User login
Genetic profile flags scleroderma patients with best HSCT responses
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).

Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).
Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
CHICAGO – Subgroup categorization of patients with severe scleroderma by their gene-expression profile correlated with responses to a newly proven treatment for the disease that involves myeloablation and autologous hematopoietic stem cell transplantation (HSCT).
Patients who fell into the “fibroproliferative” scleroderma subgroup, roughly one-third of patients enrolled in the treatment study, showed a high level of benefit from myeloablation and autologous HSCT, Michael L. Whitfield, PhD, said at the annual meeting of the American College of Rheumatology.
In contrast, the roughly one-third of patients in the study with a gene-expression profile that placed them into the “normal-like” subgroup had outcomes that closely matched the normal-like patients in the control group, who were treated with cyclophosphamide, which suggests that the normal-like patients are probably not good candidates for HSCT, said Dr. Whitfield, a professor of molecular and systems biology at the Geisel School of Medicine at Dartmouth in Hanover, N.H.
This study starts to get at the question of “How do you do personalized medicine in a disease like scleroderma?” he explained. “HSCT may be a game changer for patients with the fibroproliferative type of scleroderma,” who did relatively poorly in the control group of the trial when they received cyclophosphamide. Categorization of patients by their gene-expression profiles is a way to find order among scleroderma patients in what is otherwise “a very heterogeneous disease, where some patients improve on a treatment and others do not,” Dr Whitfield said.
The study used data collected in the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial, which enrolled 75 patients with severe scleroderma at any of 26 sites in the United States and Canada. SCOT compared the safety and efficacy of myeloablation followed by autologous HSCT with that of treatment with cyclophosphamide, and it followed patients for a median of 54 months. The results showed that overall HSCT was superior for the primary endpoint and several secondary endpoints, including event-free survival, which was 79% after HSCT and 50% in control patients by the end of follow-up (N Engl J Med. 2018 Jan 4;378[1]:35-47).
Dr. Whitfield’s group took peripheral blood cells from 30 of the patients treated by HSCT and from 33 of the patients treated with cyclophosphamide per protocol and analyzed the gene-expression profiles of the cells to categorize patients into the gene-expression subtypes of scleroderma that had been previously defined by Dr. Whitfield and his associates: fibroproliferative, inflammatory, limited, or normal-like (PLOS One. 2008 Jul 18;3[7]:e2696). The gene-expression analysis, which now looks at the activity of about 1,300 genes, showed that the 33 cyclophosphamide-treated patients included 12 with an inflammatory profile, 12 with a normal-like profile, and 9 with a fibroproliferative profile. Among the 30 patients treated with HSCT, 11 were in the fibroproliferative group, 11 were normal-like, and 8 had an inflammatory pattern.
Analysis of event-free survival out to 6 years following enrollment showed that, in the fibroproliferative subgroup, roughly 90% of patients treated with HSCT remained alive and event free, compared with about 35% of the cyclophosphamide patients, a highly statistically significant difference. In the inflammatory subgroup, event-free survival persisted in about 90% in the HSCT recipients, compared with about 50% of those in the control arm, a difference that did not reach statistical significance. Among patients with normal-like gene expression, the event-free survival rate was about the same regardless of treatment, about 60% in each treatment arm. The results suggest that patients with normal-like disease “are probably not good candidates for a treatment as intensive as HSCT,” Dr. Whitfield said in an interview.
Although the HSCT and cyclophosphamide treatment groups included relatively small numbers of patients, when the researchers subdivided the trial cohort into three different scleroderma types, the analysis remained “powered well enough to see a difference; the difference was very clearly statistically significant,” Dr. Whitfield declared.
“Now that we have a treatment [HSCT] to tie to the [gene-expression analysis], we can think about using this in routine practice,” he concluded.
Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
SOURCE: Franks J et al. Arthritis Rheumatol. 2018;70(suppl 10): Abstract 1876.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point:
Major finding: About 90% of fibroproliferative scleroderma patients had prolonged event-free survival after stem cell transplant, compared with about 35% of controls.
Study details: The study used data collected in the SCOT trial of 75 patients with severe scleroderma.
Disclosures: Dr. Whitfield is a cofounder of Celdara, and he has been a consultant to Bristol-Myers Squibb, Corbus, UCB, and Third Rock Ventures.
Source: Franks J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1876.
Dueling SLE classification criteria: And the winner is...
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.

“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.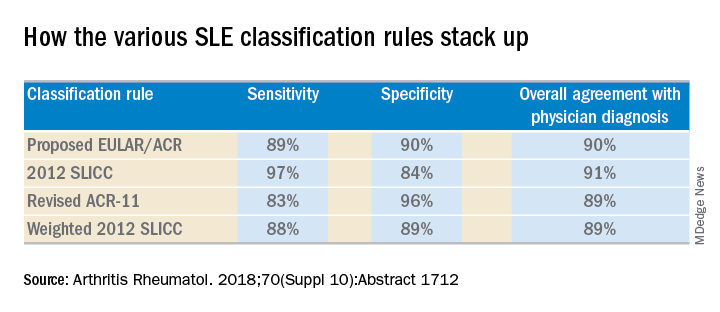
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.
“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
CHICAGO – Newer isn’t necessarily better – especially when it comes to the plethora of SLE classification criteria, according to Michelle A. Petri, MD, professor of medicine and director of the Hopkins Lupus Center at Johns Hopkins University, Baltimore.
“We have an embarrassment of criteria for lupus right now, and everyone wants to know if one is better than the others,” the rheumatologist said at the annual meeting of the American College of Rheumatology.
She and several coinvestigators who are members of the Systemic Lupus International Collaborating Clinics (SLICC) set out to learn the answer. They developed a new, modified, weighted version of the 2012 SLICC criteria and compared its sensitivity and specificity for SLE diagnosis by 690 physicians with three other major classification systems: the 1997 update to ACR-11 criteria, the nonweighted SLICC 2012 criteria, and the proposed EULAR/ACR criteria, which uses a differentially weighted approach in which the various possible disease manifestations are each assigned a different point score. In contrast, the revised ACR-11 and SLICC 2012 criteria count each SLE manifestation equally.
Long story short: “The two newly derived weighted classification rules did not perform better than the existing list-based rules in terms of overall agreement,” according to Dr. Petri.
“We don’t think that weighting all the criteria, which is what the EULAR/ACR and weighted SLICC 2012 rules do, adds to the performance of the criteria set, and in fact it makes it much more difficult for clinicians to use when there’s a complicated weighting system, unless it’s web-based or there’s an app for it. And to be honest, clinicians are so busy that they’re probably not going to take time out in a clinic visit to go use the web or an app. Our criteria need to be user friendly,” she continued.
So which of the four classification systems is most user friendly? The EULAR/ACR criteria can be dismissed on that score because they are supposed to be used only for research, according to the rheumatologist.
“I think the SLICC 2012 criteria are very useful for clinicians because they have the highest sensitivity. And what a clinician wants is not to miss a diagnosis and to start treatment early,” Dr. Petri said.
To develop the weighted SLICC criteria, whose future at this point doesn’t look bright, she and her coinvestigators redeployed the same physician-rated patient scenarios used to develop the nonweighted 2012 SLICC classification criteria and assigned each of the potential manifestations of SLE a specific point score. For example, acute cutaneous manifestations received 16 points, serositis 9, oral ulcers 16, thrombocytopenia 15, and so forth. Under this system, a patient with a score of at least 56 points, or lupus nephritis, or least one clinical and one immunologic component of SLE was classified as having the disease.
Dr. Petri reported having no financial conflicts regarding her study, supported by the National Institutes of Health.
SOURCE: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: The 2012 SLICC SLE classification criteria are best suited for clinical practice.
Major finding:
Study details: This study compared the sensitivity, specificity, and overall agreement with physician diagnosis of four different sets of SLE classification criteria.
Disclosures: The study was supported by the National Institutes of Health. The presenter reported having no financial conflicts.
Source: Petri MA et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1712.
ACR and EULAR draft classification criteria for IgG4-related disease
CHICAGO – A joint American College of Rheumatology and European League Against Rheumatism panel has written the first-ever classification criteria for immunoglobulin G4-related disease (IgG4-RD), and the draft version of the criteria identified the disorder with 99.2% specificity and 85.5% sensitivity when compared with expert case opinions.

“We’ve come a long way” to write these criteria 17 years after the first case report, and about a decade after IgG4-RD first became part of routine rheumatology practice, John H. Stone, MD, said at the annual meeting of the American College of Rheumatology. He cited one estimate that about 185,000 Americans currently have IgG4-RD.
Approval of the draft criteria by both the ACR and EULAR remains pending.
The working group assembled by the American College of Rheumatology and the European League Against Rheumatism to write the classification criteria included 89 members, and the draft document they produced combined inclusion and exclusion criteria, “the first ACR and EULAR classification criteria to include specific exclusions, to my knowledge,” said Dr. Stone professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital in Boston. The exclusions reflect the many other disorders that can mimic IgG4-RD, including cancers and several rheumatologic diseases, especially granulomatosis with polyangiitis and Sjögren’s syndrome.
The writing panel used 487 case reports from 272 patients diagnosed with IgG4-RD and 215 patients diagnosed with a different, mimic disease to derive the classification criteria, and then used 908 case reports – 493 from IgG4-RD patients and 415 reports from mimic cases – to test and validate the criteria.
The first step in classifying a patient with IgG4-RD is to identify involvement of at least one organ from the list the panel compiled of 10 organs where involvement typifies the disease: pancreas, bile ducts, orbits, lacrimal glands, major salivary glands, retroperitoneum, kidney, aorta, pachymeninges, and thyroid gland (Riedel’s thyroiditis, but not Hashimoto’s disease). Patients who do not have disease involvement in at least one of these organs don’t qualify as having IgG4-RD.
The next step is to rule out patients who have at least one exclusion criterion from a list of 21 exclusions the panel cited, divided into four categories based on the test that finds each exclusion: clinical examination, serology, radiology, or pathology.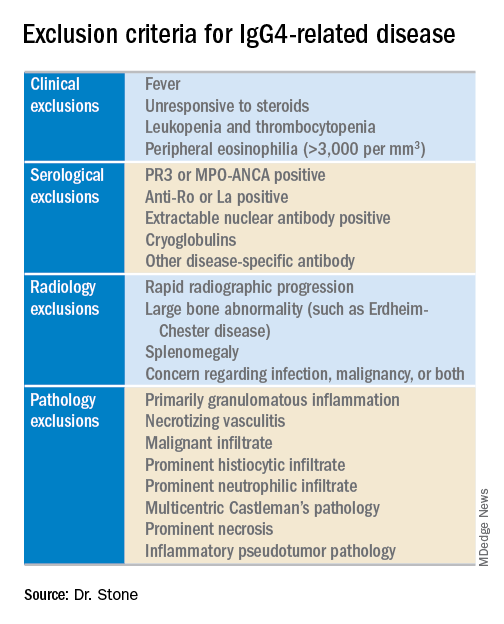
The last step is to identify enough individual classification hallmarks in the patient so that collectively they definitively identify IgG4-RD. The writing panel endorsed seven inclusion-criteria domains that each contain at least two different disease manifestations that confer points if fulfilled. To qualify for IgG4-RD classification, a patient needs to have enough manifestations to tally at least 19 points.
