User login
Black nonsmokers still at high risk for secondhand smoke exposure
Despite 30+ years of antismoking public policies and dramatic overall decline in secondhand smoke (SHS) exposure, .

No risk-free SHS exposure
Surendranath S. Shastri, MD, of MD Anderson Cancer Center, Houston, and colleagues underscored the U.S. Surgeon General’s determination that there is no risk-free level of SHS exposure in a recent JAMA Internal Medicine Research Letter.
“With the outbreak of the coronavirus disease 2019, which affects lung function, improving smoke-free policies to enhance air quality should be a growing priority,”they wrote.
Dr. Shastri and colleagues looked at 2011-2018 data from the National Health and Nutrition Examination Survey (NHANES), which detailed prevalence of SHS exposure in the U.S. population aged 3 years and older using interviews and biological specimens to test for cotinine levels. For the survey, nonsmokers having serum cotinine levels of 0.05 to 10 ng/mL were considered to have SHS exposure.
While the prevalence of SHS exposure among nonsmokers declined from 87.5% to 25.3% between 1988 and 2012, levels have stagnated since 2012 and racial and economic disparities are evident. Higher smoking rates, less knowledge about health risks, higher workplace exposure, greater likelihood of living in low-income, multi-unit housing, plus having their communities targeted by tobacco companies, may all help explain higher serum levels of cotinine in populations with lower socioeconomic status.
“Multivariable logistic regression identified younger age (odds ratio [OR], 1.88, for 12-19 years, and OR, 2.29, for 3-11 years), non-Hispanic Black race/ethnicity (OR, 2.75), less than high school education (OR, 1.59), and living below the poverty level (OR, 2.61) as risk factors for SHSe in the 2017-2018 cycle, with little change across all data cycles,” the researchers wrote.
Disparities in SHS exposure
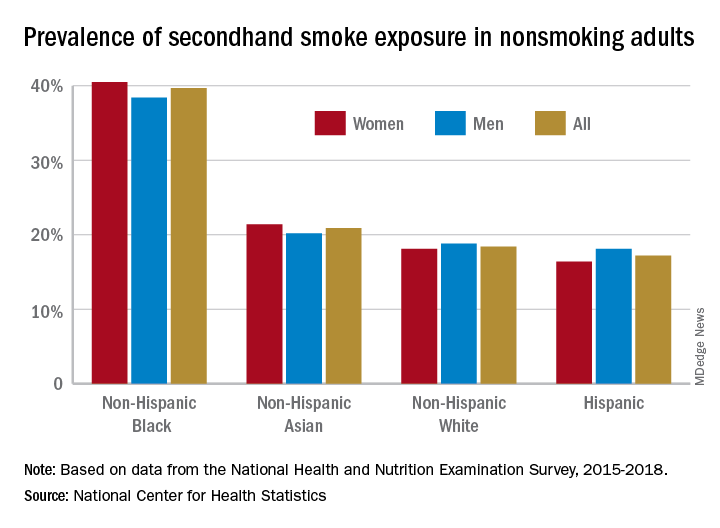
A second report from NHANES data for 2015-2018, published in a National Center for Health Statistics Data Brief (No. 396, February 2021) showed that 20.8% of nonsmoking U.S. adults had SHS exposure, again with greater prevalence among non-Hispanic Black adults (39.7%), than for non-Hispanic White (18.4%), non-Hispanic Asian (20.9%), and Hispanic (17.2%) adults. Exposure was also greater in the younger age groups, with SHS rates for adults aged 18-39 years, 40-59 years, and ≥60 years at 25.6%, 19.1%, and 17.6%, respectively. Lower education (high school or less vs. some college education) and lower income levels were also associated with higher levels of SHS exposure. The investigators noted that among households with smokers, non-Hispanic Black adults are less likely to have complete smoking bans in homes, and among Medicaid or uninsured parents of any race or ethnicity, bans on smoking in family vehicles are less likely.
Overall, the prevalence of SHS exposure declined from 27.7% to 20.7% from 2009 to 2018, but the decreases were mediated by race and income.
SHS exposure in private spaces
A research brief from the Centers for Disease Control and Prevention on SHS exposure in homes and vehicles in the U.S. among middle and high school students also found a general decline in SHS exposure over 2011-2018 in homes (26.8%-20.9%; P < .001) and vehicles (30.2%-19.8%; P < .001). The findings, derived from the National Youth Tobacco Survey for 2011-2019, showed that no reduction occurred in homes among non-Hispanic Black students. Overall, a significant difference in home SHS exposure was observed by race/ethnicity: non-Hispanic Black (28.4%) and non-Hispanic White (27.4%) students both had a higher prevalence compared with Hispanic (20.0%) and non-Hispanic other (20.2%) students (P < .001).
Progress in reducing SHS exposure in public spaces has been made over the last 2 decades, with 27 states and more than 1,000 municipalities implementing comprehensive smoke-free laws that prohibit smoking in indoor public places, including workplaces, restaurants, and bars. While the prevalence of voluntary smoke-free home (83.7%) and vehicle (78.1%) rules has increased over time, private settings remain major sources of SHS exposure for many people, including youths. “Although SHS exposures have declined,” the authors wrote, “more than 6 million young people remain exposed to SHS in these private settings.”

In reviewing the data, Mary Cataletto, MD, FCCP, clinical professor of pediatrics at NYU Long Island School of Medicine, stated that these studies “highlight the need for implementation of smoke-free policies to reduce exposure to secondhand smoke, especially in homes and cars and with focused advocacy efforts in highly affected communities.”
Panagis Galiatsatos, MD, MHS, assistant professor of medicine at Johns Hopkins University, Baltimore, emphasized implementation of smoke-free policies but also treatment for smokers. “I’m not at all surprised by these statistics,” he noted in an interview. “Public health policies have helped us to get to where we are now, but there’s a reason that we have plateaued over the last decade. It’s hard to mitigate secondhand smoke exposure because the ones who are smoking now are the most refractory, challenging cases. ... You need good clinical interventions with counseling supported by pharmacological agents to help them if you want to stop secondhand smoke exposure.” He added, “You have to look at current smokers no differently than you look at patients with stage IV cancer – a group that requires a lot of resources to help them get through. Remember, all of them want to quit, but the promise of well-designed, precision-medicine strategies to help them quit has not been kept. Public health policy isn’t going to do it. We need to manage these patients clinically.”
