User login
FDA panel to reassess the fate of paclitaxel-coated PAD devices
The Food and Drug Administration (FDA) announced that the Circulatory System Devices Panel of the Medical Devices Advisory Committee will meet June 19-20, 2019, at the Gaithersburg Holiday Inn, Gaithersburg, Md., to “discuss and make recommendations on information related to recent observations of increased compared to patients treated with uncoated comparator devices.”

The FDA is requesting the panel’s input “regarding the presence and magnitude of the signal and potential causes.” In addition, the FDA will seek input “regarding appropriate regulatory actions associated with the findings.”
In a Letter to Healthcare Providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
Their recommendation was: “Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending the above announced ongoing safety review.
The FDA intends to make background material available to the public no later than 2 business days before the meeting on its website at the appropriate advisory committee meeting link.
The Food and Drug Administration (FDA) announced that the Circulatory System Devices Panel of the Medical Devices Advisory Committee will meet June 19-20, 2019, at the Gaithersburg Holiday Inn, Gaithersburg, Md., to “discuss and make recommendations on information related to recent observations of increased compared to patients treated with uncoated comparator devices.”
The FDA is requesting the panel’s input “regarding the presence and magnitude of the signal and potential causes.” In addition, the FDA will seek input “regarding appropriate regulatory actions associated with the findings.”
In a Letter to Healthcare Providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
Their recommendation was: “Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending the above announced ongoing safety review.
The FDA intends to make background material available to the public no later than 2 business days before the meeting on its website at the appropriate advisory committee meeting link.
The Food and Drug Administration (FDA) announced that the Circulatory System Devices Panel of the Medical Devices Advisory Committee will meet June 19-20, 2019, at the Gaithersburg Holiday Inn, Gaithersburg, Md., to “discuss and make recommendations on information related to recent observations of increased compared to patients treated with uncoated comparator devices.”
The FDA is requesting the panel’s input “regarding the presence and magnitude of the signal and potential causes.” In addition, the FDA will seek input “regarding appropriate regulatory actions associated with the findings.”
In a Letter to Healthcare Providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
Their recommendation was: “Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending the above announced ongoing safety review.
The FDA intends to make background material available to the public no later than 2 business days before the meeting on its website at the appropriate advisory committee meeting link.
U.S. measles total sees smallest increase in 2 months
according to the Centers for Disease Control and Prevention.
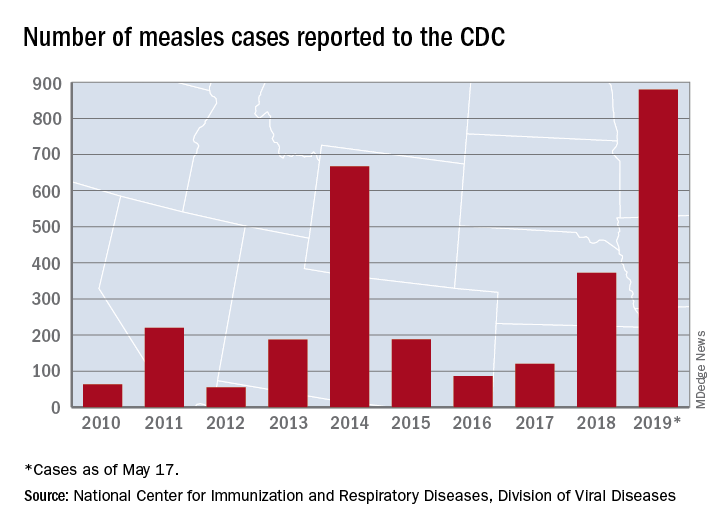
That weekly increase of 41 cases is the smallest since the week ending March 14, when the total rose by 40. The largest 1-week rise of the year came during the week ending April 11, when there were 90 new cases, CDC data show.
A case that has been reported by the media in the last week but not officially through the CDC would make New Mexico the 25th state with a measles case this year. The state’s health department has confirmed measles in a 1-year-old from Sierra County, which is New Mexico’s first case since 2014, the Las Cruces Sun News reported, adding that 4,441 school-aged children had an exemption for vaccination filed with the state in 2018.
Making a return appearance to the CDC’s list of outbreaks is Washington State, which reported six new cases last week in three Puget Sound counties (King, Pierce, and Snohomish). The most likely location and date of exposure was at Seattle-Tacoma International Airport on April 25, the Washington State Department of Health said. In February and March, there were 71 cases in Clark County on the state’s border with Oregon.
The ongoing outbreak in Michigan had been quiet since April, but the state’s Department of Health and Human Services confirmed a measles case in St. Clair County on May 17, bringing the total to 44 for the year. The new case, which is not related to an earlier outbreak that occurred mainly in Oakland County, involves an international traveler visiting Michigan.
according to the Centers for Disease Control and Prevention.
