User login
FDA approves new immunotherapy for endometrial cancer
Use limited to patients with biomarker
Usage of the new checkpoint inhibitor is limited to patients who have progressed on or following prior treatment with a platinum-containing chemotherapy. Eligibility must also be determined by an FDA-approved test for the dMMR biomarker. Approximately 25%-30% of patients with advanced endometrial cancer have dMMR tumors, according to the FDA.
The approval is “evidence of the FDA’s progress in applying precision medicine to expand treatment options for patients with cancer,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
He explained that the immunotherapy was “specifically studied to target dMMR endometrial cancer and leverages scientific knowledge surrounding the mechanism of immunotherapy response.”
The new drug also addresses an unmet medical need, as there are limited therapeutic options in this setting following frontline standard treatment with a platinum-containing chemotherapy.
The approval is based on safety and efficacy data from a single-arm, multicohort clinical trial. Of the 71 patients with dMMR recurrent or advanced endometrial cancer who received dostarlimab, 42.3% had a response. For 93% of that group, the response lasted 6 months or longer.
The drug’s maker, GlaxoSmithKline, is currently conducting additional, larger trials in more patients with dMMR endometrial tumors to verify and further describe clinical benefits.
Common side effects of dostarlimab include fatigue, nausea, diarrhea, anemia, and constipation. Like other checkpoint inhibitors, the new drug can cause immune-mediated side effects such as pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
Dostarlimab is contraindicated in women who are pregnant or breastfeeding because it may cause harm to a developing fetus or newborn baby.
The FDA approval comes a month after the Committee for Medicinal Products for Human Use of the European Medicines Agency recommended granting conditional marketing authorization for dostarlimab for use as monotherapy in this same patient group.
In the United States, dostarlimab received Priority Review designation and Breakthrough Therapy designation for this indication.
A version of this article first appeared on Medscape.com.
Use limited to patients with biomarker
Use limited to patients with biomarker
Usage of the new checkpoint inhibitor is limited to patients who have progressed on or following prior treatment with a platinum-containing chemotherapy. Eligibility must also be determined by an FDA-approved test for the dMMR biomarker. Approximately 25%-30% of patients with advanced endometrial cancer have dMMR tumors, according to the FDA.
The approval is “evidence of the FDA’s progress in applying precision medicine to expand treatment options for patients with cancer,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
He explained that the immunotherapy was “specifically studied to target dMMR endometrial cancer and leverages scientific knowledge surrounding the mechanism of immunotherapy response.”
The new drug also addresses an unmet medical need, as there are limited therapeutic options in this setting following frontline standard treatment with a platinum-containing chemotherapy.
The approval is based on safety and efficacy data from a single-arm, multicohort clinical trial. Of the 71 patients with dMMR recurrent or advanced endometrial cancer who received dostarlimab, 42.3% had a response. For 93% of that group, the response lasted 6 months or longer.
The drug’s maker, GlaxoSmithKline, is currently conducting additional, larger trials in more patients with dMMR endometrial tumors to verify and further describe clinical benefits.
Common side effects of dostarlimab include fatigue, nausea, diarrhea, anemia, and constipation. Like other checkpoint inhibitors, the new drug can cause immune-mediated side effects such as pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
Dostarlimab is contraindicated in women who are pregnant or breastfeeding because it may cause harm to a developing fetus or newborn baby.
The FDA approval comes a month after the Committee for Medicinal Products for Human Use of the European Medicines Agency recommended granting conditional marketing authorization for dostarlimab for use as monotherapy in this same patient group.
In the United States, dostarlimab received Priority Review designation and Breakthrough Therapy designation for this indication.
A version of this article first appeared on Medscape.com.
Usage of the new checkpoint inhibitor is limited to patients who have progressed on or following prior treatment with a platinum-containing chemotherapy. Eligibility must also be determined by an FDA-approved test for the dMMR biomarker. Approximately 25%-30% of patients with advanced endometrial cancer have dMMR tumors, according to the FDA.
The approval is “evidence of the FDA’s progress in applying precision medicine to expand treatment options for patients with cancer,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
He explained that the immunotherapy was “specifically studied to target dMMR endometrial cancer and leverages scientific knowledge surrounding the mechanism of immunotherapy response.”
The new drug also addresses an unmet medical need, as there are limited therapeutic options in this setting following frontline standard treatment with a platinum-containing chemotherapy.
The approval is based on safety and efficacy data from a single-arm, multicohort clinical trial. Of the 71 patients with dMMR recurrent or advanced endometrial cancer who received dostarlimab, 42.3% had a response. For 93% of that group, the response lasted 6 months or longer.
The drug’s maker, GlaxoSmithKline, is currently conducting additional, larger trials in more patients with dMMR endometrial tumors to verify and further describe clinical benefits.
Common side effects of dostarlimab include fatigue, nausea, diarrhea, anemia, and constipation. Like other checkpoint inhibitors, the new drug can cause immune-mediated side effects such as pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
Dostarlimab is contraindicated in women who are pregnant or breastfeeding because it may cause harm to a developing fetus or newborn baby.
The FDA approval comes a month after the Committee for Medicinal Products for Human Use of the European Medicines Agency recommended granting conditional marketing authorization for dostarlimab for use as monotherapy in this same patient group.
In the United States, dostarlimab received Priority Review designation and Breakthrough Therapy designation for this indication.
A version of this article first appeared on Medscape.com.
Oral contraceptive with new estrogen earns approval
The Food and Drug Administration has approved a new estrogen for the first time in more than 50 years.

The novel combined oral contraceptive, marketed as Nextstellis, contains 3 mg drospirenone (DRSP) and 14.2 mg of estetrol (E4) in tablet form. Estetrol is an estrogen that is naturally produced during pregnancy, but will now be produced from a plant source; it has not previously been used in oral contraceptives.
Approval of the unique estetrol/drospirenone combination was based on data from a pair of phase 3 clinical trials including 3,725 women. Overall, Nextstellis was safe and effective while meeting its primary endpoint of pregnancy prevention, according to a company press release. Participants also reported favorable results on secondary endpoints including cycle control, bleeding profile, safety, and tolerability.
Although many women take short-acting contraceptives containing estrogen and progestin, concerns persist about side effects, said Mitchell Creinin, MD, of the University of California, in the press release. In addition to providing effective contraception, the drug showed minimal impact on specific markers of concern, including triglycerides, cholesterol, and glucose, as well as weight and endocrine markers, Dr. Creinin said.
Nextstellis was developed by the Belgian biotech company Mithra Pharmaceuticals, and the drug is licensed for distribution in Australia and the United States by Mayne Pharma, with an expected launch at the end of June 2021.
The Food and Drug Administration has approved a new estrogen for the first time in more than 50 years.
The novel combined oral contraceptive, marketed as Nextstellis, contains 3 mg drospirenone (DRSP) and 14.2 mg of estetrol (E4) in tablet form. Estetrol is an estrogen that is naturally produced during pregnancy, but will now be produced from a plant source; it has not previously been used in oral contraceptives.
Approval of the unique estetrol/drospirenone combination was based on data from a pair of phase 3 clinical trials including 3,725 women. Overall, Nextstellis was safe and effective while meeting its primary endpoint of pregnancy prevention, according to a company press release. Participants also reported favorable results on secondary endpoints including cycle control, bleeding profile, safety, and tolerability.
Although many women take short-acting contraceptives containing estrogen and progestin, concerns persist about side effects, said Mitchell Creinin, MD, of the University of California, in the press release. In addition to providing effective contraception, the drug showed minimal impact on specific markers of concern, including triglycerides, cholesterol, and glucose, as well as weight and endocrine markers, Dr. Creinin said.
Nextstellis was developed by the Belgian biotech company Mithra Pharmaceuticals, and the drug is licensed for distribution in Australia and the United States by Mayne Pharma, with an expected launch at the end of June 2021.
The Food and Drug Administration has approved a new estrogen for the first time in more than 50 years.
The novel combined oral contraceptive, marketed as Nextstellis, contains 3 mg drospirenone (DRSP) and 14.2 mg of estetrol (E4) in tablet form. Estetrol is an estrogen that is naturally produced during pregnancy, but will now be produced from a plant source; it has not previously been used in oral contraceptives.
Approval of the unique estetrol/drospirenone combination was based on data from a pair of phase 3 clinical trials including 3,725 women. Overall, Nextstellis was safe and effective while meeting its primary endpoint of pregnancy prevention, according to a company press release. Participants also reported favorable results on secondary endpoints including cycle control, bleeding profile, safety, and tolerability.
