User login
FDA approves voclosporin for lupus nephritis
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.

Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.
Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
The Food and Drug Administration has approved voclosporin (Lupkynis) for the treatment of lupus nephritis, according to a Jan. 22 press release from manufacturer Aurinia Pharmaceuticals.
Lupkynis is a calcineurin-inhibitor immunosuppressant, and is the first oral medication to show effectiveness in lupus nephritis, according to the company. The drug is indicated for the treatment of adult patients with active lupus nephritis in combination with a background immunosuppressive therapy regimen, according to the drug label, which also has a boxed warning describing the increased risk of infections and malignancies, including lymphoma.
The approval of voclosporin was based on data from two studies, the AURORA phase 3 study and the AURA-LV phase 2 study. The studies included 533 adults with lupus nephritis who were randomized to 23.7 mg or placebo of voclosporin twice daily in the form of oral capsules, or placebo capsules, in addition to standard of care (mycophenolate mofetil plus low-dose glucocorticoids).
In the AURORA phase 3 study of 357 patients, close to twice as many patients in the treatment group showed a complete renal response, compared with the placebo group after 1 year (40.8% vs. 22.5%). In addition, patients treated with voclosporin more quickly achieved a significant reduction in urine protein to creatinine ratio, compared with the placebo patients (169 days vs. 372 days).
Severe adverse events were similar between the groups, including the most common complication of infection (10.1% and 11.2% for voclosporin and control groups, respectively). Other adverse reactions reported in at least 3% of the study participants included a decrease in glomerular filtration rate, hypertension, diarrhea, headache, anemia, cough, urinary tract infection, upper abdominal pain, dyspepsia, alopecia, renal impairment, abdominal pain, mouth ulceration, fatigue, tremor, acute kidney injury, and decreased appetite, according to the company press release.
Full clinical trial information for the AURORA study is available here.
Anaphylaxis cases after COVID-19 vaccine rising but still rare: CDC
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
Health care providers should be ready to treat rare cases of anaphylaxis following administration of COVID-19 vaccines, federal medical officials have urged. The officials also stressed the importance of continuing vaccinations, despite reports of the rare side effect.
There have been 29 cases of anaphylaxis to date following administration of a COVID-19 vaccine, officials from the Centers for Disease Control and Prevention said in a call with reporters on Jan. 6.
The severe allergic reaction, which appears to be rare, can happen with either the Pfizer-BioNTech vaccine or the rival Moderna product. The Food and Drug Administration granted emergency use authorizations for these two vaccines in December.
Even with the cases seen to date, the COVID-19 vaccines remain a “good value proposition,” Nancy Messonnier, MD, director of the CDC’s National Center for Immunization, said in the call.
There have been about 11.1 cases of anaphylaxis per million doses with the Pfizer-BioNTech COVID-19 vaccine, which is higher than the estimated 1.3 cases per million doses with influenza vaccines, she said. But the low risk of anaphylaxis must be balanced against the threat of COVID-19, which currently claims about 2,000 lives a day in the United States, she said. In addition, many people are reporting long-term complications with COVID-19 even if they recover.
Kept in context, the data on anaphylaxis should not scare people away from getting a COVID-19 vaccine, she added.
“Their risk from COVID and poor outcomes is still more than the risk of a severe outcome from the vaccine,” Dr. Messonnier said. “And fortunately, we know how to treat anaphylaxis.”
Dr. Messonnier urged health care workers administering COVID-19 vaccines to be prepared.
“Anybody administering vaccines needs not just to have the EpiPen available, but frankly, to know how to use it,” Dr. Messonnier said.
MMWR details
The CDC on Jan. 6 also provided an update on anaphylaxis in Morbidity and Mortality Weekly Report (MMWR).
The information included in the report was based on cases reported with the Pfizer-BioNTech vaccine – the first to get emergency use authorization from the FDA. On the call with reporters, CDC officials confirmed there have been additional reports since then and anaphylaxis has been reported with both the Pfizer-BioNTech and Moderna vaccines. CDC officials said they could not give a breakdown of how many cases were linked to each of these products at this time.
Between Dec. 14 and 23, 2020, monitoring by the Vaccine Adverse Event Reporting System detected 21 cases of anaphylaxis after administration of a reported 1,893,360 first doses of the Pfizer-BioNTech COVID-19 vaccine. Most reactions – 71% – occurred within 15 minutes of vaccination.
A version of this article first appeared on Medscape.com.
FDA warns about risk for false negatives from Curative COVID test
which is being used in Los Angeles and other large metropolitan areas in the United States.
The real-time reverse transcription polymerase chain reaction (PCR) test was developed by Menlo Park, Calif.–based health care start-up Curative. Results are analyzed by the company’s clinical lab, KorvaLabs. The test, which is authorized for prescription use only, received emergency-use authorization from the FDA on April 16, 2020. By Nov. 9, the company had processed 6 million test results, according to the company.
The FDA alert cautions that false negative results from any COVID-19 test can lead to delays in or the lack of supportive treatment and increase the risk for viral spread.
To mitigate the risk for false negatives, the agency advises clinicians to perform the Curative test as described in the product’s Fact Sheet for Healthcare Providers. This includes limiting its use to people who have had COVID-19 symptoms for 14 days or less. “Consider retesting your patients using a different test if you suspect an inaccurate result was given recently by the Curative SARS-Cov-2 test,” the FDA alert stated. “If testing was performed more than 2 weeks ago, and there is no reason to suspect current SARS-CoV-2 infection, it is not necessary to retest.”
The alert also notes that a negative result from the Curative PCR test “does not rule out COVID-19 and should not be used as the sole basis for treatment or patient management decisions. A negative result does not exclude the possibility of COVID-19.”
According to a press release issued by Curative on Oct. 7, its PCR test is being used by the Department of Defense, as well as the states of Alaska, California, Colorado, Delaware, Florida, Georgia (Atlanta and Savannah), Illinois (Chicago), Louisiana, Texas, and Wyoming. The company also operates Clinical Laboratory Improvement Amendments–certified laboratories in San Dimas, Calif.; Washington, D.C.; and Pflugerville, Tex.
A version of this article first appeared on Medscape.com.
which is being used in Los Angeles and other large metropolitan areas in the United States.
The real-time reverse transcription polymerase chain reaction (PCR) test was developed by Menlo Park, Calif.–based health care start-up Curative. Results are analyzed by the company’s clinical lab, KorvaLabs. The test, which is authorized for prescription use only, received emergency-use authorization from the FDA on April 16, 2020. By Nov. 9, the company had processed 6 million test results, according to the company.
The FDA alert cautions that false negative results from any COVID-19 test can lead to delays in or the lack of supportive treatment and increase the risk for viral spread.
To mitigate the risk for false negatives, the agency advises clinicians to perform the Curative test as described in the product’s Fact Sheet for Healthcare Providers. This includes limiting its use to people who have had COVID-19 symptoms for 14 days or less. “Consider retesting your patients using a different test if you suspect an inaccurate result was given recently by the Curative SARS-Cov-2 test,” the FDA alert stated. “If testing was performed more than 2 weeks ago, and there is no reason to suspect current SARS-CoV-2 infection, it is not necessary to retest.”
The alert also notes that a negative result from the Curative PCR test “does not rule out COVID-19 and should not be used as the sole basis for treatment or patient management decisions. A negative result does not exclude the possibility of COVID-19.”
According to a press release issued by Curative on Oct. 7, its PCR test is being used by the Department of Defense, as well as the states of Alaska, California, Colorado, Delaware, Florida, Georgia (Atlanta and Savannah), Illinois (Chicago), Louisiana, Texas, and Wyoming. The company also operates Clinical Laboratory Improvement Amendments–certified laboratories in San Dimas, Calif.; Washington, D.C.; and Pflugerville, Tex.
A version of this article first appeared on Medscape.com.
which is being used in Los Angeles and other large metropolitan areas in the United States.
The real-time reverse transcription polymerase chain reaction (PCR) test was developed by Menlo Park, Calif.–based health care start-up Curative. Results are analyzed by the company’s clinical lab, KorvaLabs. The test, which is authorized for prescription use only, received emergency-use authorization from the FDA on April 16, 2020. By Nov. 9, the company had processed 6 million test results, according to the company.
The FDA alert cautions that false negative results from any COVID-19 test can lead to delays in or the lack of supportive treatment and increase the risk for viral spread.
To mitigate the risk for false negatives, the agency advises clinicians to perform the Curative test as described in the product’s Fact Sheet for Healthcare Providers. This includes limiting its use to people who have had COVID-19 symptoms for 14 days or less. “Consider retesting your patients using a different test if you suspect an inaccurate result was given recently by the Curative SARS-Cov-2 test,” the FDA alert stated. “If testing was performed more than 2 weeks ago, and there is no reason to suspect current SARS-CoV-2 infection, it is not necessary to retest.”
