User login
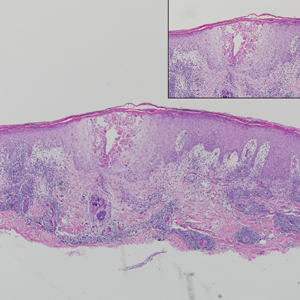
Ulcerated Nodule on the Scalp
The Diagnosis: Proliferating Pilar Tumor
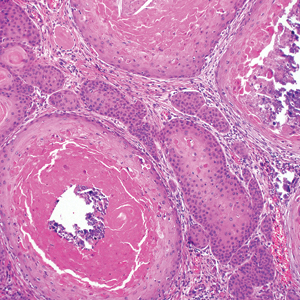
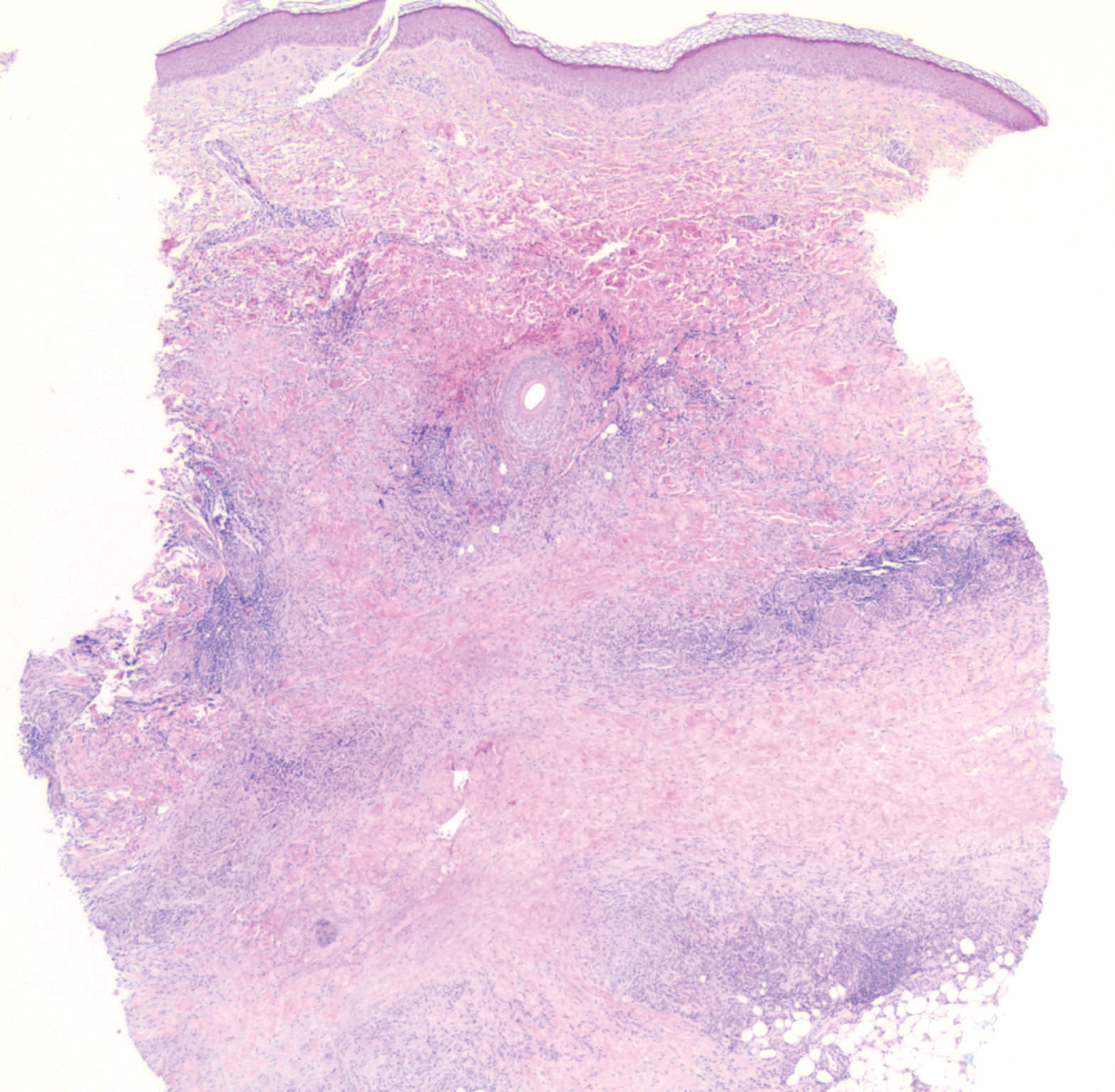
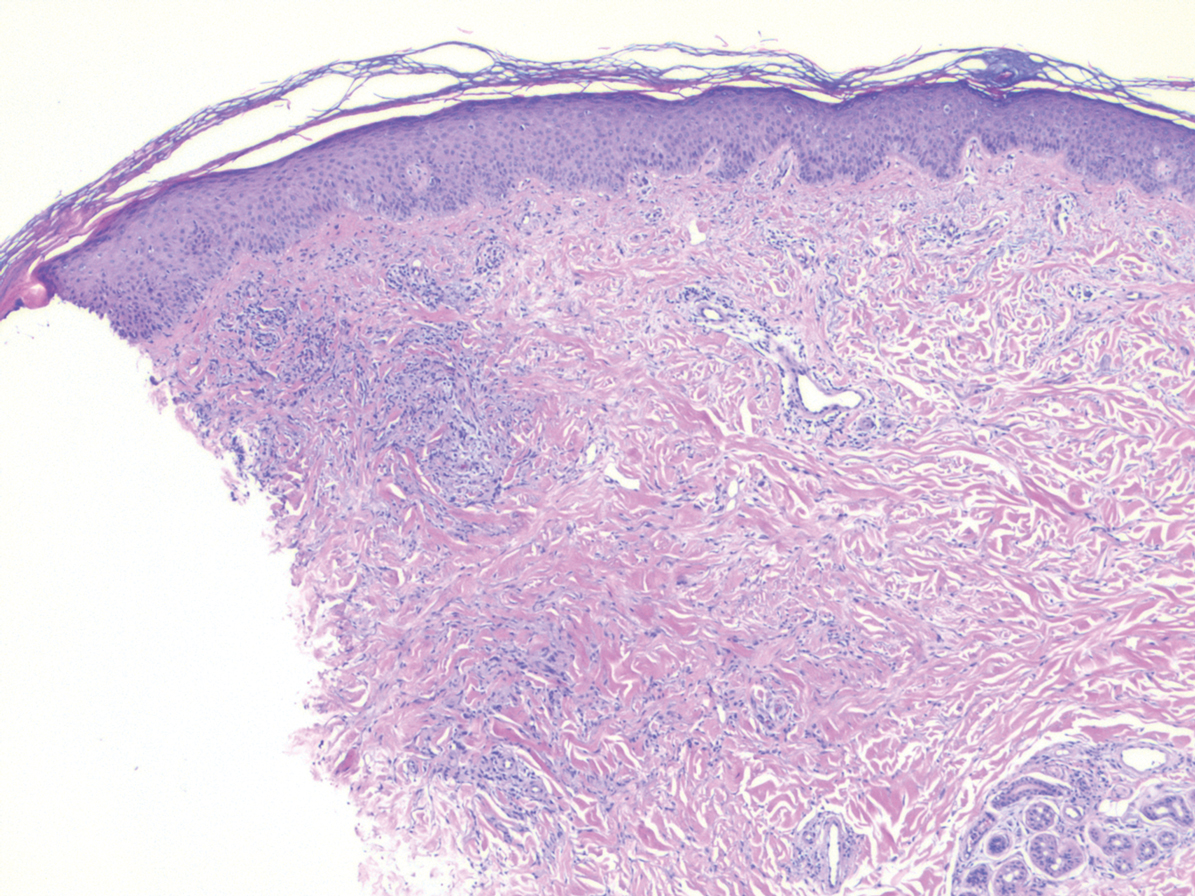
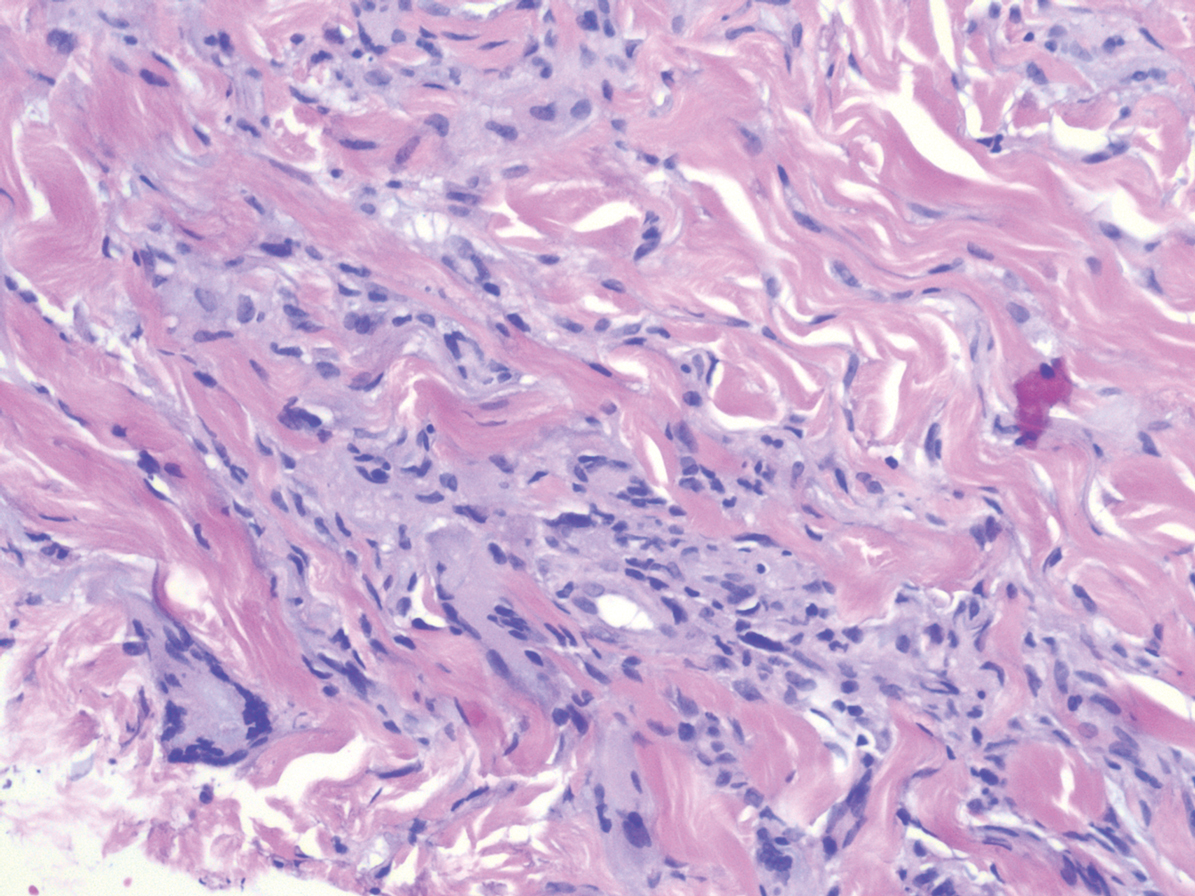
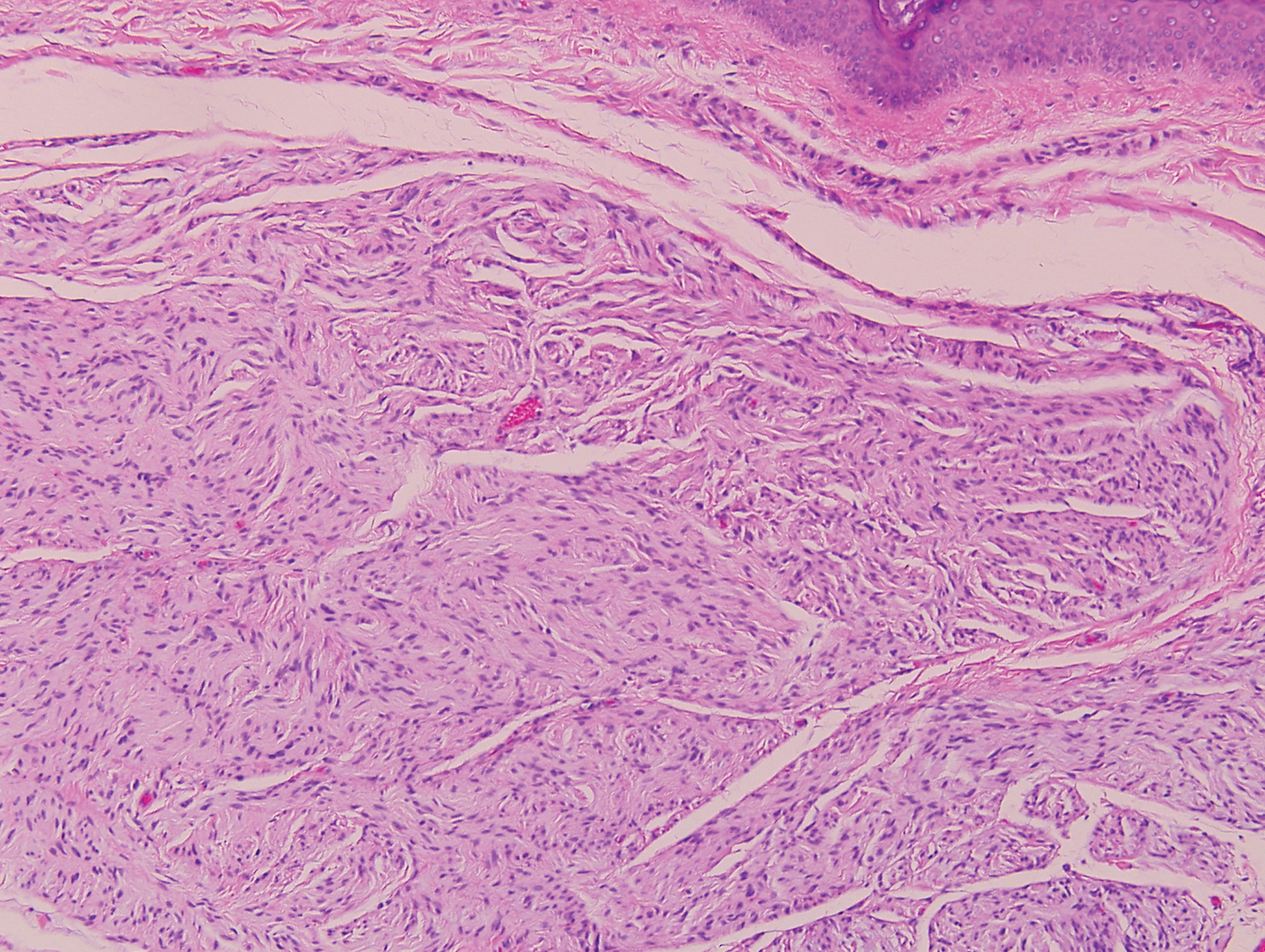
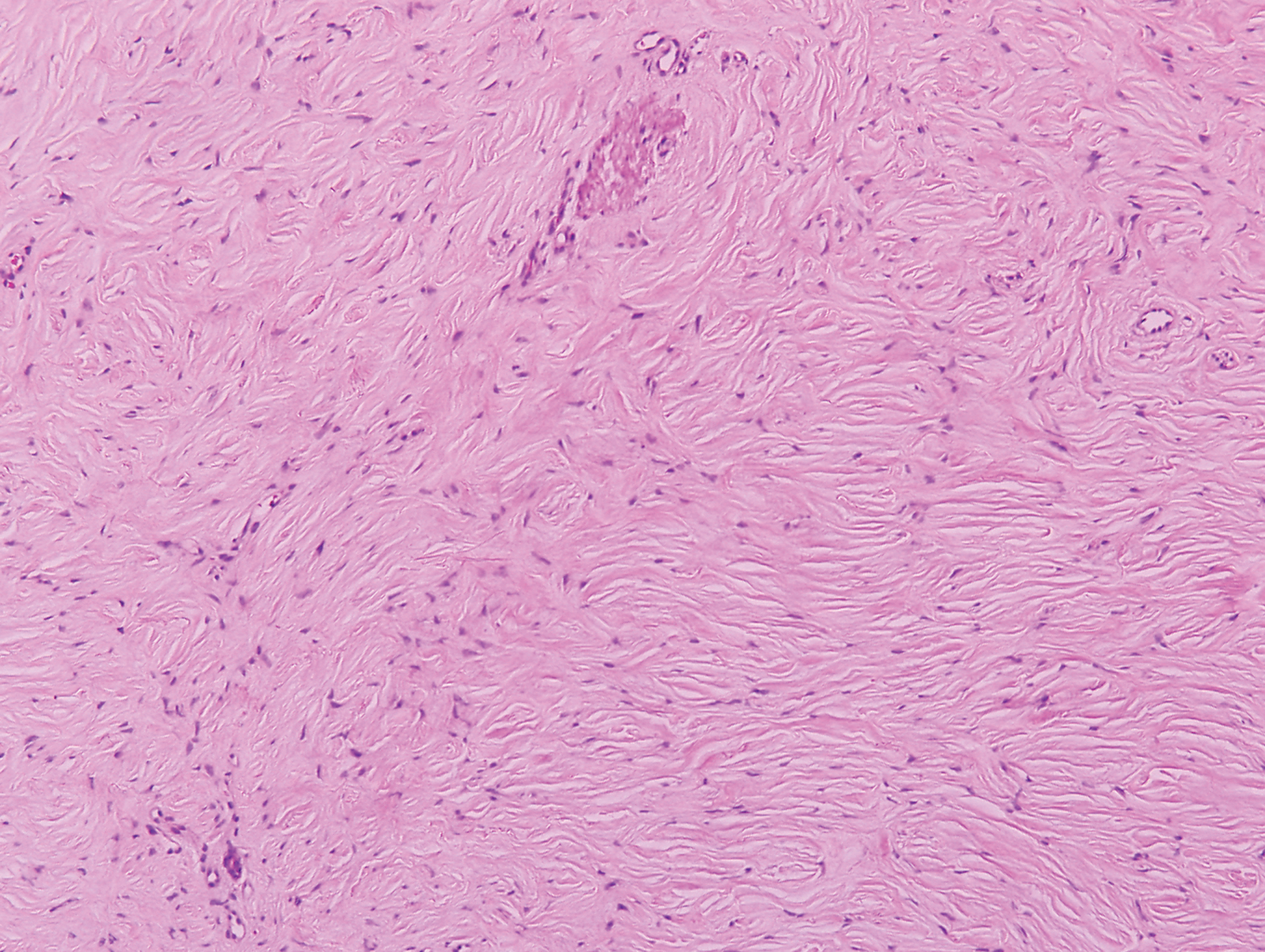
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
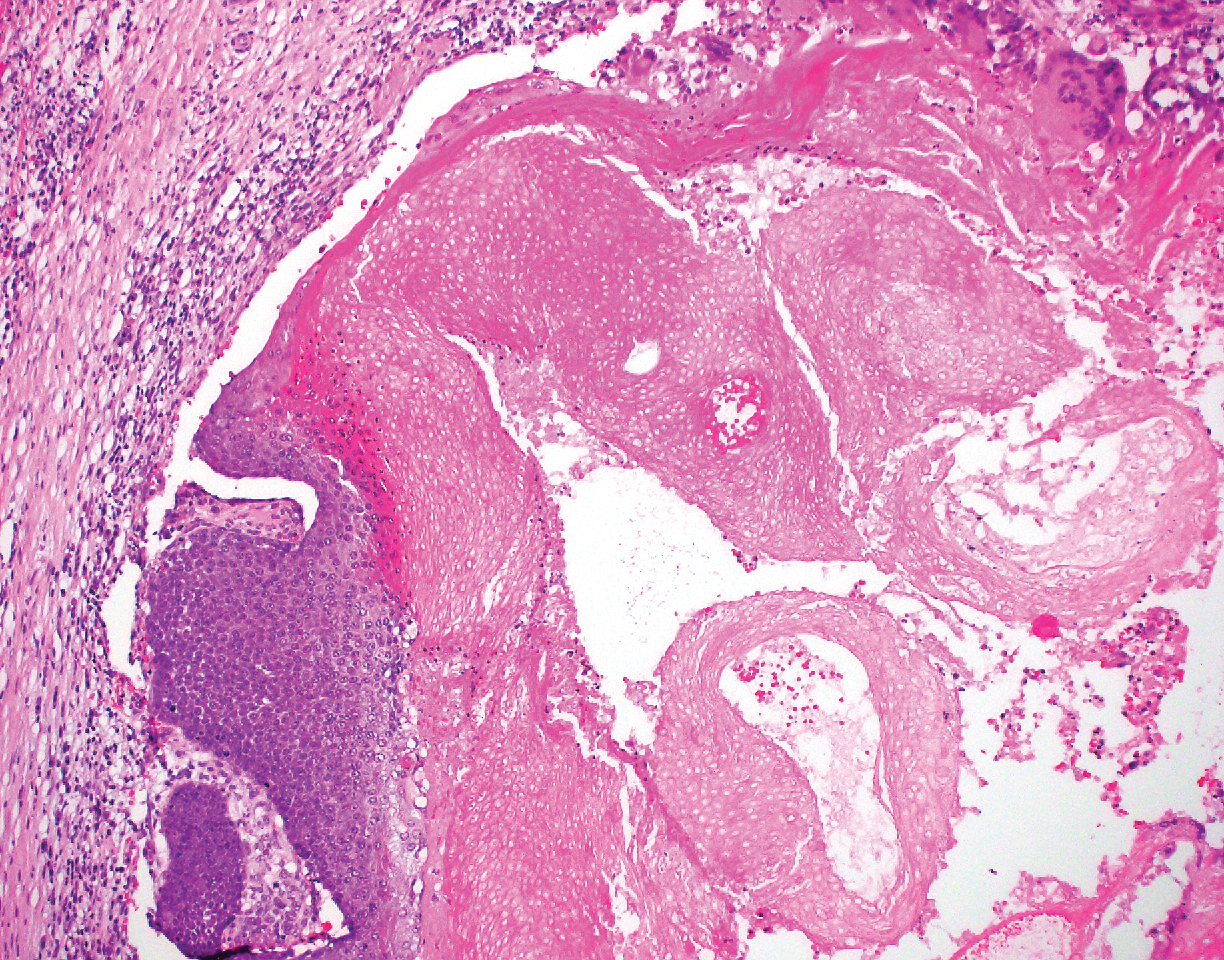
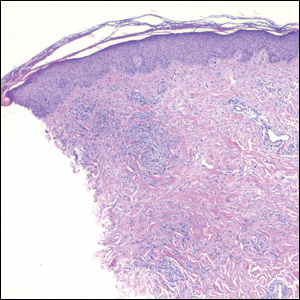
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
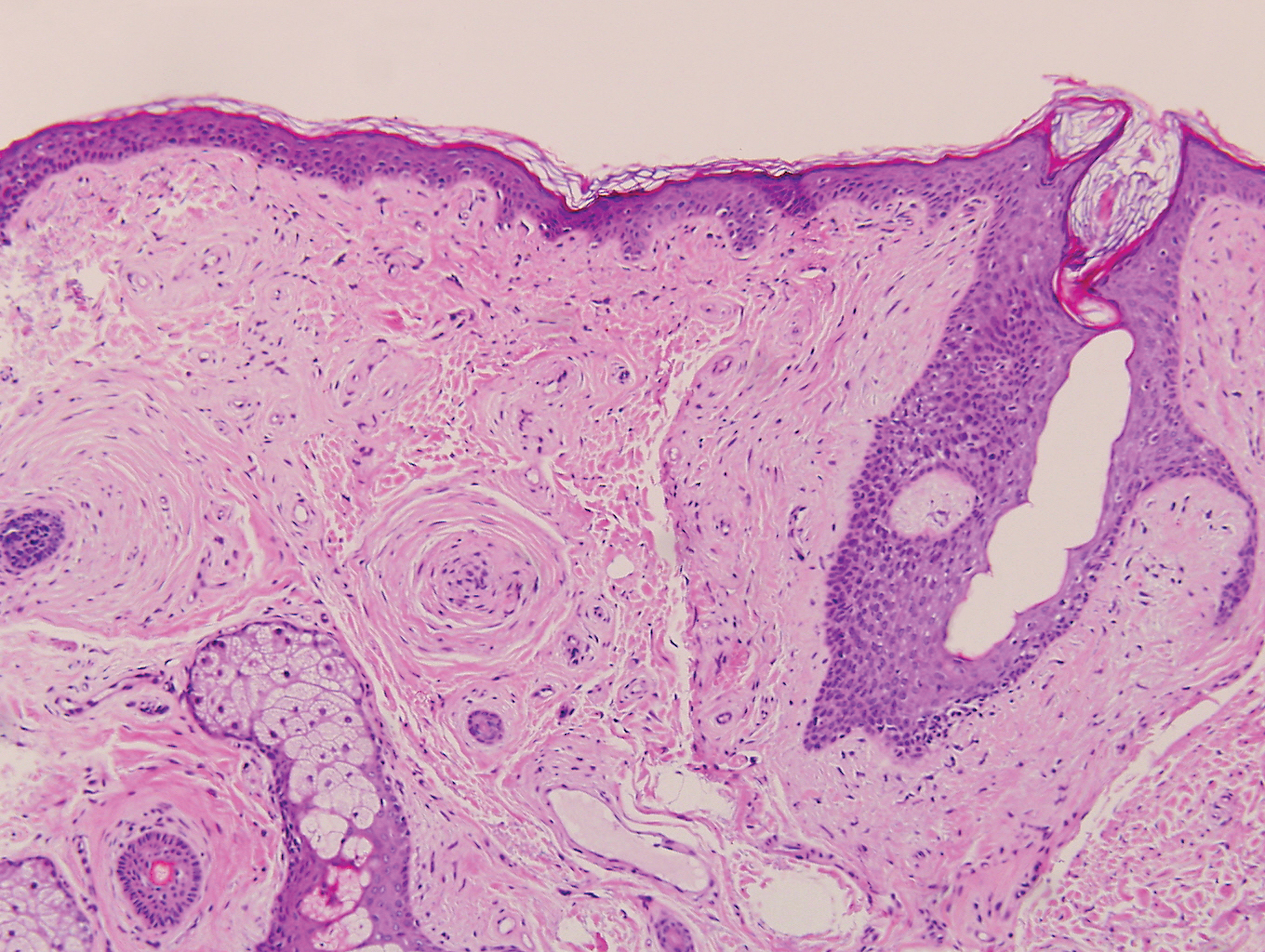
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
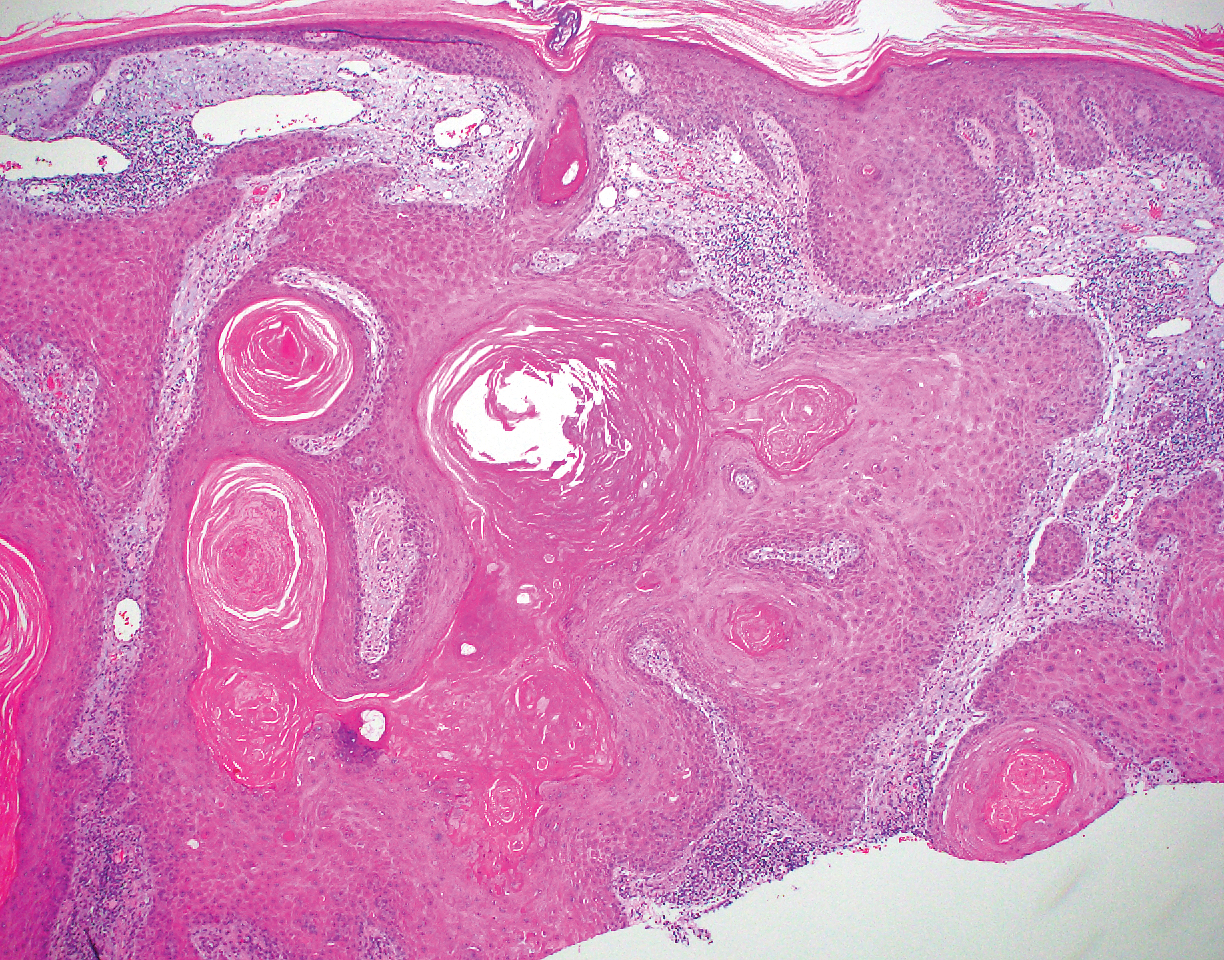
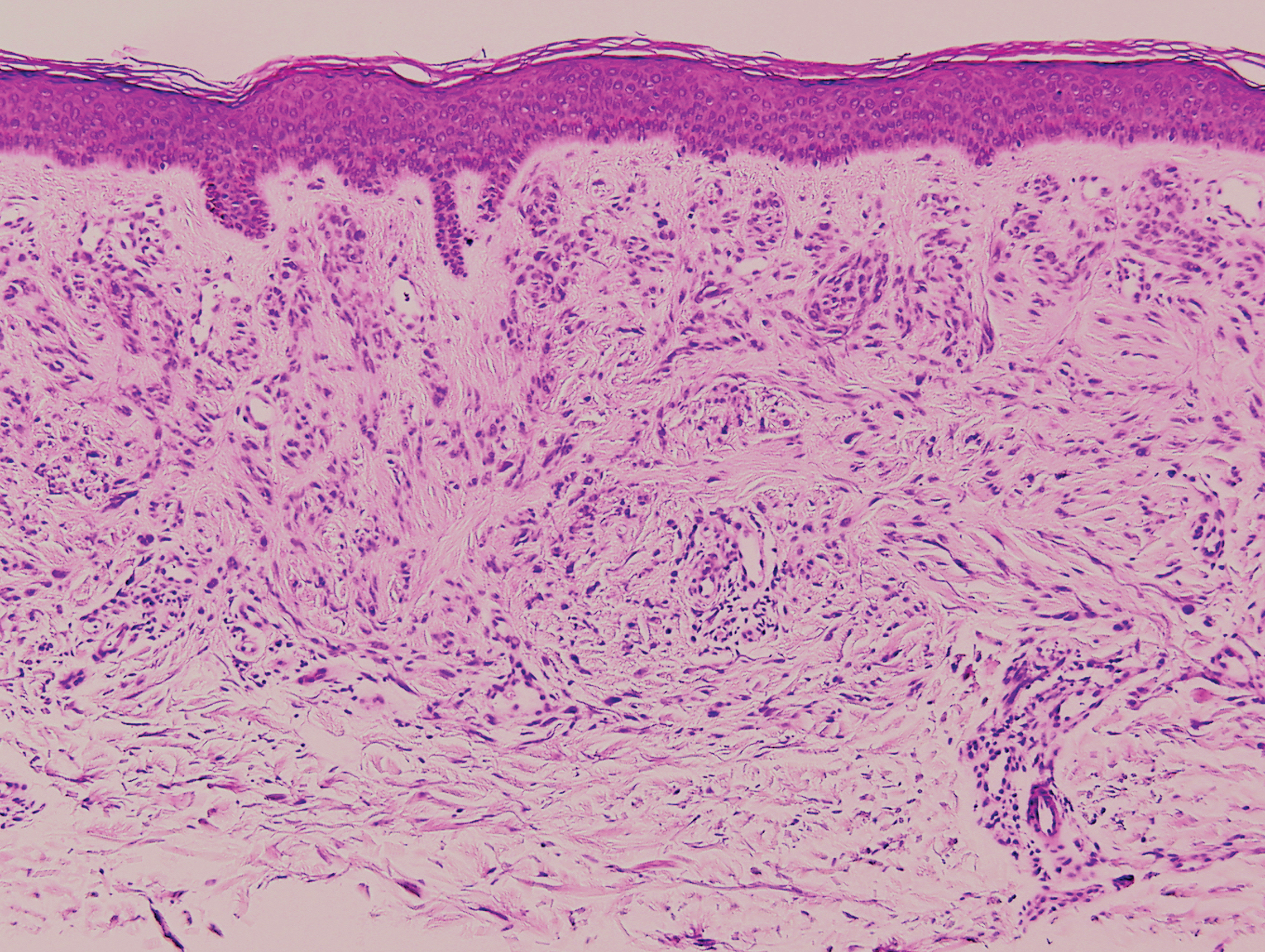
