User login
Tick-borne disease has become a national issue
Pennsylvania had more reported cases of tick-borne disease from 2004 to 2016 than any other state, but these diseases are becoming a national threat, according to the Centers for Disease Control and Prevention.
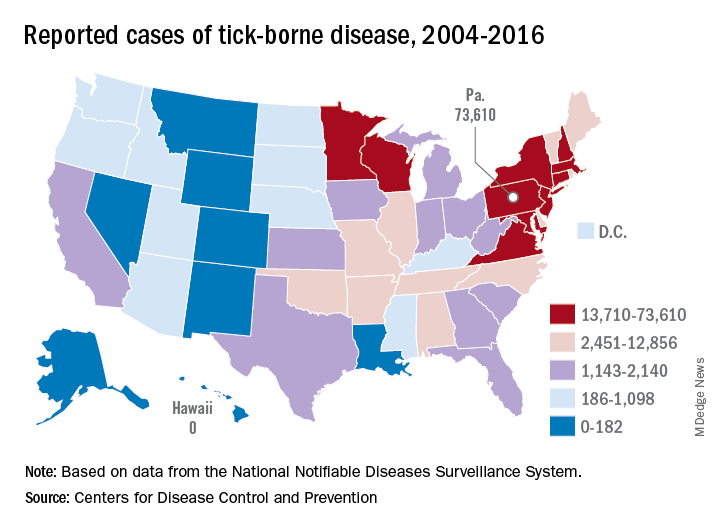
There were 73,000 cases reported in Pennsylvania over that period, and tick-borne diseases, including Lyme disease, anaplasmosis/ehrlichiosis, spotted fever rickettsiosis, babesiosis, tularemia, and Powassan virus, among others, affected almost 492,000 people nationwide, with Lyme disease representing the majority of cases, the CDC said in a Vital Signs report.
Although it’s no surprise that Pennsylvania, New York, and Connecticut were tick-borne disease hot spots, non-Northeastern states like Virginia, Wisconsin, and Minnesota also were among the top 10 in cases. States even further away from the Northeast can be found in the next 10: Arkansas had more than 7,000 cases in 13 years, and Oklahoma had over 4,600 cases, data from the National Notifiable Diseases Surveillance System show.
Nationally, the number of cases more than doubled from 23,000 in 2004 to 49,000 in 2016, and tick-borne disease hit every state except Hawaii. Over that same time, seven new tick-borne pathogens were discovered or introduced into the United States, the CDC reported.
“Local and state health departments and vector control organizations face increasing demands to respond to these threats,” the CDC said, but “more than 80% of vector control organizations report needing improvement in one or more of five core competencies, such as testing for pesticide resistance [and using] data to drive local decisions about vector control.”
Pennsylvania had more reported cases of tick-borne disease from 2004 to 2016 than any other state, but these diseases are becoming a national threat, according to the Centers for Disease Control and Prevention.
There were 73,000 cases reported in Pennsylvania over that period, and tick-borne diseases, including Lyme disease, anaplasmosis/ehrlichiosis, spotted fever rickettsiosis, babesiosis, tularemia, and Powassan virus, among others, affected almost 492,000 people nationwide, with Lyme disease representing the majority of cases, the CDC said in a Vital Signs report.
Although it’s no surprise that Pennsylvania, New York, and Connecticut were tick-borne disease hot spots, non-Northeastern states like Virginia, Wisconsin, and Minnesota also were among the top 10 in cases. States even further away from the Northeast can be found in the next 10: Arkansas had more than 7,000 cases in 13 years, and Oklahoma had over 4,600 cases, data from the National Notifiable Diseases Surveillance System show.
Nationally, the number of cases more than doubled from 23,000 in 2004 to 49,000 in 2016, and tick-borne disease hit every state except Hawaii. Over that same time, seven new tick-borne pathogens were discovered or introduced into the United States, the CDC reported.
“Local and state health departments and vector control organizations face increasing demands to respond to these threats,” the CDC said, but “more than 80% of vector control organizations report needing improvement in one or more of five core competencies, such as testing for pesticide resistance [and using] data to drive local decisions about vector control.”
Pennsylvania had more reported cases of tick-borne disease from 2004 to 2016 than any other state, but these diseases are becoming a national threat, according to the Centers for Disease Control and Prevention.
There were 73,000 cases reported in Pennsylvania over that period, and tick-borne diseases, including Lyme disease, anaplasmosis/ehrlichiosis, spotted fever rickettsiosis, babesiosis, tularemia, and Powassan virus, among others, affected almost 492,000 people nationwide, with Lyme disease representing the majority of cases, the CDC said in a Vital Signs report.
