User login
Salmonella-related mycotic pseudoaneurysm
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
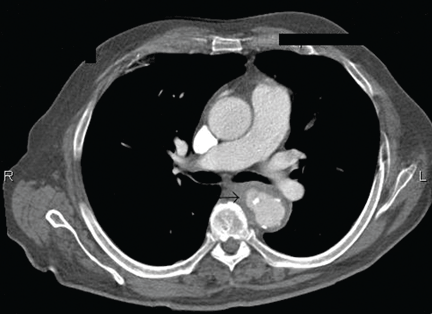
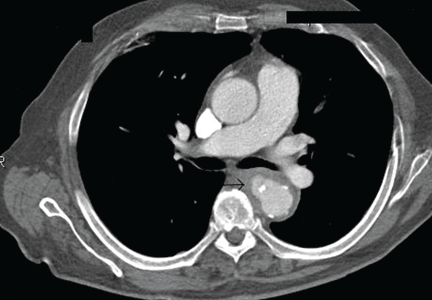
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
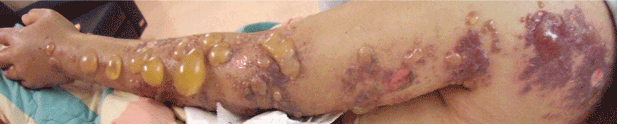
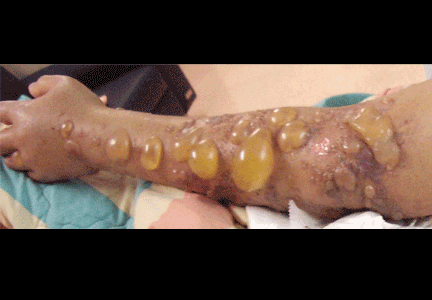
Multiple huge bullae after renal transplant
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
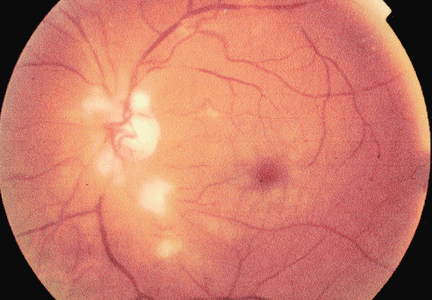
Unilateral cotton wool spots: An important clue
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
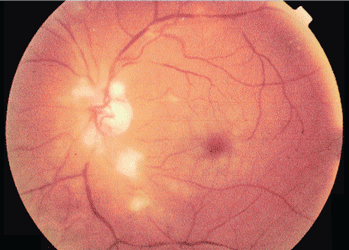
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
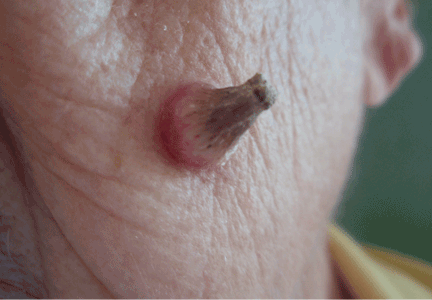
A facial cutaneous horn
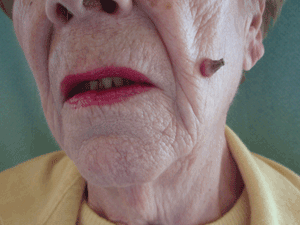
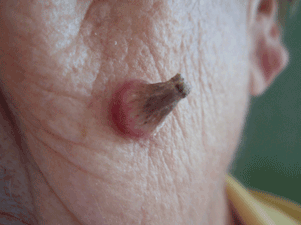
The patient says she has had intense sunlight exposure during her life, due to agricultural work.
Q: What is the most likely diagnosis?
- Basal cell carcinoma
- Cutaneous leishmaniasis
- Squamous cell carcinoma
- Viral wart
- Pyoderma gangrenosum
A: Squamous cell carcinoma is the most likely diagnosis. It is the second most common form of skin cancer (after basal cell carcinoma) that arises on the sun-exposed skin of elderly individuals.
Basal cell carcinoma usually presents as a slow-growing, pearly nodule with telangiectasias over the surface, but without a keratotic component. Cutaneous leishmaniasis is caused by a protozoan and is transmitted by the bite of a sandfly. It presents as a crusted papule or ulcer at the site of the bite, only rarely presenting as a cutaneous horn. Viral warts may present as horns (filiform warts), but the horn is slender and is usually around the lips, eyelids, or nares. Although warts can occur at any age, they are more frequently seen among school-aged children, and they peak between ages 12 and 16. Pyoderma gangrenosum is considered a reactive dermatosis sometimes secondary to a systemic disease such as inflammatory bowel disease. It usually presents as a small, red papule or pustule that progresses to a larger ulcerative lesion.
SQUAMOUS CELL CARCINOMA: CAUSES, RISK FACTORS
Exposure to ultraviolet radiation is the most common cause of squamous cell carcinoma. Other risk factors include exposure to ionizing radiation, human papilloma virus, thermal injury, burns, immunosuppression, exposure to chemical carcinogens, and diseases such as xeroderma pigmentosum, oculocutaneous albinism, and junctional epidermolysis bullosa.1,2 Organ transplantation is another significant risk factor, and cutaneous squamous cell carcinoma is the most common malignancy in renal transplant recipients.3
People born in areas that receive high amounts of ultraviolet radiation from the sun have a risk of squamous cell carcinoma three times higher than people who move to those areas in adulthood.1 Occupational exposure to ultraviolet radiation is also implicated in this type of cancer.
CLINICAL APPEARANCE
Clinically, squamous cell carcinoma of the skin may manifest as a variety of primary morphologies with or without associated symptoms. The characteristic invasive squamous cell carcinoma is a raised, firm, pink or flesh-colored keratotic papule or plaque arising on sun-exposed skin. Approximately 70% of all squamous cell carcinomas occur on the head and neck, with an additional 15% found on the upper extremities. Surface changes may include scaling, ulceration, crusting, or the presence of a cutaneous horn. Squamous cell carcinoma can also present as an eczematous patch that does not resolve with emollient or topical steroids. Less commonly, it may manifest as a pink cutaneous nodule with no overlying surface changes. Often, the patient has a background of severely sun-damaged skin, including solar elastosis, mottled dyspigmentation, telangiectasia, and multiple actinic keratoses.
MAKING THE DIAGNOSIS
Skin biopsy usually confirms the clinical diagnosis. Histologic examination shows neoplastic proliferation of atypical keratinocytes extending into the dermis, with large hyperchromatic and pleomorphic nuclei.
Imaging is not routinely indicated for diagnosing cutaneous squamous cell carcinoma. However, radiologic imaging should be obtained in patients with regional lymphadenopathy or neurologic symptoms suggestive of perineural involvement.
Other lesions found at the base of a cutaneous horn include actinic keratosis, keratoacanthoma, basal cell carcinoma, and benign lesions such as seborrheic keratosis and verruca vulgaris.4,5
MANAGEMENT
Most cutaneous squamous cell carcinomas are easily treated, and the rate of cure is high. Treatment options include topical chemotherapy, cryosurgery, electrodesiccation and curettage, surgical excision, Mohs micrographic surgery, radiotherapy, and photodynamic therapy. Fractionated radiation treatment may be preferred for patients who are unable to tolerate surgery or who have inoperable tumors, and it may provide favorable functional and cosmetic results.1
Photodynamic therapy and topical chemotherapy with imiquimod are currently not recommended for invasive squamous cell carcinoma.
Low-risk tumors are usually cured with surgical therapy. However, patients who develop one squamous cell carcinoma have a 40% risk of developing additional squamous cell carcinomas within the next 2 years. This risk is likely even greater as more time elapses. Thus, patients with a history of this cancer should be evaluated with a complete skin examination every 6 to 12 months.
OUR PATIENT’S TREATMENT
Our patient underwent total surgical excision of the lesion with a safety margin of 0.5 cm. Histologic results confirmed the diagnosis of squamous cell carcinoma with tumor-free margins. No clinical relapse was observed after 1 year of follow-up.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Mallipeddi R, Keane FM, Mcgrath JA, Mayou BJ, Eady RA. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol 2004; 18:521–526.
- Rubel JR, Milford EL, Abdi R. Cutaneous neoplasms in renal transplant recipients. Eur J Dermatol 2002; 12:532–535.
- Aydogan K, Ozbek S, Balaban Adim S, Tokgöz N. Irritated seborrhoeic keratosis presenting as a cutaneous horn. J Eur Acad Dermatol Venereol 2006; 20:626–628.
- Vañó-Galván S, Sanchez-Olaso A. Images in clinical medicine. Squamous-cell carcinoma manifesting as a cutaneous horn. N Engl J Med 2008; 359:e10.
The patient says she has had intense sunlight exposure during her life, due to agricultural work.
Q: What is the most likely diagnosis?
- Basal cell carcinoma
- Cutaneous leishmaniasis
- Squamous cell carcinoma
- Viral wart
- Pyoderma gangrenosum
A: Squamous cell carcinoma is the most likely diagnosis. It is the second most common form of skin cancer (after basal cell carcinoma) that arises on the sun-exposed skin of elderly individuals.
Basal cell carcinoma usually presents as a slow-growing, pearly nodule with telangiectasias over the surface, but without a keratotic component. Cutaneous leishmaniasis is caused by a protozoan and is transmitted by the bite of a sandfly. It presents as a crusted papule or ulcer at the site of the bite, only rarely presenting as a cutaneous horn. Viral warts may present as horns (filiform warts), but the horn is slender and is usually around the lips, eyelids, or nares. Although warts can occur at any age, they are more frequently seen among school-aged children, and they peak between ages 12 and 16. Pyoderma gangrenosum is considered a reactive dermatosis sometimes secondary to a systemic disease such as inflammatory bowel disease. It usually presents as a small, red papule or pustule that progresses to a larger ulcerative lesion.
SQUAMOUS CELL CARCINOMA: CAUSES, RISK FACTORS
Exposure to ultraviolet radiation is the most common cause of squamous cell carcinoma. Other risk factors include exposure to ionizing radiation, human papilloma virus, thermal injury, burns, immunosuppression, exposure to chemical carcinogens, and diseases such as xeroderma pigmentosum, oculocutaneous albinism, and junctional epidermolysis bullosa.1,2 Organ transplantation is another significant risk factor, and cutaneous squamous cell carcinoma is the most common malignancy in renal transplant recipients.3
People born in areas that receive high amounts of ultraviolet radiation from the sun have a risk of squamous cell carcinoma three times higher than people who move to those areas in adulthood.1 Occupational exposure to ultraviolet radiation is also implicated in this type of cancer.
