User login
Bipolar moving target: Draw a bead on rapid cycling with type-specific therapies
Rapid-cycling bipolar disorder is a moving target, with treatment-resistant depression recurring frequently and alternating with hypomanic/manic episodes (Box).1,2 Can one medication adequately treat these complicated patients, or is combination therapy necessary? If more than one medication is needed, are some combinations more effective than others?
This article attempts to answer these questions by:
- discussing recent treatment trial results
- suggesting an algorithm for managing hypomanic/manic and depressive episodes in rapid-cycling patients with bipolar disorder types I or II.
CLINICAL CHARACTERISTICS
Rapid cycling is associated most consistently with female gender and bipolar II disorder2 (Table); why these two groups are primarily affected is unknown. Results of studies linking rapid cycling with hypothyroidism, gonadal steroid effects, family history, and substance use have been inconsistent and contradictory.2
Age of onset. Recent studies examining bipolar disorder’s age of onset have contradicted earlier rapid-cycling literature. In two large studies, Schneck et al3 and Coryell et al4 found rapid cycling associated with early onset of bipolar illness. The authors note that high rates of rapid cycling in children and adolescents resemble adult rapid cycling and speculate that early-onset bipolar illness might lead to rapid cycling vulnerability.5
Rapid cycling—defined in DSM-IV-TR as four or more depressive, manic, hypomanic, or mixed episodes in the previous 12 months—is considered a longitudinal course specifier for bipolar I or II disorder.1 Episodes must be demarcated by:
- full or partial remission lasting at least 2 months
- or a switch to a mood state of opposite polarity.
Cycling variations include ultra-rapid (1 day to 1 week), ultra-ultra rapid or ultradian (<24 hours), and continuous (no euthymic periods between mood episodes). Rapid cycling occurs in an estimated 15% to 25% of patients with bipolar disorder,2 though psychiatrists in specialty and tertiary referral centers see higher percentages because of the illness’ refractory nature.
Transient vs persistent state. Rapid cycling is thought to be either a transient state in long-term bipolar illness or a more chronic expression of the illness. Several studies6,7 have described rapid cycling as a transient phenomenon, whereas others8-11 have found a more persistent rapid cycling course during follow-up. Interestingly, a recent study11 suggested the mood-cycle pattern may be the most important predictor of rapid cycling. Patients with a depression–hypomania/mania-euthymia course demonstrated more-persistent rapid cycling than did those with a hypomania/mania-depression-euthymia course.
Antidepressants. Antidepressants’ role in initiating or exacerbating rapid cycling also remains unclear. Wehr et al8 found that discontinuing antidepressants contributed to cycling cessation or slowing. However, two prospective studies by Coryell et al4 that controlled for major depression found no association between antidepressant use and rapid cycling.
More recently, Yildiz and Sachs12 found a possible gender-specific relationship between antidepressants and rapid cycling. Women exposed to antidepressants before their first hypomanic/manic episode were more likely to develop rapid cycling than women who were not so exposed. This association was not evident in men.
NO DEFINITIVE CHOICES
Any discussion of treating rapid-cycling bipolar disorder is based on limited data, as few prospective studies of this exclusive cohort exist. Many studies report on mixed cohorts of refractory bipolar patients that include rapid cyclers, but separate analyses of rapid-cycling subgroups are not usually reported. Notable exceptions are recent studies by Calabrese et al, which are discussed below.
Lithium. Dunner and Fieve13 were the first to suggest that rapid-cycling bipolar patients respond poorly to lithium maintenance monotherapy. Later studies, however, suggested that lithium could benefit rapid cyclers, primarily in reducing hypomanic or manic episodes.
Baldessarini et al10 found that lithium was less effective for rapid than nonrapid cyclers only in reducing recurrence of depressive episodes. Kukopulos et al14 reported that lithium response in rapid cyclers increased from 16% to 78% after antidepressants were stopped, suggesting that a positive response to lithium may require more limited antidepressant use (or patients not having been exposed to antidepressants at all).
Thus, lithium prophylaxis has at least partial efficacy in many rapid cyclers, especially when antidepressants are avoided.
Divalproex. As with lithium, divalproex sodium appears more effective in treating and preventing hypomanic/manic episodes than depressive episodes in bipolar patients with rapid-cycling illness. Six open studies showed that patients who had not responded to lithium tended to do better with divalproex.15
Calabrese et al then tested the hypothesis that rapid cycling predicts nonresponse to lithium and positive response to divalproex.16 In a randomized controlled trial, they enrolled 254 recently hypomanic/manic rapid-cycling outpatients in an open-label stabilization phase involving combination lithium and divalproex therapy. Stabilized patients were then randomized to monotherapy with lithium, serum level ≥ 0.8 mEq/L, or divalproex, serum level ≥ 50 mcg/mL. Only 60 patients (24%) met stability criteria for randomization, achieving a persistent bimodal response as measured by continuous weeks of:
- Hamilton depression scale (24-item) score ≤ 20
- Young Mania Rating Scale score ≤ 12.5
- Global Assessment Scale score ≥ 51.
Most nonresponse was attributed to refractory depression.
After 20 months of maintenance therapy, about one-half of patients relapsed on either monotherapy. In the survival analysis, the median time to any mood episode was 45 weeks with divalproex monotherapy and 18 weeks with lithium monotherapy, although this difference was not statistically significant. The small sample size and high dropout rate may have created a false-negative error in this study.
Thus, these data did not show divalproex monotherapy to be more effective than lithium monotherapy in managing rapid-cycling bipolar disorder. The combination proved more effective in treating mania than depression and superior to monotherapy. This finding underscores combination therapy’s importance and the need to use mood stabilizers that also treat the depressed phase of bipolar disorder in rapid cyclers.
Table
Clinical characteristics of rapid cycling
| Prevalence approximately 15% to 25% in patients with bipolar disorder |
| More common in women than men |
| More common with type II than type I bipolar disorder |
| Primarily a depressive disease |
| Low treatment response rates and high recurrence risk |
| Associated with antidepressant use in some cases |
Carbamazepine. Recent data refute earlier reports suggesting that rapid cycling predicted positive response to carbamazepine. Multiple open studies and four controlled studies suggest that carbamazepine—like lithium and divalproex—possesses moderate to marked efficacy in the hypomanic/manic phase but poor to moderate efficacy in the depressed phase of rapid-cycling bipolar disorder.17
Lamotrigine. Lamotrigine is the first mood-stabilizing agent that has shown efficacy in maintenance treatment of bipolar depression and rapid cycling. In a double-blind, prospective, placebo-controlled trial, Calabrese et al18 enrolled 324 rapid-cycling patients with bipolar disorder type I or II in an open-label stabilization phase with lamotrigine. The 182 stabilized patients were then randomly assigned to receive either lamotrigine (mean 288 +/- 94 mg/d) or placebo.
For 6 months, 41% of patients receiving lamotrigine and 26% of those receiving placebo remained stable without relapse (P = 0.03), although the difference was statistically significant only for the bipolar II subtype. Lamotrigine appeared most effective in patients with the biphasic pattern of depression-hypomania/mania-euthymia.
Topiramate. Most studies of topiramate in rapid cycling have been retrospective and/or small add-on studies to existing mood stabilizers, with topiramate use associated with moderately or markedly improved manic symptoms.19 Evidence supports further controlled investigations, particularly because topiramate’s weight-loss effects may help overweight or obese patients.
Gabapentin. Gabapentin’s efficacy in rapid cycling has not been established. Although open-label studies showed a 67% response rate when gabapentin was used as adjunctive therapy, two double-blind, placebocontrolled studies of bipolar patients failed to show efficacy.20,21
Atypical antipsychotics. Five atypical antipsychotics—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for treating acute mania. Olanzapine is also indicated for bipolar maintenance treatment and has the most data showing efficacy in rapid cycling:
- In a 3-week, placebo-controlled study of 139 patients with bipolar I acute mania, olanzapine (median modal 15 mg/d) reduced manic symptoms to a statistically significantly extent in the 45 rapid cyclers.22
- A long-term prospective study followed 23 patients—30% of whom were rapid cyclers—who used olanzapine (mean 8.2 mg/d) as an adjunct to mood stabilizers. Manic and depressive symptoms were reduced significantly in the cohort, which was followed for a mean of 303 days.23
- An 8-week, double-blind, placebo-controlled study24 compared olanzapine monotherapy with the olanzapine/fluoxetine combination (OFC) in 833 depressed bipolar I patients, of whom 315 (37%) had rapid cycling. Mean olanzapine dosage was 9.7 mg in monotherapy and 7.4 mg in combination therapy; mean fluoxetine dosage was 39.3 mg.
A follow-up analysis25 showed that rapid cyclers’ depressive symptoms improved rapidly, and this improvement was sustained with OFC but not olanzapine monotherapy. Nonrapid-cycling patients responded to both treatments.
Other atypicals have shown partial efficacy in rapid-cycling bipolar disorder, although the studies have had methodologic limitations. Suppes et al26 conducted the first controlled trial using clozapine as add-on therapy in a 1-year, randomized evaluation of 38 patients with treatment-refractory bipolar disorder. The 21 rapid cyclers received a mean peak of 234 mg/d. Brief Psychiatric Rating Scale and Clinical Global Improvement scores improved significantly overall, but data specific to the rapid-cycling patients were not reported.
Small, open-label studies using risperidone and quetiapine as adjuncts to mood stabilizers have shown modest efficacy in rapid cycling, usually in treating manic symptoms. A recent 8-week, double-blind, placebo-controlled trial of quetiapine in bipolar depression showed promising results, though its efficacy in rapid cycling was not reported.27
RECOMMENDED TREATMENT
Because coincidental cycling may give the false appearance of efficacy in the short term, we recommend that you treat rapid cyclers methodically and judge outcomes over several months or cycle-lengths. A general approach includes:
- identify and treat underlying medical illnesses, such as hypothyroidism
- identify and treat comorbid alcohol/drug abuse
- taper or discontinue cycle-inducing agents such as antidepressants or sympathomimetics
- use standard mood stabilizers and/or atypical antipsychotics alone or in combination (Algorithm).
Algorithm Managing manic and depressive phases of rapid-cycling bipolar disorder
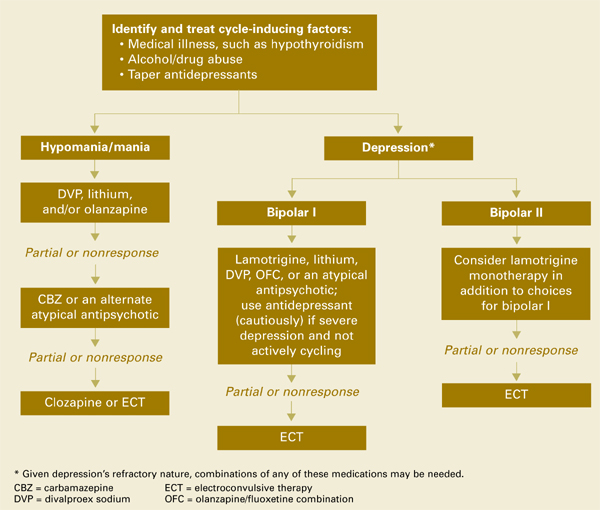
Treating acute mania in rapid-cycling patients is similar to managing this phase in nonrapid cyclers. First-tier therapy includes established mood stabilizers such as lithium, divalproex, or atypical antipsychotics. Carbamazepine is usually considered second-tier because of its effects on other medications via cytochrome P-450 system induction, and limited data exist on oxcarbazepine’s efficacy. Lamotrigine has not been proven effective in acute mania. If monotherapy is ineffective, try combinations of mood stabilizers and/or atypical antipsychotics.
Treating the depressed phase in rapid cyclers is far more difficult than treating acute mania and may depend on bipolar subtype:
- Bipolar I patients likely will require one or more mood stabilizers (such as lithium, divalproex, olanzapine) plus add-on lamotrigine.
- Bipolar II patients may benefit from lamotrigine alone.
- Atypical antipsychotics that have putative antidepressant effects without apparent cycle-accelerating effects may also be considered. At this time, olanzapine has the most data.
Given depression’s refractory nature in rapid-cycling bipolar illness, you may need to combine any of the above medications, try electroconvulsive therapy, or use more-experimental strategies such as:
- omega-3 fatty acids
- donepezil
- pramipexole
- high-dose levothyroxine/T4.
Antidepressants. Before using antidepressants to treat bipolar depression, consider carefully the risk of initiating or exacerbating rapid cycling. No definitive evidence is available to guide your decision.
Likewise, the optimal duration of antidepressant treatment is unclear, although tapering the antidepressant as tolerated may be prudent after depressive symptoms are in remission.
Psychosocial interventions. Finally, don’t overlook psychosocial interventions. Bipolar-specific psychotherapies can enhance compliance, lessen depression, and improve treatment response.28
CONCLUSION
Standard mood stabilizers appear to show partial efficacy in rapid cycling’s hypomanic/manic phase but only modest efficacy in the depressed phase. Lamotrigine appears more-promising in treating depressive than acute manic episodes and may be particularly effective for bipolar II patients. Evidence is growing that atypical antipsychotics also have partial efficacy in treating rapid cyclers, though whether this effect is phase-specific is unclear. As no single agent provides ideal bimodal treatment, combination therapy is recommended.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org. Accessed Oct. 14, 2004.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
- Marneros A, Goodwin FK (eds). Bipolar disorders: Mixed states, rapid cycling and atypical bipolar disorder. Cambridge, UK: Cambridge University Press (in press).
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Donepezil • Aricept
- Divalproex sodium • Depakote
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Levothyroxine • Synthroid, others
- Lithium • Lithobid, others
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Oxcarbazepine • Trileptal
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Ziprasidone • Geodon
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. Schneck is a consultant to AstraZeneca Pharmaceuticals, UCB Pharma, and Bristol-Myers Squibb Co. and a speaker for AstraZeneca Pharmaceuticals.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association, 2000.
2. Kupka RW, Luckenbaugh DA, Post RM, et al. Rapid and non-rapid cycling bipolar disorder: a meta-analysis of clinical studies. J Clin Psychiatry 2003;64(12):1483-94.
3. Schneck CD, Miklowitz DJ, Calabrese JR, et al. Phenomenology of rapid cycling bipolar disorder: data from the first 500 participants in the Systematic Treatment Enhancement Program. Am J Psychiatry 2004;161(10):1902-8.
4. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry 2003;60(9):914-20.
5. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric comorbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3:202-10.
6. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder. Demographics, diagnosis, family history, and course. Arch Gen Psychiatry 1992;49:126-31.
7. Maj M, Magliano L, Pirozzi R, et al. Validity of rapid cycling as a course specifier for bipolar disorder. Am J Psychiatry 1994;151:1015-19.
8. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
9. Bauer MS, Calabrese J, Dunner DL, et al. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
10. Baldessarini RJ, Tondo L, Floris G, Hennen J. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord 2000;61:13-22.
11. Koukopoulos A, Sani G, Koukopoulos AE, et al. Duration and stability of the rapid-cycling course: a long-term personal follow-up of 109 patients. J Affect Disord 2003;73:75-85.
12. Yildiz A, Sachs GS. Do antidepressants induce rapid cycling? A gender-specific association. J Clin Psychiatry 2003;64:814-18.
13. Dunner DL, Fieve RR. Clinical factors in lithium carbonate prophylaxis failure. Arch Gen Psychiatry 1974;30:229-33.
14. Kukopulos A, Reginaldi D, Laddomada P, et al. Course of the manic-depressive cycle and changes caused by treatments. Pharmakopsychiatr Neuropsychopharmakol 1980;13:156-67.
15. Calabrese JR, Woyshville MJ, Kimmel SE, Rapport DJ. Predictors of valproate response in bipolar rapid cycling. J Clin Psychopharmacol 1993;13:280-3.
16. Calabrese JR, Shelton M, Rapport DJ, et al. A double-blind 20 month maintenance study of lithium vs. divalproex in rapid-cycling bipolar disorder [presentation]. Pittsburgh, PA: Fifth International Conference on Bipolar Disorder, June 12-14, 2003.
17. Calabrese JR, Bowden C, Woyshville MJ. Lithium and anticonvulsants in the treatment of bipolar disorders. In: Bloom E, Kupfer D (eds). Psychopharmacology: The third generation of progress. New York: Raven Press, 1995;1099-1112.
18. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. J Clin Psychiatry 2000;61(11):841-50.
19. Marcotte D. Use of topiramate, a new anti-epileptic as a mood stabilizer. J Affect Disord 1998;50(2-3):245-51.
20. Pande AC, Crockatt JG, Janney CA, et al. Gabapentin in bipolar disorder: a placebo-controlled trial of adjunctive therapy. Gabapentin Bipolar Disorder Study Group. Bipolar Disord 2000;2(3 pt 2):249-55.
21. Frye MA, Ketter TA, Kimbrell TA, et al. A placebo-controlled study of lamotrigine and gabapentin monotherapy in refractory mood disorders. J Clin Psychopharmacol 2000;20(6):607-14.
22. Sanger TM, Tohen M, Vieta E, et al. Olanzapine in the acute treatment of bipolar I disorder with a history of rapid cycling. J Affect Disord 2003;73:155-61.
23. Calabrese JR, Kasper S, Johnson G, et al. International consensus group on bipolar I depression treatment guidelines. J Clin Psychiatry 2004;65:569-79.
24. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
25. Keck P, Corya S, Andersen SW, et al. Analysis of olanzapine/fluoxetine combination in the treatment of rapid-cycling bipolar depression [presentation]. Boca Raton, FL: New Clinical Drug Evaluation Unit, 2003.
26. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry 1999;156:1164-9.
27. Calabrese JR, Macfadden W, McCoy R, et al. Double-blind, placebo-controlled study of quetiapine in bipolar depression [presentation]. Phoenix, AZ: New Clinical Drug Evaluation Unit, 2004.
28. Craighead WE, Miklowitz DJ. Psychosocial interventions for bipolar disorder. J Clin Psychiatry 2000;61(suppl 13):58-64.
Rapid-cycling bipolar disorder is a moving target, with treatment-resistant depression recurring frequently and alternating with hypomanic/manic episodes (Box).1,2 Can one medication adequately treat these complicated patients, or is combination therapy necessary? If more than one medication is needed, are some combinations more effective than others?
This article attempts to answer these questions by:
- discussing recent treatment trial results
- suggesting an algorithm for managing hypomanic/manic and depressive episodes in rapid-cycling patients with bipolar disorder types I or II.
CLINICAL CHARACTERISTICS
Rapid cycling is associated most consistently with female gender and bipolar II disorder2 (Table); why these two groups are primarily affected is unknown. Results of studies linking rapid cycling with hypothyroidism, gonadal steroid effects, family history, and substance use have been inconsistent and contradictory.2
Age of onset. Recent studies examining bipolar disorder’s age of onset have contradicted earlier rapid-cycling literature. In two large studies, Schneck et al3 and Coryell et al4 found rapid cycling associated with early onset of bipolar illness. The authors note that high rates of rapid cycling in children and adolescents resemble adult rapid cycling and speculate that early-onset bipolar illness might lead to rapid cycling vulnerability.5
Rapid cycling—defined in DSM-IV-TR as four or more depressive, manic, hypomanic, or mixed episodes in the previous 12 months—is considered a longitudinal course specifier for bipolar I or II disorder.1 Episodes must be demarcated by:
- full or partial remission lasting at least 2 months
- or a switch to a mood state of opposite polarity.
Cycling variations include ultra-rapid (1 day to 1 week), ultra-ultra rapid or ultradian (<24 hours), and continuous (no euthymic periods between mood episodes). Rapid cycling occurs in an estimated 15% to 25% of patients with bipolar disorder,2 though psychiatrists in specialty and tertiary referral centers see higher percentages because of the illness’ refractory nature.
Transient vs persistent state. Rapid cycling is thought to be either a transient state in long-term bipolar illness or a more chronic expression of the illness. Several studies6,7 have described rapid cycling as a transient phenomenon, whereas others8-11 have found a more persistent rapid cycling course during follow-up. Interestingly, a recent study11 suggested the mood-cycle pattern may be the most important predictor of rapid cycling. Patients with a depression–hypomania/mania-euthymia course demonstrated more-persistent rapid cycling than did those with a hypomania/mania-depression-euthymia course.
Antidepressants. Antidepressants’ role in initiating or exacerbating rapid cycling also remains unclear. Wehr et al8 found that discontinuing antidepressants contributed to cycling cessation or slowing. However, two prospective studies by Coryell et al4 that controlled for major depression found no association between antidepressant use and rapid cycling.
More recently, Yildiz and Sachs12 found a possible gender-specific relationship between antidepressants and rapid cycling. Women exposed to antidepressants before their first hypomanic/manic episode were more likely to develop rapid cycling than women who were not so exposed. This association was not evident in men.
NO DEFINITIVE CHOICES
Any discussion of treating rapid-cycling bipolar disorder is based on limited data, as few prospective studies of this exclusive cohort exist. Many studies report on mixed cohorts of refractory bipolar patients that include rapid cyclers, but separate analyses of rapid-cycling subgroups are not usually reported. Notable exceptions are recent studies by Calabrese et al, which are discussed below.
Lithium. Dunner and Fieve13 were the first to suggest that rapid-cycling bipolar patients respond poorly to lithium maintenance monotherapy. Later studies, however, suggested that lithium could benefit rapid cyclers, primarily in reducing hypomanic or manic episodes.
Baldessarini et al10 found that lithium was less effective for rapid than nonrapid cyclers only in reducing recurrence of depressive episodes. Kukopulos et al14 reported that lithium response in rapid cyclers increased from 16% to 78% after antidepressants were stopped, suggesting that a positive response to lithium may require more limited antidepressant use (or patients not having been exposed to antidepressants at all).
Thus, lithium prophylaxis has at least partial efficacy in many rapid cyclers, especially when antidepressants are avoided.
Divalproex. As with lithium, divalproex sodium appears more effective in treating and preventing hypomanic/manic episodes than depressive episodes in bipolar patients with rapid-cycling illness. Six open studies showed that patients who had not responded to lithium tended to do better with divalproex.15
Calabrese et al then tested the hypothesis that rapid cycling predicts nonresponse to lithium and positive response to divalproex.16 In a randomized controlled trial, they enrolled 254 recently hypomanic/manic rapid-cycling outpatients in an open-label stabilization phase involving combination lithium and divalproex therapy. Stabilized patients were then randomized to monotherapy with lithium, serum level ≥ 0.8 mEq/L, or divalproex, serum level ≥ 50 mcg/mL. Only 60 patients (24%) met stability criteria for randomization, achieving a persistent bimodal response as measured by continuous weeks of:
- Hamilton depression scale (24-item) score ≤ 20
- Young Mania Rating Scale score ≤ 12.5
- Global Assessment Scale score ≥ 51.
Most nonresponse was attributed to refractory depression.
After 20 months of maintenance therapy, about one-half of patients relapsed on either monotherapy. In the survival analysis, the median time to any mood episode was 45 weeks with divalproex monotherapy and 18 weeks with lithium monotherapy, although this difference was not statistically significant. The small sample size and high dropout rate may have created a false-negative error in this study.
Thus, these data did not show divalproex monotherapy to be more effective than lithium monotherapy in managing rapid-cycling bipolar disorder. The combination proved more effective in treating mania than depression and superior to monotherapy. This finding underscores combination therapy’s importance and the need to use mood stabilizers that also treat the depressed phase of bipolar disorder in rapid cyclers.
Table
Clinical characteristics of rapid cycling
| Prevalence approximately 15% to 25% in patients with bipolar disorder |
| More common in women than men |
| More common with type II than type I bipolar disorder |
| Primarily a depressive disease |
| Low treatment response rates and high recurrence risk |
| Associated with antidepressant use in some cases |
Carbamazepine. Recent data refute earlier reports suggesting that rapid cycling predicted positive response to carbamazepine. Multiple open studies and four controlled studies suggest that carbamazepine—like lithium and divalproex—possesses moderate to marked efficacy in the hypomanic/manic phase but poor to moderate efficacy in the depressed phase of rapid-cycling bipolar disorder.17
Lamotrigine. Lamotrigine is the first mood-stabilizing agent that has shown efficacy in maintenance treatment of bipolar depression and rapid cycling. In a double-blind, prospective, placebo-controlled trial, Calabrese et al18 enrolled 324 rapid-cycling patients with bipolar disorder type I or II in an open-label stabilization phase with lamotrigine. The 182 stabilized patients were then randomly assigned to receive either lamotrigine (mean 288 +/- 94 mg/d) or placebo.
For 6 months, 41% of patients receiving lamotrigine and 26% of those receiving placebo remained stable without relapse (P = 0.03), although the difference was statistically significant only for the bipolar II subtype. Lamotrigine appeared most effective in patients with the biphasic pattern of depression-hypomania/mania-euthymia.
Topiramate. Most studies of topiramate in rapid cycling have been retrospective and/or small add-on studies to existing mood stabilizers, with topiramate use associated with moderately or markedly improved manic symptoms.19 Evidence supports further controlled investigations, particularly because topiramate’s weight-loss effects may help overweight or obese patients.
Gabapentin. Gabapentin’s efficacy in rapid cycling has not been established. Although open-label studies showed a 67% response rate when gabapentin was used as adjunctive therapy, two double-blind, placebocontrolled studies of bipolar patients failed to show efficacy.20,21
Atypical antipsychotics. Five atypical antipsychotics—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for treating acute mania. Olanzapine is also indicated for bipolar maintenance treatment and has the most data showing efficacy in rapid cycling:
- In a 3-week, placebo-controlled study of 139 patients with bipolar I acute mania, olanzapine (median modal 15 mg/d) reduced manic symptoms to a statistically significantly extent in the 45 rapid cyclers.22
- A long-term prospective study followed 23 patients—30% of whom were rapid cyclers—who used olanzapine (mean 8.2 mg/d) as an adjunct to mood stabilizers. Manic and depressive symptoms were reduced significantly in the cohort, which was followed for a mean of 303 days.23
- An 8-week, double-blind, placebo-controlled study24 compared olanzapine monotherapy with the olanzapine/fluoxetine combination (OFC) in 833 depressed bipolar I patients, of whom 315 (37%) had rapid cycling. Mean olanzapine dosage was 9.7 mg in monotherapy and 7.4 mg in combination therapy; mean fluoxetine dosage was 39.3 mg.
A follow-up analysis25 showed that rapid cyclers’ depressive symptoms improved rapidly, and this improvement was sustained with OFC but not olanzapine monotherapy. Nonrapid-cycling patients responded to both treatments.
Other atypicals have shown partial efficacy in rapid-cycling bipolar disorder, although the studies have had methodologic limitations. Suppes et al26 conducted the first controlled trial using clozapine as add-on therapy in a 1-year, randomized evaluation of 38 patients with treatment-refractory bipolar disorder. The 21 rapid cyclers received a mean peak of 234 mg/d. Brief Psychiatric Rating Scale and Clinical Global Improvement scores improved significantly overall, but data specific to the rapid-cycling patients were not reported.
Small, open-label studies using risperidone and quetiapine as adjuncts to mood stabilizers have shown modest efficacy in rapid cycling, usually in treating manic symptoms. A recent 8-week, double-blind, placebo-controlled trial of quetiapine in bipolar depression showed promising results, though its efficacy in rapid cycling was not reported.27
RECOMMENDED TREATMENT
Because coincidental cycling may give the false appearance of efficacy in the short term, we recommend that you treat rapid cyclers methodically and judge outcomes over several months or cycle-lengths. A general approach includes:
- identify and treat underlying medical illnesses, such as hypothyroidism
- identify and treat comorbid alcohol/drug abuse
- taper or discontinue cycle-inducing agents such as antidepressants or sympathomimetics
- use standard mood stabilizers and/or atypical antipsychotics alone or in combination (Algorithm).
Algorithm Managing manic and depressive phases of rapid-cycling bipolar disorder
Treating acute mania in rapid-cycling patients is similar to managing this phase in nonrapid cyclers. First-tier therapy includes established mood stabilizers such as lithium, divalproex, or atypical antipsychotics. Carbamazepine is usually considered second-tier because of its effects on other medications via cytochrome P-450 system induction, and limited data exist on oxcarbazepine’s efficacy. Lamotrigine has not been proven effective in acute mania. If monotherapy is ineffective, try combinations of mood stabilizers and/or atypical antipsychotics.
Treating the depressed phase in rapid cyclers is far more difficult than treating acute mania and may depend on bipolar subtype:
- Bipolar I patients likely will require one or more mood stabilizers (such as lithium, divalproex, olanzapine) plus add-on lamotrigine.
- Bipolar II patients may benefit from lamotrigine alone.
- Atypical antipsychotics that have putative antidepressant effects without apparent cycle-accelerating effects may also be considered. At this time, olanzapine has the most data.
Given depression’s refractory nature in rapid-cycling bipolar illness, you may need to combine any of the above medications, try electroconvulsive therapy, or use more-experimental strategies such as:
- omega-3 fatty acids
- donepezil
- pramipexole
- high-dose levothyroxine/T4.
Antidepressants. Before using antidepressants to treat bipolar depression, consider carefully the risk of initiating or exacerbating rapid cycling. No definitive evidence is available to guide your decision.
Likewise, the optimal duration of antidepressant treatment is unclear, although tapering the antidepressant as tolerated may be prudent after depressive symptoms are in remission.
Psychosocial interventions. Finally, don’t overlook psychosocial interventions. Bipolar-specific psychotherapies can enhance compliance, lessen depression, and improve treatment response.28
CONCLUSION
Standard mood stabilizers appear to show partial efficacy in rapid cycling’s hypomanic/manic phase but only modest efficacy in the depressed phase. Lamotrigine appears more-promising in treating depressive than acute manic episodes and may be particularly effective for bipolar II patients. Evidence is growing that atypical antipsychotics also have partial efficacy in treating rapid cyclers, though whether this effect is phase-specific is unclear. As no single agent provides ideal bimodal treatment, combination therapy is recommended.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org. Accessed Oct. 14, 2004.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
- Marneros A, Goodwin FK (eds). Bipolar disorders: Mixed states, rapid cycling and atypical bipolar disorder. Cambridge, UK: Cambridge University Press (in press).
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Donepezil • Aricept
- Divalproex sodium • Depakote
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Levothyroxine • Synthroid, others
- Lithium • Lithobid, others
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Oxcarbazepine • Trileptal
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Ziprasidone • Geodon
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. Schneck is a consultant to AstraZeneca Pharmaceuticals, UCB Pharma, and Bristol-Myers Squibb Co. and a speaker for AstraZeneca Pharmaceuticals.
Rapid-cycling bipolar disorder is a moving target, with treatment-resistant depression recurring frequently and alternating with hypomanic/manic episodes (Box).1,2 Can one medication adequately treat these complicated patients, or is combination therapy necessary? If more than one medication is needed, are some combinations more effective than others?
This article attempts to answer these questions by:
- discussing recent treatment trial results
- suggesting an algorithm for managing hypomanic/manic and depressive episodes in rapid-cycling patients with bipolar disorder types I or II.
CLINICAL CHARACTERISTICS
Rapid cycling is associated most consistently with female gender and bipolar II disorder2 (Table); why these two groups are primarily affected is unknown. Results of studies linking rapid cycling with hypothyroidism, gonadal steroid effects, family history, and substance use have been inconsistent and contradictory.2
Age of onset. Recent studies examining bipolar disorder’s age of onset have contradicted earlier rapid-cycling literature. In two large studies, Schneck et al3 and Coryell et al4 found rapid cycling associated with early onset of bipolar illness. The authors note that high rates of rapid cycling in children and adolescents resemble adult rapid cycling and speculate that early-onset bipolar illness might lead to rapid cycling vulnerability.5
Rapid cycling—defined in DSM-IV-TR as four or more depressive, manic, hypomanic, or mixed episodes in the previous 12 months—is considered a longitudinal course specifier for bipolar I or II disorder.1 Episodes must be demarcated by:
- full or partial remission lasting at least 2 months
- or a switch to a mood state of opposite polarity.
Cycling variations include ultra-rapid (1 day to 1 week), ultra-ultra rapid or ultradian (<24 hours), and continuous (no euthymic periods between mood episodes). Rapid cycling occurs in an estimated 15% to 25% of patients with bipolar disorder,2 though psychiatrists in specialty and tertiary referral centers see higher percentages because of the illness’ refractory nature.
Transient vs persistent state. Rapid cycling is thought to be either a transient state in long-term bipolar illness or a more chronic expression of the illness. Several studies6,7 have described rapid cycling as a transient phenomenon, whereas others8-11 have found a more persistent rapid cycling course during follow-up. Interestingly, a recent study11 suggested the mood-cycle pattern may be the most important predictor of rapid cycling. Patients with a depression–hypomania/mania-euthymia course demonstrated more-persistent rapid cycling than did those with a hypomania/mania-depression-euthymia course.
Antidepressants. Antidepressants’ role in initiating or exacerbating rapid cycling also remains unclear. Wehr et al8 found that discontinuing antidepressants contributed to cycling cessation or slowing. However, two prospective studies by Coryell et al4 that controlled for major depression found no association between antidepressant use and rapid cycling.
More recently, Yildiz and Sachs12 found a possible gender-specific relationship between antidepressants and rapid cycling. Women exposed to antidepressants before their first hypomanic/manic episode were more likely to develop rapid cycling than women who were not so exposed. This association was not evident in men.
NO DEFINITIVE CHOICES
Any discussion of treating rapid-cycling bipolar disorder is based on limited data, as few prospective studies of this exclusive cohort exist. Many studies report on mixed cohorts of refractory bipolar patients that include rapid cyclers, but separate analyses of rapid-cycling subgroups are not usually reported. Notable exceptions are recent studies by Calabrese et al, which are discussed below.
Lithium. Dunner and Fieve13 were the first to suggest that rapid-cycling bipolar patients respond poorly to lithium maintenance monotherapy. Later studies, however, suggested that lithium could benefit rapid cyclers, primarily in reducing hypomanic or manic episodes.
Baldessarini et al10 found that lithium was less effective for rapid than nonrapid cyclers only in reducing recurrence of depressive episodes. Kukopulos et al14 reported that lithium response in rapid cyclers increased from 16% to 78% after antidepressants were stopped, suggesting that a positive response to lithium may require more limited antidepressant use (or patients not having been exposed to antidepressants at all).
Thus, lithium prophylaxis has at least partial efficacy in many rapid cyclers, especially when antidepressants are avoided.
Divalproex. As with lithium, divalproex sodium appears more effective in treating and preventing hypomanic/manic episodes than depressive episodes in bipolar patients with rapid-cycling illness. Six open studies showed that patients who had not responded to lithium tended to do better with divalproex.15
Calabrese et al then tested the hypothesis that rapid cycling predicts nonresponse to lithium and positive response to divalproex.16 In a randomized controlled trial, they enrolled 254 recently hypomanic/manic rapid-cycling outpatients in an open-label stabilization phase involving combination lithium and divalproex therapy. Stabilized patients were then randomized to monotherapy with lithium, serum level ≥ 0.8 mEq/L, or divalproex, serum level ≥ 50 mcg/mL. Only 60 patients (24%) met stability criteria for randomization, achieving a persistent bimodal response as measured by continuous weeks of:
- Hamilton depression scale (24-item) score ≤ 20
- Young Mania Rating Scale score ≤ 12.5
- Global Assessment Scale score ≥ 51.
Most nonresponse was attributed to refractory depression.
After 20 months of maintenance therapy, about one-half of patients relapsed on either monotherapy. In the survival analysis, the median time to any mood episode was 45 weeks with divalproex monotherapy and 18 weeks with lithium monotherapy, although this difference was not statistically significant. The small sample size and high dropout rate may have created a false-negative error in this study.
Thus, these data did not show divalproex monotherapy to be more effective than lithium monotherapy in managing rapid-cycling bipolar disorder. The combination proved more effective in treating mania than depression and superior to monotherapy. This finding underscores combination therapy’s importance and the need to use mood stabilizers that also treat the depressed phase of bipolar disorder in rapid cyclers.
Table
Clinical characteristics of rapid cycling
| Prevalence approximately 15% to 25% in patients with bipolar disorder |
| More common in women than men |
| More common with type II than type I bipolar disorder |
| Primarily a depressive disease |
| Low treatment response rates and high recurrence risk |
| Associated with antidepressant use in some cases |
Carbamazepine. Recent data refute earlier reports suggesting that rapid cycling predicted positive response to carbamazepine. Multiple open studies and four controlled studies suggest that carbamazepine—like lithium and divalproex—possesses moderate to marked efficacy in the hypomanic/manic phase but poor to moderate efficacy in the depressed phase of rapid-cycling bipolar disorder.17
Lamotrigine. Lamotrigine is the first mood-stabilizing agent that has shown efficacy in maintenance treatment of bipolar depression and rapid cycling. In a double-blind, prospective, placebo-controlled trial, Calabrese et al18 enrolled 324 rapid-cycling patients with bipolar disorder type I or II in an open-label stabilization phase with lamotrigine. The 182 stabilized patients were then randomly assigned to receive either lamotrigine (mean 288 +/- 94 mg/d) or placebo.
For 6 months, 41% of patients receiving lamotrigine and 26% of those receiving placebo remained stable without relapse (P = 0.03), although the difference was statistically significant only for the bipolar II subtype. Lamotrigine appeared most effective in patients with the biphasic pattern of depression-hypomania/mania-euthymia.
Topiramate. Most studies of topiramate in rapid cycling have been retrospective and/or small add-on studies to existing mood stabilizers, with topiramate use associated with moderately or markedly improved manic symptoms.19 Evidence supports further controlled investigations, particularly because topiramate’s weight-loss effects may help overweight or obese patients.
Gabapentin. Gabapentin’s efficacy in rapid cycling has not been established. Although open-label studies showed a 67% response rate when gabapentin was used as adjunctive therapy, two double-blind, placebocontrolled studies of bipolar patients failed to show efficacy.20,21
Atypical antipsychotics. Five atypical antipsychotics—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for treating acute mania. Olanzapine is also indicated for bipolar maintenance treatment and has the most data showing efficacy in rapid cycling:
- In a 3-week, placebo-controlled study of 139 patients with bipolar I acute mania, olanzapine (median modal 15 mg/d) reduced manic symptoms to a statistically significantly extent in the 45 rapid cyclers.22
- A long-term prospective study followed 23 patients—30% of whom were rapid cyclers—who used olanzapine (mean 8.2 mg/d) as an adjunct to mood stabilizers. Manic and depressive symptoms were reduced significantly in the cohort, which was followed for a mean of 303 days.23
- An 8-week, double-blind, placebo-controlled study24 compared olanzapine monotherapy with the olanzapine/fluoxetine combination (OFC) in 833 depressed bipolar I patients, of whom 315 (37%) had rapid cycling. Mean olanzapine dosage was 9.7 mg in monotherapy and 7.4 mg in combination therapy; mean fluoxetine dosage was 39.3 mg.
A follow-up analysis25 showed that rapid cyclers’ depressive symptoms improved rapidly, and this improvement was sustained with OFC but not olanzapine monotherapy. Nonrapid-cycling patients responded to both treatments.
Other atypicals have shown partial efficacy in rapid-cycling bipolar disorder, although the studies have had methodologic limitations. Suppes et al26 conducted the first controlled trial using clozapine as add-on therapy in a 1-year, randomized evaluation of 38 patients with treatment-refractory bipolar disorder. The 21 rapid cyclers received a mean peak of 234 mg/d. Brief Psychiatric Rating Scale and Clinical Global Improvement scores improved significantly overall, but data specific to the rapid-cycling patients were not reported.