Although many women take short-acting contraceptives containing estrogen and progestin, concerns persist about side effects, said Mitchell Creinin, MD, of the University of California, in the press release. In addition to providing effective contraception, the drug showed minimal impact on specific markers of concern, including triglycerides, cholesterol, and glucose, as well as weight and endocrine markers, Dr. Creinin said.
Nextstellis was developed by the Belgian biotech company Mithra Pharmaceuticals, and the drug is licensed for distribution in Australia and the United States by Mayne Pharma, with an expected launch at the end of June 2021.
FDA approves frontline immunotherapy for gastric cancers
This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
This is the first immunotherapy approved for the frontline treatment of gastric cancers, the agency says in a press release.
The approval comes after nivolumab received Priority Review and Orphan Drug designations for this indication. There are approximately 28,000 new diagnoses of gastric cancer annually in the United States, and overall survival is generally poor with currently available therapy, points out the FDA.
“Today’s approval is the first treatment in more than a decade to show a survival benefit for patients with advanced or metastatic gastric cancer who are being treated for the first time,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, states in an FDA press release.
Efficacy in the gastric cancer setting was demonstrated in the randomized, phase 3, open-label CheckMate 649 study of 1,518 untreated patients. Median survival was 13.8 months among those treated with nivolumab, compared with 11.6 months with chemotherapy alone (hazard ratio, 0.80; P = .0002).
Common side effects experienced by patients in the nivolumab group included peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Nivolumab is also approved for numerous other cancers. Other known adverse effects include immune-mediated inflammation of the lungs, colon, liver, endocrine glands, and kidneys.
“Patients should tell their health care providers if they have immune system problems, lung or breathing problems, liver problems, have had an organ transplant, or are pregnant or plan to become pregnant before starting treatment,” the FDA states.
A version of this article first appeared on Medscape.com.
CDC: STI rates rise for sixth year in a row
Annual cases of sexually transmitted infections in the United States jumped for the sixth year in a row in 2019, according to a new Centers for Disease Control and Prevention report that highlights an increase in congenital syphilis and rising rates of syphilis, chlamydia, and gonorrhea in men, especially men who have sex with men (MSM).
The report says nothing about STI rates during the COVID-19 pandemic, when both casual sex and disease screening and surveillance declined significantly, at least in the early months. But epidemiologist Patricia Kissinger, PhD, MPH, from Tulane University School, New Orleans, said in an interview that the findings reflect how “a confluence of factors” drove up rates before the age of COVID. Those factors include online dating, the opioid epidemic, the decline in condom use in the MSM community as HIV became more preventable, and indifference among policy makers and the community at large.
The CDC report, based on data from local health departments, says there were 129,813 cases of syphilis in 2019, up 74% since 2015. Almost 2,000 cases of congenital syphilis were reported, up 279% since 2015, and 128 infants died.
“There’s no reason for us to have congenital syphilis,” said Dr. Kissinger, who noted that the disease can cause birth defects and meningitis in addition to death. “Women should be screened, and it’s relatively easy to treat via penicillin injections.”
Indeed, medical guidelines suggest that pregnant women be routinely tested for syphilis. But that doesn’t always happen because “it falls through the cracks,” Dr. Kissinger said. Or, she added, women might not be tested enough times during their pregnancies: “You have to screen women in the third trimester. You can’t just do it in the first trimester because people do have sex when they’re pregnant.”
Rising congenital syphilis numbers have convinced at least one health system to take action. As of June 1, the University of California, San Diego, will routinely test pregnant women in the emergency department for syphilis in addition to HIV and hepatitis C, Martin Hoenigl, MD, a UCSF infectious disease specialist, said in an interview.
The CDC report also notes 1.8 million cases of chlamydia in 2019, a jump of 19% in 4 years, and a 56% increase in gonorrhea in that time period, to a total of 616,392 cases.
The report says increasing gonorrhea and chlamydia cases in men, especially MSM, could be caused by increased testing/screening, increased transmission, or both. Although women are generally diagnosed with chlamydia more often than men, the report says, numbers among men grew by 32% from 2015 to 2019. And since 2013, rates of gonorrhea among men have risen at a much faster clip than among women.
MSM accounted for most male cases of primary and secondary syphilis in 2019, although the report said the apparent long-term rise in these cases might be slowing.
Many MSM no longer use condoms because they’re using pre-exposure prophylaxis (PrEP) or have undetectable levels of HIV because of treatment, said Jeffrey Klausner, MD, MPH, an STI specialist at the University of Southern California in Los Angeles, said in an interview.
Many MSM might be getting screened much more often for STIs than in the past because frequent screening is required for those on PrEP. However, Dr. Kissinger said some clinics weren’t able to test at times during the pandemic because of a swab shortage. In addition, patients of all types avoided routine medical care during the pandemic, and some medical professionals in the infectious disease field were redirected to COVID care.
Clinical trials have been investigating a possible preventive STI strategy in MSM who don’t wear condoms – prophylaxis, either before or after exposure, with the antibiotic doxycycline. “That’s a very good solution,” Dr. Klausner said, but he believes bigger challenges remain. According to him, the existence of the report itself – which offers statistics from 2 years ago instead of more relevant recent numbers – is evidence of how the federal government isn’t doing enough to fight STIs. “If we’re taking the STD epidemic seriously, there should be timely and regular reporting.” Dr. Klausner said he likes the idea of monthly reports, as well as more funding for prevention.
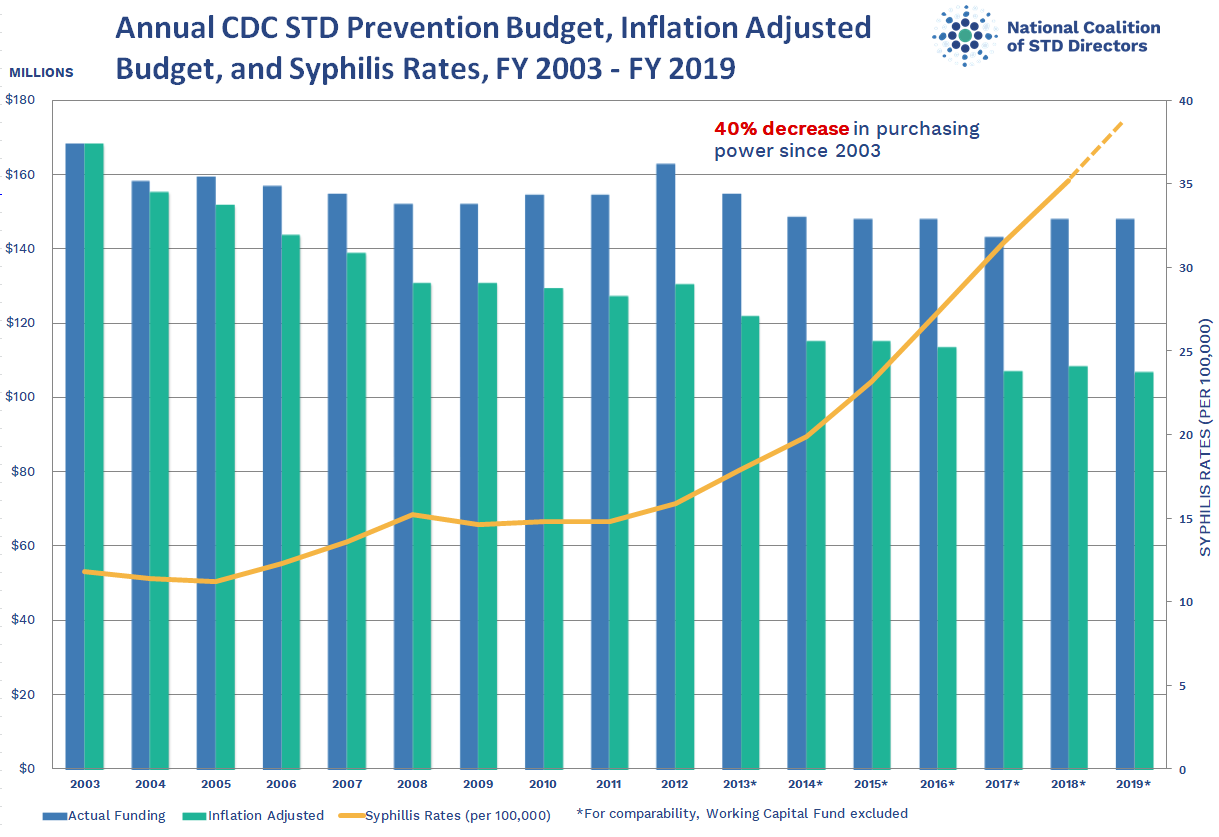
Instead, he noted, the federal government cut STI prevention funding by 40% in inflation-adjusted dollars from 2002-2003 to 2018-2019, according to the National Coalition of STD Directors. “Burying your head in the sand and hoping the problem goes away is not an effective strategy,” he said.