The alert also notes that a negative result from the Curative PCR test “does not rule out COVID-19 and should not be used as the sole basis for treatment or patient management decisions. A negative result does not exclude the possibility of COVID-19.”
According to a press release issued by Curative on Oct. 7, its PCR test is being used by the Department of Defense, as well as the states of Alaska, California, Colorado, Delaware, Florida, Georgia (Atlanta and Savannah), Illinois (Chicago), Louisiana, Texas, and Wyoming. The company also operates Clinical Laboratory Improvement Amendments–certified laboratories in San Dimas, Calif.; Washington, D.C.; and Pflugerville, Tex.
A version of this article first appeared on Medscape.com.
CDC issues COVID-19 vaccine guidance for underlying conditions
The Centers for Disease Control and Prevention has issued updated guidance for people with underlying medical conditions who are considering getting the coronavirus vaccine.
“Adults of any age with certain underlying medical conditions are at increased risk for severe illness from the virus that causes COVID-19,” the CDC said in the guidance, posted on Dec. 26. “mRNA COVID-19 vaccines may be administered to people with underlying medical conditions provided they have not had a severe allergic reaction to any of the ingredients in the vaccine.”
Both the Pfizer and Moderna vaccines use mRNA, or messenger RNA.
The CDC guidance had specific information for people with HIV, weakened immune systems, and autoimmune conditions such as Guillain-Barré syndrome (GBS) and Bell’s palsy who are thinking of getting the vaccine.
People with HIV and weakened immune systems “may receive a COVID-19 vaccine. However, they should be aware of the limited safety data,” the CDC said.
There’s no information available yet about the safety of the vaccines for people with weakened immune systems. People with HIV were included in clinical trials, but “safety data specific to this group are not yet available at this time,” the CDC said.
Cases of Bell’s palsy, a temporary facial paralysis, were reported in people receiving the Pfizer and Moderna vaccines in clinical trials, the Food and Drug Administration said Dec. 17.
But the new CDC guidance said that the FDA “does not consider these to be above the rate expected in the general population. They have not concluded these cases were caused by vaccination. Therefore, persons who have previously had Bell’s palsy may receive an mRNA COVID-19 vaccine.”
Researchers have determined the vaccines are safe for people with GBS, a rare autoimmune disorder in which the body’s immune system attacks nerves just as they leave the spinal cord, the CDC said.
“To date, no cases of GBS have been reported following vaccination among participants in the mRNA COVID-19 vaccine clinical trials,” the CDC guidance said. “With few exceptions, the independent Advisory Committee on Immunization Practices general best practice guidelines for immunization do not include a history of GBS as a precaution to vaccination with other vaccines.”
For months, the CDC and other health authorities have said that people with certain medical conditions are at an increased risk of developing severe cases of COVID-19.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention has issued updated guidance for people with underlying medical conditions who are considering getting the coronavirus vaccine.
“Adults of any age with certain underlying medical conditions are at increased risk for severe illness from the virus that causes COVID-19,” the CDC said in the guidance, posted on Dec. 26. “mRNA COVID-19 vaccines may be administered to people with underlying medical conditions provided they have not had a severe allergic reaction to any of the ingredients in the vaccine.”
Both the Pfizer and Moderna vaccines use mRNA, or messenger RNA.
The CDC guidance had specific information for people with HIV, weakened immune systems, and autoimmune conditions such as Guillain-Barré syndrome (GBS) and Bell’s palsy who are thinking of getting the vaccine.
People with HIV and weakened immune systems “may receive a COVID-19 vaccine. However, they should be aware of the limited safety data,” the CDC said.
There’s no information available yet about the safety of the vaccines for people with weakened immune systems. People with HIV were included in clinical trials, but “safety data specific to this group are not yet available at this time,” the CDC said.
Cases of Bell’s palsy, a temporary facial paralysis, were reported in people receiving the Pfizer and Moderna vaccines in clinical trials, the Food and Drug Administration said Dec. 17.
But the new CDC guidance said that the FDA “does not consider these to be above the rate expected in the general population. They have not concluded these cases were caused by vaccination. Therefore, persons who have previously had Bell’s palsy may receive an mRNA COVID-19 vaccine.”
Researchers have determined the vaccines are safe for people with GBS, a rare autoimmune disorder in which the body’s immune system attacks nerves just as they leave the spinal cord, the CDC said.
“To date, no cases of GBS have been reported following vaccination among participants in the mRNA COVID-19 vaccine clinical trials,” the CDC guidance said. “With few exceptions, the independent Advisory Committee on Immunization Practices general best practice guidelines for immunization do not include a history of GBS as a precaution to vaccination with other vaccines.”
For months, the CDC and other health authorities have said that people with certain medical conditions are at an increased risk of developing severe cases of COVID-19.
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention has issued updated guidance for people with underlying medical conditions who are considering getting the coronavirus vaccine.
“Adults of any age with certain underlying medical conditions are at increased risk for severe illness from the virus that causes COVID-19,” the CDC said in the guidance, posted on Dec. 26. “mRNA COVID-19 vaccines may be administered to people with underlying medical conditions provided they have not had a severe allergic reaction to any of the ingredients in the vaccine.”
Both the Pfizer and Moderna vaccines use mRNA, or messenger RNA.
The CDC guidance had specific information for people with HIV, weakened immune systems, and autoimmune conditions such as Guillain-Barré syndrome (GBS) and Bell’s palsy who are thinking of getting the vaccine.
People with HIV and weakened immune systems “may receive a COVID-19 vaccine. However, they should be aware of the limited safety data,” the CDC said.
There’s no information available yet about the safety of the vaccines for people with weakened immune systems. People with HIV were included in clinical trials, but “safety data specific to this group are not yet available at this time,” the CDC said.
Cases of Bell’s palsy, a temporary facial paralysis, were reported in people receiving the Pfizer and Moderna vaccines in clinical trials, the Food and Drug Administration said Dec. 17.
But the new CDC guidance said that the FDA “does not consider these to be above the rate expected in the general population. They have not concluded these cases were caused by vaccination. Therefore, persons who have previously had Bell’s palsy may receive an mRNA COVID-19 vaccine.”
Researchers have determined the vaccines are safe for people with GBS, a rare autoimmune disorder in which the body’s immune system attacks nerves just as they leave the spinal cord, the CDC said.
“To date, no cases of GBS have been reported following vaccination among participants in the mRNA COVID-19 vaccine clinical trials,” the CDC guidance said. “With few exceptions, the independent Advisory Committee on Immunization Practices general best practice guidelines for immunization do not include a history of GBS as a precaution to vaccination with other vaccines.”
For months, the CDC and other health authorities have said that people with certain medical conditions are at an increased risk of developing severe cases of COVID-19.
A version of this article first appeared on Medscape.com.
Moderna’s COVID-19 vaccine deemed ‘highly effective,’ but further studies needed
The Food and Drug Administration’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) evaluated
The panel acknowledged that further studies will be required post issuance of an Emergency Use Authorization (EUA) to collect additional data on the safety and effectiveness of the vaccine. A briefing document released by the FDA on Dec. 17, 2020, summarized interim results and included recommendations from VRBPAC on use of Moderna’s mRNA-1273 COVID-19 vaccine.
“On November 30, 2020, ModernaTX (the Sponsor) submitted an EUA request to FDA for an investigational COVID-19 vaccine (mRNA-1273) intended to prevent COVID-19,” the committee wrote.
The mRNA-1273 vaccine trial
Among 30,351 individuals aged 18 years and older, the efficacy, safety, and immunogenicity of the mRNA-1273 vaccine candidate was evaluated in a randomized, stratified, observer-blind, placebo-controlled phase 3 study. Participants were randomly assigned (1:1) to receive two injections of either 100 mcg of mRNA-1273 (n = 15,181) or saline placebo (n = 15,170) administered intramuscularly on day 1 and day 29.
The primary efficacy endpoint was efficacy of mRNA-1273 against PCR-confirmed COVID-19 with onset at least 14 days following the second dose. The primary safety endpoint was to characterize the safety of the vaccine following one or two doses.
Efficacy
Among 27,817 subjects included in the first interim analysis (data cutoff: Nov. 7, 2020), 5 cases of COVID-19 with onset at least 14 days after the second dose occurred among vaccine recipients and 90 case occurred among placebo recipients, corresponding to 94.5% vaccine efficacy (95% confidence interval, 86.5%-97.8%).