Fulfilling the inclusion criteria is the key step in classification, but the exclusion criteria also play a role in helping to rule the disease in or out, Dr. Stone noted. Without the exclusion criteria, the remaining classification criteria identified the 1,395 total cases and mimics studied with an increased sensitivity of 90% (compared with 85.5% when the exclusion criteria also apply), but with reduced specificity of 88.5% (compared with 99.2%). High specificity is a key aim. The criteria are supposed to give greater uniformity to patient selection for studies and ensure that enrolled patients actually have IgG4-RD. “Our goal was criteria that would prevent enrollment of patients without IgG4-RD,” he said.
Although IgG4 level is one of the seven inclusion domains and can give a patient as many as 10.8 points toward classification when the level exceeds five times the upper limit of normal, the criteria solidify the notion that “we have greatly overemphasized IgG4” in past considerations of the disease, said Dr. Stone. Elevation of IgG4 is one of several disease markers in most patients, but it’s not essential to classification and is missing in nearly a third of patients. While the cause of IgG4-RD remains unknown, it appears to involve an abnormal interaction between B cells and a CD4+ cytotoxic T lymphocyte, an understanding that has led to testing investigational therapies that target B cells including rituximab (Rituxan) and an agent called XmAb5871. “Rituximab works very well,” Dr. Stone said. The absence of a known cause is a reason why classification is so complex.
Dr. Stone also reminded his audience that IgG4-RD is an indolent disease that can produce symptoms for months or years before getting diagnosed. It often is accompanied by significant weight loss of 20 or more pounds, but without fever, and often features a dissociation between a high erythrocyte sedimentation rate but a relatively low level of C-reactive protein. “It’s astonishing how much weight patients lose,” he said.
Though barely more than a decade on the scene, awareness of IgG4-RD among rheumatologists has become widespread, though awareness has probably lagged among many primary care physicians, Dr. Stone said in an interview. The estimated prevalence of about 185,000 U.S. residents with IgG4-RD is probably an underestimate, he added. His group at Massachusetts General Hospital in Boston averages 3-5 patients evaluated each week as possibly having IgG4-RD, and this one group is now following more than 350 patients who have been diagnosed with the disease. “It’s probably more common than a lot of other conditions that rheumatologists treat, more common than scleroderma or ANCA-associated vasculitis,” Dr. Stone said. “The new criteria will help further raise awareness.”
Dr. Stone has been a consultant to and has received research funding from Genentech, Roche, and Xencor.
CHICAGO – A joint American College of Rheumatology and European League Against Rheumatism panel has written the first-ever classification criteria for immunoglobulin G4-related disease (IgG4-RD), and the draft version of the criteria identified the disorder with 99.2% specificity and 85.5% sensitivity when compared with expert case opinions.
“We’ve come a long way” to write these criteria 17 years after the first case report, and about a decade after IgG4-RD first became part of routine rheumatology practice, John H. Stone, MD, said at the annual meeting of the American College of Rheumatology. He cited one estimate that about 185,000 Americans currently have IgG4-RD.
Approval of the draft criteria by both the ACR and EULAR remains pending.
The working group assembled by the American College of Rheumatology and the European League Against Rheumatism to write the classification criteria included 89 members, and the draft document they produced combined inclusion and exclusion criteria, “the first ACR and EULAR classification criteria to include specific exclusions, to my knowledge,” said Dr. Stone professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital in Boston. The exclusions reflect the many other disorders that can mimic IgG4-RD, including cancers and several rheumatologic diseases, especially granulomatosis with polyangiitis and Sjögren’s syndrome.
The writing panel used 487 case reports from 272 patients diagnosed with IgG4-RD and 215 patients diagnosed with a different, mimic disease to derive the classification criteria, and then used 908 case reports – 493 from IgG4-RD patients and 415 reports from mimic cases – to test and validate the criteria.
The first step in classifying a patient with IgG4-RD is to identify involvement of at least one organ from the list the panel compiled of 10 organs where involvement typifies the disease: pancreas, bile ducts, orbits, lacrimal glands, major salivary glands, retroperitoneum, kidney, aorta, pachymeninges, and thyroid gland (Riedel’s thyroiditis, but not Hashimoto’s disease). Patients who do not have disease involvement in at least one of these organs don’t qualify as having IgG4-RD.
The next step is to rule out patients who have at least one exclusion criterion from a list of 21 exclusions the panel cited, divided into four categories based on the test that finds each exclusion: clinical examination, serology, radiology, or pathology.
The last step is to identify enough individual classification hallmarks in the patient so that collectively they definitively identify IgG4-RD. The writing panel endorsed seven inclusion-criteria domains that each contain at least two different disease manifestations that confer points if fulfilled. To qualify for IgG4-RD classification, a patient needs to have enough manifestations to tally at least 19 points.
Fulfilling the inclusion criteria is the key step in classification, but the exclusion criteria also play a role in helping to rule the disease in or out, Dr. Stone noted. Without the exclusion criteria, the remaining classification criteria identified the 1,395 total cases and mimics studied with an increased sensitivity of 90% (compared with 85.5% when the exclusion criteria also apply), but with reduced specificity of 88.5% (compared with 99.2%). High specificity is a key aim. The criteria are supposed to give greater uniformity to patient selection for studies and ensure that enrolled patients actually have IgG4-RD. “Our goal was criteria that would prevent enrollment of patients without IgG4-RD,” he said.
Although IgG4 level is one of the seven inclusion domains and can give a patient as many as 10.8 points toward classification when the level exceeds five times the upper limit of normal, the criteria solidify the notion that “we have greatly overemphasized IgG4” in past considerations of the disease, said Dr. Stone. Elevation of IgG4 is one of several disease markers in most patients, but it’s not essential to classification and is missing in nearly a third of patients. While the cause of IgG4-RD remains unknown, it appears to involve an abnormal interaction between B cells and a CD4+ cytotoxic T lymphocyte, an understanding that has led to testing investigational therapies that target B cells including rituximab (Rituxan) and an agent called XmAb5871. “Rituximab works very well,” Dr. Stone said. The absence of a known cause is a reason why classification is so complex.
Dr. Stone also reminded his audience that IgG4-RD is an indolent disease that can produce symptoms for months or years before getting diagnosed. It often is accompanied by significant weight loss of 20 or more pounds, but without fever, and often features a dissociation between a high erythrocyte sedimentation rate but a relatively low level of C-reactive protein. “It’s astonishing how much weight patients lose,” he said.
Though barely more than a decade on the scene, awareness of IgG4-RD among rheumatologists has become widespread, though awareness has probably lagged among many primary care physicians, Dr. Stone said in an interview. The estimated prevalence of about 185,000 U.S. residents with IgG4-RD is probably an underestimate, he added. His group at Massachusetts General Hospital in Boston averages 3-5 patients evaluated each week as possibly having IgG4-RD, and this one group is now following more than 350 patients who have been diagnosed with the disease. “It’s probably more common than a lot of other conditions that rheumatologists treat, more common than scleroderma or ANCA-associated vasculitis,” Dr. Stone said. “The new criteria will help further raise awareness.”
Dr. Stone has been a consultant to and has received research funding from Genentech, Roche, and Xencor.
CHICAGO – A joint American College of Rheumatology and European League Against Rheumatism panel has written the first-ever classification criteria for immunoglobulin G4-related disease (IgG4-RD), and the draft version of the criteria identified the disorder with 99.2% specificity and 85.5% sensitivity when compared with expert case opinions.
“We’ve come a long way” to write these criteria 17 years after the first case report, and about a decade after IgG4-RD first became part of routine rheumatology practice, John H. Stone, MD, said at the annual meeting of the American College of Rheumatology. He cited one estimate that about 185,000 Americans currently have IgG4-RD.
Approval of the draft criteria by both the ACR and EULAR remains pending.
The working group assembled by the American College of Rheumatology and the European League Against Rheumatism to write the classification criteria included 89 members, and the draft document they produced combined inclusion and exclusion criteria, “the first ACR and EULAR classification criteria to include specific exclusions, to my knowledge,” said Dr. Stone professor of medicine at Harvard Medical School and director of clinical rheumatology at Massachusetts General Hospital in Boston. The exclusions reflect the many other disorders that can mimic IgG4-RD, including cancers and several rheumatologic diseases, especially granulomatosis with polyangiitis and Sjögren’s syndrome.
The writing panel used 487 case reports from 272 patients diagnosed with IgG4-RD and 215 patients diagnosed with a different, mimic disease to derive the classification criteria, and then used 908 case reports – 493 from IgG4-RD patients and 415 reports from mimic cases – to test and validate the criteria.
The first step in classifying a patient with IgG4-RD is to identify involvement of at least one organ from the list the panel compiled of 10 organs where involvement typifies the disease: pancreas, bile ducts, orbits, lacrimal glands, major salivary glands, retroperitoneum, kidney, aorta, pachymeninges, and thyroid gland (Riedel’s thyroiditis, but not Hashimoto’s disease). Patients who do not have disease involvement in at least one of these organs don’t qualify as having IgG4-RD.
The next step is to rule out patients who have at least one exclusion criterion from a list of 21 exclusions the panel cited, divided into four categories based on the test that finds each exclusion: clinical examination, serology, radiology, or pathology.
The last step is to identify enough individual classification hallmarks in the patient so that collectively they definitively identify IgG4-RD. The writing panel endorsed seven inclusion-criteria domains that each contain at least two different disease manifestations that confer points if fulfilled. To qualify for IgG4-RD classification, a patient needs to have enough manifestations to tally at least 19 points.
Fulfilling the inclusion criteria is the key step in classification, but the exclusion criteria also play a role in helping to rule the disease in or out, Dr. Stone noted. Without the exclusion criteria, the remaining classification criteria identified the 1,395 total cases and mimics studied with an increased sensitivity of 90% (compared with 85.5% when the exclusion criteria also apply), but with reduced specificity of 88.5% (compared with 99.2%). High specificity is a key aim. The criteria are supposed to give greater uniformity to patient selection for studies and ensure that enrolled patients actually have IgG4-RD. “Our goal was criteria that would prevent enrollment of patients without IgG4-RD,” he said.
