User login
Ocular complications of dermatologic treatments: Advice from a pediatric ophthalmologist
ASHEVILLE, N.C. – The, according to one of several clinical messages from a pediatric ophthalmologist who spoke at the annual meeting of the Society for Pediatric Dermatology.
“There is a lot of steroid fear out there, which you can argue is actually harmful in itself, because not treating periorbital eczema is related to a lot of eye problems, including chronic discomfort and the eye rubbing that can cause corneal abrasions and keratoconus,” said Sara Grace, MD, a pediatric ophthalmologist who is on the clinical staff at Duke University, Durham, N.C. She maintains a practice at North Carolina Eye, Ear, Nose, and Throat in Durham.
Although the risks of periorbital steroid absorption are real, a limited course of low potency topical steroids is generally adequate for common periorbital indications, and these appear to be safe.
“There is insufficient evidence to link weak periocular topical corticosteroids such as desonide or hydrocortisone with ocular complications,” said Dr. Grace, suggesting that pediatric dermatologists can be reassured when using these medications at low concentrations.
“Potent periocular steroids have been associated with ocular complications, but this has typically involved exposures over months to years,” Dr. Grace specified.
When topical corticosteroids are applied at high concentrations on the face away from the periorbital area, glaucoma and other feared ophthalmic complications cannot be entirely ruled out, but, again, the risk is low in the absence of “very large quantities” of potent topical agents applied for lengthy periods of time, according to Dr. Grace, basing this observation on case studies.
In children, as in adults, the potential exception is a child with existing ocular disease. In such cases, or in children with risk factors for ocular disease, Dr. Grace recommends referral to an ophthalmologist for a baseline examination prior to a course of topical corticosteroids with the potential of periocular absorption. With a baseline assessment, adverse effects are more easily documented if exposure is prolonged.
The message, although not identical, is similar for use of dupilumab (Dupixent) or other biologics that target the interleukin-13 (IL-13) pathway. The potential for complications cannot be ignored but these are often time-limited and the benefit is likely to exceed the risk in children who have severe atopic dermatitis or other skin conditions for which these treatments are effective.
There are several potential mechanisms by which biologics targeting IL-13 might increase risk of ocular complications, one of which is the role that IL-13 plays in ocular mucus production, regulation of conjunctival goblet cells, and tear production, according to several published reports.
“Up to 30% of children will get some type of eye complication but, fortunately, most of them will not have to stop therapy,” Dr. Grace said. These side effects include conjunctivitis, blepharitis, keratitis, dry eye, and itching, but they are typically manageable. Topical steroids or calcineurin inhibitors can be offered if needed, but many of these conditions will self-resolve. Dr. Grace estimated that less than 1% of patients need to stop treatment because of ophthalmic side effects.
Lesions that obstruct vision
Dr. Grace urged pediatric dermatologists to be aware of the risk for amblyopia in young children with lesions that obstruct vision in one eye. In early development, prolonged obstruction of vision in one eye can alter neural communication with the brain, producing permanent vision impairment.
She explained that clearing the obstructed vision, whether from a capillary hemangioma or any periorbital growth, should be considered urgent to avoid irreversible damage.
Similarly, periorbital port-wine stains associated with Sturge-Weber syndrome, which is primarily a vascular disorder that predisposes children to glaucoma, represents a condition that requires prompt attention. Sturge-Weber syndrome is often but not always identified at birth, but it is a condition for which evaluation and treatment should involve the participation of an ophthalmologist.
Meibomian gland disease is another disorder that is often seen first by a pediatric dermatologist but also requires collaborative management. The challenge is sorting out the underlying cause or causes and initiating a therapy that unclogs the gland without having to resort to incision and drainage.
“Drainage is hard to do and is not necessarily effective,” explained Dr. Grace. While scrubs, warmth, and massage frequently are adequate to unclog the gland – which secretes meibum, a complex of lipids that perform several functions in protecting the eye – therapies specific to the cause, such as Demodex-related blepharitis, chalazions, and styes, might be needed.
Dr. Grace indicated that patience is often needed. The process of unclogging these glands often takes time, but she emphasized that a first-line conservative approach is always appropriate to avoid the difficulty and potential problems of incisions.
In general, these messages are not novel, but they provide a refresher for pediatric dermatologists who do not regularly confront complications that involve the eyes. According to session moderator, Elizabeth Neiman, MD, assistant professor of pediatric dermatology, University of North Carolina at Chapel Hill, the messages regarding topical steroids on the face and the eyes are “important” and worth emphasizing.
“It’s useful to reinforce the point that corticosteroids should be used when needed in the periorbital area [to control skin diseases] if they are used in low concentrations,” Dr. Neiman told this news organization.
Similarly, conjunctivitis and other ocular complications of dupilumab are a source of concern for parents as well as dermatologists. Dr. Neiman indicated that a review of the benefit-to-risk ratio is important when considering these treatments in patients with indications for severe skin disorders.
Dr. Grace and Dr. Nieman have no potential financial conflicts related to this topic.
A version of this article first appeared on Medscape.com.
ASHEVILLE, N.C. – The, according to one of several clinical messages from a pediatric ophthalmologist who spoke at the annual meeting of the Society for Pediatric Dermatology.
“There is a lot of steroid fear out there, which you can argue is actually harmful in itself, because not treating periorbital eczema is related to a lot of eye problems, including chronic discomfort and the eye rubbing that can cause corneal abrasions and keratoconus,” said Sara Grace, MD, a pediatric ophthalmologist who is on the clinical staff at Duke University, Durham, N.C. She maintains a practice at North Carolina Eye, Ear, Nose, and Throat in Durham.
Although the risks of periorbital steroid absorption are real, a limited course of low potency topical steroids is generally adequate for common periorbital indications, and these appear to be safe.
“There is insufficient evidence to link weak periocular topical corticosteroids such as desonide or hydrocortisone with ocular complications,” said Dr. Grace, suggesting that pediatric dermatologists can be reassured when using these medications at low concentrations.
“Potent periocular steroids have been associated with ocular complications, but this has typically involved exposures over months to years,” Dr. Grace specified.
When topical corticosteroids are applied at high concentrations on the face away from the periorbital area, glaucoma and other feared ophthalmic complications cannot be entirely ruled out, but, again, the risk is low in the absence of “very large quantities” of potent topical agents applied for lengthy periods of time, according to Dr. Grace, basing this observation on case studies.
In children, as in adults, the potential exception is a child with existing ocular disease. In such cases, or in children with risk factors for ocular disease, Dr. Grace recommends referral to an ophthalmologist for a baseline examination prior to a course of topical corticosteroids with the potential of periocular absorption. With a baseline assessment, adverse effects are more easily documented if exposure is prolonged.
The message, although not identical, is similar for use of dupilumab (Dupixent) or other biologics that target the interleukin-13 (IL-13) pathway. The potential for complications cannot be ignored but these are often time-limited and the benefit is likely to exceed the risk in children who have severe atopic dermatitis or other skin conditions for which these treatments are effective.
There are several potential mechanisms by which biologics targeting IL-13 might increase risk of ocular complications, one of which is the role that IL-13 plays in ocular mucus production, regulation of conjunctival goblet cells, and tear production, according to several published reports.
“Up to 30% of children will get some type of eye complication but, fortunately, most of them will not have to stop therapy,” Dr. Grace said. These side effects include conjunctivitis, blepharitis, keratitis, dry eye, and itching, but they are typically manageable. Topical steroids or calcineurin inhibitors can be offered if needed, but many of these conditions will self-resolve. Dr. Grace estimated that less than 1% of patients need to stop treatment because of ophthalmic side effects.
Lesions that obstruct vision
Dr. Grace urged pediatric dermatologists to be aware of the risk for amblyopia in young children with lesions that obstruct vision in one eye. In early development, prolonged obstruction of vision in one eye can alter neural communication with the brain, producing permanent vision impairment.
She explained that clearing the obstructed vision, whether from a capillary hemangioma or any periorbital growth, should be considered urgent to avoid irreversible damage.
Similarly, periorbital port-wine stains associated with Sturge-Weber syndrome, which is primarily a vascular disorder that predisposes children to glaucoma, represents a condition that requires prompt attention. Sturge-Weber syndrome is often but not always identified at birth, but it is a condition for which evaluation and treatment should involve the participation of an ophthalmologist.
Meibomian gland disease is another disorder that is often seen first by a pediatric dermatologist but also requires collaborative management. The challenge is sorting out the underlying cause or causes and initiating a therapy that unclogs the gland without having to resort to incision and drainage.
“Drainage is hard to do and is not necessarily effective,” explained Dr. Grace. While scrubs, warmth, and massage frequently are adequate to unclog the gland – which secretes meibum, a complex of lipids that perform several functions in protecting the eye – therapies specific to the cause, such as Demodex-related blepharitis, chalazions, and styes, might be needed.
Dr. Grace indicated that patience is often needed. The process of unclogging these glands often takes time, but she emphasized that a first-line conservative approach is always appropriate to avoid the difficulty and potential problems of incisions.
In general, these messages are not novel, but they provide a refresher for pediatric dermatologists who do not regularly confront complications that involve the eyes. According to session moderator, Elizabeth Neiman, MD, assistant professor of pediatric dermatology, University of North Carolina at Chapel Hill, the messages regarding topical steroids on the face and the eyes are “important” and worth emphasizing.
“It’s useful to reinforce the point that corticosteroids should be used when needed in the periorbital area [to control skin diseases] if they are used in low concentrations,” Dr. Neiman told this news organization.
Similarly, conjunctivitis and other ocular complications of dupilumab are a source of concern for parents as well as dermatologists. Dr. Neiman indicated that a review of the benefit-to-risk ratio is important when considering these treatments in patients with indications for severe skin disorders.
Dr. Grace and Dr. Nieman have no potential financial conflicts related to this topic.
A version of this article first appeared on Medscape.com.
ASHEVILLE, N.C. – The, according to one of several clinical messages from a pediatric ophthalmologist who spoke at the annual meeting of the Society for Pediatric Dermatology.
“There is a lot of steroid fear out there, which you can argue is actually harmful in itself, because not treating periorbital eczema is related to a lot of eye problems, including chronic discomfort and the eye rubbing that can cause corneal abrasions and keratoconus,” said Sara Grace, MD, a pediatric ophthalmologist who is on the clinical staff at Duke University, Durham, N.C. She maintains a practice at North Carolina Eye, Ear, Nose, and Throat in Durham.
Although the risks of periorbital steroid absorption are real, a limited course of low potency topical steroids is generally adequate for common periorbital indications, and these appear to be safe.
“There is insufficient evidence to link weak periocular topical corticosteroids such as desonide or hydrocortisone with ocular complications,” said Dr. Grace, suggesting that pediatric dermatologists can be reassured when using these medications at low concentrations.
“Potent periocular steroids have been associated with ocular complications, but this has typically involved exposures over months to years,” Dr. Grace specified.
When topical corticosteroids are applied at high concentrations on the face away from the periorbital area, glaucoma and other feared ophthalmic complications cannot be entirely ruled out, but, again, the risk is low in the absence of “very large quantities” of potent topical agents applied for lengthy periods of time, according to Dr. Grace, basing this observation on case studies.
In children, as in adults, the potential exception is a child with existing ocular disease. In such cases, or in children with risk factors for ocular disease, Dr. Grace recommends referral to an ophthalmologist for a baseline examination prior to a course of topical corticosteroids with the potential of periocular absorption. With a baseline assessment, adverse effects are more easily documented if exposure is prolonged.
The message, although not identical, is similar for use of dupilumab (Dupixent) or other biologics that target the interleukin-13 (IL-13) pathway. The potential for complications cannot be ignored but these are often time-limited and the benefit is likely to exceed the risk in children who have severe atopic dermatitis or other skin conditions for which these treatments are effective.
There are several potential mechanisms by which biologics targeting IL-13 might increase risk of ocular complications, one of which is the role that IL-13 plays in ocular mucus production, regulation of conjunctival goblet cells, and tear production, according to several published reports.
“Up to 30% of children will get some type of eye complication but, fortunately, most of them will not have to stop therapy,” Dr. Grace said. These side effects include conjunctivitis, blepharitis, keratitis, dry eye, and itching, but they are typically manageable. Topical steroids or calcineurin inhibitors can be offered if needed, but many of these conditions will self-resolve. Dr. Grace estimated that less than 1% of patients need to stop treatment because of ophthalmic side effects.
Lesions that obstruct vision
Dr. Grace urged pediatric dermatologists to be aware of the risk for amblyopia in young children with lesions that obstruct vision in one eye. In early development, prolonged obstruction of vision in one eye can alter neural communication with the brain, producing permanent vision impairment.
She explained that clearing the obstructed vision, whether from a capillary hemangioma or any periorbital growth, should be considered urgent to avoid irreversible damage.
Similarly, periorbital port-wine stains associated with Sturge-Weber syndrome, which is primarily a vascular disorder that predisposes children to glaucoma, represents a condition that requires prompt attention. Sturge-Weber syndrome is often but not always identified at birth, but it is a condition for which evaluation and treatment should involve the participation of an ophthalmologist.
Meibomian gland disease is another disorder that is often seen first by a pediatric dermatologist but also requires collaborative management. The challenge is sorting out the underlying cause or causes and initiating a therapy that unclogs the gland without having to resort to incision and drainage.
“Drainage is hard to do and is not necessarily effective,” explained Dr. Grace. While scrubs, warmth, and massage frequently are adequate to unclog the gland – which secretes meibum, a complex of lipids that perform several functions in protecting the eye – therapies specific to the cause, such as Demodex-related blepharitis, chalazions, and styes, might be needed.
Dr. Grace indicated that patience is often needed. The process of unclogging these glands often takes time, but she emphasized that a first-line conservative approach is always appropriate to avoid the difficulty and potential problems of incisions.
In general, these messages are not novel, but they provide a refresher for pediatric dermatologists who do not regularly confront complications that involve the eyes. According to session moderator, Elizabeth Neiman, MD, assistant professor of pediatric dermatology, University of North Carolina at Chapel Hill, the messages regarding topical steroids on the face and the eyes are “important” and worth emphasizing.
“It’s useful to reinforce the point that corticosteroids should be used when needed in the periorbital area [to control skin diseases] if they are used in low concentrations,” Dr. Neiman told this news organization.
Similarly, conjunctivitis and other ocular complications of dupilumab are a source of concern for parents as well as dermatologists. Dr. Neiman indicated that a review of the benefit-to-risk ratio is important when considering these treatments in patients with indications for severe skin disorders.
Dr. Grace and Dr. Nieman have no potential financial conflicts related to this topic.
A version of this article first appeared on Medscape.com.
AT SPD 2023
Case report describes pediatric RIME triggered by norovirus
, according to a newly published case report.
Lead author Anna Yasmine Kirkorian, MD, chief of dermatology at Children’s National Hospital in Washington, said she wanted to get the word out in part because it seems like RIME is occurring more frequently. “I do feel like we’re seeing more cases and from a more diverse number of pathogens,” Dr. Kirkorian told this news organization.