Small, open-label studies using risperidone and quetiapine as adjuncts to mood stabilizers have shown modest efficacy in rapid cycling, usually in treating manic symptoms. A recent 8-week, double-blind, placebo-controlled trial of quetiapine in bipolar depression showed promising results, though its efficacy in rapid cycling was not reported.27
RECOMMENDED TREATMENT
Because coincidental cycling may give the false appearance of efficacy in the short term, we recommend that you treat rapid cyclers methodically and judge outcomes over several months or cycle-lengths. A general approach includes:
- identify and treat underlying medical illnesses, such as hypothyroidism
- identify and treat comorbid alcohol/drug abuse
- taper or discontinue cycle-inducing agents such as antidepressants or sympathomimetics
- use standard mood stabilizers and/or atypical antipsychotics alone or in combination (Algorithm).
Algorithm Managing manic and depressive phases of rapid-cycling bipolar disorder
Treating acute mania in rapid-cycling patients is similar to managing this phase in nonrapid cyclers. First-tier therapy includes established mood stabilizers such as lithium, divalproex, or atypical antipsychotics. Carbamazepine is usually considered second-tier because of its effects on other medications via cytochrome P-450 system induction, and limited data exist on oxcarbazepine’s efficacy. Lamotrigine has not been proven effective in acute mania. If monotherapy is ineffective, try combinations of mood stabilizers and/or atypical antipsychotics.
Treating the depressed phase in rapid cyclers is far more difficult than treating acute mania and may depend on bipolar subtype:
- Bipolar I patients likely will require one or more mood stabilizers (such as lithium, divalproex, olanzapine) plus add-on lamotrigine.
- Bipolar II patients may benefit from lamotrigine alone.
- Atypical antipsychotics that have putative antidepressant effects without apparent cycle-accelerating effects may also be considered. At this time, olanzapine has the most data.
Given depression’s refractory nature in rapid-cycling bipolar illness, you may need to combine any of the above medications, try electroconvulsive therapy, or use more-experimental strategies such as:
- omega-3 fatty acids
- donepezil
- pramipexole
- high-dose levothyroxine/T4.
Antidepressants. Before using antidepressants to treat bipolar depression, consider carefully the risk of initiating or exacerbating rapid cycling. No definitive evidence is available to guide your decision.
Likewise, the optimal duration of antidepressant treatment is unclear, although tapering the antidepressant as tolerated may be prudent after depressive symptoms are in remission.
Psychosocial interventions. Finally, don’t overlook psychosocial interventions. Bipolar-specific psychotherapies can enhance compliance, lessen depression, and improve treatment response.28
CONCLUSION
Standard mood stabilizers appear to show partial efficacy in rapid cycling’s hypomanic/manic phase but only modest efficacy in the depressed phase. Lamotrigine appears more-promising in treating depressive than acute manic episodes and may be particularly effective for bipolar II patients. Evidence is growing that atypical antipsychotics also have partial efficacy in treating rapid cyclers, though whether this effect is phase-specific is unclear. As no single agent provides ideal bimodal treatment, combination therapy is recommended.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org. Accessed Oct. 14, 2004.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
- Marneros A, Goodwin FK (eds). Bipolar disorders: Mixed states, rapid cycling and atypical bipolar disorder. Cambridge, UK: Cambridge University Press (in press).
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Donepezil • Aricept
- Divalproex sodium • Depakote
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Levothyroxine • Synthroid, others
- Lithium • Lithobid, others
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Oxcarbazepine • Trileptal
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Ziprasidone • Geodon
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
Dr. Schneck is a consultant to AstraZeneca Pharmaceuticals, UCB Pharma, and Bristol-Myers Squibb Co. and a speaker for AstraZeneca Pharmaceuticals.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association, 2000.
2. Kupka RW, Luckenbaugh DA, Post RM, et al. Rapid and non-rapid cycling bipolar disorder: a meta-analysis of clinical studies. J Clin Psychiatry 2003;64(12):1483-94.
3. Schneck CD, Miklowitz DJ, Calabrese JR, et al. Phenomenology of rapid cycling bipolar disorder: data from the first 500 participants in the Systematic Treatment Enhancement Program. Am J Psychiatry 2004;161(10):1902-8.
4. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry 2003;60(9):914-20.
5. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric comorbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3:202-10.
6. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder. Demographics, diagnosis, family history, and course. Arch Gen Psychiatry 1992;49:126-31.
7. Maj M, Magliano L, Pirozzi R, et al. Validity of rapid cycling as a course specifier for bipolar disorder. Am J Psychiatry 1994;151:1015-19.
8. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
9. Bauer MS, Calabrese J, Dunner DL, et al. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
10. Baldessarini RJ, Tondo L, Floris G, Hennen J. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord 2000;61:13-22.
11. Koukopoulos A, Sani G, Koukopoulos AE, et al. Duration and stability of the rapid-cycling course: a long-term personal follow-up of 109 patients. J Affect Disord 2003;73:75-85.
12. Yildiz A, Sachs GS. Do antidepressants induce rapid cycling? A gender-specific association. J Clin Psychiatry 2003;64:814-18.
13. Dunner DL, Fieve RR. Clinical factors in lithium carbonate prophylaxis failure. Arch Gen Psychiatry 1974;30:229-33.
14. Kukopulos A, Reginaldi D, Laddomada P, et al. Course of the manic-depressive cycle and changes caused by treatments. Pharmakopsychiatr Neuropsychopharmakol 1980;13:156-67.
15. Calabrese JR, Woyshville MJ, Kimmel SE, Rapport DJ. Predictors of valproate response in bipolar rapid cycling. J Clin Psychopharmacol 1993;13:280-3.
16. Calabrese JR, Shelton M, Rapport DJ, et al. A double-blind 20 month maintenance study of lithium vs. divalproex in rapid-cycling bipolar disorder [presentation]. Pittsburgh, PA: Fifth International Conference on Bipolar Disorder, June 12-14, 2003.
17. Calabrese JR, Bowden C, Woyshville MJ. Lithium and anticonvulsants in the treatment of bipolar disorders. In: Bloom E, Kupfer D (eds). Psychopharmacology: The third generation of progress. New York: Raven Press, 1995;1099-1112.
18. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. J Clin Psychiatry 2000;61(11):841-50.
19. Marcotte D. Use of topiramate, a new anti-epileptic as a mood stabilizer. J Affect Disord 1998;50(2-3):245-51.
20. Pande AC, Crockatt JG, Janney CA, et al. Gabapentin in bipolar disorder: a placebo-controlled trial of adjunctive therapy. Gabapentin Bipolar Disorder Study Group. Bipolar Disord 2000;2(3 pt 2):249-55.
21. Frye MA, Ketter TA, Kimbrell TA, et al. A placebo-controlled study of lamotrigine and gabapentin monotherapy in refractory mood disorders. J Clin Psychopharmacol 2000;20(6):607-14.
22. Sanger TM, Tohen M, Vieta E, et al. Olanzapine in the acute treatment of bipolar I disorder with a history of rapid cycling. J Affect Disord 2003;73:155-61.
23. Calabrese JR, Kasper S, Johnson G, et al. International consensus group on bipolar I depression treatment guidelines. J Clin Psychiatry 2004;65:569-79.
24. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
25. Keck P, Corya S, Andersen SW, et al. Analysis of olanzapine/fluoxetine combination in the treatment of rapid-cycling bipolar depression [presentation]. Boca Raton, FL: New Clinical Drug Evaluation Unit, 2003.
26. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry 1999;156:1164-9.
27. Calabrese JR, Macfadden W, McCoy R, et al. Double-blind, placebo-controlled study of quetiapine in bipolar depression [presentation]. Phoenix, AZ: New Clinical Drug Evaluation Unit, 2004.
28. Craighead WE, Miklowitz DJ. Psychosocial interventions for bipolar disorder. J Clin Psychiatry 2000;61(suppl 13):58-64.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association, 2000.
2. Kupka RW, Luckenbaugh DA, Post RM, et al. Rapid and non-rapid cycling bipolar disorder: a meta-analysis of clinical studies. J Clin Psychiatry 2003;64(12):1483-94.
3. Schneck CD, Miklowitz DJ, Calabrese JR, et al. Phenomenology of rapid cycling bipolar disorder: data from the first 500 participants in the Systematic Treatment Enhancement Program. Am J Psychiatry 2004;161(10):1902-8.
4. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry 2003;60(9):914-20.
5. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric comorbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3:202-10.
6. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder. Demographics, diagnosis, family history, and course. Arch Gen Psychiatry 1992;49:126-31.
7. Maj M, Magliano L, Pirozzi R, et al. Validity of rapid cycling as a course specifier for bipolar disorder. Am J Psychiatry 1994;151:1015-19.
8. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
9. Bauer MS, Calabrese J, Dunner DL, et al. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
10. Baldessarini RJ, Tondo L, Floris G, Hennen J. Effects of rapid cycling on response to lithium maintenance treatment in 360 bipolar I and II disorder patients. J Affect Disord 2000;61:13-22.
11. Koukopoulos A, Sani G, Koukopoulos AE, et al. Duration and stability of the rapid-cycling course: a long-term personal follow-up of 109 patients. J Affect Disord 2003;73:75-85.
12. Yildiz A, Sachs GS. Do antidepressants induce rapid cycling? A gender-specific association. J Clin Psychiatry 2003;64:814-18.
13. Dunner DL, Fieve RR. Clinical factors in lithium carbonate prophylaxis failure. Arch Gen Psychiatry 1974;30:229-33.
14. Kukopulos A, Reginaldi D, Laddomada P, et al. Course of the manic-depressive cycle and changes caused by treatments. Pharmakopsychiatr Neuropsychopharmakol 1980;13:156-67.
15. Calabrese JR, Woyshville MJ, Kimmel SE, Rapport DJ. Predictors of valproate response in bipolar rapid cycling. J Clin Psychopharmacol 1993;13:280-3.
16. Calabrese JR, Shelton M, Rapport DJ, et al. A double-blind 20 month maintenance study of lithium vs. divalproex in rapid-cycling bipolar disorder [presentation]. Pittsburgh, PA: Fifth International Conference on Bipolar Disorder, June 12-14, 2003.
17. Calabrese JR, Bowden C, Woyshville MJ. Lithium and anticonvulsants in the treatment of bipolar disorders. In: Bloom E, Kupfer D (eds). Psychopharmacology: The third generation of progress. New York: Raven Press, 1995;1099-1112.
18. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. J Clin Psychiatry 2000;61(11):841-50.
19. Marcotte D. Use of topiramate, a new anti-epileptic as a mood stabilizer. J Affect Disord 1998;50(2-3):245-51.
20. Pande AC, Crockatt JG, Janney CA, et al. Gabapentin in bipolar disorder: a placebo-controlled trial of adjunctive therapy. Gabapentin Bipolar Disorder Study Group. Bipolar Disord 2000;2(3 pt 2):249-55.
21. Frye MA, Ketter TA, Kimbrell TA, et al. A placebo-controlled study of lamotrigine and gabapentin monotherapy in refractory mood disorders. J Clin Psychopharmacol 2000;20(6):607-14.
22. Sanger TM, Tohen M, Vieta E, et al. Olanzapine in the acute treatment of bipolar I disorder with a history of rapid cycling. J Affect Disord 2003;73:155-61.
23. Calabrese JR, Kasper S, Johnson G, et al. International consensus group on bipolar I depression treatment guidelines. J Clin Psychiatry 2004;65:569-79.
24. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
25. Keck P, Corya S, Andersen SW, et al. Analysis of olanzapine/fluoxetine combination in the treatment of rapid-cycling bipolar depression [presentation]. Boca Raton, FL: New Clinical Drug Evaluation Unit, 2003.
26. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry 1999;156:1164-9.
27. Calabrese JR, Macfadden W, McCoy R, et al. Double-blind, placebo-controlled study of quetiapine in bipolar depression [presentation]. Phoenix, AZ: New Clinical Drug Evaluation Unit, 2004.
28. Craighead WE, Miklowitz DJ. Psychosocial interventions for bipolar disorder. J Clin Psychiatry 2000;61(suppl 13):58-64.
Risk taking adolescents: When and how to intervene
Boys will be boys” and other platitudes may condone adolescent reckless driving, substance use, or sexual promiscuity—but to write off dangerous behavior as normal would be a mistake. Because adolescent impulsivity and sensation-seeking may have physiologic as well as emotional causes,1,2 excessive risk taking may be treatable.
This article discusses the neurobiology of adolescent risk taking, suggests how to determine when such behavior may be pathologic, and offers a treatment approach for at-risk teens and their parents.
CASE: ‘WHAT’S WRONG WITH OUR SON?’
Josh, age 17, is brought to the adolescent psychiatry clinic by his distraught parents, who report their son has undergone a “personality change” over the past 2 years. They recall that he was respectful, studious, and soft-spoken until age 15. Since then, he has been skipping school, staying out late at night with his friends, and “obsessed” with TV poker games.
His parents recently discovered he has been gambling for money, which greatly upsets them. They also found a pack of cigarettes in their son’s car and are concerned that he might be using other substances. What finally prompted the psychiatric visit was Josh’s recent traffic citation for driving 25 miles over the speed limit.
CAUSES OF RISK TAKING
Normal development. In the absence of psychopathology, adolescent risk taking appears to be a normal development stage that is vital to successful transition to adulthood. This assumes that adolescents such as Josh learn to moderate their behavior and avoid long-term negative consequences.
Impulsivity and sensation seeking are recognized as key factors in adolescent risk taking Box 1.1-4 Apparently, these traits result primarily from incomplete neural circuit maturation. Adolescent brain regions involved in impulsivity and risk taking are also involved in reward, and these centers exhibit an exaggerated response to stimuli.5 This amplified response may help explain an adolescent’s propensity for risky behavior.
Despite potential hazards, adolescent risk taking may confer benefits. In taking risks, adolescents:
- explore adult behavior
- learn to accomplish increasingly difficult developmental tasks
- reinforce their self-esteem.
Adolescent risk-takers have been found to be more self-confident, to feel more accepted, and to be better liked than their more-cautious peers.6
Psychiatric comorbidity. Excessive risk taking can be associated with psychiatric illness, including bipolar mania, psychosis, substance abuse, and impulse control disorders. Individuals with borderline personality and other cluster B disorders have marked impulsivity and thus are prone to risky behavior.
Teens with attention-deficit/hyperactivity disorder (ADHD), conduct disorder, and oppositional-defiant disorder (ODD) also tend to exhibit high impulsivity.
Alcohol. 40% of adult alcoholics report having had their first alcoholism-related symptoms between ages 15 and 19.1
Gambling. 10% to 14% of adolescents engage in problem or pathologic gambling, and gambling typically begins at age 12.2
Automobile crashes are the leading cause of death among North American adolescents; both sexes ages 16 to 20 are at least twice as likely to be in a motor vehicle accident as are drivers ages 20 to 50.3
STDs. Each year, 3 million U.S. adolescents contract a sexually transmitted disease (STD). HIV infection is the seventh leading cause of death for Americans ages 13 to 24.4
Sexual activity. Adolescents are more likely than adults to engage in impulsive sexual behavior, to have multiple partners, and to fail to use contraceptives. Younger teens (ages 12 to 14) are more likely to engage in risky sexual practices than older teens (ages 16 to 19).4
CASE CONTINUED: JUST ‘HAVING FUN’
When interviewed alone, Josh admits to “occasional” truancy, which he attributes to being “bored” with school and wanting to spend time with his friends “doing fun stuff, like going to the beach.” He admits to gambling for money and smoking a half-pack of cigarettes daily, as well as drinking beer and smoking marijuana “a few times a week.”
Josh says he engages in these activities “because they’re fun,” and states he is annoyed by his parents’ concern. He blames the speeding ticket on “not paying attention.” He admits to drinking and driving but claims he always feels “in control.”
He also reports he has been sexually active since age 16 and often has had unprotected intercourse. When asked if he is concerned that he might contract a sexually transmitted disease or impregnate his partner, Josh appears ambivalent.
IMPULSIVITY IN ADOLESCENCE
Josh is engaging in numerous impulsive behaviors. Adolescents generally are more impulsive than adults, as demonstrated by their significantly higher impulsivity scores on standardized tests.7 Furthermore, as measured by improved response inhibition (go/no go tasks), the level of adolescent impulsivity is inversely related to age.8
Problem behavior syndrome. High impulsivity is predictive of problem gambling, drug use, and risky driving and sexual practices later in life.1,2,9-11 Adolescents with what some authors describe as a “problem behavior syndrome” engage in behaviors—such as substance use, risky sexual behavior,12 gambling,13 and reckless driving14—that share a common trend toward impulsivity.
Impaired data processing. Decision making has been proposed as a three-part cognitive process:
- accumulating sensory input
- processing this input and formulating a behavioral response appropriate to the situation
- planning and implementing the resultant motor output.2
Impulsivity is believed to result from impaired ability of the brain to process accumulated information or to formulate a response to it—or both. Impulsive individuals thus experience impaired data processing, in which they:
- misjudge the likely risk of a given action or overestimate their ability to accomplish a task
- show impaired response inhibition and thus find it difficult to resist an impulse to participate in a given activity.
Sensation seeking. Adolescents who exhibit risk-taking behavior may wish to experience the thrill of the behavior (sensation or novelty seeking). Alcoholic or drug-dependent individuals and those who engage in pathologic gambling or take chances while driving also demonstrate significantly impaired decision making.15-17 Adolescents who engage in these and other problem behaviors have similarly scored high on sensation-seeking scales.10,18
DECISION-MAKING BIOLOGY
At least four neural circuits process decisions, weighing the risks and benefits of a given situation and formulating a response. These circuits are:
- prefrontal cortices, including orbitofrontal, dorsolateral, and ventromedial
- ventral striatum, including the nucleus accumbens
- thalamus
- monoaminergic brainstem nuclei (ventral tegmental area [VTA] and raphe nuclei).19
Functional imaging studies—including MRI and PET, EEG, and electrophysiology—have confirmed that these four brain regions are integral to response inhibition and show abnormal activity in impulsive individuals.20,21 Indeed, prefrontal cortex damage has been extensively documented to cause marked impulsivity, poor decision making, and an increased propensity for substance abuse and dependency.1
Functional imaging studies also have shown that adolescents appear to use these neural regions inefficiently during decision making. Extensive areas of the involved brain regions are activated in individuals ages 8 to 20, whereas only focal activation occurs in adults.22
Dopamine. The nuclear accumbens (NA) plays an important role in processing afferent excitatory glutamatergic projections and then instigating the given response.23 Dopamine is released in the NA in response to a long list of stimuli, including:
- exposure to substances
- natural rewards such as food or sex
- stimulating situations, such as playing video games, gambling, or thrill seeking.2
Novel experiences and rewards that are delivered erratically cause an elevated dopamine release in the NA. This may explain, in part, the excitement one gains from activities with unpredictable outcomes, such as gambling, bungee jumping, parachuting, white-water rafting, or taking risks while driving.
As rewarding stimuli are re-experienced, dopamine response accelerates in magnitude, and the reward becomes progressively stronger as the experience is repeated. This repeated dopamine release in the NA changes the cellular proteins involved in signaling pathways thought to be associated with the transition from impulsive to compulsive behavior.2 Therefore, addiction may be caused by neurocircuitry changes induced by repeated dopamine release. Similarly, persons who engage in impulsive behavior may have hypersensitive dopamine-related reward circuitry, which may, in part, explain their predisposition to addictive behavior.
Serotonin. Serotonergic projections originate mainly in the midbrain’s raphe nuclei and are transmitted to the ventral tegmental area, NA, prefrontal cortex, amygdala, and hippocampus.1 Abnormal serotonin levels have been implicated in impaired impulse control2 and decreased CNS serotonin in impulsive behavior.24
Functional brain imaging studies have shown reduced serotonin neurotransmission in highly impulsive individuals, compared with normal controls.25 Administering serotonergic agents seems to markedly decrease impulsive behavior.26
Activity within this network is modulated by excitatory glutamatergic transmission and inhibitory GABAergic transmission within the cortices and by dopaminergic and serotonergic transmission within the VTA and raphe nuclei, respectively.2,20 Although all of these neurotransmitters have been implicated in impulsivity, dopamine and serotonin have been studied most extensively (box 2).1,2,23-26
CASE CONTINUED: PSYCHIATRIC WORKUP
Josh clearly is engaged in worrisome behavior with potential long-term consequences. To evaluate him for underlying psychopathology, the psychiatrist used a structured psychiatric exam, Minnesota Multi-phasic Personality Inventory (MMPI), and SNAP-IV Rating Scale for ADHD (see Related resources). Josh endorsed some depressive symptoms—which were also evident on the MMPI—but did not meet DSM-IVTR criteria for major depressive disorder. Neither were his symptoms diagnostic for any other Axis I or Axis II disorder.
Given the risk of harm and likelihood of worsening behavior over time, the psychiatrist schedules Josh for weekly psychotherapy and possible medication.
Psychosocial interventions are discussed with Josh’s parents, including monitoring his activities, restricting access to peers who have been a poor influence, reinforcing good behavior, and enlisting help from teachers and his friends’ parents. The effect of these interventions is to be explored in follow-up visits.
After months or years of conflict with their child, the parents of an adolescent with severe risk-taking behavior are often distraught and frustrated. You can comfort them by explaining:
- the biology of adolescent risk taking
- how you will treat such behavior in their adolescent
- and their role in the treatment plan.
Often the child’s behaviors have weakened their marriage, given adolescents’ tendency to divide and manipulate their parents. To help them set and maintain limits in the face of their child’s hostility:
Educate them to communicate with each other, to maintain a united front, and to set firm limits for their adolescent. For example, recommend that they:
- forbid cell phone use while the adolescent is driving
- limit the number of passengers allowed in the adolescent’s car to reduce distractions
- reduce the amount of money and free time available to the adolescent.
Counsel them that they are unlikely to receive the child’s respect or affection in the short term. Reassure them, however, that the child will thank them for their firm guidance after he or she matures to adulthood.
DEFINING DEGREES OF RISK
Although no criteria differentiate “normal” from “pathologic” risk taking, the definition of taking a risk implies potential adverse consequences. In evaluating the impulsive adolescent, it is important to determine which behaviors:
- can be instructive and promote maturation
- fall outside normal adolescent behavior and/or carry potentially severe outcomes.
Acceptable. Risk taking is acceptable if the potential adverse outcome is relatively benign and the adolescent is likely to learn from the experience. For example, driving 10 miles over the speed limit and receiving a ticket can lead to stricter observance of the speed limit.
Pathologic. Josh clearly exhibits risky behaviors that one would reasonably consider “pathologic,” as they carry potentially severe consequences that exceed any possible developmental gain. For example, drinking and driving can result in a DUI citation and/or a motor vehicle accident with physical injuries or death.
TREATMENT OPTIONS
Psychiatric comorbidity. When you evaluate an adolescent engaged in excessive risk taking, consider Axis I and II disorders characterized by marked impulsivity. If the patient meets diagnostic criteria for a psychopathology such as bipolar disorder or ADHD, treating the underlying condition will likely improve impulsivity.
Recommended approach. Even without an Axis I or Axis II disorder, adolescents who engage in pathologic risky behavior may benefit from psychosocial interventions (Box 3), psychotherapy, and perhaps medication.
Because very little evidence supports using psychotropics to treat pathologically impulsive adolescents, we recommend that you:
- first try psychosocial interventions and psychotherapy
- reserve medications for patients who do not respond adequately to nondrug approaches and engage in impulsive behaviors that pose a high risk for grave consequences.
Psychotherapy can be effective once the adolescent and clinician form a therapeutic alliance. Because Josh—like other such teens—will likely view his psychiatrist as “just another adult lecturing me on what to do,” focus first on establishing rapport by:
- getting to know him
- helping him feel at ease
- showing interest in his thoughts and empathy towards his concerns and complaints
- discussing anything but the reason his parents brought him to your office.
After you establish an alliance, focus therapy on helping the adolescent gain insight into his or her dangerous behaviors and their consequences. To illustrate to Josh the potential consequences of his behaviors, for example, you might introduce him to:
- someone disabled in a motor vehicle accident
- an HIV-positive activist
- a recovering alcoholic
- a long-time smoker with severe chronic obstructive pulmonary disease.
At-risk adolescents also could be encouraged to complete an educational program that teaches alternate activities for sensation seeking (such as skiing instead of high-speed driving).
Medication. Although the monoaminergic systems are known to modulate impulsive behavior, few studies have examined using medications to treat risk-taking adolescents, and no drugs are FDA-approved for this indication.
SSRIs. Selective serotonin reuptake inhibitors such as fluoxetine, sertraline, or escitalopram might be useful for treating excessive adolescent risk taking. A preliminary study with paroxetine—an SSRI not recommended for children and adolescents—suggests this class of antidepressants may help reduce impulsivity.26 In the absence of data specific to risk-taking behavior, we recommend using SSRI dosages similar to those used to treat mood disorders in adolescents.
Clomipramine acts mainly on the serotonin receptor, preventing serotonin reuptake in a manner similar to an SSRI. Because it has the greatest serotonergic effect in its drug class, clomipramine is the only tricyclic proven effective in obsessive-compulsive disorder.27 Although no data have shown that clomipramine affects impulsivity, it theoretically could be effective because of its effect on serotonin.
Divalproex sodium has been shown to effectively treat impulsivity, particularly in patients with autism spectrum disorders, intermittent explosive disorder, schizophrenia, borderline personality disorder, and bipolar disorder.27-30 As an off-label use, one could consider trying this agent in an adolescent with pathologic risk-taking behavior. Use the same dosages and obtain routine labs as indicated for adolescents with other disorders.
Adherence. Like Josh, adolescents who engage in high-risk behaviors often do not recognize their pathology and resist psychiatric intervention. Getting them to take medication or participate in psychotherapy can be quite difficult.
Adolescents are far more likely to adhere to treatment if you develop a rapport with them and they trust you. As psychotherapy and psychosocial interventions progress, patients become more likely to gain insight into their conditions and become more adherent.
Other options to encourage adherence include having the parent:
- administer the medication and ensure that the patient swallows it
- use rewards to reinforce the adolescent’s good behavior and adherence to treatment.
Follow up weekly with patients such as Josh who exhibit high-risk behaviors and require psychotherapy and medication. Follow less-acute patients 2 weeks after the initial evaluation, then monthly if they are responding to medication.
- Strauch B. The primal teen: what the new discoveries about the teenage brain tell us about our kids. New York: Doubleday, 2004.
- SNAP-IV Rating Scale to screen for attention-deficit/hyperactivity disorder. Is your child really ADD/ADHD? www.drbiofeedback.com. Accessed Sept. 8, 2004.
- Focus Adolescent Services. Resources and information for families with adolescent behavior problems, including high-risk behavior. http://www.focusas.com/BehavioralDisorders.html. Accessed Aug. 26, 2004.
Drug brand names
- Clomipramine • Anafranil
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
Dr. Husted reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr Shapira receives grant or research support from Abbott Laboratories, Janssen Pharmaceutica, Ortho-McNeil Pharmaceutical, Bristol-Myers Squibb Co., Eli Lilly and Co., and Pfizer Inc. He is a speaker for AstraZeneca Pharmaceuticals, Forest Laboratories, and Ortho-McNeil Pharmaceutical, Inc.
1. Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry 2003;160:1041-52.
2. Chambers RA, Potenza MN. Neurodevelopment, impulsivity, and adolescent gambling. J Gambl Stud 2003;19:53-84.
3. Turner C, McClure R. Age and gender differences in risk taking behavior as an explanation for high incidence of motor vehicle crashes as a driver in young males. Inj Control Saf Promot 2003;10:123-30.
4. Bachanas PJ, Morris MK, Lewis-Gess JK, et al. Psychological adjustment, substance use, HIV knowledge, and risky sexual behavior in at-risk minority females: developmental differences during adolescence. J Pediatr Psych 2002;27:373-84.
5. Goldstein RZ, Volkow ND. Drug addiction and its underlying neuro- biological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 2002;159:1642-52.
6. Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev 2000;24:417-63.
7. Clayton R. Transitions in drug use: risk and protective factors. In: Glantz M, Pickens R (eds). Vulnerability to drug abuse. Washington, DC: American Psychological Association, 1992;15-52.
8. Tamm L, Menon V, Reiss AL. Maturation of brain function associated with response inhibition. J Am Acad Child Adolesc Psychiatry 2002;41:1231-8.
9. Jonah BA. Sensation seeking and risky driving: a review and synthesis of the literature. Accid Anal Prev 1997;29:651-6.
10. Vitaro F, Arseneault L, Tremblay RE. Dispositional predictors of problem gambling in male adolescents. Am J Psychiatry 1997;154:1769-70.
11. Malow RM, Devieux JG, Jennings T, et al. Substance-abusing adolescents at varying levels of HIV risk: psychosocial characteristics, drug use, and sexual behavior. J Subst Abuse 2001;13:103-17.
12. Donovan JE, Jessor R, Costa FM. Syndrome of problem behavior in adolescence: a replication. J Consult Clin Psychol 1988;56:762-5.
13. Vitaro F, Brendgen M, Ladouceur R, Tremblay RE. Gambling, delinquency, and drug use during adolescence: mutual influences and common risk factors. J Gambl Stud 2001;17:171-90.
14. Shope JT, Bingham CR. Drinking-driving as a component of problem driving and problem behavior in young adults. J Stud Alcohol 2002;63(1):24-33.
15. Potenza MN. The neurobiology of pathological gambling. Semin Clin Neuropsychiatry 2001;6:217-26.
16. Petry NM. Substance abuse, pathological gambling, and impulsiveness. Drug Alcohol Depend 2001;63:29-38.
17. Bechara A. Neurobiology of decision-making: risk and reward. Semin Clin Neuropsychiatry 2001;6:205-16.
18. Zuckerman M. Sensation seeking: the balance between risk and reward. In: Lipsitt LP, Mitnick LL (eds). Self-regulatory behavior and risk taking. Norwood, NJ: Ablex Publishing; 1992;143-52.
19. Masterman DL, Cummings JL. Frontal-subcortical circuits: the anatomical basis of executive, social and motivational behaviors. J Psychopharmacol 1997;11:107-14.
20. Horn NR, Dolan M, Elliot R, et al. Response inhibition and impulsivity: an fMRI study. Neuropsychologia 2003;41:1959-66.
21. Fallgatter AJ, Herrmann MJ. Electrophysiological assessment of impulsive behavior in healthy subjects. Neuropsychologia 2001;39:328-33.
22. Booth JR, Burman DD, Meyer JR, et al. Neural development of selective attention and response inhibition. Neuroimage 2003;20:737-51.
23. O’Donnell P, Greene J, Pabello N, et al. Modulation of cell firing in the nucleus accumbens. Ann NY Acad Sci 1999;877:157-75.
24. Nordin C, Eklundh T. Altered CSF 5-HIAA disposition in pathologic male gamblers. CNS Spectrums 1999;4:25-33.
25. Leyton M, Okazawa H, Diksic M, et al. Brain regional alpha-[11C] methyl-L-tryptophan trapping in impulsive subjects with borderline personality disorder. Am J Psychiatry 2001;158:775-82.
26. Cherek DR, Lane SD, Pietras CJ, Steinberg JL. Effects of chronic paroxetine administration on measures of aggressive and impulsive responses of adult males with a history of conduct disorder. Psychopharmacology (Berl) 2002;159:266-74.
27. Geller DA, Biederman J, Stewart SE, et al. Which SSRI? A meta-analysis of pharmacotherapy trials in pediatric obsessive-compulsive disorder. Am J Psychiatry 2003;160:1919-28.
28. Hollander E, Allen A, Lopez RP, et al. A preliminary double-blind, placebo-controlled trial of divalproex sodium in borderline personality disorder. J Clin Psychiatry 2001;62(3):199-203.
29. Swann AC. Treatment of aggression in patients with bipolar disorder. J Clin Psychiatry 1999;60(suppl 15):25-8.
30. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
Boys will be boys” and other platitudes may condone adolescent reckless driving, substance use, or sexual promiscuity—but to write off dangerous behavior as normal would be a mistake. Because adolescent impulsivity and sensation-seeking may have physiologic as well as emotional causes,1,2 excessive risk taking may be treatable.
This article discusses the neurobiology of adolescent risk taking, suggests how to determine when such behavior may be pathologic, and offers a treatment approach for at-risk teens and their parents.
CASE: ‘WHAT’S WRONG WITH OUR SON?’
Josh, age 17, is brought to the adolescent psychiatry clinic by his distraught parents, who report their son has undergone a “personality change” over the past 2 years. They recall that he was respectful, studious, and soft-spoken until age 15. Since then, he has been skipping school, staying out late at night with his friends, and “obsessed” with TV poker games.
His parents recently discovered he has been gambling for money, which greatly upsets them. They also found a pack of cigarettes in their son’s car and are concerned that he might be using other substances. What finally prompted the psychiatric visit was Josh’s recent traffic citation for driving 25 miles over the speed limit.
CAUSES OF RISK TAKING
Normal development. In the absence of psychopathology, adolescent risk taking appears to be a normal development stage that is vital to successful transition to adulthood. This assumes that adolescents such as Josh learn to moderate their behavior and avoid long-term negative consequences.
Impulsivity and sensation seeking are recognized as key factors in adolescent risk taking Box 1.1-4 Apparently, these traits result primarily from incomplete neural circuit maturation. Adolescent brain regions involved in impulsivity and risk taking are also involved in reward, and these centers exhibit an exaggerated response to stimuli.5 This amplified response may help explain an adolescent’s propensity for risky behavior.
Despite potential hazards, adolescent risk taking may confer benefits. In taking risks, adolescents:
- explore adult behavior
- learn to accomplish increasingly difficult developmental tasks
- reinforce their self-esteem.
Adolescent risk-takers have been found to be more self-confident, to feel more accepted, and to be better liked than their more-cautious peers.6
Psychiatric comorbidity. Excessive risk taking can be associated with psychiatric illness, including bipolar mania, psychosis, substance abuse, and impulse control disorders. Individuals with borderline personality and other cluster B disorders have marked impulsivity and thus are prone to risky behavior.
Teens with attention-deficit/hyperactivity disorder (ADHD), conduct disorder, and oppositional-defiant disorder (ODD) also tend to exhibit high impulsivity.
Alcohol. 40% of adult alcoholics report having had their first alcoholism-related symptoms between ages 15 and 19.1
Gambling. 10% to 14% of adolescents engage in problem or pathologic gambling, and gambling typically begins at age 12.2
Automobile crashes are the leading cause of death among North American adolescents; both sexes ages 16 to 20 are at least twice as likely to be in a motor vehicle accident as are drivers ages 20 to 50.3
STDs. Each year, 3 million U.S. adolescents contract a sexually transmitted disease (STD). HIV infection is the seventh leading cause of death for Americans ages 13 to 24.4
Sexual activity. Adolescents are more likely than adults to engage in impulsive sexual behavior, to have multiple partners, and to fail to use contraceptives. Younger teens (ages 12 to 14) are more likely to engage in risky sexual practices than older teens (ages 16 to 19).4
CASE CONTINUED: JUST ‘HAVING FUN’
When interviewed alone, Josh admits to “occasional” truancy, which he attributes to being “bored” with school and wanting to spend time with his friends “doing fun stuff, like going to the beach.” He admits to gambling for money and smoking a half-pack of cigarettes daily, as well as drinking beer and smoking marijuana “a few times a week.”
Josh says he engages in these activities “because they’re fun,” and states he is annoyed by his parents’ concern. He blames the speeding ticket on “not paying attention.” He admits to drinking and driving but claims he always feels “in control.”
He also reports he has been sexually active since age 16 and often has had unprotected intercourse. When asked if he is concerned that he might contract a sexually transmitted disease or impregnate his partner, Josh appears ambivalent.
IMPULSIVITY IN ADOLESCENCE
Josh is engaging in numerous impulsive behaviors. Adolescents generally are more impulsive than adults, as demonstrated by their significantly higher impulsivity scores on standardized tests.7 Furthermore, as measured by improved response inhibition (go/no go tasks), the level of adolescent impulsivity is inversely related to age.8
Problem behavior syndrome. High impulsivity is predictive of problem gambling, drug use, and risky driving and sexual practices later in life.1,2,9-11 Adolescents with what some authors describe as a “problem behavior syndrome” engage in behaviors—such as substance use, risky sexual behavior,12 gambling,13 and reckless driving14—that share a common trend toward impulsivity.
Impaired data processing. Decision making has been proposed as a three-part cognitive process:
- accumulating sensory input
- processing this input and formulating a behavioral response appropriate to the situation
- planning and implementing the resultant motor output.2
Impulsivity is believed to result from impaired ability of the brain to process accumulated information or to formulate a response to it—or both. Impulsive individuals thus experience impaired data processing, in which they:
- misjudge the likely risk of a given action or overestimate their ability to accomplish a task
- show impaired response inhibition and thus find it difficult to resist an impulse to participate in a given activity.
Sensation seeking. Adolescents who exhibit risk-taking behavior may wish to experience the thrill of the behavior (sensation or novelty seeking). Alcoholic or drug-dependent individuals and those who engage in pathologic gambling or take chances while driving also demonstrate significantly impaired decision making.15-17 Adolescents who engage in these and other problem behaviors have similarly scored high on sensation-seeking scales.10,18
DECISION-MAKING BIOLOGY
At least four neural circuits process decisions, weighing the risks and benefits of a given situation and formulating a response. These circuits are:
- prefrontal cortices, including orbitofrontal, dorsolateral, and ventromedial
- ventral striatum, including the nucleus accumbens
- thalamus
- monoaminergic brainstem nuclei (ventral tegmental area [VTA] and raphe nuclei).19
Functional imaging studies—including MRI and PET, EEG, and electrophysiology—have confirmed that these four brain regions are integral to response inhibition and show abnormal activity in impulsive individuals.20,21 Indeed, prefrontal cortex damage has been extensively documented to cause marked impulsivity, poor decision making, and an increased propensity for substance abuse and dependency.1
Functional imaging studies also have shown that adolescents appear to use these neural regions inefficiently during decision making. Extensive areas of the involved brain regions are activated in individuals ages 8 to 20, whereas only focal activation occurs in adults.22
Dopamine. The nuclear accumbens (NA) plays an important role in processing afferent excitatory glutamatergic projections and then instigating the given response.23 Dopamine is released in the NA in response to a long list of stimuli, including:
- exposure to substances
- natural rewards such as food or sex
- stimulating situations, such as playing video games, gambling, or thrill seeking.2
Novel experiences and rewards that are delivered erratically cause an elevated dopamine release in the NA. This may explain, in part, the excitement one gains from activities with unpredictable outcomes, such as gambling, bungee jumping, parachuting, white-water rafting, or taking risks while driving.
As rewarding stimuli are re-experienced, dopamine response accelerates in magnitude, and the reward becomes progressively stronger as the experience is repeated. This repeated dopamine release in the NA changes the cellular proteins involved in signaling pathways thought to be associated with the transition from impulsive to compulsive behavior.2 Therefore, addiction may be caused by neurocircuitry changes induced by repeated dopamine release. Similarly, persons who engage in impulsive behavior may have hypersensitive dopamine-related reward circuitry, which may, in part, explain their predisposition to addictive behavior.
