User login
FDA’s Woodcock: Give biosimilars a chance
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.

She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.

“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM
Biosimilars face barriers to broader acceptance
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.

But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.
But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
BOSTON – For rheumatologists, the primary selling points of biosimilar agents are their presumed efficacy and safety, and their promised cost savings, compared with the reference product, an original biologic agent.
But market forces, patient worries, and provider concerns threaten to file off at least some of biosimilars’ presumed competitive edge, says a rheumatologist involved in the development of biosimilars to treat rheumatic diseases.
“The availability of lower-priced biosimilars, if they’re truly lower priced, should decrease the cost of treating patients; if they’re not lower cost, then this advantage is not holding true,” said Jonathan Kay, MD, professor of medicine at UMass Memorial Medical Center in Worcester.
But the world is far from perfect, and there are a host of educational, financial, and societal barriers to more widespread acceptance of these agents, he said.
Misperceptions
A common concern among patients and uninformed providers is that biosimilars are the same as biomimics – cheap, usually foreign-made knockoffs of existing drugs that have not undergone regulatory scrutiny and may be neither effective nor safe.
“Biomimics scare patients and scare physicians,” he said, “and misunderstanding of the regulatory approval process, or lack of understanding of the regulatory review or approval process, also intimidates individuals about biosimilars.”
Acceptance of biosimilars is also hampered by a lack of long-term, real-world efficacy and safety data. This is due, in part, to the “inverted pyramid” nature of the biosimilar development and approval process, which starts with myriad analytical, chemical, and functional characterization studies; nonclinical studies; and pharmacokinetic and pharmacodynamic analyses, but only one or two clinical trials. In contrast, the development process for innovator biologics starts with molecular characterization, then moves through the traditional drug development stages of laboratory and preclinical studies, phase I and II trials, and finally phase III registration trials and post-marketing studies.
Because most of the drug development for biosimilars happens behind the scenes, patients and their physicians are often leery about the finished product, no matter how rigorously it was developed. Similarly, there is a paucity of data on long-term immunogenicity of biosimilars, Dr. Kay noted.
Extrapolating about extrapolation
The idea of extrapolation of indications for biosimilars is also an alien concept to many. Simply put, if a reference product is approved for rheumatoid arthritis and has additional indications for treatment of psoriatic arthritis, the approved biosimilar to the original can be extrapolated to be effective against the same indications, even if it has only been tested against one of the indications.
“We have a tremendous amount of clinical experience not only in randomized, controlled clinical trials but also years of marketing experience showing that the reference product is effective and safe for each of the indications for which it’s been approved, and knowing that the biosimilar is essentially like another [manufacturing] run of the reference product allows us to extrapolate indications. But until you become comfortable with accepting that concept, it is certainly a barrier to biosimilars,” he said.
Patients are also scared by the issue of interchangeability, which would allow pharmacists to substitute one product for another without approval of either the patient or the prescribing physician. However, there are currently no biosimilar agents approved as interchangeable by the Food and Drug Administration.
What cost savings?
In the United States, the price of marketed biosimilars has been only 15% lower than that of the reference product, Dr. Kay noted, and multiple discount programs and rebates offered by marketers of the reference product make it difficult to figure out the actual costs of the drugs to payers and whether biosimilars are actually any cheaper.
Additionally, clinicians may be reluctant to adopt biosimilars because of a Centers for Medicare & Medicaid Services billing quirk that currently offers only a single “J” code with a blended reimbursement for all biosimilar agents of a reference product, regardless of cost differences.
Nordic competition model
Dr. Kay pointed to the Norwegian Tender System as a model for using biosimilars to drive down drug costs.
The Norwegian Drug Procurement Cooperation (LIS) annually procures drugs for all publicly funded hospitals, which ensures that prices offered to hospitals for patent-protected medicines are lower than those offered for distribution to wholesalers or pharmacies. Under this system, cost is the driving factor, assuming that drugs in a similar class have similar efficacy and safety, he said.
In 2015, when Norway was purchasing infliximab, it chose Remsima, the biosimilar, over the reference product Remicade, resulting in a 69% cost saving.
The potential interchangeability of Remsima and Remicade in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis is currently being explored in the NOR-SWITCH trial.
Dr. Kay disclosed receiving institutional grants from AbbVie, Genentech, Roche Laboratories, and UCB, and serving as a consultant for those companies.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM
Intravitreal sirolimus proves effective in reducing noninfectious uveitis inflammation
Intravitreal sirolimus 440 mcg or 880 mcg administered on days 1, 60, and 120, was shown to significantly improve ocular inflammation with preservation of best-corrected visual acuity in patients with noninfectious uveitis of the posterior segment, a phase III study has shown.
In the multinational SAKURA (Study Assessing Double-masked Uveitis Treatment) study, 346 study eyes were analyzed in this randomly assigned, double-masked, actively controlled study. In the study arm given intravitreal sirolimus 440 mcg, 22.8% (P = .025) met the primary endpoint of no vitreous haze (VH) at month 5 in the study eye without the aid of rescue therapy. In the group given intravitreal sirolimus 880 mcg, 16.4% (P = .182) met the primary endpoint, compared with 10.3% of active controls who were given 44 mcg intravitreal sirolimus.
For the secondary outcome, 52.6% (P = .008) of the intravitreal sirolimus 440 mcg arm had VH scores of 0 or a 0.5+ response rate at month 5. In the intravitreal sirolimus 880 mcg, 43.1% (P = .228) achieved a VH score of 0 or a 0.5+ response rate, compared with 35% of the 44 mcg active control group. Mean best-corrected visual acuity was maintained throughout the study in each study arm, with 76.9% of those who received corticosteroids at baseline in the 440 mcg study arm successfully tapering them to 5 mg per day or less by month 5, and 66.7% of those receiving corticosteroids in the 880 mcg group doing so. This was in comparison with 63.6% of those using corticosteroids in the active control group. Adverse events were similar across the study, and all doses were well tolerated. The study was funded by Santen.
Read the full study in Ophthalmology (2016;23[11]:2413-23).
[email protected]
On Twitter @whitneymcknight
Intravitreal sirolimus 440 mcg or 880 mcg administered on days 1, 60, and 120, was shown to significantly improve ocular inflammation with preservation of best-corrected visual acuity in patients with noninfectious uveitis of the posterior segment, a phase III study has shown.
In the multinational SAKURA (Study Assessing Double-masked Uveitis Treatment) study, 346 study eyes were analyzed in this randomly assigned, double-masked, actively controlled study. In the study arm given intravitreal sirolimus 440 mcg, 22.8% (P = .025) met the primary endpoint of no vitreous haze (VH) at month 5 in the study eye without the aid of rescue therapy. In the group given intravitreal sirolimus 880 mcg, 16.4% (P = .182) met the primary endpoint, compared with 10.3% of active controls who were given 44 mcg intravitreal sirolimus.
For the secondary outcome, 52.6% (P = .008) of the intravitreal sirolimus 440 mcg arm had VH scores of 0 or a 0.5+ response rate at month 5. In the intravitreal sirolimus 880 mcg, 43.1% (P = .228) achieved a VH score of 0 or a 0.5+ response rate, compared with 35% of the 44 mcg active control group. Mean best-corrected visual acuity was maintained throughout the study in each study arm, with 76.9% of those who received corticosteroids at baseline in the 440 mcg study arm successfully tapering them to 5 mg per day or less by month 5, and 66.7% of those receiving corticosteroids in the 880 mcg group doing so. This was in comparison with 63.6% of those using corticosteroids in the active control group. Adverse events were similar across the study, and all doses were well tolerated. The study was funded by Santen.
Read the full study in Ophthalmology (2016;23[11]:2413-23).
[email protected]
On Twitter @whitneymcknight
Intravitreal sirolimus 440 mcg or 880 mcg administered on days 1, 60, and 120, was shown to significantly improve ocular inflammation with preservation of best-corrected visual acuity in patients with noninfectious uveitis of the posterior segment, a phase III study has shown.
In the multinational SAKURA (Study Assessing Double-masked Uveitis Treatment) study, 346 study eyes were analyzed in this randomly assigned, double-masked, actively controlled study. In the study arm given intravitreal sirolimus 440 mcg, 22.8% (P = .025) met the primary endpoint of no vitreous haze (VH) at month 5 in the study eye without the aid of rescue therapy. In the group given intravitreal sirolimus 880 mcg, 16.4% (P = .182) met the primary endpoint, compared with 10.3% of active controls who were given 44 mcg intravitreal sirolimus.
For the secondary outcome, 52.6% (P = .008) of the intravitreal sirolimus 440 mcg arm had VH scores of 0 or a 0.5+ response rate at month 5. In the intravitreal sirolimus 880 mcg, 43.1% (P = .228) achieved a VH score of 0 or a 0.5+ response rate, compared with 35% of the 44 mcg active control group. Mean best-corrected visual acuity was maintained throughout the study in each study arm, with 76.9% of those who received corticosteroids at baseline in the 440 mcg study arm successfully tapering them to 5 mg per day or less by month 5, and 66.7% of those receiving corticosteroids in the 880 mcg group doing so. This was in comparison with 63.6% of those using corticosteroids in the active control group. Adverse events were similar across the study, and all doses were well tolerated. The study was funded by Santen.