“Subgroup analyses of the primary efficacy endpoint showed similar efficacy point estimates across age groups, genders, racial and ethnic groups, and participants with medical comorbidities associated with high risk of severe COVID-19,” they reported.
Data from the final scheduled analysis of the primary efficacy endpoint (data cutoff: Nov. 21, 2020; median follow-up of >2 months after dose 2), demonstrated 94.1% vaccine efficacy (95% confidence interval, 89.3%-96.8%), corresponding to 11 cases of COVID-19 in the vaccine group and 185 cases in the placebo group.
When stratified by age, the vaccine efficacy was 95.6% (95% CI, 90.6%-97.9%) for individuals 18-64 years of age and 86.4% (95% CI, 61.4%-95.5%) for those 65 years of age or older.
In addition, results from secondary analyses indicated benefit for mRNA-1273 in preventing severe COVID-19 cases, COVID-19 in those with prior SARS-CoV-2 infection, and infection after the first dose, but these data were not conclusive.
Safety
Among 30,350 subjects included in the first interim analysis (data cutoff: Nov. 11, 2020; median follow-up of 7 weeks post second dose), no specific safety concerns were observed that would prevent issuance of an EUA.
Additional safety data (data cutoff: Nov. 25, 2020; median follow-up of 9 weeks post second dose) were provided on Dec. 7, 2020, but did not change the conclusions from the first interim analysis.
The most common vaccine-related adverse reactions were injection site pain (91.6%), fatigue (68.5%), headache (63.0%), muscle pain (59.6%), joint pain (44.8%), and chills (43.4%).
“The frequency of serious adverse events (SAEs) was low (1.0% in the mRNA-1273 arm and 1.0% in the placebo arm), without meaningful imbalances between study arms,” they reported.
Myocardial infarction (0.03%), nephrolithiasis (0.02%), and cholecystitis (0.02%) were the most common SAEs that were numerically greater in the vaccine arm than the placebo arm; however, the small number of cases does not infer a casual relationship.
“The 2-dose vaccination regimen was highly effective in preventing PCR-confirmed COVID-19 occurring at least 14 days after receipt of the second dose,” the committee wrote. “[However], it is critical to continue to gather data about the vaccine even after it is made available under EUA.”
The associated phase 3 study was sponsored by ModernaTX.
SOURCE: FDA Briefing Document: Moderna COVID-19 Vaccine. FDA Vaccines and Related Biological Products Advisory Committee. Published Dec. 17, 2020.
The Food and Drug Administration’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) evaluated
The panel acknowledged that further studies will be required post issuance of an Emergency Use Authorization (EUA) to collect additional data on the safety and effectiveness of the vaccine. A briefing document released by the FDA on Dec. 17, 2020, summarized interim results and included recommendations from VRBPAC on use of Moderna’s mRNA-1273 COVID-19 vaccine.
“On November 30, 2020, ModernaTX (the Sponsor) submitted an EUA request to FDA for an investigational COVID-19 vaccine (mRNA-1273) intended to prevent COVID-19,” the committee wrote.
The mRNA-1273 vaccine trial
Among 30,351 individuals aged 18 years and older, the efficacy, safety, and immunogenicity of the mRNA-1273 vaccine candidate was evaluated in a randomized, stratified, observer-blind, placebo-controlled phase 3 study. Participants were randomly assigned (1:1) to receive two injections of either 100 mcg of mRNA-1273 (n = 15,181) or saline placebo (n = 15,170) administered intramuscularly on day 1 and day 29.
The primary efficacy endpoint was efficacy of mRNA-1273 against PCR-confirmed COVID-19 with onset at least 14 days following the second dose. The primary safety endpoint was to characterize the safety of the vaccine following one or two doses.
Efficacy
Among 27,817 subjects included in the first interim analysis (data cutoff: Nov. 7, 2020), 5 cases of COVID-19 with onset at least 14 days after the second dose occurred among vaccine recipients and 90 case occurred among placebo recipients, corresponding to 94.5% vaccine efficacy (95% confidence interval, 86.5%-97.8%).
“Subgroup analyses of the primary efficacy endpoint showed similar efficacy point estimates across age groups, genders, racial and ethnic groups, and participants with medical comorbidities associated with high risk of severe COVID-19,” they reported.
Data from the final scheduled analysis of the primary efficacy endpoint (data cutoff: Nov. 21, 2020; median follow-up of >2 months after dose 2), demonstrated 94.1% vaccine efficacy (95% confidence interval, 89.3%-96.8%), corresponding to 11 cases of COVID-19 in the vaccine group and 185 cases in the placebo group.
When stratified by age, the vaccine efficacy was 95.6% (95% CI, 90.6%-97.9%) for individuals 18-64 years of age and 86.4% (95% CI, 61.4%-95.5%) for those 65 years of age or older.
In addition, results from secondary analyses indicated benefit for mRNA-1273 in preventing severe COVID-19 cases, COVID-19 in those with prior SARS-CoV-2 infection, and infection after the first dose, but these data were not conclusive.
Safety
Among 30,350 subjects included in the first interim analysis (data cutoff: Nov. 11, 2020; median follow-up of 7 weeks post second dose), no specific safety concerns were observed that would prevent issuance of an EUA.
Additional safety data (data cutoff: Nov. 25, 2020; median follow-up of 9 weeks post second dose) were provided on Dec. 7, 2020, but did not change the conclusions from the first interim analysis.
The most common vaccine-related adverse reactions were injection site pain (91.6%), fatigue (68.5%), headache (63.0%), muscle pain (59.6%), joint pain (44.8%), and chills (43.4%).
“The frequency of serious adverse events (SAEs) was low (1.0% in the mRNA-1273 arm and 1.0% in the placebo arm), without meaningful imbalances between study arms,” they reported.
Myocardial infarction (0.03%), nephrolithiasis (0.02%), and cholecystitis (0.02%) were the most common SAEs that were numerically greater in the vaccine arm than the placebo arm; however, the small number of cases does not infer a casual relationship.
“The 2-dose vaccination regimen was highly effective in preventing PCR-confirmed COVID-19 occurring at least 14 days after receipt of the second dose,” the committee wrote. “[However], it is critical to continue to gather data about the vaccine even after it is made available under EUA.”
The associated phase 3 study was sponsored by ModernaTX.
SOURCE: FDA Briefing Document: Moderna COVID-19 Vaccine. FDA Vaccines and Related Biological Products Advisory Committee. Published Dec. 17, 2020.
The Food and Drug Administration’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) evaluated
The panel acknowledged that further studies will be required post issuance of an Emergency Use Authorization (EUA) to collect additional data on the safety and effectiveness of the vaccine. A briefing document released by the FDA on Dec. 17, 2020, summarized interim results and included recommendations from VRBPAC on use of Moderna’s mRNA-1273 COVID-19 vaccine.
“On November 30, 2020, ModernaTX (the Sponsor) submitted an EUA request to FDA for an investigational COVID-19 vaccine (mRNA-1273) intended to prevent COVID-19,” the committee wrote.
The mRNA-1273 vaccine trial
Among 30,351 individuals aged 18 years and older, the efficacy, safety, and immunogenicity of the mRNA-1273 vaccine candidate was evaluated in a randomized, stratified, observer-blind, placebo-controlled phase 3 study. Participants were randomly assigned (1:1) to receive two injections of either 100 mcg of mRNA-1273 (n = 15,181) or saline placebo (n = 15,170) administered intramuscularly on day 1 and day 29.
The primary efficacy endpoint was efficacy of mRNA-1273 against PCR-confirmed COVID-19 with onset at least 14 days following the second dose. The primary safety endpoint was to characterize the safety of the vaccine following one or two doses.
Efficacy
Among 27,817 subjects included in the first interim analysis (data cutoff: Nov. 7, 2020), 5 cases of COVID-19 with onset at least 14 days after the second dose occurred among vaccine recipients and 90 case occurred among placebo recipients, corresponding to 94.5% vaccine efficacy (95% confidence interval, 86.5%-97.8%).
“Subgroup analyses of the primary efficacy endpoint showed similar efficacy point estimates across age groups, genders, racial and ethnic groups, and participants with medical comorbidities associated with high risk of severe COVID-19,” they reported.
Data from the final scheduled analysis of the primary efficacy endpoint (data cutoff: Nov. 21, 2020; median follow-up of >2 months after dose 2), demonstrated 94.1% vaccine efficacy (95% confidence interval, 89.3%-96.8%), corresponding to 11 cases of COVID-19 in the vaccine group and 185 cases in the placebo group.
When stratified by age, the vaccine efficacy was 95.6% (95% CI, 90.6%-97.9%) for individuals 18-64 years of age and 86.4% (95% CI, 61.4%-95.5%) for those 65 years of age or older.
