User login
Dyspnea, arthralgias, and muscle weakness
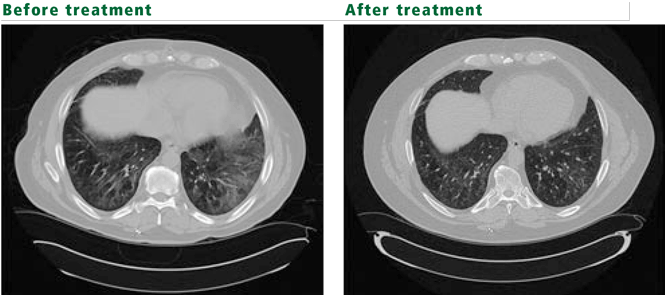
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
Q: Which condition is most likely?
- Rheumatoid arthritis with pulmonary involvement
- Hypertrophic pulmonary osteoarthropathy
- Polymyositis-dermatomyositis with pulmonary involvement
- Systemic lupus erythematosus with pulmonary involvement
A: The patient’s symptoms and physical findings suggest polymyositis-dermatomyositis with associated interstitial lung disease.
Rheumatoid arthritis can also cause lung disease and proximal myopathy, but early physical findings in the hands would include symmetrical joint effusions and soft tissue swelling of the metacarpophalangeal joints.
Patients with hypertrophic pulmonary osteoarthropathy present with arthralgias without weakness. Radiographic findings such as osteophytosis and tufting of terminal processes in the hands would support its diagnosis.
A small number of patients with systemic lupus erythematosus develop deforming arthritis with hand involvement that is either erosive (rhupus hand) or nonerosive (Jaccoud arthropathy, or lupus hand), but interstitial lung disease is rare in lupus, making this combination unlikely.
MULTIPLE PATHS TO DIAGNOSIS
Physical examination, review of systems, laboratory screening, radiographic findings, lung biopsy, electromyography, and muscle biopsy may be used in conjunction.
The criteria of Bohan and Peter are often used to diagnose polymyositis-dermatomyositis: symmetric proximal muscle weakness, elevated muscle enzymes, electromyographic changes consistent with myopathy, and compatible histologic findings on muscle biopsy, with or without the characteristic dermatologic manifestations.1,2 However, the diagnosis can be made in the typical clinical setting on the basis of characteristic levels of anti-Jo-1 antinuclear antibody and elevated serum muscle enzyme.
Depending on the criteria used, the incidence of interstitial lung disease in various studies of polymyositis-dermatomyositis ranged from 5% to 46%.3 Pulmonary involvement can present in one of three forms:
- Sudden onset of dyspnea and fever with alveolar infiltrates on chest radiography and ground-glass opacities on high-resolution chest CT
- Progressive dyspnea with radiographic findings of chronic interstitial lung disease
- No clinical symptoms, but with abnormal findings on chest radiography.4
In a minority of patients, lung disease precedes the onset of muscle or skin disease. Much more commonly, patients present with skin and muscle involvement first. In these patients, pulmonary involvement is typically seen 2 to 5 years after the diagnosis.2 Patients with interstitial lung disease are more likely to have arthralgias and arthritis than are those without lung involvement. Interestingly, the finding of microangiopathy on nail fold capillaroscopy strongly suggests pulmonary disease.3
Laboratory findings
Creatine kinase elevation is a marker of disease activity in the muscles. Aldolase, aspartate aminotransferase, and alanine aminotransferase levels may also be elevated but are not muscle-specific. Anti-Jo-1 antinuclear antibody is characteristic, although it can be negative in some patients.
Pulmonary function testing
Restrictive lung physiology with impaired diffusing capacity is the predominant pattern noted.
Lung biopsy findings
Polymyositis-dermatomyositis-associated interstitial lung disease is not limited to one histologic pattern. Nonspecific interstitial pneumonitis is the most common, but usual interstitial pneumonia, organizing pneumonia, and diffuse alveolar damage are also described.2 Patients with nonspecific interstitial pneumonitis and organizing pneumonia are suspected to have a better response to immunosuppression and better survival, although controlled studies are absent.
CT appearance
Polymyositis-dermatomyositis complicated by interstitial lung disease does not have a distinct appearance on high-resolution CT. However, the radiographic changes in most cases will suggest the underlying pathology, and this can be used to guide therapy. Most common are bibasilar subpleural ground-glass and reticular opacities that curiously spare the immediate 1 to 2 mm of subpleural parenchyma.1,3 This pattern is very suggestive of fibrotic nonspecific interstitial pneumonitis. Patchy consolidations with air bronchograms suggest organizing pneumonia. Bibasilar, subpleural honeycomb cystic changes and traction bronchiectasis are noted in usual interstitial pneumonia and suggest fibrosis, which will not improve with therapy. In patients whose disease is progressive, the areas of consolidation often evolve into honeycomb cystic changes.2,4
TREATMENT IS WITH STEROIDS AND OTHER IMMUNOSUPPRESSIVES
Oral corticosteroids in dosages of 0.5 to 1 mg/kg are first-line therapy. Clinically, muscle disease often improves before lung disease, and treatment may be extended to several months. The histologic pattern suggested by CT or by pathologic study of surgical lung biopsy specimens is a better predictor of treatment response than clinical presentation. Nonspecific interstitial pneumonitis and organizing pneumonia have the highest steroid response rates.3 However, many patients do not respond to steroids alone—only 44% in one study.5 Furthermore, treatment response does not indicate recovery, as the disease may relapse.