It’s not clear whether STI rates are on the decline because of pandemic restrictions and stay-at-home orders. Surveys suggest that a dip in casual sex early in pandemic – when much of society shut down – was only temporary, Dr. Klausner said.
Dr. Kissinger disclosed no relevant financial relationships. Dr. Hoenigl reported receiving research funding via his university from Gilead. Dr. Klausner has recently provided consulting services to Danaher, Cepheid, Roche, GlaxoSmithKline, Talis Bio, SpeeDx, and Visby Medical, all manufacturers of diagnostic assays for STIs.
{kind=link}
A version of this article first appeared on Medscape.com.
Annual cases of sexually transmitted infections in the United States jumped for the sixth year in a row in 2019, according to a new Centers for Disease Control and Prevention report that highlights an increase in congenital syphilis and rising rates of syphilis, chlamydia, and gonorrhea in men, especially men who have sex with men (MSM).
The report says nothing about STI rates during the COVID-19 pandemic, when both casual sex and disease screening and surveillance declined significantly, at least in the early months. But epidemiologist Patricia Kissinger, PhD, MPH, from Tulane University School, New Orleans, said in an interview that the findings reflect how “a confluence of factors” drove up rates before the age of COVID. Those factors include online dating, the opioid epidemic, the decline in condom use in the MSM community as HIV became more preventable, and indifference among policy makers and the community at large.
The CDC report, based on data from local health departments, says there were 129,813 cases of syphilis in 2019, up 74% since 2015. Almost 2,000 cases of congenital syphilis were reported, up 279% since 2015, and 128 infants died.
“There’s no reason for us to have congenital syphilis,” said Dr. Kissinger, who noted that the disease can cause birth defects and meningitis in addition to death. “Women should be screened, and it’s relatively easy to treat via penicillin injections.”
Indeed, medical guidelines suggest that pregnant women be routinely tested for syphilis. But that doesn’t always happen because “it falls through the cracks,” Dr. Kissinger said. Or, she added, women might not be tested enough times during their pregnancies: “You have to screen women in the third trimester. You can’t just do it in the first trimester because people do have sex when they’re pregnant.”
Rising congenital syphilis numbers have convinced at least one health system to take action. As of June 1, the University of California, San Diego, will routinely test pregnant women in the emergency department for syphilis in addition to HIV and hepatitis C, Martin Hoenigl, MD, a UCSF infectious disease specialist, said in an interview.
The CDC report also notes 1.8 million cases of chlamydia in 2019, a jump of 19% in 4 years, and a 56% increase in gonorrhea in that time period, to a total of 616,392 cases.
The report says increasing gonorrhea and chlamydia cases in men, especially MSM, could be caused by increased testing/screening, increased transmission, or both. Although women are generally diagnosed with chlamydia more often than men, the report says, numbers among men grew by 32% from 2015 to 2019. And since 2013, rates of gonorrhea among men have risen at a much faster clip than among women.
MSM accounted for most male cases of primary and secondary syphilis in 2019, although the report said the apparent long-term rise in these cases might be slowing.
Many MSM no longer use condoms because they’re using pre-exposure prophylaxis (PrEP) or have undetectable levels of HIV because of treatment, said Jeffrey Klausner, MD, MPH, an STI specialist at the University of Southern California in Los Angeles, said in an interview.
Many MSM might be getting screened much more often for STIs than in the past because frequent screening is required for those on PrEP. However, Dr. Kissinger said some clinics weren’t able to test at times during the pandemic because of a swab shortage. In addition, patients of all types avoided routine medical care during the pandemic, and some medical professionals in the infectious disease field were redirected to COVID care.
Clinical trials have been investigating a possible preventive STI strategy in MSM who don’t wear condoms – prophylaxis, either before or after exposure, with the antibiotic doxycycline. “That’s a very good solution,” Dr. Klausner said, but he believes bigger challenges remain. According to him, the existence of the report itself – which offers statistics from 2 years ago instead of more relevant recent numbers – is evidence of how the federal government isn’t doing enough to fight STIs. “If we’re taking the STD epidemic seriously, there should be timely and regular reporting.” Dr. Klausner said he likes the idea of monthly reports, as well as more funding for prevention.
Instead, he noted, the federal government cut STI prevention funding by 40% in inflation-adjusted dollars from 2002-2003 to 2018-2019, according to the National Coalition of STD Directors. “Burying your head in the sand and hoping the problem goes away is not an effective strategy,” he said.
It’s not clear whether STI rates are on the decline because of pandemic restrictions and stay-at-home orders. Surveys suggest that a dip in casual sex early in pandemic – when much of society shut down – was only temporary, Dr. Klausner said.
Dr. Kissinger disclosed no relevant financial relationships. Dr. Hoenigl reported receiving research funding via his university from Gilead. Dr. Klausner has recently provided consulting services to Danaher, Cepheid, Roche, GlaxoSmithKline, Talis Bio, SpeeDx, and Visby Medical, all manufacturers of diagnostic assays for STIs.
A version of this article first appeared on Medscape.com.
Annual cases of sexually transmitted infections in the United States jumped for the sixth year in a row in 2019, according to a new Centers for Disease Control and Prevention report that highlights an increase in congenital syphilis and rising rates of syphilis, chlamydia, and gonorrhea in men, especially men who have sex with men (MSM).
The report says nothing about STI rates during the COVID-19 pandemic, when both casual sex and disease screening and surveillance declined significantly, at least in the early months. But epidemiologist Patricia Kissinger, PhD, MPH, from Tulane University School, New Orleans, said in an interview that the findings reflect how “a confluence of factors” drove up rates before the age of COVID. Those factors include online dating, the opioid epidemic, the decline in condom use in the MSM community as HIV became more preventable, and indifference among policy makers and the community at large.
The CDC report, based on data from local health departments, says there were 129,813 cases of syphilis in 2019, up 74% since 2015. Almost 2,000 cases of congenital syphilis were reported, up 279% since 2015, and 128 infants died.
“There’s no reason for us to have congenital syphilis,” said Dr. Kissinger, who noted that the disease can cause birth defects and meningitis in addition to death. “Women should be screened, and it’s relatively easy to treat via penicillin injections.”
Indeed, medical guidelines suggest that pregnant women be routinely tested for syphilis. But that doesn’t always happen because “it falls through the cracks,” Dr. Kissinger said. Or, she added, women might not be tested enough times during their pregnancies: “You have to screen women in the third trimester. You can’t just do it in the first trimester because people do have sex when they’re pregnant.”
Rising congenital syphilis numbers have convinced at least one health system to take action. As of June 1, the University of California, San Diego, will routinely test pregnant women in the emergency department for syphilis in addition to HIV and hepatitis C, Martin Hoenigl, MD, a UCSF infectious disease specialist, said in an interview.
The CDC report also notes 1.8 million cases of chlamydia in 2019, a jump of 19% in 4 years, and a 56% increase in gonorrhea in that time period, to a total of 616,392 cases.
The report says increasing gonorrhea and chlamydia cases in men, especially MSM, could be caused by increased testing/screening, increased transmission, or both. Although women are generally diagnosed with chlamydia more often than men, the report says, numbers among men grew by 32% from 2015 to 2019. And since 2013, rates of gonorrhea among men have risen at a much faster clip than among women.
MSM accounted for most male cases of primary and secondary syphilis in 2019, although the report said the apparent long-term rise in these cases might be slowing.
Many MSM no longer use condoms because they’re using pre-exposure prophylaxis (PrEP) or have undetectable levels of HIV because of treatment, said Jeffrey Klausner, MD, MPH, an STI specialist at the University of Southern California in Los Angeles, said in an interview.
Many MSM might be getting screened much more often for STIs than in the past because frequent screening is required for those on PrEP. However, Dr. Kissinger said some clinics weren’t able to test at times during the pandemic because of a swab shortage. In addition, patients of all types avoided routine medical care during the pandemic, and some medical professionals in the infectious disease field were redirected to COVID care.
Clinical trials have been investigating a possible preventive STI strategy in MSM who don’t wear condoms – prophylaxis, either before or after exposure, with the antibiotic doxycycline. “That’s a very good solution,” Dr. Klausner said, but he believes bigger challenges remain. According to him, the existence of the report itself – which offers statistics from 2 years ago instead of more relevant recent numbers – is evidence of how the federal government isn’t doing enough to fight STIs. “If we’re taking the STD epidemic seriously, there should be timely and regular reporting.” Dr. Klausner said he likes the idea of monthly reports, as well as more funding for prevention.
