User login
What Should We Do About Proteinuria?
ACE inhibitors and ARBs: One or the other—not both—for high-risk patients
Avoid prescribing an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) for patients at high risk of vascular events or renal dysfunction. The combination does not reduce poor outcomes, and leads to more adverse drug-related events than an ACE inhibitor or ARB alone.1
Strength of recommendation
B: 1 large, high-quality randomized controlled trial (RCT).
The ONTARGET investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
ILLUSTRATIVE CASE
A 56-year-old patient with well-controlled type 2 diabetes and hypertension comes to see you for routine follow up. His blood pressure is controlled with lisinopril 40 mg/d. But his albumin-to-creatinine ratio is 75 mg/g, and your records reveal that his albuminuria is getting progressively worse.
You’re aware of the potential benefits of a dual angiotensin blockade, and are considering adding an angiotensin receptor blocker (ARB) to your patient’s medication regimen. You wonder whether the combination of an angiotensin-converting enzyme (ACE) inhibitor and an ARB will slow the decline of renal function. You also wonder whether the combination will reduce your patient’s cardiovascular risk.
ACE inhibitors are known to reduce cardiovascular morbidity and mortality, as well as proteinuria in patients with vascular disease or diabetes, whether or not they have heart failure.2 But few studies have compared the effects of ACE inhibitors and ARBs in high-risk patients without heart failure. Nor has there been a definitive study of the effects of an ACE inhibitor–ARB combination on proteinuria and cardiovascular risk.
Are 2 drugs better than 1?
In a recent meta-analysis, researchers reported that combination therapy had a beneficial effect on proteinuria.3 But that observation was based on a small number of patients (N=309 from 10 studies), short follow up, and a lack of data on key clinical end points such as decline of the glomerular filtration rate (GFR) and the onset of dialysis.
Other evidence comes from a study of 199 patients with diabetes and microalbuminuria, in which the ACE inhibitor-ARB combination reduced proteinuria more than either agent alone.4 And in a study of 336 patients with nondiabetic nephropathy, the 2-drug combination slowed the decline in renal function more than monotherapy.5
Small studies raise hopes. These preliminary findings, along with the theoretical benefits of dual angiotensin blockade, suggested that the benefits of taking both agents together could be significant. A large, well-done randomized controlled trial (RCT) was needed to determine the following: (1) whether an ARB is as effective as an ACE inhibitor in reducing morbidity and mortality in high-risk patients who don’t have heart failure, and (2) whether the ACE inhibitor–ARB combination is better than monotherapy for patients at high risk.
The ONTARGET study:
- established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in high-risk patients without heart failure.
- found that either drug alone is more effective than combination therapy for this patient population.
- cast fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction.
STUDY SUMMARY: Vascular outcomes same for ACE inhibitors, ARBs
The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET), a multi-year study of thousands of patients, addressed both of those questions. The researchers compared the effects of both telmisartan (Micardis, an ARB) alone and a telmisartan + ramipril (Altace, an ACE inhibitor) combination with the effects of the ACE inhibitor alone in patients ≥55 years of age with established atherosclerotic vascular disease or diabetes with end-organ damage.1 Exclusion criteria included major renal artery stenosis, uncorrected volume or sodium depletion, a serum creatinine concentration of ≥3 mg/dL, and uncontrolled hypertension (>160 mm Hg systolic or >100 mm Hg diastolic).
After a 3-week run-in period to eliminate those who were unable to tolerate either medication or were nonadherent, a total of 25,620 patients remained. They were randomly assigned to take ramipril 10 mg/d, telmisartan 80 mg/d, or both the ACE inhibitor and the ARB. The researchers followed the patients for a median of 56 months.
The primary composite outcome was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure;1 the main renal outcome was a composite of first dialysis, doubling of serum creatinine, or death.6
The percentage of patients with the primary outcome was the same in all 3 groups (~16.5%). This finding was somewhat surprising because the blood pressure of patients in the combination therapy group was 2 to 3 mm Hg lower overall (both systolic and diastolic) than the blood pressure of patients on monotherapy—a difference that in other studies has been associated with an estimated 4% to 5% reduction in risk.1,2 Patients in the combination group had more hypotensive symptoms compared with those in the ramipril group (4.8% vs 1.7%, number needed to harm [NNH]=32, P<.001).
Renal dysfunction was highest in dual therapy group
Patients in the combination therapy group had higher rates of renal dysfunction than either the ramipril group (13.5% vs 10.2%, NNH=30, P<.001) or the telmisartan group (10.6%), despite a decrease in proteinuria among those on dual therapy. Patients taking the 2-drug combination also had higher rates of hyperkalemia.
While telmisartan proved to be equal to ramipril in reducing vascular events in high-risk patients, patients taking the ACE inhibitor experienced more cough (NNH=32, P<.001) and angioedema (NNH=500, P=.01). In both monotherapy groups, the rates of adverse drug reactions were probably lower than what we typically see in clinical practice because after the run-in period, only patients who were better able to tolerate both medications remained.
WHAT’S NEW: Combination causes renal impairment
This study established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in patients without heart failure. In addition, as the largest RCT to explore the effects of a dual blockade of the renin-angiotensin system with an ACE inhibitor and an ARB, it casts fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction. The reduction in proteinuria seen in patients in the combination therapy group came at a cost of increased renal impairment.
CAVEATS: Findings do not apply to heart failure patients
More than 11% of potential subjects were excluded from this study during the run-in period. This suggests that physicians in practice are likely to find a significant number of patients who are unable to tolerate (or fail to adhere to) monotherapy with ACE inhibitors or ARBs.
At baseline, only a small subgroup—13%—had overt diabetic nephropathy, the hallmark for a substantial continuous decline of GFR. However, 38% of the study group had diabetes, and almost 30% of these diabetes patients had microalbuminuria. Subgroup analysis found results consistent with the overall group, and the large sample size reduces the likelihood that these findings were due to low power. The overall rate of dialysis and doubling of serum creatinine was low, but still statistically significant, due to the large size of this study.