CLINICAL APPEARANCE
Clinically, squamous cell carcinoma of the skin may manifest as a variety of primary morphologies with or without associated symptoms. The characteristic invasive squamous cell carcinoma is a raised, firm, pink or flesh-colored keratotic papule or plaque arising on sun-exposed skin. Approximately 70% of all squamous cell carcinomas occur on the head and neck, with an additional 15% found on the upper extremities. Surface changes may include scaling, ulceration, crusting, or the presence of a cutaneous horn. Squamous cell carcinoma can also present as an eczematous patch that does not resolve with emollient or topical steroids. Less commonly, it may manifest as a pink cutaneous nodule with no overlying surface changes. Often, the patient has a background of severely sun-damaged skin, including solar elastosis, mottled dyspigmentation, telangiectasia, and multiple actinic keratoses.
MAKING THE DIAGNOSIS
Skin biopsy usually confirms the clinical diagnosis. Histologic examination shows neoplastic proliferation of atypical keratinocytes extending into the dermis, with large hyperchromatic and pleomorphic nuclei.
Imaging is not routinely indicated for diagnosing cutaneous squamous cell carcinoma. However, radiologic imaging should be obtained in patients with regional lymphadenopathy or neurologic symptoms suggestive of perineural involvement.
Other lesions found at the base of a cutaneous horn include actinic keratosis, keratoacanthoma, basal cell carcinoma, and benign lesions such as seborrheic keratosis and verruca vulgaris.4,5
MANAGEMENT
Most cutaneous squamous cell carcinomas are easily treated, and the rate of cure is high. Treatment options include topical chemotherapy, cryosurgery, electrodesiccation and curettage, surgical excision, Mohs micrographic surgery, radiotherapy, and photodynamic therapy. Fractionated radiation treatment may be preferred for patients who are unable to tolerate surgery or who have inoperable tumors, and it may provide favorable functional and cosmetic results.1
Photodynamic therapy and topical chemotherapy with imiquimod are currently not recommended for invasive squamous cell carcinoma.
Low-risk tumors are usually cured with surgical therapy. However, patients who develop one squamous cell carcinoma have a 40% risk of developing additional squamous cell carcinomas within the next 2 years. This risk is likely even greater as more time elapses. Thus, patients with a history of this cancer should be evaluated with a complete skin examination every 6 to 12 months.
OUR PATIENT’S TREATMENT
Our patient underwent total surgical excision of the lesion with a safety margin of 0.5 cm. Histologic results confirmed the diagnosis of squamous cell carcinoma with tumor-free margins. No clinical relapse was observed after 1 year of follow-up.
The patient says she has had intense sunlight exposure during her life, due to agricultural work.
Q: What is the most likely diagnosis?
- Basal cell carcinoma
- Cutaneous leishmaniasis
- Squamous cell carcinoma
- Viral wart
- Pyoderma gangrenosum
A: Squamous cell carcinoma is the most likely diagnosis. It is the second most common form of skin cancer (after basal cell carcinoma) that arises on the sun-exposed skin of elderly individuals.
Basal cell carcinoma usually presents as a slow-growing, pearly nodule with telangiectasias over the surface, but without a keratotic component. Cutaneous leishmaniasis is caused by a protozoan and is transmitted by the bite of a sandfly. It presents as a crusted papule or ulcer at the site of the bite, only rarely presenting as a cutaneous horn. Viral warts may present as horns (filiform warts), but the horn is slender and is usually around the lips, eyelids, or nares. Although warts can occur at any age, they are more frequently seen among school-aged children, and they peak between ages 12 and 16. Pyoderma gangrenosum is considered a reactive dermatosis sometimes secondary to a systemic disease such as inflammatory bowel disease. It usually presents as a small, red papule or pustule that progresses to a larger ulcerative lesion.
SQUAMOUS CELL CARCINOMA: CAUSES, RISK FACTORS
Exposure to ultraviolet radiation is the most common cause of squamous cell carcinoma. Other risk factors include exposure to ionizing radiation, human papilloma virus, thermal injury, burns, immunosuppression, exposure to chemical carcinogens, and diseases such as xeroderma pigmentosum, oculocutaneous albinism, and junctional epidermolysis bullosa.1,2 Organ transplantation is another significant risk factor, and cutaneous squamous cell carcinoma is the most common malignancy in renal transplant recipients.3
People born in areas that receive high amounts of ultraviolet radiation from the sun have a risk of squamous cell carcinoma three times higher than people who move to those areas in adulthood.1 Occupational exposure to ultraviolet radiation is also implicated in this type of cancer.
CLINICAL APPEARANCE
Clinically, squamous cell carcinoma of the skin may manifest as a variety of primary morphologies with or without associated symptoms. The characteristic invasive squamous cell carcinoma is a raised, firm, pink or flesh-colored keratotic papule or plaque arising on sun-exposed skin. Approximately 70% of all squamous cell carcinomas occur on the head and neck, with an additional 15% found on the upper extremities. Surface changes may include scaling, ulceration, crusting, or the presence of a cutaneous horn. Squamous cell carcinoma can also present as an eczematous patch that does not resolve with emollient or topical steroids. Less commonly, it may manifest as a pink cutaneous nodule with no overlying surface changes. Often, the patient has a background of severely sun-damaged skin, including solar elastosis, mottled dyspigmentation, telangiectasia, and multiple actinic keratoses.
MAKING THE DIAGNOSIS
Skin biopsy usually confirms the clinical diagnosis. Histologic examination shows neoplastic proliferation of atypical keratinocytes extending into the dermis, with large hyperchromatic and pleomorphic nuclei.
Imaging is not routinely indicated for diagnosing cutaneous squamous cell carcinoma. However, radiologic imaging should be obtained in patients with regional lymphadenopathy or neurologic symptoms suggestive of perineural involvement.
Other lesions found at the base of a cutaneous horn include actinic keratosis, keratoacanthoma, basal cell carcinoma, and benign lesions such as seborrheic keratosis and verruca vulgaris.4,5
MANAGEMENT
Most cutaneous squamous cell carcinomas are easily treated, and the rate of cure is high. Treatment options include topical chemotherapy, cryosurgery, electrodesiccation and curettage, surgical excision, Mohs micrographic surgery, radiotherapy, and photodynamic therapy. Fractionated radiation treatment may be preferred for patients who are unable to tolerate surgery or who have inoperable tumors, and it may provide favorable functional and cosmetic results.1
Photodynamic therapy and topical chemotherapy with imiquimod are currently not recommended for invasive squamous cell carcinoma.
Low-risk tumors are usually cured with surgical therapy. However, patients who develop one squamous cell carcinoma have a 40% risk of developing additional squamous cell carcinomas within the next 2 years. This risk is likely even greater as more time elapses. Thus, patients with a history of this cancer should be evaluated with a complete skin examination every 6 to 12 months.
OUR PATIENT’S TREATMENT
Our patient underwent total surgical excision of the lesion with a safety margin of 0.5 cm. Histologic results confirmed the diagnosis of squamous cell carcinoma with tumor-free margins. No clinical relapse was observed after 1 year of follow-up.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Mallipeddi R, Keane FM, Mcgrath JA, Mayou BJ, Eady RA. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol 2004; 18:521–526.
- Rubel JR, Milford EL, Abdi R. Cutaneous neoplasms in renal transplant recipients. Eur J Dermatol 2002; 12:532–535.
- Aydogan K, Ozbek S, Balaban Adim S, Tokgöz N. Irritated seborrhoeic keratosis presenting as a cutaneous horn. J Eur Acad Dermatol Venereol 2006; 20:626–628.
- Vañó-Galván S, Sanchez-Olaso A. Images in clinical medicine. Squamous-cell carcinoma manifesting as a cutaneous horn. N Engl J Med 2008; 359:e10.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med 2001; 344:975–983.
- Mallipeddi R, Keane FM, Mcgrath JA, Mayou BJ, Eady RA. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol 2004; 18:521–526.
- Rubel JR, Milford EL, Abdi R. Cutaneous neoplasms in renal transplant recipients. Eur J Dermatol 2002; 12:532–535.
- Aydogan K, Ozbek S, Balaban Adim S, Tokgöz N. Irritated seborrhoeic keratosis presenting as a cutaneous horn. J Eur Acad Dermatol Venereol 2006; 20:626–628.
- Vañó-Galván S, Sanchez-Olaso A. Images in clinical medicine. Squamous-cell carcinoma manifesting as a cutaneous horn. N Engl J Med 2008; 359:e10.
A persistently swollen lip
A 44-year-old man is referred for evaluation of asymptomatic swelling of the lower lip that has persisted for 10 months. He has been treated unsuccessfully with oral antihistamines for suspected chronic angioedema. He has no other symptoms and appears to be well otherwise. He has no history of applied irritants or local trauma, and his medical history is unremarkable.
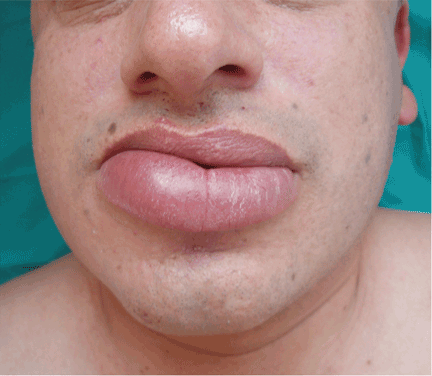
Results of the laboratory evaluation, including serum angiotensin-converting enzyme level, are normal. Patch tests to detect contact sensitivity to food additives are negative. Biopsy of the affected lip reveals dense infiltrate of the submucosal connective tissue with focal nonnecrotizing granulomas. Imaging and endoscopic studies show no evidence of sarcoidosis or Crohn disease.
Q: Given what we know so far, which of the following is the most likely diagnosis of the persistent lip swelling?
- Melkersson-Rosenthal syndrome
- Amyloidosis
- Quincke edema
- Cheilitis granulomatosa
- Cutaneous tuberculosis
A: From what we know so far, the correct answer is cheilitis granulomatosa. While this rare condition may be a feature of Melkersson-Rosenthal syndrome and amyloidosis, at this point in the evaluation these have not been confirmed. Quincke edema (ie, angioedema) is unlikely, given the ineffectiveness of previous treatment with oral antihistamines. Cutaneous tuberculosis usually presents as “lupus vulgaris,” which is characterized by solitary, small, sharply marginated, red-brown papules of gelatinous consistency (“apple-jelly nodules”), mainly on the head and neck.