There was a decrease in RIME during the early stages of the COVID-19 pandemic when people were isolating more, Dr. Kirkorian said. SARS-CoV-2 has been a trigger for some cases, but she did not find that remarkable, given that respiratory viruses are known RIME precursors. The question is why RIME is being triggered more frequently now that people have essentially gone back to their normal lives, she said.
Dr. Kirkorian and colleagues at Children’s National Hospital and George Washington University, Washington, wrote about a 5-year-old boy with norovirus-triggered RIME in a case report published in Pediatric Dermatology.
RIME – previously known as Mycoplasma pneumoniae–induced rash and mucositis (MIRM) – tends to arise after a viral infection, with upper respiratory viruses such as mycoplasma and Chlamydophila pneumoniae, influenza, and enterovirus among the common triggers. “We think this is actually your own immune system overreacting to a pathogen,” Dr. Kirkorian said in an interview, adding that the mechanism of RIME is still not understood.
While the norovirus discovery was a surprise, it shows that much is still unknown about this rare condition. “I don’t think we know what is usual and what is unusual,” Dr. Kirkorian said.
In this case, the boy swiftly declined, with progressive conjunctivitis, high fever, and rapidly developing mucositis. By the time the 5-year-old got to Children’s National Hospital, he had a spreading, painful rash, including tense vesicles and bullae involving more than 30% of his total body surface area, and areas of denuded skin on both cheeks and the back of his neck.
He had hemorrhagic mucositis of the lips, a large erosion at the urethral meatus, and hemorrhagic conjunctivitis of both eyes with thick yellow crusting on the eyelids.
The clinicians intubated the boy and admitted him to the intensive care unit. He was given a one-time injection of etanercept (25 mg) followed by 8 days of intravenous cyclosporine at a dose of 5 mg per kilogram, divided twice daily, which helped calm the mucositis and stopped the rash from progressing. There is not an accepted protocol or list of evidence-based therapeutics for RIME, Dr. Kirkorian noted.
The severe eye damage required amniotic membrane grafts. The patient was extubated after 9 days but remained in the hospital for a total of 26 days because he needed to receive nutritional support (the mucositis kept him from eating), and for pain control and weaning of sedation.
As the clinicians searched for a potential triggering virus, they came up empty. Results were negative for adenovirus, Epstein Barr virus, cytomegalovirus, herpes simplex, and varicella zoster. But they noted that the child’s household contacts had all been sick a week before with presumed viral gastroenteritis. They decided to run a stool screen and the polymerase chain reaction for norovirus was positive. The boy never had GI symptoms.
Dr. Kirkorian said in the interview that she has seen other RIME cases where a child did not have symptoms associated with the original virus but did have a sudden onset of mucositis.
Although the definition of RIME is evolving, it is defined in part by mucositis in at least two of three areas: the mouth, eyes, and genitals. “Once you have the inflammation of the mucous membranes you should be on alert to think about more serious conditions,” like RIME, said Dr. Kirkorian. “Why does it manifest with the mucositis? I don’t think we know that,” she added.
RIME recurrence has also been vexing for patients, families and clinicians. In May, at the annual Atlantic Dermatology Conference, held in Baltimore, Dr. Kirkorian also discussed an 11-year-old patient who had RIME after SARS-CoV-2 infection early in the pandemic, resulting in a 22-day hospitalization and placement of a peripherally inserted central catheter and a feeding tube. He improved with cyclosporine and was discharged on systemic tacrolimus.
He was fine for several years, until another COVID infection. He again responded to medication. But not long after, an undetermined viral infection triggered another episode of RIME.
Dr. Kirkorian said there is no way to predict recurrence – making a devastating condition all the more worrisome. “Knowing that it might come back and it’s totally haphazard as to what might make it come back – that is very stressful for families,” she said in the interview.
“Some of the most perplexing patients with RIME are those with recurrent disease,” wrote Warren R. Heymann, MD, professor of dermatology and pediatrics at Rowan University, Camden, N.J., wrote in an online column on RIME in the American Academy of Dermatology’s “Dermatology World Insights and Inquiries”.
“Recurrent RIME is of particular interest, given that we could potentially intervene and prevent additional disease,” wrote Camille Introcaso, MD, associate professor of medicine at Rowan University, in response to Dr. Heymann’s remarks. “Although multiple possible mechanisms for the clinical findings of RIME have been proposed, including molecular mimicry between infectious agent proteins and keratinocyte antigens, immune complex deposition, and combinations of medication and infection, the pathophysiology is unknown,” she added.
In the interview, Dr. Kirkorian said that she and colleagues in the Pediatric Dermatology Research Alliance (PeDRA) are trying to assemble more multicenter trials to assess the underlying pathology of RIME, effectiveness of various treatments, and to “find some predictive factors.” Given that RIME is an acute-onset emergency, it is not easy to conduct randomized controlled trials, she added.
Dr. Kirkorian, Dr. Heymann, and Dr. Introcaso report no relevant financial relationships.
, according to a newly published case report.
Lead author Anna Yasmine Kirkorian, MD, chief of dermatology at Children’s National Hospital in Washington, said she wanted to get the word out in part because it seems like RIME is occurring more frequently. “I do feel like we’re seeing more cases and from a more diverse number of pathogens,” Dr. Kirkorian told this news organization.
There was a decrease in RIME during the early stages of the COVID-19 pandemic when people were isolating more, Dr. Kirkorian said. SARS-CoV-2 has been a trigger for some cases, but she did not find that remarkable, given that respiratory viruses are known RIME precursors. The question is why RIME is being triggered more frequently now that people have essentially gone back to their normal lives, she said.
Dr. Kirkorian and colleagues at Children’s National Hospital and George Washington University, Washington, wrote about a 5-year-old boy with norovirus-triggered RIME in a case report published in Pediatric Dermatology.
RIME – previously known as Mycoplasma pneumoniae–induced rash and mucositis (MIRM) – tends to arise after a viral infection, with upper respiratory viruses such as mycoplasma and Chlamydophila pneumoniae, influenza, and enterovirus among the common triggers. “We think this is actually your own immune system overreacting to a pathogen,” Dr. Kirkorian said in an interview, adding that the mechanism of RIME is still not understood.
While the norovirus discovery was a surprise, it shows that much is still unknown about this rare condition. “I don’t think we know what is usual and what is unusual,” Dr. Kirkorian said.
In this case, the boy swiftly declined, with progressive conjunctivitis, high fever, and rapidly developing mucositis. By the time the 5-year-old got to Children’s National Hospital, he had a spreading, painful rash, including tense vesicles and bullae involving more than 30% of his total body surface area, and areas of denuded skin on both cheeks and the back of his neck.
He had hemorrhagic mucositis of the lips, a large erosion at the urethral meatus, and hemorrhagic conjunctivitis of both eyes with thick yellow crusting on the eyelids.
The clinicians intubated the boy and admitted him to the intensive care unit. He was given a one-time injection of etanercept (25 mg) followed by 8 days of intravenous cyclosporine at a dose of 5 mg per kilogram, divided twice daily, which helped calm the mucositis and stopped the rash from progressing. There is not an accepted protocol or list of evidence-based therapeutics for RIME, Dr. Kirkorian noted.
The severe eye damage required amniotic membrane grafts. The patient was extubated after 9 days but remained in the hospital for a total of 26 days because he needed to receive nutritional support (the mucositis kept him from eating), and for pain control and weaning of sedation.
As the clinicians searched for a potential triggering virus, they came up empty. Results were negative for adenovirus, Epstein Barr virus, cytomegalovirus, herpes simplex, and varicella zoster. But they noted that the child’s household contacts had all been sick a week before with presumed viral gastroenteritis. They decided to run a stool screen and the polymerase chain reaction for norovirus was positive. The boy never had GI symptoms.
Dr. Kirkorian said in the interview that she has seen other RIME cases where a child did not have symptoms associated with the original virus but did have a sudden onset of mucositis.
Although the definition of RIME is evolving, it is defined in part by mucositis in at least two of three areas: the mouth, eyes, and genitals. “Once you have the inflammation of the mucous membranes you should be on alert to think about more serious conditions,” like RIME, said Dr. Kirkorian. “Why does it manifest with the mucositis? I don’t think we know that,” she added.
RIME recurrence has also been vexing for patients, families and clinicians. In May, at the annual Atlantic Dermatology Conference, held in Baltimore, Dr. Kirkorian also discussed an 11-year-old patient who had RIME after SARS-CoV-2 infection early in the pandemic, resulting in a 22-day hospitalization and placement of a peripherally inserted central catheter and a feeding tube. He improved with cyclosporine and was discharged on systemic tacrolimus.
He was fine for several years, until another COVID infection. He again responded to medication. But not long after, an undetermined viral infection triggered another episode of RIME.
Dr. Kirkorian said there is no way to predict recurrence – making a devastating condition all the more worrisome. “Knowing that it might come back and it’s totally haphazard as to what might make it come back – that is very stressful for families,” she said in the interview.
“Some of the most perplexing patients with RIME are those with recurrent disease,” wrote Warren R. Heymann, MD, professor of dermatology and pediatrics at Rowan University, Camden, N.J., wrote in an online column on RIME in the American Academy of Dermatology’s “Dermatology World Insights and Inquiries”.
“Recurrent RIME is of particular interest, given that we could potentially intervene and prevent additional disease,” wrote Camille Introcaso, MD, associate professor of medicine at Rowan University, in response to Dr. Heymann’s remarks. “Although multiple possible mechanisms for the clinical findings of RIME have been proposed, including molecular mimicry between infectious agent proteins and keratinocyte antigens, immune complex deposition, and combinations of medication and infection, the pathophysiology is unknown,” she added.
In the interview, Dr. Kirkorian said that she and colleagues in the Pediatric Dermatology Research Alliance (PeDRA) are trying to assemble more multicenter trials to assess the underlying pathology of RIME, effectiveness of various treatments, and to “find some predictive factors.” Given that RIME is an acute-onset emergency, it is not easy to conduct randomized controlled trials, she added.
Dr. Kirkorian, Dr. Heymann, and Dr. Introcaso report no relevant financial relationships.
, according to a newly published case report.
Lead author Anna Yasmine Kirkorian, MD, chief of dermatology at Children’s National Hospital in Washington, said she wanted to get the word out in part because it seems like RIME is occurring more frequently. “I do feel like we’re seeing more cases and from a more diverse number of pathogens,” Dr. Kirkorian told this news organization.
There was a decrease in RIME during the early stages of the COVID-19 pandemic when people were isolating more, Dr. Kirkorian said. SARS-CoV-2 has been a trigger for some cases, but she did not find that remarkable, given that respiratory viruses are known RIME precursors. The question is why RIME is being triggered more frequently now that people have essentially gone back to their normal lives, she said.
Dr. Kirkorian and colleagues at Children’s National Hospital and George Washington University, Washington, wrote about a 5-year-old boy with norovirus-triggered RIME in a case report published in Pediatric Dermatology.
RIME – previously known as Mycoplasma pneumoniae–induced rash and mucositis (MIRM) – tends to arise after a viral infection, with upper respiratory viruses such as mycoplasma and Chlamydophila pneumoniae, influenza, and enterovirus among the common triggers. “We think this is actually your own immune system overreacting to a pathogen,” Dr. Kirkorian said in an interview, adding that the mechanism of RIME is still not understood.
While the norovirus discovery was a surprise, it shows that much is still unknown about this rare condition. “I don’t think we know what is usual and what is unusual,” Dr. Kirkorian said.
In this case, the boy swiftly declined, with progressive conjunctivitis, high fever, and rapidly developing mucositis. By the time the 5-year-old got to Children’s National Hospital, he had a spreading, painful rash, including tense vesicles and bullae involving more than 30% of his total body surface area, and areas of denuded skin on both cheeks and the back of his neck.
He had hemorrhagic mucositis of the lips, a large erosion at the urethral meatus, and hemorrhagic conjunctivitis of both eyes with thick yellow crusting on the eyelids.
The clinicians intubated the boy and admitted him to the intensive care unit. He was given a one-time injection of etanercept (25 mg) followed by 8 days of intravenous cyclosporine at a dose of 5 mg per kilogram, divided twice daily, which helped calm the mucositis and stopped the rash from progressing. There is not an accepted protocol or list of evidence-based therapeutics for RIME, Dr. Kirkorian noted.
The severe eye damage required amniotic membrane grafts. The patient was extubated after 9 days but remained in the hospital for a total of 26 days because he needed to receive nutritional support (the mucositis kept him from eating), and for pain control and weaning of sedation.
As the clinicians searched for a potential triggering virus, they came up empty. Results were negative for adenovirus, Epstein Barr virus, cytomegalovirus, herpes simplex, and varicella zoster. But they noted that the child’s household contacts had all been sick a week before with presumed viral gastroenteritis. They decided to run a stool screen and the polymerase chain reaction for norovirus was positive. The boy never had GI symptoms.
Dr. Kirkorian said in the interview that she has seen other RIME cases where a child did not have symptoms associated with the original virus but did have a sudden onset of mucositis.
Although the definition of RIME is evolving, it is defined in part by mucositis in at least two of three areas: the mouth, eyes, and genitals. “Once you have the inflammation of the mucous membranes you should be on alert to think about more serious conditions,” like RIME, said Dr. Kirkorian. “Why does it manifest with the mucositis? I don’t think we know that,” she added.
RIME recurrence has also been vexing for patients, families and clinicians. In May, at the annual Atlantic Dermatology Conference, held in Baltimore, Dr. Kirkorian also discussed an 11-year-old patient who had RIME after SARS-CoV-2 infection early in the pandemic, resulting in a 22-day hospitalization and placement of a peripherally inserted central catheter and a feeding tube. He improved with cyclosporine and was discharged on systemic tacrolimus.
He was fine for several years, until another COVID infection. He again responded to medication. But not long after, an undetermined viral infection triggered another episode of RIME.
Dr. Kirkorian said there is no way to predict recurrence – making a devastating condition all the more worrisome. “Knowing that it might come back and it’s totally haphazard as to what might make it come back – that is very stressful for families,” she said in the interview.
“Some of the most perplexing patients with RIME are those with recurrent disease,” wrote Warren R. Heymann, MD, professor of dermatology and pediatrics at Rowan University, Camden, N.J., wrote in an online column on RIME in the American Academy of Dermatology’s “Dermatology World Insights and Inquiries”.
“Recurrent RIME is of particular interest, given that we could potentially intervene and prevent additional disease,” wrote Camille Introcaso, MD, associate professor of medicine at Rowan University, in response to Dr. Heymann’s remarks. “Although multiple possible mechanisms for the clinical findings of RIME have been proposed, including molecular mimicry between infectious agent proteins and keratinocyte antigens, immune complex deposition, and combinations of medication and infection, the pathophysiology is unknown,” she added.
In the interview, Dr. Kirkorian said that she and colleagues in the Pediatric Dermatology Research Alliance (PeDRA) are trying to assemble more multicenter trials to assess the underlying pathology of RIME, effectiveness of various treatments, and to “find some predictive factors.” Given that RIME is an acute-onset emergency, it is not easy to conduct randomized controlled trials, she added.
Dr. Kirkorian, Dr. Heymann, and Dr. Introcaso report no relevant financial relationships.