In addition, results from secondary analyses indicated benefit for mRNA-1273 in preventing severe COVID-19 cases, COVID-19 in those with prior SARS-CoV-2 infection, and infection after the first dose, but these data were not conclusive.
Safety
Among 30,350 subjects included in the first interim analysis (data cutoff: Nov. 11, 2020; median follow-up of 7 weeks post second dose), no specific safety concerns were observed that would prevent issuance of an EUA.
Additional safety data (data cutoff: Nov. 25, 2020; median follow-up of 9 weeks post second dose) were provided on Dec. 7, 2020, but did not change the conclusions from the first interim analysis.
The most common vaccine-related adverse reactions were injection site pain (91.6%), fatigue (68.5%), headache (63.0%), muscle pain (59.6%), joint pain (44.8%), and chills (43.4%).
“The frequency of serious adverse events (SAEs) was low (1.0% in the mRNA-1273 arm and 1.0% in the placebo arm), without meaningful imbalances between study arms,” they reported.
Myocardial infarction (0.03%), nephrolithiasis (0.02%), and cholecystitis (0.02%) were the most common SAEs that were numerically greater in the vaccine arm than the placebo arm; however, the small number of cases does not infer a casual relationship.
“The 2-dose vaccination regimen was highly effective in preventing PCR-confirmed COVID-19 occurring at least 14 days after receipt of the second dose,” the committee wrote. “[However], it is critical to continue to gather data about the vaccine even after it is made available under EUA.”
The associated phase 3 study was sponsored by ModernaTX.
SOURCE: FDA Briefing Document: Moderna COVID-19 Vaccine. FDA Vaccines and Related Biological Products Advisory Committee. Published Dec. 17, 2020.
Key clinical point: The FDA’s Vaccines and Related Biological Products Advisory Committee regarded Moderna’s COVID-19 vaccine as highly effective with a favorable safety profile, based on interim phase 3 results.
Major finding: The two-dose vaccine regimen had a low frequency of serious adverse events (1.0% each in the mRNA-1273 and placebo arms, respectively) and demonstrated 94.1% (95% CI, 89.3%-96.8%) vaccine efficacy.
Study details: A briefing document summarized interim data and recommendations from the FDA’s VRBPAC on Moderna’s mRNA-1273 COVID-19 vaccine.
Disclosures: The associated phase 3 study was sponsored by ModernaTX.
Source: FDA Briefing Document: Moderna COVID-19 Vaccine. FDA Vaccines and Related Biological Products Advisory Committee. Published Dec. 17, 2020.
Child abuse visits to EDs declined in 2020, but not admissions
but the visits in 2020 were significantly more likely to result in hospitalization, based on analysis of a national ED database.
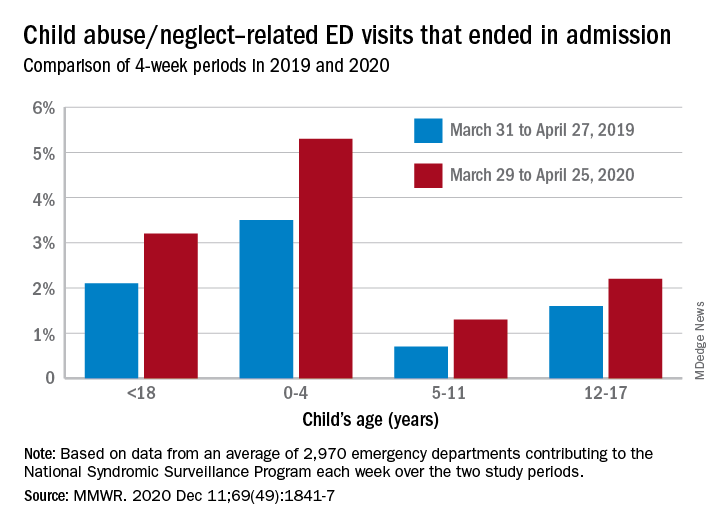
The number of ED visits involving child abuse and neglect was down by 53% during the 4-week period from March 29 to April 25, 2020, compared with the 4 weeks from March 31 to April 27, 2019. The proportion of those ED visits that ended in hospitalizations, however, increased from 2.1% in 2019 to 3.2% in 2020, Elizabeth Swedo, MD, and associates at the Centers for Disease Control and Prevention said in the Morbidity and Mortality Weekly Report.
“ED visits related to suspected or confirmed child abuse and neglect decreased beginning the week of March 15, 2020, coinciding with the declaration of a national emergency related to COVID-19 and implementation of community mitigation measures,” they wrote.
An earlier study involving the same database (the National Syndromic Surveillance Program) showed that, over the two same 4-week periods, the volume of all ED visits in 2020 was down 72% for children aged 10 years and younger and 71% for those aged 11-14 years.
In the current study, however, all age subgroups had significant increases in hospital admissions. The proportion of ED visits related to child abuse and neglect that resulted in hospitalization rose from 3.5% in 2019 to 5.3% in 2020 among ages 0-4 years, 0.7% to 1.3% for ages 5-11 years, and 1.6% to 2.2% for adolescents aged 12-17, Dr. Swedo and associates reported.
The absence of a corresponding drop in hospitalizations may be tied to risk factors related to the pandemic, “such as loss of income, increased stress related to parental child care and schooling responsibilities, and increased substance use and mental health conditions among adults,” the investigators added.
The National Syndromic Surveillance Program receives daily data from 3,310 EDs in 47 states, but the number of facilities meeting the investigators’ criteria averaged 2,970 a week for the 8 weeks of the study period.
SOURCE: Swedo E et al. MMWR. 2020 Dec. 11;69(49):1841-7.
but the visits in 2020 were significantly more likely to result in hospitalization, based on analysis of a national ED database.
The number of ED visits involving child abuse and neglect was down by 53% during the 4-week period from March 29 to April 25, 2020, compared with the 4 weeks from March 31 to April 27, 2019. The proportion of those ED visits that ended in hospitalizations, however, increased from 2.1% in 2019 to 3.2% in 2020, Elizabeth Swedo, MD, and associates at the Centers for Disease Control and Prevention said in the Morbidity and Mortality Weekly Report.
“ED visits related to suspected or confirmed child abuse and neglect decreased beginning the week of March 15, 2020, coinciding with the declaration of a national emergency related to COVID-19 and implementation of community mitigation measures,” they wrote.
An earlier study involving the same database (the National Syndromic Surveillance Program) showed that, over the two same 4-week periods, the volume of all ED visits in 2020 was down 72% for children aged 10 years and younger and 71% for those aged 11-14 years.
In the current study, however, all age subgroups had significant increases in hospital admissions. The proportion of ED visits related to child abuse and neglect that resulted in hospitalization rose from 3.5% in 2019 to 5.3% in 2020 among ages 0-4 years, 0.7% to 1.3% for ages 5-11 years, and 1.6% to 2.2% for adolescents aged 12-17, Dr. Swedo and associates reported.
The absence of a corresponding drop in hospitalizations may be tied to risk factors related to the pandemic, “such as loss of income, increased stress related to parental child care and schooling responsibilities, and increased substance use and mental health conditions among adults,” the investigators added.
The National Syndromic Surveillance Program receives daily data from 3,310 EDs in 47 states, but the number of facilities meeting the investigators’ criteria averaged 2,970 a week for the 8 weeks of the study period.
SOURCE: Swedo E et al. MMWR. 2020 Dec. 11;69(49):1841-7.
but the visits in 2020 were significantly more likely to result in hospitalization, based on analysis of a national ED database.
The number of ED visits involving child abuse and neglect was down by 53% during the 4-week period from March 29 to April 25, 2020, compared with the 4 weeks from March 31 to April 27, 2019. The proportion of those ED visits that ended in hospitalizations, however, increased from 2.1% in 2019 to 3.2% in 2020, Elizabeth Swedo, MD, and associates at the Centers for Disease Control and Prevention said in the Morbidity and Mortality Weekly Report.
“ED visits related to suspected or confirmed child abuse and neglect decreased beginning the week of March 15, 2020, coinciding with the declaration of a national emergency related to COVID-19 and implementation of community mitigation measures,” they wrote.
An earlier study involving the same database (the National Syndromic Surveillance Program) showed that, over the two same 4-week periods, the volume of all ED visits in 2020 was down 72% for children aged 10 years and younger and 71% for those aged 11-14 years.
In the current study, however, all age subgroups had significant increases in hospital admissions. The proportion of ED visits related to child abuse and neglect that resulted in hospitalization rose from 3.5% in 2019 to 5.3% in 2020 among ages 0-4 years, 0.7% to 1.3% for ages 5-11 years, and 1.6% to 2.2% for adolescents aged 12-17, Dr. Swedo and associates reported.
