User login
Prenatal Antibiotics May Increase Seborrheic Dermatitis Risk in Babies
, but this association was not as strong for childhood-onset SD.
The findings come from a large analysis of data from the United Kingdom that was presented during a late-breaking abstract session at the annual meeting of the Society for Investigative Dermatology.
SD is a common skin disease “that shares similarities with atopic dermatitis or atopic eczema as both are prevalent inflammatory skin diseases that can present with a chronic relapsing, remitting course,” the study’s corresponding author Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, said in an interview. “Like atopic dermatitis, the pathophysiology of seborrheic dermatitis is thought to be complex and involves an interplay between genetics, immune dysregulation, and alterations in lipid composition and the skin microbiome, among others.”

In a previous study, she and colleagues showed that exposure to antibiotics both in utero and during the first 90 days of life increases the risk for atopic dermatitis (AD) in children, with risk being highest with exposure to penicillin even among children whose mothers did not have a history of AD.
For the current study, the researchers drew from a large electronic medical records database in the United Kingdom to perform a prospective cohort analysis of mother-child pairs that used proportional hazards models to examine the association between maternal in utero antibiotic exposure and SD in the child. The population included 1,023,140 children with linked maternal data who were followed for a mean of 10.2 years, which amounts to more than 10-million-person years of data. At baseline, the mean age of mothers was 28 years, 3% had SD, 14% had AD, and 51% of the children were male.
In unadjusted analyses, mothers with SD were more likely to receive an antibiotic during pregnancy than were those who did not have SD (odds ratio [OR], 1.42; 95% CI, 1.39-1.46). In addition, maternal in utero exposure to any antibiotic was associated with an increased risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) but less for childhood-onset SD (OR, 1.26; 95% CI, 1.20-1.32). “This effect changed little after adjustment and was still observed if mothers with SD and their babies were excluded,” the authors wrote in their poster abstract.
Any penicillin exposure during pregnancy increased the likelihood of a child having SD (OR, 1.54; 95% CI, 1.50-1.59), with the greater risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) than for childhood-onset SD (OR, 1.25; 95% CI, 1.18-1.32). “The trimester of the in utero penicillin exposure did not seem to affect the association with SD,” the authors wrote. The risk was also increased with cephalosporin exposure but was less for sulfonamides and not for childhood-onset SD.
“We observed that antibiotic exposure in utero was primarily associated with an increased risk of infantile SD regardless of the mother’s history of SD, but this association was not as strong for childhood-onset SD,” Dr. Chiesa Fuxench said. “This would suggest that in utero exposure to antibiotics, particularly penicillin, may have its greatest effect on the colonization of skin microbiota in the newborn period leading to the development of infantile SD. Aside from seeking to improve our understanding of the pathophysiology of SD, our findings also suggest that infantile SD and childhood-onset SD may be separate entities with different risk factors, a hypothesis that needs to be further studied.”
She acknowledged certain limitations of the analysis, including the potential for unrecorded diagnoses of SD or misclassified cases in the database. For example, AD and psoriasis “may appear clinically like SD,” she said, although they performed sensitivity analysis excluding patients with these diagnoses and found similar results. In addition, there is the possibility that not all antibiotic exposures were captured in this database, and data on antibiotic exposure may be missing, she added.
Dr. Chiesa Fuxench disclosed that she received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, Vanda, and Incyte for work related to AD and from Menlo Therapeutics and Galderma for work related to prurigo nodularis. She has served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer and from Beiersdorf for work related to skin cancer and sun protection.
A version of this article appeared on Medscape.com .
, but this association was not as strong for childhood-onset SD.
The findings come from a large analysis of data from the United Kingdom that was presented during a late-breaking abstract session at the annual meeting of the Society for Investigative Dermatology.
SD is a common skin disease “that shares similarities with atopic dermatitis or atopic eczema as both are prevalent inflammatory skin diseases that can present with a chronic relapsing, remitting course,” the study’s corresponding author Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, said in an interview. “Like atopic dermatitis, the pathophysiology of seborrheic dermatitis is thought to be complex and involves an interplay between genetics, immune dysregulation, and alterations in lipid composition and the skin microbiome, among others.”
In a previous study, she and colleagues showed that exposure to antibiotics both in utero and during the first 90 days of life increases the risk for atopic dermatitis (AD) in children, with risk being highest with exposure to penicillin even among children whose mothers did not have a history of AD.
For the current study, the researchers drew from a large electronic medical records database in the United Kingdom to perform a prospective cohort analysis of mother-child pairs that used proportional hazards models to examine the association between maternal in utero antibiotic exposure and SD in the child. The population included 1,023,140 children with linked maternal data who were followed for a mean of 10.2 years, which amounts to more than 10-million-person years of data. At baseline, the mean age of mothers was 28 years, 3% had SD, 14% had AD, and 51% of the children were male.
In unadjusted analyses, mothers with SD were more likely to receive an antibiotic during pregnancy than were those who did not have SD (odds ratio [OR], 1.42; 95% CI, 1.39-1.46). In addition, maternal in utero exposure to any antibiotic was associated with an increased risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) but less for childhood-onset SD (OR, 1.26; 95% CI, 1.20-1.32). “This effect changed little after adjustment and was still observed if mothers with SD and their babies were excluded,” the authors wrote in their poster abstract.
Any penicillin exposure during pregnancy increased the likelihood of a child having SD (OR, 1.54; 95% CI, 1.50-1.59), with the greater risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) than for childhood-onset SD (OR, 1.25; 95% CI, 1.18-1.32). “The trimester of the in utero penicillin exposure did not seem to affect the association with SD,” the authors wrote. The risk was also increased with cephalosporin exposure but was less for sulfonamides and not for childhood-onset SD.
“We observed that antibiotic exposure in utero was primarily associated with an increased risk of infantile SD regardless of the mother’s history of SD, but this association was not as strong for childhood-onset SD,” Dr. Chiesa Fuxench said. “This would suggest that in utero exposure to antibiotics, particularly penicillin, may have its greatest effect on the colonization of skin microbiota in the newborn period leading to the development of infantile SD. Aside from seeking to improve our understanding of the pathophysiology of SD, our findings also suggest that infantile SD and childhood-onset SD may be separate entities with different risk factors, a hypothesis that needs to be further studied.”
She acknowledged certain limitations of the analysis, including the potential for unrecorded diagnoses of SD or misclassified cases in the database. For example, AD and psoriasis “may appear clinically like SD,” she said, although they performed sensitivity analysis excluding patients with these diagnoses and found similar results. In addition, there is the possibility that not all antibiotic exposures were captured in this database, and data on antibiotic exposure may be missing, she added.
Dr. Chiesa Fuxench disclosed that she received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, Vanda, and Incyte for work related to AD and from Menlo Therapeutics and Galderma for work related to prurigo nodularis. She has served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer and from Beiersdorf for work related to skin cancer and sun protection.
A version of this article appeared on Medscape.com .
, but this association was not as strong for childhood-onset SD.
The findings come from a large analysis of data from the United Kingdom that was presented during a late-breaking abstract session at the annual meeting of the Society for Investigative Dermatology.
SD is a common skin disease “that shares similarities with atopic dermatitis or atopic eczema as both are prevalent inflammatory skin diseases that can present with a chronic relapsing, remitting course,” the study’s corresponding author Zelma C. Chiesa Fuxench, MD, MSCE, assistant professor of dermatology at the University of Pennsylvania, Philadelphia, said in an interview. “Like atopic dermatitis, the pathophysiology of seborrheic dermatitis is thought to be complex and involves an interplay between genetics, immune dysregulation, and alterations in lipid composition and the skin microbiome, among others.”
In a previous study, she and colleagues showed that exposure to antibiotics both in utero and during the first 90 days of life increases the risk for atopic dermatitis (AD) in children, with risk being highest with exposure to penicillin even among children whose mothers did not have a history of AD.
For the current study, the researchers drew from a large electronic medical records database in the United Kingdom to perform a prospective cohort analysis of mother-child pairs that used proportional hazards models to examine the association between maternal in utero antibiotic exposure and SD in the child. The population included 1,023,140 children with linked maternal data who were followed for a mean of 10.2 years, which amounts to more than 10-million-person years of data. At baseline, the mean age of mothers was 28 years, 3% had SD, 14% had AD, and 51% of the children were male.
In unadjusted analyses, mothers with SD were more likely to receive an antibiotic during pregnancy than were those who did not have SD (odds ratio [OR], 1.42; 95% CI, 1.39-1.46). In addition, maternal in utero exposure to any antibiotic was associated with an increased risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) but less for childhood-onset SD (OR, 1.26; 95% CI, 1.20-1.32). “This effect changed little after adjustment and was still observed if mothers with SD and their babies were excluded,” the authors wrote in their poster abstract.
Any penicillin exposure during pregnancy increased the likelihood of a child having SD (OR, 1.54; 95% CI, 1.50-1.59), with the greater risk for infantile SD (OR, 1.70; 95% CI, 1.65-1.76) than for childhood-onset SD (OR, 1.25; 95% CI, 1.18-1.32). “The trimester of the in utero penicillin exposure did not seem to affect the association with SD,” the authors wrote. The risk was also increased with cephalosporin exposure but was less for sulfonamides and not for childhood-onset SD.
“We observed that antibiotic exposure in utero was primarily associated with an increased risk of infantile SD regardless of the mother’s history of SD, but this association was not as strong for childhood-onset SD,” Dr. Chiesa Fuxench said. “This would suggest that in utero exposure to antibiotics, particularly penicillin, may have its greatest effect on the colonization of skin microbiota in the newborn period leading to the development of infantile SD. Aside from seeking to improve our understanding of the pathophysiology of SD, our findings also suggest that infantile SD and childhood-onset SD may be separate entities with different risk factors, a hypothesis that needs to be further studied.”
She acknowledged certain limitations of the analysis, including the potential for unrecorded diagnoses of SD or misclassified cases in the database. For example, AD and psoriasis “may appear clinically like SD,” she said, although they performed sensitivity analysis excluding patients with these diagnoses and found similar results. In addition, there is the possibility that not all antibiotic exposures were captured in this database, and data on antibiotic exposure may be missing, she added.
Dr. Chiesa Fuxench disclosed that she received research grants from Lilly, LEO Pharma, Regeneron, Sanofi, Tioga, Vanda, and Incyte for work related to AD and from Menlo Therapeutics and Galderma for work related to prurigo nodularis. She has served as a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, AbbVie, Incyte Corporation, and Pfizer and received honoraria for CME work in AD sponsored by education grants from Regeneron/Sanofi and Pfizer and from Beiersdorf for work related to skin cancer and sun protection.
A version of this article appeared on Medscape.com .
FROM SID 2024
Frontal Fibrosing Alopecia: Study Finds Oral Contraceptive Use Modulates Risk In Women with Genetic Variant
TOPLINE:
Investigators found that .
METHODOLOGY:
- OC use has been considered a possible factor behind the increased incidence of FFA because it was first documented in 1994, and a recent genome-wide association study of FFA identified a signal for an association with a variant in CYP1B1.
- The same researchers conducted a gene-environment interaction study with a case-control design involving 489 White female patients (mean age, 65.8 years) with FFA and 34,254 controls, matched for age and genetic ancestry.
- Data were collected from July 2015 to September 2017 and analyzed from October 2022 to December 2023.
- The study aimed to investigate the modulatory effect of OC use on the CYP1B1 variant’s impact on FFA risk, using logistic regression models for analysis.
TAKEAWAY:
- The use of OCs was associated with a 1.9 times greater risk for FFA in individuals with the specific CYP1B1 genetic variant, but there was no association among those with no history of OC use.
- The study suggests a significant gene-environment interaction, indicating that OC use may influence FFA risk in genetically predisposed individuals.
IN PRACTICE:
“This gene-environment interaction analysis suggests that the protective effect of the CYPIB1 missense variant on FFA risk might be mediated by exposure” to OCs, the authors wrote. The study, they added, “underscores the importance of considering genetic predispositions and environmental factors, such as oral contraceptive use, in understanding and managing frontal fibrosing alopecia.”