Although IgG4 level is one of the seven inclusion domains and can give a patient as many as 10.8 points toward classification when the level exceeds five times the upper limit of normal, the criteria solidify the notion that “we have greatly overemphasized IgG4” in past considerations of the disease, said Dr. Stone. Elevation of IgG4 is one of several disease markers in most patients, but it’s not essential to classification and is missing in nearly a third of patients. While the cause of IgG4-RD remains unknown, it appears to involve an abnormal interaction between B cells and a CD4+ cytotoxic T lymphocyte, an understanding that has led to testing investigational therapies that target B cells including rituximab (Rituxan) and an agent called XmAb5871. “Rituximab works very well,” Dr. Stone said. The absence of a known cause is a reason why classification is so complex.
Dr. Stone also reminded his audience that IgG4-RD is an indolent disease that can produce symptoms for months or years before getting diagnosed. It often is accompanied by significant weight loss of 20 or more pounds, but without fever, and often features a dissociation between a high erythrocyte sedimentation rate but a relatively low level of C-reactive protein. “It’s astonishing how much weight patients lose,” he said.
Though barely more than a decade on the scene, awareness of IgG4-RD among rheumatologists has become widespread, though awareness has probably lagged among many primary care physicians, Dr. Stone said in an interview. The estimated prevalence of about 185,000 U.S. residents with IgG4-RD is probably an underestimate, he added. His group at Massachusetts General Hospital in Boston averages 3-5 patients evaluated each week as possibly having IgG4-RD, and this one group is now following more than 350 patients who have been diagnosed with the disease. “It’s probably more common than a lot of other conditions that rheumatologists treat, more common than scleroderma or ANCA-associated vasculitis,” Dr. Stone said. “The new criteria will help further raise awareness.”
Dr. Stone has been a consultant to and has received research funding from Genentech, Roche, and Xencor.
REPORTING FROM THE ACR ANNUAL MEETING
Complications cluster in inflammatory arthritis patients after total knee replacement
CHICAGO – Patients with an inflammatory arthritis had significantly higher rates of infections, transfusions, and readmissions following total knee replacement than did patients without inflammatory arthritis in a study of more than 137,000 Americans who underwent this surgery.

A sampling of U.S. patients who underwent total knee arthroplasty (TKA) during 2007-2016 showed that among the small percentage of these patients who had an inflammatory arthritis (IA), the rate of periprosthetic joint or wound infection while hospitalized or out to 30 days after surgery was a statistically significant 64% higher relative to patients without inflammatory arthritis, after adjustment for several demographic and clinical confounders, including recent glucocorticoid treatment, Susan M. Goodman, MD, said at the annual meeting of the American College of Rheumatology. The analysis also showed a statistically significant 46% higher relative rate of hospital readmission for any cause during the 90 days after surgery, and a significant 39% relative increase in blood transfusions during the 30 days after TKA in the IA patients.
“These results have important implications for evolving bundled payment models” for TKA, said Dr. Goodman, a rheumatologist at the Hospital for Special Surgery in New York. “Hospitals should receive commensurate resources to maintain access to total TKA for patients with IA.”
For this analysis, Dr. Goodman and her associates classified IA as a patient with a recorded diagnosis of rheumatoid arthritis, spondyloarthritis, or systemic lupus erythematosus if the patient had also received treatment during the year before surgery with a disease-modifying antirheumatic drug, a biologic agent, or a drug that treats systemic lupus erythematosus.
Complications following TKA became a particular concern to hospitals starting in 2013 when the Centers for Medicare & Medicaid Services began a program that penalized hospitals for outcomes such as excessive readmissions following selected types of hospitalizations and also with recent steps to bundle TKA reimbursement with related 90-day outcomes.
“My concern is to ensure that patients with IA aren’t penalized and can maintain access” to TKA despite recent policy moves by the CMS. Faced with potential disincentives to treat patients with an IA, “hospitals might cherry pick patients,” Dr. Goodman said in an interview. The new findings “are a reason for administrators to argue for patients with IA to come out of the cost bundle.”
Dr. Goodman expressed hope that future policies will better reflect the higher levels of risk faced by patients with an IA undergoing TKA. CMS “is pretty responsive,” she said.
The study used data collected by Humana for about 25 million American health insurance beneficiaries during 2007-2016, which included 137,550 people who underwent a TKA. Of these, 3,067 (2%) met the study’s definition for IA, and 134,483 did not. Most of those who did not meet the definition likely had osteoarthritis, Dr. Goodman said. This low percentage of U.S. TKA patients with IA was consistent with numbers in prior reports.
The researchers calculated the relative risk of the IA patients, compared with all the others, for nine potential complications, including acute MI, pneumonia, sepsis, pulmonary embolism, and death. The complications with significantly higher rates among the IA patients after confounder adjustment were 30-day infections, 30-day transfusions, and 90-day readmissions.
Dr. Goodman had no relevant disclosures.
SOURCE: Richardson S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1932.
CHICAGO – Patients with an inflammatory arthritis had significantly higher rates of infections, transfusions, and readmissions following total knee replacement than did patients without inflammatory arthritis in a study of more than 137,000 Americans who underwent this surgery.
A sampling of U.S. patients who underwent total knee arthroplasty (TKA) during 2007-2016 showed that among the small percentage of these patients who had an inflammatory arthritis (IA), the rate of periprosthetic joint or wound infection while hospitalized or out to 30 days after surgery was a statistically significant 64% higher relative to patients without inflammatory arthritis, after adjustment for several demographic and clinical confounders, including recent glucocorticoid treatment, Susan M. Goodman, MD, said at the annual meeting of the American College of Rheumatology. The analysis also showed a statistically significant 46% higher relative rate of hospital readmission for any cause during the 90 days after surgery, and a significant 39% relative increase in blood transfusions during the 30 days after TKA in the IA patients.
“These results have important implications for evolving bundled payment models” for TKA, said Dr. Goodman, a rheumatologist at the Hospital for Special Surgery in New York. “Hospitals should receive commensurate resources to maintain access to total TKA for patients with IA.”
For this analysis, Dr. Goodman and her associates classified IA as a patient with a recorded diagnosis of rheumatoid arthritis, spondyloarthritis, or systemic lupus erythematosus if the patient had also received treatment during the year before surgery with a disease-modifying antirheumatic drug, a biologic agent, or a drug that treats systemic lupus erythematosus.
Complications following TKA became a particular concern to hospitals starting in 2013 when the Centers for Medicare & Medicaid Services began a program that penalized hospitals for outcomes such as excessive readmissions following selected types of hospitalizations and also with recent steps to bundle TKA reimbursement with related 90-day outcomes.
“My concern is to ensure that patients with IA aren’t penalized and can maintain access” to TKA despite recent policy moves by the CMS. Faced with potential disincentives to treat patients with an IA, “hospitals might cherry pick patients,” Dr. Goodman said in an interview. The new findings “are a reason for administrators to argue for patients with IA to come out of the cost bundle.”
Dr. Goodman expressed hope that future policies will better reflect the higher levels of risk faced by patients with an IA undergoing TKA. CMS “is pretty responsive,” she said.
The study used data collected by Humana for about 25 million American health insurance beneficiaries during 2007-2016, which included 137,550 people who underwent a TKA. Of these, 3,067 (2%) met the study’s definition for IA, and 134,483 did not. Most of those who did not meet the definition likely had osteoarthritis, Dr. Goodman said. This low percentage of U.S. TKA patients with IA was consistent with numbers in prior reports.
The researchers calculated the relative risk of the IA patients, compared with all the others, for nine potential complications, including acute MI, pneumonia, sepsis, pulmonary embolism, and death. The complications with significantly higher rates among the IA patients after confounder adjustment were 30-day infections, 30-day transfusions, and 90-day readmissions.
Dr. Goodman had no relevant disclosures.
SOURCE: Richardson S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1932.
CHICAGO – Patients with an inflammatory arthritis had significantly higher rates of infections, transfusions, and readmissions following total knee replacement than did patients without inflammatory arthritis in a study of more than 137,000 Americans who underwent this surgery.
A sampling of U.S. patients who underwent total knee arthroplasty (TKA) during 2007-2016 showed that among the small percentage of these patients who had an inflammatory arthritis (IA), the rate of periprosthetic joint or wound infection while hospitalized or out to 30 days after surgery was a statistically significant 64% higher relative to patients without inflammatory arthritis, after adjustment for several demographic and clinical confounders, including recent glucocorticoid treatment, Susan M. Goodman, MD, said at the annual meeting of the American College of Rheumatology. The analysis also showed a statistically significant 46% higher relative rate of hospital readmission for any cause during the 90 days after surgery, and a significant 39% relative increase in blood transfusions during the 30 days after TKA in the IA patients.
“These results have important implications for evolving bundled payment models” for TKA, said Dr. Goodman, a rheumatologist at the Hospital for Special Surgery in New York. “Hospitals should receive commensurate resources to maintain access to total TKA for patients with IA.”
For this analysis, Dr. Goodman and her associates classified IA as a patient with a recorded diagnosis of rheumatoid arthritis, spondyloarthritis, or systemic lupus erythematosus if the patient had also received treatment during the year before surgery with a disease-modifying antirheumatic drug, a biologic agent, or a drug that treats systemic lupus erythematosus.
Complications following TKA became a particular concern to hospitals starting in 2013 when the Centers for Medicare & Medicaid Services began a program that penalized hospitals for outcomes such as excessive readmissions following selected types of hospitalizations and also with recent steps to bundle TKA reimbursement with related 90-day outcomes.
“My concern is to ensure that patients with IA aren’t penalized and can maintain access” to TKA despite recent policy moves by the CMS. Faced with potential disincentives to treat patients with an IA, “hospitals might cherry pick patients,” Dr. Goodman said in an interview. The new findings “are a reason for administrators to argue for patients with IA to come out of the cost bundle.”
Dr. Goodman expressed hope that future policies will better reflect the higher levels of risk faced by patients with an IA undergoing TKA. CMS “is pretty responsive,” she said.
The study used data collected by Humana for about 25 million American health insurance beneficiaries during 2007-2016, which included 137,550 people who underwent a TKA. Of these, 3,067 (2%) met the study’s definition for IA, and 134,483 did not. Most of those who did not meet the definition likely had osteoarthritis, Dr. Goodman said. This low percentage of U.S. TKA patients with IA was consistent with numbers in prior reports.
The researchers calculated the relative risk of the IA patients, compared with all the others, for nine potential complications, including acute MI, pneumonia, sepsis, pulmonary embolism, and death. The complications with significantly higher rates among the IA patients after confounder adjustment were 30-day infections, 30-day transfusions, and 90-day readmissions.
Dr. Goodman had no relevant disclosures.