The absence of a corresponding drop in hospitalizations may be tied to risk factors related to the pandemic, “such as loss of income, increased stress related to parental child care and schooling responsibilities, and increased substance use and mental health conditions among adults,” the investigators added.
The National Syndromic Surveillance Program receives daily data from 3,310 EDs in 47 states, but the number of facilities meeting the investigators’ criteria averaged 2,970 a week for the 8 weeks of the study period.
SOURCE: Swedo E et al. MMWR. 2020 Dec. 11;69(49):1841-7.
FROM MMWR
FDA OKs osimertinib as first adjuvant drug for NSCLC
Osimertinib was first approved in the US in 2018 for the first-line treatment of patients with metastatic EGFR-mutated NSCLC.
With this new indication, “patients may be treated with this targeted therapy in an earlier and potentially more curative stage of non-small cell lung cancer,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The expanded indication is based on results of the ADAURA clinical trial, which compared osimertinib with placebo following complete resection of localized or locally advanced NSCLC with negative margins.
In the trial, adjuvant osimertinib reduced the relative risk of disease recurrence or death by 83% in patients with stage II and IIIA disease (hazard ratio [HR], 0.17; 95% CI, 0.12 - 0.23; P < .0001).
Disease-free survival (DFS) in the overall trial population of patients with stage IB-IIIA disease showed osimertinib reduced the risk of disease recurrence or death by 80% (HR, 0.20; 95% CI, 0.15 - 0.27; P < .0001).
At 2 years, 89% of patients treated with the targeted agent remained alive and disease free vs 52% on placebo after surgery. The safety and tolerability of osimertinib in the adjuvant setting was consistent with previous trials in the metastatic setting.
The trial of 682 patients was unblinded early and halted on the recommendation of the independent data-monitoring committee, because of the efficacy of osimertinib.
“If I were on the committee, I would have done the same thing. These are extraordinary results,” study investigator Roy S. Herbst, MD, PhD, chief of medical oncology at the Yale Cancer Center, New Haven, Connecticut, said at a press briefing prior to the study presentation at the American Society of Clinical Oncology’s (ASCO) virtual scientific program last spring.
In a Medscape commentary, Mark Kris, MD, of Memorial Sloan Kettering Cancer Center in New York City, said the data with osimertinib in the adjuvant setting are “important and practice-changing.”
“The potential for this drug to improve outcomes has been there for a long time. This phase 3 randomized trial presented at the plenary session of ASCO showed a more than doubling of disease-free survival at 2 years. It shows that we can use therapies in the earlier stages of disease,” Kris noted.
“This approval dispels the notion that treatment is over after surgery and chemotherapy, as the ADAURA results show that Tagrisso can dramatically change the course of this disease,” Dave Fredrickson, executive vice president, AstraZeneca oncology business unit, said in a news release.
Osimertinib had orphan drug status and breakthrough therapy designation for treatment of EGFR mutation-positive NSCLC.
A version of this article first appeared on Medscape.com.
Osimertinib was first approved in the US in 2018 for the first-line treatment of patients with metastatic EGFR-mutated NSCLC.
With this new indication, “patients may be treated with this targeted therapy in an earlier and potentially more curative stage of non-small cell lung cancer,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The expanded indication is based on results of the ADAURA clinical trial, which compared osimertinib with placebo following complete resection of localized or locally advanced NSCLC with negative margins.
In the trial, adjuvant osimertinib reduced the relative risk of disease recurrence or death by 83% in patients with stage II and IIIA disease (hazard ratio [HR], 0.17; 95% CI, 0.12 - 0.23; P < .0001).
Disease-free survival (DFS) in the overall trial population of patients with stage IB-IIIA disease showed osimertinib reduced the risk of disease recurrence or death by 80% (HR, 0.20; 95% CI, 0.15 - 0.27; P < .0001).
At 2 years, 89% of patients treated with the targeted agent remained alive and disease free vs 52% on placebo after surgery. The safety and tolerability of osimertinib in the adjuvant setting was consistent with previous trials in the metastatic setting.
The trial of 682 patients was unblinded early and halted on the recommendation of the independent data-monitoring committee, because of the efficacy of osimertinib.
“If I were on the committee, I would have done the same thing. These are extraordinary results,” study investigator Roy S. Herbst, MD, PhD, chief of medical oncology at the Yale Cancer Center, New Haven, Connecticut, said at a press briefing prior to the study presentation at the American Society of Clinical Oncology’s (ASCO) virtual scientific program last spring.
In a Medscape commentary, Mark Kris, MD, of Memorial Sloan Kettering Cancer Center in New York City, said the data with osimertinib in the adjuvant setting are “important and practice-changing.”
“The potential for this drug to improve outcomes has been there for a long time. This phase 3 randomized trial presented at the plenary session of ASCO showed a more than doubling of disease-free survival at 2 years. It shows that we can use therapies in the earlier stages of disease,” Kris noted.
“This approval dispels the notion that treatment is over after surgery and chemotherapy, as the ADAURA results show that Tagrisso can dramatically change the course of this disease,” Dave Fredrickson, executive vice president, AstraZeneca oncology business unit, said in a news release.
Osimertinib had orphan drug status and breakthrough therapy designation for treatment of EGFR mutation-positive NSCLC.
A version of this article first appeared on Medscape.com.
Osimertinib was first approved in the US in 2018 for the first-line treatment of patients with metastatic EGFR-mutated NSCLC.
With this new indication, “patients may be treated with this targeted therapy in an earlier and potentially more curative stage of non-small cell lung cancer,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The expanded indication is based on results of the ADAURA clinical trial, which compared osimertinib with placebo following complete resection of localized or locally advanced NSCLC with negative margins.
In the trial, adjuvant osimertinib reduced the relative risk of disease recurrence or death by 83% in patients with stage II and IIIA disease (hazard ratio [HR], 0.17; 95% CI, 0.12 - 0.23; P < .0001).
Disease-free survival (DFS) in the overall trial population of patients with stage IB-IIIA disease showed osimertinib reduced the risk of disease recurrence or death by 80% (HR, 0.20; 95% CI, 0.15 - 0.27; P < .0001).
At 2 years, 89% of patients treated with the targeted agent remained alive and disease free vs 52% on placebo after surgery. The safety and tolerability of osimertinib in the adjuvant setting was consistent with previous trials in the metastatic setting.
The trial of 682 patients was unblinded early and halted on the recommendation of the independent data-monitoring committee, because of the efficacy of osimertinib.
“If I were on the committee, I would have done the same thing. These are extraordinary results,” study investigator Roy S. Herbst, MD, PhD, chief of medical oncology at the Yale Cancer Center, New Haven, Connecticut, said at a press briefing prior to the study presentation at the American Society of Clinical Oncology’s (ASCO) virtual scientific program last spring.
In a Medscape commentary, Mark Kris, MD, of Memorial Sloan Kettering Cancer Center in New York City, said the data with osimertinib in the adjuvant setting are “important and practice-changing.”
“The potential for this drug to improve outcomes has been there for a long time. This phase 3 randomized trial presented at the plenary session of ASCO showed a more than doubling of disease-free survival at 2 years. It shows that we can use therapies in the earlier stages of disease,” Kris noted.
“This approval dispels the notion that treatment is over after surgery and chemotherapy, as the ADAURA results show that Tagrisso can dramatically change the course of this disease,” Dave Fredrickson, executive vice president, AstraZeneca oncology business unit, said in a news release.
Osimertinib had orphan drug status and breakthrough therapy designation for treatment of EGFR mutation-positive NSCLC.
A version of this article first appeared on Medscape.com.
FDA OKs first oral hormone therapy for advanced prostate cancer
Relugolix is an oral gonadotropin-releasing hormone antagonist. The new pill form may mean fewer clinic visits for patients, an added benefit during the COVID-19 pandemic, said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a press statement.
“Today’s approval marks the first oral drug in this class, and it may eliminate some patients’ need to visit the clinic for treatments that require administration by a health care provider,” he commented.
Relugolix works by preventing the pituitary gland from making luteinizing hormone and follicle-stimulating hormone, thus reducing the amount of testosterone the testicles can make.
In the open-label HERO trial, 930 patients with locally advanced or metastatic prostate cancer were randomly assigned to receive either once-daily oral relugolix or leuprolide injection every 3 months for 48 weeks.
The study met its primary endpoint, demonstrating that castration at 48 weeks was maintained in 96.7% of men receiving relugolix vs. 88.8% for patients receiving leuprolide; castration levels of testosterone had to be reached by day 29 and then sustained through the end of the treatment course.