Read the full study in Ophthalmology (2016;23[11]:2413-23).
[email protected]
On Twitter @whitneymcknight
TNF inhibitors’ effect on ankylosing spondylitis progression may be greatest after 6 years
The benefits of treatment with TNF-alpha inhibitors in reducing spinal radiographic progression in ankylosing spondylitis became most evident 6-8 years after the biologic therapy was initiated, according to findings from a prospective observational cohort study.
The study enrolled 210 consecutive patients from the Groningen Leeuwarden AS (GLAS) cohort who initiated treatment with TNF-alpha inhibitors during 2004-2012 and who received baseline and biannual radiographs over the 8-year follow-up.
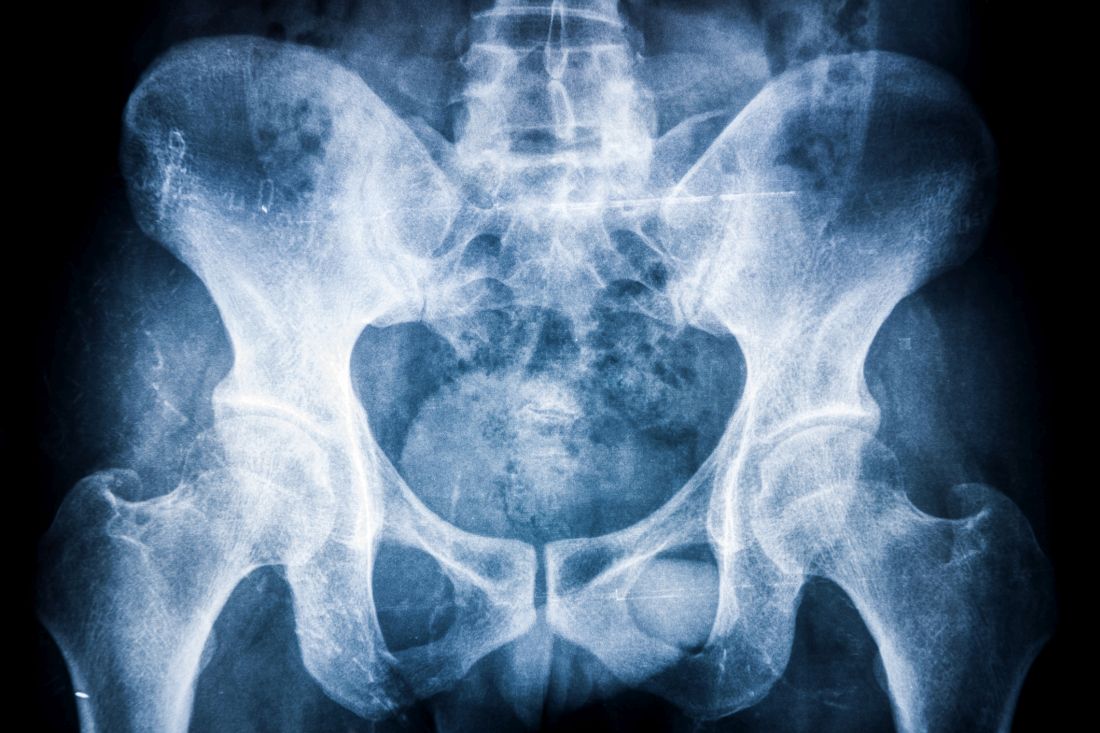
In patients with complete modified Stoke AS Spine Score (mSASSS) data over 8 years of follow-up, the estimated mean spinal radiographic progression was 2.3 points during the first 2 years of treatment and then declined steadily to 1.4 in years 2-4, 1.0 in years 4-6 and 0.8 in years 6-8. This decrease was seen even after adjusting for baseline mSASSS, the presence of syndesmophytes, sex, HLA-B27 status, age, symptom duration, smoking duration, body mass index, disease activity, and NSAID use.
Patients with longer follow-up also showed more use of NSAIDs, higher C-reactive protein levels, and more spinal radiographic damage at baseline. There were, however, significant improvements in all disease activity measures as soon as patients began treatment, and patients also showed a rapid decrease in NSAID use over time, said Fiona Maas of the University Medical Center Groningen (Netherlands) and her associates.
Multiple studies have been conducted into the impact of TNF-alpha inhibitors on spinal radiographic progression in ankylosing spondylitis, but the results have been subject to some debate, the investigators noted.
“It is known that radiographic progression in AS is overall slow and highly variable between patients,” they wrote. “Therefore, differences in patient numbers at the different time points during follow-up can affect the outcome measure of interest, in this case radiographic progression.”
In this study, researchers saw a straightforward linear progression of disease in the first 4 years after treatment was initiated but a deflection from linear progression in years 6 and 8.
“These results may refer to a delayed effect of TNF-alpha inhibitors on radiographic progression and support the TNF brake hypothesis,” they wrote, suggesting that the long-term inhibition of inflammation with TNF-alpha inhibitors diminishes new bone formation over time in patients with longstanding disease.
The GLAS cohort was supported Pfizer. Four authors declared research grants and consulting fees from pharmaceutical companies including Pfizer. No other conflicts of interest were declared.
The benefits of treatment with TNF-alpha inhibitors in reducing spinal radiographic progression in ankylosing spondylitis became most evident 6-8 years after the biologic therapy was initiated, according to findings from a prospective observational cohort study.
The study enrolled 210 consecutive patients from the Groningen Leeuwarden AS (GLAS) cohort who initiated treatment with TNF-alpha inhibitors during 2004-2012 and who received baseline and biannual radiographs over the 8-year follow-up.
In patients with complete modified Stoke AS Spine Score (mSASSS) data over 8 years of follow-up, the estimated mean spinal radiographic progression was 2.3 points during the first 2 years of treatment and then declined steadily to 1.4 in years 2-4, 1.0 in years 4-6 and 0.8 in years 6-8. This decrease was seen even after adjusting for baseline mSASSS, the presence of syndesmophytes, sex, HLA-B27 status, age, symptom duration, smoking duration, body mass index, disease activity, and NSAID use.
Patients with longer follow-up also showed more use of NSAIDs, higher C-reactive protein levels, and more spinal radiographic damage at baseline. There were, however, significant improvements in all disease activity measures as soon as patients began treatment, and patients also showed a rapid decrease in NSAID use over time, said Fiona Maas of the University Medical Center Groningen (Netherlands) and her associates.
Multiple studies have been conducted into the impact of TNF-alpha inhibitors on spinal radiographic progression in ankylosing spondylitis, but the results have been subject to some debate, the investigators noted.
“It is known that radiographic progression in AS is overall slow and highly variable between patients,” they wrote. “Therefore, differences in patient numbers at the different time points during follow-up can affect the outcome measure of interest, in this case radiographic progression.”
In this study, researchers saw a straightforward linear progression of disease in the first 4 years after treatment was initiated but a deflection from linear progression in years 6 and 8.
“These results may refer to a delayed effect of TNF-alpha inhibitors on radiographic progression and support the TNF brake hypothesis,” they wrote, suggesting that the long-term inhibition of inflammation with TNF-alpha inhibitors diminishes new bone formation over time in patients with longstanding disease.
The GLAS cohort was supported Pfizer. Four authors declared research grants and consulting fees from pharmaceutical companies including Pfizer. No other conflicts of interest were declared.
The benefits of treatment with TNF-alpha inhibitors in reducing spinal radiographic progression in ankylosing spondylitis became most evident 6-8 years after the biologic therapy was initiated, according to findings from a prospective observational cohort study.
The study enrolled 210 consecutive patients from the Groningen Leeuwarden AS (GLAS) cohort who initiated treatment with TNF-alpha inhibitors during 2004-2012 and who received baseline and biannual radiographs over the 8-year follow-up.
In patients with complete modified Stoke AS Spine Score (mSASSS) data over 8 years of follow-up, the estimated mean spinal radiographic progression was 2.3 points during the first 2 years of treatment and then declined steadily to 1.4 in years 2-4, 1.0 in years 4-6 and 0.8 in years 6-8. This decrease was seen even after adjusting for baseline mSASSS, the presence of syndesmophytes, sex, HLA-B27 status, age, symptom duration, smoking duration, body mass index, disease activity, and NSAID use.
Patients with longer follow-up also showed more use of NSAIDs, higher C-reactive protein levels, and more spinal radiographic damage at baseline. There were, however, significant improvements in all disease activity measures as soon as patients began treatment, and patients also showed a rapid decrease in NSAID use over time, said Fiona Maas of the University Medical Center Groningen (Netherlands) and her associates.
Multiple studies have been conducted into the impact of TNF-alpha inhibitors on spinal radiographic progression in ankylosing spondylitis, but the results have been subject to some debate, the investigators noted.
“It is known that radiographic progression in AS is overall slow and highly variable between patients,” they wrote. “Therefore, differences in patient numbers at the different time points during follow-up can affect the outcome measure of interest, in this case radiographic progression.”
In this study, researchers saw a straightforward linear progression of disease in the first 4 years after treatment was initiated but a deflection from linear progression in years 6 and 8.
“These results may refer to a delayed effect of TNF-alpha inhibitors on radiographic progression and support the TNF brake hypothesis,” they wrote, suggesting that the long-term inhibition of inflammation with TNF-alpha inhibitors diminishes new bone formation over time in patients with longstanding disease.
