User login
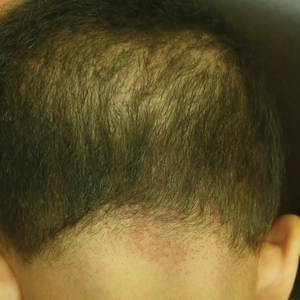
Sparse Hair on the Scalp
The Diagnosis: Monilethrix
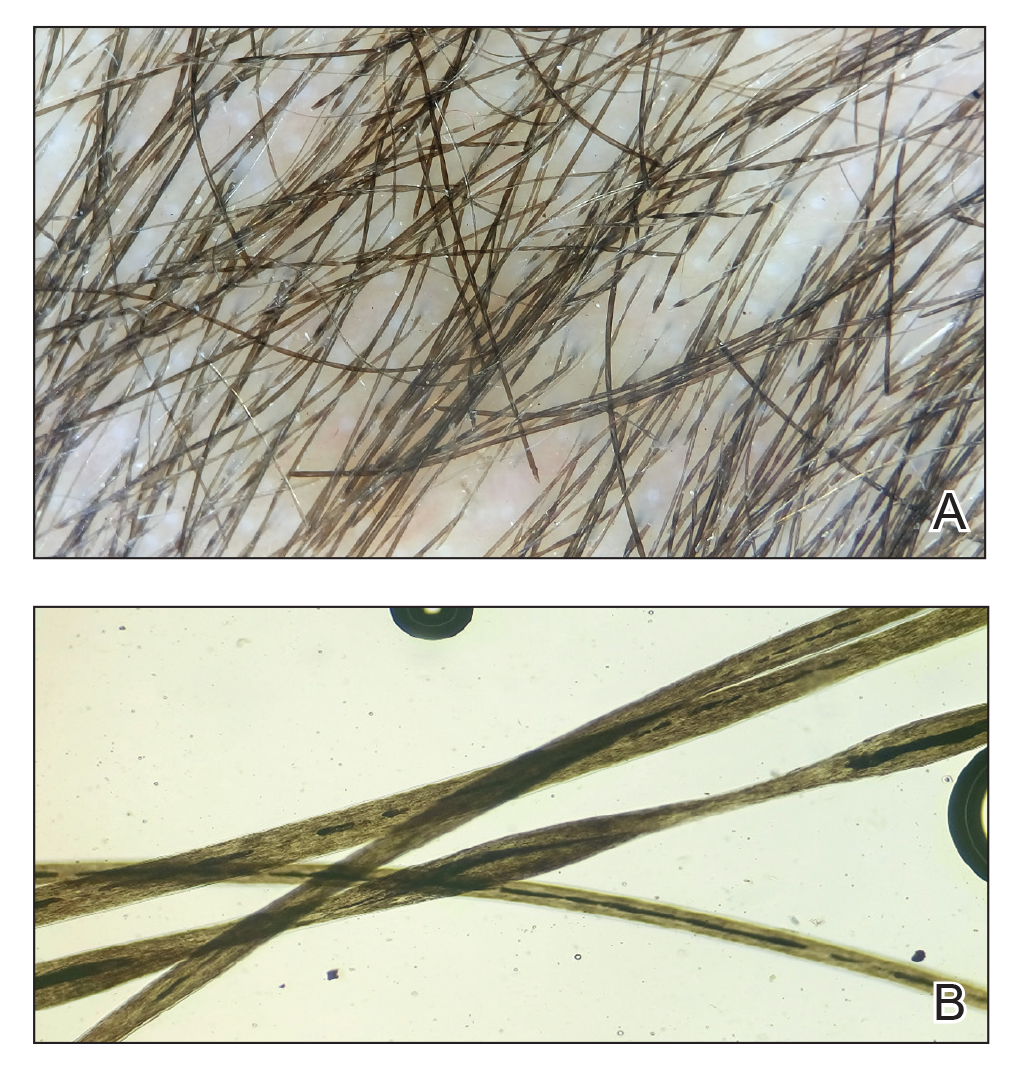
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
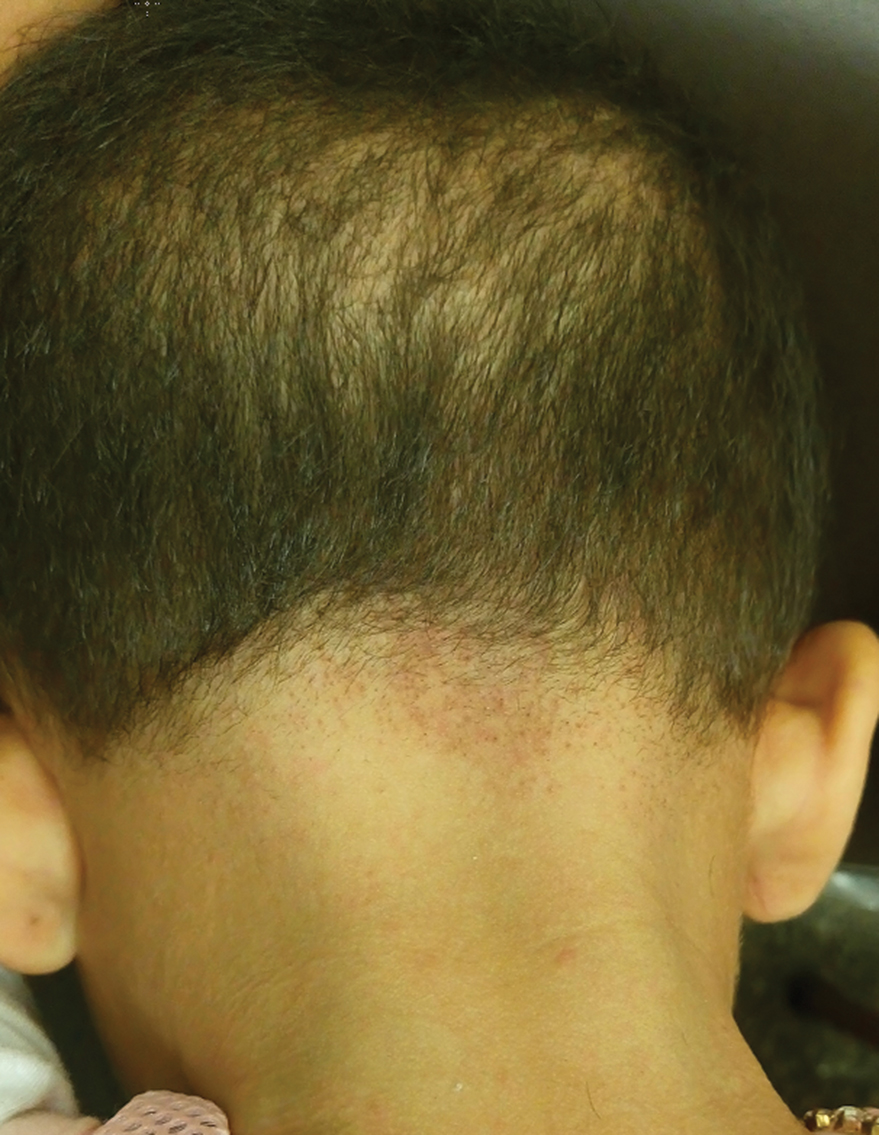
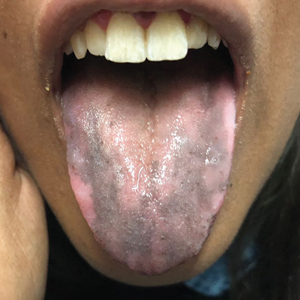
A 5-year-old girl presented to our clinic with sparse scalp hair. Her mother reported thinning of the hair and breakage that appeared shortly after birth. She also reported that the patient’s hair was dull, dry, and unable to be grown long. The patient was otherwise healthy. She was born to nonconsanguineous parents, and her family history was unremarkable. Physical examination revealed dry, brittle, and short hairs. The hair was sparser on the occipital area of the scalp, and multiple keratotic papules were noted in this area. No abnormalities were detected on the teeth or nails, and a review of systems was unremarkable. Trichoscopy and light microscopy were performed.
Hemorrhagic Papular Eruption on the Dorsal Hands
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
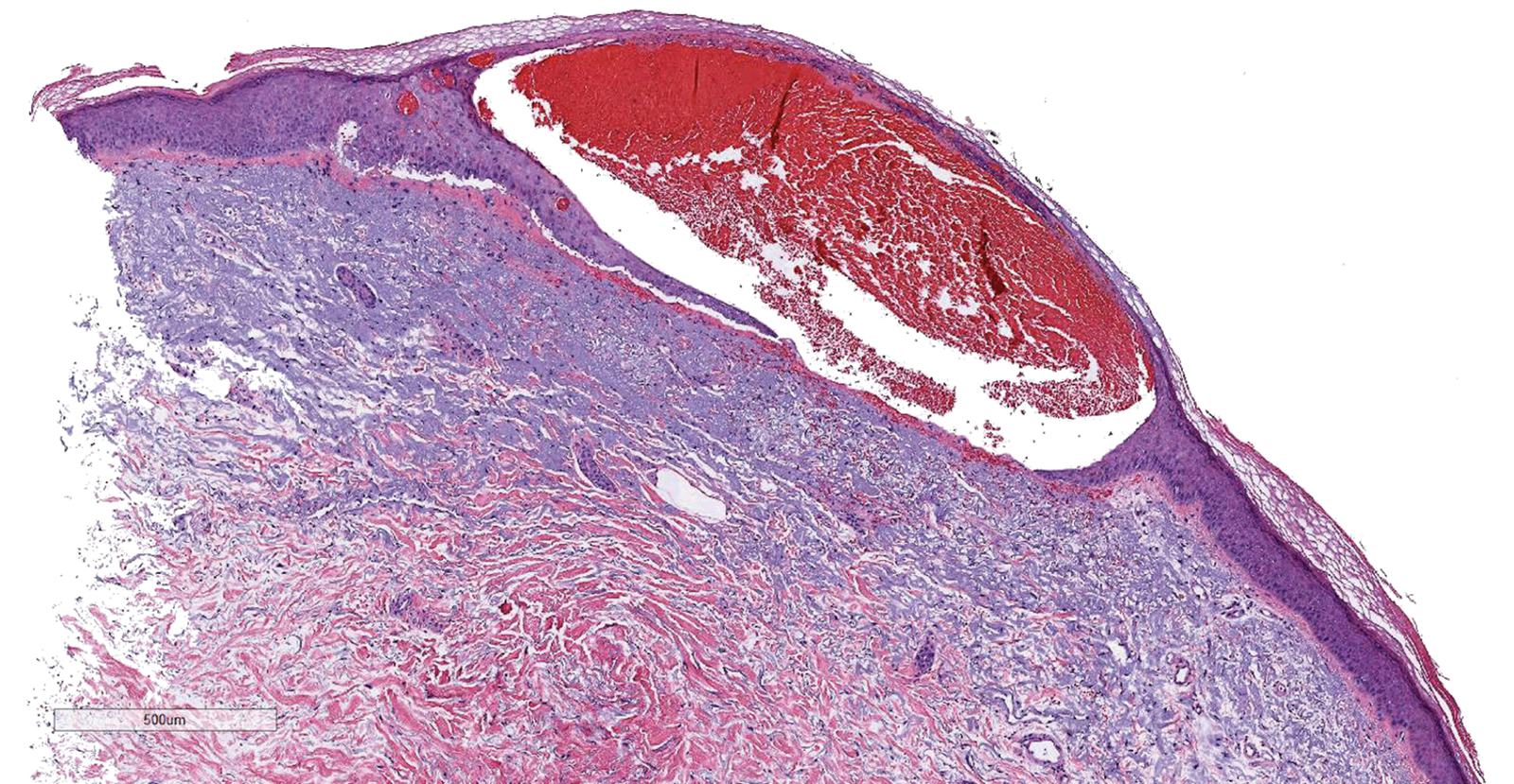
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
The Diagnosis: Heparin-Induced Bullous Hemorrhagic Dermatosis
Results of a punch biopsy of one of the hemorrhagic papules revealed a subcorneal hemorrhagic vesicle without underlying vasculitis, vasculopathy, inflammation, or viral changes (Figure). Tissue and blood cultures were sterile. Heparin and platelet factor 4 antibody testing was negative. The patient was diagnosed with heparin induced bullous hemorrhagic dermatosis (BHD). After chest imaging ruled out a pulmonary embolism, anticoagulation therapy was discontinued. Respiratory symptoms improved on antibiotics, and the skin lesions resolved completely within 2 weeks.
Bullous hemorrhagic dermatosis is an uncommon and underrecognized reaction to various anticoagulants. Bullous hemorrhagic dermatosis presents with painless, noninflammatory, hemorrhagic vesicles and bullae occurring at sites distant from anticoagulant administration. The condition was first characterized in 2006 by Perrinaud et al,1 who presented 3 cases in patients treated with heparin and low-molecular-weight heparin. Since then, there have been at least 90 cases reported in the international literature, with elderly men found to be the more affected demographic (male to female ratio, 1.9:1).2 Typically, BHD presents within 1 week of administration of an anticoagulant, but delayed onset has been reported.2 Bullous hemorrhagic dermatosis is most commonly observed with enoxaparin use but also has been described in association with unfractionated heparin, low-molecular-weight heparin products, and warfarin.2
The noninflammatory-appearing hemorrhagic papules and small plaques of BHD generally are seen on the extremities but can occur anywhere on the body including the oral mucosa.3 The differential diagnosis of BHD may include autoimmune vesiculobullous conditions, bullous drug eruptions, herpetic infection, supratherapeutic anticoagulation, porphyria cutanea tarda, amyloidosis, leukocytoclastic vasculitis, angioinvasive infections, and heparin necrosis. Diagnosis of BHD can be made clinically, but a biopsy is useful to exclude other conditions.
Histologically, BHD is characterized by the presence of intraepidermal hemorrhagic bullae without thrombotic, inflammatory, or vasculitic changes. Although heparinrelated skin lesions have been attributed to various mechanisms, including immune-mediated thrombocytopenia, type IV hypersensitivity reactions, type I allergic hypersensitivity reactions, pustulosis, and skin necrosis, the pathogenesis of BHD remains poorly understood.4 The condition has demonstrated koebnerization in some cases.5
In our patient, the absence of histologic inflammation, viral changes, vasculitis, and amyloid deposition helped rule out the other entities in the differential. The absence of heparin and platelet factor 4 antibodies helped exclude heparin necrosis. Direct immunofluorescence testing was not obtained in our patient but may be used to evaluate for an immunobullous etiology.
Management strategies for BHD are variable, and associated evidence is lacking. Treatment of BHD should be considered in the clinical context based on the necessity for anticoagulation and the severity of the eruption. Discontinuation of anticoagulation therapy, if possible, may prevent morbidity in some cases.6 If it is necessary to continue anticoagulation therapy, changing the drug or decreasing the dose are reasonable options. Skin lesions may resolve even if anticoagulation therapy is continued at the same dose.7,8 Concurrent supportive wound care is beneficial.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
- Perrinaud A, Jacobi D, Machet MC, et al. Bullous hemorrhagic dermatosis occurring at sites distant from subcutaneous injections of heparin: three cases. J Am Acad Dermatol. 2006;54(suppl):S5-S7.
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose low-molecular weight heparin: a case report and review of the literature [published online July 6, 2018]. Exp Hematol. 2018;7:15.
- Harris HB, Kurth BJ, Lam TK, et al. Heparin-induced bullous hemorrhagic dermatosis confined to the oral mucosa. Cutis. 2019;103:365-366, 370.
- Schindewolf M, Schwaner S, Wolter M, et al. Incidence and causes of heparin-induced skin lesions. CMAJ. 2009;181:477-481.
- Gargallo V, Romero FT, Rodríguez-Peralto JL, et al. Heparin induced bullous hemorrhagic dermatosis at a site distant from the injection. a report of five cases. An Bras Dermatol. 2016;91:857-859.
- Choudhry S, Fishman PM, Hernandez C. Heparin-induced bullous hemorrhagic dermatosis. Cutis. 2013;91:93-98.
- Maldonado Cid P, Moreno Alonso de Celada R, Herranz Pinto P, et al. Bullous hemorrhagic dermatosis at sites distant from subcutaneous injections of heparin: a report of 5 cases. J Am Acad Dermatol. 2012;67:E220-E222.
- Snow SC, Pearson DR, Fathi R, et al. Heparin-induced haemorrhagic bullous dermatosis. Clin Exp Dermatol. 2018;43:393-398.