Although it’s no surprise that Pennsylvania, New York, and Connecticut were tick-borne disease hot spots, non-Northeastern states like Virginia, Wisconsin, and Minnesota also were among the top 10 in cases. States even further away from the Northeast can be found in the next 10: Arkansas had more than 7,000 cases in 13 years, and Oklahoma had over 4,600 cases, data from the National Notifiable Diseases Surveillance System show.
Nationally, the number of cases more than doubled from 23,000 in 2004 to 49,000 in 2016, and tick-borne disease hit every state except Hawaii. Over that same time, seven new tick-borne pathogens were discovered or introduced into the United States, the CDC reported.
“Local and state health departments and vector control organizations face increasing demands to respond to these threats,” the CDC said, but “more than 80% of vector control organizations report needing improvement in one or more of five core competencies, such as testing for pesticide resistance [and using] data to drive local decisions about vector control.”
FDA approves new treatment for hospital-acquired, ventilator-associated bacterial pneumonia
authorizing it for the treatment of both hospital-acquired and ventilator-associated bacterial pneumonia.

The new indication is for patients 18 years and older. It was based on results of a multinational, double-blind study that compared Zerbaxa with a different antibacterial drug in 726 patients hospitalized with hospital-acquired/ventilator-associated bacterial pneumonia. Mortality and cure rates were similar in the Zerbaxa and comparator groups.
The most common adverse events observed in the trial were elevated liver enzyme levels, renal impairment or failure, and diarrhea. Patients with hypersensitivity to beta-lactam drugs should not be receive Zerbaxa.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections. Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. ... That’s why, among our other efforts to address antimicrobial resistance, we’re focused on facilitating the development of safe and effective new treatments to give patients more options to fight life-threatening infections,” said Amy Abernethy, MD, PhD, the FDA’s principal deputy commissioner.
Zerbaxa was initially approved in 2014 for treatment of complicated intra-abdominal and urinary tract infections.
Find the full press release on the FDA website.
authorizing it for the treatment of both hospital-acquired and ventilator-associated bacterial pneumonia.
The new indication is for patients 18 years and older. It was based on results of a multinational, double-blind study that compared Zerbaxa with a different antibacterial drug in 726 patients hospitalized with hospital-acquired/ventilator-associated bacterial pneumonia. Mortality and cure rates were similar in the Zerbaxa and comparator groups.
The most common adverse events observed in the trial were elevated liver enzyme levels, renal impairment or failure, and diarrhea. Patients with hypersensitivity to beta-lactam drugs should not be receive Zerbaxa.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections. Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. ... That’s why, among our other efforts to address antimicrobial resistance, we’re focused on facilitating the development of safe and effective new treatments to give patients more options to fight life-threatening infections,” said Amy Abernethy, MD, PhD, the FDA’s principal deputy commissioner.
Zerbaxa was initially approved in 2014 for treatment of complicated intra-abdominal and urinary tract infections.
Find the full press release on the FDA website.
authorizing it for the treatment of both hospital-acquired and ventilator-associated bacterial pneumonia.
The new indication is for patients 18 years and older. It was based on results of a multinational, double-blind study that compared Zerbaxa with a different antibacterial drug in 726 patients hospitalized with hospital-acquired/ventilator-associated bacterial pneumonia. Mortality and cure rates were similar in the Zerbaxa and comparator groups.
The most common adverse events observed in the trial were elevated liver enzyme levels, renal impairment or failure, and diarrhea. Patients with hypersensitivity to beta-lactam drugs should not be receive Zerbaxa.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections. Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. ... That’s why, among our other efforts to address antimicrobial resistance, we’re focused on facilitating the development of safe and effective new treatments to give patients more options to fight life-threatening infections,” said Amy Abernethy, MD, PhD, the FDA’s principal deputy commissioner.
Zerbaxa was initially approved in 2014 for treatment of complicated intra-abdominal and urinary tract infections.
Find the full press release on the FDA website.
FDA announces clearance of modified endoscope connector
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
AGA Center for GI Innovation and Technology will continue to monitor this issue and encourages all GIs to follow the most up-to-date FDA guidance.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
AGA Center for GI Innovation and Technology will continue to monitor this issue and encourages all GIs to follow the most up-to-date FDA guidance.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
AGA Center for GI Innovation and Technology will continue to monitor this issue and encourages all GIs to follow the most up-to-date FDA guidance.