The addition of immunosuppressive therapy with cyclophosphamide (Cytoxan) may halt deterioration in patients with polymyositis-dermatomyositis-associated interstitial lung disease who are steroid-resistant or may be useful as a steroid-sparing agent in recurrent disease after initial steroid withdrawal. In some cases, therapy with cyclophosphamide improved oxygenation and led to resolution of abnormalities on chest radiography.6,7
Azathioprine (Imuran), methotrexate, and hydroxychloroquine (Plaquenil) have all been used as part of a steroid-sparing regimen.1 Tacrolimus (FK 506; Prograf) and rituximab (Rituxan) are emerging therapies, especially for patients who cannot tolerate cytotoxic immunosuppressive agents or who progress despite them.8,9
PROGNOSIS IS WORSE IF LUNG DISEASE IS PRESENT
The presence of interstitial lung disease increases the risk of death in polymyositis-dermatomyositis. Additionally, clinicians must assess interstitial lung disease separately from muscle or skin disease, as there does not have to be correlation between the activity in the separate organs. Fortunately, the treatment for lung, muscle, and skin involvement is often the same.
Several elements suggest poor prognosis. An acute and aggressive presentation often heralds a poor outcome.1 Neutrophil alveolitis on bronchoalveolar lavage and a very low diffusing capacity (< 45%) have both been associated with a poorer prognosis.3 The histologic pattern not only predicts treatment response but also prognosis. Patients whose lung biopsies reveal nonspecific interstitial pneumonitis or organizing pneumonia have a better outcome than do patients with usual interstitial pneumonia or diffuse alveolar damage.
In one study,1 36 patients with polymyositis-dermatomyositis and interstitial lung disease were followed for 5 years. Resolution was noted in 19.4%, improvement in 55.6%, and deterioration in 25%. Overall, the survival rate was 86.5% at 5 years, and the death rate attributable to pulmonary complications was 13.9% in patients with interstitial lung disease.1
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
- Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001; 164:1182–1185.
- Marie I, Hatron PY, Hachulla E, Wallaert B, Michon-Pasturel U, Devulder B. Pulmonary involvement in polymyositis and in dermatomyositis. J Rheumatol 1998; 25:1336–1343.
- Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47:614–622.
- Akira M, Hara H, Sakatani M. Interstitial lung disease in association with polymyositis-dermatomyositis: long-term follow-up CT evaluation in seven patients. Radiology 1999; 210:333–338.
- Nawata Y, Kurasawa K, Takabayashi K, et al. Corticosteroid resistant interstitial pneumonitis in dermatomyositis/polymyositis: prediction and treatment with cyclosporine. J Rheumatol 1999; 26:1527–1533.
- Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arthritis Rheum 1998; 41:1215–1220.
- Shinohara T, Hidaka T, Matsuki Y, et al. Rapidly progressive interstitial lung disease associated with dermatomyositis responding to intravenous cyclophosphamide pulse therapy. Intern Med 1997; 36:519–523.
- Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52:2439–2446.
- Ytterberg SR. Treatment of refractory polymyositis and dermatomyositis. Curr Rheumatol Rep 2006; 8:167–173.
Generalized pruritus after a beach vacation
A 25-year-old man presents with a 2-month history of generalized itching that began 5 weeks after returning from a trip to a beach in Brazil, during which he had sexual relations without protection. He has already been treated with topical steroids and antihistamines, with no improvement.
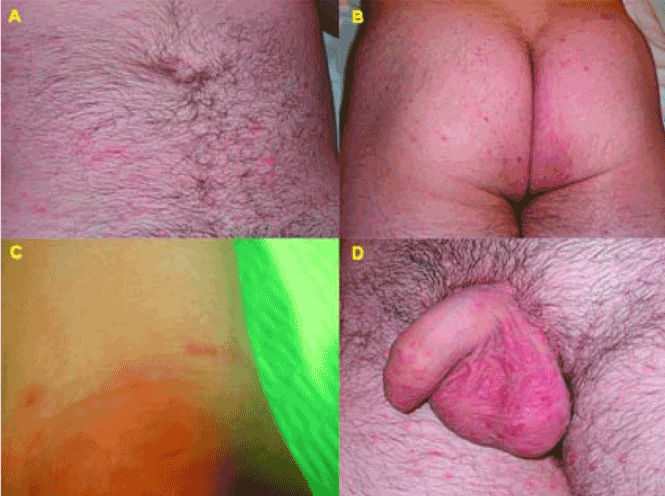
Q: What is the most likely diagnosis?
- Herpes simplex type 2
- Acute trypanosomiasis
- Syphilis
- Scabies
- Papular urticaria
A: The most likely diagnosis is scabies, an intensely pruritic skin infestation caused by the host-specific mite Sarcoptes scabiei var hominis.1 Herpes simplex type 2 infection presents with clustered vesicles, not nodules, on an erythematous base in the genital area, without generalized pruritus. Acute trypanosomiasis involves malaise, fever, vomiting, diarrhea, anorexia, rash, tachycardia, and even generalized lymphadenopathy and meningeal irritation, but generalized itching is not typical. The location and morphology of the patient’s lesions are not consistent with syphilis. Topical steroids and oral antihistamines should have improved the pruritus of papular urticaria.
A GLOBAL PUBLIC HEALTH PROBLEM
Scabies is a worldwide public health problem, affecting people of all ages, races, and socioeconomic groups.1 Overcrowding, delayed diagnosis and treatment, and poor public education contribute to the prevalence of scabies in both industrialized and nonindustrialized nations.2 Prevalence rates are higher in children and people who are sexually active.3
Sexual transmission is by close skin-to-skin contact. Poor sensory perception in conditions such as leprosy and compromised immunity due to organ transplantation, human immunodeficiency virus infection, or old age increase the risk for the crusted variant of scabies.2 Patients with the crusted variant tend to present with clinically atypical lesions, and because of this they are often misdiag-nosed, thus delaying treatment and elevating the risk of local epidemics.