That weekly increase of 41 cases is the smallest since the week ending March 14, when the total rose by 40. The largest 1-week rise of the year came during the week ending April 11, when there were 90 new cases, CDC data show.
A case that has been reported by the media in the last week but not officially through the CDC would make New Mexico the 25th state with a measles case this year. The state’s health department has confirmed measles in a 1-year-old from Sierra County, which is New Mexico’s first case since 2014, the Las Cruces Sun News reported, adding that 4,441 school-aged children had an exemption for vaccination filed with the state in 2018.
Making a return appearance to the CDC’s list of outbreaks is Washington State, which reported six new cases last week in three Puget Sound counties (King, Pierce, and Snohomish). The most likely location and date of exposure was at Seattle-Tacoma International Airport on April 25, the Washington State Department of Health said. In February and March, there were 71 cases in Clark County on the state’s border with Oregon.
The ongoing outbreak in Michigan had been quiet since April, but the state’s Department of Health and Human Services confirmed a measles case in St. Clair County on May 17, bringing the total to 44 for the year. The new case, which is not related to an earlier outbreak that occurred mainly in Oakland County, involves an international traveler visiting Michigan.
according to the Centers for Disease Control and Prevention.
That weekly increase of 41 cases is the smallest since the week ending March 14, when the total rose by 40. The largest 1-week rise of the year came during the week ending April 11, when there were 90 new cases, CDC data show.
A case that has been reported by the media in the last week but not officially through the CDC would make New Mexico the 25th state with a measles case this year. The state’s health department has confirmed measles in a 1-year-old from Sierra County, which is New Mexico’s first case since 2014, the Las Cruces Sun News reported, adding that 4,441 school-aged children had an exemption for vaccination filed with the state in 2018.
Making a return appearance to the CDC’s list of outbreaks is Washington State, which reported six new cases last week in three Puget Sound counties (King, Pierce, and Snohomish). The most likely location and date of exposure was at Seattle-Tacoma International Airport on April 25, the Washington State Department of Health said. In February and March, there were 71 cases in Clark County on the state’s border with Oregon.
The ongoing outbreak in Michigan had been quiet since April, but the state’s Department of Health and Human Services confirmed a measles case in St. Clair County on May 17, bringing the total to 44 for the year. The new case, which is not related to an earlier outbreak that occurred mainly in Oakland County, involves an international traveler visiting Michigan.
FDA approves venetoclax/obinutuzumab combo for CLL
The Food and Drug Administration has approved the combination of venetoclax (Venclexta) plus obinutuzumab (Gazyva) for patients with previously untreated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma.

The approval provides a chemotherapy-free, fixed duration treatment. The FDA based the approval on the results of the phase 3 CLL14 trial, which will be presented at the 2019 annual meeting of the American Society of Clinical Oncology.
Researchers randomized 432 patients to either a 12-month duration of venetoclax with a 6-month duration of obinutuzumab or to a 6-month duration of obinutuzumab plus chlorambucil and another 6-month duration of chlorambucil.
The newly approved combination reduced the risk of disease progression or death (progression-free survival as assessed by an independent review committee) by 67%, compared with obinutuzumab/chlorambucil (hazard ratio, 0.33; P less than .0001).
Venetoclax/obinutuzumab also had a higher rate of minimal residual disease negativity in bone marrow and peripheral blood, compared to the other combination, according to Genentech.
The most common adverse reactions of any grade reported for venetoclax/obinutuzumab were neutropenia, diarrhea, fatigue, nausea, anemia, and upper respiratory tract infection.
The Food and Drug Administration has approved the combination of venetoclax (Venclexta) plus obinutuzumab (Gazyva) for patients with previously untreated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma.
The approval provides a chemotherapy-free, fixed duration treatment. The FDA based the approval on the results of the phase 3 CLL14 trial, which will be presented at the 2019 annual meeting of the American Society of Clinical Oncology.
Researchers randomized 432 patients to either a 12-month duration of venetoclax with a 6-month duration of obinutuzumab or to a 6-month duration of obinutuzumab plus chlorambucil and another 6-month duration of chlorambucil.
The newly approved combination reduced the risk of disease progression or death (progression-free survival as assessed by an independent review committee) by 67%, compared with obinutuzumab/chlorambucil (hazard ratio, 0.33; P less than .0001).
Venetoclax/obinutuzumab also had a higher rate of minimal residual disease negativity in bone marrow and peripheral blood, compared to the other combination, according to Genentech.
The most common adverse reactions of any grade reported for venetoclax/obinutuzumab were neutropenia, diarrhea, fatigue, nausea, anemia, and upper respiratory tract infection.
The Food and Drug Administration has approved the combination of venetoclax (Venclexta) plus obinutuzumab (Gazyva) for patients with previously untreated chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma.