The investigators had no conflict disclosures.
Despite 30+ years of antismoking public policies and dramatic overall decline in secondhand smoke (SHS) exposure, .
No risk-free SHS exposure
Surendranath S. Shastri, MD, of MD Anderson Cancer Center, Houston, and colleagues underscored the U.S. Surgeon General’s determination that there is no risk-free level of SHS exposure in a recent JAMA Internal Medicine Research Letter.
“With the outbreak of the coronavirus disease 2019, which affects lung function, improving smoke-free policies to enhance air quality should be a growing priority,”they wrote.
Dr. Shastri and colleagues looked at 2011-2018 data from the National Health and Nutrition Examination Survey (NHANES), which detailed prevalence of SHS exposure in the U.S. population aged 3 years and older using interviews and biological specimens to test for cotinine levels. For the survey, nonsmokers having serum cotinine levels of 0.05 to 10 ng/mL were considered to have SHS exposure.
While the prevalence of SHS exposure among nonsmokers declined from 87.5% to 25.3% between 1988 and 2012, levels have stagnated since 2012 and racial and economic disparities are evident. Higher smoking rates, less knowledge about health risks, higher workplace exposure, greater likelihood of living in low-income, multi-unit housing, plus having their communities targeted by tobacco companies, may all help explain higher serum levels of cotinine in populations with lower socioeconomic status.
“Multivariable logistic regression identified younger age (odds ratio [OR], 1.88, for 12-19 years, and OR, 2.29, for 3-11 years), non-Hispanic Black race/ethnicity (OR, 2.75), less than high school education (OR, 1.59), and living below the poverty level (OR, 2.61) as risk factors for SHSe in the 2017-2018 cycle, with little change across all data cycles,” the researchers wrote.
Disparities in SHS exposure
A second report from NHANES data for 2015-2018, published in a National Center for Health Statistics Data Brief (No. 396, February 2021) showed that 20.8% of nonsmoking U.S. adults had SHS exposure, again with greater prevalence among non-Hispanic Black adults (39.7%), than for non-Hispanic White (18.4%), non-Hispanic Asian (20.9%), and Hispanic (17.2%) adults. Exposure was also greater in the younger age groups, with SHS rates for adults aged 18-39 years, 40-59 years, and ≥60 years at 25.6%, 19.1%, and 17.6%, respectively. Lower education (high school or less vs. some college education) and lower income levels were also associated with higher levels of SHS exposure. The investigators noted that among households with smokers, non-Hispanic Black adults are less likely to have complete smoking bans in homes, and among Medicaid or uninsured parents of any race or ethnicity, bans on smoking in family vehicles are less likely.
Overall, the prevalence of SHS exposure declined from 27.7% to 20.7% from 2009 to 2018, but the decreases were mediated by race and income.
SHS exposure in private spaces
A research brief from the Centers for Disease Control and Prevention on SHS exposure in homes and vehicles in the U.S. among middle and high school students also found a general decline in SHS exposure over 2011-2018 in homes (26.8%-20.9%; P < .001) and vehicles (30.2%-19.8%; P < .001). The findings, derived from the National Youth Tobacco Survey for 2011-2019, showed that no reduction occurred in homes among non-Hispanic Black students. Overall, a significant difference in home SHS exposure was observed by race/ethnicity: non-Hispanic Black (28.4%) and non-Hispanic White (27.4%) students both had a higher prevalence compared with Hispanic (20.0%) and non-Hispanic other (20.2%) students (P < .001).
Progress in reducing SHS exposure in public spaces has been made over the last 2 decades, with 27 states and more than 1,000 municipalities implementing comprehensive smoke-free laws that prohibit smoking in indoor public places, including workplaces, restaurants, and bars. While the prevalence of voluntary smoke-free home (83.7%) and vehicle (78.1%) rules has increased over time, private settings remain major sources of SHS exposure for many people, including youths. “Although SHS exposures have declined,” the authors wrote, “more than 6 million young people remain exposed to SHS in these private settings.”
In reviewing the data, Mary Cataletto, MD, FCCP, clinical professor of pediatrics at NYU Long Island School of Medicine, stated that these studies “highlight the need for implementation of smoke-free policies to reduce exposure to secondhand smoke, especially in homes and cars and with focused advocacy efforts in highly affected communities.”
Panagis Galiatsatos, MD, MHS, assistant professor of medicine at Johns Hopkins University, Baltimore, emphasized implementation of smoke-free policies but also treatment for smokers. “I’m not at all surprised by these statistics,” he noted in an interview. “Public health policies have helped us to get to where we are now, but there’s a reason that we have plateaued over the last decade. It’s hard to mitigate secondhand smoke exposure because the ones who are smoking now are the most refractory, challenging cases. ... You need good clinical interventions with counseling supported by pharmacological agents to help them if you want to stop secondhand smoke exposure.” He added, “You have to look at current smokers no differently than you look at patients with stage IV cancer – a group that requires a lot of resources to help them get through. Remember, all of them want to quit, but the promise of well-designed, precision-medicine strategies to help them quit has not been kept. Public health policy isn’t going to do it. We need to manage these patients clinically.”
The investigators had no conflict disclosures.
Despite 30+ years of antismoking public policies and dramatic overall decline in secondhand smoke (SHS) exposure, .
No risk-free SHS exposure
Surendranath S. Shastri, MD, of MD Anderson Cancer Center, Houston, and colleagues underscored the U.S. Surgeon General’s determination that there is no risk-free level of SHS exposure in a recent JAMA Internal Medicine Research Letter.
“With the outbreak of the coronavirus disease 2019, which affects lung function, improving smoke-free policies to enhance air quality should be a growing priority,”they wrote.
Dr. Shastri and colleagues looked at 2011-2018 data from the National Health and Nutrition Examination Survey (NHANES), which detailed prevalence of SHS exposure in the U.S. population aged 3 years and older using interviews and biological specimens to test for cotinine levels. For the survey, nonsmokers having serum cotinine levels of 0.05 to 10 ng/mL were considered to have SHS exposure.