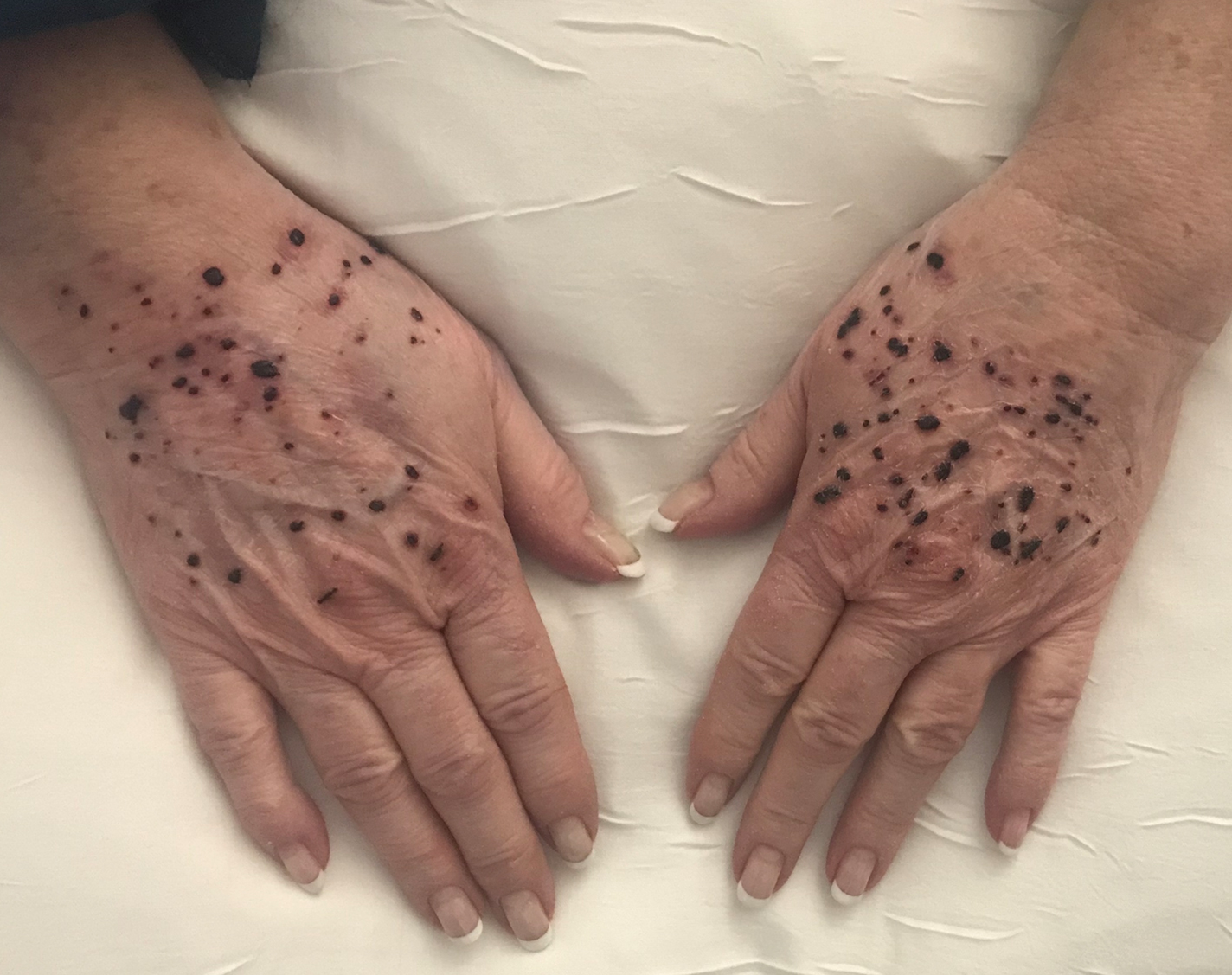
A 66-year-old woman with a history of granulomatous lung disease managed with methotrexate and prednisone, diabetes mellitus, hypertension, and Grave disease was admitted to the hospital for hypoxic respiratory failure. At admission, treatment was empirically initiated for pneumonia with intravenous ceftriaxone and azithromycin. Given the concern of a pulmonary embolism, intravenous heparin also was initiated. Dermatology was consulted for multiple painless blood blisters that erupted on the hands within 24 hours of admission. Physical examination revealed numerous firm hemorrhagic papules on the dorsal hands. Laboratory workup revealed a slightly elevated white blood cell count (11,800/µL [reference range, 4500–11,000/µL]), a normal stable platelet count (231,000/µL [reference range, 150,000– 350,000/µL]), and a normal international normalized ratio.
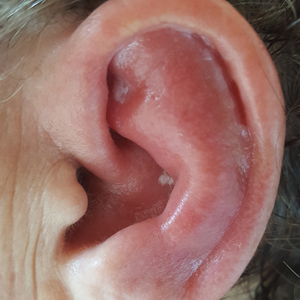
Red, Swollen, Tender Ear
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
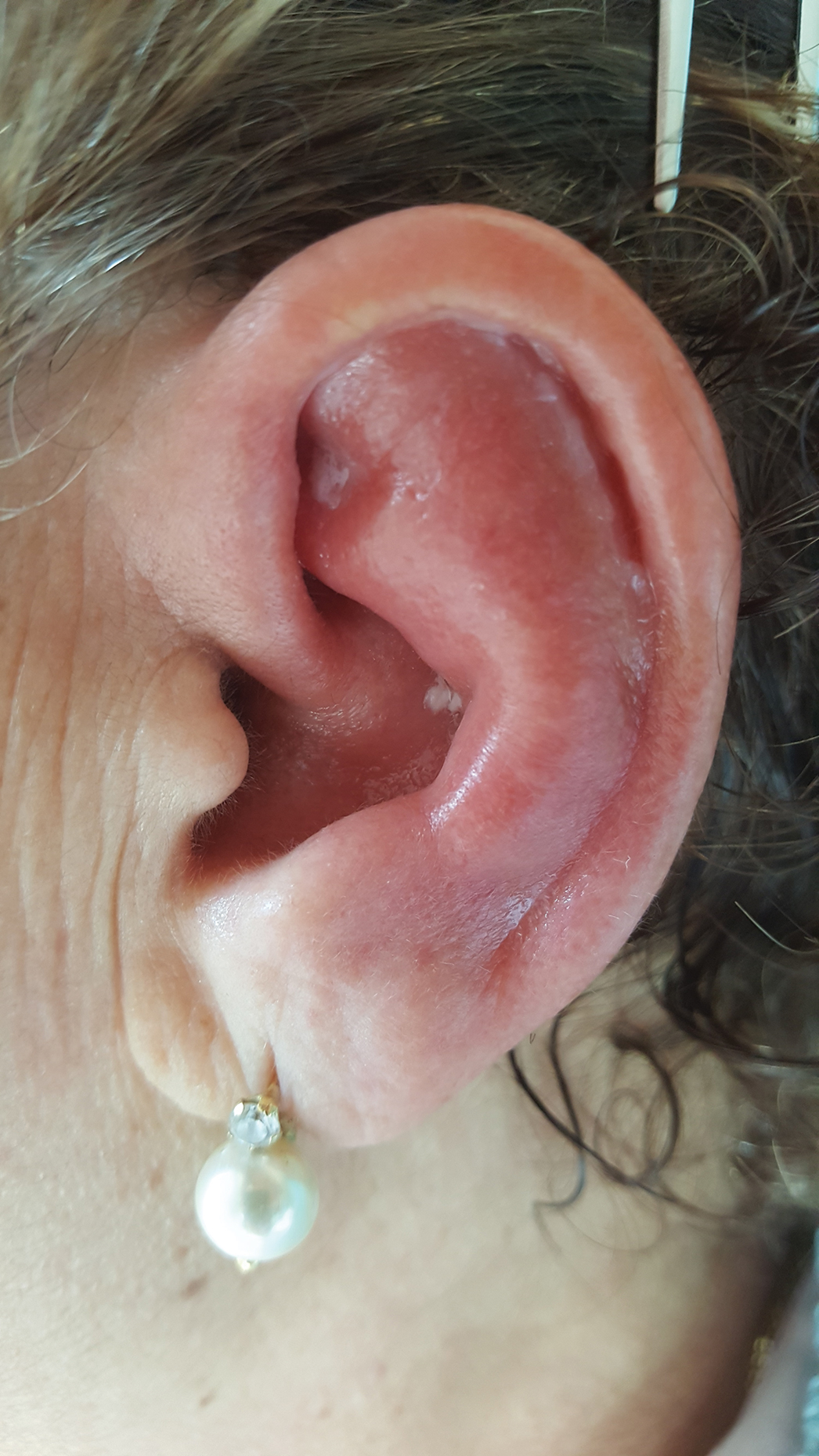
A 67-year-old woman presented with severe pain of the left external ear. She explained that similar episodes had occurred 2 years prior and affected the right ear and the nose. Her general practitioner prescribed topical and systemic antibiotic treatment, but there was no improvement. The patient also reported diffuse small joint pain without any radiologic sign of erosive arthritis. Physical examination revealed a red swollen external ear that was tender to the touch from the helix to the antitragus; conversely, the earlobe did not present any sign of inflammation. Redness of the left eye also was noticed, and a slit-lamp examination confirmed our suspect of scleritis. Results from routine blood tests, including an autoimmune panel, were within reference range, except for a nonspecific increase of inflammatory markers (erythrocyte sedimentation rate, 43 mm/h [reference range, 0–20 mm/h]; C-reactive protein, 5.65 mg/L [reference range, 0.08–3.1 mg/L]).
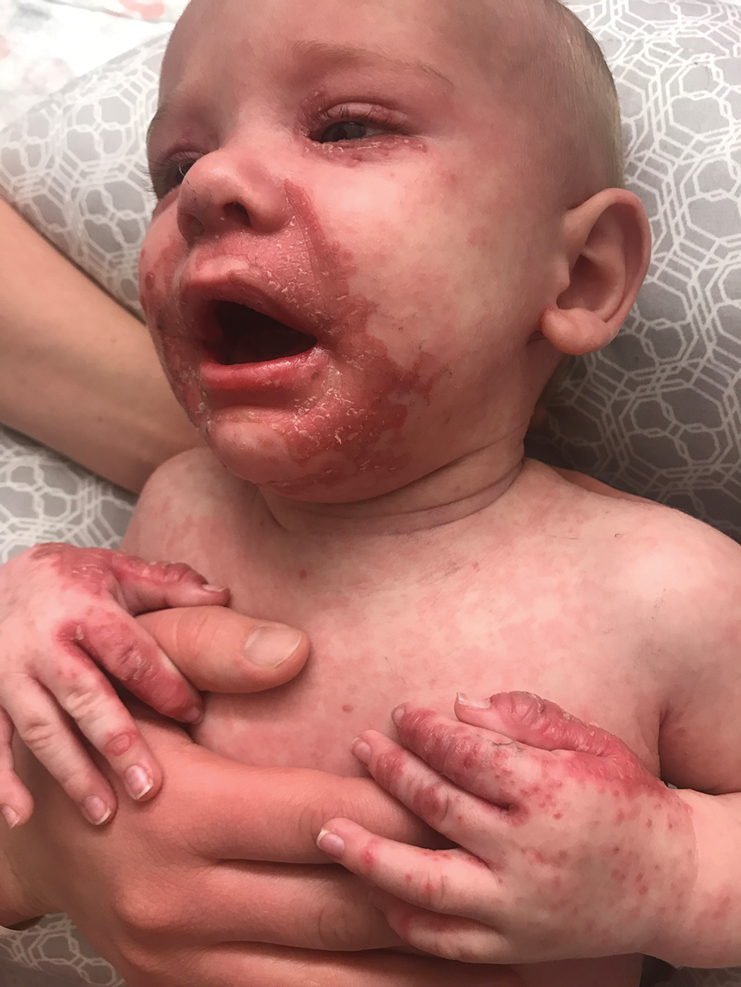
Irritable Baby With Weight Loss and a Periorificial and Truncal Rash
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
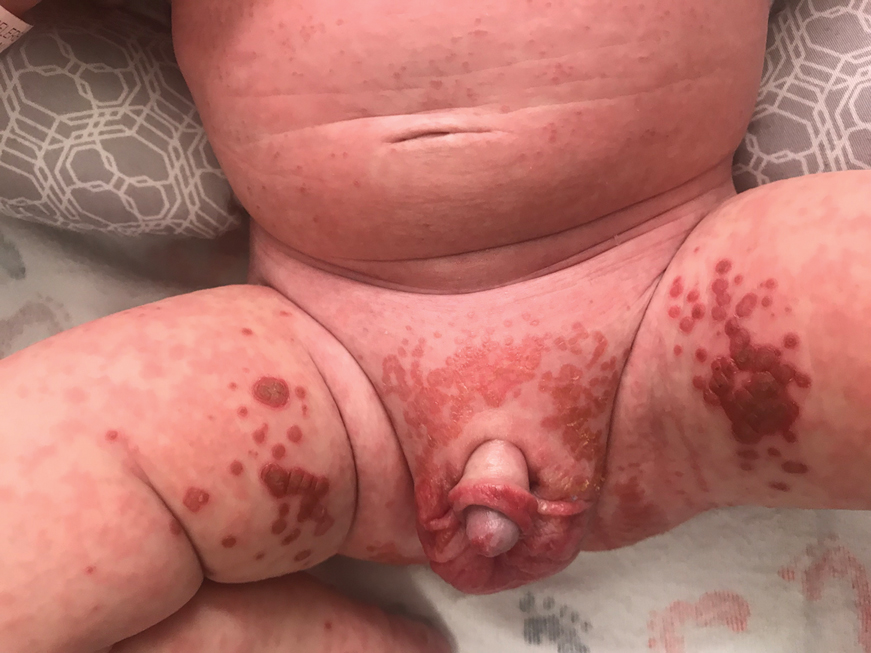
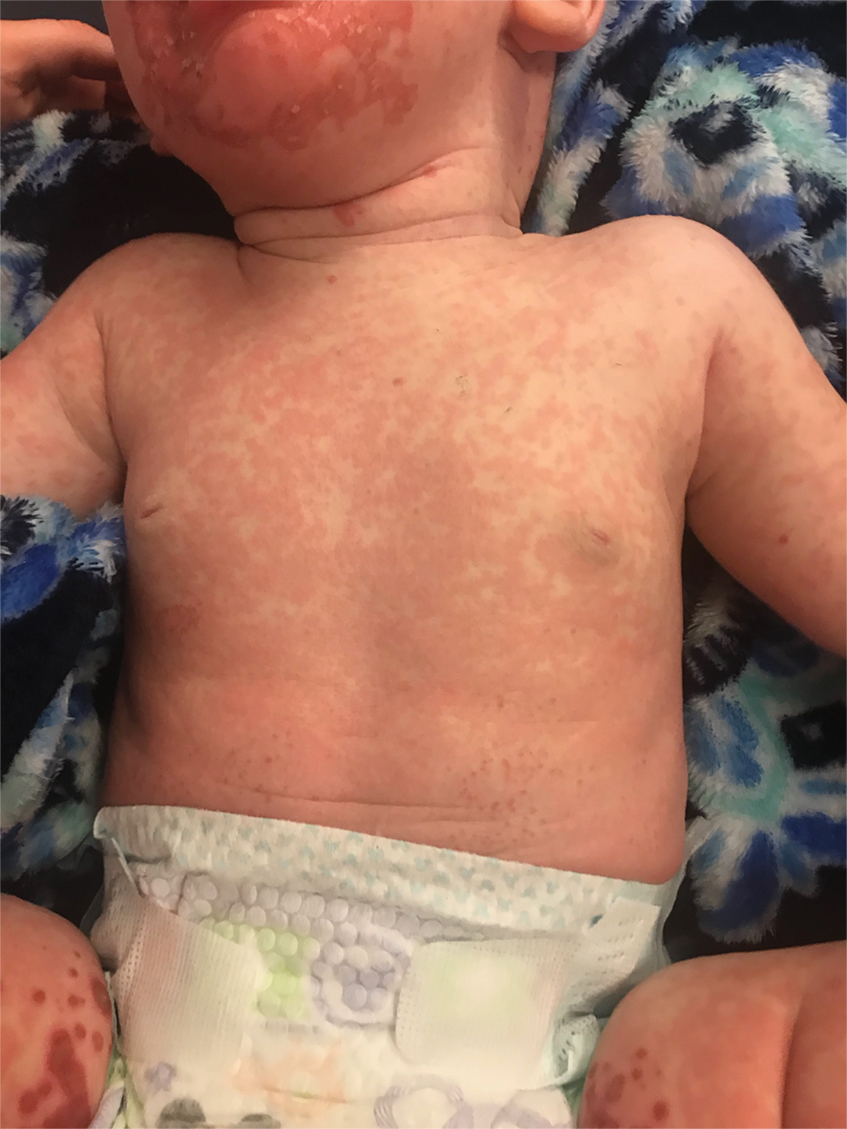
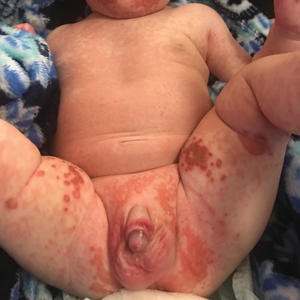
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.