Serotonin. Serotonergic projections originate mainly in the midbrain’s raphe nuclei and are transmitted to the ventral tegmental area, NA, prefrontal cortex, amygdala, and hippocampus.1 Abnormal serotonin levels have been implicated in impaired impulse control2 and decreased CNS serotonin in impulsive behavior.24
Functional brain imaging studies have shown reduced serotonin neurotransmission in highly impulsive individuals, compared with normal controls.25 Administering serotonergic agents seems to markedly decrease impulsive behavior.26
Activity within this network is modulated by excitatory glutamatergic transmission and inhibitory GABAergic transmission within the cortices and by dopaminergic and serotonergic transmission within the VTA and raphe nuclei, respectively.2,20 Although all of these neurotransmitters have been implicated in impulsivity, dopamine and serotonin have been studied most extensively (box 2).1,2,23-26
CASE CONTINUED: PSYCHIATRIC WORKUP
Josh clearly is engaged in worrisome behavior with potential long-term consequences. To evaluate him for underlying psychopathology, the psychiatrist used a structured psychiatric exam, Minnesota Multi-phasic Personality Inventory (MMPI), and SNAP-IV Rating Scale for ADHD (see Related resources). Josh endorsed some depressive symptoms—which were also evident on the MMPI—but did not meet DSM-IVTR criteria for major depressive disorder. Neither were his symptoms diagnostic for any other Axis I or Axis II disorder.
Given the risk of harm and likelihood of worsening behavior over time, the psychiatrist schedules Josh for weekly psychotherapy and possible medication.
Psychosocial interventions are discussed with Josh’s parents, including monitoring his activities, restricting access to peers who have been a poor influence, reinforcing good behavior, and enlisting help from teachers and his friends’ parents. The effect of these interventions is to be explored in follow-up visits.
After months or years of conflict with their child, the parents of an adolescent with severe risk-taking behavior are often distraught and frustrated. You can comfort them by explaining:
- the biology of adolescent risk taking
- how you will treat such behavior in their adolescent
- and their role in the treatment plan.
Often the child’s behaviors have weakened their marriage, given adolescents’ tendency to divide and manipulate their parents. To help them set and maintain limits in the face of their child’s hostility:
Educate them to communicate with each other, to maintain a united front, and to set firm limits for their adolescent. For example, recommend that they:
- forbid cell phone use while the adolescent is driving
- limit the number of passengers allowed in the adolescent’s car to reduce distractions
- reduce the amount of money and free time available to the adolescent.
Counsel them that they are unlikely to receive the child’s respect or affection in the short term. Reassure them, however, that the child will thank them for their firm guidance after he or she matures to adulthood.
DEFINING DEGREES OF RISK
Although no criteria differentiate “normal” from “pathologic” risk taking, the definition of taking a risk implies potential adverse consequences. In evaluating the impulsive adolescent, it is important to determine which behaviors:
- can be instructive and promote maturation
- fall outside normal adolescent behavior and/or carry potentially severe outcomes.
Acceptable. Risk taking is acceptable if the potential adverse outcome is relatively benign and the adolescent is likely to learn from the experience. For example, driving 10 miles over the speed limit and receiving a ticket can lead to stricter observance of the speed limit.
Pathologic. Josh clearly exhibits risky behaviors that one would reasonably consider “pathologic,” as they carry potentially severe consequences that exceed any possible developmental gain. For example, drinking and driving can result in a DUI citation and/or a motor vehicle accident with physical injuries or death.
TREATMENT OPTIONS
Psychiatric comorbidity. When you evaluate an adolescent engaged in excessive risk taking, consider Axis I and II disorders characterized by marked impulsivity. If the patient meets diagnostic criteria for a psychopathology such as bipolar disorder or ADHD, treating the underlying condition will likely improve impulsivity.
Recommended approach. Even without an Axis I or Axis II disorder, adolescents who engage in pathologic risky behavior may benefit from psychosocial interventions (Box 3), psychotherapy, and perhaps medication.
Because very little evidence supports using psychotropics to treat pathologically impulsive adolescents, we recommend that you:
- first try psychosocial interventions and psychotherapy
- reserve medications for patients who do not respond adequately to nondrug approaches and engage in impulsive behaviors that pose a high risk for grave consequences.
Psychotherapy can be effective once the adolescent and clinician form a therapeutic alliance. Because Josh—like other such teens—will likely view his psychiatrist as “just another adult lecturing me on what to do,” focus first on establishing rapport by:
- getting to know him
- helping him feel at ease
- showing interest in his thoughts and empathy towards his concerns and complaints
- discussing anything but the reason his parents brought him to your office.
After you establish an alliance, focus therapy on helping the adolescent gain insight into his or her dangerous behaviors and their consequences. To illustrate to Josh the potential consequences of his behaviors, for example, you might introduce him to:
- someone disabled in a motor vehicle accident
- an HIV-positive activist
- a recovering alcoholic
- a long-time smoker with severe chronic obstructive pulmonary disease.
At-risk adolescents also could be encouraged to complete an educational program that teaches alternate activities for sensation seeking (such as skiing instead of high-speed driving).
Medication. Although the monoaminergic systems are known to modulate impulsive behavior, few studies have examined using medications to treat risk-taking adolescents, and no drugs are FDA-approved for this indication.
SSRIs. Selective serotonin reuptake inhibitors such as fluoxetine, sertraline, or escitalopram might be useful for treating excessive adolescent risk taking. A preliminary study with paroxetine—an SSRI not recommended for children and adolescents—suggests this class of antidepressants may help reduce impulsivity.26 In the absence of data specific to risk-taking behavior, we recommend using SSRI dosages similar to those used to treat mood disorders in adolescents.
Clomipramine acts mainly on the serotonin receptor, preventing serotonin reuptake in a manner similar to an SSRI. Because it has the greatest serotonergic effect in its drug class, clomipramine is the only tricyclic proven effective in obsessive-compulsive disorder.27 Although no data have shown that clomipramine affects impulsivity, it theoretically could be effective because of its effect on serotonin.
Divalproex sodium has been shown to effectively treat impulsivity, particularly in patients with autism spectrum disorders, intermittent explosive disorder, schizophrenia, borderline personality disorder, and bipolar disorder.27-30 As an off-label use, one could consider trying this agent in an adolescent with pathologic risk-taking behavior. Use the same dosages and obtain routine labs as indicated for adolescents with other disorders.
Adherence. Like Josh, adolescents who engage in high-risk behaviors often do not recognize their pathology and resist psychiatric intervention. Getting them to take medication or participate in psychotherapy can be quite difficult.
Adolescents are far more likely to adhere to treatment if you develop a rapport with them and they trust you. As psychotherapy and psychosocial interventions progress, patients become more likely to gain insight into their conditions and become more adherent.
Other options to encourage adherence include having the parent:
- administer the medication and ensure that the patient swallows it
- use rewards to reinforce the adolescent’s good behavior and adherence to treatment.
Follow up weekly with patients such as Josh who exhibit high-risk behaviors and require psychotherapy and medication. Follow less-acute patients 2 weeks after the initial evaluation, then monthly if they are responding to medication.
- Strauch B. The primal teen: what the new discoveries about the teenage brain tell us about our kids. New York: Doubleday, 2004.
- SNAP-IV Rating Scale to screen for attention-deficit/hyperactivity disorder. Is your child really ADD/ADHD? www.drbiofeedback.com. Accessed Sept. 8, 2004.
- Focus Adolescent Services. Resources and information for families with adolescent behavior problems, including high-risk behavior. http://www.focusas.com/BehavioralDisorders.html. Accessed Aug. 26, 2004.
Drug brand names
- Clomipramine • Anafranil
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
Dr. Husted reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr Shapira receives grant or research support from Abbott Laboratories, Janssen Pharmaceutica, Ortho-McNeil Pharmaceutical, Bristol-Myers Squibb Co., Eli Lilly and Co., and Pfizer Inc. He is a speaker for AstraZeneca Pharmaceuticals, Forest Laboratories, and Ortho-McNeil Pharmaceutical, Inc.
Boys will be boys” and other platitudes may condone adolescent reckless driving, substance use, or sexual promiscuity—but to write off dangerous behavior as normal would be a mistake. Because adolescent impulsivity and sensation-seeking may have physiologic as well as emotional causes,1,2 excessive risk taking may be treatable.
This article discusses the neurobiology of adolescent risk taking, suggests how to determine when such behavior may be pathologic, and offers a treatment approach for at-risk teens and their parents.
CASE: ‘WHAT’S WRONG WITH OUR SON?’
Josh, age 17, is brought to the adolescent psychiatry clinic by his distraught parents, who report their son has undergone a “personality change” over the past 2 years. They recall that he was respectful, studious, and soft-spoken until age 15. Since then, he has been skipping school, staying out late at night with his friends, and “obsessed” with TV poker games.
His parents recently discovered he has been gambling for money, which greatly upsets them. They also found a pack of cigarettes in their son’s car and are concerned that he might be using other substances. What finally prompted the psychiatric visit was Josh’s recent traffic citation for driving 25 miles over the speed limit.
CAUSES OF RISK TAKING
Normal development. In the absence of psychopathology, adolescent risk taking appears to be a normal development stage that is vital to successful transition to adulthood. This assumes that adolescents such as Josh learn to moderate their behavior and avoid long-term negative consequences.
Impulsivity and sensation seeking are recognized as key factors in adolescent risk taking Box 1.1-4 Apparently, these traits result primarily from incomplete neural circuit maturation. Adolescent brain regions involved in impulsivity and risk taking are also involved in reward, and these centers exhibit an exaggerated response to stimuli.5 This amplified response may help explain an adolescent’s propensity for risky behavior.
Despite potential hazards, adolescent risk taking may confer benefits. In taking risks, adolescents:
- explore adult behavior
- learn to accomplish increasingly difficult developmental tasks
- reinforce their self-esteem.
Adolescent risk-takers have been found to be more self-confident, to feel more accepted, and to be better liked than their more-cautious peers.6
Psychiatric comorbidity. Excessive risk taking can be associated with psychiatric illness, including bipolar mania, psychosis, substance abuse, and impulse control disorders. Individuals with borderline personality and other cluster B disorders have marked impulsivity and thus are prone to risky behavior.
Teens with attention-deficit/hyperactivity disorder (ADHD), conduct disorder, and oppositional-defiant disorder (ODD) also tend to exhibit high impulsivity.
Alcohol. 40% of adult alcoholics report having had their first alcoholism-related symptoms between ages 15 and 19.1
Gambling. 10% to 14% of adolescents engage in problem or pathologic gambling, and gambling typically begins at age 12.2
Automobile crashes are the leading cause of death among North American adolescents; both sexes ages 16 to 20 are at least twice as likely to be in a motor vehicle accident as are drivers ages 20 to 50.3
STDs. Each year, 3 million U.S. adolescents contract a sexually transmitted disease (STD). HIV infection is the seventh leading cause of death for Americans ages 13 to 24.4
Sexual activity. Adolescents are more likely than adults to engage in impulsive sexual behavior, to have multiple partners, and to fail to use contraceptives. Younger teens (ages 12 to 14) are more likely to engage in risky sexual practices than older teens (ages 16 to 19).4
CASE CONTINUED: JUST ‘HAVING FUN’
When interviewed alone, Josh admits to “occasional” truancy, which he attributes to being “bored” with school and wanting to spend time with his friends “doing fun stuff, like going to the beach.” He admits to gambling for money and smoking a half-pack of cigarettes daily, as well as drinking beer and smoking marijuana “a few times a week.”
Josh says he engages in these activities “because they’re fun,” and states he is annoyed by his parents’ concern. He blames the speeding ticket on “not paying attention.” He admits to drinking and driving but claims he always feels “in control.”
He also reports he has been sexually active since age 16 and often has had unprotected intercourse. When asked if he is concerned that he might contract a sexually transmitted disease or impregnate his partner, Josh appears ambivalent.
IMPULSIVITY IN ADOLESCENCE
Josh is engaging in numerous impulsive behaviors. Adolescents generally are more impulsive than adults, as demonstrated by their significantly higher impulsivity scores on standardized tests.7 Furthermore, as measured by improved response inhibition (go/no go tasks), the level of adolescent impulsivity is inversely related to age.8
Problem behavior syndrome. High impulsivity is predictive of problem gambling, drug use, and risky driving and sexual practices later in life.1,2,9-11 Adolescents with what some authors describe as a “problem behavior syndrome” engage in behaviors—such as substance use, risky sexual behavior,12 gambling,13 and reckless driving14—that share a common trend toward impulsivity.
Impaired data processing. Decision making has been proposed as a three-part cognitive process:
- accumulating sensory input
- processing this input and formulating a behavioral response appropriate to the situation
- planning and implementing the resultant motor output.2
Impulsivity is believed to result from impaired ability of the brain to process accumulated information or to formulate a response to it—or both. Impulsive individuals thus experience impaired data processing, in which they:
- misjudge the likely risk of a given action or overestimate their ability to accomplish a task
- show impaired response inhibition and thus find it difficult to resist an impulse to participate in a given activity.
Sensation seeking. Adolescents who exhibit risk-taking behavior may wish to experience the thrill of the behavior (sensation or novelty seeking). Alcoholic or drug-dependent individuals and those who engage in pathologic gambling or take chances while driving also demonstrate significantly impaired decision making.15-17 Adolescents who engage in these and other problem behaviors have similarly scored high on sensation-seeking scales.10,18
DECISION-MAKING BIOLOGY
At least four neural circuits process decisions, weighing the risks and benefits of a given situation and formulating a response. These circuits are:
- prefrontal cortices, including orbitofrontal, dorsolateral, and ventromedial
- ventral striatum, including the nucleus accumbens
- thalamus
- monoaminergic brainstem nuclei (ventral tegmental area [VTA] and raphe nuclei).19
Functional imaging studies—including MRI and PET, EEG, and electrophysiology—have confirmed that these four brain regions are integral to response inhibition and show abnormal activity in impulsive individuals.20,21 Indeed, prefrontal cortex damage has been extensively documented to cause marked impulsivity, poor decision making, and an increased propensity for substance abuse and dependency.1
Functional imaging studies also have shown that adolescents appear to use these neural regions inefficiently during decision making. Extensive areas of the involved brain regions are activated in individuals ages 8 to 20, whereas only focal activation occurs in adults.22
Dopamine. The nuclear accumbens (NA) plays an important role in processing afferent excitatory glutamatergic projections and then instigating the given response.23 Dopamine is released in the NA in response to a long list of stimuli, including:
- exposure to substances
- natural rewards such as food or sex
- stimulating situations, such as playing video games, gambling, or thrill seeking.2
Novel experiences and rewards that are delivered erratically cause an elevated dopamine release in the NA. This may explain, in part, the excitement one gains from activities with unpredictable outcomes, such as gambling, bungee jumping, parachuting, white-water rafting, or taking risks while driving.
As rewarding stimuli are re-experienced, dopamine response accelerates in magnitude, and the reward becomes progressively stronger as the experience is repeated. This repeated dopamine release in the NA changes the cellular proteins involved in signaling pathways thought to be associated with the transition from impulsive to compulsive behavior.2 Therefore, addiction may be caused by neurocircuitry changes induced by repeated dopamine release. Similarly, persons who engage in impulsive behavior may have hypersensitive dopamine-related reward circuitry, which may, in part, explain their predisposition to addictive behavior.
Serotonin. Serotonergic projections originate mainly in the midbrain’s raphe nuclei and are transmitted to the ventral tegmental area, NA, prefrontal cortex, amygdala, and hippocampus.1 Abnormal serotonin levels have been implicated in impaired impulse control2 and decreased CNS serotonin in impulsive behavior.24
Functional brain imaging studies have shown reduced serotonin neurotransmission in highly impulsive individuals, compared with normal controls.25 Administering serotonergic agents seems to markedly decrease impulsive behavior.26
Activity within this network is modulated by excitatory glutamatergic transmission and inhibitory GABAergic transmission within the cortices and by dopaminergic and serotonergic transmission within the VTA and raphe nuclei, respectively.2,20 Although all of these neurotransmitters have been implicated in impulsivity, dopamine and serotonin have been studied most extensively (box 2).1,2,23-26
CASE CONTINUED: PSYCHIATRIC WORKUP
Josh clearly is engaged in worrisome behavior with potential long-term consequences. To evaluate him for underlying psychopathology, the psychiatrist used a structured psychiatric exam, Minnesota Multi-phasic Personality Inventory (MMPI), and SNAP-IV Rating Scale for ADHD (see Related resources). Josh endorsed some depressive symptoms—which were also evident on the MMPI—but did not meet DSM-IVTR criteria for major depressive disorder. Neither were his symptoms diagnostic for any other Axis I or Axis II disorder.
Given the risk of harm and likelihood of worsening behavior over time, the psychiatrist schedules Josh for weekly psychotherapy and possible medication.
Psychosocial interventions are discussed with Josh’s parents, including monitoring his activities, restricting access to peers who have been a poor influence, reinforcing good behavior, and enlisting help from teachers and his friends’ parents. The effect of these interventions is to be explored in follow-up visits.
After months or years of conflict with their child, the parents of an adolescent with severe risk-taking behavior are often distraught and frustrated. You can comfort them by explaining:
- the biology of adolescent risk taking
- how you will treat such behavior in their adolescent
- and their role in the treatment plan.
Often the child’s behaviors have weakened their marriage, given adolescents’ tendency to divide and manipulate their parents. To help them set and maintain limits in the face of their child’s hostility:
Educate them to communicate with each other, to maintain a united front, and to set firm limits for their adolescent. For example, recommend that they:
- forbid cell phone use while the adolescent is driving
- limit the number of passengers allowed in the adolescent’s car to reduce distractions
- reduce the amount of money and free time available to the adolescent.
Counsel them that they are unlikely to receive the child’s respect or affection in the short term. Reassure them, however, that the child will thank them for their firm guidance after he or she matures to adulthood.
DEFINING DEGREES OF RISK
Although no criteria differentiate “normal” from “pathologic” risk taking, the definition of taking a risk implies potential adverse consequences. In evaluating the impulsive adolescent, it is important to determine which behaviors:
- can be instructive and promote maturation
- fall outside normal adolescent behavior and/or carry potentially severe outcomes.
Acceptable. Risk taking is acceptable if the potential adverse outcome is relatively benign and the adolescent is likely to learn from the experience. For example, driving 10 miles over the speed limit and receiving a ticket can lead to stricter observance of the speed limit.
Pathologic. Josh clearly exhibits risky behaviors that one would reasonably consider “pathologic,” as they carry potentially severe consequences that exceed any possible developmental gain. For example, drinking and driving can result in a DUI citation and/or a motor vehicle accident with physical injuries or death.
TREATMENT OPTIONS
Psychiatric comorbidity. When you evaluate an adolescent engaged in excessive risk taking, consider Axis I and II disorders characterized by marked impulsivity. If the patient meets diagnostic criteria for a psychopathology such as bipolar disorder or ADHD, treating the underlying condition will likely improve impulsivity.
Recommended approach. Even without an Axis I or Axis II disorder, adolescents who engage in pathologic risky behavior may benefit from psychosocial interventions (Box 3), psychotherapy, and perhaps medication.
Because very little evidence supports using psychotropics to treat pathologically impulsive adolescents, we recommend that you:
- first try psychosocial interventions and psychotherapy
- reserve medications for patients who do not respond adequately to nondrug approaches and engage in impulsive behaviors that pose a high risk for grave consequences.
Psychotherapy can be effective once the adolescent and clinician form a therapeutic alliance. Because Josh—like other such teens—will likely view his psychiatrist as “just another adult lecturing me on what to do,” focus first on establishing rapport by:
- getting to know him
- helping him feel at ease
- showing interest in his thoughts and empathy towards his concerns and complaints
- discussing anything but the reason his parents brought him to your office.
After you establish an alliance, focus therapy on helping the adolescent gain insight into his or her dangerous behaviors and their consequences. To illustrate to Josh the potential consequences of his behaviors, for example, you might introduce him to:
- someone disabled in a motor vehicle accident
- an HIV-positive activist
- a recovering alcoholic
- a long-time smoker with severe chronic obstructive pulmonary disease.
At-risk adolescents also could be encouraged to complete an educational program that teaches alternate activities for sensation seeking (such as skiing instead of high-speed driving).
Medication. Although the monoaminergic systems are known to modulate impulsive behavior, few studies have examined using medications to treat risk-taking adolescents, and no drugs are FDA-approved for this indication.
SSRIs. Selective serotonin reuptake inhibitors such as fluoxetine, sertraline, or escitalopram might be useful for treating excessive adolescent risk taking. A preliminary study with paroxetine—an SSRI not recommended for children and adolescents—suggests this class of antidepressants may help reduce impulsivity.26 In the absence of data specific to risk-taking behavior, we recommend using SSRI dosages similar to those used to treat mood disorders in adolescents.
Clomipramine acts mainly on the serotonin receptor, preventing serotonin reuptake in a manner similar to an SSRI. Because it has the greatest serotonergic effect in its drug class, clomipramine is the only tricyclic proven effective in obsessive-compulsive disorder.27 Although no data have shown that clomipramine affects impulsivity, it theoretically could be effective because of its effect on serotonin.
Divalproex sodium has been shown to effectively treat impulsivity, particularly in patients with autism spectrum disorders, intermittent explosive disorder, schizophrenia, borderline personality disorder, and bipolar disorder.27-30 As an off-label use, one could consider trying this agent in an adolescent with pathologic risk-taking behavior. Use the same dosages and obtain routine labs as indicated for adolescents with other disorders.
Adherence. Like Josh, adolescents who engage in high-risk behaviors often do not recognize their pathology and resist psychiatric intervention. Getting them to take medication or participate in psychotherapy can be quite difficult.
Adolescents are far more likely to adhere to treatment if you develop a rapport with them and they trust you. As psychotherapy and psychosocial interventions progress, patients become more likely to gain insight into their conditions and become more adherent.
Other options to encourage adherence include having the parent:
- administer the medication and ensure that the patient swallows it
- use rewards to reinforce the adolescent’s good behavior and adherence to treatment.
Follow up weekly with patients such as Josh who exhibit high-risk behaviors and require psychotherapy and medication. Follow less-acute patients 2 weeks after the initial evaluation, then monthly if they are responding to medication.
- Strauch B. The primal teen: what the new discoveries about the teenage brain tell us about our kids. New York: Doubleday, 2004.
- SNAP-IV Rating Scale to screen for attention-deficit/hyperactivity disorder. Is your child really ADD/ADHD? www.drbiofeedback.com. Accessed Sept. 8, 2004.
- Focus Adolescent Services. Resources and information for families with adolescent behavior problems, including high-risk behavior. http://www.focusas.com/BehavioralDisorders.html. Accessed Aug. 26, 2004.
Drug brand names
- Clomipramine • Anafranil
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Sertraline • Zoloft
Disclosure
Dr. Husted reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr Shapira receives grant or research support from Abbott Laboratories, Janssen Pharmaceutica, Ortho-McNeil Pharmaceutical, Bristol-Myers Squibb Co., Eli Lilly and Co., and Pfizer Inc. He is a speaker for AstraZeneca Pharmaceuticals, Forest Laboratories, and Ortho-McNeil Pharmaceutical, Inc.
1. Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry 2003;160:1041-52.
2. Chambers RA, Potenza MN. Neurodevelopment, impulsivity, and adolescent gambling. J Gambl Stud 2003;19:53-84.
3. Turner C, McClure R. Age and gender differences in risk taking behavior as an explanation for high incidence of motor vehicle crashes as a driver in young males. Inj Control Saf Promot 2003;10:123-30.
4. Bachanas PJ, Morris MK, Lewis-Gess JK, et al. Psychological adjustment, substance use, HIV knowledge, and risky sexual behavior in at-risk minority females: developmental differences during adolescence. J Pediatr Psych 2002;27:373-84.
5. Goldstein RZ, Volkow ND. Drug addiction and its underlying neuro- biological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 2002;159:1642-52.
6. Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev 2000;24:417-63.
7. Clayton R. Transitions in drug use: risk and protective factors. In: Glantz M, Pickens R (eds). Vulnerability to drug abuse. Washington, DC: American Psychological Association, 1992;15-52.
8. Tamm L, Menon V, Reiss AL. Maturation of brain function associated with response inhibition. J Am Acad Child Adolesc Psychiatry 2002;41:1231-8.
9. Jonah BA. Sensation seeking and risky driving: a review and synthesis of the literature. Accid Anal Prev 1997;29:651-6.
10. Vitaro F, Arseneault L, Tremblay RE. Dispositional predictors of problem gambling in male adolescents. Am J Psychiatry 1997;154:1769-70.
11. Malow RM, Devieux JG, Jennings T, et al. Substance-abusing adolescents at varying levels of HIV risk: psychosocial characteristics, drug use, and sexual behavior. J Subst Abuse 2001;13:103-17.
12. Donovan JE, Jessor R, Costa FM. Syndrome of problem behavior in adolescence: a replication. J Consult Clin Psychol 1988;56:762-5.
13. Vitaro F, Brendgen M, Ladouceur R, Tremblay RE. Gambling, delinquency, and drug use during adolescence: mutual influences and common risk factors. J Gambl Stud 2001;17:171-90.
14. Shope JT, Bingham CR. Drinking-driving as a component of problem driving and problem behavior in young adults. J Stud Alcohol 2002;63(1):24-33.
15. Potenza MN. The neurobiology of pathological gambling. Semin Clin Neuropsychiatry 2001;6:217-26.
16. Petry NM. Substance abuse, pathological gambling, and impulsiveness. Drug Alcohol Depend 2001;63:29-38.
17. Bechara A. Neurobiology of decision-making: risk and reward. Semin Clin Neuropsychiatry 2001;6:205-16.
18. Zuckerman M. Sensation seeking: the balance between risk and reward. In: Lipsitt LP, Mitnick LL (eds). Self-regulatory behavior and risk taking. Norwood, NJ: Ablex Publishing; 1992;143-52.
19. Masterman DL, Cummings JL. Frontal-subcortical circuits: the anatomical basis of executive, social and motivational behaviors. J Psychopharmacol 1997;11:107-14.
20. Horn NR, Dolan M, Elliot R, et al. Response inhibition and impulsivity: an fMRI study. Neuropsychologia 2003;41:1959-66.
21. Fallgatter AJ, Herrmann MJ. Electrophysiological assessment of impulsive behavior in healthy subjects. Neuropsychologia 2001;39:328-33.
22. Booth JR, Burman DD, Meyer JR, et al. Neural development of selective attention and response inhibition. Neuroimage 2003;20:737-51.
23. O’Donnell P, Greene J, Pabello N, et al. Modulation of cell firing in the nucleus accumbens. Ann NY Acad Sci 1999;877:157-75.
24. Nordin C, Eklundh T. Altered CSF 5-HIAA disposition in pathologic male gamblers. CNS Spectrums 1999;4:25-33.
25. Leyton M, Okazawa H, Diksic M, et al. Brain regional alpha-[11C] methyl-L-tryptophan trapping in impulsive subjects with borderline personality disorder. Am J Psychiatry 2001;158:775-82.
26. Cherek DR, Lane SD, Pietras CJ, Steinberg JL. Effects of chronic paroxetine administration on measures of aggressive and impulsive responses of adult males with a history of conduct disorder. Psychopharmacology (Berl) 2002;159:266-74.
27. Geller DA, Biederman J, Stewart SE, et al. Which SSRI? A meta-analysis of pharmacotherapy trials in pediatric obsessive-compulsive disorder. Am J Psychiatry 2003;160:1919-28.
28. Hollander E, Allen A, Lopez RP, et al. A preliminary double-blind, placebo-controlled trial of divalproex sodium in borderline personality disorder. J Clin Psychiatry 2001;62(3):199-203.
29. Swann AC. Treatment of aggression in patients with bipolar disorder. J Clin Psychiatry 1999;60(suppl 15):25-8.
30. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
1. Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry 2003;160:1041-52.
2. Chambers RA, Potenza MN. Neurodevelopment, impulsivity, and adolescent gambling. J Gambl Stud 2003;19:53-84.
3. Turner C, McClure R. Age and gender differences in risk taking behavior as an explanation for high incidence of motor vehicle crashes as a driver in young males. Inj Control Saf Promot 2003;10:123-30.
4. Bachanas PJ, Morris MK, Lewis-Gess JK, et al. Psychological adjustment, substance use, HIV knowledge, and risky sexual behavior in at-risk minority females: developmental differences during adolescence. J Pediatr Psych 2002;27:373-84.
5. Goldstein RZ, Volkow ND. Drug addiction and its underlying neuro- biological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 2002;159:1642-52.
6. Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev 2000;24:417-63.
7. Clayton R. Transitions in drug use: risk and protective factors. In: Glantz M, Pickens R (eds). Vulnerability to drug abuse. Washington, DC: American Psychological Association, 1992;15-52.
8. Tamm L, Menon V, Reiss AL. Maturation of brain function associated with response inhibition. J Am Acad Child Adolesc Psychiatry 2002;41:1231-8.
9. Jonah BA. Sensation seeking and risky driving: a review and synthesis of the literature. Accid Anal Prev 1997;29:651-6.
10. Vitaro F, Arseneault L, Tremblay RE. Dispositional predictors of problem gambling in male adolescents. Am J Psychiatry 1997;154:1769-70.
11. Malow RM, Devieux JG, Jennings T, et al. Substance-abusing adolescents at varying levels of HIV risk: psychosocial characteristics, drug use, and sexual behavior. J Subst Abuse 2001;13:103-17.
12. Donovan JE, Jessor R, Costa FM. Syndrome of problem behavior in adolescence: a replication. J Consult Clin Psychol 1988;56:762-5.
13. Vitaro F, Brendgen M, Ladouceur R, Tremblay RE. Gambling, delinquency, and drug use during adolescence: mutual influences and common risk factors. J Gambl Stud 2001;17:171-90.
14. Shope JT, Bingham CR. Drinking-driving as a component of problem driving and problem behavior in young adults. J Stud Alcohol 2002;63(1):24-33.
15. Potenza MN. The neurobiology of pathological gambling. Semin Clin Neuropsychiatry 2001;6:217-26.
16. Petry NM. Substance abuse, pathological gambling, and impulsiveness. Drug Alcohol Depend 2001;63:29-38.
17. Bechara A. Neurobiology of decision-making: risk and reward. Semin Clin Neuropsychiatry 2001;6:205-16.
18. Zuckerman M. Sensation seeking: the balance between risk and reward. In: Lipsitt LP, Mitnick LL (eds). Self-regulatory behavior and risk taking. Norwood, NJ: Ablex Publishing; 1992;143-52.
19. Masterman DL, Cummings JL. Frontal-subcortical circuits: the anatomical basis of executive, social and motivational behaviors. J Psychopharmacol 1997;11:107-14.
20. Horn NR, Dolan M, Elliot R, et al. Response inhibition and impulsivity: an fMRI study. Neuropsychologia 2003;41:1959-66.
21. Fallgatter AJ, Herrmann MJ. Electrophysiological assessment of impulsive behavior in healthy subjects. Neuropsychologia 2001;39:328-33.
22. Booth JR, Burman DD, Meyer JR, et al. Neural development of selective attention and response inhibition. Neuroimage 2003;20:737-51.
23. O’Donnell P, Greene J, Pabello N, et al. Modulation of cell firing in the nucleus accumbens. Ann NY Acad Sci 1999;877:157-75.
24. Nordin C, Eklundh T. Altered CSF 5-HIAA disposition in pathologic male gamblers. CNS Spectrums 1999;4:25-33.
25. Leyton M, Okazawa H, Diksic M, et al. Brain regional alpha-[11C] methyl-L-tryptophan trapping in impulsive subjects with borderline personality disorder. Am J Psychiatry 2001;158:775-82.
26. Cherek DR, Lane SD, Pietras CJ, Steinberg JL. Effects of chronic paroxetine administration on measures of aggressive and impulsive responses of adult males with a history of conduct disorder. Psychopharmacology (Berl) 2002;159:266-74.
27. Geller DA, Biederman J, Stewart SE, et al. Which SSRI? A meta-analysis of pharmacotherapy trials in pediatric obsessive-compulsive disorder. Am J Psychiatry 2003;160:1919-28.
28. Hollander E, Allen A, Lopez RP, et al. A preliminary double-blind, placebo-controlled trial of divalproex sodium in borderline personality disorder. J Clin Psychiatry 2001;62(3):199-203.
29. Swann AC. Treatment of aggression in patients with bipolar disorder. J Clin Psychiatry 1999;60(suppl 15):25-8.
30. Hollander E, Dolgoff-Kaspar R, Cartwright C, et al. An open trial of divalproex sodium in autism spectrum disorders. J Clin Psychiatry 2001;62:530-4.
Taking the mystery out of missing persons
A patient with schizophrenia or Alzheimer’s disease can function at home, but his family fears he would suddenly “run off”—potentially harming himself or others—unless he is watched around the clock. Can this patient avoid more-restrictive, institutionalized care without burdening the family?
Enter global positioning and radio frequency tracking technologies that are gaining wider acceptance and could one day play a role in caring for the chronically mentally ill.
Tracking technologies
The global positioning system (GPS) was created in 1993, when the United States Air Force launched the 24 th Navstar satellite.1 The system contains both military and civilian signals, but civilian accuracy initially was limited to 100 feet compared with 60 feet for the military signals. This limitation has since been lifted from civilian devices.
At first, GPS technology was used to navigate vehicles; later, specialized handheld devices provide navigation for hikers.
Today, GPS receivers are available for personal digital assistants, as are specialized wrist devices. Wherify Wireless offers GPS wrist devices, including GPS Locator for Kids, which allows parents or guardians to locate children within minutes and relay a message telling them to come home. GPS devices are increasingly popular and are used in the game of Geocaching,2 where players use GPS technology to hunt for a cache.
Radio frequency identification devices (RFIDs) are microchips the size of a grain of rice that allow retailers to track goods from warehouse to retail shelf. The chip contains no power source, but utilizes the energy within the initial radio signal.
RFID tags do not harbor information other than an identification number, which can be linked to a medical record or other database. Unlike bar codes, which require direct exposure to scan, hidden RFIDs broadcast themselves when activated by the radio signal, making transactions faster and more convenient.
RFIDs have caught on. Cards with RFID tags have been used at the Academy Awards ceremony to control access. Guards at jails throughout the United States use RFID tags to verify inmates’ whereabouts. Customers at Exxon and Mobil gasoline stations use RFID devices (called Speedpass) to facilitate purchases at the pump or register.
In 2002, RFID implants became available at medical clinics3 and are beginning to reach the mainstream. At the Baja Beach nightclub in Barcelona, for example, guests with the implant in their arms gain access to VIP areas once they pass through the scanner (which automatically assesses the cover charge to their tab).4
Approximately 1,000 persons have received VeriChip RFID implants over 2 years.5
How GPS, RFID can help caretakers
GPS. Caretakers can use GPS tracking devices to track a patient with a chronic mental disorder.
For example, the patient can wear a Wherify Wireless GPS locator wristwatch. To track the patient, the caretaker would log on to Wherify’s demo Web site (http://www.wherifywireless.com/demo.htm) and enter the device’s ID number. After clicking on the “locate” button, the locator device is contacted and its position and time of position is displayed on a map. Alternately, subscribers can call an 800 number and ask the operator to relay the locator address.
The wristwatch or locator phone costs about $200, and a monthly subscription ranges from $20 to $45 depending on number of location queries. The wristwatch comes with a remote-activated safety lock feature to prevent the patient from taking it off.
RFID. VeriChip implants are geared for indoor use, but can supplement a GPS device to track a chronically mentally ill patient.
The 11-millimeter chip, commonly used for standard security applications, is injected into the fatty tissue of the right tricep. When the recipient is near a VeriChip scanning device, the chip radios an ID number to the scanner. If the number matches an ID number in the database, the person with the implant can enter a secured room or complete a financial transaction.5
The FDA is reviewing whether hospitals can use RFID implants to identify patients and allow staff access to medical records without violating patients’ privacy.5
RFID tags located in wristbands could be used to identify hospitalized patients and prevent medication errors. RFID-tagged identification cards could help authenticate staff for access to the electronic medical record. Tags embedded in the bottle cap could measure medication compliance,6 and blister packs reveal when the medication was last taken.7 Special probes also could determine if the medication has expired or been stored properly.
Inviting ‘Big Brother’?
Critics, however, say use of GPS or RFID technology threatens privacy. The group Consumers Against Supermarket Privacy Invasion and Numbering (CASPIAN) complains that unique ID tags identify who purchased which product and where. Because the monitoring is passive via radio waves, these tags can be hidden and read at a distance—meaning that people can be monitored without their knowledge or consent. Critics also fear that the radio waves may pose a health hazard.8
Accuracy is another concern. Although GPS technology provides location information, the locator device still depends on cellular phone technology to transmit the information. Poor cellular coverage areas may decrease the device’s usefulness. More importantly, because GPS devices require a clear view of the sky to access the satellites, they do not work indoors. Thus, a patient must have escaped the house for the device to work.
Related Resources
- World Research Group: Leveraging RFID for hospitals. http://www.worldrg.com/HW455/agenda.asp
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
(accessed Sept. 20, 2004)
1. National Academy of Sciences. The global positioning system: The role of atomic clocks-introduction. http://www.beyonddiscovery.org/content/view.page.asp?I=1275
2. Groundspeak Inc.—Geocaching. http://www.geocaching.com
3. ABC News: Meet the Chipsons. http://abcnews.go.com/sections/scitech/TechTV/techtv_chipfamily020510.html
4. VIP microchip launched in Baja Beach Club, Barcelona. http://www.niburu.nl/index.php?showarticle.php?articleID=2638
5. Kanellos M. Under-the-skin ID chips move toward U.S. hospitals. CNET news.com July 27, 2004. http://news.com.com/Under-the-kin+ID+chips+move+toward+U.S.+hospital+use/2100-7337_3-5285815.html?tag=alert
6. Information Mediary Corp. Med-ic eCap Compliance Monitor. http://informationmediary.com/ecap/
7. Ibid. http://informationmediary.com/certiscan.htm
8. CASPIAN. Stop RFID. www.stoprfid.org
A patient with schizophrenia or Alzheimer’s disease can function at home, but his family fears he would suddenly “run off”—potentially harming himself or others—unless he is watched around the clock. Can this patient avoid more-restrictive, institutionalized care without burdening the family?
Enter global positioning and radio frequency tracking technologies that are gaining wider acceptance and could one day play a role in caring for the chronically mentally ill.
Tracking technologies
The global positioning system (GPS) was created in 1993, when the United States Air Force launched the 24 th Navstar satellite.1 The system contains both military and civilian signals, but civilian accuracy initially was limited to 100 feet compared with 60 feet for the military signals. This limitation has since been lifted from civilian devices.
At first, GPS technology was used to navigate vehicles; later, specialized handheld devices provide navigation for hikers.
Today, GPS receivers are available for personal digital assistants, as are specialized wrist devices. Wherify Wireless offers GPS wrist devices, including GPS Locator for Kids, which allows parents or guardians to locate children within minutes and relay a message telling them to come home. GPS devices are increasingly popular and are used in the game of Geocaching,2 where players use GPS technology to hunt for a cache.
Radio frequency identification devices (RFIDs) are microchips the size of a grain of rice that allow retailers to track goods from warehouse to retail shelf. The chip contains no power source, but utilizes the energy within the initial radio signal.
RFID tags do not harbor information other than an identification number, which can be linked to a medical record or other database. Unlike bar codes, which require direct exposure to scan, hidden RFIDs broadcast themselves when activated by the radio signal, making transactions faster and more convenient.
RFIDs have caught on. Cards with RFID tags have been used at the Academy Awards ceremony to control access. Guards at jails throughout the United States use RFID tags to verify inmates’ whereabouts. Customers at Exxon and Mobil gasoline stations use RFID devices (called Speedpass) to facilitate purchases at the pump or register.
