User login
FDA orders Juul to stop selling E-cigarettes
The marketing denial order covers all the company’s products in the United States, which means Juul must stop distributing the products and remove everything on the market. That includes the Juul device and flavor replacement pods in the tobacco and menthol flavors.
“Today’s action is further progress on the FDA’s commitment to ensuring that all e-cigarette and electronic nicotine delivery system products currently being marketed to consumers meet our public health standards,” Robert Califf, MD, the FDA commissioner, said in the announcement.
“The agency has dedicated significant resources to review products from the companies that account for most of the U.S. market,” he said. “We recognize these make up a significant part of the available products and many have played a disproportionate role in the rise in youth vaping.”
The marketing denial order covers only the commercial distribution and retail sale of Juul’s products and doesn’t restrict consumer possession or use. The FDA “cannot and will not” enforce actions against consumers, the agency said.
The order comes after a 2-year review of the company’s application seeking authorization to continue selling non–fruit-flavored products, such as menthol and tobacco. The FDA determined the application “lacked sufficient evidence regarding the toxicological profile of the products to demonstrate that marketing of the products would be appropriate for the protection of the public health.”
Some of Juul’s study findings raised concerns because of “insufficient and conflicting data,” the FDA said, including potentially harmful chemicals leaching from the Juul liquid replacement pods.
“To date, the FDA has not received clinical information to suggest an immediate hazard associated with the use of the JUUL device or JUUL pods,” the agency said. “However, the [orders] issued today reflect FDA’s determination that there is insufficient evidence to assess the potential toxicological risks of using the JUUL products.”
Juul is expected to appeal the FDA’s decision, according to The New York Times.
In recent years, the FDA has reviewed marketing applications from Juul and other e-cigarette companies as anti-tobacco groups have called for new rules to limit products that led to a surge in youth vaping during the past decade. At the same time, advocates of e-cigarettes and nicotine-delivery devices have said the products help adult smokers to quit cigarettes and other tobacco products.
Juul, in particular, has been blamed for fueling the surge in underage vaping due to fruity flavors and hip marketing, according to The Wall Street Journal. The company removed sweet and fruity flavors from shelves in 2019 and has been trying to repair its reputation by limiting its marketing and focusing on adult cigarette smokers.
In 2020, all e-cigarette manufacturers in the United States were required to submit their products for FDA review to stay on the market, the newspaper reported. The agency has been weighing the potential benefits for adult cigarette smokers against the harms for young people.
The FDA banned the sale of fruit- and mint-flavored cartridges and juice pods in 2020, but menthol and tobacco-flavored products were left on the market, according to USA Today. In September 2021, the agency also banned the sale of hundreds of thousands of vaping and e-cigarette products but didn’t rule on Juul.
Meanwhile, the FDA has cleared Reynolds American and NJOY Holdings – two of Juul’s biggest rivals – to keep tobacco-flavored products on the market. Industry experts expected Juul to receive similar clearance, the Journal reported.
Juul, which was at the top of the U.S. e-cigarette market in 2018, has moved to second place behind Reynolds’s Vuse brand, the newspaper reported. The United States represents most of the company’s revenue, though its products are also available in Canada, the United Kingdom, France, Italy, and the Philippines.
Underage vaping has fallen in the United States since federal restrictions raised the legal purchase age for tobacco products to 21 and banned the sale of sweet and fruity cartridges, according to the Journal. Juul’s popularity has also dropped among youth, with other products such as Puff Bar, Vuse, and Smok becoming more popular among e-cigarette users in high school.
In a separate decision announced this week, the FDA is also moving forward with a plan to reduce the amount of nicotine in cigarettes. The decision, which has been years in the making, is aimed at prompting millions of cigarette users to quit smoking or switch to alternatives such as e-cigarettes, as well as limit the number of users who pick up smoking at an early age.
A version of this article first appeared on WebMD.com .
The marketing denial order covers all the company’s products in the United States, which means Juul must stop distributing the products and remove everything on the market. That includes the Juul device and flavor replacement pods in the tobacco and menthol flavors.
“Today’s action is further progress on the FDA’s commitment to ensuring that all e-cigarette and electronic nicotine delivery system products currently being marketed to consumers meet our public health standards,” Robert Califf, MD, the FDA commissioner, said in the announcement.
“The agency has dedicated significant resources to review products from the companies that account for most of the U.S. market,” he said. “We recognize these make up a significant part of the available products and many have played a disproportionate role in the rise in youth vaping.”
The marketing denial order covers only the commercial distribution and retail sale of Juul’s products and doesn’t restrict consumer possession or use. The FDA “cannot and will not” enforce actions against consumers, the agency said.
The order comes after a 2-year review of the company’s application seeking authorization to continue selling non–fruit-flavored products, such as menthol and tobacco. The FDA determined the application “lacked sufficient evidence regarding the toxicological profile of the products to demonstrate that marketing of the products would be appropriate for the protection of the public health.”
Some of Juul’s study findings raised concerns because of “insufficient and conflicting data,” the FDA said, including potentially harmful chemicals leaching from the Juul liquid replacement pods.
“To date, the FDA has not received clinical information to suggest an immediate hazard associated with the use of the JUUL device or JUUL pods,” the agency said. “However, the [orders] issued today reflect FDA’s determination that there is insufficient evidence to assess the potential toxicological risks of using the JUUL products.”
Juul is expected to appeal the FDA’s decision, according to The New York Times.
In recent years, the FDA has reviewed marketing applications from Juul and other e-cigarette companies as anti-tobacco groups have called for new rules to limit products that led to a surge in youth vaping during the past decade. At the same time, advocates of e-cigarettes and nicotine-delivery devices have said the products help adult smokers to quit cigarettes and other tobacco products.
Juul, in particular, has been blamed for fueling the surge in underage vaping due to fruity flavors and hip marketing, according to The Wall Street Journal. The company removed sweet and fruity flavors from shelves in 2019 and has been trying to repair its reputation by limiting its marketing and focusing on adult cigarette smokers.
In 2020, all e-cigarette manufacturers in the United States were required to submit their products for FDA review to stay on the market, the newspaper reported. The agency has been weighing the potential benefits for adult cigarette smokers against the harms for young people.
The FDA banned the sale of fruit- and mint-flavored cartridges and juice pods in 2020, but menthol and tobacco-flavored products were left on the market, according to USA Today. In September 2021, the agency also banned the sale of hundreds of thousands of vaping and e-cigarette products but didn’t rule on Juul.
Meanwhile, the FDA has cleared Reynolds American and NJOY Holdings – two of Juul’s biggest rivals – to keep tobacco-flavored products on the market. Industry experts expected Juul to receive similar clearance, the Journal reported.
Juul, which was at the top of the U.S. e-cigarette market in 2018, has moved to second place behind Reynolds’s Vuse brand, the newspaper reported. The United States represents most of the company’s revenue, though its products are also available in Canada, the United Kingdom, France, Italy, and the Philippines.
Underage vaping has fallen in the United States since federal restrictions raised the legal purchase age for tobacco products to 21 and banned the sale of sweet and fruity cartridges, according to the Journal. Juul’s popularity has also dropped among youth, with other products such as Puff Bar, Vuse, and Smok becoming more popular among e-cigarette users in high school.
In a separate decision announced this week, the FDA is also moving forward with a plan to reduce the amount of nicotine in cigarettes. The decision, which has been years in the making, is aimed at prompting millions of cigarette users to quit smoking or switch to alternatives such as e-cigarettes, as well as limit the number of users who pick up smoking at an early age.
A version of this article first appeared on WebMD.com .
The marketing denial order covers all the company’s products in the United States, which means Juul must stop distributing the products and remove everything on the market. That includes the Juul device and flavor replacement pods in the tobacco and menthol flavors.
“Today’s action is further progress on the FDA’s commitment to ensuring that all e-cigarette and electronic nicotine delivery system products currently being marketed to consumers meet our public health standards,” Robert Califf, MD, the FDA commissioner, said in the announcement.
“The agency has dedicated significant resources to review products from the companies that account for most of the U.S. market,” he said. “We recognize these make up a significant part of the available products and many have played a disproportionate role in the rise in youth vaping.”
The marketing denial order covers only the commercial distribution and retail sale of Juul’s products and doesn’t restrict consumer possession or use. The FDA “cannot and will not” enforce actions against consumers, the agency said.
The order comes after a 2-year review of the company’s application seeking authorization to continue selling non–fruit-flavored products, such as menthol and tobacco. The FDA determined the application “lacked sufficient evidence regarding the toxicological profile of the products to demonstrate that marketing of the products would be appropriate for the protection of the public health.”
Some of Juul’s study findings raised concerns because of “insufficient and conflicting data,” the FDA said, including potentially harmful chemicals leaching from the Juul liquid replacement pods.
“To date, the FDA has not received clinical information to suggest an immediate hazard associated with the use of the JUUL device or JUUL pods,” the agency said. “However, the [orders] issued today reflect FDA’s determination that there is insufficient evidence to assess the potential toxicological risks of using the JUUL products.”
Juul is expected to appeal the FDA’s decision, according to The New York Times.
In recent years, the FDA has reviewed marketing applications from Juul and other e-cigarette companies as anti-tobacco groups have called for new rules to limit products that led to a surge in youth vaping during the past decade. At the same time, advocates of e-cigarettes and nicotine-delivery devices have said the products help adult smokers to quit cigarettes and other tobacco products.
Juul, in particular, has been blamed for fueling the surge in underage vaping due to fruity flavors and hip marketing, according to The Wall Street Journal. The company removed sweet and fruity flavors from shelves in 2019 and has been trying to repair its reputation by limiting its marketing and focusing on adult cigarette smokers.
In 2020, all e-cigarette manufacturers in the United States were required to submit their products for FDA review to stay on the market, the newspaper reported. The agency has been weighing the potential benefits for adult cigarette smokers against the harms for young people.
The FDA banned the sale of fruit- and mint-flavored cartridges and juice pods in 2020, but menthol and tobacco-flavored products were left on the market, according to USA Today. In September 2021, the agency also banned the sale of hundreds of thousands of vaping and e-cigarette products but didn’t rule on Juul.
Meanwhile, the FDA has cleared Reynolds American and NJOY Holdings – two of Juul’s biggest rivals – to keep tobacco-flavored products on the market. Industry experts expected Juul to receive similar clearance, the Journal reported.
Juul, which was at the top of the U.S. e-cigarette market in 2018, has moved to second place behind Reynolds’s Vuse brand, the newspaper reported. The United States represents most of the company’s revenue, though its products are also available in Canada, the United Kingdom, France, Italy, and the Philippines.
Underage vaping has fallen in the United States since federal restrictions raised the legal purchase age for tobacco products to 21 and banned the sale of sweet and fruity cartridges, according to the Journal. Juul’s popularity has also dropped among youth, with other products such as Puff Bar, Vuse, and Smok becoming more popular among e-cigarette users in high school.