In determining treatment for high-risk patients with vascular disease or diabetes, it is important to keep the study population in mind. Studies of patients with poorly controlled congestive heart failure (CHF) have shown potential benefits from an ACE inhibitor–ARB combination.7 The ONTARGET trial specifically excluded individuals with CHF, and its findings—and recommendations to avoid combination therapy—should not be applied to heart failure patients.
CHALLENGES TO IMPLEMENTATION: Best microalbuminuria Tx remains elusive
Although albuminuria has been considered an early sign of the onset of diabetic nephropathy, the ONTARGET study demonstrated that combination therapy may cause further reduction in albuminuria but still adversely affect renal function. Thus, this study raises important questions about the best treatment for patients with diabetes who have microalbuminuria and are already on either an ACE inhibitor or an ARB. We wonder, too, whether we should continue to test for microalbuminuria in patients who are taking one of these agents, given the lack of guidance regarding further treatment.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
1. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
2. Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145-153.
3. Jennings DL, Kalus JS, Coleman CI, et al. Combination therapy with an ACE inhibitor and an angiotensin receptor blocker for diabetic nephropathy: a meta-analysis. Diabet Med. 2007;24:486-493.
4. Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321:1440-1444.
5. Nakao N, Yoshimura A, Morita H, et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003;361:117-124.
6. Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547-553.
7. Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667-1675.
Avoid prescribing an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) for patients at high risk of vascular events or renal dysfunction. The combination does not reduce poor outcomes, and leads to more adverse drug-related events than an ACE inhibitor or ARB alone.1
Strength of recommendation
B: 1 large, high-quality randomized controlled trial (RCT).
The ONTARGET investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
ILLUSTRATIVE CASE
A 56-year-old patient with well-controlled type 2 diabetes and hypertension comes to see you for routine follow up. His blood pressure is controlled with lisinopril 40 mg/d. But his albumin-to-creatinine ratio is 75 mg/g, and your records reveal that his albuminuria is getting progressively worse.
You’re aware of the potential benefits of a dual angiotensin blockade, and are considering adding an angiotensin receptor blocker (ARB) to your patient’s medication regimen. You wonder whether the combination of an angiotensin-converting enzyme (ACE) inhibitor and an ARB will slow the decline of renal function. You also wonder whether the combination will reduce your patient’s cardiovascular risk.
ACE inhibitors are known to reduce cardiovascular morbidity and mortality, as well as proteinuria in patients with vascular disease or diabetes, whether or not they have heart failure.2 But few studies have compared the effects of ACE inhibitors and ARBs in high-risk patients without heart failure. Nor has there been a definitive study of the effects of an ACE inhibitor–ARB combination on proteinuria and cardiovascular risk.
Are 2 drugs better than 1?
In a recent meta-analysis, researchers reported that combination therapy had a beneficial effect on proteinuria.3 But that observation was based on a small number of patients (N=309 from 10 studies), short follow up, and a lack of data on key clinical end points such as decline of the glomerular filtration rate (GFR) and the onset of dialysis.
Other evidence comes from a study of 199 patients with diabetes and microalbuminuria, in which the ACE inhibitor-ARB combination reduced proteinuria more than either agent alone.4 And in a study of 336 patients with nondiabetic nephropathy, the 2-drug combination slowed the decline in renal function more than monotherapy.5
Small studies raise hopes. These preliminary findings, along with the theoretical benefits of dual angiotensin blockade, suggested that the benefits of taking both agents together could be significant. A large, well-done randomized controlled trial (RCT) was needed to determine the following: (1) whether an ARB is as effective as an ACE inhibitor in reducing morbidity and mortality in high-risk patients who don’t have heart failure, and (2) whether the ACE inhibitor–ARB combination is better than monotherapy for patients at high risk.
The ONTARGET study:
- established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in high-risk patients without heart failure.
- found that either drug alone is more effective than combination therapy for this patient population.
- cast fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction.
STUDY SUMMARY: Vascular outcomes same for ACE inhibitors, ARBs
The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET), a multi-year study of thousands of patients, addressed both of those questions. The researchers compared the effects of both telmisartan (Micardis, an ARB) alone and a telmisartan + ramipril (Altace, an ACE inhibitor) combination with the effects of the ACE inhibitor alone in patients ≥55 years of age with established atherosclerotic vascular disease or diabetes with end-organ damage.1 Exclusion criteria included major renal artery stenosis, uncorrected volume or sodium depletion, a serum creatinine concentration of ≥3 mg/dL, and uncontrolled hypertension (>160 mm Hg systolic or >100 mm Hg diastolic).
After a 3-week run-in period to eliminate those who were unable to tolerate either medication or were nonadherent, a total of 25,620 patients remained. They were randomly assigned to take ramipril 10 mg/d, telmisartan 80 mg/d, or both the ACE inhibitor and the ARB. The researchers followed the patients for a median of 56 months.
The primary composite outcome was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure;1 the main renal outcome was a composite of first dialysis, doubling of serum creatinine, or death.6
The percentage of patients with the primary outcome was the same in all 3 groups (~16.5%). This finding was somewhat surprising because the blood pressure of patients in the combination therapy group was 2 to 3 mm Hg lower overall (both systolic and diastolic) than the blood pressure of patients on monotherapy—a difference that in other studies has been associated with an estimated 4% to 5% reduction in risk.1,2 Patients in the combination group had more hypotensive symptoms compared with those in the ramipril group (4.8% vs 1.7%, number needed to harm [NNH]=32, P<.001).
Renal dysfunction was highest in dual therapy group
Patients in the combination therapy group had higher rates of renal dysfunction than either the ramipril group (13.5% vs 10.2%, NNH=30, P<.001) or the telmisartan group (10.6%), despite a decrease in proteinuria among those on dual therapy. Patients taking the 2-drug combination also had higher rates of hyperkalemia.