SOURCE:
Tuntas Rayinda, MD, MSc, PhD, of St. John’s Institute of Dermatology, King’s College London, led the study, which was published online May 29, 2024, in JAMA Dermatology.
LIMITATIONS:
The study’s reliance on self-reported OC use may have introduced recall and differences in ascertainment of OC use between patient and control groups and could have affected the study’s findings. The study also did not collect information on the type of OC used, which could have influenced the observed interaction.
DISCLOSURES:
The study was supported by the British Skin Foundation Young Investigator Award. One investigator reported being a subinvestigator on an alopecia areata study funded by Pfizer. No other disclosures were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com.
TOPLINE:
Investigators found that .
METHODOLOGY:
- OC use has been considered a possible factor behind the increased incidence of FFA because it was first documented in 1994, and a recent genome-wide association study of FFA identified a signal for an association with a variant in CYP1B1.
- The same researchers conducted a gene-environment interaction study with a case-control design involving 489 White female patients (mean age, 65.8 years) with FFA and 34,254 controls, matched for age and genetic ancestry.
- Data were collected from July 2015 to September 2017 and analyzed from October 2022 to December 2023.
- The study aimed to investigate the modulatory effect of OC use on the CYP1B1 variant’s impact on FFA risk, using logistic regression models for analysis.
TAKEAWAY:
- The use of OCs was associated with a 1.9 times greater risk for FFA in individuals with the specific CYP1B1 genetic variant, but there was no association among those with no history of OC use.
- The study suggests a significant gene-environment interaction, indicating that OC use may influence FFA risk in genetically predisposed individuals.
IN PRACTICE:
“This gene-environment interaction analysis suggests that the protective effect of the CYPIB1 missense variant on FFA risk might be mediated by exposure” to OCs, the authors wrote. The study, they added, “underscores the importance of considering genetic predispositions and environmental factors, such as oral contraceptive use, in understanding and managing frontal fibrosing alopecia.”
SOURCE:
Tuntas Rayinda, MD, MSc, PhD, of St. John’s Institute of Dermatology, King’s College London, led the study, which was published online May 29, 2024, in JAMA Dermatology.
LIMITATIONS:
The study’s reliance on self-reported OC use may have introduced recall and differences in ascertainment of OC use between patient and control groups and could have affected the study’s findings. The study also did not collect information on the type of OC used, which could have influenced the observed interaction.
DISCLOSURES:
The study was supported by the British Skin Foundation Young Investigator Award. One investigator reported being a subinvestigator on an alopecia areata study funded by Pfizer. No other disclosures were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com.
TOPLINE:
Investigators found that .
METHODOLOGY:
- OC use has been considered a possible factor behind the increased incidence of FFA because it was first documented in 1994, and a recent genome-wide association study of FFA identified a signal for an association with a variant in CYP1B1.
- The same researchers conducted a gene-environment interaction study with a case-control design involving 489 White female patients (mean age, 65.8 years) with FFA and 34,254 controls, matched for age and genetic ancestry.
- Data were collected from July 2015 to September 2017 and analyzed from October 2022 to December 2023.
- The study aimed to investigate the modulatory effect of OC use on the CYP1B1 variant’s impact on FFA risk, using logistic regression models for analysis.
TAKEAWAY:
- The use of OCs was associated with a 1.9 times greater risk for FFA in individuals with the specific CYP1B1 genetic variant, but there was no association among those with no history of OC use.
- The study suggests a significant gene-environment interaction, indicating that OC use may influence FFA risk in genetically predisposed individuals.
IN PRACTICE:
“This gene-environment interaction analysis suggests that the protective effect of the CYPIB1 missense variant on FFA risk might be mediated by exposure” to OCs, the authors wrote. The study, they added, “underscores the importance of considering genetic predispositions and environmental factors, such as oral contraceptive use, in understanding and managing frontal fibrosing alopecia.”
SOURCE:
Tuntas Rayinda, MD, MSc, PhD, of St. John’s Institute of Dermatology, King’s College London, led the study, which was published online May 29, 2024, in JAMA Dermatology.
LIMITATIONS:
The study’s reliance on self-reported OC use may have introduced recall and differences in ascertainment of OC use between patient and control groups and could have affected the study’s findings. The study also did not collect information on the type of OC used, which could have influenced the observed interaction.
DISCLOSURES:
The study was supported by the British Skin Foundation Young Investigator Award. One investigator reported being a subinvestigator on an alopecia areata study funded by Pfizer. No other disclosures were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com.
Plantar Hyperpigmentation
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyper-pigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key Clinical Features in Individuals With Darker Skin Tones
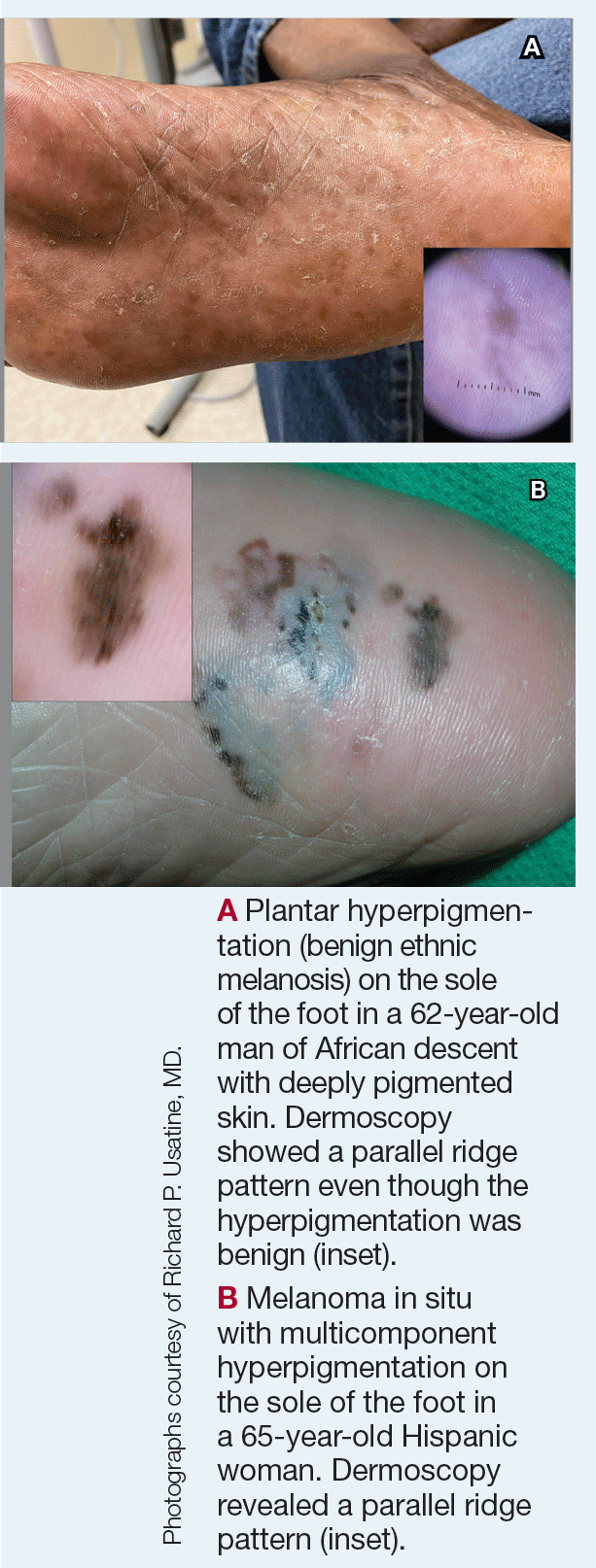
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
• Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
• Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
• Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
1. Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
2. Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
3. Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
4. Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
5. Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
6. Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
7. Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
8. Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
9. Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285. doi:10.12788/cutis.0513.
10. Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
11. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
12. Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
13. Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
14. Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
15. Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dematol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
16. Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
17. Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyper-pigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key Clinical Features in Individuals With Darker Skin Tones
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
• Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
• Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
• Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyper-pigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key Clinical Features in Individuals With Darker Skin Tones
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
• Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
• Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
• Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
1. Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
2. Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
3. Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
4. Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
5. Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
6. Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
7. Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
8. Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
9. Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285. doi:10.12788/cutis.0513.
10. Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
11. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
12. Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
13. Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
14. Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
15. Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dematol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
16. Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
17. Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
1. Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
2. Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
3. Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
4. Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
5. Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
6. Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
7. Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
8. Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
9. Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285. doi:10.12788/cutis.0513.
10. Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
11. Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
12. Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
13. Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
14. Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
15. Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dematol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
16. Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
17. Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
EASI, Other Instruments Recommended to Evaluate Patients With Atopic Dermatitis
recommended.
These include the Eczema Area and Severity Index (EASI), the Validated Investigator Global Assessment for AD (vIGAAD), and the Investigator’s Global Assessment (IGA) multiplied by or measured concurrently with a body surface area (BSA) assessment.
The recommendations are part of a consensus statement based on an updated systematic review conducted by the Harmonizing Outcome Measures for Eczema Clinical Practice (HOME-CP) initiative, whose goal is to identify validated, feasible outcome instruments designed to measure AD in the clinical setting. In the statement, which was published in JAMA Dermatology on May 22, 2024, corresponding author Eric L. Simpson, MD, MCR, professor of dermatology at Oregon Health & Science University, Portland, and coauthors described HOME-CP as “a ‘pick-and-choose’ list of valid and feasible OMIs [outcome measure instruments] that can be incorporated into the practice setting depending on the particular need of that clinic or health system.”
For the effort, the authors implemented a mixed methods design and incorporated systematic reviews and qualitative consensus methods modeled after the HOME core outcome set initiative, which developed a set of consensus-based core outcome sets for clinical trials and clinical practice. In October of 2022, a daylong in-person consensus exercise was held in Montreal, Canada, where attendees met to reach consensus on recommended instruments to measure AD clinical signs in clinical practice, based on an updated systematic review evaluating the validity of clinical signs instruments.
The review included 22 studies describing 16 instruments that assessed AD clinical signs and an additional 12 variants of instruments. The meeting was attended by 34 individuals from 13 countries, including patient and patient advocate research partners, health care professionals, researchers, methodologists, and industry representatives. Consensus was defined as less than 30% disagreement.
Following their daylong consensus exercise, the stakeholders reached consensus on recommendations to use the EASI, the vIGAAD, and an IGA multiplied or measured alongside a BSA measurement to measure the domain of clinical signs of AD in the clinical practice setting. “The use of multiple IGAs, most with insufficient validation, and the diverse methods used to assess BSA prevented participants from making specific recommendations for the exact IGA/BSA instrument,” the authors wrote. “We recommend that clinicians include at least one of the recommended instruments in their clinical practices and in documentation.”
They explained that the ideal method of measuring BSA was difficult to assess “because multiple techniques exist for its measurement, including regional percentages, the Rule of Nines, or the handprint method. Most studies did not report which method was performed, and to our knowledge, no studies have been performed in patients with AD that have formally compared them.”
During the consensus exercise, the authors noted, several clinicians “expressed concern whether the EASI was feasible for universal use in clinical practice given its complexity, long completion time, and documentation/calculation requirements.” But clinicians who commonly perform the EASI in clinical practice said that the time it takes to complete this measure “has dropped substantially and now is not a considerable burden,” they wrote, adding that, “studies have shown that with trained investigators, EASI completion times can be as low as nearly 2 minutes.”
The authors acknowledged certain limitations of their recommendations, including the lack of input from primary care clinicians. “It is unknown whether ClinROMs [clinician-reported outcome measures] for AD clinical signs are used in the primary care setting, especially given the large amount of conditions that are managed simultaneously and the ever-increasing number of primary care documentation requirements,” they wrote.
Robert Sidbury, MD, MPH, chief of the division of dermatology at Seattle Children’s Hospital, who was asked to comment on the consensus statement, said that with the advent of new, improved, and more expensive medications for AD, “it is ever more important that [the clinical] assessment is reliable and reproducible.”
Insurers “are understandably less willing to rubber-stamp approval of more expensive medications without a reliable standard by which to justify such decisions,” he added. “This is even more important in a disease state like atopic dermatitis that lacks a reliable biomarker. Therefore, one or several practical, reliable, validated severity metrics will help standardize and improve AD care.”