SOURCE: Richardson S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1932.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: Complications were more common after total knee arthroplasty in patients with an inflammatory arthritis.
Major finding: Inflammatory arthritis patients had a 64% higher rate of infections after total knee arthroplasty, compared with patients without inflammatory arthritis.
Study details: Data analysis for 137,550 Americans who underwent total knee arthroplasty during 2007-2016.
Disclosures: Dr. Goodman had no relevant disclosures.
Source: Richardson S et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 1932.
Childhood abuse linked with tripled adult SLE incidence
CHICAGO – , in a study of more than 67,000 American nurses.
The results also suggested that development of depression and post-traumatic stress disorder (PTSD) may have been intermediary steps between episodes of childhood abuse and later development of systemic lupus erythematosus (SLE), Candace H. Feldman, MD, said at the annual meeting of the American College of Rheumatology.
These findings suggest the “importance of screening for childhood abuse exposures as well as for depression and PTSD in routine practice,” although Dr. Feldman acknowledged that interventions aimed at treating depression and PTSD have as of now no proven role for mitigating SLE.
The analysis Dr. Feldman and her associates ran on data collected in the Nurses Health Study II also documented a “striking” number of the enrolled women who completed the survey in 2001 and reported a history of abuse when they were 11 years old or younger: 30% of the 67,516 respondents reported a moderate level of abuse, and 24% reported a high level of abuse. An additional 22% reported either no or a very low level of abuse. These numbers suggest that abuse of girls “is very common and probably underreported,” she said in a video interview.
The Nurses Health Study II enrolled more than 116,429 U.S. women in 1989 who were 25-42 years old and had no history of SLE. Recording of incident SLE cases began in 1991 and for this analysis continued for 24 years, through 2015, during which time 94 women developed SLE that was confirmed in a review by two rheumatologists applying the 1997 SLE classification criteria (Arthritis Rheum. 1997 Sept;40[9]:1725. The incidence of SLE was 2.57-fold more common among women who reported a high level of abuse, compared with those who had no or very low abuse, after adjustment for several demographic and clinical confounders, reported Dr. Feldman, a rheumatologist at Brigham and Women’s Hospital in Boston.
“To our knowledge this is the first study to prospectively look at exposure to different forms of childhood abuse and SLE incidence in a general population of women,” she said.
To make the analysis more prospective the researchers also ran a calculation that considered only SLE cases that appeared after completion of the 2001 abuse survey. Using this criterion the incidence was 3.11-fold higher among women who reported a high level of childhood abuse. Further analyses showed that statistically a diagnosis of PTSD accounted for about 23% of the risk for developing SLE, and depression appeared responsible for about 17% of the risk. The analysis also showed no statistically significant link between sexual abuse in childhood or as a teenager and later onset of SLE.
The findings are consistent with prior reports that linked stress to development of various autoimmune diseases, Dr. Feldman noted. She speculated that high childhood stress could cause changes in inflammation, immune function, epigenetics, the autonomic nervous system, and endocrine pathways that could play a role in triggering depression or PTSD, and eventually SLE.
[email protected]
On Twitter @mitchelzoler
SOURCE:Feldman C et al. Arthritis Rheumatol. 2018;70(suppl 10) Abstract 2807.
CHICAGO – , in a study of more than 67,000 American nurses.
The results also suggested that development of depression and post-traumatic stress disorder (PTSD) may have been intermediary steps between episodes of childhood abuse and later development of systemic lupus erythematosus (SLE), Candace H. Feldman, MD, said at the annual meeting of the American College of Rheumatology.
These findings suggest the “importance of screening for childhood abuse exposures as well as for depression and PTSD in routine practice,” although Dr. Feldman acknowledged that interventions aimed at treating depression and PTSD have as of now no proven role for mitigating SLE.
The analysis Dr. Feldman and her associates ran on data collected in the Nurses Health Study II also documented a “striking” number of the enrolled women who completed the survey in 2001 and reported a history of abuse when they were 11 years old or younger: 30% of the 67,516 respondents reported a moderate level of abuse, and 24% reported a high level of abuse. An additional 22% reported either no or a very low level of abuse. These numbers suggest that abuse of girls “is very common and probably underreported,” she said in a video interview.
The Nurses Health Study II enrolled more than 116,429 U.S. women in 1989 who were 25-42 years old and had no history of SLE. Recording of incident SLE cases began in 1991 and for this analysis continued for 24 years, through 2015, during which time 94 women developed SLE that was confirmed in a review by two rheumatologists applying the 1997 SLE classification criteria (Arthritis Rheum. 1997 Sept;40[9]:1725. The incidence of SLE was 2.57-fold more common among women who reported a high level of abuse, compared with those who had no or very low abuse, after adjustment for several demographic and clinical confounders, reported Dr. Feldman, a rheumatologist at Brigham and Women’s Hospital in Boston.
“To our knowledge this is the first study to prospectively look at exposure to different forms of childhood abuse and SLE incidence in a general population of women,” she said.
To make the analysis more prospective the researchers also ran a calculation that considered only SLE cases that appeared after completion of the 2001 abuse survey. Using this criterion the incidence was 3.11-fold higher among women who reported a high level of childhood abuse. Further analyses showed that statistically a diagnosis of PTSD accounted for about 23% of the risk for developing SLE, and depression appeared responsible for about 17% of the risk. The analysis also showed no statistically significant link between sexual abuse in childhood or as a teenager and later onset of SLE.
The findings are consistent with prior reports that linked stress to development of various autoimmune diseases, Dr. Feldman noted. She speculated that high childhood stress could cause changes in inflammation, immune function, epigenetics, the autonomic nervous system, and endocrine pathways that could play a role in triggering depression or PTSD, and eventually SLE.
[email protected]
On Twitter @mitchelzoler
SOURCE:Feldman C et al. Arthritis Rheumatol. 2018;70(suppl 10) Abstract 2807.
CHICAGO – , in a study of more than 67,000 American nurses.
The results also suggested that development of depression and post-traumatic stress disorder (PTSD) may have been intermediary steps between episodes of childhood abuse and later development of systemic lupus erythematosus (SLE), Candace H. Feldman, MD, said at the annual meeting of the American College of Rheumatology.
These findings suggest the “importance of screening for childhood abuse exposures as well as for depression and PTSD in routine practice,” although Dr. Feldman acknowledged that interventions aimed at treating depression and PTSD have as of now no proven role for mitigating SLE.
The analysis Dr. Feldman and her associates ran on data collected in the Nurses Health Study II also documented a “striking” number of the enrolled women who completed the survey in 2001 and reported a history of abuse when they were 11 years old or younger: 30% of the 67,516 respondents reported a moderate level of abuse, and 24% reported a high level of abuse. An additional 22% reported either no or a very low level of abuse. These numbers suggest that abuse of girls “is very common and probably underreported,” she said in a video interview.
The Nurses Health Study II enrolled more than 116,429 U.S. women in 1989 who were 25-42 years old and had no history of SLE. Recording of incident SLE cases began in 1991 and for this analysis continued for 24 years, through 2015, during which time 94 women developed SLE that was confirmed in a review by two rheumatologists applying the 1997 SLE classification criteria (Arthritis Rheum. 1997 Sept;40[9]:1725. The incidence of SLE was 2.57-fold more common among women who reported a high level of abuse, compared with those who had no or very low abuse, after adjustment for several demographic and clinical confounders, reported Dr. Feldman, a rheumatologist at Brigham and Women’s Hospital in Boston.
“To our knowledge this is the first study to prospectively look at exposure to different forms of childhood abuse and SLE incidence in a general population of women,” she said.
To make the analysis more prospective the researchers also ran a calculation that considered only SLE cases that appeared after completion of the 2001 abuse survey. Using this criterion the incidence was 3.11-fold higher among women who reported a high level of childhood abuse. Further analyses showed that statistically a diagnosis of PTSD accounted for about 23% of the risk for developing SLE, and depression appeared responsible for about 17% of the risk. The analysis also showed no statistically significant link between sexual abuse in childhood or as a teenager and later onset of SLE.
The findings are consistent with prior reports that linked stress to development of various autoimmune diseases, Dr. Feldman noted. She speculated that high childhood stress could cause changes in inflammation, immune function, epigenetics, the autonomic nervous system, and endocrine pathways that could play a role in triggering depression or PTSD, and eventually SLE.
[email protected]
On Twitter @mitchelzoler
SOURCE:Feldman C et al. Arthritis Rheumatol. 2018;70(suppl 10) Abstract 2807.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: A history of high childhood abuse linked with a nearly three-fold higher incidence of systemic lupus erythematosus during adulthood.
Major finding: The incidence of systemic lupus erythematosus was 2.57-fold higher among women with high childhood abuse compared with unabused women.
Study details: Data from 67,516 women enrolled in the Nurses Health Study II.
Disclosures: Dr. Feldman had no disclosures.
Source: Feldman C et al. Arthritis Rheumatol. 2018;70(suppl 10) Abstract 2807.

Vascular ultrasound reasonable for first-line imaging of large-vessel GCA
CHICAGO – Vascular ultrasound showed high sensitivity and specificity for diagnosing large-vessel giant-cell arteritis (LV-GCA) in a prospective study of patients with suspected new-onset disease.

The findings highlight the value of vascular ultrasound – in the hands of experienced sonographers – as a first-line imaging test in this setting, Berit Dalsgaard Nielsen, MD, reported at the annual meeting of the American College of Rheumatology.
Of 41 control subjects without LV-GCA, none had a positive ultrasound, whereas 36 of 45 LV-GCA patients had a positive ultrasound, which gives the test a specificity of 100% and sensitivity of 80%, Dr. Nielsen of Aarhus (Denmark) University Hospital said during a press briefing at the meeting.
Ultrasound was performed on the carotid artery in the neck and axillary arteries under the arm, which are easily accessible by ultrasound.
“These patients also had temporal arteries evaluated, and if we included this evaluation in the diagnostic performance, it showed a sensitivity of 91%,” she noted, explaining that temporal artery ultrasound alone conferred 71% sensitivity. “So it actually helped us identify more GCA patients.”
The study subjects were adults with suspected GCA. Inclusion criteria included age of at least 50 years, C-reactive protein of more than 15 mg/L or erythrocyte sedimentation rate of more than 40 mm, and either cranial symptoms, new-onset limb claudication, protracted constitutional symptoms, or polymyalgia rheumatica (PMR) symptoms. Patients were excluded if they had recent or ongoing glucocorticoid or disease-modifying antirheumatic drug treatment, a previous GCA or PMR diagnosis, or a large vessel inflammation that mimicked LV-GCA.
Clinical evaluations and imaging tests were performed prior to treatment initiation. The reference diagnosis was a clinical diagnosis of GCA and a positive 18F-FDG PET/CT scan, Dr Nielsen said, adding that ultrasound examinations were performed by experienced sonographers who were blinded to the PET/CT results.