In 2002, RFID implants became available at medical clinics3 and are beginning to reach the mainstream. At the Baja Beach nightclub in Barcelona, for example, guests with the implant in their arms gain access to VIP areas once they pass through the scanner (which automatically assesses the cover charge to their tab).4
Approximately 1,000 persons have received VeriChip RFID implants over 2 years.5
How GPS, RFID can help caretakers
GPS. Caretakers can use GPS tracking devices to track a patient with a chronic mental disorder.
For example, the patient can wear a Wherify Wireless GPS locator wristwatch. To track the patient, the caretaker would log on to Wherify’s demo Web site (http://www.wherifywireless.com/demo.htm) and enter the device’s ID number. After clicking on the “locate” button, the locator device is contacted and its position and time of position is displayed on a map. Alternately, subscribers can call an 800 number and ask the operator to relay the locator address.
The wristwatch or locator phone costs about $200, and a monthly subscription ranges from $20 to $45 depending on number of location queries. The wristwatch comes with a remote-activated safety lock feature to prevent the patient from taking it off.
RFID. VeriChip implants are geared for indoor use, but can supplement a GPS device to track a chronically mentally ill patient.
The 11-millimeter chip, commonly used for standard security applications, is injected into the fatty tissue of the right tricep. When the recipient is near a VeriChip scanning device, the chip radios an ID number to the scanner. If the number matches an ID number in the database, the person with the implant can enter a secured room or complete a financial transaction.5
The FDA is reviewing whether hospitals can use RFID implants to identify patients and allow staff access to medical records without violating patients’ privacy.5
RFID tags located in wristbands could be used to identify hospitalized patients and prevent medication errors. RFID-tagged identification cards could help authenticate staff for access to the electronic medical record. Tags embedded in the bottle cap could measure medication compliance,6 and blister packs reveal when the medication was last taken.7 Special probes also could determine if the medication has expired or been stored properly.
Inviting ‘Big Brother’?
Critics, however, say use of GPS or RFID technology threatens privacy. The group Consumers Against Supermarket Privacy Invasion and Numbering (CASPIAN) complains that unique ID tags identify who purchased which product and where. Because the monitoring is passive via radio waves, these tags can be hidden and read at a distance—meaning that people can be monitored without their knowledge or consent. Critics also fear that the radio waves may pose a health hazard.8
Accuracy is another concern. Although GPS technology provides location information, the locator device still depends on cellular phone technology to transmit the information. Poor cellular coverage areas may decrease the device’s usefulness. More importantly, because GPS devices require a clear view of the sky to access the satellites, they do not work indoors. Thus, a patient must have escaped the house for the device to work.
Related Resources
- World Research Group: Leveraging RFID for hospitals. http://www.worldrg.com/HW455/agenda.asp
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
A patient with schizophrenia or Alzheimer’s disease can function at home, but his family fears he would suddenly “run off”—potentially harming himself or others—unless he is watched around the clock. Can this patient avoid more-restrictive, institutionalized care without burdening the family?
Enter global positioning and radio frequency tracking technologies that are gaining wider acceptance and could one day play a role in caring for the chronically mentally ill.
Tracking technologies
The global positioning system (GPS) was created in 1993, when the United States Air Force launched the 24 th Navstar satellite.1 The system contains both military and civilian signals, but civilian accuracy initially was limited to 100 feet compared with 60 feet for the military signals. This limitation has since been lifted from civilian devices.
At first, GPS technology was used to navigate vehicles; later, specialized handheld devices provide navigation for hikers.
Today, GPS receivers are available for personal digital assistants, as are specialized wrist devices. Wherify Wireless offers GPS wrist devices, including GPS Locator for Kids, which allows parents or guardians to locate children within minutes and relay a message telling them to come home. GPS devices are increasingly popular and are used in the game of Geocaching,2 where players use GPS technology to hunt for a cache.
Radio frequency identification devices (RFIDs) are microchips the size of a grain of rice that allow retailers to track goods from warehouse to retail shelf. The chip contains no power source, but utilizes the energy within the initial radio signal.
RFID tags do not harbor information other than an identification number, which can be linked to a medical record or other database. Unlike bar codes, which require direct exposure to scan, hidden RFIDs broadcast themselves when activated by the radio signal, making transactions faster and more convenient.
RFIDs have caught on. Cards with RFID tags have been used at the Academy Awards ceremony to control access. Guards at jails throughout the United States use RFID tags to verify inmates’ whereabouts. Customers at Exxon and Mobil gasoline stations use RFID devices (called Speedpass) to facilitate purchases at the pump or register.
In 2002, RFID implants became available at medical clinics3 and are beginning to reach the mainstream. At the Baja Beach nightclub in Barcelona, for example, guests with the implant in their arms gain access to VIP areas once they pass through the scanner (which automatically assesses the cover charge to their tab).4
Approximately 1,000 persons have received VeriChip RFID implants over 2 years.5
How GPS, RFID can help caretakers
GPS. Caretakers can use GPS tracking devices to track a patient with a chronic mental disorder.
For example, the patient can wear a Wherify Wireless GPS locator wristwatch. To track the patient, the caretaker would log on to Wherify’s demo Web site (http://www.wherifywireless.com/demo.htm) and enter the device’s ID number. After clicking on the “locate” button, the locator device is contacted and its position and time of position is displayed on a map. Alternately, subscribers can call an 800 number and ask the operator to relay the locator address.
The wristwatch or locator phone costs about $200, and a monthly subscription ranges from $20 to $45 depending on number of location queries. The wristwatch comes with a remote-activated safety lock feature to prevent the patient from taking it off.
RFID. VeriChip implants are geared for indoor use, but can supplement a GPS device to track a chronically mentally ill patient.
The 11-millimeter chip, commonly used for standard security applications, is injected into the fatty tissue of the right tricep. When the recipient is near a VeriChip scanning device, the chip radios an ID number to the scanner. If the number matches an ID number in the database, the person with the implant can enter a secured room or complete a financial transaction.5
The FDA is reviewing whether hospitals can use RFID implants to identify patients and allow staff access to medical records without violating patients’ privacy.5
RFID tags located in wristbands could be used to identify hospitalized patients and prevent medication errors. RFID-tagged identification cards could help authenticate staff for access to the electronic medical record. Tags embedded in the bottle cap could measure medication compliance,6 and blister packs reveal when the medication was last taken.7 Special probes also could determine if the medication has expired or been stored properly.
Inviting ‘Big Brother’?
Critics, however, say use of GPS or RFID technology threatens privacy. The group Consumers Against Supermarket Privacy Invasion and Numbering (CASPIAN) complains that unique ID tags identify who purchased which product and where. Because the monitoring is passive via radio waves, these tags can be hidden and read at a distance—meaning that people can be monitored without their knowledge or consent. Critics also fear that the radio waves may pose a health hazard.8
Accuracy is another concern. Although GPS technology provides location information, the locator device still depends on cellular phone technology to transmit the information. Poor cellular coverage areas may decrease the device’s usefulness. More importantly, because GPS devices require a clear view of the sky to access the satellites, they do not work indoors. Thus, a patient must have escaped the house for the device to work.
Related Resources
- World Research Group: Leveraging RFID for hospitals. http://www.worldrg.com/HW455/agenda.asp
Disclosure
Dr. Luo reports no financial relationship with any company whose products are mentioned in this article. The opinions expressed by Dr. Luo in this column are his own and do not necessarily reflect those of CURRENT PSYCHIATRY.
(accessed Sept. 20, 2004)
1. National Academy of Sciences. The global positioning system: The role of atomic clocks-introduction. http://www.beyonddiscovery.org/content/view.page.asp?I=1275
2. Groundspeak Inc.—Geocaching. http://www.geocaching.com
3. ABC News: Meet the Chipsons. http://abcnews.go.com/sections/scitech/TechTV/techtv_chipfamily020510.html
4. VIP microchip launched in Baja Beach Club, Barcelona. http://www.niburu.nl/index.php?showarticle.php?articleID=2638
5. Kanellos M. Under-the-skin ID chips move toward U.S. hospitals. CNET news.com July 27, 2004. http://news.com.com/Under-the-kin+ID+chips+move+toward+U.S.+hospital+use/2100-7337_3-5285815.html?tag=alert
6. Information Mediary Corp. Med-ic eCap Compliance Monitor. http://informationmediary.com/ecap/
7. Ibid. http://informationmediary.com/certiscan.htm
8. CASPIAN. Stop RFID. www.stoprfid.org
(accessed Sept. 20, 2004)
1. National Academy of Sciences. The global positioning system: The role of atomic clocks-introduction. http://www.beyonddiscovery.org/content/view.page.asp?I=1275
2. Groundspeak Inc.—Geocaching. http://www.geocaching.com
3. ABC News: Meet the Chipsons. http://abcnews.go.com/sections/scitech/TechTV/techtv_chipfamily020510.html
4. VIP microchip launched in Baja Beach Club, Barcelona. http://www.niburu.nl/index.php?showarticle.php?articleID=2638
5. Kanellos M. Under-the-skin ID chips move toward U.S. hospitals. CNET news.com July 27, 2004. http://news.com.com/Under-the-kin+ID+chips+move+toward+U.S.+hospital+use/2100-7337_3-5285815.html?tag=alert
6. Information Mediary Corp. Med-ic eCap Compliance Monitor. http://informationmediary.com/ecap/
7. Ibid. http://informationmediary.com/certiscan.htm
8. CASPIAN. Stop RFID. www.stoprfid.org
Secondary amenorrhea: Don’t dismiss it as ‘normal’
A young or middle-aged patient who stops menstruating may be pregnant or have an underlying medical problem that, left undiagnosed, could cause obesity, sexual dysfunction, infertility, osteoporosis, endometrial hyperplasia, or endometrial cancer.
Yet clinicians too often dismiss secondary amenorrhea as a “normal” result of a mental disorder or psychotropic. Psychiatrists need to:
- identify when a psychiatric disorder or drug disrupts menses
- diagnose medical causes, including thyroid dysfunction, pituitary adenomas, and polycystic ovary syndrome (PCOS).
This article outlines the most common and serious causes of secondary amenorrhea among psychiatric patients, and offers an algorithm for ruling out medical problems in nonpregnant women of child-bearing age who have stopped menstruating for 3 months. The diagnostic approach described here does not apply to women with primary amenorrhea (have never menstruated).
Table 1
Psychotropics that may cause amenorrhea
| Effect | Drug/class |
|---|---|
| Prolactin elevation | Antipsychotics (chlorpromazine, haloperidol, risperidone) SSRIs (citalopram, escitalopram, fluoxetine) |
| Sex hormone-binding globulin elevation | Carbamazepine |
| Association with PCOS unknown mechanism | Valproic acid |
| SSRIs: Selective serotonin reuptake inhibitors | |
| PCOS: Polycystic ovary syndrome | |
CASE REPORT: NO PREGNANCY, NO PERIOD
Two years ago Ms. J, age 28, was diagnosed with depression. Her psychiatrist prescribed fluoxetine, 20 mg/d titrated across 4 weeks to 40 mg/d. About 4 months later, she experienced her first manic episode. The psychiatrist changed the diagnosis to bipolar I disorder and added risperidone, 2 mg/d, to manage her mania.
Ms. J’s bipolar disorder has been under control for 1 year, but she reports that her menstruation stopped 6 months ago. She is sexually active; she and her partner use spermicide-coated condoms. She does not want to be pregnant now but might want to bear a child within the next year. Several home pregnancy tests across 6 months were negative.
The patient is obese (5 feet, 5 inches, 186 lbs, body mass index 31) and has gained about 30 pounds during the past year. Vital signs are normal; psychiatric examination indicates normal mood and affect. Skin exam reveals mild papular acne on her face and back and increased hair growth on her chin. Other physical findings—including cardiac, lung, and neurologic examinations—are normal.
Laboratory evaluation reveals a prolactin level of 105 ng/mL, a negative serum ß-Hcg reading, and normal TSH, FSH, DHEA-S and testosterone levels.
Discussion. Ms. J’s history, physical examination, and laboratory tests suggest several possible causes of secondary amenorrhea:
- Are psychotropics or a prolactin-secreting tumor elevating her prolactin level?
- Does she have PCOS, as her weight gain, hirsutism, and acne might indicate?
- Is her bipolar disorder a factor? Consider psychiatric illness, medication side effects, and medical causes when evaluating secondary amenorrhea.
PSYCHIATRIC ILLNESS
Patients with high emotional stress may have amenorrhea or menstrual irregularities related to hypothalamic dysfunction.1 Also:
Anorexia nervosa has been shown to cause hypothalamic dysfunction, leading to amenorrhea.2 A correlation exists between weight loss and menses cessation, and between regain of weight and menses resumption.2
Depression. Estradiol levels are lower in depressed women than in euthymic women, probably because of altered hypothalamic-pituitary axis (HPA) function. Also, physical distress is correlated with menses disruption.3
In a 3-year study of women ages 36 to 45,4 those with a history of depression exhibited 1.2 times the rate of perimenopause as nondepressed women. Subjects with Hamilton Rating Scale for Depression scores >8 at enrollment had twice the rate of perimenopause after 3 years compared with nondepressed women. The findings suggest that depression might increase a woman’s risk of ceasing ovarian function in her 30s or 40s. Natural menopause on average begins at age 51.5
In another study,6 16 of 32 women with PCOS had Center for Epidemiological Studies-Depression Rating Scale scores indicating depression (≥16). The study suggests a high prevalence of depression among women with PCOS, but was limited by possible selection bias, no further diagnostic evaluation for depression, small sample size, and lack of an age-matched control group.
Bipolar disorder. High rates of menstrual disturbances have been reported among women with bipolar disorder.7 Although the mechanism has not been ascertained, disruption of HPA function similar to that seen in depression is likely.7
MEDICATIONS AND AMENORRHEA
Medications can cause amenorrhea, primarily through hyperprolactinemia—although other mechanisms may be involved (Table 1). Prolactin suppresses hypothalamic luteinizing hormone-releasing hormone (LHRH) production, leading to decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH), thus reducing circulating estrogen. Prolactin-secreting pituitary tumors and drug side effects mostly commonly cause hyperprolactinemia.
Antipsychotics. Phenothiazines such as chlorpromazine, butyrophenones such as haloperidol, and the atypical antipsychotic risperidone raise prolactin levels via dopamine-receptor antagonism.
Other atypical antipsychotics—including aripiprazole, clozapine, olanzapine, quetiapine, and ziprasidone—are associated with lower serum prolactin levels than risperidone.8,9 Preliminary studies suggest, for example, that switching patients from risperidone to quetiapine may help resume menstruation without worsening psychotic symptoms,10 and that amenorrhea often resolves after the patient is switched to another atypical antipsychotic.11
SSRIs. All selective serotonin reuptake inhibitors except sertraline are associated with hyperprolactinemia and can lead to amenorrhea in some patients.12
Table 2
Differential diagnosis of secondary amenorrhea
Ovarian causes
|
Hypothalamic causes
|
Hyperprolactinemia
|
Uterine causes
|
| * Turner’s syndrome: A rare chromosomal disorder characterized by short stature, lack of sexual development at puberty. |
| † Asherman’s syndrome: Endometrial adhesions, scar tissue that develop after uterine curettage or infections. |
Anticonvulsants used as mood stabilizers to treat bipolar disorder may cause menstrual irregularities, although most data relate to women with seizure disorders.
Valproic acid has been associated with PCOS in patients with epilepsy,13 although it is unknown whether the agent’s androgenizing effects vary with age. Carbamazepine, which increases sex hormone-binding globulin, may also lead to menstrual disorders by decreasing bioavailability of circulating estrogen.14 Consider switching a patient with disrupted menses to lithium, lamotrigine, or oxcarbazepine, which have not been associated with menstrual dysfunction.
MEDICAL CAUSES
Pregnancy is the most common cause of menses cessation, followed by ovarian, hypothalamic, pituitary, or uterine dysfunction (Table 2). Hypothalamic and pituitary dysfunction often cause amenorrhea in psychiatric patients, whereas ovarian causes are common among all patients with secondary amenorrhea.15
Ovarian. In PCOS, the ovaries and sometimes the adrenal glands produce excess androgens, leading to infrequent or light periods (oligomenorrhea) or amenorrhea.
Patients with depression are prone to ovarian failure in their 30s or 40s, possibly because of chronic HPA disruption.4 Premature ovarian failure also is common among patients with Turner’s syndrome, a rare chromosomal disorder characterized by short stature and lack of sexual development at puberty. Ovarian failure also can occur spontaneously.
Hypothalamic. Functional hypothalamic amenorrhea occurs in mood and eating disorders. Emotional stress, excessive physical exercise, and nutritional deficiencies reduce LHRH secretion by the hypothalamus, which interrupts the reproductive cycle. Cardiovascular disease, respiratory disease, cancer, and other acute and chronic medical illnesses can cause significant physiologic stress, thus leading to HPA dysfunction. Hypothalamic amenorrhea is treated by targeting the underlying psychiatric or medical condition.
Pituitary. Prolactin-secreting pituitary tumors, such as a pituitary adenoma, must be ruled out in patients whose prolactin levels remain high after a medication change.15 Hypothyroidism also can trigger hyperprolactinemia by causing pituitary gland hyperplasia.
Uterine. Women who have had uterine curettage or infections can develop adhesions and scar tissue that ablate the endometrial lining. This condition, called Asherman’s syndrome, is the most common uterine cause of menstrual disruption.
EVALUATING SECONDARY AMENORRHEA
When a patient presents with secondary amenorrhea, immediately rule out pregnancy because psychiatric disorders often are managed differently in pregnant than in nonpregnant women.16
Next, take a thorough patient history to determine whether referral is necessary. Ask about weight loss (intentional or unintentional), increased stressors, or a medical illness that may point to functional hypothalamic amenorrhea. Galactorrhea or vision changes—particularly loss of peripheral vision—could suggest a pituitary tumor. Skin changes, cold intolerance, fatigue, or constipation could indicate hypothyroidism.
Menopausal symptoms such as hot flashes and vaginal dryness could point to premature ovarian failure. Galactorrhea may indicate high prolactin levels. Obesity, hirsutism, or acne could point to PCOS. Consider Asherman’s syndrome in patients with endometritis or who have had a uterine dilation and curettage.
Laboratory testing. Once pregnancy is ruled out, measure prolactin. If it exceeds 25 ng/mL by 15 ng/mL or more, do a confirmative second prolactin test. If a patient is taking a prolactin-raising medication and her prolactin was not gauged before treatment, change to a prolactin-sparing agent, then measure her prolactin 2 weeks later.17
When to refer. If prolactin persistently exceeds 50 ng/mL even after changing medications, refer the patient for brain MRI to rule out a pituitary tumor.
Tests for other underlying medical causes of secondary amenorrhea—and when to perform them—are shown in the algorithm. Psychiatrists can give these tests or refer the patient to her primary care physician.
Algorithm Laboratory evaluation of secondary amenorrhea
Communication between care team members is key to determining treatment. If a medical problem arises during psychiatric treatment, call the patient’s primary care physician or send a letter describing the problem. Also send the referring physician available lab reports.
CASE CONTINUED: TREATMENT CHANGE
Ms. J’s psychiatrist tapered risperidone to 1 mg/d for 2 weeks, then switched to olanzapine, 5 mg/d. Three weeks later, her prolactin decreased to 25 ng/mL. She continued fluoxetine, 40 mg/d, and tolerated the change in antipsychotics.
Ms. J’s bipolar disorder remains well-controlled, but menses had not resumed for another 2 months, so the psychiatrist referred Ms. J back to her primary care physician. Androgenizing and pituitary tumors were ruled out based on normal TSH, prolactin, and testosterone levels. Ms. J was diagnosed as having PCOS based on her constellation of signs and symptoms. She was started on metformin, an insulin sensitizer used to treat PCOS, and was referred to a dietitian to help her lose weight.
One year later, Ms. J still struggles with weight control, but menstruation is back to normal.
Related resources
- Lean M, De Smedt G. Schizophrenia and osteoporosis. Int Clin Psychopharmacol 2004;19:31-5.
- Berga SL, Marcus MD, Loucks TL, et al. Recovery of ovarian activity in women with functional hypothalamic amenorrhea who were treated with cognitive behavior therapy. Fertil Steril 2003;80:976-81.
- Carr BR, Bradshaw KD. Disturbances of menstruation and other common gynecologic complaints in women. In: Braunwald E, Hauser SL, Fauci AS, et al (eds). Harrison’s principles of internal medicine(15th ed). New York: McGraw-Hill, 2001:Chapter 52.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Metformin • Glucovance, others
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kaplan JR, Manuck SB. Ovarian dysfunction, stress, and disease: a primate continuum. ILAR J 2004;45(2):89-115.
2. Mitan LA. Menstrual dysfunction in anorexia nervosa. J Pediatr Adolesc Gynecol 2004;17:81-85.
3. Young EA, Korzun A. The hypothalamic-pituitary-gonadal axis in mood disorders. Endocrinol Metab Clin North Am 2002;31(1):63-78.
4. Harlow BL, Wise LA, Otto MW, et al. Depression and its influence on reproductive endocrine and menstrual cycle markers associated with perimenopause. The Harvard Study of Moods and Cycles. Arch Gen Psychiatry 2003;60:29-36.
5. Brizendine L. Minding menopause. Psychotropics vs. estrogen? What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Rasgon NL, Rao RC, Hwang S, et al. Depression in women with polycystic ovary syndrome: clinical and biochemical correlates. J Affect Disord 2003;74(3):299-304.
7. Rasgon NL, Altshuler LL, Gundeman D, et al. Medication status and polycystic ovary syndrome in women with bipolar disorder: a preliminary report. J Clin Psychiatry 2000;61(3):173-8.
8. Kim KS, Pae CU, Chae JH, et al. Effects of olanzapine on prolactin levels of female patients with schizophrenia treated with risperidone. J Clin Psychiatry 2002;63(5):408-13.
9. Goodnick PJ, Rodriguez L, Santana O. Antipsychotics: impact on prolactin levels. Expert Opin Pharmacother 2002;3(10):1381-91.
10. Takahashi H, Higuchi H, Kamata M, et al. Effectiveness of switching to quetiapine for neuroleptic-induced amenorrhea. J Neuropsychiatry Clin Neurosci 2003;15:375-7.
11. Knegtering H, van der Moolen AE, Castelien S, et al. What are the effects of antipsychotics on sexual dysfunctions and endocrine functioning? Psychoneuroendocrinology 2003;28(suppl 2):109-23.
12. Goodnick PJ, Chaudry T, Artadi J, Arcey S. Women’s issues in mood disorders. Expert Opin Pharmacother 2000;1(5):903-16.
13. Isojarvi JI, Laatikainen TJ, Pakarinen AJ, et al. Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. N Engl J Med 1993;329(19):1383-8.
14. Isojarvi JI. Reproductive dysfunction in women with epilepsy. Neurology 2003;61(6 Suppl 2):S27-S34.
15. Reindollar RH, Novak M, Tho SP, McDonough PG. Adult-onset amenorrhea: a study of 262 patients. Am J Obstet Gynecol 1986;155(3):531-43.
16. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry 2003;2(7):14-26.
17. Barbieri RL. Etiology, diagnosis and treatment of secondary amenorrhea. UpToDate 2003;12:1.-
A young or middle-aged patient who stops menstruating may be pregnant or have an underlying medical problem that, left undiagnosed, could cause obesity, sexual dysfunction, infertility, osteoporosis, endometrial hyperplasia, or endometrial cancer.
Yet clinicians too often dismiss secondary amenorrhea as a “normal” result of a mental disorder or psychotropic. Psychiatrists need to:
- identify when a psychiatric disorder or drug disrupts menses
- diagnose medical causes, including thyroid dysfunction, pituitary adenomas, and polycystic ovary syndrome (PCOS).
This article outlines the most common and serious causes of secondary amenorrhea among psychiatric patients, and offers an algorithm for ruling out medical problems in nonpregnant women of child-bearing age who have stopped menstruating for 3 months. The diagnostic approach described here does not apply to women with primary amenorrhea (have never menstruated).
Table 1
Psychotropics that may cause amenorrhea
| Effect | Drug/class |
|---|---|
| Prolactin elevation | Antipsychotics (chlorpromazine, haloperidol, risperidone) SSRIs (citalopram, escitalopram, fluoxetine) |
| Sex hormone-binding globulin elevation | Carbamazepine |
| Association with PCOS unknown mechanism | Valproic acid |
| SSRIs: Selective serotonin reuptake inhibitors | |
| PCOS: Polycystic ovary syndrome | |
CASE REPORT: NO PREGNANCY, NO PERIOD
Two years ago Ms. J, age 28, was diagnosed with depression. Her psychiatrist prescribed fluoxetine, 20 mg/d titrated across 4 weeks to 40 mg/d. About 4 months later, she experienced her first manic episode. The psychiatrist changed the diagnosis to bipolar I disorder and added risperidone, 2 mg/d, to manage her mania.
Ms. J’s bipolar disorder has been under control for 1 year, but she reports that her menstruation stopped 6 months ago. She is sexually active; she and her partner use spermicide-coated condoms. She does not want to be pregnant now but might want to bear a child within the next year. Several home pregnancy tests across 6 months were negative.
The patient is obese (5 feet, 5 inches, 186 lbs, body mass index 31) and has gained about 30 pounds during the past year. Vital signs are normal; psychiatric examination indicates normal mood and affect. Skin exam reveals mild papular acne on her face and back and increased hair growth on her chin. Other physical findings—including cardiac, lung, and neurologic examinations—are normal.
Laboratory evaluation reveals a prolactin level of 105 ng/mL, a negative serum ß-Hcg reading, and normal TSH, FSH, DHEA-S and testosterone levels.
Discussion. Ms. J’s history, physical examination, and laboratory tests suggest several possible causes of secondary amenorrhea:
- Are psychotropics or a prolactin-secreting tumor elevating her prolactin level?
- Does she have PCOS, as her weight gain, hirsutism, and acne might indicate?
- Is her bipolar disorder a factor? Consider psychiatric illness, medication side effects, and medical causes when evaluating secondary amenorrhea.
PSYCHIATRIC ILLNESS
Patients with high emotional stress may have amenorrhea or menstrual irregularities related to hypothalamic dysfunction.1 Also:
Anorexia nervosa has been shown to cause hypothalamic dysfunction, leading to amenorrhea.2 A correlation exists between weight loss and menses cessation, and between regain of weight and menses resumption.2
Depression. Estradiol levels are lower in depressed women than in euthymic women, probably because of altered hypothalamic-pituitary axis (HPA) function. Also, physical distress is correlated with menses disruption.3
In a 3-year study of women ages 36 to 45,4 those with a history of depression exhibited 1.2 times the rate of perimenopause as nondepressed women. Subjects with Hamilton Rating Scale for Depression scores >8 at enrollment had twice the rate of perimenopause after 3 years compared with nondepressed women. The findings suggest that depression might increase a woman’s risk of ceasing ovarian function in her 30s or 40s. Natural menopause on average begins at age 51.5
In another study,6 16 of 32 women with PCOS had Center for Epidemiological Studies-Depression Rating Scale scores indicating depression (≥16). The study suggests a high prevalence of depression among women with PCOS, but was limited by possible selection bias, no further diagnostic evaluation for depression, small sample size, and lack of an age-matched control group.
Bipolar disorder. High rates of menstrual disturbances have been reported among women with bipolar disorder.7 Although the mechanism has not been ascertained, disruption of HPA function similar to that seen in depression is likely.7
MEDICATIONS AND AMENORRHEA
Medications can cause amenorrhea, primarily through hyperprolactinemia—although other mechanisms may be involved (Table 1). Prolactin suppresses hypothalamic luteinizing hormone-releasing hormone (LHRH) production, leading to decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH), thus reducing circulating estrogen. Prolactin-secreting pituitary tumors and drug side effects mostly commonly cause hyperprolactinemia.
Antipsychotics. Phenothiazines such as chlorpromazine, butyrophenones such as haloperidol, and the atypical antipsychotic risperidone raise prolactin levels via dopamine-receptor antagonism.
Other atypical antipsychotics—including aripiprazole, clozapine, olanzapine, quetiapine, and ziprasidone—are associated with lower serum prolactin levels than risperidone.8,9 Preliminary studies suggest, for example, that switching patients from risperidone to quetiapine may help resume menstruation without worsening psychotic symptoms,10 and that amenorrhea often resolves after the patient is switched to another atypical antipsychotic.11
SSRIs. All selective serotonin reuptake inhibitors except sertraline are associated with hyperprolactinemia and can lead to amenorrhea in some patients.12
Table 2
Differential diagnosis of secondary amenorrhea
Ovarian causes
|
Hypothalamic causes
|
Hyperprolactinemia
|
Uterine causes
|
| * Turner’s syndrome: A rare chromosomal disorder characterized by short stature, lack of sexual development at puberty. |
| † Asherman’s syndrome: Endometrial adhesions, scar tissue that develop after uterine curettage or infections. |
Anticonvulsants used as mood stabilizers to treat bipolar disorder may cause menstrual irregularities, although most data relate to women with seizure disorders.
Valproic acid has been associated with PCOS in patients with epilepsy,13 although it is unknown whether the agent’s androgenizing effects vary with age. Carbamazepine, which increases sex hormone-binding globulin, may also lead to menstrual disorders by decreasing bioavailability of circulating estrogen.14 Consider switching a patient with disrupted menses to lithium, lamotrigine, or oxcarbazepine, which have not been associated with menstrual dysfunction.
MEDICAL CAUSES
Pregnancy is the most common cause of menses cessation, followed by ovarian, hypothalamic, pituitary, or uterine dysfunction (Table 2). Hypothalamic and pituitary dysfunction often cause amenorrhea in psychiatric patients, whereas ovarian causes are common among all patients with secondary amenorrhea.15
Ovarian. In PCOS, the ovaries and sometimes the adrenal glands produce excess androgens, leading to infrequent or light periods (oligomenorrhea) or amenorrhea.
Patients with depression are prone to ovarian failure in their 30s or 40s, possibly because of chronic HPA disruption.4 Premature ovarian failure also is common among patients with Turner’s syndrome, a rare chromosomal disorder characterized by short stature and lack of sexual development at puberty. Ovarian failure also can occur spontaneously.
Hypothalamic. Functional hypothalamic amenorrhea occurs in mood and eating disorders. Emotional stress, excessive physical exercise, and nutritional deficiencies reduce LHRH secretion by the hypothalamus, which interrupts the reproductive cycle. Cardiovascular disease, respiratory disease, cancer, and other acute and chronic medical illnesses can cause significant physiologic stress, thus leading to HPA dysfunction. Hypothalamic amenorrhea is treated by targeting the underlying psychiatric or medical condition.
Pituitary. Prolactin-secreting pituitary tumors, such as a pituitary adenoma, must be ruled out in patients whose prolactin levels remain high after a medication change.15 Hypothyroidism also can trigger hyperprolactinemia by causing pituitary gland hyperplasia.
Uterine. Women who have had uterine curettage or infections can develop adhesions and scar tissue that ablate the endometrial lining. This condition, called Asherman’s syndrome, is the most common uterine cause of menstrual disruption.
EVALUATING SECONDARY AMENORRHEA
When a patient presents with secondary amenorrhea, immediately rule out pregnancy because psychiatric disorders often are managed differently in pregnant than in nonpregnant women.16
Next, take a thorough patient history to determine whether referral is necessary. Ask about weight loss (intentional or unintentional), increased stressors, or a medical illness that may point to functional hypothalamic amenorrhea. Galactorrhea or vision changes—particularly loss of peripheral vision—could suggest a pituitary tumor. Skin changes, cold intolerance, fatigue, or constipation could indicate hypothyroidism.
Menopausal symptoms such as hot flashes and vaginal dryness could point to premature ovarian failure. Galactorrhea may indicate high prolactin levels. Obesity, hirsutism, or acne could point to PCOS. Consider Asherman’s syndrome in patients with endometritis or who have had a uterine dilation and curettage.
Laboratory testing. Once pregnancy is ruled out, measure prolactin. If it exceeds 25 ng/mL by 15 ng/mL or more, do a confirmative second prolactin test. If a patient is taking a prolactin-raising medication and her prolactin was not gauged before treatment, change to a prolactin-sparing agent, then measure her prolactin 2 weeks later.17
When to refer. If prolactin persistently exceeds 50 ng/mL even after changing medications, refer the patient for brain MRI to rule out a pituitary tumor.
Tests for other underlying medical causes of secondary amenorrhea—and when to perform them—are shown in the algorithm. Psychiatrists can give these tests or refer the patient to her primary care physician.
Algorithm Laboratory evaluation of secondary amenorrhea
Communication between care team members is key to determining treatment. If a medical problem arises during psychiatric treatment, call the patient’s primary care physician or send a letter describing the problem. Also send the referring physician available lab reports.
CASE CONTINUED: TREATMENT CHANGE
Ms. J’s psychiatrist tapered risperidone to 1 mg/d for 2 weeks, then switched to olanzapine, 5 mg/d. Three weeks later, her prolactin decreased to 25 ng/mL. She continued fluoxetine, 40 mg/d, and tolerated the change in antipsychotics.
Ms. J’s bipolar disorder remains well-controlled, but menses had not resumed for another 2 months, so the psychiatrist referred Ms. J back to her primary care physician. Androgenizing and pituitary tumors were ruled out based on normal TSH, prolactin, and testosterone levels. Ms. J was diagnosed as having PCOS based on her constellation of signs and symptoms. She was started on metformin, an insulin sensitizer used to treat PCOS, and was referred to a dietitian to help her lose weight.
One year later, Ms. J still struggles with weight control, but menstruation is back to normal.
Related resources
- Lean M, De Smedt G. Schizophrenia and osteoporosis. Int Clin Psychopharmacol 2004;19:31-5.
- Berga SL, Marcus MD, Loucks TL, et al. Recovery of ovarian activity in women with functional hypothalamic amenorrhea who were treated with cognitive behavior therapy. Fertil Steril 2003;80:976-81.
- Carr BR, Bradshaw KD. Disturbances of menstruation and other common gynecologic complaints in women. In: Braunwald E, Hauser SL, Fauci AS, et al (eds). Harrison’s principles of internal medicine(15th ed). New York: McGraw-Hill, 2001:Chapter 52.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Metformin • Glucovance, others
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
A young or middle-aged patient who stops menstruating may be pregnant or have an underlying medical problem that, left undiagnosed, could cause obesity, sexual dysfunction, infertility, osteoporosis, endometrial hyperplasia, or endometrial cancer.
Yet clinicians too often dismiss secondary amenorrhea as a “normal” result of a mental disorder or psychotropic. Psychiatrists need to:
- identify when a psychiatric disorder or drug disrupts menses
- diagnose medical causes, including thyroid dysfunction, pituitary adenomas, and polycystic ovary syndrome (PCOS).
This article outlines the most common and serious causes of secondary amenorrhea among psychiatric patients, and offers an algorithm for ruling out medical problems in nonpregnant women of child-bearing age who have stopped menstruating for 3 months. The diagnostic approach described here does not apply to women with primary amenorrhea (have never menstruated).
Table 1
Psychotropics that may cause amenorrhea
| Effect | Drug/class |
|---|---|
| Prolactin elevation | Antipsychotics (chlorpromazine, haloperidol, risperidone) SSRIs (citalopram, escitalopram, fluoxetine) |
| Sex hormone-binding globulin elevation | Carbamazepine |
| Association with PCOS unknown mechanism | Valproic acid |
| SSRIs: Selective serotonin reuptake inhibitors | |
| PCOS: Polycystic ovary syndrome | |
CASE REPORT: NO PREGNANCY, NO PERIOD
Two years ago Ms. J, age 28, was diagnosed with depression. Her psychiatrist prescribed fluoxetine, 20 mg/d titrated across 4 weeks to 40 mg/d. About 4 months later, she experienced her first manic episode. The psychiatrist changed the diagnosis to bipolar I disorder and added risperidone, 2 mg/d, to manage her mania.
Ms. J’s bipolar disorder has been under control for 1 year, but she reports that her menstruation stopped 6 months ago. She is sexually active; she and her partner use spermicide-coated condoms. She does not want to be pregnant now but might want to bear a child within the next year. Several home pregnancy tests across 6 months were negative.
The patient is obese (5 feet, 5 inches, 186 lbs, body mass index 31) and has gained about 30 pounds during the past year. Vital signs are normal; psychiatric examination indicates normal mood and affect. Skin exam reveals mild papular acne on her face and back and increased hair growth on her chin. Other physical findings—including cardiac, lung, and neurologic examinations—are normal.
Laboratory evaluation reveals a prolactin level of 105 ng/mL, a negative serum ß-Hcg reading, and normal TSH, FSH, DHEA-S and testosterone levels.
Discussion. Ms. J’s history, physical examination, and laboratory tests suggest several possible causes of secondary amenorrhea:
- Are psychotropics or a prolactin-secreting tumor elevating her prolactin level?
- Does she have PCOS, as her weight gain, hirsutism, and acne might indicate?
- Is her bipolar disorder a factor? Consider psychiatric illness, medication side effects, and medical causes when evaluating secondary amenorrhea.
PSYCHIATRIC ILLNESS
Patients with high emotional stress may have amenorrhea or menstrual irregularities related to hypothalamic dysfunction.1 Also:
Anorexia nervosa has been shown to cause hypothalamic dysfunction, leading to amenorrhea.2 A correlation exists between weight loss and menses cessation, and between regain of weight and menses resumption.2
Depression. Estradiol levels are lower in depressed women than in euthymic women, probably because of altered hypothalamic-pituitary axis (HPA) function. Also, physical distress is correlated with menses disruption.3
In a 3-year study of women ages 36 to 45,4 those with a history of depression exhibited 1.2 times the rate of perimenopause as nondepressed women. Subjects with Hamilton Rating Scale for Depression scores >8 at enrollment had twice the rate of perimenopause after 3 years compared with nondepressed women. The findings suggest that depression might increase a woman’s risk of ceasing ovarian function in her 30s or 40s. Natural menopause on average begins at age 51.5
In another study,6 16 of 32 women with PCOS had Center for Epidemiological Studies-Depression Rating Scale scores indicating depression (≥16). The study suggests a high prevalence of depression among women with PCOS, but was limited by possible selection bias, no further diagnostic evaluation for depression, small sample size, and lack of an age-matched control group.
Bipolar disorder. High rates of menstrual disturbances have been reported among women with bipolar disorder.7 Although the mechanism has not been ascertained, disruption of HPA function similar to that seen in depression is likely.7
MEDICATIONS AND AMENORRHEA
Medications can cause amenorrhea, primarily through hyperprolactinemia—although other mechanisms may be involved (Table 1). Prolactin suppresses hypothalamic luteinizing hormone-releasing hormone (LHRH) production, leading to decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH), thus reducing circulating estrogen. Prolactin-secreting pituitary tumors and drug side effects mostly commonly cause hyperprolactinemia.
Antipsychotics. Phenothiazines such as chlorpromazine, butyrophenones such as haloperidol, and the atypical antipsychotic risperidone raise prolactin levels via dopamine-receptor antagonism.