Instead, he noted, the federal government cut STI prevention funding by 40% in inflation-adjusted dollars from 2002-2003 to 2018-2019, according to the National Coalition of STD Directors. “Burying your head in the sand and hoping the problem goes away is not an effective strategy,” he said.
It’s not clear whether STI rates are on the decline because of pandemic restrictions and stay-at-home orders. Surveys suggest that a dip in casual sex early in pandemic – when much of society shut down – was only temporary, Dr. Klausner said.
Dr. Kissinger disclosed no relevant financial relationships. Dr. Hoenigl reported receiving research funding via his university from Gilead. Dr. Klausner has recently provided consulting services to Danaher, Cepheid, Roche, GlaxoSmithKline, Talis Bio, SpeeDx, and Visby Medical, all manufacturers of diagnostic assays for STIs.
A version of this article first appeared on Medscape.com.
FDA panel supports islet cell treatment for type 1 diabetes
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.
On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.
On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
A Food and Drug Administration advisory panel has endorsed a pancreatic islet cell transplant therapy for the treatment of people with type 1 diabetes that can’t be managed with current therapies.
On April 15, the FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee voted 12 to 4 in favor of approval of donislecel (Lantidra). There was one abstention. The panel regarded the drug as having “an overall favorable benefit-risk profile for some patients with type 1 diabetes.” The product consists of purified allogeneic pancreatic islets of Langerhans derived from cadaveric donors and is infused into the portal vein of the liver.
Benefits of the treatment include the potential for insulin independence and elimination of severe hypoglycemia. Risks are those associated with the surgical procedure and with long-term immunosuppression.
The therapy is manufactured by CellTrans. According to Jose Oberholzer, MD, the founder of CellTrans, the proposed indication is for adults with “brittle” type 1 diabetes who meet the American Diabetes Association’s (ADA) criteria for whole-organ pancreas-alone transplant (i.e., transplant of pancreas but not kidney).
The ADA criteria include the following: frequent, severe hypoglycemia, hyperglycemia, and/or ketoacidosis that requires medical attention; clinical or emotional problems regarding the use of exogenous insulin; and consistent failure of insulin-based management to prevent acute diabetes complications.
Success in two-thirds of patients in small studies
Dr. Oberholzer presented data from two single-arm open-label studies: a phase 1/2 trial initiated in 2004 with 10 patients, and a phase 3 study with 20 patients that began in 2007. The inclusion criteria differed somewhat between the two studies, but all 30 patients had hypoglycemic unawareness. Mean follow-up was 7.8 years for the phase 1/2 trial and 4.7 years for the phase 3 trial.
For all of the patients, C-peptide levels were positive after transplant. The composite endpoint for success – an A1c level of ≤ 6.5% and the absence of severe hypoglycemic episodes for 1 year – was met by 19 patients (63.3%). For five patients (16.7%), the target A1c level was not achieved, and seven patients (23.3%) experienced a severe episode of hypoglycemia.
Twenty of the 30 patients achieved insulin independence for at least 1 year.
Improvements were also seen at 1 year in mixed meal test outcomes, fasting blood glucose levels, and overall glycemic control. Graft survival 10 years post transplant was achieved by 60% of patients, Dr. Oberholzer said.
Adverse events not unexpected, but still of concern
Two patients died, one as a result of fulminant sepsis at 20 months post transplant, and the other as a result of severe dementia 9 years post transplant. Three patients experienced four serious procedure-related events, including one liver laceration and two hepatic hematomas. Elevations in portal pressure occurred in two patients.
Most adverse events were associated with immunosuppression. These included 178 infections in 26 of the 30 patients. The most common of these were herpes virus infections, Epstein-Barr virus infections, oral candidiasis, and cytomegalovirus infections. Twelve infections were severe. Renal function declined persistently in two patients (20%), and six (20%) experienced new-onset proteinuria at 1 year.
The adverse events related to the procedure and the problems associated with immunosuppression were not unexpected and were consistent with those described for patients receiving whole pancreas transplants, FDA reviewer Patricia Beaston, MD, said in her review of the CellTrans data.
Panel members support treatment for a small group of patients
During the discussion, several panel members pointed out that the target patient population for this treatment will likely be smaller today than it was when the two studies were initiated, given advances in diabetes care. Those advances include continuous glucose monitoring devices with alarms and closed-loop insulin delivery systems – the “artificial pancreas” that automatically suspends insulin delivery to prevent hypoglycemia.
Panel chair Lisa Butterfield, PhD, a surgeon and immunologist at the University of California, San Francisco, voted in favor of approval. But, she added, “I do support postapproval gathering of data to learn more about the product. ... I don’t know how many patients will really benefit, but I think it’s to be determined.”
Christopher K. Breuer, MD, a general and pediatric surgeon at the Center for Regenerative Medicine, Nationwide Children’s Hospital, Columbus, Ohio, said he supported approval for “two very small subpopulations where it would provide the only viable therapy”: those who are eligible for pancreas transplant but cannot tolerate a major operation, and those who already use the latest automated insulin delivery systems and still do not achieve acceptable glycemic control.
Temporary voting member David Harlan, MD, director of the University of Massachusetts Diabetes Center of Excellence, Worcester, Mass., voted no.
He noted that only about 100 whole pancreas-only transplants are performed annually in the United States and that such transplants are “very effective, so we’re talking about patients who aren’t pancreas transplant candidates who might get this.”
Moreover, Dr. Harlan said, “I’ve seen the awful things that can happen in posttransplant recipients. It’s really hard to get that informed consent from someone when you’re asking them to consider a future that they don’t know. When it works, it’s great. When it doesn’t work, it can be catastrophic. I just worry about opening Pandora’s box.”
The only other diabetes specialist on the panel, temporary voting member Ellen Leschek, MD, said she “reluctantly voted yes because a few people could benefit, but I think it’s a much smaller number than the company may believe.”
Dr. Leschek, of the National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, Md., said she’s concerned that “if it’s approved, too many people will get treated this way, when in fact, for a lot of those people, the risks will outweigh the benefits.”
Sandy Feng, MD, PhD, of the department of surgery at the University of California, San Francisco, pointed out that with regard to immunosuppressive therapy, “We’re concerned about the toxicity of what we currently use, but there are additional therapies being developed that might mitigate those toxicities that would be beneficial to this population.”
Dr. Feng, who voted yes, also said, “I do pancreas transplants. I can tell you that there is nothing that [patients with type 1 diabetes] like more than the freedom from dealing with the entire insulin issue. That has made a large impression on me over the last 20-plus years of clinical practice, so I do think this can help some people and will be incredibly meaningful to those people.”
FDA advisory panel members are vetted for conflicts of interest, and special waivers are granted if necessary. No such waivers were granted for this meeting.
A version of this article first appeared on Medscape.com.
How some COVID-19 vaccines could cause rare blood clots
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
on April 14, 2021, after the CDC and Food and Drug Administration recommended that states hold off on using it pending a detailed review of six cases of the same kind of rare but serious event – a blood clot in the vessels that drain blood from the brain combined with a large drop in platelets, which increases the risk for bleeding.
This combination can lead to severe strokes that can lead to brain damage or death. Among the six cases reported, which came to light over the past 3 weeks, one person died, according to the CDC. All six were women and ranged in age from 18 to 48 years.
According to a report from the Vaccine Adverse Event Reporting System (VAERS), which is maintained by the Department of Health & Human Services, the woman who died was 45. She developed a gradually worsening headache about a week after receiving the Johnson & Johnson vaccine.
On March 17, the day she came to the hospital, she was dry heaving. Her headache had suddenly gotten much worse, and the left side of her body was weak, which are signs of a stroke. A CT scan revealed both bleeding in her brain and a clot in her cortical vein. She died the following day.
In addition to VAERS, which accepts reports from anyone, the CDC and FDA are monitoring at least eight other safety systems maintained by hospitals, research centers, long-term care facilities, and insurance companies for signs of trouble with the vaccines. VAERS data is searchable and open to the public. Most of these systems are not publicly available to protect patient privacy. It’s unclear which systems detected the six cases cited by federal regulators.
“These are very serious and potentially fatal problems occurring in a healthy young adult. It’s serious and we need to get to the bottom of it,” said Ed Belongia, MD, director of the Center for Clinical Epidemiology and Population Health at the Marshfield (Wis.) Clinic Research Institute. Dr. Belongia leads a research team that helps the CDC monitor vaccine safety and effectiveness.
“Safety is always the highest priority, and I think what we’ve seen here in the past 24 hours is our vaccine safety monitoring system is working,” he said.
Others agree. “I think what CDC and FDA have detected is a rare, but likely real adverse event associated with this vaccine,” said Paul Offit, MD, director of vaccine education at Children’s Hospital of Philadelphia.