While telmisartan proved to be equal to ramipril in reducing vascular events in high-risk patients, patients taking the ACE inhibitor experienced more cough (NNH=32, P<.001) and angioedema (NNH=500, P=.01). In both monotherapy groups, the rates of adverse drug reactions were probably lower than what we typically see in clinical practice because after the run-in period, only patients who were better able to tolerate both medications remained.
WHAT’S NEW: Combination causes renal impairment
This study established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in patients without heart failure. In addition, as the largest RCT to explore the effects of a dual blockade of the renin-angiotensin system with an ACE inhibitor and an ARB, it casts fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction. The reduction in proteinuria seen in patients in the combination therapy group came at a cost of increased renal impairment.
CAVEATS: Findings do not apply to heart failure patients
More than 11% of potential subjects were excluded from this study during the run-in period. This suggests that physicians in practice are likely to find a significant number of patients who are unable to tolerate (or fail to adhere to) monotherapy with ACE inhibitors or ARBs.
At baseline, only a small subgroup—13%—had overt diabetic nephropathy, the hallmark for a substantial continuous decline of GFR. However, 38% of the study group had diabetes, and almost 30% of these diabetes patients had microalbuminuria. Subgroup analysis found results consistent with the overall group, and the large sample size reduces the likelihood that these findings were due to low power. The overall rate of dialysis and doubling of serum creatinine was low, but still statistically significant, due to the large size of this study.
In determining treatment for high-risk patients with vascular disease or diabetes, it is important to keep the study population in mind. Studies of patients with poorly controlled congestive heart failure (CHF) have shown potential benefits from an ACE inhibitor–ARB combination.7 The ONTARGET trial specifically excluded individuals with CHF, and its findings—and recommendations to avoid combination therapy—should not be applied to heart failure patients.
CHALLENGES TO IMPLEMENTATION: Best microalbuminuria Tx remains elusive
Although albuminuria has been considered an early sign of the onset of diabetic nephropathy, the ONTARGET study demonstrated that combination therapy may cause further reduction in albuminuria but still adversely affect renal function. Thus, this study raises important questions about the best treatment for patients with diabetes who have microalbuminuria and are already on either an ACE inhibitor or an ARB. We wonder, too, whether we should continue to test for microalbuminuria in patients who are taking one of these agents, given the lack of guidance regarding further treatment.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
Avoid prescribing an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) for patients at high risk of vascular events or renal dysfunction. The combination does not reduce poor outcomes, and leads to more adverse drug-related events than an ACE inhibitor or ARB alone.1
Strength of recommendation
B: 1 large, high-quality randomized controlled trial (RCT).
The ONTARGET investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
ILLUSTRATIVE CASE
A 56-year-old patient with well-controlled type 2 diabetes and hypertension comes to see you for routine follow up. His blood pressure is controlled with lisinopril 40 mg/d. But his albumin-to-creatinine ratio is 75 mg/g, and your records reveal that his albuminuria is getting progressively worse.
You’re aware of the potential benefits of a dual angiotensin blockade, and are considering adding an angiotensin receptor blocker (ARB) to your patient’s medication regimen. You wonder whether the combination of an angiotensin-converting enzyme (ACE) inhibitor and an ARB will slow the decline of renal function. You also wonder whether the combination will reduce your patient’s cardiovascular risk.
ACE inhibitors are known to reduce cardiovascular morbidity and mortality, as well as proteinuria in patients with vascular disease or diabetes, whether or not they have heart failure.2 But few studies have compared the effects of ACE inhibitors and ARBs in high-risk patients without heart failure. Nor has there been a definitive study of the effects of an ACE inhibitor–ARB combination on proteinuria and cardiovascular risk.
Are 2 drugs better than 1?
In a recent meta-analysis, researchers reported that combination therapy had a beneficial effect on proteinuria.3 But that observation was based on a small number of patients (N=309 from 10 studies), short follow up, and a lack of data on key clinical end points such as decline of the glomerular filtration rate (GFR) and the onset of dialysis.
Other evidence comes from a study of 199 patients with diabetes and microalbuminuria, in which the ACE inhibitor-ARB combination reduced proteinuria more than either agent alone.4 And in a study of 336 patients with nondiabetic nephropathy, the 2-drug combination slowed the decline in renal function more than monotherapy.5
Small studies raise hopes. These preliminary findings, along with the theoretical benefits of dual angiotensin blockade, suggested that the benefits of taking both agents together could be significant. A large, well-done randomized controlled trial (RCT) was needed to determine the following: (1) whether an ARB is as effective as an ACE inhibitor in reducing morbidity and mortality in high-risk patients who don’t have heart failure, and (2) whether the ACE inhibitor–ARB combination is better than monotherapy for patients at high risk.
The ONTARGET study:
- established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in high-risk patients without heart failure.
- found that either drug alone is more effective than combination therapy for this patient population.
- cast fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction.
STUDY SUMMARY: Vascular outcomes same for ACE inhibitors, ARBs
The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET), a multi-year study of thousands of patients, addressed both of those questions. The researchers compared the effects of both telmisartan (Micardis, an ARB) alone and a telmisartan + ramipril (Altace, an ACE inhibitor) combination with the effects of the ACE inhibitor alone in patients ≥55 years of age with established atherosclerotic vascular disease or diabetes with end-organ damage.1 Exclusion criteria included major renal artery stenosis, uncorrected volume or sodium depletion, a serum creatinine concentration of ≥3 mg/dL, and uncontrolled hypertension (>160 mm Hg systolic or >100 mm Hg diastolic).
After a 3-week run-in period to eliminate those who were unable to tolerate either medication or were nonadherent, a total of 25,620 patients remained. They were randomly assigned to take ramipril 10 mg/d, telmisartan 80 mg/d, or both the ACE inhibitor and the ARB. The researchers followed the patients for a median of 56 months.