Humira biosimilars: Five things to know
The best-selling drug Humira (adalimumab) now faces competition in the United States after a 20-year monopoly. The first adalimumab biosimilar, Amjevita, launched in the United States on January 31, and in July, seven additional biosimilars became available. These drugs have the potential to lower prescription drug prices, but when and by how much remains to be seen.
Here’s what you need to know about adalimumab biosimilars.
What Humira biosimilars are now available?
Eight different biosimilars have launched in 2023 with discounts as large at 85% from Humira’s list price of $6,922. A few companies also offer two price points.
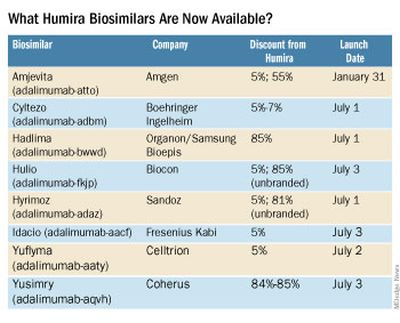
Three of these biosimilars – Hadlima, Hyrimoz, and Yuflyma – are available in high concentration formulations. This high concentration formulation makes up 85% of Humira prescriptions, according to a report from Goodroot, a collection of companies focused on lowering health care costs.
Cyltezo is currently the only adalimumab biosimilar with an interchangeability designation, meaning that a pharmacist can substitute the biosimilar for an equivalent Humira prescription without the intervention of a clinician. A total of 47 states allow for these substitutions without prior approval from a clinician, according to Goodroot, and the clinician must be notified of the switch within a certain time frame. A total of 40 states require that patients be notified of the switch before substitution.
However, it’s not clear if this interchangeability designation will prove an advantage for Cyltezo, as it is interchangeable with the lower concentration version of Humira that makes up just 15% of prescriptions.
Most of the companies behind these biosimilars are pursuing interchangeability designations for their drugs, except for Fresenius Kabi (Idacio) and Coherus (Yusimry).
A ninth biosimilar, Pfizer’s adalimumab-afzb (Abrilada), is not yet on the market and is currently awaiting an approval decision from the Food and Drug Administration to add an interchangeability designation to its prior approval for a low-concentration formulation.
Why are they priced differently?
The two price points offer different deals to payers. Pharmacy benefit managers make confidential agreements with drug manufacturers to get a discount – called a rebate – to get the drug on the PBM’s formulary. The PBM keeps a portion of that rebate, and the rest is passed on to the insurance company and patients. Biosimilars at a higher price point will likely offer larger rebates. Biosimilars offered at lower price points incorporate this discount up front in their list pricing and likely will not offer large rebates.
Will biosimilars be covered by payers?
Currently, biosimilars are being offered on formularies at parity with Humira, meaning they are on the same tier. The PBM companies OptumRx and Cigna Group’s Express Scripts will offer Amjevita (at both price points), Cyltezo, and Hyrimoz (at both price points).
“This decision allows our clients flexibility to provide access to the lower list price, so members in high-deductible plans and benefit designs with coinsurance can experience lower out-of-pocket costs,” said OptumRx spokesperson Isaac Sorensen in an email.
Mark Cuban Cost Plus Drug Company, which uses a direct-to-consumer model, will offer Yusimry for $567.27 on its website. SmithRx, a PBM based in San Francisco, announced it would partner with Cost Plus Drugs to offer Yusimry, adding that SmithRx members can use their insurance benefits to further reduce out-of-pocket costs. RxPreferred, another PBM, will also offer Yusimry through its partnership with Cuban’s company.
The news website Formulary Watch previously reported that CVS Caremark, another of the biggest PBMs, will be offering Amjevita, but as a nonpreferred brand, while Humira remains the preferred brand. CVS Caremark did not respond to a request for comment.
Will patients pay less?
Biosimilars have been touted as a potential solution to lower spending on biologic drugs, but it’s unknown if patients will ultimately benefit with lower out-of-pocket costs. It’s “impossible to predict” if the discount that third-party payers pay will be passed on to consumers, said Mark Fendrick, MD, who directs the University of Michigan Center for Value-based Insurance Design in Ann Arbor.