In a separate decision announced this week, the FDA is also moving forward with a plan to reduce the amount of nicotine in cigarettes. The decision, which has been years in the making, is aimed at prompting millions of cigarette users to quit smoking or switch to alternatives such as e-cigarettes, as well as limit the number of users who pick up smoking at an early age.
A version of this article first appeared on WebMD.com .
FDA panel rejects pimavanserin for Alzheimer’s psychosis
In a 9-3 vote, the Psychopharmacologic Drugs Advisory Committee (PDAC) found that the drug’s manufacturer failed to offer convincing evidence of its efficacy in patients with ADP.
The June 17 rejection was the second rejection in as many years for a new indication for pimavanserin, which was approved in 2016 for Parkinson’s disease psychosis (PDP).
In April 2021, the FDA denied Acadia’s supplemental new drug application to expand the drug’s indication to include the treatment of all dementia-related psychosis, regardless of the underlying cause of dementia, citing issues with two studies the company presented as evidence of efficacy.

For the current application, Acadia submitted some new analysis of those studies but limited its application to ADP, which affects up to 30% of patients with Alzheimer’s disease (AD) and currently has no approved treatment.
Committee members who opposed the application were moved by testimony from caregivers and clinicians who treat patients with ADP but ultimately decided the evidence offered by Acadia once again failed to meet the threshold needed to demonstrate efficacy for an expanded indication.
“Sometimes I struggle with a decision on an advisory committee, but not today,” Dean Follmann, PhD, assistant director for biostatistics, National Institute of Allergy and Infectious Diseases, Bethesda, Md., said of his “no” vote.
Lack of efficacy
Pimavanserin is a selective serotonin inverse agonist and antagonist preferentially targeting 5-HT2A receptors, which are thought to play an important role in psychosis, schizophrenia, depression, and other neuropsychiatric disorders.
When it rejected Acadia’s original, broader application for pimavanserin for all dementia-related psychosis, the FDA found that the HARMONY phase 3 trial, previously covered by this news organization, was underpowered to assess efficacy in specific dementia patient subgroups and lacked statistical significance of efficacy in patients with AD. In addition, it noted that overall findings appeared to be driven by results in patients with Parkinson’s disease dementia, a condition already covered by the approved indication.
The FDA found that the second study, referred to in the June 17 hearing as Study 019, which was also previously reported by this news organization, was not “an adequate and well-controlled study.”
Specifically, the agency raised concerns about “protocol deviations,” such as the inclusion of patients who lacked clear documentation that psychotic symptoms developed after an AD diagnosis had been established and patients who received exclusionary medications at the time of randomization.
Discussions between Acadia and the FDA continued over the past year, with the company submitting new analyses and responses. An FDA briefing document published in advance of the committee meeting seemed to suggest the agency was satisfied with Acadia’s response.
Lack of diversity
The advisory committee disagreed, pointing to the same concerns raised last year. Members raised concerns about patient diversity in the HARMONY trial, which included an almost entirely White and mostly male study population.
In addition, although the findings at 26 weeks did demonstrate a marked improvement in psychosis symptoms overall, committee members noted that, again, those findings were largely driven by efficacy in patients with Parkinson’s disease dementia, for which the drug is already approved.
When discussing the phase 2 Study 019, the committee noted that while the study met the primary outcome of improvement in psychosis at 6 weeks, those positive responses were not found at any other timepoint in the 12-week study.
“While it might have had a positive numerical effect in the study, the evidence is really not there to support it,” Dr. Follmann said.
Dr. Follmann and other committee members called for additional trials that focus on patients with Alzheimer’s disease, have a longer follow-up, and include more gender and racial diversity in the study population. They also called for more information about any off-label use of pimavanserin for ADP since it was approved for PDP in 2016.
An unmet need
Most individuals who testified during the public comment period pleaded with the committee to vote in favor of the new indication, sharing stories of family members and patients with ADP.
“I have been caring for and studying patients with Alzheimer’s disease and other dementias for more than 30 years, and I can tell you very simply that if left untreated, psychosis has significant and sometimes devastating consequences for our patients,” said Pierre Tariot, MD, director of the Banner Alzheimer’s Institute and a research professor of psychiatry at the University of Arizona College of Medicine, Tucson, and an investigator on the HARMONY trial.
Those on the committee who voted against the application were quick to agree that lack of an approved treatment for ADP presents a hardship.
“I’m a neurologist who has cared for patients for more than 20 years,” said Madhav R. Thambisetty, MD, PhD, senior investigator for the National Institute on Aging and an adjunct professor of neurology at Johns Hopkins University School of Medicine, Baltimore. “I recognize the unmet need in the field, I just think that the unmet need should not be a justification to cut corners.”
The committee did not focus on drug safety or unmet need in its deliberations, although information on both were presented during the meeting.
Commenting on his “no” vote, PDAC member Walter S. Dunn, MD, PhD, assistant clinical professor of psychiatry at the University of California, Los Angeles, and director of Interventional Psychiatry Service at West Los Angeles Veterans Affairs Medical Center, said he hopes that the FDA will consider those issues more broadly as they complete their review.
“The questions before the committee have been narrow and precise, so I trust the agency will take a broader approach in their final decision about approval,” Dr. Dunn said.
Commenting on the decision, Howard Fillit, MD, cofounder and chief science officer, Alzheimer’s Drug Discovery Foundation, called the news disappointing, “but while the unmet need for a treatment for ADP is clear, it is vital that approved treatments meet stringent safety and efficacy criteria so we can offer patients medications with clear benefits.”
The FDA will make its final decision by August 4.
A version of this article first appeared on Medscape.com.
In a 9-3 vote, the Psychopharmacologic Drugs Advisory Committee (PDAC) found that the drug’s manufacturer failed to offer convincing evidence of its efficacy in patients with ADP.
The June 17 rejection was the second rejection in as many years for a new indication for pimavanserin, which was approved in 2016 for Parkinson’s disease psychosis (PDP).
In April 2021, the FDA denied Acadia’s supplemental new drug application to expand the drug’s indication to include the treatment of all dementia-related psychosis, regardless of the underlying cause of dementia, citing issues with two studies the company presented as evidence of efficacy.
For the current application, Acadia submitted some new analysis of those studies but limited its application to ADP, which affects up to 30% of patients with Alzheimer’s disease (AD) and currently has no approved treatment.
Committee members who opposed the application were moved by testimony from caregivers and clinicians who treat patients with ADP but ultimately decided the evidence offered by Acadia once again failed to meet the threshold needed to demonstrate efficacy for an expanded indication.
“Sometimes I struggle with a decision on an advisory committee, but not today,” Dean Follmann, PhD, assistant director for biostatistics, National Institute of Allergy and Infectious Diseases, Bethesda, Md., said of his “no” vote.
Lack of efficacy
Pimavanserin is a selective serotonin inverse agonist and antagonist preferentially targeting 5-HT2A receptors, which are thought to play an important role in psychosis, schizophrenia, depression, and other neuropsychiatric disorders.
When it rejected Acadia’s original, broader application for pimavanserin for all dementia-related psychosis, the FDA found that the HARMONY phase 3 trial, previously covered by this news organization, was underpowered to assess efficacy in specific dementia patient subgroups and lacked statistical significance of efficacy in patients with AD. In addition, it noted that overall findings appeared to be driven by results in patients with Parkinson’s disease dementia, a condition already covered by the approved indication.
The FDA found that the second study, referred to in the June 17 hearing as Study 019, which was also previously reported by this news organization, was not “an adequate and well-controlled study.”
Specifically, the agency raised concerns about “protocol deviations,” such as the inclusion of patients who lacked clear documentation that psychotic symptoms developed after an AD diagnosis had been established and patients who received exclusionary medications at the time of randomization.
Discussions between Acadia and the FDA continued over the past year, with the company submitting new analyses and responses. An FDA briefing document published in advance of the committee meeting seemed to suggest the agency was satisfied with Acadia’s response.
Lack of diversity
The advisory committee disagreed, pointing to the same concerns raised last year. Members raised concerns about patient diversity in the HARMONY trial, which included an almost entirely White and mostly male study population.
In addition, although the findings at 26 weeks did demonstrate a marked improvement in psychosis symptoms overall, committee members noted that, again, those findings were largely driven by efficacy in patients with Parkinson’s disease dementia, for which the drug is already approved.
When discussing the phase 2 Study 019, the committee noted that while the study met the primary outcome of improvement in psychosis at 6 weeks, those positive responses were not found at any other timepoint in the 12-week study.
“While it might have had a positive numerical effect in the study, the evidence is really not there to support it,” Dr. Follmann said.
Dr. Follmann and other committee members called for additional trials that focus on patients with Alzheimer’s disease, have a longer follow-up, and include more gender and racial diversity in the study population. They also called for more information about any off-label use of pimavanserin for ADP since it was approved for PDP in 2016.
An unmet need
Most individuals who testified during the public comment period pleaded with the committee to vote in favor of the new indication, sharing stories of family members and patients with ADP.
“I have been caring for and studying patients with Alzheimer’s disease and other dementias for more than 30 years, and I can tell you very simply that if left untreated, psychosis has significant and sometimes devastating consequences for our patients,” said Pierre Tariot, MD, director of the Banner Alzheimer’s Institute and a research professor of psychiatry at the University of Arizona College of Medicine, Tucson, and an investigator on the HARMONY trial.
Those on the committee who voted against the application were quick to agree that lack of an approved treatment for ADP presents a hardship.
“I’m a neurologist who has cared for patients for more than 20 years,” said Madhav R. Thambisetty, MD, PhD, senior investigator for the National Institute on Aging and an adjunct professor of neurology at Johns Hopkins University School of Medicine, Baltimore. “I recognize the unmet need in the field, I just think that the unmet need should not be a justification to cut corners.”
The committee did not focus on drug safety or unmet need in its deliberations, although information on both were presented during the meeting.
Commenting on his “no” vote, PDAC member Walter S. Dunn, MD, PhD, assistant clinical professor of psychiatry at the University of California, Los Angeles, and director of Interventional Psychiatry Service at West Los Angeles Veterans Affairs Medical Center, said he hopes that the FDA will consider those issues more broadly as they complete their review.
“The questions before the committee have been narrow and precise, so I trust the agency will take a broader approach in their final decision about approval,” Dr. Dunn said.
Commenting on the decision, Howard Fillit, MD, cofounder and chief science officer, Alzheimer’s Drug Discovery Foundation, called the news disappointing, “but while the unmet need for a treatment for ADP is clear, it is vital that approved treatments meet stringent safety and efficacy criteria so we can offer patients medications with clear benefits.”
The FDA will make its final decision by August 4.
A version of this article first appeared on Medscape.com.