In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
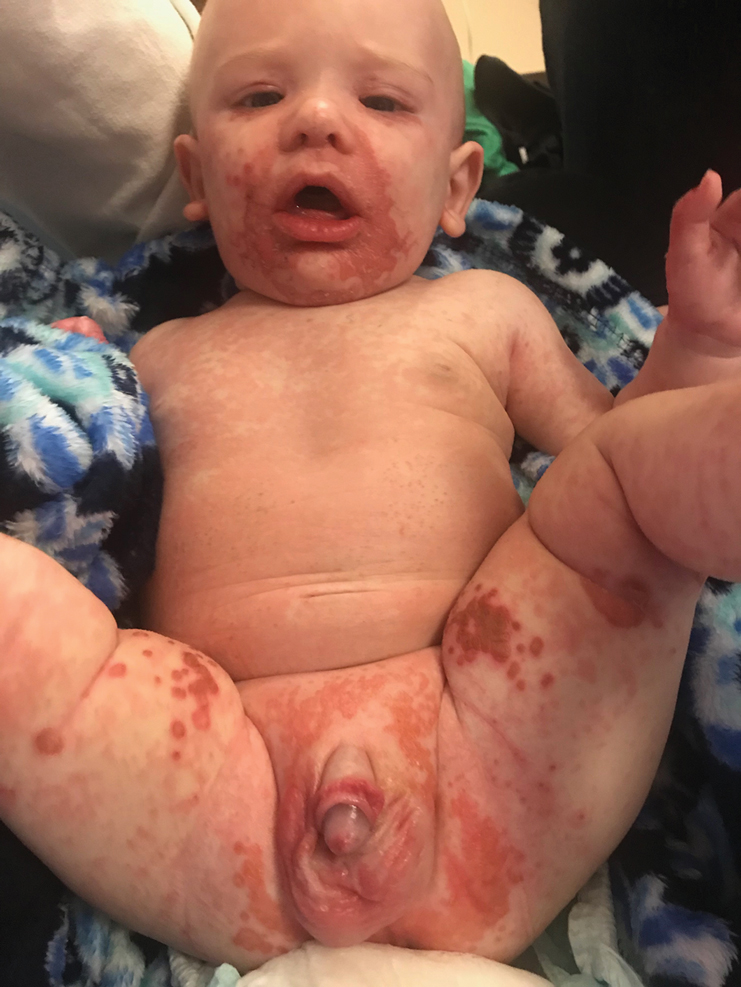
A 4-month-old infant boy presented to the pediatric hospital unit with a rash, fever, and failure to thrive. Prior to admission, the patient was treated for impetigo by a community dermatologist. After not responding to treatment, he was admitted and given intravenous acyclovir for 1 day by the pediatric hospitalist, and the dermatology service was consulted. The parents reported the patient had diarrhea for 1 month and a worsening rash over the last 2 weeks. The mother was breastfeeding. Physical examination revealed a fever (temperature, 38.9°C [102°F]) and an irritable infant whose growth curve had fallen from the 50th to 15th percentile since the 2-month well-baby examination. He had a fine, red, papular truncal rash with confluent plaques in a periorificial distribution that spared the inguinal skin folds, with some vesicles in a herpetiform presentation on the thighs as well as inflammation on the feet and hands. A complete blood cell count was within reference range, but the alkaline phosphatase level was low at 53 U/L (reference range, 72–307 U/L). A herpes simplex virus test was negative. A human immunodeficiency virus test and skin biopsy were performed.
Paronychia and Target Lesions After Hematopoietic Cell Transplant
The Diagnosis: Fusariosis
A periodic acid-Schiff stain of the seropurulent drainage from a skin nodule revealed neutrophils and scarce branching hyaline hyphae. Skin and blood cultures grew a white cottony colony. Microscopic examination showed sickle-shaped macroconidia and septate hyaline hyphae with branching acute angles (Figure). Molecular analysis by polymerase chain reaction yielded Fusarium solani species complex. Histopathology as well as culture and molecular findings were consistent with a diagnosis of disseminated fusariosis. Amphotericin B was started with rapid clinical improvement. The patient was asymptomatic upon discharge with voriconazole 200 mg twice daily.
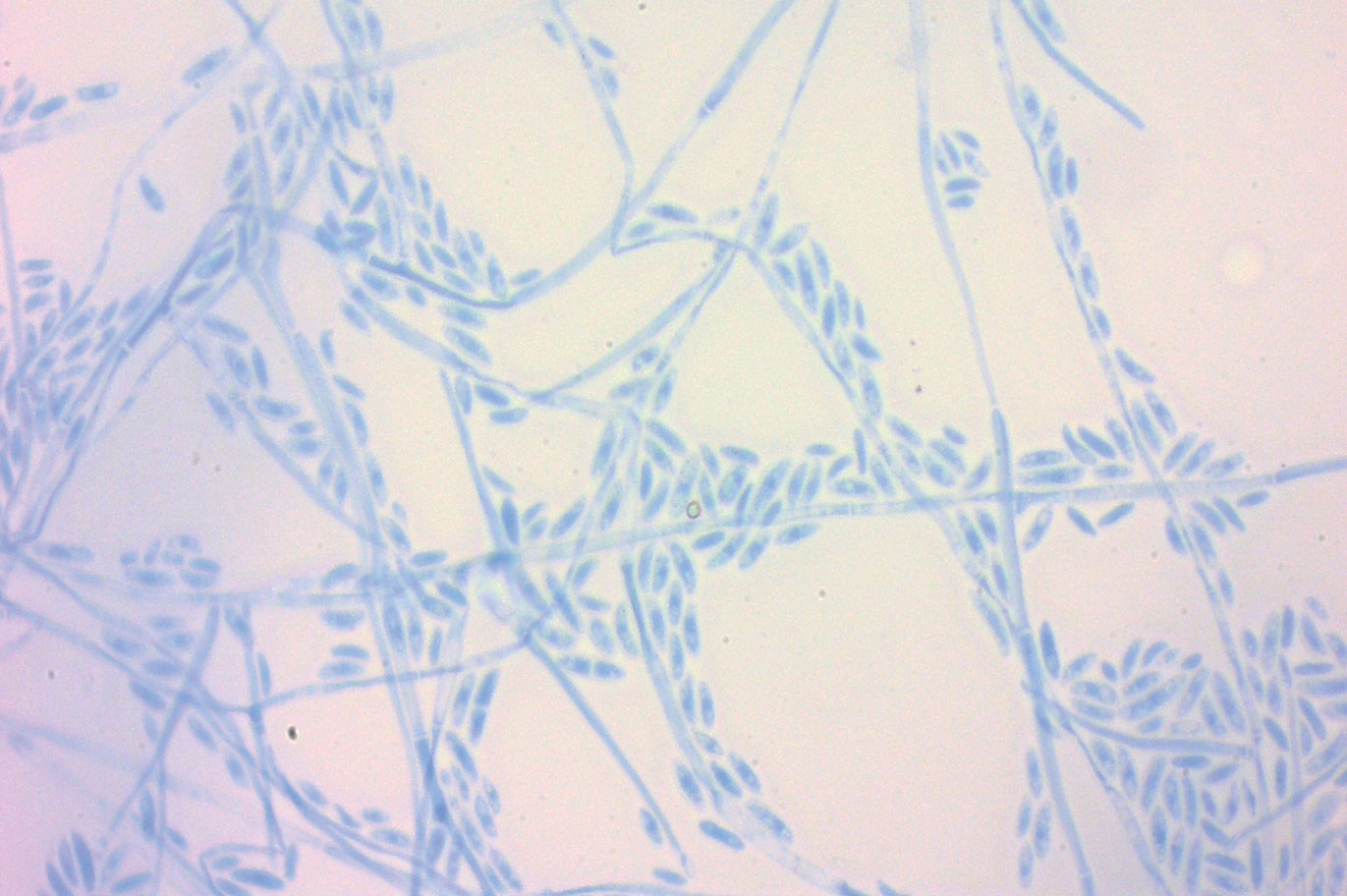
Fusariosis is an emerging, opportunistic, and life-threatening mycosis. In immunocompetent patients it may cause onychomycosis and keratitis.1 Invasive fusariosis predominantly is caused by the F solani species complex and affects immunocompromised patients, especially those with neutropenia or acute leukemia or hematopoietic stem cell transplant recipients.2
Before invasion, the infection frequently may begin by affecting the nail apparatus as onychomycosis or paronychia of the skin. As in our case, trauma or manipulation of the nail favors dissemination.3 Skin manifestations include erythematous to violaceous papules, macules, and nodules with central necrosis or crust; some may exhibit target morphology. Other organs may be affected, including the sinuses, lungs, liver, spleen, and kidneys. A comprehensive clinical examination before hematopoietic cell transplant and during fever and neutropenia may opportunely identify these potential infective foci.3,4
The differential diagnosis of disseminated fusariosis includes bacterial infections, especially Staphylococcus aureus and Pseudomonas aeruginosa, and other invasive fungal infections, particularly aspergillosis, mucormycosis, and candidiasis.5 Symptom persistence after broad-spectrum antibiotic initiation should raise diagnostic suspicion of systemic mycosis or mycobacterial infection. Mucormycosis and candidiasis have histopathologic profiles that differ from fusariosis, presenting with broad ribbonlike hyphae with 90° angulation and pseudohyphae with budding yeast cells, respectively. Differentiation of disseminated fusariosis and aspergillosis in neutropenic patients is difficult. Hyphae cannot be differentiated from those of Aspergillus species on histology.6 Furthermore, serologic assays, such as galactomannan and (1,3)-β-D-glucan, cross-react with both genera. Clinically, Fusarium species exhibit metastatic skin lesions, cellulitis, and positive blood cultures due to adventitious sporulation more frequently than Aspergillus species. Patients with aspergillosis more commonly present with sinusitis, pneumonia, and pulmonary macronodules with the halo sign.6 Although nocardiosis presents with disseminated subcutaneous nodules with pulmonary affection in immunocompromised patients, its morphology is very different from fusariosis. Nocardia presents with a gram-positive bacillus with the microscopic appearance of branching filaments. Yeastlike microorganisms with morphology ranging from oval to sausagelike are found in talaromycosis, an uncommon fungal infection predominantly caused by Talaromyces marneffei. Fusarium species culture reveals white cottony colonies with characteristic hyaline, canoe-shaped or sickle-shaped (banana-shaped), multicellular macroconidia, and microconidia. Precise species identification requires molecular analyses such as polymerase chain reaction.
Mortality is high, ranging from 50% to 70% of cases.5 Voriconazole or lipid-based amphotericin B are considered first-line treatments. Posaconazole may be employed as a second-line alternative. Surgical debridement of infected tissues and removal of colonized venous catheters is recommended. Secondary prophylaxis should be considered with agents such as voriconazole, posaconazole, or amphotericin B.5 Resolution of immunosuppression and neutropenia is an important factor to reduce the mortality rate.
- Ranawaka RR, Nagahawatte A, Gunasekara TA. Fusarium onychomycosis: prevalence, clinical presentations, response toitraconazole and terbinafine pulse therapy, and 1-year follow-up in nine cases. Int J Dermatol. 2015;54:1275-1282.
- Nucci F, Nouer SA, Capone D, et al. Fusariosis. Semin Respir Crit Care Med. 2015;36:706-714.
- Varon AG, Nouer SA, Barreiros G, et al. Superficial skin lesions positive for Fusarium are associated with subsequent development of invasive fusariosis. J Infect. 2014;68:85-89.
- Hay RJ. Fusarium infections of the skin. Curr Opin Infect Dis. 2007;20:115-117.
- Tortorano AM, Richardson M, Roilides E, et al. ESCMID and ECMM joint guidelines on diagnosis and management of hyalohyphomycosis: Fusarium spp., Scedosporium spp. and others. Clin Microbiol Infect. 2014;20:27-46.
- Nucci F, Nouer SA, Capone D, et al. Invasive mould disease in haematologic patients: comparison between fusariosis and aspergillosis. Clin Microbiol Infect. 2018;24:1105.e1-1105.e4.
The Diagnosis: Fusariosis
A periodic acid-Schiff stain of the seropurulent drainage from a skin nodule revealed neutrophils and scarce branching hyaline hyphae. Skin and blood cultures grew a white cottony colony. Microscopic examination showed sickle-shaped macroconidia and septate hyaline hyphae with branching acute angles (Figure). Molecular analysis by polymerase chain reaction yielded Fusarium solani species complex. Histopathology as well as culture and molecular findings were consistent with a diagnosis of disseminated fusariosis. Amphotericin B was started with rapid clinical improvement. The patient was asymptomatic upon discharge with voriconazole 200 mg twice daily.
Fusariosis is an emerging, opportunistic, and life-threatening mycosis. In immunocompetent patients it may cause onychomycosis and keratitis.1 Invasive fusariosis predominantly is caused by the F solani species complex and affects immunocompromised patients, especially those with neutropenia or acute leukemia or hematopoietic stem cell transplant recipients.2
Before invasion, the infection frequently may begin by affecting the nail apparatus as onychomycosis or paronychia of the skin. As in our case, trauma or manipulation of the nail favors dissemination.3 Skin manifestations include erythematous to violaceous papules, macules, and nodules with central necrosis or crust; some may exhibit target morphology. Other organs may be affected, including the sinuses, lungs, liver, spleen, and kidneys. A comprehensive clinical examination before hematopoietic cell transplant and during fever and neutropenia may opportunely identify these potential infective foci.3,4
The differential diagnosis of disseminated fusariosis includes bacterial infections, especially Staphylococcus aureus and Pseudomonas aeruginosa, and other invasive fungal infections, particularly aspergillosis, mucormycosis, and candidiasis.5 Symptom persistence after broad-spectrum antibiotic initiation should raise diagnostic suspicion of systemic mycosis or mycobacterial infection. Mucormycosis and candidiasis have histopathologic profiles that differ from fusariosis, presenting with broad ribbonlike hyphae with 90° angulation and pseudohyphae with budding yeast cells, respectively. Differentiation of disseminated fusariosis and aspergillosis in neutropenic patients is difficult. Hyphae cannot be differentiated from those of Aspergillus species on histology.6 Furthermore, serologic assays, such as galactomannan and (1,3)-β-D-glucan, cross-react with both genera. Clinically, Fusarium species exhibit metastatic skin lesions, cellulitis, and positive blood cultures due to adventitious sporulation more frequently than Aspergillus species. Patients with aspergillosis more commonly present with sinusitis, pneumonia, and pulmonary macronodules with the halo sign.6 Although nocardiosis presents with disseminated subcutaneous nodules with pulmonary affection in immunocompromised patients, its morphology is very different from fusariosis. Nocardia presents with a gram-positive bacillus with the microscopic appearance of branching filaments. Yeastlike microorganisms with morphology ranging from oval to sausagelike are found in talaromycosis, an uncommon fungal infection predominantly caused by Talaromyces marneffei. Fusarium species culture reveals white cottony colonies with characteristic hyaline, canoe-shaped or sickle-shaped (banana-shaped), multicellular macroconidia, and microconidia. Precise species identification requires molecular analyses such as polymerase chain reaction.
Mortality is high, ranging from 50% to 70% of cases.5 Voriconazole or lipid-based amphotericin B are considered first-line treatments. Posaconazole may be employed as a second-line alternative. Surgical debridement of infected tissues and removal of colonized venous catheters is recommended. Secondary prophylaxis should be considered with agents such as voriconazole, posaconazole, or amphotericin B.5 Resolution of immunosuppression and neutropenia is an important factor to reduce the mortality rate.
The Diagnosis: Fusariosis
A periodic acid-Schiff stain of the seropurulent drainage from a skin nodule revealed neutrophils and scarce branching hyaline hyphae. Skin and blood cultures grew a white cottony colony. Microscopic examination showed sickle-shaped macroconidia and septate hyaline hyphae with branching acute angles (Figure). Molecular analysis by polymerase chain reaction yielded Fusarium solani species complex. Histopathology as well as culture and molecular findings were consistent with a diagnosis of disseminated fusariosis. Amphotericin B was started with rapid clinical improvement. The patient was asymptomatic upon discharge with voriconazole 200 mg twice daily.
Fusariosis is an emerging, opportunistic, and life-threatening mycosis. In immunocompetent patients it may cause onychomycosis and keratitis.1 Invasive fusariosis predominantly is caused by the F solani species complex and affects immunocompromised patients, especially those with neutropenia or acute leukemia or hematopoietic stem cell transplant recipients.2
Before invasion, the infection frequently may begin by affecting the nail apparatus as onychomycosis or paronychia of the skin. As in our case, trauma or manipulation of the nail favors dissemination.3 Skin manifestations include erythematous to violaceous papules, macules, and nodules with central necrosis or crust; some may exhibit target morphology. Other organs may be affected, including the sinuses, lungs, liver, spleen, and kidneys. A comprehensive clinical examination before hematopoietic cell transplant and during fever and neutropenia may opportunely identify these potential infective foci.3,4
The differential diagnosis of disseminated fusariosis includes bacterial infections, especially Staphylococcus aureus and Pseudomonas aeruginosa, and other invasive fungal infections, particularly aspergillosis, mucormycosis, and candidiasis.5 Symptom persistence after broad-spectrum antibiotic initiation should raise diagnostic suspicion of systemic mycosis or mycobacterial infection. Mucormycosis and candidiasis have histopathologic profiles that differ from fusariosis, presenting with broad ribbonlike hyphae with 90° angulation and pseudohyphae with budding yeast cells, respectively. Differentiation of disseminated fusariosis and aspergillosis in neutropenic patients is difficult. Hyphae cannot be differentiated from those of Aspergillus species on histology.6 Furthermore, serologic assays, such as galactomannan and (1,3)-β-D-glucan, cross-react with both genera. Clinically, Fusarium species exhibit metastatic skin lesions, cellulitis, and positive blood cultures due to adventitious sporulation more frequently than Aspergillus species. Patients with aspergillosis more commonly present with sinusitis, pneumonia, and pulmonary macronodules with the halo sign.6 Although nocardiosis presents with disseminated subcutaneous nodules with pulmonary affection in immunocompromised patients, its morphology is very different from fusariosis. Nocardia presents with a gram-positive bacillus with the microscopic appearance of branching filaments. Yeastlike microorganisms with morphology ranging from oval to sausagelike are found in talaromycosis, an uncommon fungal infection predominantly caused by Talaromyces marneffei. Fusarium species culture reveals white cottony colonies with characteristic hyaline, canoe-shaped or sickle-shaped (banana-shaped), multicellular macroconidia, and microconidia. Precise species identification requires molecular analyses such as polymerase chain reaction.
