User login
FDA approves new treatment for Factor X deficiency
The U.S. Food and Drug Administration has provided its first approval of a coagulation factor replacement drug specifically for patients with hereditary Factor X deficiency who are aged 12 years or older, the FDA said in a written statement. Typically, patients with this inherited disorder are treated with plasma-derived prothrombin complex concentrates.
Coagadex, the new coagulation factor replacement drug, was effective at controlling bleeding episodes in individuals with moderate to severe hereditary Factor X deficiency, in a 16-patient study. Prior to being treated with the purified Factor X concentrate, the study’s participants had the following types of bleeding episodes: spontaneous; traumatic; or heavy menstrual.
Researchers also conducted a smaller study of the drug’s effectiveness on patients who were undergoing surgery. In this research project, Coagadex was successful at controlling blood loss during and after surgery, in those patients with mild hereditary Factor X deficiency.
“The approval of Coagadex is a significant advancement for patients who suffer from this rare but serious disease,” said Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, in the release.
Bio Products Laboratory manufactured the new drug.
The U.S. Food and Drug Administration has provided its first approval of a coagulation factor replacement drug specifically for patients with hereditary Factor X deficiency who are aged 12 years or older, the FDA said in a written statement. Typically, patients with this inherited disorder are treated with plasma-derived prothrombin complex concentrates.
Coagadex, the new coagulation factor replacement drug, was effective at controlling bleeding episodes in individuals with moderate to severe hereditary Factor X deficiency, in a 16-patient study. Prior to being treated with the purified Factor X concentrate, the study’s participants had the following types of bleeding episodes: spontaneous; traumatic; or heavy menstrual.
Researchers also conducted a smaller study of the drug’s effectiveness on patients who were undergoing surgery. In this research project, Coagadex was successful at controlling blood loss during and after surgery, in those patients with mild hereditary Factor X deficiency.
“The approval of Coagadex is a significant advancement for patients who suffer from this rare but serious disease,” said Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, in the release.
Bio Products Laboratory manufactured the new drug.
The U.S. Food and Drug Administration has provided its first approval of a coagulation factor replacement drug specifically for patients with hereditary Factor X deficiency who are aged 12 years or older, the FDA said in a written statement. Typically, patients with this inherited disorder are treated with plasma-derived prothrombin complex concentrates.
Coagadex, the new coagulation factor replacement drug, was effective at controlling bleeding episodes in individuals with moderate to severe hereditary Factor X deficiency, in a 16-patient study. Prior to being treated with the purified Factor X concentrate, the study’s participants had the following types of bleeding episodes: spontaneous; traumatic; or heavy menstrual.
Researchers also conducted a smaller study of the drug’s effectiveness on patients who were undergoing surgery. In this research project, Coagadex was successful at controlling blood loss during and after surgery, in those patients with mild hereditary Factor X deficiency.
“The approval of Coagadex is a significant advancement for patients who suffer from this rare but serious disease,” said Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, in the release.
Bio Products Laboratory manufactured the new drug.
FDA expands Promacta approval to include pediatric patients
The Food and Drug Administration has extended its approval of Promacta (eltrombopag) to include pediatric patients with chronic immune thrombocytopenic purpura, the agency announced Aug. 24.
Approved in 2008 for adults with immune thrombocytopenic purpura (ITP), eltrombopag is now approved for the treatment of the rare blood disorder in patients aged 1 year and older. The drug may be used in children who have not responded to other ITP medications or spleen surgery, the FDA said in a statement.
Eltrombopag’s efficacy and safety in children aged 1-17 years was established in two placebo-controlled trials comprising 159 patients. Findings from the first trial found that over 7 weeks, 62% of patients given eltrombopag had improved platelet counts without rescue therapy between weeks 1 and 6, compared with 32% in the placebo group.
In the second trial, which lasted 13 weeks, 41% of patients taking eltrombopag experienced increased platelet counts for at least 6 out of 8 weeks between weeks 5 and 12, compared with 3% of patients in the placebo group, the FDA reported.
“In both trials, patients taking Promacta also had less need for other treatments to increase their platelet counts, such as corticosteroids or platelet transfusions,” the FDA said. “Among patients taking one or more ITP medications at the start of the trials, about half were able to reduce or discontinue their use of these medications, primarily corticosteroids.”
Eltrombopag may be taken once daily in tablet form, or as a powder mixed with liquid for children aged 1-5 years. It should be used only in ITP patients with an increased risk of bleeding.
The most common side effects in children were infections of the upper respiratory tract or nose and throat, diarrhea, abdominal pain, rash, and increase in liver enzymes.
Promacta is manufactured by Novartis in East Hanover, N.J.
The Food and Drug Administration has extended its approval of Promacta (eltrombopag) to include pediatric patients with chronic immune thrombocytopenic purpura, the agency announced Aug. 24.
Approved in 2008 for adults with immune thrombocytopenic purpura (ITP), eltrombopag is now approved for the treatment of the rare blood disorder in patients aged 1 year and older. The drug may be used in children who have not responded to other ITP medications or spleen surgery, the FDA said in a statement.
Eltrombopag’s efficacy and safety in children aged 1-17 years was established in two placebo-controlled trials comprising 159 patients. Findings from the first trial found that over 7 weeks, 62% of patients given eltrombopag had improved platelet counts without rescue therapy between weeks 1 and 6, compared with 32% in the placebo group.
In the second trial, which lasted 13 weeks, 41% of patients taking eltrombopag experienced increased platelet counts for at least 6 out of 8 weeks between weeks 5 and 12, compared with 3% of patients in the placebo group, the FDA reported.
“In both trials, patients taking Promacta also had less need for other treatments to increase their platelet counts, such as corticosteroids or platelet transfusions,” the FDA said. “Among patients taking one or more ITP medications at the start of the trials, about half were able to reduce or discontinue their use of these medications, primarily corticosteroids.”
Eltrombopag may be taken once daily in tablet form, or as a powder mixed with liquid for children aged 1-5 years. It should be used only in ITP patients with an increased risk of bleeding.
The most common side effects in children were infections of the upper respiratory tract or nose and throat, diarrhea, abdominal pain, rash, and increase in liver enzymes.
Promacta is manufactured by Novartis in East Hanover, N.J.
The Food and Drug Administration has extended its approval of Promacta (eltrombopag) to include pediatric patients with chronic immune thrombocytopenic purpura, the agency announced Aug. 24.
Approved in 2008 for adults with immune thrombocytopenic purpura (ITP), eltrombopag is now approved for the treatment of the rare blood disorder in patients aged 1 year and older. The drug may be used in children who have not responded to other ITP medications or spleen surgery, the FDA said in a statement.
Eltrombopag’s efficacy and safety in children aged 1-17 years was established in two placebo-controlled trials comprising 159 patients. Findings from the first trial found that over 7 weeks, 62% of patients given eltrombopag had improved platelet counts without rescue therapy between weeks 1 and 6, compared with 32% in the placebo group.
In the second trial, which lasted 13 weeks, 41% of patients taking eltrombopag experienced increased platelet counts for at least 6 out of 8 weeks between weeks 5 and 12, compared with 3% of patients in the placebo group, the FDA reported.