In a 9-3 vote, the Psychopharmacologic Drugs Advisory Committee (PDAC) found that the drug’s manufacturer failed to offer convincing evidence of its efficacy in patients with ADP.
The June 17 rejection was the second rejection in as many years for a new indication for pimavanserin, which was approved in 2016 for Parkinson’s disease psychosis (PDP).
In April 2021, the FDA denied Acadia’s supplemental new drug application to expand the drug’s indication to include the treatment of all dementia-related psychosis, regardless of the underlying cause of dementia, citing issues with two studies the company presented as evidence of efficacy.
For the current application, Acadia submitted some new analysis of those studies but limited its application to ADP, which affects up to 30% of patients with Alzheimer’s disease (AD) and currently has no approved treatment.
Committee members who opposed the application were moved by testimony from caregivers and clinicians who treat patients with ADP but ultimately decided the evidence offered by Acadia once again failed to meet the threshold needed to demonstrate efficacy for an expanded indication.
“Sometimes I struggle with a decision on an advisory committee, but not today,” Dean Follmann, PhD, assistant director for biostatistics, National Institute of Allergy and Infectious Diseases, Bethesda, Md., said of his “no” vote.
Lack of efficacy
Pimavanserin is a selective serotonin inverse agonist and antagonist preferentially targeting 5-HT2A receptors, which are thought to play an important role in psychosis, schizophrenia, depression, and other neuropsychiatric disorders.
When it rejected Acadia’s original, broader application for pimavanserin for all dementia-related psychosis, the FDA found that the HARMONY phase 3 trial, previously covered by this news organization, was underpowered to assess efficacy in specific dementia patient subgroups and lacked statistical significance of efficacy in patients with AD. In addition, it noted that overall findings appeared to be driven by results in patients with Parkinson’s disease dementia, a condition already covered by the approved indication.
The FDA found that the second study, referred to in the June 17 hearing as Study 019, which was also previously reported by this news organization, was not “an adequate and well-controlled study.”
Specifically, the agency raised concerns about “protocol deviations,” such as the inclusion of patients who lacked clear documentation that psychotic symptoms developed after an AD diagnosis had been established and patients who received exclusionary medications at the time of randomization.
Discussions between Acadia and the FDA continued over the past year, with the company submitting new analyses and responses. An FDA briefing document published in advance of the committee meeting seemed to suggest the agency was satisfied with Acadia’s response.
Lack of diversity
The advisory committee disagreed, pointing to the same concerns raised last year. Members raised concerns about patient diversity in the HARMONY trial, which included an almost entirely White and mostly male study population.
In addition, although the findings at 26 weeks did demonstrate a marked improvement in psychosis symptoms overall, committee members noted that, again, those findings were largely driven by efficacy in patients with Parkinson’s disease dementia, for which the drug is already approved.
When discussing the phase 2 Study 019, the committee noted that while the study met the primary outcome of improvement in psychosis at 6 weeks, those positive responses were not found at any other timepoint in the 12-week study.
“While it might have had a positive numerical effect in the study, the evidence is really not there to support it,” Dr. Follmann said.
Dr. Follmann and other committee members called for additional trials that focus on patients with Alzheimer’s disease, have a longer follow-up, and include more gender and racial diversity in the study population. They also called for more information about any off-label use of pimavanserin for ADP since it was approved for PDP in 2016.
An unmet need
Most individuals who testified during the public comment period pleaded with the committee to vote in favor of the new indication, sharing stories of family members and patients with ADP.
“I have been caring for and studying patients with Alzheimer’s disease and other dementias for more than 30 years, and I can tell you very simply that if left untreated, psychosis has significant and sometimes devastating consequences for our patients,” said Pierre Tariot, MD, director of the Banner Alzheimer’s Institute and a research professor of psychiatry at the University of Arizona College of Medicine, Tucson, and an investigator on the HARMONY trial.
Those on the committee who voted against the application were quick to agree that lack of an approved treatment for ADP presents a hardship.
“I’m a neurologist who has cared for patients for more than 20 years,” said Madhav R. Thambisetty, MD, PhD, senior investigator for the National Institute on Aging and an adjunct professor of neurology at Johns Hopkins University School of Medicine, Baltimore. “I recognize the unmet need in the field, I just think that the unmet need should not be a justification to cut corners.”
The committee did not focus on drug safety or unmet need in its deliberations, although information on both were presented during the meeting.
Commenting on his “no” vote, PDAC member Walter S. Dunn, MD, PhD, assistant clinical professor of psychiatry at the University of California, Los Angeles, and director of Interventional Psychiatry Service at West Los Angeles Veterans Affairs Medical Center, said he hopes that the FDA will consider those issues more broadly as they complete their review.
“The questions before the committee have been narrow and precise, so I trust the agency will take a broader approach in their final decision about approval,” Dr. Dunn said.
Commenting on the decision, Howard Fillit, MD, cofounder and chief science officer, Alzheimer’s Drug Discovery Foundation, called the news disappointing, “but while the unmet need for a treatment for ADP is clear, it is vital that approved treatments meet stringent safety and efficacy criteria so we can offer patients medications with clear benefits.”
The FDA will make its final decision by August 4.
A version of this article first appeared on Medscape.com.

FDA approves risankizumab (Skyrizi) for Crohn’s disease
The U.S. Food and Drug Administration – making it the first specific anti–interleukin-23 monoclonal antibody indicated for Crohn’s disease.
The safety and efficacy of risankizumab in Crohn’s disease is supported by data from two induction clinical trials (ADVANCE and MOTIVATE) and one maintenance clinical trial (FORTIFY).
Results of the three studies were presented at the annual scientific meeting of the American College of Gastroenterology in 2021.
“In both the induction and maintenance clinical trials, a significantly greater number of adult patients saw few or no symptoms and a meaningful reduction of visible signs of intestinal inflammation, compared to placebo,” Marla Dubinsky, MD, gastroenterologist with the Mount Sinai Health System and codirector of the IBD Center at Mount Sinai, New York, said in a news release from AbbVie.
“This approval provides health care professionals with a greatly needed additional option for treating the disruptive symptoms of Crohn’s disease,” Dr. Dubinsky said.
For the treatment of Crohn’s disease, risankizumab is dosed at 600 mg administered by intravenous infusion over at least 1 hour at week 0, 4, and 8, followed by 360 mg self-administered by subcutaneous injection at week 12, and every 8 weeks thereafter.
Risankizumab is already approved in the United States for the treatment of adults with active psoriatic arthritis and moderate to severe plaque psoriasis.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration – making it the first specific anti–interleukin-23 monoclonal antibody indicated for Crohn’s disease.
The safety and efficacy of risankizumab in Crohn’s disease is supported by data from two induction clinical trials (ADVANCE and MOTIVATE) and one maintenance clinical trial (FORTIFY).
Results of the three studies were presented at the annual scientific meeting of the American College of Gastroenterology in 2021.
“In both the induction and maintenance clinical trials, a significantly greater number of adult patients saw few or no symptoms and a meaningful reduction of visible signs of intestinal inflammation, compared to placebo,” Marla Dubinsky, MD, gastroenterologist with the Mount Sinai Health System and codirector of the IBD Center at Mount Sinai, New York, said in a news release from AbbVie.
“This approval provides health care professionals with a greatly needed additional option for treating the disruptive symptoms of Crohn’s disease,” Dr. Dubinsky said.
For the treatment of Crohn’s disease, risankizumab is dosed at 600 mg administered by intravenous infusion over at least 1 hour at week 0, 4, and 8, followed by 360 mg self-administered by subcutaneous injection at week 12, and every 8 weeks thereafter.
Risankizumab is already approved in the United States for the treatment of adults with active psoriatic arthritis and moderate to severe plaque psoriasis.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration – making it the first specific anti–interleukin-23 monoclonal antibody indicated for Crohn’s disease.
The safety and efficacy of risankizumab in Crohn’s disease is supported by data from two induction clinical trials (ADVANCE and MOTIVATE) and one maintenance clinical trial (FORTIFY).
Results of the three studies were presented at the annual scientific meeting of the American College of Gastroenterology in 2021.
“In both the induction and maintenance clinical trials, a significantly greater number of adult patients saw few or no symptoms and a meaningful reduction of visible signs of intestinal inflammation, compared to placebo,” Marla Dubinsky, MD, gastroenterologist with the Mount Sinai Health System and codirector of the IBD Center at Mount Sinai, New York, said in a news release from AbbVie.
“This approval provides health care professionals with a greatly needed additional option for treating the disruptive symptoms of Crohn’s disease,” Dr. Dubinsky said.
For the treatment of Crohn’s disease, risankizumab is dosed at 600 mg administered by intravenous infusion over at least 1 hour at week 0, 4, and 8, followed by 360 mg self-administered by subcutaneous injection at week 12, and every 8 weeks thereafter.
Risankizumab is already approved in the United States for the treatment of adults with active psoriatic arthritis and moderate to severe plaque psoriasis.
A version of this article first appeared on Medscape.com.
FDA authorizes COVID vaccines in kids as young as 6 months
, one of the final steps in a long-awaited authorization process to extend protection to the youngest of Americans.
The agency’s move comes after a closely watched FDA advisory group vote earlier this week, which resulted in a unanimous vote in favor of the FDA authorizing both vaccines in this age group.
“The FDA’s evaluation and analysis of the safety, effectiveness, and manufacturing data of these vaccines was rigorous and comprehensive, supporting the EUAs,” the agency said in a news release.
The data show that the “known and potential benefits” of the vaccines outweigh any potential risks, the agency said.
The Moderna vaccine is authorized as a two-dose primary series in children 6 months to 17 years of age. The Pfizer vaccine is now authorized as a three-dose primary series in children 6 months up to 4 years of age. Pfizer’s vaccine was already authorized in children 5 years old and older.
Now all eyes are on the Centers for Disease Control and Prevention, which is expected to decide on the final regulatory hurdle at a meeting June 18. The CDC’s Advisory Committee on Immunization Practices has scheduled a vote on whether to give the vaccines the green light.
If ACIP gives the OK, CDC Director Rochelle Walensky, MD, MPH, is expected to issue recommendations for use shortly thereafter.
Following these final regulatory steps, parents could start bringing their children to pediatricians, family doctors, or local pharmacies for vaccination as early as June 20.
A version of this article first appeared on WebMD.com.
, one of the final steps in a long-awaited authorization process to extend protection to the youngest of Americans.
The agency’s move comes after a closely watched FDA advisory group vote earlier this week, which resulted in a unanimous vote in favor of the FDA authorizing both vaccines in this age group.
“The FDA’s evaluation and analysis of the safety, effectiveness, and manufacturing data of these vaccines was rigorous and comprehensive, supporting the EUAs,” the agency said in a news release.
