, the diagnosis is based on clinical findings, but in some cases histologic features on biopsy study confirm the diagnosis.
Lesions of lupus pernio (sarcoidosis) remain unaltered with changes in temperature, and biopsy study usually shows granulomas without caseous necrosis with little inflammatory infiltrate at the periphery.
Rosacea usually gets worse with heat and with alcohol consumption, although it can be exacerbated by cold. Biopsy study shows a nonspecific perivascular and perifollicular lymphohistiocytic infiltrate accompanied occasionally by multinucleated cells.
Seborrheic dermatitis is a papulosquamous disorder characterized by greasy scaling over inflamed skin on the scalp, face, and trunk. Disease activity is increased in winter and spring, with remissions commonly occurring in summer. The histologic features of seborrheic dermatitis are nonspecific; in this case, the histologic features were compatible with chilblain lupus without changes of seborrheic dermatitis.
Lupus vulgaris is a chronic form of cutaneous tuberculosis characterized by redbrown papules with central atrophy. The nose and ears are usually affected. Histologically, granulomatous tubercles with epithelioid cells and caseation necrosis are usually found.
CHILBLAIN LUPUS ERYTHEMATOSUS
Pernio, or chilblain, is a localized inflammatory lesion of the skin resulting from an abnormal response to cold.1 The cutaneous lesions of chilblain may be classified as idiopathic, autoimmune-related (as in systemic lupus erythematosus, subacute cutaneous lupus), and induced by drugs such as terbinafine (Lamisil)2 or infliximab (Remicade).,3
Chilblain lupus is a rare form of cutaneous lupus erythematosus and should not be confused with lupus pernio, which is a misleading name used for a type of cutaneous sarcoidosis.4
Chilblain lupus is characterized by reddish-purple plaques in acral areas (more often the hands and feet, but also the nose and ears) that are induced by exposure to cold—unlike other lesions of lupus erythematosus, which worsen with exposure to sunlight. The main difference from the cutaneous variety of sarcoidosis (lupus pernio) is the histopathologic appearance. In patients with chilblain lupus, epidermal atrophy, perivascular and periadnexal inflammatory infiltrates, and degeneration of the basal layer are found, whereas in lupus pernio (sarcoidosis), we observe granulomas without caseous necrosis, but with few inflammatory infiltrates on the periphery.
PROPOSED DIAGNOSTIC CRITERIA
Su et al5 have proposed diagnostic criteria for chilblain lupus. Their two major criteria are skin lesions in acral locations induced by exposure to cold or a drop in temperature, and evidence of lupus erythematosus in the skin lesions by histopathologic examination or immunofluorescence study. Both of these criteria must be met, plus one of three minor criteria: the coexistence of systemic lupus erythematosus or of skin lesions of discoid lupus erythematosus; response to lupus therapy; and negative results of testing for cryoglobulin and cold agglutinins.
CHILBLAIN LUPUS VS SYSTEMIC LUPUS
Chilblain lupus is an uncommon manifestation of systemic lupus erythematosus, and it is reported to occur in about 20% of patients with that condition.6 Often, the onset of chilblain lupus precedes the systemic disease. Patients with systemic lupus erythematosus and chilblain lupus do not usually present with renal disease, mucosal lesions, or central nervous system involvement. However, Raynaud phenomenon and photosensitivity have been reported to be more frequently associated with chilblain lupus.7
A disorder of peripheral circulation could be involved in the pathogenesis of chilblain lupus, and the association with Raynaud phenomenon, livedo reticularis, antiphospholipid syndrome, and changes in nailfold capillaries supports this hypothesis. Antinuclear antibody and anti-Ro/SS-A antibody are commonly detected in the serum of patients with chilblain lupus, and anti-Ro/SS-A antibody seems to be a major serologic marker of chilblain lupus in patients with systemic lupus erythematosus.7
TREATMENT
Protection from cold by physical measures is very important, as well as the use of topical or oral antibiotics if the lesions are infected. In severe cases unresponsive to topical corticosteroids, a calcium channel blocker is a good therapeutic option; antimalarials, commonly used in the treatment of lupus erythematosus, can also have a positive effect in patients with chilblain lupus.
CASE CONCLUDED
Our patient was advised to protect herself from the cold. Topical corticosteroids and oral hydroxychloroquine (200 mg/day) were prescribed, and they produced a good response. In severe cases, oral corticosteroids, etretinate (Tegison), mycophenolate (CellCept), or thalidomide (Thalomid) may be used.8
References
Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics2005; 116:e472–e475.
Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol2001; 44:925–931.
Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol2005; 32:760–761.
Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest2009; 136:946–947.
Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis1994; 54:395–399.
Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol1996; 135:355–362.
Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus1999; 8:215–219.
Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus2007; 16:163–167.
References
Simon TD, Soep JB, Hollister JR. Pernio in pediatrics. Pediatrics2005; 116:e472–e475.
Bonsmann G, Schiller M, Luger TA, Ständer S. Terbinafine-induced subacute cutaneous lupus erythematosus. J Am Acad Dermatol2001; 44:925–931.
Richez C, Dumoulin C, Schaeverbeke T. Infliximab induced chilblain lupus in a patient with rheumatoid arthritis. J Rheumatol2005; 32:760–761.
Arias-Santiago SA, Girón-Prieto MS, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N. Lupus pernio or chilblain lupus?: two different entities. Chest2009; 136:946–947.
Su WP, Perniciaro C, Rogers RS, White JW. Chilblain lupus erythematosus (lupus pernio): clinical review of the Mayo Clinic experience and proposal of diagnostic criteria. Cutis1994; 54:395–399.
Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol1996; 135:355–362.
Franceschini F, Calzavara-Pinton P, Quinzanini M, et al. Chilblain lupus erythematosus is associated with antibodies to SSA/Ro. Lupus1999; 8:215–219.
Bouaziz JD, Barete S, Le Pelletier F, Amoura Z, Piette JC, Francès C. Cutaneous lesions of the digits in systemic lupus erythematosus: 50 cases. Lupus2007; 16:163–167.
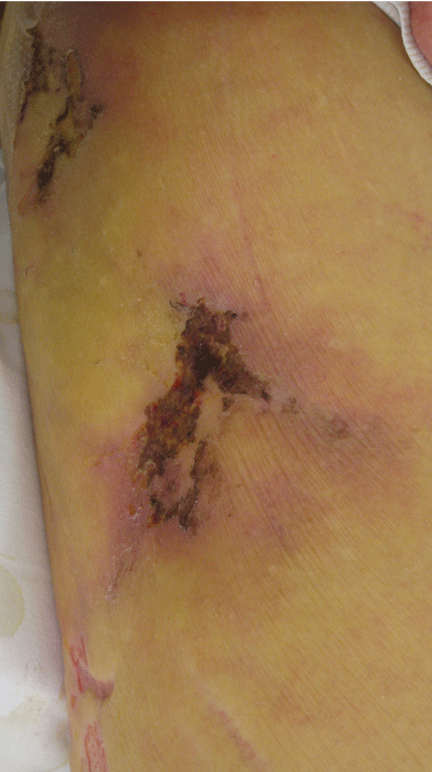
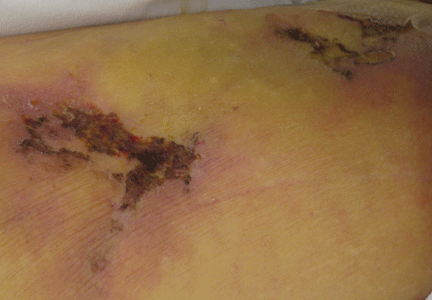
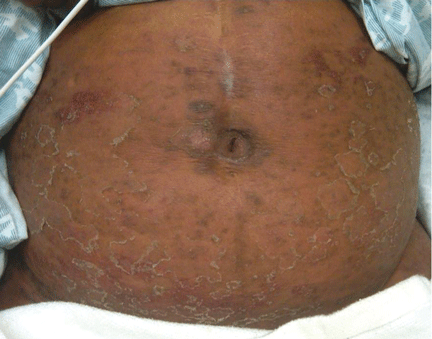
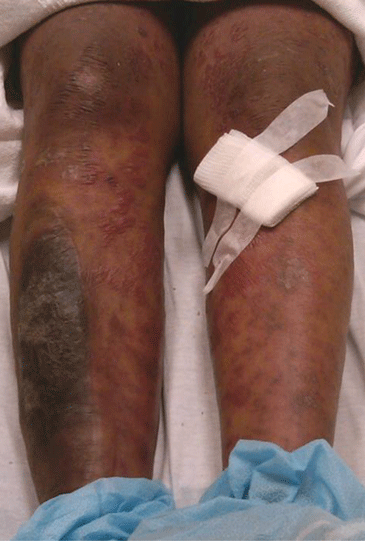
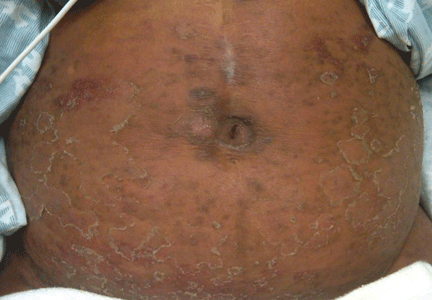
Figure 1. The patient’s right lateral thigh shows the classic features of calciphylaxis: ischemia and necrosis in an area of increased adipose tissue.
A 44-year-old woman with end-stage liver disease presents with a painful, ischemic, necrotic lesion on her right lateral and medial thigh (Figure 1). Several months ago, while being evaluated in the hospital for liver transplantation, she developed bacteremia, anion-gap metabolic acidosis, hepatorenal syndrome, and acute renal failure. She began continuous hemodialysis, which lasted for about 1 month, ending 35 days after the renal failure resolved.
Current laboratory values:
Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
Phosphorus 6.4 mg/dL (2.5–4.5)
Corrected calcium-phosphorus product 55
Parathyroid hormone 275 pg/mL (10–60)
25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
Necrotizing fasciitis
Calciphylaxis
Disseminated intravascular coagulation
Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
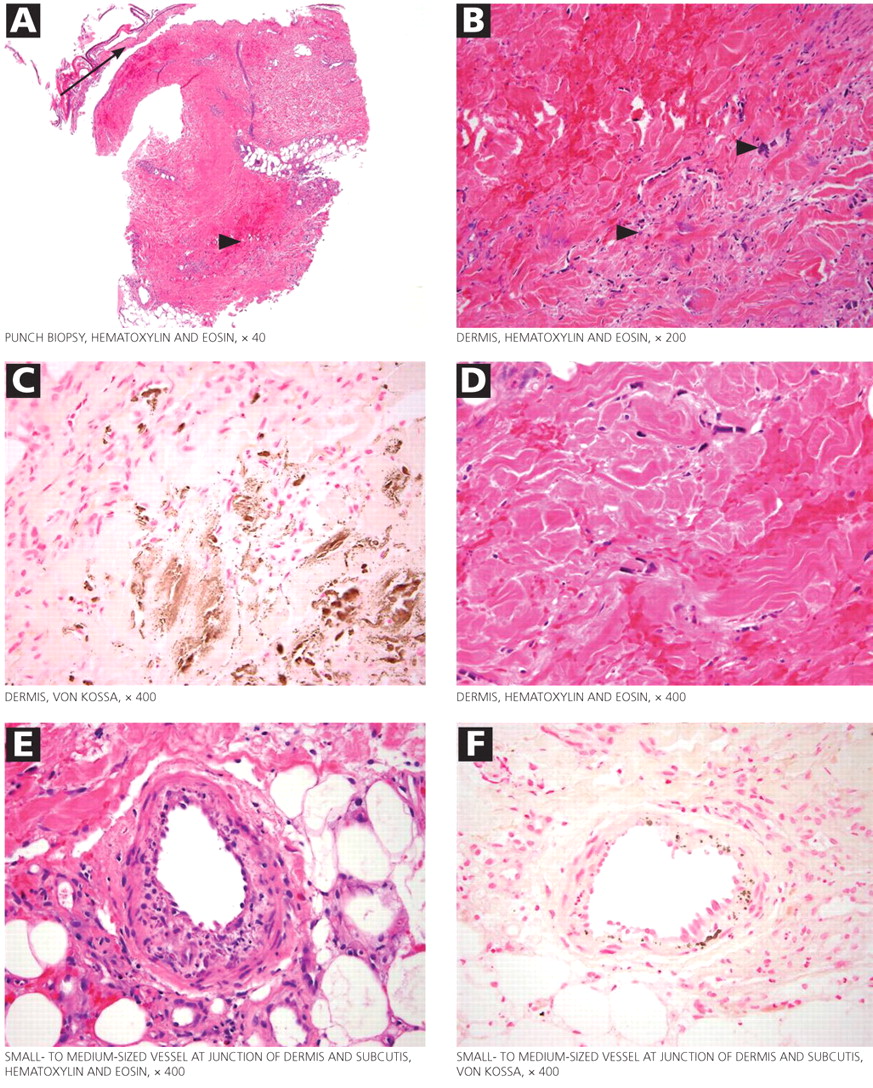
Figure 2. Histologic study of the biopsied skin lesions. (A) A low-power image of the punch biopsy shows necrotic epidermis (arrow) that has physically separated from the underlying unhealthy hemorrhagic dermis (arrowhead). (B) A higher-power view of the hemorrhagic dermis shows scattered foci of deeply basophilic material (arrowheads). A reasonable differential diagnosis for this finding is atypical hyperchromatic fibroblastic and endothelial nuclei vs calcium deposits. (C) Von Kossa stain was performed to evaluate for the presence of calcium deposits; brown-staining areas indicate calcium deposition. (D) A section of the same tissue seen in C. (E and F) Calcium deposits within the wall of the centrally placed small- to medium-sized vessel.
The skin lesions are characteristically erythematous and tender, with mottling of the skin early in the course. As the lesions progress, they develop central necrosis and deep ulcerations with eschar formation. The ulcers have irregular borders and do not heal. Histopathologic study typically shows epidermis with ischemic necrosis and calcium deposition along elastic fibers on Von Kossa calcium stains (Figure 2).