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion. 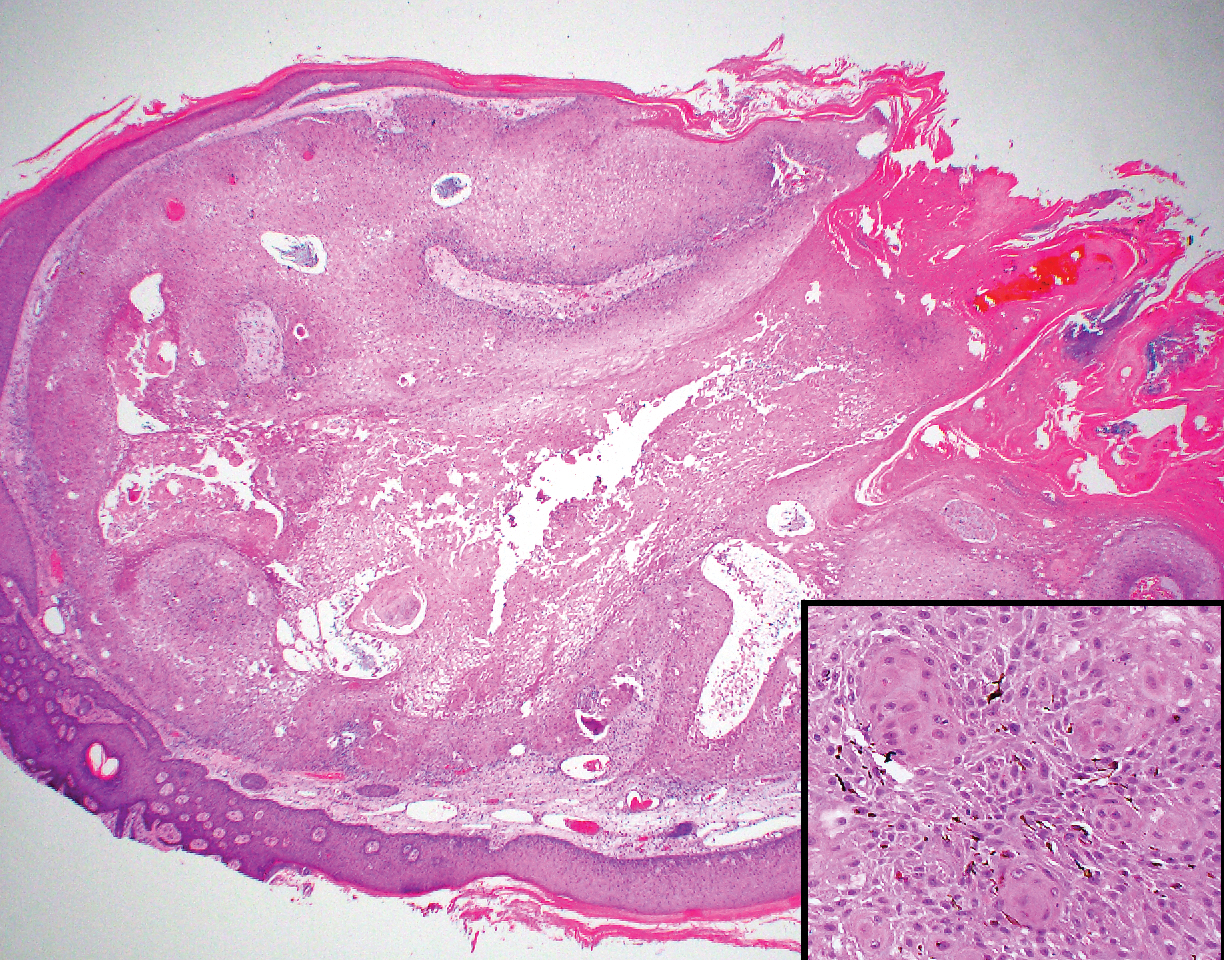
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29

Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
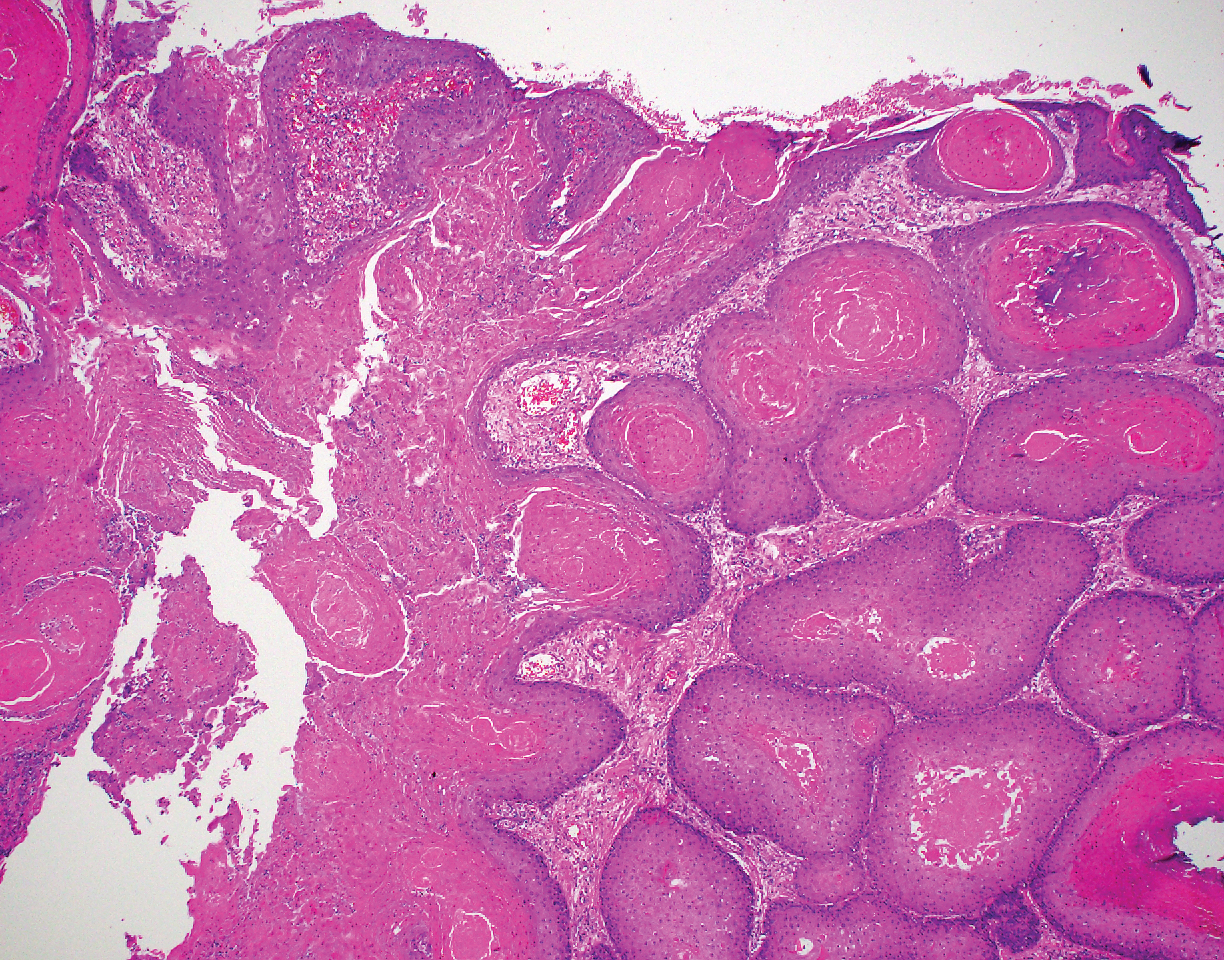
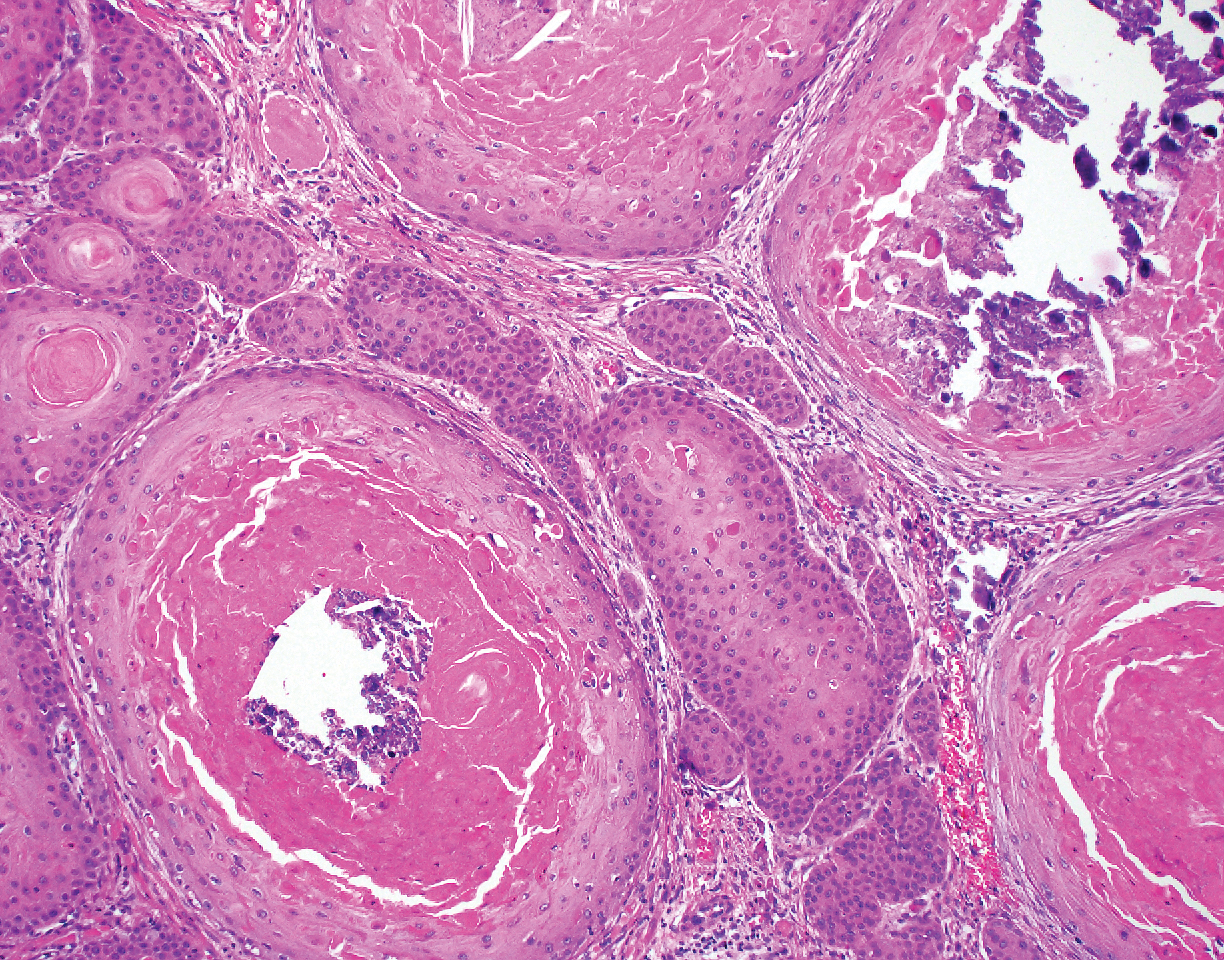
A 66-year-old woman presented to the dermatology clinic with a rapidly enlarging, draining lesion on the scalp. The lesion seemed to enlarge over the last 3 months from a lesion that had been there for years. Physical examination revealed a 2.2-cm ulcerated nodule on the right parietal scalp. A shave biopsy was obtained.
Tender Papules on the Bilateral Dorsal Hands
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
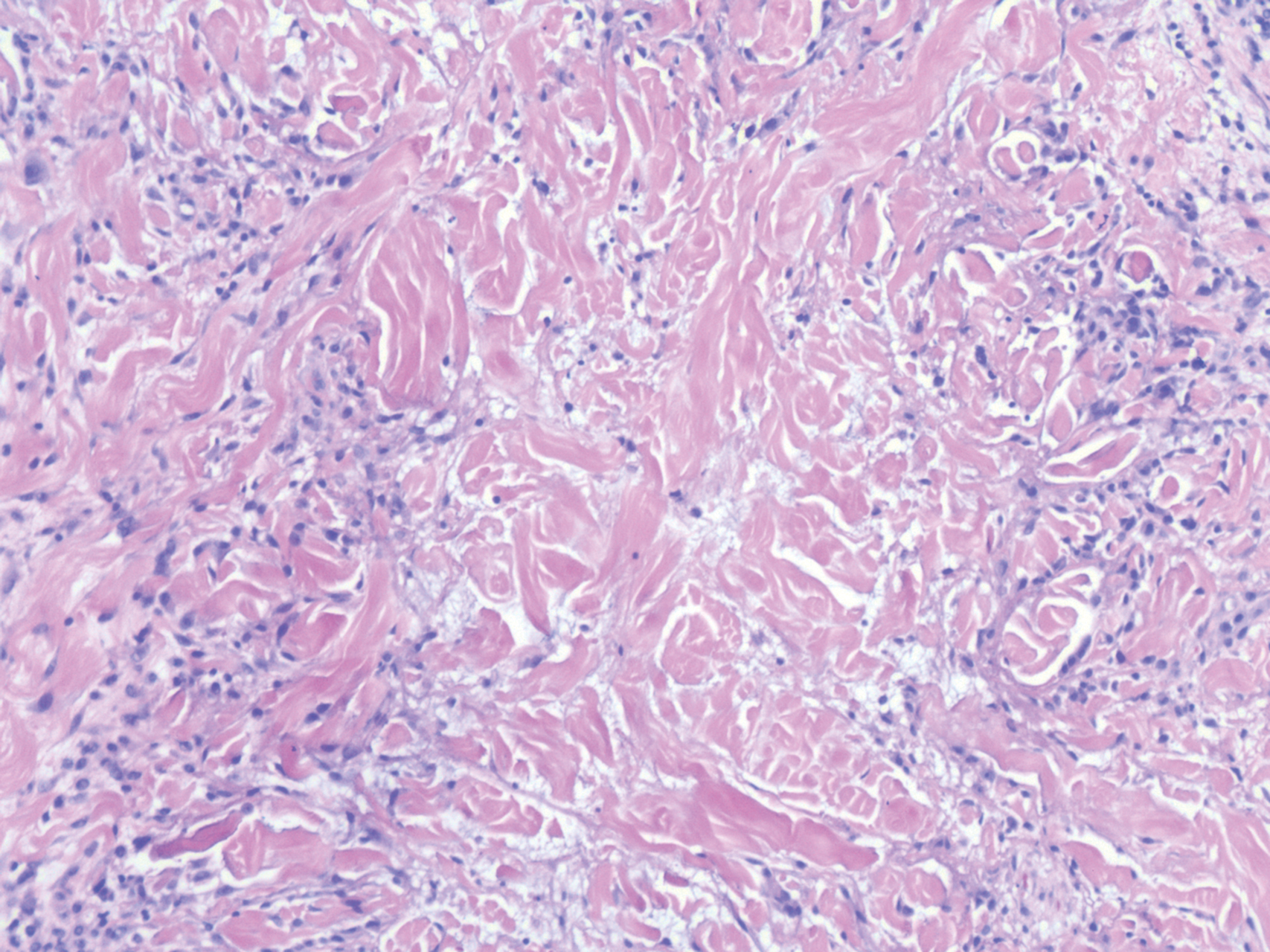