The data show that the “known and potential benefits” of the vaccines outweigh any potential risks, the agency said.
The Moderna vaccine is authorized as a two-dose primary series in children 6 months to 17 years of age. The Pfizer vaccine is now authorized as a three-dose primary series in children 6 months up to 4 years of age. Pfizer’s vaccine was already authorized in children 5 years old and older.
Now all eyes are on the Centers for Disease Control and Prevention, which is expected to decide on the final regulatory hurdle at a meeting June 18. The CDC’s Advisory Committee on Immunization Practices has scheduled a vote on whether to give the vaccines the green light.
If ACIP gives the OK, CDC Director Rochelle Walensky, MD, MPH, is expected to issue recommendations for use shortly thereafter.
Following these final regulatory steps, parents could start bringing their children to pediatricians, family doctors, or local pharmacies for vaccination as early as June 20.
A version of this article first appeared on WebMD.com.
, one of the final steps in a long-awaited authorization process to extend protection to the youngest of Americans.
The agency’s move comes after a closely watched FDA advisory group vote earlier this week, which resulted in a unanimous vote in favor of the FDA authorizing both vaccines in this age group.
“The FDA’s evaluation and analysis of the safety, effectiveness, and manufacturing data of these vaccines was rigorous and comprehensive, supporting the EUAs,” the agency said in a news release.
The data show that the “known and potential benefits” of the vaccines outweigh any potential risks, the agency said.
The Moderna vaccine is authorized as a two-dose primary series in children 6 months to 17 years of age. The Pfizer vaccine is now authorized as a three-dose primary series in children 6 months up to 4 years of age. Pfizer’s vaccine was already authorized in children 5 years old and older.
Now all eyes are on the Centers for Disease Control and Prevention, which is expected to decide on the final regulatory hurdle at a meeting June 18. The CDC’s Advisory Committee on Immunization Practices has scheduled a vote on whether to give the vaccines the green light.
If ACIP gives the OK, CDC Director Rochelle Walensky, MD, MPH, is expected to issue recommendations for use shortly thereafter.
Following these final regulatory steps, parents could start bringing their children to pediatricians, family doctors, or local pharmacies for vaccination as early as June 20.
A version of this article first appeared on WebMD.com.
FDA panel votes unanimously for COVID shots for youngest kids
Federal advisers to the U.S. Food and Drug Administration voted unanimously June 15 to recommend the use of the Moderna and Pfizer-BioNTech COVID-19 vaccines in infants and young children.
The Vaccines and Related Biological Products Advisory Committee (VRBPAC) of the FDA voted 21-0 to say that benefits of a two-dose series of Moderna’s mRNA vaccine outweigh risk for use in infants and children 6 months through 5 years of age.
The panel then voted 21-0 to say that benefits of a three-dose series of the Pfizer-BioNTech mRNA vaccine outweigh risk for use in infants and children 6 months through 4 years of age.
The FDA is not bound to follow the suggestions of its advisory committees, but it often does. Moderna and Pfizer are seeking to expand emergency use authorization (EUA) for their vaccines. EUAs are special clearances used to allow use of products in connection with public health crises such as the pandemic.
The Pfizer vaccine has standard, nonemergency FDA approval for use in people 16 years of age and older. The FDA also has granted EUA clearance for use of the shot in people ages 5 to 15.
The VRBPAC on June 15 recommended granting EUA clearance for Moderna’s COVID-19 vaccine for people ages 6 to 17. The Moderna vaccine already has full approval for use in people 18 years of age and older.
Many parents have been waiting for a clearance of COVID vaccines for their infants and young children, seeking protection for them at a time of continued spread of the virus.
The White House on June 9 outlined plans for making 10 million doses of COVID vaccines available for children under the age of 5 in the coming weeks.
The Centers for Disease Control and Prevention (CDC) has scheduled a June 18 meeting of its Advisory Committee on Immunization Practices, where members of that panel will vote on recommendations about use of the Moderna and Pfizer-BioNTech vaccines in infants and young children. The last step in the approval process to get shots into arms will be endorsement by the CDC director if the committee votes in favor of the vaccines.
For and against
During the public session during the June 15 FDA meeting, speakers offered varied opinions.
Some urged the panel to vote against the EUA expansion, citing concerns about risks of COVID vaccines in general.
But at the close of the meeting, top FDA vaccine official Peter Marks, MD, PhD, urged the public to be cautious about drawing conclusions from reading incident reports of side effects.
He said he has seen a “Twitter storm” during the day about claims of side effects. but stressed that the FDA has reported to the public on the rare side effects linked to the COVID vaccines, such as myocarditis, with advisories based on a review of reports of side effects. But many of these reports, gathered from the Vaccine Adverse Event Reporting System (VAERS) system, will turn out on further inspection not to be related to vaccination.
Many other speakers urged members of the panel to support expanded use of the vaccines for infants and young children. These speakers emphasized how lack of a vaccine to date has isolated young children who remain unprotected, even with about 83% of those age 5 and older in the United States having received at least one COVID shot.
Dr. Marks noted that there have been 442 deaths from COVID among children under 4 years of age during the pandemic, a number that he compared with the 78 deaths reported in the H1N1 flu. He urged the panel “to be careful that we don’t become numb to the number of pediatric deaths because of the overwhelming number of older deaths here.”
Panelist H. Cody Meissner, MD, a pediatric infectious disease specialist from Tufts University, said the vaccine should be made available -- particularly for children considered to be at high risk for complications from COVID --but health officials need to present a clear picture of the relatively low risks to children of harm from the vaccines-- and from COVID.
“That has to be communicated clearly to parents so that they can participate in the decision about vaccinating a child in this age group,” Dr. Meissner said.
The results presented June 15 from studies of the shots in younger children were less impressive than those from the initial COVID vaccine trials done in adults. This was not a surprise to panelists given the rise of the omicron variant and the evolution of the pandemic, but it still led to comments about the need for further continued study of the vaccines in young children even if they are authorized.
Consider that in 2020, Pfizer won the first EUA for a COVID vaccine of any kind with data that pegged the shot’s efficacy rate at 95%. Statisticians estimated a likely possible range, or 95% confidence interval, for the vaccine efficacy rate at 90.3% to 97.6%.
Those estimates were based on finding eight cases of COVID reported among 18,198 study participants who got the Pfizer-BioNTech shot, compared with 162 cases among the 18,325 people in the placebo group, according to the FDA review of Pifzer’s initial application.
Study data
But on June 15, FDA advisers had to consider an EUA application for which the data did not make as strong a case for the vaccine’s benefit among younger patients.
Pfizer presented what the FDA called a “preliminary descriptive analysis” of vaccine efficacy among participants in Study C4591007 who received three study vaccinations, following accrual of 10 total confirmed COVID-19 cases occurring at least 7 days after the third dose.
Looking at results for study participants ages 6 to 23 months of age, there was one case in the group that got the Pfizer-BioNTech shot and two in the placebo group, pegged as a 75.6% vaccine efficacy rate -- but one with caveats to the small numbers of cases. The 95% confidence interval for this vaccine efficacy rate was reported as-369.1% to 99.6% according to the FDA staff review.
For participants 2-4 years of age with and without evidence of prior SARS-CoV-
2 infection, there were two cases in the group that got the shot and five in the placebo group showing a vaccine efficacy rate of 82.4%, with a 95% confidence interval estimated ranging between -7.6% and 98.3%. For the combined analysis of both age groups, the efficacy rate was estimated at 80.4%, with a 95% confidence interval of 14.1% and 96.7%.
Doran Fink, MD, PhD, a top official in the FDA’s vaccines division, noted that the current EUA application for expanded pediatric use involved “some very preliminary” results that involved “a small number of cases and limited follow up time.”
But he stressed that the evidence gathered to date for the Pifzer application for use of its COVID shot in infants and young children met the threshold for conditional clearance during a crisis.
“We do feel very confident that the evidentiary standard for benefit for an EUA has been met here,” but added that more data would be needed to address questions about the efficacy of the vaccine beyond a third dose and whether an additional dose may be needed.
Pfizer also used a comparison known as “immunobridging” in support of the application. This looked at SARS- CoV-2 50% neutralizing antibody titers for the children in the age group covered by the EUA application and compared them to a randomly selected subset of 16-25-year-old participants in another study,
Key data for the pending Moderna EUA for use of its shot in infants and young children came from study P204. In it, Moderna found 51 cases of COVID among 1,511 children ages 6 months to 23 months who got the vaccines, versus 34 cases among 513 children who received a placebo, according to an FDA staff review.
That resulted in a vaccine efficacy rate pegged at 50.6%, with a 95% confidence interval of 21.4% to 68.6%.
Looking at the children ages 2 to 5 years in the P204 study, there were 119 cases out of 2,594 participants who got the shot, versus 61 cases of 858 in the placebo arm, or 7.1%. That translated to a 36.8% vaccine efficacy rate, with a confidence interval 12.5% to 54.0%.
Panelist Jay Portnoy, MD, of Children’s Mercy Hospital in Kansas City said all of the pediatricians he knows are waiting for the FDA to authorize the new uses of these vaccines in infants and young children.
“The death rate from COVID in young children may not be extremely high, but it’s absolutely terrifying to parents to have their child be sick, have to go to the hospital or even go to the emergency room or their primary care doctor because they’re sick and having trouble breathing,” said Dr. Portnoy, who served as the panel’s consumer representative.
A version of this article first appeared on WebMD.com.
This article was updated on 6/16/22.
Federal advisers to the U.S. Food and Drug Administration voted unanimously June 15 to recommend the use of the Moderna and Pfizer-BioNTech COVID-19 vaccines in infants and young children.
The Vaccines and Related Biological Products Advisory Committee (VRBPAC) of the FDA voted 21-0 to say that benefits of a two-dose series of Moderna’s mRNA vaccine outweigh risk for use in infants and children 6 months through 5 years of age.
The panel then voted 21-0 to say that benefits of a three-dose series of the Pfizer-BioNTech mRNA vaccine outweigh risk for use in infants and children 6 months through 4 years of age.
The FDA is not bound to follow the suggestions of its advisory committees, but it often does. Moderna and Pfizer are seeking to expand emergency use authorization (EUA) for their vaccines. EUAs are special clearances used to allow use of products in connection with public health crises such as the pandemic.
The Pfizer vaccine has standard, nonemergency FDA approval for use in people 16 years of age and older. The FDA also has granted EUA clearance for use of the shot in people ages 5 to 15.
The VRBPAC on June 15 recommended granting EUA clearance for Moderna’s COVID-19 vaccine for people ages 6 to 17. The Moderna vaccine already has full approval for use in people 18 years of age and older.
Many parents have been waiting for a clearance of COVID vaccines for their infants and young children, seeking protection for them at a time of continued spread of the virus.