Of the 86 patients included, 45 had LV-GCA with or without concomitant cranial GCA, 10 had isolated cranial GCA, 21 had PMR, and 10 were diagnosed with other diseases. The patients found to not have LV-GCA were considered control subjects.
The findings are notable because, while PET is considered the gold standard, it is very expensive and not always readily available, Dr. Nielsen said.
Additionally, while cranial-GCA patients generally present with symptoms such as headache, jaw claudication, and visual disturbances that are considered typical for GCA, LV-GCA patients rarely present with these symptoms.
Rather, these LV-GCA patients tend to present with constitutional symptoms mimicking infection or cancer, and they undergo extensive examination programs before the diagnosis is established. For this reason, diagnosis is often delayed for several months in LV-GCA patients until late in the disease course.
“During this time they often experience a decline in physical ability,” she said. “So in this disease subset of patients with GCA, there’s an unmet need for earlier recognition and earlier diagnosis.”
New recommendations from the European League Against Rheumatism call for early diagnostic imaging in all cases of suspected GCA, she added, noting that, for cranial-GCA symptoms, temporal artery ultrasound is recommended first line, but for those who present without cranial symptoms, no particular imaging modality is recommended because of a lack of comparative and diagnostic accuracy data in LV-GCA.
Biopsy has traditionally been used in these cases, but now imaging can be substituted – and vascular ultrasound is an attractive first-line option given its affordability and availability.
Indeed, the current findings support its use in this setting, she said.
“We think that these results indicate that ultrasound should not only be the first-line imaging test in patients presenting with cranial symptoms, but also in patients suspected of GCA presenting with constitutional symptoms, and if this examination is included in the standard examinations in fast-track clinics, it may overcome the delay in diagnosis and the patients can be treated earlier. It may also spare the unneeded examinations performed in these patients,” she concluded.
Dr. Nielsen disclosed a relationship with Roche.
SOURCE: Nielsen B et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2905.
CHICAGO – Vascular ultrasound showed high sensitivity and specificity for diagnosing large-vessel giant-cell arteritis (LV-GCA) in a prospective study of patients with suspected new-onset disease.
The findings highlight the value of vascular ultrasound – in the hands of experienced sonographers – as a first-line imaging test in this setting, Berit Dalsgaard Nielsen, MD, reported at the annual meeting of the American College of Rheumatology.
Of 41 control subjects without LV-GCA, none had a positive ultrasound, whereas 36 of 45 LV-GCA patients had a positive ultrasound, which gives the test a specificity of 100% and sensitivity of 80%, Dr. Nielsen of Aarhus (Denmark) University Hospital said during a press briefing at the meeting.
Ultrasound was performed on the carotid artery in the neck and axillary arteries under the arm, which are easily accessible by ultrasound.
“These patients also had temporal arteries evaluated, and if we included this evaluation in the diagnostic performance, it showed a sensitivity of 91%,” she noted, explaining that temporal artery ultrasound alone conferred 71% sensitivity. “So it actually helped us identify more GCA patients.”
The study subjects were adults with suspected GCA. Inclusion criteria included age of at least 50 years, C-reactive protein of more than 15 mg/L or erythrocyte sedimentation rate of more than 40 mm, and either cranial symptoms, new-onset limb claudication, protracted constitutional symptoms, or polymyalgia rheumatica (PMR) symptoms. Patients were excluded if they had recent or ongoing glucocorticoid or disease-modifying antirheumatic drug treatment, a previous GCA or PMR diagnosis, or a large vessel inflammation that mimicked LV-GCA.
Clinical evaluations and imaging tests were performed prior to treatment initiation. The reference diagnosis was a clinical diagnosis of GCA and a positive 18F-FDG PET/CT scan, Dr Nielsen said, adding that ultrasound examinations were performed by experienced sonographers who were blinded to the PET/CT results.
Of the 86 patients included, 45 had LV-GCA with or without concomitant cranial GCA, 10 had isolated cranial GCA, 21 had PMR, and 10 were diagnosed with other diseases. The patients found to not have LV-GCA were considered control subjects.
The findings are notable because, while PET is considered the gold standard, it is very expensive and not always readily available, Dr. Nielsen said.
Additionally, while cranial-GCA patients generally present with symptoms such as headache, jaw claudication, and visual disturbances that are considered typical for GCA, LV-GCA patients rarely present with these symptoms.
Rather, these LV-GCA patients tend to present with constitutional symptoms mimicking infection or cancer, and they undergo extensive examination programs before the diagnosis is established. For this reason, diagnosis is often delayed for several months in LV-GCA patients until late in the disease course.
“During this time they often experience a decline in physical ability,” she said. “So in this disease subset of patients with GCA, there’s an unmet need for earlier recognition and earlier diagnosis.”
New recommendations from the European League Against Rheumatism call for early diagnostic imaging in all cases of suspected GCA, she added, noting that, for cranial-GCA symptoms, temporal artery ultrasound is recommended first line, but for those who present without cranial symptoms, no particular imaging modality is recommended because of a lack of comparative and diagnostic accuracy data in LV-GCA.
Biopsy has traditionally been used in these cases, but now imaging can be substituted – and vascular ultrasound is an attractive first-line option given its affordability and availability.
Indeed, the current findings support its use in this setting, she said.
“We think that these results indicate that ultrasound should not only be the first-line imaging test in patients presenting with cranial symptoms, but also in patients suspected of GCA presenting with constitutional symptoms, and if this examination is included in the standard examinations in fast-track clinics, it may overcome the delay in diagnosis and the patients can be treated earlier. It may also spare the unneeded examinations performed in these patients,” she concluded.
Dr. Nielsen disclosed a relationship with Roche.
SOURCE: Nielsen B et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2905.
CHICAGO – Vascular ultrasound showed high sensitivity and specificity for diagnosing large-vessel giant-cell arteritis (LV-GCA) in a prospective study of patients with suspected new-onset disease.
The findings highlight the value of vascular ultrasound – in the hands of experienced sonographers – as a first-line imaging test in this setting, Berit Dalsgaard Nielsen, MD, reported at the annual meeting of the American College of Rheumatology.
Of 41 control subjects without LV-GCA, none had a positive ultrasound, whereas 36 of 45 LV-GCA patients had a positive ultrasound, which gives the test a specificity of 100% and sensitivity of 80%, Dr. Nielsen of Aarhus (Denmark) University Hospital said during a press briefing at the meeting.
Ultrasound was performed on the carotid artery in the neck and axillary arteries under the arm, which are easily accessible by ultrasound.
“These patients also had temporal arteries evaluated, and if we included this evaluation in the diagnostic performance, it showed a sensitivity of 91%,” she noted, explaining that temporal artery ultrasound alone conferred 71% sensitivity. “So it actually helped us identify more GCA patients.”
The study subjects were adults with suspected GCA. Inclusion criteria included age of at least 50 years, C-reactive protein of more than 15 mg/L or erythrocyte sedimentation rate of more than 40 mm, and either cranial symptoms, new-onset limb claudication, protracted constitutional symptoms, or polymyalgia rheumatica (PMR) symptoms. Patients were excluded if they had recent or ongoing glucocorticoid or disease-modifying antirheumatic drug treatment, a previous GCA or PMR diagnosis, or a large vessel inflammation that mimicked LV-GCA.
Clinical evaluations and imaging tests were performed prior to treatment initiation. The reference diagnosis was a clinical diagnosis of GCA and a positive 18F-FDG PET/CT scan, Dr Nielsen said, adding that ultrasound examinations were performed by experienced sonographers who were blinded to the PET/CT results.
Of the 86 patients included, 45 had LV-GCA with or without concomitant cranial GCA, 10 had isolated cranial GCA, 21 had PMR, and 10 were diagnosed with other diseases. The patients found to not have LV-GCA were considered control subjects.
The findings are notable because, while PET is considered the gold standard, it is very expensive and not always readily available, Dr. Nielsen said.
Additionally, while cranial-GCA patients generally present with symptoms such as headache, jaw claudication, and visual disturbances that are considered typical for GCA, LV-GCA patients rarely present with these symptoms.
Rather, these LV-GCA patients tend to present with constitutional symptoms mimicking infection or cancer, and they undergo extensive examination programs before the diagnosis is established. For this reason, diagnosis is often delayed for several months in LV-GCA patients until late in the disease course.
“During this time they often experience a decline in physical ability,” she said. “So in this disease subset of patients with GCA, there’s an unmet need for earlier recognition and earlier diagnosis.”
New recommendations from the European League Against Rheumatism call for early diagnostic imaging in all cases of suspected GCA, she added, noting that, for cranial-GCA symptoms, temporal artery ultrasound is recommended first line, but for those who present without cranial symptoms, no particular imaging modality is recommended because of a lack of comparative and diagnostic accuracy data in LV-GCA.
Biopsy has traditionally been used in these cases, but now imaging can be substituted – and vascular ultrasound is an attractive first-line option given its affordability and availability.
Indeed, the current findings support its use in this setting, she said.
“We think that these results indicate that ultrasound should not only be the first-line imaging test in patients presenting with cranial symptoms, but also in patients suspected of GCA presenting with constitutional symptoms, and if this examination is included in the standard examinations in fast-track clinics, it may overcome the delay in diagnosis and the patients can be treated earlier. It may also spare the unneeded examinations performed in these patients,” she concluded.
Dr. Nielsen disclosed a relationship with Roche.
SOURCE: Nielsen B et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2905.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: Vascular ultrasound is reasonable for first-line maging of suspected LV-GCA.
Major finding: Vascular ultrasound had 100% specificity and 80% sensitivity.
Study details: A prospective study of 86 patients.
Disclosures: Dr. Nielsen disclosed a relationship with Roche.
Source: Nielsen BD et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 2905.
Leflunomide-hydroxychloroquine combo shows promise in primary Sjögren’s pilot study
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
CHICAGO – Combination therapy with leflunomide and hydroxychloroquine met all goals for efficacy, safety, and tolerability among patients with primary Sjögren’s syndrome in a randomized, placebo-controlled pilot study, lending support to evidence suggesting the two drugs have additive benefits.
The combined treatment was associated with a statistically significant decrease in the EULAR Sjögren’s syndrome disease activity index (ESSDAI) over 24 weeks – the primary endpoint of the study – in 21 patients in the treatment group. The ESSDAI score on combination treatment dropped from about 10 at baseline to about 6 at 24 weeks, compared with no change from a baseline of about 10 in eight patients in the placebo group. An ESSDAI decrease of 3 or more points occurred in 11 patients in the combination therapy group, compared with none in the placebo group, Joel A.G. van Roon, PhD, a researcher in the Laboratory of Translational Immunology at the University Medical Center Utrecht, the Netherlands, reported in a late-breaking poster at the annual meeting of the American College of Rheumatology.