Although much is still unknown about these events, they follow a similar pattern of blood clots reported with the AstraZeneca vaccine in Europe. That vaccine is now sold under the brand name Vaxzevria.
This has experts questioning whether all vaccines of this type may cause these rare clots.
“I think it’s likely a class effect,” said Dr. Offit, who was a member of the FDA advisory committee that reviewed clinical trial data on the J&J vaccine before it was authorized for use.
Adenovirus vaccines scrutinized
Both the Johnson & Johnson and Vaxzevria vaccines use an adenovirus to ferry genetic instructions for making the coronaviruses spike protein into our cells.
Adenoviruses are common, relatively simple viruses that normally cause mild cold or flu symptoms. The ones used in the vaccine are disabled so they can’t make us sick. They’re more like Trojan horses.
Once inside our cells, they release the DNA instructions they carry to make the spike protein of the new coronavirus. Those cells then crank out copies of the spike protein, which then get displayed on the outer surface of the cell membrane where they are recognized by the immune system.
The immune system then makes antibodies and other defenses against the spike so that, when the real coronavirus comes along, our bodies are ready to fight the infection.
There’s no question the vaccine works. In clinical trials, the Johnson & Johnson vaccine was 66% percent effective at preventing against moderate to severe COVID-19 infection, and none of the patients who got COVID-19 after vaccination had to be admitted to the hospital or died.
The idea behind using adenoviruses in vaccines isn’t a new one. In a kind of fight-fire-with-fire approach, the idea is to use a virus, which is good at infecting us, to fight a different kind of virus.
Researchers have been working on the concept for about 10 years, but the COVID-19 vaccines that use this technology are some of the first adenovirus-vector vaccines deployed in humans.
Only one other adenovirus vaccine, for Ebola, has been approved for use in humans. It was approved in Europe last year. Before the Johnson & Johnson vaccine, no other adenovirus vector has been available for use in humans in the United States.
There are six adenovirus-vector vaccines for COVID-19. In addition to AstraZeneca and Johnson & Johnson, there’s the Russian-developed vaccine Sputnik V, along with CanSino from China, and the Covishield vaccine in India.
Adenovirus vaccines are more stable than the mRNA vaccines. That makes them easier to store and transport.
But they have a significant downside, too. Because adenoviruses infect humans out in the world, we already make antibodies against them. So there’s always a danger that our immune systems might recognize and react to the vaccine, rendering it ineffective. For that reason, scientists try to carefully select the adenovirus vectors, or carriers, they use.
The two vaccines under investigation for blood clots are slightly different. The Johnson & Johnson vaccine uses the vector AD26, because most of the population lacks preexisting immunity to it. Vaxzevria uses an adenovirus that infects chimpanzees, called ChAdOx1.
Vaxzevria has been widely used in Europe but has not yet been authorized in the United States.
On April 7, the European Medicines Agency, Europe’s counterpart to the FDA, ruled that unusual blood clots with low blood platelets should be listed as rare side effects on the Vaxzevria vaccine.
The decision came after reviewing 62 cases of cerebral venous sinus thrombosis (CVST) linked to the vaccine and 25 cases of another rare type of clot, called a splanchnic vein thrombosis. Splanchnic veins drain blood from the major organs in the digestive system, including the stomach, liver, and intestines; 18 of those events were fatal.
The reports were culled from reporting in Europe and the United Kingdom, where around 25 million people have received the Vaxzevria vaccine, making these clots exceptionally rare, but serious.
So far, six cases of CVST have been reported in the United States, after more than 7 million doses of the Johnson & Johnson vaccines have been administered.
A key question for U.S. regulators will be the background rate for these types of rare combinations of clots and deplenished platelets. The background rate is the number of events that would be expected to occur naturally in a population of unvaccinated people. On a press call on April 13, Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, was asked about the frequency of this dangerous combination. He said the combination of low platelets and clots was so rare that it was hard to pinpoint, but might be somewhere between 2 and 14 cases per million people over the course of a year.
The first Johnson & Johnson doses were given in early March. That means the six cases came to light within the first few weeks of use of the vaccine in the United States, a very short amount of time.
“These were six cases per million people for 2 weeks, which is the same thing as 25 million per year, so it’s clearly above the background rate,” Dr. Offit said.
Studies suggest possible mechanism
On April 9, the New England Journal of Medicine published a detailed evaluation of the 11 patients in Germany and Austria who developed the rare clots after their Vaxzevria vaccines.
The study detected rare antibodies to a signaling protein called platelet factor 4, which helps to coordinate clot formation.
These same type of antibodies form in some people given the blood thinning drug heparin. In those reactions, which are also exceptionally rare, the same type of syndrome develops, leading to large, devastating clots that consume circulating platelets.
It’s not yet clear whether people who develop reactions to the vaccines already have some platelet factor 4 antibodies before they are vaccinated, or whether the vaccines somehow spur the body to make these antibodies, which then launch a kind of autoimmune attack.
The researchers on the paper gave the syndrome a name, vaccine-induced thrombotic thrombocytopenia (VITT).
It’s also not clear why more cases seem to be in women than in men. Andrew Eisenberger, MD, an associate professor of hematology and oncology at Columbia University, New York, said the most common causes of cerebral venous sinus thrombosis have to do with conditions that raise estrogen levels, like pregnancy and hormonal contraception.
“Estrogen naturally leads to changes in several clotting proteins in the blood that may predispose to abnormal blood clotting in a few different sites in the body,” he said. “The clotting changes we are encountering with some of COVID-19 vaccines are likely to be synergistic with the effects of estrogen on the blood.”
No matter the cause, the CDC on April 13 alerted doctors to keep a high index of suspicion for VITT in patients who have received the Johnson & Johnson vaccination within the last 2 weeks. In those patients, the usual course of treatment with blood thinning drugs like heparin may be harmful.
Symptoms to watch for include severe headache or backache, new neurologic symptoms, severe abdominal pain, shortness of breath, leg swelling, tiny red spots on the skin, or easy bruising.
Grappling with evidence
The CDC’s Advisory Committee on Immunization Practices will meet today in an emergency session to review the cases and see if any changes are needed to use of the J&J vaccine in the United States.
Last week, for example, the United Kingdom restricted the use of the AstraZeneca vaccine in people aged younger than 30 years, saying the risks and benefits of vaccination are “more finely balanced” for this age group.
With cases of COVID-19 rising again in the United States, and the Johnson & Johnson vaccine currently the most convenient form of protection against the virus, the committee will have to weigh the risks of that infection against the risk of rare clots caused by vaccination.
They will also likely have to rule out whether any of the cases had COVID. At least one study has reported CVST clots in three patients with confirmed COVID infections. In Europe, COVID infection did not seem to play a role in the formation of the clots with low platelets.
Hilda Bastian, PhD, a clinical trials expert who cofounded the Cochrane Collaboration, said it won’t be an easy task. Much will depend on how certain the committee members feel they know about all the events linked to the vaccine.
“That’s the really, really hard issue from my point of view for them right this moment. Have we missed any? Or how many are we likely to have missed?” asked Dr. Bastian, who lives in Australia.
“In a country that size with that fragmented [of] a health care system, how sure can you be that you know them all? That’s going to be a really difficult situation for them to grapple with, the quality of information that they’ve got,” she said.
A version of this article first appeared on Medscape.com.
FDA to recommend limits on heavy metals in baby foods
The Food and Drug Administration has responded to congressional criticism and launched a multiyear plan to reduce the amount of heavy metals such as mercury and arsenic found in baby food.

Called “Closer to Zero,” the FDA plan calls for continued scientific investigation, establishes acceptable levels of heavy metals, sets up a way to monitor manufacturers’ compliance, and sets “action levels.”
“Although the FDA’s testing shows that children are not at an immediate health risk from exposure to toxic elements at the levels found in foods, we are starting the plan’s work immediately, with both short- and long-term goals for achieving continued improvements in reducing levels of toxic elements in these foods over time,” the FDA said.
However, Closer to Zero will only make recommendations on heavy metal levels.
“Although action levels are not binding, we have seen that, over the years, our guidance on action levels and other actions have contributed to significant reductions of toxic elements in food,” an FDA spokeswoman wrote in a statement, according to the Washington Post.
A congressional panel said in February 2021 that major brands of commercial baby food routinely have high levels of toxic heavy metals. The House Oversight Committee said this leaves babies at risk for serious developmental and neurologic problems.
The committee sharply criticized the FDA for not taking action.