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
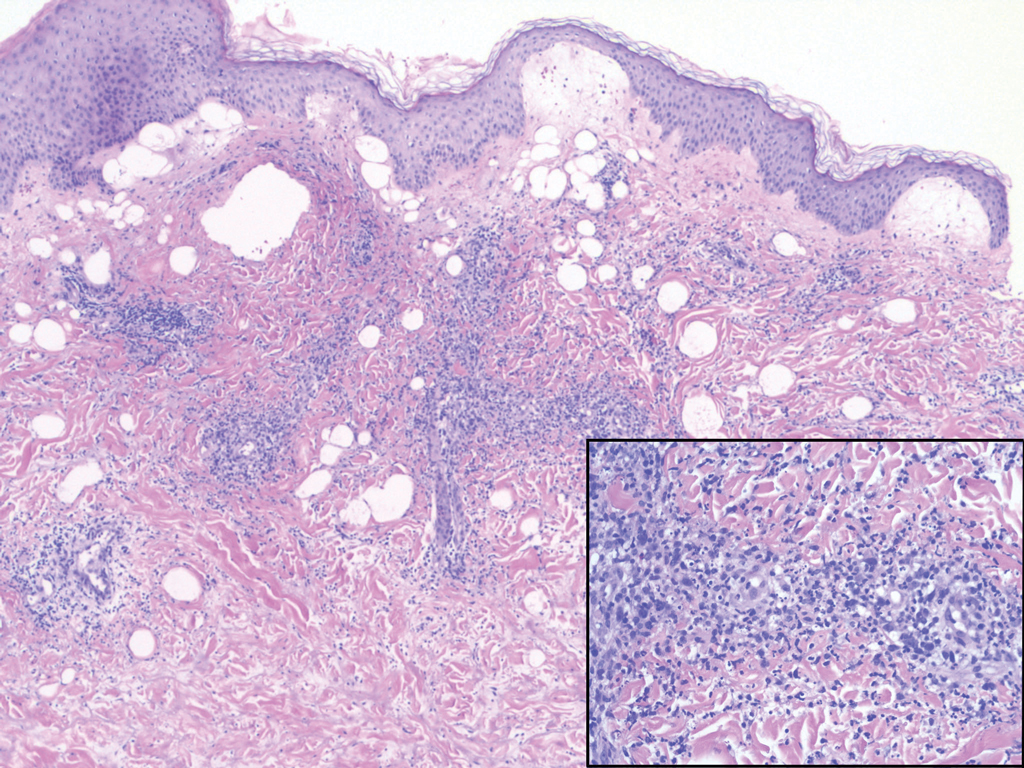
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
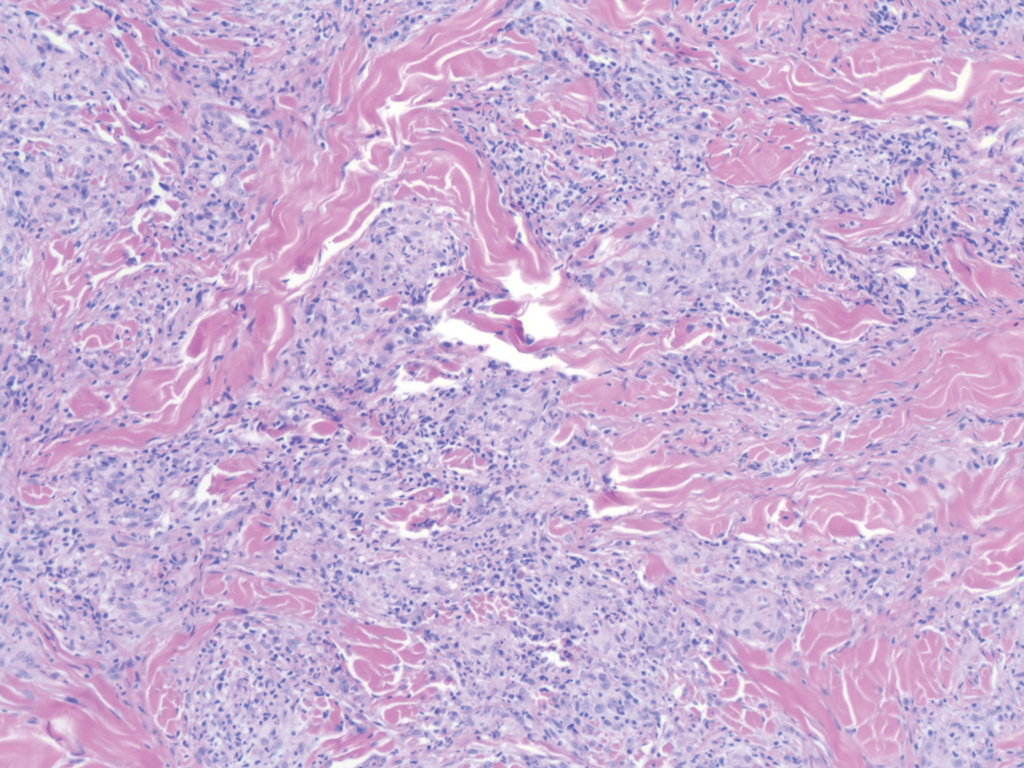
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
A 58-year-old woman with a medical history of asthma, hypertension, hypothyroidism, and hyperlipidemia presented with a painful rash of 10 days' duration. The rash was associated with fever at home (temperature, 38.5.2 °C), and a review of systems was positive for joint pain. Physical examination revealed numerous 8- to 10-mm, erythematous, discus-shaped papules on the bilateral dorsal hands, bilateral palms, right knee, and right dorsal foot with slight tenderness to palpation. A papule on the right dorsal hand was biopsied.
Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
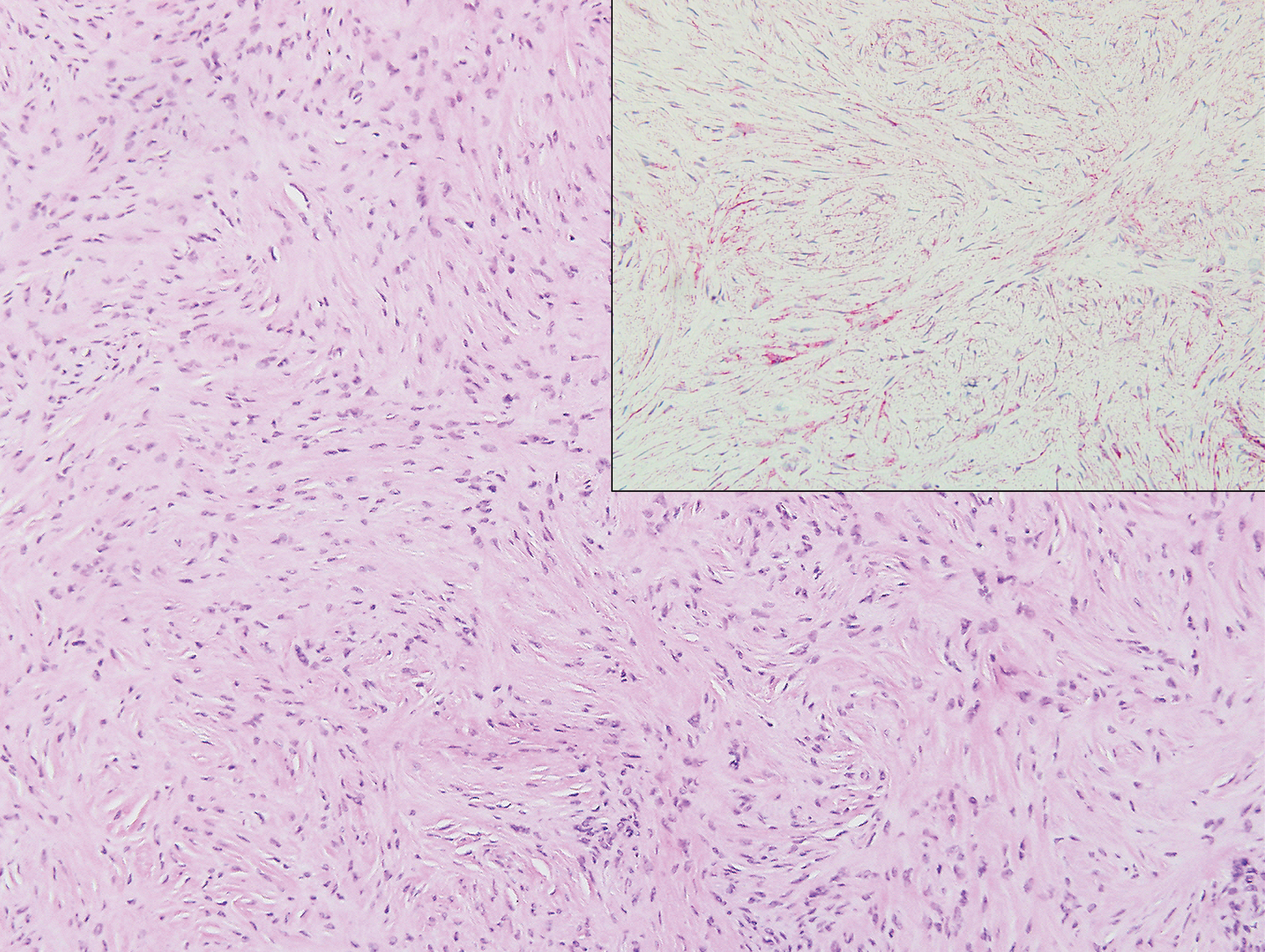
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
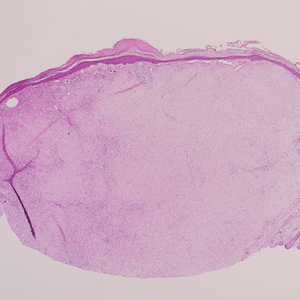
Erythematous Papules on the Scrotum, Trunk, and Extremities
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
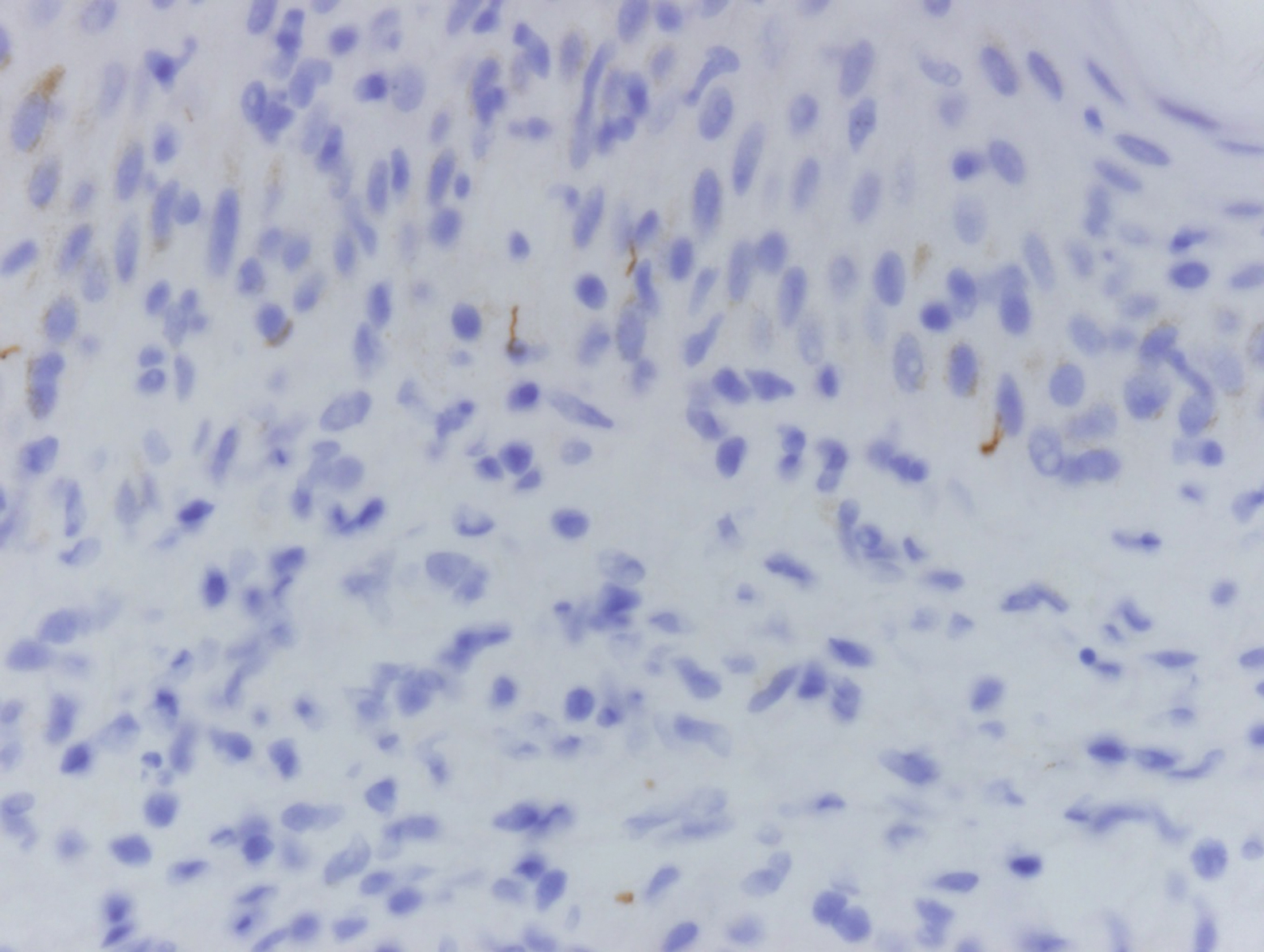
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19

Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
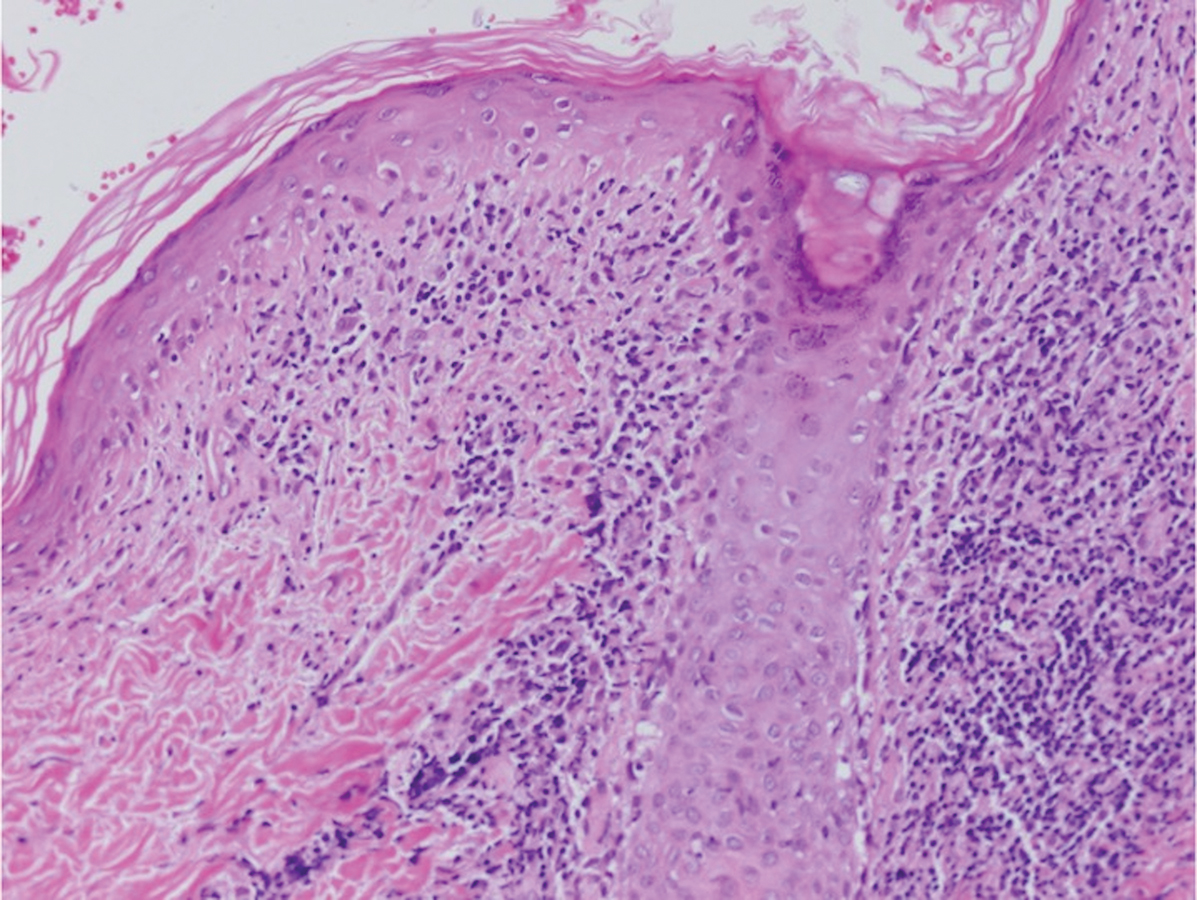
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
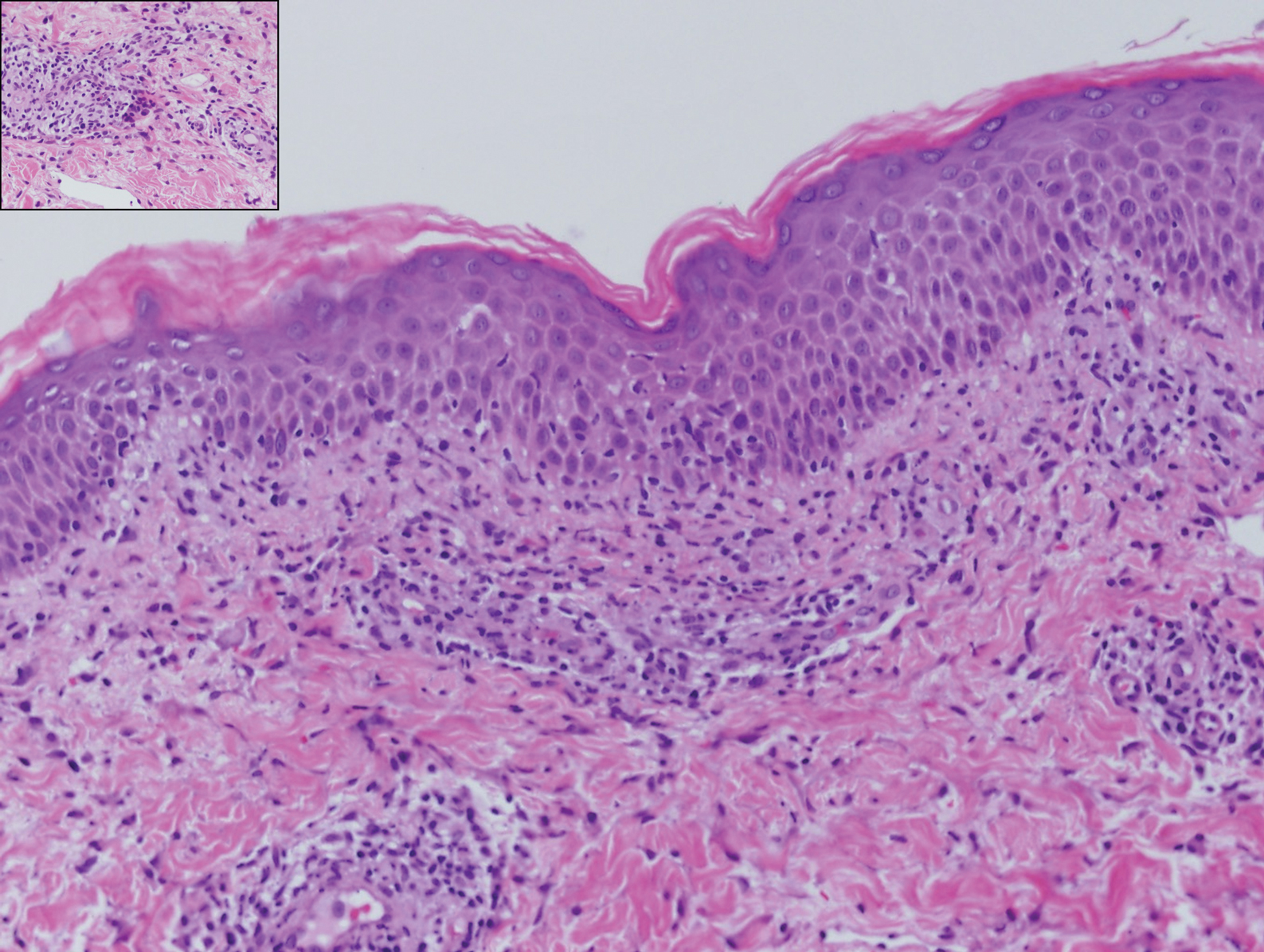
A 54-year-old man presented with painful, nonpruritic, erythematous papules that began on the scrotum. The eruption progressed to involve the trunk, arms, and legs.
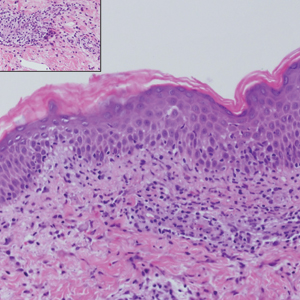
Solitary Papule on the Leg
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
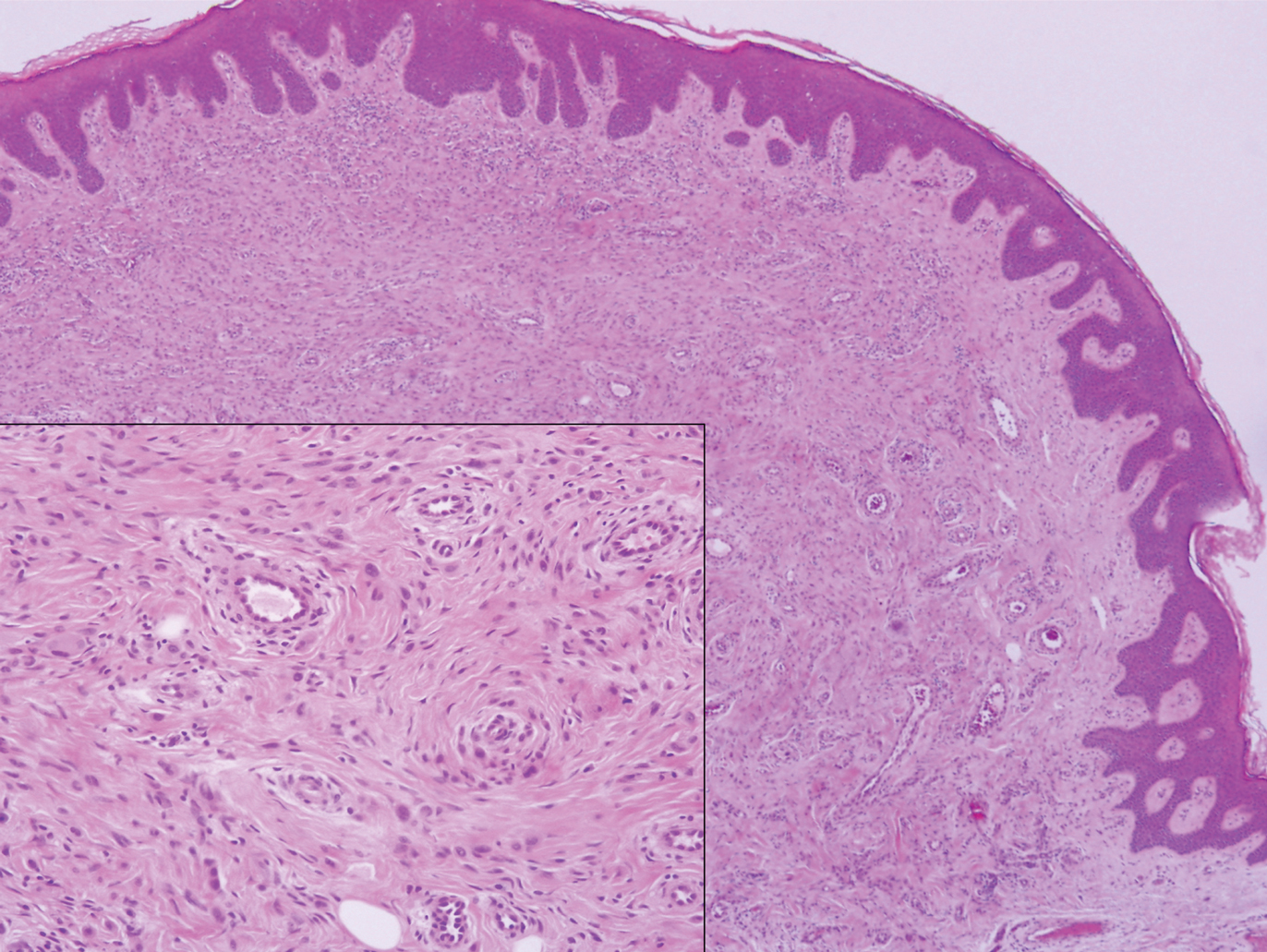
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18

Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22

Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
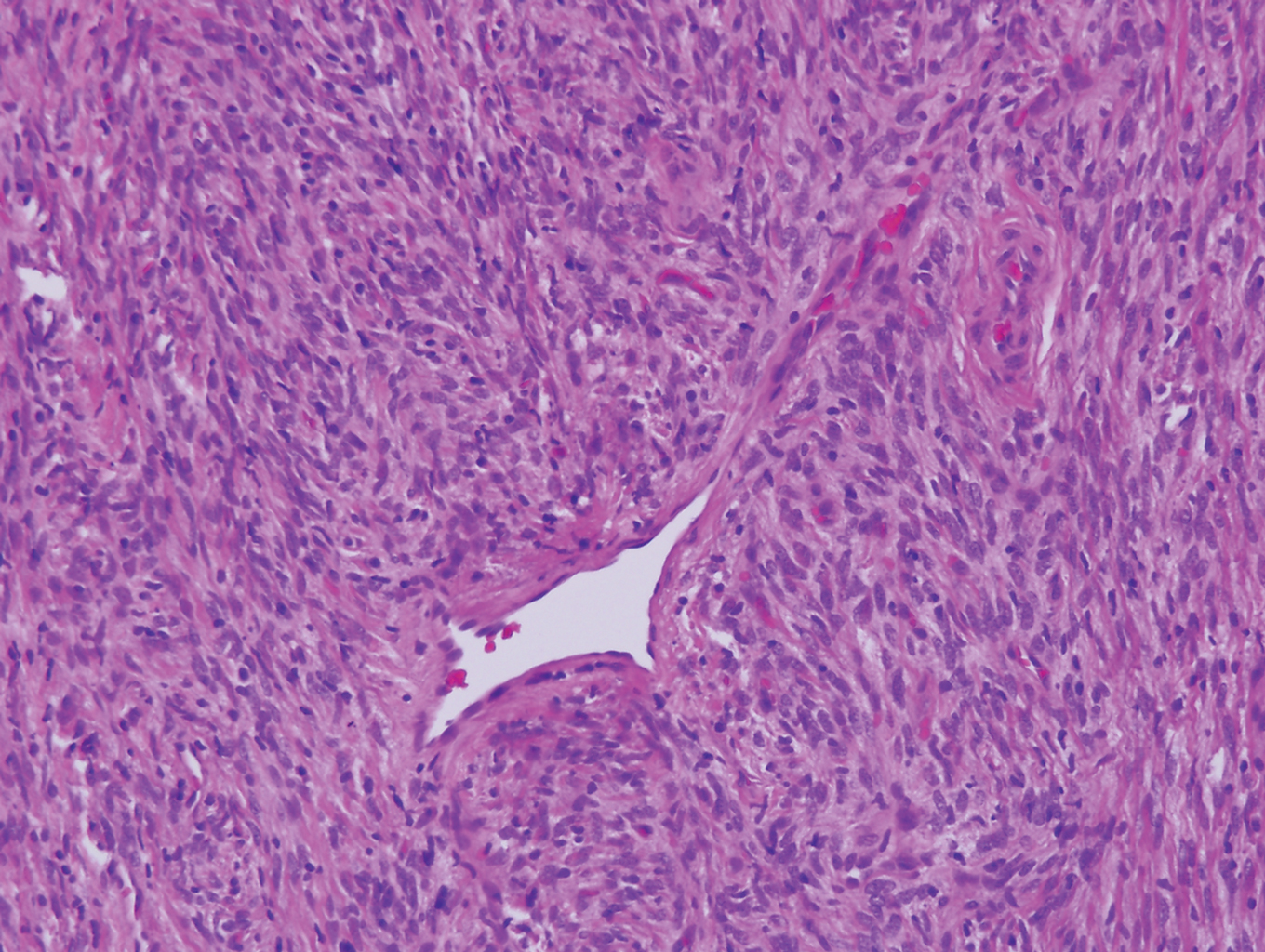
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
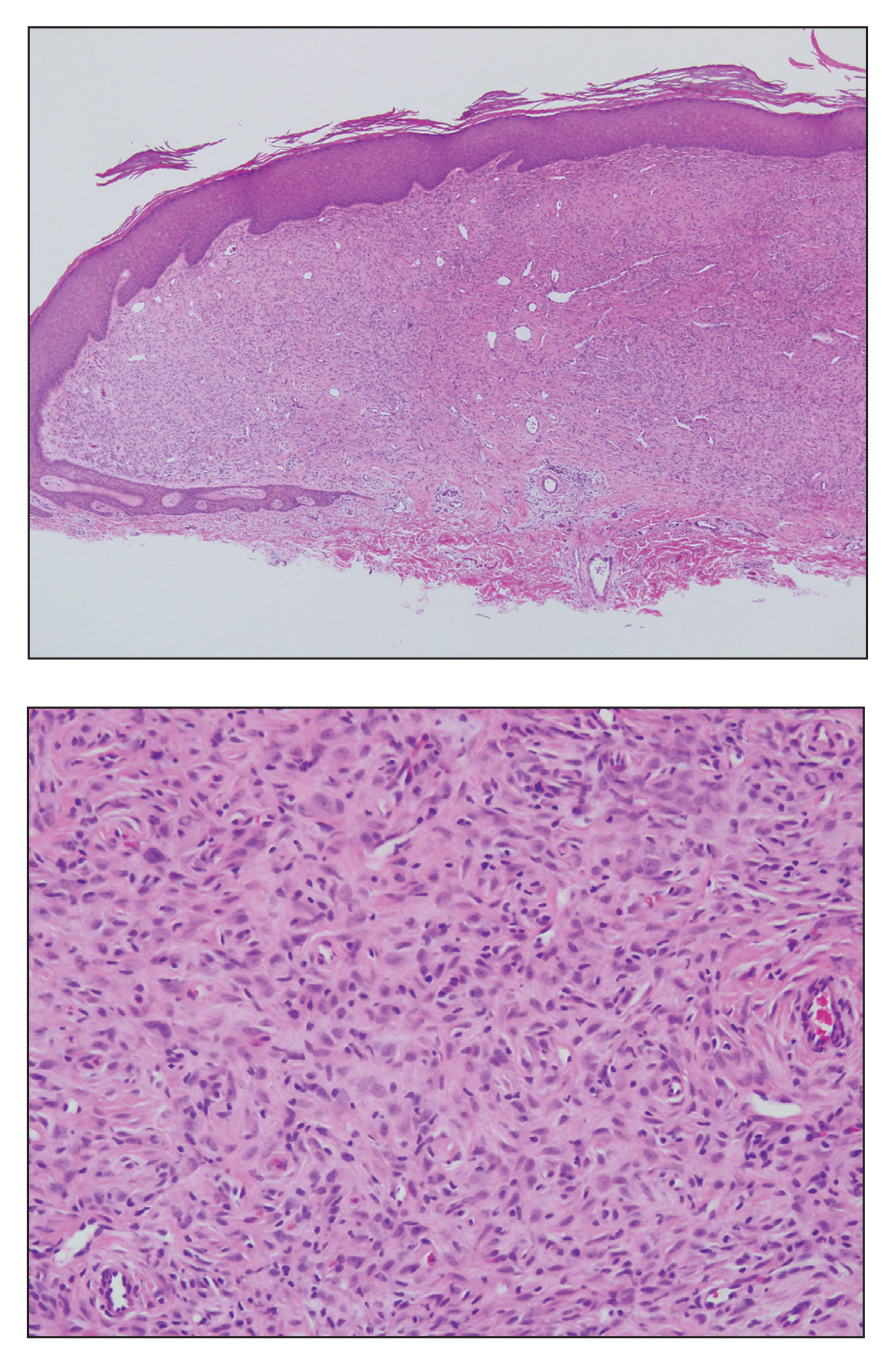
A 28-year-old man presented with a growing asymptomatic papule on the right leg.