While the prevalence of SHS exposure among nonsmokers declined from 87.5% to 25.3% between 1988 and 2012, levels have stagnated since 2012 and racial and economic disparities are evident. Higher smoking rates, less knowledge about health risks, higher workplace exposure, greater likelihood of living in low-income, multi-unit housing, plus having their communities targeted by tobacco companies, may all help explain higher serum levels of cotinine in populations with lower socioeconomic status.
“Multivariable logistic regression identified younger age (odds ratio [OR], 1.88, for 12-19 years, and OR, 2.29, for 3-11 years), non-Hispanic Black race/ethnicity (OR, 2.75), less than high school education (OR, 1.59), and living below the poverty level (OR, 2.61) as risk factors for SHSe in the 2017-2018 cycle, with little change across all data cycles,” the researchers wrote.
Disparities in SHS exposure
A second report from NHANES data for 2015-2018, published in a National Center for Health Statistics Data Brief (No. 396, February 2021) showed that 20.8% of nonsmoking U.S. adults had SHS exposure, again with greater prevalence among non-Hispanic Black adults (39.7%), than for non-Hispanic White (18.4%), non-Hispanic Asian (20.9%), and Hispanic (17.2%) adults. Exposure was also greater in the younger age groups, with SHS rates for adults aged 18-39 years, 40-59 years, and ≥60 years at 25.6%, 19.1%, and 17.6%, respectively. Lower education (high school or less vs. some college education) and lower income levels were also associated with higher levels of SHS exposure. The investigators noted that among households with smokers, non-Hispanic Black adults are less likely to have complete smoking bans in homes, and among Medicaid or uninsured parents of any race or ethnicity, bans on smoking in family vehicles are less likely.
Overall, the prevalence of SHS exposure declined from 27.7% to 20.7% from 2009 to 2018, but the decreases were mediated by race and income.
SHS exposure in private spaces
A research brief from the Centers for Disease Control and Prevention on SHS exposure in homes and vehicles in the U.S. among middle and high school students also found a general decline in SHS exposure over 2011-2018 in homes (26.8%-20.9%; P < .001) and vehicles (30.2%-19.8%; P < .001). The findings, derived from the National Youth Tobacco Survey for 2011-2019, showed that no reduction occurred in homes among non-Hispanic Black students. Overall, a significant difference in home SHS exposure was observed by race/ethnicity: non-Hispanic Black (28.4%) and non-Hispanic White (27.4%) students both had a higher prevalence compared with Hispanic (20.0%) and non-Hispanic other (20.2%) students (P < .001).
Progress in reducing SHS exposure in public spaces has been made over the last 2 decades, with 27 states and more than 1,000 municipalities implementing comprehensive smoke-free laws that prohibit smoking in indoor public places, including workplaces, restaurants, and bars. While the prevalence of voluntary smoke-free home (83.7%) and vehicle (78.1%) rules has increased over time, private settings remain major sources of SHS exposure for many people, including youths. “Although SHS exposures have declined,” the authors wrote, “more than 6 million young people remain exposed to SHS in these private settings.”
In reviewing the data, Mary Cataletto, MD, FCCP, clinical professor of pediatrics at NYU Long Island School of Medicine, stated that these studies “highlight the need for implementation of smoke-free policies to reduce exposure to secondhand smoke, especially in homes and cars and with focused advocacy efforts in highly affected communities.”
Panagis Galiatsatos, MD, MHS, assistant professor of medicine at Johns Hopkins University, Baltimore, emphasized implementation of smoke-free policies but also treatment for smokers. “I’m not at all surprised by these statistics,” he noted in an interview. “Public health policies have helped us to get to where we are now, but there’s a reason that we have plateaued over the last decade. It’s hard to mitigate secondhand smoke exposure because the ones who are smoking now are the most refractory, challenging cases. ... You need good clinical interventions with counseling supported by pharmacological agents to help them if you want to stop secondhand smoke exposure.” He added, “You have to look at current smokers no differently than you look at patients with stage IV cancer – a group that requires a lot of resources to help them get through. Remember, all of them want to quit, but the promise of well-designed, precision-medicine strategies to help them quit has not been kept. Public health policy isn’t going to do it. We need to manage these patients clinically.”
The investigators had no conflict disclosures.
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
CDC data strengthen link between obesity and severe COVID
Officials have previously linked being overweight or obese to a greater risk for more severe COVID-19. A report today from the U.S. Centers for Disease Control and Prevention adds numbers and some nuance to the association.
Data from nearly 150,000 U.S. adults hospitalized with COVID-19 nationwide indicate that risk for more severe disease outcomes increases along with body mass index (BMI). The risk of COVID-19–related hospitalization and death associated with obesity was particularly high among people younger than 65.
“As clinicians develop care plans for COVID-19 patients, they should consider the risk for severe outcomes in patients with higher BMIs, especially for those with severe obesity,” the researchers note. They add that their findings suggest “progressively intensive management of COVID-19 might be needed for patients with more severe obesity.”
People with COVID-19 close to the border between a healthy and overweight BMI – from 23.7 kg/m2 to 25.9 kg/m2 – had the lowest risks for adverse outcomes.
The study was published online today in Morbidity and Mortality Weekly Report.
Greater need for critical care
The risk of ICU admission was particularly associated with severe obesity. For example, those with a BMI in the 40-44.9 kg/m2 category had a 6% increased risk, which jumped to 16% higher among those with a BMI of 45 or greater.
Compared to people with a healthy BMI, the need for invasive mechanical ventilation was 12% more likely among overweight adults with a BMI of 25-29.2. The risked jumped to 108% greater among the most obese people, those with a BMI of 45 or greater, lead CDC researcher Lyudmyla Kompaniyets, PhD, and colleagues reported.
Moreover, the risks for hospitalization and death increased in a dose-response relationship with obesity.
For example, risks of being hospitalized were 7% greater for adults with a BMI between 30 and 34.9 and climbed to 33% greater for those with a BMI of 45. Risks were calculated as adjusted relative risks compared with people with a healthy BMI between 18.5 and 24.9.