Both leflunomide and hydroxychloroquine have been shown to inhibit B-cell hyperactivity, but the clinical benefits have been modest and not statistically significant. Since the two agents have complementary inhibitory properties on different immune cells – including B and T cells and plasmacytoid dendritic cells, and based on in vitro findings of additive benefits with respect to inhibition of T- and B-cell activation and CXCL13 production, Dr. van Roon and his colleagues conducted this double-blind, single-center, proof-of-concept pilot study (REPURpSS-1) to assess the efficacy, safety, and tolerability of combined treatment in primary Sjögren’s syndrome.
In all, 29 patients with clinically active disease, defined by ESSDAI of 5 or greater, were randomized 2:1 to receive either 20 mg of leflunomide daily plus 400 mg of hydroxychloroquine daily or placebo/placebo for 24 weeks.
Secondary endpoints such as oral dryness also improved significantly in the treatment group versus the placebo group. Stimulated whole saliva flow increased from about 800 mcL/5 min to about 1,400 mcL/5 min and decreased from about 1,250 to about 1,000 mcL/5 min in the groups, respectively. Median EULAR Sjögren’s syndrome patient reported index (ESSPRI), ESSPRI pain, and ESSPRI fatigue scores, as well as Physician’s and Patient’s Global Assessment scores each improved significantly in the treatment group (at least P less than .05 in all cases) but not in the placebo groups, said Dr. van Roon.
Additionally, serum IgG, IgM rheumatoid factor, and chemokine CXCL13 – a marker for lymphoid neogenesis – decreased significantly, and complement components 3 and 4 (C3 and C4) increased significantly by 24 weeks in the treatment group, but not in the placebo group. B-cell hyperactivity as measured by serum IgG decreased from about 20 g/L to about 14 g/L versus no change from about 15 g/L at baseline in the placebo group, he noted.
“Overall, combination leflunomide and hydroxychloroquine was safe and well tolerated, but larger randomized, controlled trials are needed to confirm the observed effects and to identify potential biomarkers for response,” he concluded.
This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
SOURCE: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: but larger randomized, controlled trials are needed to confirm the observed effects.
Major finding: Combined treatment was associated with a decline in EULAR Sjögren’s syndrome disease activity index score from about 10 at baseline to about 6 at 24 weeks.
Study details: A randomized, placebo-controlled pilot study of 29 patients.
Disclosures: This study was supported by ZonMw (the Netherlands Organization for Health Research and Development). Dr. van Roon reported having no relevant disclosures.
Source: van Roon JAG et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract L10.
Fever, intestinal symptoms may delay diagnosis of Kawasaki disease in children
Symptoms of gastrointestinal involvement such as abdominal pain and vomiting may delay diagnosis of Kawasaki disease in pediatric patients.
“The clinical onset of Kawasaki disease with gastrointestinal involvement often leads to diagnostic and therapeutic delays – a risk factor for the development of coronary complications,” Claudia Colomba, MD, from the department of sciences for health promotion and mother and child care at the University of Palermo (Italy), and her colleagues wrote in the Journal of Pediatrics.
After caring for a boy aged 14 years at their center who presented with these symptoms, Dr. Colomba and her colleagues performed a search of the PubMed and Scopus databases and identified 33 articles with 48 total cases of Kawasaki disease with intestinal involvement between 1979 and 2017.
There were 40 cases of fever (82%), 34 cases of abdominal pain (69%), and 24 cases of vomiting (49%) at disease onset, with diarrhea occurring in 14 cases (29%) and jaundice in 1 case (2%), the researchers noted. Cardiac involvement occurred in 21 cases (43%). With regard to imaging, 38 cases of pseudo-obstruction (77%) were identified by plain radiograph, ultrasonography, and CT. Over half of the cases required surgery; of these 25 cases (51%), 8 cases involved a resection of the restricted loop and included a temporary colostomy (16%), 5 cases were exploratory laparotomy (10%), and there was 1 case with enterolysis (2%).
A total of 45 patients received medical treatment, with 12 patients (25%) receiving intravenous immunoglobulin and 18 (37%) receiving intravenous immunoglobulin plus aspirin. One patient had cyanosis and hand and foot gangrene. There were three patients who died, with massive liver necrosis identified during the autopsy of one patient. Of the other two who died, one did so 2 days after exploratory laparotomy and the other died because of Pseudomonas septic shock.
The researchers reported a good outcome in 28 patients (57%), which included 3 cases where there was no treatment.
“The diagnosis of Kawasaki disease should be considered in all children with fever, abdominal pain, and radiologic signs of pseudo-obstruction, even in the absence of typical symptoms and signs,” Dr. Colomba and her colleagues wrote. “A more comprehensive analysis including all clinical forms of Kawasaki disease would be useful to correlate intestinal involvement with worse outcomes for cardiac complications, as well as to clues to more rapid diagnosis and avoidance of unnecessary invasive procedures.”
The authors reported no relevant conflicts of interest.
SOURCE: Colomba C et al. J Pediatr. 2018 Jul 17. doi: 10.1016/j.jpeds.2018.06.034.
Adding abdominal pain–first presentation to Kawasaki disease is not unprecedented considering lymph node–first presentation was first introduced as a concept in the Journal of Pediatrics in 2013, Sarah S. Long, MD, wrote in a related editorial.
“It should not be too surprising that intestinal vasculitis could be significant in some cases,” Dr. Long said. “Might it not suggest an intestinal portal of microbe or super antigen entry, as might cervical lymphadenitis a respiratory tract portal of entry?”
Dr. Long noted diagnostic and reporting bias was most likely the cause of the 43% rate of coronary artery aneurysms reported in the study by Colomba et al, but said that “it behooves us all to consider Kawasaki disease in the differential when a child has high fever and abdominal pain.”
Dr. Long is a professor of pediatrics at Drexel University, Philadelphia. She made her comments regarding the article by Colomba et al. in the Journal of Pediatrics (2018 Jul 17. doi: 10.1016/j.jpeds.2018.09.018 ). Dr. Long is on the editorial board of the journal, served as the chief editor on and receives royalties from “Principles and Practice of Pediatric Infectious Diseases,” and serves as the associate editor of the Red Book Report of the American Academy of Pediatrics Committee on Infectious Diseases.
Adding abdominal pain–first presentation to Kawasaki disease is not unprecedented considering lymph node–first presentation was first introduced as a concept in the Journal of Pediatrics in 2013, Sarah S. Long, MD, wrote in a related editorial.
“It should not be too surprising that intestinal vasculitis could be significant in some cases,” Dr. Long said. “Might it not suggest an intestinal portal of microbe or super antigen entry, as might cervical lymphadenitis a respiratory tract portal of entry?”
Dr. Long noted diagnostic and reporting bias was most likely the cause of the 43% rate of coronary artery aneurysms reported in the study by Colomba et al, but said that “it behooves us all to consider Kawasaki disease in the differential when a child has high fever and abdominal pain.”
Dr. Long is a professor of pediatrics at Drexel University, Philadelphia. She made her comments regarding the article by Colomba et al. in the Journal of Pediatrics (2018 Jul 17. doi: 10.1016/j.jpeds.2018.09.018 ). Dr. Long is on the editorial board of the journal, served as the chief editor on and receives royalties from “Principles and Practice of Pediatric Infectious Diseases,” and serves as the associate editor of the Red Book Report of the American Academy of Pediatrics Committee on Infectious Diseases.
Adding abdominal pain–first presentation to Kawasaki disease is not unprecedented considering lymph node–first presentation was first introduced as a concept in the Journal of Pediatrics in 2013, Sarah S. Long, MD, wrote in a related editorial.
“It should not be too surprising that intestinal vasculitis could be significant in some cases,” Dr. Long said. “Might it not suggest an intestinal portal of microbe or super antigen entry, as might cervical lymphadenitis a respiratory tract portal of entry?”
Dr. Long noted diagnostic and reporting bias was most likely the cause of the 43% rate of coronary artery aneurysms reported in the study by Colomba et al, but said that “it behooves us all to consider Kawasaki disease in the differential when a child has high fever and abdominal pain.”
Dr. Long is a professor of pediatrics at Drexel University, Philadelphia. She made her comments regarding the article by Colomba et al. in the Journal of Pediatrics (2018 Jul 17. doi: 10.1016/j.jpeds.2018.09.018 ). Dr. Long is on the editorial board of the journal, served as the chief editor on and receives royalties from “Principles and Practice of Pediatric Infectious Diseases,” and serves as the associate editor of the Red Book Report of the American Academy of Pediatrics Committee on Infectious Diseases.
Symptoms of gastrointestinal involvement such as abdominal pain and vomiting may delay diagnosis of Kawasaki disease in pediatric patients.
“The clinical onset of Kawasaki disease with gastrointestinal involvement often leads to diagnostic and therapeutic delays – a risk factor for the development of coronary complications,” Claudia Colomba, MD, from the department of sciences for health promotion and mother and child care at the University of Palermo (Italy), and her colleagues wrote in the Journal of Pediatrics.
After caring for a boy aged 14 years at their center who presented with these symptoms, Dr. Colomba and her colleagues performed a search of the PubMed and Scopus databases and identified 33 articles with 48 total cases of Kawasaki disease with intestinal involvement between 1979 and 2017.
There were 40 cases of fever (82%), 34 cases of abdominal pain (69%), and 24 cases of vomiting (49%) at disease onset, with diarrhea occurring in 14 cases (29%) and jaundice in 1 case (2%), the researchers noted. Cardiac involvement occurred in 21 cases (43%). With regard to imaging, 38 cases of pseudo-obstruction (77%) were identified by plain radiograph, ultrasonography, and CT. Over half of the cases required surgery; of these 25 cases (51%), 8 cases involved a resection of the restricted loop and included a temporary colostomy (16%), 5 cases were exploratory laparotomy (10%), and there was 1 case with enterolysis (2%).
A total of 45 patients received medical treatment, with 12 patients (25%) receiving intravenous immunoglobulin and 18 (37%) receiving intravenous immunoglobulin plus aspirin. One patient had cyanosis and hand and foot gangrene. There were three patients who died, with massive liver necrosis identified during the autopsy of one patient. Of the other two who died, one did so 2 days after exploratory laparotomy and the other died because of Pseudomonas septic shock.
The researchers reported a good outcome in 28 patients (57%), which included 3 cases where there was no treatment.
“The diagnosis of Kawasaki disease should be considered in all children with fever, abdominal pain, and radiologic signs of pseudo-obstruction, even in the absence of typical symptoms and signs,” Dr. Colomba and her colleagues wrote. “A more comprehensive analysis including all clinical forms of Kawasaki disease would be useful to correlate intestinal involvement with worse outcomes for cardiac complications, as well as to clues to more rapid diagnosis and avoidance of unnecessary invasive procedures.”
