User login
FDA denies petition to disqualify researchers over controversial ketamine studies
The U.S. Food and Drug Administration has declined to take further action against a group of investigators at Hennepin County Medical Center/Hennepin Healthcare (HCMC) who conducted controversial studies involving ketamine and other sedatives on agitated persons without their consent.
A citizen petition filed by Public Citizen, a consumer advocacy group, had asked the FDA to initiate clinical-investigator disqualification proceedings against Jon Cole, MD, and Lauren Klein, MD, along with other researchers who participated in the studies, for “repeatedly and deliberately initiating and conducting clinical investigations of investigational drug products” without having submitted or having in effect the investigational new drug applications (INDs) required by the FDA.
In certain situations, wherein the FDA alleges that a clinical investigator has violated applicable regulations, the agency may initiate clinical investigator disqualification proceedings. The names of the disqualified researchers are then added to a federal database.
The petition, which was filed in November 2021, also requested that the FDA initiate disqualification proceedings against the institutional review board (IRB) at HCMC for repeatedly failing to comply with federal regulations that adversely affected the rights and welfare of the individuals who were enrolled in the study without their consent.
Of note, Public Citizen stated that the FDA should have required the hospital to contact the more than 1,700 patients who “were unwittingly enrolled in unethical experiments” and inform them that their rights had been violated and their health potentially endangered by the research team.
Michael A. Carome, MD, director of Public Citizen’s Health Research Group, told this news organization that it is uncommon for the FDA to disqualify researchers. “It should be more common than it is,” he said. “I think that FDA is just reluctant to take more action.”
The actions of the Hennepin investigators were “repetitive and appeared to be in deliberate violation of regulations,” he added. “The case for the FDA disqualifying the HCMC researchers is overwhelming. The FDA’s slap-on-the-wrist approach to such appalling regulatory and ethical violations risks emboldening other researchers to disregard the rights and welfare of human subjects.”
Carl Elliott, MD, PhD, a bioethicist at the University of Minnesota, Minneapolis, agrees that the researcher from HCMC should be disqualified. “They didn’t just conduct risky, exploitative studies – they conducted them after the FDA had warned them not to proceed,” he said. “The message sent by this slap on the wrist is that investigators can do whatever they want to nonconsenting subjects, and the FDA will look the other way.”
Initial complaint
Public Citizen initially filed a complaint with the FDA in 2018, after learning that researchers affiliated with HCMC were conducting high-risk clinical trials involving ketamine to control agitation outside of the hospital setting. The complaint was cosigned by 64 doctors, bioethicists, and academic researchers and was also submitted to the Office for Human Research Protections.
The FDA typically allows investigational drugs to be used in emergency situation without obtaining informed consent if the therapies are known to carry a minimal risk. The IRB at HCMC had determined that this was the case with ketamine and approved the trials.
But according to Public Citizen’s complaint, prior research had suggested that ketamine could cause more complications and severe adverse events, compared with other sedatives.
The trials were conducted between 2014 and 2018, and in its letter, Public Citizen alleged that the investigators and the IRB had allowed these trials to proceed without obtaining informed consent from patients. The goal was to evaluate how well ketamine worked, compared with other drugs in calming agitated individuals: “The patients were given either ketamine or haloperidol for agitation by paramedics who responded to medical emergencies, and the goal was to see which drug worked faster,” said Dr. Carome. “Patients were only notified afterwards that they had received a sedative. Informed consent had been waived by IRB.”
In the first clinical trial conducted by HCMC, published in 2016, the researchers had hypothesized that 5 mg/kg of intramuscular ketamine would be superior to 10 mg of intramuscular haloperidol for severe prehospital agitation. Time to adequate sedation was the primary outcome measure. The study included 146 people; 64 received ketamine and 82 received haloperidol. They found that ketamine worked far more quickly than haloperidol (5 minutes vs. 17 minutes) but that the risk for complications was much higher. Complications occurred in 49% of patients receiving ketamine, compared with 5%.
“There was a 10-fold risk of adverse events,” said Dr. Carome. “And 39% of patients given ketamine had respiratory problems requiring intubation, compared to 4% who received haloperidol.”
A second study was launched in 2017, wherein ketamine was compared with midazolam in agitated patients. During the first 6-month period of the study, individuals would receive a ketamine-based protocol for prehospital agitation, and during the second 6 months, that would switch to midazolam. However, the study was halted in June 2018 after the local newspaper, the Star Tribune, reported that the city police had encouraged medical personnel to sedate agitated patients. This included individuals who had already been physically restrained.
The report stated that “in many cases, the individual being detained or arrested was not only handcuffed but strapped down on a stretcher in an ambulance before receiving ketamine,” and that it raised a “concerning question” over why these people were given the drug before they were transported to the hospital, “given the immediate effects on breathing and heart function that the drug induces.”
Along with halting the trial, HCMC asked for a review of cases involving its paramedics; an independent investigation led by former U.S. Deputy Attorney General Sally Yates was initiated to assess whether the Minneapolis police had crossed a line and urged paramedics to use ketamine.
“The decision to use ketamine was based on the study’s timeline and not on clinical judgment,” said Dr. Carome.
The FDA acknowledged receipt of the complaint and inspected the IRB records and the clinical trial data. Preliminary reports received by Public Citizen confirmed their allegations. “There were not appropriate protections for vulnerable subjects,” he said. “In 2019, the FDA did further investigations, and those reports had similar findings.”
FDA letters
The FDA had sent warning letters to Dr. Cole and Dr. Klein, citing them for ignoring federal safety laws in experimental research on the public. In their investigations, the FDA cited “objectionable conditions” for the studies led by Dr. Cole and Dr. Klein, according to the letters. Both researchers seemingly ignored FDA regulations and used practices that subjected patients to “significantly increased risk,” and the hospital defended its research with “factually incorrect” statements.
In a letter to Dr. Cole, the FDA noted that he never filed INDs for the trials with the FDA, as required by law, and that he also failed to write appropriate protocols to ensure that children and pregnant women were not enrolled in the research. Individuals under the influence of intoxicants also were not excluded, though the use of ketamine is cautioned in this population.
“Administration of the investigational drugs to these subjects placed them at significantly increased risk of the adverse events associated with the investigational products and decreased the acceptability of those risks,” the FDA said in its letter. “Your failure to exclude, and the lack of any precautions for, subjects under the influence of various intoxicants significantly increased the risks and/or decreased the acceptability of the risks associated with the investigational drugs.”
However, Dr. Cole conducted both studies in the prehospital setting and failed to initiate any specific measures to protect study participants, according to the FDA.
Petition denied
Dr. Carome noted that the researchers had committed repetitive egregious regulatory violations over a 4-year period, which were documented by the FDA in their warning letters to Dr. Cole and Dr. Klein. “We felt that they were so egregious that we need to send a signal to the community that this sort of behavior will not be tolerated,” he said. “The FDA denied our petition, and we think that sends the wrong signal to the research community.”
In their response, the FDA noted that as with judicial enforcement, “the Agency makes decisions regarding whether to pursue administrative enforcement action, including disqualification proceedings, on a case-by-case basis, considering all relevant facts and circumstances.” They added that at this time, they would not be taking further action against Dr. Cole and Dr. Klein.
“However, we intend to continue to consider all the options available to the Agency as we determine whether to pursue additional compliance actions related to this matter,” the FDA concluded.
The FDA declined to comment further on their decision.
Dr. Cole also declined to comment, but Hennepin Healthcare told this news organization that the “decision by the FDA to deny the petition validates the changes we made to strengthen and improve the clinical research program across the institution since the closing of the studies in 2018. We look forward to continuing to work with the FDA to ensure full compliance with the standards in place to protect research subjects.”
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has declined to take further action against a group of investigators at Hennepin County Medical Center/Hennepin Healthcare (HCMC) who conducted controversial studies involving ketamine and other sedatives on agitated persons without their consent.
A citizen petition filed by Public Citizen, a consumer advocacy group, had asked the FDA to initiate clinical-investigator disqualification proceedings against Jon Cole, MD, and Lauren Klein, MD, along with other researchers who participated in the studies, for “repeatedly and deliberately initiating and conducting clinical investigations of investigational drug products” without having submitted or having in effect the investigational new drug applications (INDs) required by the FDA.
In certain situations, wherein the FDA alleges that a clinical investigator has violated applicable regulations, the agency may initiate clinical investigator disqualification proceedings. The names of the disqualified researchers are then added to a federal database.
The petition, which was filed in November 2021, also requested that the FDA initiate disqualification proceedings against the institutional review board (IRB) at HCMC for repeatedly failing to comply with federal regulations that adversely affected the rights and welfare of the individuals who were enrolled in the study without their consent.
Of note, Public Citizen stated that the FDA should have required the hospital to contact the more than 1,700 patients who “were unwittingly enrolled in unethical experiments” and inform them that their rights had been violated and their health potentially endangered by the research team.
Michael A. Carome, MD, director of Public Citizen’s Health Research Group, told this news organization that it is uncommon for the FDA to disqualify researchers. “It should be more common than it is,” he said. “I think that FDA is just reluctant to take more action.”
The actions of the Hennepin investigators were “repetitive and appeared to be in deliberate violation of regulations,” he added. “The case for the FDA disqualifying the HCMC researchers is overwhelming. The FDA’s slap-on-the-wrist approach to such appalling regulatory and ethical violations risks emboldening other researchers to disregard the rights and welfare of human subjects.”
Carl Elliott, MD, PhD, a bioethicist at the University of Minnesota, Minneapolis, agrees that the researcher from HCMC should be disqualified. “They didn’t just conduct risky, exploitative studies – they conducted them after the FDA had warned them not to proceed,” he said. “The message sent by this slap on the wrist is that investigators can do whatever they want to nonconsenting subjects, and the FDA will look the other way.”
Initial complaint
Public Citizen initially filed a complaint with the FDA in 2018, after learning that researchers affiliated with HCMC were conducting high-risk clinical trials involving ketamine to control agitation outside of the hospital setting. The complaint was cosigned by 64 doctors, bioethicists, and academic researchers and was also submitted to the Office for Human Research Protections.
The FDA typically allows investigational drugs to be used in emergency situation without obtaining informed consent if the therapies are known to carry a minimal risk. The IRB at HCMC had determined that this was the case with ketamine and approved the trials.
But according to Public Citizen’s complaint, prior research had suggested that ketamine could cause more complications and severe adverse events, compared with other sedatives.
The trials were conducted between 2014 and 2018, and in its letter, Public Citizen alleged that the investigators and the IRB had allowed these trials to proceed without obtaining informed consent from patients. The goal was to evaluate how well ketamine worked, compared with other drugs in calming agitated individuals: “The patients were given either ketamine or haloperidol for agitation by paramedics who responded to medical emergencies, and the goal was to see which drug worked faster,” said Dr. Carome. “Patients were only notified afterwards that they had received a sedative. Informed consent had been waived by IRB.”
In the first clinical trial conducted by HCMC, published in 2016, the researchers had hypothesized that 5 mg/kg of intramuscular ketamine would be superior to 10 mg of intramuscular haloperidol for severe prehospital agitation. Time to adequate sedation was the primary outcome measure. The study included 146 people; 64 received ketamine and 82 received haloperidol. They found that ketamine worked far more quickly than haloperidol (5 minutes vs. 17 minutes) but that the risk for complications was much higher. Complications occurred in 49% of patients receiving ketamine, compared with 5%.