The White House on June 9 outlined plans for making 10 million doses of COVID vaccines available for children under the age of 5 in the coming weeks.
The Centers for Disease Control and Prevention (CDC) has scheduled a June 18 meeting of its Advisory Committee on Immunization Practices, where members of that panel will vote on recommendations about use of the Moderna and Pfizer-BioNTech vaccines in infants and young children. The last step in the approval process to get shots into arms will be endorsement by the CDC director if the committee votes in favor of the vaccines.
For and against
During the public session during the June 15 FDA meeting, speakers offered varied opinions.
Some urged the panel to vote against the EUA expansion, citing concerns about risks of COVID vaccines in general.
But at the close of the meeting, top FDA vaccine official Peter Marks, MD, PhD, urged the public to be cautious about drawing conclusions from reading incident reports of side effects.
He said he has seen a “Twitter storm” during the day about claims of side effects. but stressed that the FDA has reported to the public on the rare side effects linked to the COVID vaccines, such as myocarditis, with advisories based on a review of reports of side effects. But many of these reports, gathered from the Vaccine Adverse Event Reporting System (VAERS) system, will turn out on further inspection not to be related to vaccination.
Many other speakers urged members of the panel to support expanded use of the vaccines for infants and young children. These speakers emphasized how lack of a vaccine to date has isolated young children who remain unprotected, even with about 83% of those age 5 and older in the United States having received at least one COVID shot.
Dr. Marks noted that there have been 442 deaths from COVID among children under 4 years of age during the pandemic, a number that he compared with the 78 deaths reported in the H1N1 flu. He urged the panel “to be careful that we don’t become numb to the number of pediatric deaths because of the overwhelming number of older deaths here.”
Panelist H. Cody Meissner, MD, a pediatric infectious disease specialist from Tufts University, said the vaccine should be made available -- particularly for children considered to be at high risk for complications from COVID --but health officials need to present a clear picture of the relatively low risks to children of harm from the vaccines-- and from COVID.
“That has to be communicated clearly to parents so that they can participate in the decision about vaccinating a child in this age group,” Dr. Meissner said.
The results presented June 15 from studies of the shots in younger children were less impressive than those from the initial COVID vaccine trials done in adults. This was not a surprise to panelists given the rise of the omicron variant and the evolution of the pandemic, but it still led to comments about the need for further continued study of the vaccines in young children even if they are authorized.
Consider that in 2020, Pfizer won the first EUA for a COVID vaccine of any kind with data that pegged the shot’s efficacy rate at 95%. Statisticians estimated a likely possible range, or 95% confidence interval, for the vaccine efficacy rate at 90.3% to 97.6%.
Those estimates were based on finding eight cases of COVID reported among 18,198 study participants who got the Pfizer-BioNTech shot, compared with 162 cases among the 18,325 people in the placebo group, according to the FDA review of Pifzer’s initial application.
Study data
But on June 15, FDA advisers had to consider an EUA application for which the data did not make as strong a case for the vaccine’s benefit among younger patients.
Pfizer presented what the FDA called a “preliminary descriptive analysis” of vaccine efficacy among participants in Study C4591007 who received three study vaccinations, following accrual of 10 total confirmed COVID-19 cases occurring at least 7 days after the third dose.
Looking at results for study participants ages 6 to 23 months of age, there was one case in the group that got the Pfizer-BioNTech shot and two in the placebo group, pegged as a 75.6% vaccine efficacy rate -- but one with caveats to the small numbers of cases. The 95% confidence interval for this vaccine efficacy rate was reported as-369.1% to 99.6% according to the FDA staff review.
For participants 2-4 years of age with and without evidence of prior SARS-CoV-
2 infection, there were two cases in the group that got the shot and five in the placebo group showing a vaccine efficacy rate of 82.4%, with a 95% confidence interval estimated ranging between -7.6% and 98.3%. For the combined analysis of both age groups, the efficacy rate was estimated at 80.4%, with a 95% confidence interval of 14.1% and 96.7%.
Doran Fink, MD, PhD, a top official in the FDA’s vaccines division, noted that the current EUA application for expanded pediatric use involved “some very preliminary” results that involved “a small number of cases and limited follow up time.”
But he stressed that the evidence gathered to date for the Pifzer application for use of its COVID shot in infants and young children met the threshold for conditional clearance during a crisis.
“We do feel very confident that the evidentiary standard for benefit for an EUA has been met here,” but added that more data would be needed to address questions about the efficacy of the vaccine beyond a third dose and whether an additional dose may be needed.
Pfizer also used a comparison known as “immunobridging” in support of the application. This looked at SARS- CoV-2 50% neutralizing antibody titers for the children in the age group covered by the EUA application and compared them to a randomly selected subset of 16-25-year-old participants in another study,
Key data for the pending Moderna EUA for use of its shot in infants and young children came from study P204. In it, Moderna found 51 cases of COVID among 1,511 children ages 6 months to 23 months who got the vaccines, versus 34 cases among 513 children who received a placebo, according to an FDA staff review.
That resulted in a vaccine efficacy rate pegged at 50.6%, with a 95% confidence interval of 21.4% to 68.6%.
Looking at the children ages 2 to 5 years in the P204 study, there were 119 cases out of 2,594 participants who got the shot, versus 61 cases of 858 in the placebo arm, or 7.1%. That translated to a 36.8% vaccine efficacy rate, with a confidence interval 12.5% to 54.0%.
Panelist Jay Portnoy, MD, of Children’s Mercy Hospital in Kansas City said all of the pediatricians he knows are waiting for the FDA to authorize the new uses of these vaccines in infants and young children.
“The death rate from COVID in young children may not be extremely high, but it’s absolutely terrifying to parents to have their child be sick, have to go to the hospital or even go to the emergency room or their primary care doctor because they’re sick and having trouble breathing,” said Dr. Portnoy, who served as the panel’s consumer representative.
A version of this article first appeared on WebMD.com.
This article was updated on 6/16/22.
Federal advisers to the U.S. Food and Drug Administration voted unanimously June 15 to recommend the use of the Moderna and Pfizer-BioNTech COVID-19 vaccines in infants and young children.
The Vaccines and Related Biological Products Advisory Committee (VRBPAC) of the FDA voted 21-0 to say that benefits of a two-dose series of Moderna’s mRNA vaccine outweigh risk for use in infants and children 6 months through 5 years of age.
The panel then voted 21-0 to say that benefits of a three-dose series of the Pfizer-BioNTech mRNA vaccine outweigh risk for use in infants and children 6 months through 4 years of age.
The FDA is not bound to follow the suggestions of its advisory committees, but it often does. Moderna and Pfizer are seeking to expand emergency use authorization (EUA) for their vaccines. EUAs are special clearances used to allow use of products in connection with public health crises such as the pandemic.
The Pfizer vaccine has standard, nonemergency FDA approval for use in people 16 years of age and older. The FDA also has granted EUA clearance for use of the shot in people ages 5 to 15.
The VRBPAC on June 15 recommended granting EUA clearance for Moderna’s COVID-19 vaccine for people ages 6 to 17. The Moderna vaccine already has full approval for use in people 18 years of age and older.
Many parents have been waiting for a clearance of COVID vaccines for their infants and young children, seeking protection for them at a time of continued spread of the virus.
The White House on June 9 outlined plans for making 10 million doses of COVID vaccines available for children under the age of 5 in the coming weeks.
The Centers for Disease Control and Prevention (CDC) has scheduled a June 18 meeting of its Advisory Committee on Immunization Practices, where members of that panel will vote on recommendations about use of the Moderna and Pfizer-BioNTech vaccines in infants and young children. The last step in the approval process to get shots into arms will be endorsement by the CDC director if the committee votes in favor of the vaccines.
For and against
During the public session during the June 15 FDA meeting, speakers offered varied opinions.
Some urged the panel to vote against the EUA expansion, citing concerns about risks of COVID vaccines in general.
But at the close of the meeting, top FDA vaccine official Peter Marks, MD, PhD, urged the public to be cautious about drawing conclusions from reading incident reports of side effects.
He said he has seen a “Twitter storm” during the day about claims of side effects. but stressed that the FDA has reported to the public on the rare side effects linked to the COVID vaccines, such as myocarditis, with advisories based on a review of reports of side effects. But many of these reports, gathered from the Vaccine Adverse Event Reporting System (VAERS) system, will turn out on further inspection not to be related to vaccination.
Many other speakers urged members of the panel to support expanded use of the vaccines for infants and young children. These speakers emphasized how lack of a vaccine to date has isolated young children who remain unprotected, even with about 83% of those age 5 and older in the United States having received at least one COVID shot.
Dr. Marks noted that there have been 442 deaths from COVID among children under 4 years of age during the pandemic, a number that he compared with the 78 deaths reported in the H1N1 flu. He urged the panel “to be careful that we don’t become numb to the number of pediatric deaths because of the overwhelming number of older deaths here.”
Panelist H. Cody Meissner, MD, a pediatric infectious disease specialist from Tufts University, said the vaccine should be made available -- particularly for children considered to be at high risk for complications from COVID --but health officials need to present a clear picture of the relatively low risks to children of harm from the vaccines-- and from COVID.
“That has to be communicated clearly to parents so that they can participate in the decision about vaccinating a child in this age group,” Dr. Meissner said.
The results presented June 15 from studies of the shots in younger children were less impressive than those from the initial COVID vaccine trials done in adults. This was not a surprise to panelists given the rise of the omicron variant and the evolution of the pandemic, but it still led to comments about the need for further continued study of the vaccines in young children even if they are authorized.
Consider that in 2020, Pfizer won the first EUA for a COVID vaccine of any kind with data that pegged the shot’s efficacy rate at 95%. Statisticians estimated a likely possible range, or 95% confidence interval, for the vaccine efficacy rate at 90.3% to 97.6%.
Those estimates were based on finding eight cases of COVID reported among 18,198 study participants who got the Pfizer-BioNTech shot, compared with 162 cases among the 18,325 people in the placebo group, according to the FDA review of Pifzer’s initial application.
Study data
But on June 15, FDA advisers had to consider an EUA application for which the data did not make as strong a case for the vaccine’s benefit among younger patients.
Pfizer presented what the FDA called a “preliminary descriptive analysis” of vaccine efficacy among participants in Study C4591007 who received three study vaccinations, following accrual of 10 total confirmed COVID-19 cases occurring at least 7 days after the third dose.
Looking at results for study participants ages 6 to 23 months of age, there was one case in the group that got the Pfizer-BioNTech shot and two in the placebo group, pegged as a 75.6% vaccine efficacy rate -- but one with caveats to the small numbers of cases. The 95% confidence interval for this vaccine efficacy rate was reported as-369.1% to 99.6% according to the FDA staff review.