The GLAS cohort was supported Pfizer. Four authors declared research grants and consulting fees from pharmaceutical companies including Pfizer. No other conflicts of interest were declared.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point: The .
Major finding: Spinal radiographic progression rates on the modified Stoke AS Spine Score in ankylosing spondylitis patients treated with TNF-alpha inhibitors dropped from 2.3 during the first 2 years of treatment to 0.8 in years 6-8 after treatment initiation.
Data source: The Groningen Leeuwarden AS (GLAS) prospective observational cohort study involving 210 patients with ankylosing spondylitis.
Disclosures: The GLAS cohort was supported by Pfizer. Four authors declared research grants and consulting fees from pharmaceutical companies, including Pfizer. No other conflicts of interest were declared.
Most children with JIA get different diagnosis as adults
Two-thirds of children diagnosed with juvenile idiopathic arthritis are classified later as having a different form of arthritis as adults, with 72% of them requiring disease-modifying medication and 13% forced into retirement, according to a cross-sectional analysis of a registry database.
But among patients with inactive disease, more than one-third are off medication, and the majority have either no or very mild disabilities, reported Filipa Oliveira-Ramos, MD, of Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisbon, and her colleagues (RMD Open 2016;2:e000304. doi: 10.1136/rmdopen-2016-000304).
The team used data from the Rheumatic Diseases Portuguese Register database to discern how rheumatic disease classifications evolve as people with JIA grow into adulthood. The analysis comprised 426 patients and examined fulfillment of adult classification criteria, function as assessed by the Health Assessment Questionnaire (HAQ), clinical disease characteristics as assessed by the Juvenile Arthritis Damage Index–articular (JADI-A) and Juvenile Arthritis Damage Index–extra-articular (JADI-E), and disease activity.
The patients were a mean of 34 years old at the time of the last visit entered into the database. The patients’ mean disease duration was 22.5 years, including 80% with at least 10 years and 24% with more than 30 years.
All had been diagnosed with JIA, at a mean age of about 10 years. Disease categories were persistent oligoarthritis (19%), extended oligoarthritis (14%), rheumatoid factor–positive polyarthritis (17%), rheumatoid factor–negative polyarthritis (18%), systemic disease (10%), enthesitis-related arthritis (19%), psoriatic arthritis (3%), and undifferentiated arthritis (1%).
A total of 72% of patients were still employed, although 13% had retired because of disease-related disability. Most (67%) still had active disease, and 72% were taking a disease-modifying antirheumatic drug.
JIA had evolved into numerous new diagnoses, the team observed. Most patients with systemic-onset JIA (92%) were classified as having adult Still’s disease – more than half (58%) with persistent systemic features and about 42% with predominately polyarticular involvement.
The majority of patients with RF-positive polyarthritis (96%) and of those with RF-negative polyarthritis as children (57%) fulfilled the adult criteria for rheumatoid arthritis.
Patients who had persistent oligoarthritis as children were, as adults, most likely be classified with spondyloarthritis (35%), although 59% remained unclassifiable.
Most of the patients with extended oligoarthritis as children were later classified as having either rheumatoid arthritis (39%) or spondyloarthritis (26%). Most patients with juvenile enthesitis-related arthritis were also reclassified as having spondyloarthritis (95%).
All of those with childhood psoriatic arthritis retained that classification as adults.
A smaller portion of patients (21%) were unclassified as adults, the investigators said. Most of these patients had RF-negative polyarticular or oligoarticular classifications as children.
In a series of multivariate analyses, the team found a number of significant associations with adult outcomes. After adjustment for International League of Associations for Rheumatology (ILAR) category, inactive adult disease was associated with shorter disease duration, less delay in diagnosis, a lower HAQ score, and less exposure to corticosteroids. A higher HAQ score was associated with a longer disease duration and exposure to biologics, while a lower HAQ was associated with the persistence of systemic disease features.
Higher JADI-A scores were associated with disability-related retirement, longer disease duration, and past or current use of biologics.
Another series of multivariate models assessed outcomes associated with inactive disease. Patients who were older at disease onset were more likely to have inactive disease as adults. A positive test for anticitrullinated protein antibodies decreased the likelihood of disease inactivity by 93%.
Finally, the investigators evaluated associations with function and clinical characteristics. Younger age at disease onset was associated with higher HAQ and JADI scores in adulthood. Patients with RF-positive polyarthritis and systemic-onset JIA were more likely to have worse JADI-A and JADI-E scores, compared with patients who had persistent oligoarthritis. Corticosteroid exposure was also predictive of worse extra-articular scores on the JADI.
“Understanding the way these juvenile diseases progress could add useful information for the ongoing discussion of a new classification capable of better unifying the language between pediatric and adult care,” the authors concluded.
None of the authors had financial disclosures.
[email protected]
On Twitter @alz_gal
This is an important article in that it highlights one of the fundamental flaws of the juvenile idiopathic arthritis classification.
The manifestations of the rheumatic diseases often evolve over time. There may be psoriasis before arthritis, but there also may be arthritis before psoriasis. Similarly, children with an ultimate diagnosis of Crohn’s disease may first present to the rheumatologist with arthritis.
The definition of JIA requires only the onset of arthritis lasting more than 3 weeks before 16 years of age with the exclusion of other obvious cause. A careful reading of the subclassification criteria quickly reveals that many children are “unclassifiable” because of family history or other factors.
The present study makes it clear that by the time they reach adulthood, a significant number of individuals who were told they had JIA will, in fact, meet criteria for a different classification. This would be of only casual interest were it not for the fact that these children will have previously been entered into databases about the natural history of JIA with erroneous classifications.
More worrisome is that fact that some will have been included in therapeutic trials with erroneous classifications as well. The pediatric rheumatology community would do well to recognize the urgent need for a reassessment of the classification criteria and nomenclature to better reflect the diversity of causes of childhood arthritis.
Thomas Lehman, MD, is chief of pediatric rheumatology at the Hospital for Special Surgery, New York. He has no relevant disclosures.
This is an important article in that it highlights one of the fundamental flaws of the juvenile idiopathic arthritis classification.
The manifestations of the rheumatic diseases often evolve over time. There may be psoriasis before arthritis, but there also may be arthritis before psoriasis. Similarly, children with an ultimate diagnosis of Crohn’s disease may first present to the rheumatologist with arthritis.
The definition of JIA requires only the onset of arthritis lasting more than 3 weeks before 16 years of age with the exclusion of other obvious cause. A careful reading of the subclassification criteria quickly reveals that many children are “unclassifiable” because of family history or other factors.
The present study makes it clear that by the time they reach adulthood, a significant number of individuals who were told they had JIA will, in fact, meet criteria for a different classification. This would be of only casual interest were it not for the fact that these children will have previously been entered into databases about the natural history of JIA with erroneous classifications.
More worrisome is that fact that some will have been included in therapeutic trials with erroneous classifications as well. The pediatric rheumatology community would do well to recognize the urgent need for a reassessment of the classification criteria and nomenclature to better reflect the diversity of causes of childhood arthritis.
Thomas Lehman, MD, is chief of pediatric rheumatology at the Hospital for Special Surgery, New York. He has no relevant disclosures.
This is an important article in that it highlights one of the fundamental flaws of the juvenile idiopathic arthritis classification.
The manifestations of the rheumatic diseases often evolve over time. There may be psoriasis before arthritis, but there also may be arthritis before psoriasis. Similarly, children with an ultimate diagnosis of Crohn’s disease may first present to the rheumatologist with arthritis.
The definition of JIA requires only the onset of arthritis lasting more than 3 weeks before 16 years of age with the exclusion of other obvious cause. A careful reading of the subclassification criteria quickly reveals that many children are “unclassifiable” because of family history or other factors.
The present study makes it clear that by the time they reach adulthood, a significant number of individuals who were told they had JIA will, in fact, meet criteria for a different classification. This would be of only casual interest were it not for the fact that these children will have previously been entered into databases about the natural history of JIA with erroneous classifications.
More worrisome is that fact that some will have been included in therapeutic trials with erroneous classifications as well. The pediatric rheumatology community would do well to recognize the urgent need for a reassessment of the classification criteria and nomenclature to better reflect the diversity of causes of childhood arthritis.
Thomas Lehman, MD, is chief of pediatric rheumatology at the Hospital for Special Surgery, New York. He has no relevant disclosures.
Two-thirds of children diagnosed with juvenile idiopathic arthritis are classified later as having a different form of arthritis as adults, with 72% of them requiring disease-modifying medication and 13% forced into retirement, according to a cross-sectional analysis of a registry database.
But among patients with inactive disease, more than one-third are off medication, and the majority have either no or very mild disabilities, reported Filipa Oliveira-Ramos, MD, of Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisbon, and her colleagues (RMD Open 2016;2:e000304. doi: 10.1136/rmdopen-2016-000304).
The team used data from the Rheumatic Diseases Portuguese Register database to discern how rheumatic disease classifications evolve as people with JIA grow into adulthood. The analysis comprised 426 patients and examined fulfillment of adult classification criteria, function as assessed by the Health Assessment Questionnaire (HAQ), clinical disease characteristics as assessed by the Juvenile Arthritis Damage Index–articular (JADI-A) and Juvenile Arthritis Damage Index–extra-articular (JADI-E), and disease activity.