Dr. Sidbury, who cochaired the 2023 American Academy of Dermatology guidelines of care for the management of AD in adults with phototherapy and systemic therapies, added that the instruments evaluated in the review “can be challenging for anyone,” not just primary care providers. “The EASI isn’t that easy, and while there is a learning curve and it ultimately does, like anything, become more efficient in the gathering, it is unclear if non-AD researchers will be willing to invest the time” to routinely use it, he said.
Dr. Simpson and several coauthors reported receiving grants and personal fees from multiple pharmaceutical companies. Dr. Sidbury reported that he serves as an investigator for Regeneron, Galderma, UCB, Castle, and Pfizer; is a consultant for LEO, Lilly, Arcutis, Dermavant, and Pierre Fabre; and a speaker for Beiersdorf.
A version of this article appeared on Medscape.com .
recommended.
These include the Eczema Area and Severity Index (EASI), the Validated Investigator Global Assessment for AD (vIGAAD), and the Investigator’s Global Assessment (IGA) multiplied by or measured concurrently with a body surface area (BSA) assessment.
The recommendations are part of a consensus statement based on an updated systematic review conducted by the Harmonizing Outcome Measures for Eczema Clinical Practice (HOME-CP) initiative, whose goal is to identify validated, feasible outcome instruments designed to measure AD in the clinical setting. In the statement, which was published in JAMA Dermatology on May 22, 2024, corresponding author Eric L. Simpson, MD, MCR, professor of dermatology at Oregon Health & Science University, Portland, and coauthors described HOME-CP as “a ‘pick-and-choose’ list of valid and feasible OMIs [outcome measure instruments] that can be incorporated into the practice setting depending on the particular need of that clinic or health system.”
For the effort, the authors implemented a mixed methods design and incorporated systematic reviews and qualitative consensus methods modeled after the HOME core outcome set initiative, which developed a set of consensus-based core outcome sets for clinical trials and clinical practice. In October of 2022, a daylong in-person consensus exercise was held in Montreal, Canada, where attendees met to reach consensus on recommended instruments to measure AD clinical signs in clinical practice, based on an updated systematic review evaluating the validity of clinical signs instruments.
The review included 22 studies describing 16 instruments that assessed AD clinical signs and an additional 12 variants of instruments. The meeting was attended by 34 individuals from 13 countries, including patient and patient advocate research partners, health care professionals, researchers, methodologists, and industry representatives. Consensus was defined as less than 30% disagreement.
Following their daylong consensus exercise, the stakeholders reached consensus on recommendations to use the EASI, the vIGAAD, and an IGA multiplied or measured alongside a BSA measurement to measure the domain of clinical signs of AD in the clinical practice setting. “The use of multiple IGAs, most with insufficient validation, and the diverse methods used to assess BSA prevented participants from making specific recommendations for the exact IGA/BSA instrument,” the authors wrote. “We recommend that clinicians include at least one of the recommended instruments in their clinical practices and in documentation.”
They explained that the ideal method of measuring BSA was difficult to assess “because multiple techniques exist for its measurement, including regional percentages, the Rule of Nines, or the handprint method. Most studies did not report which method was performed, and to our knowledge, no studies have been performed in patients with AD that have formally compared them.”
During the consensus exercise, the authors noted, several clinicians “expressed concern whether the EASI was feasible for universal use in clinical practice given its complexity, long completion time, and documentation/calculation requirements.” But clinicians who commonly perform the EASI in clinical practice said that the time it takes to complete this measure “has dropped substantially and now is not a considerable burden,” they wrote, adding that, “studies have shown that with trained investigators, EASI completion times can be as low as nearly 2 minutes.”
The authors acknowledged certain limitations of their recommendations, including the lack of input from primary care clinicians. “It is unknown whether ClinROMs [clinician-reported outcome measures] for AD clinical signs are used in the primary care setting, especially given the large amount of conditions that are managed simultaneously and the ever-increasing number of primary care documentation requirements,” they wrote.
Robert Sidbury, MD, MPH, chief of the division of dermatology at Seattle Children’s Hospital, who was asked to comment on the consensus statement, said that with the advent of new, improved, and more expensive medications for AD, “it is ever more important that [the clinical] assessment is reliable and reproducible.”
Insurers “are understandably less willing to rubber-stamp approval of more expensive medications without a reliable standard by which to justify such decisions,” he added. “This is even more important in a disease state like atopic dermatitis that lacks a reliable biomarker. Therefore, one or several practical, reliable, validated severity metrics will help standardize and improve AD care.”
Dr. Sidbury, who cochaired the 2023 American Academy of Dermatology guidelines of care for the management of AD in adults with phototherapy and systemic therapies, added that the instruments evaluated in the review “can be challenging for anyone,” not just primary care providers. “The EASI isn’t that easy, and while there is a learning curve and it ultimately does, like anything, become more efficient in the gathering, it is unclear if non-AD researchers will be willing to invest the time” to routinely use it, he said.
Dr. Simpson and several coauthors reported receiving grants and personal fees from multiple pharmaceutical companies. Dr. Sidbury reported that he serves as an investigator for Regeneron, Galderma, UCB, Castle, and Pfizer; is a consultant for LEO, Lilly, Arcutis, Dermavant, and Pierre Fabre; and a speaker for Beiersdorf.
A version of this article appeared on Medscape.com .
recommended.
These include the Eczema Area and Severity Index (EASI), the Validated Investigator Global Assessment for AD (vIGAAD), and the Investigator’s Global Assessment (IGA) multiplied by or measured concurrently with a body surface area (BSA) assessment.
The recommendations are part of a consensus statement based on an updated systematic review conducted by the Harmonizing Outcome Measures for Eczema Clinical Practice (HOME-CP) initiative, whose goal is to identify validated, feasible outcome instruments designed to measure AD in the clinical setting. In the statement, which was published in JAMA Dermatology on May 22, 2024, corresponding author Eric L. Simpson, MD, MCR, professor of dermatology at Oregon Health & Science University, Portland, and coauthors described HOME-CP as “a ‘pick-and-choose’ list of valid and feasible OMIs [outcome measure instruments] that can be incorporated into the practice setting depending on the particular need of that clinic or health system.”
For the effort, the authors implemented a mixed methods design and incorporated systematic reviews and qualitative consensus methods modeled after the HOME core outcome set initiative, which developed a set of consensus-based core outcome sets for clinical trials and clinical practice. In October of 2022, a daylong in-person consensus exercise was held in Montreal, Canada, where attendees met to reach consensus on recommended instruments to measure AD clinical signs in clinical practice, based on an updated systematic review evaluating the validity of clinical signs instruments.
The review included 22 studies describing 16 instruments that assessed AD clinical signs and an additional 12 variants of instruments. The meeting was attended by 34 individuals from 13 countries, including patient and patient advocate research partners, health care professionals, researchers, methodologists, and industry representatives. Consensus was defined as less than 30% disagreement.
Following their daylong consensus exercise, the stakeholders reached consensus on recommendations to use the EASI, the vIGAAD, and an IGA multiplied or measured alongside a BSA measurement to measure the domain of clinical signs of AD in the clinical practice setting. “The use of multiple IGAs, most with insufficient validation, and the diverse methods used to assess BSA prevented participants from making specific recommendations for the exact IGA/BSA instrument,” the authors wrote. “We recommend that clinicians include at least one of the recommended instruments in their clinical practices and in documentation.”
They explained that the ideal method of measuring BSA was difficult to assess “because multiple techniques exist for its measurement, including regional percentages, the Rule of Nines, or the handprint method. Most studies did not report which method was performed, and to our knowledge, no studies have been performed in patients with AD that have formally compared them.”
During the consensus exercise, the authors noted, several clinicians “expressed concern whether the EASI was feasible for universal use in clinical practice given its complexity, long completion time, and documentation/calculation requirements.” But clinicians who commonly perform the EASI in clinical practice said that the time it takes to complete this measure “has dropped substantially and now is not a considerable burden,” they wrote, adding that, “studies have shown that with trained investigators, EASI completion times can be as low as nearly 2 minutes.”
The authors acknowledged certain limitations of their recommendations, including the lack of input from primary care clinicians. “It is unknown whether ClinROMs [clinician-reported outcome measures] for AD clinical signs are used in the primary care setting, especially given the large amount of conditions that are managed simultaneously and the ever-increasing number of primary care documentation requirements,” they wrote.
Robert Sidbury, MD, MPH, chief of the division of dermatology at Seattle Children’s Hospital, who was asked to comment on the consensus statement, said that with the advent of new, improved, and more expensive medications for AD, “it is ever more important that [the clinical] assessment is reliable and reproducible.”
Insurers “are understandably less willing to rubber-stamp approval of more expensive medications without a reliable standard by which to justify such decisions,” he added. “This is even more important in a disease state like atopic dermatitis that lacks a reliable biomarker. Therefore, one or several practical, reliable, validated severity metrics will help standardize and improve AD care.”
Dr. Sidbury, who cochaired the 2023 American Academy of Dermatology guidelines of care for the management of AD in adults with phototherapy and systemic therapies, added that the instruments evaluated in the review “can be challenging for anyone,” not just primary care providers. “The EASI isn’t that easy, and while there is a learning curve and it ultimately does, like anything, become more efficient in the gathering, it is unclear if non-AD researchers will be willing to invest the time” to routinely use it, he said.
Dr. Simpson and several coauthors reported receiving grants and personal fees from multiple pharmaceutical companies. Dr. Sidbury reported that he serves as an investigator for Regeneron, Galderma, UCB, Castle, and Pfizer; is a consultant for LEO, Lilly, Arcutis, Dermavant, and Pierre Fabre; and a speaker for Beiersdorf.
A version of this article appeared on Medscape.com .
FROM JAMA DERMATOLOGY
Parental e-Cigarette Use Linked to Atopic Dermatitis Risk in Children
TOPLINE:
METHODOLOGY:
- AD is one of the most common inflammatory conditions in children and is linked to environmental risk factors, such as exposure to secondhand smoke and prenatal exposure to tobacco.
- To address the effect of e-cigarettes use on children, researchers conducted a cross-sectional analysis of data from the 2014-2018 National Health Interview Survey, a nationally representative sample of the US population.
- The analysis included 48,637,111 individuals (mean age, 8.4 years), with 6,354,515 (13%) indicating a history of AD (mean age, 8 years).
TAKEAWAY:
- The prevalence of parental e-cigarette use was 18.0% among individuals with AD, compared with 14.4% among those without AD.
- This corresponded to a 24% higher risk for AD associated with parental e-cigarette use (adjusted odds ratio, 1.24; 95% CI, 1.08-1.42).
- The association between e-cigarette use and AD in children held regardless of parent’s sex.
IN PRACTICE:
“Our results suggest that parental e-cigarette use was associated with pediatric AD,” the authors concluded. They noted that the authors of a previous study that associated e-cigarette use with AD in adults postulated that the cause was “the inflammatory state created by” e-cigarettes.
SOURCE:
This study, led by Gun Min Youn, Department of Dermatology, Stanford University School of Medicine, Stanford, California, was published online in JAMA Dermatology.
LIMITATIONS:
The cross-sectional survey design limited the ability to draw causal inferences. Defining e-cigarette use as a single past instance could affect the strength of the findings. Only past-year e-cigarette use was considered. Furthermore, data on pediatric cigarette or e-cigarette use, a potential confounder, were unavailable.
DISCLOSURES:
The study did not disclose funding information. One author reported receiving consultation fees outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- AD is one of the most common inflammatory conditions in children and is linked to environmental risk factors, such as exposure to secondhand smoke and prenatal exposure to tobacco.
- To address the effect of e-cigarettes use on children, researchers conducted a cross-sectional analysis of data from the 2014-2018 National Health Interview Survey, a nationally representative sample of the US population.
- The analysis included 48,637,111 individuals (mean age, 8.4 years), with 6,354,515 (13%) indicating a history of AD (mean age, 8 years).
TAKEAWAY:
- The prevalence of parental e-cigarette use was 18.0% among individuals with AD, compared with 14.4% among those without AD.
- This corresponded to a 24% higher risk for AD associated with parental e-cigarette use (adjusted odds ratio, 1.24; 95% CI, 1.08-1.42).