“There was a 10-fold risk of adverse events,” said Dr. Carome. “And 39% of patients given ketamine had respiratory problems requiring intubation, compared to 4% who received haloperidol.”
A second study was launched in 2017, wherein ketamine was compared with midazolam in agitated patients. During the first 6-month period of the study, individuals would receive a ketamine-based protocol for prehospital agitation, and during the second 6 months, that would switch to midazolam. However, the study was halted in June 2018 after the local newspaper, the Star Tribune, reported that the city police had encouraged medical personnel to sedate agitated patients. This included individuals who had already been physically restrained.
The report stated that “in many cases, the individual being detained or arrested was not only handcuffed but strapped down on a stretcher in an ambulance before receiving ketamine,” and that it raised a “concerning question” over why these people were given the drug before they were transported to the hospital, “given the immediate effects on breathing and heart function that the drug induces.”
Along with halting the trial, HCMC asked for a review of cases involving its paramedics; an independent investigation led by former U.S. Deputy Attorney General Sally Yates was initiated to assess whether the Minneapolis police had crossed a line and urged paramedics to use ketamine.
“The decision to use ketamine was based on the study’s timeline and not on clinical judgment,” said Dr. Carome.
The FDA acknowledged receipt of the complaint and inspected the IRB records and the clinical trial data. Preliminary reports received by Public Citizen confirmed their allegations. “There were not appropriate protections for vulnerable subjects,” he said. “In 2019, the FDA did further investigations, and those reports had similar findings.”
FDA letters
The FDA had sent warning letters to Dr. Cole and Dr. Klein, citing them for ignoring federal safety laws in experimental research on the public. In their investigations, the FDA cited “objectionable conditions” for the studies led by Dr. Cole and Dr. Klein, according to the letters. Both researchers seemingly ignored FDA regulations and used practices that subjected patients to “significantly increased risk,” and the hospital defended its research with “factually incorrect” statements.
In a letter to Dr. Cole, the FDA noted that he never filed INDs for the trials with the FDA, as required by law, and that he also failed to write appropriate protocols to ensure that children and pregnant women were not enrolled in the research. Individuals under the influence of intoxicants also were not excluded, though the use of ketamine is cautioned in this population.
“Administration of the investigational drugs to these subjects placed them at significantly increased risk of the adverse events associated with the investigational products and decreased the acceptability of those risks,” the FDA said in its letter. “Your failure to exclude, and the lack of any precautions for, subjects under the influence of various intoxicants significantly increased the risks and/or decreased the acceptability of the risks associated with the investigational drugs.”
However, Dr. Cole conducted both studies in the prehospital setting and failed to initiate any specific measures to protect study participants, according to the FDA.
Petition denied
Dr. Carome noted that the researchers had committed repetitive egregious regulatory violations over a 4-year period, which were documented by the FDA in their warning letters to Dr. Cole and Dr. Klein. “We felt that they were so egregious that we need to send a signal to the community that this sort of behavior will not be tolerated,” he said. “The FDA denied our petition, and we think that sends the wrong signal to the research community.”
In their response, the FDA noted that as with judicial enforcement, “the Agency makes decisions regarding whether to pursue administrative enforcement action, including disqualification proceedings, on a case-by-case basis, considering all relevant facts and circumstances.” They added that at this time, they would not be taking further action against Dr. Cole and Dr. Klein.
“However, we intend to continue to consider all the options available to the Agency as we determine whether to pursue additional compliance actions related to this matter,” the FDA concluded.
The FDA declined to comment further on their decision.
Dr. Cole also declined to comment, but Hennepin Healthcare told this news organization that the “decision by the FDA to deny the petition validates the changes we made to strengthen and improve the clinical research program across the institution since the closing of the studies in 2018. We look forward to continuing to work with the FDA to ensure full compliance with the standards in place to protect research subjects.”
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has declined to take further action against a group of investigators at Hennepin County Medical Center/Hennepin Healthcare (HCMC) who conducted controversial studies involving ketamine and other sedatives on agitated persons without their consent.
A citizen petition filed by Public Citizen, a consumer advocacy group, had asked the FDA to initiate clinical-investigator disqualification proceedings against Jon Cole, MD, and Lauren Klein, MD, along with other researchers who participated in the studies, for “repeatedly and deliberately initiating and conducting clinical investigations of investigational drug products” without having submitted or having in effect the investigational new drug applications (INDs) required by the FDA.
In certain situations, wherein the FDA alleges that a clinical investigator has violated applicable regulations, the agency may initiate clinical investigator disqualification proceedings. The names of the disqualified researchers are then added to a federal database.
The petition, which was filed in November 2021, also requested that the FDA initiate disqualification proceedings against the institutional review board (IRB) at HCMC for repeatedly failing to comply with federal regulations that adversely affected the rights and welfare of the individuals who were enrolled in the study without their consent.
Of note, Public Citizen stated that the FDA should have required the hospital to contact the more than 1,700 patients who “were unwittingly enrolled in unethical experiments” and inform them that their rights had been violated and their health potentially endangered by the research team.
Michael A. Carome, MD, director of Public Citizen’s Health Research Group, told this news organization that it is uncommon for the FDA to disqualify researchers. “It should be more common than it is,” he said. “I think that FDA is just reluctant to take more action.”
The actions of the Hennepin investigators were “repetitive and appeared to be in deliberate violation of regulations,” he added. “The case for the FDA disqualifying the HCMC researchers is overwhelming. The FDA’s slap-on-the-wrist approach to such appalling regulatory and ethical violations risks emboldening other researchers to disregard the rights and welfare of human subjects.”
Carl Elliott, MD, PhD, a bioethicist at the University of Minnesota, Minneapolis, agrees that the researcher from HCMC should be disqualified. “They didn’t just conduct risky, exploitative studies – they conducted them after the FDA had warned them not to proceed,” he said. “The message sent by this slap on the wrist is that investigators can do whatever they want to nonconsenting subjects, and the FDA will look the other way.”
Initial complaint
Public Citizen initially filed a complaint with the FDA in 2018, after learning that researchers affiliated with HCMC were conducting high-risk clinical trials involving ketamine to control agitation outside of the hospital setting. The complaint was cosigned by 64 doctors, bioethicists, and academic researchers and was also submitted to the Office for Human Research Protections.
The FDA typically allows investigational drugs to be used in emergency situation without obtaining informed consent if the therapies are known to carry a minimal risk. The IRB at HCMC had determined that this was the case with ketamine and approved the trials.
But according to Public Citizen’s complaint, prior research had suggested that ketamine could cause more complications and severe adverse events, compared with other sedatives.
The trials were conducted between 2014 and 2018, and in its letter, Public Citizen alleged that the investigators and the IRB had allowed these trials to proceed without obtaining informed consent from patients. The goal was to evaluate how well ketamine worked, compared with other drugs in calming agitated individuals: “The patients were given either ketamine or haloperidol for agitation by paramedics who responded to medical emergencies, and the goal was to see which drug worked faster,” said Dr. Carome. “Patients were only notified afterwards that they had received a sedative. Informed consent had been waived by IRB.”
In the first clinical trial conducted by HCMC, published in 2016, the researchers had hypothesized that 5 mg/kg of intramuscular ketamine would be superior to 10 mg of intramuscular haloperidol for severe prehospital agitation. Time to adequate sedation was the primary outcome measure. The study included 146 people; 64 received ketamine and 82 received haloperidol. They found that ketamine worked far more quickly than haloperidol (5 minutes vs. 17 minutes) but that the risk for complications was much higher. Complications occurred in 49% of patients receiving ketamine, compared with 5%.
“There was a 10-fold risk of adverse events,” said Dr. Carome. “And 39% of patients given ketamine had respiratory problems requiring intubation, compared to 4% who received haloperidol.”
A second study was launched in 2017, wherein ketamine was compared with midazolam in agitated patients. During the first 6-month period of the study, individuals would receive a ketamine-based protocol for prehospital agitation, and during the second 6 months, that would switch to midazolam. However, the study was halted in June 2018 after the local newspaper, the Star Tribune, reported that the city police had encouraged medical personnel to sedate agitated patients. This included individuals who had already been physically restrained.
The report stated that “in many cases, the individual being detained or arrested was not only handcuffed but strapped down on a stretcher in an ambulance before receiving ketamine,” and that it raised a “concerning question” over why these people were given the drug before they were transported to the hospital, “given the immediate effects on breathing and heart function that the drug induces.”
Along with halting the trial, HCMC asked for a review of cases involving its paramedics; an independent investigation led by former U.S. Deputy Attorney General Sally Yates was initiated to assess whether the Minneapolis police had crossed a line and urged paramedics to use ketamine.
“The decision to use ketamine was based on the study’s timeline and not on clinical judgment,” said Dr. Carome.
The FDA acknowledged receipt of the complaint and inspected the IRB records and the clinical trial data. Preliminary reports received by Public Citizen confirmed their allegations. “There were not appropriate protections for vulnerable subjects,” he said. “In 2019, the FDA did further investigations, and those reports had similar findings.”
FDA letters
The FDA had sent warning letters to Dr. Cole and Dr. Klein, citing them for ignoring federal safety laws in experimental research on the public. In their investigations, the FDA cited “objectionable conditions” for the studies led by Dr. Cole and Dr. Klein, according to the letters. Both researchers seemingly ignored FDA regulations and used practices that subjected patients to “significantly increased risk,” and the hospital defended its research with “factually incorrect” statements.
In a letter to Dr. Cole, the FDA noted that he never filed INDs for the trials with the FDA, as required by law, and that he also failed to write appropriate protocols to ensure that children and pregnant women were not enrolled in the research. Individuals under the influence of intoxicants also were not excluded, though the use of ketamine is cautioned in this population.
“Administration of the investigational drugs to these subjects placed them at significantly increased risk of the adverse events associated with the investigational products and decreased the acceptability of those risks,” the FDA said in its letter. “Your failure to exclude, and the lack of any precautions for, subjects under the influence of various intoxicants significantly increased the risks and/or decreased the acceptability of the risks associated with the investigational drugs.”
However, Dr. Cole conducted both studies in the prehospital setting and failed to initiate any specific measures to protect study participants, according to the FDA.
Petition denied
Dr. Carome noted that the researchers had committed repetitive egregious regulatory violations over a 4-year period, which were documented by the FDA in their warning letters to Dr. Cole and Dr. Klein. “We felt that they were so egregious that we need to send a signal to the community that this sort of behavior will not be tolerated,” he said. “The FDA denied our petition, and we think that sends the wrong signal to the research community.”
In their response, the FDA noted that as with judicial enforcement, “the Agency makes decisions regarding whether to pursue administrative enforcement action, including disqualification proceedings, on a case-by-case basis, considering all relevant facts and circumstances.” They added that at this time, they would not be taking further action against Dr. Cole and Dr. Klein.
“However, we intend to continue to consider all the options available to the Agency as we determine whether to pursue additional compliance actions related to this matter,” the FDA concluded.
The FDA declined to comment further on their decision.
Dr. Cole also declined to comment, but Hennepin Healthcare told this news organization that the “decision by the FDA to deny the petition validates the changes we made to strengthen and improve the clinical research program across the institution since the closing of the studies in 2018. We look forward to continuing to work with the FDA to ensure full compliance with the standards in place to protect research subjects.”
A version of this article first appeared on Medscape.com.