Interestingly, being underweight was associated with elevated risk for COVID-19 hospitalization as well. For example, people with a BMI of less than 18.5 had a 20% greater chance of admission vs. people in the healthy BMI range. Unknown underlying medical conditions or issues related to nutrition or immune function could be contributing factors, the researchers note.
Elevated risk of dying
The risk of death in adults with obesity ranged from 8% higher in the 30-34.9 range up to 61% greater for those with a BMI of 45.
Chronic inflammation or impaired lung function from excess weight are possible reasons that higher BMI imparts greater risk, the researchers note.
The CDC researchers evaluated 148,494 adults from 238 hospitals participating in PHD-SR database. Because the study was limited to people hospitalized with COVID-19, the findings may not apply to all adults with COVID-19.
Another potential limitation is that investigators were unable to calculate BMI for all patients in the database because about 28% of participating hospitals did not report height and weight.
The study authors had no relevant financial relationships to disclose.
A version of this article first appeared on Medscape.com.
Officials have previously linked being overweight or obese to a greater risk for more severe COVID-19. A report today from the U.S. Centers for Disease Control and Prevention adds numbers and some nuance to the association.
Data from nearly 150,000 U.S. adults hospitalized with COVID-19 nationwide indicate that risk for more severe disease outcomes increases along with body mass index (BMI). The risk of COVID-19–related hospitalization and death associated with obesity was particularly high among people younger than 65.
“As clinicians develop care plans for COVID-19 patients, they should consider the risk for severe outcomes in patients with higher BMIs, especially for those with severe obesity,” the researchers note. They add that their findings suggest “progressively intensive management of COVID-19 might be needed for patients with more severe obesity.”
People with COVID-19 close to the border between a healthy and overweight BMI – from 23.7 kg/m2 to 25.9 kg/m2 – had the lowest risks for adverse outcomes.
The study was published online today in Morbidity and Mortality Weekly Report.
Greater need for critical care
The risk of ICU admission was particularly associated with severe obesity. For example, those with a BMI in the 40-44.9 kg/m2 category had a 6% increased risk, which jumped to 16% higher among those with a BMI of 45 or greater.
Compared to people with a healthy BMI, the need for invasive mechanical ventilation was 12% more likely among overweight adults with a BMI of 25-29.2. The risked jumped to 108% greater among the most obese people, those with a BMI of 45 or greater, lead CDC researcher Lyudmyla Kompaniyets, PhD, and colleagues reported.
Moreover, the risks for hospitalization and death increased in a dose-response relationship with obesity.
For example, risks of being hospitalized were 7% greater for adults with a BMI between 30 and 34.9 and climbed to 33% greater for those with a BMI of 45. Risks were calculated as adjusted relative risks compared with people with a healthy BMI between 18.5 and 24.9.
Interestingly, being underweight was associated with elevated risk for COVID-19 hospitalization as well. For example, people with a BMI of less than 18.5 had a 20% greater chance of admission vs. people in the healthy BMI range. Unknown underlying medical conditions or issues related to nutrition or immune function could be contributing factors, the researchers note.
Elevated risk of dying
The risk of death in adults with obesity ranged from 8% higher in the 30-34.9 range up to 61% greater for those with a BMI of 45.
Chronic inflammation or impaired lung function from excess weight are possible reasons that higher BMI imparts greater risk, the researchers note.
The CDC researchers evaluated 148,494 adults from 238 hospitals participating in PHD-SR database. Because the study was limited to people hospitalized with COVID-19, the findings may not apply to all adults with COVID-19.
Another potential limitation is that investigators were unable to calculate BMI for all patients in the database because about 28% of participating hospitals did not report height and weight.
The study authors had no relevant financial relationships to disclose.
A version of this article first appeared on Medscape.com.
Officials have previously linked being overweight or obese to a greater risk for more severe COVID-19. A report today from the U.S. Centers for Disease Control and Prevention adds numbers and some nuance to the association.
Data from nearly 150,000 U.S. adults hospitalized with COVID-19 nationwide indicate that risk for more severe disease outcomes increases along with body mass index (BMI). The risk of COVID-19–related hospitalization and death associated with obesity was particularly high among people younger than 65.
“As clinicians develop care plans for COVID-19 patients, they should consider the risk for severe outcomes in patients with higher BMIs, especially for those with severe obesity,” the researchers note. They add that their findings suggest “progressively intensive management of COVID-19 might be needed for patients with more severe obesity.”
People with COVID-19 close to the border between a healthy and overweight BMI – from 23.7 kg/m2 to 25.9 kg/m2 – had the lowest risks for adverse outcomes.
The study was published online today in Morbidity and Mortality Weekly Report.
Greater need for critical care
The risk of ICU admission was particularly associated with severe obesity. For example, those with a BMI in the 40-44.9 kg/m2 category had a 6% increased risk, which jumped to 16% higher among those with a BMI of 45 or greater.
Compared to people with a healthy BMI, the need for invasive mechanical ventilation was 12% more likely among overweight adults with a BMI of 25-29.2. The risked jumped to 108% greater among the most obese people, those with a BMI of 45 or greater, lead CDC researcher Lyudmyla Kompaniyets, PhD, and colleagues reported.
Moreover, the risks for hospitalization and death increased in a dose-response relationship with obesity.
For example, risks of being hospitalized were 7% greater for adults with a BMI between 30 and 34.9 and climbed to 33% greater for those with a BMI of 45. Risks were calculated as adjusted relative risks compared with people with a healthy BMI between 18.5 and 24.9.
Interestingly, being underweight was associated with elevated risk for COVID-19 hospitalization as well. For example, people with a BMI of less than 18.5 had a 20% greater chance of admission vs. people in the healthy BMI range. Unknown underlying medical conditions or issues related to nutrition or immune function could be contributing factors, the researchers note.
Elevated risk of dying
The risk of death in adults with obesity ranged from 8% higher in the 30-34.9 range up to 61% greater for those with a BMI of 45.
Chronic inflammation or impaired lung function from excess weight are possible reasons that higher BMI imparts greater risk, the researchers note.
The CDC researchers evaluated 148,494 adults from 238 hospitals participating in PHD-SR database. Because the study was limited to people hospitalized with COVID-19, the findings may not apply to all adults with COVID-19.