Generally, a consumer’s copay is a percentage of a drug’s list price, so it stands to reason that a low drug price would result in lower out-of-pocket payments. While this is mostly true, Humira has a successful copay assistance program to lower prescription costs for consumers. According to a 2022 IQVIA report, 82% of commercial prescriptions cost patients less than $10 for Humira because of this program.
To appeal to patients, biosimilar companies will need to offer similar savings, Dr. Fendrick added. “There will be some discontent if patients are actually asked to pay more out-of-pocket for a less expensive drug,” he said.
All eight companies behind these biosimilars are offering or will be launching copay saving programs, many which advertise copays as low as $0 per month for eligible patients.
How will Humira respond?
Marta Wosińska, PhD, a health care economist at the Brookings Institute, Washington, predicts payers will use these lower biosimilar prices to negotiate better deals with AbbVie, Humira’s manufacturer. “We have a lot of players coming into [the market] right now, so the competition is really fierce,” she said. In response, AbbVie will need to increase rebates on Humira and/or lower its price to compete with these biosimilars.
“The ball is in AbbVie’s court,” she said. “If [the company] is not willing to drop price sufficiently, then payers will start switching to biosimilars.”
Dr. Fendrick reported past financial relationships and consulting arrangements with AbbVie, Amgen, Arnold Ventures, Bayer, CareFirst, BlueCross BlueShield, and many other companies. Dr. Wosińska has received funding from Arnold Ventures and serves as an expert witness on antitrust cases involving generic medication.
A version of this article first appeared on Medscape.com.
The best-selling drug Humira (adalimumab) now faces competition in the United States after a 20-year monopoly. The first adalimumab biosimilar, Amjevita, launched in the United States on January 31, and in July, seven additional biosimilars became available. These drugs have the potential to lower prescription drug prices, but when and by how much remains to be seen.
Here’s what you need to know about adalimumab biosimilars.
What Humira biosimilars are now available?
Eight different biosimilars have launched in 2023 with discounts as large at 85% from Humira’s list price of $6,922. A few companies also offer two price points.
Three of these biosimilars – Hadlima, Hyrimoz, and Yuflyma – are available in high concentration formulations. This high concentration formulation makes up 85% of Humira prescriptions, according to a report from Goodroot, a collection of companies focused on lowering health care costs.
Cyltezo is currently the only adalimumab biosimilar with an interchangeability designation, meaning that a pharmacist can substitute the biosimilar for an equivalent Humira prescription without the intervention of a clinician. A total of 47 states allow for these substitutions without prior approval from a clinician, according to Goodroot, and the clinician must be notified of the switch within a certain time frame. A total of 40 states require that patients be notified of the switch before substitution.
However, it’s not clear if this interchangeability designation will prove an advantage for Cyltezo, as it is interchangeable with the lower concentration version of Humira that makes up just 15% of prescriptions.
Most of the companies behind these biosimilars are pursuing interchangeability designations for their drugs, except for Fresenius Kabi (Idacio) and Coherus (Yusimry).
A ninth biosimilar, Pfizer’s adalimumab-afzb (Abrilada), is not yet on the market and is currently awaiting an approval decision from the Food and Drug Administration to add an interchangeability designation to its prior approval for a low-concentration formulation.
Why are they priced differently?
The two price points offer different deals to payers. Pharmacy benefit managers make confidential agreements with drug manufacturers to get a discount – called a rebate – to get the drug on the PBM’s formulary. The PBM keeps a portion of that rebate, and the rest is passed on to the insurance company and patients. Biosimilars at a higher price point will likely offer larger rebates. Biosimilars offered at lower price points incorporate this discount up front in their list pricing and likely will not offer large rebates.
Will biosimilars be covered by payers?
Currently, biosimilars are being offered on formularies at parity with Humira, meaning they are on the same tier. The PBM companies OptumRx and Cigna Group’s Express Scripts will offer Amjevita (at both price points), Cyltezo, and Hyrimoz (at both price points).
“This decision allows our clients flexibility to provide access to the lower list price, so members in high-deductible plans and benefit designs with coinsurance can experience lower out-of-pocket costs,” said OptumRx spokesperson Isaac Sorensen in an email.
Mark Cuban Cost Plus Drug Company, which uses a direct-to-consumer model, will offer Yusimry for $567.27 on its website. SmithRx, a PBM based in San Francisco, announced it would partner with Cost Plus Drugs to offer Yusimry, adding that SmithRx members can use their insurance benefits to further reduce out-of-pocket costs. RxPreferred, another PBM, will also offer Yusimry through its partnership with Cuban’s company.
The news website Formulary Watch previously reported that CVS Caremark, another of the biggest PBMs, will be offering Amjevita, but as a nonpreferred brand, while Humira remains the preferred brand. CVS Caremark did not respond to a request for comment.
Will patients pay less?
Biosimilars have been touted as a potential solution to lower spending on biologic drugs, but it’s unknown if patients will ultimately benefit with lower out-of-pocket costs. It’s “impossible to predict” if the discount that third-party payers pay will be passed on to consumers, said Mark Fendrick, MD, who directs the University of Michigan Center for Value-based Insurance Design in Ann Arbor.
Generally, a consumer’s copay is a percentage of a drug’s list price, so it stands to reason that a low drug price would result in lower out-of-pocket payments. While this is mostly true, Humira has a successful copay assistance program to lower prescription costs for consumers. According to a 2022 IQVIA report, 82% of commercial prescriptions cost patients less than $10 for Humira because of this program.
To appeal to patients, biosimilar companies will need to offer similar savings, Dr. Fendrick added. “There will be some discontent if patients are actually asked to pay more out-of-pocket for a less expensive drug,” he said.
All eight companies behind these biosimilars are offering or will be launching copay saving programs, many which advertise copays as low as $0 per month for eligible patients.
How will Humira respond?
Marta Wosińska, PhD, a health care economist at the Brookings Institute, Washington, predicts payers will use these lower biosimilar prices to negotiate better deals with AbbVie, Humira’s manufacturer. “We have a lot of players coming into [the market] right now, so the competition is really fierce,” she said. In response, AbbVie will need to increase rebates on Humira and/or lower its price to compete with these biosimilars.
“The ball is in AbbVie’s court,” she said. “If [the company] is not willing to drop price sufficiently, then payers will start switching to biosimilars.”
Dr. Fendrick reported past financial relationships and consulting arrangements with AbbVie, Amgen, Arnold Ventures, Bayer, CareFirst, BlueCross BlueShield, and many other companies. Dr. Wosińska has received funding from Arnold Ventures and serves as an expert witness on antitrust cases involving generic medication.
A version of this article first appeared on Medscape.com.
The best-selling drug Humira (adalimumab) now faces competition in the United States after a 20-year monopoly. The first adalimumab biosimilar, Amjevita, launched in the United States on January 31, and in July, seven additional biosimilars became available. These drugs have the potential to lower prescription drug prices, but when and by how much remains to be seen.
Here’s what you need to know about adalimumab biosimilars.
What Humira biosimilars are now available?
Eight different biosimilars have launched in 2023 with discounts as large at 85% from Humira’s list price of $6,922. A few companies also offer two price points.
Three of these biosimilars – Hadlima, Hyrimoz, and Yuflyma – are available in high concentration formulations. This high concentration formulation makes up 85% of Humira prescriptions, according to a report from Goodroot, a collection of companies focused on lowering health care costs.
Cyltezo is currently the only adalimumab biosimilar with an interchangeability designation, meaning that a pharmacist can substitute the biosimilar for an equivalent Humira prescription without the intervention of a clinician. A total of 47 states allow for these substitutions without prior approval from a clinician, according to Goodroot, and the clinician must be notified of the switch within a certain time frame. A total of 40 states require that patients be notified of the switch before substitution.
However, it’s not clear if this interchangeability designation will prove an advantage for Cyltezo, as it is interchangeable with the lower concentration version of Humira that makes up just 15% of prescriptions.
Most of the companies behind these biosimilars are pursuing interchangeability designations for their drugs, except for Fresenius Kabi (Idacio) and Coherus (Yusimry).
A ninth biosimilar, Pfizer’s adalimumab-afzb (Abrilada), is not yet on the market and is currently awaiting an approval decision from the Food and Drug Administration to add an interchangeability designation to its prior approval for a low-concentration formulation.
Why are they priced differently?
The two price points offer different deals to payers. Pharmacy benefit managers make confidential agreements with drug manufacturers to get a discount – called a rebate – to get the drug on the PBM’s formulary. The PBM keeps a portion of that rebate, and the rest is passed on to the insurance company and patients. Biosimilars at a higher price point will likely offer larger rebates. Biosimilars offered at lower price points incorporate this discount up front in their list pricing and likely will not offer large rebates.
Will biosimilars be covered by payers?
Currently, biosimilars are being offered on formularies at parity with Humira, meaning they are on the same tier. The PBM companies OptumRx and Cigna Group’s Express Scripts will offer Amjevita (at both price points), Cyltezo, and Hyrimoz (at both price points).
“This decision allows our clients flexibility to provide access to the lower list price, so members in high-deductible plans and benefit designs with coinsurance can experience lower out-of-pocket costs,” said OptumRx spokesperson Isaac Sorensen in an email.
Mark Cuban Cost Plus Drug Company, which uses a direct-to-consumer model, will offer Yusimry for $567.27 on its website. SmithRx, a PBM based in San Francisco, announced it would partner with Cost Plus Drugs to offer Yusimry, adding that SmithRx members can use their insurance benefits to further reduce out-of-pocket costs. RxPreferred, another PBM, will also offer Yusimry through its partnership with Cuban’s company.
The news website Formulary Watch previously reported that CVS Caremark, another of the biggest PBMs, will be offering Amjevita, but as a nonpreferred brand, while Humira remains the preferred brand. CVS Caremark did not respond to a request for comment.
Will patients pay less?
Biosimilars have been touted as a potential solution to lower spending on biologic drugs, but it’s unknown if patients will ultimately benefit with lower out-of-pocket costs. It’s “impossible to predict” if the discount that third-party payers pay will be passed on to consumers, said Mark Fendrick, MD, who directs the University of Michigan Center for Value-based Insurance Design in Ann Arbor.
Generally, a consumer’s copay is a percentage of a drug’s list price, so it stands to reason that a low drug price would result in lower out-of-pocket payments. While this is mostly true, Humira has a successful copay assistance program to lower prescription costs for consumers. According to a 2022 IQVIA report, 82% of commercial prescriptions cost patients less than $10 for Humira because of this program.
To appeal to patients, biosimilar companies will need to offer similar savings, Dr. Fendrick added. “There will be some discontent if patients are actually asked to pay more out-of-pocket for a less expensive drug,” he said.
All eight companies behind these biosimilars are offering or will be launching copay saving programs, many which advertise copays as low as $0 per month for eligible patients.
How will Humira respond?
Marta Wosińska, PhD, a health care economist at the Brookings Institute, Washington, predicts payers will use these lower biosimilar prices to negotiate better deals with AbbVie, Humira’s manufacturer. “We have a lot of players coming into [the market] right now, so the competition is really fierce,” she said. In response, AbbVie will need to increase rebates on Humira and/or lower its price to compete with these biosimilars.
“The ball is in AbbVie’s court,” she said. “If [the company] is not willing to drop price sufficiently, then payers will start switching to biosimilars.”
Dr. Fendrick reported past financial relationships and consulting arrangements with AbbVie, Amgen, Arnold Ventures, Bayer, CareFirst, BlueCross BlueShield, and many other companies. Dr. Wosińska has received funding from Arnold Ventures and serves as an expert witness on antitrust cases involving generic medication.
A version of this article first appeared on Medscape.com.
Remote teams offer chance to improve difficult-to-treat PsA
DUBLIN – according to presenters at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
In the same session at the meeting, GRAPPA also announced a new initiative to define difficult-to-treat PsA.
Deepak Jadon, MBBCh, PhD, a rheumatologist with Cambridge (England) University Hospitals NHS Foundation Trust, described his experience of running a clinic for patients with difficult-to-treat PsA in eastern England, covering a catchment area of approximately 6 million people between six and seven hospitals. He discussed how the MDT in his region operates to discuss the management of such patients, whose treatment options may also have indications for comorbidities such as inflammatory bowel disease or uveitis, or have complicating factors such as metabolic syndrome.
“You have to have an interested and engaged colleague to form that collaboration,” Dr. Jadon said. “If you are working in isolation, without your colleagues in the same building, that becomes harder. We have been running remote multispecialty meetings without the patient being present, and I have had the good fortune of having medical students brought into our practice. We discussed approximately 220 patients, initially in our psoriasis-spondyloarthritis MDT and subsequently in our inflammatory bowel disease–spondyloarthritis MDT.”
There are also MDTs with hepatologist colleagues carried out on an ad hoc basis to discuss patients with nonalcoholic fatty liver disease, as well as patients with hepatitis or a transplanted liver, who have psoriatic disease.
This difficult-to-treat cohort is discussed in MDT meetings conducted on Zoom. At MDT meetings, carried out with frequencies ranging from monthly to bimonthly, Dr. Jadon said there would be two dermatologists, two rheumatologists, one to four dermatology and rheumatology trainees and fellows, one to four specialist nurses, one to three research nurses, and one biologics pharmacist. They record the meetings and discuss anywhere from 4 to 18 patients, reviewing items in their electronic medical record, calling or writing patients and/or their primary care clinician as needed. They take about an hour to meet, with a half hour of prep time and another 1.5 hours to undertake necessary actions.
“Generally, the question is, how can we change treatment to best cover the domains of disease?” Dr. Jadon said. “Progressively, more patients are being put onto biologics as a result of these conversations, and I do feel that it has helped our patients and us to consolidate their management plan. Naturally, as all clinicians do, we doubt ourselves and wonder if we are missing something. Is there an aspect of the disease [being missed]? Is there a treatment that I haven’t been using? [The meetings have] been reassuring in that regard. I also learn from my colleagues who have earlier access to treatments, especially in dermatology.”
In a small number of patients, some combinations of advanced therapies, such as combining a Janus kinase inhibitor with a biologic, have been used as a result of these collaborations, “and to discuss this in an MDT has been reassuring, including from a medico-legal perspective,” Dr. Jadon said. “One of the main things we found to be useful is having a brief referral pro forma. Usually, by the time patients reach this forum, they have used a lot of treatments, and it can be difficult to remember that on the spot. It is also important to focus on what the actual question is. Naturally, in these discussions, where you talk about the complexities and various facets of disease, you can get a bit lost and sometimes you actually don’t address the original question.”
He also said it has been very beneficial to use screen sharing in the remote MDTs so that different disciplines can review images together, such as with radiology colleagues. “There are varying skill sets among our colleagues, especially in radiology, and it has been quite nice to review their peripheral imaging, their axial imaging, laboratory markers, and skin lesions together.”
New GRAPPA project to provide clarity
A new GRAPPA project has been devised to help physicians identify and define difficult-to-treat and difficult-to-manage PsA in order to help physicians to categorize and treat these patients.
“We have a growing treatment armamentarium ... but we still do not reach all the patients that we would like to,” said Fabian Proft, MD, of Charité University Medicine, Berlin. “We set our targets, but we see in the real world that we are only reaching them in 40% or 50% of our patients. So, we need to do better, and in order to do better, we need to understand better.”
“We should not only make a definition of difficult-to-treat PsA, which is nonresponse to treatment with objective signs of inflammation, but also we need to address and acknowledge difficult-to-manage [patients],” Dr. Proft said. “We should not stop as soon as we come up with a definition. This will be a working definition and will need to be validated.”
The speakers reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
DUBLIN – according to presenters at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
In the same session at the meeting, GRAPPA also announced a new initiative to define difficult-to-treat PsA.
Deepak Jadon, MBBCh, PhD, a rheumatologist with Cambridge (England) University Hospitals NHS Foundation Trust, described his experience of running a clinic for patients with difficult-to-treat PsA in eastern England, covering a catchment area of approximately 6 million people between six and seven hospitals. He discussed how the MDT in his region operates to discuss the management of such patients, whose treatment options may also have indications for comorbidities such as inflammatory bowel disease or uveitis, or have complicating factors such as metabolic syndrome.
“You have to have an interested and engaged colleague to form that collaboration,” Dr. Jadon said. “If you are working in isolation, without your colleagues in the same building, that becomes harder. We have been running remote multispecialty meetings without the patient being present, and I have had the good fortune of having medical students brought into our practice. We discussed approximately 220 patients, initially in our psoriasis-spondyloarthritis MDT and subsequently in our inflammatory bowel disease–spondyloarthritis MDT.”
There are also MDTs with hepatologist colleagues carried out on an ad hoc basis to discuss patients with nonalcoholic fatty liver disease, as well as patients with hepatitis or a transplanted liver, who have psoriatic disease.
This difficult-to-treat cohort is discussed in MDT meetings conducted on Zoom. At MDT meetings, carried out with frequencies ranging from monthly to bimonthly, Dr. Jadon said there would be two dermatologists, two rheumatologists, one to four dermatology and rheumatology trainees and fellows, one to four specialist nurses, one to three research nurses, and one biologics pharmacist. They record the meetings and discuss anywhere from 4 to 18 patients, reviewing items in their electronic medical record, calling or writing patients and/or their primary care clinician as needed. They take about an hour to meet, with a half hour of prep time and another 1.5 hours to undertake necessary actions.
“Generally, the question is, how can we change treatment to best cover the domains of disease?” Dr. Jadon said. “Progressively, more patients are being put onto biologics as a result of these conversations, and I do feel that it has helped our patients and us to consolidate their management plan. Naturally, as all clinicians do, we doubt ourselves and wonder if we are missing something. Is there an aspect of the disease [being missed]? Is there a treatment that I haven’t been using? [The meetings have] been reassuring in that regard. I also learn from my colleagues who have earlier access to treatments, especially in dermatology.”
In a small number of patients, some combinations of advanced therapies, such as combining a Janus kinase inhibitor with a biologic, have been used as a result of these collaborations, “and to discuss this in an MDT has been reassuring, including from a medico-legal perspective,” Dr. Jadon said. “One of the main things we found to be useful is having a brief referral pro forma. Usually, by the time patients reach this forum, they have used a lot of treatments, and it can be difficult to remember that on the spot. It is also important to focus on what the actual question is. Naturally, in these discussions, where you talk about the complexities and various facets of disease, you can get a bit lost and sometimes you actually don’t address the original question.”
He also said it has been very beneficial to use screen sharing in the remote MDTs so that different disciplines can review images together, such as with radiology colleagues. “There are varying skill sets among our colleagues, especially in radiology, and it has been quite nice to review their peripheral imaging, their axial imaging, laboratory markers, and skin lesions together.”
New GRAPPA project to provide clarity
A new GRAPPA project has been devised to help physicians identify and define difficult-to-treat and difficult-to-manage PsA in order to help physicians to categorize and treat these patients.
“We have a growing treatment armamentarium ... but we still do not reach all the patients that we would like to,” said Fabian Proft, MD, of Charité University Medicine, Berlin. “We set our targets, but we see in the real world that we are only reaching them in 40% or 50% of our patients. So, we need to do better, and in order to do better, we need to understand better.”
“We should not only make a definition of difficult-to-treat PsA, which is nonresponse to treatment with objective signs of inflammation, but also we need to address and acknowledge difficult-to-manage [patients],” Dr. Proft said. “We should not stop as soon as we come up with a definition. This will be a working definition and will need to be validated.”
The speakers reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
DUBLIN – according to presenters at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
In the same session at the meeting, GRAPPA also announced a new initiative to define difficult-to-treat PsA.
Deepak Jadon, MBBCh, PhD, a rheumatologist with Cambridge (England) University Hospitals NHS Foundation Trust, described his experience of running a clinic for patients with difficult-to-treat PsA in eastern England, covering a catchment area of approximately 6 million people between six and seven hospitals. He discussed how the MDT in his region operates to discuss the management of such patients, whose treatment options may also have indications for comorbidities such as inflammatory bowel disease or uveitis, or have complicating factors such as metabolic syndrome.
“You have to have an interested and engaged colleague to form that collaboration,” Dr. Jadon said. “If you are working in isolation, without your colleagues in the same building, that becomes harder. We have been running remote multispecialty meetings without the patient being present, and I have had the good fortune of having medical students brought into our practice. We discussed approximately 220 patients, initially in our psoriasis-spondyloarthritis MDT and subsequently in our inflammatory bowel disease–spondyloarthritis MDT.”
There are also MDTs with hepatologist colleagues carried out on an ad hoc basis to discuss patients with nonalcoholic fatty liver disease, as well as patients with hepatitis or a transplanted liver, who have psoriatic disease.
This difficult-to-treat cohort is discussed in MDT meetings conducted on Zoom. At MDT meetings, carried out with frequencies ranging from monthly to bimonthly, Dr. Jadon said there would be two dermatologists, two rheumatologists, one to four dermatology and rheumatology trainees and fellows, one to four specialist nurses, one to three research nurses, and one biologics pharmacist. They record the meetings and discuss anywhere from 4 to 18 patients, reviewing items in their electronic medical record, calling or writing patients and/or their primary care clinician as needed. They take about an hour to meet, with a half hour of prep time and another 1.5 hours to undertake necessary actions.
“Generally, the question is, how can we change treatment to best cover the domains of disease?” Dr. Jadon said. “Progressively, more patients are being put onto biologics as a result of these conversations, and I do feel that it has helped our patients and us to consolidate their management plan. Naturally, as all clinicians do, we doubt ourselves and wonder if we are missing something. Is there an aspect of the disease [being missed]? Is there a treatment that I haven’t been using? [The meetings have] been reassuring in that regard. I also learn from my colleagues who have earlier access to treatments, especially in dermatology.”
In a small number of patients, some combinations of advanced therapies, such as combining a Janus kinase inhibitor with a biologic, have been used as a result of these collaborations, “and to discuss this in an MDT has been reassuring, including from a medico-legal perspective,” Dr. Jadon said. “One of the main things we found to be useful is having a brief referral pro forma. Usually, by the time patients reach this forum, they have used a lot of treatments, and it can be difficult to remember that on the spot. It is also important to focus on what the actual question is. Naturally, in these discussions, where you talk about the complexities and various facets of disease, you can get a bit lost and sometimes you actually don’t address the original question.”
He also said it has been very beneficial to use screen sharing in the remote MDTs so that different disciplines can review images together, such as with radiology colleagues. “There are varying skill sets among our colleagues, especially in radiology, and it has been quite nice to review their peripheral imaging, their axial imaging, laboratory markers, and skin lesions together.”
New GRAPPA project to provide clarity
A new GRAPPA project has been devised to help physicians identify and define difficult-to-treat and difficult-to-manage PsA in order to help physicians to categorize and treat these patients.
“We have a growing treatment armamentarium ... but we still do not reach all the patients that we would like to,” said Fabian Proft, MD, of Charité University Medicine, Berlin. “We set our targets, but we see in the real world that we are only reaching them in 40% or 50% of our patients. So, we need to do better, and in order to do better, we need to understand better.”
“We should not only make a definition of difficult-to-treat PsA, which is nonresponse to treatment with objective signs of inflammation, but also we need to address and acknowledge difficult-to-manage [patients],” Dr. Proft said. “We should not stop as soon as we come up with a definition. This will be a working definition and will need to be validated.”
The speakers reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
AT GRAPPA 2023
Keep depression, anxiety screening top of mind in patients with psoriatic disease
DUBLIN – , warranting routine screening and having community contacts for mental health professional referrals, Elizabeth Wallace, MD, said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
Dr. Wallace, of Cherry Hills Dermatology, Englewood, Colo., discussed the complex interactions between mental illness and psoriatic disease and the potential pitfalls of this comorbidity for these patients.