Children & COVID: Rise in new cases slows
New cases of COVID-19 in children climbed for the seventh consecutive week, but the latest increase was the smallest of the seven, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Since the weekly total bottomed out at just under 26,000 in early April, the new-case count has risen by 28.0%, 11.8%, 43.5%, 17.4%, 50%, 14.6%, and 5.0%, based on data from the AAP/CHA weekly COVID-19 report.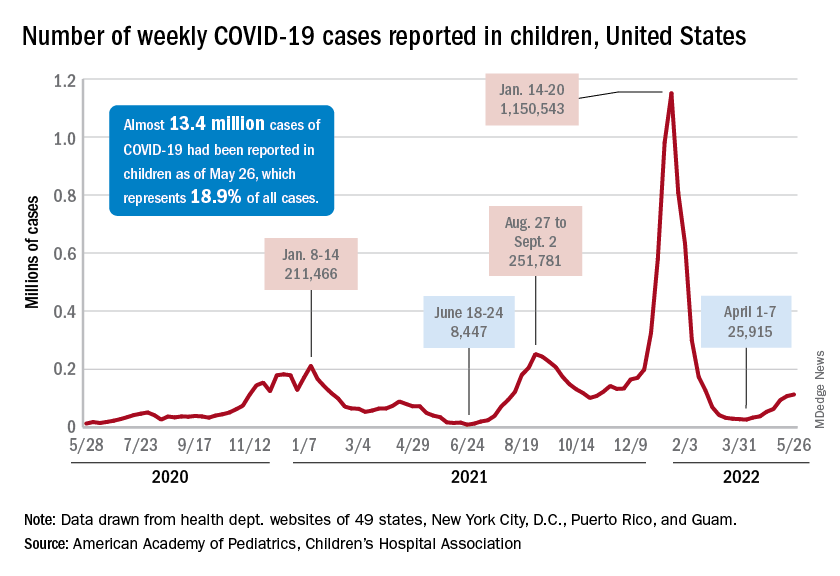
The cumulative number of pediatric cases is almost 13.4 million since the pandemic began, and those infected children represent 18.9% of all cases, the AAP and CHA said based on data from 49 states, New York City, the District of Columbia, Puerto Rico, and Guam.
That 18.9% is noteworthy because it marks the first decline in that particular measure since the AAP and CHA started keeping track in April of 2020. Children’s share of the overall COVID burden had been holding at 19.0% for 14 straight weeks, the AAP/CHA data show.
Regionally, new cases were up in the South and the West, where recent rising trends continued, and down in the Midwest and Northeast, where the recent rising trends were reversed for the first time. At the state/territory level, Puerto Rico had the largest percent increase over the last 2 weeks, followed by Maryland and Delaware, the organizations noted in their joint report.
Hospital admissions in children aged 0-17 have changed little in the last week, with the Centers for Disease Control and Prevention reporting rates of 0.25 per 100,000 population on May 23 and 0.25 per 100,000 on May 29, the latest date available. There was, however, a move up to 0.26 per 100,000 from May 24 to May 28, and the CDC acknowledges a possible reporting delay over the most recent 7-day period.
Emergency department visits have dipped slightly in recent days, with children aged 0-11 years at a 7-day average of 2.0% of ED visits with diagnosed COVID on May 28, down from a 5-day stretch at 2.2% from May 19 to May 23. Children aged 12-15 years were at 1.8% on May 28, compared with 2.0% on May 23-24, and 15- to 17-year-olds were at 2.0% on May 28, down from the 2.1% reached over the previous 2 days, the CDC reported on its COVID Data Tracker.
New cases of COVID-19 in children climbed for the seventh consecutive week, but the latest increase was the smallest of the seven, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Since the weekly total bottomed out at just under 26,000 in early April, the new-case count has risen by 28.0%, 11.8%, 43.5%, 17.4%, 50%, 14.6%, and 5.0%, based on data from the AAP/CHA weekly COVID-19 report.
The cumulative number of pediatric cases is almost 13.4 million since the pandemic began, and those infected children represent 18.9% of all cases, the AAP and CHA said based on data from 49 states, New York City, the District of Columbia, Puerto Rico, and Guam.
That 18.9% is noteworthy because it marks the first decline in that particular measure since the AAP and CHA started keeping track in April of 2020. Children’s share of the overall COVID burden had been holding at 19.0% for 14 straight weeks, the AAP/CHA data show.
Regionally, new cases were up in the South and the West, where recent rising trends continued, and down in the Midwest and Northeast, where the recent rising trends were reversed for the first time. At the state/territory level, Puerto Rico had the largest percent increase over the last 2 weeks, followed by Maryland and Delaware, the organizations noted in their joint report.
Hospital admissions in children aged 0-17 have changed little in the last week, with the Centers for Disease Control and Prevention reporting rates of 0.25 per 100,000 population on May 23 and 0.25 per 100,000 on May 29, the latest date available. There was, however, a move up to 0.26 per 100,000 from May 24 to May 28, and the CDC acknowledges a possible reporting delay over the most recent 7-day period.
Emergency department visits have dipped slightly in recent days, with children aged 0-11 years at a 7-day average of 2.0% of ED visits with diagnosed COVID on May 28, down from a 5-day stretch at 2.2% from May 19 to May 23. Children aged 12-15 years were at 1.8% on May 28, compared with 2.0% on May 23-24, and 15- to 17-year-olds were at 2.0% on May 28, down from the 2.1% reached over the previous 2 days, the CDC reported on its COVID Data Tracker.
New cases of COVID-19 in children climbed for the seventh consecutive week, but the latest increase was the smallest of the seven, according to the American Academy of Pediatrics and the Children’s Hospital Association.
Since the weekly total bottomed out at just under 26,000 in early April, the new-case count has risen by 28.0%, 11.8%, 43.5%, 17.4%, 50%, 14.6%, and 5.0%, based on data from the AAP/CHA weekly COVID-19 report.
The cumulative number of pediatric cases is almost 13.4 million since the pandemic began, and those infected children represent 18.9% of all cases, the AAP and CHA said based on data from 49 states, New York City, the District of Columbia, Puerto Rico, and Guam.
That 18.9% is noteworthy because it marks the first decline in that particular measure since the AAP and CHA started keeping track in April of 2020. Children’s share of the overall COVID burden had been holding at 19.0% for 14 straight weeks, the AAP/CHA data show.
Regionally, new cases were up in the South and the West, where recent rising trends continued, and down in the Midwest and Northeast, where the recent rising trends were reversed for the first time. At the state/territory level, Puerto Rico had the largest percent increase over the last 2 weeks, followed by Maryland and Delaware, the organizations noted in their joint report.
Hospital admissions in children aged 0-17 have changed little in the last week, with the Centers for Disease Control and Prevention reporting rates of 0.25 per 100,000 population on May 23 and 0.25 per 100,000 on May 29, the latest date available. There was, however, a move up to 0.26 per 100,000 from May 24 to May 28, and the CDC acknowledges a possible reporting delay over the most recent 7-day period.
Emergency department visits have dipped slightly in recent days, with children aged 0-11 years at a 7-day average of 2.0% of ED visits with diagnosed COVID on May 28, down from a 5-day stretch at 2.2% from May 19 to May 23. Children aged 12-15 years were at 1.8% on May 28, compared with 2.0% on May 23-24, and 15- to 17-year-olds were at 2.0% on May 28, down from the 2.1% reached over the previous 2 days, the CDC reported on its COVID Data Tracker.
FDA withdraws lymphoma drug approval after investigation
Umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
But safety concerns began to emerge in the phase 3 UNITY-CLL trial, which evaluated the drug in a related cancer type: chronic lymphocytic leukemia.
Last February, the FDA said it was investigating a possible increased risk of death associated with umbralisib.
Five months later, the results are in.
“Updated findings from the UNITY-CLL clinical trial continued to show a possible increased risk of death in patients receiving Ukoniq. As a result, we determined the risks of treatment with Ukoniq outweigh its benefits,” the FDA wrote in a drug safety communication published June 1.
In April, the drug manufacturer, TG Therapeutics, announced it was voluntarily withdrawing umbralisib from the market for its approved uses in marginal zone lymphoma and follicular lymphoma.
The FDA’s safety notice includes instructions for physicians and patients. The FDA urges health care professionals to “stop prescribing Ukoniq and switch patients to alternative treatments” and to “inform patients currently taking Ukoniq of the increased risk of death seen in the clinical trial and advise them to stop taking the medicine.”
In special instances in which a patient may be benefiting from the drug, the company plans to make umbralisib available under expanded access.
The FDA also recommends that patients who discontinue taking the drug dispose of unused umbralisib using a drug take-back location, such as a pharmacy, or throwing it away in the household trash after placing it in a sealed bag mixed with dirt or cat litter and removing personal identification information.
A version of this article first appeared on Medscape.com.
Umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
But safety concerns began to emerge in the phase 3 UNITY-CLL trial, which evaluated the drug in a related cancer type: chronic lymphocytic leukemia.
Last February, the FDA said it was investigating a possible increased risk of death associated with umbralisib.
Five months later, the results are in.
“Updated findings from the UNITY-CLL clinical trial continued to show a possible increased risk of death in patients receiving Ukoniq. As a result, we determined the risks of treatment with Ukoniq outweigh its benefits,” the FDA wrote in a drug safety communication published June 1.
In April, the drug manufacturer, TG Therapeutics, announced it was voluntarily withdrawing umbralisib from the market for its approved uses in marginal zone lymphoma and follicular lymphoma.
The FDA’s safety notice includes instructions for physicians and patients. The FDA urges health care professionals to “stop prescribing Ukoniq and switch patients to alternative treatments” and to “inform patients currently taking Ukoniq of the increased risk of death seen in the clinical trial and advise them to stop taking the medicine.”
In special instances in which a patient may be benefiting from the drug, the company plans to make umbralisib available under expanded access.
The FDA also recommends that patients who discontinue taking the drug dispose of unused umbralisib using a drug take-back location, such as a pharmacy, or throwing it away in the household trash after placing it in a sealed bag mixed with dirt or cat litter and removing personal identification information.
A version of this article first appeared on Medscape.com.
Umbralisib had received accelerated approval in February 2021 to treat adults with relapsed or refractory marginal zone lymphoma following at least one prior therapy and those with relapsed or refractory follicular lymphoma who had received at least three prior therapies.
But safety concerns began to emerge in the phase 3 UNITY-CLL trial, which evaluated the drug in a related cancer type: chronic lymphocytic leukemia.
Last February, the FDA said it was investigating a possible increased risk of death associated with umbralisib.
Five months later, the results are in.
“Updated findings from the UNITY-CLL clinical trial continued to show a possible increased risk of death in patients receiving Ukoniq. As a result, we determined the risks of treatment with Ukoniq outweigh its benefits,” the FDA wrote in a drug safety communication published June 1.
In April, the drug manufacturer, TG Therapeutics, announced it was voluntarily withdrawing umbralisib from the market for its approved uses in marginal zone lymphoma and follicular lymphoma.
The FDA’s safety notice includes instructions for physicians and patients. The FDA urges health care professionals to “stop prescribing Ukoniq and switch patients to alternative treatments” and to “inform patients currently taking Ukoniq of the increased risk of death seen in the clinical trial and advise them to stop taking the medicine.”
In special instances in which a patient may be benefiting from the drug, the company plans to make umbralisib available under expanded access.