Another potential limitation is that investigators were unable to calculate BMI for all patients in the database because about 28% of participating hospitals did not report height and weight.
The study authors had no relevant financial relationships to disclose.
A version of this article first appeared on Medscape.com.
FDA authorizes first molecular at-home, OTC COVID-19 test
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has granted emergency use authorization (EUA) for the Cue COVID-19 Test for Home and Over The Counter Use (Cue OTC Test, Cue Health).
The Cue OTC Test is the first molecular diagnostic test available to consumers without a prescription.
The test detects genetic material from SARS-CoV-2 present in the nostrils and delivers results in about 20 minutes to the user’s mobile smart device via the Cue Health app.
In testing, the Cue OTC Test correctly identified 96% of positive nasal swab samples from individuals known to have symptoms and correctly identified 100% of positive samples from individuals without symptoms.
The test is intended for use in people aged 2 years and older with and without symptoms.
“With this authorization, consumers can purchase and self-administer one of the easiest, fastest, and most accurate tests without a prescription,” Clint Sever, cofounder and chief product officer of Cue Health, said in a news release.
“This FDA authorization will help us improve patient outcomes with a solution that provides the accuracy of central lab tests, with the speed and accessibility required to address emergent global health issues,” he said.
Cue Health expects to produce more than 100,000 single-use test kits per day by this summer. Dena Cook, the company’s chief communications officer, told this news organization that the company hasn’t announced pricing information yet, but the price will be “comparable” to other price points and other products on the market.
“The FDA continues to prioritize the availability of more at-home testing options in response to the pandemic,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
“Cue COVID-19 Test for Home and Over-the-Counter Use provides access to accurate and reliable testing at home, without a prescription. The FDA will continue to work collaboratively with test developers to advance effective testing options for doctors, clinicians, and the public,” he said.
In June, the FDA granted an EUA to Cue Health’s COVID-19 test for use in clinical and point-of-care settings.
The test is currently being used in hospitals, physicians’ offices, and dental clinics, as well as schools, essential businesses, nursing homes, and other congregate-care facilities. The test is also being distributed through a program led by the U.S. Department of Defense and the U.S. Department of Health & Human Services across several states.
A version of this article first appeared on Medscape.com.
Tocilizumab (Actemra) scores FDA approval for systemic sclerosis–associated interstitial lung disease
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.

Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.
Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
The Food and Drug Administration has approved subcutaneously-injected tocilizumab (Actemra) to reduce the rate of pulmonary function decline in systemic sclerosis–associated interstitial lung disease (SSc-ILD) patients, according to a press release from manufacturer Genentech.
Tocilizumab is the first biologic to be approved by the agency for adults with SSc-ILD, a rare and potentially life-threatening condition that may affect up to 80% of SSc patients and lead to lung inflammation and scarring.
The approval was based primarily on data from a phase 3 randomized, double-blind, placebo-controlled clinical trial (the focuSSced trial) that included 212 adults with SSc. Although that study failed to meet its primary endpoint of change from baseline to 48 weeks in the modified Rodnan Skin Score, the researchers observed a significantly reduced lung function decline as measured by forced vital capacity (FVC) and percent predicted forced vital capacity (ppFVC) among tocilizumab-treated patients, compared with those who received placebo. A total of 68 patients (65%) in the tocilizumab group and 68 patients (64%) in the placebo group had SSc-ILD at baseline.
In a subgroup analysis, patients taking tocilizumab had a smaller decline in mean ppFVC, compared with placebo patients (0.07% vs. –6.4%; mean difference, 6.47%), and a smaller decline in FVC (mean change –14 mL vs. –255 mL with placebo; mean difference, 241 mL).
The mean change from baseline to week 48 in modified Rodnan Skin Score was –5.88 for patients on tocilizumab and –3.77 with placebo.
Safety data were similar between tocilizumab and placebo groups through 48 weeks, and similar for patients with and without SSc-ILD. In general, tocilizumab side effects include increased susceptibility to infections, and serious side effects may include stomach tears, hepatotoxicity, and increased risk of cancer and hepatitis B, according to the prescribing information. However, the most common side effects are upper respiratory tract infections, headache, hypertension, and injection-site reactions.
Tocilizumab, an interleukin-6 receptor antagonist, is already approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis, as well as for adult patients with giant cell arteritis; patients aged 2 years and older with active polyarticular juvenile idiopathic arthritis or active systemic juvenile idiopathic arthritis; and adults and pediatric patients 2 years of age and older with chimeric antigen receptor T-cell–induced severe or life-threatening cytokine release syndrome.
Prescribing information is available here.
U.S. suicide rate in 2019 took first downturn in 14 years
In 2019, the U.S. suicide rate dropped for the first time in 14 years, driven largely by a significant decline in firearm-related deaths, according to a new analysis of National Vital Statistics System data.
Since firearms are the “most common and most lethal” mechanism of suicide, the drop in deaths is “particularly encouraging,” Deborah M. Stone, ScD, MSW, MPH, and associates wrote in the Morbidity and Mortality Weekly Report.
The national suicide rate decreased from 14.2 per 100,000 population in 2018 to 13.9 per 100,000 in 2019, a statistically significant drop of 2.1% that reversed a 20-year trend that saw the rate increase by 33% since 1999, they said.
The rate for firearm use, which is involved in half of all suicides, declined from 7.0 per 100,000 to 6.8, for a significant change of 2.9%, said Dr. Stone and associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control.
The only other method with a drop in suicide rate from 2018 to 2019 was suffocation – the second most common mechanism of injury – but the relative change of 2.3% was not significant, they noted.
Significant declines also occurred in several subgroups: Whites; those aged 15-24, 55-64, and 65-74 years; and those living in counties classified as large fringe metropolitan or micropolitan (urban cluster of ≥ 10,000 but less than 50,000 population), they said, based on data from the National Vital Statistics System.
the investigators wrote.
The states with significant increases were Hawaii (30.3%) and Nebraska (20.1%), while declines in the suicide rate were significant in five states – Idaho, Indiana, Massachusetts, North Carolina, and Virginia, Dr. Stone and associates reported. Altogether, the rate fell in 31 states, increased in 18, and did not change in 2.