The primary composite outcome was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure;1 the main renal outcome was a composite of first dialysis, doubling of serum creatinine, or death.6
The percentage of patients with the primary outcome was the same in all 3 groups (~16.5%). This finding was somewhat surprising because the blood pressure of patients in the combination therapy group was 2 to 3 mm Hg lower overall (both systolic and diastolic) than the blood pressure of patients on monotherapy—a difference that in other studies has been associated with an estimated 4% to 5% reduction in risk.1,2 Patients in the combination group had more hypotensive symptoms compared with those in the ramipril group (4.8% vs 1.7%, number needed to harm [NNH]=32, P<.001).
Renal dysfunction was highest in dual therapy group
Patients in the combination therapy group had higher rates of renal dysfunction than either the ramipril group (13.5% vs 10.2%, NNH=30, P<.001) or the telmisartan group (10.6%), despite a decrease in proteinuria among those on dual therapy. Patients taking the 2-drug combination also had higher rates of hyperkalemia.
While telmisartan proved to be equal to ramipril in reducing vascular events in high-risk patients, patients taking the ACE inhibitor experienced more cough (NNH=32, P<.001) and angioedema (NNH=500, P=.01). In both monotherapy groups, the rates of adverse drug reactions were probably lower than what we typically see in clinical practice because after the run-in period, only patients who were better able to tolerate both medications remained.
WHAT’S NEW: Combination causes renal impairment
This study established that telmisartan, an ARB, is not inferior to ramipril, an ACE inhibitor, in reducing cardiovascular and renal events in patients without heart failure. In addition, as the largest RCT to explore the effects of a dual blockade of the renin-angiotensin system with an ACE inhibitor and an ARB, it casts fresh doubt on the assumption that proteinuria is an accurate surrogate marker for progressive renal dysfunction. The reduction in proteinuria seen in patients in the combination therapy group came at a cost of increased renal impairment.
CAVEATS: Findings do not apply to heart failure patients
More than 11% of potential subjects were excluded from this study during the run-in period. This suggests that physicians in practice are likely to find a significant number of patients who are unable to tolerate (or fail to adhere to) monotherapy with ACE inhibitors or ARBs.
At baseline, only a small subgroup—13%—had overt diabetic nephropathy, the hallmark for a substantial continuous decline of GFR. However, 38% of the study group had diabetes, and almost 30% of these diabetes patients had microalbuminuria. Subgroup analysis found results consistent with the overall group, and the large sample size reduces the likelihood that these findings were due to low power. The overall rate of dialysis and doubling of serum creatinine was low, but still statistically significant, due to the large size of this study.
In determining treatment for high-risk patients with vascular disease or diabetes, it is important to keep the study population in mind. Studies of patients with poorly controlled congestive heart failure (CHF) have shown potential benefits from an ACE inhibitor–ARB combination.7 The ONTARGET trial specifically excluded individuals with CHF, and its findings—and recommendations to avoid combination therapy—should not be applied to heart failure patients.
CHALLENGES TO IMPLEMENTATION: Best microalbuminuria Tx remains elusive
Although albuminuria has been considered an early sign of the onset of diabetic nephropathy, the ONTARGET study demonstrated that combination therapy may cause further reduction in albuminuria but still adversely affect renal function. Thus, this study raises important questions about the best treatment for patients with diabetes who have microalbuminuria and are already on either an ACE inhibitor or an ARB. We wonder, too, whether we should continue to test for microalbuminuria in patients who are taking one of these agents, given the lack of guidance regarding further treatment.
Acknowledgements
The PURLs Surveillance System is supported in part by Grant Number UL1RR024999 from the National Center for Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
PURLs methodology
This study was selected and evaluated using FPIN’s Priority Updates from the Research Literature (PURL) Surveillance System methodology. The criteria and findings leading to the selection of this study as a PURL can be accessed at www.jfponline.com/purls.
Click here to view PURL METHODOLOGY
1. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
2. Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145-153.
3. Jennings DL, Kalus JS, Coleman CI, et al. Combination therapy with an ACE inhibitor and an angiotensin receptor blocker for diabetic nephropathy: a meta-analysis. Diabet Med. 2007;24:486-493.
4. Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321:1440-1444.
5. Nakao N, Yoshimura A, Morita H, et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003;361:117-124.
6. Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547-553.
7. Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667-1675.
1. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
2. Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145-153.
3. Jennings DL, Kalus JS, Coleman CI, et al. Combination therapy with an ACE inhibitor and an angiotensin receptor blocker for diabetic nephropathy: a meta-analysis. Diabet Med. 2007;24:486-493.
4. Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321:1440-1444.
5. Nakao N, Yoshimura A, Morita H, et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003;361:117-124.
6. Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547-553.
7. Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667-1675.
Copyright © 2009 The Family Physicians Inquiries Network.
All rights reserved.
Preventing renal disease progression: Can complete renin-angiotensin-aldosterone blockade work?
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
An update on proteinuric chronic kidney disease: The dual-goal approach
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7

Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status

For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone

Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.

A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7
Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status
For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone
Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.
A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
When angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) were introduced, we hoped that these drugs would slow or stop the inexorable progression of chronic kidney disease. This hasn’t come to pass: the incidence of end-stage renal disease continued to increase throughout the 1990s, and although it may have finally reached a plateau, it remains unacceptably high.1 One reason may be that, used singly, drugs that block the renin-angiotensin-aldosterone system are only moderately successful, as approximately 20% to 40% of patients still reach unfavorable renal end points such as doubling of the serum creatinine level or dialysis.2–7
In view of these disappointing results, some experts are advocating a new strategy in which they advise that both blood pressure and urinary albumin excretion be lowered to specific goals. To achieve these goals, we will generally have to give higher doses of ACE inhibitors and ARBs alone or use a combination of these and other drugs that block the renin-angiotensin-aldosterone system at various sites.
This article describes how the dual-goal approach, with a focus on renin-angiotensin-aldosterone system inhibition, may be applied in the therapy of proteinuric chronic kidney disease. This appears to be a reasonable approach, based on current evidence, to address the epidemic of renal failure. However, further studies are needed to establish the effectiveness of this approach, and the risk of hyperkalemia following aggressive inhibition of the renin-angiotensin-aldosterone system poses a significant management problem.