Cheilitis granulomatosa is a rare inflammatory disorder1 that primarily affects young adults. Its key feature is recurrent or persistent painless swelling of one or both lips. It may occur without other signs of disease, but it is also a manifestation of Melkersson-Rosenthal syndrome and it may be a presenting symptom of Crohn disease or, rarely, sarcoidosis.2 The term “orofacial granulomatosis” was introduced to encompass the broad spectrum of nonnecrotizing granulomatous inflammation in the orofacial region, including cheilitis granulomatosa, the complete Melkersson-Rosenthal syndrome, sarcoidosis, Crohn disease, and infectious disorders such as tuberculosis.1
The cause of cheilitis granulomatosa is unknown. Specific T-cell clonality has been identified in several patients with orofacial granulomatosis, suggesting a delayed hypersensitivity response. Moreover, the HLA haplotypes HLA-A2 and HLA-A11 have been found in 25% of patients with orofacial granulomatosis, suggesting a viral etiology. A genetic predisposition may exist in Melkersson- Rosenthal syndrome: siblings have been affected, and otherwise unaffected relatives may have a fissured tongue (lingua plicata).
Melkersson-Rosenthal syndrome, a rare condition, is characterized by a classic triad of recurrent swelling of the lips or face (or both), fissured tongue, and relapsing peripheral facial nerve paralysis. It is an unusual cause of facial swelling that can be confused with angioedema.3 This syndrome can be ruled out in this patient because he has only one of the three classic signs. Contact antigens are sometimes implicated.
DIFFERENTIAL DIAGNOSIS OF CHEILITIS GRANULOMATOSA
The differential diagnosis of cheilitis granulomatosa is extensive and includes amyloidosis, cheilitis glandularis, sarcoidosis, Crohn disease, actinic cheilitis, neoplasms, and infections, such as tuberculosis, syphilis, and leprosy.1
As many as 11% of patients with Crohn disease may develop mucocutaneous lesions. Oral lesions of Crohn disease include apthae, cobblestoning of the buccal mucosa, swelling of one or both lips (soft or rubbery), vertical clefts of the lips, or hypertrophic gingivitis. Only 5% of patients with Crohn disease ever develop cheilitis granulomatosa, though most cases occur in children.
Ultimately, the diagnosis of cheilitis granulomatosa is made by correlating the patient’s history and clinical features, usually supported by histopathologic findings of nonnecrotic granulomas extending into the deep dermis, composed of histiocytes and giant cells and associated with a lymphomonocytic infiltrate.
TREATMENT
Treatment of cheilitis granulomatosa is difficult because the cause is unknown and the rate of recurrence is high. Response to treatment is often late and unpredictable. Corticosteroids, clofazimine (Lamprene), and surgical intervention such as cheiloplasty have been described as treatment options. Other treatment options include thalidomide (Thalomid), sulfasalazine (Sulfazine), erythromycin, azathioprine (Imuran), and cyclosporine (Sandimmune). Infliximab (Remicade) has been recently reported as a new alternative treatment, in particular for Melkersson-Rosenthal syndrome.4
In our patient, twice-monthly injections of 1 mL of triamcinolone acetonide 10 mg/mL into the affected lip brought acceptable improvement at 3 months. The patient is on maintenance treatment with twice-monthly triamcinolone injections and has had no relapses after 2 years.
- van der Waal RI, Schulten EA, van de Scheur MR, Wauters IM, Starink TM, van der Waal I. Cheilitis granulomatosa. J Eur Acad Dermatol Venereol 2001; 15:519–523.
- van der Waal RI, Schulten EA, van der Meij EH, van de Scheur MR, Starink TM, van der Waal I. Cheilitis granulomatosa: overview of 13 patients with long-term follow-up—results of management. Int J Dermatol 2002; 41:225–229.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol 2007; 99:185–189.
- Ratzinger G, Sepp N, Vogetseder W, Tilg H. Cheilitis granulomatosa and Melkersson-Rosenthal syndrome: evaluation of gastrointestinal involvement and therapeutic regimens in a series of 14 patients. J Eur Acad Dermatol Venereol 2007; 21:1065–1070.
A 44-year-old man is referred for evaluation of asymptomatic swelling of the lower lip that has persisted for 10 months. He has been treated unsuccessfully with oral antihistamines for suspected chronic angioedema. He has no other symptoms and appears to be well otherwise. He has no history of applied irritants or local trauma, and his medical history is unremarkable.
Results of the laboratory evaluation, including serum angiotensin-converting enzyme level, are normal. Patch tests to detect contact sensitivity to food additives are negative. Biopsy of the affected lip reveals dense infiltrate of the submucosal connective tissue with focal nonnecrotizing granulomas. Imaging and endoscopic studies show no evidence of sarcoidosis or Crohn disease.
Q: Given what we know so far, which of the following is the most likely diagnosis of the persistent lip swelling?
- Melkersson-Rosenthal syndrome
- Amyloidosis
- Quincke edema
- Cheilitis granulomatosa
- Cutaneous tuberculosis
A: From what we know so far, the correct answer is cheilitis granulomatosa. While this rare condition may be a feature of Melkersson-Rosenthal syndrome and amyloidosis, at this point in the evaluation these have not been confirmed. Quincke edema (ie, angioedema) is unlikely, given the ineffectiveness of previous treatment with oral antihistamines. Cutaneous tuberculosis usually presents as “lupus vulgaris,” which is characterized by solitary, small, sharply marginated, red-brown papules of gelatinous consistency (“apple-jelly nodules”), mainly on the head and neck.
Cheilitis granulomatosa is a rare inflammatory disorder1 that primarily affects young adults. Its key feature is recurrent or persistent painless swelling of one or both lips. It may occur without other signs of disease, but it is also a manifestation of Melkersson-Rosenthal syndrome and it may be a presenting symptom of Crohn disease or, rarely, sarcoidosis.2 The term “orofacial granulomatosis” was introduced to encompass the broad spectrum of nonnecrotizing granulomatous inflammation in the orofacial region, including cheilitis granulomatosa, the complete Melkersson-Rosenthal syndrome, sarcoidosis, Crohn disease, and infectious disorders such as tuberculosis.1
The cause of cheilitis granulomatosa is unknown. Specific T-cell clonality has been identified in several patients with orofacial granulomatosis, suggesting a delayed hypersensitivity response. Moreover, the HLA haplotypes HLA-A2 and HLA-A11 have been found in 25% of patients with orofacial granulomatosis, suggesting a viral etiology. A genetic predisposition may exist in Melkersson- Rosenthal syndrome: siblings have been affected, and otherwise unaffected relatives may have a fissured tongue (lingua plicata).
Melkersson-Rosenthal syndrome, a rare condition, is characterized by a classic triad of recurrent swelling of the lips or face (or both), fissured tongue, and relapsing peripheral facial nerve paralysis. It is an unusual cause of facial swelling that can be confused with angioedema.3 This syndrome can be ruled out in this patient because he has only one of the three classic signs. Contact antigens are sometimes implicated.
DIFFERENTIAL DIAGNOSIS OF CHEILITIS GRANULOMATOSA
The differential diagnosis of cheilitis granulomatosa is extensive and includes amyloidosis, cheilitis glandularis, sarcoidosis, Crohn disease, actinic cheilitis, neoplasms, and infections, such as tuberculosis, syphilis, and leprosy.1
As many as 11% of patients with Crohn disease may develop mucocutaneous lesions. Oral lesions of Crohn disease include apthae, cobblestoning of the buccal mucosa, swelling of one or both lips (soft or rubbery), vertical clefts of the lips, or hypertrophic gingivitis. Only 5% of patients with Crohn disease ever develop cheilitis granulomatosa, though most cases occur in children.
Ultimately, the diagnosis of cheilitis granulomatosa is made by correlating the patient’s history and clinical features, usually supported by histopathologic findings of nonnecrotic granulomas extending into the deep dermis, composed of histiocytes and giant cells and associated with a lymphomonocytic infiltrate.
TREATMENT
Treatment of cheilitis granulomatosa is difficult because the cause is unknown and the rate of recurrence is high. Response to treatment is often late and unpredictable. Corticosteroids, clofazimine (Lamprene), and surgical intervention such as cheiloplasty have been described as treatment options. Other treatment options include thalidomide (Thalomid), sulfasalazine (Sulfazine), erythromycin, azathioprine (Imuran), and cyclosporine (Sandimmune). Infliximab (Remicade) has been recently reported as a new alternative treatment, in particular for Melkersson-Rosenthal syndrome.4
In our patient, twice-monthly injections of 1 mL of triamcinolone acetonide 10 mg/mL into the affected lip brought acceptable improvement at 3 months. The patient is on maintenance treatment with twice-monthly triamcinolone injections and has had no relapses after 2 years.
A 44-year-old man is referred for evaluation of asymptomatic swelling of the lower lip that has persisted for 10 months. He has been treated unsuccessfully with oral antihistamines for suspected chronic angioedema. He has no other symptoms and appears to be well otherwise. He has no history of applied irritants or local trauma, and his medical history is unremarkable.
Results of the laboratory evaluation, including serum angiotensin-converting enzyme level, are normal. Patch tests to detect contact sensitivity to food additives are negative. Biopsy of the affected lip reveals dense infiltrate of the submucosal connective tissue with focal nonnecrotizing granulomas. Imaging and endoscopic studies show no evidence of sarcoidosis or Crohn disease.
Q: Given what we know so far, which of the following is the most likely diagnosis of the persistent lip swelling?
- Melkersson-Rosenthal syndrome
- Amyloidosis
- Quincke edema
- Cheilitis granulomatosa
- Cutaneous tuberculosis
A: From what we know so far, the correct answer is cheilitis granulomatosa. While this rare condition may be a feature of Melkersson-Rosenthal syndrome and amyloidosis, at this point in the evaluation these have not been confirmed. Quincke edema (ie, angioedema) is unlikely, given the ineffectiveness of previous treatment with oral antihistamines. Cutaneous tuberculosis usually presents as “lupus vulgaris,” which is characterized by solitary, small, sharply marginated, red-brown papules of gelatinous consistency (“apple-jelly nodules”), mainly on the head and neck.