The authors reported no relevant conflicts of interest.
SOURCE: Colomba C et al. J Pediatr. 2018 Jul 17. doi: 10.1016/j.jpeds.2018.06.034.
Symptoms of gastrointestinal involvement such as abdominal pain and vomiting may delay diagnosis of Kawasaki disease in pediatric patients.
“The clinical onset of Kawasaki disease with gastrointestinal involvement often leads to diagnostic and therapeutic delays – a risk factor for the development of coronary complications,” Claudia Colomba, MD, from the department of sciences for health promotion and mother and child care at the University of Palermo (Italy), and her colleagues wrote in the Journal of Pediatrics.
After caring for a boy aged 14 years at their center who presented with these symptoms, Dr. Colomba and her colleagues performed a search of the PubMed and Scopus databases and identified 33 articles with 48 total cases of Kawasaki disease with intestinal involvement between 1979 and 2017.
There were 40 cases of fever (82%), 34 cases of abdominal pain (69%), and 24 cases of vomiting (49%) at disease onset, with diarrhea occurring in 14 cases (29%) and jaundice in 1 case (2%), the researchers noted. Cardiac involvement occurred in 21 cases (43%). With regard to imaging, 38 cases of pseudo-obstruction (77%) were identified by plain radiograph, ultrasonography, and CT. Over half of the cases required surgery; of these 25 cases (51%), 8 cases involved a resection of the restricted loop and included a temporary colostomy (16%), 5 cases were exploratory laparotomy (10%), and there was 1 case with enterolysis (2%).
A total of 45 patients received medical treatment, with 12 patients (25%) receiving intravenous immunoglobulin and 18 (37%) receiving intravenous immunoglobulin plus aspirin. One patient had cyanosis and hand and foot gangrene. There were three patients who died, with massive liver necrosis identified during the autopsy of one patient. Of the other two who died, one did so 2 days after exploratory laparotomy and the other died because of Pseudomonas septic shock.
The researchers reported a good outcome in 28 patients (57%), which included 3 cases where there was no treatment.
“The diagnosis of Kawasaki disease should be considered in all children with fever, abdominal pain, and radiologic signs of pseudo-obstruction, even in the absence of typical symptoms and signs,” Dr. Colomba and her colleagues wrote. “A more comprehensive analysis including all clinical forms of Kawasaki disease would be useful to correlate intestinal involvement with worse outcomes for cardiac complications, as well as to clues to more rapid diagnosis and avoidance of unnecessary invasive procedures.”
The authors reported no relevant conflicts of interest.
SOURCE: Colomba C et al. J Pediatr. 2018 Jul 17. doi: 10.1016/j.jpeds.2018.06.034.
FROM THE JOURNAL OF PEDIATRICS
Key clinical point: Abdominal pain, fever, and radiologically identified pseudo-obstruction may delay diagnosis of Kawasaki disease in children and should be considered for these patients.
Major finding: Fever, abdominal pain, and vomiting were the most common symptoms that appeared prior to traditional Kawasaki disease symptoms.
Study details: A systematic review of 48 cases of Kawasaki disease patients with intestinal involvement.
Disclosures: The authors reported no relevant conflicts of interest.
Source: Colomba C et al. J Pediatr. 2018 Jul 17. doi: 10.1016/j.jpeds.2018.06.034.
Antiphospholipid antibodies are surprisingly common in first-MI patients
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
CHICAGO – Patients with a first MI were nearly nine times more likely to have detectable IgG antiphospholipid antibodies than were matched controls in a cross-sectional cohort study, Elisabet Svenungsson, MD, PhD, reported at the annual meeting of the American College of Rheumatology.
Her case-control study included 805 Swedish patients tested for antiphospholipid antibodies 6-10 weeks after experiencing their first MI and an equal number of age-, sex-, and location-matched controls. Prior to their MIs, none of the patients had been diagnosed with antiphospholipid syndrome, which requires both positive antiphospholipid antibodies and a vascular thrombotic event or obstetric morbidity.
A positive test for IgG anti-cardiolipin antibody was present in 10.9% of the first-MI patients, compared with 0.9% of controls. Similarly, 10.4% of acute MI patients and 0.9% of controls were positive for anti-beta2-glycoprotein-1 antibodies. Most patients who tested positive for one were positive for both. Thus, it’s possible that IgG antiphospholipid antibody positivity is an important silent risk factor that’s present in 1 in 10 MI patients, according to Dr. Svenungsson, professor of rheumatology at the Karolinska Institute in Stockholm.
If these results are confirmed and expanded upon in additional studies, testing for antiphospholipid antibodies could become part of the routine care in patients with an acute MI. Those who test positive would meet the criteria for antiphospholipid syndrome and qualify for long-term oral anticoagulation to reduce their elevated risk of further vascular events, she explained in this video interview.
The study was published in Annals of Internal Medicine simultaneously with the presentation at the ACR annual meeting (Ann Int Med. 2018 Oct 23. doi: 10.7326/M18-2130).
SOURCE: Grosso G et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 855.
REPORTING FROM THE ACR ANNUAL MEETING

ACR readies first-ever guidelines on managing reproductive health in rheumatology
CHICAGO – Help is on the way for rheumatologists who may feel out of their depth regarding reproductive health issues in their patients.

for internal review in draft form. Lisa R. Sammaritano, MD, a leader of the expert panel that developed the evidence-based recommendations, shared highlights of the forthcoming guidelines at the annual meeting of the American College of Rheumatology.
“Our patients, fortunately, are pursuing pregnancy more often now than in years past. One of the key messages of the guidelines is that patients really do want to discuss these topics with their rheumatologist, even though that often does not happen now. What patients told us [in the guideline-development process] is their rheumatologist knows them better than their gynecologist or any of their other doctors because we have followed them for a long period of time and we understand their disease and their symptoms. They really want our input on questions about contraception, when to plan a pregnancy, and medication use,” said Dr. Sammaritano of the Hospital for Special Surgery and Cornell University in New York.
The guidelines were created over the course of a year and a half with extensive input from ob.gyns., as well as a patient panel. The project included a systematic review of more than 300 published studies in which guideline panelists attempt to find answers to an initial list of 370 questions. Dr. Sammaritano predicted that the guidelines will prove to be useful not only for rheumatologists, but for their colleagues in ob.gyn. as well. Just as rheumatologists likely haven’t kept up with the sea changes that have occurred in ob.gyn. since their medical school days, most ob.gyns. know little about rheumatic diseases.
“There’s room for education on both sides,” she observed in an interview. “I have had to write letters to gynecologists to get them to put my patients with antiphospholipid antibodies on a contraceptive that includes a progestin because the labeling says, ‘May increase risk of thrombosis.’ And yet if you look at the literature, most of the progestins do not increase the risk of thrombosis, even in patients who are already at increased risk because of a genetic prothrombotic abnormality. I practically had to sign my life away to get a gynecologist to put a progestin-containing IUD in my patient, whereas the risk of thrombosis to my patient with an unplanned pregnancy would have been 10-fold or 100-fold higher. Unplanned pregnancy is dangerous for patients with our diseases.”
And yet, she noted, half of all pregnancies in the United States are unplanned. Among women with rheumatic diseases, the proportion may well be even higher in light of their documented low rate of utilization of effective contraception.
A publication date for the guidelines won’t be set until the review is completed, but the plan is to issue three separate documents. One will address reproductive health outside of pregnancy, with key topics to include contraception, fertility preservation, menopause, and hormone replacement therapy. The second document will focus on pregnancy management, with special attention devoted to women with lupus or antiphospholipid antibodies because they are at particularly high risk of adverse pregnancy outcomes. The third document will be devoted to medications, covering issues including which medications can be continued during pregnancy and when to safely stop the ones that can’t. This section will address both maternal and paternal use of rheumatologic medications, the latter being a topic below the radar of ob.gyns.
The three medications whose paternal use in pregnancy generate the most questions in clinical practice are methotrexate, cyclophosphamide, and sulfasalazine.
“I cannot tell you how many times I’ve been asked whether male patients with rheumatic diseases need to stop their methotrexate before they plan to father a child – that’s been a big one. The answer is they don’t need to stop, but that’s a conditional recommendation because the product label still says to stop it 3 months before. But that’s based on theoretical concerns, and all the data support a lack of teratogenicity for men using methotrexate prior to and during pregnancy,” Dr. Sammaritano said.
Men on cyclophosphamide absolutely have to stop the drug 3 months before pregnancy because the drug causes DNA fragmentation in the sperm. Sulfasalazine is known to impair male fertility. The ACR guidelines will recommend that men continue the drug, but if pregnancy doesn’t occur within a reasonable time, then it’s appropriate to get a semen analysis rather than stopping sulfasalazine unnecessarily.
American College of Obstetricians and Gynecologists guidelines now recommend long-acting reversible contraception, including IUDs and progestin implants, as first-line contraception for all women. The ACR draft guidelines strongly recommend the same.
“That is new. The use of this form of contraception in women with rheumatic diseases is quite low. In general, our patients don’t use contraception as often as other women, and when they do, they don’t use effective contraception. There are many theories as to why that may be: perhaps it’s a focus on the more immediate issues of their rheumatic disease that doesn’t allow their rheumatologist to get to the point of discussing contraception,” according to Dr. Sammaritano.
Many rheumatologists will be pleasantly surprised to learn that the problem of increased risk of pelvic inflammatory disease associated with earlier-generation IUDs is no longer an issue with the current devices. And contrary to a misconception among some ob.gyns., autoimmune disease will not cause a woman to reject her IUD.
The ACR guidelines recommend continuing hydroxychloroquine in lupus patients during pregnancy – and considering starting the drug in those not already on it – because of strong evidence supporting both safety and benefit for mother and baby.
“We are recommending the use of low-dose aspirin for patients with lupus and antiphospholipid antibodies because those two conditions increase the risk for preeclampsia, and the ob.gyns. routinely use low-dose aspirin starting toward the end of the first trimester as preventive therapy. Large studies show that it reduces the risk,” she continued.
Dr. Sammaritano cautioned that the literature on the use of rheumatologic medications in pregnancy and breast feeding is generally weak – and in the case of the new oral small molecule JAK inhibitors, essentially nonexistent.
“A lot of our recommendations are conditional because we did not feel that the data support a strong recommendation. But you have to do something. As long as you communicate the idea that we’re doing the best we can with what information is available, I think patients will respond to that,” the rheumatologist said.
She reported having no financial conflicts regarding her presentation.