CLINICAL ASPECTS
Scabies can mimic a broad range of skin diseases. Patients present with intense itching that is worse at night. The face and neck are rarely affected. The pathognomonic signs of scabies are burrows, erythematous papules, and generalized pruritus (also on non-infested skin) with nocturnal predominance.3 Reddish to brownish extremely pruritic nodules of 2 to 20 mm in diameter may be also present on the genitalia (more commonly in males than in females), buttocks, groin, and axillary regions. Patients usually have secondary papules, pustules, vesicles, and excoriations.
DIAGNOSIS
Every patient with intense pruritus should be suspected of having scabies, but especially if a family member reports similar symptoms.3 A diagnosis can be made clinically if a burrow is detected at a typical predilection site and if the lesion itches severely. In this case, even a single burrow is pathognomonic.2 The diagnosis is confirmed by light-microscopic identification of mites, larvae, ova, or scybala (fecal pellets) in skin scrapings.1
TREATMENT
Treatment includes a scabicidal agent, an antipruritic agent such as a sedating antihistamine, and an appropriate antimicrobial agent in cases of secondary infection. Permethrin (Acticin), a 5% synthetic pyrethroid cream, is an excellent scabicide and is the preferred treatment.1 All family members and close contacts must be evaluated and treated, even if they do not have symptoms.1
- Chosidow O. Clinical practices. Scabies. N Engl J Med 2006; 354:1718–1727.
- Heukelbach J, Feldmeier H. Scabies. Lancet 2006; 367:1767–1774.
- Hengge UR, Currie BJ, Jäger G, Lupi O, Schwartz RA. Scabies: a ubiquitous neglected skin disease. Lancet Infect Dis 2006; 6:769–779.
A 25-year-old man presents with a 2-month history of generalized itching that began 5 weeks after returning from a trip to a beach in Brazil, during which he had sexual relations without protection. He has already been treated with topical steroids and antihistamines, with no improvement.
Q: What is the most likely diagnosis?
- Herpes simplex type 2
- Acute trypanosomiasis
- Syphilis
- Scabies
- Papular urticaria
A: The most likely diagnosis is scabies, an intensely pruritic skin infestation caused by the host-specific mite Sarcoptes scabiei var hominis.1 Herpes simplex type 2 infection presents with clustered vesicles, not nodules, on an erythematous base in the genital area, without generalized pruritus. Acute trypanosomiasis involves malaise, fever, vomiting, diarrhea, anorexia, rash, tachycardia, and even generalized lymphadenopathy and meningeal irritation, but generalized itching is not typical. The location and morphology of the patient’s lesions are not consistent with syphilis. Topical steroids and oral antihistamines should have improved the pruritus of papular urticaria.
A GLOBAL PUBLIC HEALTH PROBLEM
Scabies is a worldwide public health problem, affecting people of all ages, races, and socioeconomic groups.1 Overcrowding, delayed diagnosis and treatment, and poor public education contribute to the prevalence of scabies in both industrialized and nonindustrialized nations.2 Prevalence rates are higher in children and people who are sexually active.3
Sexual transmission is by close skin-to-skin contact. Poor sensory perception in conditions such as leprosy and compromised immunity due to organ transplantation, human immunodeficiency virus infection, or old age increase the risk for the crusted variant of scabies.2 Patients with the crusted variant tend to present with clinically atypical lesions, and because of this they are often misdiag-nosed, thus delaying treatment and elevating the risk of local epidemics.
CLINICAL ASPECTS
Scabies can mimic a broad range of skin diseases. Patients present with intense itching that is worse at night. The face and neck are rarely affected. The pathognomonic signs of scabies are burrows, erythematous papules, and generalized pruritus (also on non-infested skin) with nocturnal predominance.3 Reddish to brownish extremely pruritic nodules of 2 to 20 mm in diameter may be also present on the genitalia (more commonly in males than in females), buttocks, groin, and axillary regions. Patients usually have secondary papules, pustules, vesicles, and excoriations.
DIAGNOSIS
Every patient with intense pruritus should be suspected of having scabies, but especially if a family member reports similar symptoms.3 A diagnosis can be made clinically if a burrow is detected at a typical predilection site and if the lesion itches severely. In this case, even a single burrow is pathognomonic.2 The diagnosis is confirmed by light-microscopic identification of mites, larvae, ova, or scybala (fecal pellets) in skin scrapings.1
TREATMENT
Treatment includes a scabicidal agent, an antipruritic agent such as a sedating antihistamine, and an appropriate antimicrobial agent in cases of secondary infection. Permethrin (Acticin), a 5% synthetic pyrethroid cream, is an excellent scabicide and is the preferred treatment.1 All family members and close contacts must be evaluated and treated, even if they do not have symptoms.1
A 25-year-old man presents with a 2-month history of generalized itching that began 5 weeks after returning from a trip to a beach in Brazil, during which he had sexual relations without protection. He has already been treated with topical steroids and antihistamines, with no improvement.
Q: What is the most likely diagnosis?
- Herpes simplex type 2
- Acute trypanosomiasis
- Syphilis
- Scabies
- Papular urticaria
A: The most likely diagnosis is scabies, an intensely pruritic skin infestation caused by the host-specific mite Sarcoptes scabiei var hominis.1 Herpes simplex type 2 infection presents with clustered vesicles, not nodules, on an erythematous base in the genital area, without generalized pruritus. Acute trypanosomiasis involves malaise, fever, vomiting, diarrhea, anorexia, rash, tachycardia, and even generalized lymphadenopathy and meningeal irritation, but generalized itching is not typical. The location and morphology of the patient’s lesions are not consistent with syphilis. Topical steroids and oral antihistamines should have improved the pruritus of papular urticaria.