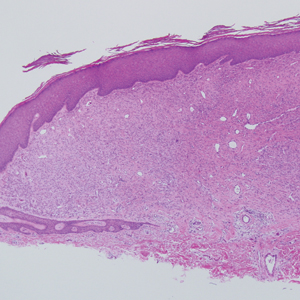
Pigmented Mass on the Shoulder
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
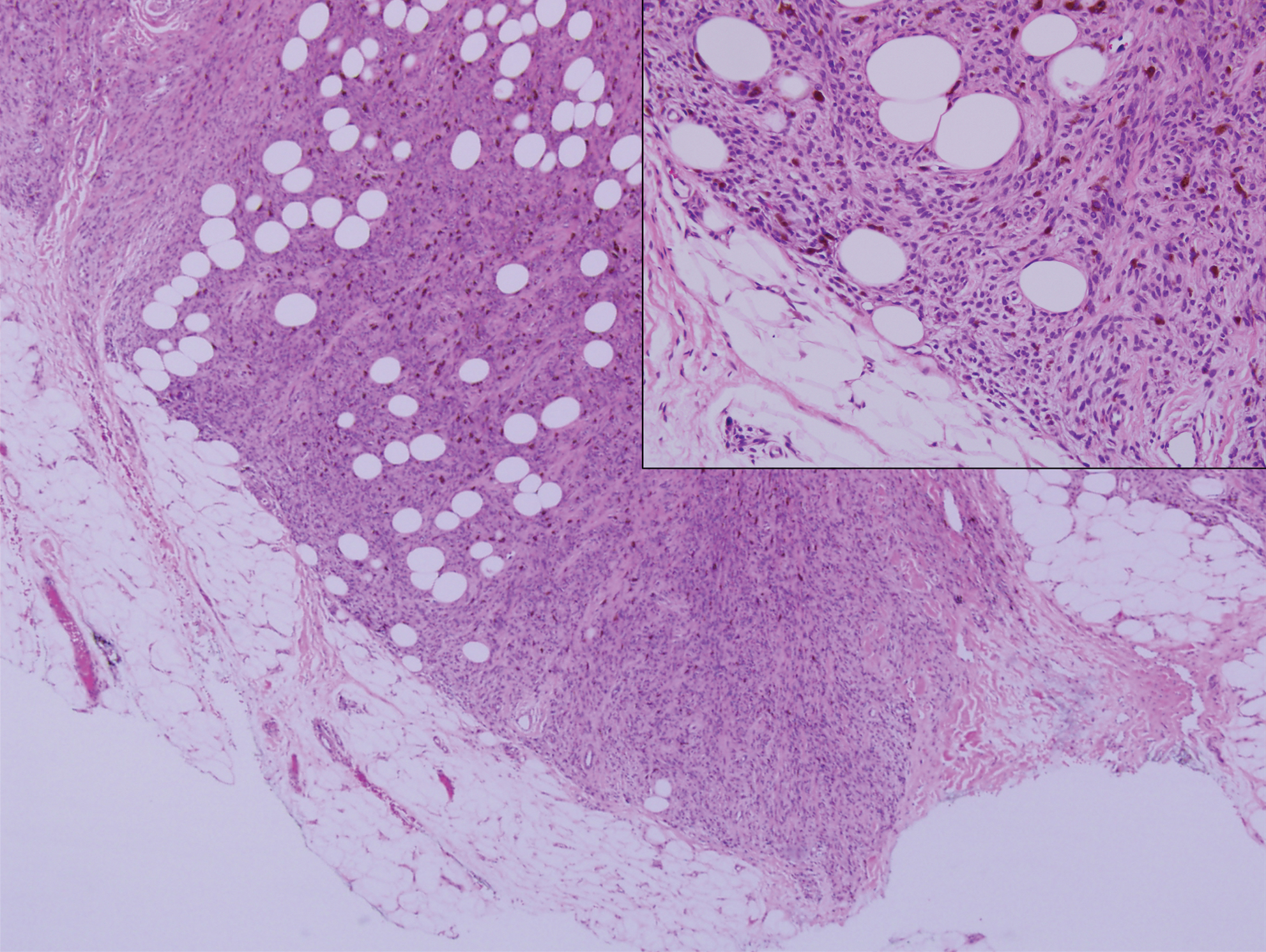
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
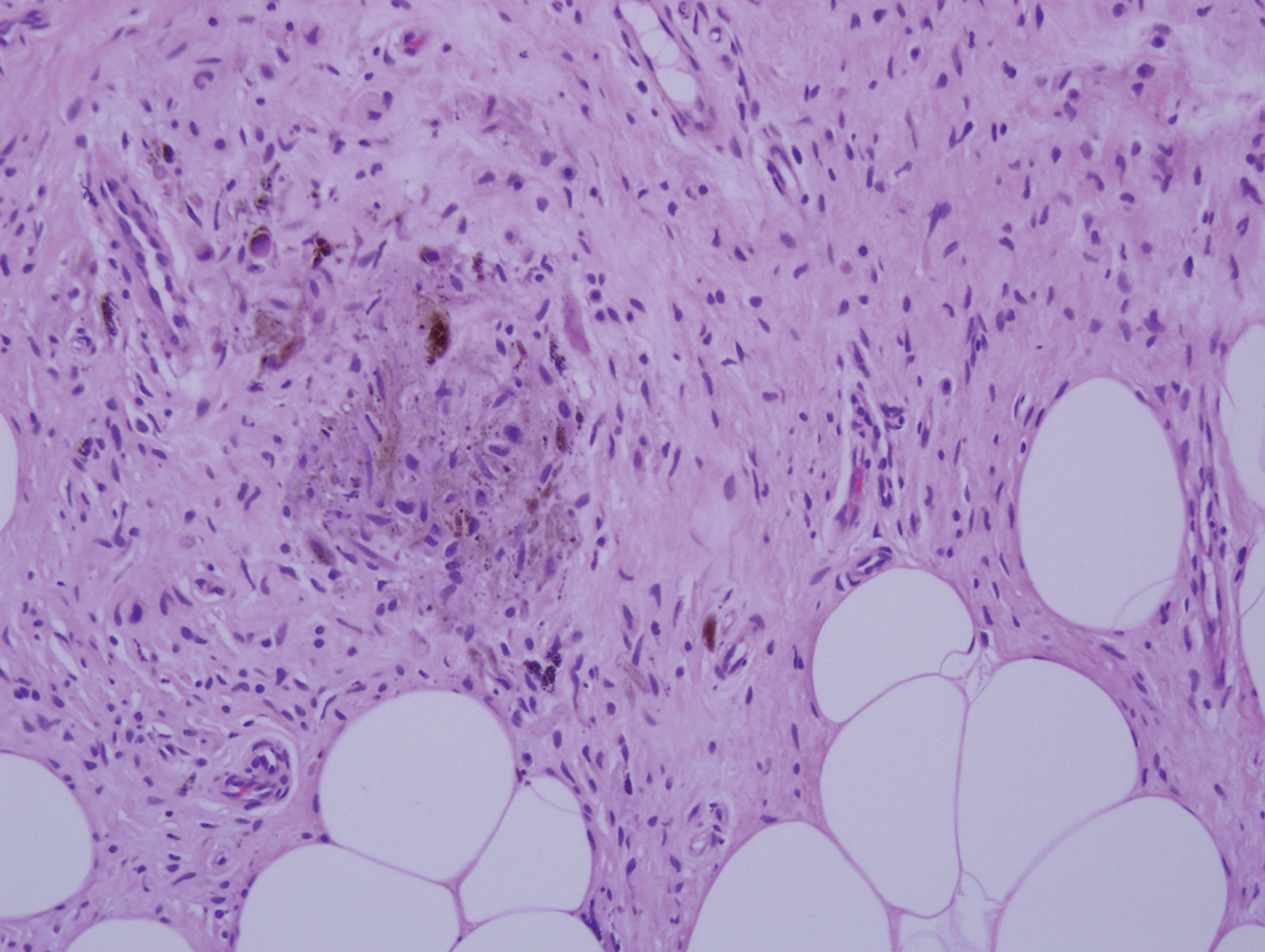
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
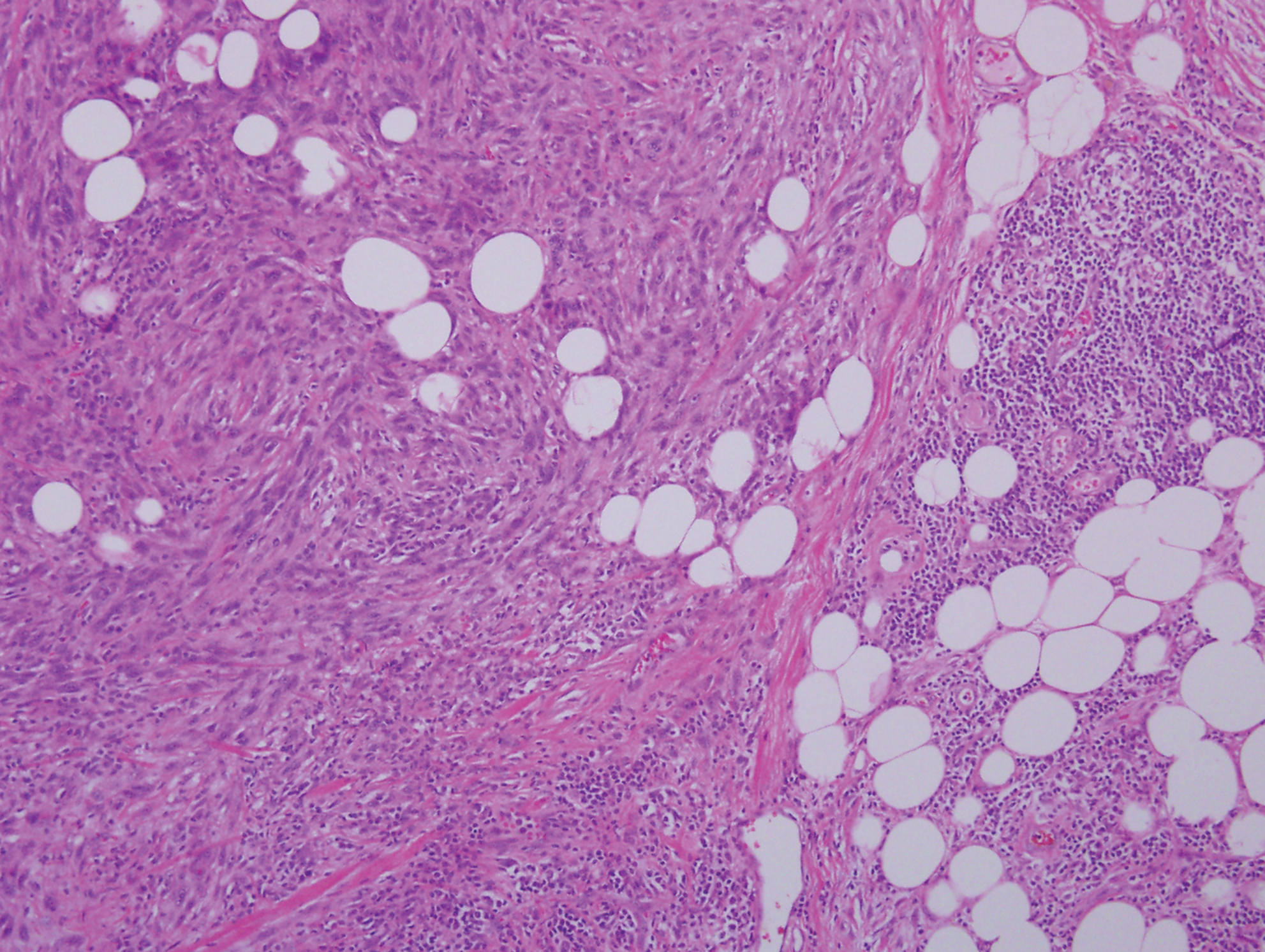
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
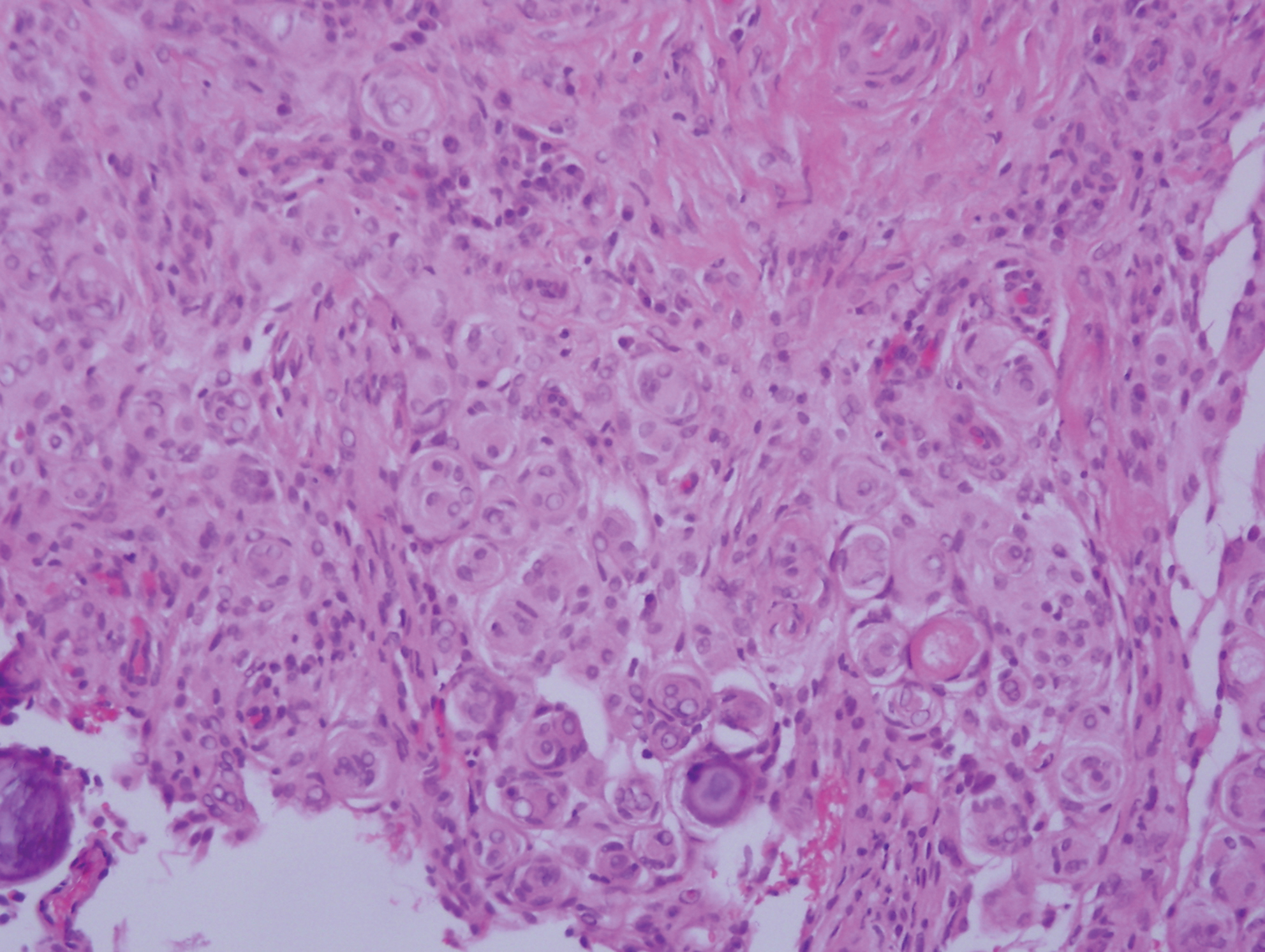
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.

A 37-year-old woman presented with an asymptomatic, indurated, pigmented, subcutaneous nodule on the right shoulder of more than 3 years' duration. The lesion had gradually increased in size with no associated symptoms. The patient had a history of endometrial adenocarcinoma and papillary thyroid carcinoma, which had been treated by hysterectomy-oophorectomy and right thyroidectomy, respectively. She had no other notable systemic abnormalities, and there was no family history of genetic disease or cancer. Physical examination demonstrated a 1.2×1.8-cm nontender, pigmented, subcutaneous nodule with a rough surface and indistinct borders. An excisional biopsy was performed.