The approval provides a chemotherapy-free, fixed duration treatment. The FDA based the approval on the results of the phase 3 CLL14 trial, which will be presented at the 2019 annual meeting of the American Society of Clinical Oncology.
Researchers randomized 432 patients to either a 12-month duration of venetoclax with a 6-month duration of obinutuzumab or to a 6-month duration of obinutuzumab plus chlorambucil and another 6-month duration of chlorambucil.
The newly approved combination reduced the risk of disease progression or death (progression-free survival as assessed by an independent review committee) by 67%, compared with obinutuzumab/chlorambucil (hazard ratio, 0.33; P less than .0001).
Venetoclax/obinutuzumab also had a higher rate of minimal residual disease negativity in bone marrow and peripheral blood, compared to the other combination, according to Genentech.
The most common adverse reactions of any grade reported for venetoclax/obinutuzumab were neutropenia, diarrhea, fatigue, nausea, anemia, and upper respiratory tract infection.
FDA panel not ready to recommend quizartinib approval for FLT3-ITD+ AML
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.
Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.
Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
SILVER SPRING, MD. – Daiichi Sankyo failed to make the case for approval of its investigational tyrosine kinase inhibitor quizartinib for patients with acute myeloid leukemia bearing the FLT3 internal tandem duplication (ITD) mutation.
Members of the Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted 8-3 not to recommend approval of the drug at this time, despite the prevailing sentiment among oncologists on the panel that, as one stated, “I need this drug. I want this drug.”
The prevailing majority of committee members agreed that the drug may have a place in the treatment of patients with FLT3-mutated AML, but that more robust data were needed to prove it.
Currently, only one agent, gilteritinib (Xospata) is approved by the FDA for the treatment of patients with relapsed or refractory FLT3-mutated AML.
QuANTUM-R
Daiichi Sankyo sought approval for quizartinib based on results of the phase 3 randomized QuANTUM-R trial. In this trial, single-agent therapy with quizartinib slightly but significantly prolonged survival – compared with salvage chemotherapy – of patients with relapsed/refractory FLT3-ITD positive AML.
Median overall survival (OS), the trial’s primary endpoint, was 6.2 months for 245 patients randomized to quizartinib, compared with 4.7 months for 122 patients assigned to salvage chemotherapy, a difference that translated into a hazard ratio (HR) for death of 0.76 (P = .0177).
The patients were randomly assigned on a 2:1 basis to receive either quizartinib or salvage chemotherapy. Quizartinib was dosed 30 mg per day for 15 days, which could be titrated upward to 60 mg daily if the corrected QT interval by Fredericia (QTcF) was 450 ms or less on day 16.
Chemotherapy was the investigator’s choice of one of three specified regimens: either low-dose cytarabine (LoDAC); mitoxantrone, etoposide, and intermediate-dose cytarabine (MEC); or fludarabine, cytarabine, and granulocyte-colony stimulating factor (G-CSF) with idarubicin (FLAG-IDA). Up to 2 cycles of MEC or FLAG-IDA were permitted; quizartinib and LoDAC were given until lack of benefit, unacceptable toxicity, or until the patient went on to hematopoietic stem cell transplant (HSCT).
Principal investigator Jorge Cortes, MD, from the University of Texas MD Anderson Cancer Center in Houston, speaking in support of the application, said that combined with the phase 2 study results, “these data support a clear and clinically meaningful benefit of quizartinib in this patient population.”
Mark Levis, MD. PhD, from the Johns Hopkins Sidney Kimmel Cancer Center in Baltimore, also spoke in support of the FLT3 inhibitor.
“I have studied both in the lab and in the clinic most FLT3 inhibitors that have been developed, including lestaurtinib, midostaurin, sorafenib and gilteritinib. Quizartinib is the most highly potent and selective FLT3 inhibitor I have ever worked with,” Dr. Levis said.
FDA: Data not up to snuff
But as FDA staff member Kunthel By, PhD, a statistical reviewer in the Office of Biostatistics, pointed out, the upper limit of the hazard ratio favoring quizartinib over chemotherapy was 0.99, and the difference in median overall survival was just 6.5 weeks.
Additionally, the trial data lacked internal consistency, showing no benefits for the drug in either event-free survival (EFS) or in complete response rates.
There were also imbalances in the number of patients with subsequent HSCT between the arms, with more patients on quizartinib undergoing HSCT despite not having a complete remission, than in the chemotherapy group. Also, there were differences in the number of patients who were randomized but not treated and in those censored early. And statistical stress tests indicated “a lack of robustness in the estimated treatment effect,” he said.
Safety issues raised in QuANTUM-R included slow potassium channel (IKs) blockade and related cardiac toxicitites, as well as the differentiation syndrome, acute febrile neutrophilic dermatosis, and cytopenias, said Aviva Krauss, MD, a clinical reviewer in the FDA’s Office of Hematology and Oncology Products.