A GLOBAL PUBLIC HEALTH PROBLEM
Scabies is a worldwide public health problem, affecting people of all ages, races, and socioeconomic groups.1 Overcrowding, delayed diagnosis and treatment, and poor public education contribute to the prevalence of scabies in both industrialized and nonindustrialized nations.2 Prevalence rates are higher in children and people who are sexually active.3
Sexual transmission is by close skin-to-skin contact. Poor sensory perception in conditions such as leprosy and compromised immunity due to organ transplantation, human immunodeficiency virus infection, or old age increase the risk for the crusted variant of scabies.2 Patients with the crusted variant tend to present with clinically atypical lesions, and because of this they are often misdiag-nosed, thus delaying treatment and elevating the risk of local epidemics.
CLINICAL ASPECTS
Scabies can mimic a broad range of skin diseases. Patients present with intense itching that is worse at night. The face and neck are rarely affected. The pathognomonic signs of scabies are burrows, erythematous papules, and generalized pruritus (also on non-infested skin) with nocturnal predominance.3 Reddish to brownish extremely pruritic nodules of 2 to 20 mm in diameter may be also present on the genitalia (more commonly in males than in females), buttocks, groin, and axillary regions. Patients usually have secondary papules, pustules, vesicles, and excoriations.
DIAGNOSIS
Every patient with intense pruritus should be suspected of having scabies, but especially if a family member reports similar symptoms.3 A diagnosis can be made clinically if a burrow is detected at a typical predilection site and if the lesion itches severely. In this case, even a single burrow is pathognomonic.2 The diagnosis is confirmed by light-microscopic identification of mites, larvae, ova, or scybala (fecal pellets) in skin scrapings.1
TREATMENT
Treatment includes a scabicidal agent, an antipruritic agent such as a sedating antihistamine, and an appropriate antimicrobial agent in cases of secondary infection. Permethrin (Acticin), a 5% synthetic pyrethroid cream, is an excellent scabicide and is the preferred treatment.1 All family members and close contacts must be evaluated and treated, even if they do not have symptoms.1
- Chosidow O. Clinical practices. Scabies. N Engl J Med 2006; 354:1718–1727.
- Heukelbach J, Feldmeier H. Scabies. Lancet 2006; 367:1767–1774.
- Hengge UR, Currie BJ, Jäger G, Lupi O, Schwartz RA. Scabies: a ubiquitous neglected skin disease. Lancet Infect Dis 2006; 6:769–779.
- Chosidow O. Clinical practices. Scabies. N Engl J Med 2006; 354:1718–1727.
- Heukelbach J, Feldmeier H. Scabies. Lancet 2006; 367:1767–1774.
- Hengge UR, Currie BJ, Jäger G, Lupi O, Schwartz RA. Scabies: a ubiquitous neglected skin disease. Lancet Infect Dis 2006; 6:769–779.
A woman with ulcerating, painful skin lesions
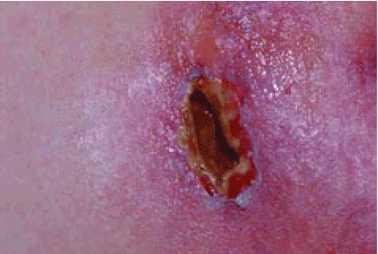
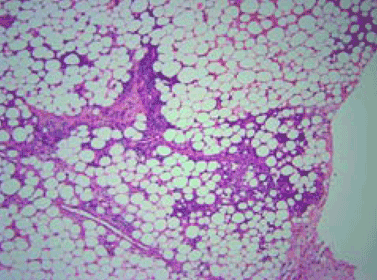
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY

Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY
Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
Q: On the basis of the skin findings, which test should be ordered to establish a diagnosis?
- Serum anti-nuclear antibody (ANA)
- Alpha-1 antitrypsin serum level
- Angiotensin-converting enzyme (ACE)
- Serum amylase
A: The lesions of a lobular, neutrophilic panniculitis should raise the possibility of alpha-1 antitrypsin deficiency. Hence, measuring the alpha-1 antitrypsin serum level is the best answer.
Many other conditions can give rise to panniculitis, including pancreatitis, lupus, and sarcoidosis, each of which is suggested by the various other test choices above. However, although the ANA level might be elevated in lupus, the ANA is quite nonspecific. The serum ACE level is frequently ordered as a screening test for sarcoidosis, although it has very little utility in its diagnosis. Panniculitis due to pancreatitis with an elevated serum amylase level would be relatively unlikely in the absence of pancreatic symptoms (eg, abdominal pain).
Panniculitis may be the only clinical manifestation of alpha-1 antitrypsin deficiency, which can also be accompanied (depending on the phenotype) by chronic obstructive pulmonary disease and cirrhosis. Since alpha-1 antitrypsin deficiency is underrecognized in general, suspecting it when patients present with panniculitis will likely enhance its detection. Similarly, national guidelines recommend testing for alpha-1 antitrypsin deficiency in patients with either symptomatic, fixed airflow obstruction or cirrhosis that is otherwise unexplained, as well as in patients with panniculitis.1
PANNICULITIS DUE TO ALPHA-1 ANTITRYPSIN DEFICIENCY
Likely due to proteolytic damage
Though incompletely understood, the panniculitis in alpha-1 antitrypsin deficiency is likely the result of unopposed proteolytic damage in the subcutaneous fat by membrane-bound serine proteases, akin to the pathogenesis of pulmonary emphysema in people with severe deficiency of alpha-1 antitrypsin. Supporting evidence for the inflammatory, proteolytic pathogenesis of panniculitis in alpha-1 anti-trypsin deficiency includes the presence of inflammatory exudates in the subcutaneous tissues, as well as the rapid improvement seen with the infusion of purified pooled human alpha-1 proteinase inhibitor.2–4
Red, painful nodules
Panniculitis due to alpha-1 antitrypsin deficiency classically presents as red, painful nodules that may break down and ooze an oily discharge.5–10 As in the patient presented here, common sites of occurrence are areas of trauma, eg, on the thighs and buttocks, abdomen, and upper extremities (Figure 1). Indeed, in a review of the 41 reported cases of panniculitis related to alpha-1 antitrypsin deficiency, Geraminejad et al11 reported that the erythe-matous plaques and nodules occurred on the thighs, hips, buttocks, or groin in 44% of cases in which the location was cited. Factors predisposing to panniculitis include trauma (cited in 35% of instances), cryosurgery, and, in the case of alpha-1 antitrypsin deficiency, extravasation of intravenous clarithromycin (Biaxin).11–13
Clinical features that distinguish the panniculitis associated with alpha-1 antitrypsin deficiency from other types of panniculitis include ulceration and an oily discharge, both of which were present in the patient discussed here.