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
References
Daudén E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008; 26:557–568.
Pliquett RU, Schwock J, Paschke R, Achenbach H. Calciphylaxis in chronic, non-dialysis-dependent renal disease. BMC Nephrol 2003; 4:8.
Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
Figure 1. The patient’s right lateral thigh shows the classic features of calciphylaxis: ischemia and necrosis in an area of increased adipose tissue.
A 44-year-old woman with end-stage liver disease presents with a painful, ischemic, necrotic lesion on her right lateral and medial thigh (Figure 1). Several months ago, while being evaluated in the hospital for liver transplantation, she developed bacteremia, anion-gap metabolic acidosis, hepatorenal syndrome, and acute renal failure. She began continuous hemodialysis, which lasted for about 1 month, ending 35 days after the renal failure resolved.
Current laboratory values:
Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
Phosphorus 6.4 mg/dL (2.5–4.5)
Corrected calcium-phosphorus product 55
Parathyroid hormone 275 pg/mL (10–60)
25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
Necrotizing fasciitis
Calciphylaxis
Disseminated intravascular coagulation
Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
Figure 2. Histologic study of the biopsied skin lesions. (A) A low-power image of the punch biopsy shows necrotic epidermis (arrow) that has physically separated from the underlying unhealthy hemorrhagic dermis (arrowhead). (B) A higher-power view of the hemorrhagic dermis shows scattered foci of deeply basophilic material (arrowheads). A reasonable differential diagnosis for this finding is atypical hyperchromatic fibroblastic and endothelial nuclei vs calcium deposits. (C) Von Kossa stain was performed to evaluate for the presence of calcium deposits; brown-staining areas indicate calcium deposition. (D) A section of the same tissue seen in C. (E and F) Calcium deposits within the wall of the centrally placed small- to medium-sized vessel.
The skin lesions are characteristically erythematous and tender, with mottling of the skin early in the course. As the lesions progress, they develop central necrosis and deep ulcerations with eschar formation. The ulcers have irregular borders and do not heal. Histopathologic study typically shows epidermis with ischemic necrosis and calcium deposition along elastic fibers on Von Kossa calcium stains (Figure 2).
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
Figure 1. The patient’s right lateral thigh shows the classic features of calciphylaxis: ischemia and necrosis in an area of increased adipose tissue.
A 44-year-old woman with end-stage liver disease presents with a painful, ischemic, necrotic lesion on her right lateral and medial thigh (Figure 1). Several months ago, while being evaluated in the hospital for liver transplantation, she developed bacteremia, anion-gap metabolic acidosis, hepatorenal syndrome, and acute renal failure. She began continuous hemodialysis, which lasted for about 1 month, ending 35 days after the renal failure resolved.
Current laboratory values:
Serum calcium concentration 7.8 mg/dL (reference range 8.5–10.5)
Phosphorus 6.4 mg/dL (2.5–4.5)
Corrected calcium-phosphorus product 55
Parathyroid hormone 275 pg/mL (10–60)
25-hydroxyvitamin D 7.4 ng/mL (31–80).
Q: Given the patient’s history, which of the following does her skin lesion likely represent?
Necrotizing fasciitis
Calciphylaxis
Disseminated intravascular coagulation
Anticoagulant-induced skin necrosis
A: Calciphylaxis, or calcific uremic arteriolopathy, is the most likely. It is rare in people with normal renal function, and still rare but somewhat less so in end-stage renal disease patients undergoing chronic hemodialysis.
WHAT CAUSED IT IN OUR PATIENT?
The cause of calciphylaxis is unknown. Theories have focused on protein C and parathyroid hormone. Putative precipitating factors include acute tubular necrosis, albumin infusion with paracentesis, deficiency of protein C or S, hyperparathyroidism, hyperphosphatemia, hypercalcemia, vitamin D supplementation, steroids, trauma, and warfarin use.
Our patient had a history of hypothyroidism, ulcerative colitis, and end-stage liver disease due to primary sclerosing cholangitis, but no previous history of renal disease.
At the time of her acute renal failure, her calcium-phosphorus level was 55, parathyroid hormone level 274 pg/mL (normal 10–60), and protein C level 26% (normal 76%–147%). At the time the skin lesions were discovered, her protein C level had dropped to 14%; her parathyroid level had returned to normal.
Her home medications included furosemide (Lasix), levothyroxine (Synthroid), mesalamine (Pentasa), azathioprine (Imuran), ursodiol (Actigall), spironolactone (Aldactone), and omeprazole (Prilosec).
NONHEALING LESIONS
Figure 2. Histologic study of the biopsied skin lesions. (A) A low-power image of the punch biopsy shows necrotic epidermis (arrow) that has physically separated from the underlying unhealthy hemorrhagic dermis (arrowhead). (B) A higher-power view of the hemorrhagic dermis shows scattered foci of deeply basophilic material (arrowheads). A reasonable differential diagnosis for this finding is atypical hyperchromatic fibroblastic and endothelial nuclei vs calcium deposits. (C) Von Kossa stain was performed to evaluate for the presence of calcium deposits; brown-staining areas indicate calcium deposition. (D) A section of the same tissue seen in C. (E and F) Calcium deposits within the wall of the centrally placed small- to medium-sized vessel.
The skin lesions are characteristically erythematous and tender, with mottling of the skin early in the course. As the lesions progress, they develop central necrosis and deep ulcerations with eschar formation. The ulcers have irregular borders and do not heal. Histopathologic study typically shows epidermis with ischemic necrosis and calcium deposition along elastic fibers on Von Kossa calcium stains (Figure 2).
The skin lesions of calciphylaxis usually occur in areas of increased adipose tissue. The lesions may not manifest until several weeks after the initial insult (ie, the elevated calcium-phosphate level). Skin biopsy is recommended if a necrotic skin lesion is identified in a patient with an elevated calcium-phosphate level or in a patient with risk factors for renal, liver, or parathyroid disease.
PROGNOSIS IS POOR
Treatment is supportive. Intensive wound care (with surgical evaluation for skin grafting), hyperbaric oxygen, and possibly tissue plasminogen activator (if there is evidence of a hypercoagulable state and occlusive vasculopathy) may be the most beneficial. Identifying the underlying cause and regulating the calcium-phosphorus product level with diet, phosphate binders, bisphosphonates, and sodium thiosulfate are also important in wound healing. Cinacalcet (Sensipar) and parathyroidectomy should be considered in cases of secondary hyperparathyroidism.
Calciphylaxis is important to recognize early in its course and may require a multidisciplinary approach to treatment. Its prognosis is poor, with death rates ranging from 40% to 60%.
Our patient developed recurrent hepatorenal syndrome and sepsis and eventually died of septic shock.
References
Daudén E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008; 26:557–568.
Pliquett RU, Schwock J, Paschke R, Achenbach H. Calciphylaxis in chronic, non-dialysis-dependent renal disease. BMC Nephrol 2003; 4:8.
Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol 2008; 3:1139–1143.
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
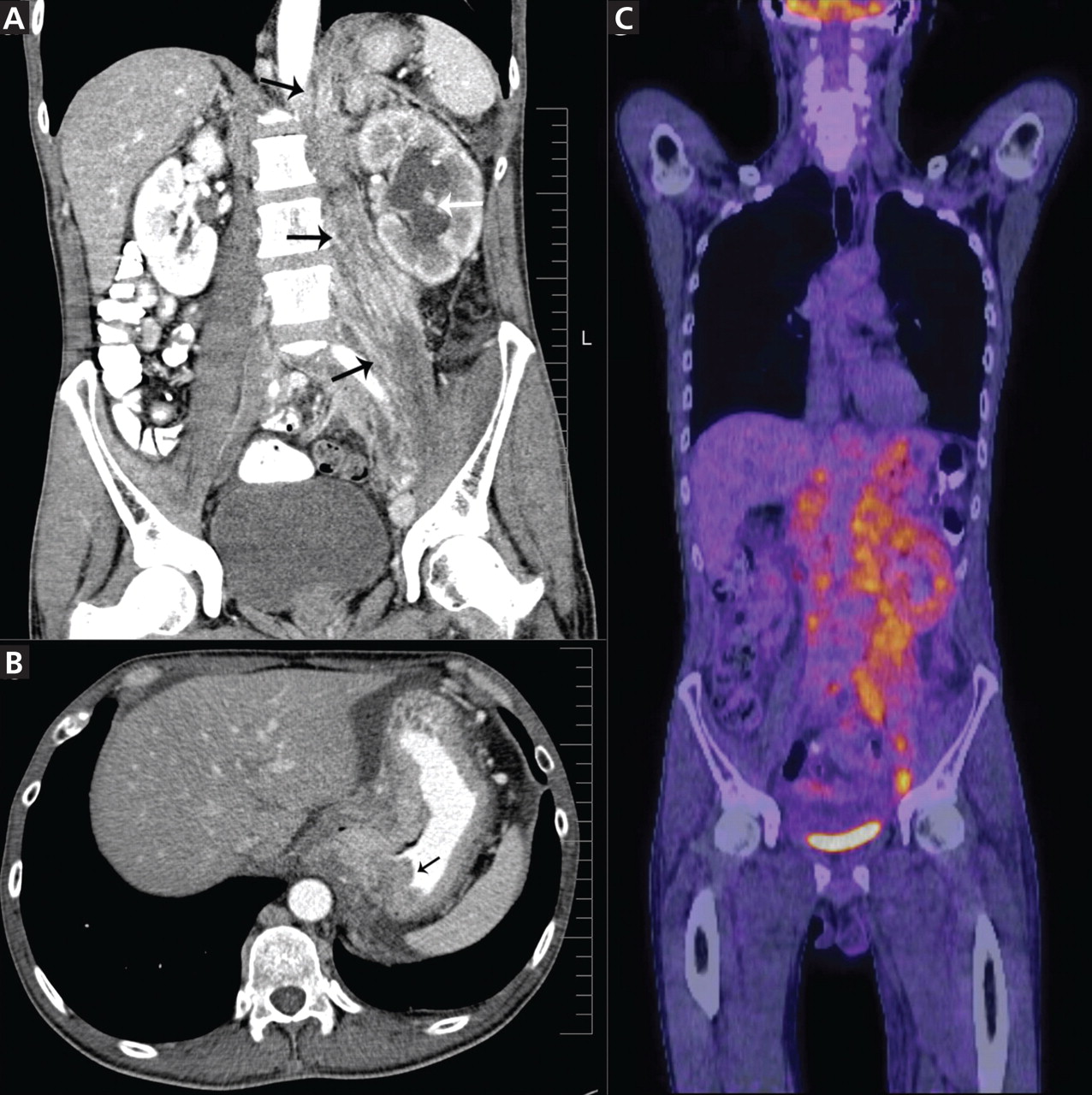
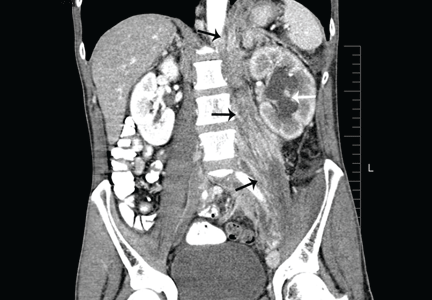
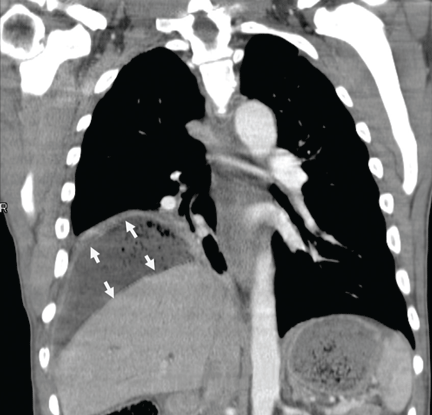
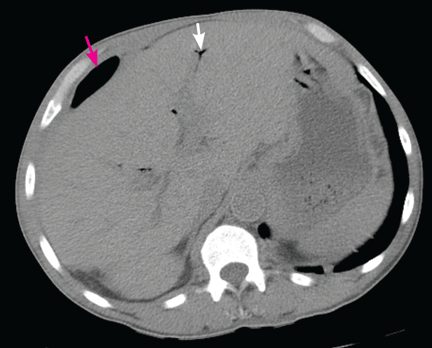
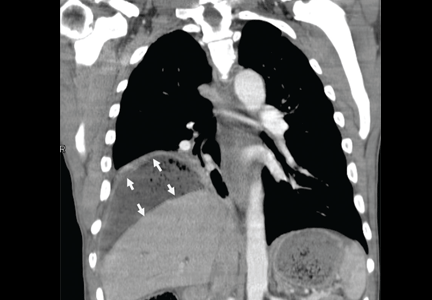
Figure 1. (A) Abdominal computed tomography reveals an extensive, heterogeneous, ill-defined infiltrative process in the retroperitoneum extending into the left pelvis and invading the left psoas, hemidiaphragm, and adrenal gland (black arrows), with associated left hydronephrosis (white arrow) related to compression of the left ureter. (B) Also visualized is stomach-wall thickening, particularly near the cardia (black arrow). (C) Positron emission tomography shows a retroperitoneal infiltrative process and shows the thickened gastric cardia to be hypermetabolic.
Computed tomography (CT) at our clinic revealed an extensive heterogeneous ill-defined infiltrative process in the retroperitoneum extending into the left pelvis, invading the left psoas, left hemidiaphragm, and left adrenal gland (Figure 1A). Also noted were left hydronephrosis, related to compression of the left ureter, and stomach-wall thickening, most marked near the cardia (Figure 1B).