The patients were a mean of 34 years old at the time of the last visit entered into the database. The patients’ mean disease duration was 22.5 years, including 80% with at least 10 years and 24% with more than 30 years.
All had been diagnosed with JIA, at a mean age of about 10 years. Disease categories were persistent oligoarthritis (19%), extended oligoarthritis (14%), rheumatoid factor–positive polyarthritis (17%), rheumatoid factor–negative polyarthritis (18%), systemic disease (10%), enthesitis-related arthritis (19%), psoriatic arthritis (3%), and undifferentiated arthritis (1%).
A total of 72% of patients were still employed, although 13% had retired because of disease-related disability. Most (67%) still had active disease, and 72% were taking a disease-modifying antirheumatic drug.
JIA had evolved into numerous new diagnoses, the team observed. Most patients with systemic-onset JIA (92%) were classified as having adult Still’s disease – more than half (58%) with persistent systemic features and about 42% with predominately polyarticular involvement.
The majority of patients with RF-positive polyarthritis (96%) and of those with RF-negative polyarthritis as children (57%) fulfilled the adult criteria for rheumatoid arthritis.
Patients who had persistent oligoarthritis as children were, as adults, most likely be classified with spondyloarthritis (35%), although 59% remained unclassifiable.
Most of the patients with extended oligoarthritis as children were later classified as having either rheumatoid arthritis (39%) or spondyloarthritis (26%). Most patients with juvenile enthesitis-related arthritis were also reclassified as having spondyloarthritis (95%).
All of those with childhood psoriatic arthritis retained that classification as adults.
A smaller portion of patients (21%) were unclassified as adults, the investigators said. Most of these patients had RF-negative polyarticular or oligoarticular classifications as children.
In a series of multivariate analyses, the team found a number of significant associations with adult outcomes. After adjustment for International League of Associations for Rheumatology (ILAR) category, inactive adult disease was associated with shorter disease duration, less delay in diagnosis, a lower HAQ score, and less exposure to corticosteroids. A higher HAQ score was associated with a longer disease duration and exposure to biologics, while a lower HAQ was associated with the persistence of systemic disease features.
Higher JADI-A scores were associated with disability-related retirement, longer disease duration, and past or current use of biologics.
Another series of multivariate models assessed outcomes associated with inactive disease. Patients who were older at disease onset were more likely to have inactive disease as adults. A positive test for anticitrullinated protein antibodies decreased the likelihood of disease inactivity by 93%.
Finally, the investigators evaluated associations with function and clinical characteristics. Younger age at disease onset was associated with higher HAQ and JADI scores in adulthood. Patients with RF-positive polyarthritis and systemic-onset JIA were more likely to have worse JADI-A and JADI-E scores, compared with patients who had persistent oligoarthritis. Corticosteroid exposure was also predictive of worse extra-articular scores on the JADI.
“Understanding the way these juvenile diseases progress could add useful information for the ongoing discussion of a new classification capable of better unifying the language between pediatric and adult care,” the authors concluded.
None of the authors had financial disclosures.
[email protected]
On Twitter @alz_gal
Two-thirds of children diagnosed with juvenile idiopathic arthritis are classified later as having a different form of arthritis as adults, with 72% of them requiring disease-modifying medication and 13% forced into retirement, according to a cross-sectional analysis of a registry database.
But among patients with inactive disease, more than one-third are off medication, and the majority have either no or very mild disabilities, reported Filipa Oliveira-Ramos, MD, of Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisbon, and her colleagues (RMD Open 2016;2:e000304. doi: 10.1136/rmdopen-2016-000304).
The team used data from the Rheumatic Diseases Portuguese Register database to discern how rheumatic disease classifications evolve as people with JIA grow into adulthood. The analysis comprised 426 patients and examined fulfillment of adult classification criteria, function as assessed by the Health Assessment Questionnaire (HAQ), clinical disease characteristics as assessed by the Juvenile Arthritis Damage Index–articular (JADI-A) and Juvenile Arthritis Damage Index–extra-articular (JADI-E), and disease activity.
The patients were a mean of 34 years old at the time of the last visit entered into the database. The patients’ mean disease duration was 22.5 years, including 80% with at least 10 years and 24% with more than 30 years.
All had been diagnosed with JIA, at a mean age of about 10 years. Disease categories were persistent oligoarthritis (19%), extended oligoarthritis (14%), rheumatoid factor–positive polyarthritis (17%), rheumatoid factor–negative polyarthritis (18%), systemic disease (10%), enthesitis-related arthritis (19%), psoriatic arthritis (3%), and undifferentiated arthritis (1%).
A total of 72% of patients were still employed, although 13% had retired because of disease-related disability. Most (67%) still had active disease, and 72% were taking a disease-modifying antirheumatic drug.
JIA had evolved into numerous new diagnoses, the team observed. Most patients with systemic-onset JIA (92%) were classified as having adult Still’s disease – more than half (58%) with persistent systemic features and about 42% with predominately polyarticular involvement.
The majority of patients with RF-positive polyarthritis (96%) and of those with RF-negative polyarthritis as children (57%) fulfilled the adult criteria for rheumatoid arthritis.
Patients who had persistent oligoarthritis as children were, as adults, most likely be classified with spondyloarthritis (35%), although 59% remained unclassifiable.
Most of the patients with extended oligoarthritis as children were later classified as having either rheumatoid arthritis (39%) or spondyloarthritis (26%). Most patients with juvenile enthesitis-related arthritis were also reclassified as having spondyloarthritis (95%).
All of those with childhood psoriatic arthritis retained that classification as adults.
A smaller portion of patients (21%) were unclassified as adults, the investigators said. Most of these patients had RF-negative polyarticular or oligoarticular classifications as children.
In a series of multivariate analyses, the team found a number of significant associations with adult outcomes. After adjustment for International League of Associations for Rheumatology (ILAR) category, inactive adult disease was associated with shorter disease duration, less delay in diagnosis, a lower HAQ score, and less exposure to corticosteroids. A higher HAQ score was associated with a longer disease duration and exposure to biologics, while a lower HAQ was associated with the persistence of systemic disease features.
Higher JADI-A scores were associated with disability-related retirement, longer disease duration, and past or current use of biologics.
Another series of multivariate models assessed outcomes associated with inactive disease. Patients who were older at disease onset were more likely to have inactive disease as adults. A positive test for anticitrullinated protein antibodies decreased the likelihood of disease inactivity by 93%.
Finally, the investigators evaluated associations with function and clinical characteristics. Younger age at disease onset was associated with higher HAQ and JADI scores in adulthood. Patients with RF-positive polyarthritis and systemic-onset JIA were more likely to have worse JADI-A and JADI-E scores, compared with patients who had persistent oligoarthritis. Corticosteroid exposure was also predictive of worse extra-articular scores on the JADI.
“Understanding the way these juvenile diseases progress could add useful information for the ongoing discussion of a new classification capable of better unifying the language between pediatric and adult care,” the authors concluded.
None of the authors had financial disclosures.
[email protected]
On Twitter @alz_gal
Key clinical point:
Major finding: The childhood diagnosis of JIA is reclassified to another form of arthritis in two-thirds of patients in adulthood.
Data source: The Rheumatic Diseases Portuguese Register.
Disclosures: None of the authors had financial disclosures.
Biosimilar version of etanercept gains FDA approval
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
A biosimilar of etanercept received clearance for marketing from the Food and Drug Administration on Aug. 30 for all of the inflammatory disease indications held by the reference originator etanercept product, Enbrel, according to an announcement from the agency.
Approval for all of Enbrel’s indications – rheumatoid arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, and polyarticular juvenile idiopathic arthritis – was initially met with skepticism by members of the agency’s Arthritis Advisory Committee at a meeting in July because the biosimilar was compared against Enbrel in patients with plaque psoriasis only, but eventually all panel members voted to recommend approval.
The approval allows the biosimilar etanercept, called etanercept-szzs, to be marketed as a biosimilar only, not as an interchangeable product. The FDA has not yet developed guidance for manufacturers to follow to get approval for interchangeability, which means that a biosimilar “may be substituted for the reference product by a pharmacist without the intervention of the health care provider who prescribed the reference product,” according to the agency.
“We carefully evaluate the structural and functional characteristics of these complex molecules. Patients and providers can have confidence that there are no clinically meaningful differences in safety and efficacy from the reference product,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency’s announcement.
Etanercept-szzs will be marketed by Sandoz as Erelzi. Erelzi’s prescribing information can be found here. The biosimilar is currently undergoing review with the European Medicines Agency.
Ankylosing spondylitis patients develop multiple comorbidities after diagnosis
DENVER – Evidence continues to mount that ankylosing spondylitis patients are at increased risk for developing various comorbidities, compared with the general adult population.
Patients newly diagnosed with ankylosing spondylitis (AS) had twice the rate of new-onset depression during the first 3 years following diagnosis, compared with matched people from the general population in a study of more than 21,000 American adults. Patients with newly diagnosed AS also had a 60% higher rate of developing a new cardiovascular disease, compared with the matched general population, Jessica A. Walsh, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network.