Neutrophils, necrosis, scarring, fibrosis
Several distinctive phases and features characterize the histology of panniculitis associated with alpha-1 antitrypsin deficiency.5,7 Initially, neutrophils briskly infiltrate the reticular dermis, splaying the collagen bundles. In the subcutaneous fat, the neutrophilic infiltrate is in a lobular pattern, affecting individual adipocytes. Rarely, a septal pattern or a mixed lobular and septal pattern can be seen. This phase is followed by dissolution of the dermal collagen, with liquefactive necrosis of the subcutaneous fat (clinically appearing as ulceration and leading to oily drainage). In the late stage, there is scarring and fibrosis with little or no inflammation.
Various treatments tried
Various therapies for panniculitis associated with alpha-1 antitrypsin deficiency have been tried, including corticosteroids, doxycycline (Vibramycin), dapsone, plasma exchange, liver transplantation, and intravenous pooled human plasma alpha-1 proteinase inhibitor (so-called augmentation therapy). Because panniculitis associated with alpha-1 antitrypsin deficiency is rare, neither controlled, blinded studies nor even large observational series have been reported. However, the limited reported experience with augmentation therapy suggests that it can confer rapid and dramatic improvement in panniculitis in patients with alpha-1 antitrypsin deficiency.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Smith KC, Pittelkow MR, Su WP. Panniculitis associated with severe alpha-1 antitrypsin deficiency. Treatment and review of the literature. Arch Dermatol 1987; 123:1655–1661.
- Furey NL, Golden RS, Potts SR. Treatment of alpha-1 antitrypsin deficiency, massive edema, and panniculitis with alpha-1 protease inhibitor [letter]. Ann Intern Med 1996; 125:699.
- O’Riordan K, Blei A, Rao MS, Abecassis M. Alpha-1 antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1 antitrypsin administration and liver transplantation. Transplantation 1997; 63:480–482.
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med 2008; 15:113–117.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha-1 antitrypsin deficiency associated with panniculitis. J Am Acad Derm 1988; 18:684–692.
- Loche F, Tremeau-Martinage C, Laplanche G, Massip P, Bazex J. Panniculitis revealing qualitative alpha-1 antitrypsine deficiency (MS variant). Eur J Dermatol 1999; 9:565–567.
- McBean J, Sable A, Maude J, Robinson-Bostom L. Alpha 1-antitrypsin deficiency panniculitis. Cutis 2003; 71:205–209.
- Pittelkow MR, Smith KC, Su WP. Alpha-1 antitrypsin deficiency and panniculitis. Perspectives on disease relationship and replacement therapy. Am J Med 1988; 84:80–86.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001; 45:325–361.
- Geraminejad P, DeBloom JR, Walling HW, Sontheimer RD, VanBeek M. Alpha-1-antitrypsin associated panniculitis: the MS variant. J Am Acad Dermatol 2004; 51:645–655.
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to alpha-1 antitrypsin deficiency induced by cryosurgery [letter]. Br J Dermatol 1998; 138:552–553.
- Parr DG, Stewart DG, Hero I, Stockley RA. Panniculitis secondary to extravasation of clarithromycin in a patient with alpha 1-antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003; 149:410–413.
Acute facial purpura in an 82-year-old woman with a respiratory tract infection
An 82-year-old woman presents with facial purpuric lesions that developed while she was hospitalized for an acute respiratory tract infection characterized by severe paroxysms of nonproductive cough and dyspnea. The lesions appeared suddenly and spontaneously and were not associated with trauma. The patient denies pruritus or pain and is otherwise well. The remainder of her physical examination is within normal limits. She has hypertension, diabetes, and hyperuricemia but has had no recent changes in her medications.
Q: What is the most likely diagnosis?
- Amyloidosis
- Idiopathic thrombocytopenic purpura
- “Cough purpura”
- Purpura fulminans
- Actinic purpura
A: The correct answer is cough purpura, a benign and nonthrombocytopenic eruption that appears to be related to a sudden rise in the venous and capillary pressure in the head and neck caused by a rise in intrathoracic pressure during coughing.1–3 Physical examination shows erythematous, nonblanching macules, smaller than 1 cm, distributed on the face and neck.
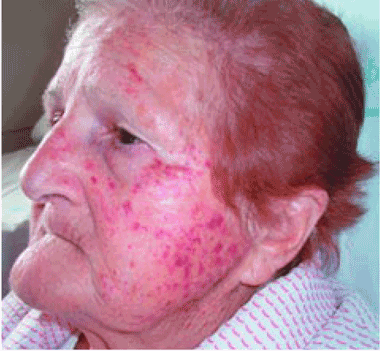
Cough purpura is one form of the “mask phenomenon,”4 which is an unusual purpura of the relatively loose tissues of the face and neck occurring after vigorous vomiting, the Valsalva maneuver, parturition, prolonged coughing, or any other exertion that raises intrathoracic or abdominal pressure.1–5 The onset is acute. A workup for a coagulation or platelet defect is usually not required. Facial localization is unusual in most other forms of purpura, so its presence in addition to coughing should suggest cough purpura.