Mortality is high, ranging from 50% to 70% of cases.5 Voriconazole or lipid-based amphotericin B are considered first-line treatments. Posaconazole may be employed as a second-line alternative. Surgical debridement of infected tissues and removal of colonized venous catheters is recommended. Secondary prophylaxis should be considered with agents such as voriconazole, posaconazole, or amphotericin B.5 Resolution of immunosuppression and neutropenia is an important factor to reduce the mortality rate.
- Ranawaka RR, Nagahawatte A, Gunasekara TA. Fusarium onychomycosis: prevalence, clinical presentations, response toitraconazole and terbinafine pulse therapy, and 1-year follow-up in nine cases. Int J Dermatol. 2015;54:1275-1282.
- Nucci F, Nouer SA, Capone D, et al. Fusariosis. Semin Respir Crit Care Med. 2015;36:706-714.
- Varon AG, Nouer SA, Barreiros G, et al. Superficial skin lesions positive for Fusarium are associated with subsequent development of invasive fusariosis. J Infect. 2014;68:85-89.
- Hay RJ. Fusarium infections of the skin. Curr Opin Infect Dis. 2007;20:115-117.
- Tortorano AM, Richardson M, Roilides E, et al. ESCMID and ECMM joint guidelines on diagnosis and management of hyalohyphomycosis: Fusarium spp., Scedosporium spp. and others. Clin Microbiol Infect. 2014;20:27-46.
- Nucci F, Nouer SA, Capone D, et al. Invasive mould disease in haematologic patients: comparison between fusariosis and aspergillosis. Clin Microbiol Infect. 2018;24:1105.e1-1105.e4.
- Ranawaka RR, Nagahawatte A, Gunasekara TA. Fusarium onychomycosis: prevalence, clinical presentations, response toitraconazole and terbinafine pulse therapy, and 1-year follow-up in nine cases. Int J Dermatol. 2015;54:1275-1282.
- Nucci F, Nouer SA, Capone D, et al. Fusariosis. Semin Respir Crit Care Med. 2015;36:706-714.
- Varon AG, Nouer SA, Barreiros G, et al. Superficial skin lesions positive for Fusarium are associated with subsequent development of invasive fusariosis. J Infect. 2014;68:85-89.
- Hay RJ. Fusarium infections of the skin. Curr Opin Infect Dis. 2007;20:115-117.
- Tortorano AM, Richardson M, Roilides E, et al. ESCMID and ECMM joint guidelines on diagnosis and management of hyalohyphomycosis: Fusarium spp., Scedosporium spp. and others. Clin Microbiol Infect. 2014;20:27-46.
- Nucci F, Nouer SA, Capone D, et al. Invasive mould disease in haematologic patients: comparison between fusariosis and aspergillosis. Clin Microbiol Infect. 2018;24:1105.e1-1105.e4.
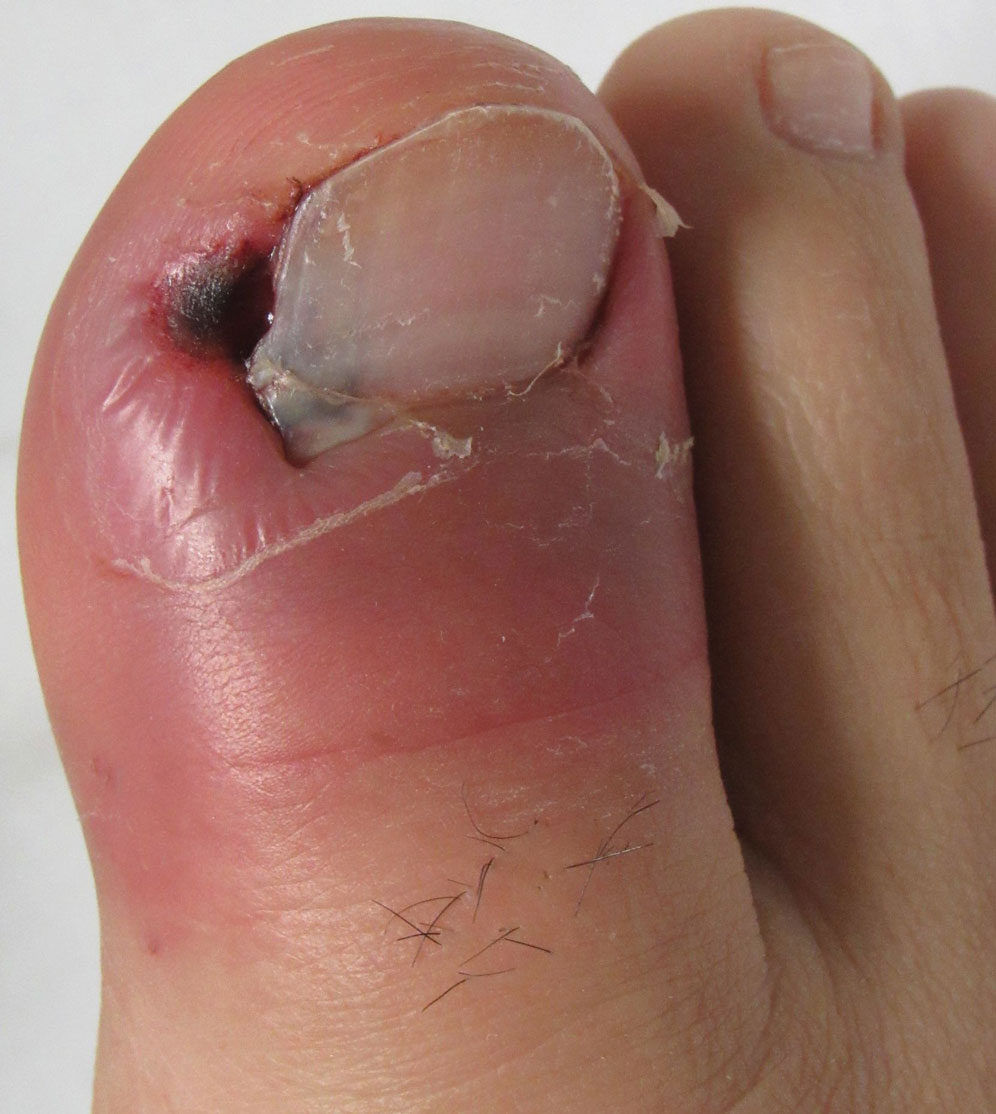
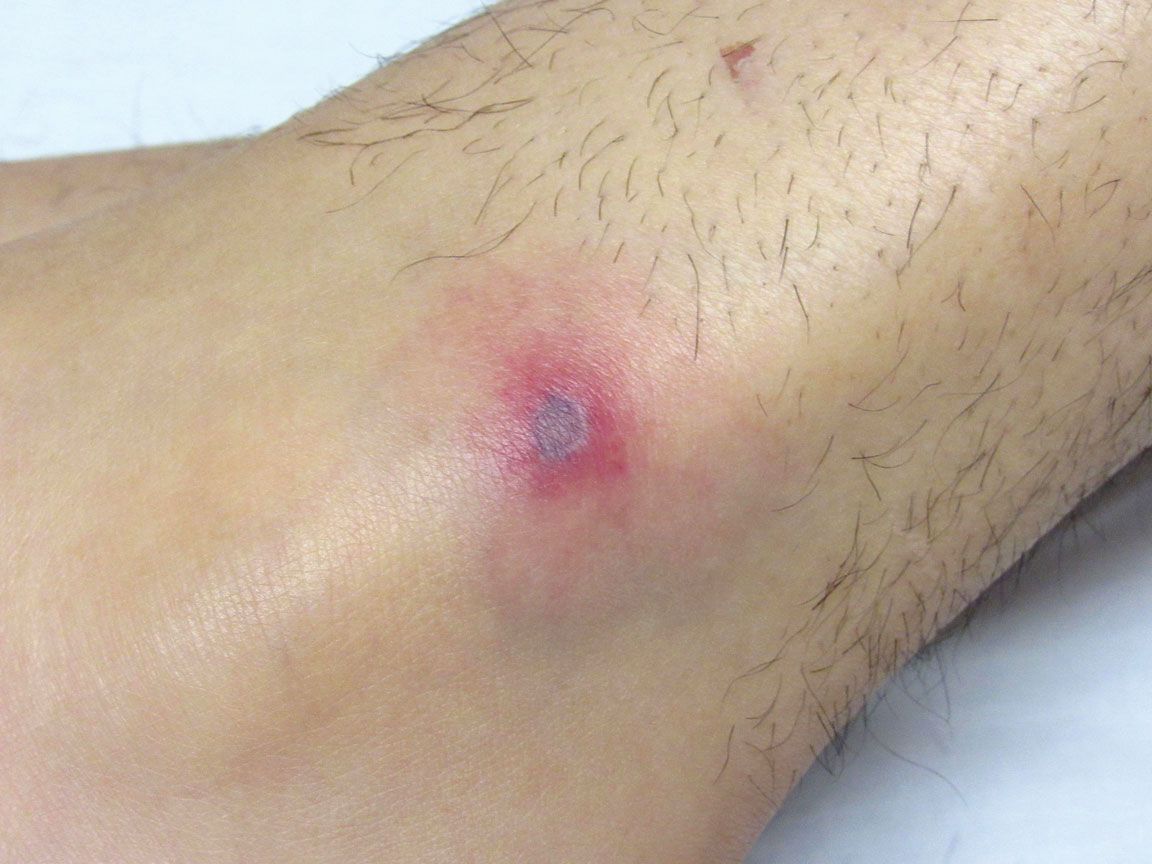
A 19-year-old man with acute lymphoblastic leukemia was admitted for an allogeneic hematopoietic cell transplant. On the 11th day of hospitalization, he experienced a right toe trauma in his hospital room and subsequently developed edema, erythema, and pain on the right hallux (top). The next day, a general surgeon performed a minor incision and drainage of the affected area. After 2 days, the patient developed a fever and a disseminated dermatosis located on the arms and legs characterized by target lesions with a necrotic center and erythematous papules and macules (bottom). On day 3, he developed severe neutropenia (0.042×109 cells/L [reference range, 2.0–6.9×109 cells/L]). Broad-spectrum antibiotics were initiated without clinical improvement. The patient developed dyspnea on day 5. Skin, nail, and blood cultures were obtained. High-resolution computed tomography of the chest displayed multiple small pulmonary nodules, ground-glass opacities, and the tree-in-bud sign.
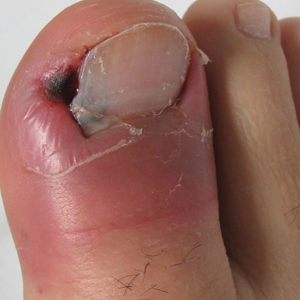
Erythematous Abdominal Nodule
The Diagnosis: Foreign Body Reaction With Sinus Tract
A delayed foreign body reaction is a rare complication of retained temporary pacing wires following cardiovascular surgery. These epicardial pacing wires are important for the management of postoperative arrhythmia and normally are removed by external traction after normal rhythm has been re-established. However, it is not uncommon for these wires to be cut at the skin surface in the setting of difficult removal, as retained pacing wires generally are viewed as benign.1 These reactions often take years after placement of the pacing wire to present themselves and most often resolve with either complete removal of the wire or resection of the distal end.2
Our patient was referred to dermatology and underwent a shave biopsy. Results were consistent with chronic inflammatory granulation tissue. Bacterial tissue culture grew Staphylococcus epidermidis. Cultures for acid-fast bacilli and fungi were negative. The patient was referred to cardiothoracic surgery. Computed tomography identified a retained temporary pacing wire extending to the base of the lesion. The lesion was excised and the distal aspect of the pacing wire was removed, which resulted in resolution of the nodule.
The differential diagnosis includes pyoderma gangrenosum, nodular basal cell carcinoma, Sister Mary Joseph nodule, and pyogenic granuloma. Pyoderma gangrenosum is a neutrophilic dermatosis that presents as a rapidly progressing, painful, necrotic ulcer. It is classically associated with inflammatory bowel disease and other systemic diseases but also can occur in isolation.3
Nodular basal cell carcinomas often develop in chronically sun-exposed areas of the body. Morphologically, they present as pink, pearl appearing papules with rolled borders and overlying arborizing telangiectasia. Nodular basal cell carcinomas may present with recurrent bleeding but typically do not have continuous drainage.4
Sister Mary Joseph nodule represents a periumbilical lymphatic metastasis from an underlying (usually intra-abdominal) malignancy. It typically presents as an umbilical or periumbilical nodule measuring 0.5 to 15 cm in diameter. The nodules often are painful and discharge a serous fluid. It is estimated that they are present in 1% to 3% of cases of abdominopelvic malignancy, but Sister Mary Joseph nodules also have been reported in several other types of solid organ tumors.5
Pyogenic granuloma is a benign vascular lesion that classically develops rapidly over the course of a few weeks. It often presents as a single red, moist, friable papule with a collarette of scale and frequently is associated with pain, bleeding, and ulceration. Keratinized skin or mucosa can be affected, and pyogenic granuloma is most common in children and young adults.6
- Chung DA, Smith EE. Delayed presentation of foreign body reaction secondary to retained pacing wires. Ann Thorac Surg. 1998;66:550-551.
- Gentry WH, Hassan AA. Complications of retained epicardial pacing wires (an unusual bronchial foreign body. Ann Thorac Surg. 1993;56:1391-1393.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Tanese K. Diagnosis and treatment of basal cell carcinoma. Curr Treat Options Oncol. 2019;20:13
- Tso S, Brockley J, Recica H, et al. Sister Mary Joseph's nodule: an unusual but important physical finding characteristic of widespread internal malignancy. Br J Gen Pract. 2013;63:551-552.
- Mashiah J, Hadj-Rabia S, Slodownik D, et al. Effectiveness of topical propranolol 4% gel in the treatment of pyogenic granuloma in children. J Dermatol. 2019;46:245-248.
The Diagnosis: Foreign Body Reaction With Sinus Tract
A delayed foreign body reaction is a rare complication of retained temporary pacing wires following cardiovascular surgery. These epicardial pacing wires are important for the management of postoperative arrhythmia and normally are removed by external traction after normal rhythm has been re-established. However, it is not uncommon for these wires to be cut at the skin surface in the setting of difficult removal, as retained pacing wires generally are viewed as benign.1 These reactions often take years after placement of the pacing wire to present themselves and most often resolve with either complete removal of the wire or resection of the distal end.2
Our patient was referred to dermatology and underwent a shave biopsy. Results were consistent with chronic inflammatory granulation tissue. Bacterial tissue culture grew Staphylococcus epidermidis. Cultures for acid-fast bacilli and fungi were negative. The patient was referred to cardiothoracic surgery. Computed tomography identified a retained temporary pacing wire extending to the base of the lesion. The lesion was excised and the distal aspect of the pacing wire was removed, which resulted in resolution of the nodule.