“We need to figure out what to do about all the comorbidities. Rheumatologists need to either screen their AS patients for comorbidities, or they need to be sure their patients are plugged in with another physician who will screen them,” said Dr. Walsh, director of the spondyloarthritis clinic at the University of Utah in Salt Lake City.
Her analysis used data from the Truven Health MarketScan databases for U.S. patients covered by Medicare or commercial health insurance, and included 6,370 patients with newly diagnosed AS and 14,988 adults matched by age and sex. The analysis only included AS patients who were free from comorbidities during the 2 years prior to their AS diagnosis. The average age of the people in the study was 52 years, and 54% were men.
During an average follow-up of 2.9 years following initial AS diagnosis, the most common comorbidity among the AS patients was uveitis, which occurred nearly 15-fold more frequently among the AS patients than in the controls. Other common incident comorbidities included inflammatory bowel disease, nearly sixfold more common among the AS patients, and osteoporosis, which was nearly threefold more common during follow-up after AS diagnosis, compared with the controls.
Other comorbidities with increased incidence in the AS patients included sleep apnea (80% more common among the AS patients during follow-up), asthma (50% more often), hypertension (44% more common), malignancy (23% more common), diabetes (20% more common), and dyslipidemia (11% more common). All these incidence rates represented statistically significant increases in the AS patients, compared with the controls.
A related analysis reported by Dr. Walsh also used data from the Truven Health databases for a somewhat larger group of AS patients, 6,679 followed for 1 year after their AS diagnosis, and 19,951 matched controls. The AS patients had a significantly higher rate of hospital admissions – 12%, compared with 6% among the controls – and a significantly higher rate of emergency department visits, at 23%, compared with 15% among the controls. The AS patients also had double the rate of physician office visits and prescribed medications.
“Obviously, the AS patients are not as healthy,” Dr. Walsh said in an interview. “We adjusted for their comorbidities, but that did not affect the hospitalization rates. We need to look into this more; I don’t know why the AS patients are being hospitalized. Typically AS itself does not lead to hospitalization, so I suspect it’s because of comorbidities, or perhaps because of adverse events from treatment.”
Dr. Walsh is a consultant to AbbVie and Novartis.
On Twitter @mitchelzoler
DENVER – Evidence continues to mount that ankylosing spondylitis patients are at increased risk for developing various comorbidities, compared with the general adult population.
Patients newly diagnosed with ankylosing spondylitis (AS) had twice the rate of new-onset depression during the first 3 years following diagnosis, compared with matched people from the general population in a study of more than 21,000 American adults. Patients with newly diagnosed AS also had a 60% higher rate of developing a new cardiovascular disease, compared with the matched general population, Jessica A. Walsh, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network.
“We need to figure out what to do about all the comorbidities. Rheumatologists need to either screen their AS patients for comorbidities, or they need to be sure their patients are plugged in with another physician who will screen them,” said Dr. Walsh, director of the spondyloarthritis clinic at the University of Utah in Salt Lake City.
Her analysis used data from the Truven Health MarketScan databases for U.S. patients covered by Medicare or commercial health insurance, and included 6,370 patients with newly diagnosed AS and 14,988 adults matched by age and sex. The analysis only included AS patients who were free from comorbidities during the 2 years prior to their AS diagnosis. The average age of the people in the study was 52 years, and 54% were men.
During an average follow-up of 2.9 years following initial AS diagnosis, the most common comorbidity among the AS patients was uveitis, which occurred nearly 15-fold more frequently among the AS patients than in the controls. Other common incident comorbidities included inflammatory bowel disease, nearly sixfold more common among the AS patients, and osteoporosis, which was nearly threefold more common during follow-up after AS diagnosis, compared with the controls.
Other comorbidities with increased incidence in the AS patients included sleep apnea (80% more common among the AS patients during follow-up), asthma (50% more often), hypertension (44% more common), malignancy (23% more common), diabetes (20% more common), and dyslipidemia (11% more common). All these incidence rates represented statistically significant increases in the AS patients, compared with the controls.
A related analysis reported by Dr. Walsh also used data from the Truven Health databases for a somewhat larger group of AS patients, 6,679 followed for 1 year after their AS diagnosis, and 19,951 matched controls. The AS patients had a significantly higher rate of hospital admissions – 12%, compared with 6% among the controls – and a significantly higher rate of emergency department visits, at 23%, compared with 15% among the controls. The AS patients also had double the rate of physician office visits and prescribed medications.
“Obviously, the AS patients are not as healthy,” Dr. Walsh said in an interview. “We adjusted for their comorbidities, but that did not affect the hospitalization rates. We need to look into this more; I don’t know why the AS patients are being hospitalized. Typically AS itself does not lead to hospitalization, so I suspect it’s because of comorbidities, or perhaps because of adverse events from treatment.”
Dr. Walsh is a consultant to AbbVie and Novartis.
On Twitter @mitchelzoler
DENVER – Evidence continues to mount that ankylosing spondylitis patients are at increased risk for developing various comorbidities, compared with the general adult population.
Patients newly diagnosed with ankylosing spondylitis (AS) had twice the rate of new-onset depression during the first 3 years following diagnosis, compared with matched people from the general population in a study of more than 21,000 American adults. Patients with newly diagnosed AS also had a 60% higher rate of developing a new cardiovascular disease, compared with the matched general population, Jessica A. Walsh, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network.
“We need to figure out what to do about all the comorbidities. Rheumatologists need to either screen their AS patients for comorbidities, or they need to be sure their patients are plugged in with another physician who will screen them,” said Dr. Walsh, director of the spondyloarthritis clinic at the University of Utah in Salt Lake City.
Her analysis used data from the Truven Health MarketScan databases for U.S. patients covered by Medicare or commercial health insurance, and included 6,370 patients with newly diagnosed AS and 14,988 adults matched by age and sex. The analysis only included AS patients who were free from comorbidities during the 2 years prior to their AS diagnosis. The average age of the people in the study was 52 years, and 54% were men.
During an average follow-up of 2.9 years following initial AS diagnosis, the most common comorbidity among the AS patients was uveitis, which occurred nearly 15-fold more frequently among the AS patients than in the controls. Other common incident comorbidities included inflammatory bowel disease, nearly sixfold more common among the AS patients, and osteoporosis, which was nearly threefold more common during follow-up after AS diagnosis, compared with the controls.
Other comorbidities with increased incidence in the AS patients included sleep apnea (80% more common among the AS patients during follow-up), asthma (50% more often), hypertension (44% more common), malignancy (23% more common), diabetes (20% more common), and dyslipidemia (11% more common). All these incidence rates represented statistically significant increases in the AS patients, compared with the controls.
A related analysis reported by Dr. Walsh also used data from the Truven Health databases for a somewhat larger group of AS patients, 6,679 followed for 1 year after their AS diagnosis, and 19,951 matched controls. The AS patients had a significantly higher rate of hospital admissions – 12%, compared with 6% among the controls – and a significantly higher rate of emergency department visits, at 23%, compared with 15% among the controls. The AS patients also had double the rate of physician office visits and prescribed medications.
“Obviously, the AS patients are not as healthy,” Dr. Walsh said in an interview. “We adjusted for their comorbidities, but that did not affect the hospitalization rates. We need to look into this more; I don’t know why the AS patients are being hospitalized. Typically AS itself does not lead to hospitalization, so I suspect it’s because of comorbidities, or perhaps because of adverse events from treatment.”
Dr. Walsh is a consultant to AbbVie and Novartis.
On Twitter @mitchelzoler
AT THE 2016 SPARTAN ANNUAL MEETING
Key clinical point: In the 3 years after initial ankylosing spondylitis diagnosis, the incidence of several comorbidities rises significantly above those in the general population.
Major finding: The incidence of new-onset depression among newly diagnosed ankylosing spondylitis patients was twice as high as it was among matched controls.
Data source: Observational data collected by Truven Health MarketScan, with a total of 21,358 patients and controls.
Disclosures: Dr. Walsh is a consultant to AbbVie and Novartis.

MRI now anchors spondyloarthritis diagnosis
DENVER – MRI of the sacroiliac joint now serves as the primary imaging driver for a diagnosis of spondyloarthritis, especially early spondyloarthritis, and has largely supplanted radiographic assessment, Walter P. Maksymowych, MD, said at an educational symposium organized by the Spondyloarthritis Research and Treatment Network.
“MRI is reliable for early diagnosis; radiographic assessment of early spondyloarthritis is problematic,” said Dr. Maksymowych, a rheumatologist and professor of medicine at the University of Alberta in Edmonton. “The EULAR recommendations now say that early diagnosis of spondyloarthritis needs to focus on MRI.”

While the recommendations from the European League Against Rheumatism (EULAR) that came out last year on using imaging for diagnosing and managing spondyloarthritis (SpA) cite radiography of the patient’s sacroiliac joint as the recommended first imaging method to use to diagnose sacroiliitis as part of a diagnosis of axial SpA, the EULAR recommendations place MRI very close behind.
The recommendations list MRI as an “alternative” first-line approach for diagnostic imaging. For patients who appear negative for axial SpA on radiographic imaging but who remain suspected of having SpA the EULAR panel “recommended” MRI of the sacroiliac joints as a backup method for imaging assessment and to definitively rule out SpA. No other imaging method received EULAR’s endorsement for SpA diagnosis aside from these two approaches.