“In both trials, patients taking Promacta also had less need for other treatments to increase their platelet counts, such as corticosteroids or platelet transfusions,” the FDA said. “Among patients taking one or more ITP medications at the start of the trials, about half were able to reduce or discontinue their use of these medications, primarily corticosteroids.”
Eltrombopag may be taken once daily in tablet form, or as a powder mixed with liquid for children aged 1-5 years. It should be used only in ITP patients with an increased risk of bleeding.
The most common side effects in children were infections of the upper respiratory tract or nose and throat, diarrhea, abdominal pain, rash, and increase in liver enzymes.
Promacta is manufactured by Novartis in East Hanover, N.J.
Novel triple therapy in ITP provides enduring responses
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
FROM BLOOD
Key clinical point: Patients with primary immune thrombocytopenia can achieve an enduring response with a novel triple drug regimen.
Major finding: Relapse-free survival was 92% at 12 months for responders and 76% at 24 months
Data source: Prospective, single-arm, phase IIb study involving 20 patients.
Disclosures: No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
Eltrombopag yields 40% response rate in pediatric immune thrombocytopenia
Treatment with the thrombopoietin receptor agonist eltrombopag led to a sustained platelet response in 40% of children and adolescents with chronic immune thrombocytopenia, compared with only 3% of the placebo group, according to a randomized multicenter trial published online in The Lancet.
Eltrombopag is approved in the United States for adults with chronic immune thrombocytopenia (CIT) who have not responded adequately to corticosteroids, immunoglobulins, or splenectomy, but few trials have assessed CIT therapies in children, said Dr. John Grainger of the Royal Manchester Children’s Hospital and the University of Manchester (England) and his associates.
Their multicenter, international study included 92 patients up to age 17 with CIT. During the 13-week double-blinded period of the study, patients received once-daily placebo or eltrombopag dosed at 0.89-1.2 mg/kg for patients aged 1-5 years and at 25-50 mg for patients aged 6-17 years. Dosing ranges were adjusted for ethnicity as well as body weight because east Asians have higher eltrombopag exposures and need lower starting doses, the investigators noted. After the double-blinded period, all patients entered 24 weeks of open-label treatment with eltrombopag (Lancet. 2015 Jul 29. doi: 10.1016/S0140-6736(15)61107-2.).
A total of 25 (40%) patients who received eltrombopag achieved platelet counts of at least 50 × 10⁹ per L for at least 6 of the last 8 weeks of the double-blinded period, compared with only one patient (3%) on placebo (odds ratio, 18; 95% confidence interval, 2.3-140.9; P = .0004), said the researchers. Based on the World Health Organization bleeding scale, the percentage of patients who experienced grade 1-4 bleeding events fell from 63% at the start of the open-label period to 24% at the end, and clinically significant (grade 2-4) bleeding events dropped from 20% to 6%. Seven of 87 patients were able to stop all other drugs they were taking for CIT without needing rescue therapy during open-label treatment, the researchers said.
Two patients stopped eltrombopag because of elevated liver aminotransferases. Rates of nasopharyngitis, rhinitis, upper respiratory tract infection, and cough were more common for eltrombopag than placebo, the investigators reported. However, serious adverse events were more common with placebo (14% vs. 8%), and there were no deaths, thrombotic events, or malignancies. Safety trends were similar during the double-blinded and open-label study periods, they added.
GlaxoSmithKline funded the study. Dr. Grainger reported receiving honoraria from GlaxoSmithKline, Amgen, and Baxter. Eleven coauthors reported financial conflicts of interest with GlaxoSmithKline and a number of other pharmaceutical companies.
Treatment with the thrombopoietin receptor agonist eltrombopag led to a sustained platelet response in 40% of children and adolescents with chronic immune thrombocytopenia, compared with only 3% of the placebo group, according to a randomized multicenter trial published online in The Lancet.
Eltrombopag is approved in the United States for adults with chronic immune thrombocytopenia (CIT) who have not responded adequately to corticosteroids, immunoglobulins, or splenectomy, but few trials have assessed CIT therapies in children, said Dr. John Grainger of the Royal Manchester Children’s Hospital and the University of Manchester (England) and his associates.
Their multicenter, international study included 92 patients up to age 17 with CIT. During the 13-week double-blinded period of the study, patients received once-daily placebo or eltrombopag dosed at 0.89-1.2 mg/kg for patients aged 1-5 years and at 25-50 mg for patients aged 6-17 years. Dosing ranges were adjusted for ethnicity as well as body weight because east Asians have higher eltrombopag exposures and need lower starting doses, the investigators noted. After the double-blinded period, all patients entered 24 weeks of open-label treatment with eltrombopag (Lancet. 2015 Jul 29. doi: 10.1016/S0140-6736(15)61107-2.).
A total of 25 (40%) patients who received eltrombopag achieved platelet counts of at least 50 × 10⁹ per L for at least 6 of the last 8 weeks of the double-blinded period, compared with only one patient (3%) on placebo (odds ratio, 18; 95% confidence interval, 2.3-140.9; P = .0004), said the researchers. Based on the World Health Organization bleeding scale, the percentage of patients who experienced grade 1-4 bleeding events fell from 63% at the start of the open-label period to 24% at the end, and clinically significant (grade 2-4) bleeding events dropped from 20% to 6%. Seven of 87 patients were able to stop all other drugs they were taking for CIT without needing rescue therapy during open-label treatment, the researchers said.
Two patients stopped eltrombopag because of elevated liver aminotransferases. Rates of nasopharyngitis, rhinitis, upper respiratory tract infection, and cough were more common for eltrombopag than placebo, the investigators reported. However, serious adverse events were more common with placebo (14% vs. 8%), and there were no deaths, thrombotic events, or malignancies. Safety trends were similar during the double-blinded and open-label study periods, they added.
GlaxoSmithKline funded the study. Dr. Grainger reported receiving honoraria from GlaxoSmithKline, Amgen, and Baxter. Eleven coauthors reported financial conflicts of interest with GlaxoSmithKline and a number of other pharmaceutical companies.
Treatment with the thrombopoietin receptor agonist eltrombopag led to a sustained platelet response in 40% of children and adolescents with chronic immune thrombocytopenia, compared with only 3% of the placebo group, according to a randomized multicenter trial published online in The Lancet.
Eltrombopag is approved in the United States for adults with chronic immune thrombocytopenia (CIT) who have not responded adequately to corticosteroids, immunoglobulins, or splenectomy, but few trials have assessed CIT therapies in children, said Dr. John Grainger of the Royal Manchester Children’s Hospital and the University of Manchester (England) and his associates.
Their multicenter, international study included 92 patients up to age 17 with CIT. During the 13-week double-blinded period of the study, patients received once-daily placebo or eltrombopag dosed at 0.89-1.2 mg/kg for patients aged 1-5 years and at 25-50 mg for patients aged 6-17 years. Dosing ranges were adjusted for ethnicity as well as body weight because east Asians have higher eltrombopag exposures and need lower starting doses, the investigators noted. After the double-blinded period, all patients entered 24 weeks of open-label treatment with eltrombopag (Lancet. 2015 Jul 29. doi: 10.1016/S0140-6736(15)61107-2.).