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
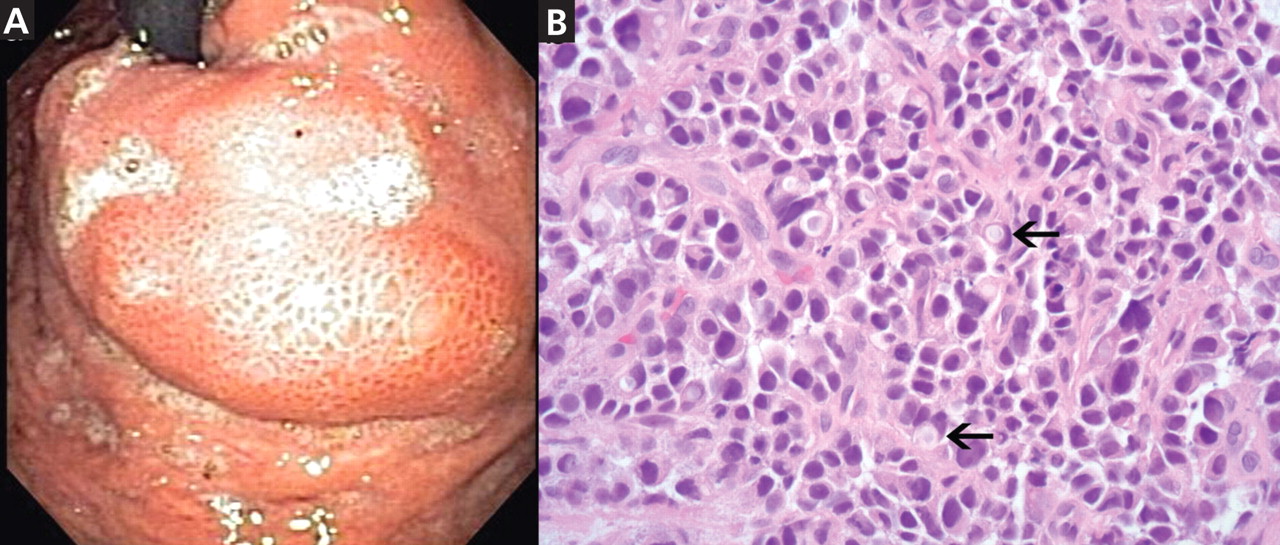
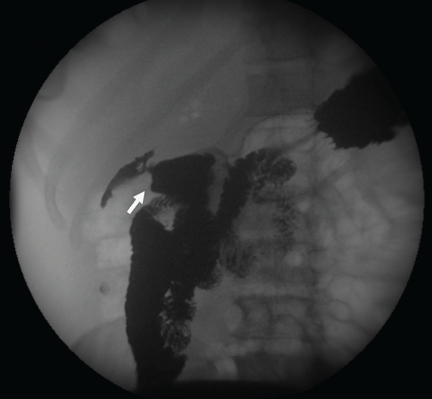
Figure 2. (A) Esophagogastroduodenoscopy shows a large, ulcerated, submucosal, nodular mass in the gastric cardia. (B) Biopsy shows poorly differentiated adenocarcinoma with scattered signet-ring cells (black arrows).
Esophagogastro- duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin2005; 55:74–108.
Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol2006; 12:354–362.
Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw2010; 8:437–447.
Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol2006; 13:221–228.
Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am2002; 86:1165–1216.
Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
Figure 1. (A) Abdominal computed tomography reveals an extensive, heterogeneous, ill-defined infiltrative process in the retroperitoneum extending into the left pelvis and invading the left psoas, hemidiaphragm, and adrenal gland (black arrows), with associated left hydronephrosis (white arrow) related to compression of the left ureter. (B) Also visualized is stomach-wall thickening, particularly near the cardia (black arrow). (C) Positron emission tomography shows a retroperitoneal infiltrative process and shows the thickened gastric cardia to be hypermetabolic.
Computed tomography (CT) at our clinic revealed an extensive heterogeneous ill-defined infiltrative process in the retroperitoneum extending into the left pelvis, invading the left psoas, left hemidiaphragm, and left adrenal gland (Figure 1A). Also noted were left hydronephrosis, related to compression of the left ureter, and stomach-wall thickening, most marked near the cardia (Figure 1B).
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
Figure 2. (A) Esophagogastroduodenoscopy shows a large, ulcerated, submucosal, nodular mass in the gastric cardia. (B) Biopsy shows poorly differentiated adenocarcinoma with scattered signet-ring cells (black arrows).
Esophagogastro- duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
A 50-year-old male Japanese immigrant with a history of smoking and occasional untreated heartburn presented with the recent onset of flank pain, weight loss, headache, syncope, and blurred vision.
Previously healthy, he began feeling moderate pain in his left flank 1 month ago; it was diagnosed as kidney stones and was treated conservatively. Two weeks later he had an episode of syncope and soon after developed blurred vision, mainly in his left eye, along with severe bifrontal headache. An eye examination and magnetic resonance imaging of the brain indicated optic neuritis, for which he was given glucocorticoids intravenously for 3 days, with moderate improvement.
As his symptoms continued over the next 2 weeks, he lost 20 lb (9.1 kg) due to the pain, loss of appetite, nausea, and occasional vomiting.
Figure 1. (A) Abdominal computed tomography reveals an extensive, heterogeneous, ill-defined infiltrative process in the retroperitoneum extending into the left pelvis and invading the left psoas, hemidiaphragm, and adrenal gland (black arrows), with associated left hydronephrosis (white arrow) related to compression of the left ureter. (B) Also visualized is stomach-wall thickening, particularly near the cardia (black arrow). (C) Positron emission tomography shows a retroperitoneal infiltrative process and shows the thickened gastric cardia to be hypermetabolic.
Computed tomography (CT) at our clinic revealed an extensive heterogeneous ill-defined infiltrative process in the retroperitoneum extending into the left pelvis, invading the left psoas, left hemidiaphragm, and left adrenal gland (Figure 1A). Also noted were left hydronephrosis, related to compression of the left ureter, and stomach-wall thickening, most marked near the cardia (Figure 1B).
Positron emission tomography showed the retroperitoneal infiltrative process and the thickened gastric cardia to be hypermetabolic (Figure 1C).
The area of retroperitoneal infiltration was biopsied under CT guidance, and pathologic study showed poorly differentiated carcinoma with signet-ring cells, a feature of gastric cancer.
The patient underwent lumbar puncture. His cerebrospinal fluid had 206 white blood cells/μL (reference range 0–5) and large numbers of poorly differentiated malignant cells, most consistent with adenocarcinoma on cytologic study.
Figure 2. (A) Esophagogastroduodenoscopy shows a large, ulcerated, submucosal, nodular mass in the gastric cardia. (B) Biopsy shows poorly differentiated adenocarcinoma with scattered signet-ring cells (black arrows).
Esophagogastro- duodenoscopy (EGD) revealed a large, ulcerated, submucosal, nodular mass in the cardia of the stomach extending to the gastroesophageal junction (Figure 2A). Biopsy of the mass again revealed poorly differentiated adenocarcinoma with scattered signet-ring cells undermining the gastric mucosa, favoring a gastric origin (Figure 2B).
THREE SUBTYPES OF GASTRIC CANCER
Worldwide, gastric cancer is the third most common type of cancer and the second most common cause of cancer-related deaths.1 In the United States, blacks and people of Asian ancestry have almost twice the risk of death, with the highest incidence and mortality rates.2,3
Most cases of gastric adenocarcinoma can be categorized as either intestinal or diffuse, but a new proximal subtype is emerging.4
Intestinal-type gastric adenocarcinoma is the most common subtype and accounts for almost all the ethnic and geographic variation in incidence.2 The lesions are often ulcerative and distal; the pathogenesis is stepwise and is initiated by chronic inflammation. Risk factors include old age, Helicobacter pylori infection, tobacco smoking, family history, and high salt intake, with an observed risk-reduction with the use of nonsteroidal anti-inflammatory drugs and with a high intake of fruits and vegetables.3
Diffuse gastric adenocarcinoma, on the other hand, has a uniform distribution worldwide, and its incidence is increasing. It typically carries a poor prognosis. Evidence thus far has shown its pathogenesis to be independent of chronic inflammation, but it has a strong tendency to be hereditary.3
Proximal gastric adenocarcinoma is observed in the gastric cardia and near the gastroesophageal junction. It is often grouped with the distal esophageal adenocarcinomas and has similar risk factors, including reflux disease, obesity, alcohol abuse, and tobacco smoking. Interestingly, however, H pylori infection does not contribute to the pathogenesis of this type, and it may even have a protective role.3
DIFFICULT TO DETECT EARLY
Gastric cancer is difficult to detect early enough in its course to be cured. Understanding its risk factors, recognizing its common symptoms, and regarding its uncommon symptoms with suspicion may lead to earlier diagnosis and more effective treatment.
Our patient’s proximal gastric cancer was diagnosed late even though he had several risk factors for it (he was Japanese, he was a smoker, and he had gastroesophageal reflux disease) because of a late and atypical presentation with misleading paraneoplastic symptoms.
Early diagnosis is difficult because most patients have no symptoms in the early stage; weight loss and abdominal pain are often late signs of tumor progression.
Screening may be justified in high-risk groups in the United States, although the issue is debatable. Diagnostic imaging is the only effective method for screening,5 with EGD considered the first-line targeted evaluation should there be suspicion of gastric cancer either from the clinical presentation or from barium swallow.6 Candidates for screening may include elderly patients with atrophic gastritis or pernicious anemia, immigrants from countries with high rates of gastric carcinoma, and people with a family history of gastrointestinal cancer.7
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin2005; 55:74–108.
Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol2006; 12:354–362.
Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw2010; 8:437–447.
Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol2006; 13:221–228.
Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am2002; 86:1165–1216.
Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
References
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin2005; 55:74–108.
Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol2006; 12:354–362.
Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw2010; 8:437–447.
Fine G, Chan K. Alimentary tract. In:Kissane JM, editor. Anderson’s Pathology. 8th ed. Saint Louis, MO: Mosby; 1985:1055–1095.
Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol2006; 13:221–228.
Cappell MS, Friedel D. The role of esophagogastroduodenoscopy in the diagnosis and management of upper gastrointestinal disorders. Med Clin North Am2002; 86:1165–1216.
Hisamuchi S, Fukao P, Sugawara N, et al. Evaluation of mass screening programme for stomach cancer in Japan. In:Miller AB, Chamberlain J, Day NE, et al, editors. Cancer Screening. Cambridge, UK: Cambridge University Press; 1991:357–372.
A 23-year-old man presents with a sore throat, dysphagia, and general malaise that began 1 week ago. He also reports a 5-pound weight loss. He has not recently taken antibiotics or inhaled glucocorticoids, and he has no history of tobacco use or trauma to his mouth. He has no personal or family history of oral cancer. He uses cocaine on occasion. He reports feeling feverish and having a decreased appetite.
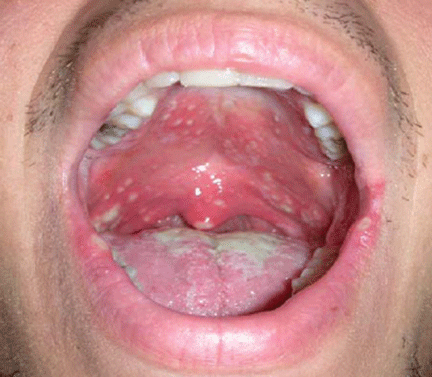
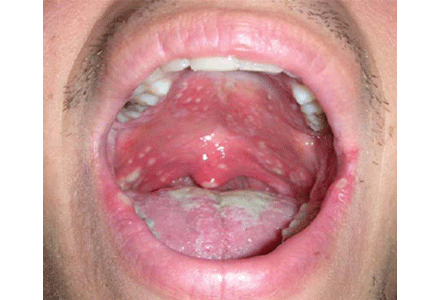
Figure 1.
An examination of his mouth reveals white plaques of varying sizes (Figure 1). The plaques are easily removed using a tongue blade, with no bleeding. No regional lymphadenopathy is noted.
Q: Based on the history, the symptoms, and the physical examination, which of the following is the most likely diagnosis in this patient?
Oral hairy leukoplakia
Squamous cell carcinoma
Oral candidiasis
Herpetic gingivostomatitis
Streptococcal pharyngitis
A: Oral candidiasis is correct.
Otherwise known as thrush, it is common in infants and in denture wearers, and it also can occur in diabetes mellitus, antibiotic therapy, chemotherapy, radiation therapy, and cellular immune deficiency states such as cancer or human immunodeficiency virus (HIV) infection.1 Patients using inhaled glucocorticoids are also at risk and should always be advised to rinse their mouth out with water after inhaled steroid use.
Although Candida albicans is the species most often responsible for candidal infections, other candidal species are increasingly responsible for infections in immunocompromised patients. Candida is part of the normal flora in many adults.
Oral hairy leukoplakia is caused by the Epstein-Barr virus and is often seen in HIV infection. It is a white, painless, corrugated lesion, typically found on the lateral aspect of the tongue, and it cannot be scraped from the adherent surfaces. It can also be found on the dorsum of the tongue, the buccal surfaces, and the floor of the mouth. In an asymptomatic patient with oral hairy leukoplakia, HIV infection with moderate immunosuppression is most likely present.2 Oral hairy leukoplakia is diagnosed by biopsy of suspected lesions. It is not a premalignant lesion, and how to best treat it is still being investigated.3
Squamous cell carcinoma of the oral cavity can present as nonhealing ulcers or masses, dental changes, or exophytic lesions with or without pain.1 They may be accompanied by cervical nodal disease. Malignancies of the oral cavity account for 14% of all head and neck cancers, with squamous cell carcinoma the predominant type.4 Alcohol and tobacco use increase the risk. Alcohol and tobacco together have a synergistic effect on the incidence of oral carcinoma.1,4 Predisposing lesions are leukoplakia, lichen planus of the erosive subtype, submucosal fibrosis, and erythroplakia. Oral infection with human papillomavirus has been shown to increase the risk of oral cancer by a factor of 14, and papillomavirus type 16 is detected in 72% of patients with oropharyngeal cancer.5
Herpetic gingivostomatitis is a manifestation of herpes simplex virus infection. The initial infection may be asymptomatic or may produce groups of vesicles that develop into shallow, painful, and superficial ulcerations on an erythematous base.1,3 If the gingiva is involved, it is erythematous, boggy, and tender.3 Infections are self-limited, lasting up to 2 weeks, but there is potential for recurrence because of the ability of herpes simplex virus to undergo latency. Recurrence is usually heralded by prodromal symptoms 24 hours before onset, with tingling, pain, or burning at the infected site. The diagnosis can be made clinically, but the Tzanck smear test, viral culture, direct fluorescent antibody test, or polymerase chain reaction test can be used to confirm the diagnosis. In patients who are immunocompromised, infections tend to be more severe and to last longer.