Cheilitis granulomatosa is a rare inflammatory disorder1 that primarily affects young adults. Its key feature is recurrent or persistent painless swelling of one or both lips. It may occur without other signs of disease, but it is also a manifestation of Melkersson-Rosenthal syndrome and it may be a presenting symptom of Crohn disease or, rarely, sarcoidosis.2 The term “orofacial granulomatosis” was introduced to encompass the broad spectrum of nonnecrotizing granulomatous inflammation in the orofacial region, including cheilitis granulomatosa, the complete Melkersson-Rosenthal syndrome, sarcoidosis, Crohn disease, and infectious disorders such as tuberculosis.1
The cause of cheilitis granulomatosa is unknown. Specific T-cell clonality has been identified in several patients with orofacial granulomatosis, suggesting a delayed hypersensitivity response. Moreover, the HLA haplotypes HLA-A2 and HLA-A11 have been found in 25% of patients with orofacial granulomatosis, suggesting a viral etiology. A genetic predisposition may exist in Melkersson- Rosenthal syndrome: siblings have been affected, and otherwise unaffected relatives may have a fissured tongue (lingua plicata).
Melkersson-Rosenthal syndrome, a rare condition, is characterized by a classic triad of recurrent swelling of the lips or face (or both), fissured tongue, and relapsing peripheral facial nerve paralysis. It is an unusual cause of facial swelling that can be confused with angioedema.3 This syndrome can be ruled out in this patient because he has only one of the three classic signs. Contact antigens are sometimes implicated.
DIFFERENTIAL DIAGNOSIS OF CHEILITIS GRANULOMATOSA
The differential diagnosis of cheilitis granulomatosa is extensive and includes amyloidosis, cheilitis glandularis, sarcoidosis, Crohn disease, actinic cheilitis, neoplasms, and infections, such as tuberculosis, syphilis, and leprosy.1
As many as 11% of patients with Crohn disease may develop mucocutaneous lesions. Oral lesions of Crohn disease include apthae, cobblestoning of the buccal mucosa, swelling of one or both lips (soft or rubbery), vertical clefts of the lips, or hypertrophic gingivitis. Only 5% of patients with Crohn disease ever develop cheilitis granulomatosa, though most cases occur in children.
Ultimately, the diagnosis of cheilitis granulomatosa is made by correlating the patient’s history and clinical features, usually supported by histopathologic findings of nonnecrotic granulomas extending into the deep dermis, composed of histiocytes and giant cells and associated with a lymphomonocytic infiltrate.
TREATMENT
Treatment of cheilitis granulomatosa is difficult because the cause is unknown and the rate of recurrence is high. Response to treatment is often late and unpredictable. Corticosteroids, clofazimine (Lamprene), and surgical intervention such as cheiloplasty have been described as treatment options. Other treatment options include thalidomide (Thalomid), sulfasalazine (Sulfazine), erythromycin, azathioprine (Imuran), and cyclosporine (Sandimmune). Infliximab (Remicade) has been recently reported as a new alternative treatment, in particular for Melkersson-Rosenthal syndrome.4
In our patient, twice-monthly injections of 1 mL of triamcinolone acetonide 10 mg/mL into the affected lip brought acceptable improvement at 3 months. The patient is on maintenance treatment with twice-monthly triamcinolone injections and has had no relapses after 2 years.
- van der Waal RI, Schulten EA, van de Scheur MR, Wauters IM, Starink TM, van der Waal I. Cheilitis granulomatosa. J Eur Acad Dermatol Venereol 2001; 15:519–523.
- van der Waal RI, Schulten EA, van der Meij EH, van de Scheur MR, Starink TM, van der Waal I. Cheilitis granulomatosa: overview of 13 patients with long-term follow-up—results of management. Int J Dermatol 2002; 41:225–229.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol 2007; 99:185–189.
- Ratzinger G, Sepp N, Vogetseder W, Tilg H. Cheilitis granulomatosa and Melkersson-Rosenthal syndrome: evaluation of gastrointestinal involvement and therapeutic regimens in a series of 14 patients. J Eur Acad Dermatol Venereol 2007; 21:1065–1070.
- van der Waal RI, Schulten EA, van de Scheur MR, Wauters IM, Starink TM, van der Waal I. Cheilitis granulomatosa. J Eur Acad Dermatol Venereol 2001; 15:519–523.
- van der Waal RI, Schulten EA, van der Meij EH, van de Scheur MR, Starink TM, van der Waal I. Cheilitis granulomatosa: overview of 13 patients with long-term follow-up—results of management. Int J Dermatol 2002; 41:225–229.
- Kakimoto C, Sparks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol 2007; 99:185–189.
- Ratzinger G, Sepp N, Vogetseder W, Tilg H. Cheilitis granulomatosa and Melkersson-Rosenthal syndrome: evaluation of gastrointestinal involvement and therapeutic regimens in a series of 14 patients. J Eur Acad Dermatol Venereol 2007; 21:1065–1070.
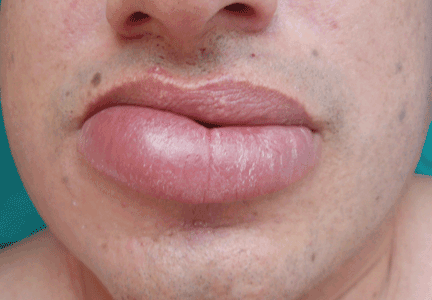
Black hairy tongue
A 71-year-old man presents for evaluation of an asymptomatic black discoloration of the tongue that he noticed several days earlier. The tongue does not itch or hurt, and the patient is otherwise well, although he is concerned about potential malignancy.
He has a history of hypertension, hyperuricemia, and type 2 diabetes treated with oral glucose-lowering drugs, and he has had no recent changes in his medications. He drinks coffee and uses tobacco. His oral hygiene is poor, with intense halitosis.
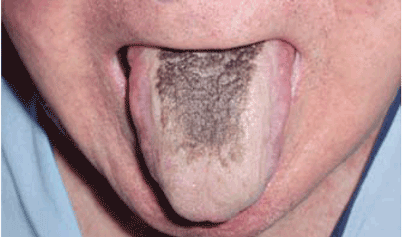
Q: What is the most likely diagnosis?
- Oral leukoplakia
- Epidermoid carcinoma of the tongue
- Malignant melanoma of the tongue
- Mucosal candidiasis
- Black hairy tongue
A: Black hairy tongue is correct. A simple treatment consisting of brushing the tongue daily with a soft toothbrush enhanced by previous application of 30% urea is recommended to the patient, and the discoloration resolves completely within 4 weeks. He is educated on correct oral hygiene and discontinues smoking, with no clinical relapses after 2 years of follow-up.
THE CAUSES AND THE COURSE
Black hairy tongue, also known as lingua villosa nigra, is a painless, benign disorder caused by defective desquamation and reactive hypertrophy of the filiform papillae of the tongue. The hairy appearance is due to elongation of keratinized filiform papillae, which may have different colors, varying from white to yellowish brown to black depending on extrinsic factors (eg, tobacco, coffee, tea, food) and intrinsic factors (ie, chromogenic organisms in normal flora).1
The exact pathogenesis is unclear. Precipitating factors include poor oral hygiene, use of the antipsychotic drug olanzapine1 (Zyprexa) or a broad-spectrum antibiotic such as erythromycin,2 and therapeutic radiation of the head and the neck. Tobacco use and drinking coffee and tea are also contributory factors. Neurologic conditions such as trigeminal neuropathy may be associated. 3 Manabe et al4 applied a panel of antikeratin probes, showing that defective desquamation of the cells in the central column of filiform papillae resulted in the formation of highly elongated, cornified spines or “hairs”—the hallmark of lingua villosa nigra.
PRESENTATION AND DIAGNOSIS
Black hairy tongue is usually asymptomatic. However, symptoms such as altered (metallic) taste, nausea, or halitosis may be noted. Most patients with hairy tongue drink coffee or tea, often in addition to tobacco use.
The diagnosis is based on filiform papillae that are elongated more than 3 mm on the dorsal surface of the tongue. Cultures may be taken to rule out a superimposed oral candidiasis or other suspected oral infection.
MANAGEMENT
Although frightening to the patient, black hairy tongue is completely harmless. In most cases, treatment does not require drugs. If fungal overgrowth is present, a topical antifungal can be used when the condition is symptomatic.
Empirical approaches such as brushing or scraping the tongue, improving oral hygiene, and eliminating potential offending factors (eg, tobacco, candies, strong mouthwashes, antibiotics) is usually sufficient to resolve the lesions.5
In our experience, educating the patient about proper oral hygiene (including discontinuing smoking) and encouraging routine tongue brushing are the best preventive and therapeutic measures.
- Tamam L, Annagur BB. Black hairy tongue associated with olanzapine treatment: a case report. Mt Sinai J Med 2006; 73:891–894.
- Pigatto PD, Spadari F, Meroni L, Guzzi G. Black hairy tongue associated with long-term oral erythromycin use. J Eur Acad Dermatol Venereol 2008; 22:1269–1270.
- Chesire WP. Unilateral black hairy tongue in trigeminal neuralgia. Headache 2004; 44:908–910.
- Manabe M, Lim HW, Winzer M, Loomis CA. Architectural organization of filiform papillae in normal and black hairy tongue epithelium: dissection of differentiation pathways in a complex human epithelium according to their patterns of keratin expression. Arch Dermatol 1999; 135:177–181.
- Sarti GM, Haddy RI, Schaffer D, Kihm J. Black hairy tongue. Am Fam Physician 1990; 41:1751–1755.
A 71-year-old man presents for evaluation of an asymptomatic black discoloration of the tongue that he noticed several days earlier. The tongue does not itch or hurt, and the patient is otherwise well, although he is concerned about potential malignancy.
He has a history of hypertension, hyperuricemia, and type 2 diabetes treated with oral glucose-lowering drugs, and he has had no recent changes in his medications. He drinks coffee and uses tobacco. His oral hygiene is poor, with intense halitosis.
Q: What is the most likely diagnosis?
- Oral leukoplakia
- Epidermoid carcinoma of the tongue
- Malignant melanoma of the tongue
- Mucosal candidiasis
- Black hairy tongue
A: Black hairy tongue is correct. A simple treatment consisting of brushing the tongue daily with a soft toothbrush enhanced by previous application of 30% urea is recommended to the patient, and the discoloration resolves completely within 4 weeks. He is educated on correct oral hygiene and discontinues smoking, with no clinical relapses after 2 years of follow-up.