For participants 2-4 years of age with and without evidence of prior SARS-CoV-
2 infection, there were two cases in the group that got the shot and five in the placebo group showing a vaccine efficacy rate of 82.4%, with a 95% confidence interval estimated ranging between -7.6% and 98.3%. For the combined analysis of both age groups, the efficacy rate was estimated at 80.4%, with a 95% confidence interval of 14.1% and 96.7%.
Doran Fink, MD, PhD, a top official in the FDA’s vaccines division, noted that the current EUA application for expanded pediatric use involved “some very preliminary” results that involved “a small number of cases and limited follow up time.”
But he stressed that the evidence gathered to date for the Pifzer application for use of its COVID shot in infants and young children met the threshold for conditional clearance during a crisis.
“We do feel very confident that the evidentiary standard for benefit for an EUA has been met here,” but added that more data would be needed to address questions about the efficacy of the vaccine beyond a third dose and whether an additional dose may be needed.
Pfizer also used a comparison known as “immunobridging” in support of the application. This looked at SARS- CoV-2 50% neutralizing antibody titers for the children in the age group covered by the EUA application and compared them to a randomly selected subset of 16-25-year-old participants in another study,
Key data for the pending Moderna EUA for use of its shot in infants and young children came from study P204. In it, Moderna found 51 cases of COVID among 1,511 children ages 6 months to 23 months who got the vaccines, versus 34 cases among 513 children who received a placebo, according to an FDA staff review.
That resulted in a vaccine efficacy rate pegged at 50.6%, with a 95% confidence interval of 21.4% to 68.6%.
Looking at the children ages 2 to 5 years in the P204 study, there were 119 cases out of 2,594 participants who got the shot, versus 61 cases of 858 in the placebo arm, or 7.1%. That translated to a 36.8% vaccine efficacy rate, with a confidence interval 12.5% to 54.0%.
Panelist Jay Portnoy, MD, of Children’s Mercy Hospital in Kansas City said all of the pediatricians he knows are waiting for the FDA to authorize the new uses of these vaccines in infants and young children.
“The death rate from COVID in young children may not be extremely high, but it’s absolutely terrifying to parents to have their child be sick, have to go to the hospital or even go to the emergency room or their primary care doctor because they’re sick and having trouble breathing,” said Dr. Portnoy, who served as the panel’s consumer representative.
A version of this article first appeared on WebMD.com.
This article was updated on 6/16/22.
Children and COVID: New cases hold steady in nonholiday week
The new-case count for the most recent reporting week – 87,644 for June 3-9 – did go up from the previous week, but by only 270 cases, the American Academy of Pediatrics and Children’s Hospital Association said in their weekly COVID report. That’s just 0.31% higher than a week ago and probably is affected by reduced testing and reporting because of Memorial Day, as the AAP and CHA noted earlier.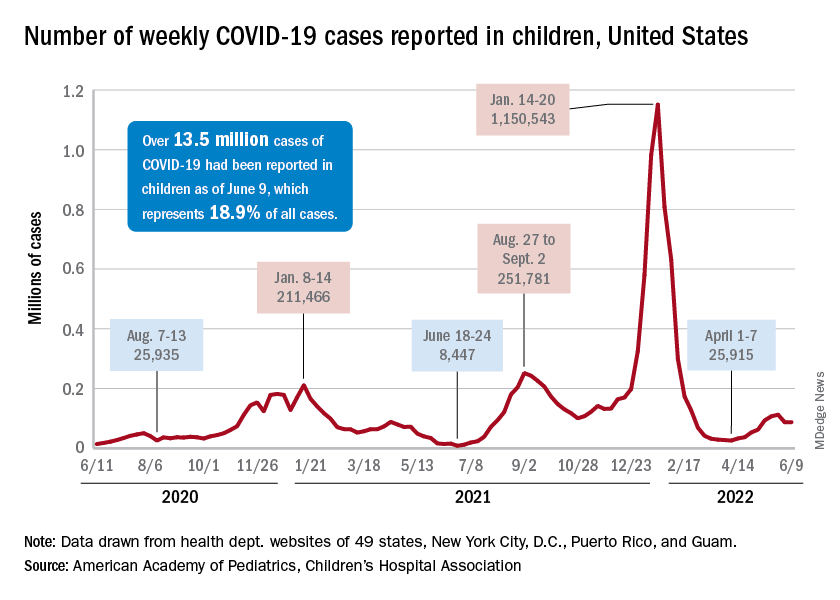
That hint of a continued decline accompanies the latest trend for new cases for all age groups: They have leveled out over the last month, with the moving 7-day daily average hovering around 100,000-110,000 since mid-May, data from the Centers for Disease Control and Prevention show.
The Food and Drug Administration, meanwhile, is in the news this week as two of its advisory panels take the next steps toward pediatric approvals of vaccines from Pfizer/BioNTtech and Moderna. The panels could advance the approvals of the Pfizer vaccine for children under the age of 5 years and the Moderna vaccine for children aged 6 months to 17 years.
Matthew Harris, MD, medical director of the COVID-19 vaccination program for Northwell Health in New Hyde Park, N.Y., emphasized the importance of vaccinations, as well as the continued challenge of convincing parents to get the shots for eligible children. “We still have a long way to go for primary vaccines and boosters for children 5 years and above,” he said in an interview.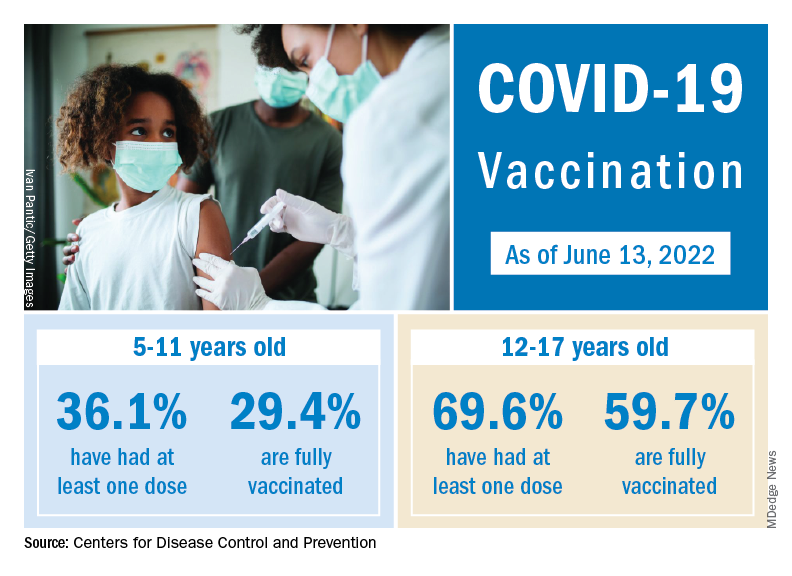
The vaccination effort against COVID-19 has stalled somewhat as interest has waned since the Omicron surge. Weekly initial vaccinations for children aged 5-11 years, which topped 100,000 as recently as mid-March, have been about 43,000 a week for the last 3 weeks, while 12- to 17-year-olds had around 27,000 or 28,000 initial vaccinations per week over that span, the AAP said in a separate report.
The latest data available from the CDC show that overall vaccine coverage levels for the younger group are only about half those of the 12- to 17-year-olds, both in terms of initial doses and completions. The 5- to 11-year-olds are not eligible for boosters yet, but 26.5% of the older children had received one as of June 13, according to the CDC’s COVID Data Tracker.
The new-case count for the most recent reporting week – 87,644 for June 3-9 – did go up from the previous week, but by only 270 cases, the American Academy of Pediatrics and Children’s Hospital Association said in their weekly COVID report. That’s just 0.31% higher than a week ago and probably is affected by reduced testing and reporting because of Memorial Day, as the AAP and CHA noted earlier.
That hint of a continued decline accompanies the latest trend for new cases for all age groups: They have leveled out over the last month, with the moving 7-day daily average hovering around 100,000-110,000 since mid-May, data from the Centers for Disease Control and Prevention show.
The Food and Drug Administration, meanwhile, is in the news this week as two of its advisory panels take the next steps toward pediatric approvals of vaccines from Pfizer/BioNTtech and Moderna. The panels could advance the approvals of the Pfizer vaccine for children under the age of 5 years and the Moderna vaccine for children aged 6 months to 17 years.
Matthew Harris, MD, medical director of the COVID-19 vaccination program for Northwell Health in New Hyde Park, N.Y., emphasized the importance of vaccinations, as well as the continued challenge of convincing parents to get the shots for eligible children. “We still have a long way to go for primary vaccines and boosters for children 5 years and above,” he said in an interview.
The vaccination effort against COVID-19 has stalled somewhat as interest has waned since the Omicron surge. Weekly initial vaccinations for children aged 5-11 years, which topped 100,000 as recently as mid-March, have been about 43,000 a week for the last 3 weeks, while 12- to 17-year-olds had around 27,000 or 28,000 initial vaccinations per week over that span, the AAP said in a separate report.
The latest data available from the CDC show that overall vaccine coverage levels for the younger group are only about half those of the 12- to 17-year-olds, both in terms of initial doses and completions. The 5- to 11-year-olds are not eligible for boosters yet, but 26.5% of the older children had received one as of June 13, according to the CDC’s COVID Data Tracker.
The new-case count for the most recent reporting week – 87,644 for June 3-9 – did go up from the previous week, but by only 270 cases, the American Academy of Pediatrics and Children’s Hospital Association said in their weekly COVID report. That’s just 0.31% higher than a week ago and probably is affected by reduced testing and reporting because of Memorial Day, as the AAP and CHA noted earlier.
That hint of a continued decline accompanies the latest trend for new cases for all age groups: They have leveled out over the last month, with the moving 7-day daily average hovering around 100,000-110,000 since mid-May, data from the Centers for Disease Control and Prevention show.
The Food and Drug Administration, meanwhile, is in the news this week as two of its advisory panels take the next steps toward pediatric approvals of vaccines from Pfizer/BioNTtech and Moderna. The panels could advance the approvals of the Pfizer vaccine for children under the age of 5 years and the Moderna vaccine for children aged 6 months to 17 years.
Matthew Harris, MD, medical director of the COVID-19 vaccination program for Northwell Health in New Hyde Park, N.Y., emphasized the importance of vaccinations, as well as the continued challenge of convincing parents to get the shots for eligible children. “We still have a long way to go for primary vaccines and boosters for children 5 years and above,” he said in an interview.
The vaccination effort against COVID-19 has stalled somewhat as interest has waned since the Omicron surge. Weekly initial vaccinations for children aged 5-11 years, which topped 100,000 as recently as mid-March, have been about 43,000 a week for the last 3 weeks, while 12- to 17-year-olds had around 27,000 or 28,000 initial vaccinations per week over that span, the AAP said in a separate report.