The FDA also recommends that patients who discontinue taking the drug dispose of unused umbralisib using a drug take-back location, such as a pharmacy, or throwing it away in the household trash after placing it in a sealed bag mixed with dirt or cat litter and removing personal identification information.
A version of this article first appeared on Medscape.com.
Immunotherapy now first line for esophageal cancer
The new approval for the drug, a programmed cell death–ligand-1 inhibitor, is for use in this patient population regardless of PD-L1 status.
The indication also specifies that nivolumab is to be used together with chemotherapy (with a fluoropyrimidine- and platinum-containing regimen) or in combination with ipilimumab (Yervoy), an immunotherapy with a different mechanism of action.
“Today’s approvals bring two first-line immunotherapy-based treatment options at once ... to newly diagnosed patients with unresectable advanced or metastatic ESCC,” commented Adam Lenkowsky, a senior vice president at Bristol-Myers Squibb, which makes both nivolumab and ipilimumab.
The approval of the new indication by the Food and Drug Administration was based on improved survival shown in the phase 3 CheckMate-648 trial, which involved nearly 1,000 patients. The trial had three arms and compared nivolumab plus chemotherapy (n = 321) and nivolumab plus ipilimumab (n = 324) with chemotherapy alone (n = 324).
The results showed improved survival with both nivolumab combinations compared with chemotherapy (fluorouracil and cisplatin) alone. Overall survival was improved both in all randomized patients (a secondary endpoint) and in patients whose tumors expressed PD-L1 (≥ 1%), the primary endpoint.
For the combination of nivolumab plus chemotherapy, median overall survival was 13.2 versus 10.7 months, compared with chemotherapy alone in all randomized patients, and 15.4 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
For the combination of nivolumab plus ipilimumab, median overall survival was 12.8 versus 10.7 months with chemotherapy alone in all randomized patients and 13.7 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
However, progression-free survival did not reach statistical significance in any group.
“Unresectable advanced or metastatic ESCC is a challenging disease, and there’s a need for additional treatment options that may extend survival in the first-line setting,” commented Jaffer A. Ajani, MD, professor of gastrointestinal medical oncology at the University of Texas MD Anderson Cancer Center, Houston. He was also the lead U.S. investigator for CheckMate-648 and, in a company press release, said the “two nivolumab-based combinations showed a survival benefit compared to chemotherapy alone, offering new treatment options regardless of PD-L1 status.”
Results from the trial were presented at the 2021 annual meeting of the American Society of Clinical Oncology. At that time, trial investigator Ian Chau, MD, a consultant medical oncologist at the Royal Marsden Hospital in Sutton, England, told attendees that “nivolumab plus chemotherapy and nivolumab plus ipilimumab each represent a new potential first-line standard of care for patients with advanced ESCC.”
Commenting on that presentation, Samuel J. Klempner, MD, a gastrointestinal medical oncologist at the Massachusetts General Hospital Cancer Center, Boston, noted that the “prospect of a chemo-free regimen for advanced ESCC with the well-studied combination of ipilimumab and nivolumab would represent a welcome addition to our treatment armamentarium.”
No new safety signals
Dr. Chau noted there were no new safety signals with either of the immunotherapies.
Nivolumab and/or chemotherapy were discontinued in 39% of patients and delayed in 71% of patients for an adverse reaction.
Nivolumab and/or ipilimumab were discontinued in 23% of patients and delayed in 46% of patients for an adverse reaction.
The manufacturer cautioned that immunotherapy with nivolumab with or without ipilimumab has been associated with severe and fatal immune-mediated adverse reactions including pneumonitis, colitis, hepatitis and hepatotoxicity, endocrinopathies, nephritis and renal dysfunction, dermatologic adverse reactions, and infusion-related reactions.
A version of this article first appeared on Medscape.com.
The new approval for the drug, a programmed cell death–ligand-1 inhibitor, is for use in this patient population regardless of PD-L1 status.
The indication also specifies that nivolumab is to be used together with chemotherapy (with a fluoropyrimidine- and platinum-containing regimen) or in combination with ipilimumab (Yervoy), an immunotherapy with a different mechanism of action.
“Today’s approvals bring two first-line immunotherapy-based treatment options at once ... to newly diagnosed patients with unresectable advanced or metastatic ESCC,” commented Adam Lenkowsky, a senior vice president at Bristol-Myers Squibb, which makes both nivolumab and ipilimumab.
The approval of the new indication by the Food and Drug Administration was based on improved survival shown in the phase 3 CheckMate-648 trial, which involved nearly 1,000 patients. The trial had three arms and compared nivolumab plus chemotherapy (n = 321) and nivolumab plus ipilimumab (n = 324) with chemotherapy alone (n = 324).
The results showed improved survival with both nivolumab combinations compared with chemotherapy (fluorouracil and cisplatin) alone. Overall survival was improved both in all randomized patients (a secondary endpoint) and in patients whose tumors expressed PD-L1 (≥ 1%), the primary endpoint.
For the combination of nivolumab plus chemotherapy, median overall survival was 13.2 versus 10.7 months, compared with chemotherapy alone in all randomized patients, and 15.4 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
For the combination of nivolumab plus ipilimumab, median overall survival was 12.8 versus 10.7 months with chemotherapy alone in all randomized patients and 13.7 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
However, progression-free survival did not reach statistical significance in any group.
“Unresectable advanced or metastatic ESCC is a challenging disease, and there’s a need for additional treatment options that may extend survival in the first-line setting,” commented Jaffer A. Ajani, MD, professor of gastrointestinal medical oncology at the University of Texas MD Anderson Cancer Center, Houston. He was also the lead U.S. investigator for CheckMate-648 and, in a company press release, said the “two nivolumab-based combinations showed a survival benefit compared to chemotherapy alone, offering new treatment options regardless of PD-L1 status.”
Results from the trial were presented at the 2021 annual meeting of the American Society of Clinical Oncology. At that time, trial investigator Ian Chau, MD, a consultant medical oncologist at the Royal Marsden Hospital in Sutton, England, told attendees that “nivolumab plus chemotherapy and nivolumab plus ipilimumab each represent a new potential first-line standard of care for patients with advanced ESCC.”
Commenting on that presentation, Samuel J. Klempner, MD, a gastrointestinal medical oncologist at the Massachusetts General Hospital Cancer Center, Boston, noted that the “prospect of a chemo-free regimen for advanced ESCC with the well-studied combination of ipilimumab and nivolumab would represent a welcome addition to our treatment armamentarium.”
No new safety signals
Dr. Chau noted there were no new safety signals with either of the immunotherapies.
Nivolumab and/or chemotherapy were discontinued in 39% of patients and delayed in 71% of patients for an adverse reaction.
Nivolumab and/or ipilimumab were discontinued in 23% of patients and delayed in 46% of patients for an adverse reaction.
The manufacturer cautioned that immunotherapy with nivolumab with or without ipilimumab has been associated with severe and fatal immune-mediated adverse reactions including pneumonitis, colitis, hepatitis and hepatotoxicity, endocrinopathies, nephritis and renal dysfunction, dermatologic adverse reactions, and infusion-related reactions.
A version of this article first appeared on Medscape.com.
The new approval for the drug, a programmed cell death–ligand-1 inhibitor, is for use in this patient population regardless of PD-L1 status.
The indication also specifies that nivolumab is to be used together with chemotherapy (with a fluoropyrimidine- and platinum-containing regimen) or in combination with ipilimumab (Yervoy), an immunotherapy with a different mechanism of action.
“Today’s approvals bring two first-line immunotherapy-based treatment options at once ... to newly diagnosed patients with unresectable advanced or metastatic ESCC,” commented Adam Lenkowsky, a senior vice president at Bristol-Myers Squibb, which makes both nivolumab and ipilimumab.
The approval of the new indication by the Food and Drug Administration was based on improved survival shown in the phase 3 CheckMate-648 trial, which involved nearly 1,000 patients. The trial had three arms and compared nivolumab plus chemotherapy (n = 321) and nivolumab plus ipilimumab (n = 324) with chemotherapy alone (n = 324).
The results showed improved survival with both nivolumab combinations compared with chemotherapy (fluorouracil and cisplatin) alone. Overall survival was improved both in all randomized patients (a secondary endpoint) and in patients whose tumors expressed PD-L1 (≥ 1%), the primary endpoint.
For the combination of nivolumab plus chemotherapy, median overall survival was 13.2 versus 10.7 months, compared with chemotherapy alone in all randomized patients, and 15.4 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
For the combination of nivolumab plus ipilimumab, median overall survival was 12.8 versus 10.7 months with chemotherapy alone in all randomized patients and 13.7 versus 9.1 months in patients whose tumors express PD-L1 (≥ 1%).
However, progression-free survival did not reach statistical significance in any group.
“Unresectable advanced or metastatic ESCC is a challenging disease, and there’s a need for additional treatment options that may extend survival in the first-line setting,” commented Jaffer A. Ajani, MD, professor of gastrointestinal medical oncology at the University of Texas MD Anderson Cancer Center, Houston. He was also the lead U.S. investigator for CheckMate-648 and, in a company press release, said the “two nivolumab-based combinations showed a survival benefit compared to chemotherapy alone, offering new treatment options regardless of PD-L1 status.”
Results from the trial were presented at the 2021 annual meeting of the American Society of Clinical Oncology. At that time, trial investigator Ian Chau, MD, a consultant medical oncologist at the Royal Marsden Hospital in Sutton, England, told attendees that “nivolumab plus chemotherapy and nivolumab plus ipilimumab each represent a new potential first-line standard of care for patients with advanced ESCC.”
Commenting on that presentation, Samuel J. Klempner, MD, a gastrointestinal medical oncologist at the Massachusetts General Hospital Cancer Center, Boston, noted that the “prospect of a chemo-free regimen for advanced ESCC with the well-studied combination of ipilimumab and nivolumab would represent a welcome addition to our treatment armamentarium.”
No new safety signals
Dr. Chau noted there were no new safety signals with either of the immunotherapies.
Nivolumab and/or chemotherapy were discontinued in 39% of patients and delayed in 71% of patients for an adverse reaction.
Nivolumab and/or ipilimumab were discontinued in 23% of patients and delayed in 46% of patients for an adverse reaction.
The manufacturer cautioned that immunotherapy with nivolumab with or without ipilimumab has been associated with severe and fatal immune-mediated adverse reactions including pneumonitis, colitis, hepatitis and hepatotoxicity, endocrinopathies, nephritis and renal dysfunction, dermatologic adverse reactions, and infusion-related reactions.
A version of this article first appeared on Medscape.com.
FDA approves first drug for eosinophilic esophagitis
The U.S. Food and Drug Administration has approved dupilumab (Dupixent, Regeneron) to treat eosinophilic esophagitis (EoE) in adults and children aged 12 years and older weighing at least 40 kg.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway. It’s the first drug to be approved by the FDA for EoE.
In a phase 3 trial, dupilumab 300 mg weekly significantly improved signs and symptoms of eosinophilic esophagitis, compared with placebo, underscoring the role of type 2 inflammation in this disease, Regeneron says in a news release.
According to the company, there are roughly 160,000 patients in the United States living with EoE who are currently using treatments not specifically approved for the disease. Of those patients, about 48,000 continue to experience symptoms despite multiple treatments.