Streptococcal pharyngitis, most often caused by group A beta-hemolytic streptococci, is the most common type of bacterial pharyngitis in the clinical setting. The bacteria incubate for 2 to 5 days. The condition mainly affects younger children.6 Patients with “strep throat” often present with a sore throat and high-grade fever. Other symptoms include chills, myalgia, headache, and nausea. Findings on examination may include petechiae of the palate, pharyngeal and tonsillar erythema and exudates, and anterior cervical adenopathy.6 Children often present with coinciding abdominal complaints. A rapid antigen detection test for streptococcal infection can be performed in the office for quick diagnosis, but if clinical suspicion is high, a throat culture is necessary to confirm the diagnosis. Treatment is to prevent complications such as rheumatic fever.6
FEATURES AND DIAGNOSIS OF ORAL CANDIDIASIS
Lesions of oral candidiasis can vary in their appearance. The pseudomembranous form is the most characteristic, with white adherent “cottage-cheese-like” plaques that wipe away, causing minimal bleeding.1,7 The erythematous or atrophic form is associated with denture use and causes a “beefy” appearance on the dorsum of the tongue or on the mucosa that supports a denture.1,7 A third form affects the angles of the mouth, causing angular cheilitis (perlèche).7,8 Chronic infection appears as localized, firmly adherent plaques with an irregular surface similar to hyperkeratosis caused by chronic frictional irritation.7
Oral candidiasis can occur in different forms at the same time. Patients often describe minimal symptoms such as dysgeusia or dry mouth.1,7 Infections causing dysphagia or odynophagia warrant suspicion for involvement of the esophagus.
The diagnosis is made empirically if the lesions resolve with anticandidal therapy. A more definitive diagnosis can be made by microscopy with a potassium hydroxide preparation showing pseudohyphae. Formal culture can also determine the yeast’s susceptibility to medication in recurrent or resistant cases.2
Oral candidiasis may be the manifesting symptom of HIV infection, and more than 90% of patients with adult immunodeficiency syndrome have an episode of thrush.8 When candidiasis is diagnosed without obvious cause, HIV testing should be offered, regardless of a patient’s lack of obvious risk factors. Other oral lesions in HIV patients are oral hairy leukoplakia, Kaposi sarcoma, periodontal and gingival infections, aphthous ulcers, herpes simplex stomatitis, and xerostomia.2 With highly active antiretroviral therapy, the incidence of oral candidiasis has decreased by about 50%.2
Our patient was diagnosed with HIV when screened after this initial presentation. Lower CD4 counts and higher viral loads increase the patient’s risk for oral candidiasis and other lesions. This patient’s initial CD4 count was 524 cells/μL, and his viral load was 11,232 copies/mL.
TREATMENT
In HIV-negative patients or in HIV-positive patients with a CD4 count greater than 200 cells/μL, the treatment of oral candidiasis involves topical antifungal agents, including a nystatin suspension (Nystat-Rx) or clotrimazole (Mycelex) troches.3,7,9 Treatment should be continued for at least 7 days after resolution of the infection. If resolution does not occur, oral fluconazole (Diflucan) 200 mg daily should be given.
For HIV patients with CD4 counts below 200 cells/μL, oral fluconazole or itraconazole (Sporanox) is recommended, with posaconazole (Noxafil) as an alternative for refractory disease.3,9 Giving fluconazole prophylactically to prevent oral candidiasis is not recommended because of the risk of adverse effects, lack of survival benefit, associated cost, and potential to develop antifungal resistance.3,9
References
Reichart PA. Clinical management of selected oral fungal and viral infections during HIV-disease. Int Dent J1999; 49:251–259.
Kim TB, Pletcher SD, Goldberg AN. Head and neck manifestations in the immunocompromised host. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:209–229(225–226).
Sciubba JJ. Oral mucosal lesions. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1229–1231).
Wein R. Malignant Neoplasms of the Oral Cavity. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1236).
D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med2007; 356:1944–1956.
Hayes CS, Williamson H. Management of group A betahemolytic streptococcal pharyngitis. Am Fam Physician2001; 63:1557–1564.
Coleman GC. Diseases of the mouth. In:Bope ET, Rakel RE, Kellerman R, editors. Conn’s Current Therapy. Philadelphia, PA: Saunders; 2010:861–867.
Habif TP. Candidiasis (moniliasis). In: Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh: Mosby; 2010:523–536.
Pappas PG, Rex JH, Sobel JD, et al; Infectious Diseases Society of America. Guidelines for treatment of candidiasis. Clin Infect Dis2004; 38:161–189.
Amber S. Tully, MD Assistant Professor, Department of Family and Community Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA
Carol Dao, MD Thomas Jefferson University, Philadelphia, PA
Address: Amber Tully, MD, Family and Community Medicine, Thomas Jefferson University Hospital, 1100 Walnut Street, Suite 603, Philadephia, PA 19107; e-mail [email protected]
Amber S. Tully, MD Assistant Professor, Department of Family and Community Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA
Carol Dao, MD Thomas Jefferson University, Philadelphia, PA
Address: Amber Tully, MD, Family and Community Medicine, Thomas Jefferson University Hospital, 1100 Walnut Street, Suite 603, Philadephia, PA 19107; e-mail [email protected]
Author and Disclosure Information
Amber S. Tully, MD Assistant Professor, Department of Family and Community Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA
Carol Dao, MD Thomas Jefferson University, Philadelphia, PA
Address: Amber Tully, MD, Family and Community Medicine, Thomas Jefferson University Hospital, 1100 Walnut Street, Suite 603, Philadephia, PA 19107; e-mail [email protected]
A 23-year-old man presents with a sore throat, dysphagia, and general malaise that began 1 week ago. He also reports a 5-pound weight loss. He has not recently taken antibiotics or inhaled glucocorticoids, and he has no history of tobacco use or trauma to his mouth. He has no personal or family history of oral cancer. He uses cocaine on occasion. He reports feeling feverish and having a decreased appetite.
Figure 1.
An examination of his mouth reveals white plaques of varying sizes (Figure 1). The plaques are easily removed using a tongue blade, with no bleeding. No regional lymphadenopathy is noted.
Q: Based on the history, the symptoms, and the physical examination, which of the following is the most likely diagnosis in this patient?
Oral hairy leukoplakia
Squamous cell carcinoma
Oral candidiasis
Herpetic gingivostomatitis
Streptococcal pharyngitis
A: Oral candidiasis is correct.
Otherwise known as thrush, it is common in infants and in denture wearers, and it also can occur in diabetes mellitus, antibiotic therapy, chemotherapy, radiation therapy, and cellular immune deficiency states such as cancer or human immunodeficiency virus (HIV) infection.1 Patients using inhaled glucocorticoids are also at risk and should always be advised to rinse their mouth out with water after inhaled steroid use.
Although Candida albicans is the species most often responsible for candidal infections, other candidal species are increasingly responsible for infections in immunocompromised patients. Candida is part of the normal flora in many adults.
Oral hairy leukoplakia is caused by the Epstein-Barr virus and is often seen in HIV infection. It is a white, painless, corrugated lesion, typically found on the lateral aspect of the tongue, and it cannot be scraped from the adherent surfaces. It can also be found on the dorsum of the tongue, the buccal surfaces, and the floor of the mouth. In an asymptomatic patient with oral hairy leukoplakia, HIV infection with moderate immunosuppression is most likely present.2 Oral hairy leukoplakia is diagnosed by biopsy of suspected lesions. It is not a premalignant lesion, and how to best treat it is still being investigated.3
Squamous cell carcinoma of the oral cavity can present as nonhealing ulcers or masses, dental changes, or exophytic lesions with or without pain.1 They may be accompanied by cervical nodal disease. Malignancies of the oral cavity account for 14% of all head and neck cancers, with squamous cell carcinoma the predominant type.4 Alcohol and tobacco use increase the risk. Alcohol and tobacco together have a synergistic effect on the incidence of oral carcinoma.1,4 Predisposing lesions are leukoplakia, lichen planus of the erosive subtype, submucosal fibrosis, and erythroplakia. Oral infection with human papillomavirus has been shown to increase the risk of oral cancer by a factor of 14, and papillomavirus type 16 is detected in 72% of patients with oropharyngeal cancer.5
Herpetic gingivostomatitis is a manifestation of herpes simplex virus infection. The initial infection may be asymptomatic or may produce groups of vesicles that develop into shallow, painful, and superficial ulcerations on an erythematous base.1,3 If the gingiva is involved, it is erythematous, boggy, and tender.3 Infections are self-limited, lasting up to 2 weeks, but there is potential for recurrence because of the ability of herpes simplex virus to undergo latency. Recurrence is usually heralded by prodromal symptoms 24 hours before onset, with tingling, pain, or burning at the infected site. The diagnosis can be made clinically, but the Tzanck smear test, viral culture, direct fluorescent antibody test, or polymerase chain reaction test can be used to confirm the diagnosis. In patients who are immunocompromised, infections tend to be more severe and to last longer.
Streptococcal pharyngitis, most often caused by group A beta-hemolytic streptococci, is the most common type of bacterial pharyngitis in the clinical setting. The bacteria incubate for 2 to 5 days. The condition mainly affects younger children.6 Patients with “strep throat” often present with a sore throat and high-grade fever. Other symptoms include chills, myalgia, headache, and nausea. Findings on examination may include petechiae of the palate, pharyngeal and tonsillar erythema and exudates, and anterior cervical adenopathy.6 Children often present with coinciding abdominal complaints. A rapid antigen detection test for streptococcal infection can be performed in the office for quick diagnosis, but if clinical suspicion is high, a throat culture is necessary to confirm the diagnosis. Treatment is to prevent complications such as rheumatic fever.6
FEATURES AND DIAGNOSIS OF ORAL CANDIDIASIS
Lesions of oral candidiasis can vary in their appearance. The pseudomembranous form is the most characteristic, with white adherent “cottage-cheese-like” plaques that wipe away, causing minimal bleeding.1,7 The erythematous or atrophic form is associated with denture use and causes a “beefy” appearance on the dorsum of the tongue or on the mucosa that supports a denture.1,7 A third form affects the angles of the mouth, causing angular cheilitis (perlèche).7,8 Chronic infection appears as localized, firmly adherent plaques with an irregular surface similar to hyperkeratosis caused by chronic frictional irritation.7
Oral candidiasis can occur in different forms at the same time. Patients often describe minimal symptoms such as dysgeusia or dry mouth.1,7 Infections causing dysphagia or odynophagia warrant suspicion for involvement of the esophagus.
The diagnosis is made empirically if the lesions resolve with anticandidal therapy. A more definitive diagnosis can be made by microscopy with a potassium hydroxide preparation showing pseudohyphae. Formal culture can also determine the yeast’s susceptibility to medication in recurrent or resistant cases.2
Oral candidiasis may be the manifesting symptom of HIV infection, and more than 90% of patients with adult immunodeficiency syndrome have an episode of thrush.8 When candidiasis is diagnosed without obvious cause, HIV testing should be offered, regardless of a patient’s lack of obvious risk factors. Other oral lesions in HIV patients are oral hairy leukoplakia, Kaposi sarcoma, periodontal and gingival infections, aphthous ulcers, herpes simplex stomatitis, and xerostomia.2 With highly active antiretroviral therapy, the incidence of oral candidiasis has decreased by about 50%.2
Our patient was diagnosed with HIV when screened after this initial presentation. Lower CD4 counts and higher viral loads increase the patient’s risk for oral candidiasis and other lesions. This patient’s initial CD4 count was 524 cells/μL, and his viral load was 11,232 copies/mL.
TREATMENT
In HIV-negative patients or in HIV-positive patients with a CD4 count greater than 200 cells/μL, the treatment of oral candidiasis involves topical antifungal agents, including a nystatin suspension (Nystat-Rx) or clotrimazole (Mycelex) troches.3,7,9 Treatment should be continued for at least 7 days after resolution of the infection. If resolution does not occur, oral fluconazole (Diflucan) 200 mg daily should be given.
For HIV patients with CD4 counts below 200 cells/μL, oral fluconazole or itraconazole (Sporanox) is recommended, with posaconazole (Noxafil) as an alternative for refractory disease.3,9 Giving fluconazole prophylactically to prevent oral candidiasis is not recommended because of the risk of adverse effects, lack of survival benefit, associated cost, and potential to develop antifungal resistance.3,9
A 23-year-old man presents with a sore throat, dysphagia, and general malaise that began 1 week ago. He also reports a 5-pound weight loss. He has not recently taken antibiotics or inhaled glucocorticoids, and he has no history of tobacco use or trauma to his mouth. He has no personal or family history of oral cancer. He uses cocaine on occasion. He reports feeling feverish and having a decreased appetite.
Figure 1.
An examination of his mouth reveals white plaques of varying sizes (Figure 1). The plaques are easily removed using a tongue blade, with no bleeding. No regional lymphadenopathy is noted.
Q: Based on the history, the symptoms, and the physical examination, which of the following is the most likely diagnosis in this patient?
Oral hairy leukoplakia
Squamous cell carcinoma
Oral candidiasis
Herpetic gingivostomatitis
Streptococcal pharyngitis
A: Oral candidiasis is correct.
Otherwise known as thrush, it is common in infants and in denture wearers, and it also can occur in diabetes mellitus, antibiotic therapy, chemotherapy, radiation therapy, and cellular immune deficiency states such as cancer or human immunodeficiency virus (HIV) infection.1 Patients using inhaled glucocorticoids are also at risk and should always be advised to rinse their mouth out with water after inhaled steroid use.