The significance of those changes varied between males and females. Declines were significant for females in Indiana, Massachusetts, and Washington, and for males in Florida, Kentucky, Massachusetts, North Carolina, and West Virginia. Minnesota was the only state with a significant increase among females, with Hawaii and Wyoming posting increases for males, they said.
As the response to the COVID-19 pandemic continues, the investigators pointed out, “prevention is more important than ever. Past research indicates that suicide rates remain stable or decline during infrastructure disruption (e.g., natural disasters), only to rise afterwards as the longer-term sequelae unfold in persons, families, and communities.”
In 2019, the U.S. suicide rate dropped for the first time in 14 years, driven largely by a significant decline in firearm-related deaths, according to a new analysis of National Vital Statistics System data.
Since firearms are the “most common and most lethal” mechanism of suicide, the drop in deaths is “particularly encouraging,” Deborah M. Stone, ScD, MSW, MPH, and associates wrote in the Morbidity and Mortality Weekly Report.
The national suicide rate decreased from 14.2 per 100,000 population in 2018 to 13.9 per 100,000 in 2019, a statistically significant drop of 2.1% that reversed a 20-year trend that saw the rate increase by 33% since 1999, they said.
The rate for firearm use, which is involved in half of all suicides, declined from 7.0 per 100,000 to 6.8, for a significant change of 2.9%, said Dr. Stone and associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control.
The only other method with a drop in suicide rate from 2018 to 2019 was suffocation – the second most common mechanism of injury – but the relative change of 2.3% was not significant, they noted.
Significant declines also occurred in several subgroups: Whites; those aged 15-24, 55-64, and 65-74 years; and those living in counties classified as large fringe metropolitan or micropolitan (urban cluster of ≥ 10,000 but less than 50,000 population), they said, based on data from the National Vital Statistics System.
the investigators wrote.
The states with significant increases were Hawaii (30.3%) and Nebraska (20.1%), while declines in the suicide rate were significant in five states – Idaho, Indiana, Massachusetts, North Carolina, and Virginia, Dr. Stone and associates reported. Altogether, the rate fell in 31 states, increased in 18, and did not change in 2.
The significance of those changes varied between males and females. Declines were significant for females in Indiana, Massachusetts, and Washington, and for males in Florida, Kentucky, Massachusetts, North Carolina, and West Virginia. Minnesota was the only state with a significant increase among females, with Hawaii and Wyoming posting increases for males, they said.
As the response to the COVID-19 pandemic continues, the investigators pointed out, “prevention is more important than ever. Past research indicates that suicide rates remain stable or decline during infrastructure disruption (e.g., natural disasters), only to rise afterwards as the longer-term sequelae unfold in persons, families, and communities.”
In 2019, the U.S. suicide rate dropped for the first time in 14 years, driven largely by a significant decline in firearm-related deaths, according to a new analysis of National Vital Statistics System data.
Since firearms are the “most common and most lethal” mechanism of suicide, the drop in deaths is “particularly encouraging,” Deborah M. Stone, ScD, MSW, MPH, and associates wrote in the Morbidity and Mortality Weekly Report.
The national suicide rate decreased from 14.2 per 100,000 population in 2018 to 13.9 per 100,000 in 2019, a statistically significant drop of 2.1% that reversed a 20-year trend that saw the rate increase by 33% since 1999, they said.
The rate for firearm use, which is involved in half of all suicides, declined from 7.0 per 100,000 to 6.8, for a significant change of 2.9%, said Dr. Stone and associates at the Centers for Disease Control and Prevention’s National Center for Injury Prevention and Control.
The only other method with a drop in suicide rate from 2018 to 2019 was suffocation – the second most common mechanism of injury – but the relative change of 2.3% was not significant, they noted.
Significant declines also occurred in several subgroups: Whites; those aged 15-24, 55-64, and 65-74 years; and those living in counties classified as large fringe metropolitan or micropolitan (urban cluster of ≥ 10,000 but less than 50,000 population), they said, based on data from the National Vital Statistics System.
the investigators wrote.
The states with significant increases were Hawaii (30.3%) and Nebraska (20.1%), while declines in the suicide rate were significant in five states – Idaho, Indiana, Massachusetts, North Carolina, and Virginia, Dr. Stone and associates reported. Altogether, the rate fell in 31 states, increased in 18, and did not change in 2.
The significance of those changes varied between males and females. Declines were significant for females in Indiana, Massachusetts, and Washington, and for males in Florida, Kentucky, Massachusetts, North Carolina, and West Virginia. Minnesota was the only state with a significant increase among females, with Hawaii and Wyoming posting increases for males, they said.
As the response to the COVID-19 pandemic continues, the investigators pointed out, “prevention is more important than ever. Past research indicates that suicide rates remain stable or decline during infrastructure disruption (e.g., natural disasters), only to rise afterwards as the longer-term sequelae unfold in persons, families, and communities.”
FROM MMWR
Pembrolizumab SCLC indication withdrawn in U.S.
The move does not affect any of the drug’s other indications. The immunotherapy is used in the treatment of many different types of cancer.
The SCLC indication had been granted an accelerated approval by the Food and Drug Administration in 2019 based on tumor response rate and durability of response data from patient cohorts in two trials. However, the anti-PD-1 therapy failed to demonstrate statistically significant improved overall survival in a confirmatory trial, which is mandated after an accelerated approval.
The FDA is conducting “an industry-wide evaluation of indications based on accelerated approvals that have not yet met their postmarketing requirements,” said Merck.
In February of 2021, an indication for durvalumab (Imfinzi) was withdrawn by AstraZeneca in concert with the FDA after the drug failed to improve overall survival in unresectable metastatic bladder cancer in a confirmatory trial, as reported by Medscape Medical News.
“We will continue to rigorously evaluate the benefits of [pembrolizumab] in small cell lung cancer and other types of cancer, in pursuit of Merck’s mission to save and improve lives,” Roy Baynes, MD, chief medical officer, Merck Research Laboratories, said in the company statement
Dr. Baynes also championed the value of accelerated approvals.