The latest data available from the CDC show that overall vaccine coverage levels for the younger group are only about half those of the 12- to 17-year-olds, both in terms of initial doses and completions. The 5- to 11-year-olds are not eligible for boosters yet, but 26.5% of the older children had received one as of June 13, according to the CDC’s COVID Data Tracker.
FDA warning released for Volara respiratory system
The Food and Drug Administration published a warning from the medical device company Baxter International, citing problems with their device used for at-home respiratory therapy. The release cautions Volara system users that using certain therapies from the device may cause a change in lung pressure and a decrease in oxygen level. This cautionary warning was issued following a single reported case of oxygen loss while using the device.
The Volara system is meant to help patients with persistent pulmonary problems who are transitioning from the hospital to the outpatient setting. It can connect to three pieces commonly used in treating the respiratory conditions – a tracheostomy tube, a mask, and an in-line ventilator. The device offers three therapies – one to expand lungs (OLE), one to shake mucus from the lungs (CHFO), and a nebulizer to deliver medication.
This particular warning is relevant only to patients who use the system with an in-line ventilator or to patients who use OLE and CHFO therapies. The concern is that a rapid change in lung pressure (barotrauma), could damage the tissue by overextending the surface of the organ. Additionally, as noted in the reported case, Volara users may be at risk for a decrease in the level of oxygen while using the device (oxygen desaturation).
If patients have been directed to use Volara by a physician, the FDA recommends they continue to use it as prescribed. But they should look out for signs of respiratory distress. These include changes in alertness, the appearance of a blue tint around the mouth, increased breathing rate, and wheezing. If a patient or caregiver sees these signs, the patient should stop using Volara immediately and should seek help if their symptoms don’t improve.
In response to these precautions, Baxter says it will update the instructions for the use of its device and will add additional warnings. The company says it will dispatch a trainer to patients’ homes to help them understand the newest guidelines.
Both the FDA and Baxter urge patients who have experienced any problems with the device to report it to the hotlines listed at the bottom of their release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration published a warning from the medical device company Baxter International, citing problems with their device used for at-home respiratory therapy. The release cautions Volara system users that using certain therapies from the device may cause a change in lung pressure and a decrease in oxygen level. This cautionary warning was issued following a single reported case of oxygen loss while using the device.
The Volara system is meant to help patients with persistent pulmonary problems who are transitioning from the hospital to the outpatient setting. It can connect to three pieces commonly used in treating the respiratory conditions – a tracheostomy tube, a mask, and an in-line ventilator. The device offers three therapies – one to expand lungs (OLE), one to shake mucus from the lungs (CHFO), and a nebulizer to deliver medication.
This particular warning is relevant only to patients who use the system with an in-line ventilator or to patients who use OLE and CHFO therapies. The concern is that a rapid change in lung pressure (barotrauma), could damage the tissue by overextending the surface of the organ. Additionally, as noted in the reported case, Volara users may be at risk for a decrease in the level of oxygen while using the device (oxygen desaturation).
If patients have been directed to use Volara by a physician, the FDA recommends they continue to use it as prescribed. But they should look out for signs of respiratory distress. These include changes in alertness, the appearance of a blue tint around the mouth, increased breathing rate, and wheezing. If a patient or caregiver sees these signs, the patient should stop using Volara immediately and should seek help if their symptoms don’t improve.
In response to these precautions, Baxter says it will update the instructions for the use of its device and will add additional warnings. The company says it will dispatch a trainer to patients’ homes to help them understand the newest guidelines.
Both the FDA and Baxter urge patients who have experienced any problems with the device to report it to the hotlines listed at the bottom of their release.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration published a warning from the medical device company Baxter International, citing problems with their device used for at-home respiratory therapy. The release cautions Volara system users that using certain therapies from the device may cause a change in lung pressure and a decrease in oxygen level. This cautionary warning was issued following a single reported case of oxygen loss while using the device.
The Volara system is meant to help patients with persistent pulmonary problems who are transitioning from the hospital to the outpatient setting. It can connect to three pieces commonly used in treating the respiratory conditions – a tracheostomy tube, a mask, and an in-line ventilator. The device offers three therapies – one to expand lungs (OLE), one to shake mucus from the lungs (CHFO), and a nebulizer to deliver medication.
This particular warning is relevant only to patients who use the system with an in-line ventilator or to patients who use OLE and CHFO therapies. The concern is that a rapid change in lung pressure (barotrauma), could damage the tissue by overextending the surface of the organ. Additionally, as noted in the reported case, Volara users may be at risk for a decrease in the level of oxygen while using the device (oxygen desaturation).
If patients have been directed to use Volara by a physician, the FDA recommends they continue to use it as prescribed. But they should look out for signs of respiratory distress. These include changes in alertness, the appearance of a blue tint around the mouth, increased breathing rate, and wheezing. If a patient or caregiver sees these signs, the patient should stop using Volara immediately and should seek help if their symptoms don’t improve.
In response to these precautions, Baxter says it will update the instructions for the use of its device and will add additional warnings. The company says it will dispatch a trainer to patients’ homes to help them understand the newest guidelines.
Both the FDA and Baxter urge patients who have experienced any problems with the device to report it to the hotlines listed at the bottom of their release.
A version of this article first appeared on Medscape.com.
FDA: Urgent device correction, recall for Philips ventilator
The U.S. Food and Drug Administration has announced a Class I recall for Philips Respironics V60 and V60 Plus ventilators, citing a power failure leading to potential oxygen deprivation. Class I recalls, the most severe, are reserved for devices that may cause serious injury or death, as noted in the FDA’s announcement. As of April 14, one death and four injuries have been associated with this device failure.
These ventilators are commonly used in hospitals or under medical supervision for patients who have difficulty regulating breathing on their own. Normally, if oxygen flow is interrupted, the device sounds alarms, alerting supervisors. The failure comes when a power fluctuation causes the device to randomly shut down, which forces the alarm system to reboot. This internal disruption is the reason for the recall.
When the device shuts down out of the blue, it may or may not sound the requisite alarm that would allow providers to intervene. If the device does not sound the alarm, patients may lose oxygen for an extended period, without a provider even knowing.
Philips was notified of these problems and began the recall process on March 10. Currently, it is estimated that 56,671 devices have been distributed throughout the United States. The FDA and Philips Respironics advise that if providers are already using these ventilators, they may continue to do so in accordance with extra set of instructions.
First, customers should connect the device to an external alarm or nurse call system. Second, they should use an external oxygen monitor and a pulse oximeter to keep track of air flow. Finally, if one is available, there should be a backup ventilator on the premises. That way, if there is an interruption in oxygen flow, someone will be alerted and can quickly intervene.
If there is a problem, the patient should be removed from the Philips ventilator and immediately placed on an alternate device. The FDA instructs customers who have experienced problems to report them to its MedWatch database.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has announced a Class I recall for Philips Respironics V60 and V60 Plus ventilators, citing a power failure leading to potential oxygen deprivation. Class I recalls, the most severe, are reserved for devices that may cause serious injury or death, as noted in the FDA’s announcement. As of April 14, one death and four injuries have been associated with this device failure.
These ventilators are commonly used in hospitals or under medical supervision for patients who have difficulty regulating breathing on their own. Normally, if oxygen flow is interrupted, the device sounds alarms, alerting supervisors. The failure comes when a power fluctuation causes the device to randomly shut down, which forces the alarm system to reboot. This internal disruption is the reason for the recall.
When the device shuts down out of the blue, it may or may not sound the requisite alarm that would allow providers to intervene. If the device does not sound the alarm, patients may lose oxygen for an extended period, without a provider even knowing.
Philips was notified of these problems and began the recall process on March 10. Currently, it is estimated that 56,671 devices have been distributed throughout the United States. The FDA and Philips Respironics advise that if providers are already using these ventilators, they may continue to do so in accordance with extra set of instructions.
First, customers should connect the device to an external alarm or nurse call system. Second, they should use an external oxygen monitor and a pulse oximeter to keep track of air flow. Finally, if one is available, there should be a backup ventilator on the premises. That way, if there is an interruption in oxygen flow, someone will be alerted and can quickly intervene.
If there is a problem, the patient should be removed from the Philips ventilator and immediately placed on an alternate device. The FDA instructs customers who have experienced problems to report them to its MedWatch database.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has announced a Class I recall for Philips Respironics V60 and V60 Plus ventilators, citing a power failure leading to potential oxygen deprivation. Class I recalls, the most severe, are reserved for devices that may cause serious injury or death, as noted in the FDA’s announcement. As of April 14, one death and four injuries have been associated with this device failure.
These ventilators are commonly used in hospitals or under medical supervision for patients who have difficulty regulating breathing on their own. Normally, if oxygen flow is interrupted, the device sounds alarms, alerting supervisors. The failure comes when a power fluctuation causes the device to randomly shut down, which forces the alarm system to reboot. This internal disruption is the reason for the recall.
When the device shuts down out of the blue, it may or may not sound the requisite alarm that would allow providers to intervene. If the device does not sound the alarm, patients may lose oxygen for an extended period, without a provider even knowing.
Philips was notified of these problems and began the recall process on March 10. Currently, it is estimated that 56,671 devices have been distributed throughout the United States. The FDA and Philips Respironics advise that if providers are already using these ventilators, they may continue to do so in accordance with extra set of instructions.
First, customers should connect the device to an external alarm or nurse call system. Second, they should use an external oxygen monitor and a pulse oximeter to keep track of air flow. Finally, if one is available, there should be a backup ventilator on the premises. That way, if there is an interruption in oxygen flow, someone will be alerted and can quickly intervene.
If there is a problem, the patient should be removed from the Philips ventilator and immediately placed on an alternate device. The FDA instructs customers who have experienced problems to report them to its MedWatch database.
A version of this article first appeared on Medscape.com.
FDA adds RA indication for Riabni rituximab biosimilar
The Food and Drug Administration has approved adding adult patients with rheumatoid arthritis to the list of indications for the rituximab biosimilar Riabni (rituximab-arrx) on the basis of results of a randomized, double-blind, comparative clinical study with the CD20-directed cytolytic antibody reference product, Rituxan, the biosimilar’s manufacturer, Amgen, announced June 6.
The RA indication is specifically for adults with moderate to severely active disease who have had an inadequate response to one or more tumor necrosis factor inhibitors. Riabni was approved in December 2020 for the treatment of adult patients with non-Hodgkin lymphoma, chronic lymphocytic leukemia, granulomatosis with polyangiitis, and microscopic polyangiitis.
The clinical study testing Riabni against Rituxan involved 311 patients with moderate to severe RA who received Riabni, Rituxan manufactured in the United States, and Rituxan manufactured in the European Union. The patients who received the U.S.-manufactured Rituxan were transitioned to receive Riabni for their second dose of rituximab, whereas patients in other groups stayed with the same treatment. The trial’s primary efficacy endpoint of the change in Disease Activity Score in 28 joints using C-reactive protein from baseline to week 24 was within the predefined equivalence margin for clinical efficacy between Riabni and Rituxan. The two products also had similar safety, pharmacokinetics, and immunogenicity profiles, according to Amgen.