ALBUMIN MAY BE TOXIC
While hypertension has long been associated with poor renal outcomes, urinary albumin has more recently been implicated by observational and experimental evidence as a tubular-interstitial toxin that may also accelerate the progression of renal disease.
For example, in both the Reduction of Endpoints in NIDDM With the Angiotensin II Antagonist Losartan (RENAAL) study8 and the Ramipril Efficacy in Nephropathy study,4 baseline proteinuria was almost linearly related to worse renal outcomes. In RENAAL, patients who excreted more than about 3 g of albumin per day had an 8.1-fold higher risk of progressing to end-stage renal disease.8 Moreover, the more that protein excretion could be reduced, the better the renal outcomes, down to a level of about 500 mg/day.8
Of importance, lowering blood pressure did not always decrease protein excretion—nearly 40% of patients had a dissociation between the two.9 In fact, prescribing a single ACE inhibitor or ARB while targeting only blood pressure has not predictably reduced protein excretion to 500 mg/day (the proposed goal).2–7
Although albumin has not been conclusively proven to be a renal toxin, targeting the reduction of proteinuria may also succeed if urinary albumin simply serves as a marker of the success of chronic kidney disease treatment and reflects prognosis.
A DUAL-GOAL APPROACH
In view of the observational and experimental evidence, many experts10,15–18 are advocating a dual-goal approach that stresses lowering both blood pressure and urinary protein (albumin) excretion. The recommended goal for systolic blood pressure is less than 120 to 125 mm Hg; the goal for proteinuria is less than 300 to 500 mg/24 hours,16,17,19 aiming to slow the decline in glomerular filtration rate to less than 2 mL/ min/year.11,20
The strategy of targeting both proteinuria and blood pressure has recently received further support. In a prospective randomized controlled study,21 nondiabetic patients with proteinuria received either an ACE inhibitor or an ARB. In one group, the dose was adjusted to lower the blood pressure to less than 130/80 mm/Hg; in the other group, the dose was adjusted to lower the blood pressure to 130/80 and to reduce protein excretion maximally. Only about half as many patients in the group with the dual-goal strategy reached the composite primary end point (doubling of serum creatinine, end-stage renal disease, or death) over a median of 3.7 years of follow-up, as compared with those treated by targeting the blood pressure alone.
In retrospect, the suboptimal success in the earlier landmark studies2–7 may have derived from the failure of ACE inhibitors and ARBs, used by themselves at moderate doses, to either lower the blood pressure to the recently advised goal (the actual results obtained varied from about 128 to about 145 mm Hg systolic) or, perhaps, to reduce proteinuria to the goal level.
Not all nephrologists currently pursue the stringent proteinuria goal of 500 mg per day—the targeted reduction of proteinuria requires further prospective evidence to support it. However, nephrologists do commonly follow the broad theme that antihypertensive therapy in proteinuric chronic kidney disease should accentuate medicines that protect the kidney beyond their antihypertensive effect (Table 1), and that proteinuria is an important metric that, at the very least, reflects the response to therapy and prognosis.
BLOCKING RENIN-ANGIOTENSIN- ALDOSTERONE MORE COMPLETELY
These issues may be addressed by more complete inhibition of the renin-angiotensin-aldosterone system, now achievable with the addition of aldosterone receptor antagonists and direct renin inhibitors to the ACE inhibitors and ARBs. Although we lack long-term studies of the relative efficacy of these medicines alone or in various combinations, the multistep sequence of the renin-angiotensin-aldosterone system allows for the possibility that more complete suppression via coordinated pharmacologic attention to multiple sites will yield beneficial results.
Combining an ACE inhibitor and an ARB
Even in the absence of ACE, angiotensin II is also produced by other kinases and therefore is not completely suppressed by an ACE inhibitor. For this and other reasons, there are theoretical advantages to adding an ARB to an ACE inhibitor.
In the Combination Treatment of Angiotensin 2 Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-Diabetic Renal Disease (COOPERATE) study,20 the combination of an ACE inhibitor and an ARB protected the kidneys better than either medicine alone, not only in terms of less protein in the urine but also in terms of significantly fewer patients progressing to the primary end points of doubling of serum creatinine or end-stage renal disease after 3 years of follow-up (11% of patients on combination therapy vs 23% on single therapy).
Aldosterone receptor antagonists or renin inhibitors plus ACE inhibitors and ARBs
Aldosterone escape is common during long-term therapy with ACE inhibitors and ARBs, and an aldosterone-receptor antagonist reduces proteinuria11–13 and stabilizes kidney function13 in a manner additive to that of ACE inhibitors and ARBs.
Direct renin inhibitors overcome the reactive rises in renin activity and in angiotensin II that complicate therapy with ACE inhibitors and ARBs, and they also reduce urinary aldosterone excretion.14
When to consider combination therapy
Inhibition of the renin-angiotensin-aldosterone system at multiple sites may be considered in cases of persistent hypertension or proteinuria, or of progression of chronic kidney disease despite single-drug therapy, or more broadly, with increasing evidence that combination therapy may preserve the glomerular filtration rate.13,20 This article suggests one way to apply the several available renin-angiotensin-aldosterone inhibitors, keeping in mind extensive interindividual variations, uncertain responses, and the absence of a linear evidence-based strategy known to be broadly successful.
INITIAL CONSIDERATION: WHAT IS THE BLOOD PRESSURE GOAL?
Determining the blood pressure goal for a patient may not be as straightforward as usually assumed. Typically, advisories suggest a discrete goal; for example, the Seventh Joint National Committee22 recommended a systolic blood pressure of 130 mm Hg or lower for patients with chronic kidney disease or diabetes. However, if we weigh the risks and benefits, we find that the situation is more nuanced. The blood pressure goal should vary among patients, depending on age, amount of proteinuria, whether the patient can tolerate the lowered blood pressure, and whether lowering the blood pressure to this goal stabilizes kidney function.
Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study demonstrated a benefit of setting the goal mean arterial pressure to less than 92 mm Hg (about 125 mm Hg systolic) regardless of proteinuria.23 In addition, a meta-analysis suggested that nondiabetic proteinuric patients benefit from even lower systolic blood pressures (110–119 mm Hg).19
In older patients
However, in the MDRD study, the goal of approximately 125 mm Hg systolic pertained only to patients no older than 60 years.23 The goal was increased to about 130 mm Hg for patients 61 to 70 years old. In addition, major clinical studies of chronic kidney disease have excluded patients older than 70 years.2–7,23
Therapy for chronic kidney disease in this older age group is essentially unstudied, and we should be cautious about extrapolating results of aggressive blood pressure-lowering (and renin-angiotensin-aldosterone inhibition) from younger patients to older patients, who may have extensive vascular disease.24,25
For patients older than 70 years, guidance is perhaps best provided by the Systolic Hypertension in the Elderly Program (SHEP), which found that lowering systolic blood pressure to an average of 143 mm Hg reduced the incidence of stroke and cardiovascular disease.26 The SHEP study does not establish the optimal blood pressure goal for preventing progressive chronic kidney disease (or even cardiovascular disease) in the older age group. However, this is the lowest systolic pressure yet shown to be generally safe and associated with any improved outcome for these patients.
Additional studies are needed to evaluate whether this blood pressure level provides the best outcomes in patients with chronic kidney disease, or whether even lower blood pressures in the elderly are safe and will further improve either renal or cardiovascular outcomes.
In younger patients
In contrast, younger patients without diabetes or vascular disease may, in theory, be candidates for even lower blood pressure. No major study of chronic kidney disease isolated patients from about 20 to 40 years old for analysis, precluding direct evidence-based guidelines for this cohort at this time.
However, some of these patients may have had premorbid systolic blood pressures of 90 to 110 mm Hg, so systolic pressures of 110 to 120 mm Hg would be “hypertensive” by 10 to 30 mm Hg for them. It is possible that some patients in this cohort will tolerate a systolic pressure lower than 110 mm Hg, and that the lower blood pressure may provide additional long-term renal protection for them. This notion is theoretical, however, and has not been verified by clinical studies.
No one pressure fits all
In summary, an initial target systolic pressure for proteinuric patients, based on available evidence, might be less than 130 mm Hg for patients 61 to 70 years old,23 less than 125 mm Hg for patients younger than 61 years,23 and perhaps as low as 110 to 119 mm Hg for non-diabetic patients.19 Caution is advised against targeting systolic blood pressure less than 140 mm Hg for patients older than 70 years.
These are only initial goals and should be reevaluated as treatment progresses. The achieved blood pressure must be clinically tolerated—symptoms of tissue hypoperfusion indicate that the blood pressure is too low for the patient. In addition, the blood pressure goal (like the proteinuria goal) is only a surrogate end point, and if kidney function declines even though the surrogate end points are attained, then those end points should be reevaluated.
Tailoring blood pressure goals to the individual patient dovetails with the recent suggestion that blood pressure should not be perceived as a rigid dichotomy of “hypertension” vs “normal.”27 There is, in general, a continuous correlation between blood pressure, beginning at low levels, and the risk of cardiorenal disease, and choosing an optimal blood pressure goal for an individual patient requires an ongoing assessment of benefits, risks, and side effects.
STARTING ANTIHYPERTENSIVE THERAPY
The question of which antihypertensive drug to try first is moot in chronic kidney disease because almost all patients need multiple medicines to reach their blood pressure goals.
The Seventh Joint National Committee recommended an ACE inhibitor for initial therapy in hypertensive patients with chronic kidney disease,22 although an ARB is a reasonable first choice for those with type 2 diabetes.5,6
Diuretics potentiate the effects of ACE inhibitors and ARBs and are generally prescribed concomitantly or as the second choice.
A beta-blocker may be recommended as a third medicine (when needed), to provide a complementary class of antihypertensive, to address the high incidence of concomitant coronary artery disease and systolic dysfunction, and because of evidence that sympathetic excess contributes to the hypertension and progression of chronic kidney disease.28,29 The National Kidney Foundation30 suggests that the dose of beta-blocker be increased if the heart rate is greater than 84.
INTENSIFYING RENIN-ANGIOTENSIN-ALDOSTERONE INHIBITION: WHICH DRUGS, AND WHEN?
When hypertension and proteinuria persist despite the use of an ACE inhibitor or an ARB, additional inhibition of the renin-angiotensin-aldosterone system is generally recommended to lower both the blood pressure and the protein excretion. Increasing the dose of ACE inhibitor or ARB,31–34 combining an ACE inhibitor and an ARB,20 or adding an aldosterone receptor antagonist to either an ACE inhibitor or an ARB11–13 have all been shown to reduce proteinuria (as a surrogate end point), and several studies have, importantly, found that these combinations preserve kidney function over time.13,20
However, lacking long-term studies that compare these options, we cannot insist upon specific treatment choices or sequences in these situations.
An approach based on serum potassium and volume status
For example, if a patient has obvious signs of volume excess (eg, edema, jugular venous distention, rales) and the serum potassium concentration is less than about 5.0 or 5.5 mEq/L, then an aldosterone receptor antagonist may logically be added or increased in dose.
Aldosterone is more than a kidney hormone
Increasing the diuretic or renin-angiotensin-aldosterone inhibition
For patients who have obvious signs of volume excess and a serum potassium level greater than 5.0 mEq/L, the dosage of kaliuretic (potassium-excreting) diuretic (usually a loop diuretic in chronic kidney disease) can be increased. Although kaliuretic diuretics do not specifically lower proteinuria, they will help control volume and blood pressure and, by lowering the serum potassium level, facilitate the subsequent augmention of renin-angiotensin-aldosterone inhibition.