“As researchers and clinicians have gained knowledge about eosinophilic esophagitis in recent years, more cases of the disorder have been recognized and diagnosed in the U.S.,” Jessica Lee, MD, director of the Division of Gastroenterology in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
The approval of dupilumab will “fulfill an important unmet need for the increasing number of patients with eosinophilic esophagitis,” Dr. Lee said.
The efficacy and safety of dupilumab in EoE was demonstrated in a randomized, double-blind, parallel-group, multicenter, placebo-controlled trial that included two 24-week treatment periods (parts A and B) that were conducted independently in separate groups of patients.
In both part A and B, patients received dupilumab 300 mg or placebo every week.
In part A of the trial, 60% of the 42 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 5% of the 39 patients who received placebo, the FDA said.
Patients who received dupilumab also experienced an average improvement of 22 points in the Dysphagia Symptom Questionnaire (DSQ) score, compared with 10 points for patients who received placebo.
In part B, 59% of the 80 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 6% of the 79 patients who received placebo.
Patients who received dupilumab also experienced an average improvement of 24 points in their DSQ score, compared with 14 points for patients who received placebo.
“Assessments incorporating the perspectives from patients with EoE supported that the DSQ score improvement in patients who received Dupixent in the clinical trial was representative of clinically meaningful improvement in dysphagia,” the FDA noted.
“Treatment for patients with eosinophilic esophagitis can be challenging, particularly with no previously approved medications,” Evan Dellon, MD, principal investigator for the phase 3 trial, said in the company news release.
“Now, patients and their doctors have a treatment option available as part of their management plan that has the potential to control symptoms, improve inflammation, and heal the changes in the esophagus caused by this progressive and burdensome disease,” added Dr. Dellon, who is professor of medicine in the division of gastroenterology and hepatology at the University of North Carolina at Chapel Hill.
The FDA granted dupilumab priority review and breakthrough therapy designations for EoE.
Dupilumab is already approved in the United States for treatment of moderate to severe atopic dermatitis in adults and children aged 6 years and older whose disease is not adequately controlled by topical prescription therapies or for whom those therapies are not advisable.
The drug is also approved as an add-on maintenance treatment for adults and children aged 6 years and older with certain types of moderate to severe asthma and as an add-on maintenance treatment for adults with inadequately controlled chronic rhinosinusitis with nasal polyposis.
A version of this article first appeared on Medscape.com .
Eosinophilic esophagitis (EoE) is a chronic disease requiring long-term treatment for both induction and maintenance of response. For decades, however, Food and Drug Administration–approved therapies for EoE have not been available. Dupilumab is the first drug to receive FDA approval to treat EoE. This human monoclonal antibody directed against the interleukin (IL)4 receptor–alpha component of the type 2 receptor inhibits signaling of IL4 and IL13. Dupilumab has shown efficacy in similar diseases, such as atopic dermatitis and eosinophilic asthma. In 2017 dupilumab was granted Orphan Drug designation for the potential treatment of EoE and in 2020 the FDA granted Breakthrough Therapy designation for EoE. Recent data from the phase 3 trial of dupilumab 300 mg weekly enrolling patients aged 12 years and older demonstrated a significantly greater reduction in disease symptoms, normalization of esophageal eosinophilia, and reduction in endoscopic findings by week 24 compared with placebo.
The highly anticipated approval of dupilumab marks a paradigm shift toward biologic medications for treatment of EoE when historical treatments have relied on proton pump–inhibitor therapy or topical swallowed steroids. As we await updates about availability and access of dupilumab for our patients, we can rest assured that a highly efficacious treatment is now approved and will fill an important treatment gap in EoE, particularly for patients not deriving adequate response with traditionally used strategies. With multiple clinical trials underway, this milestone likely represents the beginning of additional effective therapies (nonbiologic and biologic) that will be available for EoE.
Rena Yadlapati, MD, MSHS, FACG, is associate professor of clinical medicine in the division of gastroenterology at the University of California, San Diego, medical director of the UCSD Center for Esophageal Diseases, and director of the GI Motility Lab. She has no relevant conflicts of interest.
Eosinophilic esophagitis (EoE) is a chronic disease requiring long-term treatment for both induction and maintenance of response. For decades, however, Food and Drug Administration–approved therapies for EoE have not been available. Dupilumab is the first drug to receive FDA approval to treat EoE. This human monoclonal antibody directed against the interleukin (IL)4 receptor–alpha component of the type 2 receptor inhibits signaling of IL4 and IL13. Dupilumab has shown efficacy in similar diseases, such as atopic dermatitis and eosinophilic asthma. In 2017 dupilumab was granted Orphan Drug designation for the potential treatment of EoE and in 2020 the FDA granted Breakthrough Therapy designation for EoE. Recent data from the phase 3 trial of dupilumab 300 mg weekly enrolling patients aged 12 years and older demonstrated a significantly greater reduction in disease symptoms, normalization of esophageal eosinophilia, and reduction in endoscopic findings by week 24 compared with placebo.
The highly anticipated approval of dupilumab marks a paradigm shift toward biologic medications for treatment of EoE when historical treatments have relied on proton pump–inhibitor therapy or topical swallowed steroids. As we await updates about availability and access of dupilumab for our patients, we can rest assured that a highly efficacious treatment is now approved and will fill an important treatment gap in EoE, particularly for patients not deriving adequate response with traditionally used strategies. With multiple clinical trials underway, this milestone likely represents the beginning of additional effective therapies (nonbiologic and biologic) that will be available for EoE.
Rena Yadlapati, MD, MSHS, FACG, is associate professor of clinical medicine in the division of gastroenterology at the University of California, San Diego, medical director of the UCSD Center for Esophageal Diseases, and director of the GI Motility Lab. She has no relevant conflicts of interest.
Eosinophilic esophagitis (EoE) is a chronic disease requiring long-term treatment for both induction and maintenance of response. For decades, however, Food and Drug Administration–approved therapies for EoE have not been available. Dupilumab is the first drug to receive FDA approval to treat EoE. This human monoclonal antibody directed against the interleukin (IL)4 receptor–alpha component of the type 2 receptor inhibits signaling of IL4 and IL13. Dupilumab has shown efficacy in similar diseases, such as atopic dermatitis and eosinophilic asthma. In 2017 dupilumab was granted Orphan Drug designation for the potential treatment of EoE and in 2020 the FDA granted Breakthrough Therapy designation for EoE. Recent data from the phase 3 trial of dupilumab 300 mg weekly enrolling patients aged 12 years and older demonstrated a significantly greater reduction in disease symptoms, normalization of esophageal eosinophilia, and reduction in endoscopic findings by week 24 compared with placebo.
The highly anticipated approval of dupilumab marks a paradigm shift toward biologic medications for treatment of EoE when historical treatments have relied on proton pump–inhibitor therapy or topical swallowed steroids. As we await updates about availability and access of dupilumab for our patients, we can rest assured that a highly efficacious treatment is now approved and will fill an important treatment gap in EoE, particularly for patients not deriving adequate response with traditionally used strategies. With multiple clinical trials underway, this milestone likely represents the beginning of additional effective therapies (nonbiologic and biologic) that will be available for EoE.
Rena Yadlapati, MD, MSHS, FACG, is associate professor of clinical medicine in the division of gastroenterology at the University of California, San Diego, medical director of the UCSD Center for Esophageal Diseases, and director of the GI Motility Lab. She has no relevant conflicts of interest.
The U.S. Food and Drug Administration has approved dupilumab (Dupixent, Regeneron) to treat eosinophilic esophagitis (EoE) in adults and children aged 12 years and older weighing at least 40 kg.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway. It’s the first drug to be approved by the FDA for EoE.
In a phase 3 trial, dupilumab 300 mg weekly significantly improved signs and symptoms of eosinophilic esophagitis, compared with placebo, underscoring the role of type 2 inflammation in this disease, Regeneron says in a news release.
According to the company, there are roughly 160,000 patients in the United States living with EoE who are currently using treatments not specifically approved for the disease. Of those patients, about 48,000 continue to experience symptoms despite multiple treatments.
“As researchers and clinicians have gained knowledge about eosinophilic esophagitis in recent years, more cases of the disorder have been recognized and diagnosed in the U.S.,” Jessica Lee, MD, director of the Division of Gastroenterology in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
The approval of dupilumab will “fulfill an important unmet need for the increasing number of patients with eosinophilic esophagitis,” Dr. Lee said.
The efficacy and safety of dupilumab in EoE was demonstrated in a randomized, double-blind, parallel-group, multicenter, placebo-controlled trial that included two 24-week treatment periods (parts A and B) that were conducted independently in separate groups of patients.
In both part A and B, patients received dupilumab 300 mg or placebo every week.
In part A of the trial, 60% of the 42 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 5% of the 39 patients who received placebo, the FDA said.
Patients who received dupilumab also experienced an average improvement of 22 points in the Dysphagia Symptom Questionnaire (DSQ) score, compared with 10 points for patients who received placebo.
In part B, 59% of the 80 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 6% of the 79 patients who received placebo.
Patients who received dupilumab also experienced an average improvement of 24 points in their DSQ score, compared with 14 points for patients who received placebo.
“Assessments incorporating the perspectives from patients with EoE supported that the DSQ score improvement in patients who received Dupixent in the clinical trial was representative of clinically meaningful improvement in dysphagia,” the FDA noted.
“Treatment for patients with eosinophilic esophagitis can be challenging, particularly with no previously approved medications,” Evan Dellon, MD, principal investigator for the phase 3 trial, said in the company news release.
“Now, patients and their doctors have a treatment option available as part of their management plan that has the potential to control symptoms, improve inflammation, and heal the changes in the esophagus caused by this progressive and burdensome disease,” added Dr. Dellon, who is professor of medicine in the division of gastroenterology and hepatology at the University of North Carolina at Chapel Hill.
The FDA granted dupilumab priority review and breakthrough therapy designations for EoE.
Dupilumab is already approved in the United States for treatment of moderate to severe atopic dermatitis in adults and children aged 6 years and older whose disease is not adequately controlled by topical prescription therapies or for whom those therapies are not advisable.
The drug is also approved as an add-on maintenance treatment for adults and children aged 6 years and older with certain types of moderate to severe asthma and as an add-on maintenance treatment for adults with inadequately controlled chronic rhinosinusitis with nasal polyposis.
A version of this article first appeared on Medscape.com .
The U.S. Food and Drug Administration has approved dupilumab (Dupixent, Regeneron) to treat eosinophilic esophagitis (EoE) in adults and children aged 12 years and older weighing at least 40 kg.
EoE is a chronic inflammatory disorder driven by type 2 inflammation that damages the esophagus and causes difficulty swallowing and eating.
Dupilumab is a monoclonal antibody that acts to inhibit part of the inflammatory pathway. It’s the first drug to be approved by the FDA for EoE.
In a phase 3 trial, dupilumab 300 mg weekly significantly improved signs and symptoms of eosinophilic esophagitis, compared with placebo, underscoring the role of type 2 inflammation in this disease, Regeneron says in a news release.
According to the company, there are roughly 160,000 patients in the United States living with EoE who are currently using treatments not specifically approved for the disease. Of those patients, about 48,000 continue to experience symptoms despite multiple treatments.
“As researchers and clinicians have gained knowledge about eosinophilic esophagitis in recent years, more cases of the disorder have been recognized and diagnosed in the U.S.,” Jessica Lee, MD, director of the Division of Gastroenterology in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
The approval of dupilumab will “fulfill an important unmet need for the increasing number of patients with eosinophilic esophagitis,” Dr. Lee said.