A total of 25 (40%) patients who received eltrombopag achieved platelet counts of at least 50 × 10⁹ per L for at least 6 of the last 8 weeks of the double-blinded period, compared with only one patient (3%) on placebo (odds ratio, 18; 95% confidence interval, 2.3-140.9; P = .0004), said the researchers. Based on the World Health Organization bleeding scale, the percentage of patients who experienced grade 1-4 bleeding events fell from 63% at the start of the open-label period to 24% at the end, and clinically significant (grade 2-4) bleeding events dropped from 20% to 6%. Seven of 87 patients were able to stop all other drugs they were taking for CIT without needing rescue therapy during open-label treatment, the researchers said.
Two patients stopped eltrombopag because of elevated liver aminotransferases. Rates of nasopharyngitis, rhinitis, upper respiratory tract infection, and cough were more common for eltrombopag than placebo, the investigators reported. However, serious adverse events were more common with placebo (14% vs. 8%), and there were no deaths, thrombotic events, or malignancies. Safety trends were similar during the double-blinded and open-label study periods, they added.
GlaxoSmithKline funded the study. Dr. Grainger reported receiving honoraria from GlaxoSmithKline, Amgen, and Baxter. Eleven coauthors reported financial conflicts of interest with GlaxoSmithKline and a number of other pharmaceutical companies.
FROM THE LANCET
Key clinical point: Eltrombopag markedly outperformed placebo and had no new safety signals in children and adolescents with chronic immune thrombocytopenia.
Major finding: Forty percent of patients achieved sustained platelet response on eltrombopag, compared with 3% of the placebo group (OR, 18.0; P = .0004).
Data source: Randomized, double-blinded, multicenter, international trial of 92 patients aged 1-17 years.
Disclosures: GlaxoSmithKline funded the study. Dr. Grainger reported receiving honoraria from GlaxoSmithKline, Amgen, and Baxter. Eleven coauthors reported financial conflicts of interest with GlaxoSmithKline and a number of other pharmaceutical companies.
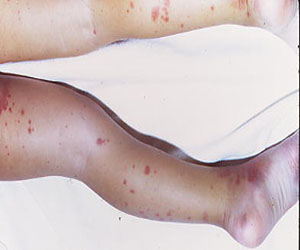
Drug-induced iron deficiency decreased congenital erythropoietic porphyria symptoms
Using deferasirox to induce iron deficiency may improve the symptoms of congenital erythropoietic porphyria.
The findings come from a study of a woman of Alaskan Native descent who was diagnosed as an infant with CEP and had compound heterozygosity for C73R and A104V mutations in uroporphyrinogen III synthase. Her disease complications included chronic hemolysis and severe photosensitivity with scarring, which were managed with sun avoidance and supportive measures including blood transfusions.
With an episode of occult gastrointestinal bleeding, the patient reported a dramatic improvement in photosensitivity and a normalization in urine color. “Markers of hemolysis also improved, with a reduction in lactate dehydrogenase (LDH) level to 138 U/L and a marked reduction in circulating nucleated red blood cells (nRBCs) to 0.2 × 103 cells/mL. Hemoglobin level increased from 6.9 to 9.4. Reticulocyte count decreased to 55 billion cells/L.”
When the GI bleeding resolved, the patient’s serum ferritin level rose to 208 ng/mL, her photosensitivity worsened, and her urine color darkened. “Similarly, her LDH level increased to 540 U/L, reticulocyte count increased markedly to 163 billion cells/L, and nRBCs increased to 4.3 x 103 cells/mL.”
The researchers initiated a trial of deferasirox initially dosed at 500 mg daily for several months and then adjusted to 500 mg three times weekly to target a ferritin level range of 10-15 ng/mL. “With deferasirox, [her] serum ferritin level dropped to 16 ng/mL at 2 months and the patient again reported an improvement in her quality of life with reduced photosensitivity.” Also, total urine porphyrins decreased from 108,364 mcg/24 hours before treatment to 5896.3 mcg/24 hours after 4 months. Concurrently, LDH normalized to a level of 135 U/L, reticulocyte count decreased to 47 billion/L, nRBCs remained between 0.14 and 0.68 × 103 cells/mL, and hemoglobin level remained between 6.8 and 9.0 g/dL without transfusional support.
Upper GI bleeding recurred, however, so deferasirox was discontinued and red blood cell transfusions were administered. “Over the ensuing 2 years, numerous repeat endoscopies showed persistent gastric erosions and ulcerations despite ongoing use of a proton pump inhibitor.” As a result of the ongoing iron losses, ferritin levels remained within normal range and symptoms and laboratory markers of hemolysis were stabilized.
When the GI bleeding resolved, the patient’s lab values and her photosensitivity suddenly worsened.
Although not known to be a complication of congenital erythropoietic porphyria, the patient developed liver disease of unknown etiology and she died at age 35 of complications of liver failure, hepatorenal syndrome, and hemolysis after nearly 3 years of reduced symptom severity “and a considerable improvement in her quality of life,” Dr. Daniel N. Egan of the University of Washington, Seattle, and his colleagues reported in the case study in Blood (2015;126:257-261).
“Microscopic examination of the liver at autopsy demonstrated areas of marked sinusoidal congestion and dilatation containing aggregates of erythroid precursors consistent with diffuse intrasinusoidal extramedullary hematopoiesis. Extensive patchy fibrosis was present, without regenerative nodules or cirrhosis. Importantly, there was no evidence of polarizable material to suggest porphyrin metabolite accumulation.”
The patient’s sister, who had the same disorder, also died “from sudden cardiac death in the setting of pulmonary hypertension.”
The researchers obtained bone marrow cells from both sisters and cultured the cells in 5% plasma and varying ratios of holo-transferrin (holo-Tf) and apo-transferrin (apo-Tf) (Sigma). The level of available iron was 1.52 mcM with 100% apo-Tf and 9.52 mcM with 100% holo-Tf.
“Erythroid cells obtained from the bone marrow of this CEP patient demonstrated improved growth and differentiation in conditions of relative iron deficiency,” the authors wrote. “The percentages of normal cells reaching stages III and IV at culture day 10 progressively decreased from 67.2% to 38.4% when available iron was reduced.”
“Iron restriction likely impedes heme synthesis upstream of uroporphyrinogen III synthase via decreasing 5-aminolevulinate synthase 2 mRNA translation. Because the tricarboxylic acid cycle enzyme aconitase contains a 4Fe-4S cluster, it also is possible that decreased availability of succinyl coenzyme A, a key substrate for the rate-limiting step in heme production, may play a role,” the researchers wrote.
Using deferasirox to induce iron deficiency may improve the symptoms of congenital erythropoietic porphyria.
The findings come from a study of a woman of Alaskan Native descent who was diagnosed as an infant with CEP and had compound heterozygosity for C73R and A104V mutations in uroporphyrinogen III synthase. Her disease complications included chronic hemolysis and severe photosensitivity with scarring, which were managed with sun avoidance and supportive measures including blood transfusions.
With an episode of occult gastrointestinal bleeding, the patient reported a dramatic improvement in photosensitivity and a normalization in urine color. “Markers of hemolysis also improved, with a reduction in lactate dehydrogenase (LDH) level to 138 U/L and a marked reduction in circulating nucleated red blood cells (nRBCs) to 0.2 × 103 cells/mL. Hemoglobin level increased from 6.9 to 9.4. Reticulocyte count decreased to 55 billion cells/L.”