The differential diagnosis includes pyoderma gangrenosum, nodular basal cell carcinoma, Sister Mary Joseph nodule, and pyogenic granuloma. Pyoderma gangrenosum is a neutrophilic dermatosis that presents as a rapidly progressing, painful, necrotic ulcer. It is classically associated with inflammatory bowel disease and other systemic diseases but also can occur in isolation.3
Nodular basal cell carcinomas often develop in chronically sun-exposed areas of the body. Morphologically, they present as pink, pearl appearing papules with rolled borders and overlying arborizing telangiectasia. Nodular basal cell carcinomas may present with recurrent bleeding but typically do not have continuous drainage.4
Sister Mary Joseph nodule represents a periumbilical lymphatic metastasis from an underlying (usually intra-abdominal) malignancy. It typically presents as an umbilical or periumbilical nodule measuring 0.5 to 15 cm in diameter. The nodules often are painful and discharge a serous fluid. It is estimated that they are present in 1% to 3% of cases of abdominopelvic malignancy, but Sister Mary Joseph nodules also have been reported in several other types of solid organ tumors.5
Pyogenic granuloma is a benign vascular lesion that classically develops rapidly over the course of a few weeks. It often presents as a single red, moist, friable papule with a collarette of scale and frequently is associated with pain, bleeding, and ulceration. Keratinized skin or mucosa can be affected, and pyogenic granuloma is most common in children and young adults.6
The Diagnosis: Foreign Body Reaction With Sinus Tract
A delayed foreign body reaction is a rare complication of retained temporary pacing wires following cardiovascular surgery. These epicardial pacing wires are important for the management of postoperative arrhythmia and normally are removed by external traction after normal rhythm has been re-established. However, it is not uncommon for these wires to be cut at the skin surface in the setting of difficult removal, as retained pacing wires generally are viewed as benign.1 These reactions often take years after placement of the pacing wire to present themselves and most often resolve with either complete removal of the wire or resection of the distal end.2
Our patient was referred to dermatology and underwent a shave biopsy. Results were consistent with chronic inflammatory granulation tissue. Bacterial tissue culture grew Staphylococcus epidermidis. Cultures for acid-fast bacilli and fungi were negative. The patient was referred to cardiothoracic surgery. Computed tomography identified a retained temporary pacing wire extending to the base of the lesion. The lesion was excised and the distal aspect of the pacing wire was removed, which resulted in resolution of the nodule.
The differential diagnosis includes pyoderma gangrenosum, nodular basal cell carcinoma, Sister Mary Joseph nodule, and pyogenic granuloma. Pyoderma gangrenosum is a neutrophilic dermatosis that presents as a rapidly progressing, painful, necrotic ulcer. It is classically associated with inflammatory bowel disease and other systemic diseases but also can occur in isolation.3
Nodular basal cell carcinomas often develop in chronically sun-exposed areas of the body. Morphologically, they present as pink, pearl appearing papules with rolled borders and overlying arborizing telangiectasia. Nodular basal cell carcinomas may present with recurrent bleeding but typically do not have continuous drainage.4
Sister Mary Joseph nodule represents a periumbilical lymphatic metastasis from an underlying (usually intra-abdominal) malignancy. It typically presents as an umbilical or periumbilical nodule measuring 0.5 to 15 cm in diameter. The nodules often are painful and discharge a serous fluid. It is estimated that they are present in 1% to 3% of cases of abdominopelvic malignancy, but Sister Mary Joseph nodules also have been reported in several other types of solid organ tumors.5
Pyogenic granuloma is a benign vascular lesion that classically develops rapidly over the course of a few weeks. It often presents as a single red, moist, friable papule with a collarette of scale and frequently is associated with pain, bleeding, and ulceration. Keratinized skin or mucosa can be affected, and pyogenic granuloma is most common in children and young adults.6
- Chung DA, Smith EE. Delayed presentation of foreign body reaction secondary to retained pacing wires. Ann Thorac Surg. 1998;66:550-551.
- Gentry WH, Hassan AA. Complications of retained epicardial pacing wires (an unusual bronchial foreign body. Ann Thorac Surg. 1993;56:1391-1393.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Tanese K. Diagnosis and treatment of basal cell carcinoma. Curr Treat Options Oncol. 2019;20:13
- Tso S, Brockley J, Recica H, et al. Sister Mary Joseph's nodule: an unusual but important physical finding characteristic of widespread internal malignancy. Br J Gen Pract. 2013;63:551-552.
- Mashiah J, Hadj-Rabia S, Slodownik D, et al. Effectiveness of topical propranolol 4% gel in the treatment of pyogenic granuloma in children. J Dermatol. 2019;46:245-248.
- Chung DA, Smith EE. Delayed presentation of foreign body reaction secondary to retained pacing wires. Ann Thorac Surg. 1998;66:550-551.
- Gentry WH, Hassan AA. Complications of retained epicardial pacing wires (an unusual bronchial foreign body. Ann Thorac Surg. 1993;56:1391-1393.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Tanese K. Diagnosis and treatment of basal cell carcinoma. Curr Treat Options Oncol. 2019;20:13
- Tso S, Brockley J, Recica H, et al. Sister Mary Joseph's nodule: an unusual but important physical finding characteristic of widespread internal malignancy. Br J Gen Pract. 2013;63:551-552.
- Mashiah J, Hadj-Rabia S, Slodownik D, et al. Effectiveness of topical propranolol 4% gel in the treatment of pyogenic granuloma in children. J Dermatol. 2019;46:245-248.
A 71-year-old man presented with an inflamed erythematous papule on the right subcostal region of 12 months’ duration. It began as a small pimplelike bump that slowly enlarged. The patient did not report any pain or pruritus, but the lesion intermittently drained purulent fluid. The patient had a pacemaker and a history of severe aortic stenosis for which he underwent bioprosthetic aortic valve repair approximately 3 years prior to presentation. His postoperative course was complicated by sternal wound infection and sepsis, prompting surgical replacement of the graft and the pacemaker. He then developed aortitis secondary to bacterial endocarditis with multiple associated septic emboli and is now on lifelong levofloxacin and minocycline therapy. Physical examination revealed a 1.5-cm, erythematous, soft, protuberant nodule with surrounding skin dimpling on the right subcostal region adjacent to a well-healed surgical scar. Approximately 1 to 2 mL of purulent fluid was expressed.
Congenital Defect of the Toenail
The Diagnosis: Onychodystrophy Secondary to Polydactyly
Radiographs of the feet demonstrated an accessory distal phalanx of the left great toe with a similar smaller accessory distal phalanx on the right great toe (Figure). The patient was referred to orthopedic surgery, and surgical intervention was recommended for only the left great toe given recurrent skin inflammation and nail complications. An excision of the left great toe polydactyly was performed. The patient healed well without complications.
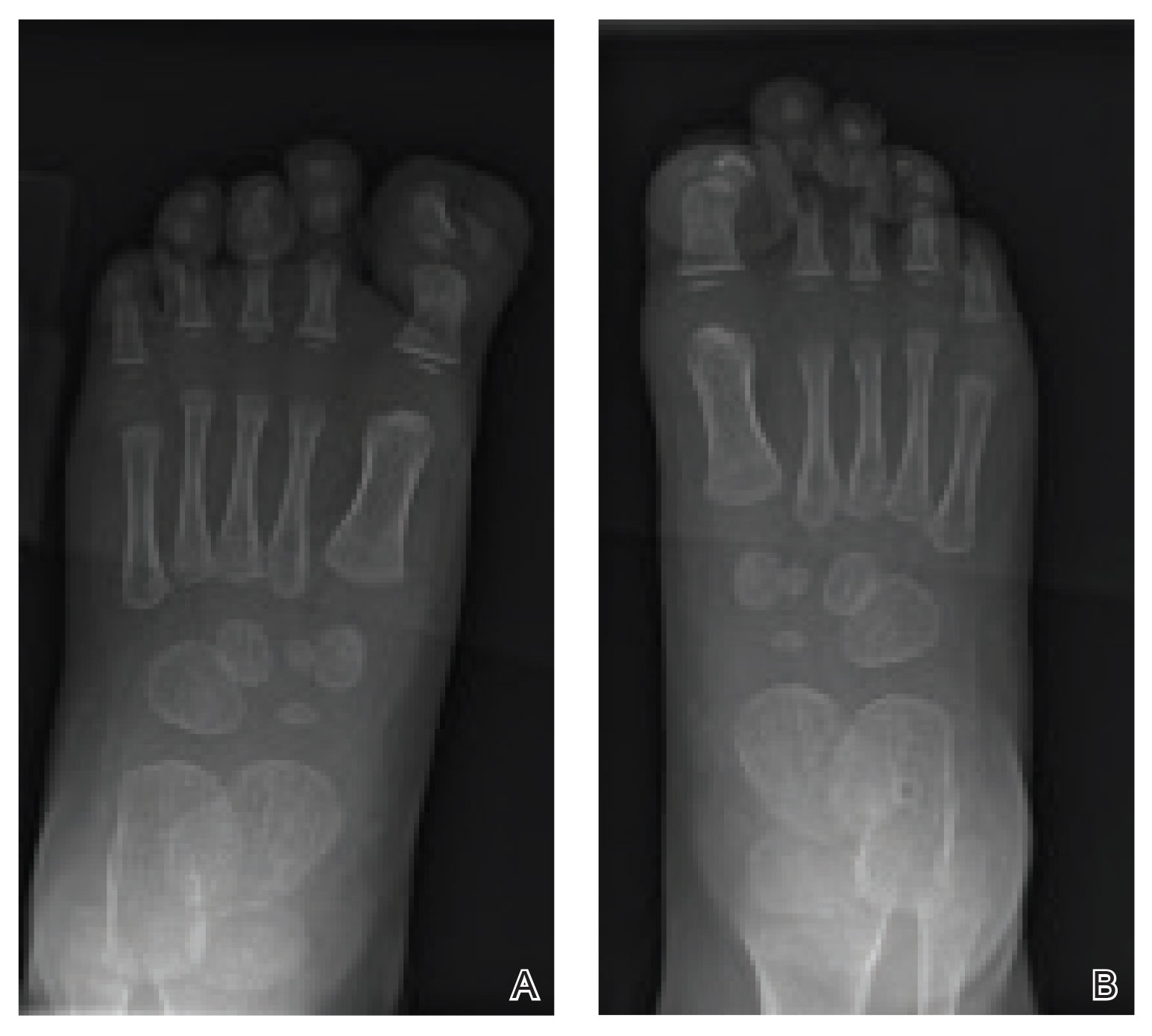
Many clinically heterogeneous phenotypes exist for polydactyly and syndactyly, which both are common entities with incidences of 1 in 700 to 1000 births and 1 in 2000 to 3000 births, respectively.1 Both polydactyly and syndactyly can be an isolated variant in newborns or present with multiple concurrent malformations as part of a genetic syndrome, with more than 300 syndromic anomalies described. The genetic basis of these conditions is equally diverse, with homeobox genes, hedgehog pathways, fibroblast growth factors, and boneand cartilage-derived morphogenetic proteins implicated in their development.1
The differential diagnosis for our patient included congenital malalignment of the great toenails, nail-patella syndrome, onychodystrophy secondary to polydactyly, and congenital hypertrophy of the lateral nail fold. Given the strong family history, polydactyly was suspected.
Congenital malalignment of the great toenails results in lateral deviation of the nail plates.2 It is an underdiagnosed condition with different etiologies hypothesized, such as genetic factors with possible autosomal-dominant transmission and extrinsic factors.3 One proposed mechanism of pathogenesis is desynchronization during growth of the nail and distal phalanx of the hallux, leading to larger nail plates that grow laterally.4 Typical features associated with this disease are nail discoloration, nail plate thickening, and transversal grooves or ridges, none of which were seen in our patient.2
Children with nail-patella syndrome have dysplastic nails and associated bony abnormalities, such as absent patellae.5 This syndrome results from an autosomaldominant mutation in the LIM homeobox transcription factor 1-beta gene, LMX1B, which is responsible for dorsal-ventral patterning of the limb, as well as patterning of the nails, patellae and long bones, and even the kidney tubule.6 As such, patients with nail-patella syndrome have associated renal abnormalities. The findings in our patient were limited to the feet, making an underlying syndrome unlikely to be the cause.
First described in 1968 by Meadow,7 fetal hydantoin syndrome is a well-documented sequela in women taking phenytoin throughout pregnancy. Multiple malformations are possible, including cardiac defects, cleft lip/palate, digit and nail hypoplasia, abnormal facial features, mental disability, and growth abnormalities.8 The teratogenicity behind phenytoin results from reactive oxygen species that alter embryonic DNA, proteins, and lipids.9 The mother of this child was not on any seizure prophylaxis, eliminating it from the differential.
Congenital hypertrophy of the lateral nail fold is a defect of the soft tissue of the hallux leading to hypertrophy of the nail fold, commonly presenting with inflammation and pain10 possibly due to dyssynchronous growth between the soft tissue and nail plate.11 With this defect, a lip covering the nail plate is common, which was not seen in our patient.
As demonstrated in our patient, family history can help guide the diagnosis. Seven of 9 nonsyndromic forms of syndactyly are inherited in an autosomal-dominant fashion and range from mild presentations, as in our patient, to more severe deformations with underlying bone fusion and functional impairment.12 Polydactyly also often is expressed in an autosomal-dominant pattern, with up to 30% of patients having a positive family history. Polydactyly traditionally is classified by the location of the supernumerary digit as preaxial (radial), central, or postaxial (ulnar), and many further morphologic variations exist within these groups. Overall, preaxial polydactyly is relatively rare and represents 15% of polydactylies, with central and postaxial comprising the other 6% and 79%, respectively.13 Delineation of the underlying anatomy may reveal ray duplications (digit and corresponding metacarpal or metatarsal bone), metatarsal variants, and duplicated phalanges that may be hypoplastic or deformed. Patients may report difficulty finding comfortable footwear, cosmetic concerns, and nail-related complications. Although not always required, surgical intervention may provide definitive treatment but can leave residual deformities in the surrounding altered anatomy; thus, orthopedic or plastic surgery consultations are critical in appropriately counseling patients.
- Ahmed H, Akbari H, Emanmi A, et al. Genetic overview of syndactyly and polydactyly. Plast Reconstr Surg Glob Open. 2017;5:e1549.
- Catalfo P, Musumeci ML, Lacarrubba F, et al. Congenital malalignment of the great toenails: a review. Skin Appendage Disord. 2018;4:230-235.
- Kus S, Tahmaz E, Gurunluoglu R, et al. Congenital malalignment of the great toenails in dizygotic twins. Pediatr Dermatol. 2005;22:434-435.
- Chaniotakis I, Bonitsis N, Stergiopoulou C, et al. Dizygotic twins with congenital malalignment of the great toenails: reappraisal of the pathogenesis. J Am Acad Dermatol. 2007;57:711-715.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Dreyer SD, Zhou G, Baldini A, et al, Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19:47-50.
- Meadow SR. Anticonvulsant drugs and congenital abnormalities. Lancet. 1968;2:1296.
- Scheinfeld N. Phenytoin in cutaneous medicine: its uses, mechanisms and side effects. Dermatol Online J. 2003;9:6.
- Winn LM, Wells PG. Phenytoin-initiated DNA oxidation in murine embryo culture, and embryo protection by the antioxidative enzymes superoxide dismutase and catalase: evidence for reactive oxygen species-mediated DNA oxidation in the molecular mechanism of phenytoin teratogenicity. Mol Pharmacol. 1995;48:112-120.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Martinet C, Pascal M, Civatte J, et al. Lateral nail-pad of the big toe in infants. apropos of 2 cases. Ann Dermatol Venereol. 1984;111:731-732.
- Malik S. Syndactyly: phenotypes, genetics and current classification. Eur J Hum Genet. 2012;20:817-824.
- Belthur MV, Linton JL, Barnes DA. The spectrum of preaxial polydactyly of the foot. J Pediatr Orthop. 2011;31:435-447.
The Diagnosis: Onychodystrophy Secondary to Polydactyly
Radiographs of the feet demonstrated an accessory distal phalanx of the left great toe with a similar smaller accessory distal phalanx on the right great toe (Figure). The patient was referred to orthopedic surgery, and surgical intervention was recommended for only the left great toe given recurrent skin inflammation and nail complications. An excision of the left great toe polydactyly was performed. The patient healed well without complications.
Many clinically heterogeneous phenotypes exist for polydactyly and syndactyly, which both are common entities with incidences of 1 in 700 to 1000 births and 1 in 2000 to 3000 births, respectively.1 Both polydactyly and syndactyly can be an isolated variant in newborns or present with multiple concurrent malformations as part of a genetic syndrome, with more than 300 syndromic anomalies described. The genetic basis of these conditions is equally diverse, with homeobox genes, hedgehog pathways, fibroblast growth factors, and boneand cartilage-derived morphogenetic proteins implicated in their development.1
The differential diagnosis for our patient included congenital malalignment of the great toenails, nail-patella syndrome, onychodystrophy secondary to polydactyly, and congenital hypertrophy of the lateral nail fold. Given the strong family history, polydactyly was suspected.
Congenital malalignment of the great toenails results in lateral deviation of the nail plates.2 It is an underdiagnosed condition with different etiologies hypothesized, such as genetic factors with possible autosomal-dominant transmission and extrinsic factors.3 One proposed mechanism of pathogenesis is desynchronization during growth of the nail and distal phalanx of the hallux, leading to larger nail plates that grow laterally.4 Typical features associated with this disease are nail discoloration, nail plate thickening, and transversal grooves or ridges, none of which were seen in our patient.2
Children with nail-patella syndrome have dysplastic nails and associated bony abnormalities, such as absent patellae.5 This syndrome results from an autosomaldominant mutation in the LIM homeobox transcription factor 1-beta gene, LMX1B, which is responsible for dorsal-ventral patterning of the limb, as well as patterning of the nails, patellae and long bones, and even the kidney tubule.6 As such, patients with nail-patella syndrome have associated renal abnormalities. The findings in our patient were limited to the feet, making an underlying syndrome unlikely to be the cause.