Relugolix has the “potential to become a new standard for ADT and advanced prostate cancer,” commented study investigator Neal D. Shore, MD, of Carolina Urologic Research Center, Myrtle Beach, South Carolina, at the 2020 annual meeting of the American Society of Clinical Oncology, where the results were first presented.
They were published simultaneously in The New England Journal of Medicine.
At the ASCO meeting, David R. Wise, MD, PhD, Perlmutter Cancer Center at NYU Langone Health, New York, agreed that the drug could be practice-changing – but claimed that it would be for a subset of patients only, specifically, patients with a significant history of cardiovascular disease who are without gastrointestinal malabsorption.
Notably, in the study, relugolix cut the risk for major adverse cardiovascular events by 54% in comparison with leuprolide, as reported by Medscape Medical News.
According to the FDA, the most common side effects of relugolix include hot flush, increased glucose levels, increased triglyceride levels, musculoskeletal pain, decreased hemoglobin, fatigue, constipation, diarrhea, and increased levels of certain liver enzymes.
Concurrent use of relugolix with drugs that inhibit P-glycoprotein is contraindicated.
Also, health care providers should consider having patients undergo periodic electrocardiographic monitoring as well as periodic monitoring of electrolyte levels. Owing to the drug’s suppression of the pituitary gonadal system, any diagnostic test results of the pituitary gonadotropic and gonadal functions conducted during and after taking relugolix may be affected.
A version of this article first appeared on Medscape.com.
Relugolix is an oral gonadotropin-releasing hormone antagonist. The new pill form may mean fewer clinic visits for patients, an added benefit during the COVID-19 pandemic, said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a press statement.
“Today’s approval marks the first oral drug in this class, and it may eliminate some patients’ need to visit the clinic for treatments that require administration by a health care provider,” he commented.
Relugolix works by preventing the pituitary gland from making luteinizing hormone and follicle-stimulating hormone, thus reducing the amount of testosterone the testicles can make.
In the open-label HERO trial, 930 patients with locally advanced or metastatic prostate cancer were randomly assigned to receive either once-daily oral relugolix or leuprolide injection every 3 months for 48 weeks.
The study met its primary endpoint, demonstrating that castration at 48 weeks was maintained in 96.7% of men receiving relugolix vs. 88.8% for patients receiving leuprolide; castration levels of testosterone had to be reached by day 29 and then sustained through the end of the treatment course.
Relugolix has the “potential to become a new standard for ADT and advanced prostate cancer,” commented study investigator Neal D. Shore, MD, of Carolina Urologic Research Center, Myrtle Beach, South Carolina, at the 2020 annual meeting of the American Society of Clinical Oncology, where the results were first presented.
They were published simultaneously in The New England Journal of Medicine.
At the ASCO meeting, David R. Wise, MD, PhD, Perlmutter Cancer Center at NYU Langone Health, New York, agreed that the drug could be practice-changing – but claimed that it would be for a subset of patients only, specifically, patients with a significant history of cardiovascular disease who are without gastrointestinal malabsorption.
Notably, in the study, relugolix cut the risk for major adverse cardiovascular events by 54% in comparison with leuprolide, as reported by Medscape Medical News.
According to the FDA, the most common side effects of relugolix include hot flush, increased glucose levels, increased triglyceride levels, musculoskeletal pain, decreased hemoglobin, fatigue, constipation, diarrhea, and increased levels of certain liver enzymes.
Concurrent use of relugolix with drugs that inhibit P-glycoprotein is contraindicated.
Also, health care providers should consider having patients undergo periodic electrocardiographic monitoring as well as periodic monitoring of electrolyte levels. Owing to the drug’s suppression of the pituitary gonadal system, any diagnostic test results of the pituitary gonadotropic and gonadal functions conducted during and after taking relugolix may be affected.
A version of this article first appeared on Medscape.com.
Relugolix is an oral gonadotropin-releasing hormone antagonist. The new pill form may mean fewer clinic visits for patients, an added benefit during the COVID-19 pandemic, said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a press statement.
“Today’s approval marks the first oral drug in this class, and it may eliminate some patients’ need to visit the clinic for treatments that require administration by a health care provider,” he commented.
Relugolix works by preventing the pituitary gland from making luteinizing hormone and follicle-stimulating hormone, thus reducing the amount of testosterone the testicles can make.
In the open-label HERO trial, 930 patients with locally advanced or metastatic prostate cancer were randomly assigned to receive either once-daily oral relugolix or leuprolide injection every 3 months for 48 weeks.
The study met its primary endpoint, demonstrating that castration at 48 weeks was maintained in 96.7% of men receiving relugolix vs. 88.8% for patients receiving leuprolide; castration levels of testosterone had to be reached by day 29 and then sustained through the end of the treatment course.
Relugolix has the “potential to become a new standard for ADT and advanced prostate cancer,” commented study investigator Neal D. Shore, MD, of Carolina Urologic Research Center, Myrtle Beach, South Carolina, at the 2020 annual meeting of the American Society of Clinical Oncology, where the results were first presented.
They were published simultaneously in The New England Journal of Medicine.
At the ASCO meeting, David R. Wise, MD, PhD, Perlmutter Cancer Center at NYU Langone Health, New York, agreed that the drug could be practice-changing – but claimed that it would be for a subset of patients only, specifically, patients with a significant history of cardiovascular disease who are without gastrointestinal malabsorption.
Notably, in the study, relugolix cut the risk for major adverse cardiovascular events by 54% in comparison with leuprolide, as reported by Medscape Medical News.
According to the FDA, the most common side effects of relugolix include hot flush, increased glucose levels, increased triglyceride levels, musculoskeletal pain, decreased hemoglobin, fatigue, constipation, diarrhea, and increased levels of certain liver enzymes.
Concurrent use of relugolix with drugs that inhibit P-glycoprotein is contraindicated.
Also, health care providers should consider having patients undergo periodic electrocardiographic monitoring as well as periodic monitoring of electrolyte levels. Owing to the drug’s suppression of the pituitary gonadal system, any diagnostic test results of the pituitary gonadotropic and gonadal functions conducted during and after taking relugolix may be affected.
A version of this article first appeared on Medscape.com.
FDA expands belimumab indication to adults with lupus nephritis
The U.S. Food and Drug Administration has expanded the indication for belimumab (Benlysta) to adults with active lupus nephritis who are receiving standard therapy.

Roughly 40% of patients with systemic lupus erythematosus (SLE) develop lupus nephritis (LN), which causes inflammation in the kidneys and can lead to end-stage kidney disease.
“Benlysta is the first medicine approved to treat systemic lupus and adults with active lupus nephritis, an important treatment advance for patients with this incurable autoimmune disease,” Hal Barron, MD, GlaxoSmithKline’s chief scientific officer and president of research and development, said in a company news release.
Belimumab IV infusion was first approved in the United States in March 2011 for adults with SLE. The FDA approved belimumab IV infusion for use in children as young as age 5 years with SLE in 2019.
Both the IV and subcutaneous formulations are now indicated in the United States for adults with SLE and LN.
Belimumab is a B-lymphocyte stimulator protein inhibitor that is thought to decrease the amount of abnormal B cells; the latter are thought to play a role in lupus.
The expanded indication for belimumab for patients with LN is based on findings from the BLISS-LN phase 3 trial, published in The New England Journal of Medicine in September.
“Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys” represents a “compelling therapeutic approach to lupus nephritis,” the lead investigator of BLISS-LN, Richard Furie, MD, told the online annual Perspectives in Rheumatic Diseases meeting recently.
“In the 4 decades I have been caring for people with lupus, we have not been able to achieve remission in more than just one-third of patients with lupus nephritis, and despite all of our efforts, 10%-30% of patients with lupus kidney disease still progress to end-stage kidney disease,” Dr. Furie, who is chief of the division of rheumatology at Northwell Health, notes in the GSK statement.
“The data from the BLISS-LN study show that Benlysta added to standard therapy not only increased response rates over 2 years, but it also prevented worsening of kidney disease in patients with active lupus nephritis, compared to standard therapy alone,” he added.
BLISS-LN study: Belimumab effect seen mostly in those on MMF
BLISS-LN enrolled 448 adults with biopsy-confirmed active LN. Half were randomly allocated to receive IV belimumab (10 mg/kg) plus standard therapy (mycophenolate mofetil for induction and maintenance or cyclophosphamide for induction followed by azathioprine for maintenance, with steroids) and half to receive placebo plus standard therapy.
At 2 years, significantly more patients in the belimumab group than in the placebo group had a primary efficacy renal response (43% vs. 32%; odds ratio, 1.6; 95% confidence interval, 1.0- 2.3; P = .03).