Other atypical antipsychotics—including aripiprazole, clozapine, olanzapine, quetiapine, and ziprasidone—are associated with lower serum prolactin levels than risperidone.8,9 Preliminary studies suggest, for example, that switching patients from risperidone to quetiapine may help resume menstruation without worsening psychotic symptoms,10 and that amenorrhea often resolves after the patient is switched to another atypical antipsychotic.11
SSRIs. All selective serotonin reuptake inhibitors except sertraline are associated with hyperprolactinemia and can lead to amenorrhea in some patients.12
Table 2
Differential diagnosis of secondary amenorrhea
Ovarian causes
|
Hypothalamic causes
|
Hyperprolactinemia
|
Uterine causes
|
| * Turner’s syndrome: A rare chromosomal disorder characterized by short stature, lack of sexual development at puberty. |
| † Asherman’s syndrome: Endometrial adhesions, scar tissue that develop after uterine curettage or infections. |
Anticonvulsants used as mood stabilizers to treat bipolar disorder may cause menstrual irregularities, although most data relate to women with seizure disorders.
Valproic acid has been associated with PCOS in patients with epilepsy,13 although it is unknown whether the agent’s androgenizing effects vary with age. Carbamazepine, which increases sex hormone-binding globulin, may also lead to menstrual disorders by decreasing bioavailability of circulating estrogen.14 Consider switching a patient with disrupted menses to lithium, lamotrigine, or oxcarbazepine, which have not been associated with menstrual dysfunction.
MEDICAL CAUSES
Pregnancy is the most common cause of menses cessation, followed by ovarian, hypothalamic, pituitary, or uterine dysfunction (Table 2). Hypothalamic and pituitary dysfunction often cause amenorrhea in psychiatric patients, whereas ovarian causes are common among all patients with secondary amenorrhea.15
Ovarian. In PCOS, the ovaries and sometimes the adrenal glands produce excess androgens, leading to infrequent or light periods (oligomenorrhea) or amenorrhea.
Patients with depression are prone to ovarian failure in their 30s or 40s, possibly because of chronic HPA disruption.4 Premature ovarian failure also is common among patients with Turner’s syndrome, a rare chromosomal disorder characterized by short stature and lack of sexual development at puberty. Ovarian failure also can occur spontaneously.
Hypothalamic. Functional hypothalamic amenorrhea occurs in mood and eating disorders. Emotional stress, excessive physical exercise, and nutritional deficiencies reduce LHRH secretion by the hypothalamus, which interrupts the reproductive cycle. Cardiovascular disease, respiratory disease, cancer, and other acute and chronic medical illnesses can cause significant physiologic stress, thus leading to HPA dysfunction. Hypothalamic amenorrhea is treated by targeting the underlying psychiatric or medical condition.
Pituitary. Prolactin-secreting pituitary tumors, such as a pituitary adenoma, must be ruled out in patients whose prolactin levels remain high after a medication change.15 Hypothyroidism also can trigger hyperprolactinemia by causing pituitary gland hyperplasia.
Uterine. Women who have had uterine curettage or infections can develop adhesions and scar tissue that ablate the endometrial lining. This condition, called Asherman’s syndrome, is the most common uterine cause of menstrual disruption.
EVALUATING SECONDARY AMENORRHEA
When a patient presents with secondary amenorrhea, immediately rule out pregnancy because psychiatric disorders often are managed differently in pregnant than in nonpregnant women.16
Next, take a thorough patient history to determine whether referral is necessary. Ask about weight loss (intentional or unintentional), increased stressors, or a medical illness that may point to functional hypothalamic amenorrhea. Galactorrhea or vision changes—particularly loss of peripheral vision—could suggest a pituitary tumor. Skin changes, cold intolerance, fatigue, or constipation could indicate hypothyroidism.
Menopausal symptoms such as hot flashes and vaginal dryness could point to premature ovarian failure. Galactorrhea may indicate high prolactin levels. Obesity, hirsutism, or acne could point to PCOS. Consider Asherman’s syndrome in patients with endometritis or who have had a uterine dilation and curettage.
Laboratory testing. Once pregnancy is ruled out, measure prolactin. If it exceeds 25 ng/mL by 15 ng/mL or more, do a confirmative second prolactin test. If a patient is taking a prolactin-raising medication and her prolactin was not gauged before treatment, change to a prolactin-sparing agent, then measure her prolactin 2 weeks later.17
When to refer. If prolactin persistently exceeds 50 ng/mL even after changing medications, refer the patient for brain MRI to rule out a pituitary tumor.
Tests for other underlying medical causes of secondary amenorrhea—and when to perform them—are shown in the algorithm. Psychiatrists can give these tests or refer the patient to her primary care physician.
Algorithm Laboratory evaluation of secondary amenorrhea
Communication between care team members is key to determining treatment. If a medical problem arises during psychiatric treatment, call the patient’s primary care physician or send a letter describing the problem. Also send the referring physician available lab reports.
CASE CONTINUED: TREATMENT CHANGE
Ms. J’s psychiatrist tapered risperidone to 1 mg/d for 2 weeks, then switched to olanzapine, 5 mg/d. Three weeks later, her prolactin decreased to 25 ng/mL. She continued fluoxetine, 40 mg/d, and tolerated the change in antipsychotics.
Ms. J’s bipolar disorder remains well-controlled, but menses had not resumed for another 2 months, so the psychiatrist referred Ms. J back to her primary care physician. Androgenizing and pituitary tumors were ruled out based on normal TSH, prolactin, and testosterone levels. Ms. J was diagnosed as having PCOS based on her constellation of signs and symptoms. She was started on metformin, an insulin sensitizer used to treat PCOS, and was referred to a dietitian to help her lose weight.
One year later, Ms. J still struggles with weight control, but menstruation is back to normal.
Related resources
- Lean M, De Smedt G. Schizophrenia and osteoporosis. Int Clin Psychopharmacol 2004;19:31-5.
- Berga SL, Marcus MD, Loucks TL, et al. Recovery of ovarian activity in women with functional hypothalamic amenorrhea who were treated with cognitive behavior therapy. Fertil Steril 2003;80:976-81.
- Carr BR, Bradshaw KD. Disturbances of menstruation and other common gynecologic complaints in women. In: Braunwald E, Hauser SL, Fauci AS, et al (eds). Harrison’s principles of internal medicine(15th ed). New York: McGraw-Hill, 2001:Chapter 52.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Metformin • Glucovance, others
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kaplan JR, Manuck SB. Ovarian dysfunction, stress, and disease: a primate continuum. ILAR J 2004;45(2):89-115.
2. Mitan LA. Menstrual dysfunction in anorexia nervosa. J Pediatr Adolesc Gynecol 2004;17:81-85.
3. Young EA, Korzun A. The hypothalamic-pituitary-gonadal axis in mood disorders. Endocrinol Metab Clin North Am 2002;31(1):63-78.
4. Harlow BL, Wise LA, Otto MW, et al. Depression and its influence on reproductive endocrine and menstrual cycle markers associated with perimenopause. The Harvard Study of Moods and Cycles. Arch Gen Psychiatry 2003;60:29-36.
5. Brizendine L. Minding menopause. Psychotropics vs. estrogen? What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Rasgon NL, Rao RC, Hwang S, et al. Depression in women with polycystic ovary syndrome: clinical and biochemical correlates. J Affect Disord 2003;74(3):299-304.
7. Rasgon NL, Altshuler LL, Gundeman D, et al. Medication status and polycystic ovary syndrome in women with bipolar disorder: a preliminary report. J Clin Psychiatry 2000;61(3):173-8.
8. Kim KS, Pae CU, Chae JH, et al. Effects of olanzapine on prolactin levels of female patients with schizophrenia treated with risperidone. J Clin Psychiatry 2002;63(5):408-13.
9. Goodnick PJ, Rodriguez L, Santana O. Antipsychotics: impact on prolactin levels. Expert Opin Pharmacother 2002;3(10):1381-91.
10. Takahashi H, Higuchi H, Kamata M, et al. Effectiveness of switching to quetiapine for neuroleptic-induced amenorrhea. J Neuropsychiatry Clin Neurosci 2003;15:375-7.
11. Knegtering H, van der Moolen AE, Castelien S, et al. What are the effects of antipsychotics on sexual dysfunctions and endocrine functioning? Psychoneuroendocrinology 2003;28(suppl 2):109-23.
12. Goodnick PJ, Chaudry T, Artadi J, Arcey S. Women’s issues in mood disorders. Expert Opin Pharmacother 2000;1(5):903-16.
13. Isojarvi JI, Laatikainen TJ, Pakarinen AJ, et al. Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. N Engl J Med 1993;329(19):1383-8.
14. Isojarvi JI. Reproductive dysfunction in women with epilepsy. Neurology 2003;61(6 Suppl 2):S27-S34.
15. Reindollar RH, Novak M, Tho SP, McDonough PG. Adult-onset amenorrhea: a study of 262 patients. Am J Obstet Gynecol 1986;155(3):531-43.
16. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry 2003;2(7):14-26.
17. Barbieri RL. Etiology, diagnosis and treatment of secondary amenorrhea. UpToDate 2003;12:1.-
1. Kaplan JR, Manuck SB. Ovarian dysfunction, stress, and disease: a primate continuum. ILAR J 2004;45(2):89-115.
2. Mitan LA. Menstrual dysfunction in anorexia nervosa. J Pediatr Adolesc Gynecol 2004;17:81-85.
3. Young EA, Korzun A. The hypothalamic-pituitary-gonadal axis in mood disorders. Endocrinol Metab Clin North Am 2002;31(1):63-78.
4. Harlow BL, Wise LA, Otto MW, et al. Depression and its influence on reproductive endocrine and menstrual cycle markers associated with perimenopause. The Harvard Study of Moods and Cycles. Arch Gen Psychiatry 2003;60:29-36.
5. Brizendine L. Minding menopause. Psychotropics vs. estrogen? What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Rasgon NL, Rao RC, Hwang S, et al. Depression in women with polycystic ovary syndrome: clinical and biochemical correlates. J Affect Disord 2003;74(3):299-304.
7. Rasgon NL, Altshuler LL, Gundeman D, et al. Medication status and polycystic ovary syndrome in women with bipolar disorder: a preliminary report. J Clin Psychiatry 2000;61(3):173-8.
8. Kim KS, Pae CU, Chae JH, et al. Effects of olanzapine on prolactin levels of female patients with schizophrenia treated with risperidone. J Clin Psychiatry 2002;63(5):408-13.
9. Goodnick PJ, Rodriguez L, Santana O. Antipsychotics: impact on prolactin levels. Expert Opin Pharmacother 2002;3(10):1381-91.
10. Takahashi H, Higuchi H, Kamata M, et al. Effectiveness of switching to quetiapine for neuroleptic-induced amenorrhea. J Neuropsychiatry Clin Neurosci 2003;15:375-7.
11. Knegtering H, van der Moolen AE, Castelien S, et al. What are the effects of antipsychotics on sexual dysfunctions and endocrine functioning? Psychoneuroendocrinology 2003;28(suppl 2):109-23.
12. Goodnick PJ, Chaudry T, Artadi J, Arcey S. Women’s issues in mood disorders. Expert Opin Pharmacother 2000;1(5):903-16.
13. Isojarvi JI, Laatikainen TJ, Pakarinen AJ, et al. Polycystic ovaries and hyperandrogenism in women taking valproate for epilepsy. N Engl J Med 1993;329(19):1383-8.
14. Isojarvi JI. Reproductive dysfunction in women with epilepsy. Neurology 2003;61(6 Suppl 2):S27-S34.
15. Reindollar RH, Novak M, Tho SP, McDonough PG. Adult-onset amenorrhea: a study of 262 patients. Am J Obstet Gynecol 1986;155(3):531-43.
16. Altshuler L, Richards M, Yonkers K. Treating bipolar disorder during pregnancy. Current Psychiatry 2003;2(7):14-26.
17. Barbieri RL. Etiology, diagnosis and treatment of secondary amenorrhea. UpToDate 2003;12:1.-
Prudent prescribing: Intelligent use of lab tests and other diagnostics
Evidence that atypical antipsychotics can increase risk of diabetes and heart disease is changing psychiatry’s approach to laboratory testing. The need for careful psychotropic prescribing—with intelligent use of diagnostic testing—has been emphasized by:
- four medical associations recommending that physicians screen and monitor patients taking atypical antipsychotics.
- FDA requiring antipsychotic labeling to describe increased risk of hyperglycemia and diabetes
- medical malpractice lawyers using television and Internet ads to seek clients who might have developed diabetes while taking antipsychotics.
This article offers information you need to detect emerging metabolic problems in patients taking atypical antipsychotics. We also discuss five other clinical situations where laboratory testing can help you:
- rule out organic illness
- perform therapeutic drug monitoring
- protect the heart when prescribing
- watch for clozapine’s side effects
- monitor for substance abuse.
Table 1
Lab testing with atypical antipsychotics*
Obtain baseline values before or as soon as possible after starting the antipsychotic:
Also note patient/family histories of obesity, diabetes, hypertension, hyperlipidemia, heart disease† |
Repeat diabetes monitoring with fasting blood glucose and/or Hb A1c after 3 months of treatment, then at least annually. More-frequent monitoring (quarterly or monthly) may be indicated for patients with:
|
Consider switching to a medication with less weight-gain liability‡ for patients:
|
| Identify patients with metabolic syndrome,§ and ensure that they are carefully monitored by a primary care clinician. Check weight (with BMI) monthly for all patients for the first 6 months, then every 3 months thereafter |
| Repeat fasting lipid profile after 3 months, then every 2 years if serum lipids are normal or every 6 months in consultation with primary care clinician if LDL >130 mg/dL |
| * Individualize to particular patients’ needs. |
| † Patients with schizophrenia are at increased risk of coronary heart disease. |
| ‡ Weight gain liability = clozapine, olanzapine > risperidone, quetiapine > aripiprazole, ziprasidone |
| § Metabolic syndrome: A proinflammatory, prothrombotic state described by a cluster of abnormalities including abdominal obesity, hypertriglyceridemia, insulin resistance, hypertension, and low HDL cholesterol. Can be exacerbated by atypical antipsychotics. |
| Source: Adapted from reference 3. |
DIABETES RISK
New monitoring standards. The American Psychiatric Association set a new standard of care by collaborating with the American Diabetes Association and others in recommending how to manage the potential for increased risk of obesity, diabetes, and lipid disorders when using atypical antipsychotics.2 The February 2004 APA/ADA report cites olanzapine and clozapine as the atypicals most likely to cause metabolic changes that increase heart disease risk. It also notes, however, that atypicals’ potential benefits to certain patients outweigh the risks.
Because of this report, psychiatrists who prescribe atypicals are now obligated to document baseline lab values and monitor patients for potential side effects (Table 1).1 We recommend that you also note patient race, as certain ethnic populations (such as African-American, Hispanic, Native American, Asian, Pacific Islander) are at elevated risk for diabetes.
Determining BMI. When starting patients on atypical antipsychotics, calculate baseline body mass index (BMI) with the simple formula in Table 2 or by using BMI tables (see Related resources).4 Determine BMI before starting a new atypical antipsychotic, at every visit for the first 6 months, and then quarterly when the dosage is stable.
A BMI increase of 1 unit warrants medical intervention, including increased weight monitoring and placing the patient in a weight-management program and switching to another antipsychotic.3
Table 2 An easy formula to calculate body mass index (BMI)

The increasing incidence of diabetes in the U.S. population makes it difficult to assess the relationship between atypical antipsychotic use and blood glucose abnormalities. Moreover, the risk of diabetes may be elevated in patients with schizophrenia, whether or not they are receiving medications. Diabetes and disturbed carbohydrate metabolism may be an integral component of schizophrenia itself.1
RULING OUT ORGANIC ILLNESS
A classic role of laboratory and diagnostic testing in psychiatry is to exclude organic illness that may be causing or exacerbating psychiatric symptoms. For a patient presenting with serious psychiatric symptoms, most sources recommend a standard battery of screening tests (Table 3).
Of course, the DSM-IV-TR “mental disorder due to a general medical condition” should be included in the differential diagnosis of any psychiatric presentation. DSM-IV-TR also calls for disease-specific tests, such as polysomnography in certain sleep disorders, CT for enlarged ventricles in schizophrenia, and electrolyte analysis in patients with anorexia nervosa.5 Order other tests as indicated, depending on patients’ medical conditions.
THERAPEUTIC DRUG MONITORING
Therapeutic drug monitoring (TDM) is used to optimize treatment with medications for which therapeutic blood levels for psychiatric disorders have been described.6 These include lithium, valproate, carbamazepine, clozapine, and tricyclic antidepressants.
Keep in mind that “therapeutic” blood levels have been determined in “usual” patients in controlled clinical trials and may not apply to the many “unusual” patients who metabolize drugs differently because of genetic variation, age, and concomitant diseases, diet, or medications.7
Lithium. A therapeutic blood level is typically 0.6 to 1.2 mEq/L, and—although the dosage must be individualized—900 to 1,200 mg/d in divided doses usually maintains this blood level. Lower levels between 0.4 mEq/L and 0.8 mEq/L have been described for the elderly.8
In uncomplicated cases, monitor lithium levels at least every 2 months during maintenance therapy. Draw blood immediately before a scheduled dose—such as 8 to 12 hours after the previous dose—when lithium concentrations are relatively stable.
Consider both clinical signs and serum levels when dosing, as patients unusually sensitive to lithium may exhibit toxic signs at <1.0 mEq/L. Elderly patients often respond to reduced dosages and may exhibit signs of toxicity—such as gastric upset and confusion—at serum levels most younger patients can tolerate.
Valproate. For seizure and bipolar disorders, the therapeutic blood level is 50 to 100 mcg/mL. Potential hematologic complications include thrombocytopenia; indigestion and nausea are common side effects. Typical practice is to obtain levels weekly for the first few weeks and then quarterly thereafter.
Carbamazepine. Plasma carbamazepine concentrations have not been correlated with response in bipolar disorder but are measured to prevent or identify toxicity. Dosages of 600 to 1,200 mg/d usually produce nontoxic levels of 4 to 12 mcg/mL. Carbamazepine interacts with many drugs that affect or are affected by hepatic metabolism. Blood dyscrasias including aplastic anemia are rare side effects.
Clozapine. Consensus is lacking on the optimal clozapine plasma level needed to achieve a therapeutic response. For some patients, it may be 200 to 350 ng/mL, which usually corresponds to 200 to 400 mg/d. Dosing must be individualized, however, because clozapine levels can vary almost 50-fold among patients taking the same dosage.9 Other studies10 and at least one recent textbook11 have reported therapeutic response most associated with clozapine levels >350 ng/mL, although adverse effects may be more likely at this higher dosage.
PROTECTING THE HEART
Before you prescribe any psychotropic with potential cardiotoxic effects, we recommend a baseline ECG for patients with cardiac risk factors, including:
- history of heart disease or ECG abnormalities
- history of syncope
- family history of sudden death before age 40, especially if both parents had sudden death
- history of prolonged QTc interval, such as congenital long QT syndrome.
Cardiotoxic effects such as QTc interval prolongation and torsades de pointes have been associated with thioridazine, mesoridazine, and pimozide. On ECG, a QTc interval >500 msec suggests an increased risk of potentially fatal arrhythmias. Do not prescribe medications associated with QTc interval prolongation to patients with this ECG finding.
Table 3
Screening tests most sources recommend for psychiatric practice
| Blood |
| Complete blood count (CBC) |
| Serum chemistry panel (“CHEM-20,” including liver function tests) |
| Lipid panels |
| Thyroid function tests (TFTs, TSH) |
| Screening tests for HIV, hepatitis C, syphilis |
| Serum B12 |
| Pregnancy tests in women of childbearing age and potential |
| Blood alcohol level in alcohol-intoxicated patient |
| Urine |
| Urine drug toxicology screen for substance abuse |
| Urinalysis |
| Cardiac |
| ECG |
| Imaging |
| Brain CT or MRI (preferred) if clinically indicated* |
| Chest radiography |
| Others |
| Serum medication levels† |
| Erythrocyte sedimentation rate or urine heavy metal screen, as indicated by medical history |
| Erythrocyte uroporphyrinogen-1-synthase |
| Urine uroporphyrins |
| EEG |
| Skull radiography |
| * Such as patient with disorientation, confusion, or abnormal neurologic exam |
| † When therapeutic/toxic blood levels are available for patient’s medications, such as theophylline, tricyclics, digoxin |
ECG is also indicated in patients who experience symptoms associated with a prolonged QT interval—such as dizziness or syncope—while taking antipsychotics. If ziprasidone is prescribed for patients with any of the risk factors described above, we recommend a baseline ECG before treatment begins, with a follow-up ECG if the patient experiences dizziness or syncope.4
Table 4
Screening tests for a patient beginning substance abuse treatment
|
WHEN USING CLOZAPINE
Clozapine is the only antipsychotic shown to improve neuroleptic-resistant symptoms12 and reduce suicidality13 in patients with schizophrenia. Unfortunately, clozapine’s potential side effects—including potentially life-threatening agranulocytosis—are legion, but careful monitoring with necessary lab testing can allow its benefits to outweigh the risks.
Agranulocytosis. Obtain white blood cell (WBC) count and differential at baseline, during treatment, and for 4 weeks after discontinuing clozapine, following the distribution program’s required schedule. Advise patients to immediately report flu-like complaints or signs that might suggest infection, such as lethargy, weakness, fever, sore throat, malaise, or mucous membrane ulceration.
Eosinophilia. In clinical trials, 1% of patients developed eosinophilia, which can be substantial in rare cases. If a differential count reveals a total eosinophil count >4,000/mm3 , stop clozapine therapy until the eosinophil count falls below 3,000/mm3 .
Myocarditis. Clozapine-treated patients are at much greater risk for developing myocarditis and of dying from it—especially during the first 6 weeks of therapy—than is the general population.3 Tachycardia can be a presenting sign.
Abnormal laboratory findings associated with clozapine-induced myocarditis may include increased WBC count, eosinophilia, increased erythrocyte sedimentation rate, and increased cardiac enzyme levels and plasma troponin. Because the mortality rate of clozapine-induced myocarditis approaches 40%, stop clozapine and refer the patient for medical evaluation as soon as possible when you suspect myocarditis.3
Endocrine and hepatic effects. Severe hyperglycemia, sometimes leading to ketoacidosis, can occur during clozapine treatment in patients without a history of hyperglycemia. Ketoacidosis symptoms include rapid breathing, nausea, vomiting, clouding of sensorium (even coma), weight loss, polyuria, polydipsia, and dehydration. Monitoring for blood glucose changes, as described in Table 1, is recommended with clozapine as with all other atypical antipsychotics.
Hepatitis during clozapine therapy has been reported in patients with baseline normal or preexisting abnormal liver function. After baseline liver function tests, we suggest follow-up LFTs:
- annually for patients with normal baseline values
- every 6 months for patients with minimally abnormal values
- every 3 months for patients with liver disease.
MONITORING SUBSTANCE ABUSE
Substance abuse is often associated with medical comorbidities that require laboratory workup and monitoring. These include overdose sequelae, sexual assault, cirrhosis, endocarditis, HIV infection, viral hepatitis, tuberculosis, and syphilis. Some testing is mandated by federal law for patients in methadone maintenance or opioid agonist therapy programs with methadone.
We recommend that new patients with substance abuse be screened for organic illness as described above, plus the workup in Table 4. Also gather a careful history for hepatitis, pancreatitis, diabetes, cirrhosis, unusual infections (cellulitis, endocarditis, atypical pneumonias, HIV), frequent hospitalizations, falls, injuries, and blackouts.
Obtain a blood alcohol level in alcohol-intoxicated patients and urine toxicology to screen for locally-available street drugs (typically marijuana, sedative/hypnotics, amphetamines, cocaine, opiates, and phencyclidine).
Confer with your laboratory staff about the capabilities and sensitivities of their drug testing methods. Marijuana may be detected for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for up to 2 to 4 days in urine.
- Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock B, Kaplan HI (eds). Comprehensive textbook of psychiatry, 6th ed. Baltimore: Lippincott, Williams & Wilkins 1995:601-19.
- National Institute of Diabetes and Digestive and Kidney Diseases. Body mass index tables. www.niddk.nih.gov/health/nutrit/pubs/statobes.htm#table.
- National Cholesterol Education Program. Guidelines for managing patients at risk for coronary heart disease. www.nhlbi.nih.gov/about/ncep/
- Food and Drug Administration. Guidelines on hyperglycemia associated with atypical antipsychotics [example]. www.fda.gov/medwatch/SAFETY/2004/Clozaril-deardoc.pdf
Drug brand names
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Lithium • Lithobid, others
- Mesoridazine • Serentil
- Pimozide • Orap
- Thioridazine • Mellaril
- Valproate • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry 2004;47(Apr):S64-S66.
2. Association of Clinical Endocrinologists, North merican Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
3. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-49.
4. Work Group on Schizophrenia. Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004, 161:2(suppl). For BMI information related to this guideline, see http://www.cdc.gov/nccdphp/dnpa/bmi/bmi-adult-formula.htm.
5. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., text rev). Washington, DC: American Psychiatric Association, 2000.
6. Preskorn SH, Fast GA. Therapeutic drug monitoring for antidepressants: efficacy, safety, and cost effectiveness. J Clin Psychiatry 1991;(52 suppl):23-33.
7. Preskorn SH. Why patients may not respond to usual recommended dosages: 3 variables to consider when prescribing antipsychotics [commentary]. Current Psychiatry 2004;3(8):38-43.
8. Price DG, Ghaemi SN. Lithium. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004:355.
9. Kronig MH, Munne RA, Szymanski S, et al. Plasma clozapine levels and clinical response for treatment-refractory schizophrenic patients. Am J Psychiatry 1995;152(2):179-82.
10. Schulte P. What is an adequate trial with clozapine? Therapeutic drug monitoring and time to response in treatment-refractory schizophrenia. Clin Pharmacokinet 2003;42(7):607-18.
11. Henderson DC, Kunkel L, Goff DC. Antipsychotic drugs. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004;338-9.
12. Meltzer HY, Alphs L, Green AI, et al. International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT). Arch Gen Psychiatry 2003;60(1):82-91.
13. Meltzer HY. Suicide in schizophrenia: risk factors and clozapine treatment. J Clin Psychiatry. 1998;59(suppl 3):15-20.
Evidence that atypical antipsychotics can increase risk of diabetes and heart disease is changing psychiatry’s approach to laboratory testing. The need for careful psychotropic prescribing—with intelligent use of diagnostic testing—has been emphasized by:
- four medical associations recommending that physicians screen and monitor patients taking atypical antipsychotics.
- FDA requiring antipsychotic labeling to describe increased risk of hyperglycemia and diabetes
- medical malpractice lawyers using television and Internet ads to seek clients who might have developed diabetes while taking antipsychotics.
This article offers information you need to detect emerging metabolic problems in patients taking atypical antipsychotics. We also discuss five other clinical situations where laboratory testing can help you:
- rule out organic illness
- perform therapeutic drug monitoring
- protect the heart when prescribing
- watch for clozapine’s side effects
- monitor for substance abuse.
Table 1
Lab testing with atypical antipsychotics*
Obtain baseline values before or as soon as possible after starting the antipsychotic:
Also note patient/family histories of obesity, diabetes, hypertension, hyperlipidemia, heart disease† |
Repeat diabetes monitoring with fasting blood glucose and/or Hb A1c after 3 months of treatment, then at least annually. More-frequent monitoring (quarterly or monthly) may be indicated for patients with:
|
Consider switching to a medication with less weight-gain liability‡ for patients:
|
| Identify patients with metabolic syndrome,§ and ensure that they are carefully monitored by a primary care clinician. Check weight (with BMI) monthly for all patients for the first 6 months, then every 3 months thereafter |
| Repeat fasting lipid profile after 3 months, then every 2 years if serum lipids are normal or every 6 months in consultation with primary care clinician if LDL >130 mg/dL |
| * Individualize to particular patients’ needs. |
| † Patients with schizophrenia are at increased risk of coronary heart disease. |
| ‡ Weight gain liability = clozapine, olanzapine > risperidone, quetiapine > aripiprazole, ziprasidone |
| § Metabolic syndrome: A proinflammatory, prothrombotic state described by a cluster of abnormalities including abdominal obesity, hypertriglyceridemia, insulin resistance, hypertension, and low HDL cholesterol. Can be exacerbated by atypical antipsychotics. |
| Source: Adapted from reference 3. |
DIABETES RISK
New monitoring standards. The American Psychiatric Association set a new standard of care by collaborating with the American Diabetes Association and others in recommending how to manage the potential for increased risk of obesity, diabetes, and lipid disorders when using atypical antipsychotics.2 The February 2004 APA/ADA report cites olanzapine and clozapine as the atypicals most likely to cause metabolic changes that increase heart disease risk. It also notes, however, that atypicals’ potential benefits to certain patients outweigh the risks.
Because of this report, psychiatrists who prescribe atypicals are now obligated to document baseline lab values and monitor patients for potential side effects (Table 1).1 We recommend that you also note patient race, as certain ethnic populations (such as African-American, Hispanic, Native American, Asian, Pacific Islander) are at elevated risk for diabetes.
Determining BMI. When starting patients on atypical antipsychotics, calculate baseline body mass index (BMI) with the simple formula in Table 2 or by using BMI tables (see Related resources).4 Determine BMI before starting a new atypical antipsychotic, at every visit for the first 6 months, and then quarterly when the dosage is stable.
A BMI increase of 1 unit warrants medical intervention, including increased weight monitoring and placing the patient in a weight-management program and switching to another antipsychotic.3
Table 2 An easy formula to calculate body mass index (BMI)
The increasing incidence of diabetes in the U.S. population makes it difficult to assess the relationship between atypical antipsychotic use and blood glucose abnormalities. Moreover, the risk of diabetes may be elevated in patients with schizophrenia, whether or not they are receiving medications. Diabetes and disturbed carbohydrate metabolism may be an integral component of schizophrenia itself.1
RULING OUT ORGANIC ILLNESS
A classic role of laboratory and diagnostic testing in psychiatry is to exclude organic illness that may be causing or exacerbating psychiatric symptoms. For a patient presenting with serious psychiatric symptoms, most sources recommend a standard battery of screening tests (Table 3).
Of course, the DSM-IV-TR “mental disorder due to a general medical condition” should be included in the differential diagnosis of any psychiatric presentation. DSM-IV-TR also calls for disease-specific tests, such as polysomnography in certain sleep disorders, CT for enlarged ventricles in schizophrenia, and electrolyte analysis in patients with anorexia nervosa.5 Order other tests as indicated, depending on patients’ medical conditions.
THERAPEUTIC DRUG MONITORING
Therapeutic drug monitoring (TDM) is used to optimize treatment with medications for which therapeutic blood levels for psychiatric disorders have been described.6 These include lithium, valproate, carbamazepine, clozapine, and tricyclic antidepressants.
Keep in mind that “therapeutic” blood levels have been determined in “usual” patients in controlled clinical trials and may not apply to the many “unusual” patients who metabolize drugs differently because of genetic variation, age, and concomitant diseases, diet, or medications.7
Lithium. A therapeutic blood level is typically 0.6 to 1.2 mEq/L, and—although the dosage must be individualized—900 to 1,200 mg/d in divided doses usually maintains this blood level. Lower levels between 0.4 mEq/L and 0.8 mEq/L have been described for the elderly.8
In uncomplicated cases, monitor lithium levels at least every 2 months during maintenance therapy. Draw blood immediately before a scheduled dose—such as 8 to 12 hours after the previous dose—when lithium concentrations are relatively stable.
Consider both clinical signs and serum levels when dosing, as patients unusually sensitive to lithium may exhibit toxic signs at <1.0 mEq/L. Elderly patients often respond to reduced dosages and may exhibit signs of toxicity—such as gastric upset and confusion—at serum levels most younger patients can tolerate.
Valproate. For seizure and bipolar disorders, the therapeutic blood level is 50 to 100 mcg/mL. Potential hematologic complications include thrombocytopenia; indigestion and nausea are common side effects. Typical practice is to obtain levels weekly for the first few weeks and then quarterly thereafter.
Carbamazepine. Plasma carbamazepine concentrations have not been correlated with response in bipolar disorder but are measured to prevent or identify toxicity. Dosages of 600 to 1,200 mg/d usually produce nontoxic levels of 4 to 12 mcg/mL. Carbamazepine interacts with many drugs that affect or are affected by hepatic metabolism. Blood dyscrasias including aplastic anemia are rare side effects.
Clozapine. Consensus is lacking on the optimal clozapine plasma level needed to achieve a therapeutic response. For some patients, it may be 200 to 350 ng/mL, which usually corresponds to 200 to 400 mg/d. Dosing must be individualized, however, because clozapine levels can vary almost 50-fold among patients taking the same dosage.9 Other studies10 and at least one recent textbook11 have reported therapeutic response most associated with clozapine levels >350 ng/mL, although adverse effects may be more likely at this higher dosage.
PROTECTING THE HEART
Before you prescribe any psychotropic with potential cardiotoxic effects, we recommend a baseline ECG for patients with cardiac risk factors, including:
- history of heart disease or ECG abnormalities
- history of syncope
- family history of sudden death before age 40, especially if both parents had sudden death
- history of prolonged QTc interval, such as congenital long QT syndrome.
Cardiotoxic effects such as QTc interval prolongation and torsades de pointes have been associated with thioridazine, mesoridazine, and pimozide. On ECG, a QTc interval >500 msec suggests an increased risk of potentially fatal arrhythmias. Do not prescribe medications associated with QTc interval prolongation to patients with this ECG finding.
Table 3
Screening tests most sources recommend for psychiatric practice
| Blood |
| Complete blood count (CBC) |
| Serum chemistry panel (“CHEM-20,” including liver function tests) |
| Lipid panels |
| Thyroid function tests (TFTs, TSH) |
| Screening tests for HIV, hepatitis C, syphilis |
| Serum B12 |
| Pregnancy tests in women of childbearing age and potential |
| Blood alcohol level in alcohol-intoxicated patient |
| Urine |
| Urine drug toxicology screen for substance abuse |
| Urinalysis |
| Cardiac |
| ECG |
| Imaging |
| Brain CT or MRI (preferred) if clinically indicated* |
| Chest radiography |
| Others |
| Serum medication levels† |
| Erythrocyte sedimentation rate or urine heavy metal screen, as indicated by medical history |
| Erythrocyte uroporphyrinogen-1-synthase |
| Urine uroporphyrins |
| EEG |
| Skull radiography |
| * Such as patient with disorientation, confusion, or abnormal neurologic exam |
| † When therapeutic/toxic blood levels are available for patient’s medications, such as theophylline, tricyclics, digoxin |
ECG is also indicated in patients who experience symptoms associated with a prolonged QT interval—such as dizziness or syncope—while taking antipsychotics. If ziprasidone is prescribed for patients with any of the risk factors described above, we recommend a baseline ECG before treatment begins, with a follow-up ECG if the patient experiences dizziness or syncope.4
Table 4
Screening tests for a patient beginning substance abuse treatment
|
WHEN USING CLOZAPINE
Clozapine is the only antipsychotic shown to improve neuroleptic-resistant symptoms12 and reduce suicidality13 in patients with schizophrenia. Unfortunately, clozapine’s potential side effects—including potentially life-threatening agranulocytosis—are legion, but careful monitoring with necessary lab testing can allow its benefits to outweigh the risks.
Agranulocytosis. Obtain white blood cell (WBC) count and differential at baseline, during treatment, and for 4 weeks after discontinuing clozapine, following the distribution program’s required schedule. Advise patients to immediately report flu-like complaints or signs that might suggest infection, such as lethargy, weakness, fever, sore throat, malaise, or mucous membrane ulceration.
Eosinophilia. In clinical trials, 1% of patients developed eosinophilia, which can be substantial in rare cases. If a differential count reveals a total eosinophil count >4,000/mm3 , stop clozapine therapy until the eosinophil count falls below 3,000/mm3 .
Myocarditis. Clozapine-treated patients are at much greater risk for developing myocarditis and of dying from it—especially during the first 6 weeks of therapy—than is the general population.3 Tachycardia can be a presenting sign.
Abnormal laboratory findings associated with clozapine-induced myocarditis may include increased WBC count, eosinophilia, increased erythrocyte sedimentation rate, and increased cardiac enzyme levels and plasma troponin. Because the mortality rate of clozapine-induced myocarditis approaches 40%, stop clozapine and refer the patient for medical evaluation as soon as possible when you suspect myocarditis.3
Endocrine and hepatic effects. Severe hyperglycemia, sometimes leading to ketoacidosis, can occur during clozapine treatment in patients without a history of hyperglycemia. Ketoacidosis symptoms include rapid breathing, nausea, vomiting, clouding of sensorium (even coma), weight loss, polyuria, polydipsia, and dehydration. Monitoring for blood glucose changes, as described in Table 1, is recommended with clozapine as with all other atypical antipsychotics.
Hepatitis during clozapine therapy has been reported in patients with baseline normal or preexisting abnormal liver function. After baseline liver function tests, we suggest follow-up LFTs:
- annually for patients with normal baseline values
- every 6 months for patients with minimally abnormal values
- every 3 months for patients with liver disease.
MONITORING SUBSTANCE ABUSE
Substance abuse is often associated with medical comorbidities that require laboratory workup and monitoring. These include overdose sequelae, sexual assault, cirrhosis, endocarditis, HIV infection, viral hepatitis, tuberculosis, and syphilis. Some testing is mandated by federal law for patients in methadone maintenance or opioid agonist therapy programs with methadone.
We recommend that new patients with substance abuse be screened for organic illness as described above, plus the workup in Table 4. Also gather a careful history for hepatitis, pancreatitis, diabetes, cirrhosis, unusual infections (cellulitis, endocarditis, atypical pneumonias, HIV), frequent hospitalizations, falls, injuries, and blackouts.
Obtain a blood alcohol level in alcohol-intoxicated patients and urine toxicology to screen for locally-available street drugs (typically marijuana, sedative/hypnotics, amphetamines, cocaine, opiates, and phencyclidine).
Confer with your laboratory staff about the capabilities and sensitivities of their drug testing methods. Marijuana may be detected for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for up to 2 to 4 days in urine.
- Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock B, Kaplan HI (eds). Comprehensive textbook of psychiatry, 6th ed. Baltimore: Lippincott, Williams & Wilkins 1995:601-19.
- National Institute of Diabetes and Digestive and Kidney Diseases. Body mass index tables. www.niddk.nih.gov/health/nutrit/pubs/statobes.htm#table.
- National Cholesterol Education Program. Guidelines for managing patients at risk for coronary heart disease. www.nhlbi.nih.gov/about/ncep/
- Food and Drug Administration. Guidelines on hyperglycemia associated with atypical antipsychotics [example]. www.fda.gov/medwatch/SAFETY/2004/Clozaril-deardoc.pdf
Drug brand names
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Lithium • Lithobid, others
- Mesoridazine • Serentil
- Pimozide • Orap
- Thioridazine • Mellaril
- Valproate • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Evidence that atypical antipsychotics can increase risk of diabetes and heart disease is changing psychiatry’s approach to laboratory testing. The need for careful psychotropic prescribing—with intelligent use of diagnostic testing—has been emphasized by:
- four medical associations recommending that physicians screen and monitor patients taking atypical antipsychotics.
- FDA requiring antipsychotic labeling to describe increased risk of hyperglycemia and diabetes
- medical malpractice lawyers using television and Internet ads to seek clients who might have developed diabetes while taking antipsychotics.
This article offers information you need to detect emerging metabolic problems in patients taking atypical antipsychotics. We also discuss five other clinical situations where laboratory testing can help you:
- rule out organic illness
- perform therapeutic drug monitoring
- protect the heart when prescribing
- watch for clozapine’s side effects
- monitor for substance abuse.