When the GI bleeding resolved, the patient’s serum ferritin level rose to 208 ng/mL, her photosensitivity worsened, and her urine color darkened. “Similarly, her LDH level increased to 540 U/L, reticulocyte count increased markedly to 163 billion cells/L, and nRBCs increased to 4.3 x 103 cells/mL.”
The researchers initiated a trial of deferasirox initially dosed at 500 mg daily for several months and then adjusted to 500 mg three times weekly to target a ferritin level range of 10-15 ng/mL. “With deferasirox, [her] serum ferritin level dropped to 16 ng/mL at 2 months and the patient again reported an improvement in her quality of life with reduced photosensitivity.” Also, total urine porphyrins decreased from 108,364 mcg/24 hours before treatment to 5896.3 mcg/24 hours after 4 months. Concurrently, LDH normalized to a level of 135 U/L, reticulocyte count decreased to 47 billion/L, nRBCs remained between 0.14 and 0.68 × 103 cells/mL, and hemoglobin level remained between 6.8 and 9.0 g/dL without transfusional support.
Upper GI bleeding recurred, however, so deferasirox was discontinued and red blood cell transfusions were administered. “Over the ensuing 2 years, numerous repeat endoscopies showed persistent gastric erosions and ulcerations despite ongoing use of a proton pump inhibitor.” As a result of the ongoing iron losses, ferritin levels remained within normal range and symptoms and laboratory markers of hemolysis were stabilized.
When the GI bleeding resolved, the patient’s lab values and her photosensitivity suddenly worsened.
Although not known to be a complication of congenital erythropoietic porphyria, the patient developed liver disease of unknown etiology and she died at age 35 of complications of liver failure, hepatorenal syndrome, and hemolysis after nearly 3 years of reduced symptom severity “and a considerable improvement in her quality of life,” Dr. Daniel N. Egan of the University of Washington, Seattle, and his colleagues reported in the case study in Blood (2015;126:257-261).
“Microscopic examination of the liver at autopsy demonstrated areas of marked sinusoidal congestion and dilatation containing aggregates of erythroid precursors consistent with diffuse intrasinusoidal extramedullary hematopoiesis. Extensive patchy fibrosis was present, without regenerative nodules or cirrhosis. Importantly, there was no evidence of polarizable material to suggest porphyrin metabolite accumulation.”
The patient’s sister, who had the same disorder, also died “from sudden cardiac death in the setting of pulmonary hypertension.”
The researchers obtained bone marrow cells from both sisters and cultured the cells in 5% plasma and varying ratios of holo-transferrin (holo-Tf) and apo-transferrin (apo-Tf) (Sigma). The level of available iron was 1.52 mcM with 100% apo-Tf and 9.52 mcM with 100% holo-Tf.
“Erythroid cells obtained from the bone marrow of this CEP patient demonstrated improved growth and differentiation in conditions of relative iron deficiency,” the authors wrote. “The percentages of normal cells reaching stages III and IV at culture day 10 progressively decreased from 67.2% to 38.4% when available iron was reduced.”
“Iron restriction likely impedes heme synthesis upstream of uroporphyrinogen III synthase via decreasing 5-aminolevulinate synthase 2 mRNA translation. Because the tricarboxylic acid cycle enzyme aconitase contains a 4Fe-4S cluster, it also is possible that decreased availability of succinyl coenzyme A, a key substrate for the rate-limiting step in heme production, may play a role,” the researchers wrote.
Using deferasirox to induce iron deficiency may improve the symptoms of congenital erythropoietic porphyria.
The findings come from a study of a woman of Alaskan Native descent who was diagnosed as an infant with CEP and had compound heterozygosity for C73R and A104V mutations in uroporphyrinogen III synthase. Her disease complications included chronic hemolysis and severe photosensitivity with scarring, which were managed with sun avoidance and supportive measures including blood transfusions.
With an episode of occult gastrointestinal bleeding, the patient reported a dramatic improvement in photosensitivity and a normalization in urine color. “Markers of hemolysis also improved, with a reduction in lactate dehydrogenase (LDH) level to 138 U/L and a marked reduction in circulating nucleated red blood cells (nRBCs) to 0.2 × 103 cells/mL. Hemoglobin level increased from 6.9 to 9.4. Reticulocyte count decreased to 55 billion cells/L.”
When the GI bleeding resolved, the patient’s serum ferritin level rose to 208 ng/mL, her photosensitivity worsened, and her urine color darkened. “Similarly, her LDH level increased to 540 U/L, reticulocyte count increased markedly to 163 billion cells/L, and nRBCs increased to 4.3 x 103 cells/mL.”
The researchers initiated a trial of deferasirox initially dosed at 500 mg daily for several months and then adjusted to 500 mg three times weekly to target a ferritin level range of 10-15 ng/mL. “With deferasirox, [her] serum ferritin level dropped to 16 ng/mL at 2 months and the patient again reported an improvement in her quality of life with reduced photosensitivity.” Also, total urine porphyrins decreased from 108,364 mcg/24 hours before treatment to 5896.3 mcg/24 hours after 4 months. Concurrently, LDH normalized to a level of 135 U/L, reticulocyte count decreased to 47 billion/L, nRBCs remained between 0.14 and 0.68 × 103 cells/mL, and hemoglobin level remained between 6.8 and 9.0 g/dL without transfusional support.
Upper GI bleeding recurred, however, so deferasirox was discontinued and red blood cell transfusions were administered. “Over the ensuing 2 years, numerous repeat endoscopies showed persistent gastric erosions and ulcerations despite ongoing use of a proton pump inhibitor.” As a result of the ongoing iron losses, ferritin levels remained within normal range and symptoms and laboratory markers of hemolysis were stabilized.
When the GI bleeding resolved, the patient’s lab values and her photosensitivity suddenly worsened.
Although not known to be a complication of congenital erythropoietic porphyria, the patient developed liver disease of unknown etiology and she died at age 35 of complications of liver failure, hepatorenal syndrome, and hemolysis after nearly 3 years of reduced symptom severity “and a considerable improvement in her quality of life,” Dr. Daniel N. Egan of the University of Washington, Seattle, and his colleagues reported in the case study in Blood (2015;126:257-261).
“Microscopic examination of the liver at autopsy demonstrated areas of marked sinusoidal congestion and dilatation containing aggregates of erythroid precursors consistent with diffuse intrasinusoidal extramedullary hematopoiesis. Extensive patchy fibrosis was present, without regenerative nodules or cirrhosis. Importantly, there was no evidence of polarizable material to suggest porphyrin metabolite accumulation.”
The patient’s sister, who had the same disorder, also died “from sudden cardiac death in the setting of pulmonary hypertension.”
The researchers obtained bone marrow cells from both sisters and cultured the cells in 5% plasma and varying ratios of holo-transferrin (holo-Tf) and apo-transferrin (apo-Tf) (Sigma). The level of available iron was 1.52 mcM with 100% apo-Tf and 9.52 mcM with 100% holo-Tf.
“Erythroid cells obtained from the bone marrow of this CEP patient demonstrated improved growth and differentiation in conditions of relative iron deficiency,” the authors wrote. “The percentages of normal cells reaching stages III and IV at culture day 10 progressively decreased from 67.2% to 38.4% when available iron was reduced.”