“Quizartinib therapy is associated with significant and unique safety concerns in the [proposed population], including the risk of fatal cardiac events that cannot be predicted with certainty using routine QTc measurements,” she said.
She noted that the events occurred in QuANTUM-R despite dose modifications and concomitant medications guidelines in the study protocol.
Reviewers recommended that should the drug receive approval, the package labeling should include contraindication for use with other QT-prolonging agents, and a recommendation for prophylactic beta blockage, although the panelists in general felt that the latter recommendation was not necessary.
‘I believe in this drug’
The ODAC meeting was convened to answer questions about whether the overall survival results were credible based on a single clinical trial and outweighed the risks of treatment with quizartinib, and to assess risk strategies for reducing risks of potentially fatal cardiac toxicities, primarily prolongation of the QT interval.
A. Michael Lincoff, MD, a cardiologist at Case Western Reserve University and the Cleveland Clinic, both in Cleveland, Ohio, voted in favor of approval.
“I’m less concerned about the risk and I do think on the balance there is benefit,” he said.
But most committee members echoed the comments of Anthony D. Sung, MD, from the division of hematologic malignancies and cellular therapy at Duke University in Durham, N.C.
“My vote is based purely on the data I’m shown, and my vote is no,” he said. “But I want the FDA to know that I believe in this drug, and I think it should get approved, and I want to use it.”
The trial was sponsored by Daiichi Sankyo. Dr. Cortes reported research funding from Daiichi Sankyo, Pfizer, Arog, Astellas Pharma and Novartis, and consulting activities for all of the same companies except Arog. Dr. Levis is a paid consultant for Daiichi Sankyo. He and Dr. Cortes stated that they had no financial interests in the outcome of the ODAC meeting.
Peanut contamination risk prompts Promacta recall
Novartis has recalled three lots of 12.5-mg eltrombopag (Promacta) for oral suspension following discovery of possible contamination with peanut flour at a third-party manufacturing site.
Tablets at doses of 12.5 mg, 25 mg, 50 mg, and 75 mg are unaffected by this recall because they are not manufactured in the same facility. The recalled lots of medication were distributed between January and April 2019, but so far, Novartis has not received any reports of adverse events related to the recall.
Oral suspension of eltrombopag is indicated for certain patients with chronic immune thrombocytopenia, hepatitis C–associated thrombocytopenia, and severe aplastic anemia.
More information on the recalled lots and instructions on how to return the product can be found in the full announcement, which is also available through the Food and Drug Administration website.
Novartis has recalled three lots of 12.5-mg eltrombopag (Promacta) for oral suspension following discovery of possible contamination with peanut flour at a third-party manufacturing site.
Tablets at doses of 12.5 mg, 25 mg, 50 mg, and 75 mg are unaffected by this recall because they are not manufactured in the same facility. The recalled lots of medication were distributed between January and April 2019, but so far, Novartis has not received any reports of adverse events related to the recall.
Oral suspension of eltrombopag is indicated for certain patients with chronic immune thrombocytopenia, hepatitis C–associated thrombocytopenia, and severe aplastic anemia.
More information on the recalled lots and instructions on how to return the product can be found in the full announcement, which is also available through the Food and Drug Administration website.
Novartis has recalled three lots of 12.5-mg eltrombopag (Promacta) for oral suspension following discovery of possible contamination with peanut flour at a third-party manufacturing site.
Tablets at doses of 12.5 mg, 25 mg, 50 mg, and 75 mg are unaffected by this recall because they are not manufactured in the same facility. The recalled lots of medication were distributed between January and April 2019, but so far, Novartis has not received any reports of adverse events related to the recall.
Oral suspension of eltrombopag is indicated for certain patients with chronic immune thrombocytopenia, hepatitis C–associated thrombocytopenia, and severe aplastic anemia.
More information on the recalled lots and instructions on how to return the product can be found in the full announcement, which is also available through the Food and Drug Administration website.
FDA issues final guidance on seeking licensure for interchangeable biologics

The action provides “clarity for developers who want to demonstrate that their proposed biological product meets the statutory interchangeability standard under the Public Health Service Act,” Norman E. Sharpless, MD, the acting commissioner of the agency, said in a press release.
Biologics deemed “interchangeable” will be able to be substituted without the involvement of the prescriber, similar to how generic drugs are now routinely substituted for brand name drugs. On March 23, 2020, the FDA will be able to license such interchangeable products.
On May 13, the agency will consider what factors need to be weighed when determining whether an insulin product is biosimilar to or interchangeable with a reference product at a public hearing: “The Future of Insulin Biosimilars: Increasing Access and Facilitating the Efficient Development of Biosimilar and Interchangeable Insulin Products.”