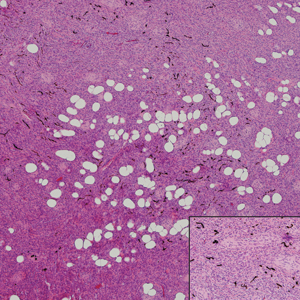
Ill-Defined Macule on the Abdomen
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
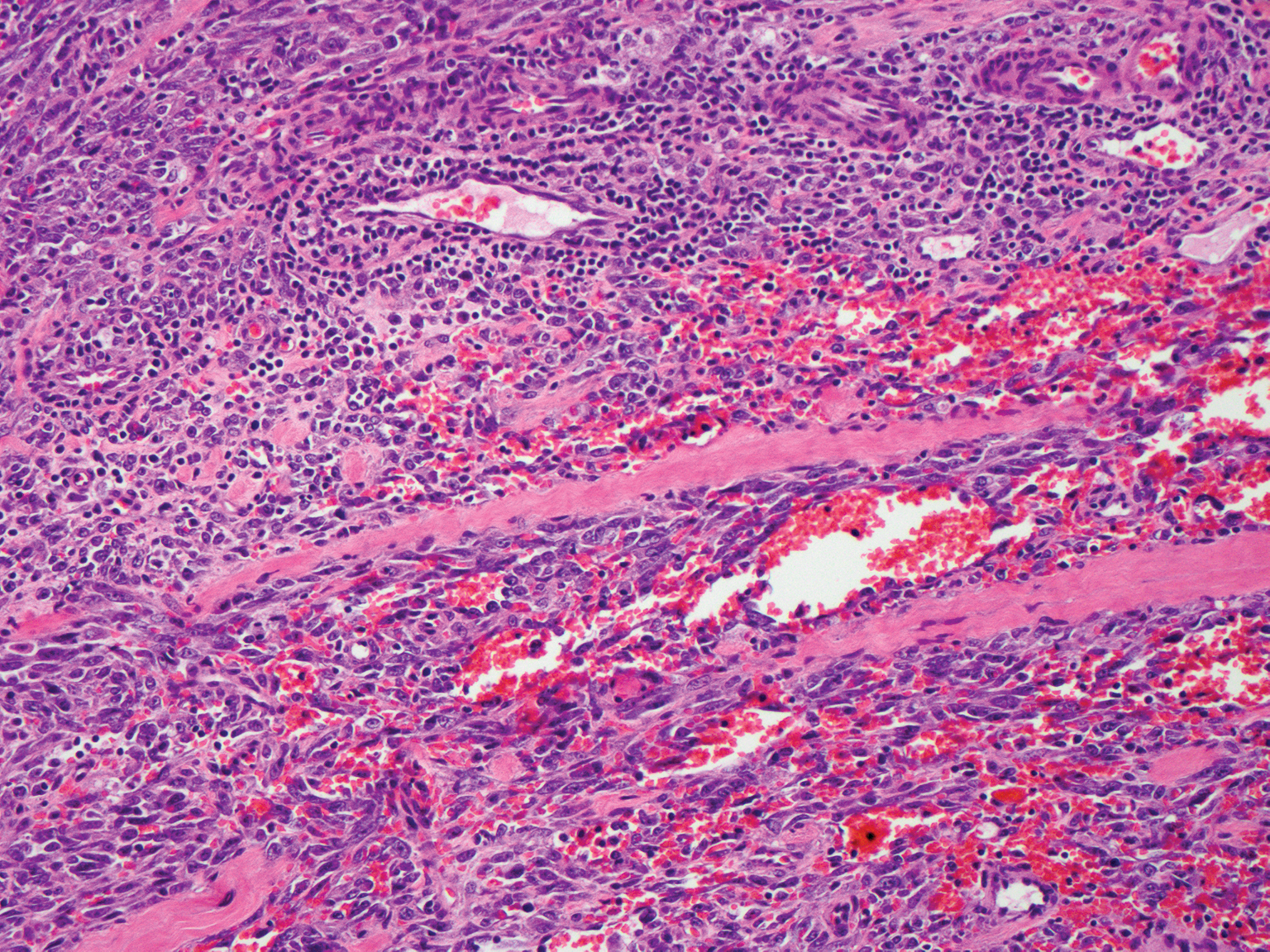
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
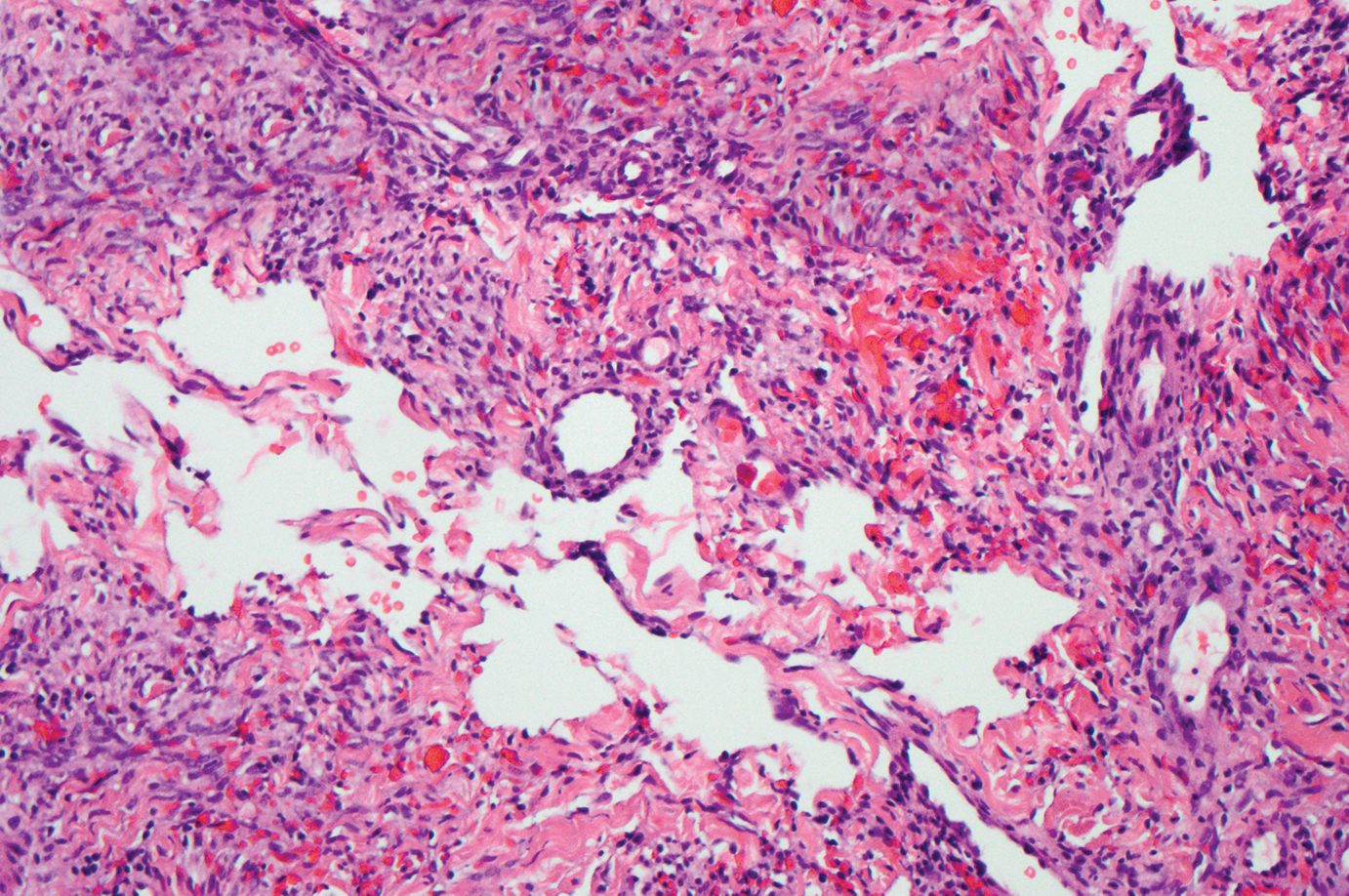
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
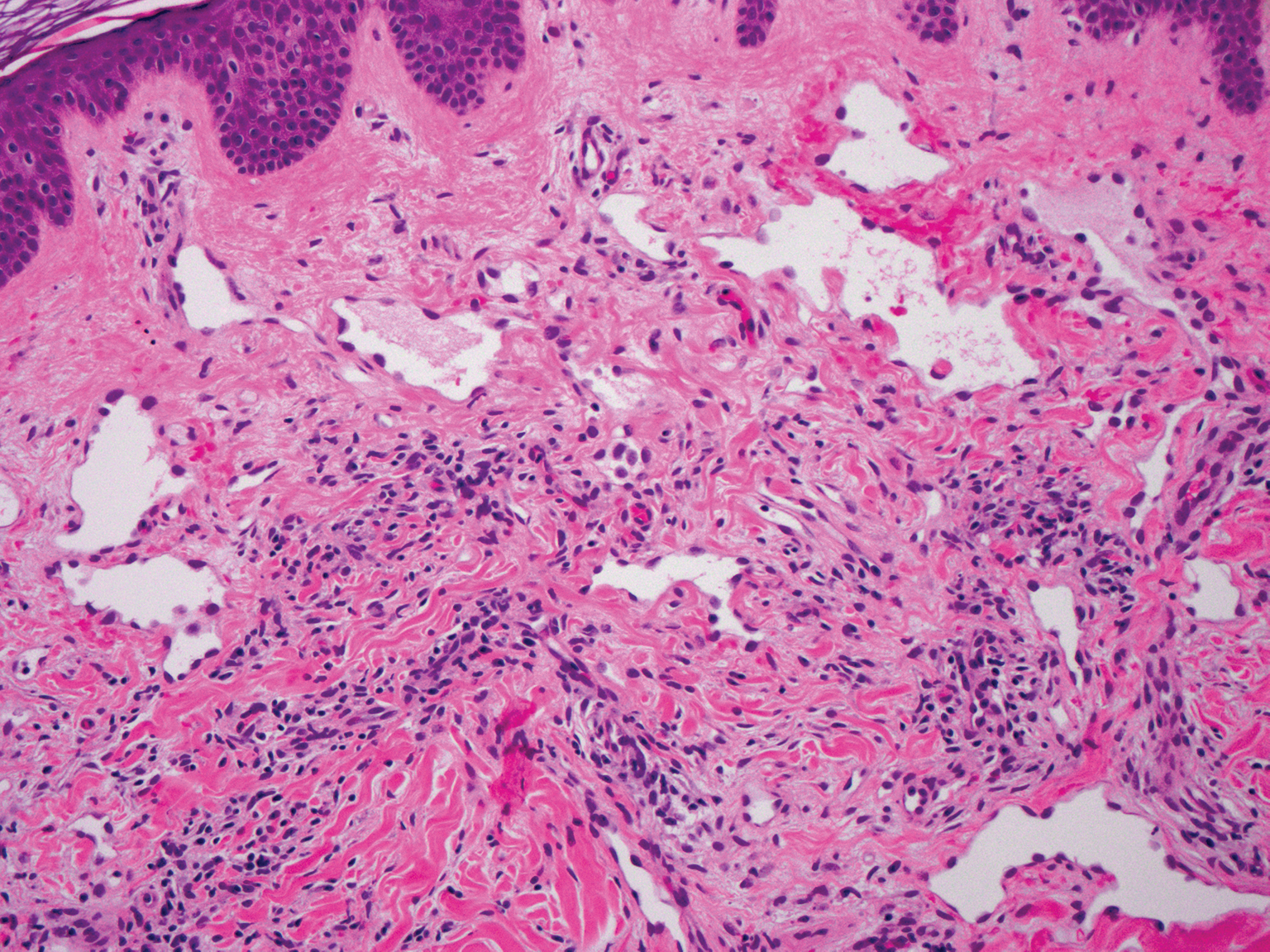
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15

- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
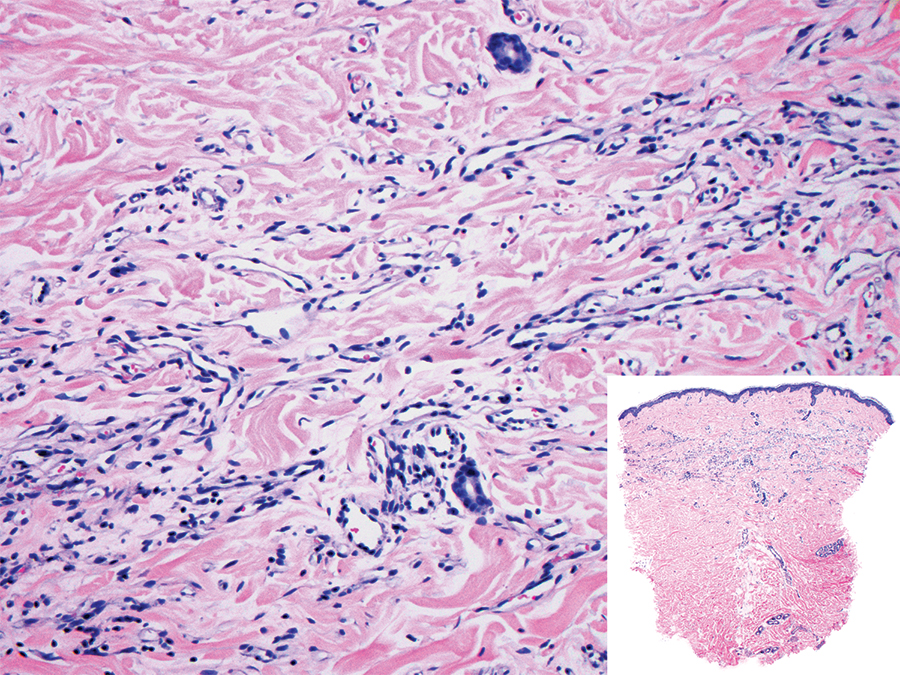
A 38-year-old woman presented with an asymptomatic lesion on the abdomen. On physical examination, there was a 5×2-mm, solitary, ill-defined pink macule on the right side of the abdomen. The patient denied recent change in size or color of the lesion, prior trauma, or a personal or family history of similar lesions. Due to the uncertain diagnostic appearance, a punch biopsy was performed.
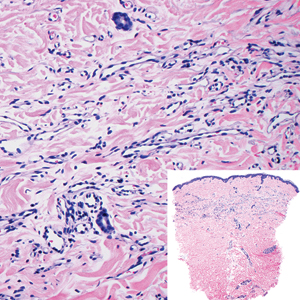
Enlarging Nodule on the Thigh
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
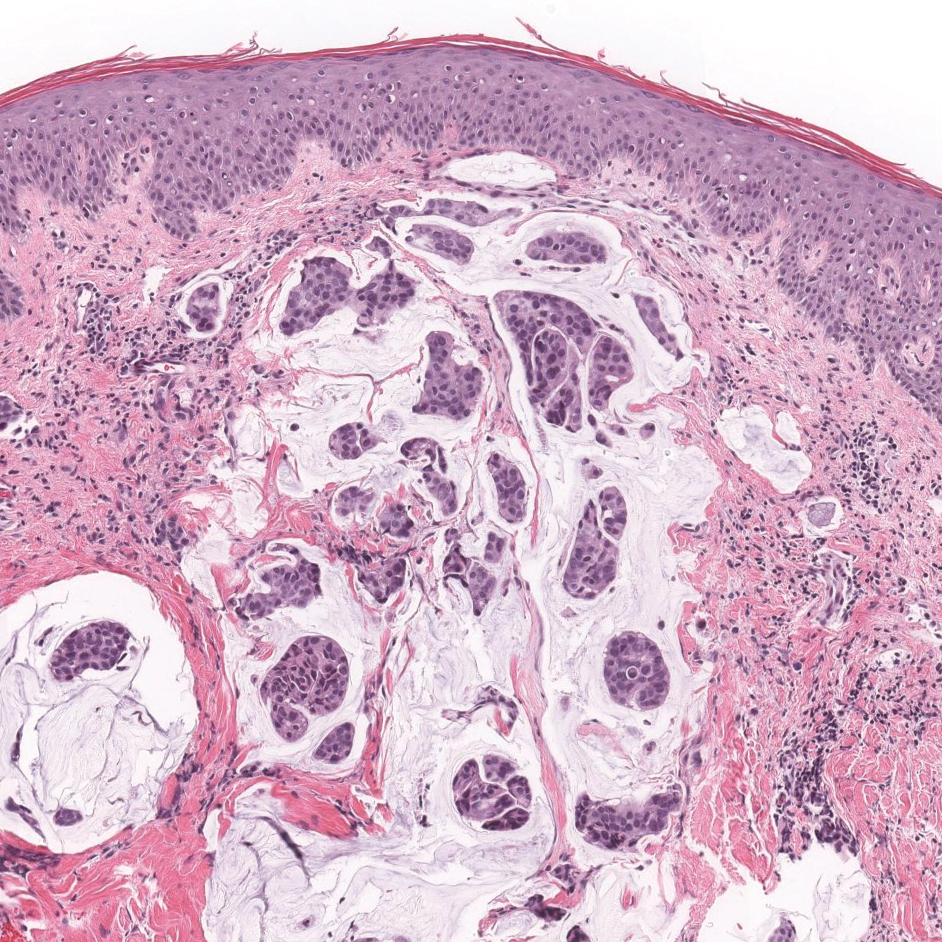
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
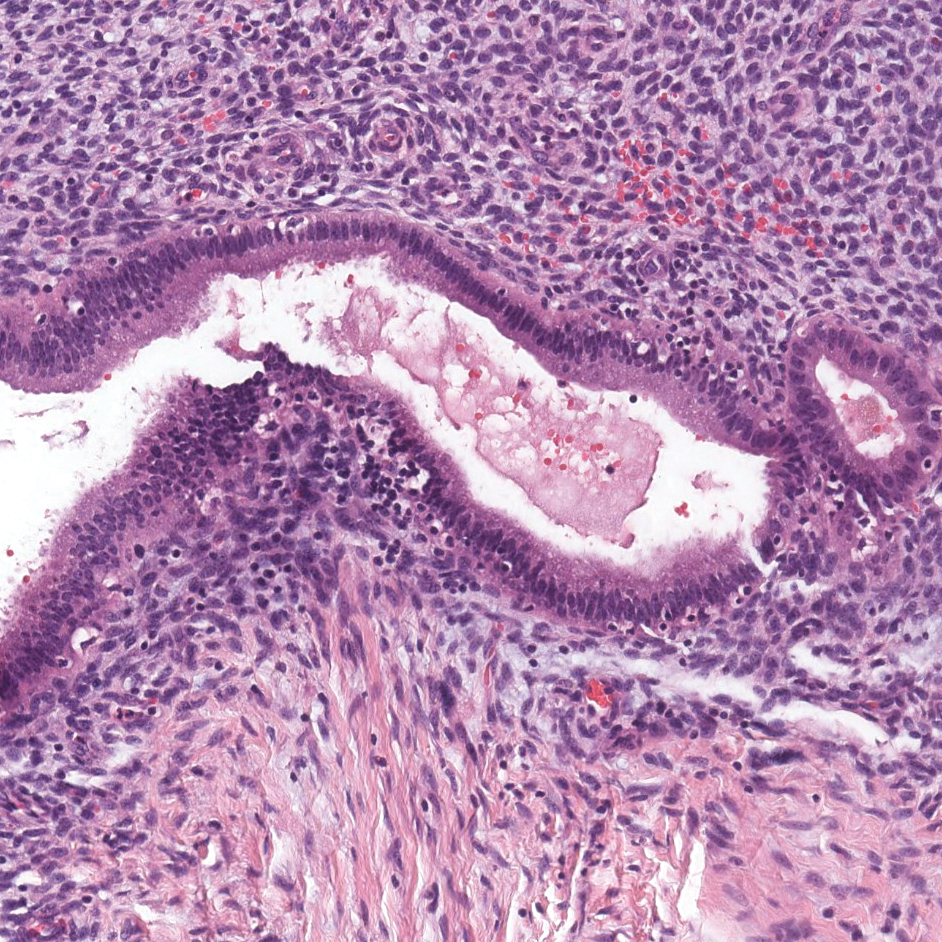
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