The recommendations direct the MRI examination to include assessment of inflammation based on bone marrow edema, and also structural abnormalities including bone erosion, new bone formation, sclerosis and fat infiltration. The 2015 EULAR recommendations also cite roles for MRI in diagnosing peripheral SpA, monitoring disease activity in axial and peripheral SpA, monioring structural changes in axial and peripheral SpA, predicting outcome and treatment effect in axial SpA, assessing spinal fracture in patients with axial SpA and to assess osteoporosis in selected axial SpA patients.
The MRI imaging sequences particularly useful for SpA diagnosis are “short tau inversion recovery” (STIR), a “water sensitive” sequence that involves suppression of the fat MRI signal, and a T1 weighted sequence that is a “fat sensitive” scan, explained Dr. Maksymowych at the symposium, also organized by the Group for the Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). He strongly encouraged clinicians to apply a standardized approach to imaging in every patient suspected of having axial SpA.
The definition of an MRI imaging result that is positive for SpA remains a consensus of expert opinion without a firm evidence base. The currently accepted MRI marker of SpA involves identifying several areas of bone marrow edema in a single T1 and STIR MRI image-slice through the sacroiliac joint, or focal bone marrow edema visible in at least two consecutive sacroiliac joint T1 and STIR image slices.
The confluence of a structural lesion at the same site as bone marrow edema provides further confirmatory information, he said. “A structural lesions at the site of bone marrow edema enhances your confidence in the diagnosis,” he said.
“The specificity of MRI for SpA is quite high,” Dr. Maksymowych noted, with specificity rates often in the range of 85%-95%. Sensitivity rates are lower, often 50%-70%. Sensitivity further improves by factoring in the presence of bone erosions. Diagnostic confidence also increases as the number of lesions visualized increases.
MRI has become so integral to the assessment of SpA that “to understand SpA you need to understand the language of MRI,” Dr. Maksymowych concluded.
Dr. Maksymowych has received honoraria from eight drug companies and research grant support from Abbvie and Pfizer.
On Twitter @mitchelzoler
DENVER – MRI of the sacroiliac joint now serves as the primary imaging driver for a diagnosis of spondyloarthritis, especially early spondyloarthritis, and has largely supplanted radiographic assessment, Walter P. Maksymowych, MD, said at an educational symposium organized by the Spondyloarthritis Research and Treatment Network.
“MRI is reliable for early diagnosis; radiographic assessment of early spondyloarthritis is problematic,” said Dr. Maksymowych, a rheumatologist and professor of medicine at the University of Alberta in Edmonton. “The EULAR recommendations now say that early diagnosis of spondyloarthritis needs to focus on MRI.”
While the recommendations from the European League Against Rheumatism (EULAR) that came out last year on using imaging for diagnosing and managing spondyloarthritis (SpA) cite radiography of the patient’s sacroiliac joint as the recommended first imaging method to use to diagnose sacroiliitis as part of a diagnosis of axial SpA, the EULAR recommendations place MRI very close behind.
The recommendations list MRI as an “alternative” first-line approach for diagnostic imaging. For patients who appear negative for axial SpA on radiographic imaging but who remain suspected of having SpA the EULAR panel “recommended” MRI of the sacroiliac joints as a backup method for imaging assessment and to definitively rule out SpA. No other imaging method received EULAR’s endorsement for SpA diagnosis aside from these two approaches.
The recommendations direct the MRI examination to include assessment of inflammation based on bone marrow edema, and also structural abnormalities including bone erosion, new bone formation, sclerosis and fat infiltration. The 2015 EULAR recommendations also cite roles for MRI in diagnosing peripheral SpA, monitoring disease activity in axial and peripheral SpA, monioring structural changes in axial and peripheral SpA, predicting outcome and treatment effect in axial SpA, assessing spinal fracture in patients with axial SpA and to assess osteoporosis in selected axial SpA patients.
The MRI imaging sequences particularly useful for SpA diagnosis are “short tau inversion recovery” (STIR), a “water sensitive” sequence that involves suppression of the fat MRI signal, and a T1 weighted sequence that is a “fat sensitive” scan, explained Dr. Maksymowych at the symposium, also organized by the Group for the Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). He strongly encouraged clinicians to apply a standardized approach to imaging in every patient suspected of having axial SpA.
The definition of an MRI imaging result that is positive for SpA remains a consensus of expert opinion without a firm evidence base. The currently accepted MRI marker of SpA involves identifying several areas of bone marrow edema in a single T1 and STIR MRI image-slice through the sacroiliac joint, or focal bone marrow edema visible in at least two consecutive sacroiliac joint T1 and STIR image slices.
The confluence of a structural lesion at the same site as bone marrow edema provides further confirmatory information, he said. “A structural lesions at the site of bone marrow edema enhances your confidence in the diagnosis,” he said.
“The specificity of MRI for SpA is quite high,” Dr. Maksymowych noted, with specificity rates often in the range of 85%-95%. Sensitivity rates are lower, often 50%-70%. Sensitivity further improves by factoring in the presence of bone erosions. Diagnostic confidence also increases as the number of lesions visualized increases.
MRI has become so integral to the assessment of SpA that “to understand SpA you need to understand the language of MRI,” Dr. Maksymowych concluded.
Dr. Maksymowych has received honoraria from eight drug companies and research grant support from Abbvie and Pfizer.
On Twitter @mitchelzoler
DENVER – MRI of the sacroiliac joint now serves as the primary imaging driver for a diagnosis of spondyloarthritis, especially early spondyloarthritis, and has largely supplanted radiographic assessment, Walter P. Maksymowych, MD, said at an educational symposium organized by the Spondyloarthritis Research and Treatment Network.
“MRI is reliable for early diagnosis; radiographic assessment of early spondyloarthritis is problematic,” said Dr. Maksymowych, a rheumatologist and professor of medicine at the University of Alberta in Edmonton. “The EULAR recommendations now say that early diagnosis of spondyloarthritis needs to focus on MRI.”
While the recommendations from the European League Against Rheumatism (EULAR) that came out last year on using imaging for diagnosing and managing spondyloarthritis (SpA) cite radiography of the patient’s sacroiliac joint as the recommended first imaging method to use to diagnose sacroiliitis as part of a diagnosis of axial SpA, the EULAR recommendations place MRI very close behind.
The recommendations list MRI as an “alternative” first-line approach for diagnostic imaging. For patients who appear negative for axial SpA on radiographic imaging but who remain suspected of having SpA the EULAR panel “recommended” MRI of the sacroiliac joints as a backup method for imaging assessment and to definitively rule out SpA. No other imaging method received EULAR’s endorsement for SpA diagnosis aside from these two approaches.
The recommendations direct the MRI examination to include assessment of inflammation based on bone marrow edema, and also structural abnormalities including bone erosion, new bone formation, sclerosis and fat infiltration. The 2015 EULAR recommendations also cite roles for MRI in diagnosing peripheral SpA, monitoring disease activity in axial and peripheral SpA, monioring structural changes in axial and peripheral SpA, predicting outcome and treatment effect in axial SpA, assessing spinal fracture in patients with axial SpA and to assess osteoporosis in selected axial SpA patients.
The MRI imaging sequences particularly useful for SpA diagnosis are “short tau inversion recovery” (STIR), a “water sensitive” sequence that involves suppression of the fat MRI signal, and a T1 weighted sequence that is a “fat sensitive” scan, explained Dr. Maksymowych at the symposium, also organized by the Group for the Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA). He strongly encouraged clinicians to apply a standardized approach to imaging in every patient suspected of having axial SpA.
The definition of an MRI imaging result that is positive for SpA remains a consensus of expert opinion without a firm evidence base. The currently accepted MRI marker of SpA involves identifying several areas of bone marrow edema in a single T1 and STIR MRI image-slice through the sacroiliac joint, or focal bone marrow edema visible in at least two consecutive sacroiliac joint T1 and STIR image slices.
The confluence of a structural lesion at the same site as bone marrow edema provides further confirmatory information, he said. “A structural lesions at the site of bone marrow edema enhances your confidence in the diagnosis,” he said.
“The specificity of MRI for SpA is quite high,” Dr. Maksymowych noted, with specificity rates often in the range of 85%-95%. Sensitivity rates are lower, often 50%-70%. Sensitivity further improves by factoring in the presence of bone erosions. Diagnostic confidence also increases as the number of lesions visualized increases.
MRI has become so integral to the assessment of SpA that “to understand SpA you need to understand the language of MRI,” Dr. Maksymowych concluded.
Dr. Maksymowych has received honoraria from eight drug companies and research grant support from Abbvie and Pfizer.
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM A SPARTAN-GRAPPA SYMPOSIUM

Revised axial spondyloarthritis classification criteria remain elusive
DENVER – The revised axial spondyloarthritis classification criteria that U.S. rheumatologists say they desperately need remain elusive, with no firm path to creation.
The 2009 classification criteria for axial spondyloarthritis (SpA) developed by the Assessment of Spondyloarthritis international Society (ASAS) was a landmark in creating a definition for axial SpA to use when enrolling patients into clinical trials and to also potentially use for diagnosis.
But the 2009 classification criteria also have several shortcomings, especially from a U.S. perspective, that have limited the utility of the criteria in U.S. practice and studies.