“We also expect to hear stakeholder feedback on whether certain insulin products – for example, those that use insulin pumps for continuous subcutaneous infusion among the approved uses – raise unique scientific considerations that we should be considering when evaluating biosimilar or interchangeable insulin products. And importantly, we’ll also be seeking input directly from patients about their experience with insulin products, and this input will inform the FDA’s approach to implementing the regulatory pathway for biosimilar and interchangeable insulin products,” Dr. Sharpless said in the statement.
The ability to substitute an interchangeable insulin product – or any chronically used biologic medication – at the pharmacy could potentially increase access and lower costs for patients.
Dr. Sharpless added that the FDA has developed and is working to implement a Biosimilars Action Plan that includes a suite of ongoing efforts to encourage innovation and competition among biologics and the development of biosimilars.
The final guidance gives an overview of important scientific considerations in demonstrating interchangeability with a reference product and explains the scientific recommendations for an application or a supplement for a proposed interchangeable product. The guidance also explains potential ways to address the Biologics Price Competition and Innovation Act requirement for interchangeability that, for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product will not be greater than the risk of using the reference product without alternating or switching.
“Our rigorous scientific standards for approval will be maintained for interchangeable biologics and should serve as assurance to health care professionals and patients that they can be confident in the safety and effectiveness of both interchangeable products and biosimilar products, just as they would be for reference products,” Dr. Sharpless said in the statement.
Medical specialty organizations have begun to weigh in on the FDA final guidance, “Considerations in Demonstrating Interchangeability with a Reference Product.”
The American College of Rheumatology applauded the FDA action.
“Specifically, we are pleased to see that the final guidance expects manufacturers to use robust switching studies. At least three switches with each switch crossing over to the alternate product will be needed to determine whether alternating between a biosimilar and its reference product impacts the safety or efficacy of the drug. The ACR believes these studies will provide an understanding of what patients are likely to experience when changing formularies in a multipayer, multistate market,” Dr. Angus Worthing, chair of the American College of Rheumatology’s Government Affairs Committee, said in a statement.
“We are also pleased to see the FDA finalize its approach to safety, immunogenicity, and efficacy for the demonstration of interchangeability. And we agree with the FDA that postmarketing safety monitoring for an interchangeable product should also have robust pharmacovigilance mechanisms in place. In order to improve clarity, the ACR suggests that FDA prescribing information for all biosimilars include statements about whether each agent is or is not interchangeable to the reference product,” he said. “The ACR shares the FDA’s goal of ensuring that more affordable treatments reach patients as quickly as possible and appreciates the agency’s measured and thoughtful approach throughout this process.”
The action provides “clarity for developers who want to demonstrate that their proposed biological product meets the statutory interchangeability standard under the Public Health Service Act,” Norman E. Sharpless, MD, the acting commissioner of the agency, said in a press release.
Biologics deemed “interchangeable” will be able to be substituted without the involvement of the prescriber, similar to how generic drugs are now routinely substituted for brand name drugs. On March 23, 2020, the FDA will be able to license such interchangeable products.
On May 13, the agency will consider what factors need to be weighed when determining whether an insulin product is biosimilar to or interchangeable with a reference product at a public hearing: “The Future of Insulin Biosimilars: Increasing Access and Facilitating the Efficient Development of Biosimilar and Interchangeable Insulin Products.”
“We also expect to hear stakeholder feedback on whether certain insulin products – for example, those that use insulin pumps for continuous subcutaneous infusion among the approved uses – raise unique scientific considerations that we should be considering when evaluating biosimilar or interchangeable insulin products. And importantly, we’ll also be seeking input directly from patients about their experience with insulin products, and this input will inform the FDA’s approach to implementing the regulatory pathway for biosimilar and interchangeable insulin products,” Dr. Sharpless said in the statement.
The ability to substitute an interchangeable insulin product – or any chronically used biologic medication – at the pharmacy could potentially increase access and lower costs for patients.
Dr. Sharpless added that the FDA has developed and is working to implement a Biosimilars Action Plan that includes a suite of ongoing efforts to encourage innovation and competition among biologics and the development of biosimilars.
The final guidance gives an overview of important scientific considerations in demonstrating interchangeability with a reference product and explains the scientific recommendations for an application or a supplement for a proposed interchangeable product. The guidance also explains potential ways to address the Biologics Price Competition and Innovation Act requirement for interchangeability that, for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product will not be greater than the risk of using the reference product without alternating or switching.
“Our rigorous scientific standards for approval will be maintained for interchangeable biologics and should serve as assurance to health care professionals and patients that they can be confident in the safety and effectiveness of both interchangeable products and biosimilar products, just as they would be for reference products,” Dr. Sharpless said in the statement.
Medical specialty organizations have begun to weigh in on the FDA final guidance, “Considerations in Demonstrating Interchangeability with a Reference Product.”
The American College of Rheumatology applauded the FDA action.