- The association between e-cigarette use and AD in children held regardless of parent’s sex.
IN PRACTICE:
“Our results suggest that parental e-cigarette use was associated with pediatric AD,” the authors concluded. They noted that the authors of a previous study that associated e-cigarette use with AD in adults postulated that the cause was “the inflammatory state created by” e-cigarettes.
SOURCE:
This study, led by Gun Min Youn, Department of Dermatology, Stanford University School of Medicine, Stanford, California, was published online in JAMA Dermatology.
LIMITATIONS:
The cross-sectional survey design limited the ability to draw causal inferences. Defining e-cigarette use as a single past instance could affect the strength of the findings. Only past-year e-cigarette use was considered. Furthermore, data on pediatric cigarette or e-cigarette use, a potential confounder, were unavailable.
DISCLOSURES:
The study did not disclose funding information. One author reported receiving consultation fees outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- AD is one of the most common inflammatory conditions in children and is linked to environmental risk factors, such as exposure to secondhand smoke and prenatal exposure to tobacco.
- To address the effect of e-cigarettes use on children, researchers conducted a cross-sectional analysis of data from the 2014-2018 National Health Interview Survey, a nationally representative sample of the US population.
- The analysis included 48,637,111 individuals (mean age, 8.4 years), with 6,354,515 (13%) indicating a history of AD (mean age, 8 years).
TAKEAWAY:
- The prevalence of parental e-cigarette use was 18.0% among individuals with AD, compared with 14.4% among those without AD.
- This corresponded to a 24% higher risk for AD associated with parental e-cigarette use (adjusted odds ratio, 1.24; 95% CI, 1.08-1.42).
- The association between e-cigarette use and AD in children held regardless of parent’s sex.
IN PRACTICE:
“Our results suggest that parental e-cigarette use was associated with pediatric AD,” the authors concluded. They noted that the authors of a previous study that associated e-cigarette use with AD in adults postulated that the cause was “the inflammatory state created by” e-cigarettes.
SOURCE:
This study, led by Gun Min Youn, Department of Dermatology, Stanford University School of Medicine, Stanford, California, was published online in JAMA Dermatology.
LIMITATIONS:
The cross-sectional survey design limited the ability to draw causal inferences. Defining e-cigarette use as a single past instance could affect the strength of the findings. Only past-year e-cigarette use was considered. Furthermore, data on pediatric cigarette or e-cigarette use, a potential confounder, were unavailable.
DISCLOSURES:
The study did not disclose funding information. One author reported receiving consultation fees outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
Cortisol Test Confirms HPA Axis Recovery from Steroid Use
TOPLINE:
An early serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) has been validated as a safe and useful screening test with 100% specificity for predicting recovery of the hypothalamic-pituitary-adrenal (HPA) axis in patients on tapering regimes from long‐term chronic glucocorticoid therapy (CGT).
METHODOLOGY:
- A retrospective review of 250-µg Synacthen test (SST) results performed in patients on tapering CGT doses from a single-center rheumatology department over 12 months.
- A total of 60 SSTs were performed in 58 patients, all in the morning (7-12 AM) after withholding CGT for 48 hours.
- Peripheral blood was sampled for cortisol at baseline, 30 minutes, and 60 minutes.
- Adrenal insufficiency (AI) was defined as a peak serum cortisol concentration.
TAKEAWAY:
- The mean duration of CGT (all prednisolone) was 63 months, prescribed primarily for giant cell arteritis/polymyalgia rheumatica (48%) and inflammatory arthritis (18%), with a mean daily dose of 3.4 mg at the time of SST.
- With the investigators’ previously reported basal serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) used to confirm an intact HPA axis, no patient with AI would have been missed, but 37 of 51 (73%) unnecessary SSTs in euadrenal patients would have been avoided.
- A basal serum cortisol concentration of > 227 nmol/L had a specificity of 100% for predicting passing the SST, while a basal serum cortisol concentration of ≤ 55 nmol/L had a 100% sensitivity for predicting failure.
- A mean daily prednisolone dosing at the time of SST in patients with AI was significantly higher than that with normal SSTs (5.7 vs 2.9 mg, respectively; P = .01).
IN PRACTICE:
“This offers a more rapid, convenient, and cost‐effective screening method for patients requiring biochemical assessment of the HPA axis with the potential for significant resource savings without any adverse impact on patient safety,” the authors wrote.
SOURCE:
The study was conducted by Ella Sharma, of the Department of Endocrinology, Royal Victoria Infirmary, Newcastle upon Tyne, UK, and colleagues and published online on May 19, 2024, as a letter in Clinical Endocrinology.
LIMITATIONS:
Not provided.
DISCLOSURES:
Not provided.
A version of this article appeared on Medscape.com.
TOPLINE:
An early serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) has been validated as a safe and useful screening test with 100% specificity for predicting recovery of the hypothalamic-pituitary-adrenal (HPA) axis in patients on tapering regimes from long‐term chronic glucocorticoid therapy (CGT).
METHODOLOGY:
- A retrospective review of 250-µg Synacthen test (SST) results performed in patients on tapering CGT doses from a single-center rheumatology department over 12 months.
- A total of 60 SSTs were performed in 58 patients, all in the morning (7-12 AM) after withholding CGT for 48 hours.
- Peripheral blood was sampled for cortisol at baseline, 30 minutes, and 60 minutes.
- Adrenal insufficiency (AI) was defined as a peak serum cortisol concentration.
TAKEAWAY:
- The mean duration of CGT (all prednisolone) was 63 months, prescribed primarily for giant cell arteritis/polymyalgia rheumatica (48%) and inflammatory arthritis (18%), with a mean daily dose of 3.4 mg at the time of SST.
- With the investigators’ previously reported basal serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) used to confirm an intact HPA axis, no patient with AI would have been missed, but 37 of 51 (73%) unnecessary SSTs in euadrenal patients would have been avoided.
- A basal serum cortisol concentration of > 227 nmol/L had a specificity of 100% for predicting passing the SST, while a basal serum cortisol concentration of ≤ 55 nmol/L had a 100% sensitivity for predicting failure.
- A mean daily prednisolone dosing at the time of SST in patients with AI was significantly higher than that with normal SSTs (5.7 vs 2.9 mg, respectively; P = .01).
IN PRACTICE:
“This offers a more rapid, convenient, and cost‐effective screening method for patients requiring biochemical assessment of the HPA axis with the potential for significant resource savings without any adverse impact on patient safety,” the authors wrote.
SOURCE:
The study was conducted by Ella Sharma, of the Department of Endocrinology, Royal Victoria Infirmary, Newcastle upon Tyne, UK, and colleagues and published online on May 19, 2024, as a letter in Clinical Endocrinology.
LIMITATIONS:
Not provided.
DISCLOSURES:
Not provided.
A version of this article appeared on Medscape.com.
TOPLINE:
An early serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) has been validated as a safe and useful screening test with 100% specificity for predicting recovery of the hypothalamic-pituitary-adrenal (HPA) axis in patients on tapering regimes from long‐term chronic glucocorticoid therapy (CGT).
METHODOLOGY:
- A retrospective review of 250-µg Synacthen test (SST) results performed in patients on tapering CGT doses from a single-center rheumatology department over 12 months.
- A total of 60 SSTs were performed in 58 patients, all in the morning (7-12 AM) after withholding CGT for 48 hours.
- Peripheral blood was sampled for cortisol at baseline, 30 minutes, and 60 minutes.
- Adrenal insufficiency (AI) was defined as a peak serum cortisol concentration.
TAKEAWAY:
- The mean duration of CGT (all prednisolone) was 63 months, prescribed primarily for giant cell arteritis/polymyalgia rheumatica (48%) and inflammatory arthritis (18%), with a mean daily dose of 3.4 mg at the time of SST.
- With the investigators’ previously reported basal serum cortisol concentration of > 237 nmol/L (> 8.6 μg/dL) used to confirm an intact HPA axis, no patient with AI would have been missed, but 37 of 51 (73%) unnecessary SSTs in euadrenal patients would have been avoided.
- A basal serum cortisol concentration of > 227 nmol/L had a specificity of 100% for predicting passing the SST, while a basal serum cortisol concentration of ≤ 55 nmol/L had a 100% sensitivity for predicting failure.
- A mean daily prednisolone dosing at the time of SST in patients with AI was significantly higher than that with normal SSTs (5.7 vs 2.9 mg, respectively; P = .01).
IN PRACTICE:
“This offers a more rapid, convenient, and cost‐effective screening method for patients requiring biochemical assessment of the HPA axis with the potential for significant resource savings without any adverse impact on patient safety,” the authors wrote.
SOURCE:
The study was conducted by Ella Sharma, of the Department of Endocrinology, Royal Victoria Infirmary, Newcastle upon Tyne, UK, and colleagues and published online on May 19, 2024, as a letter in Clinical Endocrinology.
LIMITATIONS:
Not provided.
DISCLOSURES:
Not provided.
A version of this article appeared on Medscape.com.
New Administration Routes for Adrenaline in Anaphylaxis
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
PARIS — While anaphylaxis requires immediate adrenaline administration through autoinjection, the use of this treatment is not optimal. Therefore, the development of new adrenaline formulations (such as for intranasal, sublingual, and transcutaneous routes) aims to facilitate the drug’s use and reduce persistent delays in administration by patients and caregivers. An overview of the research was presented at the 19th French-speaking Congress of Allergology.
Anaphylaxis is a severe and potentially fatal immediate hypersensitivity reaction with highly variable and dynamic clinical presentations. It requires prompt recognition for immediate treatment with intramuscular (IM) adrenaline (at the anterolateral aspect of the mid-thigh).
One might think that this reflex is acquired, but in France, while the number of prescribed adrenaline autoinjection (AAI) devices has been increasing for a decade, reaching 965,944 units in 2022, this first-line treatment is underused. Anapen (150, 300, and 500 µg), EpiPen (150 and 300 µg), Jext (150 µg and 300 µg), and Emerade (150, 300, and 500 µg) are the four products marketed in France in 2024.
“Only 17.3% of individuals presenting to the emergency department in the Lorraine region used it in 2015,” said Catherine Neukirch, MD, a pneumologist at Hôpital Bichat–Claude Bernard in Paris, France, with rates of 11.3% for children and 20.3% for adults.
Anaphylaxis Incidence Increasing
Approximately 0.3% (95% CI, 0.1-0.5) of the population will experience an anaphylaxis episode in their lifetime. Incidence in Europe, across all causes, is estimated between 1.5 and 7.9 cases per 100,000 inhabitants per year. Although anaphylaxis is on the rise, its associated mortality remains low, ranging between 0.05 and 0.51 per million per year for drugs, between 0.03 and 0.32 per million per year for foods, and between 0.09 and 0.13 per million per year for hymenopteran venoms.
Data from the European Anaphylaxis Registry indicate that anaphylaxis manifests rapidly after allergen exposure: 55% of cases occur within 10 minutes and 80% within 30 minutes. In addition, a biphasic reaction, which can occur up to 72 hours after exposure, is observed in < 5% of cases.
While a delay in adrenaline use is associated with risk for increased morbidity and mortality, AAI significantly reduces error rates compared with manual treatments involving ampoules, needles, and syringes. It also reduces the associated panic risks. However, there are multiple barriers to adrenaline use. The clinical symptoms of anaphylaxis may be misleading, especially if it occurs without cutaneous and urticarial manifestations but with only acute bronchospasm. It may present as isolated laryngeal edema without digestive involvement, hypotension, or other respiratory problems.
Other limitations to adrenaline use include technical difficulties and the possibility of incorrect administration, the need for appropriate needle sizes for patients with obesity, needle phobia, potential adverse effects of adrenaline injections, failure to carry two autoinjectors, constraints related to storage and bulky transport, as well as the need for training and practice.
“These factors contribute to underuse of adrenaline by patients and caregivers,” said Dr. Neukirch, which results in delays in necessary administration.
Adrenaline Treatment Criteria?