Several years of discussions among North American rheumatologists about the classification criteria have produced consensus about what is wrong with them: The clinical elements of the criteria are not specific enough with sensitivity and specificity rates that are each about 80%-85%; the imaging component of the criteria is not totally up to date and as one example only considers osteitis and not other MRI abnormalities such as fatty metaplasia and T1 structural lesions; the criteria were entirely based on results from studies with Europeans, making their applicability to other patient types uncertain; and some terminology in the criteria are confusing, Liron Caplan, MD, said during a discussion of the criteria at the annual meeting of the of the Spondyloarthritis Research and Treatment Network.
But while the SpA experts who gathered at the meeting and who have also been discussing this issue since 2013 are of one mind on the problems, agreement on exactly how to solve them remains unresolved.
“Everything is still on the table,” said Dr. Caplan, a rheumatologist and director of the SpA program at the University of Colorado in Aurora.
A year ago, at SPARTAN’s prior annual meeting, circumstances looked on track for a quicker resolution. In a vote of the membership at the 2015 gathering, 64% backed creation of new classification criteria, and Dr. Caplan proposed quickly preparing a research proposal to present in early 2016 to the American College of Rheumatology and the European League Against Rheumatism for funding.

But in the ensuing year, the effort got mired in discussions on how to perform the study needed to gather the evidence base for criteria revisions. Some progress occurred, most notably an agreement between SPARTAN officials and the ASAS leadership to work together on the revision, said Atul A. Deodhar, MD, professor of medicine and medical director of the rheumatology clinics at the Oregon Health & Science University in Portland.
The SPARTAN and ASAS representatives also agreed on several specifics of what’s needed, including a weighting formula for the 10 different clinical criteria of the SpA classification. Currently, in individuals with low back pain lasting 3 months or longer at an age of onset younger than 45 years, any one of these 10 clinical criteria can create a case of SpA either if combined with MRI or radiographic evidence of sacroiliitis, or without any imaging if the individual is HLA-B27 positive and fulfills two items from the 10-item list of clinical criteria.
It’s the lack of radiographic imaging confirmation, so-called “nonradiographic SpA,” that generates the greatest concern about below-par specificity and that may be helped by a weighting adjustment that varies the classification power of each of the 10 clinical items. A more ideal balance might be criteria with sensitivity reduced from the current level to about 70% and with specificity rising to about 90%, Dr. Caplan suggested.
The keystone of any revision will be a large, new study with SpA patients from various locations. Dr. Caplan emphasized that “no firm decisions have yet been made” regarding the study's name or research focus.
On Twitter @mitchelzoler
This article was updated August 11, 2016.
DENVER – The revised axial spondyloarthritis classification criteria that U.S. rheumatologists say they desperately need remain elusive, with no firm path to creation.
The 2009 classification criteria for axial spondyloarthritis (SpA) developed by the Assessment of Spondyloarthritis international Society (ASAS) was a landmark in creating a definition for axial SpA to use when enrolling patients into clinical trials and to also potentially use for diagnosis.
But the 2009 classification criteria also have several shortcomings, especially from a U.S. perspective, that have limited the utility of the criteria in U.S. practice and studies.
Several years of discussions among North American rheumatologists about the classification criteria have produced consensus about what is wrong with them: The clinical elements of the criteria are not specific enough with sensitivity and specificity rates that are each about 80%-85%; the imaging component of the criteria is not totally up to date and as one example only considers osteitis and not other MRI abnormalities such as fatty metaplasia and T1 structural lesions; the criteria were entirely based on results from studies with Europeans, making their applicability to other patient types uncertain; and some terminology in the criteria are confusing, Liron Caplan, MD, said during a discussion of the criteria at the annual meeting of the of the Spondyloarthritis Research and Treatment Network.
But while the SpA experts who gathered at the meeting and who have also been discussing this issue since 2013 are of one mind on the problems, agreement on exactly how to solve them remains unresolved.
“Everything is still on the table,” said Dr. Caplan, a rheumatologist and director of the SpA program at the University of Colorado in Aurora.
A year ago, at SPARTAN’s prior annual meeting, circumstances looked on track for a quicker resolution. In a vote of the membership at the 2015 gathering, 64% backed creation of new classification criteria, and Dr. Caplan proposed quickly preparing a research proposal to present in early 2016 to the American College of Rheumatology and the European League Against Rheumatism for funding.
But in the ensuing year, the effort got mired in discussions on how to perform the study needed to gather the evidence base for criteria revisions. Some progress occurred, most notably an agreement between SPARTAN officials and the ASAS leadership to work together on the revision, said Atul A. Deodhar, MD, professor of medicine and medical director of the rheumatology clinics at the Oregon Health & Science University in Portland.
The SPARTAN and ASAS representatives also agreed on several specifics of what’s needed, including a weighting formula for the 10 different clinical criteria of the SpA classification. Currently, in individuals with low back pain lasting 3 months or longer at an age of onset younger than 45 years, any one of these 10 clinical criteria can create a case of SpA either if combined with MRI or radiographic evidence of sacroiliitis, or without any imaging if the individual is HLA-B27 positive and fulfills two items from the 10-item list of clinical criteria.
It’s the lack of radiographic imaging confirmation, so-called “nonradiographic SpA,” that generates the greatest concern about below-par specificity and that may be helped by a weighting adjustment that varies the classification power of each of the 10 clinical items. A more ideal balance might be criteria with sensitivity reduced from the current level to about 70% and with specificity rising to about 90%, Dr. Caplan suggested.
The keystone of any revision will be a large, new study with SpA patients from various locations. Dr. Caplan emphasized that “no firm decisions have yet been made” regarding the study's name or research focus.
On Twitter @mitchelzoler
This article was updated August 11, 2016.
DENVER – The revised axial spondyloarthritis classification criteria that U.S. rheumatologists say they desperately need remain elusive, with no firm path to creation.
The 2009 classification criteria for axial spondyloarthritis (SpA) developed by the Assessment of Spondyloarthritis international Society (ASAS) was a landmark in creating a definition for axial SpA to use when enrolling patients into clinical trials and to also potentially use for diagnosis.
But the 2009 classification criteria also have several shortcomings, especially from a U.S. perspective, that have limited the utility of the criteria in U.S. practice and studies.
Several years of discussions among North American rheumatologists about the classification criteria have produced consensus about what is wrong with them: The clinical elements of the criteria are not specific enough with sensitivity and specificity rates that are each about 80%-85%; the imaging component of the criteria is not totally up to date and as one example only considers osteitis and not other MRI abnormalities such as fatty metaplasia and T1 structural lesions; the criteria were entirely based on results from studies with Europeans, making their applicability to other patient types uncertain; and some terminology in the criteria are confusing, Liron Caplan, MD, said during a discussion of the criteria at the annual meeting of the of the Spondyloarthritis Research and Treatment Network.
But while the SpA experts who gathered at the meeting and who have also been discussing this issue since 2013 are of one mind on the problems, agreement on exactly how to solve them remains unresolved.
“Everything is still on the table,” said Dr. Caplan, a rheumatologist and director of the SpA program at the University of Colorado in Aurora.
A year ago, at SPARTAN’s prior annual meeting, circumstances looked on track for a quicker resolution. In a vote of the membership at the 2015 gathering, 64% backed creation of new classification criteria, and Dr. Caplan proposed quickly preparing a research proposal to present in early 2016 to the American College of Rheumatology and the European League Against Rheumatism for funding.
But in the ensuing year, the effort got mired in discussions on how to perform the study needed to gather the evidence base for criteria revisions. Some progress occurred, most notably an agreement between SPARTAN officials and the ASAS leadership to work together on the revision, said Atul A. Deodhar, MD, professor of medicine and medical director of the rheumatology clinics at the Oregon Health & Science University in Portland.
The SPARTAN and ASAS representatives also agreed on several specifics of what’s needed, including a weighting formula for the 10 different clinical criteria of the SpA classification. Currently, in individuals with low back pain lasting 3 months or longer at an age of onset younger than 45 years, any one of these 10 clinical criteria can create a case of SpA either if combined with MRI or radiographic evidence of sacroiliitis, or without any imaging if the individual is HLA-B27 positive and fulfills two items from the 10-item list of clinical criteria.
It’s the lack of radiographic imaging confirmation, so-called “nonradiographic SpA,” that generates the greatest concern about below-par specificity and that may be helped by a weighting adjustment that varies the classification power of each of the 10 clinical items. A more ideal balance might be criteria with sensitivity reduced from the current level to about 70% and with specificity rising to about 90%, Dr. Caplan suggested.
The keystone of any revision will be a large, new study with SpA patients from various locations. Dr. Caplan emphasized that “no firm decisions have yet been made” regarding the study's name or research focus.
On Twitter @mitchelzoler
This article was updated August 11, 2016.
EXPERT ANALYSIS FROM THE 2016 SPARTAN ANNUAL MEETING

Concurrent fibromyalgia intensifies ankylosing spondylitis symptoms
DENVER – Fibromyalgia syndrome commonly occurred in patients with ankylosing spondylitis who were reviewed at one U.S. center, and when the two coexisted ankylosing spondylitis disease severity significantly increased.