The efficacy and safety of dupilumab in EoE was demonstrated in a randomized, double-blind, parallel-group, multicenter, placebo-controlled trial that included two 24-week treatment periods (parts A and B) that were conducted independently in separate groups of patients.
In both part A and B, patients received dupilumab 300 mg or placebo every week.
In part A of the trial, 60% of the 42 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 5% of the 39 patients who received placebo, the FDA said.
Patients who received dupilumab also experienced an average improvement of 22 points in the Dysphagia Symptom Questionnaire (DSQ) score, compared with 10 points for patients who received placebo.
In part B, 59% of the 80 patients who received dupilumab achieved the predetermined level of reduction of eosinophils in the esophagus, compared with 6% of the 79 patients who received placebo.
Patients who received dupilumab also experienced an average improvement of 24 points in their DSQ score, compared with 14 points for patients who received placebo.
“Assessments incorporating the perspectives from patients with EoE supported that the DSQ score improvement in patients who received Dupixent in the clinical trial was representative of clinically meaningful improvement in dysphagia,” the FDA noted.
“Treatment for patients with eosinophilic esophagitis can be challenging, particularly with no previously approved medications,” Evan Dellon, MD, principal investigator for the phase 3 trial, said in the company news release.
“Now, patients and their doctors have a treatment option available as part of their management plan that has the potential to control symptoms, improve inflammation, and heal the changes in the esophagus caused by this progressive and burdensome disease,” added Dr. Dellon, who is professor of medicine in the division of gastroenterology and hepatology at the University of North Carolina at Chapel Hill.
The FDA granted dupilumab priority review and breakthrough therapy designations for EoE.
Dupilumab is already approved in the United States for treatment of moderate to severe atopic dermatitis in adults and children aged 6 years and older whose disease is not adequately controlled by topical prescription therapies or for whom those therapies are not advisable.
The drug is also approved as an add-on maintenance treatment for adults and children aged 6 years and older with certain types of moderate to severe asthma and as an add-on maintenance treatment for adults with inadequately controlled chronic rhinosinusitis with nasal polyposis.
A version of this article first appeared on Medscape.com .
FDA authorizes Pfizer’s COVID booster for kids ages 5 to 11
emergency use authorization (EUA), allowing the Pfizer-BioNTech COVID-19 booster shot for children ages 5 to 11 who are at least 5 months out from their first vaccine series.
According to the most recent data from the Centers for Disease Control and Prevention, 28.6% of children in this age group have received both initial doses of Pfizer’s COVID-19 vaccine, and 35.3% have received their first dose.
Pfizer’s vaccine trial involving 4,500 children showed few side effects among children younger than 12 who received a booster, or third dose, according to a company statement.
Pfizer asked the FDA for an amended authorization in April, after submitting data showing that a third dose in children between 5 and 11 raised antibodies targeting the Omicron variant by 36 times.
“While it has largely been the case that COVID-19 tends to be less severe in children than adults, the omicron wave has seen more kids getting sick with the disease and being hospitalized, and children may also experience longer-term effects, even following initially mild disease,” FDA Commissioner Robert M. Califf, MD, said in a news release.
A study done by the New York State Department of Health showed the effectiveness of Pfizer’s two-dose vaccine series fell from 68% to 12% 4-5 months after the second dose was given to children 5 to 11 during the Omicron surge. A CDC study published in March also showed that the Pfizer shot reduced the risk of Omicron by 31% in children 5 to 11, a significantly lower rate than for kids 12 to 15, who had a 59% risk reduction after receiving two doses.
To some experts, this data suggest an even greater need for children under 12 to be eligible for a third dose.
“Since authorizing the vaccine for children down to 5 years of age in October 2021, emerging data suggest that vaccine effectiveness against COVID-19 wanes after the second dose of the vaccine in all authorized populations,” says Peter Marks, MD, PhD, the director of the FDA’s Center for Biologics Evaluation and Research.
The CDC still needs to sign off on the shots before they can be allowed. The agency’s Advisory Committee on Immunization Practices is set to meet on May 19 to discuss boosters in this age group.
FDA advisory panels plan to meet next month to discuss allowing Pfizer’s and Moderna’s COVID-19 vaccines for children under 6 years old.
A version of this article first appeared on WebMD.com.
emergency use authorization (EUA), allowing the Pfizer-BioNTech COVID-19 booster shot for children ages 5 to 11 who are at least 5 months out from their first vaccine series.
According to the most recent data from the Centers for Disease Control and Prevention, 28.6% of children in this age group have received both initial doses of Pfizer’s COVID-19 vaccine, and 35.3% have received their first dose.
Pfizer’s vaccine trial involving 4,500 children showed few side effects among children younger than 12 who received a booster, or third dose, according to a company statement.
Pfizer asked the FDA for an amended authorization in April, after submitting data showing that a third dose in children between 5 and 11 raised antibodies targeting the Omicron variant by 36 times.
“While it has largely been the case that COVID-19 tends to be less severe in children than adults, the omicron wave has seen more kids getting sick with the disease and being hospitalized, and children may also experience longer-term effects, even following initially mild disease,” FDA Commissioner Robert M. Califf, MD, said in a news release.
A study done by the New York State Department of Health showed the effectiveness of Pfizer’s two-dose vaccine series fell from 68% to 12% 4-5 months after the second dose was given to children 5 to 11 during the Omicron surge. A CDC study published in March also showed that the Pfizer shot reduced the risk of Omicron by 31% in children 5 to 11, a significantly lower rate than for kids 12 to 15, who had a 59% risk reduction after receiving two doses.
To some experts, this data suggest an even greater need for children under 12 to be eligible for a third dose.
“Since authorizing the vaccine for children down to 5 years of age in October 2021, emerging data suggest that vaccine effectiveness against COVID-19 wanes after the second dose of the vaccine in all authorized populations,” says Peter Marks, MD, PhD, the director of the FDA’s Center for Biologics Evaluation and Research.
The CDC still needs to sign off on the shots before they can be allowed. The agency’s Advisory Committee on Immunization Practices is set to meet on May 19 to discuss boosters in this age group.
FDA advisory panels plan to meet next month to discuss allowing Pfizer’s and Moderna’s COVID-19 vaccines for children under 6 years old.
A version of this article first appeared on WebMD.com.
emergency use authorization (EUA), allowing the Pfizer-BioNTech COVID-19 booster shot for children ages 5 to 11 who are at least 5 months out from their first vaccine series.
According to the most recent data from the Centers for Disease Control and Prevention, 28.6% of children in this age group have received both initial doses of Pfizer’s COVID-19 vaccine, and 35.3% have received their first dose.
Pfizer’s vaccine trial involving 4,500 children showed few side effects among children younger than 12 who received a booster, or third dose, according to a company statement.
Pfizer asked the FDA for an amended authorization in April, after submitting data showing that a third dose in children between 5 and 11 raised antibodies targeting the Omicron variant by 36 times.
“While it has largely been the case that COVID-19 tends to be less severe in children than adults, the omicron wave has seen more kids getting sick with the disease and being hospitalized, and children may also experience longer-term effects, even following initially mild disease,” FDA Commissioner Robert M. Califf, MD, said in a news release.
A study done by the New York State Department of Health showed the effectiveness of Pfizer’s two-dose vaccine series fell from 68% to 12% 4-5 months after the second dose was given to children 5 to 11 during the Omicron surge. A CDC study published in March also showed that the Pfizer shot reduced the risk of Omicron by 31% in children 5 to 11, a significantly lower rate than for kids 12 to 15, who had a 59% risk reduction after receiving two doses.
To some experts, this data suggest an even greater need for children under 12 to be eligible for a third dose.
“Since authorizing the vaccine for children down to 5 years of age in October 2021, emerging data suggest that vaccine effectiveness against COVID-19 wanes after the second dose of the vaccine in all authorized populations,” says Peter Marks, MD, PhD, the director of the FDA’s Center for Biologics Evaluation and Research.
The CDC still needs to sign off on the shots before they can be allowed. The agency’s Advisory Committee on Immunization Practices is set to meet on May 19 to discuss boosters in this age group.
FDA advisory panels plan to meet next month to discuss allowing Pfizer’s and Moderna’s COVID-19 vaccines for children under 6 years old.
A version of this article first appeared on WebMD.com.
CDC updates guidelines for hepatitis outbreak among children
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention updated its recommendations for doctors and public health officials regarding the unusual outbreak of acute hepatitis among children.
As of May 5, the CDC and state health departments are investigating 109 children with hepatitis of unknown origin across 25 states and territories.
More than half have tested positive for adenovirus, the CDC said. More than 90% have been hospitalized, and 14% have had liver transplants. Five deaths are under investigation.
This week’s CDC alert provides updated recommendations for testing, given the potential association between adenovirus infection and pediatric hepatitis, or liver inflammation.
“Clinicians are recommended to consider adenovirus testing for patients with hepatitis of unknown etiology and to report such cases to their state or jurisdictional public health authorities,” the CDC said.
Doctors should also consider collecting a blood sample, respiratory sample, and stool sample. They may also collect liver tissue if a biopsy occurred or an autopsy is available.
In November 2021, clinicians at a large children’s hospital in Alabama notified the CDC about five pediatric patients with significant liver injury, including three with acute liver failure, who also tested positive for adenovirus. All children were previously healthy, and none had COVID-19, according to a CDC alert in April.
Four additional pediatric patients with hepatitis and adenovirus infection were identified. After lab testing found adenovirus infection in all nine patients in the initial cluster, public health officials began investigating a possible association between pediatric hepatitis and adenovirus. Among the five specimens that could be sequenced, they were all adenovirus type 41.
Unexplained hepatitis cases have been reported in children worldwide, reaching 450 cases and 11 deaths, according to the latest update from the European Centre for Disease Prevention and Control.
The cases have been reported in more than two dozen countries around the world, with 14 countries reporting more than five cases. The United Kingdom and the United States have reported the largest case counts so far.
In the United Kingdom, officials have identified 163 cases in children under age 16 years, including 11 that required liver transplants.
In the European Union, 14 countries have reported 106 cases collectively, with Italy reporting 35 cases and Spain reporting 22 cases. Outside of the European Union, Brazil has reported 16, Indonesia has reported 15, and Israel has reported 12.
Among the 11 deaths reported globally, the Uniyed States has reported five, Indonesia has reported five, and Palestine has reported one.
The cause of severe hepatitis remains a mystery, according to Ars Technica. Some cases have been identified retrospectively, dating back to the beginning of October 2021.
About 70% of the cases that have been tested for an adenovirus have tested positive, and subtype testing continues to show adenovirus type 41. The cases don’t appear to be linked to common causes, such as hepatitis viruses A, B, C, D, or E, which can cause liver inflammation and injury.
Adenoviruses aren’t known to cause hepatitis in healthy children, though the viruses have been linked to liver damage in children with compromised immune systems, according to Ars Technica. Adenoviruses typically cause respiratory infections in children, although type 41 tends to cause gastrointestinal illness.
“At present, the leading hypotheses remain those which involve adenovirus,” Philippa Easterbrook, a senior scientist at the WHO, said May 10 during a press briefing.
“I think [there’s] also still an important consideration about the role of COVID as well, either as a co-infection or as a past infection,” she said.