“Specifically, we are pleased to see that the final guidance expects manufacturers to use robust switching studies. At least three switches with each switch crossing over to the alternate product will be needed to determine whether alternating between a biosimilar and its reference product impacts the safety or efficacy of the drug. The ACR believes these studies will provide an understanding of what patients are likely to experience when changing formularies in a multipayer, multistate market,” Dr. Angus Worthing, chair of the American College of Rheumatology’s Government Affairs Committee, said in a statement.
“We are also pleased to see the FDA finalize its approach to safety, immunogenicity, and efficacy for the demonstration of interchangeability. And we agree with the FDA that postmarketing safety monitoring for an interchangeable product should also have robust pharmacovigilance mechanisms in place. In order to improve clarity, the ACR suggests that FDA prescribing information for all biosimilars include statements about whether each agent is or is not interchangeable to the reference product,” he said. “The ACR shares the FDA’s goal of ensuring that more affordable treatments reach patients as quickly as possible and appreciates the agency’s measured and thoughtful approach throughout this process.”
The action provides “clarity for developers who want to demonstrate that their proposed biological product meets the statutory interchangeability standard under the Public Health Service Act,” Norman E. Sharpless, MD, the acting commissioner of the agency, said in a press release.
Biologics deemed “interchangeable” will be able to be substituted without the involvement of the prescriber, similar to how generic drugs are now routinely substituted for brand name drugs. On March 23, 2020, the FDA will be able to license such interchangeable products.
On May 13, the agency will consider what factors need to be weighed when determining whether an insulin product is biosimilar to or interchangeable with a reference product at a public hearing: “The Future of Insulin Biosimilars: Increasing Access and Facilitating the Efficient Development of Biosimilar and Interchangeable Insulin Products.”
“We also expect to hear stakeholder feedback on whether certain insulin products – for example, those that use insulin pumps for continuous subcutaneous infusion among the approved uses – raise unique scientific considerations that we should be considering when evaluating biosimilar or interchangeable insulin products. And importantly, we’ll also be seeking input directly from patients about their experience with insulin products, and this input will inform the FDA’s approach to implementing the regulatory pathway for biosimilar and interchangeable insulin products,” Dr. Sharpless said in the statement.
The ability to substitute an interchangeable insulin product – or any chronically used biologic medication – at the pharmacy could potentially increase access and lower costs for patients.
Dr. Sharpless added that the FDA has developed and is working to implement a Biosimilars Action Plan that includes a suite of ongoing efforts to encourage innovation and competition among biologics and the development of biosimilars.
The final guidance gives an overview of important scientific considerations in demonstrating interchangeability with a reference product and explains the scientific recommendations for an application or a supplement for a proposed interchangeable product. The guidance also explains potential ways to address the Biologics Price Competition and Innovation Act requirement for interchangeability that, for a biological product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product will not be greater than the risk of using the reference product without alternating or switching.
“Our rigorous scientific standards for approval will be maintained for interchangeable biologics and should serve as assurance to health care professionals and patients that they can be confident in the safety and effectiveness of both interchangeable products and biosimilar products, just as they would be for reference products,” Dr. Sharpless said in the statement.
Medical specialty organizations have begun to weigh in on the FDA final guidance, “Considerations in Demonstrating Interchangeability with a Reference Product.”
The American College of Rheumatology applauded the FDA action.
“Specifically, we are pleased to see that the final guidance expects manufacturers to use robust switching studies. At least three switches with each switch crossing over to the alternate product will be needed to determine whether alternating between a biosimilar and its reference product impacts the safety or efficacy of the drug. The ACR believes these studies will provide an understanding of what patients are likely to experience when changing formularies in a multipayer, multistate market,” Dr. Angus Worthing, chair of the American College of Rheumatology’s Government Affairs Committee, said in a statement.
“We are also pleased to see the FDA finalize its approach to safety, immunogenicity, and efficacy for the demonstration of interchangeability. And we agree with the FDA that postmarketing safety monitoring for an interchangeable product should also have robust pharmacovigilance mechanisms in place. In order to improve clarity, the ACR suggests that FDA prescribing information for all biosimilars include statements about whether each agent is or is not interchangeable to the reference product,” he said. “The ACR shares the FDA’s goal of ensuring that more affordable treatments reach patients as quickly as possible and appreciates the agency’s measured and thoughtful approach throughout this process.”
U.S. measles cases climb to over 800 for the year
according to the Centers for Disease Control and Prevention.