CHICAGO – Help is on the way for rheumatologists who may feel out of their depth regarding reproductive health issues in their patients.
for internal review in draft form. Lisa R. Sammaritano, MD, a leader of the expert panel that developed the evidence-based recommendations, shared highlights of the forthcoming guidelines at the annual meeting of the American College of Rheumatology.
“Our patients, fortunately, are pursuing pregnancy more often now than in years past. One of the key messages of the guidelines is that patients really do want to discuss these topics with their rheumatologist, even though that often does not happen now. What patients told us [in the guideline-development process] is their rheumatologist knows them better than their gynecologist or any of their other doctors because we have followed them for a long period of time and we understand their disease and their symptoms. They really want our input on questions about contraception, when to plan a pregnancy, and medication use,” said Dr. Sammaritano of the Hospital for Special Surgery and Cornell University in New York.
The guidelines were created over the course of a year and a half with extensive input from ob.gyns., as well as a patient panel. The project included a systematic review of more than 300 published studies in which guideline panelists attempt to find answers to an initial list of 370 questions. Dr. Sammaritano predicted that the guidelines will prove to be useful not only for rheumatologists, but for their colleagues in ob.gyn. as well. Just as rheumatologists likely haven’t kept up with the sea changes that have occurred in ob.gyn. since their medical school days, most ob.gyns. know little about rheumatic diseases.
“There’s room for education on both sides,” she observed in an interview. “I have had to write letters to gynecologists to get them to put my patients with antiphospholipid antibodies on a contraceptive that includes a progestin because the labeling says, ‘May increase risk of thrombosis.’ And yet if you look at the literature, most of the progestins do not increase the risk of thrombosis, even in patients who are already at increased risk because of a genetic prothrombotic abnormality. I practically had to sign my life away to get a gynecologist to put a progestin-containing IUD in my patient, whereas the risk of thrombosis to my patient with an unplanned pregnancy would have been 10-fold or 100-fold higher. Unplanned pregnancy is dangerous for patients with our diseases.”
And yet, she noted, half of all pregnancies in the United States are unplanned. Among women with rheumatic diseases, the proportion may well be even higher in light of their documented low rate of utilization of effective contraception.
A publication date for the guidelines won’t be set until the review is completed, but the plan is to issue three separate documents. One will address reproductive health outside of pregnancy, with key topics to include contraception, fertility preservation, menopause, and hormone replacement therapy. The second document will focus on pregnancy management, with special attention devoted to women with lupus or antiphospholipid antibodies because they are at particularly high risk of adverse pregnancy outcomes. The third document will be devoted to medications, covering issues including which medications can be continued during pregnancy and when to safely stop the ones that can’t. This section will address both maternal and paternal use of rheumatologic medications, the latter being a topic below the radar of ob.gyns.
The three medications whose paternal use in pregnancy generate the most questions in clinical practice are methotrexate, cyclophosphamide, and sulfasalazine.
“I cannot tell you how many times I’ve been asked whether male patients with rheumatic diseases need to stop their methotrexate before they plan to father a child – that’s been a big one. The answer is they don’t need to stop, but that’s a conditional recommendation because the product label still says to stop it 3 months before. But that’s based on theoretical concerns, and all the data support a lack of teratogenicity for men using methotrexate prior to and during pregnancy,” Dr. Sammaritano said.
Men on cyclophosphamide absolutely have to stop the drug 3 months before pregnancy because the drug causes DNA fragmentation in the sperm. Sulfasalazine is known to impair male fertility. The ACR guidelines will recommend that men continue the drug, but if pregnancy doesn’t occur within a reasonable time, then it’s appropriate to get a semen analysis rather than stopping sulfasalazine unnecessarily.
American College of Obstetricians and Gynecologists guidelines now recommend long-acting reversible contraception, including IUDs and progestin implants, as first-line contraception for all women. The ACR draft guidelines strongly recommend the same.
“That is new. The use of this form of contraception in women with rheumatic diseases is quite low. In general, our patients don’t use contraception as often as other women, and when they do, they don’t use effective contraception. There are many theories as to why that may be: perhaps it’s a focus on the more immediate issues of their rheumatic disease that doesn’t allow their rheumatologist to get to the point of discussing contraception,” according to Dr. Sammaritano.
Many rheumatologists will be pleasantly surprised to learn that the problem of increased risk of pelvic inflammatory disease associated with earlier-generation IUDs is no longer an issue with the current devices. And contrary to a misconception among some ob.gyns., autoimmune disease will not cause a woman to reject her IUD.
The ACR guidelines recommend continuing hydroxychloroquine in lupus patients during pregnancy – and considering starting the drug in those not already on it – because of strong evidence supporting both safety and benefit for mother and baby.
“We are recommending the use of low-dose aspirin for patients with lupus and antiphospholipid antibodies because those two conditions increase the risk for preeclampsia, and the ob.gyns. routinely use low-dose aspirin starting toward the end of the first trimester as preventive therapy. Large studies show that it reduces the risk,” she continued.
Dr. Sammaritano cautioned that the literature on the use of rheumatologic medications in pregnancy and breast feeding is generally weak – and in the case of the new oral small molecule JAK inhibitors, essentially nonexistent.
“A lot of our recommendations are conditional because we did not feel that the data support a strong recommendation. But you have to do something. As long as you communicate the idea that we’re doing the best we can with what information is available, I think patients will respond to that,” the rheumatologist said.
She reported having no financial conflicts regarding her presentation.
CHICAGO – Help is on the way for rheumatologists who may feel out of their depth regarding reproductive health issues in their patients.
for internal review in draft form. Lisa R. Sammaritano, MD, a leader of the expert panel that developed the evidence-based recommendations, shared highlights of the forthcoming guidelines at the annual meeting of the American College of Rheumatology.
“Our patients, fortunately, are pursuing pregnancy more often now than in years past. One of the key messages of the guidelines is that patients really do want to discuss these topics with their rheumatologist, even though that often does not happen now. What patients told us [in the guideline-development process] is their rheumatologist knows them better than their gynecologist or any of their other doctors because we have followed them for a long period of time and we understand their disease and their symptoms. They really want our input on questions about contraception, when to plan a pregnancy, and medication use,” said Dr. Sammaritano of the Hospital for Special Surgery and Cornell University in New York.
The guidelines were created over the course of a year and a half with extensive input from ob.gyns., as well as a patient panel. The project included a systematic review of more than 300 published studies in which guideline panelists attempt to find answers to an initial list of 370 questions. Dr. Sammaritano predicted that the guidelines will prove to be useful not only for rheumatologists, but for their colleagues in ob.gyn. as well. Just as rheumatologists likely haven’t kept up with the sea changes that have occurred in ob.gyn. since their medical school days, most ob.gyns. know little about rheumatic diseases.
“There’s room for education on both sides,” she observed in an interview. “I have had to write letters to gynecologists to get them to put my patients with antiphospholipid antibodies on a contraceptive that includes a progestin because the labeling says, ‘May increase risk of thrombosis.’ And yet if you look at the literature, most of the progestins do not increase the risk of thrombosis, even in patients who are already at increased risk because of a genetic prothrombotic abnormality. I practically had to sign my life away to get a gynecologist to put a progestin-containing IUD in my patient, whereas the risk of thrombosis to my patient with an unplanned pregnancy would have been 10-fold or 100-fold higher. Unplanned pregnancy is dangerous for patients with our diseases.”
And yet, she noted, half of all pregnancies in the United States are unplanned. Among women with rheumatic diseases, the proportion may well be even higher in light of their documented low rate of utilization of effective contraception.
A publication date for the guidelines won’t be set until the review is completed, but the plan is to issue three separate documents. One will address reproductive health outside of pregnancy, with key topics to include contraception, fertility preservation, menopause, and hormone replacement therapy. The second document will focus on pregnancy management, with special attention devoted to women with lupus or antiphospholipid antibodies because they are at particularly high risk of adverse pregnancy outcomes. The third document will be devoted to medications, covering issues including which medications can be continued during pregnancy and when to safely stop the ones that can’t. This section will address both maternal and paternal use of rheumatologic medications, the latter being a topic below the radar of ob.gyns.
The three medications whose paternal use in pregnancy generate the most questions in clinical practice are methotrexate, cyclophosphamide, and sulfasalazine.
“I cannot tell you how many times I’ve been asked whether male patients with rheumatic diseases need to stop their methotrexate before they plan to father a child – that’s been a big one. The answer is they don’t need to stop, but that’s a conditional recommendation because the product label still says to stop it 3 months before. But that’s based on theoretical concerns, and all the data support a lack of teratogenicity for men using methotrexate prior to and during pregnancy,” Dr. Sammaritano said.
Men on cyclophosphamide absolutely have to stop the drug 3 months before pregnancy because the drug causes DNA fragmentation in the sperm. Sulfasalazine is known to impair male fertility. The ACR guidelines will recommend that men continue the drug, but if pregnancy doesn’t occur within a reasonable time, then it’s appropriate to get a semen analysis rather than stopping sulfasalazine unnecessarily.
American College of Obstetricians and Gynecologists guidelines now recommend long-acting reversible contraception, including IUDs and progestin implants, as first-line contraception for all women. The ACR draft guidelines strongly recommend the same.
“That is new. The use of this form of contraception in women with rheumatic diseases is quite low. In general, our patients don’t use contraception as often as other women, and when they do, they don’t use effective contraception. There are many theories as to why that may be: perhaps it’s a focus on the more immediate issues of their rheumatic disease that doesn’t allow their rheumatologist to get to the point of discussing contraception,” according to Dr. Sammaritano.
Many rheumatologists will be pleasantly surprised to learn that the problem of increased risk of pelvic inflammatory disease associated with earlier-generation IUDs is no longer an issue with the current devices. And contrary to a misconception among some ob.gyns., autoimmune disease will not cause a woman to reject her IUD.
The ACR guidelines recommend continuing hydroxychloroquine in lupus patients during pregnancy – and considering starting the drug in those not already on it – because of strong evidence supporting both safety and benefit for mother and baby.
“We are recommending the use of low-dose aspirin for patients with lupus and antiphospholipid antibodies because those two conditions increase the risk for preeclampsia, and the ob.gyns. routinely use low-dose aspirin starting toward the end of the first trimester as preventive therapy. Large studies show that it reduces the risk,” she continued.
Dr. Sammaritano cautioned that the literature on the use of rheumatologic medications in pregnancy and breast feeding is generally weak – and in the case of the new oral small molecule JAK inhibitors, essentially nonexistent.
“A lot of our recommendations are conditional because we did not feel that the data support a strong recommendation. But you have to do something. As long as you communicate the idea that we’re doing the best we can with what information is available, I think patients will respond to that,” the rheumatologist said.
She reported having no financial conflicts regarding her presentation.
REPORTING FROM THE ACR ANNUAL MEETING