First described in 1968 by Meadow,7 fetal hydantoin syndrome is a well-documented sequela in women taking phenytoin throughout pregnancy. Multiple malformations are possible, including cardiac defects, cleft lip/palate, digit and nail hypoplasia, abnormal facial features, mental disability, and growth abnormalities.8 The teratogenicity behind phenytoin results from reactive oxygen species that alter embryonic DNA, proteins, and lipids.9 The mother of this child was not on any seizure prophylaxis, eliminating it from the differential.
Congenital hypertrophy of the lateral nail fold is a defect of the soft tissue of the hallux leading to hypertrophy of the nail fold, commonly presenting with inflammation and pain10 possibly due to dyssynchronous growth between the soft tissue and nail plate.11 With this defect, a lip covering the nail plate is common, which was not seen in our patient.
As demonstrated in our patient, family history can help guide the diagnosis. Seven of 9 nonsyndromic forms of syndactyly are inherited in an autosomal-dominant fashion and range from mild presentations, as in our patient, to more severe deformations with underlying bone fusion and functional impairment.12 Polydactyly also often is expressed in an autosomal-dominant pattern, with up to 30% of patients having a positive family history. Polydactyly traditionally is classified by the location of the supernumerary digit as preaxial (radial), central, or postaxial (ulnar), and many further morphologic variations exist within these groups. Overall, preaxial polydactyly is relatively rare and represents 15% of polydactylies, with central and postaxial comprising the other 6% and 79%, respectively.13 Delineation of the underlying anatomy may reveal ray duplications (digit and corresponding metacarpal or metatarsal bone), metatarsal variants, and duplicated phalanges that may be hypoplastic or deformed. Patients may report difficulty finding comfortable footwear, cosmetic concerns, and nail-related complications. Although not always required, surgical intervention may provide definitive treatment but can leave residual deformities in the surrounding altered anatomy; thus, orthopedic or plastic surgery consultations are critical in appropriately counseling patients.
The Diagnosis: Onychodystrophy Secondary to Polydactyly
Radiographs of the feet demonstrated an accessory distal phalanx of the left great toe with a similar smaller accessory distal phalanx on the right great toe (Figure). The patient was referred to orthopedic surgery, and surgical intervention was recommended for only the left great toe given recurrent skin inflammation and nail complications. An excision of the left great toe polydactyly was performed. The patient healed well without complications.
Many clinically heterogeneous phenotypes exist for polydactyly and syndactyly, which both are common entities with incidences of 1 in 700 to 1000 births and 1 in 2000 to 3000 births, respectively.1 Both polydactyly and syndactyly can be an isolated variant in newborns or present with multiple concurrent malformations as part of a genetic syndrome, with more than 300 syndromic anomalies described. The genetic basis of these conditions is equally diverse, with homeobox genes, hedgehog pathways, fibroblast growth factors, and boneand cartilage-derived morphogenetic proteins implicated in their development.1
The differential diagnosis for our patient included congenital malalignment of the great toenails, nail-patella syndrome, onychodystrophy secondary to polydactyly, and congenital hypertrophy of the lateral nail fold. Given the strong family history, polydactyly was suspected.
Congenital malalignment of the great toenails results in lateral deviation of the nail plates.2 It is an underdiagnosed condition with different etiologies hypothesized, such as genetic factors with possible autosomal-dominant transmission and extrinsic factors.3 One proposed mechanism of pathogenesis is desynchronization during growth of the nail and distal phalanx of the hallux, leading to larger nail plates that grow laterally.4 Typical features associated with this disease are nail discoloration, nail plate thickening, and transversal grooves or ridges, none of which were seen in our patient.2
Children with nail-patella syndrome have dysplastic nails and associated bony abnormalities, such as absent patellae.5 This syndrome results from an autosomaldominant mutation in the LIM homeobox transcription factor 1-beta gene, LMX1B, which is responsible for dorsal-ventral patterning of the limb, as well as patterning of the nails, patellae and long bones, and even the kidney tubule.6 As such, patients with nail-patella syndrome have associated renal abnormalities. The findings in our patient were limited to the feet, making an underlying syndrome unlikely to be the cause.
First described in 1968 by Meadow,7 fetal hydantoin syndrome is a well-documented sequela in women taking phenytoin throughout pregnancy. Multiple malformations are possible, including cardiac defects, cleft lip/palate, digit and nail hypoplasia, abnormal facial features, mental disability, and growth abnormalities.8 The teratogenicity behind phenytoin results from reactive oxygen species that alter embryonic DNA, proteins, and lipids.9 The mother of this child was not on any seizure prophylaxis, eliminating it from the differential.
Congenital hypertrophy of the lateral nail fold is a defect of the soft tissue of the hallux leading to hypertrophy of the nail fold, commonly presenting with inflammation and pain10 possibly due to dyssynchronous growth between the soft tissue and nail plate.11 With this defect, a lip covering the nail plate is common, which was not seen in our patient.
As demonstrated in our patient, family history can help guide the diagnosis. Seven of 9 nonsyndromic forms of syndactyly are inherited in an autosomal-dominant fashion and range from mild presentations, as in our patient, to more severe deformations with underlying bone fusion and functional impairment.12 Polydactyly also often is expressed in an autosomal-dominant pattern, with up to 30% of patients having a positive family history. Polydactyly traditionally is classified by the location of the supernumerary digit as preaxial (radial), central, or postaxial (ulnar), and many further morphologic variations exist within these groups. Overall, preaxial polydactyly is relatively rare and represents 15% of polydactylies, with central and postaxial comprising the other 6% and 79%, respectively.13 Delineation of the underlying anatomy may reveal ray duplications (digit and corresponding metacarpal or metatarsal bone), metatarsal variants, and duplicated phalanges that may be hypoplastic or deformed. Patients may report difficulty finding comfortable footwear, cosmetic concerns, and nail-related complications. Although not always required, surgical intervention may provide definitive treatment but can leave residual deformities in the surrounding altered anatomy; thus, orthopedic or plastic surgery consultations are critical in appropriately counseling patients.
- Ahmed H, Akbari H, Emanmi A, et al. Genetic overview of syndactyly and polydactyly. Plast Reconstr Surg Glob Open. 2017;5:e1549.
- Catalfo P, Musumeci ML, Lacarrubba F, et al. Congenital malalignment of the great toenails: a review. Skin Appendage Disord. 2018;4:230-235.
- Kus S, Tahmaz E, Gurunluoglu R, et al. Congenital malalignment of the great toenails in dizygotic twins. Pediatr Dermatol. 2005;22:434-435.
- Chaniotakis I, Bonitsis N, Stergiopoulou C, et al. Dizygotic twins with congenital malalignment of the great toenails: reappraisal of the pathogenesis. J Am Acad Dermatol. 2007;57:711-715.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Dreyer SD, Zhou G, Baldini A, et al, Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19:47-50.
- Meadow SR. Anticonvulsant drugs and congenital abnormalities. Lancet. 1968;2:1296.
- Scheinfeld N. Phenytoin in cutaneous medicine: its uses, mechanisms and side effects. Dermatol Online J. 2003;9:6.
- Winn LM, Wells PG. Phenytoin-initiated DNA oxidation in murine embryo culture, and embryo protection by the antioxidative enzymes superoxide dismutase and catalase: evidence for reactive oxygen species-mediated DNA oxidation in the molecular mechanism of phenytoin teratogenicity. Mol Pharmacol. 1995;48:112-120.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Martinet C, Pascal M, Civatte J, et al. Lateral nail-pad of the big toe in infants. apropos of 2 cases. Ann Dermatol Venereol. 1984;111:731-732.
- Malik S. Syndactyly: phenotypes, genetics and current classification. Eur J Hum Genet. 2012;20:817-824.
- Belthur MV, Linton JL, Barnes DA. The spectrum of preaxial polydactyly of the foot. J Pediatr Orthop. 2011;31:435-447.
- Ahmed H, Akbari H, Emanmi A, et al. Genetic overview of syndactyly and polydactyly. Plast Reconstr Surg Glob Open. 2017;5:e1549.
- Catalfo P, Musumeci ML, Lacarrubba F, et al. Congenital malalignment of the great toenails: a review. Skin Appendage Disord. 2018;4:230-235.
- Kus S, Tahmaz E, Gurunluoglu R, et al. Congenital malalignment of the great toenails in dizygotic twins. Pediatr Dermatol. 2005;22:434-435.
- Chaniotakis I, Bonitsis N, Stergiopoulou C, et al. Dizygotic twins with congenital malalignment of the great toenails: reappraisal of the pathogenesis. J Am Acad Dermatol. 2007;57:711-715.
- Witzgall R. Nail-patella syndrome. Pflugers Arch. 2017;469:927-936.
- Dreyer SD, Zhou G, Baldini A, et al, Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet. 1998;19:47-50.
- Meadow SR. Anticonvulsant drugs and congenital abnormalities. Lancet. 1968;2:1296.
- Scheinfeld N. Phenytoin in cutaneous medicine: its uses, mechanisms and side effects. Dermatol Online J. 2003;9:6.
- Winn LM, Wells PG. Phenytoin-initiated DNA oxidation in murine embryo culture, and embryo protection by the antioxidative enzymes superoxide dismutase and catalase: evidence for reactive oxygen species-mediated DNA oxidation in the molecular mechanism of phenytoin teratogenicity. Mol Pharmacol. 1995;48:112-120.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Martinet C, Pascal M, Civatte J, et al. Lateral nail-pad of the big toe in infants. apropos of 2 cases. Ann Dermatol Venereol. 1984;111:731-732.
- Malik S. Syndactyly: phenotypes, genetics and current classification. Eur J Hum Genet. 2012;20:817-824.
- Belthur MV, Linton JL, Barnes DA. The spectrum of preaxial polydactyly of the foot. J Pediatr Orthop. 2011;31:435-447.
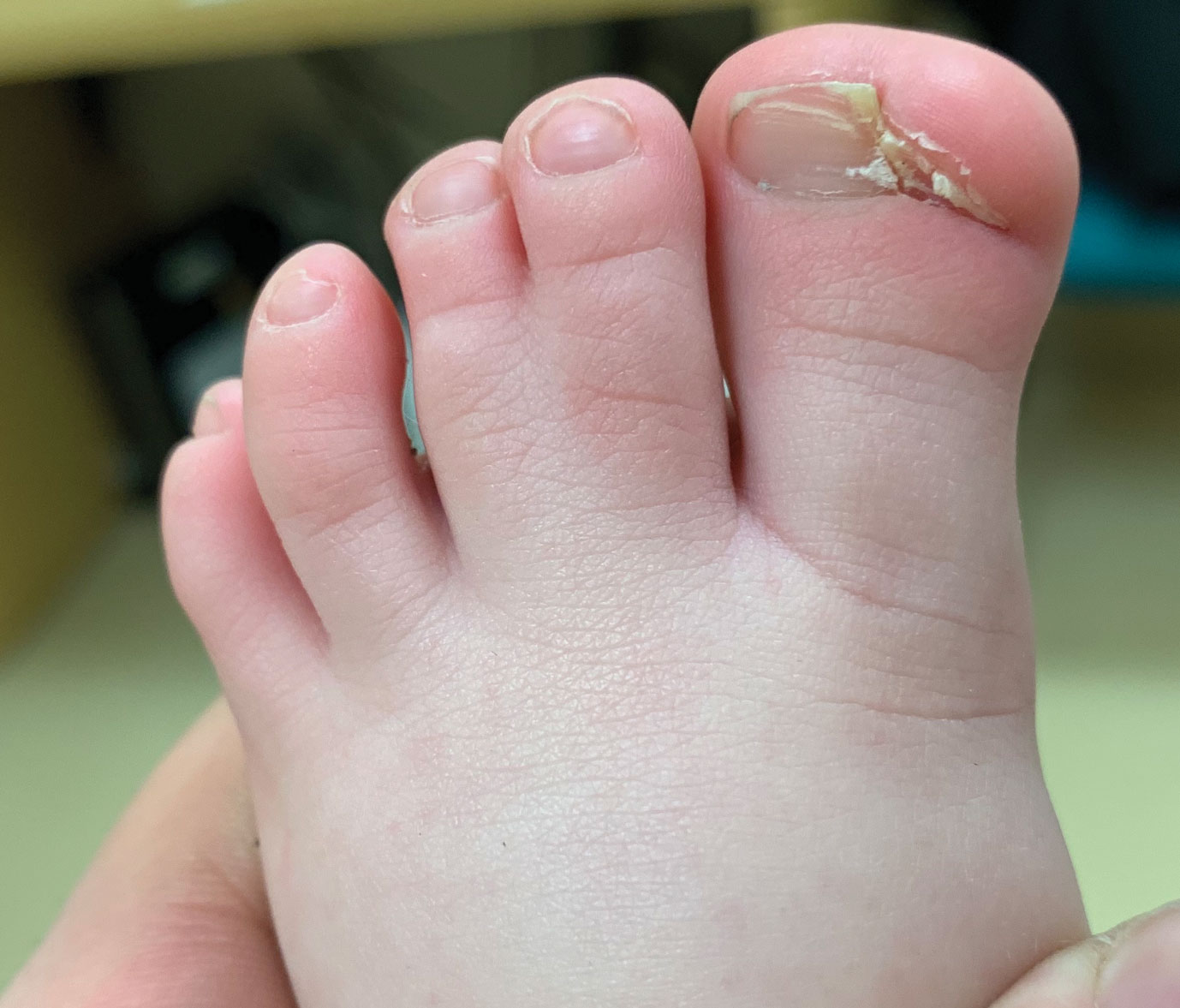
An 18-month-old girl presented for evaluation of nail dystrophy. The patient’s parents stated that the left great toenail had been dystrophic since birth, leading to skin irritation and “snagging” of the toenail on socks and footwear. Additional history revealed that the patient also had webbed toes, and there was a paternal family history of polydactyly and syndactyly. Physical examination revealed webbing of the second and third toes to the distal interphalangeal joints on both feet, marked nail plate dystrophy on the left big toe, and an irregularly shaped nail plate on the right big toe. The patient had no similar findings on the hands.
Pruritic Axillary Plaques
The Diagnosis: Granular Parakeratosis
Microscopic examination of a punch biopsy from the left axilla revealed verruciform epidermal hyperplasia with overlying parakeratosis and retention of keratohyalin granules in the stratum corneum (Figure). There was no evidence of acantholysis, dyskeratosis, epidermal neutrophils, or neutrophilic microabscesses.
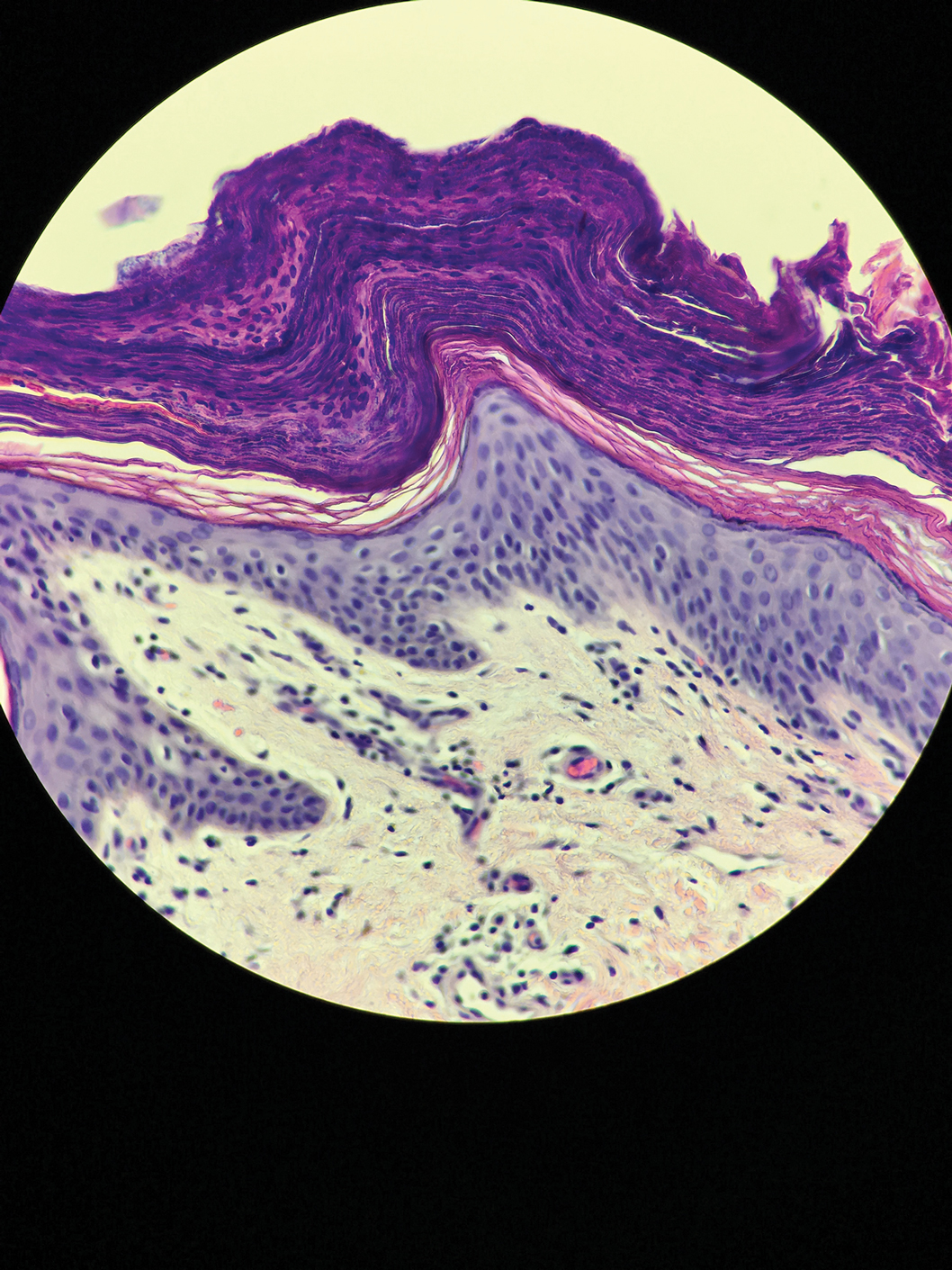
The patient's history and characteristic histopathologic findings confirmed the diagnosis of granular parakeratosis (GP). He was instructed to discontinue his current antiperspirant and began treatment with topical fluocinolone oil 0.01% every morning and urea cream 20% every night. Complete resolution was achieved within 2 weeks, and he reported no recurrence at a 2-year follow-up visit.