Recognizing cough purpura can help avoid misdiagnoses such thrombocytopenic purpura or purpura fulminans that could lead to ordering unnecessary tests, frightening the patient, and unnecessary confusion. The purpura fades spontaneously within 24 to 72 hours, and no treatment is needed.
- Kravitz P. The clinical picture of “cough purpura,” benign and non-thrombocytopenic eruption. Va Med 1979; 106:373–374.
- Pierson JC, Suh PS. Powerlifter’s purpura: a Valsalva-associated phenomenon. Cutis 2002; 70:93–94.
- Santiago Sánchez-Mateos JL, Aldanondo Fernández de la Mora I, Harto Castaño A, Jaén Olasolo P. Facial-cervical purpura. Rev Clin Esp 2007; 207:530–532.
- Alcalay J, Ingber A, Sandbank M. Mask phenomenon: postemesis facial purpura. Cutis 1986; 38:28.
- Wilkin JK. Benign parturient purpura. JAMA 1978; 239:930.
An 82-year-old woman presents with facial purpuric lesions that developed while she was hospitalized for an acute respiratory tract infection characterized by severe paroxysms of nonproductive cough and dyspnea. The lesions appeared suddenly and spontaneously and were not associated with trauma. The patient denies pruritus or pain and is otherwise well. The remainder of her physical examination is within normal limits. She has hypertension, diabetes, and hyperuricemia but has had no recent changes in her medications.
Q: What is the most likely diagnosis?
- Amyloidosis
- Idiopathic thrombocytopenic purpura
- “Cough purpura”
- Purpura fulminans
- Actinic purpura
A: The correct answer is cough purpura, a benign and nonthrombocytopenic eruption that appears to be related to a sudden rise in the venous and capillary pressure in the head and neck caused by a rise in intrathoracic pressure during coughing.1–3 Physical examination shows erythematous, nonblanching macules, smaller than 1 cm, distributed on the face and neck.
Cough purpura is one form of the “mask phenomenon,”4 which is an unusual purpura of the relatively loose tissues of the face and neck occurring after vigorous vomiting, the Valsalva maneuver, parturition, prolonged coughing, or any other exertion that raises intrathoracic or abdominal pressure.1–5 The onset is acute. A workup for a coagulation or platelet defect is usually not required. Facial localization is unusual in most other forms of purpura, so its presence in addition to coughing should suggest cough purpura.
Recognizing cough purpura can help avoid misdiagnoses such thrombocytopenic purpura or purpura fulminans that could lead to ordering unnecessary tests, frightening the patient, and unnecessary confusion. The purpura fades spontaneously within 24 to 72 hours, and no treatment is needed.
An 82-year-old woman presents with facial purpuric lesions that developed while she was hospitalized for an acute respiratory tract infection characterized by severe paroxysms of nonproductive cough and dyspnea. The lesions appeared suddenly and spontaneously and were not associated with trauma. The patient denies pruritus or pain and is otherwise well. The remainder of her physical examination is within normal limits. She has hypertension, diabetes, and hyperuricemia but has had no recent changes in her medications.
Q: What is the most likely diagnosis?
- Amyloidosis
- Idiopathic thrombocytopenic purpura
- “Cough purpura”
- Purpura fulminans
- Actinic purpura
A: The correct answer is cough purpura, a benign and nonthrombocytopenic eruption that appears to be related to a sudden rise in the venous and capillary pressure in the head and neck caused by a rise in intrathoracic pressure during coughing.1–3 Physical examination shows erythematous, nonblanching macules, smaller than 1 cm, distributed on the face and neck.
Cough purpura is one form of the “mask phenomenon,”4 which is an unusual purpura of the relatively loose tissues of the face and neck occurring after vigorous vomiting, the Valsalva maneuver, parturition, prolonged coughing, or any other exertion that raises intrathoracic or abdominal pressure.1–5 The onset is acute. A workup for a coagulation or platelet defect is usually not required. Facial localization is unusual in most other forms of purpura, so its presence in addition to coughing should suggest cough purpura.
Recognizing cough purpura can help avoid misdiagnoses such thrombocytopenic purpura or purpura fulminans that could lead to ordering unnecessary tests, frightening the patient, and unnecessary confusion. The purpura fades spontaneously within 24 to 72 hours, and no treatment is needed.
- Kravitz P. The clinical picture of “cough purpura,” benign and non-thrombocytopenic eruption. Va Med 1979; 106:373–374.
- Pierson JC, Suh PS. Powerlifter’s purpura: a Valsalva-associated phenomenon. Cutis 2002; 70:93–94.
- Santiago Sánchez-Mateos JL, Aldanondo Fernández de la Mora I, Harto Castaño A, Jaén Olasolo P. Facial-cervical purpura. Rev Clin Esp 2007; 207:530–532.
- Alcalay J, Ingber A, Sandbank M. Mask phenomenon: postemesis facial purpura. Cutis 1986; 38:28.
- Wilkin JK. Benign parturient purpura. JAMA 1978; 239:930.
- Kravitz P. The clinical picture of “cough purpura,” benign and non-thrombocytopenic eruption. Va Med 1979; 106:373–374.
- Pierson JC, Suh PS. Powerlifter’s purpura: a Valsalva-associated phenomenon. Cutis 2002; 70:93–94.