WHO officials expect data within a week from U.K. cases, Ms. Easterbrook said, which may indicate whether the adenovirus is an incidental infection or a more direct cause.
A version of this article first appeared on Medscape.com.
FDA-cleared panties could reduce STI risk during oral sex
.
The underwear, sold as Lorals for Protection, are single-use, vanilla-scented, natural latex panties that cover the genitals and anus and block the transfer of bodily fluids during oral sex, according to the company website. They sell in packages of four for $25.
The FDA didn’t run human clinical trials but granted authorization after the company gave it data about the product, The New York Times reported.
“The FDA’s authorization of this product gives people another option to protect against STIs during oral sex,” said Courtney Lias, PhD, director of the FDA office that led the review of the underwear.
Previously, the FDA authorized oral dams to prevent the spread of STIs during oral sex. Oral dams, sometimes called oral sex condoms, are thin latex barriers that go between one partner’s mouth and the other person’s genitals. The dams haven’t been widely used, partly because a person has to hold the dam in place during sex, unlike the panties.
“They’re extremely unpopular,” Jeanne Marrazzo, MD, director of the division of infectious diseases at the University of Alabama at Birmingham, told the Times. “I mean, honestly, could there be anything less sexy than a dental dam?”Melanie Cristol said she came up with the idea for the panties after discovering on her 2014 honeymoon that she had an infection that could be sexually transmitted.
“I wanted to feel sexy and confident and use something that was made with my body and actual sex in mind,” she told the Times.
The panties are made of material about as thin as a condom and form a seal on the thigh to keep fluids inside, she said.
Dr. Marrazzo said the panties are an advancement because there are few options for safe oral sex. She noted that some teenagers have their first sexual experience with oral sex and that the panties could reduce anxiety for people of all ages.
A version of this article first appeared on WebMD.com.
.
The underwear, sold as Lorals for Protection, are single-use, vanilla-scented, natural latex panties that cover the genitals and anus and block the transfer of bodily fluids during oral sex, according to the company website. They sell in packages of four for $25.
The FDA didn’t run human clinical trials but granted authorization after the company gave it data about the product, The New York Times reported.
“The FDA’s authorization of this product gives people another option to protect against STIs during oral sex,” said Courtney Lias, PhD, director of the FDA office that led the review of the underwear.
Previously, the FDA authorized oral dams to prevent the spread of STIs during oral sex. Oral dams, sometimes called oral sex condoms, are thin latex barriers that go between one partner’s mouth and the other person’s genitals. The dams haven’t been widely used, partly because a person has to hold the dam in place during sex, unlike the panties.
“They’re extremely unpopular,” Jeanne Marrazzo, MD, director of the division of infectious diseases at the University of Alabama at Birmingham, told the Times. “I mean, honestly, could there be anything less sexy than a dental dam?”Melanie Cristol said she came up with the idea for the panties after discovering on her 2014 honeymoon that she had an infection that could be sexually transmitted.
“I wanted to feel sexy and confident and use something that was made with my body and actual sex in mind,” she told the Times.
The panties are made of material about as thin as a condom and form a seal on the thigh to keep fluids inside, she said.
Dr. Marrazzo said the panties are an advancement because there are few options for safe oral sex. She noted that some teenagers have their first sexual experience with oral sex and that the panties could reduce anxiety for people of all ages.
A version of this article first appeared on WebMD.com.
.
The underwear, sold as Lorals for Protection, are single-use, vanilla-scented, natural latex panties that cover the genitals and anus and block the transfer of bodily fluids during oral sex, according to the company website. They sell in packages of four for $25.
The FDA didn’t run human clinical trials but granted authorization after the company gave it data about the product, The New York Times reported.
“The FDA’s authorization of this product gives people another option to protect against STIs during oral sex,” said Courtney Lias, PhD, director of the FDA office that led the review of the underwear.
Previously, the FDA authorized oral dams to prevent the spread of STIs during oral sex. Oral dams, sometimes called oral sex condoms, are thin latex barriers that go between one partner’s mouth and the other person’s genitals. The dams haven’t been widely used, partly because a person has to hold the dam in place during sex, unlike the panties.
“They’re extremely unpopular,” Jeanne Marrazzo, MD, director of the division of infectious diseases at the University of Alabama at Birmingham, told the Times. “I mean, honestly, could there be anything less sexy than a dental dam?”Melanie Cristol said she came up with the idea for the panties after discovering on her 2014 honeymoon that she had an infection that could be sexually transmitted.
“I wanted to feel sexy and confident and use something that was made with my body and actual sex in mind,” she told the Times.
The panties are made of material about as thin as a condom and form a seal on the thigh to keep fluids inside, she said.
Dr. Marrazzo said the panties are an advancement because there are few options for safe oral sex. She noted that some teenagers have their first sexual experience with oral sex and that the panties could reduce anxiety for people of all ages.
A version of this article first appeared on WebMD.com.
Children and COVID: New cases climb slowly but steadily
The current sustained increase in COVID-19 has brought the total number of cases in children to over 13 million since the start of the pandemic, according to the American Academy of Pediatrics and the Children’s Hospital Association.
, when cases dropped to their lowest point since last summer. The cumulative number of cases in children is 13,052,988, which accounts for 19.0% of all cases reported in the United States, the AAP and CHA said in their weekly COVID-19 report.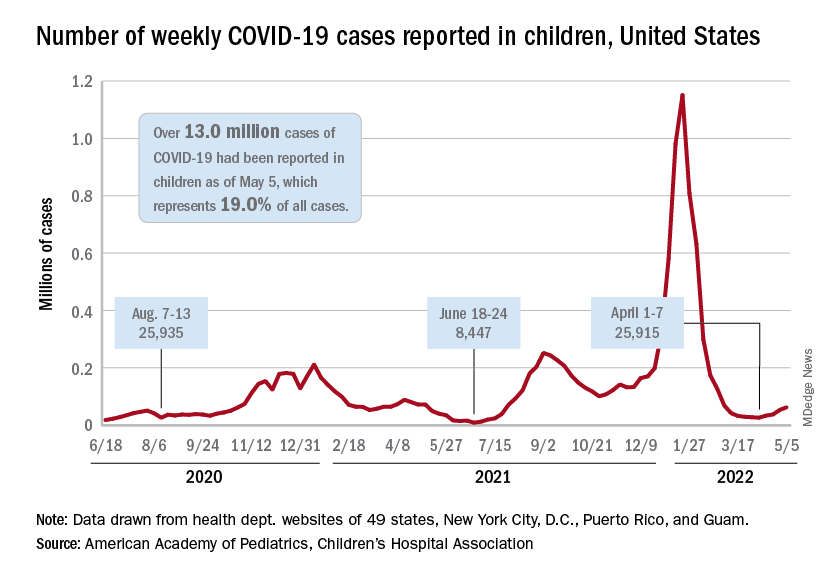
Other measures of incidence show the same steady rise. The rate of new admissions of children aged 0-17 with confirmed COVID-19, which had dipped as low as 0.13 per 100,000 population on April 11, was up to 0.19 per 100,000 on May 6, and the 7-day average for total admissions was 136 per day for May 1-7, compared with 118 for the last week of April, according to the Centers for Disease Control and Prevention.
At the state level, new admission rates for May 6 show wide variation, even regionally. Rhode Island came in with a 0.00 per 100,000 on that day, while Vermont recorded 0.88 admissions per 100,000, the highest of any state and lower only than the District of Columbia’s 1.23 per 100,000. Connecticut (0.45) and Massachusetts (0.33) also were in the highest group (see map), while Maine was in the lowest, CDC data show.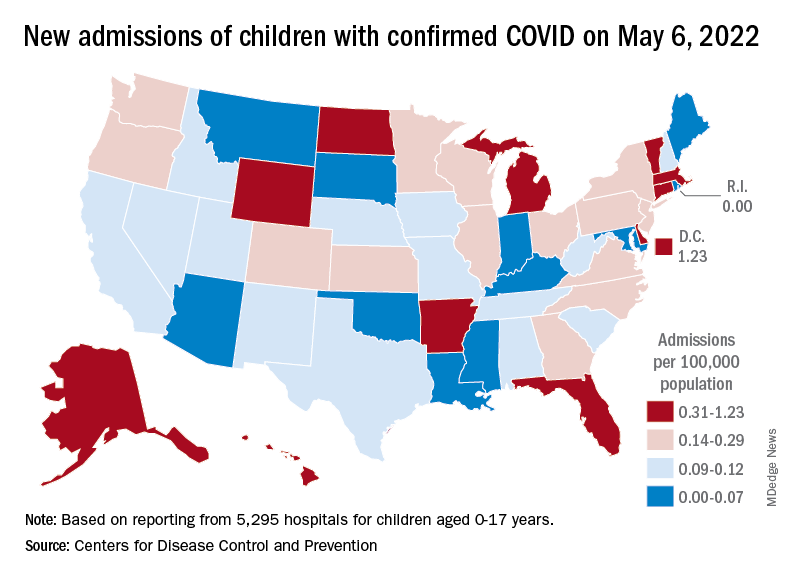
Nationally, emergency department visits also have been rising over the last month or so. Children aged 0-11 years, who were down to a 7-day average of 0.5% of ED visits with diagnosed COVID-19 in early April, saw that number rise to 1.4% on May 5. Children aged 12-15 years went from a rate of 0.3% in late March to the current 1.2%, as did 16- to 17-year-olds, the CDC said on its COVID Data Tracker.
The vaccination effort, meanwhile, continues to lose steam, at least among children who are currently eligible. Initial vaccinations in those aged 5-11 slipped to their lowest-ever 1-week total, 47,000 for April 28 to May 4, while children aged 16-17 continued a long-term slide that has the weekly count down to just 29,000, the AAP said in its weekly vaccination report.
Here’s how those latest recipients changed the populations of vaccinated children in the last week: 35.4% of all 5- to 11-year-olds had received at least one dose as of May 4, compared with 35.3% on April 27, with increases from 67.4% to 67.5% for 12- to 15-year-olds and 72.7% to 72.8% among those aged 16-17, the CDC reported.
The current sustained increase in COVID-19 has brought the total number of cases in children to over 13 million since the start of the pandemic, according to the American Academy of Pediatrics and the Children’s Hospital Association.
, when cases dropped to their lowest point since last summer. The cumulative number of cases in children is 13,052,988, which accounts for 19.0% of all cases reported in the United States, the AAP and CHA said in their weekly COVID-19 report.
Other measures of incidence show the same steady rise. The rate of new admissions of children aged 0-17 with confirmed COVID-19, which had dipped as low as 0.13 per 100,000 population on April 11, was up to 0.19 per 100,000 on May 6, and the 7-day average for total admissions was 136 per day for May 1-7, compared with 118 for the last week of April, according to the Centers for Disease Control and Prevention.
At the state level, new admission rates for May 6 show wide variation, even regionally. Rhode Island came in with a 0.00 per 100,000 on that day, while Vermont recorded 0.88 admissions per 100,000, the highest of any state and lower only than the District of Columbia’s 1.23 per 100,000. Connecticut (0.45) and Massachusetts (0.33) also were in the highest group (see map), while Maine was in the lowest, CDC data show.