“Iron restriction likely impedes heme synthesis upstream of uroporphyrinogen III synthase via decreasing 5-aminolevulinate synthase 2 mRNA translation. Because the tricarboxylic acid cycle enzyme aconitase contains a 4Fe-4S cluster, it also is possible that decreased availability of succinyl coenzyme A, a key substrate for the rate-limiting step in heme production, may play a role,” the researchers wrote.
FROM BLOOD
Dicloxacillin may cut INR levels in warfarin users
The antibiotic dicloxacillin appears to markedly decrease INR levels in patients taking warfarin, reducing the mean INR to subtherapeutic ranges in the majority who take both drugs concomitantly, according to a research letter to the editor published online July 20 in JAMA.
Adverse interactions between warfarin and other drugs are often suspected, but solid data are lacking. Case reports have suggested that the commonly used antibiotic dicloxacillin reduces warfarin’s anticoagulant effects, but no studies have examined the issue, said Anton Pottegård, Ph.D., of the department of clinical pharmacology, University of Southern Denmark, Odense, and his associates (JAMA 2015;314:296-7).
To further investigate that possibility, the investigators analyzed information in an anticoagulant database covering 7,400 patients treated by three outpatient clinics and 50 general practitioners during a 15-year period. They focused on weekly INR levels recorded for 236 patients (median age, 68 years), most of whom took warfarin because of atrial fibrillation or heart valve replacement.
The mean INR level before dicloxacillin exposure was 2.59, compared with 1.97 after dicloxacillin exposure (P < .001). A total of 144 patients (61%) had subtherapeutic INR levels (< 2.0) during the 2-4 weeks following a course of dicloxacillin, Dr. Pottegård and his associates said.
A similar but less drastic decrease was observed among the 64 patients taking a different anticoagulant, phenprocoumon, who were given dicloxacillin. Mean INR levels dropped from 2.61 before exposure to 2.30 afterward (P = .003), and 41% of the group had subtherapeutic INR levels after taking the antibiotic.
No sponsor was reported for this study. Dr. Pottegård and his associates reported having no relevant financial disclosures.
The antibiotic dicloxacillin appears to markedly decrease INR levels in patients taking warfarin, reducing the mean INR to subtherapeutic ranges in the majority who take both drugs concomitantly, according to a research letter to the editor published online July 20 in JAMA.
Adverse interactions between warfarin and other drugs are often suspected, but solid data are lacking. Case reports have suggested that the commonly used antibiotic dicloxacillin reduces warfarin’s anticoagulant effects, but no studies have examined the issue, said Anton Pottegård, Ph.D., of the department of clinical pharmacology, University of Southern Denmark, Odense, and his associates (JAMA 2015;314:296-7).
To further investigate that possibility, the investigators analyzed information in an anticoagulant database covering 7,400 patients treated by three outpatient clinics and 50 general practitioners during a 15-year period. They focused on weekly INR levels recorded for 236 patients (median age, 68 years), most of whom took warfarin because of atrial fibrillation or heart valve replacement.
The mean INR level before dicloxacillin exposure was 2.59, compared with 1.97 after dicloxacillin exposure (P < .001). A total of 144 patients (61%) had subtherapeutic INR levels (< 2.0) during the 2-4 weeks following a course of dicloxacillin, Dr. Pottegård and his associates said.
A similar but less drastic decrease was observed among the 64 patients taking a different anticoagulant, phenprocoumon, who were given dicloxacillin. Mean INR levels dropped from 2.61 before exposure to 2.30 afterward (P = .003), and 41% of the group had subtherapeutic INR levels after taking the antibiotic.
No sponsor was reported for this study. Dr. Pottegård and his associates reported having no relevant financial disclosures.
The antibiotic dicloxacillin appears to markedly decrease INR levels in patients taking warfarin, reducing the mean INR to subtherapeutic ranges in the majority who take both drugs concomitantly, according to a research letter to the editor published online July 20 in JAMA.
Adverse interactions between warfarin and other drugs are often suspected, but solid data are lacking. Case reports have suggested that the commonly used antibiotic dicloxacillin reduces warfarin’s anticoagulant effects, but no studies have examined the issue, said Anton Pottegård, Ph.D., of the department of clinical pharmacology, University of Southern Denmark, Odense, and his associates (JAMA 2015;314:296-7).
To further investigate that possibility, the investigators analyzed information in an anticoagulant database covering 7,400 patients treated by three outpatient clinics and 50 general practitioners during a 15-year period. They focused on weekly INR levels recorded for 236 patients (median age, 68 years), most of whom took warfarin because of atrial fibrillation or heart valve replacement.
The mean INR level before dicloxacillin exposure was 2.59, compared with 1.97 after dicloxacillin exposure (P < .001). A total of 144 patients (61%) had subtherapeutic INR levels (< 2.0) during the 2-4 weeks following a course of dicloxacillin, Dr. Pottegård and his associates said.
A similar but less drastic decrease was observed among the 64 patients taking a different anticoagulant, phenprocoumon, who were given dicloxacillin. Mean INR levels dropped from 2.61 before exposure to 2.30 afterward (P = .003), and 41% of the group had subtherapeutic INR levels after taking the antibiotic.
No sponsor was reported for this study. Dr. Pottegård and his associates reported having no relevant financial disclosures.
FROM JAMA
Key clinical point: The antibiotic dicloxacillin appears to markedly decrease INR levels in patients using warfarin.
Major finding: 144 patients taking warfarin (61%) had subtherapeutic international normalized ratio levels during the 2-4 weeks following a course of dicloxacillin.
Data source: An analysis of INR levels before and after antibiotic use from a Danish database of 7,400 patients taking anticoagulants.
Disclosures: No sponsor was reported for this study. Dr. Pottegard and his associates reported having no relevant financial disclosures.
World Federation of Hemophilia Seeks Proposals
The World Federation of Hemophilia is seeking proposals by August 31 for international clinical research projects that address inherited bleeding disorders. For more Information: http://www.wfh.org/en/our-work/crgp-application-process-and-deadlines
The World Federation of Hemophilia is seeking proposals by August 31 for international clinical research projects that address inherited bleeding disorders. For more Information: http://www.wfh.org/en/our-work/crgp-application-process-and-deadlines
The World Federation of Hemophilia is seeking proposals by August 31 for international clinical research projects that address inherited bleeding disorders. For more Information: http://www.wfh.org/en/our-work/crgp-application-process-and-deadlines

Surgical bleeding risk ‘remarkable’ with platelet disorders
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.

Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.
Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
VIENNA – Inherited platelet function disorders are associated with a significant bleeding risk during surgery, a retrospective, multicenter study shows.
“The frequency of excessive bleeding assessed by one of three criteria at surgery is pretty remarkable because we had rates ranging from 13% to about 19% depending on the assessment criteria,” Dr. Paolo Gresele said at the annual congress of the European Hematology Association.
Any excessive bleeding by any criteria took place in about 23% of patients.
Although conventionally considered rare, inherited platelet function disorders (IPFD) are more frequent than previously thought, and recent advances in their diagnosis have expanded the forms and number of reported patients.
Only a handful of case reports or small case series have evaluated the bleeding risk associated with surgery in patients with IPFD. As a result, the exact bleeding risk and most appropriate management options in IPFD are not known, Dr. Gresele of University of Perugia (Italy) said.