“Despite the well-known risks of harm to babies from toxic heavy metals, FDA has not taken adequate steps to decrease their presence in baby foods,” the committee said. “FDA has not issued thresholds for the vast majority of toxic heavy metals in baby foods and does not require warning labels on any baby food products.”
A version of this article first appeared on WebMD.com.
The Food and Drug Administration has responded to congressional criticism and launched a multiyear plan to reduce the amount of heavy metals such as mercury and arsenic found in baby food.
Called “Closer to Zero,” the FDA plan calls for continued scientific investigation, establishes acceptable levels of heavy metals, sets up a way to monitor manufacturers’ compliance, and sets “action levels.”
“Although the FDA’s testing shows that children are not at an immediate health risk from exposure to toxic elements at the levels found in foods, we are starting the plan’s work immediately, with both short- and long-term goals for achieving continued improvements in reducing levels of toxic elements in these foods over time,” the FDA said.
However, Closer to Zero will only make recommendations on heavy metal levels.
“Although action levels are not binding, we have seen that, over the years, our guidance on action levels and other actions have contributed to significant reductions of toxic elements in food,” an FDA spokeswoman wrote in a statement, according to the Washington Post.
A congressional panel said in February 2021 that major brands of commercial baby food routinely have high levels of toxic heavy metals. The House Oversight Committee said this leaves babies at risk for serious developmental and neurologic problems.
The committee sharply criticized the FDA for not taking action.
“Despite the well-known risks of harm to babies from toxic heavy metals, FDA has not taken adequate steps to decrease their presence in baby foods,” the committee said. “FDA has not issued thresholds for the vast majority of toxic heavy metals in baby foods and does not require warning labels on any baby food products.”
A version of this article first appeared on WebMD.com.
The Food and Drug Administration has responded to congressional criticism and launched a multiyear plan to reduce the amount of heavy metals such as mercury and arsenic found in baby food.
Called “Closer to Zero,” the FDA plan calls for continued scientific investigation, establishes acceptable levels of heavy metals, sets up a way to monitor manufacturers’ compliance, and sets “action levels.”
“Although the FDA’s testing shows that children are not at an immediate health risk from exposure to toxic elements at the levels found in foods, we are starting the plan’s work immediately, with both short- and long-term goals for achieving continued improvements in reducing levels of toxic elements in these foods over time,” the FDA said.
However, Closer to Zero will only make recommendations on heavy metal levels.
“Although action levels are not binding, we have seen that, over the years, our guidance on action levels and other actions have contributed to significant reductions of toxic elements in food,” an FDA spokeswoman wrote in a statement, according to the Washington Post.
A congressional panel said in February 2021 that major brands of commercial baby food routinely have high levels of toxic heavy metals. The House Oversight Committee said this leaves babies at risk for serious developmental and neurologic problems.
The committee sharply criticized the FDA for not taking action.
“Despite the well-known risks of harm to babies from toxic heavy metals, FDA has not taken adequate steps to decrease their presence in baby foods,” the committee said. “FDA has not issued thresholds for the vast majority of toxic heavy metals in baby foods and does not require warning labels on any baby food products.”
A version of this article first appeared on WebMD.com.
FDA lifts in-person dispensing requirement for mifepristone
The Food and Drug Administration has lifted in-person dispensing requirements for mifepristone when used for medical termination of early pregnancy.
In an April 12, 2021, letter to the American College of Obstetricians and Gynecologists and the Society of Maternal-Fetal Medicine, acting commissioner of food and drugs Janet Woodcock stated that the FDA would exercise discretion to permit the dispensing of mifepristone through the mail when done by or under the supervision of a certified prescriber; or through a mail-order pharmacy under the supervision of a certified prescriber.
The decision follows a trial period of suspension of the in-person dispensing requirement in response to safety concerns for patients as well as providers associated with in-person clinic visits during the COVID-19 pandemic. The Center for Drug Evaluation and Research reviewed safety and clinical outcomes data on mifepristone use when prescriptions were handled by mail or mail-order pharmacy and found that "the small number of adverse events reported to FDA during the COVID-19 public health emergency [PHE] provide no indication that any program deviation or noncompliance with the mifepristone [Risk Evaluation and Mitigation Strategy] program contributed to the reported adverse events," according to the letter. The analysis covers Mifeprex and the approved generic, mifepristone tablets, both 200-mg doses.
As long as other mifepristone REMS criteria are met, the FDA will continue to permit mail and mail-order prescriptions, according to the letter.
"By halting enforcement of the in-person dispensing requirement during the COVID-19 pandemic, the FDA is recognizing and responding to the available evidence - which has clearly and definitively demonstrated that the in-person dispensing requirement for mifepristone is unnecessary and restrictive," Maureen G. Phipps, MD, MPH, CEO of ACOG, said in a statement in response to the FDA decision.
ACOG petitioned the FDA to suspend the in-person requirement to reduce the risk of transmission in the wake of the COVID-19 pandemic, given safety concerns and the potential impact on hard-hit communities, particularly communities of color, Dr. Phipps emphasized. Data from a review period with a suspension of the in-person requirement yielded no additional safety concerns with mifepristone use, and contributed to the FDA decision to lift the requirement.
"Thanks to the FDA's intent to exercise discretion in enforcing the in-person dispensing requirement, those in need of an abortion or miscarriage management will be able to do so safety and effectively by acquiring mifepristone though the mail - just as they would any other medication with a similarly strong safety profile," said Dr. Phipps. "We are pleased to see mifepristone regulated on the basis of the scientific evidence during the pandemic, rather than political bias against comprehensive reproductive health care, and we look forward to working with policy makers to ensure this principle governs postpandemic care."
CDER is communicating the decision to all approved application holders subject to the mifepristone REMS program, according to the letter.
[email protected]
The Food and Drug Administration has lifted in-person dispensing requirements for mifepristone when used for medical termination of early pregnancy.
In an April 12, 2021, letter to the American College of Obstetricians and Gynecologists and the Society of Maternal-Fetal Medicine, acting commissioner of food and drugs Janet Woodcock stated that the FDA would exercise discretion to permit the dispensing of mifepristone through the mail when done by or under the supervision of a certified prescriber; or through a mail-order pharmacy under the supervision of a certified prescriber.
The decision follows a trial period of suspension of the in-person dispensing requirement in response to safety concerns for patients as well as providers associated with in-person clinic visits during the COVID-19 pandemic. The Center for Drug Evaluation and Research reviewed safety and clinical outcomes data on mifepristone use when prescriptions were handled by mail or mail-order pharmacy and found that "the small number of adverse events reported to FDA during the COVID-19 public health emergency [PHE] provide no indication that any program deviation or noncompliance with the mifepristone [Risk Evaluation and Mitigation Strategy] program contributed to the reported adverse events," according to the letter. The analysis covers Mifeprex and the approved generic, mifepristone tablets, both 200-mg doses.
As long as other mifepristone REMS criteria are met, the FDA will continue to permit mail and mail-order prescriptions, according to the letter.
"By halting enforcement of the in-person dispensing requirement during the COVID-19 pandemic, the FDA is recognizing and responding to the available evidence - which has clearly and definitively demonstrated that the in-person dispensing requirement for mifepristone is unnecessary and restrictive," Maureen G. Phipps, MD, MPH, CEO of ACOG, said in a statement in response to the FDA decision.
ACOG petitioned the FDA to suspend the in-person requirement to reduce the risk of transmission in the wake of the COVID-19 pandemic, given safety concerns and the potential impact on hard-hit communities, particularly communities of color, Dr. Phipps emphasized. Data from a review period with a suspension of the in-person requirement yielded no additional safety concerns with mifepristone use, and contributed to the FDA decision to lift the requirement.
"Thanks to the FDA's intent to exercise discretion in enforcing the in-person dispensing requirement, those in need of an abortion or miscarriage management will be able to do so safety and effectively by acquiring mifepristone though the mail - just as they would any other medication with a similarly strong safety profile," said Dr. Phipps. "We are pleased to see mifepristone regulated on the basis of the scientific evidence during the pandemic, rather than political bias against comprehensive reproductive health care, and we look forward to working with policy makers to ensure this principle governs postpandemic care."
CDER is communicating the decision to all approved application holders subject to the mifepristone REMS program, according to the letter.
[email protected]
The Food and Drug Administration has lifted in-person dispensing requirements for mifepristone when used for medical termination of early pregnancy.
In an April 12, 2021, letter to the American College of Obstetricians and Gynecologists and the Society of Maternal-Fetal Medicine, acting commissioner of food and drugs Janet Woodcock stated that the FDA would exercise discretion to permit the dispensing of mifepristone through the mail when done by or under the supervision of a certified prescriber; or through a mail-order pharmacy under the supervision of a certified prescriber.