When a hypertensive patient does not seem to have excess volume or tachycardia and the serum potassium level is less than about 5.5 mEq/L, then additional renin-angiotensin-aldosterone inhibition is indicated.16 This may be accomplished either by increasing the ACE inhibitor or the ARB to its maximal antihypertensive dose or by starting combination therapy.
Starting a calcium channel blocker
When the serum potassium level is higher than about 5.5 mEq/L, further inhibition of the renin-angiotensin-aldosterone system is contraindicated, and a nondihydropyridine calcium channel blocker can be added for its anti-hypertensive and antiproteinuric effects.16,36
When nondihydropyridine calcium channel blockers are contraindicated due to their antiinotropic effect, an attractive alternative may be to cautiously increase the dose of kaliuretic diuretics. Given the high prevalence of (often covert) volume excess in chronic kidney disease, empiric diuresis may lower blood pressure, particularly in patients already receiving several vasodilators.37 Moreover, as mentioned, by reducing serum potassium, kaliuretic diuretics help allow for a subsequent increase in renin-angiotensin-aldosterone inhibition.
IF BLOOD PRESSURE IS NORMAL, BUT PROTEINURIA PERSISTS
Because lowering blood pressure does not necessarily reduce protein excretion, some patients achieve their blood pressure goal but still have excessive proteinuria. Proponents of the dual-goal approach suggest that these patients require further treatment modifications to reach the proteinuria goal and their optimal renal prognosis.
A cautious increase in renin-angiotensin-aldosterone inhibition is possible but is likely to be limited by low blood pressure. When applicable, any nonessential antihypertensive drug that does not specifically reduce proteinuria (ie, dihydropyridine calcium channel blockers and central and direct vasodilators) should first be discontinued. This allows additional renin-angiotensin-aldosterone inhibition to reduce proteinuria without causing hypotension.
In addition, “ultra-high” doses of these drugs—two or more times the maximal antihypertensive dose—appear to reduce proteinuria without further reducing blood pressure.31–34
Various combinations of an ACE inhibitor, an ARB, and an aldosterone receptor antagonist (and possibly a renin inhibitor) may also be prescribed, striving for more complete suppression of the renin-angiotensin-aldosterone system, with dose adjustments to prevent hypotension.
KEEPING SERUM POTASSIUM AT SAFE LEVELS
Intensive inhibition of the renin-angiotensin-aldosterone system, via higher doses or combination therapy, increases the risk of hyperkalemia. This risk must be addressed energetically to prevent a potentially life-threatening complication.
When prescribed by nephrologists in clinical studies, renin-angiotensin-aldosterone inhibition has proven safe, with minimal adverse events (including hyperkalemia), even with high doses,32–34 in stage 4 chronic kidney disease (ie, with a glomerular filtration rate of 15 to 29 mL/min/1.73m2, inclusively)7 and with combination therapy.11–13,20
However, the increased incidence of hyperkalemia reported with spironolactone in patients with congestive heart failure following publication of the Randomized Aldactone Evaluation Study38 suggests that safety in clinical studies should not be extrapolated to mean safety in routine, community use. Patients with chronic kidney disease should not be given high doses or combinations of these drugs unless the treating physician is experienced in the prevention and treatment of hyperkalemia; typically such therapy should be guided by a nephrologist.
When serum potassium levels exceed 5.6 mEq/L, renin-angiotensin-aldosterone inhibitors should be decreased in dose or discontinued.39 Ideally, the drug or drugs should be restarted (to provide the potential benefits of these classes of drugs) when hyperkalemia has resolved, but this requires not only resolution of hyperkalemia but also steps to prevent this serious problem from recurring. The serum potassium level should be checked frequently, particularly after any increase in renin-angiotensin-aldosterone inhibition.
Treating hyperkalemia
Potential treatments for hyperkalemia include dietary restriction, sodium bicarbonate,39 fludrocortisone (Florinef),40 kaliuretic diuretics, and sodium polystyrene sulfonate (Kayexalate). Nonsteroidal anti-inflammatory drugs should be avoided.
Dietary restriction should be particularly emphasized: if potassium intake is decreased to the same extent as renin-angiotensin-aldosterone inhibitors reduce its excretion, then the serum potassium level will remain acceptable. All dietary supplements whose contents are not precisely known should be proscribed. A list of high-potassium foods to avoid should be given with the initial prescription for the drug. If briefly reviewed at each visit, with feedback given based on measured serum potassium levels, dietary treatment is typically effective (personal observation).
Fludrocortisone is an option when dietary potassium restriction fails.
An increase in the dose of diuretic is typically required with fludrocortisone to prevent sodium retention. The combination of dietary potassium restriction, fludrocortisone (0.1 mg/day, 3–5 days a week), and furosemide (Lasix) allowed high doses of an ACE inhibitor or a combination of an ACE inhibitor and an ARB to be given in 132 patients with chronic kidney disease.40 Over several years, their mean peak potassium level was 4.87 mEq/L, and no instance of acute hyperkalemia requiried stopping the ACE inhibitor or ARB.
However, fludrocortisone is an aldosterone analogue with potentially long-term aldosterone-mediated injurious effects on heart and renal function, even though only low doses were required in the three-pronged approach to hyperkalemia.40 The long-term effect of a regimen of an ACE inhibitor plus an ARB plus fludrocortisone on cardiac and renal outcomes is unknown and of concern.
Therefore, fludrocortisone should probably be avoided in patients with systolic heart dysfunction and should be used cautiously in general. Its use might be limited to patients with proteinuric chronic kidney disease that progresses despite therapy, particularly when that progression is in the context of inability to give significant renin-angiotensin-aldosterone inhibition because of hyperkalemia.
MORE STUDY NEEDED
Chronic kidney disease treatment is becoming increasingly complex, with a lengthening list of potentially effective drugs, difficult-to-reach goals, and a less structured approach. This complexity is magnified by issues of potassium homeostasis and interindividual variations in response to renin-angiotensin-aldosterone inhibition.