He reported on a retrospective study involving patients with IPFD enrolled at 42 centers from 16 countries. The median age of the 205 patients was 35 years, and 56% were women; 89 had Glanzmann thrombasthenia, 46 had primary secretion defect, 14 had combined alpha-delta granule deficiency, 11 had Hermansky-Pudlak syndrome, 9 had gray platelet syndrome, and 36 had other forms of IPFD.
Data were collected from 389 procedures, of which 54.5% were surgeries, 30% were dental procedures, and 15.5% were invasive procedures. The most frequent surgeries were otolaryngologic, abdominal, orthopedic, gynecologic, urologic, and dermatologic.
The frequency of excessive bleeding at surgery was about 19.7% by the Bleeding Academic Research Consortium (BARC) bleeding classification, 17% by subjective evaluation by the surgeon or patient, and 13% based on duration of bleeding at surgery of at least 6 or more hours, Dr. Gresele said.
The World Health Organization (WHO) bleeding scale before surgery was quite predictive of excessive bleeding at surgery, with patients at grade 3 and 4 having an odds ratio for excessive bleeding of 9.22 and 28, respectively, compared with patients at grade 0, he said.
Somewhat unexpectedly, the most serious bleeders based on a BARC of at least 2 or more at surgery were patients with Hermansky-Pudlak syndrome, Glanzmann thrombasthenia, and gray platelet syndrome. Those with primary secretion defect and combined alpha-delta granule deficiency had a relatively low risk of bleeding at surgery, he said.
The procedures associated with more excessive bleeding were urologic surgery, gynecologic surgery, and abdominal surgery.
Preparation for surgery was made in 82% of procedures and included prophylactic platelet transfusions in 38.5%, desmopressin (DDAVP) in about 19%, antifibrinolytic agents in about 12%, factor VIIa in less than 10%, and other interventions such as cryoprecipitate, fibrinogen, fresh frozen plasma, intravenous immunoglobulin, suture, and local hemostatic agent in less than 5%.
Generally all measures had a good ability to prevent excessive bleeding, bringing rates down to about 20% or less, Dr. Gresele said. The two exceptions were no prophylaxis and “other” interventions, where the bleeding rate reached a high of about 45% for the latter. In contrast, bleeding risk was significantly reduced when any prophylaxis (odds ratio, 0.38) or DDAVP (OR, 0.24) were used.
In 87 cases, however, emergency treatment for excessive bleeding was required, he said. Platelet transfusions were the most common treatment, given in roughly half of patients, followed by antifibrinolytic agents. The success rate was about 80% for all measures, except for “other” interventions, which had a success rate of about 65%.
“Treatment of excessive bleeding is made mostly with platelet transfusions or antifibrinolytic agents and is able to stop bleeding in many, but not all cases,” Dr. Gresele concluded.
On Twitter @pwendl
AT THE EHA CONGRESS
Key clinical point: Inherited platelet function disorders are associated with a significant surgical bleeding risk.
Major finding: The frequency of excessive bleeding ranged from about 13% to about 19%.
Data source: Retrospective, multicenter study in 205 patients with inherited platelet function disorders and 389 procedures.
Disclosures: Dr. Gesele reported no financial disclosures.

Gene therapy trial underway in hemophilia B
TORONTO – Researchers may be making some headway in the search for a gene therapy strategy to treat hemophilia B.
Preliminary findings from an ongoing phase 1-2 trial of an investigational gene therapy treatment in patients with hemophilia B were generally positive, based on results from the first seven patients.
Dr. Paul E. Monahan of the University of North Carolina Gene Therapy Center and Lineberger Comprehensive Cancer Center, Chapel Hill, reported that BAX 335 consists of a codon-optimized hyperactive FIX transgene driven by the liver-specific transthyretin (TTR) promoter in a nonintegrating adeno-associated viral (AAV) vector carrying a blood-clotting factor IX (FIX) gene.

The trial, the first in-human study of BAX 335, aims to evaluate the safety and kinetics of this gene therapy candidate. Dr. Monahan presented results from a combined follow-up of nearly 100 months on the first seven subjects, who were given one of three dosing schedules.
The adult male patients had hemophilia B and more than three hemorrhages per year requiring treatment with FIX, plasma FIX activity less than or equal to 2%, and a negative screen for hepatitis C. Patients with a family history of inhibitor to FIX protein or an allergic reaction to any FIX product were excluded, as were those with evidence of hepatic inflammation or cirrhosis.
The results for these seven subjects were generally positive. The BAX 335 doses were Cohort 1 (2 subjects): 1 x 1012 vg kg-1; Cohort 2 (3 subjects): 2 x 1012 vg kg-1; Cohort 3 (2 subjects): 3 x 1012 vg kg-1.
In the cohort given the lowest dose, some FIX expression was observed. In the intermediate dose cohort, two out of three study subjects remain free of spontaneous hemorrhage without regular infusions of FIX, and one of these patients has had sustained expression levels of 20% for 12 months. In the highest dose cohort, both subjects have achieved expression levels above 40%, but they also experienced an immune response with variable neutralizing antibody titers to the AAV8 capsid. One study participant has returned to regular FIX infusions.
Previous research efforts to use gene therapy to deliver DNA carrying the genetic sequence encoding FIX have been frustrated by the immune response that these therapies provoke. The overall findings to date “underscore the difficulty of thinking that we can easily interrupt an immune response once the process has begun but, thankfully, there have been no inhibitors observed against FIX or against the variant used in this vector,” Dr. Monahan concluded.
An additional five patients will be included in this study.
The trial is sponsored by Baxalta. Dr. Monahan disclosed that he has consulted for and received research support from Baxalta.
On Twitter @dougbrunk
TORONTO – Researchers may be making some headway in the search for a gene therapy strategy to treat hemophilia B.
Preliminary findings from an ongoing phase 1-2 trial of an investigational gene therapy treatment in patients with hemophilia B were generally positive, based on results from the first seven patients.
Dr. Paul E. Monahan of the University of North Carolina Gene Therapy Center and Lineberger Comprehensive Cancer Center, Chapel Hill, reported that BAX 335 consists of a codon-optimized hyperactive FIX transgene driven by the liver-specific transthyretin (TTR) promoter in a nonintegrating adeno-associated viral (AAV) vector carrying a blood-clotting factor IX (FIX) gene.
The trial, the first in-human study of BAX 335, aims to evaluate the safety and kinetics of this gene therapy candidate. Dr. Monahan presented results from a combined follow-up of nearly 100 months on the first seven subjects, who were given one of three dosing schedules.
The adult male patients had hemophilia B and more than three hemorrhages per year requiring treatment with FIX, plasma FIX activity less than or equal to 2%, and a negative screen for hepatitis C. Patients with a family history of inhibitor to FIX protein or an allergic reaction to any FIX product were excluded, as were those with evidence of hepatic inflammation or cirrhosis.
The results for these seven subjects were generally positive. The BAX 335 doses were Cohort 1 (2 subjects): 1 x 1012 vg kg-1; Cohort 2 (3 subjects): 2 x 1012 vg kg-1; Cohort 3 (2 subjects): 3 x 1012 vg kg-1.