“The accelerated pathways created by the FDA have been integral to the remarkable progress in oncology care over the past 5 years and have helped many cancer patients with advanced disease, including small cell lung cancer, access new treatments,” he said.
However, in the past, the FDA has been criticized for approving new cancer drugs based on surrogate markers such as response rates because, in many cases, subsequent studies often show that the drug fails to improve overall survival.
For example, a 2015 study found that 36 (67%) of 54 cancer drug approvals from 2008 to 2012 were made on the basis of surrogate markers – either tumor response rate or progression-free survival. Over a median follow-up period of 4.4 years, only 5 of those 36 drugs were shown in randomized studies to improve overall survival, as reported by Medscape Medical News.
The FDA says that it instituted the accelerated approval program to “allow for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint.” The program was started in 1992, in the midst of the HIV/AIDS epidemic.
In 2020, the nonprofit Friends of Cancer Research issued a white paper calling for reform in the accelerated approval process, which included a proposal to add risk assessment to surrogate endpoints that would factor in variables such as toxicity.
A version of this article first appeared on Medscape.com.
The move does not affect any of the drug’s other indications. The immunotherapy is used in the treatment of many different types of cancer.
The SCLC indication had been granted an accelerated approval by the Food and Drug Administration in 2019 based on tumor response rate and durability of response data from patient cohorts in two trials. However, the anti-PD-1 therapy failed to demonstrate statistically significant improved overall survival in a confirmatory trial, which is mandated after an accelerated approval.
The FDA is conducting “an industry-wide evaluation of indications based on accelerated approvals that have not yet met their postmarketing requirements,” said Merck.
In February of 2021, an indication for durvalumab (Imfinzi) was withdrawn by AstraZeneca in concert with the FDA after the drug failed to improve overall survival in unresectable metastatic bladder cancer in a confirmatory trial, as reported by Medscape Medical News.
“We will continue to rigorously evaluate the benefits of [pembrolizumab] in small cell lung cancer and other types of cancer, in pursuit of Merck’s mission to save and improve lives,” Roy Baynes, MD, chief medical officer, Merck Research Laboratories, said in the company statement
Dr. Baynes also championed the value of accelerated approvals.
“The accelerated pathways created by the FDA have been integral to the remarkable progress in oncology care over the past 5 years and have helped many cancer patients with advanced disease, including small cell lung cancer, access new treatments,” he said.
However, in the past, the FDA has been criticized for approving new cancer drugs based on surrogate markers such as response rates because, in many cases, subsequent studies often show that the drug fails to improve overall survival.
For example, a 2015 study found that 36 (67%) of 54 cancer drug approvals from 2008 to 2012 were made on the basis of surrogate markers – either tumor response rate or progression-free survival. Over a median follow-up period of 4.4 years, only 5 of those 36 drugs were shown in randomized studies to improve overall survival, as reported by Medscape Medical News.
The FDA says that it instituted the accelerated approval program to “allow for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint.” The program was started in 1992, in the midst of the HIV/AIDS epidemic.
In 2020, the nonprofit Friends of Cancer Research issued a white paper calling for reform in the accelerated approval process, which included a proposal to add risk assessment to surrogate endpoints that would factor in variables such as toxicity.
A version of this article first appeared on Medscape.com.
The move does not affect any of the drug’s other indications. The immunotherapy is used in the treatment of many different types of cancer.
The SCLC indication had been granted an accelerated approval by the Food and Drug Administration in 2019 based on tumor response rate and durability of response data from patient cohorts in two trials. However, the anti-PD-1 therapy failed to demonstrate statistically significant improved overall survival in a confirmatory trial, which is mandated after an accelerated approval.
The FDA is conducting “an industry-wide evaluation of indications based on accelerated approvals that have not yet met their postmarketing requirements,” said Merck.
In February of 2021, an indication for durvalumab (Imfinzi) was withdrawn by AstraZeneca in concert with the FDA after the drug failed to improve overall survival in unresectable metastatic bladder cancer in a confirmatory trial, as reported by Medscape Medical News.
“We will continue to rigorously evaluate the benefits of [pembrolizumab] in small cell lung cancer and other types of cancer, in pursuit of Merck’s mission to save and improve lives,” Roy Baynes, MD, chief medical officer, Merck Research Laboratories, said in the company statement
Dr. Baynes also championed the value of accelerated approvals.
“The accelerated pathways created by the FDA have been integral to the remarkable progress in oncology care over the past 5 years and have helped many cancer patients with advanced disease, including small cell lung cancer, access new treatments,” he said.
However, in the past, the FDA has been criticized for approving new cancer drugs based on surrogate markers such as response rates because, in many cases, subsequent studies often show that the drug fails to improve overall survival.
For example, a 2015 study found that 36 (67%) of 54 cancer drug approvals from 2008 to 2012 were made on the basis of surrogate markers – either tumor response rate or progression-free survival. Over a median follow-up period of 4.4 years, only 5 of those 36 drugs were shown in randomized studies to improve overall survival, as reported by Medscape Medical News.
The FDA says that it instituted the accelerated approval program to “allow for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need based on a surrogate endpoint.” The program was started in 1992, in the midst of the HIV/AIDS epidemic.
In 2020, the nonprofit Friends of Cancer Research issued a white paper calling for reform in the accelerated approval process, which included a proposal to add risk assessment to surrogate endpoints that would factor in variables such as toxicity.
A version of this article first appeared on Medscape.com.
Bladder cancer indication withdrawn for durvalumab
The change does not affect this indication outside the United States, nor does it affect other approved durvalumab indications within the United States.
For example, durvalumab remains approved by the Food and Drug Administration in the curative-intent setting of unresectable, stage III non–small cell lung cancer after chemoradiotherapy and for the treatment of extensive-stage small cell lung cancer.
AstraZeneca is continuing with clinical trials of durvalumab in various combinations for the treatment of bladder cancer.
Granted accelerated approval
Durvalumab was granted accelerated approval in May 2017 by the FDA specifically for the treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing chemotherapy or who experience disease progression within 12 months of neoadjuvant or adjuvant treatment with that chemotherapy.