Although Candida albicans is the species most often responsible for candidal infections, other candidal species are increasingly responsible for infections in immunocompromised patients. Candida is part of the normal flora in many adults.
Oral hairy leukoplakia is caused by the Epstein-Barr virus and is often seen in HIV infection. It is a white, painless, corrugated lesion, typically found on the lateral aspect of the tongue, and it cannot be scraped from the adherent surfaces. It can also be found on the dorsum of the tongue, the buccal surfaces, and the floor of the mouth. In an asymptomatic patient with oral hairy leukoplakia, HIV infection with moderate immunosuppression is most likely present.2 Oral hairy leukoplakia is diagnosed by biopsy of suspected lesions. It is not a premalignant lesion, and how to best treat it is still being investigated.3
Squamous cell carcinoma of the oral cavity can present as nonhealing ulcers or masses, dental changes, or exophytic lesions with or without pain.1 They may be accompanied by cervical nodal disease. Malignancies of the oral cavity account for 14% of all head and neck cancers, with squamous cell carcinoma the predominant type.4 Alcohol and tobacco use increase the risk. Alcohol and tobacco together have a synergistic effect on the incidence of oral carcinoma.1,4 Predisposing lesions are leukoplakia, lichen planus of the erosive subtype, submucosal fibrosis, and erythroplakia. Oral infection with human papillomavirus has been shown to increase the risk of oral cancer by a factor of 14, and papillomavirus type 16 is detected in 72% of patients with oropharyngeal cancer.5
Herpetic gingivostomatitis is a manifestation of herpes simplex virus infection. The initial infection may be asymptomatic or may produce groups of vesicles that develop into shallow, painful, and superficial ulcerations on an erythematous base.1,3 If the gingiva is involved, it is erythematous, boggy, and tender.3 Infections are self-limited, lasting up to 2 weeks, but there is potential for recurrence because of the ability of herpes simplex virus to undergo latency. Recurrence is usually heralded by prodromal symptoms 24 hours before onset, with tingling, pain, or burning at the infected site. The diagnosis can be made clinically, but the Tzanck smear test, viral culture, direct fluorescent antibody test, or polymerase chain reaction test can be used to confirm the diagnosis. In patients who are immunocompromised, infections tend to be more severe and to last longer.
Streptococcal pharyngitis, most often caused by group A beta-hemolytic streptococci, is the most common type of bacterial pharyngitis in the clinical setting. The bacteria incubate for 2 to 5 days. The condition mainly affects younger children.6 Patients with “strep throat” often present with a sore throat and high-grade fever. Other symptoms include chills, myalgia, headache, and nausea. Findings on examination may include petechiae of the palate, pharyngeal and tonsillar erythema and exudates, and anterior cervical adenopathy.6 Children often present with coinciding abdominal complaints. A rapid antigen detection test for streptococcal infection can be performed in the office for quick diagnosis, but if clinical suspicion is high, a throat culture is necessary to confirm the diagnosis. Treatment is to prevent complications such as rheumatic fever.6
FEATURES AND DIAGNOSIS OF ORAL CANDIDIASIS
Lesions of oral candidiasis can vary in their appearance. The pseudomembranous form is the most characteristic, with white adherent “cottage-cheese-like” plaques that wipe away, causing minimal bleeding.1,7 The erythematous or atrophic form is associated with denture use and causes a “beefy” appearance on the dorsum of the tongue or on the mucosa that supports a denture.1,7 A third form affects the angles of the mouth, causing angular cheilitis (perlèche).7,8 Chronic infection appears as localized, firmly adherent plaques with an irregular surface similar to hyperkeratosis caused by chronic frictional irritation.7
Oral candidiasis can occur in different forms at the same time. Patients often describe minimal symptoms such as dysgeusia or dry mouth.1,7 Infections causing dysphagia or odynophagia warrant suspicion for involvement of the esophagus.
The diagnosis is made empirically if the lesions resolve with anticandidal therapy. A more definitive diagnosis can be made by microscopy with a potassium hydroxide preparation showing pseudohyphae. Formal culture can also determine the yeast’s susceptibility to medication in recurrent or resistant cases.2
Oral candidiasis may be the manifesting symptom of HIV infection, and more than 90% of patients with adult immunodeficiency syndrome have an episode of thrush.8 When candidiasis is diagnosed without obvious cause, HIV testing should be offered, regardless of a patient’s lack of obvious risk factors. Other oral lesions in HIV patients are oral hairy leukoplakia, Kaposi sarcoma, periodontal and gingival infections, aphthous ulcers, herpes simplex stomatitis, and xerostomia.2 With highly active antiretroviral therapy, the incidence of oral candidiasis has decreased by about 50%.2
Our patient was diagnosed with HIV when screened after this initial presentation. Lower CD4 counts and higher viral loads increase the patient’s risk for oral candidiasis and other lesions. This patient’s initial CD4 count was 524 cells/μL, and his viral load was 11,232 copies/mL.
TREATMENT
In HIV-negative patients or in HIV-positive patients with a CD4 count greater than 200 cells/μL, the treatment of oral candidiasis involves topical antifungal agents, including a nystatin suspension (Nystat-Rx) or clotrimazole (Mycelex) troches.3,7,9 Treatment should be continued for at least 7 days after resolution of the infection. If resolution does not occur, oral fluconazole (Diflucan) 200 mg daily should be given.
For HIV patients with CD4 counts below 200 cells/μL, oral fluconazole or itraconazole (Sporanox) is recommended, with posaconazole (Noxafil) as an alternative for refractory disease.3,9 Giving fluconazole prophylactically to prevent oral candidiasis is not recommended because of the risk of adverse effects, lack of survival benefit, associated cost, and potential to develop antifungal resistance.3,9
References
Reichart PA. Clinical management of selected oral fungal and viral infections during HIV-disease. Int Dent J1999; 49:251–259.
Kim TB, Pletcher SD, Goldberg AN. Head and neck manifestations in the immunocompromised host. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:209–229(225–226).
Sciubba JJ. Oral mucosal lesions. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1229–1231).
Wein R. Malignant Neoplasms of the Oral Cavity. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1236).
D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med2007; 356:1944–1956.
Hayes CS, Williamson H. Management of group A betahemolytic streptococcal pharyngitis. Am Fam Physician2001; 63:1557–1564.
Coleman GC. Diseases of the mouth. In:Bope ET, Rakel RE, Kellerman R, editors. Conn’s Current Therapy. Philadelphia, PA: Saunders; 2010:861–867.
Habif TP. Candidiasis (moniliasis). In: Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh: Mosby; 2010:523–536.
Pappas PG, Rex JH, Sobel JD, et al; Infectious Diseases Society of America. Guidelines for treatment of candidiasis. Clin Infect Dis2004; 38:161–189.
References
Reichart PA. Clinical management of selected oral fungal and viral infections during HIV-disease. Int Dent J1999; 49:251–259.
Kim TB, Pletcher SD, Goldberg AN. Head and neck manifestations in the immunocompromised host. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:209–229(225–226).
Sciubba JJ. Oral mucosal lesions. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1229–1231).
Wein R. Malignant Neoplasms of the Oral Cavity. In:Flint PW, Haughey BH, Lund VJ, et al, editors. Cummings Otolaryngology: Head and Neck Surgery. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:1222–1244(1236).
D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med2007; 356:1944–1956.
Hayes CS, Williamson H. Management of group A betahemolytic streptococcal pharyngitis. Am Fam Physician2001; 63:1557–1564.
Coleman GC. Diseases of the mouth. In:Bope ET, Rakel RE, Kellerman R, editors. Conn’s Current Therapy. Philadelphia, PA: Saunders; 2010:861–867.
Habif TP. Candidiasis (moniliasis). In: Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh: Mosby; 2010:523–536.
Pappas PG, Rex JH, Sobel JD, et al; Infectious Diseases Society of America. Guidelines for treatment of candidiasis. Clin Infect Dis2004; 38:161–189.
A 72-year-old man with a 15-year history of a heart murmur presents to his cardiologist with shortness of breath on exertion over the past 12 months. He otherwise feels well and reports no chest discomfort, palpitations, or swelling of his legs or feet. He is not taking any cardiac drugs, and his health has previously been excellent.
Figure 1. The patient’s 12-lead electrocardiogram shows normal sinus rhythm and a rate of 55 beats per minute. The frontal-plane QRS complex vector is deviated leftward. Left atrial abnormality is present, given the terminally negative P wave in lead V1 and the bifid P wave in lead II. Left ventricular volume overload is supported by the following findings: increased QRS complex voltage, best seen in the precordial (chest) leads, indicative of increased left ventricular mass; prominent septal depolarization, as reflected by Q waves in leads V4 to V6; the absence of an ST-segment or T-wave abnormality; and negative U waves in leads V4 to V6 (arrows).
On physical examination, his pulse rate is regular at 55 beats per minute, and his blood pressure is 178/66 mm Hg. Cardiac auscultation reveals an early- to mid-diastolic murmur heard best with the patient seated and leaning forward, and located at the left sternal border; the murmur is consistent with aortic regurgitation. Standard 12-lead electrocardiography (ECG) is performed (Figure 1), followed shortly by transthoracic echocardiography, which confirms severe aortic regurgitation, moderate left ventricular hypertrophy, an enlarged left ventricular end-diastolic diameter, and normal left ventricular systolic function, with an estimated left ventricular ejection fraction of 60%. Coronary angiography shows normal coronary arteries.
Q: Which of the following findings on 12-lead ECG is not commonly reported in chronic severe aortic regurgitation?
Left ventricular hypertrophy
QRS complex left-axis deviation
A negative U wave
Atrial fibrillation
A: The correct answer is a negative U wave.
In long-standing left ventricular volume overload, such as in chronic aortic regurgitation, characteristic findings on ECG include lateral precordial narrow Q waves and left ventricular hypertrophy. The ST segment and T wave are often normal or nearly normal. The QRS complex vector may demonstrate left-axis deviation, but this is not absolute. In contrast, pressure overload conditions such as aortic stenosis and systemic hypertension commonly manifest as left ventricular hypertrophy with strain pattern of ST depression in lateral precordial leads and asymmetric T-wave inversion.
A negative U wave, best identified in leads V4 to V6, is a common finding in left ventricular volume overload. A negative U wave represents a negative deflection of small amplitude (normally < 0.1 to 3 mV) immediately following the T wave. Although not routinely reported, the negative U wave is an indicator of underlying structural heart disease.1
Q: A negative U wave has been associated with which of the following conditions?
Aortic or mitral regurgitation
Myocardial ischemia
Hypertension
All of the above
A: The correct answer is all of the above.
Negative U waves have been identified in regurgitant valvular heart disease with left ventricular volume overload, in myocardial ischemia, 2,3 and in hypertension.4 During exercise stress testing, the transient appearance of negative U waves strongly suggests flow-limiting coronary artery disease. Moreover, changes in the U wave during exercise stress testing may be a sign of well-developed coronary collaterals.5 Therefore, it is prudent to note their presence on resting ECG and to investigate further with cardiac stress testing and imaging.
The pathogenesis of the negative U wave remains unclear. Of the various hypotheses put forth, a mechano-electric phenomenon may best explain its diverse pathology.
References
Correale E, Battista R, Ricciardiello V, Martone A. The negative U wave: a pathogenetic enigma but a useful, often overlooked bedside diagnostic and prognostic clue in ischemic heart disease. Clin Cardiol2004; 27:674–677.
Rimmerman CM. A 62-year-old man with an abnormal electrocardiogram. Cleve Clin J Med2001; 68:975–976.
Gerson MC, Phillips JF, Morris SN, McHenry PL. Exercise-induced U-wave inversion as a marker of stenosis of the left anterior descending coronary artery. Circulation1979; 60:1014–1020.
Lambert J. Clinical study of the abnormalities of the terminal complex TU-U of the electrocardiogram. Circulation1957; 15:102–104.
Miwa K, Nakagawa K, Hirai T, Inoue H. Exercise-induced U-wave alterations as a marker of well-developed and well-functioning collateral vessels in patients with effort angina. J Am Coll Cardiol2000; 35:757–763.
A 72-year-old man with a 15-year history of a heart murmur presents to his cardiologist with shortness of breath on exertion over the past 12 months. He otherwise feels well and reports no chest discomfort, palpitations, or swelling of his legs or feet. He is not taking any cardiac drugs, and his health has previously been excellent.
Figure 1. The patient’s 12-lead electrocardiogram shows normal sinus rhythm and a rate of 55 beats per minute. The frontal-plane QRS complex vector is deviated leftward. Left atrial abnormality is present, given the terminally negative P wave in lead V1 and the bifid P wave in lead II. Left ventricular volume overload is supported by the following findings: increased QRS complex voltage, best seen in the precordial (chest) leads, indicative of increased left ventricular mass; prominent septal depolarization, as reflected by Q waves in leads V4 to V6; the absence of an ST-segment or T-wave abnormality; and negative U waves in leads V4 to V6 (arrows).
On physical examination, his pulse rate is regular at 55 beats per minute, and his blood pressure is 178/66 mm Hg. Cardiac auscultation reveals an early- to mid-diastolic murmur heard best with the patient seated and leaning forward, and located at the left sternal border; the murmur is consistent with aortic regurgitation. Standard 12-lead electrocardiography (ECG) is performed (Figure 1), followed shortly by transthoracic echocardiography, which confirms severe aortic regurgitation, moderate left ventricular hypertrophy, an enlarged left ventricular end-diastolic diameter, and normal left ventricular systolic function, with an estimated left ventricular ejection fraction of 60%. Coronary angiography shows normal coronary arteries.
Q: Which of the following findings on 12-lead ECG is not commonly reported in chronic severe aortic regurgitation?
Left ventricular hypertrophy
QRS complex left-axis deviation
A negative U wave
Atrial fibrillation
A: The correct answer is a negative U wave.