In the cohort given the lowest dose, some FIX expression was observed. In the intermediate dose cohort, two out of three study subjects remain free of spontaneous hemorrhage without regular infusions of FIX, and one of these patients has had sustained expression levels of 20% for 12 months. In the highest dose cohort, both subjects have achieved expression levels above 40%, but they also experienced an immune response with variable neutralizing antibody titers to the AAV8 capsid. One study participant has returned to regular FIX infusions.
Previous research efforts to use gene therapy to deliver DNA carrying the genetic sequence encoding FIX have been frustrated by the immune response that these therapies provoke. The overall findings to date “underscore the difficulty of thinking that we can easily interrupt an immune response once the process has begun but, thankfully, there have been no inhibitors observed against FIX or against the variant used in this vector,” Dr. Monahan concluded.
An additional five patients will be included in this study.
The trial is sponsored by Baxalta. Dr. Monahan disclosed that he has consulted for and received research support from Baxalta.
On Twitter @dougbrunk
TORONTO – Researchers may be making some headway in the search for a gene therapy strategy to treat hemophilia B.
Preliminary findings from an ongoing phase 1-2 trial of an investigational gene therapy treatment in patients with hemophilia B were generally positive, based on results from the first seven patients.
Dr. Paul E. Monahan of the University of North Carolina Gene Therapy Center and Lineberger Comprehensive Cancer Center, Chapel Hill, reported that BAX 335 consists of a codon-optimized hyperactive FIX transgene driven by the liver-specific transthyretin (TTR) promoter in a nonintegrating adeno-associated viral (AAV) vector carrying a blood-clotting factor IX (FIX) gene.
The trial, the first in-human study of BAX 335, aims to evaluate the safety and kinetics of this gene therapy candidate. Dr. Monahan presented results from a combined follow-up of nearly 100 months on the first seven subjects, who were given one of three dosing schedules.
The adult male patients had hemophilia B and more than three hemorrhages per year requiring treatment with FIX, plasma FIX activity less than or equal to 2%, and a negative screen for hepatitis C. Patients with a family history of inhibitor to FIX protein or an allergic reaction to any FIX product were excluded, as were those with evidence of hepatic inflammation or cirrhosis.
The results for these seven subjects were generally positive. The BAX 335 doses were Cohort 1 (2 subjects): 1 x 1012 vg kg-1; Cohort 2 (3 subjects): 2 x 1012 vg kg-1; Cohort 3 (2 subjects): 3 x 1012 vg kg-1.
In the cohort given the lowest dose, some FIX expression was observed. In the intermediate dose cohort, two out of three study subjects remain free of spontaneous hemorrhage without regular infusions of FIX, and one of these patients has had sustained expression levels of 20% for 12 months. In the highest dose cohort, both subjects have achieved expression levels above 40%, but they also experienced an immune response with variable neutralizing antibody titers to the AAV8 capsid. One study participant has returned to regular FIX infusions.
Previous research efforts to use gene therapy to deliver DNA carrying the genetic sequence encoding FIX have been frustrated by the immune response that these therapies provoke. The overall findings to date “underscore the difficulty of thinking that we can easily interrupt an immune response once the process has begun but, thankfully, there have been no inhibitors observed against FIX or against the variant used in this vector,” Dr. Monahan concluded.
An additional five patients will be included in this study.
The trial is sponsored by Baxalta. Dr. Monahan disclosed that he has consulted for and received research support from Baxalta.
On Twitter @dougbrunk
AT 2015 ISTH
Key clinical point: BAX 335, a new gene therapy candidate, is now in a phase I-II trial in hemophilia B patients.
Major finding: In the intermediate-dose cohort, two out of three study subjects remain free of spontaneous hemorrhage without regular infusions of FIX, and one of these patients has had sustained expression levels of 20% for 12 months.
Data source: Pilot study of seven adults.
Disclosures: The trial is sponsored by Baxalta. Dr. Monahan disclosed that he has consulted for and received research support from Baxalta.

Study establishes protocol for perioperative dabigatran discontinuation
TORONTO – In atrial fibrillation (AF) patients who must discontinue dabigatran for elective surgery, the risk of both stroke and major bleeding can be reduced to low levels using a formalized strategy for stopping and then restarting anticoagulation, according to results of a prospective study presented at the International Society on Thrombosis and Haemostasis Congress.
Among key findings presented at a press conference at the ISTH 2015 Congress, no strokes were recorded in more than 500 patients managed with the protocol, and the major bleeding rate was less than 2%, reported the study’s principal investigator, Dr. Sam Schulman, professor of hematology and thromboembolism, McMaster University, Hamilton, Ont.
Data from this study (Circulation 2015) were reported at the press conference alongside a second study of perioperative warfarin management. Both studies are potentially practice changing, because they supply evidence-based guidance for anticoagulation in patients with AF.

Based on the findings from these two studies, “it is important to get this message out” that there are now data available on which to base clinical decisions, reported Dr. Schulman, who is also president of the ISTH 2015 Congress. His data were presented alongside a study that found no benefit from heparin bridging in AF patients when warfarin was stopped 5 days in advance of surgery.
In the study presented by Dr. Schulman, 542 patients with AF who were on dabigatran and scheduled for elective surgery were managed on a prespecified protocol for risk assessment. The protocol provided a time for stopping dabigatran before surgery based on such factors as renal function and procedure-related bleeding risk. Dabigatran was restarted after surgery on prespecified measures of surgery complexity and severity of consequences if bleeding occurred.
The primary outcome evaluated in the study was major bleeding in the first 30 days. Other outcomes of interest included thromboembolic complications, death and minor bleeding.
Major bleeding was observed in 1.8% of patients, a rate that Dr. Schulman characterized as “low and acceptable” in the context of expected background bleeding rates. There were four deaths, but all were unrelated to either bleeding or arterial thromboembolism. The only thromboembolic complication was a single transient ischemic attack. Minor bleeding occurred in 5.2%.
On the basis of the protocol, about half of the patients discontinued dabigatran 24 hours before surgery. No patient discontinued therapy more than 96 hours prior to surgery. The median time to resumption of dabigatran after surgery was 1 day, but the point at which it was restarted ranged between hours and 2 days. Bridging, which describes the injection of heparin for short-term anticoagulation, was not employed preoperatively but was used in 1.7% of cases postoperatively.
At the press conference, data also were reported from the BRIDGE study. That study, published online in the New England Journal of Medicine (2015 June 22; epub ahead of print ), found that bridging was not an effective strategy in AF patients who discontinue warfarin prior to elective surgery. In the press conference, Dr. Thomas L. Ortel, hematology/oncology division, Duke University Medical Center, Durham, N.C., agreed with Dr. Schulman that this is an area where evidence is needed to guide care.
In the absence of data, “physicians do whatever they think is best,” Dr. Schulman noted at the press conference. Referring to strategies for stopping anticoagulants for surgery in patients with AF, Dr. Schulman said, “some of them stop the blood thinner too early because they are afraid that the patient is going to bleed during surgery and instead the patient can have a stroke. Some stop too late, and the patient can have bleeding.”
The data presented at the meeting provide an evidence base for clinical decisions. Dr. Schulman suggested that these data are meaningful for guiding care.
Dr. Ortel disclosed grant/research support from Eisai and Pfizer. Dr. Schulman had no disclosures.