THE CAUSES AND THE COURSE
Black hairy tongue, also known as lingua villosa nigra, is a painless, benign disorder caused by defective desquamation and reactive hypertrophy of the filiform papillae of the tongue. The hairy appearance is due to elongation of keratinized filiform papillae, which may have different colors, varying from white to yellowish brown to black depending on extrinsic factors (eg, tobacco, coffee, tea, food) and intrinsic factors (ie, chromogenic organisms in normal flora).1
The exact pathogenesis is unclear. Precipitating factors include poor oral hygiene, use of the antipsychotic drug olanzapine1 (Zyprexa) or a broad-spectrum antibiotic such as erythromycin,2 and therapeutic radiation of the head and the neck. Tobacco use and drinking coffee and tea are also contributory factors. Neurologic conditions such as trigeminal neuropathy may be associated. 3 Manabe et al4 applied a panel of antikeratin probes, showing that defective desquamation of the cells in the central column of filiform papillae resulted in the formation of highly elongated, cornified spines or “hairs”—the hallmark of lingua villosa nigra.
PRESENTATION AND DIAGNOSIS
Black hairy tongue is usually asymptomatic. However, symptoms such as altered (metallic) taste, nausea, or halitosis may be noted. Most patients with hairy tongue drink coffee or tea, often in addition to tobacco use.
The diagnosis is based on filiform papillae that are elongated more than 3 mm on the dorsal surface of the tongue. Cultures may be taken to rule out a superimposed oral candidiasis or other suspected oral infection.
MANAGEMENT
Although frightening to the patient, black hairy tongue is completely harmless. In most cases, treatment does not require drugs. If fungal overgrowth is present, a topical antifungal can be used when the condition is symptomatic.
Empirical approaches such as brushing or scraping the tongue, improving oral hygiene, and eliminating potential offending factors (eg, tobacco, candies, strong mouthwashes, antibiotics) is usually sufficient to resolve the lesions.5
In our experience, educating the patient about proper oral hygiene (including discontinuing smoking) and encouraging routine tongue brushing are the best preventive and therapeutic measures.
A 71-year-old man presents for evaluation of an asymptomatic black discoloration of the tongue that he noticed several days earlier. The tongue does not itch or hurt, and the patient is otherwise well, although he is concerned about potential malignancy.
He has a history of hypertension, hyperuricemia, and type 2 diabetes treated with oral glucose-lowering drugs, and he has had no recent changes in his medications. He drinks coffee and uses tobacco. His oral hygiene is poor, with intense halitosis.
Q: What is the most likely diagnosis?
- Oral leukoplakia
- Epidermoid carcinoma of the tongue
- Malignant melanoma of the tongue
- Mucosal candidiasis
- Black hairy tongue
A: Black hairy tongue is correct. A simple treatment consisting of brushing the tongue daily with a soft toothbrush enhanced by previous application of 30% urea is recommended to the patient, and the discoloration resolves completely within 4 weeks. He is educated on correct oral hygiene and discontinues smoking, with no clinical relapses after 2 years of follow-up.
THE CAUSES AND THE COURSE
Black hairy tongue, also known as lingua villosa nigra, is a painless, benign disorder caused by defective desquamation and reactive hypertrophy of the filiform papillae of the tongue. The hairy appearance is due to elongation of keratinized filiform papillae, which may have different colors, varying from white to yellowish brown to black depending on extrinsic factors (eg, tobacco, coffee, tea, food) and intrinsic factors (ie, chromogenic organisms in normal flora).1
The exact pathogenesis is unclear. Precipitating factors include poor oral hygiene, use of the antipsychotic drug olanzapine1 (Zyprexa) or a broad-spectrum antibiotic such as erythromycin,2 and therapeutic radiation of the head and the neck. Tobacco use and drinking coffee and tea are also contributory factors. Neurologic conditions such as trigeminal neuropathy may be associated. 3 Manabe et al4 applied a panel of antikeratin probes, showing that defective desquamation of the cells in the central column of filiform papillae resulted in the formation of highly elongated, cornified spines or “hairs”—the hallmark of lingua villosa nigra.
PRESENTATION AND DIAGNOSIS
Black hairy tongue is usually asymptomatic. However, symptoms such as altered (metallic) taste, nausea, or halitosis may be noted. Most patients with hairy tongue drink coffee or tea, often in addition to tobacco use.
The diagnosis is based on filiform papillae that are elongated more than 3 mm on the dorsal surface of the tongue. Cultures may be taken to rule out a superimposed oral candidiasis or other suspected oral infection.
MANAGEMENT
Although frightening to the patient, black hairy tongue is completely harmless. In most cases, treatment does not require drugs. If fungal overgrowth is present, a topical antifungal can be used when the condition is symptomatic.
Empirical approaches such as brushing or scraping the tongue, improving oral hygiene, and eliminating potential offending factors (eg, tobacco, candies, strong mouthwashes, antibiotics) is usually sufficient to resolve the lesions.5
In our experience, educating the patient about proper oral hygiene (including discontinuing smoking) and encouraging routine tongue brushing are the best preventive and therapeutic measures.
- Tamam L, Annagur BB. Black hairy tongue associated with olanzapine treatment: a case report. Mt Sinai J Med 2006; 73:891–894.
- Pigatto PD, Spadari F, Meroni L, Guzzi G. Black hairy tongue associated with long-term oral erythromycin use. J Eur Acad Dermatol Venereol 2008; 22:1269–1270.
- Chesire WP. Unilateral black hairy tongue in trigeminal neuralgia. Headache 2004; 44:908–910.
- Manabe M, Lim HW, Winzer M, Loomis CA. Architectural organization of filiform papillae in normal and black hairy tongue epithelium: dissection of differentiation pathways in a complex human epithelium according to their patterns of keratin expression. Arch Dermatol 1999; 135:177–181.
- Sarti GM, Haddy RI, Schaffer D, Kihm J. Black hairy tongue. Am Fam Physician 1990; 41:1751–1755.
- Tamam L, Annagur BB. Black hairy tongue associated with olanzapine treatment: a case report. Mt Sinai J Med 2006; 73:891–894.
- Pigatto PD, Spadari F, Meroni L, Guzzi G. Black hairy tongue associated with long-term oral erythromycin use. J Eur Acad Dermatol Venereol 2008; 22:1269–1270.
- Chesire WP. Unilateral black hairy tongue in trigeminal neuralgia. Headache 2004; 44:908–910.
- Manabe M, Lim HW, Winzer M, Loomis CA. Architectural organization of filiform papillae in normal and black hairy tongue epithelium: dissection of differentiation pathways in a complex human epithelium according to their patterns of keratin expression. Arch Dermatol 1999; 135:177–181.
- Sarti GM, Haddy RI, Schaffer D, Kihm J. Black hairy tongue. Am Fam Physician 1990; 41:1751–1755.
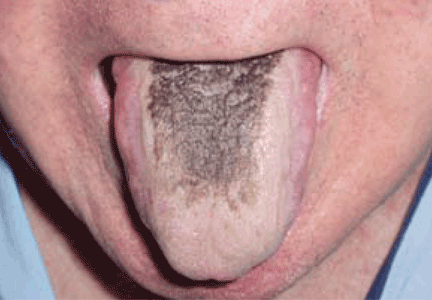
A 90-year-old woman with an asymptomatic spot on her cheek
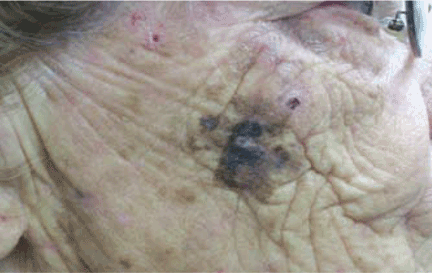
Q: Which is the best diagnosis?
- Seborrheic keratosis
- Pigmented basal cell carcinoma
- Congenital nevus
- Lentigo maligna melanoma
- Actinic purpura
A: The answer is lentigo maligna melanoma. Biopsy revealed the spread of atypical melanocytes along the dermal-epidermal junction and in irregularly shaped and confluent nests. Focal invasion into the papillary dermis was noted when definitive excision was performed.
Lentigo maligna melanoma develops on sun-damaged skin.1 Although most common in older people, lentigo maligna melanoma is now sometimes encountered in younger people. In either case, it is identified using the ABCDE rule of asymmetry, irregular border, color variations, diameter larger than 6 mm, and evolution.2
Seborrheic keratosis often has a rough surface and looks “stuck on.” Basal cell carcinoma usually has a waxy texture, may have a “rolled” border, and usually has telangiectatic vessels at the margins. A congenital nevus is typically stable and long-standing. Many nevi have more uniform pigmentation and lack the different colors and hues typical of melanoma. Purpura usually resolves after a few weeks.
An adequate biopsy is essential for diagnosis and excludes the other entities mentioned in the differential diagnosis.
Surgical treatment with adequate margin control is the cornerstone of therapy.3,4 Topical imiquimod cream (Aldara) may be of value for precursor lentigo maligna lesions proven to be in situ,5 but this is not advocated for invasive disease. Since treatment of smaller lesions is much less difficult and disfiguring, clinicians should be suspicious of any persistent, evolving pigmentary abnormality on sun-damaged skin. Biopsy clarifies the diagnosis.
SUGGESTED READING
- Cohen PJ, Lambert WC, Hill GJ, Schwartz RA. Melanoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management. New York: Springer-Verlag; 1988:104–105.
- Brodell RT, Helms SE. The changing mole. Additional warning signs of malignant melanoma. Postgrad Med 1998; 104:145–148.
- Zitelli JA, Brown CD, Hanusa BH. Surgical margins for excision of primary cutaneous melanoma. J Am Acad Dermatol 1997; 37:422–429.
- Bub JL, Berg D, Slee A, Odland B. Management of lentigo maligna and lentigo maligna melanoma with staged excision: a 5-year follow-up. Arch Dermatol 2004; 140:552–558.
- Hopson B, richey D, Sajben FP. Treatment of lentigo maligna with imiquimod 5% cream. J Drugs Dermatol 2007; 6:1037–1040.
Q: Which is the best diagnosis?
- Seborrheic keratosis
- Pigmented basal cell carcinoma
- Congenital nevus
- Lentigo maligna melanoma
- Actinic purpura
A: The answer is lentigo maligna melanoma. Biopsy revealed the spread of atypical melanocytes along the dermal-epidermal junction and in irregularly shaped and confluent nests. Focal invasion into the papillary dermis was noted when definitive excision was performed.
Lentigo maligna melanoma develops on sun-damaged skin.1 Although most common in older people, lentigo maligna melanoma is now sometimes encountered in younger people. In either case, it is identified using the ABCDE rule of asymmetry, irregular border, color variations, diameter larger than 6 mm, and evolution.2
Seborrheic keratosis often has a rough surface and looks “stuck on.” Basal cell carcinoma usually has a waxy texture, may have a “rolled” border, and usually has telangiectatic vessels at the margins. A congenital nevus is typically stable and long-standing. Many nevi have more uniform pigmentation and lack the different colors and hues typical of melanoma. Purpura usually resolves after a few weeks.