The topic of mental health is “consistently at the top of our patients’ minds, and certainly our minds too,” said session comoderator and GRAPPA president-elect Joseph F. Merola, MD, MMSc.
“In the U.S., around 17% of people with psoriasis have depression vs. 9% in those without psoriasis,” Dr. Wallace explained. “Psoriasis patients are twice as likely to have depression, compared to those without psoriasis, and psoriasis patients are 33% more likely to attempt suicide and 20% more likely to complete suicide, compared to those without psoriasis.” More severe psoriasis and younger age of onset are also associated with a greater likelihood of suicidality, she added.
Mediators of depression
“The inflammatory mechanisms driving PsD can drive depression and anxiety, and vice-versa,” she said. “There are often also genetic links, for example genetic variations in serotonin receptors, and psychological issues in psoriatic disease are predictably worsened by feelings of stigmatization, embarrassment, and social isolation.”
There are also efforts underway in clinics to “normalize” screening for anxiety and depression among this patient cohort, Dr. Wallace said. “We know that our psoriasis patients face social stigma from the visibility of their disease, and that stress can lead to flares of their condition,” she told the attendees. “We also know that patients who experience stigma also have an increased risk of depressive symptoms. We all know now that psoriasis has well-established pathways with upregulated proinflammatory cytokines.
“Increased cytokines stimulate indoleamine 2,3-dioxygenase, which converts tryptophan to kynurenine. Kynurenine is metabolized to quinolinic acid, which is neurotoxic.” She explained that because serotonin derives from tryptophan, decreases in tryptophan lead to reduced serotonin, and therefore increased risk of depression.
Interleukin-6 is known to be upregulated in depression and downregulated with the use of antidepressant medications, Dr. Wallace said. Mouse models in research have shown that deletion of the IL-6 gene produces antidepressant effects, and studies in humans have shown that IL-6, more than any other serum cytokine, is found at higher levels in humans with depression and psoriatic disease.
IL-17 is also implicated in psoriatic disease and mental health problems, Dr. Wallace said. “With stress, you get upregulation of the Tc17 cells, which produce IL-17,” she explained. “IL-17, along with other inflammatory markers, can actually make the blood-brain barrier more permeable, and when you get more permeability to the blood-brain barrier, you get these cytokines that can cross from the periphery and into the brain.
“With this crossing into the brain, you get further activation of more Th17 [cells] and that, on neurons, leads to increased potassium production, which is directly neurotoxic, so you get neuron destruction.”
Talking about depression
“So, what can we share with our patients?” Dr. Wallace asked. “We can discuss with them that psoriatic patients in general are more likely to be depressed or to have higher rates of suicide. The literature consistently shows that patients whose psoriasis is successfully treated experience reduced depression, and we can provide an understandable review of systemic medications, with warnings on depression and/or suicidality.”
Dr. Wallace advised to screen for depression with the Patient Health Questionnaire-2 (PHQ-2), a validated, two-item tool that asks, “Over the past 2 weeks, how often have you been bothered by having little interest or pleasure in doing things?” and “Over the past 2 weeks, how often have you been bothered by feeling down, depressed, or hopeless?”
She presented a case study illustrative of the type of presentation she sees in her clinic. It involved a 32-year-old man with plaque psoriasis and a high degree of body surface affected. “It’s now July in Colorado, it’s getting warm, people want to wear their shorts and T-shirts, but he said he could no longer hide his psoriasis,” said Dr. Wallace. “Further, it’s in areas that he cannot hide, such as his scalp, his beard, and he also has nail disease. Often, these patients don’t want to shake hands with their bosses or their colleagues and that’s very embarrassing for them.”
Dr. Wallace explained that this patient had seen advertisements for biologic drugs and requested to commence a treatment course. “During the exam, and now that you are developing some rapport with him, you discover that he is feeling down, is embarrassed at work, and has started to avoid social situations.” This is illustrative of a patient who should be screened for mental health conditions, specifically using PHQ-2, she said.
“You can be the person at the front line to screen these patients for mental health conditions, and, specifically for depression, with PHQ-2,” she said. PHQ-2 scores range from 0 to 6, and a score of 3 or higher is considered a positive screen.
“This is where your relationship with another health provider who is most qualified to care for these patients and validate them for their mental health condition can be absolutely critical,” Dr. Wallace said.
Successful PsD treatment lessens the risk for mental health comorbidities, and this is also seen in psoriatic arthritis, Dr. Wallace pointed out. Patient education is critical regarding their increased risk for depression and potential suicidal ideation, she added.
“It’s our job as clinicians to provide patients with an understandable, easy-to-digest review of systemic medications and warnings on depression and suicidality so that they can be aware of these factors.”
Perspective from Dr. Merola
In an interview, Dr. Merola, a double board-certified dermatologist and rheumatologist at Brigham and Women’s Hospital, Boston, discussed the interactions between mental and physical illness.
“One of the things we are learning is that it’s very much a multifactorial issue, in that skin and joints contribute, in some obvious ways, to anxiety and depression, like the fact that somebody doesn’t feel good about their appearance, or they can’t complete daily activities,” he said. “Those are the more obvious ones. But there is data and evidence that there is a biology behind that as well – inflammatory cytokines that drive skin disease probably also have a direct impact on the CNS and probably also drive anxiety and depression.
“We know that disordered sleep contributes to anxiety – think about how we feel if we get a horrible night’s sleep ... it’s hard to pick apart: ‘Am I depressed, am I anxious because I am having too much coffee? Because I am fatigued?’ So, we get into these circles, but the point is, we have to break these cycles, and we have to do it in multiple places. Yes, we have to fix the skin and the joints, but we also have to have interventions and think about how to screen for anxiety and depression. We also have to think about identifying disordered sleep, and how we intervene there as well.”
These challenges require a collaborative approach among physicians. “We can help patients to build their team that gets them help for their skin, for their joints, for their anxiety or depression, their disordered sleep, for their nutritional disorders, their obesity, and so on. So, we are trying to pick apart and unpack those complexities,” he said.
In regard to the potential impacts of this holistic strategy on physician workloads, Dr. Merola acknowledged it is important to consider physician wellness. “There’s no question that we want to be doing the best we can for our colleagues, but we don’t want to overload our colleagues by saying, ‘By the way, not only should we be treating their skin and joints,’ which of course we should be doing, but ‘could you also manage their diabetes, their obesity, their disordered sleep, their anxiety, their depression, difficulties with insurance, getting access to treatments, etc.’
“This is where effective collaboration between physicians becomes important,” he stressed. “We can’t manage every single piece, but we can make sure our patients are informed, are aware, and assist them to get the help that they need.”
In the United States, there “is a real issue” with access to mental health care and greater awareness needs to be created around this issue, he added.
Dr. Wallace and Dr. Merola report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
DUBLIN – , warranting routine screening and having community contacts for mental health professional referrals, Elizabeth Wallace, MD, said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
Dr. Wallace, of Cherry Hills Dermatology, Englewood, Colo., discussed the complex interactions between mental illness and psoriatic disease and the potential pitfalls of this comorbidity for these patients.
The topic of mental health is “consistently at the top of our patients’ minds, and certainly our minds too,” said session comoderator and GRAPPA president-elect Joseph F. Merola, MD, MMSc.
“In the U.S., around 17% of people with psoriasis have depression vs. 9% in those without psoriasis,” Dr. Wallace explained. “Psoriasis patients are twice as likely to have depression, compared to those without psoriasis, and psoriasis patients are 33% more likely to attempt suicide and 20% more likely to complete suicide, compared to those without psoriasis.” More severe psoriasis and younger age of onset are also associated with a greater likelihood of suicidality, she added.
Mediators of depression
“The inflammatory mechanisms driving PsD can drive depression and anxiety, and vice-versa,” she said. “There are often also genetic links, for example genetic variations in serotonin receptors, and psychological issues in psoriatic disease are predictably worsened by feelings of stigmatization, embarrassment, and social isolation.”
There are also efforts underway in clinics to “normalize” screening for anxiety and depression among this patient cohort, Dr. Wallace said. “We know that our psoriasis patients face social stigma from the visibility of their disease, and that stress can lead to flares of their condition,” she told the attendees. “We also know that patients who experience stigma also have an increased risk of depressive symptoms. We all know now that psoriasis has well-established pathways with upregulated proinflammatory cytokines.
“Increased cytokines stimulate indoleamine 2,3-dioxygenase, which converts tryptophan to kynurenine. Kynurenine is metabolized to quinolinic acid, which is neurotoxic.” She explained that because serotonin derives from tryptophan, decreases in tryptophan lead to reduced serotonin, and therefore increased risk of depression.
Interleukin-6 is known to be upregulated in depression and downregulated with the use of antidepressant medications, Dr. Wallace said. Mouse models in research have shown that deletion of the IL-6 gene produces antidepressant effects, and studies in humans have shown that IL-6, more than any other serum cytokine, is found at higher levels in humans with depression and psoriatic disease.
IL-17 is also implicated in psoriatic disease and mental health problems, Dr. Wallace said. “With stress, you get upregulation of the Tc17 cells, which produce IL-17,” she explained. “IL-17, along with other inflammatory markers, can actually make the blood-brain barrier more permeable, and when you get more permeability to the blood-brain barrier, you get these cytokines that can cross from the periphery and into the brain.
“With this crossing into the brain, you get further activation of more Th17 [cells] and that, on neurons, leads to increased potassium production, which is directly neurotoxic, so you get neuron destruction.”
Talking about depression
“So, what can we share with our patients?” Dr. Wallace asked. “We can discuss with them that psoriatic patients in general are more likely to be depressed or to have higher rates of suicide. The literature consistently shows that patients whose psoriasis is successfully treated experience reduced depression, and we can provide an understandable review of systemic medications, with warnings on depression and/or suicidality.”
Dr. Wallace advised to screen for depression with the Patient Health Questionnaire-2 (PHQ-2), a validated, two-item tool that asks, “Over the past 2 weeks, how often have you been bothered by having little interest or pleasure in doing things?” and “Over the past 2 weeks, how often have you been bothered by feeling down, depressed, or hopeless?”
She presented a case study illustrative of the type of presentation she sees in her clinic. It involved a 32-year-old man with plaque psoriasis and a high degree of body surface affected. “It’s now July in Colorado, it’s getting warm, people want to wear their shorts and T-shirts, but he said he could no longer hide his psoriasis,” said Dr. Wallace. “Further, it’s in areas that he cannot hide, such as his scalp, his beard, and he also has nail disease. Often, these patients don’t want to shake hands with their bosses or their colleagues and that’s very embarrassing for them.”
Dr. Wallace explained that this patient had seen advertisements for biologic drugs and requested to commence a treatment course. “During the exam, and now that you are developing some rapport with him, you discover that he is feeling down, is embarrassed at work, and has started to avoid social situations.” This is illustrative of a patient who should be screened for mental health conditions, specifically using PHQ-2, she said.
“You can be the person at the front line to screen these patients for mental health conditions, and, specifically for depression, with PHQ-2,” she said. PHQ-2 scores range from 0 to 6, and a score of 3 or higher is considered a positive screen.
“This is where your relationship with another health provider who is most qualified to care for these patients and validate them for their mental health condition can be absolutely critical,” Dr. Wallace said.
Successful PsD treatment lessens the risk for mental health comorbidities, and this is also seen in psoriatic arthritis, Dr. Wallace pointed out. Patient education is critical regarding their increased risk for depression and potential suicidal ideation, she added.
“It’s our job as clinicians to provide patients with an understandable, easy-to-digest review of systemic medications and warnings on depression and suicidality so that they can be aware of these factors.”
Perspective from Dr. Merola
In an interview, Dr. Merola, a double board-certified dermatologist and rheumatologist at Brigham and Women’s Hospital, Boston, discussed the interactions between mental and physical illness.
“One of the things we are learning is that it’s very much a multifactorial issue, in that skin and joints contribute, in some obvious ways, to anxiety and depression, like the fact that somebody doesn’t feel good about their appearance, or they can’t complete daily activities,” he said. “Those are the more obvious ones. But there is data and evidence that there is a biology behind that as well – inflammatory cytokines that drive skin disease probably also have a direct impact on the CNS and probably also drive anxiety and depression.
“We know that disordered sleep contributes to anxiety – think about how we feel if we get a horrible night’s sleep ... it’s hard to pick apart: ‘Am I depressed, am I anxious because I am having too much coffee? Because I am fatigued?’ So, we get into these circles, but the point is, we have to break these cycles, and we have to do it in multiple places. Yes, we have to fix the skin and the joints, but we also have to have interventions and think about how to screen for anxiety and depression. We also have to think about identifying disordered sleep, and how we intervene there as well.”
These challenges require a collaborative approach among physicians. “We can help patients to build their team that gets them help for their skin, for their joints, for their anxiety or depression, their disordered sleep, for their nutritional disorders, their obesity, and so on. So, we are trying to pick apart and unpack those complexities,” he said.
In regard to the potential impacts of this holistic strategy on physician workloads, Dr. Merola acknowledged it is important to consider physician wellness. “There’s no question that we want to be doing the best we can for our colleagues, but we don’t want to overload our colleagues by saying, ‘By the way, not only should we be treating their skin and joints,’ which of course we should be doing, but ‘could you also manage their diabetes, their obesity, their disordered sleep, their anxiety, their depression, difficulties with insurance, getting access to treatments, etc.’
“This is where effective collaboration between physicians becomes important,” he stressed. “We can’t manage every single piece, but we can make sure our patients are informed, are aware, and assist them to get the help that they need.”
In the United States, there “is a real issue” with access to mental health care and greater awareness needs to be created around this issue, he added.
Dr. Wallace and Dr. Merola report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
DUBLIN – , warranting routine screening and having community contacts for mental health professional referrals, Elizabeth Wallace, MD, said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis.
Dr. Wallace, of Cherry Hills Dermatology, Englewood, Colo., discussed the complex interactions between mental illness and psoriatic disease and the potential pitfalls of this comorbidity for these patients.
The topic of mental health is “consistently at the top of our patients’ minds, and certainly our minds too,” said session comoderator and GRAPPA president-elect Joseph F. Merola, MD, MMSc.
“In the U.S., around 17% of people with psoriasis have depression vs. 9% in those without psoriasis,” Dr. Wallace explained. “Psoriasis patients are twice as likely to have depression, compared to those without psoriasis, and psoriasis patients are 33% more likely to attempt suicide and 20% more likely to complete suicide, compared to those without psoriasis.” More severe psoriasis and younger age of onset are also associated with a greater likelihood of suicidality, she added.
Mediators of depression
“The inflammatory mechanisms driving PsD can drive depression and anxiety, and vice-versa,” she said. “There are often also genetic links, for example genetic variations in serotonin receptors, and psychological issues in psoriatic disease are predictably worsened by feelings of stigmatization, embarrassment, and social isolation.”
There are also efforts underway in clinics to “normalize” screening for anxiety and depression among this patient cohort, Dr. Wallace said. “We know that our psoriasis patients face social stigma from the visibility of their disease, and that stress can lead to flares of their condition,” she told the attendees. “We also know that patients who experience stigma also have an increased risk of depressive symptoms. We all know now that psoriasis has well-established pathways with upregulated proinflammatory cytokines.
“Increased cytokines stimulate indoleamine 2,3-dioxygenase, which converts tryptophan to kynurenine. Kynurenine is metabolized to quinolinic acid, which is neurotoxic.” She explained that because serotonin derives from tryptophan, decreases in tryptophan lead to reduced serotonin, and therefore increased risk of depression.
Interleukin-6 is known to be upregulated in depression and downregulated with the use of antidepressant medications, Dr. Wallace said. Mouse models in research have shown that deletion of the IL-6 gene produces antidepressant effects, and studies in humans have shown that IL-6, more than any other serum cytokine, is found at higher levels in humans with depression and psoriatic disease.
IL-17 is also implicated in psoriatic disease and mental health problems, Dr. Wallace said. “With stress, you get upregulation of the Tc17 cells, which produce IL-17,” she explained. “IL-17, along with other inflammatory markers, can actually make the blood-brain barrier more permeable, and when you get more permeability to the blood-brain barrier, you get these cytokines that can cross from the periphery and into the brain.
“With this crossing into the brain, you get further activation of more Th17 [cells] and that, on neurons, leads to increased potassium production, which is directly neurotoxic, so you get neuron destruction.”
Talking about depression
“So, what can we share with our patients?” Dr. Wallace asked. “We can discuss with them that psoriatic patients in general are more likely to be depressed or to have higher rates of suicide. The literature consistently shows that patients whose psoriasis is successfully treated experience reduced depression, and we can provide an understandable review of systemic medications, with warnings on depression and/or suicidality.”
Dr. Wallace advised to screen for depression with the Patient Health Questionnaire-2 (PHQ-2), a validated, two-item tool that asks, “Over the past 2 weeks, how often have you been bothered by having little interest or pleasure in doing things?” and “Over the past 2 weeks, how often have you been bothered by feeling down, depressed, or hopeless?”
She presented a case study illustrative of the type of presentation she sees in her clinic. It involved a 32-year-old man with plaque psoriasis and a high degree of body surface affected. “It’s now July in Colorado, it’s getting warm, people want to wear their shorts and T-shirts, but he said he could no longer hide his psoriasis,” said Dr. Wallace. “Further, it’s in areas that he cannot hide, such as his scalp, his beard, and he also has nail disease. Often, these patients don’t want to shake hands with their bosses or their colleagues and that’s very embarrassing for them.”
Dr. Wallace explained that this patient had seen advertisements for biologic drugs and requested to commence a treatment course. “During the exam, and now that you are developing some rapport with him, you discover that he is feeling down, is embarrassed at work, and has started to avoid social situations.” This is illustrative of a patient who should be screened for mental health conditions, specifically using PHQ-2, she said.
“You can be the person at the front line to screen these patients for mental health conditions, and, specifically for depression, with PHQ-2,” she said. PHQ-2 scores range from 0 to 6, and a score of 3 or higher is considered a positive screen.
“This is where your relationship with another health provider who is most qualified to care for these patients and validate them for their mental health condition can be absolutely critical,” Dr. Wallace said.
Successful PsD treatment lessens the risk for mental health comorbidities, and this is also seen in psoriatic arthritis, Dr. Wallace pointed out. Patient education is critical regarding their increased risk for depression and potential suicidal ideation, she added.
“It’s our job as clinicians to provide patients with an understandable, easy-to-digest review of systemic medications and warnings on depression and suicidality so that they can be aware of these factors.”
Perspective from Dr. Merola
In an interview, Dr. Merola, a double board-certified dermatologist and rheumatologist at Brigham and Women’s Hospital, Boston, discussed the interactions between mental and physical illness.
“One of the things we are learning is that it’s very much a multifactorial issue, in that skin and joints contribute, in some obvious ways, to anxiety and depression, like the fact that somebody doesn’t feel good about their appearance, or they can’t complete daily activities,” he said. “Those are the more obvious ones. But there is data and evidence that there is a biology behind that as well – inflammatory cytokines that drive skin disease probably also have a direct impact on the CNS and probably also drive anxiety and depression.
“We know that disordered sleep contributes to anxiety – think about how we feel if we get a horrible night’s sleep ... it’s hard to pick apart: ‘Am I depressed, am I anxious because I am having too much coffee? Because I am fatigued?’ So, we get into these circles, but the point is, we have to break these cycles, and we have to do it in multiple places. Yes, we have to fix the skin and the joints, but we also have to have interventions and think about how to screen for anxiety and depression. We also have to think about identifying disordered sleep, and how we intervene there as well.”
These challenges require a collaborative approach among physicians. “We can help patients to build their team that gets them help for their skin, for their joints, for their anxiety or depression, their disordered sleep, for their nutritional disorders, their obesity, and so on. So, we are trying to pick apart and unpack those complexities,” he said.
In regard to the potential impacts of this holistic strategy on physician workloads, Dr. Merola acknowledged it is important to consider physician wellness. “There’s no question that we want to be doing the best we can for our colleagues, but we don’t want to overload our colleagues by saying, ‘By the way, not only should we be treating their skin and joints,’ which of course we should be doing, but ‘could you also manage their diabetes, their obesity, their disordered sleep, their anxiety, their depression, difficulties with insurance, getting access to treatments, etc.’
“This is where effective collaboration between physicians becomes important,” he stressed. “We can’t manage every single piece, but we can make sure our patients are informed, are aware, and assist them to get the help that they need.”
In the United States, there “is a real issue” with access to mental health care and greater awareness needs to be created around this issue, he added.
Dr. Wallace and Dr. Merola report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT GRAPPA 2023
Camp Discovery: A place for children to be comfortable in their own skin
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.

Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.

The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.

“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”

Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.

“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.

Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.
Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.
The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.
“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”
Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.
“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.
Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.
Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.
The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.
“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”
Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.
“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.
Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
Clothing provides Dx clue

A close examination of the patient’s scalp and hair was unhelpful, but a close look at the weave and seams of her dress revealed multiple nits and lice, consistent with a diagnosis of body lice.
Head lice and body lice are 2 different ecotypes of the species Pediculus humanus and occupy different environments on the body. They differ slightly in body shape caused by variable expression of the same genes.1 Body lice primarily live and lay eggs on clothing, especially along seams and within knit weaves. They travel to the skin to feed, causing significant itching in the host from the inflammatory and allergic effects of their saliva and feces. Additionally, body lice are vectors of several serious diseases including epidemic typhus (Rickettsia prowasekii), trench fever (Bartonella quintana), and relapsing fever (Borrelia recurrentis).1
A diagnosis of body lice is a sign of severe lack of access to basic human needs—uncrowded shelter, clean clothes, and clean water for bathing. A patient who has been given this diagnosis should be offered and receive a bath or shower with generous soap and warm water. Clothes should be cleaned with hot water (up to 149 °F) or discarded. Patients also may be treated once with topical permethrin 5% cream applied from the top of the neck to the toes in the event that mites survived bathing by attaching to body hairs. Any systemic illness or fever should be evaluated for the above epidemic pathogens. Patients should also be put in touch with social services and mental health services, as appropriate.
This patient received all of the above treatments and had already accessed social services. That said, she continued to struggle with housing instability.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571. doi: 10.1016/j.pt.2012.09.003
A close examination of the patient’s scalp and hair was unhelpful, but a close look at the weave and seams of her dress revealed multiple nits and lice, consistent with a diagnosis of body lice.
Head lice and body lice are 2 different ecotypes of the species Pediculus humanus and occupy different environments on the body. They differ slightly in body shape caused by variable expression of the same genes.1 Body lice primarily live and lay eggs on clothing, especially along seams and within knit weaves. They travel to the skin to feed, causing significant itching in the host from the inflammatory and allergic effects of their saliva and feces. Additionally, body lice are vectors of several serious diseases including epidemic typhus (Rickettsia prowasekii), trench fever (Bartonella quintana), and relapsing fever (Borrelia recurrentis).1
A diagnosis of body lice is a sign of severe lack of access to basic human needs—uncrowded shelter, clean clothes, and clean water for bathing. A patient who has been given this diagnosis should be offered and receive a bath or shower with generous soap and warm water. Clothes should be cleaned with hot water (up to 149 °F) or discarded. Patients also may be treated once with topical permethrin 5% cream applied from the top of the neck to the toes in the event that mites survived bathing by attaching to body hairs. Any systemic illness or fever should be evaluated for the above epidemic pathogens. Patients should also be put in touch with social services and mental health services, as appropriate.
This patient received all of the above treatments and had already accessed social services. That said, she continued to struggle with housing instability.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
A close examination of the patient’s scalp and hair was unhelpful, but a close look at the weave and seams of her dress revealed multiple nits and lice, consistent with a diagnosis of body lice.
Head lice and body lice are 2 different ecotypes of the species Pediculus humanus and occupy different environments on the body. They differ slightly in body shape caused by variable expression of the same genes.1 Body lice primarily live and lay eggs on clothing, especially along seams and within knit weaves. They travel to the skin to feed, causing significant itching in the host from the inflammatory and allergic effects of their saliva and feces. Additionally, body lice are vectors of several serious diseases including epidemic typhus (Rickettsia prowasekii), trench fever (Bartonella quintana), and relapsing fever (Borrelia recurrentis).1
A diagnosis of body lice is a sign of severe lack of access to basic human needs—uncrowded shelter, clean clothes, and clean water for bathing. A patient who has been given this diagnosis should be offered and receive a bath or shower with generous soap and warm water. Clothes should be cleaned with hot water (up to 149 °F) or discarded. Patients also may be treated once with topical permethrin 5% cream applied from the top of the neck to the toes in the event that mites survived bathing by attaching to body hairs. Any systemic illness or fever should be evaluated for the above epidemic pathogens. Patients should also be put in touch with social services and mental health services, as appropriate.
This patient received all of the above treatments and had already accessed social services. That said, she continued to struggle with housing instability.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571. doi: 10.1016/j.pt.2012.09.003
1. Veracx A, Raoult D. Biology and genetics of human head and body lice. Trends Parasitol. 2012;28:563-571. doi: 10.1016/j.pt.2012.09.003

Intensely itchy normal skin

Severe itching should prompt suspicion for scabies and the hands are the highest-yield location. In this patient’s case, there weren’t findings in the web spaces and, in general, skin findings were largely absent; dermoscopy confirmed the diagnosis of scabies.
Sarcoptes scabiei, is a parasitic mite that lives and reproduces in and on human skin and is transmitted by very close contact, either skin-to-skin or by living within a household or institution with shared linens and furnishings. After infection, itching develops within days to weeks from both the physical movement and burrowing of mites within the skin and from the allergic and inflammatory response to mite bodies and their waste.1 Symptoms and infections may persist for years in the absence of treatment.
Sometimes (as in this case), burrows are few and very subtle. More often, there are widespread burrows and excoriated papules over the hands, trunk, extremities, and genitals. A burrowed mite is often adjacent to, but not directly in, an excoriation. Dermoscopy has transformed the ability to diagnose this condition quickly by enabling clinicians to visualize the triangular shape of the head and front legs of a mite (called the “delta sign”). This localization allows easy microscopic confirmation by paring the mite from the skin with a small scalpel blade. (A #11 or #15 blade works very well.)
Topical permethrin 5% cream is highly curative. The cream should be applied from the top of the neck to the tips of the patient’s toes and left on for 8 hours; the process should be repeated a week later. Very close contacts (eg, symptomatic household members or sexual partners) should be treated concurrently. A 60 g tube will treat 1 adult twice. (A 60 g tube of permethrin with a refill, therefore, will treat 2 adults twice.) Oral ivermectin 3 mg dosed at 200 mcg/kg in a single dose repeated in 1 to 2 weeks is an alternative.
Outbreaks in an institutional setting present a significant challenge and require population-based control and often the assistance of infection control specialists or local public health officials. Often this involves weekly treatment with ivermectin for all potentially affected individuals for 3 to 4 weeks and surveillance for follow-up. While there is some resistance to ivermectin, many failures relate more to reinfection from unidentified sources.
This patient received topical permethrin 5% cream dosed as noted above. Itching can be expected to persist for 3 to 4 weeks, so topical triamcinolone 0.1% cream was prescribed as needed for itching on days when permethrin wasn’t applied. At 6 weeks, this patient’s symptoms had resolved.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Richards RN. Scabies: diagnostic and therapeutic update. J Cutan Med Surg. 2021;25:95-101. doi: 10.1177/1203475420960446
Severe itching should prompt suspicion for scabies and the hands are the highest-yield location. In this patient’s case, there weren’t findings in the web spaces and, in general, skin findings were largely absent; dermoscopy confirmed the diagnosis of scabies.
Sarcoptes scabiei, is a parasitic mite that lives and reproduces in and on human skin and is transmitted by very close contact, either skin-to-skin or by living within a household or institution with shared linens and furnishings. After infection, itching develops within days to weeks from both the physical movement and burrowing of mites within the skin and from the allergic and inflammatory response to mite bodies and their waste.1 Symptoms and infections may persist for years in the absence of treatment.
Sometimes (as in this case), burrows are few and very subtle. More often, there are widespread burrows and excoriated papules over the hands, trunk, extremities, and genitals. A burrowed mite is often adjacent to, but not directly in, an excoriation. Dermoscopy has transformed the ability to diagnose this condition quickly by enabling clinicians to visualize the triangular shape of the head and front legs of a mite (called the “delta sign”). This localization allows easy microscopic confirmation by paring the mite from the skin with a small scalpel blade. (A #11 or #15 blade works very well.)
Topical permethrin 5% cream is highly curative. The cream should be applied from the top of the neck to the tips of the patient’s toes and left on for 8 hours; the process should be repeated a week later. Very close contacts (eg, symptomatic household members or sexual partners) should be treated concurrently. A 60 g tube will treat 1 adult twice. (A 60 g tube of permethrin with a refill, therefore, will treat 2 adults twice.) Oral ivermectin 3 mg dosed at 200 mcg/kg in a single dose repeated in 1 to 2 weeks is an alternative.
Outbreaks in an institutional setting present a significant challenge and require population-based control and often the assistance of infection control specialists or local public health officials. Often this involves weekly treatment with ivermectin for all potentially affected individuals for 3 to 4 weeks and surveillance for follow-up. While there is some resistance to ivermectin, many failures relate more to reinfection from unidentified sources.
This patient received topical permethrin 5% cream dosed as noted above. Itching can be expected to persist for 3 to 4 weeks, so topical triamcinolone 0.1% cream was prescribed as needed for itching on days when permethrin wasn’t applied. At 6 weeks, this patient’s symptoms had resolved.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Severe itching should prompt suspicion for scabies and the hands are the highest-yield location. In this patient’s case, there weren’t findings in the web spaces and, in general, skin findings were largely absent; dermoscopy confirmed the diagnosis of scabies.
Sarcoptes scabiei, is a parasitic mite that lives and reproduces in and on human skin and is transmitted by very close contact, either skin-to-skin or by living within a household or institution with shared linens and furnishings. After infection, itching develops within days to weeks from both the physical movement and burrowing of mites within the skin and from the allergic and inflammatory response to mite bodies and their waste.1 Symptoms and infections may persist for years in the absence of treatment.
Sometimes (as in this case), burrows are few and very subtle. More often, there are widespread burrows and excoriated papules over the hands, trunk, extremities, and genitals. A burrowed mite is often adjacent to, but not directly in, an excoriation. Dermoscopy has transformed the ability to diagnose this condition quickly by enabling clinicians to visualize the triangular shape of the head and front legs of a mite (called the “delta sign”). This localization allows easy microscopic confirmation by paring the mite from the skin with a small scalpel blade. (A #11 or #15 blade works very well.)
Topical permethrin 5% cream is highly curative. The cream should be applied from the top of the neck to the tips of the patient’s toes and left on for 8 hours; the process should be repeated a week later. Very close contacts (eg, symptomatic household members or sexual partners) should be treated concurrently. A 60 g tube will treat 1 adult twice. (A 60 g tube of permethrin with a refill, therefore, will treat 2 adults twice.) Oral ivermectin 3 mg dosed at 200 mcg/kg in a single dose repeated in 1 to 2 weeks is an alternative.
Outbreaks in an institutional setting present a significant challenge and require population-based control and often the assistance of infection control specialists or local public health officials. Often this involves weekly treatment with ivermectin for all potentially affected individuals for 3 to 4 weeks and surveillance for follow-up. While there is some resistance to ivermectin, many failures relate more to reinfection from unidentified sources.
This patient received topical permethrin 5% cream dosed as noted above. Itching can be expected to persist for 3 to 4 weeks, so topical triamcinolone 0.1% cream was prescribed as needed for itching on days when permethrin wasn’t applied. At 6 weeks, this patient’s symptoms had resolved.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Richards RN. Scabies: diagnostic and therapeutic update. J Cutan Med Surg. 2021;25:95-101. doi: 10.1177/1203475420960446
1. Richards RN. Scabies: diagnostic and therapeutic update. J Cutan Med Surg. 2021;25:95-101. doi: 10.1177/1203475420960446

Oral IL-23 receptor antagonist for psoriasis promising: Phase 2b study
SINGAPORE – across all doses, compared with placebo, according to results of the FRONTIER 1 trial.
In the 16-week phase 2b study, 255 adults with moderate to severe plaque psoriasis were randomly assigned into six treatment groups: placebo (n = 43), JNJ-2113 25 mg daily (n = 43), 25 mg twice daily (n = 41), 50 mg daily (n = 43), 100 mg daily (n = 43), or 100 mg twice daily (n = 42).
Of those who took the placebo, only 9.3% achieved the study’s primary endpoint of a 75% or greater improvement in the Psoriasis Area and Severity Index (PASI-75) by week 16. This was compared with 78.6% in the group that took the highest dose.