An analysis published in 2023 based on pharmacovigilance data from 30 regional French centers from 1984 to 2022 included 42 reported cases (average age, 33 years; 26% children) of reactions to AAI, which probably is an underestimate. About 40% of AAI uses occurred during anaphylaxis. The remaining 60% were triggered outside of reactions. The main reasons were accidental injections, mainly in the fingers, and cases of not triggering the autoinjector, underlining the importance of patient education.
In 2015, the European Medicines Agency required pharmacological studies for injectable adrenaline on healthy volunteers. These studies include ultrasound measurements of bolus injection, pharmacokinetics (ie, absorption, distribution, metabolism, and excretion), and pharmacodynamics (ie, the effect of the drug and the mechanism of action in the body), with precise evaluation of cardiovascular effects (eg, systolic and diastolic blood pressures and heart rate).
Among the information collected with the different products, ultrasound studies have shown a different localization of the adrenaline bolus (ie, in muscle in patients with normal BMI and mostly in adipose tissue in patients with BMI indicating overweight and obesity). The consequences of this finding are still unknown.
In a study with 500 µg Anapen, women with overweight or obesity showed different pharmacokinetic or pharmacodynamic profiles from those in men with normal weight, with an increase in the area under the curve (0-240 min) and marked changes in the heart rate time curve.
IM administration of 0.5 mg produces rapid pharmacokinetic effects in patients with normal weight, overweight, or obesity, with a delay for the second peak in the latter case. This delay perhaps results from initial local vasoconstriction due to adrenaline.
The early peak plasma concentration occurs at 5-10 minutes for AAI, with a faster speed for Anapen and EpiPen.
Moreover, needle size is not the most important factor. Rather, it is the strength and speed of injection, which can vary depending on the AAI.
Also, the optimal plasma concentration of adrenaline to treat anaphylaxis is not known; studies cannot be conducted during anaphylaxis. In terms of pharmacokinetics, a small series discovered that increased skin or muscle thickness delays the absorption of EpiPen AAI.
Intranasal Adrenaline
To facilitate rapid adrenaline use and convince reluctant patients to carry and use adrenaline, intranasal, sublingual, or transcutaneous forms are under development.
Three intranasal forms of adrenaline are already well advanced, including Neffy from ARS Pharma, epinephrine sprays from Bryn Pharma and Hikma, and Oxero from Oragoo, which contains dry powder.
A comparison of intranasal adrenaline Neffy and AAI shows that the former has satisfactory pharmacokinetic and pharmacodynamic effects.
In a phase 1 randomized crossover study of 42 healthy adults comparing the pharmacokinetic effects of Neffy adrenaline (2 mg) and EpiPen (0.3 mg), as well as IM epinephrine 0.3 mg, several observations were made. For a single dose, the maximum concentration (Cmax) of Neffy was lower than that of EpiPen.
However, with repeated doses administered 10 minutes apart, the Cmax of Neffy was higher than that of EpiPen. At this stage, pharmacodynamic responses to intranasal products are at least comparable with those of approved injectable products.
A comparison of the pharmacodynamic effects, such as systolic and diastolic blood pressures and heart rate, of Neffy adrenaline and AAI concluded that the profile of Neffy is comparable with that of EpiPen and superior to that of IM epinephrine.
In patients with a history of allergic rhinitis, adrenaline Cmax appears to be increased, while time to peak plasma concentration (Tmax) is reduced. Low blood pressure does not prevent Neffy absorption. Neffy is currently under review by the American and European health authorities.
Intranasal absorption of dry powder adrenaline appears to be faster than that of EpiPen, thus offering a clinical advantage in the short therapeutic window for anaphylaxis treatment.
In an open-label trial conducted on 12 adults with seasonal allergic rhinitis without asthma, the pharmacokinetics, pharmacodynamics, and safety of adrenaline were compared between FMXIN002 (1.6 and 3.2 mg), which was administered intranasally with or without nasal allergen challenge, and IM EpiPen 0.3 mg. Pharmacokinetics varied by patient. Nevertheless, nasal FMXIN002 had a shorter Tmax, a doubled Cmax after the allergen challenge peak, and a higher area under the curve in the 8 hours following administration compared with EpiPen. Pharmacodynamic effects comparable with those of EpiPen were noted at 15 minutes to 4 hours after administration. The tolerance was good, with mild and local side effects. The powder seems to deposit slightly better in the nasal cavity. It remains stable for 6 months at a temperature of 40 °C and relative humidity of 75% and for 2 years at a temperature of 25 °C and relative humidity of 60%.
Sublingual Adrenaline Film
AQST-109 is a sublingual film that is intended to allow rapid administration of epinephrine 1, which is a prodrug of adrenaline. The product is the size of a postage stamp, weighs < 30 g, and dissolves on contact with the tongue.
The EPIPHAST II study was a phase 1, multiperiod, crossover study conducted on 24 healthy adults (age, 24-49 years) who were randomly assigned to receive either 12 or 0.3 mg of AQST-109 of manual IM adrenaline in the first two periods. All participants received 0.3 mg of EpiPen in the last period.
EpiPen 0.3 mg resulted in a higher Cmax than AQST-109 12 mg. AQST-109 12 mg had the fastest median Tmax of 12 minutes. The areas under the curve of AQST-109 12 mg fell between those of EpiPen 0.3 mg and manual IM adrenaline 0.3 mg.
Early increases in systolic blood pressure, diastolic blood pressure, and heart rate were observed with AQST-109 12 mg. Changes were more pronounced with AQST-109 12 mg despite a higher Cmax with EpiPen 0.3 mg.
Part 3 of the EPIPHAST study evaluated the impact of food exposure (ie, a peanut butter sandwich) on the pharmacokinetics of AQST-109 12 mg in 24 healthy adults. Oral food residues did not significantly affect pharmacodynamic parameters, and no treatment-related adverse events were reported.
Researchers concluded that AQST-109 12 mg absorption would not be altered by “real” situations if used during meals. “These results suggest that the sublingual adrenaline film could be promising in real situations,” said Dr. Neukirch, especially in cases of food allergy with recent ingestion of the allergenic food.
Transcutaneous Adrenaline
A transcutaneous form of adrenaline that uses the Zeneo device developed by Crossject, a company based in Dijon, France, comes in the form of an AAI that requires no needle. This project, funded by the European Union, uses a gas generator to propel the drug at very high speed through the skin in 50 milliseconds. This method allows for extended drug storage.
Dr. Neukirch reported financial relationships with Viatris, Stallergènes, ALK, Astrazeneca, Sanofi, GSK, and Novartis.
This story was translated from the Medscape French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Study Finds Immunosuppression Affects Risk for Poor Outcomes in Patients with cSCC
PHOENIX — Immunosuppression is an independent risk factor for poorer outcomes in patients with cutaneous squamous cell carcinoma (cSCC), according to new research that was presented at the American College of Mohs Surgery (ACMS) 2024 annual meeting.
Even though immunosuppression is strongly associated with an increased risk for cSCC, studies to date have generally not shown it to be an independent risk factor for metastasis and disease-specific death (DSD), after accounting for primary tumor stage.
“Solid organ transplant puts patients at risk for developing cutaneous squamous cell carcinoma, and it’s more likely to have aggressive features,” said study author Jason Klein, MD, PhD, a dermatology resident at University of Texas Southwestern Medical Center, Dallas. “But it’s still not known if immunosuppression is an independent risk factor.”
Other groups “have tried to tackle this, but they have all primarily been single-institution data,” he noted, adding that “results so far have been tipping the scale towards immunosuppression not being an independent risk factor” for worse outcomes.
Immunosuppressed individuals face a greater risk for cSCC than the general population and often present with more aggressive, multifocal disease. However, Dr. Klein explained that a previous retrospective study comprising a cohort of approximately 7600 tumors from two centers reported that immunosuppression was not an independent risk factor for both tumor metastasis and cancer-specific death after adjusting for tumor characteristics.
Tipped the Scale
Therefore, the goal of the current study was to repeat this analysis but in a much larger retrospective cohort. Dr. Klein and his colleagues pooled cSCC data from 12 dermatology centers (11 academic and one private) that were located in the United States, Spain, and Brazil. The cohort included 4392 patients (3769 immunocompetent patients and 623 immunosuppressed patients) with 19,237 tumors (15,191 immunocompetent and 4046 immunosuppressed). Study endpoints included local recurrence, metastasis (nodal, satellite/in-transit, and distant), DSD, and “major poor outcomes” (defined as metastasis and DSD combined).
About 30% of the immunosuppressed patients were organ transplant recipients (OTR) and 10% had chronic lymphocytic leukemia (CLL). Half of the immunocompetent patients (50.3%) underwent Mohs surgery as the primary treatment, as did 58.2% of the immunosuppressed patients.
On multivariable analysis, significant predictors of “major poor outcomes” included immunosuppression (subdistribution hazard ratio [SHR], 1.3; P = .04), Brigham and Women’s Hospital tumor stage (SHR 6.7 for T2a, 18.1 for T2b, and 37.2 for T3; P < .001 for all), location on the head/neck (SHR, 2.1; P < .001), and adjuvant radiation (SHR, 1.6; P < .001).
But when metastasis and DSD were evaluated separately, immunosuppression was only predictive of DSD (SHR, 1.7; P = .008) but not metastasis (SHR, 1.2; P = .21). Dr. Klein explained that they also conducted a separate subanalysis limited to OTR and patients with CLL, which demonstrated that immunosuppression was no longer a significant predictor of “major poor outcomes” (SHR, 0.9; P = .66 for OTR; SHR, 1.4; P = .25 for CLL).
“Organ transplant status and CLL were not independent risk factors for major poor outcomes,” he said. “But in summary, we may be tipping the scale to immunosuppression being a risk factor.”
Asked to comment on the findings, Naissan O. Wesley, MD, director of Mohs surgery, Skin Care and Laser Physicians of Beverly Hills, Los Angeles, California, stated that “this larger scale study presented at this meeting was important to further confirm what we see in everyday practice, that immunosuppression may lead to poorer outcomes in patients with cutaneous squamous cell carcinoma.”
Also weighing in on the data, Jesse M. Lewin, MD, chief of Mohs micrographic and dermatologic surgery, at the Icahn School of Medicine at Mount Sinai, New York City, noted that the treatment of cSCC in high-risk patients has been challenging because of the historical lack of data and large studies to guide management.
“The authors provide a large cohort to help stratify which patients are most at risk for poor outcomes, which can inform our decision to refer for neoadjuvant or adjuvant treatment and multi-disciplinary management,” he said. “This is the first step in being able to optimize cure in these patients.”
The study was independently supported. Dr. Klein, Dr. Lewin, and Dr. Wesley reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
PHOENIX — Immunosuppression is an independent risk factor for poorer outcomes in patients with cutaneous squamous cell carcinoma (cSCC), according to new research that was presented at the American College of Mohs Surgery (ACMS) 2024 annual meeting.
Even though immunosuppression is strongly associated with an increased risk for cSCC, studies to date have generally not shown it to be an independent risk factor for metastasis and disease-specific death (DSD), after accounting for primary tumor stage.
“Solid organ transplant puts patients at risk for developing cutaneous squamous cell carcinoma, and it’s more likely to have aggressive features,” said study author Jason Klein, MD, PhD, a dermatology resident at University of Texas Southwestern Medical Center, Dallas. “But it’s still not known if immunosuppression is an independent risk factor.”
Other groups “have tried to tackle this, but they have all primarily been single-institution data,” he noted, adding that “results so far have been tipping the scale towards immunosuppression not being an independent risk factor” for worse outcomes.
Immunosuppressed individuals face a greater risk for cSCC than the general population and often present with more aggressive, multifocal disease. However, Dr. Klein explained that a previous retrospective study comprising a cohort of approximately 7600 tumors from two centers reported that immunosuppression was not an independent risk factor for both tumor metastasis and cancer-specific death after adjusting for tumor characteristics.
Tipped the Scale
Therefore, the goal of the current study was to repeat this analysis but in a much larger retrospective cohort. Dr. Klein and his colleagues pooled cSCC data from 12 dermatology centers (11 academic and one private) that were located in the United States, Spain, and Brazil. The cohort included 4392 patients (3769 immunocompetent patients and 623 immunosuppressed patients) with 19,237 tumors (15,191 immunocompetent and 4046 immunosuppressed). Study endpoints included local recurrence, metastasis (nodal, satellite/in-transit, and distant), DSD, and “major poor outcomes” (defined as metastasis and DSD combined).