Measles cases now at highest level since 1992
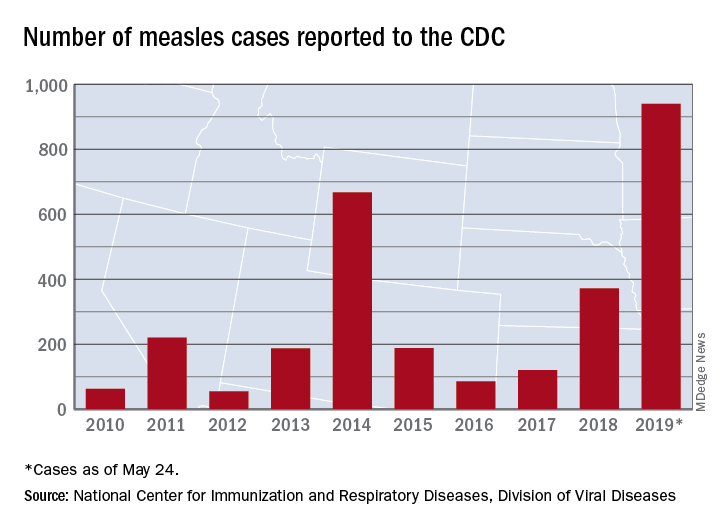
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.
FDA approves first drug for steroid-refractory acute GVHD
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
FDA approves lenalidomide/rituximab for previously treated FL, MZL
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
Measles count for 2019 now over 900 cases
according to the Centers for Disease Control and Prevention.
The CDC received reports of 60 new measles cases last week – up from 41 the previous week – bringing the U.S. total to 940 for the year as of May 24. The CDC is currently tracking 10 outbreaks in seven states: California (3), Georgia, Maryland, Michigan, New York (2), Pennsylvania, and Washington.
The Maine Center for Disease Control and Prevention confirmed the state’s first case on May 20. The school-aged child from Somerset County had been vaccinated and is fully recovered from the disease. It’s not yet known where the child was exposed to measles, but sporadic cases are not unexpected, the Maine CDC said.
New Mexico’s first measles case of the year, a 1-year-old in Sierra County, has at least one state lawmaker considering changes to the state’s immunization exemption laws, the Farmington Daily Times reported.
according to the Centers for Disease Control and Prevention.
The CDC received reports of 60 new measles cases last week – up from 41 the previous week – bringing the U.S. total to 940 for the year as of May 24. The CDC is currently tracking 10 outbreaks in seven states: California (3), Georgia, Maryland, Michigan, New York (2), Pennsylvania, and Washington.
The Maine Center for Disease Control and Prevention confirmed the state’s first case on May 20. The school-aged child from Somerset County had been vaccinated and is fully recovered from the disease. It’s not yet known where the child was exposed to measles, but sporadic cases are not unexpected, the Maine CDC said.
New Mexico’s first measles case of the year, a 1-year-old in Sierra County, has at least one state lawmaker considering changes to the state’s immunization exemption laws, the Farmington Daily Times reported.
according to the Centers for Disease Control and Prevention.
The CDC received reports of 60 new measles cases last week – up from 41 the previous week – bringing the U.S. total to 940 for the year as of May 24. The CDC is currently tracking 10 outbreaks in seven states: California (3), Georgia, Maryland, Michigan, New York (2), Pennsylvania, and Washington.
The Maine Center for Disease Control and Prevention confirmed the state’s first case on May 20. The school-aged child from Somerset County had been vaccinated and is fully recovered from the disease. It’s not yet known where the child was exposed to measles, but sporadic cases are not unexpected, the Maine CDC said.
New Mexico’s first measles case of the year, a 1-year-old in Sierra County, has at least one state lawmaker considering changes to the state’s immunization exemption laws, the Farmington Daily Times reported.
FDA approves PI3K inhibitor alpelisib for breast cancer
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
FDA announces clearance of modified endoscope connector
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
FDA: Faulty hematology analyzers face class I recall
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.
The Food and Drug Administration is alerting laboratories and providers to a class I recall on Beckman Coulter hematology analyzers because of the potential for inaccurate platelet count results.
A class I recall indicates reasonable probability of serious adverse health consequences or death associated with use, according to the FDA.
The recall is related to the devices’ platelet analyzing function; among other uses, these devices help assess patients fitness for surgery, so a faulty reading on platelet counts could result in increased risk for life-threatening bleeding during a procedure in patients who have unidentified severe thrombocytopenia, according to a statement from the agency.
“Because this may cause serious injury, or even death, to a patient, we are urging health care professionals to be aware of the potential for inaccurate diagnostic results with these analyzers and to take appropriate actions including the use of alternative diagnostic testing or confirming analyzer results with manual scanning or estimate of platelets,” Tim Stenzel, MD, PhD, director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the statement.
The recall applies to the UniCel DxH 800 Coulter Cellular Analysis System, UniCel DxH 600 Coulter Cellular Analysis System, and UniCel DxH 900 Coulter Cellular Analysis System. The faulty devices were first identified in 2018, and the manufacturer released an urgent medical device correction letter at that time. The company has more recently released a software patch for the devices, but the FDA has not yet assessed whether it resolves the problem. The agency has released detailed actions and recommendations related to these devices.
At this time, the FDA is unaware of any serious adverse events that have been directly linked to these devices, but the agency recommends that any events be reported through its MedWatch reporting system.