Nationally, emergency department visits also have been rising over the last month or so. Children aged 0-11 years, who were down to a 7-day average of 0.5% of ED visits with diagnosed COVID-19 in early April, saw that number rise to 1.4% on May 5. Children aged 12-15 years went from a rate of 0.3% in late March to the current 1.2%, as did 16- to 17-year-olds, the CDC said on its COVID Data Tracker.
The vaccination effort, meanwhile, continues to lose steam, at least among children who are currently eligible. Initial vaccinations in those aged 5-11 slipped to their lowest-ever 1-week total, 47,000 for April 28 to May 4, while children aged 16-17 continued a long-term slide that has the weekly count down to just 29,000, the AAP said in its weekly vaccination report.
Here’s how those latest recipients changed the populations of vaccinated children in the last week: 35.4% of all 5- to 11-year-olds had received at least one dose as of May 4, compared with 35.3% on April 27, with increases from 67.4% to 67.5% for 12- to 15-year-olds and 72.7% to 72.8% among those aged 16-17, the CDC reported.
The current sustained increase in COVID-19 has brought the total number of cases in children to over 13 million since the start of the pandemic, according to the American Academy of Pediatrics and the Children’s Hospital Association.
, when cases dropped to their lowest point since last summer. The cumulative number of cases in children is 13,052,988, which accounts for 19.0% of all cases reported in the United States, the AAP and CHA said in their weekly COVID-19 report.
Other measures of incidence show the same steady rise. The rate of new admissions of children aged 0-17 with confirmed COVID-19, which had dipped as low as 0.13 per 100,000 population on April 11, was up to 0.19 per 100,000 on May 6, and the 7-day average for total admissions was 136 per day for May 1-7, compared with 118 for the last week of April, according to the Centers for Disease Control and Prevention.
At the state level, new admission rates for May 6 show wide variation, even regionally. Rhode Island came in with a 0.00 per 100,000 on that day, while Vermont recorded 0.88 admissions per 100,000, the highest of any state and lower only than the District of Columbia’s 1.23 per 100,000. Connecticut (0.45) and Massachusetts (0.33) also were in the highest group (see map), while Maine was in the lowest, CDC data show.
Nationally, emergency department visits also have been rising over the last month or so. Children aged 0-11 years, who were down to a 7-day average of 0.5% of ED visits with diagnosed COVID-19 in early April, saw that number rise to 1.4% on May 5. Children aged 12-15 years went from a rate of 0.3% in late March to the current 1.2%, as did 16- to 17-year-olds, the CDC said on its COVID Data Tracker.
The vaccination effort, meanwhile, continues to lose steam, at least among children who are currently eligible. Initial vaccinations in those aged 5-11 slipped to their lowest-ever 1-week total, 47,000 for April 28 to May 4, while children aged 16-17 continued a long-term slide that has the weekly count down to just 29,000, the AAP said in its weekly vaccination report.
Here’s how those latest recipients changed the populations of vaccinated children in the last week: 35.4% of all 5- to 11-year-olds had received at least one dose as of May 4, compared with 35.3% on April 27, with increases from 67.4% to 67.5% for 12- to 15-year-olds and 72.7% to 72.8% among those aged 16-17, the CDC reported.
FDA approves two vonoprazan therapies for H. pylori eradication
: Voquezna Triple Pak (vonoprazan, amoxicillin, clarithromycin) and Voquezna Dual Pak (vonoprazan, amoxicillin), both from Phathom Pharmaceuticals.
Vonoprazan is an oral potassium-competitive acid blocker and “the first innovative acid suppressant from a new drug class approved in the United States in over 30 years,” the company said in a news release announcing the approval.
“The approval of Voquezna treatment regimens offers physicians and patients two therapeutic options that showed superior eradication rates compared to proton pump inhibitor-based (PPI) lansoprazole triple therapy in the overall patient population in a pivotal trial,” Terrie Curran, president and CEO of Phathom Pharmaceuticals, said in the release.
“H. pylori eradication rates continue to decline in part due to antibiotic resistance, inadequate acid suppression, and complex treatment regimens, resulting in treatment failures and complications for patients,” Ms. Curran noted.
“New therapies that have the potential to address the limitations of current treatments are needed, and we look forward to bringing these innovative vonoprazan-based treatment options to the millions of H pylori sufferers in the United States,” Ms. Curran said.
FDA approval of vonoprazan triple and dual therapy was based on safety and efficacy data from the phase 3 PHALCON-HP trial involving 1,046 patients.
As earlier reported, both treatment regimens were noninferior to PPI-based triple therapy (lansoprazole with amoxicillin and clarithromycin) in patients with H. pylori strains that were not resistant to clarithromycin or amoxicillin at baseline.
In this analysis, the eradication rate was 78.8% with PPI-based triple therapy, compared with 84.7% with vonoprazan triple therapy and 78.5% with vonoprazan dual therapy.
Vonoprazan triple and dual therapy were both superior to PPI-based triple therapy among all patients, including patients with clarithromycin-resistant H. pylori.
Among patients with clarithromycin-resistant H. pylori, 31.9% achieved eradication with PPI triple therapy, compared with 65.8% with vonoprazan triple therapy and 69.6% with vonoprazan dual therapy.
Among all patients, 68.5% achieved eradication with PPI triple therapy, 80.8% with vonoprazan triple therapy and 77.2% with vonoprazan dual therapy.
Adverse event rates for the vonoprazan-based regimens were comparable to lansoprazole triple therapy. Full prescribing information is available online.
“As a practicing physician, I am excited about the potential of two novel, first-line H. pylori treatment options,” William D. Chey, MD, chief of gastroenterology & hepatology at the University of Michigan, Ann Arbor, said in the news release.
“I believe the added flexibility of having two additional effective therapies, including a dual therapy option that does not contain clarithromycin, offers the potential to improve clinical outcomes in patients with H. pylori infection,” Dr. Chey added.
The company expects to launch both products in the third quarter of 2022. Both treatment regimens will be supplied in convenient blister packs to help promote compliance.
A version of this article first appeared on Medscape.com.
: Voquezna Triple Pak (vonoprazan, amoxicillin, clarithromycin) and Voquezna Dual Pak (vonoprazan, amoxicillin), both from Phathom Pharmaceuticals.
Vonoprazan is an oral potassium-competitive acid blocker and “the first innovative acid suppressant from a new drug class approved in the United States in over 30 years,” the company said in a news release announcing the approval.
“The approval of Voquezna treatment regimens offers physicians and patients two therapeutic options that showed superior eradication rates compared to proton pump inhibitor-based (PPI) lansoprazole triple therapy in the overall patient population in a pivotal trial,” Terrie Curran, president and CEO of Phathom Pharmaceuticals, said in the release.
“H. pylori eradication rates continue to decline in part due to antibiotic resistance, inadequate acid suppression, and complex treatment regimens, resulting in treatment failures and complications for patients,” Ms. Curran noted.
“New therapies that have the potential to address the limitations of current treatments are needed, and we look forward to bringing these innovative vonoprazan-based treatment options to the millions of H pylori sufferers in the United States,” Ms. Curran said.
FDA approval of vonoprazan triple and dual therapy was based on safety and efficacy data from the phase 3 PHALCON-HP trial involving 1,046 patients.
As earlier reported, both treatment regimens were noninferior to PPI-based triple therapy (lansoprazole with amoxicillin and clarithromycin) in patients with H. pylori strains that were not resistant to clarithromycin or amoxicillin at baseline.
In this analysis, the eradication rate was 78.8% with PPI-based triple therapy, compared with 84.7% with vonoprazan triple therapy and 78.5% with vonoprazan dual therapy.
Vonoprazan triple and dual therapy were both superior to PPI-based triple therapy among all patients, including patients with clarithromycin-resistant H. pylori.
Among patients with clarithromycin-resistant H. pylori, 31.9% achieved eradication with PPI triple therapy, compared with 65.8% with vonoprazan triple therapy and 69.6% with vonoprazan dual therapy.
Among all patients, 68.5% achieved eradication with PPI triple therapy, 80.8% with vonoprazan triple therapy and 77.2% with vonoprazan dual therapy.
Adverse event rates for the vonoprazan-based regimens were comparable to lansoprazole triple therapy. Full prescribing information is available online.
“As a practicing physician, I am excited about the potential of two novel, first-line H. pylori treatment options,” William D. Chey, MD, chief of gastroenterology & hepatology at the University of Michigan, Ann Arbor, said in the news release.
“I believe the added flexibility of having two additional effective therapies, including a dual therapy option that does not contain clarithromycin, offers the potential to improve clinical outcomes in patients with H. pylori infection,” Dr. Chey added.
The company expects to launch both products in the third quarter of 2022. Both treatment regimens will be supplied in convenient blister packs to help promote compliance.
A version of this article first appeared on Medscape.com.
: Voquezna Triple Pak (vonoprazan, amoxicillin, clarithromycin) and Voquezna Dual Pak (vonoprazan, amoxicillin), both from Phathom Pharmaceuticals.
Vonoprazan is an oral potassium-competitive acid blocker and “the first innovative acid suppressant from a new drug class approved in the United States in over 30 years,” the company said in a news release announcing the approval.
“The approval of Voquezna treatment regimens offers physicians and patients two therapeutic options that showed superior eradication rates compared to proton pump inhibitor-based (PPI) lansoprazole triple therapy in the overall patient population in a pivotal trial,” Terrie Curran, president and CEO of Phathom Pharmaceuticals, said in the release.
“H. pylori eradication rates continue to decline in part due to antibiotic resistance, inadequate acid suppression, and complex treatment regimens, resulting in treatment failures and complications for patients,” Ms. Curran noted.
“New therapies that have the potential to address the limitations of current treatments are needed, and we look forward to bringing these innovative vonoprazan-based treatment options to the millions of H pylori sufferers in the United States,” Ms. Curran said.
FDA approval of vonoprazan triple and dual therapy was based on safety and efficacy data from the phase 3 PHALCON-HP trial involving 1,046 patients.
As earlier reported, both treatment regimens were noninferior to PPI-based triple therapy (lansoprazole with amoxicillin and clarithromycin) in patients with H. pylori strains that were not resistant to clarithromycin or amoxicillin at baseline.
In this analysis, the eradication rate was 78.8% with PPI-based triple therapy, compared with 84.7% with vonoprazan triple therapy and 78.5% with vonoprazan dual therapy.
Vonoprazan triple and dual therapy were both superior to PPI-based triple therapy among all patients, including patients with clarithromycin-resistant H. pylori.
Among patients with clarithromycin-resistant H. pylori, 31.9% achieved eradication with PPI triple therapy, compared with 65.8% with vonoprazan triple therapy and 69.6% with vonoprazan dual therapy.
Among all patients, 68.5% achieved eradication with PPI triple therapy, 80.8% with vonoprazan triple therapy and 77.2% with vonoprazan dual therapy.
Adverse event rates for the vonoprazan-based regimens were comparable to lansoprazole triple therapy. Full prescribing information is available online.
“As a practicing physician, I am excited about the potential of two novel, first-line H. pylori treatment options,” William D. Chey, MD, chief of gastroenterology & hepatology at the University of Michigan, Ann Arbor, said in the news release.
“I believe the added flexibility of having two additional effective therapies, including a dual therapy option that does not contain clarithromycin, offers the potential to improve clinical outcomes in patients with H. pylori infection,” Dr. Chey added.
The company expects to launch both products in the third quarter of 2022. Both treatment regimens will be supplied in convenient blister packs to help promote compliance.
A version of this article first appeared on Medscape.com.