That accelerated approval was based on the surrogate markers of tumor response rate and duration of response from Study 1108, a phase 1/2 trial. In this trial, the overall response rate was 17.8% in a cohort of 191 patients with locally advanced or metastatic urothelial cancer that had progressed during or after a platinum-based regimen.
However, in the confirmatory phase 3 DANUBE trial in patients with unresectable metastatic bladder cancer, neither durvalumab nor durvalumab plus tremelimumab met the primary endpoint of improving overall survival in comparison with standard-of-care chemotherapy.
“While the withdrawal in previously treated metastatic bladder cancer is disappointing, we respect the principles FDA set out when the accelerated approval pathway was founded,” Dave Fredrickson, executive vice president, Oncology Business Unit, AstraZeneca, said in a company press statement.
A version of this article first appeared on Medscape.com.
The change does not affect this indication outside the United States, nor does it affect other approved durvalumab indications within the United States.
For example, durvalumab remains approved by the Food and Drug Administration in the curative-intent setting of unresectable, stage III non–small cell lung cancer after chemoradiotherapy and for the treatment of extensive-stage small cell lung cancer.
AstraZeneca is continuing with clinical trials of durvalumab in various combinations for the treatment of bladder cancer.
Granted accelerated approval
Durvalumab was granted accelerated approval in May 2017 by the FDA specifically for the treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing chemotherapy or who experience disease progression within 12 months of neoadjuvant or adjuvant treatment with that chemotherapy.
That accelerated approval was based on the surrogate markers of tumor response rate and duration of response from Study 1108, a phase 1/2 trial. In this trial, the overall response rate was 17.8% in a cohort of 191 patients with locally advanced or metastatic urothelial cancer that had progressed during or after a platinum-based regimen.
However, in the confirmatory phase 3 DANUBE trial in patients with unresectable metastatic bladder cancer, neither durvalumab nor durvalumab plus tremelimumab met the primary endpoint of improving overall survival in comparison with standard-of-care chemotherapy.
“While the withdrawal in previously treated metastatic bladder cancer is disappointing, we respect the principles FDA set out when the accelerated approval pathway was founded,” Dave Fredrickson, executive vice president, Oncology Business Unit, AstraZeneca, said in a company press statement.
A version of this article first appeared on Medscape.com.
The change does not affect this indication outside the United States, nor does it affect other approved durvalumab indications within the United States.
For example, durvalumab remains approved by the Food and Drug Administration in the curative-intent setting of unresectable, stage III non–small cell lung cancer after chemoradiotherapy and for the treatment of extensive-stage small cell lung cancer.
AstraZeneca is continuing with clinical trials of durvalumab in various combinations for the treatment of bladder cancer.
Granted accelerated approval
Durvalumab was granted accelerated approval in May 2017 by the FDA specifically for the treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing chemotherapy or who experience disease progression within 12 months of neoadjuvant or adjuvant treatment with that chemotherapy.
That accelerated approval was based on the surrogate markers of tumor response rate and duration of response from Study 1108, a phase 1/2 trial. In this trial, the overall response rate was 17.8% in a cohort of 191 patients with locally advanced or metastatic urothelial cancer that had progressed during or after a platinum-based regimen.
However, in the confirmatory phase 3 DANUBE trial in patients with unresectable metastatic bladder cancer, neither durvalumab nor durvalumab plus tremelimumab met the primary endpoint of improving overall survival in comparison with standard-of-care chemotherapy.
“While the withdrawal in previously treated metastatic bladder cancer is disappointing, we respect the principles FDA set out when the accelerated approval pathway was founded,” Dave Fredrickson, executive vice president, Oncology Business Unit, AstraZeneca, said in a company press statement.
A version of this article first appeared on Medscape.com.
J&J COVID-19 vaccine wins unanimous backing of FDA panel

The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.
FDA approves first targeted treatment for rare DMD mutation
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.
, the agency has announced.
This particular mutation of the DMD gene “is amenable to exon 45 skipping,” the FDA noted in a press release. The agency added that this is its first approval of a targeted treatment for patients with the mutation.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” Eric Bastings, MD, deputy director of the Office of Neuroscience at the FDA’s Center for Drug Evaluation and Research, said in a statement.
The approval was based on results from a 43-person randomized controlled trial. Patients who received casimersen had a greater increase in production of the muscle-fiber protein dystrophin compared with their counterparts who received placebo.
Approved – with cautions
The FDA noted that DMD prevalence worldwide is about 1 in 3,600 boys – although it can also affect girls in rare cases. Symptoms of the disorder are commonly first observed around age 3 years but worsen steadily over time. DMD gene mutations lead to a decrease in dystrophin.
As reported by Medscape Medical News in August, the FDA approved viltolarsen (Viltepso, NS Pharma) for the treatment of DMD in patients with a confirmed mutation amenable to exon 53 skipping, following approval of golodirsen injection (Vyondys 53, Sarepta Therapeutics) for the same indication in December 2019.
The DMD gene mutation that is amenable to exon 45 skipping is present in about 8% of patients with DMD.
The trial that carried weight with the FDA included 43 male participants with DMD aged 7-20 years. All were confirmed to have the exon 45-skipping gene mutation and all were randomly assigned 2:1 to received IV casimersen 30 mg/kg or matching placebo.
Results showed that, between baseline and 48 weeks post treatment, the casimersen group showed a significantly higher increase in levels of dystrophin protein than in the placebo group.
Upper respiratory tract infections, fever, joint and throat pain, headache, and cough were the most common adverse events experienced by the active-treatment group.
Although the clinical studies assessing casimersen did not show any reports of kidney toxicity, the adverse event was observed in some nonclinical studies. Therefore, clinicians should monitor kidney function in any patient receiving this treatment, the FDA recommended.
Overall, “the FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit” in this patient population, the agency said in its press release.
However, it noted that definitive clinical benefits such as improved motor function were not “established.”
“In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of [other] available therapy,” the agency said.
It added that the manufacturer is currently conducting a multicenter study focused on the safety and efficacy of the drug in ambulatory patients with DMD.
The FDA approved casimersen using its Accelerated Approval pathway, granted Fast Track and Priority Review designations to its applications, and gave the treatment Orphan Drug designation.
A version of this article first appeared on Medscape.com.