In long-standing left ventricular volume overload, such as in chronic aortic regurgitation, characteristic findings on ECG include lateral precordial narrow Q waves and left ventricular hypertrophy. The ST segment and T wave are often normal or nearly normal. The QRS complex vector may demonstrate left-axis deviation, but this is not absolute. In contrast, pressure overload conditions such as aortic stenosis and systemic hypertension commonly manifest as left ventricular hypertrophy with strain pattern of ST depression in lateral precordial leads and asymmetric T-wave inversion.
A negative U wave, best identified in leads V4 to V6, is a common finding in left ventricular volume overload. A negative U wave represents a negative deflection of small amplitude (normally < 0.1 to 3 mV) immediately following the T wave. Although not routinely reported, the negative U wave is an indicator of underlying structural heart disease.1
Q: A negative U wave has been associated with which of the following conditions?
Aortic or mitral regurgitation
Myocardial ischemia
Hypertension
All of the above
A: The correct answer is all of the above.
Negative U waves have been identified in regurgitant valvular heart disease with left ventricular volume overload, in myocardial ischemia, 2,3 and in hypertension.4 During exercise stress testing, the transient appearance of negative U waves strongly suggests flow-limiting coronary artery disease. Moreover, changes in the U wave during exercise stress testing may be a sign of well-developed coronary collaterals.5 Therefore, it is prudent to note their presence on resting ECG and to investigate further with cardiac stress testing and imaging.
The pathogenesis of the negative U wave remains unclear. Of the various hypotheses put forth, a mechano-electric phenomenon may best explain its diverse pathology.
A 72-year-old man with a 15-year history of a heart murmur presents to his cardiologist with shortness of breath on exertion over the past 12 months. He otherwise feels well and reports no chest discomfort, palpitations, or swelling of his legs or feet. He is not taking any cardiac drugs, and his health has previously been excellent.
Figure 1. The patient’s 12-lead electrocardiogram shows normal sinus rhythm and a rate of 55 beats per minute. The frontal-plane QRS complex vector is deviated leftward. Left atrial abnormality is present, given the terminally negative P wave in lead V1 and the bifid P wave in lead II. Left ventricular volume overload is supported by the following findings: increased QRS complex voltage, best seen in the precordial (chest) leads, indicative of increased left ventricular mass; prominent septal depolarization, as reflected by Q waves in leads V4 to V6; the absence of an ST-segment or T-wave abnormality; and negative U waves in leads V4 to V6 (arrows).
On physical examination, his pulse rate is regular at 55 beats per minute, and his blood pressure is 178/66 mm Hg. Cardiac auscultation reveals an early- to mid-diastolic murmur heard best with the patient seated and leaning forward, and located at the left sternal border; the murmur is consistent with aortic regurgitation. Standard 12-lead electrocardiography (ECG) is performed (Figure 1), followed shortly by transthoracic echocardiography, which confirms severe aortic regurgitation, moderate left ventricular hypertrophy, an enlarged left ventricular end-diastolic diameter, and normal left ventricular systolic function, with an estimated left ventricular ejection fraction of 60%. Coronary angiography shows normal coronary arteries.
Q: Which of the following findings on 12-lead ECG is not commonly reported in chronic severe aortic regurgitation?
Left ventricular hypertrophy
QRS complex left-axis deviation
A negative U wave
Atrial fibrillation
A: The correct answer is a negative U wave.
In long-standing left ventricular volume overload, such as in chronic aortic regurgitation, characteristic findings on ECG include lateral precordial narrow Q waves and left ventricular hypertrophy. The ST segment and T wave are often normal or nearly normal. The QRS complex vector may demonstrate left-axis deviation, but this is not absolute. In contrast, pressure overload conditions such as aortic stenosis and systemic hypertension commonly manifest as left ventricular hypertrophy with strain pattern of ST depression in lateral precordial leads and asymmetric T-wave inversion.
A negative U wave, best identified in leads V4 to V6, is a common finding in left ventricular volume overload. A negative U wave represents a negative deflection of small amplitude (normally < 0.1 to 3 mV) immediately following the T wave. Although not routinely reported, the negative U wave is an indicator of underlying structural heart disease.1
Q: A negative U wave has been associated with which of the following conditions?
Aortic or mitral regurgitation
Myocardial ischemia
Hypertension
All of the above
A: The correct answer is all of the above.
Negative U waves have been identified in regurgitant valvular heart disease with left ventricular volume overload, in myocardial ischemia, 2,3 and in hypertension.4 During exercise stress testing, the transient appearance of negative U waves strongly suggests flow-limiting coronary artery disease. Moreover, changes in the U wave during exercise stress testing may be a sign of well-developed coronary collaterals.5 Therefore, it is prudent to note their presence on resting ECG and to investigate further with cardiac stress testing and imaging.
The pathogenesis of the negative U wave remains unclear. Of the various hypotheses put forth, a mechano-electric phenomenon may best explain its diverse pathology.
References
Correale E, Battista R, Ricciardiello V, Martone A. The negative U wave: a pathogenetic enigma but a useful, often overlooked bedside diagnostic and prognostic clue in ischemic heart disease. Clin Cardiol2004; 27:674–677.
Rimmerman CM. A 62-year-old man with an abnormal electrocardiogram. Cleve Clin J Med2001; 68:975–976.
Gerson MC, Phillips JF, Morris SN, McHenry PL. Exercise-induced U-wave inversion as a marker of stenosis of the left anterior descending coronary artery. Circulation1979; 60:1014–1020.
Lambert J. Clinical study of the abnormalities of the terminal complex TU-U of the electrocardiogram. Circulation1957; 15:102–104.
Miwa K, Nakagawa K, Hirai T, Inoue H. Exercise-induced U-wave alterations as a marker of well-developed and well-functioning collateral vessels in patients with effort angina. J Am Coll Cardiol2000; 35:757–763.
References
Correale E, Battista R, Ricciardiello V, Martone A. The negative U wave: a pathogenetic enigma but a useful, often overlooked bedside diagnostic and prognostic clue in ischemic heart disease. Clin Cardiol2004; 27:674–677.
Rimmerman CM. A 62-year-old man with an abnormal electrocardiogram. Cleve Clin J Med2001; 68:975–976.
Gerson MC, Phillips JF, Morris SN, McHenry PL. Exercise-induced U-wave inversion as a marker of stenosis of the left anterior descending coronary artery. Circulation1979; 60:1014–1020.
Lambert J. Clinical study of the abnormalities of the terminal complex TU-U of the electrocardiogram. Circulation1957; 15:102–104.
Miwa K, Nakagawa K, Hirai T, Inoue H. Exercise-induced U-wave alterations as a marker of well-developed and well-functioning collateral vessels in patients with effort angina. J Am Coll Cardiol2000; 35:757–763.
A 54-year-old woman with hepatitis C virus infection presents with generalized rash, pruritus, and fever over the past week. The rash appeared on her left arm after she received her fifth weekly injection of pegylated interferon alfa 2b, in combination with daily oral ribavirin (Copegus, Rebetol). Over the course of 3 days, it spread to her face and the rest of her body.
Figure 1. Desquamation began on day 7 after the rash first appeared.
She has no other known medical conditions, has no history of eczema or atopy, and is not taking any other drugs.
Figure 2. She developed vesiculobullae on her lower extremities, including the left medial thigh. No vesiculobullae were noted on the upper extremities.
A diffuse erythematous macular rash now covers most of her body, with areas of desquamation (Figure 1). The rash spares her mucous membranes (oral cavity, genitalia, eyes). In addition, there are scattered vesiculobullae (Figures 2 and 3) and nonblanching purpuric lesions on the front of her legs.
Figure 3. Punch biopsy samples were taken from her left leg. The black patch on the right leg was from a prior skin graft, unrelated to the current presentation.
Her total white blood cell count is newly elevated at 18.5 × 109/L (reference range 4.0–11.0), and the differential count is “shifted to the left,” with 83% neutrophils (reference range 50%–75%) but no eosinophils. The C-reactive protein level and erythrocyte sedimentation rate are elevated: the C-reactive protein is 1.1 mg/dL (reference range 0.0–1.0), and the sedimentation rate is 70 mm/ hour (reference range for women 0–15). All other laboratory results, including aminotransferase and alkaline phosphatase levels, electrolyte levels, and coagulation studies, are normal. Additional tests for immunoglobulin A (IgA), IgM, IgG, complements C3 and C4, rheumatoid factor, antinuclear antibodies, and cryoglobulins are normal. Chest radiography is normal.
Q: What is the most likely clinical diagnosis?
Stevens-Johnson syndrome
Mixed cryoglobulinemia
Acute eczematous drug eruption
Lichen planus
A: Acute eczematous drug eruption is the most likely diagnosis.
The clinical presentation and laboratory findings suggest (the latter by exclusion) that our patient had an allergic drug reaction to the interferon or to the ribavirin therapy, or to both. Although this combination is a standard treatment for chronic hepatitis C, some patients experience adverse reactions that lead to its discontinuation. Local injection-site reactions are the most prevalent, affecting up to 12% of patients, whereas eczematous dermatoses manifest less commonly, occurring in up to 5% of patients.1
While awaiting the results of skin biopsy, a careful evaluation of the clinical features of the physical examination and an appropriate laboratory evaluation can rule out other important conditions in the differential diagnosis.
The absence of mucous membrane involvement steers the diagnosis away from Stevens-Johnson syndrome, a life-threatening hypersensitivity condition often triggered by drugs, malignant tumors, and viral infections, which may also affect internal organs. In this condition, skin biopsy specimens would be distinguished by subepidermal bullae and epidermal cell necrosis—neither of which was seen in our patient.
Mixed cryoglobulinemia should always be considered in hepatitis C patients because of the strong association between this infection and the development of cryoglobulins. The rash usually is purpuric, but it may be pleomorphic.2,3 This vasculitis often manifests with excess cryoglobulins, elevated rheumatoid factor, and low titers of complement in the blood due to consumption by immune complexes. Tissue biopsy would usually show typical vascular changes if performed on fresh lesions.4,5 The normal levels of these components in our patient coupled with the appearance of her skin makes cryoglobulinemia a less likely cause.
Furthermore, hepatitis C infection, whether or not treated with interferon and ribavirin, can cause an onset or recurrence of other dermatologic conditions, notably lichen planus, psoriasis, vitiligo, and systemic lupus erythematosus.1–4
In lichen planus, the rash is often described as flat-topped, pruritic, and violaceous. It may involve the extremities, the genitalia, and the oral cavity.4,5 The difference in quality of the rash compared with the rash in our patient makes lichen planus less likely.
Exclusion of the other conditions in the differential diagnosis, in addition to results from a definitive punch biopsy, solidified the diagnosis in our patient. Skin biopsy of the patient’s lower-extremity lesions revealed spongiotic dermatitis with lymphocytes, neutrophils, and few eosinophils—a finding characteristic of an acute eczematous drug eruption. Improvement of her rash after discontinuation of interferon and ribavirin further supported this conclusion, although it was unclear whether one or both agents were responsible.
OUTCOME
Management of acute eczematous drug eruption entails stopping the offending drug and alleviating the symptoms. Our patient’s non-life-threatening rash improved dramatically with cessation of interferon and ribavirin. She received a single dose of a systemic corticosteroid initially, out of concern for a severe medication-induced reaction (ie, Stevens-Johnson syndrome), but she was otherwise maintained with diphenhydramine (Benadryl) and a multivitamin ointment for the rash throughout her 9-day hospital stay. Her pruritus was well controlled with hydroxyzine (Atarax, Vistaril). At discharge, she was referred back to her hepatologist for further treatment of her hepatitis C, possibly with interferon and ribavirin again.
TAKE-HOME MESSAGE
Adverse reactions to interferon and ribavirin treatment in hepatitis C patients can manifest dermatologically, and the combination therapy should be discontinued to prevent further insult. A broad variety of conditions in the differential diagnosis should be taken into account, but dermatologic conditions that occur or recur specifically in hepatitis C patients should be considered as well.
References
Dereure O, Raison-Peyron N, Larrey D, Blanc F, Guilhou JJ. Diffuse inflammatory lesions in patients treated with interferon alfa and ribavirin for hepatitis C: a series of 20 patients. Br J Dermatol2002; 147:1142–1146.
Faurie P, Broussolle C, Zoulim F, Trepo C, Sève P. Sarcoidosis and hepatitis C: clinical description of 11 cases. Eur J Gastroenterol Hepatol2010; 22:967–972.
Shengyuan L, Songpo Y, Wen W, Wenjing T, Haitao Z, Binyou W. Hepatitis C virus and lichen planus: a reciprocal association determined by a meta-analysis. Arch Dermatol2009; 145:1040–1047.
Aamir S, Ullah Z, Iqbal Z, Khan AA, Yaqub F, Malik K. Cutaneous manifestations of interferon alfa and ribavirin for hepatitis C. J Pak Assoc Dermatol2008; 18:14–20.
Derek M. Tang, MD Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Lawrence Ward, MD, MPH Associate Professor of Medicine, Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Address: Derek M. Tang, MD, Department of Medicine, Temple University School of Medicine, 3401 North Broad Street, Parkinson Pavilion 812, Philadelphia, PA 19140; e-mail [email protected]
Derek M. Tang, MD Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Lawrence Ward, MD, MPH Associate Professor of Medicine, Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Address: Derek M. Tang, MD, Department of Medicine, Temple University School of Medicine, 3401 North Broad Street, Parkinson Pavilion 812, Philadelphia, PA 19140; e-mail [email protected]
Author and Disclosure Information
Derek M. Tang, MD Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Lawrence Ward, MD, MPH Associate Professor of Medicine, Department of Medicine, Temple University School of Medicine, Philadelphia, PA
Address: Derek M. Tang, MD, Department of Medicine, Temple University School of Medicine, 3401 North Broad Street, Parkinson Pavilion 812, Philadelphia, PA 19140; e-mail [email protected]
A 54-year-old woman with hepatitis C virus infection presents with generalized rash, pruritus, and fever over the past week. The rash appeared on her left arm after she received her fifth weekly injection of pegylated interferon alfa 2b, in combination with daily oral ribavirin (Copegus, Rebetol). Over the course of 3 days, it spread to her face and the rest of her body.