About 30% of the immunosuppressed patients were organ transplant recipients (OTR) and 10% had chronic lymphocytic leukemia (CLL). Half of the immunocompetent patients (50.3%) underwent Mohs surgery as the primary treatment, as did 58.2% of the immunosuppressed patients.
On multivariable analysis, significant predictors of “major poor outcomes” included immunosuppression (subdistribution hazard ratio [SHR], 1.3; P = .04), Brigham and Women’s Hospital tumor stage (SHR 6.7 for T2a, 18.1 for T2b, and 37.2 for T3; P < .001 for all), location on the head/neck (SHR, 2.1; P < .001), and adjuvant radiation (SHR, 1.6; P < .001).
But when metastasis and DSD were evaluated separately, immunosuppression was only predictive of DSD (SHR, 1.7; P = .008) but not metastasis (SHR, 1.2; P = .21). Dr. Klein explained that they also conducted a separate subanalysis limited to OTR and patients with CLL, which demonstrated that immunosuppression was no longer a significant predictor of “major poor outcomes” (SHR, 0.9; P = .66 for OTR; SHR, 1.4; P = .25 for CLL).
“Organ transplant status and CLL were not independent risk factors for major poor outcomes,” he said. “But in summary, we may be tipping the scale to immunosuppression being a risk factor.”
Asked to comment on the findings, Naissan O. Wesley, MD, director of Mohs surgery, Skin Care and Laser Physicians of Beverly Hills, Los Angeles, California, stated that “this larger scale study presented at this meeting was important to further confirm what we see in everyday practice, that immunosuppression may lead to poorer outcomes in patients with cutaneous squamous cell carcinoma.”
Also weighing in on the data, Jesse M. Lewin, MD, chief of Mohs micrographic and dermatologic surgery, at the Icahn School of Medicine at Mount Sinai, New York City, noted that the treatment of cSCC in high-risk patients has been challenging because of the historical lack of data and large studies to guide management.
“The authors provide a large cohort to help stratify which patients are most at risk for poor outcomes, which can inform our decision to refer for neoadjuvant or adjuvant treatment and multi-disciplinary management,” he said. “This is the first step in being able to optimize cure in these patients.”
The study was independently supported. Dr. Klein, Dr. Lewin, and Dr. Wesley reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
PHOENIX — Immunosuppression is an independent risk factor for poorer outcomes in patients with cutaneous squamous cell carcinoma (cSCC), according to new research that was presented at the American College of Mohs Surgery (ACMS) 2024 annual meeting.
Even though immunosuppression is strongly associated with an increased risk for cSCC, studies to date have generally not shown it to be an independent risk factor for metastasis and disease-specific death (DSD), after accounting for primary tumor stage.
“Solid organ transplant puts patients at risk for developing cutaneous squamous cell carcinoma, and it’s more likely to have aggressive features,” said study author Jason Klein, MD, PhD, a dermatology resident at University of Texas Southwestern Medical Center, Dallas. “But it’s still not known if immunosuppression is an independent risk factor.”
Other groups “have tried to tackle this, but they have all primarily been single-institution data,” he noted, adding that “results so far have been tipping the scale towards immunosuppression not being an independent risk factor” for worse outcomes.
Immunosuppressed individuals face a greater risk for cSCC than the general population and often present with more aggressive, multifocal disease. However, Dr. Klein explained that a previous retrospective study comprising a cohort of approximately 7600 tumors from two centers reported that immunosuppression was not an independent risk factor for both tumor metastasis and cancer-specific death after adjusting for tumor characteristics.
Tipped the Scale
Therefore, the goal of the current study was to repeat this analysis but in a much larger retrospective cohort. Dr. Klein and his colleagues pooled cSCC data from 12 dermatology centers (11 academic and one private) that were located in the United States, Spain, and Brazil. The cohort included 4392 patients (3769 immunocompetent patients and 623 immunosuppressed patients) with 19,237 tumors (15,191 immunocompetent and 4046 immunosuppressed). Study endpoints included local recurrence, metastasis (nodal, satellite/in-transit, and distant), DSD, and “major poor outcomes” (defined as metastasis and DSD combined).
About 30% of the immunosuppressed patients were organ transplant recipients (OTR) and 10% had chronic lymphocytic leukemia (CLL). Half of the immunocompetent patients (50.3%) underwent Mohs surgery as the primary treatment, as did 58.2% of the immunosuppressed patients.
On multivariable analysis, significant predictors of “major poor outcomes” included immunosuppression (subdistribution hazard ratio [SHR], 1.3; P = .04), Brigham and Women’s Hospital tumor stage (SHR 6.7 for T2a, 18.1 for T2b, and 37.2 for T3; P < .001 for all), location on the head/neck (SHR, 2.1; P < .001), and adjuvant radiation (SHR, 1.6; P < .001).
But when metastasis and DSD were evaluated separately, immunosuppression was only predictive of DSD (SHR, 1.7; P = .008) but not metastasis (SHR, 1.2; P = .21). Dr. Klein explained that they also conducted a separate subanalysis limited to OTR and patients with CLL, which demonstrated that immunosuppression was no longer a significant predictor of “major poor outcomes” (SHR, 0.9; P = .66 for OTR; SHR, 1.4; P = .25 for CLL).
“Organ transplant status and CLL were not independent risk factors for major poor outcomes,” he said. “But in summary, we may be tipping the scale to immunosuppression being a risk factor.”
Asked to comment on the findings, Naissan O. Wesley, MD, director of Mohs surgery, Skin Care and Laser Physicians of Beverly Hills, Los Angeles, California, stated that “this larger scale study presented at this meeting was important to further confirm what we see in everyday practice, that immunosuppression may lead to poorer outcomes in patients with cutaneous squamous cell carcinoma.”
Also weighing in on the data, Jesse M. Lewin, MD, chief of Mohs micrographic and dermatologic surgery, at the Icahn School of Medicine at Mount Sinai, New York City, noted that the treatment of cSCC in high-risk patients has been challenging because of the historical lack of data and large studies to guide management.
“The authors provide a large cohort to help stratify which patients are most at risk for poor outcomes, which can inform our decision to refer for neoadjuvant or adjuvant treatment and multi-disciplinary management,” he said. “This is the first step in being able to optimize cure in these patients.”
The study was independently supported. Dr. Klein, Dr. Lewin, and Dr. Wesley reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM ACMS 2024
Recent Evidence for Home Phototherapy Benefits May Improve Access for Patients with Psoriasis
Supporters of home phototherapy for patients with plaque and guttate psoriasis had plenty to cheer about at the annual meeting of the American Academy of Dermatology (AAD) in March. There, Joel M. Gelfand, MD, professor of dermatology and epidemiology at the University of Pennsylvania in Philadelphia, presented results from the LITE study, a trial that tested the hypothesis that narrowband ultraviolet B phototherapy of psoriasis at home is noninferior to office treatment, based on outcomes that matter to patients, clinicians, and payers. While smaller studies have drawn similar conclusions,
The co-primary outcomes in the LITE study were a Physician’s Global Assessment (PGA) score of 0/1 (clear, almost clear) and a Dermatology Life Quality Index (DLQI) score of 5 or less (small, no effect on health-related quality of life).
Dr. Gelfand and colleagues at 42 sites in the United States enrolled 783 patients aged 12 years and older who had plaque or guttate psoriasis and were candidates for phototherapy at home or in an office setting. Following 12 weeks of treatment, 25.6% of patients in the office-based phototherapy group achieved a PGA score of 0/1 compared with 32.8% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data). Similarly, 33.6% of patients in the office-based phototherapy group achieved a DLQI score of 5 or less compared with 52.4% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data).

A Safe and Effective Option
“I think that it’s important for physicians, insurance companies, and patients with psoriasis to understand that this is a very safe and effective form of therapy,” Craig A. Elmets, MD, professor of dermatology at The University of Alabama at Birmingham, said in an interview. “For people who are not interested in systemic medications or who have contraindications to systemic medications, phototherapy would be ideal,” added Dr. Elmets, first author of the joint AAD–National Psoriasis Foundation (NPF) guidelines for the management and treatment of psoriasis with phototherapy, published in 2019.
Factors beyond efficacy support the role of home phototherapy, Dr. Gelfand said, including the fact that it costs 10-100 times less than biologics for psoriasis and that office-based phototherapy is not available in 90% of counties in the United States. However, insurance coverage of home phototherapy “is highly variable because until the LITE study, there was no large-scale US data to support its use,” he told this news organization.
“Also, insurance companies are broken up into two parts: Durable medical goods and the medical side such as pharmacy costs, and they are siloed. The durable medical goods side views phototherapy as expensive, while the pharmacy side views it as dirt cheap. This is part of the problem with our health system. A lot of things are siloed and don’t make any sense,” said Dr. Gelfand, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania. By working with the NPF and payers, he added, “we’re hoping ... to transform the way insurance companies think about covering home phototherapy.”
In the meantime, he and Dr. Elmets shared practical ways to optimize access to home phototherapy for psoriasis patients:
Have the discussion. Patients “rarely bring this up as an option,” Dr. Elmets said, so the onus is on clinicians to talk about it. In his view, the ideal candidate “is averse to using systemic agents but whose disease is beyond the point where topical medicines alone will work. One of the advantages of phototherapy is that it doesn’t have immunosuppressive effects.”

Clinicians and patients can learn about the efficacy and safety of phototherapy for psoriasis, including home-based options, on the NPF’s web site and by reading the 2019 joint AAD-NPF guidelines.
Shared decision-making is key. “When a patient comes in, I’ll discuss what their treatment options are and [we] will decide upon a course of action based on their unique needs and preferences [and] if it’s medically appropriate, meaning they have the type of psoriasis likely to respond to phototherapy,” Dr. Gelfand said. A patient with psoriasis mainly on the fingernails or genitals “is not a good candidate for phototherapy. If it’s on the trunk or extremities, that patient would be a good candidate.”
Home phototherapy candidates also must be willing and able to operate a machine and have dedicated space in their dwelling for it (most units are about the size of a door). Patients also have to be reliable, follow directions, and come back in person for follow-up appointments “so we can assess their response to treatment and fine-tune things as necessary and make sure they’re not developing any skin damage,” Dr. Gelfand said.
Educate yourself about existing options. Home phototherapy units from manufacturers such as Daavlin, National Biological Corporation, and SolRx range between $1200 and $6000 in cost, Dr. Gelfand said. He and his colleagues used the Daavlin 7 series in the LITE study. That unit features an integrated dosimetry system that delivers the correct dose of energy based on parameters that the prescribing clinician recommends. Settings are based on the patient’s skin type and how much the prescriber wants to increase the dose for each treatment. “The machine does the rest,” he said. “It knows what dose to give, so they get the same dosing as they would in an office situation.”
Smaller home-based phototherapy units designed to treat the hands and feet are available. So are handheld units to treat the scalp. “These can be a nice option for patients who have a few spots, but if the disease is moderate to severe, then it’s going to be pretty laborious to [use them],” Dr. Elmets said.
Remember that phototherapy is not a cure-all. According to the joint AAD-NPF guidelines, most phototherapy regimens require treatments two to three times per week for 10-14 weeks. Once patients achieve their home phototherapy treatment goal, Dr. Elmets often recommends treatments one to two times per week for maintenance.
“Patients with psoriasis have a lifetime condition,” he noted. “There are certainly cases where people have gone on phototherapy, cleared, and then stopped for a period of time. If they flare up, they can always go back to phototherapy. Usually, people who are on phototherapy use some type of topical agents to touch up areas that are resistant.”
Expect pushback from insurers on coverage. While Medicare and some integrated health plans cover home phototherapy, expect to spend time writing letters or placing phone calls to insurance companies to convince them why they should cover home phototherapy for candidate psoriasis patients. “Usually there’s a lot of letter writing and a long delay in getting approval,” Dr. Elmets said.