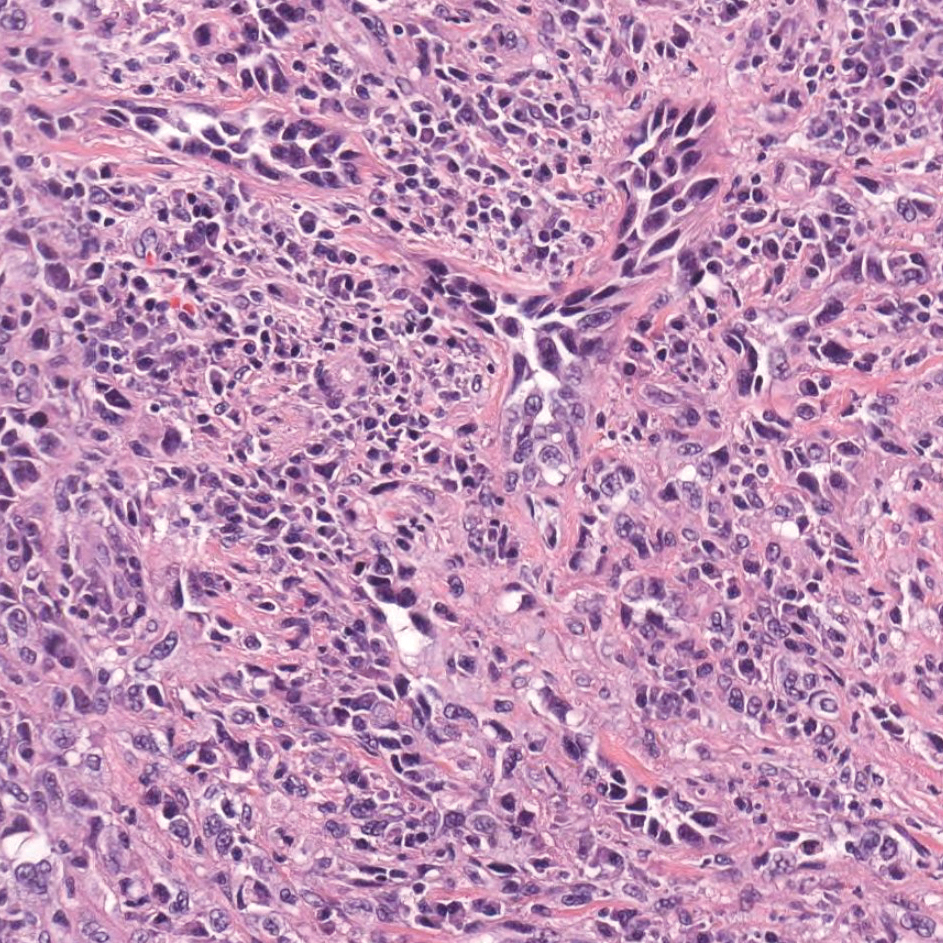
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
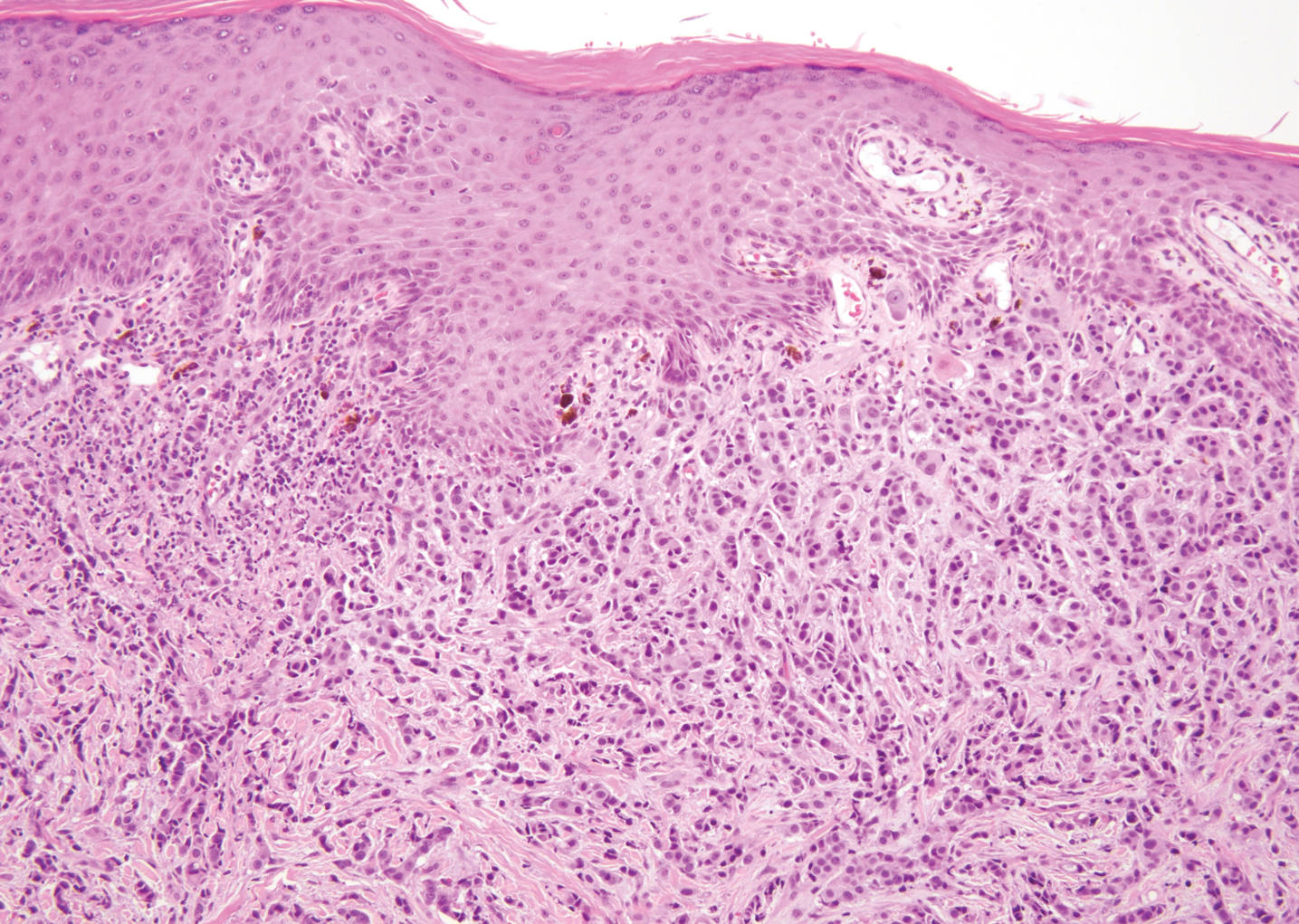
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
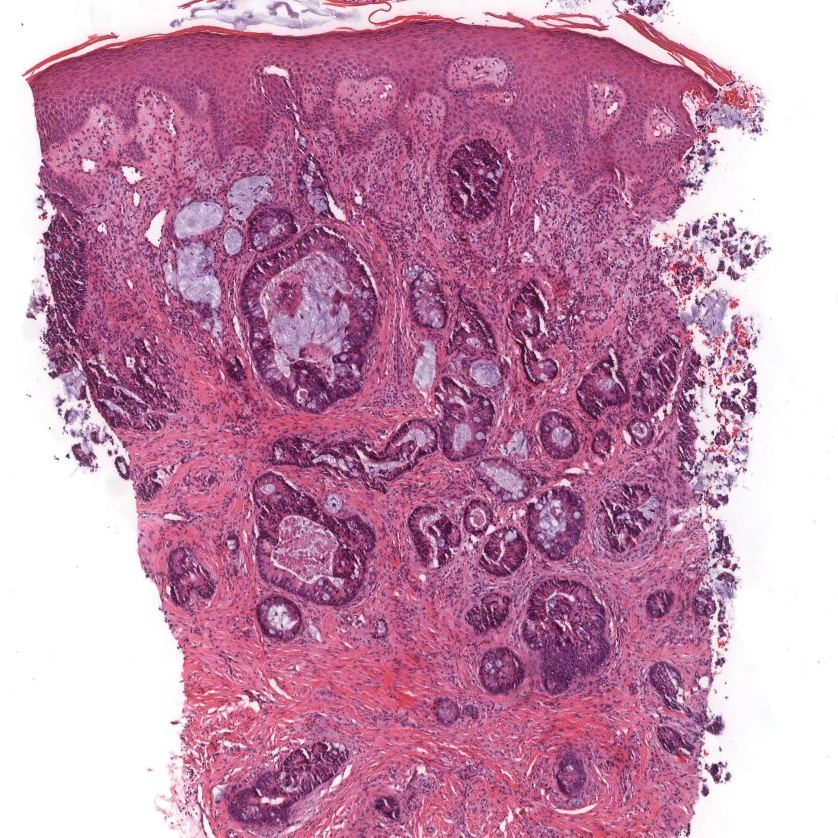
A 68-year-old patient presented with an enlarging flesh-colored nodule on the thigh that was positive for cytokeratin 20 and negative for cytokeratin 7.
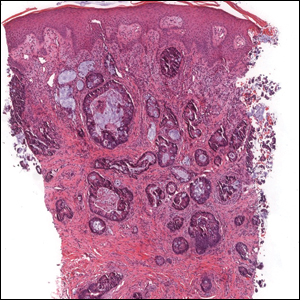
Unilateral Facial Papules and Plaques
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15

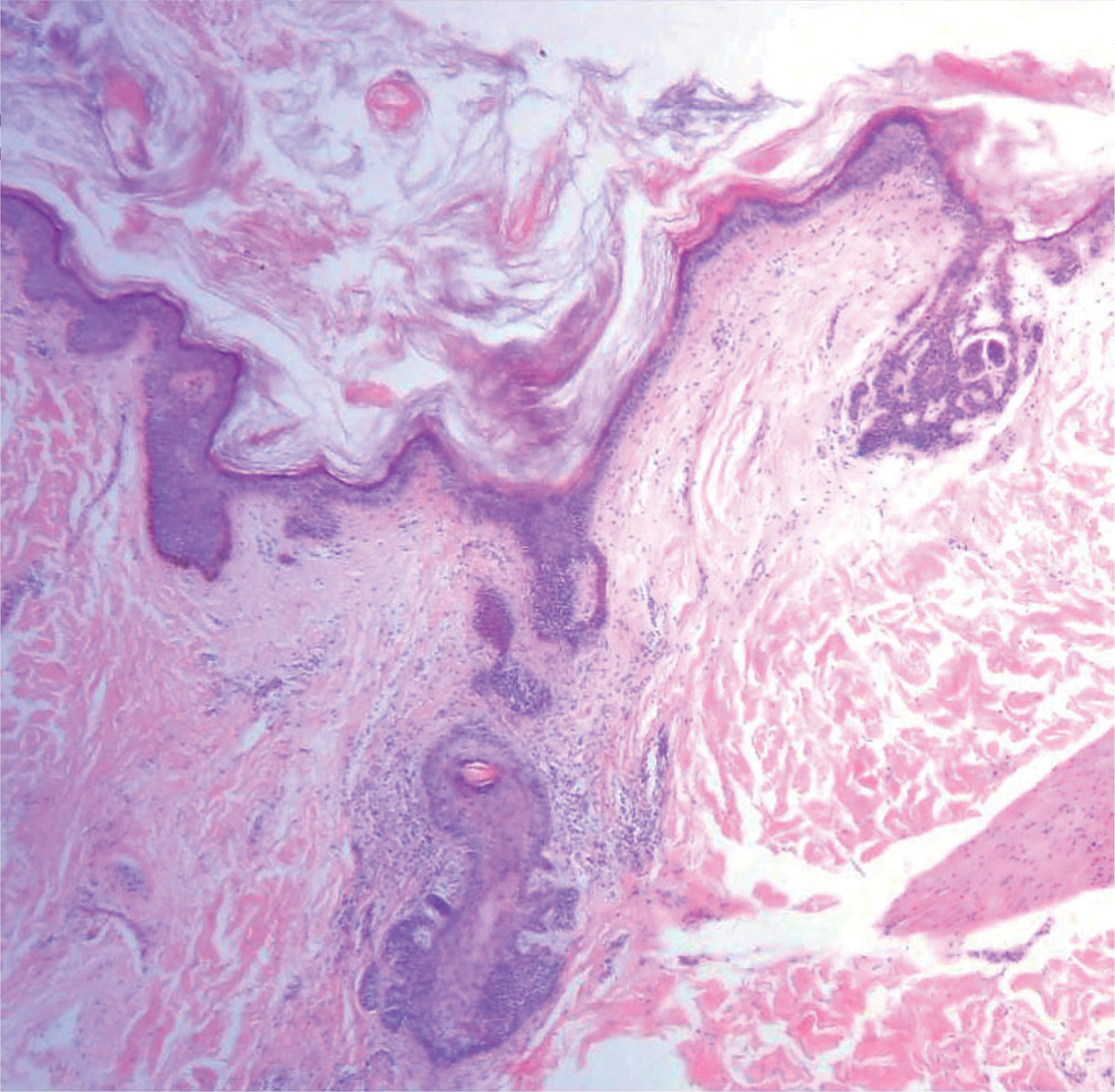

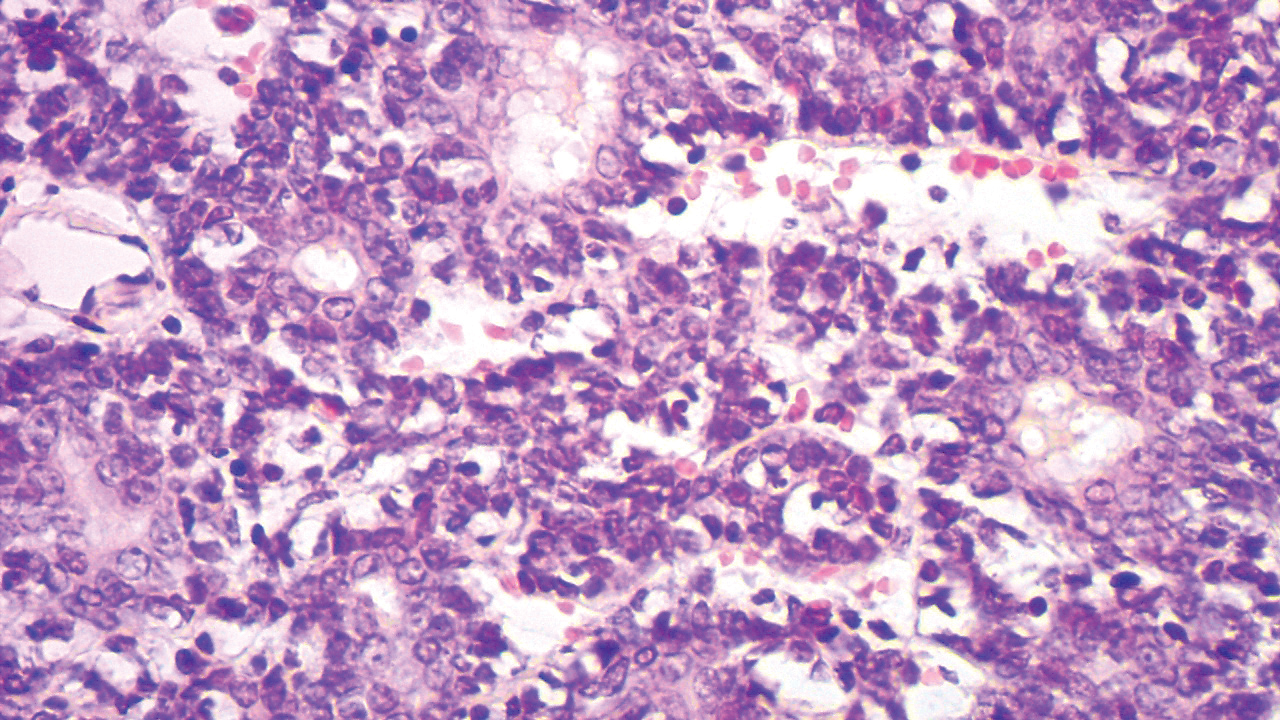
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.

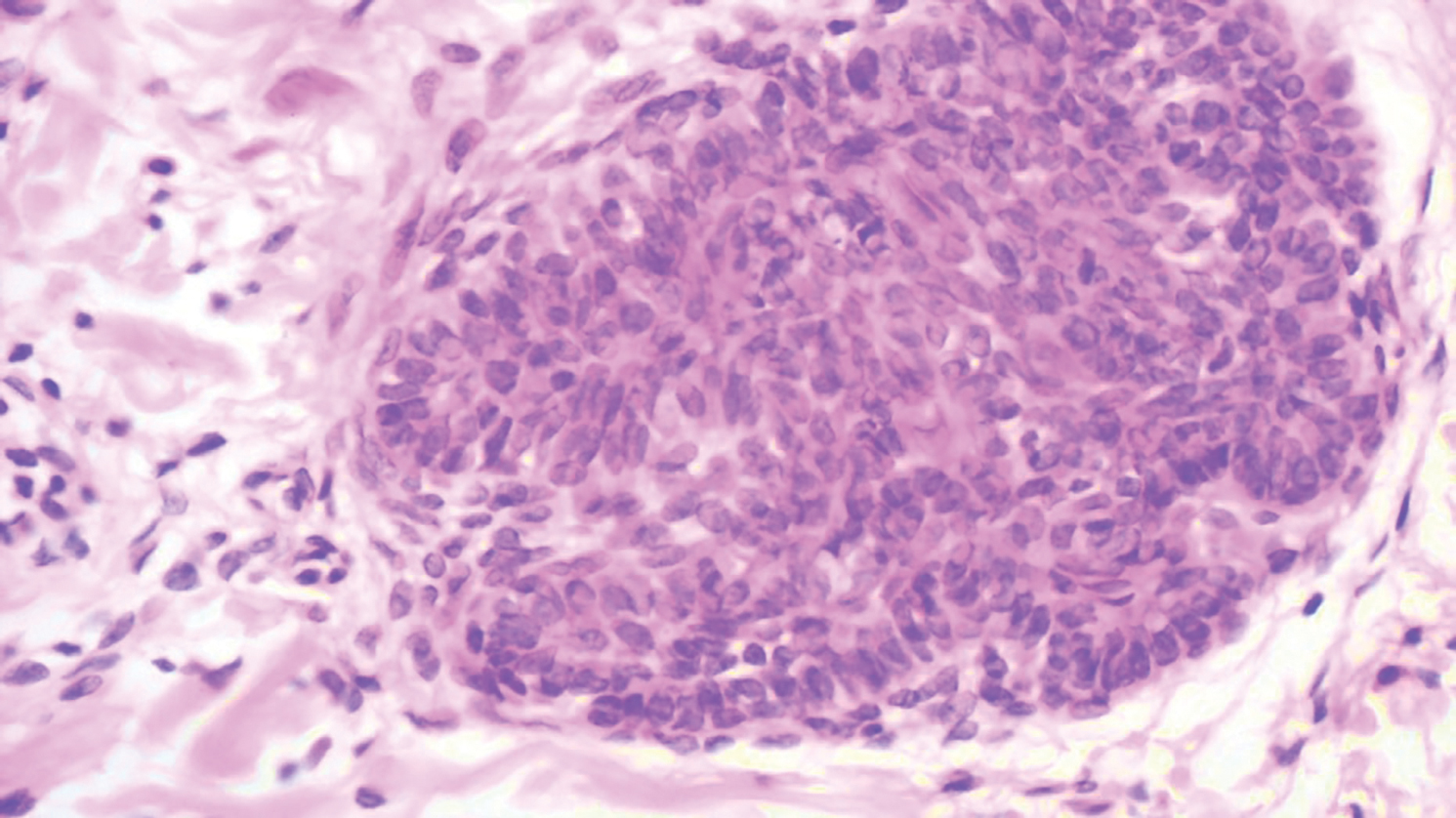
A 9-year-old boy presented with a slowly progressive lesion of 5 years’ duration affecting only the left side of the face in a dermatomal pattern. The patient denied any symptoms and had no other anomalies or family history of similar lesions. On physical examination the lesion was found to span a 12×7-cm area of the lateral half of the left cheek and was composed of multiple variable-sized, pinkish to flesh-colored papules that coalesced in some areas to form small plaques. Few milialike cysts were present. One papule was biopsied.