This primary endpoint was defined as a ratio of urinary protein to creatinine of ≤0.7, an estimated glomerular filtration rate that was no worse than 20% below the value before the renal flare or ≥60 mL per minute per 1.73 m2 of body surface area, without use of rescue therapy.
The risk for a renal-related event or death was also significantly lower among patients who received belimumab than among those who received placebo (hazard ratio, 0.51; P = .001). The safety profile of belimumab was consistent with that observed in prior studies.
But in a commentary that accompanied the publication of BLISS-LN, editorialists noted that “most of the treatment effect was seen in patients who had received mycophenolate mofetil. No benefit was present in the subgroup of patients who received cyclophosphamide-azathioprine.”
In addition, induction treatment was not randomly assigned, editorialists Michael Ward, MD, MPH, and Maria Tektonidou, MD, PhD, noted.
“If patients with more severe nephritis were preferentially treated with cyclophosphamide, a likely inclination among most physicians, the trial may be telling us that belimumab enhances responses only among less severely affected patients,” observed Dr. Ward, who is with the National Institutes of Health, and Dr. Tektonidou, of the National and Kopodistrian University, in Athens.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has expanded the indication for belimumab (Benlysta) to adults with active lupus nephritis who are receiving standard therapy.
Roughly 40% of patients with systemic lupus erythematosus (SLE) develop lupus nephritis (LN), which causes inflammation in the kidneys and can lead to end-stage kidney disease.
“Benlysta is the first medicine approved to treat systemic lupus and adults with active lupus nephritis, an important treatment advance for patients with this incurable autoimmune disease,” Hal Barron, MD, GlaxoSmithKline’s chief scientific officer and president of research and development, said in a company news release.
Belimumab IV infusion was first approved in the United States in March 2011 for adults with SLE. The FDA approved belimumab IV infusion for use in children as young as age 5 years with SLE in 2019.
Both the IV and subcutaneous formulations are now indicated in the United States for adults with SLE and LN.
Belimumab is a B-lymphocyte stimulator protein inhibitor that is thought to decrease the amount of abnormal B cells; the latter are thought to play a role in lupus.
The expanded indication for belimumab for patients with LN is based on findings from the BLISS-LN phase 3 trial, published in The New England Journal of Medicine in September.
“Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys” represents a “compelling therapeutic approach to lupus nephritis,” the lead investigator of BLISS-LN, Richard Furie, MD, told the online annual Perspectives in Rheumatic Diseases meeting recently.
“In the 4 decades I have been caring for people with lupus, we have not been able to achieve remission in more than just one-third of patients with lupus nephritis, and despite all of our efforts, 10%-30% of patients with lupus kidney disease still progress to end-stage kidney disease,” Dr. Furie, who is chief of the division of rheumatology at Northwell Health, notes in the GSK statement.
“The data from the BLISS-LN study show that Benlysta added to standard therapy not only increased response rates over 2 years, but it also prevented worsening of kidney disease in patients with active lupus nephritis, compared to standard therapy alone,” he added.
BLISS-LN study: Belimumab effect seen mostly in those on MMF
BLISS-LN enrolled 448 adults with biopsy-confirmed active LN. Half were randomly allocated to receive IV belimumab (10 mg/kg) plus standard therapy (mycophenolate mofetil for induction and maintenance or cyclophosphamide for induction followed by azathioprine for maintenance, with steroids) and half to receive placebo plus standard therapy.
At 2 years, significantly more patients in the belimumab group than in the placebo group had a primary efficacy renal response (43% vs. 32%; odds ratio, 1.6; 95% confidence interval, 1.0- 2.3; P = .03).
This primary endpoint was defined as a ratio of urinary protein to creatinine of ≤0.7, an estimated glomerular filtration rate that was no worse than 20% below the value before the renal flare or ≥60 mL per minute per 1.73 m2 of body surface area, without use of rescue therapy.
The risk for a renal-related event or death was also significantly lower among patients who received belimumab than among those who received placebo (hazard ratio, 0.51; P = .001). The safety profile of belimumab was consistent with that observed in prior studies.
But in a commentary that accompanied the publication of BLISS-LN, editorialists noted that “most of the treatment effect was seen in patients who had received mycophenolate mofetil. No benefit was present in the subgroup of patients who received cyclophosphamide-azathioprine.”
In addition, induction treatment was not randomly assigned, editorialists Michael Ward, MD, MPH, and Maria Tektonidou, MD, PhD, noted.
“If patients with more severe nephritis were preferentially treated with cyclophosphamide, a likely inclination among most physicians, the trial may be telling us that belimumab enhances responses only among less severely affected patients,” observed Dr. Ward, who is with the National Institutes of Health, and Dr. Tektonidou, of the National and Kopodistrian University, in Athens.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration has expanded the indication for belimumab (Benlysta) to adults with active lupus nephritis who are receiving standard therapy.
Roughly 40% of patients with systemic lupus erythematosus (SLE) develop lupus nephritis (LN), which causes inflammation in the kidneys and can lead to end-stage kidney disease.
“Benlysta is the first medicine approved to treat systemic lupus and adults with active lupus nephritis, an important treatment advance for patients with this incurable autoimmune disease,” Hal Barron, MD, GlaxoSmithKline’s chief scientific officer and president of research and development, said in a company news release.
Belimumab IV infusion was first approved in the United States in March 2011 for adults with SLE. The FDA approved belimumab IV infusion for use in children as young as age 5 years with SLE in 2019.
Both the IV and subcutaneous formulations are now indicated in the United States for adults with SLE and LN.
Belimumab is a B-lymphocyte stimulator protein inhibitor that is thought to decrease the amount of abnormal B cells; the latter are thought to play a role in lupus.
The expanded indication for belimumab for patients with LN is based on findings from the BLISS-LN phase 3 trial, published in The New England Journal of Medicine in September.
“Neutralizing B-cell activating factor and down-regulating autoreactive B-cell function in kidneys” represents a “compelling therapeutic approach to lupus nephritis,” the lead investigator of BLISS-LN, Richard Furie, MD, told the online annual Perspectives in Rheumatic Diseases meeting recently.
“In the 4 decades I have been caring for people with lupus, we have not been able to achieve remission in more than just one-third of patients with lupus nephritis, and despite all of our efforts, 10%-30% of patients with lupus kidney disease still progress to end-stage kidney disease,” Dr. Furie, who is chief of the division of rheumatology at Northwell Health, notes in the GSK statement.
“The data from the BLISS-LN study show that Benlysta added to standard therapy not only increased response rates over 2 years, but it also prevented worsening of kidney disease in patients with active lupus nephritis, compared to standard therapy alone,” he added.
BLISS-LN study: Belimumab effect seen mostly in those on MMF
BLISS-LN enrolled 448 adults with biopsy-confirmed active LN. Half were randomly allocated to receive IV belimumab (10 mg/kg) plus standard therapy (mycophenolate mofetil for induction and maintenance or cyclophosphamide for induction followed by azathioprine for maintenance, with steroids) and half to receive placebo plus standard therapy.
At 2 years, significantly more patients in the belimumab group than in the placebo group had a primary efficacy renal response (43% vs. 32%; odds ratio, 1.6; 95% confidence interval, 1.0- 2.3; P = .03).
This primary endpoint was defined as a ratio of urinary protein to creatinine of ≤0.7, an estimated glomerular filtration rate that was no worse than 20% below the value before the renal flare or ≥60 mL per minute per 1.73 m2 of body surface area, without use of rescue therapy.
The risk for a renal-related event or death was also significantly lower among patients who received belimumab than among those who received placebo (hazard ratio, 0.51; P = .001). The safety profile of belimumab was consistent with that observed in prior studies.
But in a commentary that accompanied the publication of BLISS-LN, editorialists noted that “most of the treatment effect was seen in patients who had received mycophenolate mofetil. No benefit was present in the subgroup of patients who received cyclophosphamide-azathioprine.”
In addition, induction treatment was not randomly assigned, editorialists Michael Ward, MD, MPH, and Maria Tektonidou, MD, PhD, noted.
“If patients with more severe nephritis were preferentially treated with cyclophosphamide, a likely inclination among most physicians, the trial may be telling us that belimumab enhances responses only among less severely affected patients,” observed Dr. Ward, who is with the National Institutes of Health, and Dr. Tektonidou, of the National and Kopodistrian University, in Athens.
A version of this article first appeared on Medscape.com.
Margetuximab approved for HER2-positive metastatic breast cancer
The new drug is indicated for use in combination with chemotherapy for the treatment of patients with metastatic HER2-positive breast cancer who have already received two or more prior anti-HER2 regimens, with at least one for metastatic disease.