An adequate biopsy is essential for diagnosis and excludes the other entities mentioned in the differential diagnosis.
Surgical treatment with adequate margin control is the cornerstone of therapy.3,4 Topical imiquimod cream (Aldara) may be of value for precursor lentigo maligna lesions proven to be in situ,5 but this is not advocated for invasive disease. Since treatment of smaller lesions is much less difficult and disfiguring, clinicians should be suspicious of any persistent, evolving pigmentary abnormality on sun-damaged skin. Biopsy clarifies the diagnosis.
Q: Which is the best diagnosis?
- Seborrheic keratosis
- Pigmented basal cell carcinoma
- Congenital nevus
- Lentigo maligna melanoma
- Actinic purpura
A: The answer is lentigo maligna melanoma. Biopsy revealed the spread of atypical melanocytes along the dermal-epidermal junction and in irregularly shaped and confluent nests. Focal invasion into the papillary dermis was noted when definitive excision was performed.
Lentigo maligna melanoma develops on sun-damaged skin.1 Although most common in older people, lentigo maligna melanoma is now sometimes encountered in younger people. In either case, it is identified using the ABCDE rule of asymmetry, irregular border, color variations, diameter larger than 6 mm, and evolution.2
Seborrheic keratosis often has a rough surface and looks “stuck on.” Basal cell carcinoma usually has a waxy texture, may have a “rolled” border, and usually has telangiectatic vessels at the margins. A congenital nevus is typically stable and long-standing. Many nevi have more uniform pigmentation and lack the different colors and hues typical of melanoma. Purpura usually resolves after a few weeks.
An adequate biopsy is essential for diagnosis and excludes the other entities mentioned in the differential diagnosis.
Surgical treatment with adequate margin control is the cornerstone of therapy.3,4 Topical imiquimod cream (Aldara) may be of value for precursor lentigo maligna lesions proven to be in situ,5 but this is not advocated for invasive disease. Since treatment of smaller lesions is much less difficult and disfiguring, clinicians should be suspicious of any persistent, evolving pigmentary abnormality on sun-damaged skin. Biopsy clarifies the diagnosis.
SUGGESTED READING
- Cohen PJ, Lambert WC, Hill GJ, Schwartz RA. Melanoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management. New York: Springer-Verlag; 1988:104–105.
- Brodell RT, Helms SE. The changing mole. Additional warning signs of malignant melanoma. Postgrad Med 1998; 104:145–148.
- Zitelli JA, Brown CD, Hanusa BH. Surgical margins for excision of primary cutaneous melanoma. J Am Acad Dermatol 1997; 37:422–429.
- Bub JL, Berg D, Slee A, Odland B. Management of lentigo maligna and lentigo maligna melanoma with staged excision: a 5-year follow-up. Arch Dermatol 2004; 140:552–558.
- Hopson B, richey D, Sajben FP. Treatment of lentigo maligna with imiquimod 5% cream. J Drugs Dermatol 2007; 6:1037–1040.
SUGGESTED READING
- Cohen PJ, Lambert WC, Hill GJ, Schwartz RA. Melanoma. In:Schwartz RA, editor. Skin Cancer: Recognition and Management. New York: Springer-Verlag; 1988:104–105.
- Brodell RT, Helms SE. The changing mole. Additional warning signs of malignant melanoma. Postgrad Med 1998; 104:145–148.
- Zitelli JA, Brown CD, Hanusa BH. Surgical margins for excision of primary cutaneous melanoma. J Am Acad Dermatol 1997; 37:422–429.
- Bub JL, Berg D, Slee A, Odland B. Management of lentigo maligna and lentigo maligna melanoma with staged excision: a 5-year follow-up. Arch Dermatol 2004; 140:552–558.
- Hopson B, richey D, Sajben FP. Treatment of lentigo maligna with imiquimod 5% cream. J Drugs Dermatol 2007; 6:1037–1040.
A judgment call
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
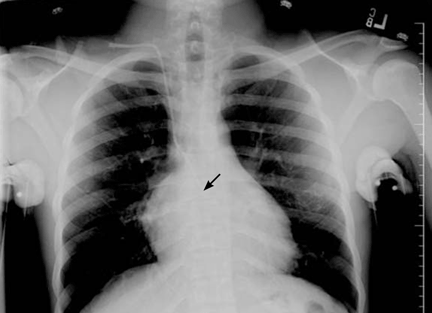
WHERE IS THE CATHETER TIP?
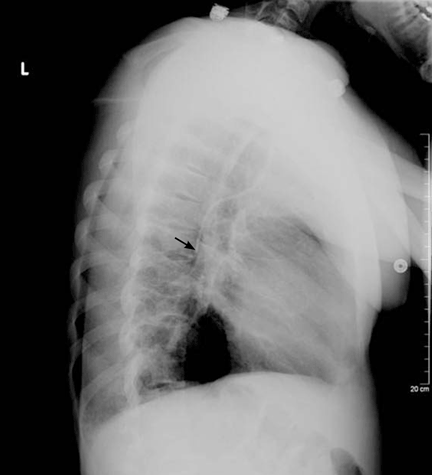
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
WHERE IS THE CATHETER TIP?
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
A 22-year-old African American man with sickle cell disease comes to the in his joints and chest—a presentation similar to those of his previous sickle cell crises. He is given intravenous fluids for dehydration and morphine sulfate for pain via a peripheral line, and he is admitted to the hospital.
Shortly afterward, the peripheral line becomes infiltrated. After failed attempts at peripheral cannulation, central venous cannulation via an internal jugular site is considered.
WHERE IS THE CATHETER TIP?
HAZARDS OF ABERRANT LINE PLACEMENT
Central venous catheters are commonly used to give various infusions (eg, parenteral nutrition), to draw blood, and to monitor the central venous pressure.1 The internal jugular vein is one of the preferred veins for central venous access.1,2 Normally, the anatomy of the jugular venous system and the design of the catheter facilitate proper insertion. Occasionally, however, despite proper technique, the tip of the catheter may not terminate at the desired level, resulting in aberrant placement in the internal thoracic vein, superior vena cava, azygos vein, accessory hemiazygos vein, or axillary vein.1–8
The use of chest radiographs to establish the correct placement of internal jugular and subclavian lines has been advocated and is routinely practiced.6,7 Obtaining a chest x-ray to confirm line placement is particularly important in patients with prior multiple and difficult catheterizations. The lateral view is seldom obtained when confirming central neck line placement, but when the anteroposterior view is not reassuring, it may be prudent to obtain this alternate view.
In a large retrospective analysis,9 cannulation of the azygos arch occurred in about 1.2% of insertions, and the rate was about seven times higher when the left jugular vein was used than when the right one was used. A smaller study gave the frequency of azygos arch cannulation as 0.7%.10
All procedure-related complications of central line insertion in the neck can also occur with aberrant azygos vein cannulation. These include infection, bacteremia, pneumothorax, hemothorax, arterial puncture, and various other mechanical complications. It should be noted that aberrant cannulation of the azygos arch is particularly hazardous, and that complication rates are typically higher. These complications are mainly due to the smaller vascular lumen and to the direction of blood flow in the azygos venous system.
KNOWING THE ANATOMY IS CRUCIAL
Knowledge of venous anatomy and its variants is crucial both for insertion and for ascertaining the correct placement of central venous lines.
The azygos vein has a much smaller lumen than the superior vena cava. Although the lumen size may vary significantly, the maximum diameter of the anterior arch of the azygos vein is about 6 to 7 mm,11 whereas the superior vena cava lumen is typically 1.5 to 2 cm in diameter.12 In addition, when a catheter is inserted into the superior vena cava, the direction of blood flow and the direction of the infusion are the same, but when the catheter is inserted into the azygos system, the directions of blood flow and infusion are opposite, contributing to local turbulence.
Both these factors increase the chance of puncturing the vein when the azygos arch is aberrantly cannulated for central venous access.9 Venous perforation has been reported in as many as 19% of cases in which the azygos arch was inadvertently cannulated. Venous perforation can lead to hemopericardium, hemomediastinum, and hemorrhagic pleural effusions, which can lead to significant morbidity and even death. Perforation, thrombosis, stenosis, and complete occlusion have been reported subsequent to catheter malposition in the azygos vein.13
Patients in whom the azygos vein is inadvertently cannulated may tolerate infusions and blood draws, but this does not mean that inadvertent azygos vein cannulation is acceptable, especially given the late complications of vascular perforation that can occur.9
The cannulation of the azygos vein in our patient was completely unintentional; nevertheless, the line was kept in and used for a short period for the initial rehydration and pain control and was subsequently removed without any complications.
WHEN IS CANNULATION OF THE AZYGOS VEIN AN OPTION?
In patients with previous multiple central vein cannulations, the rates of thrombosis and of fibrotic changes in these veins are high. In patients with thrombosis of both the superior vena cava and the inferior vena cava, direct insertion of a catheter into the azygos vein has been suggested as an alternate route to obtain access for dialysis.8 This approach has also been used successfully to administer total parenteral nutrition for a prolonged time in pediatric patients.14 In short, the azygos vein is never preferred for central venous access, but it can occasionally serve as an alternate route.5–9
TAKE-HOME POINTS
The radiographic assessment of an internal jugular or subclavian line may occasionally be deceptive if based solely on the anteroposterior view; confirmation may require a lateral view. We found no guidelines for using the azygos vein for central venous access. The options in cases of aberrant cannulation include leaving the line in, removing and reinserting it at the same or another site under fluoroscopy, and attempting to reposition it after changing the catheter over a guidewire.
The use of central lines found to be in an aberrant position should be driven by clinical judgment based on the urgency of the need of access, the availability or feasibility of other access options, and the intended use. The use of the azygos vein is fraught with procedural complications, as well as postprocedural complications related to vascular perforation. If the position of the catheter tip on the anteroposterior radiographic view is not satisfactory, obtaining a lateral view should be considered.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
- McGee DC, Goud MK. Preventing complications of central venous catheterization. N Engl J Med. 2003; 348:1123–1133.
- Pittiruti M, Malerba M, Carriero C, Tazza L, Gui D. Which is the easiest and safest technique for central venous access? A retrospective survey of more than 5,400 cases. J Vasc Access. 2000; 1:100–107.