Table 1
Lab testing with atypical antipsychotics*
Obtain baseline values before or as soon as possible after starting the antipsychotic:
Also note patient/family histories of obesity, diabetes, hypertension, hyperlipidemia, heart disease† |
Repeat diabetes monitoring with fasting blood glucose and/or Hb A1c after 3 months of treatment, then at least annually. More-frequent monitoring (quarterly or monthly) may be indicated for patients with:
|
Consider switching to a medication with less weight-gain liability‡ for patients:
|
| Identify patients with metabolic syndrome,§ and ensure that they are carefully monitored by a primary care clinician. Check weight (with BMI) monthly for all patients for the first 6 months, then every 3 months thereafter |
| Repeat fasting lipid profile after 3 months, then every 2 years if serum lipids are normal or every 6 months in consultation with primary care clinician if LDL >130 mg/dL |
| * Individualize to particular patients’ needs. |
| † Patients with schizophrenia are at increased risk of coronary heart disease. |
| ‡ Weight gain liability = clozapine, olanzapine > risperidone, quetiapine > aripiprazole, ziprasidone |
| § Metabolic syndrome: A proinflammatory, prothrombotic state described by a cluster of abnormalities including abdominal obesity, hypertriglyceridemia, insulin resistance, hypertension, and low HDL cholesterol. Can be exacerbated by atypical antipsychotics. |
| Source: Adapted from reference 3. |
DIABETES RISK
New monitoring standards. The American Psychiatric Association set a new standard of care by collaborating with the American Diabetes Association and others in recommending how to manage the potential for increased risk of obesity, diabetes, and lipid disorders when using atypical antipsychotics.2 The February 2004 APA/ADA report cites olanzapine and clozapine as the atypicals most likely to cause metabolic changes that increase heart disease risk. It also notes, however, that atypicals’ potential benefits to certain patients outweigh the risks.
Because of this report, psychiatrists who prescribe atypicals are now obligated to document baseline lab values and monitor patients for potential side effects (Table 1).1 We recommend that you also note patient race, as certain ethnic populations (such as African-American, Hispanic, Native American, Asian, Pacific Islander) are at elevated risk for diabetes.
Determining BMI. When starting patients on atypical antipsychotics, calculate baseline body mass index (BMI) with the simple formula in Table 2 or by using BMI tables (see Related resources).4 Determine BMI before starting a new atypical antipsychotic, at every visit for the first 6 months, and then quarterly when the dosage is stable.
A BMI increase of 1 unit warrants medical intervention, including increased weight monitoring and placing the patient in a weight-management program and switching to another antipsychotic.3
Table 2 An easy formula to calculate body mass index (BMI)
The increasing incidence of diabetes in the U.S. population makes it difficult to assess the relationship between atypical antipsychotic use and blood glucose abnormalities. Moreover, the risk of diabetes may be elevated in patients with schizophrenia, whether or not they are receiving medications. Diabetes and disturbed carbohydrate metabolism may be an integral component of schizophrenia itself.1
RULING OUT ORGANIC ILLNESS
A classic role of laboratory and diagnostic testing in psychiatry is to exclude organic illness that may be causing or exacerbating psychiatric symptoms. For a patient presenting with serious psychiatric symptoms, most sources recommend a standard battery of screening tests (Table 3).
Of course, the DSM-IV-TR “mental disorder due to a general medical condition” should be included in the differential diagnosis of any psychiatric presentation. DSM-IV-TR also calls for disease-specific tests, such as polysomnography in certain sleep disorders, CT for enlarged ventricles in schizophrenia, and electrolyte analysis in patients with anorexia nervosa.5 Order other tests as indicated, depending on patients’ medical conditions.
THERAPEUTIC DRUG MONITORING
Therapeutic drug monitoring (TDM) is used to optimize treatment with medications for which therapeutic blood levels for psychiatric disorders have been described.6 These include lithium, valproate, carbamazepine, clozapine, and tricyclic antidepressants.
Keep in mind that “therapeutic” blood levels have been determined in “usual” patients in controlled clinical trials and may not apply to the many “unusual” patients who metabolize drugs differently because of genetic variation, age, and concomitant diseases, diet, or medications.7
Lithium. A therapeutic blood level is typically 0.6 to 1.2 mEq/L, and—although the dosage must be individualized—900 to 1,200 mg/d in divided doses usually maintains this blood level. Lower levels between 0.4 mEq/L and 0.8 mEq/L have been described for the elderly.8
In uncomplicated cases, monitor lithium levels at least every 2 months during maintenance therapy. Draw blood immediately before a scheduled dose—such as 8 to 12 hours after the previous dose—when lithium concentrations are relatively stable.
Consider both clinical signs and serum levels when dosing, as patients unusually sensitive to lithium may exhibit toxic signs at <1.0 mEq/L. Elderly patients often respond to reduced dosages and may exhibit signs of toxicity—such as gastric upset and confusion—at serum levels most younger patients can tolerate.
Valproate. For seizure and bipolar disorders, the therapeutic blood level is 50 to 100 mcg/mL. Potential hematologic complications include thrombocytopenia; indigestion and nausea are common side effects. Typical practice is to obtain levels weekly for the first few weeks and then quarterly thereafter.
Carbamazepine. Plasma carbamazepine concentrations have not been correlated with response in bipolar disorder but are measured to prevent or identify toxicity. Dosages of 600 to 1,200 mg/d usually produce nontoxic levels of 4 to 12 mcg/mL. Carbamazepine interacts with many drugs that affect or are affected by hepatic metabolism. Blood dyscrasias including aplastic anemia are rare side effects.
Clozapine. Consensus is lacking on the optimal clozapine plasma level needed to achieve a therapeutic response. For some patients, it may be 200 to 350 ng/mL, which usually corresponds to 200 to 400 mg/d. Dosing must be individualized, however, because clozapine levels can vary almost 50-fold among patients taking the same dosage.9 Other studies10 and at least one recent textbook11 have reported therapeutic response most associated with clozapine levels >350 ng/mL, although adverse effects may be more likely at this higher dosage.
PROTECTING THE HEART
Before you prescribe any psychotropic with potential cardiotoxic effects, we recommend a baseline ECG for patients with cardiac risk factors, including:
- history of heart disease or ECG abnormalities
- history of syncope
- family history of sudden death before age 40, especially if both parents had sudden death
- history of prolonged QTc interval, such as congenital long QT syndrome.
Cardiotoxic effects such as QTc interval prolongation and torsades de pointes have been associated with thioridazine, mesoridazine, and pimozide. On ECG, a QTc interval >500 msec suggests an increased risk of potentially fatal arrhythmias. Do not prescribe medications associated with QTc interval prolongation to patients with this ECG finding.
Table 3
Screening tests most sources recommend for psychiatric practice
| Blood |
| Complete blood count (CBC) |
| Serum chemistry panel (“CHEM-20,” including liver function tests) |
| Lipid panels |
| Thyroid function tests (TFTs, TSH) |
| Screening tests for HIV, hepatitis C, syphilis |
| Serum B12 |
| Pregnancy tests in women of childbearing age and potential |
| Blood alcohol level in alcohol-intoxicated patient |
| Urine |
| Urine drug toxicology screen for substance abuse |
| Urinalysis |
| Cardiac |
| ECG |
| Imaging |
| Brain CT or MRI (preferred) if clinically indicated* |
| Chest radiography |
| Others |
| Serum medication levels† |
| Erythrocyte sedimentation rate or urine heavy metal screen, as indicated by medical history |
| Erythrocyte uroporphyrinogen-1-synthase |
| Urine uroporphyrins |
| EEG |
| Skull radiography |
| * Such as patient with disorientation, confusion, or abnormal neurologic exam |
| † When therapeutic/toxic blood levels are available for patient’s medications, such as theophylline, tricyclics, digoxin |
ECG is also indicated in patients who experience symptoms associated with a prolonged QT interval—such as dizziness or syncope—while taking antipsychotics. If ziprasidone is prescribed for patients with any of the risk factors described above, we recommend a baseline ECG before treatment begins, with a follow-up ECG if the patient experiences dizziness or syncope.4
Table 4
Screening tests for a patient beginning substance abuse treatment
|
WHEN USING CLOZAPINE
Clozapine is the only antipsychotic shown to improve neuroleptic-resistant symptoms12 and reduce suicidality13 in patients with schizophrenia. Unfortunately, clozapine’s potential side effects—including potentially life-threatening agranulocytosis—are legion, but careful monitoring with necessary lab testing can allow its benefits to outweigh the risks.
Agranulocytosis. Obtain white blood cell (WBC) count and differential at baseline, during treatment, and for 4 weeks after discontinuing clozapine, following the distribution program’s required schedule. Advise patients to immediately report flu-like complaints or signs that might suggest infection, such as lethargy, weakness, fever, sore throat, malaise, or mucous membrane ulceration.
Eosinophilia. In clinical trials, 1% of patients developed eosinophilia, which can be substantial in rare cases. If a differential count reveals a total eosinophil count >4,000/mm3 , stop clozapine therapy until the eosinophil count falls below 3,000/mm3 .
Myocarditis. Clozapine-treated patients are at much greater risk for developing myocarditis and of dying from it—especially during the first 6 weeks of therapy—than is the general population.3 Tachycardia can be a presenting sign.
Abnormal laboratory findings associated with clozapine-induced myocarditis may include increased WBC count, eosinophilia, increased erythrocyte sedimentation rate, and increased cardiac enzyme levels and plasma troponin. Because the mortality rate of clozapine-induced myocarditis approaches 40%, stop clozapine and refer the patient for medical evaluation as soon as possible when you suspect myocarditis.3
Endocrine and hepatic effects. Severe hyperglycemia, sometimes leading to ketoacidosis, can occur during clozapine treatment in patients without a history of hyperglycemia. Ketoacidosis symptoms include rapid breathing, nausea, vomiting, clouding of sensorium (even coma), weight loss, polyuria, polydipsia, and dehydration. Monitoring for blood glucose changes, as described in Table 1, is recommended with clozapine as with all other atypical antipsychotics.
Hepatitis during clozapine therapy has been reported in patients with baseline normal or preexisting abnormal liver function. After baseline liver function tests, we suggest follow-up LFTs:
- annually for patients with normal baseline values
- every 6 months for patients with minimally abnormal values
- every 3 months for patients with liver disease.
MONITORING SUBSTANCE ABUSE
Substance abuse is often associated with medical comorbidities that require laboratory workup and monitoring. These include overdose sequelae, sexual assault, cirrhosis, endocarditis, HIV infection, viral hepatitis, tuberculosis, and syphilis. Some testing is mandated by federal law for patients in methadone maintenance or opioid agonist therapy programs with methadone.
We recommend that new patients with substance abuse be screened for organic illness as described above, plus the workup in Table 4. Also gather a careful history for hepatitis, pancreatitis, diabetes, cirrhosis, unusual infections (cellulitis, endocarditis, atypical pneumonias, HIV), frequent hospitalizations, falls, injuries, and blackouts.
Obtain a blood alcohol level in alcohol-intoxicated patients and urine toxicology to screen for locally-available street drugs (typically marijuana, sedative/hypnotics, amphetamines, cocaine, opiates, and phencyclidine).
Confer with your laboratory staff about the capabilities and sensitivities of their drug testing methods. Marijuana may be detected for 3 days to 4 weeks, depending on level of use. Cocaine can be detected for up to 2 to 4 days in urine.
- Rosse RB, Deutsch LH, Deutsch SI. Medical assessment and laboratory testing in psychiatry. In: Sadock B, Kaplan HI (eds). Comprehensive textbook of psychiatry, 6th ed. Baltimore: Lippincott, Williams & Wilkins 1995:601-19.
- National Institute of Diabetes and Digestive and Kidney Diseases. Body mass index tables. www.niddk.nih.gov/health/nutrit/pubs/statobes.htm#table.
- National Cholesterol Education Program. Guidelines for managing patients at risk for coronary heart disease. www.nhlbi.nih.gov/about/ncep/
- Food and Drug Administration. Guidelines on hyperglycemia associated with atypical antipsychotics [example]. www.fda.gov/medwatch/SAFETY/2004/Clozaril-deardoc.pdf
Drug brand names
- Carbamazepine • Carbatrol, others
- Clozapine • Clozaril
- Lithium • Lithobid, others
- Mesoridazine • Serentil
- Pimozide • Orap
- Thioridazine • Mellaril
- Valproate • Depakote, Depakene
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry 2004;47(Apr):S64-S66.
2. Association of Clinical Endocrinologists, North merican Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
3. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-49.
4. Work Group on Schizophrenia. Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004, 161:2(suppl). For BMI information related to this guideline, see http://www.cdc.gov/nccdphp/dnpa/bmi/bmi-adult-formula.htm.
5. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., text rev). Washington, DC: American Psychiatric Association, 2000.
6. Preskorn SH, Fast GA. Therapeutic drug monitoring for antidepressants: efficacy, safety, and cost effectiveness. J Clin Psychiatry 1991;(52 suppl):23-33.
7. Preskorn SH. Why patients may not respond to usual recommended dosages: 3 variables to consider when prescribing antipsychotics [commentary]. Current Psychiatry 2004;3(8):38-43.
8. Price DG, Ghaemi SN. Lithium. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004:355.
9. Kronig MH, Munne RA, Szymanski S, et al. Plasma clozapine levels and clinical response for treatment-refractory schizophrenic patients. Am J Psychiatry 1995;152(2):179-82.
10. Schulte P. What is an adequate trial with clozapine? Therapeutic drug monitoring and time to response in treatment-refractory schizophrenia. Clin Pharmacokinet 2003;42(7):607-18.
11. Henderson DC, Kunkel L, Goff DC. Antipsychotic drugs. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004;338-9.
12. Meltzer HY, Alphs L, Green AI, et al. International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT). Arch Gen Psychiatry 2003;60(1):82-91.
13. Meltzer HY. Suicide in schizophrenia: risk factors and clozapine treatment. J Clin Psychiatry. 1998;59(suppl 3):15-20.
1. Kohen D. Diabetes mellitus and schizophrenia: historical perspective. Br J Psychiatry 2004;47(Apr):S64-S66.
2. Association of Clinical Endocrinologists, North merican Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care 2004;27:596-601.
3. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161:1334-49.
4. Work Group on Schizophrenia. Practice guideline for the treatment of patients with schizophrenia (2nd ed). Am J Psychiatry 2004, 161:2(suppl). For BMI information related to this guideline, see http://www.cdc.gov/nccdphp/dnpa/bmi/bmi-adult-formula.htm.
5. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., text rev). Washington, DC: American Psychiatric Association, 2000.
6. Preskorn SH, Fast GA. Therapeutic drug monitoring for antidepressants: efficacy, safety, and cost effectiveness. J Clin Psychiatry 1991;(52 suppl):23-33.
7. Preskorn SH. Why patients may not respond to usual recommended dosages: 3 variables to consider when prescribing antipsychotics [commentary]. Current Psychiatry 2004;3(8):38-43.
8. Price DG, Ghaemi SN. Lithium. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004:355.
9. Kronig MH, Munne RA, Szymanski S, et al. Plasma clozapine levels and clinical response for treatment-refractory schizophrenic patients. Am J Psychiatry 1995;152(2):179-82.
10. Schulte P. What is an adequate trial with clozapine? Therapeutic drug monitoring and time to response in treatment-refractory schizophrenia. Clin Pharmacokinet 2003;42(7):607-18.
11. Henderson DC, Kunkel L, Goff DC. Antipsychotic drugs. In: Stern TA., Herman JB (eds). The Massachusetts General Hospital psychiatry update and board preparation (2nd ed). New York: McGraw-Hill, 2004;338-9.
12. Meltzer HY, Alphs L, Green AI, et al. International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT). Arch Gen Psychiatry 2003;60(1):82-91.
13. Meltzer HY. Suicide in schizophrenia: risk factors and clozapine treatment. J Clin Psychiatry. 1998;59(suppl 3):15-20.
Commentary: Clinical perspective on pediatric depression
With “black box” warnings expected, prescribing antidepressants to children and adolescents is changing. In the past year, information from previously unpublished studies has shown the drugs’ risks to be greater and benefits less in pediatric patients than doctors had believed.
As this article went to press, the FDA said it would adopt tougher labeling for antidepressants, as recommended by its Psychopharmacologic Drugs and Pediatric advisory committees. The advisors voted 15 to 8 at a Sept. 14 hearing in favor of a “black box” for all antidepressants, warning of increased risk of suicidality in pediatric patients.
We reported on the FDA’s Feb. 2 public hearing on increased risk of suicidality with antidepressants (CURRENT PSYCHIATRY, March 2004).1 This commentary provides a follow-up perspective on:
- the Columbia group’s report on classifying suicidality in SSRI clinical trial data
- how undisclosed clinical trial data tipped the SSRI risk-benefit balance in pediatric patients
- new data on using SSRIs plus psychotherapy for depressed adolescents.
WHAT THE COLUMBIA GROUP FOUND
In March, the FDA requested a warning label on SSRIs and related antidepressants that all patients be “monitored closely for worsening depression or the emergence of suicidality.” The advisory’s text and supporting information is available on the FDA’s Web site.2
The FDA also contracted with Columbia University to classify SSRI clinical trial events—first analyzed by FDA senior epidemiologist Dr. Andrew D. Mosholder—that might represent suicidality. Dr. Mosholder had reviewed pharmaceutical industry data from 22 placebo-controlled trials involving 4,250 pediatric patients and found that youths given antidepressants were nearly twice as likely to become suicidal as those given placebo (Box 1). Suicidality has historically been attributed to depressive illness rather than antidepressant use. Therefore, FDA officials cancelled Dr. Mosholder’s scheduled testimony at the Feb. 2 hearing—a decision that triggered congressional investigations—to allow for further analysis.
Nearly 2 years ago, FDA senior epidemiologist Dr. Andrew Mosholder requested that paroxetine’s manufacturer analyze suicidal behaviors in its pediatric clinical trial database. In July 2003, the same analysis was requested for eight other antidepressants (bupropion, mirtazapine, fluoxetine, nefazodone, fluvoxamine, sertraline, citalopram, venlafaxine).
The pharmaceutical manufacturers subsequently analyzed data from 22 short-term, placebo-controlled trials involving 4,250 youths—2,298 treated with antidepressants and 1,952 given placebo. Dr. Mosholder reviewed the analyses in September 2003 and found that youths taking antidepressants were nearly twice as likely to become suicidal as those taking placebo. Statistically, the risk of suicide-related events was significantly higher with venlafaxine and paroxetine than with placebo, and data for citalopram approached statistical significance on one measure.
Relative risks for suicide-related events were 0.9 with fluoxetine and 0.5 with mirtazapine, suggesting a possible protective effect (although mirtazapine’s analysis was based on a very small number of events). For all other drugs, relative-risk estimates were >1 or undefined because of lack of events. This association between suicide-related events and active drug treatment was observed only in major depressive disorder treatment trials.
The analyses had limitations; the trials reflected short-term antidepressant use, and each sponsor analyzed its data separately. Based on the evidence, Dr. Mosholder recommended that the FDA discourage use of antidepressants other than fluoxetine in children.
As of Aug. 21, the Columbia group had analyzed data from 25 studies and reviewed 423 adverse events that occurred during the trials’ randomized double-blind phase and/or within 30 days of the last dose of randomized treatment.3 These events included intentional self-injury, suicidal ideation, suicide attempts, accidental injuries, and accidental overdose.
The preliminary evidence suggests that young antidepressant users were 1.8 times more likely to have suicidal thoughts or behaviors compared with patients given placebo4—the same conclusion Dr. Mosholder reached nearly 1 year earlier.
RISK VERSUS BENEFIT
Are SSRIs safe in children? In the United Kingdom, a review by the Medicines and Healthcare Products Regulatory Agency (MHRA) of data submitted by paroxetine’s manufacturer revealed an unfavorable risk-to-benefit ratio in children and adolescents. Review of other data on other antidepressants soon followed.
Last December, the MHRA’s Committee on Safety of Medicines and its Expert Working Group on SSRIs advised that the risks and benefits of treating major depressive disorder in patients younger than age 18 were unfavorable for sertraline, citalopram, paroxetine, and escitalopram, and could not be assessed for fluvoxamine.5 The MHRA warned British physicians against prescribing paroxetine to depressed patients younger than age 18 and ordered labeling changes for paroxetine contraindicating its use in pediatric major depression.
Fluoxetine is the only SSRI for which the committee considers the risk-benefit balance to be favorable. It cautions British physicians, however, that fluoxetine may benefit only an estimated 1 in 10 pediatric patients.5
Are SSRIs effective in children? To be labeled for treating depression in children and adolescents, an SSRI must have proven efficacy (statistically and clinically significant improvement) in two independently conducted, double-blind, placebo-controlled trials. Five trials have met this standard—fluoxetine (2),6,7 sertraline (1),8 paroxetine (1-adolescents only),9 and citalopram (1)10—and three trials have not—paroxetine (2) and citalopram (1).11 Thus, only fluoxetine is FDA-approved for treating depressed children and adolescents.
However, lacking two positive trials does not necessarily indicate that a medication is not effective, especially when only two trials were conducted.12 Also, unpublished data now becoming available show inconsistencies with the published data.13
PUBLISHED VS. UNPUBLISHED DATA
In a meta-analysis by Whittington et al,13 data from five published, randomized, controlled trials of SSRIs (fluoxetine, paroxetine, sertraline and venlafaxine) were compared with data from unpublished reports found in the United Kingdom’s Committee on Safety of Medicines’ review. In the unpublished data, for example, paroxetine had a significantly lower response rate and more-pronounced placebo effect than the published data indicated.
As a result, these investigators concluded that the favorable risk-benefit profiles of paroxetine, sertraline, and venlafaxine for children and adolescents should be switched to unfavorable. They recommended against using these three antidepressants in youth because of possible increased risk of suicidal ideation and serious adverse events—findings that corresponded to the MHRA’s 2003 decisions.
Tipping the balance? Discrepancies between published and unpublished data raise alarms about nonreporting of negative trials. Except for one paroxetine trial, one early fluoxetine trial, and one more-recent fluoxetine trial funded by the National Institute of Mental Health (NIMH),14 the FDA’s “pediatric rule” of 1997 has produced all emerging data on SSRIs in children and adolescents. This rule gives pharmaceutical companies an additional 6 months of patent protection (which translates to millions of dollars) for conducting minimal research to collect data on medications’ safety in pediatric populations.
The subsequent Pediatric Research Equity Act of 2003 (PREA) requires pharmaceutical companies to conduct pediatric studies as part of nearly every new drug application filed since Jan. 1, 1999. Unfortunately, PREA does not regulate the quality of that research nor require that negative studies be disclosed.
TADS: FLUOXETINE PLUS CBT
The recently reported Treatment for Adolescents with Depression Study (TADS)14—funded by the NIMH—showed the benefit of combining fluoxetine with cognitive-behavioral therapy (CBT) for depressed children and adolescents. In the 12-week, multi-site, double-blind, placebo-controlled trial, 439 adolescents ages 12 to 17 diagnosed with major depressive disorder received fluoxetine, 10 to 40 mg/d; CBT alone; CBT with fluoxetine, 10 to 40 mg/d; or placebo. Response rates were:
- fluoxetine alone, 61% (95% confidence interval [CI], 51-70%)
- CBT alone, 43% (95% CI, 34-52%)
- fluoxetine with CBT, 71% (95% CI, 62- 80%)
- placebo, 34.8% (95% CI, 26-44%).
The two treatments containing fluoxetine were statistically more effective than CBT alone or placebo, as measured by the Clinical Global Impression scale. Clinically significant suicidal thinking—in 29% of the adolescents at baseline—improved significantly in all treatment groups, with fluoxetine plus CBT showing the greatest reduction (P = 0.02). Seven of 439 patients (1.6%) attempted suicide; there were no completed suicides.
An association between SSRIs and suicidal ideation in children and adolescents was first reported in the early 1990s.15 In theory, agitation and nervousness that occur in some children treated with SSRIs might increase their risk of self-injury or of harming others. Agitation, hyperkinesia, mania, and hypomania tend to be more frequent among patients treated with SSRIs (including fluoxetine) than among those receiving placebo (1 to 6% vs 0 to 4%).16
Clinicians should watch carefully for activation during SSRI treatment. The following symptoms may occur in activation syndromes: anxiety, agitation, panic attacks, hostility, impulsivity, akathisia (severe restlessness), insomnia, hypomania, irritability, or mania.17
On the other hand, no evidence has shown that increased agitation with SSRIs is synonymous with suicidal behavior, and no suicides have occurred in more than 4,000 children and adolescents studied in SSRI clinical trials. In fact, increased SSRI prescribing for children ages 10 to 19 appears to parallel a significant decrease in suicide in this population. With each 1% increase in SSRI use among adolescents, the number of suicides has declined by 0.23 per 100,000 adolescents per year.18 continued
WHAT ARE CLINICIANS TO DO?
Depression is a known risk factor for suicidal ideation or behavior, and subjects with serious suicidal ideation or suicide attempts are always excluded from clinical trials of antidepressant therapy. Suicide is also relatively rare. Thus, a strong association between SSRI treatment and suicide is difficult to demonstrate. Dozens of controlled trials with thousands of pediatric subjects would be required to show definitively that suicide is associated with antidepressant use.
Recently, a panel of psychiatrists and primary care physicians discussed the FDA’s earlier advisory and its effect on depression treatment.15 Overall, the FDA findings seemed not to have convinced these clinicians of a link between suicide and SSRIs. They commented that:
- the FDA has not established a “firm causal connection” between suicide and SSRIs but uses the term “activation syndrome” (Box 2)15-18
- “activation” may give some depressed patients “the energy to carry out things they have been somewhat inhibited from doing”
- “antidepressant jitteriness syndrome” has been observed more frequently in patients diagnosed with panic disorder or somatizing anxiety than with major depressive disorder, and very little evidence exists to link this syndrome with suicide risk.
Recommendations. As this dialogue continues, how should clinicians care for pediatric patients with major depressive disorder? We suggest the following approach:
- For patients taking antidepressants, recommend that they not stop the medication abruptly, as this may result in severe withdrawal syndrome and increase the risk of depressive relapse. If you discontinue SSRI therapy, taper the dosage over 1 to 2 weeks while monitoring for risky and suicidal behavior.
- For patients newly diagnosed with severe depression, fluoxetine remains an option to use with caution. This includes making an accurate diagnosis, monitoring for suicidality, minimizing side effects, and preventing drug interactions.1
Children with depression often exhibit somatic symptoms such as abdominal pain, headaches, or irritability. Adolescents are more similar to adults, exhibiting sad mood, boredom, apathy, lack of energy, and vegetative signs. Girls and boys are equally at risk for depression until puberty, when prevalence rates for girls begin to rise above those for boys.
Up to 6% of teens meet criteria for major depressive disorder, and up to 25% are affected by it by late adolescence.19 Untreated pediatric depression is associated with substantial morbidity, reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life.20,21 Depression is a major risk factor for suicide, the third-leading cause of death among U.S. teenagers.
Because treating bipolar depression with antidepressants can cause switching to mania, rule out bipolar depression and mixed episodes before prescribing antidepressants. Bipolar illness may be characterized by marked irritability—also seen in depressed children and adolescents (Box 3).19,21
Informed consent. Inform the patient and parents of antidepressants’ labeling and side effects. Discuss the possibility of disinhibition and impulsivity during initial therapy, which may increase the risk of suicidal ideation or suicide attempts.
Dosing. Although SSRIs do not show a clear dose-response relationship, their side effects are considered dose-dependent.22 Therefore, start children on lower dosages than are used in adolescents and adults, and monitor very closely.
Nondrug intervention. CBT and other psychotherapies have shown short-term benefits for depressed children.23 Therefore, to improve SSRIs’ risk-benefit ratio, you may wish to reserve antidepressants for youths:
- with moderate to severe depression, recurrent depression, or a three-generation family history of depression
- who are unlikely to respond to psychotherapy alone, behavioral or environmental change, or general emotional support.
CONCLUSION
Deciding to start, continue, or discontinue SSRIs and other antidepressants in depressed children and adolescents is difficult for clinicians, patients, and their families. Despite data showing increased suicidal behavior in some pediatric patients, SSRIs—when used with caution—remain an important depression treatment in this population.
Related resources
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Elizabeth Weller receives research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis Pharmaceuticals Corp., AstraZeneca Pharmaceuticals, and Otsuka Pharmaceutical.
Joon Kang reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
1. Sood AB, Weller EB, Weller RA. SSRIs in children and adolescents: where do we stand? Current Psychiatry 2004;3(3):83-9.
2. Food and Drug Administration. Center for Drug Evaluation and Research. Antidepressant use in children, adolescents, and adults. Available at: http://www.fda.gov/cder/drug/antidepressants/default.htm. Accessed Sept. 2, 2004.
3. Department of Health and Human Services. Public Health Service. Report of the audit of the Columbia suicidality classification methodology [memorandum]. Aug. 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-09-TAB07-Iyasu-Audit_report.htm. Accessed Sept. 2, 2004.
4. Neergaard L. Suicide risk may prompt antidepressant warnings. Associated Press Aug. 21, 2004. Available at: http://chron.com (search archive). Accessed Sept. 15, 2004.
5. Medicines and Healthcare Products Regulatory Agency (UK). Use of selective serotonin reuptake inhibitors (SSRIs) in children and adolescents with major depressive disorder (MDD). Dec. 10, 2003. Available at: www.mhra.gov.uk/news/2003.htm#ssri. Accessed Sept. 2, 2004.
6. Emslie GJ, Rush AJ, Weinberg WA, et al. A double-blind, randomized, placebo controlled trial of fluoxetine in children and adolescents with depression. Arch Gen Psychiatry 1997;54(11):1031-7
7. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
8. Wagner KD, Ambrosini P, Rynn M, et al. Efficacy of sertraline in the treatment of children and adolescents with major depressive disorder: two randomized controlled trials. JAMA 2003;290(8):1033-41.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Wagner KD, Robb AS, Findling RL, et al. A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescents. Am J Psychiatry 2004;161(6):1079-83.
11. Laughren T. Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee (PDAC) and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee (PedsAC). Available at: http://www.fda.gov/ohrms/dockets/ac/04/brief-ing/4006B1_03_Background Memo 01-05-04.doc. Accessed Sept. 15, 2004.
12. Emslie GJ. Making sense of the research puzzle. AACAP News 2004;35(2):
13. Whittington CJ, Kendall T, Fonagy P, et al. Selective serotonin reuptake inhibitors in childhood depression: Systematic review of published versus unpublished data. Lancet 2004;363:1341-5.Also available at: http://www.thelancet.com/journal/vol363/iss9418 (scroll to article title). Accessed Sept. 2, 2004.
14. Treatment for Adolescents with Depression Study (TADS) team. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression, JAMA 2004;922(7):807-20.
15. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adol Psychiatry 2001;40:1364-5.
16. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee and the Anti-Infective Drugs Advisory Committee. Briefing information for public hearing Feb. 2, 2004. Available at: www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm. Accessed Sept. 2, 2004.
17. Culpepper L, Davidson JR, Dietrich AJ, et al. Suicidality as a possible side effect of antidepressant treatment. Primary Care Companion: J Clin Psychiatry 2004;6(2):79-86.
18. Olfson M, Gameroff MJ, Marcus SC, Waslic BD. Outpatient treatment of child and adolescent depression in the United States. Arch Gen Psychiatry 2003;60:1236-42.
19. Kressler RC, Avenevoli S, Merikangas KR. Mood disorders in children and adolescents: an epidemiological perspective. Biol Psychiatry 2001;49:1002-14.
20. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule for Children, Version 2.3 (DISC-2.3): description, acceptability, prevalence rates, and performance in the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996;35:865-77.
21. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression. III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
22. Preskorn SH. Outpatient management of depression: a guide for the practitioner(2nd ed). Caddo, OK: Professional Publications, 1999.
23. Lewinsohn PM, Clarke GN. Psychosocial treatments for adolescent depression. Clin Psychol Rev 1999;19:329-42.
With “black box” warnings expected, prescribing antidepressants to children and adolescents is changing. In the past year, information from previously unpublished studies has shown the drugs’ risks to be greater and benefits less in pediatric patients than doctors had believed.
As this article went to press, the FDA said it would adopt tougher labeling for antidepressants, as recommended by its Psychopharmacologic Drugs and Pediatric advisory committees. The advisors voted 15 to 8 at a Sept. 14 hearing in favor of a “black box” for all antidepressants, warning of increased risk of suicidality in pediatric patients.
We reported on the FDA’s Feb. 2 public hearing on increased risk of suicidality with antidepressants (CURRENT PSYCHIATRY, March 2004).1 This commentary provides a follow-up perspective on:
- the Columbia group’s report on classifying suicidality in SSRI clinical trial data
- how undisclosed clinical trial data tipped the SSRI risk-benefit balance in pediatric patients
- new data on using SSRIs plus psychotherapy for depressed adolescents.
WHAT THE COLUMBIA GROUP FOUND
In March, the FDA requested a warning label on SSRIs and related antidepressants that all patients be “monitored closely for worsening depression or the emergence of suicidality.” The advisory’s text and supporting information is available on the FDA’s Web site.2
The FDA also contracted with Columbia University to classify SSRI clinical trial events—first analyzed by FDA senior epidemiologist Dr. Andrew D. Mosholder—that might represent suicidality. Dr. Mosholder had reviewed pharmaceutical industry data from 22 placebo-controlled trials involving 4,250 pediatric patients and found that youths given antidepressants were nearly twice as likely to become suicidal as those given placebo (Box 1). Suicidality has historically been attributed to depressive illness rather than antidepressant use. Therefore, FDA officials cancelled Dr. Mosholder’s scheduled testimony at the Feb. 2 hearing—a decision that triggered congressional investigations—to allow for further analysis.
Nearly 2 years ago, FDA senior epidemiologist Dr. Andrew Mosholder requested that paroxetine’s manufacturer analyze suicidal behaviors in its pediatric clinical trial database. In July 2003, the same analysis was requested for eight other antidepressants (bupropion, mirtazapine, fluoxetine, nefazodone, fluvoxamine, sertraline, citalopram, venlafaxine).
The pharmaceutical manufacturers subsequently analyzed data from 22 short-term, placebo-controlled trials involving 4,250 youths—2,298 treated with antidepressants and 1,952 given placebo. Dr. Mosholder reviewed the analyses in September 2003 and found that youths taking antidepressants were nearly twice as likely to become suicidal as those taking placebo. Statistically, the risk of suicide-related events was significantly higher with venlafaxine and paroxetine than with placebo, and data for citalopram approached statistical significance on one measure.
Relative risks for suicide-related events were 0.9 with fluoxetine and 0.5 with mirtazapine, suggesting a possible protective effect (although mirtazapine’s analysis was based on a very small number of events). For all other drugs, relative-risk estimates were >1 or undefined because of lack of events. This association between suicide-related events and active drug treatment was observed only in major depressive disorder treatment trials.
The analyses had limitations; the trials reflected short-term antidepressant use, and each sponsor analyzed its data separately. Based on the evidence, Dr. Mosholder recommended that the FDA discourage use of antidepressants other than fluoxetine in children.
As of Aug. 21, the Columbia group had analyzed data from 25 studies and reviewed 423 adverse events that occurred during the trials’ randomized double-blind phase and/or within 30 days of the last dose of randomized treatment.3 These events included intentional self-injury, suicidal ideation, suicide attempts, accidental injuries, and accidental overdose.
The preliminary evidence suggests that young antidepressant users were 1.8 times more likely to have suicidal thoughts or behaviors compared with patients given placebo4—the same conclusion Dr. Mosholder reached nearly 1 year earlier.
RISK VERSUS BENEFIT
Are SSRIs safe in children? In the United Kingdom, a review by the Medicines and Healthcare Products Regulatory Agency (MHRA) of data submitted by paroxetine’s manufacturer revealed an unfavorable risk-to-benefit ratio in children and adolescents. Review of other data on other antidepressants soon followed.
Last December, the MHRA’s Committee on Safety of Medicines and its Expert Working Group on SSRIs advised that the risks and benefits of treating major depressive disorder in patients younger than age 18 were unfavorable for sertraline, citalopram, paroxetine, and escitalopram, and could not be assessed for fluvoxamine.5 The MHRA warned British physicians against prescribing paroxetine to depressed patients younger than age 18 and ordered labeling changes for paroxetine contraindicating its use in pediatric major depression.
Fluoxetine is the only SSRI for which the committee considers the risk-benefit balance to be favorable. It cautions British physicians, however, that fluoxetine may benefit only an estimated 1 in 10 pediatric patients.5
Are SSRIs effective in children? To be labeled for treating depression in children and adolescents, an SSRI must have proven efficacy (statistically and clinically significant improvement) in two independently conducted, double-blind, placebo-controlled trials. Five trials have met this standard—fluoxetine (2),6,7 sertraline (1),8 paroxetine (1-adolescents only),9 and citalopram (1)10—and three trials have not—paroxetine (2) and citalopram (1).11 Thus, only fluoxetine is FDA-approved for treating depressed children and adolescents.
However, lacking two positive trials does not necessarily indicate that a medication is not effective, especially when only two trials were conducted.12 Also, unpublished data now becoming available show inconsistencies with the published data.13
PUBLISHED VS. UNPUBLISHED DATA
In a meta-analysis by Whittington et al,13 data from five published, randomized, controlled trials of SSRIs (fluoxetine, paroxetine, sertraline and venlafaxine) were compared with data from unpublished reports found in the United Kingdom’s Committee on Safety of Medicines’ review. In the unpublished data, for example, paroxetine had a significantly lower response rate and more-pronounced placebo effect than the published data indicated.
As a result, these investigators concluded that the favorable risk-benefit profiles of paroxetine, sertraline, and venlafaxine for children and adolescents should be switched to unfavorable. They recommended against using these three antidepressants in youth because of possible increased risk of suicidal ideation and serious adverse events—findings that corresponded to the MHRA’s 2003 decisions.
Tipping the balance? Discrepancies between published and unpublished data raise alarms about nonreporting of negative trials. Except for one paroxetine trial, one early fluoxetine trial, and one more-recent fluoxetine trial funded by the National Institute of Mental Health (NIMH),14 the FDA’s “pediatric rule” of 1997 has produced all emerging data on SSRIs in children and adolescents. This rule gives pharmaceutical companies an additional 6 months of patent protection (which translates to millions of dollars) for conducting minimal research to collect data on medications’ safety in pediatric populations.
The subsequent Pediatric Research Equity Act of 2003 (PREA) requires pharmaceutical companies to conduct pediatric studies as part of nearly every new drug application filed since Jan. 1, 1999. Unfortunately, PREA does not regulate the quality of that research nor require that negative studies be disclosed.
TADS: FLUOXETINE PLUS CBT
The recently reported Treatment for Adolescents with Depression Study (TADS)14—funded by the NIMH—showed the benefit of combining fluoxetine with cognitive-behavioral therapy (CBT) for depressed children and adolescents. In the 12-week, multi-site, double-blind, placebo-controlled trial, 439 adolescents ages 12 to 17 diagnosed with major depressive disorder received fluoxetine, 10 to 40 mg/d; CBT alone; CBT with fluoxetine, 10 to 40 mg/d; or placebo. Response rates were:
- fluoxetine alone, 61% (95% confidence interval [CI], 51-70%)
- CBT alone, 43% (95% CI, 34-52%)
- fluoxetine with CBT, 71% (95% CI, 62- 80%)
- placebo, 34.8% (95% CI, 26-44%).
The two treatments containing fluoxetine were statistically more effective than CBT alone or placebo, as measured by the Clinical Global Impression scale. Clinically significant suicidal thinking—in 29% of the adolescents at baseline—improved significantly in all treatment groups, with fluoxetine plus CBT showing the greatest reduction (P = 0.02). Seven of 439 patients (1.6%) attempted suicide; there were no completed suicides.