Granular parakeratosis is a rare idiopathic skin condition characterized by hyperkeratotic papules and plaques, most often in intertriginous areas. Described by Northcutt et al1 as a contact reaction to antiperspirant in the axillae, GP also has been reported in the submammary and inguinal creases2 and rarely in nonintertriginous sites such as the abdomen.3 Although deodorants and antiperspirants in roll-on or stick form classically are implicated in GP, the condition also has been observed with exposure to laundry detergents containing benzalkonium chloride.4 Lesions with GP histology also have been incidentally observed in association with dermatophytosis,5 dermatomyositis,6 molluscum contagiosum,7 and carcinomas.8 Ding et al9 proposed that GP be reclassified as a reaction pattern observed in the skin as opposed to being a distinct disease entity.
Clinically, GP presents as pruritic intertriginous papules and coalescent plaques that most commonly are seen in the axillae but also may involve the groin or other sites.2,3 Both pruritus and disease burden can be aggravated by heat, sweating, or friction. There may be a history of a new irritant exposure prior to symptom onset, but GP has been observed in the absence of identifiable exposures and in the setting of long-term antiperspirant or deodorant use.3 Although a family history may be helpful, it can be difficult to distinguish GP from entities such as Hailey-Hailey disease or Darier disease based on history and examination alone; a biopsy often is necessary for definitive diagnosis.
Histologically, GP demonstrates acanthosis with parakeratosis and retention of keratohyalin granules in the stratum corneum.1 The stratum granulosum is preserved. On cursory examination, GP may resemble a psoriasiform dermatosis as can be seen in inverse psoriasis; however, neutrophilic microabscesses and infiltrates are not seen. Absence of acantholysis and dyskeratosis further differentiates GP from the clinically similar Hailey-Hailey disease and Darier disease. Spongiosis that is prominently found in allergic contact dermatitis also is absent.
Although a benign disorder, GP warrants treatment to achieve symptomatic relief. A mainstay of treatment is to eliminate exposure to suspected aggravating or inciting factors such as antiperspirants or deodorants. A variety of treatments including laser therapy, corticosteroids, isotretinoin, and vitamin D analogs such as calcipotriene and calcitriol have been reported to be effective treatments of GP in case studies and series.3,10 Large-scale clinical trials are not available because of the rarity of this condition. Our patient's clinical course suggests topical fluocinolone and urea in combination can be considered to achieve rapid resolution.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Samrao A, Reis M, Neidt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Robinson AJ, Foster RS, Halbert AR, et al. Granular parakeratosis induced by benzalkonium chloride exposure from laundry rinse aids. Australas J Dermatol. 2017;58:E138-E140.
- Resnik KS, Kantor GR, DiLeonardo M. Dermatophyte-related granular parakeratosis. Am J Dermatopathol. 2004;26:70-71.
- Pock L, Hercogová J. Incidental granular parakeratosis associated with dermatomyositis. Am J Dermatopathol. 2006;28:147-149.
- Pock L, Cermáková A, Zipfelová J, et al. Incidental granular parakeratosis associated with molluscum contagiosum. Am J Dermatopathol. 2006;28:45-47.
- Resnik KS, DiLeonardo M. Incidental granular parakeratotic cornification in carcinomas. Am J Dermatopathol. 2007;29:264-269.
- Ding CY, Liu H, Khachemoune A. Granular parakeratosis: a comprehensive review and a critical reappraisal. Am J Clin Dermatol. 2015;16:495-500.
- Patel U, Patel T, Skinner RB. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
The Diagnosis: Granular Parakeratosis
Microscopic examination of a punch biopsy from the left axilla revealed verruciform epidermal hyperplasia with overlying parakeratosis and retention of keratohyalin granules in the stratum corneum (Figure). There was no evidence of acantholysis, dyskeratosis, epidermal neutrophils, or neutrophilic microabscesses.
The patient's history and characteristic histopathologic findings confirmed the diagnosis of granular parakeratosis (GP). He was instructed to discontinue his current antiperspirant and began treatment with topical fluocinolone oil 0.01% every morning and urea cream 20% every night. Complete resolution was achieved within 2 weeks, and he reported no recurrence at a 2-year follow-up visit.
Granular parakeratosis is a rare idiopathic skin condition characterized by hyperkeratotic papules and plaques, most often in intertriginous areas. Described by Northcutt et al1 as a contact reaction to antiperspirant in the axillae, GP also has been reported in the submammary and inguinal creases2 and rarely in nonintertriginous sites such as the abdomen.3 Although deodorants and antiperspirants in roll-on or stick form classically are implicated in GP, the condition also has been observed with exposure to laundry detergents containing benzalkonium chloride.4 Lesions with GP histology also have been incidentally observed in association with dermatophytosis,5 dermatomyositis,6 molluscum contagiosum,7 and carcinomas.8 Ding et al9 proposed that GP be reclassified as a reaction pattern observed in the skin as opposed to being a distinct disease entity.
Clinically, GP presents as pruritic intertriginous papules and coalescent plaques that most commonly are seen in the axillae but also may involve the groin or other sites.2,3 Both pruritus and disease burden can be aggravated by heat, sweating, or friction. There may be a history of a new irritant exposure prior to symptom onset, but GP has been observed in the absence of identifiable exposures and in the setting of long-term antiperspirant or deodorant use.3 Although a family history may be helpful, it can be difficult to distinguish GP from entities such as Hailey-Hailey disease or Darier disease based on history and examination alone; a biopsy often is necessary for definitive diagnosis.
Histologically, GP demonstrates acanthosis with parakeratosis and retention of keratohyalin granules in the stratum corneum.1 The stratum granulosum is preserved. On cursory examination, GP may resemble a psoriasiform dermatosis as can be seen in inverse psoriasis; however, neutrophilic microabscesses and infiltrates are not seen. Absence of acantholysis and dyskeratosis further differentiates GP from the clinically similar Hailey-Hailey disease and Darier disease. Spongiosis that is prominently found in allergic contact dermatitis also is absent.
Although a benign disorder, GP warrants treatment to achieve symptomatic relief. A mainstay of treatment is to eliminate exposure to suspected aggravating or inciting factors such as antiperspirants or deodorants. A variety of treatments including laser therapy, corticosteroids, isotretinoin, and vitamin D analogs such as calcipotriene and calcitriol have been reported to be effective treatments of GP in case studies and series.3,10 Large-scale clinical trials are not available because of the rarity of this condition. Our patient's clinical course suggests topical fluocinolone and urea in combination can be considered to achieve rapid resolution.
The Diagnosis: Granular Parakeratosis
Microscopic examination of a punch biopsy from the left axilla revealed verruciform epidermal hyperplasia with overlying parakeratosis and retention of keratohyalin granules in the stratum corneum (Figure). There was no evidence of acantholysis, dyskeratosis, epidermal neutrophils, or neutrophilic microabscesses.
The patient's history and characteristic histopathologic findings confirmed the diagnosis of granular parakeratosis (GP). He was instructed to discontinue his current antiperspirant and began treatment with topical fluocinolone oil 0.01% every morning and urea cream 20% every night. Complete resolution was achieved within 2 weeks, and he reported no recurrence at a 2-year follow-up visit.
Granular parakeratosis is a rare idiopathic skin condition characterized by hyperkeratotic papules and plaques, most often in intertriginous areas. Described by Northcutt et al1 as a contact reaction to antiperspirant in the axillae, GP also has been reported in the submammary and inguinal creases2 and rarely in nonintertriginous sites such as the abdomen.3 Although deodorants and antiperspirants in roll-on or stick form classically are implicated in GP, the condition also has been observed with exposure to laundry detergents containing benzalkonium chloride.4 Lesions with GP histology also have been incidentally observed in association with dermatophytosis,5 dermatomyositis,6 molluscum contagiosum,7 and carcinomas.8 Ding et al9 proposed that GP be reclassified as a reaction pattern observed in the skin as opposed to being a distinct disease entity.
Clinically, GP presents as pruritic intertriginous papules and coalescent plaques that most commonly are seen in the axillae but also may involve the groin or other sites.2,3 Both pruritus and disease burden can be aggravated by heat, sweating, or friction. There may be a history of a new irritant exposure prior to symptom onset, but GP has been observed in the absence of identifiable exposures and in the setting of long-term antiperspirant or deodorant use.3 Although a family history may be helpful, it can be difficult to distinguish GP from entities such as Hailey-Hailey disease or Darier disease based on history and examination alone; a biopsy often is necessary for definitive diagnosis.
Histologically, GP demonstrates acanthosis with parakeratosis and retention of keratohyalin granules in the stratum corneum.1 The stratum granulosum is preserved. On cursory examination, GP may resemble a psoriasiform dermatosis as can be seen in inverse psoriasis; however, neutrophilic microabscesses and infiltrates are not seen. Absence of acantholysis and dyskeratosis further differentiates GP from the clinically similar Hailey-Hailey disease and Darier disease. Spongiosis that is prominently found in allergic contact dermatitis also is absent.
Although a benign disorder, GP warrants treatment to achieve symptomatic relief. A mainstay of treatment is to eliminate exposure to suspected aggravating or inciting factors such as antiperspirants or deodorants. A variety of treatments including laser therapy, corticosteroids, isotretinoin, and vitamin D analogs such as calcipotriene and calcitriol have been reported to be effective treatments of GP in case studies and series.3,10 Large-scale clinical trials are not available because of the rarity of this condition. Our patient's clinical course suggests topical fluocinolone and urea in combination can be considered to achieve rapid resolution.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Samrao A, Reis M, Neidt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Robinson AJ, Foster RS, Halbert AR, et al. Granular parakeratosis induced by benzalkonium chloride exposure from laundry rinse aids. Australas J Dermatol. 2017;58:E138-E140.
- Resnik KS, Kantor GR, DiLeonardo M. Dermatophyte-related granular parakeratosis. Am J Dermatopathol. 2004;26:70-71.
- Pock L, Hercogová J. Incidental granular parakeratosis associated with dermatomyositis. Am J Dermatopathol. 2006;28:147-149.
- Pock L, Cermáková A, Zipfelová J, et al. Incidental granular parakeratosis associated with molluscum contagiosum. Am J Dermatopathol. 2006;28:45-47.
- Resnik KS, DiLeonardo M. Incidental granular parakeratotic cornification in carcinomas. Am J Dermatopathol. 2007;29:264-269.
- Ding CY, Liu H, Khachemoune A. Granular parakeratosis: a comprehensive review and a critical reappraisal. Am J Clin Dermatol. 2015;16:495-500.
- Patel U, Patel T, Skinner RB. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
- Northcutt AD, Nelson DM, Tschen JA. Axillary granular parakeratosis. J Am Acad Dermatol. 1991;24:541-544.
- Burford C. Granular parakeratosis of multiple intertriginous areas. Australas J Dermatol. 2008;49:35-38.
- Samrao A, Reis M, Neidt G, et al. Granular parakeratosis: response to calcipotriene and brief review of current therapeutic options. Skinmed. 2010;8:357-359.
- Robinson AJ, Foster RS, Halbert AR, et al. Granular parakeratosis induced by benzalkonium chloride exposure from laundry rinse aids. Australas J Dermatol. 2017;58:E138-E140.
- Resnik KS, Kantor GR, DiLeonardo M. Dermatophyte-related granular parakeratosis. Am J Dermatopathol. 2004;26:70-71.
- Pock L, Hercogová J. Incidental granular parakeratosis associated with dermatomyositis. Am J Dermatopathol. 2006;28:147-149.
- Pock L, Cermáková A, Zipfelová J, et al. Incidental granular parakeratosis associated with molluscum contagiosum. Am J Dermatopathol. 2006;28:45-47.
- Resnik KS, DiLeonardo M. Incidental granular parakeratotic cornification in carcinomas. Am J Dermatopathol. 2007;29:264-269.
- Ding CY, Liu H, Khachemoune A. Granular parakeratosis: a comprehensive review and a critical reappraisal. Am J Clin Dermatol. 2015;16:495-500.
- Patel U, Patel T, Skinner RB. Resolution of granular parakeratosis with topical calcitriol. Arch Dermatol. 2011;147:997-998.
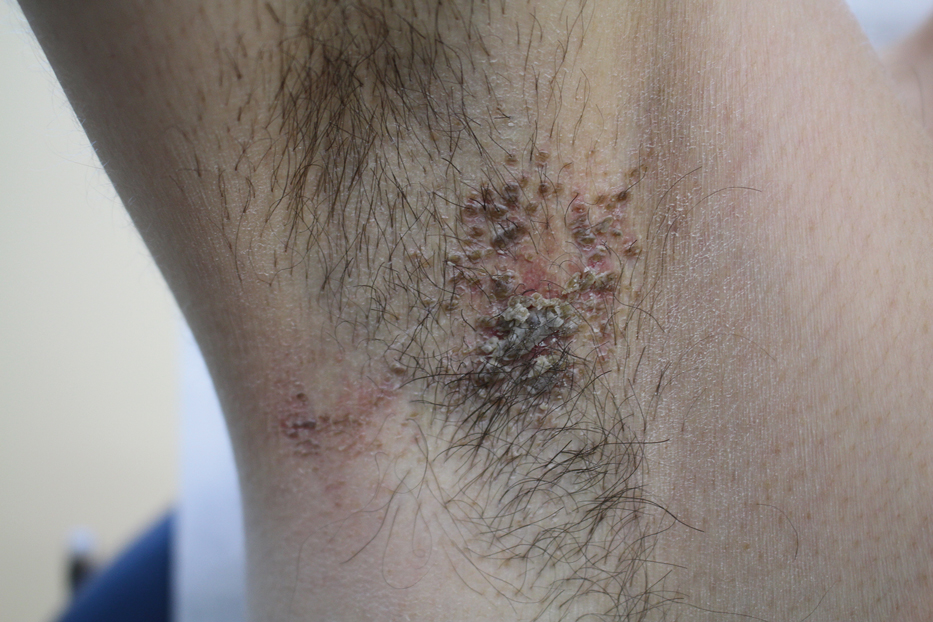
A 42-year-old man presented with pruritic axillary plaques of 6 months’ duration that were exacerbated by heat and friction. He maintained a very active lifestyle and used an antiperspirant regularly. He denied any family history of similar lesions. Thick emollients provided no relief. Physical examination demonstrated numerous soft, hyperkeratotic, waxy, yellowish brown papules coalescing into plaques localized to the bilateral axillary vaults, affecting the right axilla more than the left. Although some papules were firmly adherent to the skin, others were friable and easily removed with a cotton-tipped applicator, revealing an underlying, faintly erythematous base.