- Towers MJ. Preventing complications of central venous catheterization. N Engl J Med 2003; 348:2684–2686; author reply 2684–2686.
- Langston CS. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Smith DC, Pop PM. Malposition of a total parenteral nutrition catheter in the accessory hemiazygos vein. JPEN J Parenter Enteral Nutr. 1983; 7:289–292.
- Abood GJ, Davis KA, Esposito TJ, Luchette FA, Gamelli RL. Comparison of routine chest radiograph versus clinician judgment to determine adequate central line placement in critically ill patients. J Trauma. 2007; 63:50–56.
- Gladwin MT, Slonim A, Landucci DL, Gutierrez DC, Cunnion RE. Cannulation of the internal jugular vein: is postprocedural chest radiography always necessary? Crit Care Med 1999; 27:1819–1823.
- Meranze SG, McLean GK, Stein EJ, Jordan HA. Catheter placement in the azygos system: an unusual approach to venous access. Am J Roentgenol. 1985; 144:1075–1076.
- Bankier AA, Mallek R, Wiesmayr MN, et al. Azygos arch cannulation by central venous catheters: radiographic detection of malposition and subsequent complications. J Thorac Imaging. 1997; 12:64–69.
- Langston CT. The aberrant central venous catheter and its complications. Radiology. 1971; 100:55–59.
- Heitzman ER. Radiologic appearance of the azygos vein in cardiovascular disease. Circulation. 1973; 47:628–634.
- McGowan AR, Pugatch RD. Partial obstruction of the superior vena cava. BrighamRAD. Available at: http://brighamrad.harvard.edu/Cases/bwh/hcache/58/full.html. Accessed 9/4/2008.
- Granata A, Figuera M, Castellino S, Logias F, Basile A. Azygos arch cannulation by central venous catheters for hemodialysis. J Vasc Access. 2006; 7:43–45.
- Malt RA, Kempster M. Direct azygos vein and superior vena cava cannulation for parenteral nutrition. JPEN J Parenter Enteral Nutr. 1983; 7:580–581.
A 51-year-old man with nodular lesions
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
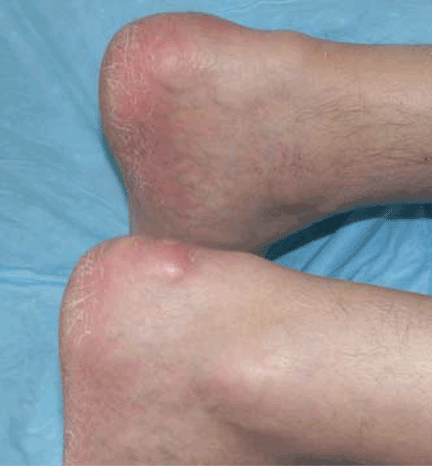
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
A 51-year-old diabetic man presents with a 1-year history of episodic pain, swelling, and stiffness in some of the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) joints of his fingers. During these episodes, he has significant morning stiffness. He says he has no other joint problems or back pain. A review of systems is otherwise unremarkable.
WHAT IS THE MOST LIKELY DIAGNOSIS?
- Gouty tophi
- Rheumatoid nodulosis
- Calcinosis cutis
- Tuberous xanthomas
GOUTY TOPHI: OUR INITIAL IMPRESSION
RHEUMATOID NODULOSIS: THE TRUE DIAGNOSIS
The patient’s nodules kept growing, and new ones kept developing, causing significant impairment of hand function. Hence, some of the larger nodules were surgically removed. The resected specimens revealed a yellow nodular cut surface on sectioning. Histopathologic analysis revealed multiple necrobiotic nodules, consistent with rheumatoid nodulosis. Urate crystals were not seen on histology, although crystals can be dissolved in some tissue preparations, and gouty tophi provoke pathologically a granulomatous inflammatory reaction. However, unlike what is expected with gouty tophi, material aspirated from the nodules did not reveal monosodium urate crystals on polarized light microscopy. A repeat rheumatoid factor test 2 years after his initial presentation became positive at 57 IU/ mL (normal < 20 IU/mL).
Comment. Rheumatoid nodules are one of the most common extra-articular manifestations of rheumatoid arthritis, seen in 20% to 25% of cases, and they are usually associated with seropositivity for rheumatoid factor and with more aggressive disease.1 Rheumatoid nodulosis, on the other hand, usually runs a more benign clinical course.2 It was first described in 1949,3 and the diagnostic criteria were developed in 1988 by Couret et al.4 Patients develop nontender subcutaneous rheumatoid nodules, usually around areas of repeated microtrauma.2 Often there is a history of attacks of palindromic rheumatism, characterized by recurrent, self-limited episodes of monoarthritis or polyarthritis without an alternative explanation, as in this patient. However, systemic manifestations of rheumatoid arthritis and radiologic evidence of joint damage are often not seen. Rheumatoid factor positivity is also not a requirement. Over time, some patients progress to full-blown rheumatoid arthritis. Methotrexate use has been associated with accelerated rheumatoid nodulosis in some rheumatoid arthritis patients.2
Rheumatoid nodulosis can be progressive and difficult to treat. Hydroxychloroquine has induced complete resolution in some cases.5 Surgical removal of the nodules may be considered if they limit joint motion.6 A placebo-controlled, double-blind trial of intralesional corticosteroid injection has demonstrated efficacy in reducing nodule size.7
In our patient, treatment with hydroxychloroquine, sulfasalazine, and methotrexate did not relieve the joint pain, nor did these drugs stop the nodules from growing. He was started on the tumor necrosis factor antagonist etanercept (Enbrel), which significantly helped the joint pain, but the nodules continued to progress relentlessly. Some of the larger nodules were later injected with triamcinolone (Kenalog), which led to significant shrinkage in nodule size.
THE OTHER DIAGNOSTIC CHOICES
The other two choices are unlikely.
Calcinosis cutis results from the cutaneous deposition of insoluble compounds of calcium (hydroxyapatite or amorphous calcium phosphate), due to local or systemic factors, or both. This can be the result either of ectopic calcification in normal tissue in the setting of hypercalcemia or hyperphosphatemia, or of dystrophic calcification in damaged tissue. They appear as multiple, firm, whitish dermal papules, plaques, nodules, or subcutaneous nodules, which can sometimes ulcerate. They are radio-opaque. On biopsy, dermal deposits of calcium are seen, with or without a surrounding foreign-body giant-cell reaction. Calcium deposition may be confirmed on Von Kossa and alizarin red stains.
Tuberous xanthomas are firm, painless, red-yellow nodules that usually develop in pressure areas such as the extensor surfaces of the knees, the elbows, and the buttocks. They can be associated with familial dysbetalipoproteinemia, familial hypercholesterolemia, and even some of the secondary dyslipidemias. Histologic study shows accumulations of vacuolated lipid-laden macrophages (foamy histiocytes) and sometimes multinucleated histiocytes (Touton giant cells). The lipid droplets are dissolved during routine histologic processing, but lipid stains on frozen sections can be useful.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
- Ziff M. The rheumatoid nodule. Arthritis Rheum 1990; 33:761–767.
- Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg 2007; 26:100–107.
- Bywaters EGL. A variant of rheumatoid arthritis characterized by recurrent digital pad nodules and palmar fasciitis, closely resembling palindromic rheumatism. Ann Rheum Dis 1949; 8:2–30.
- Couret M, Combe B, Chuong VT, et al. Rheumatoid nodulosis: report of two new cases and discussion of diagnostic criteria. J Rheumatol 1988; 15:1427–1430.
- McCarty DJ. Complete reversal of rheumatoid nodulosis. J Rheumatol 1991; 18:736–737.
- Kai Y, Anzai S, Shibuya H, et al. A case of rheumatoid nodulosis successfully treated with surgery. J Dermatol 2004; 31:910–915.
- Ching DW, Petrie JP, Klemp P, Jones JG. Injection therapy of superficial rheumatoid nodules. Br J Rheumatol 1992; 31:775–777.
Sudden hair loss associated with trachyonychia
A 30-year-old woman has a 6-month history of episodes of sudden loss of hair in well-demarcated, localized areas of her scalp. She is otherwise well and has had no pruritus or pain. She has had atopic dermatitis since childhood, occasionally treated with topical corticosteroids, and localized facial vitiligo for the past 8 years. She takes no systemic drugs. She has no family history of similar lesions.
Q: What is the most likely diagnosis?
- Telogen effluvium
- Syphilitic alopecia
- Alopecia areata
- Trichotillomania
- Androgenetic alopecia
A: The correct answer is alopecia areata, a disease characterized by recurrent episodes of sudden hair loss and nail changes such as trachyonychia. It can affect any hair-bearing area, but the scalp is the most common site (90% of cases).1 The condition can range from a single patch of hair loss to complete loss of hair on the scalp (alopecia totalis) or the entire body (alopecia universalis).
The characteristic lesion is a round or oval, totally bald, smooth patch, which may have a mild peachy hue. A frequent feature is “exclamation-mark” hairs at the margin of the patch, appearing as broad distal hair shafts, narrow proximally. Active alopecia areata can be confirmed with a positive hair-pull test.
Nail involvement is common in alopecia areata, particularly in severe forms. Pitting is the most common finding, but other abnormalities such as trachyonychia have been reported.1 Nail plate pitting tends to be more shallow and regularly spaced than in psori-atic nail pitting. Trachyonychia, also known as “twenty-nail dystrophy,” is the term used to describe nail plate roughness, pitting, and ridging, affecting 1 to 20 nails. It is associated with a number of skin conditions, including alopecia areata, psoriasis, lichen planus, atopic dermatitis, and ichthyosis vulgaris.4
AN AUTOIMMUNE DISEASE
The precise cause of this common disorder has not been elucidated, but substantial evidence suggests that it is an organ-specific autoimmune disease that targets hair follicles.1–3
The current hypothesis is that the hair follicle is a site of immune privilege. In alopecia areata, increased expression of major histocompatibility complex class I molecules and down-regulation of immunosuppressants allows the immune system to recognize the immune-privileged follicle antigens.2 Several autoimmune diseases, such as thyroid disorders, vitiligo, pernicious anemia, diabetes mellitus, lupus erythematosus, myasthenia gravis, lichen planus, autoimmune polyendocrine syndrome type I, and celiac disease, have been reported in association with alopecia areata1; thyroid disorders and vitiligo have the strongest relationship. Other diseases reported to be associated with alopecia areata at a higher rate than in the general population are atopic dermatitis and Down syndrome.1
NO CURE, BUT TREATABLE
The choice of treatment depends on the patient’s age and the extent of alopecia activity.5 No cure or preventive treatment has been found, so treatments are directed toward halting disease activity.