Margetuximab-cmkb is also the first HER2-targeted therapy shown to improve progression-free survival (PFS) as compared with the first-ever HER2-targeted agent, trastuzumab in a head-to-head, phase 3 clinical trial (known as SOPHIA).
“Early detection and treatment have had a positive impact on the survival of patients with breast cancer, but the prognosis for people diagnosed with metastatic breast cancer remains poor, and additional treatments are needed,” said Hope S. Rugo, MD, director of breast oncology and clinical trials education, University of California, San Francisco, Diller Family Comprehensive Cancer Center, in a press release.
“As the only HER2-targeted agent to have shown a PFS improvement versus trastuzumab in a head-to-head phase 3 clinical trial, margetuximab with chemotherapy represents the newest treatment option for patients who have progressed on available HER2-directed therapies,” said Dr. Rugo, who is an investigator in the SOPHIA trial.
Like trastuzumab, margetuximab-cmkb binds HER2 with high specificity and affinity and disrupts signaling that drives cell proliferation and survival, but margetuximab binds with elevated affinity to both the lower- and higher-affinity forms of CD16A, an Fc gamma receptor important for antibody dependent cell-mediated cytotoxicity against tumor cells, according to the manufacturer, MacroGenics.
Details of the pivotal trial
The SOPHIA trial was a randomized, open-label, phase 3 clinical trial that compared margetuximab-cmkb plus chemotherapy with trastuzumab plus chemotherapy in both arms in patients with HER2-positive metastatic breast cancer who had previously been treated with anti–HER2-targeted therapies.
All patients in the cohort had previously received trastuzumab, all but one patient had previously also received pertuzumab, and most of the patients (91%) had also been treated with ado-trastuzumab emtansine, or T-DM1.
The trial randomly assigned 536 patients to receive either margetuximab-cmkb (n = 266) given intravenously at 15 mg/kg every 3 weeks or trastuzumab (n = 270) given intravenously at 6 mg/kg (or 8 mg/kg for loading dose) every 3 weeks in combination with either capecitabine, eribulin, gemcitabine, or vinorelbine, given at the standard doses.
As compared with trastuzumab, margetuximab plus chemotherapy led to a significant 24% reduction in the risk for progression or death (hazard ratio, 0.76).
The median PFS also favored margetuximab (5.8 months vs. 4.9 months), as did the overall response rate (22% vs. 16%).
The final overall survival analysis is expected in the second half of 2021.
Common adverse events associated with the margetuximab regimen included fatigue/asthenia (57%), nausea (33%), diarrhea (25%), and vomiting (21%). Infusion-related reactions occurred in 13% of patients receiving margetuximab, and almost all were grade 1 or 2, with only 1.5% at grade 3.
The product also carries a boxed warning for left ventricular dysfunction and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.
The new drug is indicated for use in combination with chemotherapy for the treatment of patients with metastatic HER2-positive breast cancer who have already received two or more prior anti-HER2 regimens, with at least one for metastatic disease.
Margetuximab-cmkb is also the first HER2-targeted therapy shown to improve progression-free survival (PFS) as compared with the first-ever HER2-targeted agent, trastuzumab in a head-to-head, phase 3 clinical trial (known as SOPHIA).
“Early detection and treatment have had a positive impact on the survival of patients with breast cancer, but the prognosis for people diagnosed with metastatic breast cancer remains poor, and additional treatments are needed,” said Hope S. Rugo, MD, director of breast oncology and clinical trials education, University of California, San Francisco, Diller Family Comprehensive Cancer Center, in a press release.
“As the only HER2-targeted agent to have shown a PFS improvement versus trastuzumab in a head-to-head phase 3 clinical trial, margetuximab with chemotherapy represents the newest treatment option for patients who have progressed on available HER2-directed therapies,” said Dr. Rugo, who is an investigator in the SOPHIA trial.
Like trastuzumab, margetuximab-cmkb binds HER2 with high specificity and affinity and disrupts signaling that drives cell proliferation and survival, but margetuximab binds with elevated affinity to both the lower- and higher-affinity forms of CD16A, an Fc gamma receptor important for antibody dependent cell-mediated cytotoxicity against tumor cells, according to the manufacturer, MacroGenics.
Details of the pivotal trial
The SOPHIA trial was a randomized, open-label, phase 3 clinical trial that compared margetuximab-cmkb plus chemotherapy with trastuzumab plus chemotherapy in both arms in patients with HER2-positive metastatic breast cancer who had previously been treated with anti–HER2-targeted therapies.
All patients in the cohort had previously received trastuzumab, all but one patient had previously also received pertuzumab, and most of the patients (91%) had also been treated with ado-trastuzumab emtansine, or T-DM1.
The trial randomly assigned 536 patients to receive either margetuximab-cmkb (n = 266) given intravenously at 15 mg/kg every 3 weeks or trastuzumab (n = 270) given intravenously at 6 mg/kg (or 8 mg/kg for loading dose) every 3 weeks in combination with either capecitabine, eribulin, gemcitabine, or vinorelbine, given at the standard doses.
As compared with trastuzumab, margetuximab plus chemotherapy led to a significant 24% reduction in the risk for progression or death (hazard ratio, 0.76).
The median PFS also favored margetuximab (5.8 months vs. 4.9 months), as did the overall response rate (22% vs. 16%).
The final overall survival analysis is expected in the second half of 2021.
Common adverse events associated with the margetuximab regimen included fatigue/asthenia (57%), nausea (33%), diarrhea (25%), and vomiting (21%). Infusion-related reactions occurred in 13% of patients receiving margetuximab, and almost all were grade 1 or 2, with only 1.5% at grade 3.
The product also carries a boxed warning for left ventricular dysfunction and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.
The new drug is indicated for use in combination with chemotherapy for the treatment of patients with metastatic HER2-positive breast cancer who have already received two or more prior anti-HER2 regimens, with at least one for metastatic disease.
Margetuximab-cmkb is also the first HER2-targeted therapy shown to improve progression-free survival (PFS) as compared with the first-ever HER2-targeted agent, trastuzumab in a head-to-head, phase 3 clinical trial (known as SOPHIA).
“Early detection and treatment have had a positive impact on the survival of patients with breast cancer, but the prognosis for people diagnosed with metastatic breast cancer remains poor, and additional treatments are needed,” said Hope S. Rugo, MD, director of breast oncology and clinical trials education, University of California, San Francisco, Diller Family Comprehensive Cancer Center, in a press release.
“As the only HER2-targeted agent to have shown a PFS improvement versus trastuzumab in a head-to-head phase 3 clinical trial, margetuximab with chemotherapy represents the newest treatment option for patients who have progressed on available HER2-directed therapies,” said Dr. Rugo, who is an investigator in the SOPHIA trial.
Like trastuzumab, margetuximab-cmkb binds HER2 with high specificity and affinity and disrupts signaling that drives cell proliferation and survival, but margetuximab binds with elevated affinity to both the lower- and higher-affinity forms of CD16A, an Fc gamma receptor important for antibody dependent cell-mediated cytotoxicity against tumor cells, according to the manufacturer, MacroGenics.
Details of the pivotal trial
The SOPHIA trial was a randomized, open-label, phase 3 clinical trial that compared margetuximab-cmkb plus chemotherapy with trastuzumab plus chemotherapy in both arms in patients with HER2-positive metastatic breast cancer who had previously been treated with anti–HER2-targeted therapies.
All patients in the cohort had previously received trastuzumab, all but one patient had previously also received pertuzumab, and most of the patients (91%) had also been treated with ado-trastuzumab emtansine, or T-DM1.
The trial randomly assigned 536 patients to receive either margetuximab-cmkb (n = 266) given intravenously at 15 mg/kg every 3 weeks or trastuzumab (n = 270) given intravenously at 6 mg/kg (or 8 mg/kg for loading dose) every 3 weeks in combination with either capecitabine, eribulin, gemcitabine, or vinorelbine, given at the standard doses.
As compared with trastuzumab, margetuximab plus chemotherapy led to a significant 24% reduction in the risk for progression or death (hazard ratio, 0.76).
The median PFS also favored margetuximab (5.8 months vs. 4.9 months), as did the overall response rate (22% vs. 16%).
The final overall survival analysis is expected in the second half of 2021.
Common adverse events associated with the margetuximab regimen included fatigue/asthenia (57%), nausea (33%), diarrhea (25%), and vomiting (21%). Infusion-related reactions occurred in 13% of patients receiving margetuximab, and almost all were grade 1 or 2, with only 1.5% at grade 3.
The product also carries a boxed warning for left ventricular dysfunction and embryo-fetal toxicity.
A version of this article first appeared on Medscape.com.