- Santiago Sánchez-Mateos JL, Aldanondo Fernández de la Mora I, Harto Castaño A, Jaén Olasolo P. Facial-cervical purpura. Rev Clin Esp 2007; 207:530–532.
- Alcalay J, Ingber A, Sandbank M. Mask phenomenon: postemesis facial purpura. Cutis 1986; 38:28.
- Wilkin JK. Benign parturient purpura. JAMA 1978; 239:930.
Dropped gallstones disguised as a liver abscess
A 67-year-old retired man presents to his internist with a 3-month history of abdominal discomfort in the right upper quadrant on deep breathing. He has no other abdominal complaints, but he mentions that he underwent laparoscopic cholecystectomy 3 months ago for gallstone pancreatitis.
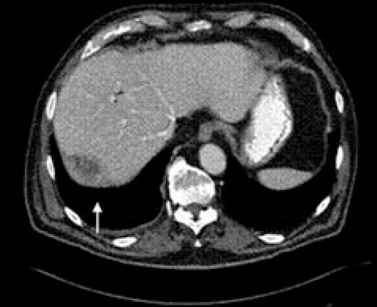
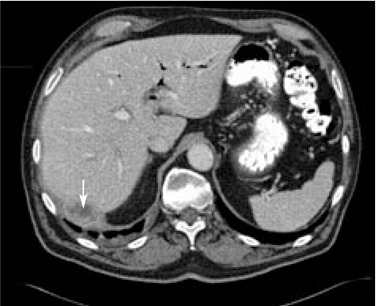
A biopsy specimen obtained with CT guidance shows chronic inflammation but is sterile on aerobic culture. There is no evidence of malignancy. Because of concern for underlying infection, the infectious disease staff recommends empirical treatment with a 4-week course of ampicillin-sulbactam (Unasyn). At completion of the antibiotic course, the patient’s symptoms have resolved.
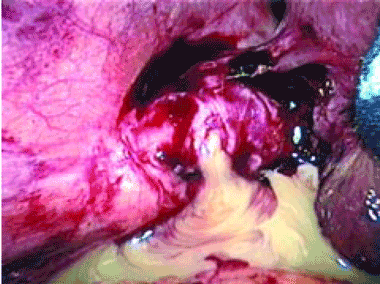
LAPAROSCOPY’S DRAWBACKS
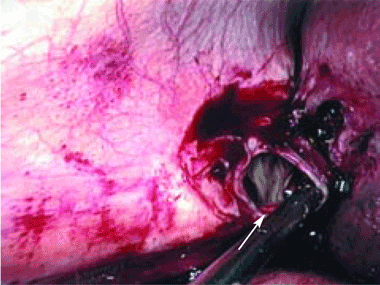
Complications of dropped stones, though rare, can include localized or systemic infection, inflammation, fibrosis, adhesion, cutaneous sinus formation, ileus, and abscess.1,6 Lohan et al1 estimated that dropped stones produce an intra-abdominal abscess in 0.6% to 2.9% of cases of dropped stones and bile spillage, based on reports by Rice et al4 and Morrin et al.7 Dropped stones should be recognized as a potential cause of intra-abdominal abscess in any cholecystectomy patient months or even years after the surgery. Also, these abscesses are not necessarily confined to the right upper quadrant: they can occur anywhere in the abdominal cavity.5,7
Given the ever-increasing popularity of laparoscopic cholecystectomy, the problem of intra-abdominal abscess due to dropped gallstones will only become a more common problem. Early diagnosis is the key to avoiding long and unnecessary treatment.
If dropped gallstones do become infected and eventually cause symptoms, they may require surgical or percutaneous removal in conjunction with antimicrobial therapy.8
- Lohan D, Walsh S, McLoughlin R, Murphy J. Imaging of the complications of laparoscopic cholecystectomy. Eur Radiol 2005; 15:904–912.
- Casillas S, Kittur DS. Late abscess formation after spilled gallstones masquerading as a liver mass. Surg Endosc 2003; 17:833.
- Tumer AR, Yuksek YN, Yasti AC, Gozalan U, Kama NA. Dropped gallstones during laparoscopic cholecystectomy: the consequences. World J Surg 2005; 29:437–440.
- Rice DC, Memon MA, Jamison RL, et al. Long-term consequences of intraoperative spillage of bile and gallstones during laparoscopic cholecystectomy. J Gastrointest Surg 1997; 1:85–91.
- Sathesh-Kumar T, Saklani AP, Vinayagam R, Blackett RL. Spilled gall stones during laparoscopic cholecystectomy: a review of the literature. Postgrad Med J 2004; 80:77–79.
- Horton M, Florence MG. Unusual abscess patterns following dropped gallstones during laparoscopic cholecystectomy. Am J Surg 1998; 175:375–379.
- Morrin MM, Kruskal JB, Hochman MG, Saldinger PF, Kane RA. Radiologic features of complications arising from dropped gallstones in laparoscopic cholecystectomy patients. AJR Am J Roentgenol 2000; 174:1441–1445.
- Akyar G, Aytac S, Yagci C, Akyar S. Abscess formation due to dropped gallstone after laparoscopic cholecystectomy. Eur J Radiol 1997; 25:242–245.
A 67-year-old retired man presents to his internist with a 3-month history of abdominal discomfort in the right upper quadrant on deep breathing. He has no other abdominal complaints, but he mentions that he underwent laparoscopic cholecystectomy 3 months ago for gallstone pancreatitis.
A biopsy specimen obtained with CT guidance shows chronic inflammation but is sterile on aerobic culture. There is no evidence of malignancy. Because of concern for underlying infection, the infectious disease staff recommends empirical treatment with a 4-week course of ampicillin-sulbactam (Unasyn). At completion of the antibiotic course, the patient’s symptoms have resolved.