Corticosteroids are currently the most popular form of treatment, and published reports support their efficacy.1 Topical and intralesional steroids are usually the first-line treatment for localized disease. Systemic steroids are rarely used, except in cases with rapid progression.
Other treatments that have been used with some success include minoxidil (Rogaine), anthralin (Dritho-Scalp), contact sensitizers (dinitrochlorobenzene, diphenylcycloprope-none), PUVA (psoralen plus ultraviolet A light), and cyclosporine (Sandimmune).1 If the alopecia is resistant to therapy, hair pros-theses can be recommended.
Indicators of poor prognosis (ie, hair that will not regrow completely) include atopy, the presence of other immune diseases, young age at onset, family history, nail dystrophy, extensive hair loss, and ophiasis (a continuous band of baldness involving the temporal and occipital margins).
Because current treatments may not bring results for 3 to 6 months, it is essential to reassure and inform the patient about the results that can be expected. Physicians should also inform patients of the chronic relapsing nature of alopecia areata and its unpredictable course. Although the condition does not threaten the patient’s general health and does not cause scarring, its psychological impact is significant. Support groups such as the National Alopecia Areata Foundation (www.naaf.org) can help.
- Wasserman D, Guzman-Sanchez DA, Scott K, McMichael A. Alopecia areata. Int J Dermatol 2007; 46:121–131.
- Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc 2003; 8:188–194.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol 2004; 14:364–370.
- Blanco FP, Scher RK. Trachyonychia: case report and review of the literature. J Drugs Dermatol 2006; 5:469–472.
- Dombrowski NC, Bergfeld WF. Alopecia areata: what to expect from current treatments. Cleve Clin J Med 2005; 72:758–768.
A 30-year-old woman has a 6-month history of episodes of sudden loss of hair in well-demarcated, localized areas of her scalp. She is otherwise well and has had no pruritus or pain. She has had atopic dermatitis since childhood, occasionally treated with topical corticosteroids, and localized facial vitiligo for the past 8 years. She takes no systemic drugs. She has no family history of similar lesions.
Q: What is the most likely diagnosis?
- Telogen effluvium
- Syphilitic alopecia
- Alopecia areata
- Trichotillomania
- Androgenetic alopecia
A: The correct answer is alopecia areata, a disease characterized by recurrent episodes of sudden hair loss and nail changes such as trachyonychia. It can affect any hair-bearing area, but the scalp is the most common site (90% of cases).1 The condition can range from a single patch of hair loss to complete loss of hair on the scalp (alopecia totalis) or the entire body (alopecia universalis).
The characteristic lesion is a round or oval, totally bald, smooth patch, which may have a mild peachy hue. A frequent feature is “exclamation-mark” hairs at the margin of the patch, appearing as broad distal hair shafts, narrow proximally. Active alopecia areata can be confirmed with a positive hair-pull test.
Nail involvement is common in alopecia areata, particularly in severe forms. Pitting is the most common finding, but other abnormalities such as trachyonychia have been reported.1 Nail plate pitting tends to be more shallow and regularly spaced than in psori-atic nail pitting. Trachyonychia, also known as “twenty-nail dystrophy,” is the term used to describe nail plate roughness, pitting, and ridging, affecting 1 to 20 nails. It is associated with a number of skin conditions, including alopecia areata, psoriasis, lichen planus, atopic dermatitis, and ichthyosis vulgaris.4
AN AUTOIMMUNE DISEASE
The precise cause of this common disorder has not been elucidated, but substantial evidence suggests that it is an organ-specific autoimmune disease that targets hair follicles.1–3
The current hypothesis is that the hair follicle is a site of immune privilege. In alopecia areata, increased expression of major histocompatibility complex class I molecules and down-regulation of immunosuppressants allows the immune system to recognize the immune-privileged follicle antigens.2 Several autoimmune diseases, such as thyroid disorders, vitiligo, pernicious anemia, diabetes mellitus, lupus erythematosus, myasthenia gravis, lichen planus, autoimmune polyendocrine syndrome type I, and celiac disease, have been reported in association with alopecia areata1; thyroid disorders and vitiligo have the strongest relationship. Other diseases reported to be associated with alopecia areata at a higher rate than in the general population are atopic dermatitis and Down syndrome.1
NO CURE, BUT TREATABLE
The choice of treatment depends on the patient’s age and the extent of alopecia activity.5 No cure or preventive treatment has been found, so treatments are directed toward halting disease activity.
Corticosteroids are currently the most popular form of treatment, and published reports support their efficacy.1 Topical and intralesional steroids are usually the first-line treatment for localized disease. Systemic steroids are rarely used, except in cases with rapid progression.
Other treatments that have been used with some success include minoxidil (Rogaine), anthralin (Dritho-Scalp), contact sensitizers (dinitrochlorobenzene, diphenylcycloprope-none), PUVA (psoralen plus ultraviolet A light), and cyclosporine (Sandimmune).1 If the alopecia is resistant to therapy, hair pros-theses can be recommended.
Indicators of poor prognosis (ie, hair that will not regrow completely) include atopy, the presence of other immune diseases, young age at onset, family history, nail dystrophy, extensive hair loss, and ophiasis (a continuous band of baldness involving the temporal and occipital margins).
Because current treatments may not bring results for 3 to 6 months, it is essential to reassure and inform the patient about the results that can be expected. Physicians should also inform patients of the chronic relapsing nature of alopecia areata and its unpredictable course. Although the condition does not threaten the patient’s general health and does not cause scarring, its psychological impact is significant. Support groups such as the National Alopecia Areata Foundation (www.naaf.org) can help.
A 30-year-old woman has a 6-month history of episodes of sudden loss of hair in well-demarcated, localized areas of her scalp. She is otherwise well and has had no pruritus or pain. She has had atopic dermatitis since childhood, occasionally treated with topical corticosteroids, and localized facial vitiligo for the past 8 years. She takes no systemic drugs. She has no family history of similar lesions.
Q: What is the most likely diagnosis?
- Telogen effluvium
- Syphilitic alopecia
- Alopecia areata
- Trichotillomania
- Androgenetic alopecia
A: The correct answer is alopecia areata, a disease characterized by recurrent episodes of sudden hair loss and nail changes such as trachyonychia. It can affect any hair-bearing area, but the scalp is the most common site (90% of cases).1 The condition can range from a single patch of hair loss to complete loss of hair on the scalp (alopecia totalis) or the entire body (alopecia universalis).
The characteristic lesion is a round or oval, totally bald, smooth patch, which may have a mild peachy hue. A frequent feature is “exclamation-mark” hairs at the margin of the patch, appearing as broad distal hair shafts, narrow proximally. Active alopecia areata can be confirmed with a positive hair-pull test.
Nail involvement is common in alopecia areata, particularly in severe forms. Pitting is the most common finding, but other abnormalities such as trachyonychia have been reported.1 Nail plate pitting tends to be more shallow and regularly spaced than in psori-atic nail pitting. Trachyonychia, also known as “twenty-nail dystrophy,” is the term used to describe nail plate roughness, pitting, and ridging, affecting 1 to 20 nails. It is associated with a number of skin conditions, including alopecia areata, psoriasis, lichen planus, atopic dermatitis, and ichthyosis vulgaris.4
AN AUTOIMMUNE DISEASE
The precise cause of this common disorder has not been elucidated, but substantial evidence suggests that it is an organ-specific autoimmune disease that targets hair follicles.1–3
The current hypothesis is that the hair follicle is a site of immune privilege. In alopecia areata, increased expression of major histocompatibility complex class I molecules and down-regulation of immunosuppressants allows the immune system to recognize the immune-privileged follicle antigens.2 Several autoimmune diseases, such as thyroid disorders, vitiligo, pernicious anemia, diabetes mellitus, lupus erythematosus, myasthenia gravis, lichen planus, autoimmune polyendocrine syndrome type I, and celiac disease, have been reported in association with alopecia areata1; thyroid disorders and vitiligo have the strongest relationship. Other diseases reported to be associated with alopecia areata at a higher rate than in the general population are atopic dermatitis and Down syndrome.1
NO CURE, BUT TREATABLE
The choice of treatment depends on the patient’s age and the extent of alopecia activity.5 No cure or preventive treatment has been found, so treatments are directed toward halting disease activity.
Corticosteroids are currently the most popular form of treatment, and published reports support their efficacy.1 Topical and intralesional steroids are usually the first-line treatment for localized disease. Systemic steroids are rarely used, except in cases with rapid progression.
Other treatments that have been used with some success include minoxidil (Rogaine), anthralin (Dritho-Scalp), contact sensitizers (dinitrochlorobenzene, diphenylcycloprope-none), PUVA (psoralen plus ultraviolet A light), and cyclosporine (Sandimmune).1 If the alopecia is resistant to therapy, hair pros-theses can be recommended.
Indicators of poor prognosis (ie, hair that will not regrow completely) include atopy, the presence of other immune diseases, young age at onset, family history, nail dystrophy, extensive hair loss, and ophiasis (a continuous band of baldness involving the temporal and occipital margins).
Because current treatments may not bring results for 3 to 6 months, it is essential to reassure and inform the patient about the results that can be expected. Physicians should also inform patients of the chronic relapsing nature of alopecia areata and its unpredictable course. Although the condition does not threaten the patient’s general health and does not cause scarring, its psychological impact is significant. Support groups such as the National Alopecia Areata Foundation (www.naaf.org) can help.
- Wasserman D, Guzman-Sanchez DA, Scott K, McMichael A. Alopecia areata. Int J Dermatol 2007; 46:121–131.
- Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc 2003; 8:188–194.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol 2004; 14:364–370.
- Blanco FP, Scher RK. Trachyonychia: case report and review of the literature. J Drugs Dermatol 2006; 5:469–472.
- Dombrowski NC, Bergfeld WF. Alopecia areata: what to expect from current treatments. Cleve Clin J Med 2005; 72:758–768.
- Wasserman D, Guzman-Sanchez DA, Scott K, McMichael A. Alopecia areata. Int J Dermatol 2007; 46:121–131.
- Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc 2003; 8:188–194.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol 2004; 14:364–370.
- Blanco FP, Scher RK. Trachyonychia: case report and review of the literature. J Drugs Dermatol 2006; 5:469–472.
- Dombrowski NC, Bergfeld WF. Alopecia areata: what to expect from current treatments. Cleve Clin J Med 2005; 72:758–768.