Figure 1. Desquamation began on day 7 after the rash first appeared.
She has no other known medical conditions, has no history of eczema or atopy, and is not taking any other drugs.
Figure 2. She developed vesiculobullae on her lower extremities, including the left medial thigh. No vesiculobullae were noted on the upper extremities.
A diffuse erythematous macular rash now covers most of her body, with areas of desquamation (Figure 1). The rash spares her mucous membranes (oral cavity, genitalia, eyes). In addition, there are scattered vesiculobullae (Figures 2 and 3) and nonblanching purpuric lesions on the front of her legs.
Figure 3. Punch biopsy samples were taken from her left leg. The black patch on the right leg was from a prior skin graft, unrelated to the current presentation.
Her total white blood cell count is newly elevated at 18.5 × 109/L (reference range 4.0–11.0), and the differential count is “shifted to the left,” with 83% neutrophils (reference range 50%–75%) but no eosinophils. The C-reactive protein level and erythrocyte sedimentation rate are elevated: the C-reactive protein is 1.1 mg/dL (reference range 0.0–1.0), and the sedimentation rate is 70 mm/ hour (reference range for women 0–15). All other laboratory results, including aminotransferase and alkaline phosphatase levels, electrolyte levels, and coagulation studies, are normal. Additional tests for immunoglobulin A (IgA), IgM, IgG, complements C3 and C4, rheumatoid factor, antinuclear antibodies, and cryoglobulins are normal. Chest radiography is normal.
Q: What is the most likely clinical diagnosis?
Stevens-Johnson syndrome
Mixed cryoglobulinemia
Acute eczematous drug eruption
Lichen planus
A: Acute eczematous drug eruption is the most likely diagnosis.
The clinical presentation and laboratory findings suggest (the latter by exclusion) that our patient had an allergic drug reaction to the interferon or to the ribavirin therapy, or to both. Although this combination is a standard treatment for chronic hepatitis C, some patients experience adverse reactions that lead to its discontinuation. Local injection-site reactions are the most prevalent, affecting up to 12% of patients, whereas eczematous dermatoses manifest less commonly, occurring in up to 5% of patients.1
While awaiting the results of skin biopsy, a careful evaluation of the clinical features of the physical examination and an appropriate laboratory evaluation can rule out other important conditions in the differential diagnosis.
The absence of mucous membrane involvement steers the diagnosis away from Stevens-Johnson syndrome, a life-threatening hypersensitivity condition often triggered by drugs, malignant tumors, and viral infections, which may also affect internal organs. In this condition, skin biopsy specimens would be distinguished by subepidermal bullae and epidermal cell necrosis—neither of which was seen in our patient.
Mixed cryoglobulinemia should always be considered in hepatitis C patients because of the strong association between this infection and the development of cryoglobulins. The rash usually is purpuric, but it may be pleomorphic.2,3 This vasculitis often manifests with excess cryoglobulins, elevated rheumatoid factor, and low titers of complement in the blood due to consumption by immune complexes. Tissue biopsy would usually show typical vascular changes if performed on fresh lesions.4,5 The normal levels of these components in our patient coupled with the appearance of her skin makes cryoglobulinemia a less likely cause.
Furthermore, hepatitis C infection, whether or not treated with interferon and ribavirin, can cause an onset or recurrence of other dermatologic conditions, notably lichen planus, psoriasis, vitiligo, and systemic lupus erythematosus.1–4
In lichen planus, the rash is often described as flat-topped, pruritic, and violaceous. It may involve the extremities, the genitalia, and the oral cavity.4,5 The difference in quality of the rash compared with the rash in our patient makes lichen planus less likely.
Exclusion of the other conditions in the differential diagnosis, in addition to results from a definitive punch biopsy, solidified the diagnosis in our patient. Skin biopsy of the patient’s lower-extremity lesions revealed spongiotic dermatitis with lymphocytes, neutrophils, and few eosinophils—a finding characteristic of an acute eczematous drug eruption. Improvement of her rash after discontinuation of interferon and ribavirin further supported this conclusion, although it was unclear whether one or both agents were responsible.
OUTCOME
Management of acute eczematous drug eruption entails stopping the offending drug and alleviating the symptoms. Our patient’s non-life-threatening rash improved dramatically with cessation of interferon and ribavirin. She received a single dose of a systemic corticosteroid initially, out of concern for a severe medication-induced reaction (ie, Stevens-Johnson syndrome), but she was otherwise maintained with diphenhydramine (Benadryl) and a multivitamin ointment for the rash throughout her 9-day hospital stay. Her pruritus was well controlled with hydroxyzine (Atarax, Vistaril). At discharge, she was referred back to her hepatologist for further treatment of her hepatitis C, possibly with interferon and ribavirin again.
TAKE-HOME MESSAGE
Adverse reactions to interferon and ribavirin treatment in hepatitis C patients can manifest dermatologically, and the combination therapy should be discontinued to prevent further insult. A broad variety of conditions in the differential diagnosis should be taken into account, but dermatologic conditions that occur or recur specifically in hepatitis C patients should be considered as well.
A 54-year-old woman with hepatitis C virus infection presents with generalized rash, pruritus, and fever over the past week. The rash appeared on her left arm after she received her fifth weekly injection of pegylated interferon alfa 2b, in combination with daily oral ribavirin (Copegus, Rebetol). Over the course of 3 days, it spread to her face and the rest of her body.
Figure 1. Desquamation began on day 7 after the rash first appeared.
She has no other known medical conditions, has no history of eczema or atopy, and is not taking any other drugs.
Figure 2. She developed vesiculobullae on her lower extremities, including the left medial thigh. No vesiculobullae were noted on the upper extremities.
A diffuse erythematous macular rash now covers most of her body, with areas of desquamation (Figure 1). The rash spares her mucous membranes (oral cavity, genitalia, eyes). In addition, there are scattered vesiculobullae (Figures 2 and 3) and nonblanching purpuric lesions on the front of her legs.
Figure 3. Punch biopsy samples were taken from her left leg. The black patch on the right leg was from a prior skin graft, unrelated to the current presentation.
Her total white blood cell count is newly elevated at 18.5 × 109/L (reference range 4.0–11.0), and the differential count is “shifted to the left,” with 83% neutrophils (reference range 50%–75%) but no eosinophils. The C-reactive protein level and erythrocyte sedimentation rate are elevated: the C-reactive protein is 1.1 mg/dL (reference range 0.0–1.0), and the sedimentation rate is 70 mm/ hour (reference range for women 0–15). All other laboratory results, including aminotransferase and alkaline phosphatase levels, electrolyte levels, and coagulation studies, are normal. Additional tests for immunoglobulin A (IgA), IgM, IgG, complements C3 and C4, rheumatoid factor, antinuclear antibodies, and cryoglobulins are normal. Chest radiography is normal.
Q: What is the most likely clinical diagnosis?
Stevens-Johnson syndrome
Mixed cryoglobulinemia
Acute eczematous drug eruption
Lichen planus
A: Acute eczematous drug eruption is the most likely diagnosis.
The clinical presentation and laboratory findings suggest (the latter by exclusion) that our patient had an allergic drug reaction to the interferon or to the ribavirin therapy, or to both. Although this combination is a standard treatment for chronic hepatitis C, some patients experience adverse reactions that lead to its discontinuation. Local injection-site reactions are the most prevalent, affecting up to 12% of patients, whereas eczematous dermatoses manifest less commonly, occurring in up to 5% of patients.1
While awaiting the results of skin biopsy, a careful evaluation of the clinical features of the physical examination and an appropriate laboratory evaluation can rule out other important conditions in the differential diagnosis.
The absence of mucous membrane involvement steers the diagnosis away from Stevens-Johnson syndrome, a life-threatening hypersensitivity condition often triggered by drugs, malignant tumors, and viral infections, which may also affect internal organs. In this condition, skin biopsy specimens would be distinguished by subepidermal bullae and epidermal cell necrosis—neither of which was seen in our patient.
Mixed cryoglobulinemia should always be considered in hepatitis C patients because of the strong association between this infection and the development of cryoglobulins. The rash usually is purpuric, but it may be pleomorphic.2,3 This vasculitis often manifests with excess cryoglobulins, elevated rheumatoid factor, and low titers of complement in the blood due to consumption by immune complexes. Tissue biopsy would usually show typical vascular changes if performed on fresh lesions.4,5 The normal levels of these components in our patient coupled with the appearance of her skin makes cryoglobulinemia a less likely cause.
Furthermore, hepatitis C infection, whether or not treated with interferon and ribavirin, can cause an onset or recurrence of other dermatologic conditions, notably lichen planus, psoriasis, vitiligo, and systemic lupus erythematosus.1–4
In lichen planus, the rash is often described as flat-topped, pruritic, and violaceous. It may involve the extremities, the genitalia, and the oral cavity.4,5 The difference in quality of the rash compared with the rash in our patient makes lichen planus less likely.
Exclusion of the other conditions in the differential diagnosis, in addition to results from a definitive punch biopsy, solidified the diagnosis in our patient. Skin biopsy of the patient’s lower-extremity lesions revealed spongiotic dermatitis with lymphocytes, neutrophils, and few eosinophils—a finding characteristic of an acute eczematous drug eruption. Improvement of her rash after discontinuation of interferon and ribavirin further supported this conclusion, although it was unclear whether one or both agents were responsible.
OUTCOME
Management of acute eczematous drug eruption entails stopping the offending drug and alleviating the symptoms. Our patient’s non-life-threatening rash improved dramatically with cessation of interferon and ribavirin. She received a single dose of a systemic corticosteroid initially, out of concern for a severe medication-induced reaction (ie, Stevens-Johnson syndrome), but she was otherwise maintained with diphenhydramine (Benadryl) and a multivitamin ointment for the rash throughout her 9-day hospital stay. Her pruritus was well controlled with hydroxyzine (Atarax, Vistaril). At discharge, she was referred back to her hepatologist for further treatment of her hepatitis C, possibly with interferon and ribavirin again.
TAKE-HOME MESSAGE
Adverse reactions to interferon and ribavirin treatment in hepatitis C patients can manifest dermatologically, and the combination therapy should be discontinued to prevent further insult. A broad variety of conditions in the differential diagnosis should be taken into account, but dermatologic conditions that occur or recur specifically in hepatitis C patients should be considered as well.
References
Dereure O, Raison-Peyron N, Larrey D, Blanc F, Guilhou JJ. Diffuse inflammatory lesions in patients treated with interferon alfa and ribavirin for hepatitis C: a series of 20 patients. Br J Dermatol2002; 147:1142–1146.
Faurie P, Broussolle C, Zoulim F, Trepo C, Sève P. Sarcoidosis and hepatitis C: clinical description of 11 cases. Eur J Gastroenterol Hepatol2010; 22:967–972.
Shengyuan L, Songpo Y, Wen W, Wenjing T, Haitao Z, Binyou W. Hepatitis C virus and lichen planus: a reciprocal association determined by a meta-analysis. Arch Dermatol2009; 145:1040–1047.
Aamir S, Ullah Z, Iqbal Z, Khan AA, Yaqub F, Malik K. Cutaneous manifestations of interferon alfa and ribavirin for hepatitis C. J Pak Assoc Dermatol2008; 18:14–20.
References
Dereure O, Raison-Peyron N, Larrey D, Blanc F, Guilhou JJ. Diffuse inflammatory lesions in patients treated with interferon alfa and ribavirin for hepatitis C: a series of 20 patients. Br J Dermatol2002; 147:1142–1146.
Faurie P, Broussolle C, Zoulim F, Trepo C, Sève P. Sarcoidosis and hepatitis C: clinical description of 11 cases. Eur J Gastroenterol Hepatol2010; 22:967–972.
Shengyuan L, Songpo Y, Wen W, Wenjing T, Haitao Z, Binyou W. Hepatitis C virus and lichen planus: a reciprocal association determined by a meta-analysis. Arch Dermatol2009; 145:1040–1047.
Aamir S, Ullah Z, Iqbal Z, Khan AA, Yaqub F, Malik K. Cutaneous manifestations of interferon alfa and ribavirin for hepatitis C. J Pak Assoc Dermatol2008; 18:14–20.
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
Figure 1. Abdominal computed tomography (coronal view) shows a subphrenic abscess (arrows).
Computed tomography (CT) (Figure 1 and Figure 2) showed a right subphrenic air-fluid collection (17 cm by 7 cm) and branching air lucencies within the liver and gallbladder fossa, signs of pneumobilia and pneumoperitoneum. The subphrenic fluid collection was drained percutaneously under fluoroscopic guidance, yielding 25 mL of purulent material. Culture of this material showed mixed flora. The percutaneous drainage catheter was left in place.
Figure 2. Pneumobilia (white arrow) and pneumoperitoneum (red arrow) are evident on this abdominal computed tomographic image (axial view).
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
Figure 3. Upper gastroduodenography with oral contrast shows contrast leaking through the perforated duodenal ulcer (arrow).
We treated him with cefepime (Maxipime), metronidazole (Flagyl), fluconazole (Diflucan), a proton pump inhibitor, and octreotide (Sandostatin) intravenously. His condition stabilized, and 2 weeks later repeat CT showed no leakage, and the abscess had resolved. He was gradually returned to a normal diet and was discharged home with oral antibiotics.
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
References
Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery2004; 135:321–325.
Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg1989; 124:830–832.
Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg1990; 56:52–56.
Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol1986; 147:1237–1240.
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
Figure 1. Abdominal computed tomography (coronal view) shows a subphrenic abscess (arrows).
Computed tomography (CT) (Figure 1 and Figure 2) showed a right subphrenic air-fluid collection (17 cm by 7 cm) and branching air lucencies within the liver and gallbladder fossa, signs of pneumobilia and pneumoperitoneum. The subphrenic fluid collection was drained percutaneously under fluoroscopic guidance, yielding 25 mL of purulent material. Culture of this material showed mixed flora. The percutaneous drainage catheter was left in place.