In the study’s 62 patients with confirmed ankylosing spondylitis (AS), 27 (44%) also met the 2010 American College of Rheumatology diagnostic criteria for fibromyalgia syndrome, Sherilyn Diomampo, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network. The fibromyalgia rate in these AS patients was substantially above prior reports of fibromyalgia prevalence rates in the range of 10%-15%, said Dr. Diomampo, a rheumatologist at MetroHealth Medical Center in Cleveland.

Patients with both disorders also had substantially higher scores across the board for all the measures of AS severity that Dr. Diomampo and her associates evaluated. For example, scores on the Bath AS Disease Activity Index averaged 6.8 in the patients with fibromyalgia and 3.8 in those without fibromyalgia. (A BASDAI score of 4.0 or higher generally suggests suboptimal disease control.) The average score of the AS Disease Activity Score using C-reactive protein (CRP) as the serum marker of inflammation was 4.2 in the subgroup with fibromyalgia and 2.8 in those without. (An ASDAS score of 3.5 or higher denotes high or very high disease activity. A score reduced by at least 1.1 indicates a clinically important improvement.) All these between-group differences were statistically significant.
Other measures of AS severity that were significantly higher with fibromyalgia included patient global self assessment, physician global assessment, average serum levels of CRP, and the average erythrocyte sedimentation rate.
The 2010 ACR diagnostic criteria for fibromyalgia syndrome used by Dr. Diomampo and her associates in their analysis require a patient to have a widespread pain index of at least 7 and symptom severity of at least 5, or alternatively, a widespread pain index of 3-6 and symptom severity of at least 9 (Arthritis Care Res. 2010 May;62[5]:600-10). In addition, for this study, the researchers stipulated that diagnosis of concomitant fibromyalgia meant patients had their fibromyalgia symptoms in place for at least 3 months, and the examining clinician could not attribute the patient’s pain to AS.
Using linear regression models with the widespread pain index and the symptom severity as the dependent variables, the researchers failed to see any statistically significant relationship between either of these two determinants of fibromyalgia severity and five different measures of AS severity, including the BASDAI and the ASDAS. In short, the assessment tools used to measure AS severity showed no ability to also measure the core characteristics of fibromyalgia, Dr. Diomampo said.
Overall, patients in the study averaged about 49 years old. The analysis showed a significantly higher rate of African-American patients in the fibromyalgia group, 63%, compared with a 37% rate of African Americans in those with AS and no fibromyalgia. The presence of acute, anterior uveitis was 27% among those with fibromyalgia and 73% of those with AS and no fibromyalgia. One notable similarity between the two subgroups was the percentage on treatment with a tumor necrosis factor inhibitor: 52% among those with concurrent fibromyalgia and 49% among those with AS alone.
Dr. Diomampo had no disclosures.
On Twitter @mitchelzoler
DENVER – Fibromyalgia syndrome commonly occurred in patients with ankylosing spondylitis who were reviewed at one U.S. center, and when the two coexisted ankylosing spondylitis disease severity significantly increased.
In the study’s 62 patients with confirmed ankylosing spondylitis (AS), 27 (44%) also met the 2010 American College of Rheumatology diagnostic criteria for fibromyalgia syndrome, Sherilyn Diomampo, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network. The fibromyalgia rate in these AS patients was substantially above prior reports of fibromyalgia prevalence rates in the range of 10%-15%, said Dr. Diomampo, a rheumatologist at MetroHealth Medical Center in Cleveland.
Patients with both disorders also had substantially higher scores across the board for all the measures of AS severity that Dr. Diomampo and her associates evaluated. For example, scores on the Bath AS Disease Activity Index averaged 6.8 in the patients with fibromyalgia and 3.8 in those without fibromyalgia. (A BASDAI score of 4.0 or higher generally suggests suboptimal disease control.) The average score of the AS Disease Activity Score using C-reactive protein (CRP) as the serum marker of inflammation was 4.2 in the subgroup with fibromyalgia and 2.8 in those without. (An ASDAS score of 3.5 or higher denotes high or very high disease activity. A score reduced by at least 1.1 indicates a clinically important improvement.) All these between-group differences were statistically significant.
Other measures of AS severity that were significantly higher with fibromyalgia included patient global self assessment, physician global assessment, average serum levels of CRP, and the average erythrocyte sedimentation rate.
The 2010 ACR diagnostic criteria for fibromyalgia syndrome used by Dr. Diomampo and her associates in their analysis require a patient to have a widespread pain index of at least 7 and symptom severity of at least 5, or alternatively, a widespread pain index of 3-6 and symptom severity of at least 9 (Arthritis Care Res. 2010 May;62[5]:600-10). In addition, for this study, the researchers stipulated that diagnosis of concomitant fibromyalgia meant patients had their fibromyalgia symptoms in place for at least 3 months, and the examining clinician could not attribute the patient’s pain to AS.
Using linear regression models with the widespread pain index and the symptom severity as the dependent variables, the researchers failed to see any statistically significant relationship between either of these two determinants of fibromyalgia severity and five different measures of AS severity, including the BASDAI and the ASDAS. In short, the assessment tools used to measure AS severity showed no ability to also measure the core characteristics of fibromyalgia, Dr. Diomampo said.
Overall, patients in the study averaged about 49 years old. The analysis showed a significantly higher rate of African-American patients in the fibromyalgia group, 63%, compared with a 37% rate of African Americans in those with AS and no fibromyalgia. The presence of acute, anterior uveitis was 27% among those with fibromyalgia and 73% of those with AS and no fibromyalgia. One notable similarity between the two subgroups was the percentage on treatment with a tumor necrosis factor inhibitor: 52% among those with concurrent fibromyalgia and 49% among those with AS alone.
Dr. Diomampo had no disclosures.
On Twitter @mitchelzoler
DENVER – Fibromyalgia syndrome commonly occurred in patients with ankylosing spondylitis who were reviewed at one U.S. center, and when the two coexisted ankylosing spondylitis disease severity significantly increased.
In the study’s 62 patients with confirmed ankylosing spondylitis (AS), 27 (44%) also met the 2010 American College of Rheumatology diagnostic criteria for fibromyalgia syndrome, Sherilyn Diomampo, MD, said at the annual meeting of the Spondyloarthritis Research and Treatment Network. The fibromyalgia rate in these AS patients was substantially above prior reports of fibromyalgia prevalence rates in the range of 10%-15%, said Dr. Diomampo, a rheumatologist at MetroHealth Medical Center in Cleveland.
Patients with both disorders also had substantially higher scores across the board for all the measures of AS severity that Dr. Diomampo and her associates evaluated. For example, scores on the Bath AS Disease Activity Index averaged 6.8 in the patients with fibromyalgia and 3.8 in those without fibromyalgia. (A BASDAI score of 4.0 or higher generally suggests suboptimal disease control.) The average score of the AS Disease Activity Score using C-reactive protein (CRP) as the serum marker of inflammation was 4.2 in the subgroup with fibromyalgia and 2.8 in those without. (An ASDAS score of 3.5 or higher denotes high or very high disease activity. A score reduced by at least 1.1 indicates a clinically important improvement.) All these between-group differences were statistically significant.
Other measures of AS severity that were significantly higher with fibromyalgia included patient global self assessment, physician global assessment, average serum levels of CRP, and the average erythrocyte sedimentation rate.
The 2010 ACR diagnostic criteria for fibromyalgia syndrome used by Dr. Diomampo and her associates in their analysis require a patient to have a widespread pain index of at least 7 and symptom severity of at least 5, or alternatively, a widespread pain index of 3-6 and symptom severity of at least 9 (Arthritis Care Res. 2010 May;62[5]:600-10). In addition, for this study, the researchers stipulated that diagnosis of concomitant fibromyalgia meant patients had their fibromyalgia symptoms in place for at least 3 months, and the examining clinician could not attribute the patient’s pain to AS.
Using linear regression models with the widespread pain index and the symptom severity as the dependent variables, the researchers failed to see any statistically significant relationship between either of these two determinants of fibromyalgia severity and five different measures of AS severity, including the BASDAI and the ASDAS. In short, the assessment tools used to measure AS severity showed no ability to also measure the core characteristics of fibromyalgia, Dr. Diomampo said.
Overall, patients in the study averaged about 49 years old. The analysis showed a significantly higher rate of African-American patients in the fibromyalgia group, 63%, compared with a 37% rate of African Americans in those with AS and no fibromyalgia. The presence of acute, anterior uveitis was 27% among those with fibromyalgia and 73% of those with AS and no fibromyalgia. One notable similarity between the two subgroups was the percentage on treatment with a tumor necrosis factor inhibitor: 52% among those with concurrent fibromyalgia and 49% among those with AS alone.
Dr. Diomampo had no disclosures.
On Twitter @mitchelzoler
AT THE 2016 SPARTAN ANNUAL MEETING
Key clinical point: Nearly half of patients with ankylosing spondylitis had concurrent fibromyalgia at one U.S. center, and patients with both had much greater AS severity.
Major finding: The average BASDAI was 6.8 in patients with ankylosing spondylitis and fibromyalgia and 3.8 in patients with AS only.
Data source: Observational data collected on 62 adults with ankylosing spondylitis at one U.S. center.
Disclosures: Dr. Diomampo had no disclosures.