“Additionally, the onset of action was fairly fast: at week 4, more than 20% of patients had achieved PASI 75,” said Robert Bissonnette, MD, CEO of Innovaderm Research in Montreal, who presented the findings during a late-breaker session at the World Congress of Dermatology.
Patients in the remaining groups demonstrated a response that corresponded to dosing level: with 37.2%, 51.2%, 58.1%, and 65.1% achieving PASI-75 in the 25 mg daily, 25 mg twice-daily, 50 mg daily, and 100 mg daily groups, respectively.
“These results are very interesting because in terms of psoriasis treatment, if this is confirmed in phase 3, it would give us an oral alternative that would be selective for IL-23,” said Dr. Bissonnette, referring to the signaling pathway that plays a critical role in the pathogenesis of several immune-mediated inflammatory diseases, including plaque psoriasis.
Although rarely life-threatening, the skin disorder is often intractable to treatment. In recent years, therapies that block IL-23 signaling and downstream inflammatory cytokine production have proven useful. “We have on the market a number of biological agents targeting IL-23 that we use on a regular basis,” said Dr. Bissonnette. “However, there are currently no orally delivered therapies.”
If successful, JNJ-2113 – a first-in-class oral IL-23 antagonist peptide developed by Janssen – could change the treatment paradigm for patients with moderate to severe plaque psoriasis. “When I was first introduced to the concept, I thought it wouldn’t work as it’s a peptide, that it would be digested by the stomach,” he told the audience. “But because of its GI stability and its potency, when you administer it orally, you can detect pharmacological activity.”
A well-tolerated alternative
Participants in the FRONTIER 1 trial were on average about 44 years old and weighed 88.9 kg (195 lb). Most had been living with psoriasis for about 18 years, with a total PASI score of 19.05. In addition, 43.1% had been treated with phototherapy in the past, 22% with biologics, and 78.4% with systemics.
PASI 90 and 100 were among some of the secondary outcomes measured. Similar to the primary outcome of PASI 75, all treatment groups demonstrated a statistically significant dose-response in PASI 90, compared with placebo. For those on the highest dose of JNJ-2113, 59.5% and 40.5% achieved PASI 90 and PASI 100, respectively, by week 16. The corresponding figures for those receiving placebo were 2.3% and 0%.
The safety profile for JNJ-2113 across all doses was similar to that of placebo, with no evidence of a dose-dependent increase in the occurrence of adverse events (AEs). The most frequently reported AEs were COVID-19 and nasopharyngitis. There were three serious AEs (COVID-19, infected cyst, suicide attempt) among those on the active drug, but the investigators assessed that they were not related to the study intervention. No deaths, major adverse cardiac events, or malignancies were reported during the study.
Approached for an independent comment, Marius-Anton Ionescu, MD, PhD, from the University Hospital Saint Louis, Paris, who specializes in psoriasis, told this news organization that the new development with JNJ-2113 “is really promising.”
Treatment for plaque psoriasis has improved to the point where some biologics, such as risankizumab (Skyrizi), only require patients to have “four shots a year,” he says. “This is the future of psoriasis treatment; it might go down to two shots a year” – a regimen that will be easier than taking an oral medication once or twice a day.
“But it’s good to have an oral option because you will always have some patients who say: ‘Shots are not for me, I’m afraid,’ ” he says.
However, Dr. Ionescu noted that if JNJ-2113 were to pass phase 3 trials, it might face stiff competition from the selective tyrosine kinase 2 (TYK2) inhibitor deucravacitinib (Sotyktu), which the U.S. Food and Drug Administration approved for use in adults with moderate to severe plaque psoriasis last September. “It has very good results and is the first oral therapy that is comparable with biologics for plaque psoriasis,” he says.
But Dr. Bissonnette remains hopeful for the future. “I think JNJ-2113 goes way beyond psoriasis because this type of strategy using oral peptide–blocking receptors could be used in other immune-mediated diseases, including atopic dermatitis and other diseases outside of dermatology.” In addition to running a phase 3 study for moderate to severe plaque psoriasis, Janssen is planning to initiate a phase 2b clinical trial of JNJ-2113 in adults with ulcerative colitis.
The study was funded by Janssen. Dr. Bissonnette reports consulting and investigating for Janssen, and being on advisory panels and receiving research funding from multiple other pharmaceutical companies. Dr. Ionescu is an investigator for Psoriasis National Register France Psobioteq (no honoraria), and an investigator and speaker for Uriage cosmetics (honoraria).
A version of this article first appeared on Medscape.com.
SINGAPORE – across all doses, compared with placebo, according to results of the FRONTIER 1 trial.
In the 16-week phase 2b study, 255 adults with moderate to severe plaque psoriasis were randomly assigned into six treatment groups: placebo (n = 43), JNJ-2113 25 mg daily (n = 43), 25 mg twice daily (n = 41), 50 mg daily (n = 43), 100 mg daily (n = 43), or 100 mg twice daily (n = 42).
Of those who took the placebo, only 9.3% achieved the study’s primary endpoint of a 75% or greater improvement in the Psoriasis Area and Severity Index (PASI-75) by week 16. This was compared with 78.6% in the group that took the highest dose.
“Additionally, the onset of action was fairly fast: at week 4, more than 20% of patients had achieved PASI 75,” said Robert Bissonnette, MD, CEO of Innovaderm Research in Montreal, who presented the findings during a late-breaker session at the World Congress of Dermatology.
Patients in the remaining groups demonstrated a response that corresponded to dosing level: with 37.2%, 51.2%, 58.1%, and 65.1% achieving PASI-75 in the 25 mg daily, 25 mg twice-daily, 50 mg daily, and 100 mg daily groups, respectively.
“These results are very interesting because in terms of psoriasis treatment, if this is confirmed in phase 3, it would give us an oral alternative that would be selective for IL-23,” said Dr. Bissonnette, referring to the signaling pathway that plays a critical role in the pathogenesis of several immune-mediated inflammatory diseases, including plaque psoriasis.
Although rarely life-threatening, the skin disorder is often intractable to treatment. In recent years, therapies that block IL-23 signaling and downstream inflammatory cytokine production have proven useful. “We have on the market a number of biological agents targeting IL-23 that we use on a regular basis,” said Dr. Bissonnette. “However, there are currently no orally delivered therapies.”
If successful, JNJ-2113 – a first-in-class oral IL-23 antagonist peptide developed by Janssen – could change the treatment paradigm for patients with moderate to severe plaque psoriasis. “When I was first introduced to the concept, I thought it wouldn’t work as it’s a peptide, that it would be digested by the stomach,” he told the audience. “But because of its GI stability and its potency, when you administer it orally, you can detect pharmacological activity.”
A well-tolerated alternative
Participants in the FRONTIER 1 trial were on average about 44 years old and weighed 88.9 kg (195 lb). Most had been living with psoriasis for about 18 years, with a total PASI score of 19.05. In addition, 43.1% had been treated with phototherapy in the past, 22% with biologics, and 78.4% with systemics.
PASI 90 and 100 were among some of the secondary outcomes measured. Similar to the primary outcome of PASI 75, all treatment groups demonstrated a statistically significant dose-response in PASI 90, compared with placebo. For those on the highest dose of JNJ-2113, 59.5% and 40.5% achieved PASI 90 and PASI 100, respectively, by week 16. The corresponding figures for those receiving placebo were 2.3% and 0%.
The safety profile for JNJ-2113 across all doses was similar to that of placebo, with no evidence of a dose-dependent increase in the occurrence of adverse events (AEs). The most frequently reported AEs were COVID-19 and nasopharyngitis. There were three serious AEs (COVID-19, infected cyst, suicide attempt) among those on the active drug, but the investigators assessed that they were not related to the study intervention. No deaths, major adverse cardiac events, or malignancies were reported during the study.
Approached for an independent comment, Marius-Anton Ionescu, MD, PhD, from the University Hospital Saint Louis, Paris, who specializes in psoriasis, told this news organization that the new development with JNJ-2113 “is really promising.”
Treatment for plaque psoriasis has improved to the point where some biologics, such as risankizumab (Skyrizi), only require patients to have “four shots a year,” he says. “This is the future of psoriasis treatment; it might go down to two shots a year” – a regimen that will be easier than taking an oral medication once or twice a day.
“But it’s good to have an oral option because you will always have some patients who say: ‘Shots are not for me, I’m afraid,’ ” he says.
However, Dr. Ionescu noted that if JNJ-2113 were to pass phase 3 trials, it might face stiff competition from the selective tyrosine kinase 2 (TYK2) inhibitor deucravacitinib (Sotyktu), which the U.S. Food and Drug Administration approved for use in adults with moderate to severe plaque psoriasis last September. “It has very good results and is the first oral therapy that is comparable with biologics for plaque psoriasis,” he says.
But Dr. Bissonnette remains hopeful for the future. “I think JNJ-2113 goes way beyond psoriasis because this type of strategy using oral peptide–blocking receptors could be used in other immune-mediated diseases, including atopic dermatitis and other diseases outside of dermatology.” In addition to running a phase 3 study for moderate to severe plaque psoriasis, Janssen is planning to initiate a phase 2b clinical trial of JNJ-2113 in adults with ulcerative colitis.
The study was funded by Janssen. Dr. Bissonnette reports consulting and investigating for Janssen, and being on advisory panels and receiving research funding from multiple other pharmaceutical companies. Dr. Ionescu is an investigator for Psoriasis National Register France Psobioteq (no honoraria), and an investigator and speaker for Uriage cosmetics (honoraria).
A version of this article first appeared on Medscape.com.
SINGAPORE – across all doses, compared with placebo, according to results of the FRONTIER 1 trial.
In the 16-week phase 2b study, 255 adults with moderate to severe plaque psoriasis were randomly assigned into six treatment groups: placebo (n = 43), JNJ-2113 25 mg daily (n = 43), 25 mg twice daily (n = 41), 50 mg daily (n = 43), 100 mg daily (n = 43), or 100 mg twice daily (n = 42).
Of those who took the placebo, only 9.3% achieved the study’s primary endpoint of a 75% or greater improvement in the Psoriasis Area and Severity Index (PASI-75) by week 16. This was compared with 78.6% in the group that took the highest dose.
“Additionally, the onset of action was fairly fast: at week 4, more than 20% of patients had achieved PASI 75,” said Robert Bissonnette, MD, CEO of Innovaderm Research in Montreal, who presented the findings during a late-breaker session at the World Congress of Dermatology.
Patients in the remaining groups demonstrated a response that corresponded to dosing level: with 37.2%, 51.2%, 58.1%, and 65.1% achieving PASI-75 in the 25 mg daily, 25 mg twice-daily, 50 mg daily, and 100 mg daily groups, respectively.
“These results are very interesting because in terms of psoriasis treatment, if this is confirmed in phase 3, it would give us an oral alternative that would be selective for IL-23,” said Dr. Bissonnette, referring to the signaling pathway that plays a critical role in the pathogenesis of several immune-mediated inflammatory diseases, including plaque psoriasis.
Although rarely life-threatening, the skin disorder is often intractable to treatment. In recent years, therapies that block IL-23 signaling and downstream inflammatory cytokine production have proven useful. “We have on the market a number of biological agents targeting IL-23 that we use on a regular basis,” said Dr. Bissonnette. “However, there are currently no orally delivered therapies.”
If successful, JNJ-2113 – a first-in-class oral IL-23 antagonist peptide developed by Janssen – could change the treatment paradigm for patients with moderate to severe plaque psoriasis. “When I was first introduced to the concept, I thought it wouldn’t work as it’s a peptide, that it would be digested by the stomach,” he told the audience. “But because of its GI stability and its potency, when you administer it orally, you can detect pharmacological activity.”
A well-tolerated alternative
Participants in the FRONTIER 1 trial were on average about 44 years old and weighed 88.9 kg (195 lb). Most had been living with psoriasis for about 18 years, with a total PASI score of 19.05. In addition, 43.1% had been treated with phototherapy in the past, 22% with biologics, and 78.4% with systemics.
PASI 90 and 100 were among some of the secondary outcomes measured. Similar to the primary outcome of PASI 75, all treatment groups demonstrated a statistically significant dose-response in PASI 90, compared with placebo. For those on the highest dose of JNJ-2113, 59.5% and 40.5% achieved PASI 90 and PASI 100, respectively, by week 16. The corresponding figures for those receiving placebo were 2.3% and 0%.
The safety profile for JNJ-2113 across all doses was similar to that of placebo, with no evidence of a dose-dependent increase in the occurrence of adverse events (AEs). The most frequently reported AEs were COVID-19 and nasopharyngitis. There were three serious AEs (COVID-19, infected cyst, suicide attempt) among those on the active drug, but the investigators assessed that they were not related to the study intervention. No deaths, major adverse cardiac events, or malignancies were reported during the study.
Approached for an independent comment, Marius-Anton Ionescu, MD, PhD, from the University Hospital Saint Louis, Paris, who specializes in psoriasis, told this news organization that the new development with JNJ-2113 “is really promising.”
Treatment for plaque psoriasis has improved to the point where some biologics, such as risankizumab (Skyrizi), only require patients to have “four shots a year,” he says. “This is the future of psoriasis treatment; it might go down to two shots a year” – a regimen that will be easier than taking an oral medication once or twice a day.
“But it’s good to have an oral option because you will always have some patients who say: ‘Shots are not for me, I’m afraid,’ ” he says.
However, Dr. Ionescu noted that if JNJ-2113 were to pass phase 3 trials, it might face stiff competition from the selective tyrosine kinase 2 (TYK2) inhibitor deucravacitinib (Sotyktu), which the U.S. Food and Drug Administration approved for use in adults with moderate to severe plaque psoriasis last September. “It has very good results and is the first oral therapy that is comparable with biologics for plaque psoriasis,” he says.
But Dr. Bissonnette remains hopeful for the future. “I think JNJ-2113 goes way beyond psoriasis because this type of strategy using oral peptide–blocking receptors could be used in other immune-mediated diseases, including atopic dermatitis and other diseases outside of dermatology.” In addition to running a phase 3 study for moderate to severe plaque psoriasis, Janssen is planning to initiate a phase 2b clinical trial of JNJ-2113 in adults with ulcerative colitis.
The study was funded by Janssen. Dr. Bissonnette reports consulting and investigating for Janssen, and being on advisory panels and receiving research funding from multiple other pharmaceutical companies. Dr. Ionescu is an investigator for Psoriasis National Register France Psobioteq (no honoraria), and an investigator and speaker for Uriage cosmetics (honoraria).
A version of this article first appeared on Medscape.com.
AT WCD 2023
JAK inhibitors efficacious for atopic dermatitis in Asian patients, study finds
SINGAPORE – conducted in Singapore has found.
“Abrocitinib and upadacitinib surprisingly appeared to have better treatment efficacy compared to baricitinib,” said study lead Yik Weng Yew, MD, PhD, MPH, deputy head of research at Singapore’s National Skin Centre (NSC), who presented the results at the 25th World Congress of Dermatology. “But overall, as a group, I think they show a very good treatment response, as well as a good effect on itch response.”