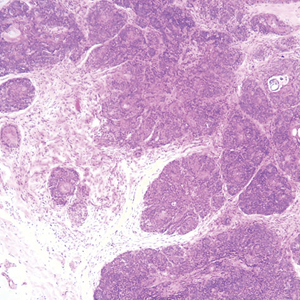
Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
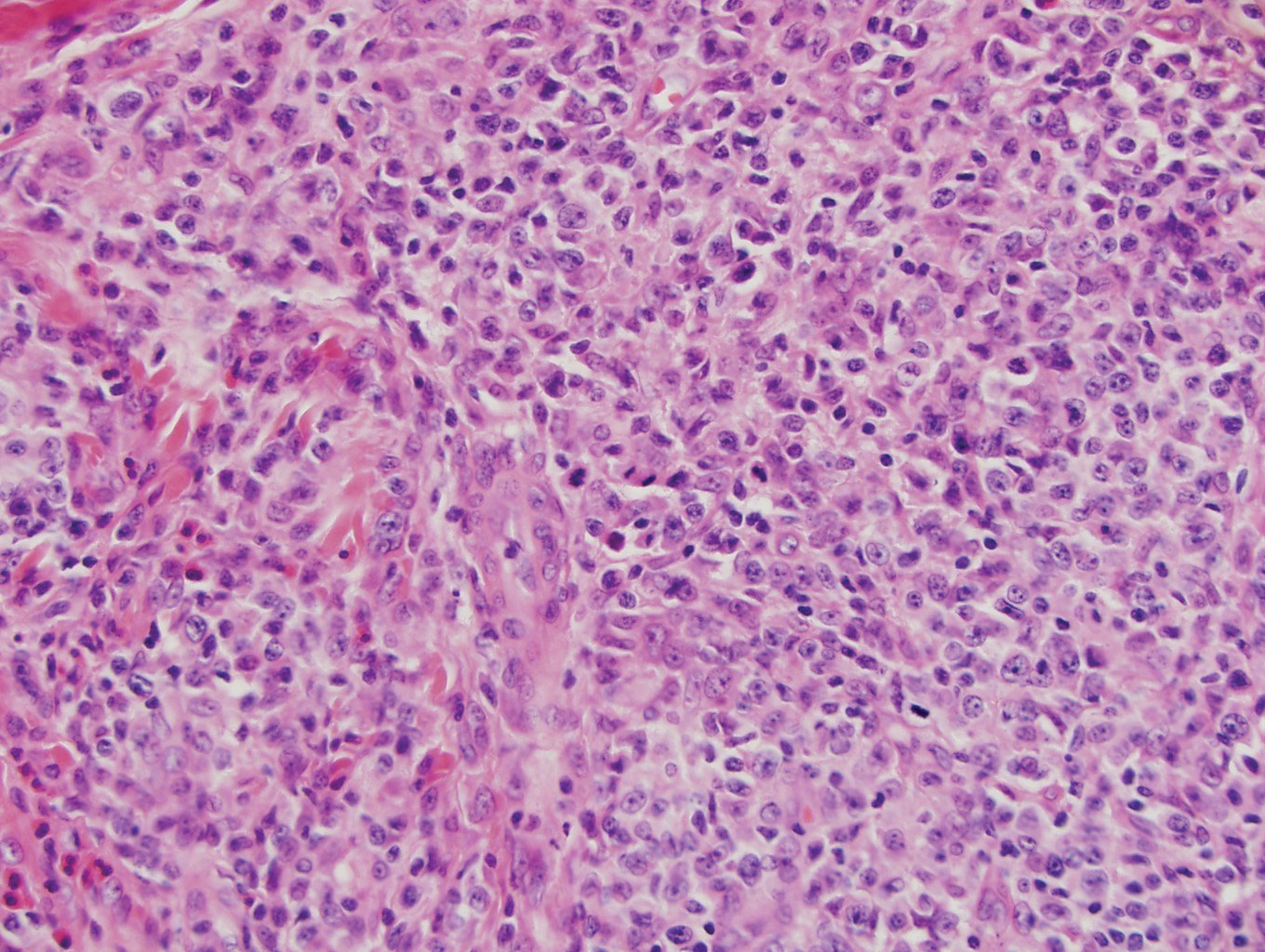
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
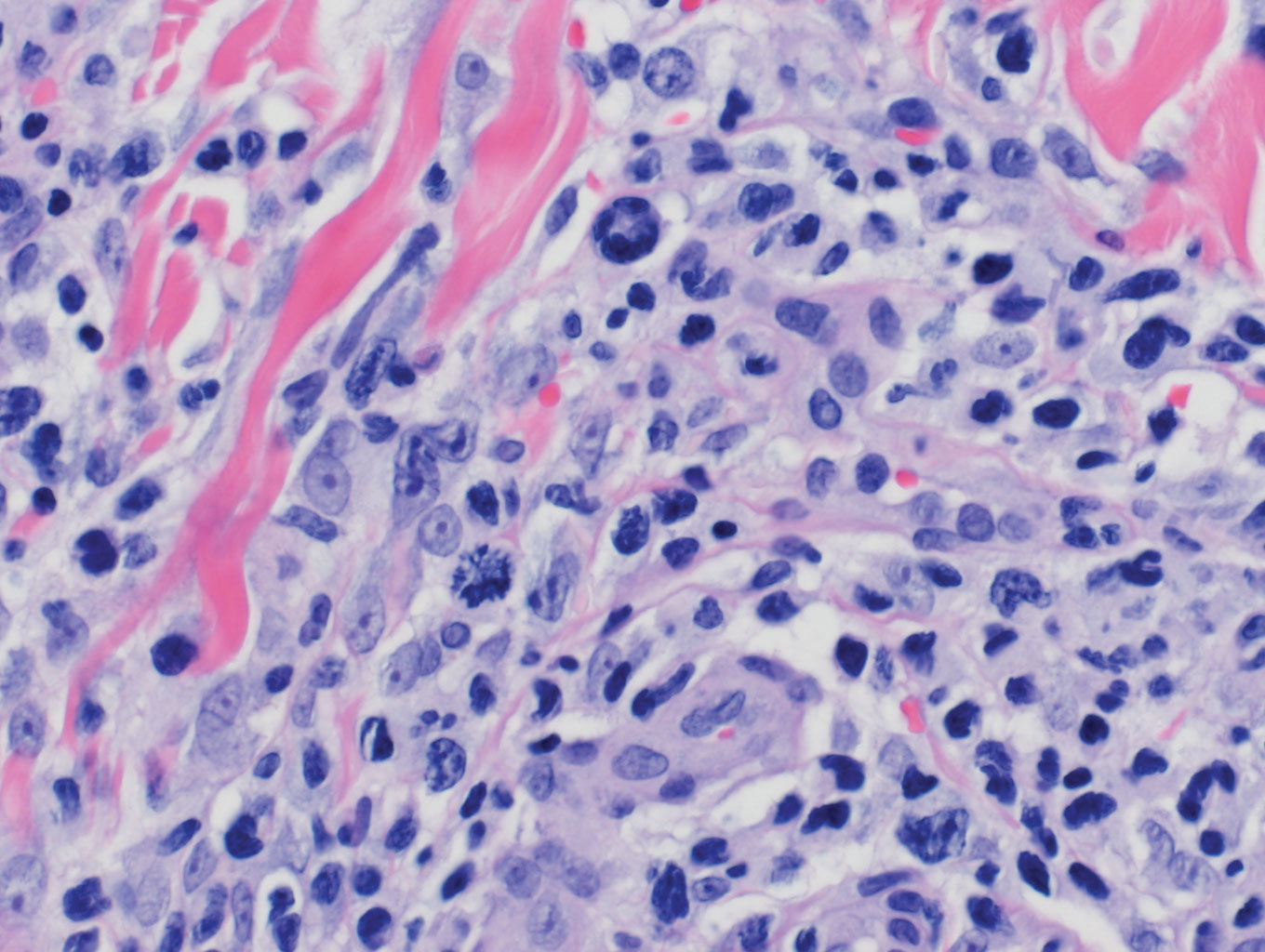
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
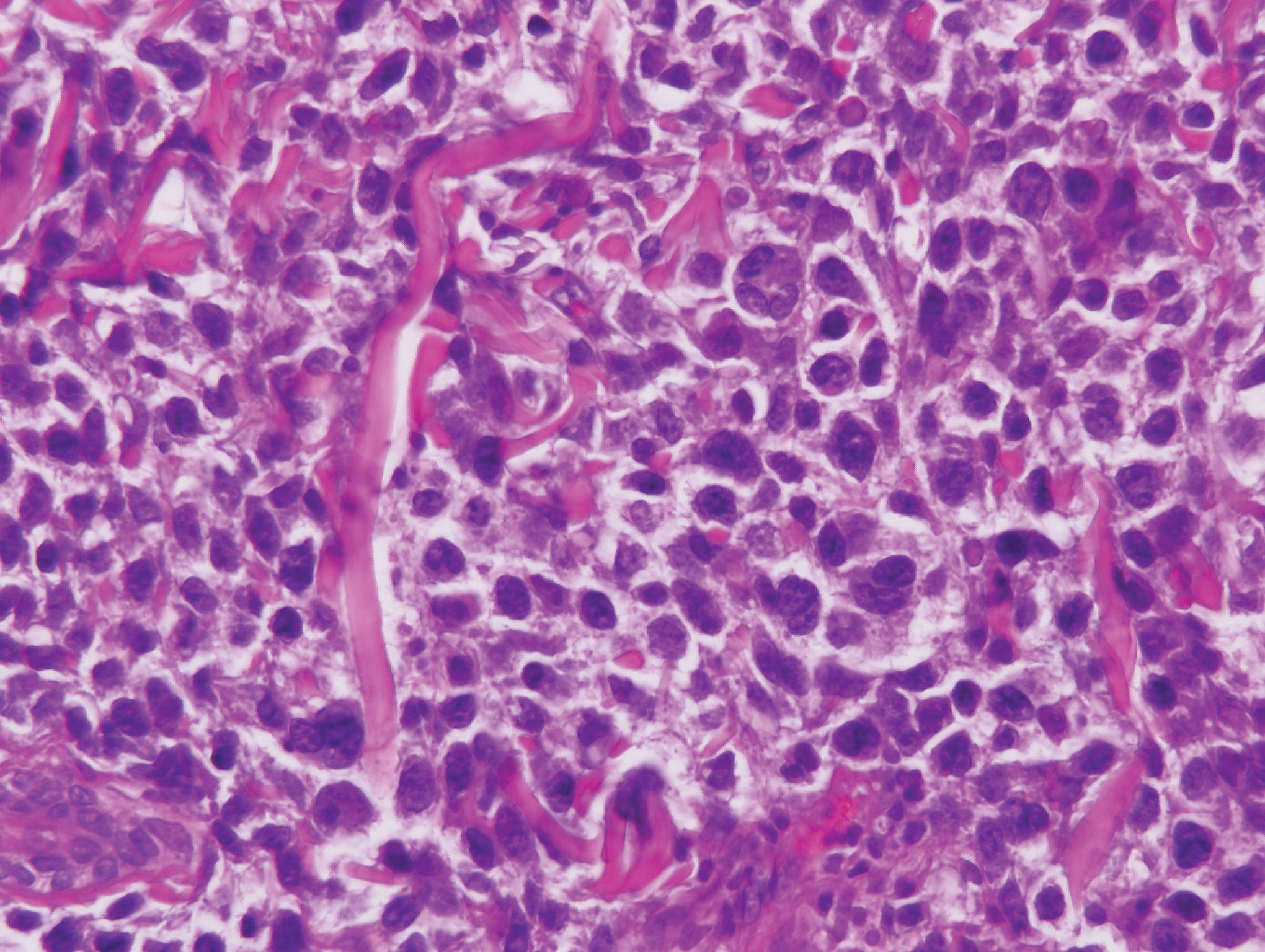
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
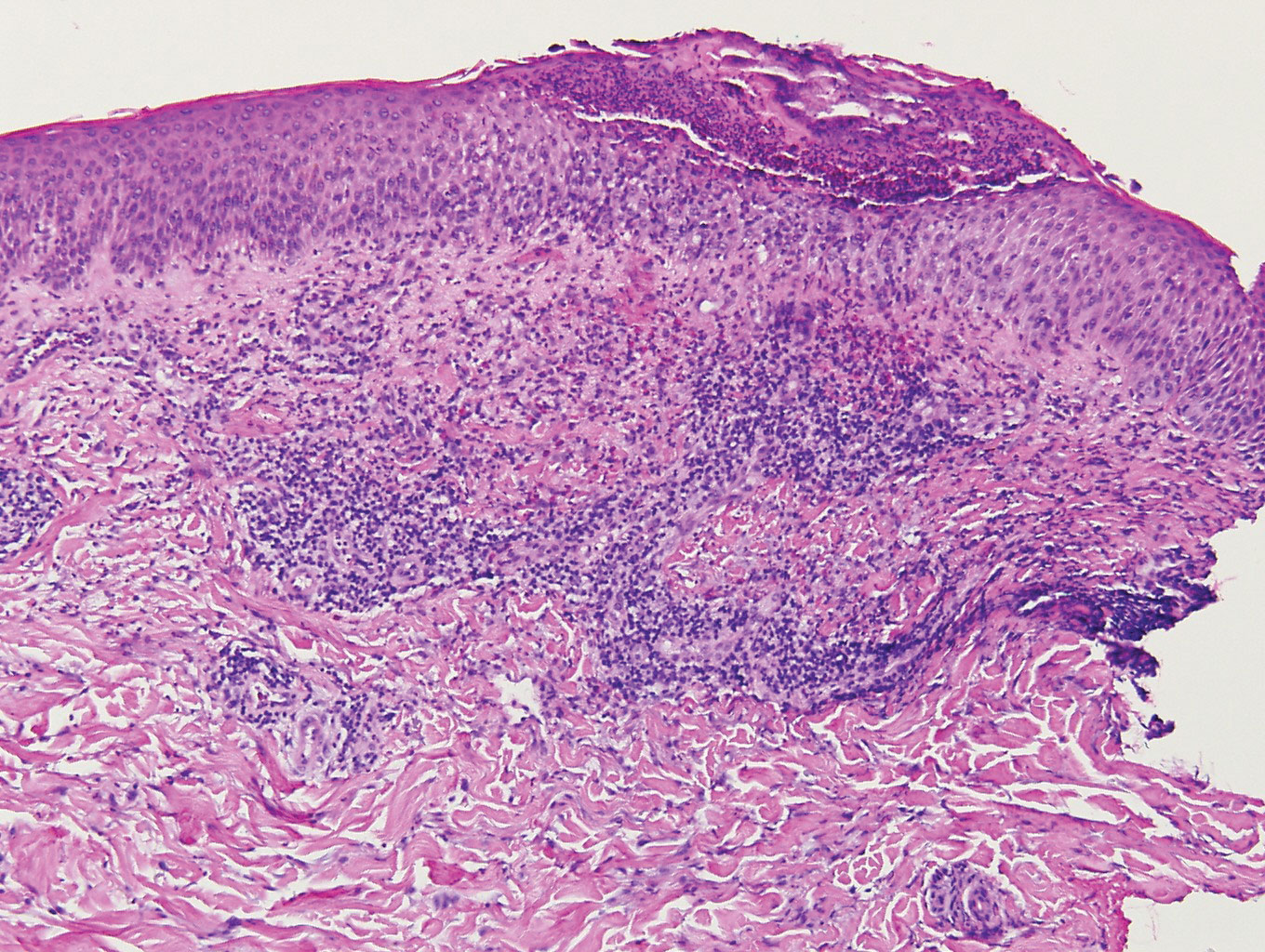
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
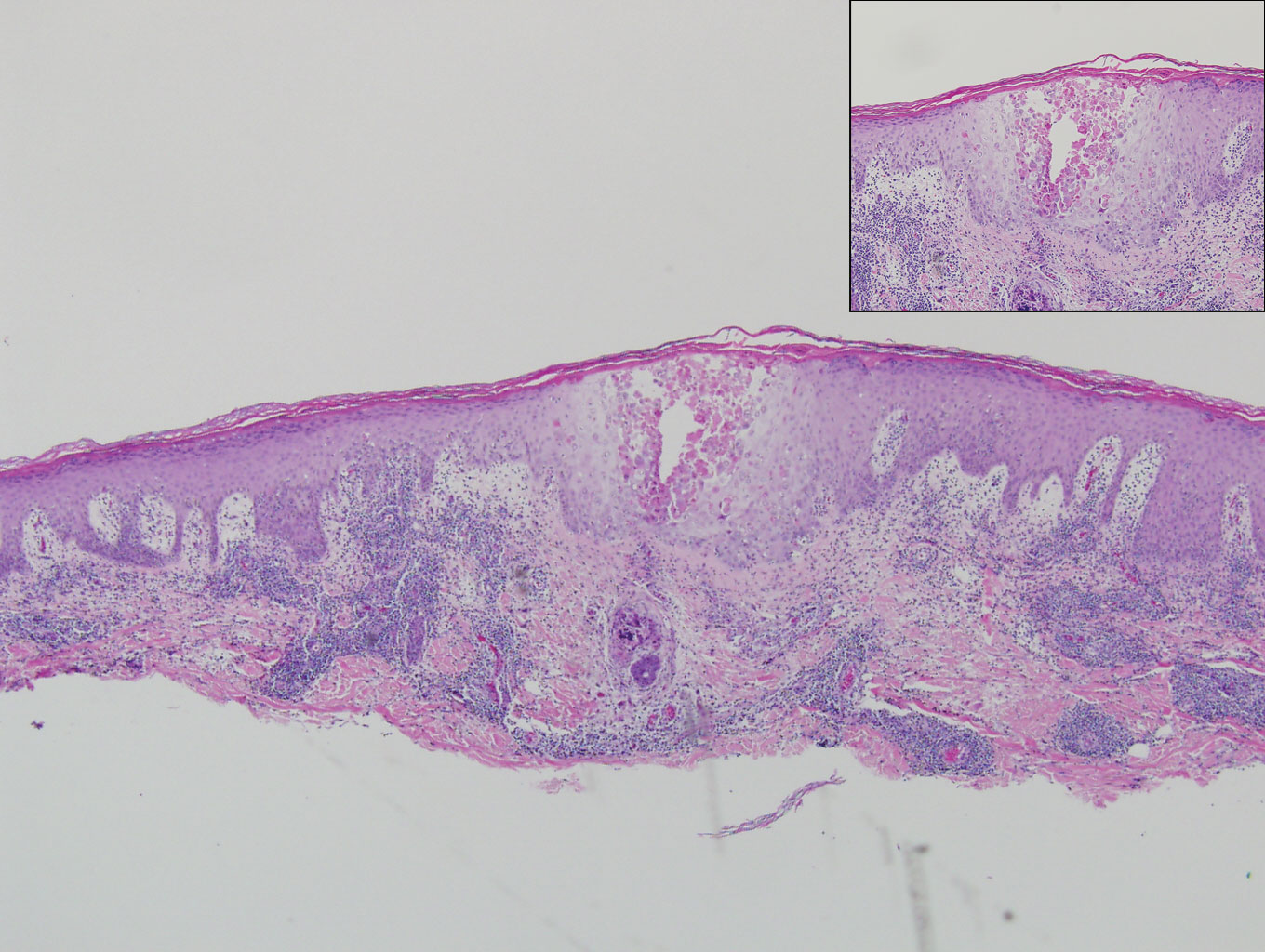
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.