Dr. Elmets and Dr. Gelfand reported no relevant financial relationships. The LITE study was funded by the Patient-Centered Outcomes Research Institute. Research partners included the National Psoriasis Foundation and Daavlin, which provided the home phototherapy machines and covered the cost of shipping the devices.
A version of this article appeared on Medscape.com.
Supporters of home phototherapy for patients with plaque and guttate psoriasis had plenty to cheer about at the annual meeting of the American Academy of Dermatology (AAD) in March. There, Joel M. Gelfand, MD, professor of dermatology and epidemiology at the University of Pennsylvania in Philadelphia, presented results from the LITE study, a trial that tested the hypothesis that narrowband ultraviolet B phototherapy of psoriasis at home is noninferior to office treatment, based on outcomes that matter to patients, clinicians, and payers. While smaller studies have drawn similar conclusions,
The co-primary outcomes in the LITE study were a Physician’s Global Assessment (PGA) score of 0/1 (clear, almost clear) and a Dermatology Life Quality Index (DLQI) score of 5 or less (small, no effect on health-related quality of life).
Dr. Gelfand and colleagues at 42 sites in the United States enrolled 783 patients aged 12 years and older who had plaque or guttate psoriasis and were candidates for phototherapy at home or in an office setting. Following 12 weeks of treatment, 25.6% of patients in the office-based phototherapy group achieved a PGA score of 0/1 compared with 32.8% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data). Similarly, 33.6% of patients in the office-based phototherapy group achieved a DLQI score of 5 or less compared with 52.4% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data).
A Safe and Effective Option
“I think that it’s important for physicians, insurance companies, and patients with psoriasis to understand that this is a very safe and effective form of therapy,” Craig A. Elmets, MD, professor of dermatology at The University of Alabama at Birmingham, said in an interview. “For people who are not interested in systemic medications or who have contraindications to systemic medications, phototherapy would be ideal,” added Dr. Elmets, first author of the joint AAD–National Psoriasis Foundation (NPF) guidelines for the management and treatment of psoriasis with phototherapy, published in 2019.
Factors beyond efficacy support the role of home phototherapy, Dr. Gelfand said, including the fact that it costs 10-100 times less than biologics for psoriasis and that office-based phototherapy is not available in 90% of counties in the United States. However, insurance coverage of home phototherapy “is highly variable because until the LITE study, there was no large-scale US data to support its use,” he told this news organization.
“Also, insurance companies are broken up into two parts: Durable medical goods and the medical side such as pharmacy costs, and they are siloed. The durable medical goods side views phototherapy as expensive, while the pharmacy side views it as dirt cheap. This is part of the problem with our health system. A lot of things are siloed and don’t make any sense,” said Dr. Gelfand, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania. By working with the NPF and payers, he added, “we’re hoping ... to transform the way insurance companies think about covering home phototherapy.”
In the meantime, he and Dr. Elmets shared practical ways to optimize access to home phototherapy for psoriasis patients:
Have the discussion. Patients “rarely bring this up as an option,” Dr. Elmets said, so the onus is on clinicians to talk about it. In his view, the ideal candidate “is averse to using systemic agents but whose disease is beyond the point where topical medicines alone will work. One of the advantages of phototherapy is that it doesn’t have immunosuppressive effects.”
Clinicians and patients can learn about the efficacy and safety of phototherapy for psoriasis, including home-based options, on the NPF’s web site and by reading the 2019 joint AAD-NPF guidelines.
Shared decision-making is key. “When a patient comes in, I’ll discuss what their treatment options are and [we] will decide upon a course of action based on their unique needs and preferences [and] if it’s medically appropriate, meaning they have the type of psoriasis likely to respond to phototherapy,” Dr. Gelfand said. A patient with psoriasis mainly on the fingernails or genitals “is not a good candidate for phototherapy. If it’s on the trunk or extremities, that patient would be a good candidate.”
Home phototherapy candidates also must be willing and able to operate a machine and have dedicated space in their dwelling for it (most units are about the size of a door). Patients also have to be reliable, follow directions, and come back in person for follow-up appointments “so we can assess their response to treatment and fine-tune things as necessary and make sure they’re not developing any skin damage,” Dr. Gelfand said.
Educate yourself about existing options. Home phototherapy units from manufacturers such as Daavlin, National Biological Corporation, and SolRx range between $1200 and $6000 in cost, Dr. Gelfand said. He and his colleagues used the Daavlin 7 series in the LITE study. That unit features an integrated dosimetry system that delivers the correct dose of energy based on parameters that the prescribing clinician recommends. Settings are based on the patient’s skin type and how much the prescriber wants to increase the dose for each treatment. “The machine does the rest,” he said. “It knows what dose to give, so they get the same dosing as they would in an office situation.”
Smaller home-based phototherapy units designed to treat the hands and feet are available. So are handheld units to treat the scalp. “These can be a nice option for patients who have a few spots, but if the disease is moderate to severe, then it’s going to be pretty laborious to [use them],” Dr. Elmets said.
Remember that phototherapy is not a cure-all. According to the joint AAD-NPF guidelines, most phototherapy regimens require treatments two to three times per week for 10-14 weeks. Once patients achieve their home phototherapy treatment goal, Dr. Elmets often recommends treatments one to two times per week for maintenance.
“Patients with psoriasis have a lifetime condition,” he noted. “There are certainly cases where people have gone on phototherapy, cleared, and then stopped for a period of time. If they flare up, they can always go back to phototherapy. Usually, people who are on phototherapy use some type of topical agents to touch up areas that are resistant.”
Expect pushback from insurers on coverage. While Medicare and some integrated health plans cover home phototherapy, expect to spend time writing letters or placing phone calls to insurance companies to convince them why they should cover home phototherapy for candidate psoriasis patients. “Usually there’s a lot of letter writing and a long delay in getting approval,” Dr. Elmets said.
Dr. Elmets and Dr. Gelfand reported no relevant financial relationships. The LITE study was funded by the Patient-Centered Outcomes Research Institute. Research partners included the National Psoriasis Foundation and Daavlin, which provided the home phototherapy machines and covered the cost of shipping the devices.
A version of this article appeared on Medscape.com.
Supporters of home phototherapy for patients with plaque and guttate psoriasis had plenty to cheer about at the annual meeting of the American Academy of Dermatology (AAD) in March. There, Joel M. Gelfand, MD, professor of dermatology and epidemiology at the University of Pennsylvania in Philadelphia, presented results from the LITE study, a trial that tested the hypothesis that narrowband ultraviolet B phototherapy of psoriasis at home is noninferior to office treatment, based on outcomes that matter to patients, clinicians, and payers. While smaller studies have drawn similar conclusions,
The co-primary outcomes in the LITE study were a Physician’s Global Assessment (PGA) score of 0/1 (clear, almost clear) and a Dermatology Life Quality Index (DLQI) score of 5 or less (small, no effect on health-related quality of life).
Dr. Gelfand and colleagues at 42 sites in the United States enrolled 783 patients aged 12 years and older who had plaque or guttate psoriasis and were candidates for phototherapy at home or in an office setting. Following 12 weeks of treatment, 25.6% of patients in the office-based phototherapy group achieved a PGA score of 0/1 compared with 32.8% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data). Similarly, 33.6% of patients in the office-based phototherapy group achieved a DLQI score of 5 or less compared with 52.4% of patients in the home-based phototherapy group (P > .0001 for noninferiority, non-response imputation for missing data).
A Safe and Effective Option
“I think that it’s important for physicians, insurance companies, and patients with psoriasis to understand that this is a very safe and effective form of therapy,” Craig A. Elmets, MD, professor of dermatology at The University of Alabama at Birmingham, said in an interview. “For people who are not interested in systemic medications or who have contraindications to systemic medications, phototherapy would be ideal,” added Dr. Elmets, first author of the joint AAD–National Psoriasis Foundation (NPF) guidelines for the management and treatment of psoriasis with phototherapy, published in 2019.
Factors beyond efficacy support the role of home phototherapy, Dr. Gelfand said, including the fact that it costs 10-100 times less than biologics for psoriasis and that office-based phototherapy is not available in 90% of counties in the United States. However, insurance coverage of home phototherapy “is highly variable because until the LITE study, there was no large-scale US data to support its use,” he told this news organization.
“Also, insurance companies are broken up into two parts: Durable medical goods and the medical side such as pharmacy costs, and they are siloed. The durable medical goods side views phototherapy as expensive, while the pharmacy side views it as dirt cheap. This is part of the problem with our health system. A lot of things are siloed and don’t make any sense,” said Dr. Gelfand, director of the Psoriasis and Phototherapy Treatment Center at the University of Pennsylvania. By working with the NPF and payers, he added, “we’re hoping ... to transform the way insurance companies think about covering home phototherapy.”
In the meantime, he and Dr. Elmets shared practical ways to optimize access to home phototherapy for psoriasis patients:
Have the discussion. Patients “rarely bring this up as an option,” Dr. Elmets said, so the onus is on clinicians to talk about it. In his view, the ideal candidate “is averse to using systemic agents but whose disease is beyond the point where topical medicines alone will work. One of the advantages of phototherapy is that it doesn’t have immunosuppressive effects.”
Clinicians and patients can learn about the efficacy and safety of phototherapy for psoriasis, including home-based options, on the NPF’s web site and by reading the 2019 joint AAD-NPF guidelines.
Shared decision-making is key. “When a patient comes in, I’ll discuss what their treatment options are and [we] will decide upon a course of action based on their unique needs and preferences [and] if it’s medically appropriate, meaning they have the type of psoriasis likely to respond to phototherapy,” Dr. Gelfand said. A patient with psoriasis mainly on the fingernails or genitals “is not a good candidate for phototherapy. If it’s on the trunk or extremities, that patient would be a good candidate.”
Home phototherapy candidates also must be willing and able to operate a machine and have dedicated space in their dwelling for it (most units are about the size of a door). Patients also have to be reliable, follow directions, and come back in person for follow-up appointments “so we can assess their response to treatment and fine-tune things as necessary and make sure they’re not developing any skin damage,” Dr. Gelfand said.
Educate yourself about existing options. Home phototherapy units from manufacturers such as Daavlin, National Biological Corporation, and SolRx range between $1200 and $6000 in cost, Dr. Gelfand said. He and his colleagues used the Daavlin 7 series in the LITE study. That unit features an integrated dosimetry system that delivers the correct dose of energy based on parameters that the prescribing clinician recommends. Settings are based on the patient’s skin type and how much the prescriber wants to increase the dose for each treatment. “The machine does the rest,” he said. “It knows what dose to give, so they get the same dosing as they would in an office situation.”
Smaller home-based phototherapy units designed to treat the hands and feet are available. So are handheld units to treat the scalp. “These can be a nice option for patients who have a few spots, but if the disease is moderate to severe, then it’s going to be pretty laborious to [use them],” Dr. Elmets said.
Remember that phototherapy is not a cure-all. According to the joint AAD-NPF guidelines, most phototherapy regimens require treatments two to three times per week for 10-14 weeks. Once patients achieve their home phototherapy treatment goal, Dr. Elmets often recommends treatments one to two times per week for maintenance.
“Patients with psoriasis have a lifetime condition,” he noted. “There are certainly cases where people have gone on phototherapy, cleared, and then stopped for a period of time. If they flare up, they can always go back to phototherapy. Usually, people who are on phototherapy use some type of topical agents to touch up areas that are resistant.”
Expect pushback from insurers on coverage. While Medicare and some integrated health plans cover home phototherapy, expect to spend time writing letters or placing phone calls to insurance companies to convince them why they should cover home phototherapy for candidate psoriasis patients. “Usually there’s a lot of letter writing and a long delay in getting approval,” Dr. Elmets said.
Dr. Elmets and Dr. Gelfand reported no relevant financial relationships. The LITE study was funded by the Patient-Centered Outcomes Research Institute. Research partners included the National Psoriasis Foundation and Daavlin, which provided the home phototherapy machines and covered the cost of shipping the devices.
A version of this article appeared on Medscape.com.
Merkel Cell: Immunotherapy Not Used for Many Patients With Metastatic Disease
PHOENIX — Immunotherapy has revolutionized outcomes for patients are better at high-volume centers.