LAPAROSCOPY’S DRAWBACKS
Complications of dropped stones, though rare, can include localized or systemic infection, inflammation, fibrosis, adhesion, cutaneous sinus formation, ileus, and abscess.1,6 Lohan et al1 estimated that dropped stones produce an intra-abdominal abscess in 0.6% to 2.9% of cases of dropped stones and bile spillage, based on reports by Rice et al4 and Morrin et al.7 Dropped stones should be recognized as a potential cause of intra-abdominal abscess in any cholecystectomy patient months or even years after the surgery. Also, these abscesses are not necessarily confined to the right upper quadrant: they can occur anywhere in the abdominal cavity.5,7
Given the ever-increasing popularity of laparoscopic cholecystectomy, the problem of intra-abdominal abscess due to dropped gallstones will only become a more common problem. Early diagnosis is the key to avoiding long and unnecessary treatment.
If dropped gallstones do become infected and eventually cause symptoms, they may require surgical or percutaneous removal in conjunction with antimicrobial therapy.8
A 67-year-old retired man presents to his internist with a 3-month history of abdominal discomfort in the right upper quadrant on deep breathing. He has no other abdominal complaints, but he mentions that he underwent laparoscopic cholecystectomy 3 months ago for gallstone pancreatitis.
A biopsy specimen obtained with CT guidance shows chronic inflammation but is sterile on aerobic culture. There is no evidence of malignancy. Because of concern for underlying infection, the infectious disease staff recommends empirical treatment with a 4-week course of ampicillin-sulbactam (Unasyn). At completion of the antibiotic course, the patient’s symptoms have resolved.
LAPAROSCOPY’S DRAWBACKS
Complications of dropped stones, though rare, can include localized or systemic infection, inflammation, fibrosis, adhesion, cutaneous sinus formation, ileus, and abscess.1,6 Lohan et al1 estimated that dropped stones produce an intra-abdominal abscess in 0.6% to 2.9% of cases of dropped stones and bile spillage, based on reports by Rice et al4 and Morrin et al.7 Dropped stones should be recognized as a potential cause of intra-abdominal abscess in any cholecystectomy patient months or even years after the surgery. Also, these abscesses are not necessarily confined to the right upper quadrant: they can occur anywhere in the abdominal cavity.5,7
Given the ever-increasing popularity of laparoscopic cholecystectomy, the problem of intra-abdominal abscess due to dropped gallstones will only become a more common problem. Early diagnosis is the key to avoiding long and unnecessary treatment.
If dropped gallstones do become infected and eventually cause symptoms, they may require surgical or percutaneous removal in conjunction with antimicrobial therapy.8
- Lohan D, Walsh S, McLoughlin R, Murphy J. Imaging of the complications of laparoscopic cholecystectomy. Eur Radiol 2005; 15:904–912.
- Casillas S, Kittur DS. Late abscess formation after spilled gallstones masquerading as a liver mass. Surg Endosc 2003; 17:833.
- Tumer AR, Yuksek YN, Yasti AC, Gozalan U, Kama NA. Dropped gallstones during laparoscopic cholecystectomy: the consequences. World J Surg 2005; 29:437–440.
- Rice DC, Memon MA, Jamison RL, et al. Long-term consequences of intraoperative spillage of bile and gallstones during laparoscopic cholecystectomy. J Gastrointest Surg 1997; 1:85–91.
- Sathesh-Kumar T, Saklani AP, Vinayagam R, Blackett RL. Spilled gall stones during laparoscopic cholecystectomy: a review of the literature. Postgrad Med J 2004; 80:77–79.
- Horton M, Florence MG. Unusual abscess patterns following dropped gallstones during laparoscopic cholecystectomy. Am J Surg 1998; 175:375–379.
- Morrin MM, Kruskal JB, Hochman MG, Saldinger PF, Kane RA. Radiologic features of complications arising from dropped gallstones in laparoscopic cholecystectomy patients. AJR Am J Roentgenol 2000; 174:1441–1445.
- Akyar G, Aytac S, Yagci C, Akyar S. Abscess formation due to dropped gallstone after laparoscopic cholecystectomy. Eur J Radiol 1997; 25:242–245.
- Lohan D, Walsh S, McLoughlin R, Murphy J. Imaging of the complications of laparoscopic cholecystectomy. Eur Radiol 2005; 15:904–912.
- Casillas S, Kittur DS. Late abscess formation after spilled gallstones masquerading as a liver mass. Surg Endosc 2003; 17:833.
- Tumer AR, Yuksek YN, Yasti AC, Gozalan U, Kama NA. Dropped gallstones during laparoscopic cholecystectomy: the consequences. World J Surg 2005; 29:437–440.
- Rice DC, Memon MA, Jamison RL, et al. Long-term consequences of intraoperative spillage of bile and gallstones during laparoscopic cholecystectomy. J Gastrointest Surg 1997; 1:85–91.
- Sathesh-Kumar T, Saklani AP, Vinayagam R, Blackett RL. Spilled gall stones during laparoscopic cholecystectomy: a review of the literature. Postgrad Med J 2004; 80:77–79.
- Horton M, Florence MG. Unusual abscess patterns following dropped gallstones during laparoscopic cholecystectomy. Am J Surg 1998; 175:375–379.
- Morrin MM, Kruskal JB, Hochman MG, Saldinger PF, Kane RA. Radiologic features of complications arising from dropped gallstones in laparoscopic cholecystectomy patients. AJR Am J Roentgenol 2000; 174:1441–1445.
- Akyar G, Aytac S, Yagci C, Akyar S. Abscess formation due to dropped gallstone after laparoscopic cholecystectomy. Eur J Radiol 1997; 25:242–245.