The decision follows a trial period of suspension of the in-person dispensing requirement in response to safety concerns for patients as well as providers associated with in-person clinic visits during the COVID-19 pandemic. The Center for Drug Evaluation and Research reviewed safety and clinical outcomes data on mifepristone use when prescriptions were handled by mail or mail-order pharmacy and found that "the small number of adverse events reported to FDA during the COVID-19 public health emergency [PHE] provide no indication that any program deviation or noncompliance with the mifepristone [Risk Evaluation and Mitigation Strategy] program contributed to the reported adverse events," according to the letter. The analysis covers Mifeprex and the approved generic, mifepristone tablets, both 200-mg doses.
As long as other mifepristone REMS criteria are met, the FDA will continue to permit mail and mail-order prescriptions, according to the letter.
"By halting enforcement of the in-person dispensing requirement during the COVID-19 pandemic, the FDA is recognizing and responding to the available evidence - which has clearly and definitively demonstrated that the in-person dispensing requirement for mifepristone is unnecessary and restrictive," Maureen G. Phipps, MD, MPH, CEO of ACOG, said in a statement in response to the FDA decision.
ACOG petitioned the FDA to suspend the in-person requirement to reduce the risk of transmission in the wake of the COVID-19 pandemic, given safety concerns and the potential impact on hard-hit communities, particularly communities of color, Dr. Phipps emphasized. Data from a review period with a suspension of the in-person requirement yielded no additional safety concerns with mifepristone use, and contributed to the FDA decision to lift the requirement.
"Thanks to the FDA's intent to exercise discretion in enforcing the in-person dispensing requirement, those in need of an abortion or miscarriage management will be able to do so safety and effectively by acquiring mifepristone though the mail - just as they would any other medication with a similarly strong safety profile," said Dr. Phipps. "We are pleased to see mifepristone regulated on the basis of the scientific evidence during the pandemic, rather than political bias against comprehensive reproductive health care, and we look forward to working with policy makers to ensure this principle governs postpandemic care."
CDER is communicating the decision to all approved application holders subject to the mifepristone REMS program, according to the letter.
[email protected]
Medtronic recall of almost 240,000 ICDs is class I, FDA says
The Food and Drug Administration has declared Medtronic’s recall of seven models of defibrillating cardiac rhythm devices, caused by a risk for premature battery depletion, as class I, which implies a potential risk for serious injury or death. A total of 444 complaints, but no deaths, have been reported in association with the 239,171 affected devices, the agency said in a statement on April 12, 2021.
Physicians were notified of the company’s recall in early February. It covered implantable cardioverter defibrillator (ICD) and cardiac resynchronization therapy–defibrillator (CRT-D) models Evera, Viva, Brava, Claria, Amplia, Compia, and Visia distributed from Aug. 31, 2012 to May 9, 2018.
The devices could be subject to “an unexpected and rapid decrease in battery life” because of a possible short circuit that could lead to a device-replacement alert “earlier than expected.” Some devices may experience full battery depletion “within as little as 1 day” after such an alert.
“If the user does not respond to the first warning, the device may stop functioning. The likelihood that this issue will occur is constant after approximately 3 years after device use,” the announcement said.
Medtronic recommends device replacement no more than 1 week after such an early warning for patients who are not pacing dependent or who have them for primary prevention, but right away for pacing-dependent patients.
A version of this article first appeared on Medscape.com
The Food and Drug Administration has declared Medtronic’s recall of seven models of defibrillating cardiac rhythm devices, caused by a risk for premature battery depletion, as class I, which implies a potential risk for serious injury or death. A total of 444 complaints, but no deaths, have been reported in association with the 239,171 affected devices, the agency said in a statement on April 12, 2021.
Physicians were notified of the company’s recall in early February. It covered implantable cardioverter defibrillator (ICD) and cardiac resynchronization therapy–defibrillator (CRT-D) models Evera, Viva, Brava, Claria, Amplia, Compia, and Visia distributed from Aug. 31, 2012 to May 9, 2018.
The devices could be subject to “an unexpected and rapid decrease in battery life” because of a possible short circuit that could lead to a device-replacement alert “earlier than expected.” Some devices may experience full battery depletion “within as little as 1 day” after such an alert.
“If the user does not respond to the first warning, the device may stop functioning. The likelihood that this issue will occur is constant after approximately 3 years after device use,” the announcement said.
Medtronic recommends device replacement no more than 1 week after such an early warning for patients who are not pacing dependent or who have them for primary prevention, but right away for pacing-dependent patients.
A version of this article first appeared on Medscape.com
The Food and Drug Administration has declared Medtronic’s recall of seven models of defibrillating cardiac rhythm devices, caused by a risk for premature battery depletion, as class I, which implies a potential risk for serious injury or death. A total of 444 complaints, but no deaths, have been reported in association with the 239,171 affected devices, the agency said in a statement on April 12, 2021.
Physicians were notified of the company’s recall in early February. It covered implantable cardioverter defibrillator (ICD) and cardiac resynchronization therapy–defibrillator (CRT-D) models Evera, Viva, Brava, Claria, Amplia, Compia, and Visia distributed from Aug. 31, 2012 to May 9, 2018.
The devices could be subject to “an unexpected and rapid decrease in battery life” because of a possible short circuit that could lead to a device-replacement alert “earlier than expected.” Some devices may experience full battery depletion “within as little as 1 day” after such an alert.
“If the user does not respond to the first warning, the device may stop functioning. The likelihood that this issue will occur is constant after approximately 3 years after device use,” the announcement said.
Medtronic recommends device replacement no more than 1 week after such an early warning for patients who are not pacing dependent or who have them for primary prevention, but right away for pacing-dependent patients.
A version of this article first appeared on Medscape.com
FDA, CDC urge pause of J&J COVID vaccine
The Food and Drug Administration and Centers for Disease Control and Prevention on April 13 recommended that use of the Johnson & Johnson COVID-19 vaccine be paused after reports of blood clots in patients receiving the shot, the agencies have announced.
In a statement, FDA said 6.8 million doses of the J&J vaccine have been administered and the agency is investigating six reported cases of a rare and severe blood clot occurring in patients who received the vaccine.
The pause is intended to give time to alert the public to this "very rare" condition, experts said during a joint CDC-FDA media briefing April 13.
"It was clear to us that we needed to alert the public," Janet Woodcock, MD, acting FDA commissioner, said. The move also will allow "time for the healthcare community to learn what they need to know about how to diagnose, treat and report" any additional cases.
The CDC will convene a meeting of the Advisory Committee on Immunization Practices on April 14 to review the cases.
"I know the information today will be very concerning to Americans who have already received the Johnson & Johnson vaccine," said Anne Schuchat, MD, principal deputy director at the CDC.
"For people who got the vaccine more than one month ago, the risk is very low at this time," she added. "For people who recently got the vaccine, in the last couple of weeks, look for symptoms."
Headache, leg pain, abdominal pain, and shortness of breath were among the reported symptoms. All six cases arose within 6 to 13 days of receipt of the Johnson & Johnson vaccine.
Traditional treatment dangerous
Importantly, treatment for traditional blood clots, such as the drug heparin, should not be used for these clots. "The issue here with these types of blood clots is that if one administers the standard treatment we give for blood clots, one can cause tremendous harm or it can be fatal," said Peter Marks, MD, director of the FDA Center for Biologics Evaluation and Research.
If health care providers see people with these symptoms along with a low platelet count or blood clots, they should ask about any recent vaccinations, Dr. Marks added.
Headache is a common side effect of COVID-19 vaccination, Dr. Marks said, but it typically happens within a day or two. In contrast, the headaches associated with these blood clots come 1 to 2 weeks later and were very severe.
Not all of the six women involved in the events had a pre-existing condition or risk factor, Dr. Schuchat said.
Severe but 'extremely rare'
To put the numbers in context, the six reported events occurred among millions of people who received the Johnson & Johnson vaccine to date.
"There have been six reports of a severe stroke-like illness due to low platelet count and more than six million doses of the Johnson & Johnson vaccine have been administered so far," Dr. Schuchat said.
"I would like to stress these events are extremely rare," Dr. Woodcock said, "but we take all reports of adverse events after vaccination very seriously."
The company response
Johnson & Johnson in a statement said, "We are aware of an extremely rare disorder involving people with blood clots in combination with low platelets in a small number of individuals who have received our COVID-19 vaccine. The United States Centers for Disease Control (CDC) and Food and Drug Administration (FDA) are reviewing data involving six reported U.S. cases out of more than 6.8 million doses administered. Out of an abundance of caution, the CDC and FDA have recommended a pause in the use of our vaccine."
The company said they are also reviewing these cases with European regulators and "we have made the decision to proactively delay the rollout of our vaccine in Europe."