JAK inhibitors are used to treat a variety of inflammatory diseases including alopecia areata, rheumatoid arthritis, and inflammatory bowel disease. Although treatment for severe eczema was previously limited to topical steroids and oral immunosuppressants, there are now two oral JAK inhibitors – abrocitinib and upadacitinib – approved in 2022 by the Food and Drug Administration for treating AD, which affects up to 2.4% of the global population. (A topical formulation of ruxolitinib, a JAK inhibitor, was approved for AD in 2021.)
The Singapore study is one of the few that have examined the safety and efficacy of JAK inhibitors for treatment of AD in a non-White population.
Chinese population
For the 12-week trial, conducted in 2022, Dr. Yew and associates recruited 35 patients from the NSC. More than half of participants (64%) were men and most (96%) were of Chinese ethnicity. Four of every five patients had previously received systemic agents: 17% had been treated with one systemic agent, 18.9% with two, 15.1% with three, 22.6% with four, and 3.8% with five. The most commonly used agents were cyclosporine (62.3%), methotrexate (47.2%), azathioprine (39.6%), and dupilumab (35.8%).
“The switch in therapy could have been a result of inadequate efficacy or cost reasons because in Singapore patients pay out of pocket for AD treatments,” said Dr. Yew.
Additionally, he offered a caveat on the profile of participants: “Perhaps they were more difficult atopic eczema patients, and therefore, the efficacy [of JAK inhibitors] might be a bit different.”
Clearer skin, less itch
Patients received one of the three study drugs: baricitinib (66%), abrocitinib (21%), and upadacitinib (13%). The distribution was “affected by reimbursement patterns and availability of the drug,” explained Dr. Yew.
They were assessed at weeks 4 and 12. By study end, the proportion of patients who self-reported an improvement in their condition was 100% for upadacitinib, 90% for abrocitinib, and 69% for baricitinib.
Scores on the Investigator Global Assessment (IGA) also improved with treatment. Patients in the baricitinib group saw their mean score fall from 4.0 to 3.0 by week 4, then to 2.0 by week 12. With upadacitinib and abrocitinib, “you can see that there is a nice decrease in IGA responses,” said Dr. Yew, referring to the larger improvement in scores experienced by patients on those two treatments. For patients on upadacitinib, IGA decreased from 3.5 to 2 at 4 weeks, then to 0.5 at 12 weeks, while those taking abrocitinib had their scores drop from 4.0 to 2.0 at 4 weeks, then to 1.0 at 12 weeks.
When it came to itch reduction, the abrocitinib group experienced the biggest reduction, with a median reduction of 5.5 points in itch score. Median reduction in itch score was 4 points for the other two groups. “Oral JAK inhibitors appear to have a good effect on itch response,” said Dr. Yew.
However, the researchers observed no significant reduction in percentage of body surface area affected, the last outcome assessed.
The most commonly reported adverse events were increased creatine kinase levels (11.3% of patients), increased LDL cholesterol levels (9.4%), and herpes zoster (9.4%). Those in the abrocitinib reported a higher number of these adverse events, compared with the other two treatment groups. (There were no herpes zoster cases among those taking baricitinib.)
For herpes zoster, Dr. Yew said “the common recommendation” is to give the inactivated shingles vaccine. “But the problem is that, number one, these patients would have probably failed multiple agents so they probably can’t wait for you to vaccinate before you initiate treatment.”
In addition, people in Singapore have to pay out-of-pocket for the two vaccine doses, “which is probably a month’s worth of medication,” he noted. “So we have a lot of resistance from patients.”
Additionally, Dr. Yew noted that contrary to what has previously been reported in the literature, there were few complaints of acne as a side effect in the Singaporean study population.
Toward greater representation
Dr. Yew pointed out that the study was limited by a few factors: neither the Eczema Area and Severity Index or Scoring of Atopic Dermatitis index data was used, and the study population was small and not representative of the real world.
Still, the new findings contribute to the overall safety and efficacy profile of JAK inhibitors in AD, which has so far been scarce in non-White populations.
“In Western studies, unfortunately, the representation of the population of skin of color or different ethnicities is underrepresented,” said Yousef Binamer, MD, chair of the dermatology department at King Faisal Specialist Hospital, Riyadh, Saudi Arabia, when approached for an independent comment on the results.
“This is now why researchers are looking into specific groups to study them,” which he pointed out, is crucial because “the immunophenotyping of AD is different for each background.”
The incidence and severity of AD tend to be higher in Asian and Middle Eastern populations, for instance, he noted. “It’s very common in Asia, and not so common in very white skin. I did my training in Canada so I see the difference,” said Dr. Binamer. “Asian people tend to be more itchy and have a tendency to scar on pigmentation.” Whereas White people “usually do not have this issue.”
“So I think real-world evidence of JAK inhibitors in the other populations is important,” he said. Studies such as the one conducted in Singapore, as well as the recently reported QUARTZ3 study, which examined the use of the JAK inhibitor ivarmacitinib in 256 Chinese patients with AD, are helping to pave the way.
The study was independently supported. Dr. Yew and Dr. Binamer have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
SINGAPORE – conducted in Singapore has found.
“Abrocitinib and upadacitinib surprisingly appeared to have better treatment efficacy compared to baricitinib,” said study lead Yik Weng Yew, MD, PhD, MPH, deputy head of research at Singapore’s National Skin Centre (NSC), who presented the results at the 25th World Congress of Dermatology. “But overall, as a group, I think they show a very good treatment response, as well as a good effect on itch response.”
JAK inhibitors are used to treat a variety of inflammatory diseases including alopecia areata, rheumatoid arthritis, and inflammatory bowel disease. Although treatment for severe eczema was previously limited to topical steroids and oral immunosuppressants, there are now two oral JAK inhibitors – abrocitinib and upadacitinib – approved in 2022 by the Food and Drug Administration for treating AD, which affects up to 2.4% of the global population. (A topical formulation of ruxolitinib, a JAK inhibitor, was approved for AD in 2021.)
The Singapore study is one of the few that have examined the safety and efficacy of JAK inhibitors for treatment of AD in a non-White population.
Chinese population
For the 12-week trial, conducted in 2022, Dr. Yew and associates recruited 35 patients from the NSC. More than half of participants (64%) were men and most (96%) were of Chinese ethnicity. Four of every five patients had previously received systemic agents: 17% had been treated with one systemic agent, 18.9% with two, 15.1% with three, 22.6% with four, and 3.8% with five. The most commonly used agents were cyclosporine (62.3%), methotrexate (47.2%), azathioprine (39.6%), and dupilumab (35.8%).
“The switch in therapy could have been a result of inadequate efficacy or cost reasons because in Singapore patients pay out of pocket for AD treatments,” said Dr. Yew.
Additionally, he offered a caveat on the profile of participants: “Perhaps they were more difficult atopic eczema patients, and therefore, the efficacy [of JAK inhibitors] might be a bit different.”
Clearer skin, less itch
Patients received one of the three study drugs: baricitinib (66%), abrocitinib (21%), and upadacitinib (13%). The distribution was “affected by reimbursement patterns and availability of the drug,” explained Dr. Yew.
They were assessed at weeks 4 and 12. By study end, the proportion of patients who self-reported an improvement in their condition was 100% for upadacitinib, 90% for abrocitinib, and 69% for baricitinib.
Scores on the Investigator Global Assessment (IGA) also improved with treatment. Patients in the baricitinib group saw their mean score fall from 4.0 to 3.0 by week 4, then to 2.0 by week 12. With upadacitinib and abrocitinib, “you can see that there is a nice decrease in IGA responses,” said Dr. Yew, referring to the larger improvement in scores experienced by patients on those two treatments. For patients on upadacitinib, IGA decreased from 3.5 to 2 at 4 weeks, then to 0.5 at 12 weeks, while those taking abrocitinib had their scores drop from 4.0 to 2.0 at 4 weeks, then to 1.0 at 12 weeks.
When it came to itch reduction, the abrocitinib group experienced the biggest reduction, with a median reduction of 5.5 points in itch score. Median reduction in itch score was 4 points for the other two groups. “Oral JAK inhibitors appear to have a good effect on itch response,” said Dr. Yew.
However, the researchers observed no significant reduction in percentage of body surface area affected, the last outcome assessed.
The most commonly reported adverse events were increased creatine kinase levels (11.3% of patients), increased LDL cholesterol levels (9.4%), and herpes zoster (9.4%). Those in the abrocitinib reported a higher number of these adverse events, compared with the other two treatment groups. (There were no herpes zoster cases among those taking baricitinib.)
For herpes zoster, Dr. Yew said “the common recommendation” is to give the inactivated shingles vaccine. “But the problem is that, number one, these patients would have probably failed multiple agents so they probably can’t wait for you to vaccinate before you initiate treatment.”
In addition, people in Singapore have to pay out-of-pocket for the two vaccine doses, “which is probably a month’s worth of medication,” he noted. “So we have a lot of resistance from patients.”
Additionally, Dr. Yew noted that contrary to what has previously been reported in the literature, there were few complaints of acne as a side effect in the Singaporean study population.
Toward greater representation
Dr. Yew pointed out that the study was limited by a few factors: neither the Eczema Area and Severity Index or Scoring of Atopic Dermatitis index data was used, and the study population was small and not representative of the real world.
Still, the new findings contribute to the overall safety and efficacy profile of JAK inhibitors in AD, which has so far been scarce in non-White populations.
“In Western studies, unfortunately, the representation of the population of skin of color or different ethnicities is underrepresented,” said Yousef Binamer, MD, chair of the dermatology department at King Faisal Specialist Hospital, Riyadh, Saudi Arabia, when approached for an independent comment on the results.
“This is now why researchers are looking into specific groups to study them,” which he pointed out, is crucial because “the immunophenotyping of AD is different for each background.”
The incidence and severity of AD tend to be higher in Asian and Middle Eastern populations, for instance, he noted. “It’s very common in Asia, and not so common in very white skin. I did my training in Canada so I see the difference,” said Dr. Binamer. “Asian people tend to be more itchy and have a tendency to scar on pigmentation.” Whereas White people “usually do not have this issue.”
“So I think real-world evidence of JAK inhibitors in the other populations is important,” he said. Studies such as the one conducted in Singapore, as well as the recently reported QUARTZ3 study, which examined the use of the JAK inhibitor ivarmacitinib in 256 Chinese patients with AD, are helping to pave the way.
The study was independently supported. Dr. Yew and Dr. Binamer have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
SINGAPORE – conducted in Singapore has found.
“Abrocitinib and upadacitinib surprisingly appeared to have better treatment efficacy compared to baricitinib,” said study lead Yik Weng Yew, MD, PhD, MPH, deputy head of research at Singapore’s National Skin Centre (NSC), who presented the results at the 25th World Congress of Dermatology. “But overall, as a group, I think they show a very good treatment response, as well as a good effect on itch response.”
JAK inhibitors are used to treat a variety of inflammatory diseases including alopecia areata, rheumatoid arthritis, and inflammatory bowel disease. Although treatment for severe eczema was previously limited to topical steroids and oral immunosuppressants, there are now two oral JAK inhibitors – abrocitinib and upadacitinib – approved in 2022 by the Food and Drug Administration for treating AD, which affects up to 2.4% of the global population. (A topical formulation of ruxolitinib, a JAK inhibitor, was approved for AD in 2021.)
The Singapore study is one of the few that have examined the safety and efficacy of JAK inhibitors for treatment of AD in a non-White population.
Chinese population
For the 12-week trial, conducted in 2022, Dr. Yew and associates recruited 35 patients from the NSC. More than half of participants (64%) were men and most (96%) were of Chinese ethnicity. Four of every five patients had previously received systemic agents: 17% had been treated with one systemic agent, 18.9% with two, 15.1% with three, 22.6% with four, and 3.8% with five. The most commonly used agents were cyclosporine (62.3%), methotrexate (47.2%), azathioprine (39.6%), and dupilumab (35.8%).
“The switch in therapy could have been a result of inadequate efficacy or cost reasons because in Singapore patients pay out of pocket for AD treatments,” said Dr. Yew.
Additionally, he offered a caveat on the profile of participants: “Perhaps they were more difficult atopic eczema patients, and therefore, the efficacy [of JAK inhibitors] might be a bit different.”
Clearer skin, less itch
Patients received one of the three study drugs: baricitinib (66%), abrocitinib (21%), and upadacitinib (13%). The distribution was “affected by reimbursement patterns and availability of the drug,” explained Dr. Yew.
They were assessed at weeks 4 and 12. By study end, the proportion of patients who self-reported an improvement in their condition was 100% for upadacitinib, 90% for abrocitinib, and 69% for baricitinib.
Scores on the Investigator Global Assessment (IGA) also improved with treatment. Patients in the baricitinib group saw their mean score fall from 4.0 to 3.0 by week 4, then to 2.0 by week 12. With upadacitinib and abrocitinib, “you can see that there is a nice decrease in IGA responses,” said Dr. Yew, referring to the larger improvement in scores experienced by patients on those two treatments. For patients on upadacitinib, IGA decreased from 3.5 to 2 at 4 weeks, then to 0.5 at 12 weeks, while those taking abrocitinib had their scores drop from 4.0 to 2.0 at 4 weeks, then to 1.0 at 12 weeks.
When it came to itch reduction, the abrocitinib group experienced the biggest reduction, with a median reduction of 5.5 points in itch score. Median reduction in itch score was 4 points for the other two groups. “Oral JAK inhibitors appear to have a good effect on itch response,” said Dr. Yew.
However, the researchers observed no significant reduction in percentage of body surface area affected, the last outcome assessed.
The most commonly reported adverse events were increased creatine kinase levels (11.3% of patients), increased LDL cholesterol levels (9.4%), and herpes zoster (9.4%). Those in the abrocitinib reported a higher number of these adverse events, compared with the other two treatment groups. (There were no herpes zoster cases among those taking baricitinib.)
For herpes zoster, Dr. Yew said “the common recommendation” is to give the inactivated shingles vaccine. “But the problem is that, number one, these patients would have probably failed multiple agents so they probably can’t wait for you to vaccinate before you initiate treatment.”
In addition, people in Singapore have to pay out-of-pocket for the two vaccine doses, “which is probably a month’s worth of medication,” he noted. “So we have a lot of resistance from patients.”
Additionally, Dr. Yew noted that contrary to what has previously been reported in the literature, there were few complaints of acne as a side effect in the Singaporean study population.
Toward greater representation
Dr. Yew pointed out that the study was limited by a few factors: neither the Eczema Area and Severity Index or Scoring of Atopic Dermatitis index data was used, and the study population was small and not representative of the real world.
Still, the new findings contribute to the overall safety and efficacy profile of JAK inhibitors in AD, which has so far been scarce in non-White populations.
“In Western studies, unfortunately, the representation of the population of skin of color or different ethnicities is underrepresented,” said Yousef Binamer, MD, chair of the dermatology department at King Faisal Specialist Hospital, Riyadh, Saudi Arabia, when approached for an independent comment on the results.
“This is now why researchers are looking into specific groups to study them,” which he pointed out, is crucial because “the immunophenotyping of AD is different for each background.”
The incidence and severity of AD tend to be higher in Asian and Middle Eastern populations, for instance, he noted. “It’s very common in Asia, and not so common in very white skin. I did my training in Canada so I see the difference,” said Dr. Binamer. “Asian people tend to be more itchy and have a tendency to scar on pigmentation.” Whereas White people “usually do not have this issue.”
“So I think real-world evidence of JAK inhibitors in the other populations is important,” he said. Studies such as the one conducted in Singapore, as well as the recently reported QUARTZ3 study, which examined the use of the JAK inhibitor ivarmacitinib in 256 Chinese patients with AD, are helping to pave the way.
The study was independently supported. Dr. Yew and Dr. Binamer have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT WCD 2023