TORONTO – In atrial fibrillation (AF) patients who must discontinue dabigatran for elective surgery, the risk of both stroke and major bleeding can be reduced to low levels using a formalized strategy for stopping and then restarting anticoagulation, according to results of a prospective study presented at the International Society on Thrombosis and Haemostasis Congress.
Among key findings presented at a press conference at the ISTH 2015 Congress, no strokes were recorded in more than 500 patients managed with the protocol, and the major bleeding rate was less than 2%, reported the study’s principal investigator, Dr. Sam Schulman, professor of hematology and thromboembolism, McMaster University, Hamilton, Ont.
Data from this study (Circulation 2015) were reported at the press conference alongside a second study of perioperative warfarin management. Both studies are potentially practice changing, because they supply evidence-based guidance for anticoagulation in patients with AF.
Based on the findings from these two studies, “it is important to get this message out” that there are now data available on which to base clinical decisions, reported Dr. Schulman, who is also president of the ISTH 2015 Congress. His data were presented alongside a study that found no benefit from heparin bridging in AF patients when warfarin was stopped 5 days in advance of surgery.
In the study presented by Dr. Schulman, 542 patients with AF who were on dabigatran and scheduled for elective surgery were managed on a prespecified protocol for risk assessment. The protocol provided a time for stopping dabigatran before surgery based on such factors as renal function and procedure-related bleeding risk. Dabigatran was restarted after surgery on prespecified measures of surgery complexity and severity of consequences if bleeding occurred.
The primary outcome evaluated in the study was major bleeding in the first 30 days. Other outcomes of interest included thromboembolic complications, death and minor bleeding.
Major bleeding was observed in 1.8% of patients, a rate that Dr. Schulman characterized as “low and acceptable” in the context of expected background bleeding rates. There were four deaths, but all were unrelated to either bleeding or arterial thromboembolism. The only thromboembolic complication was a single transient ischemic attack. Minor bleeding occurred in 5.2%.
On the basis of the protocol, about half of the patients discontinued dabigatran 24 hours before surgery. No patient discontinued therapy more than 96 hours prior to surgery. The median time to resumption of dabigatran after surgery was 1 day, but the point at which it was restarted ranged between hours and 2 days. Bridging, which describes the injection of heparin for short-term anticoagulation, was not employed preoperatively but was used in 1.7% of cases postoperatively.
At the press conference, data also were reported from the BRIDGE study. That study, published online in the New England Journal of Medicine (2015 June 22; epub ahead of print ), found that bridging was not an effective strategy in AF patients who discontinue warfarin prior to elective surgery. In the press conference, Dr. Thomas L. Ortel, hematology/oncology division, Duke University Medical Center, Durham, N.C., agreed with Dr. Schulman that this is an area where evidence is needed to guide care.
In the absence of data, “physicians do whatever they think is best,” Dr. Schulman noted at the press conference. Referring to strategies for stopping anticoagulants for surgery in patients with AF, Dr. Schulman said, “some of them stop the blood thinner too early because they are afraid that the patient is going to bleed during surgery and instead the patient can have a stroke. Some stop too late, and the patient can have bleeding.”
The data presented at the meeting provide an evidence base for clinical decisions. Dr. Schulman suggested that these data are meaningful for guiding care.
Dr. Ortel disclosed grant/research support from Eisai and Pfizer. Dr. Schulman had no disclosures.
TORONTO – In atrial fibrillation (AF) patients who must discontinue dabigatran for elective surgery, the risk of both stroke and major bleeding can be reduced to low levels using a formalized strategy for stopping and then restarting anticoagulation, according to results of a prospective study presented at the International Society on Thrombosis and Haemostasis Congress.
Among key findings presented at a press conference at the ISTH 2015 Congress, no strokes were recorded in more than 500 patients managed with the protocol, and the major bleeding rate was less than 2%, reported the study’s principal investigator, Dr. Sam Schulman, professor of hematology and thromboembolism, McMaster University, Hamilton, Ont.
Data from this study (Circulation 2015) were reported at the press conference alongside a second study of perioperative warfarin management. Both studies are potentially practice changing, because they supply evidence-based guidance for anticoagulation in patients with AF.
Based on the findings from these two studies, “it is important to get this message out” that there are now data available on which to base clinical decisions, reported Dr. Schulman, who is also president of the ISTH 2015 Congress. His data were presented alongside a study that found no benefit from heparin bridging in AF patients when warfarin was stopped 5 days in advance of surgery.
In the study presented by Dr. Schulman, 542 patients with AF who were on dabigatran and scheduled for elective surgery were managed on a prespecified protocol for risk assessment. The protocol provided a time for stopping dabigatran before surgery based on such factors as renal function and procedure-related bleeding risk. Dabigatran was restarted after surgery on prespecified measures of surgery complexity and severity of consequences if bleeding occurred.
The primary outcome evaluated in the study was major bleeding in the first 30 days. Other outcomes of interest included thromboembolic complications, death and minor bleeding.
Major bleeding was observed in 1.8% of patients, a rate that Dr. Schulman characterized as “low and acceptable” in the context of expected background bleeding rates. There were four deaths, but all were unrelated to either bleeding or arterial thromboembolism. The only thromboembolic complication was a single transient ischemic attack. Minor bleeding occurred in 5.2%.
On the basis of the protocol, about half of the patients discontinued dabigatran 24 hours before surgery. No patient discontinued therapy more than 96 hours prior to surgery. The median time to resumption of dabigatran after surgery was 1 day, but the point at which it was restarted ranged between hours and 2 days. Bridging, which describes the injection of heparin for short-term anticoagulation, was not employed preoperatively but was used in 1.7% of cases postoperatively.
At the press conference, data also were reported from the BRIDGE study. That study, published online in the New England Journal of Medicine (2015 June 22; epub ahead of print ), found that bridging was not an effective strategy in AF patients who discontinue warfarin prior to elective surgery. In the press conference, Dr. Thomas L. Ortel, hematology/oncology division, Duke University Medical Center, Durham, N.C., agreed with Dr. Schulman that this is an area where evidence is needed to guide care.
In the absence of data, “physicians do whatever they think is best,” Dr. Schulman noted at the press conference. Referring to strategies for stopping anticoagulants for surgery in patients with AF, Dr. Schulman said, “some of them stop the blood thinner too early because they are afraid that the patient is going to bleed during surgery and instead the patient can have a stroke. Some stop too late, and the patient can have bleeding.”
The data presented at the meeting provide an evidence base for clinical decisions. Dr. Schulman suggested that these data are meaningful for guiding care.
Dr. Ortel disclosed grant/research support from Eisai and Pfizer. Dr. Schulman had no disclosures.
AT 2015 ISTH CONGRESS
Key clinical point: The risk of stroke and major bleeding can be reduced to low levels using a formalized strategy for stopping and then restarting dabigatran.
Major finding: The protocol developed provided a time for stopping dabigatran before surgery based on such factors as renal function and procedure-related bleeding risk. Dabigatran was restarted after surgery on prespecified measures of surgery complexity and severity of consequences if bleeding occurred.
Data source: 542 patients with AF who were on dabigatran and scheduled for elective surgery were managed on a prespecified protocol for risk assessment.
Disclosures: Dr. Ortel disclosed grant/research support from Eisai and Pfizer. Dr. Schulman had no disclosures.