Overall vaccinations continuing apace
"This announcement will not have a significant impact on our vaccination plan. Johnson & Johnson vaccine makes up less than 5% of the recorded shots in arms in the United States to date," Jeff Zients, White House COVID-19 Response Coordinator, said in a statement.
"Based on actions taken by the president earlier this year, the United States has secured enough Pfizer and Moderna doses for 300 million Americans. We are working now with our state and federal partners to get anyone scheduled for a J&J vaccine quickly rescheduled for a Pfizer or Moderna vaccine," he added.
The likely duration of the pause remains unclear.
"I know this has been a long and difficult pandemic, and people are tired of the steps they have to take," Dr. Schuchat said. "Steps taken today make sure the health care system is ready to diagnose, treat and report [any additional cases] and the public has the information necessary to stay safe."
A version of this article first appeared on WebMD.com.
This article was updated 4/13/21.
The Food and Drug Administration and Centers for Disease Control and Prevention on April 13 recommended that use of the Johnson & Johnson COVID-19 vaccine be paused after reports of blood clots in patients receiving the shot, the agencies have announced.
In a statement, FDA said 6.8 million doses of the J&J vaccine have been administered and the agency is investigating six reported cases of a rare and severe blood clot occurring in patients who received the vaccine.
The pause is intended to give time to alert the public to this "very rare" condition, experts said during a joint CDC-FDA media briefing April 13.
"It was clear to us that we needed to alert the public," Janet Woodcock, MD, acting FDA commissioner, said. The move also will allow "time for the healthcare community to learn what they need to know about how to diagnose, treat and report" any additional cases.
The CDC will convene a meeting of the Advisory Committee on Immunization Practices on April 14 to review the cases.
"I know the information today will be very concerning to Americans who have already received the Johnson & Johnson vaccine," said Anne Schuchat, MD, principal deputy director at the CDC.
"For people who got the vaccine more than one month ago, the risk is very low at this time," she added. "For people who recently got the vaccine, in the last couple of weeks, look for symptoms."
Headache, leg pain, abdominal pain, and shortness of breath were among the reported symptoms. All six cases arose within 6 to 13 days of receipt of the Johnson & Johnson vaccine.
Traditional treatment dangerous
Importantly, treatment for traditional blood clots, such as the drug heparin, should not be used for these clots. "The issue here with these types of blood clots is that if one administers the standard treatment we give for blood clots, one can cause tremendous harm or it can be fatal," said Peter Marks, MD, director of the FDA Center for Biologics Evaluation and Research.
If health care providers see people with these symptoms along with a low platelet count or blood clots, they should ask about any recent vaccinations, Dr. Marks added.
Headache is a common side effect of COVID-19 vaccination, Dr. Marks said, but it typically happens within a day or two. In contrast, the headaches associated with these blood clots come 1 to 2 weeks later and were very severe.
Not all of the six women involved in the events had a pre-existing condition or risk factor, Dr. Schuchat said.
Severe but 'extremely rare'
To put the numbers in context, the six reported events occurred among millions of people who received the Johnson & Johnson vaccine to date.
"There have been six reports of a severe stroke-like illness due to low platelet count and more than six million doses of the Johnson & Johnson vaccine have been administered so far," Dr. Schuchat said.
"I would like to stress these events are extremely rare," Dr. Woodcock said, "but we take all reports of adverse events after vaccination very seriously."
The company response
Johnson & Johnson in a statement said, "We are aware of an extremely rare disorder involving people with blood clots in combination with low platelets in a small number of individuals who have received our COVID-19 vaccine. The United States Centers for Disease Control (CDC) and Food and Drug Administration (FDA) are reviewing data involving six reported U.S. cases out of more than 6.8 million doses administered. Out of an abundance of caution, the CDC and FDA have recommended a pause in the use of our vaccine."
The company said they are also reviewing these cases with European regulators and "we have made the decision to proactively delay the rollout of our vaccine in Europe."
Overall vaccinations continuing apace
"This announcement will not have a significant impact on our vaccination plan. Johnson & Johnson vaccine makes up less than 5% of the recorded shots in arms in the United States to date," Jeff Zients, White House COVID-19 Response Coordinator, said in a statement.
"Based on actions taken by the president earlier this year, the United States has secured enough Pfizer and Moderna doses for 300 million Americans. We are working now with our state and federal partners to get anyone scheduled for a J&J vaccine quickly rescheduled for a Pfizer or Moderna vaccine," he added.
The likely duration of the pause remains unclear.
"I know this has been a long and difficult pandemic, and people are tired of the steps they have to take," Dr. Schuchat said. "Steps taken today make sure the health care system is ready to diagnose, treat and report [any additional cases] and the public has the information necessary to stay safe."
A version of this article first appeared on WebMD.com.
This article was updated 4/13/21.
The Food and Drug Administration and Centers for Disease Control and Prevention on April 13 recommended that use of the Johnson & Johnson COVID-19 vaccine be paused after reports of blood clots in patients receiving the shot, the agencies have announced.
In a statement, FDA said 6.8 million doses of the J&J vaccine have been administered and the agency is investigating six reported cases of a rare and severe blood clot occurring in patients who received the vaccine.
The pause is intended to give time to alert the public to this "very rare" condition, experts said during a joint CDC-FDA media briefing April 13.
"It was clear to us that we needed to alert the public," Janet Woodcock, MD, acting FDA commissioner, said. The move also will allow "time for the healthcare community to learn what they need to know about how to diagnose, treat and report" any additional cases.
The CDC will convene a meeting of the Advisory Committee on Immunization Practices on April 14 to review the cases.
"I know the information today will be very concerning to Americans who have already received the Johnson & Johnson vaccine," said Anne Schuchat, MD, principal deputy director at the CDC.
"For people who got the vaccine more than one month ago, the risk is very low at this time," she added. "For people who recently got the vaccine, in the last couple of weeks, look for symptoms."
Headache, leg pain, abdominal pain, and shortness of breath were among the reported symptoms. All six cases arose within 6 to 13 days of receipt of the Johnson & Johnson vaccine.
Traditional treatment dangerous
Importantly, treatment for traditional blood clots, such as the drug heparin, should not be used for these clots. "The issue here with these types of blood clots is that if one administers the standard treatment we give for blood clots, one can cause tremendous harm or it can be fatal," said Peter Marks, MD, director of the FDA Center for Biologics Evaluation and Research.
If health care providers see people with these symptoms along with a low platelet count or blood clots, they should ask about any recent vaccinations, Dr. Marks added.
Headache is a common side effect of COVID-19 vaccination, Dr. Marks said, but it typically happens within a day or two. In contrast, the headaches associated with these blood clots come 1 to 2 weeks later and were very severe.
Not all of the six women involved in the events had a pre-existing condition or risk factor, Dr. Schuchat said.
Severe but 'extremely rare'
To put the numbers in context, the six reported events occurred among millions of people who received the Johnson & Johnson vaccine to date.
"There have been six reports of a severe stroke-like illness due to low platelet count and more than six million doses of the Johnson & Johnson vaccine have been administered so far," Dr. Schuchat said.
"I would like to stress these events are extremely rare," Dr. Woodcock said, "but we take all reports of adverse events after vaccination very seriously."
The company response
Johnson & Johnson in a statement said, "We are aware of an extremely rare disorder involving people with blood clots in combination with low platelets in a small number of individuals who have received our COVID-19 vaccine. The United States Centers for Disease Control (CDC) and Food and Drug Administration (FDA) are reviewing data involving six reported U.S. cases out of more than 6.8 million doses administered. Out of an abundance of caution, the CDC and FDA have recommended a pause in the use of our vaccine."
The company said they are also reviewing these cases with European regulators and "we have made the decision to proactively delay the rollout of our vaccine in Europe."
Overall vaccinations continuing apace
"This announcement will not have a significant impact on our vaccination plan. Johnson & Johnson vaccine makes up less than 5% of the recorded shots in arms in the United States to date," Jeff Zients, White House COVID-19 Response Coordinator, said in a statement.
"Based on actions taken by the president earlier this year, the United States has secured enough Pfizer and Moderna doses for 300 million Americans. We are working now with our state and federal partners to get anyone scheduled for a J&J vaccine quickly rescheduled for a Pfizer or Moderna vaccine," he added.
The likely duration of the pause remains unclear.
"I know this has been a long and difficult pandemic, and people are tired of the steps they have to take," Dr. Schuchat said. "Steps taken today make sure the health care system is ready to diagnose, treat and report [any additional cases] and the public has the information necessary to stay safe."
A version of this article first appeared on WebMD.com.
This article was updated 4/13/21.