The study has important implications, said study author Shayan Cheraghlou, MD, an incoming fellow in Mohs surgery at New York University, New York City. “We can see that in a real-world setting, these agents have an impact on survival,” he said. “We also found high-volume centers were significantly more likely to use the agents than low-volume centers.” He presented the findings at the annual meeting of the American College of Mohs Surgery.
MCC is a neuroendocrine skin cancer with a high rate of mortality, and even though it remains relatively rare, its incidence has been rising rapidly since the late 1990s and continues to increase. There were no approved treatments available until 2017, when the US Food and Drug Administration (FDA) approved the immunotherapy drug avelumab (Bavencio) to treat advanced MCC. Two years later, pembrolizumab (Keytruda) also received regulatory approval for MCC, and these two agents have revolutionized outcomes.
“In clinical trial settings, these agents led to significant and durable responses, and they are now the recommended treatments in guidelines for metastatic Merkel cell carcinoma,” said Dr. Cheraghlou. “However, we don’t have data as to how they are being used in the real-world setting and if survival outcomes are similar.”
Real World vs Clinical Trials
Real-world outcomes can differ from clinical trial data, and the adoption of novel therapeutics can be gradual. The goal of this study was to see if clinical trial data matched what was being observed in actual clinical use and if the agents were being used uniformly in centers across the United States.
The authors used data from the National Cancer Database that included patients diagnosed with cancer from 2004 to 2019 and identified 1017 adult cases of metastatic MCC. They then looked at the association of a variety of patient characteristics, tumors, and system factors with the likelihood of receiving systemic treatment for their disease.
“Our first finding was maybe the least surprising,” he said. “Patients who received these therapeutic agents had significantly improved survival compared to those who have not.”
Those who received immunotherapy had a 35% decrease in the risk for death per year compared with those who did not. The 1-, 3-, and 5-year survival rates were 47.2%, 21.8%, and 16.5%, respectively, for patients who did not receive immunotherapy compared with 62.7%, 34.4%, and 23.6%, respectively, for those who were treated with these agents.
Dr. Cheraghlou noted that they started to get some “surprising” findings when they looked at utilization data. “While it has been increasing over time, it is not as high as it should be,” he emphasized.
From 2017 to 2019, 54.2% of patients with metastatic MCC received immunotherapy. The data also showed an increase in use from 45.1% in 2017 to 63.0% in 2019. “This is an effective treatment for aggressive malignancy, so we have to ask why more patients aren’t getting them,” said Dr. Cheraghlou.
Their findings did suggest one possible reason, and that was that high-volume centers were significantly more likely to use the agents than low-volume centers. Centers that were in the top percentile for MCC case volume were three times as likely to use immunotherapy for MCC compared with other institutions. “So, if you have metastatic Merkel cell carcinoma and go to a low volume center, you may be less likely to get potential lifesaving treatment,” he noted.
Implications Going Forward
Dr. Cheraghlou concluded his presentation by pointing out that this study has important implications. The data showed that in a real-world setting, these agents have an impact on survival, but all eligible patients do not have access. “In other countries, there are established referral patterns for all patients with aggressive rare malignancies and really all cancers,” he added. “But in the US, cancer care is more decentralized. Studies like this and others show that high-volume centers have much better outcomes for aggressive rare malignancies, and we should be looking at why this is the case and mitigating these disparities and outcomes.”
Commenting on the study results, Jeffrey M. Farma, MD, co-director of the Melanoma and Skin Cancer Program and professor of surgical oncology at Fox Chase Cancer Center, Philadelphia, referred to the two immunotherapies that have been approved for MCC since 2017, which have demonstrated a survival benefit and improved outcomes in patients with metastatic MCC.

“In their study, immunotherapy was associated with improved outcomes,” said Dr. Farma. “This study highlights the continued lag of implementation of guidelines when new therapies are approved, and that for rare cancers like Merkel cell carcinoma, being treated at high-volume centers and the regionalization of care can lead to improved outcomes for patients.”
Dr. Cheraghlou and Dr. Farma had no disclosures.
A version of this article appeared on Medscape.com.
PHOENIX — Immunotherapy has revolutionized outcomes for patients are better at high-volume centers.
The study has important implications, said study author Shayan Cheraghlou, MD, an incoming fellow in Mohs surgery at New York University, New York City. “We can see that in a real-world setting, these agents have an impact on survival,” he said. “We also found high-volume centers were significantly more likely to use the agents than low-volume centers.” He presented the findings at the annual meeting of the American College of Mohs Surgery.
MCC is a neuroendocrine skin cancer with a high rate of mortality, and even though it remains relatively rare, its incidence has been rising rapidly since the late 1990s and continues to increase. There were no approved treatments available until 2017, when the US Food and Drug Administration (FDA) approved the immunotherapy drug avelumab (Bavencio) to treat advanced MCC. Two years later, pembrolizumab (Keytruda) also received regulatory approval for MCC, and these two agents have revolutionized outcomes.
“In clinical trial settings, these agents led to significant and durable responses, and they are now the recommended treatments in guidelines for metastatic Merkel cell carcinoma,” said Dr. Cheraghlou. “However, we don’t have data as to how they are being used in the real-world setting and if survival outcomes are similar.”
Real World vs Clinical Trials
Real-world outcomes can differ from clinical trial data, and the adoption of novel therapeutics can be gradual. The goal of this study was to see if clinical trial data matched what was being observed in actual clinical use and if the agents were being used uniformly in centers across the United States.
The authors used data from the National Cancer Database that included patients diagnosed with cancer from 2004 to 2019 and identified 1017 adult cases of metastatic MCC. They then looked at the association of a variety of patient characteristics, tumors, and system factors with the likelihood of receiving systemic treatment for their disease.
“Our first finding was maybe the least surprising,” he said. “Patients who received these therapeutic agents had significantly improved survival compared to those who have not.”
Those who received immunotherapy had a 35% decrease in the risk for death per year compared with those who did not. The 1-, 3-, and 5-year survival rates were 47.2%, 21.8%, and 16.5%, respectively, for patients who did not receive immunotherapy compared with 62.7%, 34.4%, and 23.6%, respectively, for those who were treated with these agents.
Dr. Cheraghlou noted that they started to get some “surprising” findings when they looked at utilization data. “While it has been increasing over time, it is not as high as it should be,” he emphasized.
From 2017 to 2019, 54.2% of patients with metastatic MCC received immunotherapy. The data also showed an increase in use from 45.1% in 2017 to 63.0% in 2019. “This is an effective treatment for aggressive malignancy, so we have to ask why more patients aren’t getting them,” said Dr. Cheraghlou.
Their findings did suggest one possible reason, and that was that high-volume centers were significantly more likely to use the agents than low-volume centers. Centers that were in the top percentile for MCC case volume were three times as likely to use immunotherapy for MCC compared with other institutions. “So, if you have metastatic Merkel cell carcinoma and go to a low volume center, you may be less likely to get potential lifesaving treatment,” he noted.
Implications Going Forward
Dr. Cheraghlou concluded his presentation by pointing out that this study has important implications. The data showed that in a real-world setting, these agents have an impact on survival, but all eligible patients do not have access. “In other countries, there are established referral patterns for all patients with aggressive rare malignancies and really all cancers,” he added. “But in the US, cancer care is more decentralized. Studies like this and others show that high-volume centers have much better outcomes for aggressive rare malignancies, and we should be looking at why this is the case and mitigating these disparities and outcomes.”
Commenting on the study results, Jeffrey M. Farma, MD, co-director of the Melanoma and Skin Cancer Program and professor of surgical oncology at Fox Chase Cancer Center, Philadelphia, referred to the two immunotherapies that have been approved for MCC since 2017, which have demonstrated a survival benefit and improved outcomes in patients with metastatic MCC.
“In their study, immunotherapy was associated with improved outcomes,” said Dr. Farma. “This study highlights the continued lag of implementation of guidelines when new therapies are approved, and that for rare cancers like Merkel cell carcinoma, being treated at high-volume centers and the regionalization of care can lead to improved outcomes for patients.”
Dr. Cheraghlou and Dr. Farma had no disclosures.
A version of this article appeared on Medscape.com.
PHOENIX — Immunotherapy has revolutionized outcomes for patients are better at high-volume centers.
The study has important implications, said study author Shayan Cheraghlou, MD, an incoming fellow in Mohs surgery at New York University, New York City. “We can see that in a real-world setting, these agents have an impact on survival,” he said. “We also found high-volume centers were significantly more likely to use the agents than low-volume centers.” He presented the findings at the annual meeting of the American College of Mohs Surgery.
MCC is a neuroendocrine skin cancer with a high rate of mortality, and even though it remains relatively rare, its incidence has been rising rapidly since the late 1990s and continues to increase. There were no approved treatments available until 2017, when the US Food and Drug Administration (FDA) approved the immunotherapy drug avelumab (Bavencio) to treat advanced MCC. Two years later, pembrolizumab (Keytruda) also received regulatory approval for MCC, and these two agents have revolutionized outcomes.
“In clinical trial settings, these agents led to significant and durable responses, and they are now the recommended treatments in guidelines for metastatic Merkel cell carcinoma,” said Dr. Cheraghlou. “However, we don’t have data as to how they are being used in the real-world setting and if survival outcomes are similar.”
Real World vs Clinical Trials
Real-world outcomes can differ from clinical trial data, and the adoption of novel therapeutics can be gradual. The goal of this study was to see if clinical trial data matched what was being observed in actual clinical use and if the agents were being used uniformly in centers across the United States.
The authors used data from the National Cancer Database that included patients diagnosed with cancer from 2004 to 2019 and identified 1017 adult cases of metastatic MCC. They then looked at the association of a variety of patient characteristics, tumors, and system factors with the likelihood of receiving systemic treatment for their disease.
“Our first finding was maybe the least surprising,” he said. “Patients who received these therapeutic agents had significantly improved survival compared to those who have not.”
Those who received immunotherapy had a 35% decrease in the risk for death per year compared with those who did not. The 1-, 3-, and 5-year survival rates were 47.2%, 21.8%, and 16.5%, respectively, for patients who did not receive immunotherapy compared with 62.7%, 34.4%, and 23.6%, respectively, for those who were treated with these agents.
Dr. Cheraghlou noted that they started to get some “surprising” findings when they looked at utilization data. “While it has been increasing over time, it is not as high as it should be,” he emphasized.
From 2017 to 2019, 54.2% of patients with metastatic MCC received immunotherapy. The data also showed an increase in use from 45.1% in 2017 to 63.0% in 2019. “This is an effective treatment for aggressive malignancy, so we have to ask why more patients aren’t getting them,” said Dr. Cheraghlou.
Their findings did suggest one possible reason, and that was that high-volume centers were significantly more likely to use the agents than low-volume centers. Centers that were in the top percentile for MCC case volume were three times as likely to use immunotherapy for MCC compared with other institutions. “So, if you have metastatic Merkel cell carcinoma and go to a low volume center, you may be less likely to get potential lifesaving treatment,” he noted.
Implications Going Forward
Dr. Cheraghlou concluded his presentation by pointing out that this study has important implications. The data showed that in a real-world setting, these agents have an impact on survival, but all eligible patients do not have access. “In other countries, there are established referral patterns for all patients with aggressive rare malignancies and really all cancers,” he added. “But in the US, cancer care is more decentralized. Studies like this and others show that high-volume centers have much better outcomes for aggressive rare malignancies, and we should be looking at why this is the case and mitigating these disparities and outcomes.”
Commenting on the study results, Jeffrey M. Farma, MD, co-director of the Melanoma and Skin Cancer Program and professor of surgical oncology at Fox Chase Cancer Center, Philadelphia, referred to the two immunotherapies that have been approved for MCC since 2017, which have demonstrated a survival benefit and improved outcomes in patients with metastatic MCC.
“In their study, immunotherapy was associated with improved outcomes,” said Dr. Farma. “This study highlights the continued lag of implementation of guidelines when new therapies are approved, and that for rare cancers like Merkel cell carcinoma, being treated at high-volume centers and the regionalization of care can lead to improved outcomes for patients.”
Dr. Cheraghlou and Dr. Farma had no disclosures.
A version of this article appeared on Medscape.com.
FROM ACMS 2024