Figure 2. Pneumobilia (white arrow) and pneumoperitoneum (red arrow) are evident on this abdominal computed tomographic image (axial view).
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
Figure 3. Upper gastroduodenography with oral contrast shows contrast leaking through the perforated duodenal ulcer (arrow).
We treated him with cefepime (Maxipime), metronidazole (Flagyl), fluconazole (Diflucan), a proton pump inhibitor, and octreotide (Sandostatin) intravenously. His condition stabilized, and 2 weeks later repeat CT showed no leakage, and the abscess had resolved. He was gradually returned to a normal diet and was discharged home with oral antibiotics.
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
A 55-year-old man presented after 3 weeks of sharp epigastric pain radiating to the right upper quadrant, fever, and generalized weakness. He had a history of significant heroin and cocaine abuse and was currently in a methadone maintenance program. He was not taking any nonsteroidal anti-inflammatory drugs.
His temperature was 38.7°C (101.7°F). He had tenderness in the right upper quadrant but no rebound tenderness.
Laboratory studies revealed an elevated white blood cell count of 14.9 × 109/L (reference range 4.5–11.0) with 13% band cells, a normal lipase level, and normal liver function tests. Urine toxicology testing was positive for cocaine.
Figure 1. Abdominal computed tomography (coronal view) shows a subphrenic abscess (arrows).
Computed tomography (CT) (Figure 1 and Figure 2) showed a right subphrenic air-fluid collection (17 cm by 7 cm) and branching air lucencies within the liver and gallbladder fossa, signs of pneumobilia and pneumoperitoneum. The subphrenic fluid collection was drained percutaneously under fluoroscopic guidance, yielding 25 mL of purulent material. Culture of this material showed mixed flora. The percutaneous drainage catheter was left in place.
Figure 2. Pneumobilia (white arrow) and pneumoperitoneum (red arrow) are evident on this abdominal computed tomographic image (axial view).
To investigate the cause of the abscess, we obtained an upper gastrointestinal radiographic series with oral contrast. This showed leaking of contrast from a perforated ulcer in the proximal duodenum (Figure 3).
Figure 3. Upper gastroduodenography with oral contrast shows contrast leaking through the perforated duodenal ulcer (arrow).
We treated him with cefepime (Maxipime), metronidazole (Flagyl), fluconazole (Diflucan), a proton pump inhibitor, and octreotide (Sandostatin) intravenously. His condition stabilized, and 2 weeks later repeat CT showed no leakage, and the abscess had resolved. He was gradually returned to a normal diet and was discharged home with oral antibiotics.
TREATING PERFORATED ULCER AND ITS COMPLICATIONS
Perforated ulcers usually present with an acute abdomen and are life-threatening unless immediately recognized and treated. A more insidious presentation, as reported by Wong et al1 and as seen in our patient, presents additional diagnostic challenges. Our patient’s presentation may have been insidious and relatively benign because the perforation was not a free perforation (ie, the bowel contents were not freely spilling into the abdominal cavity), and because the spillage and inflammation were confined to an abscess cavity. Also, he was taking methadone 60 mg per day, which may have further masked the symptoms.
Perforated duodenal ulcers are usually treated surgically. Nonoperative management—ie, fluid resuscitation, nasogastric tube aspiration, and intravenous antibiotics—has been described,2 but the hemodynamic status and abdominal findings need to be continually monitored to detect any deterioration in the patient’s condition.
Initial management of the abscess with early percutaneous drainage and empiric intravenous antibiotics to cover enteric flora and anaerobes may suffice if there is no fistula.3 Also, in a series of 62 patients with subphrenic abscess managed by percutaneous drainage, Mueller et al4 found that this method was successful in most of the patients, noting that small-bowel perforation required the longest drainage period, generally more than 10 days.4
Helicobacter pylori infection and nonsteroidal anti-inflammatory drugs are responsible for the vast majority of duodenal ulcers. Serum H pylori antibody testing should be done, and if it is positive, treatment should be started. This patient did not have H pylori infection as an inpatient. He received metronidazole to cover the anaerobes and a proton pump inhibitor to treat the ulcer.
Although upper endoscopy was contraindicated in our patient because of his perforation, it was warranted for follow-up. Medical management with a proton pump inhibitor in high doses helped ulcer healing by reducing acid and gastric secretions. Octreotide had similar benefits, including reducing pancreatic and biliary secretion. This conservative management resulted in healing of the ulcer and early closure of the enteric fistula.
References
Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery2004; 135:321–325.
Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg1989; 124:830–832.
Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg1990; 56:52–56.
Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol1986; 147:1237–1240.
References
Wong CH, Chow PK, Ong HS, Chan WH, Khin LW, Soo KC. Posterior perforation of peptic ulcers: presentation and outcome of an uncommon surgical emergency. Surgery2004; 135:321–325.
Berne TV, Donovan AJ. Nonoperative treatment of perforated duodenal ulcer. Arch Surg1989; 124:830–832.
Flancbaum L, Nosher JL, Brolin RE. Percutaneous catheter drainage of abdominal abscesses associated with perforated viscus. Am Surg1990; 56:52–56.
Mueller PR, Simeone JF, Butch RJ, et al. Percutaneous drainage of subphrenic abscess: a review of 62 patients. AJR Am J Roentgenol1986; 147:1237–1240.
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Giulia Rech, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Riccardo Balestri, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Federico Bardazzi, MD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Bianca Maria Piraccini, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
Annalisa Patrizi, MD, PhD Internal Medicine, Aging and Nephrologic Disease Department, Dermatology Division, Ospedale Sant’Orsola-Malpighi, Università degli Studi di Bologna, Italy
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
A 52-year-old woman is referred to our dermatology clinic by her primary care physician for swelling and redness of seven old scars. The swelling began 3 months ago.
She is on no regular medications. She has never smoked. She underwent liposuction 7 years ago and appendectomy at age 15.
Figure 1. Indurated and painful brownish-red plaques localized on old scars: (A) lower trunk, (B) left side, (C) right side.
Physical examination reveals indurated and painful brownish-red plaques on the thighs and lower trunk, extending beyond the borders of six scars from the liposuction surgery and one scar from the appendectomy (Figure 1). She reports no dyspnea, night sweats, weight loss, fever, or other constitutional symptoms, but she has a dry cough that began 2 months after the onset of the skin symptoms. Her primary care physician has managed the cough symptomatically.
Laboratory testing shows mild leukopenia, with a white blood cell count of 3.0 × 109/L (reference range 4.2–9.0). Other routine laboratory values are normal, including antinuclear antibody, extractable nuclear antibody, anti-double-stranded DNA, rheumatoid factor, urinalysis, erythrocyte sedimentation rate, C-reactive protein, serum calcium concentration, and liver and renal function tests.
Q: What is the next most appropriate diagnostic procedure?
Skin biopsy
High-resolution computed tomography (CT) of the chest
QuantiFERON-TB Gold test
Ventilatory function tests
Serum angiotensin-converting enzyme (ACE) level
A: All are appropriate at this point. In this case, skin biopsy and high-resolution CT were performed. Histopathologic examination of one of the scars showed multiple well-demarcated, large, noncaseating epitheloid granulomas with histiocytes and multinucleated giant cells. High-resolution CT confirmed bilateral hilar and mediastinal lymphadenopathy and revealed micronodular densities with a bronchovascular and subpleural distribution.
An interferon-gamma-release assay for tuberculosis—QuantiFERON-TB Gold (Cellestis, Carnegie, Australia)—was negative. Ventilatory function tests showed a normal pattern, while the serum ACE level, electrocardiography, and an eye examination revealed no pathologic findings.
Q: What is the diagnosis?
Keloids
Scar sarcoidosis
Paraneoplastic sign
Dermatofibrosarcoma protuberans
Tubercolosis
A: Based on the data outlined above, we made the diagnosis of scar sarcoidosis with involvement of hilar and mediastinal lymph nodes. The patient began systemic treatment with oral prednisone 1 mg/kg/day for 6 weeks, which was then gradually withdrawn, until the skin and hilar lesions resolved completely.
SCAR SARCOIDOSIS
Sarcoidosis is a multisystem disorder of unknown cause characterized by the formation of noncaseating granulomas in the affected organs. Patients may present with symptoms related to the specific organ affected, but they may have no symptoms or only general symptoms such as fever or general malaise.
The skin is involved in 25% of cases and presents so many polymorphous manifestations that sarcoidosis has become known as one of the “great imitators” in dermatology.1,2
Although sarcoidosis on liposuction scars has not been reported previously, the reactivation of old scars is well known on sites of previous injections, tattoos, herpes zoster, and burns.2,3
Figure 2. Noncaseating granuloma seen on skin biopsy study.
The finding of granuloma is not specific for sarcoidosis (Figure 2). The histologic differential diagnosis of sarcoidosis includes tuberculosis, atypical mycobacteriosis, fungal infection, reaction to a foreign body, rheumatoid nodules, leishmaniasis, Crohn disease, and necrobiosis lipoidica diabeticorum.
The diagnosis of scar sarcoidosis is confirmed only by excluding other conditions via a comprehensive evaluation of clinical manifestations, histology, history, and radiologic and laboratory findings.
It has been suggested that the most satisfying therapy for the patient and physician in sarcoidosis is no treatment at all,4 and in fact sarcoidosis often remits spontaneously. Currently, the choice of treatment depends on the degree of systemic involvement, and the oral corticosteroid prednisone remains the first-line treatment. If the condition does not respond, the use of other systemic agents has been reported, but their effectiveness has not been evaluated in controlled clinical trials.
Recurrence is common after the suspension of treatment; therefore, treatment may need to be continued for several years, with frequent checkups.
Skin lesions are a visible clue to the diagnosis. Reactivation of old scars may be the single manifestation of cutaneous sarcoidosis, but it may also precede or accompany systemic involvement, often representing the main sign of an exacerbation or a relapse of systemic sarcoidosis, as in our patient.5
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
Tchernev G. Cutaneous sarcoidosis: the “great imitator”: etiopathogenesis, morphology, differential diagnosis, and clinical management. Am J Clin Dermatol2006; 7:375–382.
Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol2007; 25:276–287.
Baughman RP, Lower EE, du Bois RM. Sarcoidosis. Lancet2003; 361:1111–1118.
Sorabjee JS, Garje R. Reactivation of old scars: inevitably sarcoid. Postgrad Med J2005; 81:60–61.
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
References
Freitag DS, Chin R. Facial granulomas with nasal destruction. Chest1988; 93:422–423.
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail [email protected]
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail [email protected]
Author and Disclosure Information
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debarbrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, 700004 Kolkata, India; e-mail [email protected]
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
A 12-year-old boy presents with painless swelling and ulceration on and around his nose that has progressed gradually over the last 6 months. The lesion has increased in size despite treatment with topical neomycin and oral erythromycin. He has no systemic symptoms.
Figure 1. Ulcerated plaque with destruction of the right nasal wing.
On examination (Figure 1), we note an indurated, nontender plaque with scarring at places on his right cheek, nose, and the vermilion border of the lip. In addition, there are two purulent ulcerations on the nose partly destroying the right nasal wing. The upper lip is also infiltrated, studded with a solitary ulceration. There is no regional lymphadenopathy. An examination of systems is normal.
Cutaneous tuberculosis occurs in many forms, and lupus vulgaris is one of the most common.1 Lupus vulgaris usually arises as a result of hematogenous spread from an endogenous source. It may also arise from exogenous inoculation or as a complication of vaccination with bacille Calmette-Guérin.2
Several morphologic variants have been described.1,2 One form is characterized by plaques, often studded with psoriasiform scales. Large plaques may show irregular areas of scarring with islands of active lupus tissue and a thickened and hyperkeratotic margin. Ulcerative and mutilating variants of lupus vulgaris are characterized by scarring, ulceration, crusts over areas of necrosis, and destruction of the deep tissues and cartilage, resulting in deformities. The vegetative form produces marked infiltration, ulceration, and necrosis, with minimal scarring. Mucous membranes and cartilages are often destroyed. Tumor-like hypertrophic lesions and multiple papular and nodular lesions may also be seen. Nasal lesions may start as nodules, which may bleed and then ulcerate, sometimes resulting in cartilage destruction.
CLINICAL FEATURES AND LABORATORY WORKUP CLINCHED THE DIAGNOSIS
A number of factors helped to confirm the diagnosis in this patient:
A strongly positive Mantoux test (22-mm induration at 48 hours)
Acid-fast bacilli on Ziehl-Neelsen staining of the smear taken from the purulent ulceration
Isolation of Mycobacterium tuberculosis from the purulent exudates via culture in Lowenstein-Jensen medium
Figure 2. Epithelioid cell granuloma and giant cells (hematoxylin and eosin, × 100).
A suggestive histopathologic picture (Figure 2)
The features on presentation
A significant clinical improvement within 2 months of starting antituberculosis therapy.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis includes all the conditions in the question above. However, the absence of respiratory and renal involvement helps rule out Wegener granulomatosis; the absence of impaired sensation and nerve thickening helps rule out Hansen disease; and the absence of a nasal septal defect helps rule out Wegener granulomatosis, midline lethal granuloma, and Hansen disease.
On the other hand, the lupoid form of cutaneous leishmaniasis usually presents as an erythematous, infiltrated plaque that often closely resembles lupus vulgaris, but these lesions are usually less destructive than lupus vulgaris. However, the laboratory workup including the microbiological and histopathologic examination clearly excluded the other potential diagnoses in this patient.
TREATMENT
Lupus vulgaris is treated with standard antituberculosis therapy.3 The first phase of a fourdrug regimen is given for 2 months—isoniazid, rifampin (Rifadin), pyrazinamide, and ethambutol (Myambutol). The second phase consists of isoniazid and rifampin for 4 months.3
Early recognition and confirmation of the diagnosis followed by treatment are of immense importance for preventing permanent disfigurement.
References
Freitag DS, Chin R. Facial granulomas with nasal destruction. Chest1988; 93:422–423.