An association between SSRIs and suicidal ideation in children and adolescents was first reported in the early 1990s.15 In theory, agitation and nervousness that occur in some children treated with SSRIs might increase their risk of self-injury or of harming others. Agitation, hyperkinesia, mania, and hypomania tend to be more frequent among patients treated with SSRIs (including fluoxetine) than among those receiving placebo (1 to 6% vs 0 to 4%).16
Clinicians should watch carefully for activation during SSRI treatment. The following symptoms may occur in activation syndromes: anxiety, agitation, panic attacks, hostility, impulsivity, akathisia (severe restlessness), insomnia, hypomania, irritability, or mania.17
On the other hand, no evidence has shown that increased agitation with SSRIs is synonymous with suicidal behavior, and no suicides have occurred in more than 4,000 children and adolescents studied in SSRI clinical trials. In fact, increased SSRI prescribing for children ages 10 to 19 appears to parallel a significant decrease in suicide in this population. With each 1% increase in SSRI use among adolescents, the number of suicides has declined by 0.23 per 100,000 adolescents per year.18 continued
WHAT ARE CLINICIANS TO DO?
Depression is a known risk factor for suicidal ideation or behavior, and subjects with serious suicidal ideation or suicide attempts are always excluded from clinical trials of antidepressant therapy. Suicide is also relatively rare. Thus, a strong association between SSRI treatment and suicide is difficult to demonstrate. Dozens of controlled trials with thousands of pediatric subjects would be required to show definitively that suicide is associated with antidepressant use.
Recently, a panel of psychiatrists and primary care physicians discussed the FDA’s earlier advisory and its effect on depression treatment.15 Overall, the FDA findings seemed not to have convinced these clinicians of a link between suicide and SSRIs. They commented that:
- the FDA has not established a “firm causal connection” between suicide and SSRIs but uses the term “activation syndrome” (Box 2)15-18
- “activation” may give some depressed patients “the energy to carry out things they have been somewhat inhibited from doing”
- “antidepressant jitteriness syndrome” has been observed more frequently in patients diagnosed with panic disorder or somatizing anxiety than with major depressive disorder, and very little evidence exists to link this syndrome with suicide risk.
Recommendations. As this dialogue continues, how should clinicians care for pediatric patients with major depressive disorder? We suggest the following approach:
- For patients taking antidepressants, recommend that they not stop the medication abruptly, as this may result in severe withdrawal syndrome and increase the risk of depressive relapse. If you discontinue SSRI therapy, taper the dosage over 1 to 2 weeks while monitoring for risky and suicidal behavior.
- For patients newly diagnosed with severe depression, fluoxetine remains an option to use with caution. This includes making an accurate diagnosis, monitoring for suicidality, minimizing side effects, and preventing drug interactions.1
Children with depression often exhibit somatic symptoms such as abdominal pain, headaches, or irritability. Adolescents are more similar to adults, exhibiting sad mood, boredom, apathy, lack of energy, and vegetative signs. Girls and boys are equally at risk for depression until puberty, when prevalence rates for girls begin to rise above those for boys.
Up to 6% of teens meet criteria for major depressive disorder, and up to 25% are affected by it by late adolescence.19 Untreated pediatric depression is associated with substantial morbidity, reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life.20,21 Depression is a major risk factor for suicide, the third-leading cause of death among U.S. teenagers.
Because treating bipolar depression with antidepressants can cause switching to mania, rule out bipolar depression and mixed episodes before prescribing antidepressants. Bipolar illness may be characterized by marked irritability—also seen in depressed children and adolescents (Box 3).19,21
Informed consent. Inform the patient and parents of antidepressants’ labeling and side effects. Discuss the possibility of disinhibition and impulsivity during initial therapy, which may increase the risk of suicidal ideation or suicide attempts.
Dosing. Although SSRIs do not show a clear dose-response relationship, their side effects are considered dose-dependent.22 Therefore, start children on lower dosages than are used in adolescents and adults, and monitor very closely.
Nondrug intervention. CBT and other psychotherapies have shown short-term benefits for depressed children.23 Therefore, to improve SSRIs’ risk-benefit ratio, you may wish to reserve antidepressants for youths:
- with moderate to severe depression, recurrent depression, or a three-generation family history of depression
- who are unlikely to respond to psychotherapy alone, behavioral or environmental change, or general emotional support.
CONCLUSION
Deciding to start, continue, or discontinue SSRIs and other antidepressants in depressed children and adolescents is difficult for clinicians, patients, and their families. Despite data showing increased suicidal behavior in some pediatric patients, SSRIs—when used with caution—remain an important depression treatment in this population.
Related resources
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Elizabeth Weller receives research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis Pharmaceuticals Corp., AstraZeneca Pharmaceuticals, and Otsuka Pharmaceutical.
Joon Kang reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
With “black box” warnings expected, prescribing antidepressants to children and adolescents is changing. In the past year, information from previously unpublished studies has shown the drugs’ risks to be greater and benefits less in pediatric patients than doctors had believed.
As this article went to press, the FDA said it would adopt tougher labeling for antidepressants, as recommended by its Psychopharmacologic Drugs and Pediatric advisory committees. The advisors voted 15 to 8 at a Sept. 14 hearing in favor of a “black box” for all antidepressants, warning of increased risk of suicidality in pediatric patients.
We reported on the FDA’s Feb. 2 public hearing on increased risk of suicidality with antidepressants (CURRENT PSYCHIATRY, March 2004).1 This commentary provides a follow-up perspective on:
- the Columbia group’s report on classifying suicidality in SSRI clinical trial data
- how undisclosed clinical trial data tipped the SSRI risk-benefit balance in pediatric patients
- new data on using SSRIs plus psychotherapy for depressed adolescents.
WHAT THE COLUMBIA GROUP FOUND
In March, the FDA requested a warning label on SSRIs and related antidepressants that all patients be “monitored closely for worsening depression or the emergence of suicidality.” The advisory’s text and supporting information is available on the FDA’s Web site.2
The FDA also contracted with Columbia University to classify SSRI clinical trial events—first analyzed by FDA senior epidemiologist Dr. Andrew D. Mosholder—that might represent suicidality. Dr. Mosholder had reviewed pharmaceutical industry data from 22 placebo-controlled trials involving 4,250 pediatric patients and found that youths given antidepressants were nearly twice as likely to become suicidal as those given placebo (Box 1). Suicidality has historically been attributed to depressive illness rather than antidepressant use. Therefore, FDA officials cancelled Dr. Mosholder’s scheduled testimony at the Feb. 2 hearing—a decision that triggered congressional investigations—to allow for further analysis.
Nearly 2 years ago, FDA senior epidemiologist Dr. Andrew Mosholder requested that paroxetine’s manufacturer analyze suicidal behaviors in its pediatric clinical trial database. In July 2003, the same analysis was requested for eight other antidepressants (bupropion, mirtazapine, fluoxetine, nefazodone, fluvoxamine, sertraline, citalopram, venlafaxine).
The pharmaceutical manufacturers subsequently analyzed data from 22 short-term, placebo-controlled trials involving 4,250 youths—2,298 treated with antidepressants and 1,952 given placebo. Dr. Mosholder reviewed the analyses in September 2003 and found that youths taking antidepressants were nearly twice as likely to become suicidal as those taking placebo. Statistically, the risk of suicide-related events was significantly higher with venlafaxine and paroxetine than with placebo, and data for citalopram approached statistical significance on one measure.
Relative risks for suicide-related events were 0.9 with fluoxetine and 0.5 with mirtazapine, suggesting a possible protective effect (although mirtazapine’s analysis was based on a very small number of events). For all other drugs, relative-risk estimates were >1 or undefined because of lack of events. This association between suicide-related events and active drug treatment was observed only in major depressive disorder treatment trials.
The analyses had limitations; the trials reflected short-term antidepressant use, and each sponsor analyzed its data separately. Based on the evidence, Dr. Mosholder recommended that the FDA discourage use of antidepressants other than fluoxetine in children.
As of Aug. 21, the Columbia group had analyzed data from 25 studies and reviewed 423 adverse events that occurred during the trials’ randomized double-blind phase and/or within 30 days of the last dose of randomized treatment.3 These events included intentional self-injury, suicidal ideation, suicide attempts, accidental injuries, and accidental overdose.
The preliminary evidence suggests that young antidepressant users were 1.8 times more likely to have suicidal thoughts or behaviors compared with patients given placebo4—the same conclusion Dr. Mosholder reached nearly 1 year earlier.
RISK VERSUS BENEFIT
Are SSRIs safe in children? In the United Kingdom, a review by the Medicines and Healthcare Products Regulatory Agency (MHRA) of data submitted by paroxetine’s manufacturer revealed an unfavorable risk-to-benefit ratio in children and adolescents. Review of other data on other antidepressants soon followed.
Last December, the MHRA’s Committee on Safety of Medicines and its Expert Working Group on SSRIs advised that the risks and benefits of treating major depressive disorder in patients younger than age 18 were unfavorable for sertraline, citalopram, paroxetine, and escitalopram, and could not be assessed for fluvoxamine.5 The MHRA warned British physicians against prescribing paroxetine to depressed patients younger than age 18 and ordered labeling changes for paroxetine contraindicating its use in pediatric major depression.
Fluoxetine is the only SSRI for which the committee considers the risk-benefit balance to be favorable. It cautions British physicians, however, that fluoxetine may benefit only an estimated 1 in 10 pediatric patients.5
Are SSRIs effective in children? To be labeled for treating depression in children and adolescents, an SSRI must have proven efficacy (statistically and clinically significant improvement) in two independently conducted, double-blind, placebo-controlled trials. Five trials have met this standard—fluoxetine (2),6,7 sertraline (1),8 paroxetine (1-adolescents only),9 and citalopram (1)10—and three trials have not—paroxetine (2) and citalopram (1).11 Thus, only fluoxetine is FDA-approved for treating depressed children and adolescents.
However, lacking two positive trials does not necessarily indicate that a medication is not effective, especially when only two trials were conducted.12 Also, unpublished data now becoming available show inconsistencies with the published data.13
PUBLISHED VS. UNPUBLISHED DATA
In a meta-analysis by Whittington et al,13 data from five published, randomized, controlled trials of SSRIs (fluoxetine, paroxetine, sertraline and venlafaxine) were compared with data from unpublished reports found in the United Kingdom’s Committee on Safety of Medicines’ review. In the unpublished data, for example, paroxetine had a significantly lower response rate and more-pronounced placebo effect than the published data indicated.
As a result, these investigators concluded that the favorable risk-benefit profiles of paroxetine, sertraline, and venlafaxine for children and adolescents should be switched to unfavorable. They recommended against using these three antidepressants in youth because of possible increased risk of suicidal ideation and serious adverse events—findings that corresponded to the MHRA’s 2003 decisions.
Tipping the balance? Discrepancies between published and unpublished data raise alarms about nonreporting of negative trials. Except for one paroxetine trial, one early fluoxetine trial, and one more-recent fluoxetine trial funded by the National Institute of Mental Health (NIMH),14 the FDA’s “pediatric rule” of 1997 has produced all emerging data on SSRIs in children and adolescents. This rule gives pharmaceutical companies an additional 6 months of patent protection (which translates to millions of dollars) for conducting minimal research to collect data on medications’ safety in pediatric populations.
The subsequent Pediatric Research Equity Act of 2003 (PREA) requires pharmaceutical companies to conduct pediatric studies as part of nearly every new drug application filed since Jan. 1, 1999. Unfortunately, PREA does not regulate the quality of that research nor require that negative studies be disclosed.
TADS: FLUOXETINE PLUS CBT
The recently reported Treatment for Adolescents with Depression Study (TADS)14—funded by the NIMH—showed the benefit of combining fluoxetine with cognitive-behavioral therapy (CBT) for depressed children and adolescents. In the 12-week, multi-site, double-blind, placebo-controlled trial, 439 adolescents ages 12 to 17 diagnosed with major depressive disorder received fluoxetine, 10 to 40 mg/d; CBT alone; CBT with fluoxetine, 10 to 40 mg/d; or placebo. Response rates were:
- fluoxetine alone, 61% (95% confidence interval [CI], 51-70%)
- CBT alone, 43% (95% CI, 34-52%)
- fluoxetine with CBT, 71% (95% CI, 62- 80%)
- placebo, 34.8% (95% CI, 26-44%).
The two treatments containing fluoxetine were statistically more effective than CBT alone or placebo, as measured by the Clinical Global Impression scale. Clinically significant suicidal thinking—in 29% of the adolescents at baseline—improved significantly in all treatment groups, with fluoxetine plus CBT showing the greatest reduction (P = 0.02). Seven of 439 patients (1.6%) attempted suicide; there were no completed suicides.
An association between SSRIs and suicidal ideation in children and adolescents was first reported in the early 1990s.15 In theory, agitation and nervousness that occur in some children treated with SSRIs might increase their risk of self-injury or of harming others. Agitation, hyperkinesia, mania, and hypomania tend to be more frequent among patients treated with SSRIs (including fluoxetine) than among those receiving placebo (1 to 6% vs 0 to 4%).16
Clinicians should watch carefully for activation during SSRI treatment. The following symptoms may occur in activation syndromes: anxiety, agitation, panic attacks, hostility, impulsivity, akathisia (severe restlessness), insomnia, hypomania, irritability, or mania.17
On the other hand, no evidence has shown that increased agitation with SSRIs is synonymous with suicidal behavior, and no suicides have occurred in more than 4,000 children and adolescents studied in SSRI clinical trials. In fact, increased SSRI prescribing for children ages 10 to 19 appears to parallel a significant decrease in suicide in this population. With each 1% increase in SSRI use among adolescents, the number of suicides has declined by 0.23 per 100,000 adolescents per year.18 continued
WHAT ARE CLINICIANS TO DO?
Depression is a known risk factor for suicidal ideation or behavior, and subjects with serious suicidal ideation or suicide attempts are always excluded from clinical trials of antidepressant therapy. Suicide is also relatively rare. Thus, a strong association between SSRI treatment and suicide is difficult to demonstrate. Dozens of controlled trials with thousands of pediatric subjects would be required to show definitively that suicide is associated with antidepressant use.
Recently, a panel of psychiatrists and primary care physicians discussed the FDA’s earlier advisory and its effect on depression treatment.15 Overall, the FDA findings seemed not to have convinced these clinicians of a link between suicide and SSRIs. They commented that:
- the FDA has not established a “firm causal connection” between suicide and SSRIs but uses the term “activation syndrome” (Box 2)15-18
- “activation” may give some depressed patients “the energy to carry out things they have been somewhat inhibited from doing”
- “antidepressant jitteriness syndrome” has been observed more frequently in patients diagnosed with panic disorder or somatizing anxiety than with major depressive disorder, and very little evidence exists to link this syndrome with suicide risk.
Recommendations. As this dialogue continues, how should clinicians care for pediatric patients with major depressive disorder? We suggest the following approach:
- For patients taking antidepressants, recommend that they not stop the medication abruptly, as this may result in severe withdrawal syndrome and increase the risk of depressive relapse. If you discontinue SSRI therapy, taper the dosage over 1 to 2 weeks while monitoring for risky and suicidal behavior.
- For patients newly diagnosed with severe depression, fluoxetine remains an option to use with caution. This includes making an accurate diagnosis, monitoring for suicidality, minimizing side effects, and preventing drug interactions.1
Children with depression often exhibit somatic symptoms such as abdominal pain, headaches, or irritability. Adolescents are more similar to adults, exhibiting sad mood, boredom, apathy, lack of energy, and vegetative signs. Girls and boys are equally at risk for depression until puberty, when prevalence rates for girls begin to rise above those for boys.
Up to 6% of teens meet criteria for major depressive disorder, and up to 25% are affected by it by late adolescence.19 Untreated pediatric depression is associated with substantial morbidity, reduced academic performance, substance abuse, interpersonal problems, social withdrawal, and a poor quality of life.20,21 Depression is a major risk factor for suicide, the third-leading cause of death among U.S. teenagers.
Because treating bipolar depression with antidepressants can cause switching to mania, rule out bipolar depression and mixed episodes before prescribing antidepressants. Bipolar illness may be characterized by marked irritability—also seen in depressed children and adolescents (Box 3).19,21
Informed consent. Inform the patient and parents of antidepressants’ labeling and side effects. Discuss the possibility of disinhibition and impulsivity during initial therapy, which may increase the risk of suicidal ideation or suicide attempts.
Dosing. Although SSRIs do not show a clear dose-response relationship, their side effects are considered dose-dependent.22 Therefore, start children on lower dosages than are used in adolescents and adults, and monitor very closely.
Nondrug intervention. CBT and other psychotherapies have shown short-term benefits for depressed children.23 Therefore, to improve SSRIs’ risk-benefit ratio, you may wish to reserve antidepressants for youths:
- with moderate to severe depression, recurrent depression, or a three-generation family history of depression
- who are unlikely to respond to psychotherapy alone, behavioral or environmental change, or general emotional support.
CONCLUSION
Deciding to start, continue, or discontinue SSRIs and other antidepressants in depressed children and adolescents is difficult for clinicians, patients, and their families. Despite data showing increased suicidal behavior in some pediatric patients, SSRIs—when used with caution—remain an important depression treatment in this population.
Related resources
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Elizabeth Weller receives research/grant support from Forest Pharmaceuticals, Organon, and Wyeth Pharmaceuticals and is a consultant to Johnson & Johnson, Novartis Pharmaceuticals Corp., AstraZeneca Pharmaceuticals, and Otsuka Pharmaceutical.
Joon Kang reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ronald Weller receives research/grant support from Wyeth Pharmaceuticals, Organon, and Forest Pharmaceuticals.
1. Sood AB, Weller EB, Weller RA. SSRIs in children and adolescents: where do we stand? Current Psychiatry 2004;3(3):83-9.
2. Food and Drug Administration. Center for Drug Evaluation and Research. Antidepressant use in children, adolescents, and adults. Available at: http://www.fda.gov/cder/drug/antidepressants/default.htm. Accessed Sept. 2, 2004.
3. Department of Health and Human Services. Public Health Service. Report of the audit of the Columbia suicidality classification methodology [memorandum]. Aug. 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-09-TAB07-Iyasu-Audit_report.htm. Accessed Sept. 2, 2004.
4. Neergaard L. Suicide risk may prompt antidepressant warnings. Associated Press Aug. 21, 2004. Available at: http://chron.com (search archive). Accessed Sept. 15, 2004.
5. Medicines and Healthcare Products Regulatory Agency (UK). Use of selective serotonin reuptake inhibitors (SSRIs) in children and adolescents with major depressive disorder (MDD). Dec. 10, 2003. Available at: www.mhra.gov.uk/news/2003.htm#ssri. Accessed Sept. 2, 2004.
6. Emslie GJ, Rush AJ, Weinberg WA, et al. A double-blind, randomized, placebo controlled trial of fluoxetine in children and adolescents with depression. Arch Gen Psychiatry 1997;54(11):1031-7
7. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
8. Wagner KD, Ambrosini P, Rynn M, et al. Efficacy of sertraline in the treatment of children and adolescents with major depressive disorder: two randomized controlled trials. JAMA 2003;290(8):1033-41.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Wagner KD, Robb AS, Findling RL, et al. A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescents. Am J Psychiatry 2004;161(6):1079-83.
11. Laughren T. Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee (PDAC) and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee (PedsAC). Available at: http://www.fda.gov/ohrms/dockets/ac/04/brief-ing/4006B1_03_Background Memo 01-05-04.doc. Accessed Sept. 15, 2004.
12. Emslie GJ. Making sense of the research puzzle. AACAP News 2004;35(2):
13. Whittington CJ, Kendall T, Fonagy P, et al. Selective serotonin reuptake inhibitors in childhood depression: Systematic review of published versus unpublished data. Lancet 2004;363:1341-5.Also available at: http://www.thelancet.com/journal/vol363/iss9418 (scroll to article title). Accessed Sept. 2, 2004.
14. Treatment for Adolescents with Depression Study (TADS) team. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression, JAMA 2004;922(7):807-20.
15. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adol Psychiatry 2001;40:1364-5.
16. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee and the Anti-Infective Drugs Advisory Committee. Briefing information for public hearing Feb. 2, 2004. Available at: www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm. Accessed Sept. 2, 2004.
17. Culpepper L, Davidson JR, Dietrich AJ, et al. Suicidality as a possible side effect of antidepressant treatment. Primary Care Companion: J Clin Psychiatry 2004;6(2):79-86.
18. Olfson M, Gameroff MJ, Marcus SC, Waslic BD. Outpatient treatment of child and adolescent depression in the United States. Arch Gen Psychiatry 2003;60:1236-42.
19. Kressler RC, Avenevoli S, Merikangas KR. Mood disorders in children and adolescents: an epidemiological perspective. Biol Psychiatry 2001;49:1002-14.
20. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule for Children, Version 2.3 (DISC-2.3): description, acceptability, prevalence rates, and performance in the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996;35:865-77.
21. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression. III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
22. Preskorn SH. Outpatient management of depression: a guide for the practitioner(2nd ed). Caddo, OK: Professional Publications, 1999.
23. Lewinsohn PM, Clarke GN. Psychosocial treatments for adolescent depression. Clin Psychol Rev 1999;19:329-42.
1. Sood AB, Weller EB, Weller RA. SSRIs in children and adolescents: where do we stand? Current Psychiatry 2004;3(3):83-9.
2. Food and Drug Administration. Center for Drug Evaluation and Research. Antidepressant use in children, adolescents, and adults. Available at: http://www.fda.gov/cder/drug/antidepressants/default.htm. Accessed Sept. 2, 2004.
3. Department of Health and Human Services. Public Health Service. Report of the audit of the Columbia suicidality classification methodology [memorandum]. Aug. 16, 2004. Available at: http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4065b1-09-TAB07-Iyasu-Audit_report.htm. Accessed Sept. 2, 2004.
4. Neergaard L. Suicide risk may prompt antidepressant warnings. Associated Press Aug. 21, 2004. Available at: http://chron.com (search archive). Accessed Sept. 15, 2004.
5. Medicines and Healthcare Products Regulatory Agency (UK). Use of selective serotonin reuptake inhibitors (SSRIs) in children and adolescents with major depressive disorder (MDD). Dec. 10, 2003. Available at: www.mhra.gov.uk/news/2003.htm#ssri. Accessed Sept. 2, 2004.
6. Emslie GJ, Rush AJ, Weinberg WA, et al. A double-blind, randomized, placebo controlled trial of fluoxetine in children and adolescents with depression. Arch Gen Psychiatry 1997;54(11):1031-7
7. Emslie GJ, Heiligenstein JH, Wagner KD, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo-controlled, randomized clinical trial. J Am Acad Child Adolesc Psychiatry 2002;41(10):1205-15.
8. Wagner KD, Ambrosini P, Rynn M, et al. Efficacy of sertraline in the treatment of children and adolescents with major depressive disorder: two randomized controlled trials. JAMA 2003;290(8):1033-41.
9. Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled study. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
10. Wagner KD, Robb AS, Findling RL, et al. A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescents. Am J Psychiatry 2004;161(6):1079-83.
11. Laughren T. Background comments for Feb. 2, 2004 meeting of Psychopharmacological Drugs Advisory Committee (PDAC) and Pediatric Subcommittee of the Anti-Infective Drugs Advisory Committee (PedsAC). Available at: http://www.fda.gov/ohrms/dockets/ac/04/brief-ing/4006B1_03_Background Memo 01-05-04.doc. Accessed Sept. 15, 2004.
12. Emslie GJ. Making sense of the research puzzle. AACAP News 2004;35(2):
13. Whittington CJ, Kendall T, Fonagy P, et al. Selective serotonin reuptake inhibitors in childhood depression: Systematic review of published versus unpublished data. Lancet 2004;363:1341-5.Also available at: http://www.thelancet.com/journal/vol363/iss9418 (scroll to article title). Accessed Sept. 2, 2004.
14. Treatment for Adolescents with Depression Study (TADS) team. Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression, JAMA 2004;922(7):807-20.
15. Vorstman J, Lahuis B, Buitelaar JK. SSRIs associated with behavioral activation and suicidal ideation. J Am Acad Child Adol Psychiatry 2001;40:1364-5.
16. Food and Drug Administration. Psychopharmacologic Drugs Advisory Committee and the Anti-Infective Drugs Advisory Committee. Briefing information for public hearing Feb. 2, 2004. Available at: www.fda.gov/ohrms/dockets/ac/04/briefing/4006b1.htm. Accessed Sept. 2, 2004.
17. Culpepper L, Davidson JR, Dietrich AJ, et al. Suicidality as a possible side effect of antidepressant treatment. Primary Care Companion: J Clin Psychiatry 2004;6(2):79-86.
18. Olfson M, Gameroff MJ, Marcus SC, Waslic BD. Outpatient treatment of child and adolescent depression in the United States. Arch Gen Psychiatry 2003;60:1236-42.
19. Kressler RC, Avenevoli S, Merikangas KR. Mood disorders in children and adolescents: an epidemiological perspective. Biol Psychiatry 2001;49:1002-14.
20. Shaffer D, Fisher P, Dulcan MK, et al. The NIMH Diagnostic Interview Schedule for Children, Version 2.3 (DISC-2.3): description, acceptability, prevalence rates, and performance in the MECA Study. Methods for the Epidemiology of Child and Adolescent Mental Disorders Study. J Am Acad Child Adolesc Psychiatry 1996;35:865-77.
21. Harrington R, Bredenkamp D, Groothues C, et al. Adult outcomes of child and adolescent depression. III: Links with suicidal behaviors. J Child Psychol Psychiatry 1994;35:1309-19.
22. Preskorn SH. Outpatient management of depression: a guide for the practitioner(2nd ed). Caddo, OK: Professional Publications, 1999.
23. Lewinsohn PM, Clarke GN. Psychosocial treatments for adolescent depression. Clin Psychol Rev 1999;19:329-42.
How to help nicotine-dependent adolescents quit smoking
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Many adolescent psychiatric patients who smoke are not getting the help they need to quit. When we asked 120 teen inpatients if they smoked and then checked their charts, we found only 6 of 47 smokers had been diagnosed as nicotine-dependent.1
Adolescents who cannot quit on their own may benefit from smoking cessation therapies. Based on evidence and our experience, we offer a practical approach to treating nicotine dependence in adolescents, using drug and behavioral therapies.
PSYCHIATRIC COMORBIDITY
Psychiatric comorbidity is highly associated with cigarette smoking in adults and adolescents. In the United States:
- 44% of cigarettes smoked are sold to someone with a mental illness.2
- Persons with mental illness are 2.7 times more likely to smoke than are those without mental illness.2
- Most smokers start before age 18,3 and starting before age 13 is linked to psychopathology in later adolescence.4
Table
Smoking likelihood by age and comorbidity among adolescent psychiatric inpatients
| Significant variable | Logistic regression odds ratio | 95% confidence interval | Significance (P value) |
|---|---|---|---|
| Age | 1.30 | 1.03, 1.64 | 0.03 |
| Depressive disorders | 4.02 | 1.267, 12.734 | 0.018 |
| Conduct disorder | 12.96 | 1.678, 100.07 | 0.014 |
| Cannabis use disorder | 24.60 | 3.7, 163.42 | 0.0009 |
| Source: Data from 120 patients admitted to an inpatient child and adolescent psychiatry program. | |||
| Adapted with permission from reference 1. | |||
Disruptive behavior disorders in adolescent smokers include oppositional defiant disorder, conduct disorder, and attention-deficit/hyperactivity disorder (ADHD). Among psychiatric disorders, conduct disorder has the strongest association with smoking in adolescents.1 ADHD is associated with smoking and perhaps with increased difficulty in quitting.5,6
Mood disorders. Major depressive disorders have a strong, consistent, bidirectional association with smoking in the young. Depression may lead to smoking, and smoking to depression.7
Substance use disorders. Alcohol use disorders are strongly associated with smoking among adolescents, and the association is both bidirectional and dosedependent.8 Cannabis use disorder is also associated with cigarette smoking among adolescents (Table).9
Anxiety disorders. Evidence is emerging that anxiety disorders—especially social phobia—may be linked to smoking among adolescents.10
Nicotine withdrawal symptoms—irritability, anxiety, decreased concentration, increased appetite, craving for cigarettes—can mimic those of other psychiatric disorders. Adolescent smokers admitted to locked psychiatric units may experience withdrawal symptoms that require nicotine replacement treatment (Box).
Effect on quit rates. Psychiatric comorbidity may reduce quit rates during smoking cessation treatment.6 When smokers are trying to quit, watch for remission, worsening, or emergence of psychiatric conditions.
ASSESSING ADOLESCENT SMOKING
Adolescents with psychiatric diagnoses can be assessed for nicotine dependence—and vice versa—although accurately gauging their smoking habits is more difficult than in adults. For example:
- Rating scales for nicotine dependence severity—such as the modified Fagerstrom Tolerance Questionnaire11—lack standard cutoff scores for adolescents.
- Unlike adults, many adolescents cannot reliably report use in “packs per day” because the number of cigarettes they smoke varies widely from day to day.
Biological markers commonly used to assess smoking in adults include expired-air carbon monoxide (CO), cotinine (nicotine metabolite), and thiocynate levels. Preliminary evidence indicates that cotinine may be a more sensitive and specific biological marker for smoking among adolescents than CO levels.12 Thiocynate has not been evaluated as a marker for smoking in adolescents.
CO levels typically reflect smoking in the previous few hours, whereas the half-life of cotinine is longer (1 day or more). Also, factors such as environmental pollution or marijuana use can inflate CO levels. Thus, cotinine levels have greater accuracy and specificity, reflecting only the amount of nicotine consumed.
Unfortunately, most laboratories do not measure cotinine levels, and the expired-air CO test (CO Breathalyzer) is relatively expensive for most clinicians. Commercially available single-use cotinine test kits are modestly priced and provide semi-quantitative (a range instead of an exact number) urine cotinine levels. These tests, however, might not be covered by third-party insurers.
Until cotinine testing becomes widely available, we recommend a combination of self-report and expired-air CO level to monitor abstinence.
Self-report monitoring. Most clinicians rely on self-report rate of smoking among adolescents, as no screening assessment has been validated in this age group. As initial prompts, we recommend asking all adolescents if they smoke cigarettes, if they smoke regularly, and if they smoke daily.
We recommend using the “time line follow-back” method13 to monitor the self-reported smoking rate. Begin by providing the patient with a 30-day calendar, starting backwards from the day of assessment. Cite anchor points, such as special holidays and school or family events, to help the patient recall his or her cigarette use. Then have the patient fill in the number of cigarettes smoked each day for 30 days.
This assessment method appears more reliable than asking an adolescent “how many cigarettes do you smoke per day?”. After the initial time line follow-back assessment, encourage adolescent smokers to keep a daily diary of how many cigarettes they smoke, and monitor the diary at each visit.
Beth, age 15, was admitted overnight to an inpatient psychiatric unit after running away from home and being taken into police custody. Her primary diagnosis was conduct disorder.
At morning rounds, the nurse reported that Beth was very irritable, had threatened the staff, and had been moved to seclusion. During routine examination, the psychiatrist discovered that Beth was a half-pack/day smoker and “really” wanted a cigarette. The psychiatrist told her hospital policy did not allow smoking, but she could try a transdermal nicotine patch (TNP) to help reduce her nicotine withdrawal symptoms. She agreed and received a 14 mg/d nicotine patch.
Beth’s irritability improved substantially with TNP, and she moved back to her regular room within 2 hours without incident.
We have found daily smoking to be a good indicator of nicotine dependence, and anyone who smokes daily would receive significant health benefits from quitting. Hence, any daily smoker who wants to quit, regardless of DSM-IV nicotine dependence status, is a candidate for treatment.
BEHAVIORAL THERAPY
Unlike adults, adolescents usually lack smoking-related medical consequences, such as heart or lung disease. Even so, most adolescent smokers report that they would like to quit but face barriers such as:
- having to inform parents they smoke
- not knowing how to get help for smoking cessation
- lack of transportation for treatment
- lack of third-party reimbursement for smoking cessation treatment.
To help adolescents, we recommend following the U.S. Public Health Service guideline for smoking cessation.14 At least provide and discuss smoking cessation brochures developed specifically for adolescents. For example, one Centers for Disease Control and Prevention brochure describes what symptoms to expect when quitting, how to cope with craving, and other topics (see Related resources).
To manage peer pressure, we counsel teens to let their friends know they are trying to quit so that friends do not smoke in front of them. If that does not work, we ask patients to avoid being around friends who smoke at least for the first 2 weeks and preferably 2 months.
Many states have free telephone quit lines that provide support and advice on how to stop smoking. Several Web sites also are available for smokers (including adolescents) wanting to quit (see Related resources).
PHARMACOLOGIC TREATMENT
For adults, first-line FDA-approved medications for smoking cessation include nicotine replacement therapies (NRT)in transdermal, gum, inhaler, and lozenge forms and sustained-release bupropion. Nortriptyline, doxapine, and clonidine have shown effectiveness for smoking cessation but are not FDA-approved for this indication.15 Selegiline and mecamylamine have shown initial efficacy and are being examined in larger clinical trials.
For adolescents, little is known about what medications might help them stop smoking. Nicotine replacement therapies and bupropion SR have been most explored in adolescent smokers. The effect of psychiatric comorbidity on the quit rate is not well-studied in adolescents.
The transdermal nicotine patch (TNP) has shown modest results in preliminary trials among adolescents. One study found 11% abstinence at 6 weeks,16 and another found a <5% quit rate.17 A third study reported an 18% abstinence rate with a combination of TNP and contingency management therapy.18 Discussion of contingency management and other behavioral therapies is beyond the scope of this article.
A recent study comparing TNP, nicotine gum, and placebo in adolescent smokers found the lowest drop-out rate and highest compliance among the TNP group. Three-month abstinence rates were 17.6% for TNP, 6.5% for nicotine gum, and 2.5% for placebo. The difference between the TNP and placebo groups’ abstinence rates was statistically significant.19
Bupropion SR. In an open-label pilot study, our group treated 16 adolescent smokers weighing >90 lbs with bupropion SR, 150 mg bid. Average age was 18, and two-thirds of patients had ADHD. The endpoint abstinence rate—as measured by self-report and CO levels—was 31%, which is similar to rates reported in adult smokers treated with this dosage of bupropion SR.20
The adolescents did not gain weight during the study, which may be important to this age group. Reported side effects were similar to those in adults, with one adolescent reporting an allergic reaction (urticaria). We are conducting a larger follow-up study using bupropion SR with and without behavioral therapy.
A PRACTICAL CLINICAL APPROACH
Smoking behavior. For treatment, we propose two categories of adolescent smokers: regular (daily) and nonregular (nondaily) (see Algorithm). We recognize that many nondaily smokers smoke frequently and may benefit from aggressive treatment. However, we propose this two-track approach as a starting point because of limited data and medication risks, such as possible seizures with bupropion SR. We suggest:
- using behavioral therapy and patient education as first-line treatment for nonregular adolescent smokers
- using medication and behavioral therapy as first-line treatment for regular smokers and medication as second-line treatment for nonregular smokers who do not respond to behavior therapy/patient education.
Algorithm Suggested smoking cessation approaches for adolescents
Offer a treatment for at least 6 to 8 weeks before considering a change in therapy. One definition of initial success is no tobacco use in a 7-day period by self-report and biological verification (such as CO levels).
Behavioral therapy is relatively low-risk and helps many adult smokers. Despite a lack of evidence, some sort of behavioral therapy in combination with pharmacologic therapy might also help adolescent smokers.
When adolescents get disheartened by a slip or relapse to smoking, be patient and encourage them to try again. Inform them that smokers often require multiple attempts before they can quit completely.
Medication. Based on the limited published evidence, we consider TNP and bupropion SR first-line medications for adolescent smokers who want to quit.
For adult smokers, clinicians often combine medication and NRT to increase success rates.15 No data suggest that combining TNP and bupropion SR may be more effective than monotherapy in adolescents, but the combination might help those who do not respond to either agent alone.
We recommend starting bupropion SR treatment at least 1 week before the patient’s quit date. Titrate the dosage based on the package insert and patient tolerance.
Start NRT according to package instructions, and titrate dosages based on response:
- increase if the patient reports substantial craving and withdrawal symptoms, such as irritability and anxiety.
- decrease in case of toxicity (such as nausea).
In our experience, adolescent smokers require slightly lower NRT dosages than adults, although this varies among individuals.
- Centers for Disease Control and Prevention. Tobacco Information and Prevention Source (TIPS). www.cdc.gov/tobacco/quit/iquit.htm
- National Institute of Drug Abuse. NIDA for Teens. The Science Behind Drug Abuse. http://teens.drugabuse.gov/index.asp
- Society for Research on Nicotine and Tobacco. www.srnt.org
Drug brand names
- Bupropion SR • Zyban
- Clonidine • Catapres
- Doxapine • Sinequan
- Mecamylamine • Inversine
- Nortriptyline • Pamelor
- Selegiline • Eldepryl
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
1. Upadhyaya HP, Brady KT, Wharton M, Liao J. Psychiatric disorders and cigarette smoking among child and adolescent psychiatry inpatients. Am J Addict 2003;12:144-52.
2. Lasser K, Boyd JW, Woolhandler S, et al. Smoking and mental illness. A population-based prevalence study. JAMA 2000;284(20):2606-10.
3. Johnston LD, O’Malley PM, Bachman JG. Teen smoking continues to decline in 2003, but declines are slowing. Ann Arbor, MI: University of Michigan News and Information Services, Dec. 19, 2003. Available at: www.monitoringthefuture.org/press.html. Accessed 08/13/04.
4. Upadhyaya HP, Deas D, Brady KT, Kruesi M. Cigarette smoking and psychiatric comorbidity in children and adolescents. J Am Acad Child Adolesc Psychiatry 2002;41(11):1294-1305.
5. Molina BS, Pelham WE. Childhood predictors of adolescent substance use in a longitudinal study of children with ADHD. J Abnorm Psychol 2003;112(3):497-507.
6. Pomerleau OF, Downey KK, Stelson FW, Pomerleau CS. Cigarette smoking in adult patients diagnosed with attention-deficit/hyperactivity disorder. J Subst Abuse 1995;7:373-8.
7. Dierker LC, Avenevoli S, Merikangas KR, et al. Association between psychiatric disorders and the progression of tobacco use behaviors. J Am Acad Child Adolesc Psychiatry 2001;40(10):1159-67.
8. Zacny J. Behavioral aspects of alcohol-tobacco interactions. Recent Dev Alcohol 1990;8:205-19.
9. Rohde P, Lewinsohn P, Kahler C, et al. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry 2001;40(1):83-90.
10. Sonntag H, Wittchen HU, Hofler M, et al. Are social fears and DSM-IV social anxiety disorder associated with smoking and nicotine dependence in adolescents and young adults? Eur Psychiatry 2000;15:67-74.
11. Prokhorov AV, Pallonen UE, Fava JL, et al. Measuring nicotine dependence among high-risk adolescent smokers. Addict Behav 1996;21:117-27.
12. McDonald P, Colwell B, Backinger CL, et al. Better practices for youth tobacco cessation: evidence of review panel. Am J Health Behav 2003;27(suppl 2):S144-S158.
13. Sobell LC, Sobell MB, Leo GI, Cancilla A. Reliability of a timeline method: assessing normal drinkers’ reports of recent drinking and a comparative evaluation across several populations. Br J Addict 1988;83(4):393-402.
14. Fiore M, Bailey W, Cohen S. Treating tobacco use and dependence: Clinical practice guideline. Rockville, MD: US Public Health Service, 2000.