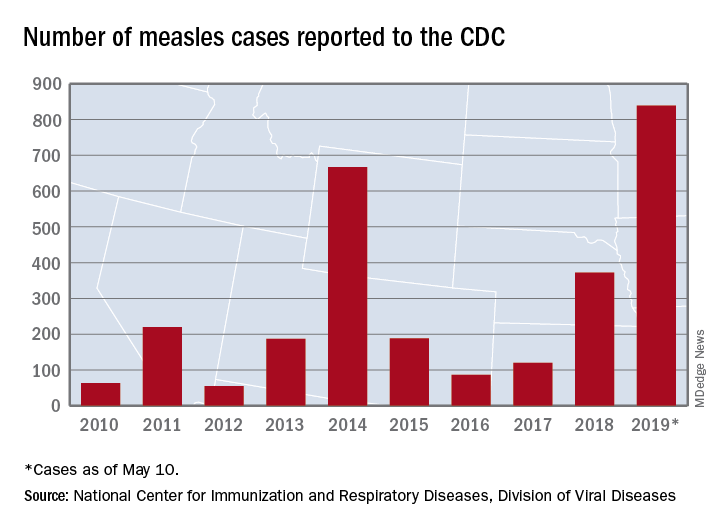
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
according to the Centers for Disease Control and Prevention.
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
according to the Centers for Disease Control and Prevention.
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
In a tight vote, FDA panel backs mannitol for CF
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).
Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).
Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).
Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
FROM AN FDA ADVISORY COMMITTEE HEARING
Insomnia meds get boxed warning from FDA
The Food and Drug Administration will now require that certain
Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
The Food and Drug Administration will now require that certain
Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
The Food and Drug Administration will now require that certain
Complex sleep behaviors have been seen with these medications in patients with and without a history of them, at low doses, and even after one dose of the medication. They’ve also been observed with and without concomitant use of alcohol or other CNS depressants.
Health care professionals should advise patients about these risks, even though they are rare. Patients should contact health care professionals if they either experience a complex sleep behavior while not fully awake on one of these medicines or have performed activities they don’t remember while taking the medicine.
More information about these risks and the safety warnings can be found in the FDA’s safety announcement. Other information is also available in a press announcement from the agency.
Most pregnancy-related deaths are preventable
according to the Centers for Disease Control and Prevention.
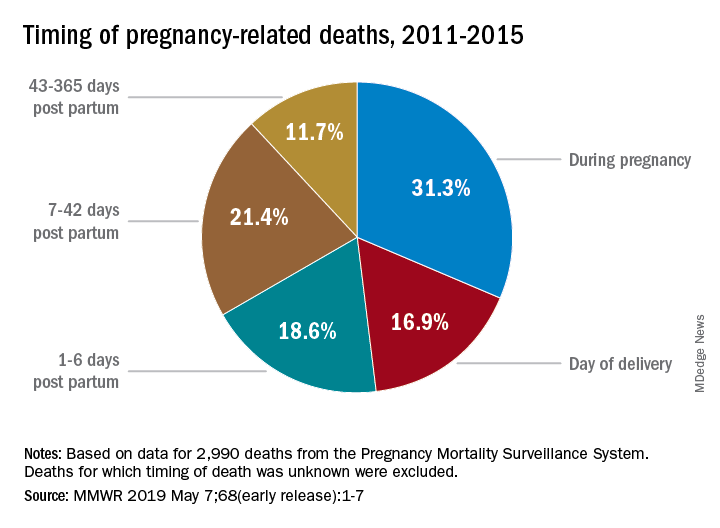
Deaths from pregnancy-related complications can occur “up to a year after delivery,” the CDC emphasized in a report released May 7. Indeed, 31% of pregnancy-related deaths happen during pregnancy, 36% happen at delivery or in the week after, and 33% happen 1 week to 1 year post partum. Yet detailed data from 13 state maternal mortality review committees (MMRCs) showed that 60% of such deaths are preventable.
There were 17 pregnancy-related deaths per 100,000 live births during 2011-2015, based on another data source: 3,410 pregnancy-related deaths (an average of 682 deaths per year) in the CDC’s Pregnancy Mortality Surveillance System. That pregnancy-related mortality ratio varied by race/ethnicity, Emily E. Peterson, MD, and the other CDC investigators reported in Morbidity and Mortality Weekly Report: Hispanic (11 deaths per 100,000), white (13), Asian/Pacific Islander (14), American Indian/Alaska Native (33), and black (43).
One aspect of the disparity was addressed by Wanda Barfield, MD, MPH, director of the CDC’s division of reproductive health and assistant surgeon general in the U.S. Public Health Service. “Recent studies have shown that racial and ethnic minority women deliver at different and lower-quality hospitals than white women and that these hospitals disproportionately care for black women at delivery,” she said at a CDC telebriefing.
Analysis of the timing of 2,990 deaths from the Pregnancy Mortality Surveillance System showed that almost a third (31.3%) occurred during pregnancy and 16.9% occurred on the day of delivery. Dr. Peterson and her CDC associates noted that more than half of pregnancy-related deaths, however, took place later: 1-6 days post partum (18.6%), 7-42 days (21.4%), and 43-365 days (11.7%).