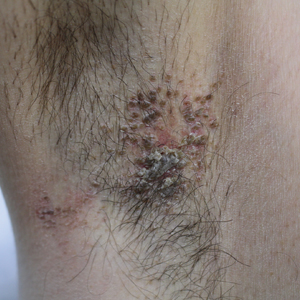
Bullae and Hyperpigmented Patches on the Legs
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
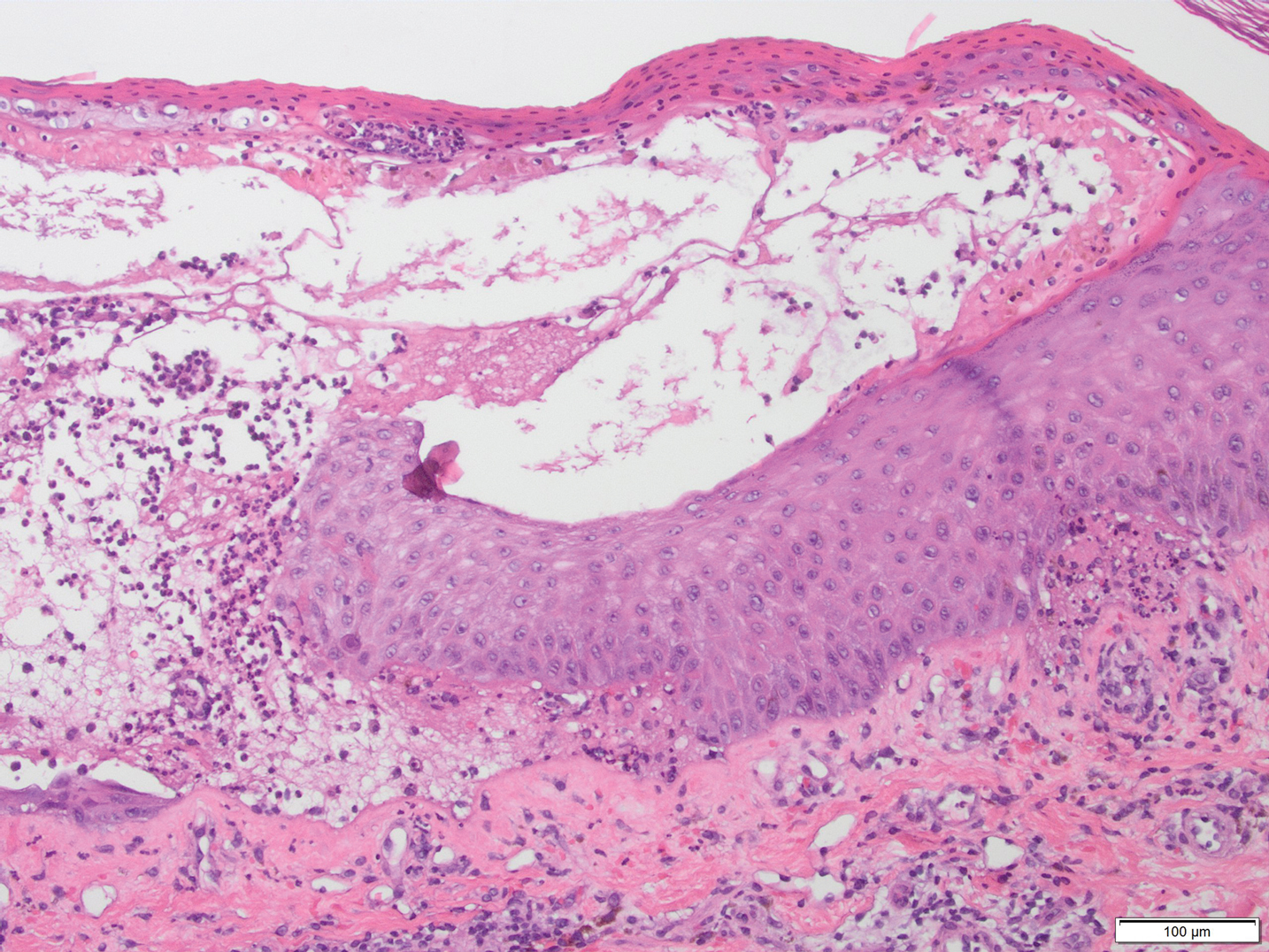
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
The Diagnosis: Lichen Planus Pemphigoides
A skin biopsy from the right thigh demonstrated subepidermal blisters containing neutrophils (Figure 1). Direct immunofluorescence revealed linear basement membrane zone staining with C3 and trace staining with IgG (Figure 2), supporting a diagnosis of lichen planus pemphigoides (LPP). Oral prednisone—starting at 60 mg daily and tapered to 40 mg for a week, 20 mg for a week, then 10 mg for a month—along with triamcinolone ointment 0.1% to affected areas led to improvement. Hydrochlorothiazide and UV light therapy were discontinued. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily prescribed as adjunctive therapy also led to improvement. The patient achieved remission with doxycycline and was doing well without prednisone; however, he experienced a flare of his disease about 6 months later and was started on mycophenolate mofetil 1 g twice daily after clearance from his gastroenterologist, given his history of hepatitis B. He has been doing well since starting mycophenolate mofetil.
Lichen planus pemphigoides is a rare autoimmune bullous dermatosis with features of both lichen planus and bullous pemphigoid.1 Violaceous papules and tense bullae may be superimposed or arise independently. The chest, abdomen, back, and upper and lower extremities typically are involved.2 Oral mucosal involvement with white reticular streaks or erosions and nail involvement have been reported.2 Histopathologic and immunologic findings establish the diagnosis. Lichen planus pemphigoides is associated with subepidermal bullae and linear deposits of IgG and C3 on the basement membrane zone.1 Autoantibodies to bullous pemphigoid (BP) antigens BP180 and BP230 are associated with LPP.3 The pathogenesis of LPP remains unclear, but there are associations with chronic diseases, medications, and certain therapies.1,4-6 Several case reports have linked LPP to chronic viral hepatitis infections, as well as malignant tumors of the skin, mucosa, and gastrointestinal tract.2 Lichen planus pemphigoides has been reported in a patient on entecavir for hepatitis B as well as in a patient treated for hepatitis C with interferon and ribavirin.1 Lichen planus pemphigoides has been described in patients treated with the angiotensin-converting enzyme inhibitors enalapril, captopril, and ramipril.4,5,7 UV phototherapy also has been associated with the development of LPP.6 Hydrochlorothiazide previously has been reported as a cause of drug-induced lichen planus.8 A PubMed search of articles indexed for MEDLINE using the terms lichen planus pemphigoides and hydrochlorothiazide revealed no reports of hydrochlorothiazide-induced LPP.
Lichen planus pemphigoides demonstrates overlap with other blistering dermatoses, such as BP, bullous lupus erythematosus, and bullous lichen planus. Although histologically and immunologically similar to BP, LPP can be differentiated clinically by the presence of violaceous papules or plaques typical of lichen planus.9 Bullous pemphigoid is more common in individuals older than 70 years, whereas LPP tends to occur in middle-aged adults.2 Bullous lupus erythematosus usually is associated with manifestations of systemic lupus erythematosus and autoantibodies to collagen type VII.10 Salt-split skin studies demonstrate immunofluorescence on the dermal side of the split. Individuals affected by bullous lupus erythematosus typically have a history of photosensitivity.10 Blisters in LPP may form de novo from unaffected skin, whereas the bullae in bullous lichen planus are limited to existing lichenoid papules.9 The autoantibodies typical of LPP are absent in bullous lichen planus. Lichen planus actinicus is a variant of lichen planus that presents with annular, dyschromic, or violaceous plaques in a photodistributed pattern without bullous lesions.9
Lichen planus pemphigoides most commonly is treated with systemic corticosteroids. Topical steroids, dapsone, erythromycin, tetracycline and nicotinamide, azathioprine, and mycophenolate mofetil have been reported as adjuncts to systemic steroid therapy.2,11 Most reports describe treatment success with resolution of blistering lesions.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
- Jang SH, Yun SJ, Lee SC, et al. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. 2015;40:868-871.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Harting MS, Hsu S. Lichen planus pemphigoides: a case report and review of the literature. Dermatol Online J. 2006;12:10.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Ben Salem C, Chengeul L, Ghariani N, et al. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. 2008;17:722-724.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Zhu YI, Fitzpatrick JE, Kornfield BW. Lichen planus pemphigoides associated with Ramipril. Int J Dermatol. 2006;45:1453-1455.
- Sin B, Miller M, Chew E. Hydrochlorothiazide induced lichen planus in the emergency department. J Pharm Pract. 2017;30:266-269.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Women Dermatol. 2015;1:140-149.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Fivenson DP, Kimbrough TL. Lichen planus pemphigoides: combination therapy with tetracycline and nicotinamide. J Am Acad Dermatol. 1997;36:638-640.
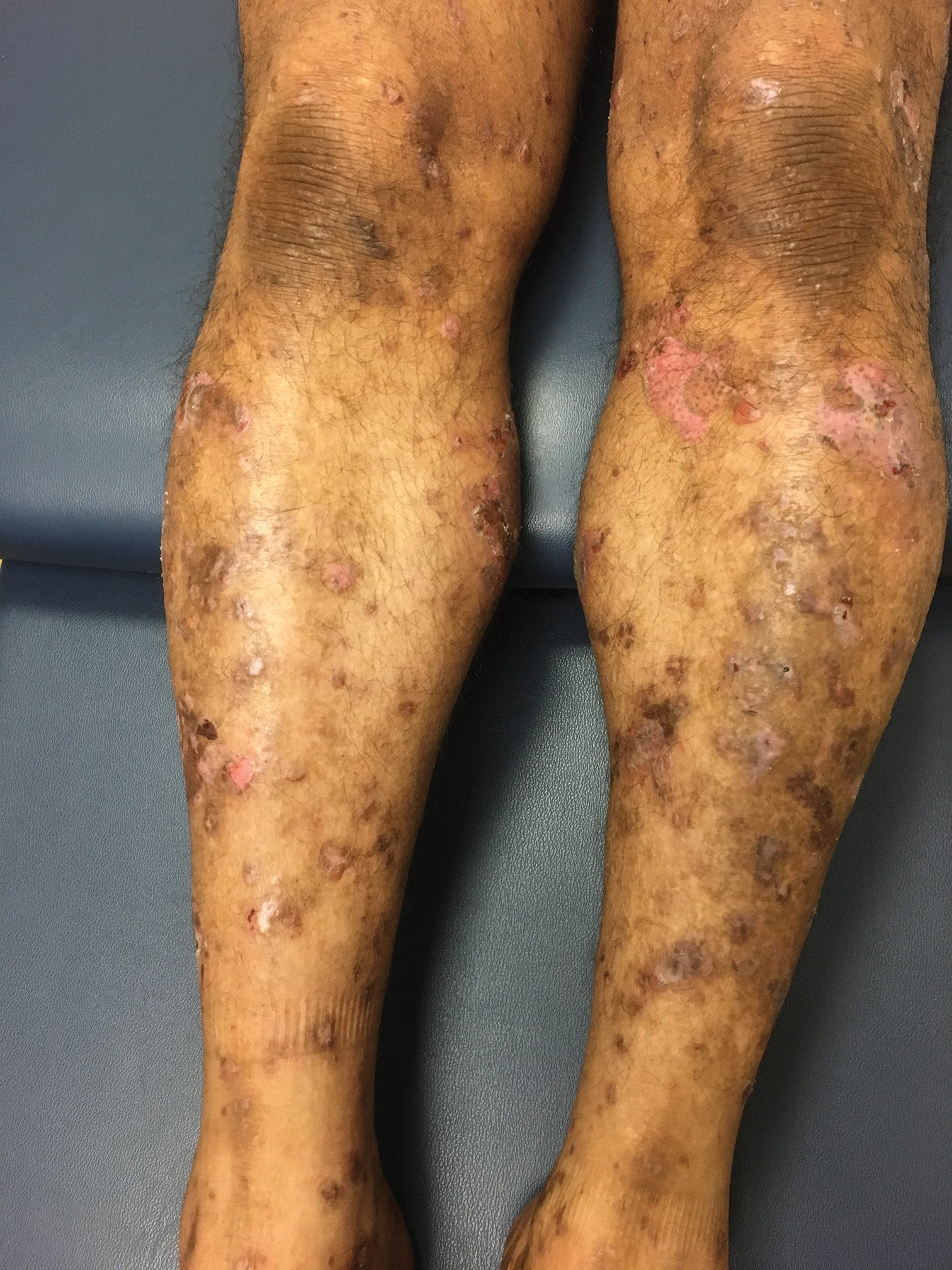
A 50-year-old man presented with a pruritic bullous dermatosis on the lower legs, arms, and back of 1 month’s duration. He had an 8-year history of lichen planus, and the lesions recently had worsened despite the addition of UVB phototherapy. His medical history was remarkable for hepatitis B treated with entecavir and the addition of hydrochlorothiazide for essential hypertension 2 weeks prior to the dramatic worsening of the rash. Physical examination revealed multiple bullae on the lower legs associated with violaceous and hyperpigmented papules and patches. He also had violaceous papules on the lower back and eroded lesions on the oral mucosa. Shave biopsies were obtained from the right thigh and mid back, and histopathologic analysis was performed for both routine histology and direct immunofluorescence.
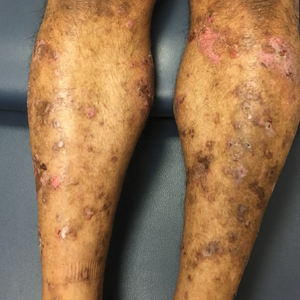
Hyperpigmentation of the Tongue
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
The Diagnosis: Addison Disease in the Context of Polyglandular Autoimmune Syndrome Type 2
The patient’s hormone levels as well as distinct clinical features led to a diagnosis of Addison disease in the context of polyglandular autoimmune syndrome type 2 (PAS-2). Approximately 50% of PAS-2 cases are familiar, and different modes of inheritance—autosomal recessive, autosomal dominant, and polygenic—have been reported. Women are affected up to 3 times more often than men.1,2 The age of onset ranges from infancy to late adulthood, with most cases occurring in early adulthood. Primary adrenal insufficiency (Addison disease) is the principal manifestation of PAS-2. It appears in approximately 50% of patients, occurring simultaneously with autoimmune thyroid disease or diabetes mellitus in 20% of patients and following them in 30% of patients.1,2 Autoimmune thyroid diseases such as chronic autoimmune thyroiditis and occasionally Graves disease as well as type 1 diabetes mellitus also are common. Polyglandular autoimmune syndrome type 2 with primary adrenal insufficiency and autoimmune thyroid disease was formerly referred to as Schmidt syndrome.3 It must be differentiated from polyglandular autoimmune syndrome type 1, a rare condition that also is referred to as autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy syndrome.1,3 As with any other cause of adrenal insufficiency, the treatment involves hormone replacement therapy up to normal levels and then tapering according to stress levels (ie, surgery or infections that require a dose increase). Our patient was diagnosed according to hormone levels and clinical features and was started on 30 mg daily of hydrocortisone and 50 μg daily of levothyroxine. No improvement in her condition was noted after 6 months of treatment. The patient is still under yearly follow-up, and the mucous hyperpigmentation faded approximately 6 months after hormonal homeostasis was achieved.
Peutz-Jeghers syndrome is inherited in an autosomal-dominant fashion. It is characterized by multiple hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and an increased risk for gastrointestinal and nongastrointestinal cancer. Mucocutaneous pigmented macules most commonly occur on the lips and perioral region, buccal mucosa, and the palms and soles. However, mucocutaneous pigmentation usually occurs during the first 1 to 2 years of life, increases in size and number over the ensuing years, and usually fades after puberty.4
Laugier-Hunziker syndrome is an acquired benign disorder presenting in adults with lentigines on the lips and buccal mucosa. It frequently is accompaniedby longitudinal melanonychia, macular pigmentation of the genitals, and involvement of the palms and soles. The diagnosis of Laugier-Hunziker syndrome is one of exclusion and is made after ruling out other causes of oral and labial hyperpigmentation, including physiologic pigmentation seen in darker-skinned individuals as well as inherited diseases associated with lentiginosis, requiring complete physical examination, endoscopy, and colonscopy.5
A wide variety of drugs and chemicals can lead to diffuse cutaneous hyperpigmentation. Increased production of melanin and/or the deposition of drug complexes or metals in the dermis is responsible for the skin discoloration. Drugs that most often cause hyperpigmentation on mucosal surfaces are hydroxychloroquine, minocycline, nicotine, silver, and some chemotherapy agents. The hyperpigmentation usually resolves with discontinuation of the offending agent, but the course may be prolonged over months to years.6
Changes in the skin and subcutaneous tissue occur in patients with Cushing syndrome. Hyperpigmentation is induced by increased secretion of adrenocorticotropic hormone, not cortisol, and occurs most often in patients with the ectopic adrenocorticotropic hormone syndrome. Hyperpigmentation may be generalized but is more intense in areas exposed to light (eg, face, neck, dorsal aspects of the hands) or to chronic mild trauma, friction, or pressure (eg, elbows, knees, spine, knuckles). Patchy pigmentation may occur on the inner surface of the lips and the buccal mucosa along the line of dental occlusion. Acanthosis nigricans also can be present in the axillae and around the neck.7
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
- Ferre EM, Rose SR, Rosenzweig SD, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy. JCI Insight. 2016;1:E88782.
- Orlova EM, Sozaeva LS, Kareva MA, et al. Expanding the phenotypic and genotypic landscape of autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2017;102:3546-3556.
- Ahonen P, Myllärniemi S, Sipilä I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-1836.
- Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136:71-82.
- Nayak RS, Kotrashetti VS, Hosmani JV. Laugier-Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245-250.
- Krause W. Drug-induced hyperpigmentation: a systematic review. J Dtsch Dermatol Ges. 2013;11:644-651.
- Newell-Price J, Trainer P, Besser M, et al. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647-672.
An otherwise healthy 17-year-old adolescent girl from Spain presented with hyperpigmentation on the tongue of several weeks’ duration. She denied licking graphite pencils or pens. Physical examination revealed pigmentation in the palmar creases and a slight generalized tan. The patient denied sun exposure. Neither melanonychia nor genital hyperpigmented lesions were noted. Blood tests showed overt hypothyroidism.