15. George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends Pharmacol Sci 2004;25(1):42-8.
16. Hurt RD, Croghan GA, Beede SD, et al. Nicotine patch therapy in 101 adolescent smokers. Efficacy, withdrawal symptom relief, and carbon monoxide and plasma cotinine levels. Arch Pediatr Adolesc Med 2000;154:31-7.
17. Smith TA, House RF, Jr, Croghan IT, et al. Nicotine patch therapy in adolescent smokers. Pediatrics 1996;98:659-67.
18. Hanson K, Allen S, Jensen S, Hatsukami D. Treatment of adolescent smokers with the nicotine patch. Nicotine Tob Res 2003;5(4):515-26.
19. Moolchan ET. Efficacy of the nicotine patch and gum for the treatment of adolescent tobacco dependence. Scottsdale, AZ: Society for Research on Nicotine and Tobacco Research annual meeting, 2004.
20. Upadhyaya HP, Brady KT, Wang W. Bupropion SR in adolescents with comorbid ADHD and nicotine dependence: a pilot study. J Am Acad Child Adolesc Psychiatry 2004;43(2):199-205.
Acute bipolar mania: Aggressive initial dosing provides faster symptom relief
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
Evidence on aggressive initial dosing of mood stabilizers and antipsychotics is changing the way acute bipolar mania is treated. To help you apply this information, we searched the literature and meeting abstracts for aggressive strategies tested to date. This article discusses how to:
- identify patients who may benefit from loading or aggressive initial dosing
- calculate mood stabilizer dosages
- dose each atypical antipsychotic
- manage potential antipsychotic side effects during maintenance therapy.
PATIENT SELECTION
Rapidly achieving therapeutic blood levels relieves patient suffering faster than standard titration. It quickly calms the hyperactivity, impulsivity, tension, hostility, and uncooperativeness that distress patients and increase the risk of harm to themselves and others.
The challenge of using higher dosages is to minimize side effects. Loading is not a one-size-fits-all approach, as antimanic drugs’ unique pharmacokinetic and pharmacotherapeutic properties influence how each agent is used.
An interesting body of literature advocates using mood stabilizers plus antipsychotics to treat mania, suggesting greater efficacy than with either agent alone.16 This strategy raises important questions, such as:
- Can two drugs be loaded simultaneously?
- Can patients taking mood stabilizers be treated with antipsychotic loading, and can those taking antipsychotics receive loading dosages of mood stabilizers?
- Would “double loading” improve bipolar mania treatment?
Answers to these questions are needed because of increased demands on clinicians to control hospital costs by rapidly and effectively treating patients with bipolar mania.
Loading doses cannot be standardized but are calculated by multiplying target steady-state plasma concentration by volume of distribution. We suggest aggressive initial schedules for divalproex sodium and atypical antipsychotics in this article with the understanding that practitioners will adjust them based on each patient’s tolerance and response.
Hospitalization. Patients with acute bipolar mania should be supervised closely in the hospital during loading or aggressive initial dosing. Monitor for cardiovascular changes, neurologic disturbances, sensorium changes, and response.
Precautions. Not all patients are candidates for aggressive initial dosing. Contraindications include age <18 or >65 years, pregnancy, breast-feeding, medical illness, and known sensitivity to the medication being given.
Higher-than-usual dosing increases the risk of excessive drug concentrations in sensitive individuals—such as those with a history of sensitivity to lower dosages of similar medications—and toxic levels of drugs with long half-lives can persist. When in doubt, consider giving a smaller amount of the loading dose early in the day, followed later by a larger amount.
DRUG SELECTION
Loading and aggressive initial dosing strategies for bipolar mania were first advanced for divalproex sodium.1 Investigators then examined loading strategies for lithium and carbamazepine, as well as the antipsychotics olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole, which are known to have antimanic properties.
Olanzapine, quetiapine, and risperidone are FDA-approved for short-term treatment of acute manic episodes associated with bipolar I disorder, and similar indications were being considered for aripiprazole and ziprasidone as this article went to press. We discuss evidence on loading and aggressive initial dosing strategies for each agent.
No studies have compared one loading strategy with another. Thus, when we choose drugs for loading, we consider what each patient needs, available formulations, tolerability, and efficacy for long-term stabilization and maintenance treatment.
LITHIUM
Lithium loading targets the therapeutic range (<1.4 mEq/L), without crossing the toxic threshold (>1.5 mEq/L). Lithium loading has shown antimanic effects, although using >30 mg/kg/d causes severe nausea and vomiting.
Moscovich et al2 reported a case series of 9 adults with acute mania who received lithium loading dosages of 4,050 mg/d. Patients tolerated lithium well at plasma drug levels of approximately 1.2 to 1.4 mEq/L. Their manic symptoms declined significantly within 4 to 5 days, as measured by Clinical Global Impression (CGI) severity of illness, Biegel-Murphy Mania State Rating Scale, and Brief Psychiatric Rating Scale scores.
Table 1
How to calculate divalproex loading for acute bipolar mania*
| Days 1 and 2 |
| Patient weight in pounds x 15 = dosage (mg/d) (Example: 150 lbs x 15 = 1,750 mg/d) |
| Days 3 to 10 |
| Patient weight in pounds x 10 = dosage (mg/d) (Example: 150 lbs. x 10 = 1,500 mg/d) |
| To avoid splitting tablets, make dosage divisible by 125 (round up for young adults, round down for older adults). Divide into bid or tid doses for improved tolerability. |
| Days 4 and 7 |
| Draw blood to monitor valproic acid levels and for other values such as liver function studies. |
| * For use with oral divalproex sodium (delayed-release or extended-release formulations). Do not use valproic acid preparations, as loading is unlikely to be well tolerated. |
| Source: Reference 5 |
Kook et al3 attempted a 30-mg/kg loading dose of slow-release lithium carbonate in 38 patients to evaluate the safety of achieving a therapeutic level in 12 hours. No patient experienced adverse effects during loading or in the 12 hours after loading was completed.
Patients who develop a manic episode while taking lithium pose a therapeutic dilemma. If they stop taking lithium—especially abruptly—manic symptoms can return in as few as 2 days. For these patients, consider increasing the usual dosage by 50% to 100% on the first day of mania treatment,4 then continue a dosage that maintains efficacy and achieves a therapeutic blood level.
To avoid GI upset, avoid any single dose of >1,500 mg; if a greater dosage is needed, divide it for improved tolerance. Obtain lithium blood levels at baseline, after 4 days, and thereafter as clinically relevant to monitor for drug interactions that may affect serum levels.
Side effects—GI irritation, tremor, muscle weakness, thirst, polyuria—are common in the first week, and weight gain may occur after prolonged treatment.
DIVALPROEX
In a study by Keck et al1 of 19 adults with acute bipolar mania, aggressive initial dosing of divalproex sodium, 20 mg/kg/d, was well-tolerated and shortened hospitalization. Side effects including sedation and nausea occurred at a rate similar to that seen with more-gradual titration.
To calculate the initial dose, the authors used a conversion factor of 20 mg/kg/d or added a zero to the patient’s weight in pounds. This amount was given in single or divided doses the first day, then continued for 4 to 7 days. Blood levels were measured on day 4, and all 15 patients who completed the study achieved serum valproate levels >50 mcg/mL.
In a double-blind study by Hirschfeld et al,5 59 adults with Young Mania Rating Scale (YMRS) scores >14 were randomly assigned to receive divalproex oral loading; divalproex nonloading (250 mg tid on days 1 and 2, followed by standard titration on days 3 to 10); or lithium carbonate (300 mg tid, followed by standard titration on days 3 to 10).
Divalproex loading—30 mg/kg/d on days 1 and 2, followed by 20 mg/kg on days 3 to 10— yielded valproate levels >50 mcg/mL in 84% of patients by day 3. Rapid loading appeared to increase antimanic efficacy, as measured by YMRS endpoint scores, without increasing adverse effects. For Table 1, we converted this study’s metric values to patient weight in pounds.
Side effects. Divalproex can inhibit metabolism of other drugs (including anticonvulsants such as lamotrigine) and increase their blood levels. Side effects such as sedation, alopecia, abdominal pain, diarrhea, and tremor may require observation and treatment. Pancreatitis, hepatitis, and allergic reactions are rare but may require discontinuation.
Recommendation. Aggressive initial dosing and loading for patients with acute mania has been reported with enteric-coated delayed-release and extended-release6 divalproex sodium tablets. With identical dosages, the extended-release form produces 11% lower serum levels than the delayed-release form.7
Aggressive dosing of oral valproic acid preparations is not recommended and is likely to be poorly tolerated.8 A pilot study evaluating IV valproate loading in acute mania found no changes in mania in the 2 hours that followed loading.9
CARBAMAZEPINE
Carbamazepine is used off-label to treat mania and mixed phases of bipolar disorder.4,9,10 Excessive absorption rates are associated with dizziness, ataxia, and nausea. Side effects may occur when the plasma level is therapeutic for epilepsy (4 to 12 mcg/mL).11
In mania, the relationship between carbamazepine levels and clinical response is not always clear. Lerer et al12 found a correlation between acute mania response and a serum level of 8.8 mcg/mL (range 4.7 to 14.0 mcg/mL).
Patients with acute mania may require 600 to 1,600 mg/d. Carbamazepine is available in immediate-release and controlled-release preparations. Because generic preparations might not be bio-equivalent,13 use the same formulation throughout treatment to maintain consistent serum levels.
Enzymatic auto-induction—in which carbamazepine gradually increases the activity of its metabolic enzyme—is likely to occur at 3 to 5 weeks of administration, often after hospital discharge. An aggressive initial rescue adjustment can be used if a patient develops mania after having been stabilized on carbamazepine for >6 weeks. Because these patients are past the point of auto-induction, a target blood level of 8.8 mcg/mL can be used and the dosage adjusted proportionally.
For example, if mania recurs in a patient who is stabilized on carbamazepine, 400 mg/d (plasma level 4.5 mcg/mL), carbamazepine can be loaded up to 800 mg/d. Measure the plasma level within 4 days; the target level would be 9.0 mcg/mL (2 times 4.5 mcg/mL), which closely approximates steady state and sets up a ratio to reach the target level of 8.8 mcg/mL.
Side effects include ataxia, diplopia, and dizziness. Complete blood counts, liver function studies, plasma levels, and serum chemistries require regular monitoring.
Recommendation. Carbamazepine is not recommended for oral loading in patients who have never taken it or for those with hyponatremia, hepatic dysfunction, or history of intolerance or agranulocytosis.
OLANZAPINE
Olanzapine has pharmacologic properties favorable for loading.4 The recommended dosage for acute mania is 15 mg/d with standard titration; Karagianis et al14 showed that initial loading doses of >20 mg/d resulted in good control of agitated patients with psychosis. Side effects were uncommon, with sedation occurring in 14% of patients in this case series. The loading dose reduced agitation more effectively than did dosages <20 mg/d given to similar patients.
A multicenter study of 148 acutely agitated inpatients with a variety of psychiatric disorders compared olanzapine rapid initial dose escalation with usual clinical practice.15 Mean aggressive dosages were 28.8 mg/d on day 1, 30.3 mg/d on day 2, and 16.1 mg/d on day 5. Usual-practice dosages were 10 mg/d, plus lorazepam, 0 to 4 mg/d for the first 2 days and 0 to 2 mg/d on days 3 to 4. Based on Positive and Negative Syndrome Scale excited component subscale scores, olanzapine loading controlled agitation more effectively than did usual practice, with similar side-effect rates.
IM olanzapine or the orally dissolving form are bioequivalent to the tablets and may be used for acute agitation associated with bipolar mania in certain clinical settings.14
Table 2
Suggested antipsychotic loading for acute bipolar mania and mixed states*
| Drug | Day(s) | Aggressive initial dosing schedule | Comment |
|---|---|---|---|
| Aripiprazole24,25 | 1 2 to 3 4+ | 30 mg once daily with food 30 mg/d with food Reduce dosage by 10 to 15 mg/d, based on tolerance and response | Nausea and vomiting may occur in first few days; adjust dosage based on tolerance and response |
| Olanzapine14,15 | 1 and 2 3 and 4 5 to 10 | 40 mg in single or divided dosage 20 to 30 mg at bedtime 15 mg once daily (may reduce to 5, 7.5, or 10 mg/d) | Adjust dosage based on tolerance and response; oral or IM formulations may be used |
| Quetiapine19,21 | 1 2 and 3 4 to 10 | 100 mg upon admission and 100 mg at bedtime 100 mg bid (or tid to qid) plus 100 to 200 mg at bedtime 200 mg bid plus 200 to 300 mg at bedtime; may adjust to 400 to 800 mg/d) | Adjust dosage based on tolerance and response |
| Risperidone16,18 | 1 2 3 and 4 | 3 mg in single or divided dosage 4 mg in single or divided dosage 5 mg in single or divided dosage | Adjust dosage by 1 mg up or down, based on tolerance and response; use tablet, rapid-dissolving tablet, or liquid form, but not long-acting IM form |
| Ziprasidone22,23 | 1 2 | 20 mg IM in single dose (may repeat for severe agitation) or 40 mg po bid with food 60 to 80 mg po bid with food | Adjust dosage based on tolerance and response |
| * For hospitalized or partially hospitalized patients, ages18. Not recommended for patients who are pregnant, breastfeeding, medically ill, age >65, or with known sensitivity to the antipsychotic being given. | |||
RISPERIDONE
Sachs et al16 studied 156 inpatients who developed an acute manic or mixed episode while receiving lithium or divalproex. These patients were randomly assigned to begin adjunctive risperidone, 2 mg/d, haloperidol, or placebo. Dosing was flexible, increasing or decreasing by 1 mg/d. Risperidone’s mean modal dosage was 3.8 mg/d across 3 weeks, with mean exposure of 17 days. Risperidone plus a mood stabilizer was more effective than a mood stabilizer alone, and the combination provided rapid, well-tolerated control of manic symptoms.
In a double-blind trial, Hirschfeld et al17 randomly assigned 279 patients with acute bipolar mania to risperidone, 1 to 6 mg/d, or placebo for 3 weeks. As early as day 3, YMRS scores were reduced significantly more with risperidone than with placebo. Somnolence was the most common side effect, and mean modal dosage was 4.1 mg/d.
Table 3
Screening schedule for antipsychotic side effects during bipolar maintenance treatment
| Baseline | |
| Side effect | Recommended screening |
| Weight gain | Weight and body mass index (BMI) monthly for first 3 months; waist circumference |
| Hypertension | Blood pressure |
| Hyperglycemia | Fasting plasma glucose, with glycosylated hemoglobin (Hb A 1c ) if hyperglycemia is detected |
| Hyperlipidemia | Fasting lipid profile |
| Tardive dyskinesia | Abnormal Involuntary Movement Scale (AIMS) or other screen |
| Ophthalmic changes | Ophthalmologic examination for patients taking quetiapine and for all with diabetes mellitus |
| Follow-up schedules | |
| 3 months | |
| Weight and BMI | |
| Blood pressure | |
| Fasting plasma glucose, with Hb A 1c if hyperglycemia is detected; Hb A 1c values may be used to measure interval changes in glucose tolerance | |
| Fasting lipid profile | |
| 6 months | |
| AIMS or other tardive dyskinesia screen | |
| Ophthalmologic examination | |
| Source: Adapted from reference 26. | |
In a multicenter, randomized, double-blind, placebo-controlled study of patients with acute bipolar mania, Khanna et al18 assigned patients to receive risperidone monotherapy (mean modal dosage 5.6 mg/d) or placebo for 3 weeks. Mania scores of patients receiving risperidone were significantly lower at weeks 1 and 2, compared with the placebo group. Risperidone was well-tolerated, with no unexpected adverse events.
Recommendation. Because of a risk of extrapyramidal symptoms (EPS) and orthostatic hypotension, initial risperidone loading dosages >4 mg on day 1 are not recommended.
QUETIAPINE
Quetiapine has shown antimanic efficacy as monotherapy and as adjunctive therapy to mood stabilizers.19,20 The effective dosage was a mean 600 mg/d (range 400 to 800 mg/d) in monotherapy and adjunctive treatment. These studies achieved 400 mg/d within the first 4 days (100 mg on day 1, 200 mg on day 2, 300 mg on day 3, and 400 mg on day 4).
These data combined with revised prescribing information suggest aggressive initial dose escalation of quetiapine within the first 4 days for selected patients. A titration study in patients with schizophrenia used a more-rapid escalation rate of 400 mg within 2 days.21 Dizziness, orthostatic hypotension, and sedation were not more frequent in this high-dose group than in the two lower-dose titration groups. In our experience, 200 to 400 mg can be given the first day of treatment.
ZIPRASIDONE
In a randomized, double-blind, controlled trial, Keck et al22 assigned 210 patients with a manic or mixed bipolar episode to 3 weeks of ziprasidone, 40 to 80 mg bid, or placebo. Ziprasidone produced rapid, sustained improvement in manic symptoms on all primary and most secondary efficacy measures, such as the YMRS and CGI.
Significant improvements seen within 2 days were maintained. Ziprasidone was well tolerated and was associated with a low EPS rate; neither weight gain nor clinically significant changes in vital signs were seen.
IM ziprasidone, which is approved for use in schizophrenia, may also have efficacy in bipolar mania.23 The recommended dose of 20 mg IM is equivalent to 120 to 160 mg orally, so a single injection may reach the target antimanic dosage.
Recommendation. Ziprasidone could be an option for aggressive initial dosing for a patient who has previously received ziprasidone IM and is not at risk for QTc prolongation.
ARIPIPRAZOLE
In a randomized controlled trial, Keck et al24 assigned 262 patients with acute bipolar mania or mixed states to aripiprazole, 30 mg/d, or placebo for 3 weeks. By day 4, manic symptoms were improved significantly more in patients receiving aripiprazole, and discontinuation rates were similar.
Similarly, in a randomized, controlled multi-center study, Sachs et al25 used 30 mg/d in 272 patients with bipolar mania or mixed states. Compared with placebo, aripiprazole produced significant improvement by day 4, with similar discontinuation rates.
Recommendation. Aggressive initial dosing of aripiprazole could be useful for a patient who does not require an IM or rapidly dissolvable medication.
Table 4
Suggested response to metabolic changes during bipolar maintenance therapy
| Metabolic change | Therapeutic action |
|---|---|
| ≥5% increase in total body weight | Consider weight-reduction strategies or medication adjustment |
| Fasting glucose: ≥126 mg/mL ≥300 mg/mL or ≤60 mg/mL | Consider evaluation for diabetes mellitus Seek immediate consultation |
| Total cholesterol ≥200 mg/dL or triglycerides ≥165 mg/dL | Consider lipid-lowering with dietary and/or medication changes |
| Source: Adapted from reference 26. | |
MAINTENANCE THERAPY
Ideally, if a medication stabilizes a patient’s acute bipolar mania, that medication is continued for further stabilization and maintenance. Aggressive initial dosing befits this approach because it establishes a therapeutic blood level and usually reveals any side effects within days. Moreover, patients often prefer to continue the medication that provided relief when they felt most distressed.
Weight gain. Long-term use of atypical antipsychotics may be associated with weight gain, dyslipidemia, and the development of metabolic syndromes and diabetes mellitus. Weight gain risk may be further elevated in patients taking both antipsychotics and lithium or valproic acid.26 When atypical antipsychotics are used for bipolar maintenance therapy, the American Diabetes Association and American Psychiatric Association recommend close monitoring (Tables 3 and 4).
Abnormal movements. Though tardive dyskinesia risk is very low with atypical antipsychotics, we recommend screening during the first year of treatment. The development of diabetes mellitus may precipitate or worsen abnormal movements.
Related resources
- American Diabetes Association. www.diabetes.org
- American Obesity Association. www.obesity.org
- Expert Consensus Treatment Guidelines for Bipolar Disorder: A Guide for Patients and Families. Task Force for the APA Practice Guideline for the Treatment of Patients with Bipolar Disorder. www.psychguides.com/pfg3.php
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine • Tegretol
- Divalproex sodium • Depakote
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Disclosures
Dr. Carroll is a consultant to Abbott Laboratories, Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Eli Lilly and Co., Janssen Pharmaceutica, and Pfizer Inc.
Dr. Fawver is a consultant to Eli Lilly and Co. and Pfizer Inc.
Dr. Thalassinos is a consultant for AstraZeneca Pharamaceuticals, Eli Lilly and Co., and Pfizer Inc.
Acknowledgment
The authors thank Donald R. Schmitt, PharmD, Christopher Thomas, PharmD, and Tina Fore, Library Service, Chillicothe VA Medical Center, for their help in identifying articles used in this review.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
1. Keck PE, Jr, McElroy SL, Tugrul KC, et al. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry. 1993;54:305-8.
2. Moscovich DG, Shapira B, Lerer B, et al. Rapid lithiumization in acute manic patients. Hum Psychopharmacol. 1992;7:343-5.
3. Kook KA, Stimmel GL, Wilkins JN, et al. Accuracy and safety of a priori lithium loading. J Clin Psychiatry. 1985;46:49-51.
4. Carroll BT, Thalassinos A, Fawver JD. Loading strategies in acute mania. CNS Spectrums. 2001;6:919-30.
5. Hirschfeld RMA, Allen MH, McEvoy J, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry. 1999;60:815-18.
6. Miller BP, et al. Oral loading of extended-release divalproex in acute mania (presentation). New Orleans: American Psychiatric Association annual meeting, 2001.
7. Thibault M, Blume WT, Saint-Hilaire JM, et al. Divalproex extended-release versus the original divalproex tablet: results of a randomized, crossover study of well-controlled epileptic patients with primary generalized seizures. Epilepsy Res. 2002;50:243-9.
8. Hirschfeld RMA, Baker JD, Wozniak P, et al. The safety and early efficacy of oral-loaded divalproex versus standard-titration divalproex, lithium, olanzapine, and placebo in the treatment of acute mania associated with bipolar disorder. J Clin Psychiatry. 2003;64:841-6.
9. Phrolov K, Applebaum J, Levine J, et al. Single-dose intravenous valproate in acute mania. J Clin Psychiatry. 2004;65:68-70.
10. Keck PE, Jr, McElroy SL, Bennet TA. Pharmacologic loading in the treatment of acute mania. Bipolar Disord. 2000;2:42-6.
11. Dunn RT, Frye MS, Tim KA, et al. The efficacy and use of anticonvulsants in mood disorders. Clin Neuropharmacol. 1998;21:215-35.
12. Lerer B, Moore N, Meyendor E, et al. Carbamazepine versus lithium in mania a double-blind study. J Clin Psychiatry. 1987;48:89-93.
13. Brown B. The use of generic mood stabilizers: carbamazepine (monograph). J Clin Psychiatry. 1997;15(4):11-17.
14. Karagianis JL, Dawe IC, Thakur A, et al. Rapid tranquilization with olanzapine in acute psychosis: a case series. J Clin Psychiatry. 2001;62(suppl 2):12-16.
15. Baker RW, Kinon BJ, Maguire GA, et al. Effectiveness of rapid initial dose escalation of up to 40 milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol. 2003;23(4):342-8.
16. Sachs GS, Grossman F, Ghaemi SN, et al. Combination of mood stabilizer with risperidone or haloperidol for the treatment of acute mania: a double-blind, placebo controlled comparison of efficacy and safety. Am J Psychiatry. 2002;159:1146-54.
17. Hirschfeld RMA, Keck PE, Jr, Karcher K, et al. Rapid antimanic effect of risperidone monotherapy: a 3-week multicenter, randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2004;161:1057-65.
18. Khanna S, Vieta E, Lyons B, et al. Risperidone monotherapy in acute bipolar mania (abstract P219). Pittsburgh: Fifth International Conference on Bipolar Disorder, 2003.
19. Jones MW, Huizar K, et al. Quetiapine monotherapy for acute mania associated with bipolar disorder (poster presentation). San Francisco: American Psychiatric Association annual meeting, 2003.
20. Sachs G, Mullen JA, Devine NA, Sweitzer DE. Quetiapine versus placebo as adjunct to mood stabilizer for the treatment of acute mania (abstract). Bipolar Disord. 2002;4(suppl 1):133.-
21. Smith MA, McCoy R, Hamer J, Brecher M. Optimal titration for quetiapine (poster presentation): Boca Raton, FL: National Clinical Drug Evaluation Unit annual meeting, 2002.
22. Keck PE, Jr, Versiani M, Potkin S, et al. Ziprasidone in the treatment of acute bipolar mania: a three-week, placebo-controlled, double-blind, randomized trial. Am J Psychiatry. 2003;160:741-8.
23. Holtzheimer PE, Neumaier JF. Treatment of acute mania. CNS Spectrums. 2003;8(12):917-28.
24. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-8.
25. Sachs G, Sanchez R, Marcus R, et al. Aripiprazole vs placebo with an acute manic or mixed episode. New York: American Psychiatric Association annual meeting, 2004.
26. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes. Diabetes Care. 2004;27:596-601.
New tool: Genotyping makes prescribing safer, more effective
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
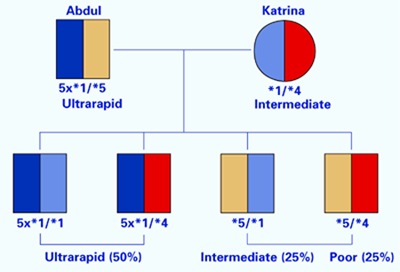
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
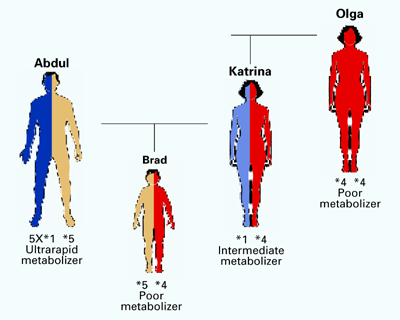
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Genotyping for cytochrome P-450 2D6 gene variations is emerging as a valuable clinical tool to help psychiatrists identify patients who:
Genetic variation has long been known to influence how individuals metabolize drugs, but only recently could we apply this information.5 Many academic medical centers and the two largest U.S. reference laboratories offer 2D6 testing at costs of $200 to $500.
Before long, psychiatrists may adopt routine genotyping before prescribing 2D6 substrate medications. This article and four vignettes illustrate the clinical benefits of psychiatric pharmacogenomics and suggest when prospective genotyping could help you select and dose medications.
Table 1
Drugs metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants | Other medications |
|---|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine | Codeine |
| Fluoxetine | Perphenazine | Dextromethorphan | |
| Nortriptyline | Risperidone | Oxycodone | |
| Paroxetine | Thioridazine | ||
| Venlafaxine | |||
| *Evidence suggests that these medications are predominantly metabolized by the 2D6 enzyme. | |||
| Be careful when prescribing these agents to patients who are poor 2D6 metabolizers. | |||
Why test for the 2D6 gene?
The 2D6 gene codes for the 2D6 enzyme, the primary enzyme required to metabolize many psychotropics Table 1.
Genetic variations. A common variation in a gene is frequently called an allele. More than 100 2D6 gene variations have been described. Consequently, the 2D6 gene’s enzyme activity also varies widely. Most mutations decrease the enzyme’s activity, but some polymorphisms change the gene’s promoter region, which can lead to upregulation and increased enzyme production.
Each 2D6 gene variation has been labeled with a standardized abbreviation (Table 2):
- *1 refers to the “normal” gene
- *2 stands for several variants with different activity levels.
- *3, *4, *6, *7, *8, *9, *10, *11, *12, *14, and *17 code for proteins with little or no activity.
- *5 indicates that the gene is deleted, and no enzyme can be produced.
Multiple copies. Another characteristic of the 2D6 gene is its unusually high propensity to accumulate in multiple copies on the 22nd chromosome. As many as 13 copies of the 2D6 gene have been shown on a single chromosome. Given that each gene can code for the 2D6 enzyme, patients with multiple copies can metabolize 2D6 substrate medications very rapidly.
Nonpsychiatric drugs. The 2D6 enzyme is also involved in metabolizing many nonpsychiatric drugs. To produce analgesia, for example, the 2D6 enzyme must metabolize the prodrug codeine to morphine. Thus, individuals with no 2D6 enzyme activity experience no analgesia with codeine. Approximately 7% of Caucasians metabolize codeine poorly. Conversely, individuals with multiple 2D6 gene copies metabolize codeine to morphine very rapidly, with potential for acute mental status changes, including psychosis.
4 metabolizer types. Based on variation in individual 2D6 genotype, a patient is usually categorized as being an ultrarapid, extensive, intermediate, or poor metabolizer (Table 3). The following case vignettes of patients in each category illustrate the clinical benefits of 2D6 genotyping.
Ultrarapid metabolizer: Extra 2D6 copies
Abdul, 49, is an Ethiopian businessman engaged in international commerce. While in the United States, he underwent a routine wisdom-tooth extraction and was treated with acetaminophen and codeine. Despite having no psychiatric history, he began to experience extreme discomfort and flashing visual hallucinations within 24 hours of taking two codeine doses. The oral surgeon instructed him to discontinue codeine, and his symptoms resolved within 24 hours.
Because of this experience, Abdul underwent genotyping for the 2D6 gene. He was found to have five active copies on one 22nd chromosome and no copies on the other (Figure 1). This genotype is unusual in western European populations but common in North Africa. Abdul then received alternate analgesics; psychiatric symptoms did not recur.
A patient such as Abdul, with multiple copies of a functional 2D6 gene, is an ultrarapid metabolizer. The 22nd chromosome—where the 2D6 gene is located—is short and contains areas of high homology. As a result, uneven crossover events occur more frequently during meiosis than is typical of larger chromosomes. Uneven crossover results in one gamete with two copies of the 2D6 gene and the other gamete with none.
2D6 enzyme activity is not essential for survival, which raises fascinating questions about this gene’s evolutionary importance. In certain geographic regions, many individuals have multiple copies of the gene. In Ethiopia—the country with the highest documented number of ultrarapid metabolizers—more than 25% of the population has one chromosome with multiple copies of the 2D6 gene.6 Because these copies produce an increased amount of 2D6 active enzyme, normal doses of 2D6 substrate medications do not benefit these individuals.
Table 2
How common 2D6 gene variations (alleles) affect 2D6 enzyme activity
| Allele label | 2D6 enzyme activity | Allele frequency (%)† |
|---|---|---|
| *1 | Normal | 37 |
| *2 | Decreased | 3.3 |
| *2P | Modestly increased | 6 |
| *3 | None | 1 |
| *4 | None | 18 |
| *5 | None (gene deletion) | 4 |
| *6 | None | 1 |
| *7 | None | <1 |
| *9 | Decreased | 3 |
| *10 | Decreased | 2 |
| *11 | None | 0 |
| *12 | None | <1 |
| *14 | Decreased | <1 |
| *17 | Decreased | <1 |
| †In Caucasian populations | ||
Table 3
Four ways patients respond to 2D6 substrate drugs
| Category | Patient characteristics | % of Caucasian population |
|---|---|---|
| Ultrarapid | Metabolize 2D6 medications rapidly resulting in poor response | 1 to 2 |
| Extensive | Metabolize 2D6 medications at a normal rate | 73 to 82 |
| Intermediate | Metabolize 2D6 medications at a slower-than-normal rate | 10 to 15 |
| Poor | Metabolize 2D6 medications very slowly with increased risk of side effects | 7 to 10 |
When treating ultrarapid metabolizers one strategy is to increase the dosage to obtain a therapeutic effect Because some substrates have complex metabolic pathways, however, high concentrations of secondary or tertiary metabolites can accumulate. Thus, when a substance’s metabolic pathway is not well-documented, a more cautious approach is to choose a medication metabolized by another pathway.
Figure 1 Genotypes and metabolizer categories of 4 illustrative patients
Extensive metabolizer: The ‘norm’
George, a 31-year-old Ethiopian architect, is Abdul’s second cousin. He developed acute depression with intense suicidal ideation and sought psychiatric consultation. He had no history of atypical drug reactions, but—because of his ethnic background—his psychiatrist was concerned that George might be a rapid metabolizer.
2D6 genotyping showed that George’s genotype was *1/*1, which meant he had two functional 2D6 copies (Figure 1). This genotype suggests that he could tolerate many antidepressants. The psychiatrist concluded—with some confidence—that George would not experience adverse effects or low serum levels when prescribed fluoxetine at usual dosages.
Extensive metabolizers have two normal 2D6 gene copies and can produce adequate active 2D6 enzyme Patients with this genotype—common in Caucasians—are generally said to have “normal” 2D6 metabolism. This means they metabolize 2D6 substrate medications at a rate within the recommended dosage ranges determined from North American or European pharmacokinetic studies.
Intermediate metabolizer: Mixed message
Katrina, 27, represents the government of her native Sweden in trade agreements. When she developed depressive symptoms (insomnia, sense of hopelessness), Katrina saw her psychiatrist. She reported that her family has a history of adverse reactions to multiple medications, but she had tolerated most medications. In fact, she had twice been successfully treated with relatively high doses of codeine.
Her psychiatrist suspected she was an intermediate 2D6 metabolizer and ordered testing. Her genotype was *1/*4, with one normal copy and one that produced no functional 2D6 enzyme (Figure 1).
Based on her clinical history and this genotypic information, the psychiatrist prescribed sertraline—metabolized by both 2D6 and 3A4 enzymes— at 50 mg/d. Because Katrina metabolized sertraline at a slower-than-usual rate, she developed a therapeutic blood level and responded well to this low dosage.
Intermediate metabolizers have a chromosome with one functional 2D6 gene copy. The other chromosome has either a copy with a defective functional polymorphism or a deletion of the gene. These patients usually tolerate 2D6 substrate drugs in low dosages.
Poor metabolizer: ‘medication-sensitive’
Olga, Katrina’s mother, has always lived in northern Sweden. She has no psychiatric history except for one psychotic episode that required hospitalization.
Her psychotic illness began on the summer solstice, during an all-night celebration. In addition to using unspecified recreational drugs, she took three 20-mg capsules of fluoxetine that her friend told her would make her feel high. She instead developed acute mania and dramatic paranoid delusions.
Figure 2 Possible genotypes of Brad, son of Abdul and Katrina
Olga was hospitalized and treated with moderate doses of haloperidol that precipitated an acute dystonic reaction. She was subsequently given ben-ztropine, and her extrapyramidal symptoms resolved. After discharge, she was treated with haloperidol and benztropine for 2 years, after which she spontaneously discontinued these drugs against medical advice. Her psychotic illness has not recurred.
Knowing her own genotype, Katrina understood that her mother had a 50% probability of having one copy of the 2D6 *4 allele. Given her mother’s history of medication intolerance, Katrina believed that her mother’s psychiatric illness might have been related to a drug reaction. She persuaded her mother to send a blood sample to a laboratory in Stockholm.
Olga’s genotype was *4/*4, indicating that she would be unlikely to tolerate even moderate doses of 2D6 substrate medications (Figure 1). Given her complete recovery and continued good health without medication, the most probable retrospective diagnosis was drug-induced psychosis. Her 2-year neuroleptic treatment probably was unnecessary.
Figure 3 Genogram for Brad, son of Abdul and Katrina
Poor metabolizers without a functional 2D6 gene copy have low tolerance for many medications and often become labeled as “medication sensitive.” When genotyping reveals that an individual is a poor metabolizer, prescribing medications that do not require 2D6 metabolism is usually prudent.
In rare cases, poor metabolizers have died from normal doses of 2D6 substrate medications.7 Far more commonly, however, they spontaneously discontinue taking these drugs because of adverse side effects.
Benefits of prospective testing
When used in clinical practice, pharmacogenomic testing’s two goals are to identify:
- ultrarapid metabolizers, who will not benefit from a medication
- poor metabolizers, who likely will have adverse responses to a medication.
The following case demonstrates the benefit of prospective 2D6 genotyping:
Brad, age 14, is the son of Abdul and Katrina, whose genotypes have been described. Brad developed a serious depression that was similar in severity and onset to an illness his mother experienced as a teen.
Brad’s parents want him to get the maximum benefit from psychopharmacologic treatment while avoiding distressing side effects. He had been healthy and had received no prescriptions other than antibiotics in the past.
How would you proceed? Without knowing Brad’s parents’ genotypes, you might reason that Brad would resemble one of them in drug response. However, when you review each parent’s genotype, you realize four scenarios are possible (Figure 2):
- Brad has a high likelihood of being an ultrarapid metabolizer because he has a 50% chance of inheriting a chromosome with five copies of the 2D6 gene from his father. He inherited the *1 or *4 form from his mother, but the effect of either will be clinically irrelevant.
- If Brad inherited the chromosome with the deletion from his father and the *1 form from his mother, he would be an intermediate metabolizer, as is his mother.
- If he inherited the chromosome with the deletion from his father and the *4 form from his mother, he would be a poor metabolizer like his grandmother, Olga. He would be at substantial risk for adverse reactions (such as intense headaches or vomiting) to 2D6 substrate medications.
On testing, Brad was found to be a poor metabolizer (Figure 3) The psychiatrist prescribed bupropion, which is metabolized by the 2B6 enzyme rather than the 2D6 enzyme.
Conclusion. To introduce the concept of genotypic testing, this review has focused on simple illustrations of variations in a single gene. However, many genes in the P-450 family play important roles in metabolizing psychotropics. In the future, genotyping of panels of these genes will likely provide more-specific guidance than can be achieved by simply testing one gene at a time.
Related resources
- Lerer B (ed). Pharmacogenetics of psychotropic drugs. Cambridge, UK: Cambridge University Press, 2002.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
- Indiana University School of Medicine, Division of Clinical Pharmacology. Drug Interactions—Defining Genetic Influences on Pharmacologic Responses. http://medicine.iupui.edu/flockhart.
Drug brand names
- Acetaminophen w/codeine phosphate • Tylenol w/codeine
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Nortriptyline • Aventyl, Pamelor
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
- Sertraline • Zoloft
- Thioridazine • Mellaril
- Venlafaxine • Effexor
Disclosure
Dr. Mrazek reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
1. Mrazek DA. Clinical genomic testing. In: Wiener J, Dulcan M (eds). Textbook of child and adolescent psychiatry (3rd ed). Washington, DC: American Psychiatric Publishing, Inc., 2001;193-203.
2. Mrazek DA. Pharmacogenomic screening for depressed children and adolescents (scientific proceedings). Miami Beach, FL: American Academy of Child and Adolescent Psychiatry annual meeting, 2003;159.-
3. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001;286(18):2270-9.
4. Shi MM, Mehrens D, Dacus K. Pharmacogenomics: Changing the health care paradigm. Modern Drug Discovery 2001;4(7):27-32.
5. Kirchheiner J, Brosen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;104:173-92.
6. Masimirembwa CM, Hasler JA. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull 1997;44(5):561-71.
7. Sallee FR, DeVane CL, Ferrell RE. Fluoxetine-related death in a child with cytochrome P-450 2D6 genetic deficiency. J Child Adolesc Psychopharmacol 2000;10(1):27-34.
8. Gaedigk A, Gotschall RR, Forbes NS, et al. Optimization of cytochrome P4502D6 (CYP2D6) phenotype assignment using a genotyping algorithm based on allele frequency data. Pharmacogenetics 1999;9(6):669-82.
Is depression neurochemical or neurodegenerative?
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
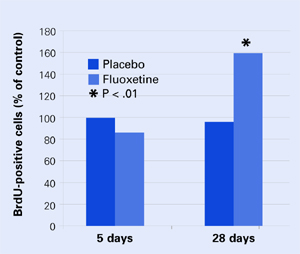
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.