The data on preventability were collected by the state MMRCs and included 232 deaths that occurred during 2013-2017. The MMRCs considered deaths preventable if they could be “averted by one or more reasonable changes to patient, community, provider, health facility, and/or system factors.”
“Our new analysis underscores the need for access to quality services, risk awareness, and early diagnosis, but it also highlights opportunities for preventing future pregnancy-related deaths,” Dr. Barfield said. “By identifying and promptly responding to warning signs not just during pregnancy, but even up to a year after delivery, we can save lives.”
according to the Centers for Disease Control and Prevention.
Deaths from pregnancy-related complications can occur “up to a year after delivery,” the CDC emphasized in a report released May 7. Indeed, 31% of pregnancy-related deaths happen during pregnancy, 36% happen at delivery or in the week after, and 33% happen 1 week to 1 year post partum. Yet detailed data from 13 state maternal mortality review committees (MMRCs) showed that 60% of such deaths are preventable.
There were 17 pregnancy-related deaths per 100,000 live births during 2011-2015, based on another data source: 3,410 pregnancy-related deaths (an average of 682 deaths per year) in the CDC’s Pregnancy Mortality Surveillance System. That pregnancy-related mortality ratio varied by race/ethnicity, Emily E. Peterson, MD, and the other CDC investigators reported in Morbidity and Mortality Weekly Report: Hispanic (11 deaths per 100,000), white (13), Asian/Pacific Islander (14), American Indian/Alaska Native (33), and black (43).
One aspect of the disparity was addressed by Wanda Barfield, MD, MPH, director of the CDC’s division of reproductive health and assistant surgeon general in the U.S. Public Health Service. “Recent studies have shown that racial and ethnic minority women deliver at different and lower-quality hospitals than white women and that these hospitals disproportionately care for black women at delivery,” she said at a CDC telebriefing.
Analysis of the timing of 2,990 deaths from the Pregnancy Mortality Surveillance System showed that almost a third (31.3%) occurred during pregnancy and 16.9% occurred on the day of delivery. Dr. Peterson and her CDC associates noted that more than half of pregnancy-related deaths, however, took place later: 1-6 days post partum (18.6%), 7-42 days (21.4%), and 43-365 days (11.7%).
The data on preventability were collected by the state MMRCs and included 232 deaths that occurred during 2013-2017. The MMRCs considered deaths preventable if they could be “averted by one or more reasonable changes to patient, community, provider, health facility, and/or system factors.”
“Our new analysis underscores the need for access to quality services, risk awareness, and early diagnosis, but it also highlights opportunities for preventing future pregnancy-related deaths,” Dr. Barfield said. “By identifying and promptly responding to warning signs not just during pregnancy, but even up to a year after delivery, we can save lives.”
according to the Centers for Disease Control and Prevention.
Deaths from pregnancy-related complications can occur “up to a year after delivery,” the CDC emphasized in a report released May 7. Indeed, 31% of pregnancy-related deaths happen during pregnancy, 36% happen at delivery or in the week after, and 33% happen 1 week to 1 year post partum. Yet detailed data from 13 state maternal mortality review committees (MMRCs) showed that 60% of such deaths are preventable.
There were 17 pregnancy-related deaths per 100,000 live births during 2011-2015, based on another data source: 3,410 pregnancy-related deaths (an average of 682 deaths per year) in the CDC’s Pregnancy Mortality Surveillance System. That pregnancy-related mortality ratio varied by race/ethnicity, Emily E. Peterson, MD, and the other CDC investigators reported in Morbidity and Mortality Weekly Report: Hispanic (11 deaths per 100,000), white (13), Asian/Pacific Islander (14), American Indian/Alaska Native (33), and black (43).
One aspect of the disparity was addressed by Wanda Barfield, MD, MPH, director of the CDC’s division of reproductive health and assistant surgeon general in the U.S. Public Health Service. “Recent studies have shown that racial and ethnic minority women deliver at different and lower-quality hospitals than white women and that these hospitals disproportionately care for black women at delivery,” she said at a CDC telebriefing.
Analysis of the timing of 2,990 deaths from the Pregnancy Mortality Surveillance System showed that almost a third (31.3%) occurred during pregnancy and 16.9% occurred on the day of delivery. Dr. Peterson and her CDC associates noted that more than half of pregnancy-related deaths, however, took place later: 1-6 days post partum (18.6%), 7-42 days (21.4%), and 43-365 days (11.7%).
The data on preventability were collected by the state MMRCs and included 232 deaths that occurred during 2013-2017. The MMRCs considered deaths preventable if they could be “averted by one or more reasonable changes to patient, community, provider, health facility, and/or system factors.”
“Our new analysis underscores the need for access to quality services, risk awareness, and early diagnosis, but it also highlights opportunities for preventing future pregnancy-related deaths,” Dr. Barfield said. “By identifying and promptly responding to warning signs not just during pregnancy, but even up to a year after delivery, we can save lives.”