Currently, Riabni and Ruxience (rituximab-pvvr) are the only two approved rituximab biosimilars in the United States. Ruxience is approved for the same indications. Rituxan alone has protected orphan drug status for the indication of adult patients with moderate to severe pemphigus vulgaris.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved adding adult patients with rheumatoid arthritis to the list of indications for the rituximab biosimilar Riabni (rituximab-arrx) on the basis of results of a randomized, double-blind, comparative clinical study with the CD20-directed cytolytic antibody reference product, Rituxan, the biosimilar’s manufacturer, Amgen, announced June 6.
The RA indication is specifically for adults with moderate to severely active disease who have had an inadequate response to one or more tumor necrosis factor inhibitors. Riabni was approved in December 2020 for the treatment of adult patients with non-Hodgkin lymphoma, chronic lymphocytic leukemia, granulomatosis with polyangiitis, and microscopic polyangiitis.
The clinical study testing Riabni against Rituxan involved 311 patients with moderate to severe RA who received Riabni, Rituxan manufactured in the United States, and Rituxan manufactured in the European Union. The patients who received the U.S.-manufactured Rituxan were transitioned to receive Riabni for their second dose of rituximab, whereas patients in other groups stayed with the same treatment. The trial’s primary efficacy endpoint of the change in Disease Activity Score in 28 joints using C-reactive protein from baseline to week 24 was within the predefined equivalence margin for clinical efficacy between Riabni and Rituxan. The two products also had similar safety, pharmacokinetics, and immunogenicity profiles, according to Amgen.
Currently, Riabni and Ruxience (rituximab-pvvr) are the only two approved rituximab biosimilars in the United States. Ruxience is approved for the same indications. Rituxan alone has protected orphan drug status for the indication of adult patients with moderate to severe pemphigus vulgaris.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved adding adult patients with rheumatoid arthritis to the list of indications for the rituximab biosimilar Riabni (rituximab-arrx) on the basis of results of a randomized, double-blind, comparative clinical study with the CD20-directed cytolytic antibody reference product, Rituxan, the biosimilar’s manufacturer, Amgen, announced June 6.
The RA indication is specifically for adults with moderate to severely active disease who have had an inadequate response to one or more tumor necrosis factor inhibitors. Riabni was approved in December 2020 for the treatment of adult patients with non-Hodgkin lymphoma, chronic lymphocytic leukemia, granulomatosis with polyangiitis, and microscopic polyangiitis.
The clinical study testing Riabni against Rituxan involved 311 patients with moderate to severe RA who received Riabni, Rituxan manufactured in the United States, and Rituxan manufactured in the European Union. The patients who received the U.S.-manufactured Rituxan were transitioned to receive Riabni for their second dose of rituximab, whereas patients in other groups stayed with the same treatment. The trial’s primary efficacy endpoint of the change in Disease Activity Score in 28 joints using C-reactive protein from baseline to week 24 was within the predefined equivalence margin for clinical efficacy between Riabni and Rituxan. The two products also had similar safety, pharmacokinetics, and immunogenicity profiles, according to Amgen.
Currently, Riabni and Ruxience (rituximab-pvvr) are the only two approved rituximab biosimilars in the United States. Ruxience is approved for the same indications. Rituxan alone has protected orphan drug status for the indication of adult patients with moderate to severe pemphigus vulgaris.
A version of this article first appeared on Medscape.com.
Children and COVID: Cases down, start of vaccinations near
The first decline in COVID-19 cases among children since early April may have been holiday related, but the shortened week also brought news about vaccination for the youngest children.
The Food and Drug Administration has accepted Pfizer’s application for a COVID-19 vaccine for children under age 5, so vaccination could begin as early as June 21, according to White House COVID-19 response coordinator Ashish Jha, MD.
“We know that many, many parents are eager to vaccinate their youngest kids and it’s important to do this right,” Dr. Jha said at a White House press briefing June 2. “We expect that vaccinations will begin in earnest as early as June 21 and really roll on throughout that week.”
Decline may just be underreporting
Over on the incidence side of the pandemic, “Testing and reporting may have been affected by the holiday weekend [since] states may change their reporting schedules, which may cause irregularities in trends,” the American Academy of Pediatrics and the Children’s Hospital association said in their latest COVID report.
The decline in new cases was not spread uniformly across the four major regions of the United States. The count actually went up in the West for the week of May 27 to June 2, while the South saw the largest decline. The Midwest and Northeast, meanwhile, saw new cases drop for the second straight week, the AAP and CHA said.
The cumulative number of COVID-19 cases in children was up to 13.45 million as of June 2, with children representing 18.9% of all cases since the start of the pandemic, according to the two organizations. The Centers for Disease Control and Prevention reported figures of 13.14 million and 17.5% on June 6.
The AAP/CHA estimates, however, are based on state data that have become increasingly hard to obtain and subject to inconsistency. “Shortages of COVID-19 tests during surges and the increasing use of COVID-19 home tests likely affect the undercounting of COVID-19 cases,” they noted, and “at times when COVID-19 transmission is low, states might reduce the frequency information is updated.”
Vaccinations held steady over the holiday
The ongoing vaccination effort in children aged 5 years and older did not show a Memorial Day drop-off, as initial vaccinations held at 43,000 in 5- to 11-year-olds and at 27,000 in 12- to 17-year-olds for a second consecutive week. That number has ranged from 34,000 to 70,000 for the younger children and from 25,000 to 47,000 for the older group since mid-March, the AAP said in a separate weekly report.
Despite weekly vaccine initiations that have been roughly double those of the older children for months, the 5- to 11-year-olds are still only at 36.0% coverage with at least one dose, compared with 69.5% for the 12- to-17-year-olds. Full vaccination for the two age groups comes in at 29.3% and 59.6%, respectively, as of June 6, according to the CDC’s COVID Data Tracker.
The first decline in COVID-19 cases among children since early April may have been holiday related, but the shortened week also brought news about vaccination for the youngest children.
The Food and Drug Administration has accepted Pfizer’s application for a COVID-19 vaccine for children under age 5, so vaccination could begin as early as June 21, according to White House COVID-19 response coordinator Ashish Jha, MD.
“We know that many, many parents are eager to vaccinate their youngest kids and it’s important to do this right,” Dr. Jha said at a White House press briefing June 2. “We expect that vaccinations will begin in earnest as early as June 21 and really roll on throughout that week.”
Decline may just be underreporting
Over on the incidence side of the pandemic, “Testing and reporting may have been affected by the holiday weekend [since] states may change their reporting schedules, which may cause irregularities in trends,” the American Academy of Pediatrics and the Children’s Hospital association said in their latest COVID report.
The decline in new cases was not spread uniformly across the four major regions of the United States. The count actually went up in the West for the week of May 27 to June 2, while the South saw the largest decline. The Midwest and Northeast, meanwhile, saw new cases drop for the second straight week, the AAP and CHA said.
The cumulative number of COVID-19 cases in children was up to 13.45 million as of June 2, with children representing 18.9% of all cases since the start of the pandemic, according to the two organizations. The Centers for Disease Control and Prevention reported figures of 13.14 million and 17.5% on June 6.
The AAP/CHA estimates, however, are based on state data that have become increasingly hard to obtain and subject to inconsistency. “Shortages of COVID-19 tests during surges and the increasing use of COVID-19 home tests likely affect the undercounting of COVID-19 cases,” they noted, and “at times when COVID-19 transmission is low, states might reduce the frequency information is updated.”
Vaccinations held steady over the holiday
The ongoing vaccination effort in children aged 5 years and older did not show a Memorial Day drop-off, as initial vaccinations held at 43,000 in 5- to 11-year-olds and at 27,000 in 12- to 17-year-olds for a second consecutive week. That number has ranged from 34,000 to 70,000 for the younger children and from 25,000 to 47,000 for the older group since mid-March, the AAP said in a separate weekly report.
Despite weekly vaccine initiations that have been roughly double those of the older children for months, the 5- to 11-year-olds are still only at 36.0% coverage with at least one dose, compared with 69.5% for the 12- to-17-year-olds. Full vaccination for the two age groups comes in at 29.3% and 59.6%, respectively, as of June 6, according to the CDC’s COVID Data Tracker.
The first decline in COVID-19 cases among children since early April may have been holiday related, but the shortened week also brought news about vaccination for the youngest children.
The Food and Drug Administration has accepted Pfizer’s application for a COVID-19 vaccine for children under age 5, so vaccination could begin as early as June 21, according to White House COVID-19 response coordinator Ashish Jha, MD.
“We know that many, many parents are eager to vaccinate their youngest kids and it’s important to do this right,” Dr. Jha said at a White House press briefing June 2. “We expect that vaccinations will begin in earnest as early as June 21 and really roll on throughout that week.”
Decline may just be underreporting
Over on the incidence side of the pandemic, “Testing and reporting may have been affected by the holiday weekend [since] states may change their reporting schedules, which may cause irregularities in trends,” the American Academy of Pediatrics and the Children’s Hospital association said in their latest COVID report.
The decline in new cases was not spread uniformly across the four major regions of the United States. The count actually went up in the West for the week of May 27 to June 2, while the South saw the largest decline. The Midwest and Northeast, meanwhile, saw new cases drop for the second straight week, the AAP and CHA said.
The cumulative number of COVID-19 cases in children was up to 13.45 million as of June 2, with children representing 18.9% of all cases since the start of the pandemic, according to the two organizations. The Centers for Disease Control and Prevention reported figures of 13.14 million and 17.5% on June 6.
The AAP/CHA estimates, however, are based on state data that have become increasingly hard to obtain and subject to inconsistency. “Shortages of COVID-19 tests during surges and the increasing use of COVID-19 home tests likely affect the undercounting of COVID-19 cases,” they noted, and “at times when COVID-19 transmission is low, states might reduce the frequency information is updated.”
Vaccinations held steady over the holiday
The ongoing vaccination effort in children aged 5 years and older did not show a Memorial Day drop-off, as initial vaccinations held at 43,000 in 5- to 11-year-olds and at 27,000 in 12- to 17-year-olds for a second consecutive week. That number has ranged from 34,000 to 70,000 for the younger children and from 25,000 to 47,000 for the older group since mid-March, the AAP said in a separate weekly report.
Despite weekly vaccine initiations that have been roughly double those of the older children for months, the 5- to 11-year-olds are still only at 36.0% coverage with at least one dose, compared with 69.5% for the 12- to-17-year-olds. Full vaccination for the two age groups comes in at 29.3% and 59.6%, respectively, as of June 6, according to the CDC’s COVID Data Tracker.