More prospective studies are needed to confirm the benefits of targeting proteinuria along with blood pressure and the metrics of the goals in tandem, but, based on available information, the dual-goal approach has been recommended for proteinuric patients,10,15–18 and evidence is accumulating for greater renal protection from larger doses of renin-angiotensin-aldosterone inhibitors and from using these drugs in combination.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
- US Renal Data System. Excerpts from the USRDS 2005 Annual Data Report. Am J Kidney Dis 2006; 47(suppl 1):S1–S286.
- Lewis E, Hunsicker L, Bain R, Rohde Rfor the Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993; 329:1456–1462.
- Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996; 334:939–945.
- The GISEN Group (Gruppo Italiano di Studi Epidemiologici in Nefrologia). Randomised placebo-controlled trial of effect of ramipril on decline in glomerular filtration rate and risk of terminal renal failure in proteinuric, non-diabetic nephropathy. Lancet. 1997; 349:1857–1863.
- Brenner B, Cooper M, De Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001; 345:861–869.
- Lewis E, Hunsicker L, Clarke W, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001; 345:851–860.
- Hou F, Zhang X, Zhang G, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006; 354:131–140.
- De Zeeuw D, Remuzzi G, Parving H-H, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004; 65:2309–2320.
- Eijkelkamp W, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotension 2 Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007; 18:1540–1546.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol. 2006; 1:229–235.
- Chrysostomou A, Pedagogoa E, MacGregor L, Becker G. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin 2 receptor blocker. Clin J Am Soc Nephrol. 2006; 1:256–262.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int. 2006; 70:536–542.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Azizi M, Menard J, Bissery A, Guyene T-T, Bura-Riviere A. Hormonal and hemodynamic effects of aliskiren and valsartan and their combinations in sodium-replete normotensive individuals Clin J Am Soc Nephrol 2007; 2:947–955.
- Hebert L, Wilmer W, Falkenhain M, Ladson-Wofford S, Nahman S, Rovin B. Renoprotection: one or many therapies? Kidney Int 2001; 59:1211–1226.
- Shieppate A, Remuzzi G. The future of renoprotection: frustration and promises. Kidney Int. 2003; 64:1947–1955.
- Zandi-Nejad K, Brenner B. Strategies to retard the progression of chronic kidney disease. Med Clin North Am. 2005; 89:489–509.
- Ritz E, Dikow R. Hypertension and antihypertensive treatment of diabetic nephropathy. Nat Clinl Pract Nephrol. 2006; 2:562–567.
- Jafar T, Stark P, Schmid C, et al., for the AIPRD Study Group. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition. A patient-level meta-analysis. Ann Intern Med. 2003; 139:244–252.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin 2 receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomized controlled trial. Lancet. 2003; 361:117–124.
- Hou F, Xie D, Zhang X, et al. Renoprotection of optimal antiproteinuric doses (ROAD) study: a randomized controlled study of benazepril and losartan in chronic renal insufficiency. J Am Soc Nephrol. 2007; 18:1889–1898.
- Chobanian AV, Bakris GL, Black HR. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003; 289:2560–2572.
- Sarnak M, Greene T, Wang X, et al. The effect of a lower target blood pressure on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease Study. Ann Intern Med. 2005; 142:342–351.
- Hemmelgarn BR, Zhang J, Manns BJ, et al. Progression of kidney dysfunction in the community-dwelling elderly. Kidney Int. 2006; 69:2155–2161.
- Locatelli F, Pozzoni P. Chronic kidney disease in the elderly: is it really a premise for overwhelming renal failure? Kidney Int 2006; 69:2118–2120.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA. 1991; 265:3255–3264.
- Forman JP, Brenner BM. ‘Hypertension’ and ‘microalbuminuria’: the bell tolls for thee. Kidney Int. 2006; 69:22–28.
- Bakris G, Hart P, Ritz E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006; 70:1905–1913.
- UKPD Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective diabetes study group. BMJ. 1998; 317:713–720.
- Bakris G, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertensive and Diabetes Executive Committees Working Group. Am J Kidney Dis. 2000; 36:646–661.
- Navis G, Kramer A, de Jong P. High-dose ACE inhibition: can it improve renoprotection? Am J Kidney Dis 2002; 40:664–666.
- Rossing K, Schjoedt K, Jensin B, Boomsma F, Parving H-H. Enhanced renoprotective effects of ultrahigh doses of irbesartan in patients with type 2 diabetes and microalbuminuria. Kidney Int. 2005; 68:1190–1198.
- Schmieder R, Klingbeil A, Fleischman E, Veelken R, Delles C. Additional antiproteinuric effect of ultrahigh dose candesartan: a double-blind, randomized, prospective study. J Am Soc Nephrol. 2005; 16:3038–3045.
- Aranda P, Segura J, Ruilope L, et al. Long-term renoprotective effects of standard versus high doses of telmisartan in hypertensive nondiabetic nephropathies. Am J Kidney Dis. 2005; 46:1074–1079.
- Calhoun D. Aldosteronism and hypertension. Clin J Am Soc Nephrol. 2006; 1:1039–1045.
- Bakris G, Weir M, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int. 2004; 65:1991–2002.
- Hirsch S. A different approach to resistant hypertension. Cleve Clin J Med 2007: 74;449–456.
- Juurling D, Mamdani M, Lee D, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004; 351:543–551.
- Palmer B. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004; 351:585–592.
- Moskowitz D. From pharmacogenomics to improved patient outcomes: angiotensin 1-converting enzyme as an example. Diabetes Tech Ther. 2002; 4:519–532.
KEY POINTS
- Evidence is emerging that urinary albumin is toxic to the kidney.
- Lowering both blood pressure and urinary albumin excretion, as a means to prevent progressive renal disease, appears to require aggressive inhibition of the renin-angiotensin-aldosterone system, often with several complementary drugs, ie, angiotensin-converting enzyme inhibitors, angiotensin II type 1 receptor blockers, aldosterone receptor antagonists, and possibly, direct renin inhibitors.
- Volume status and potassium levels may help suggest which of several available drugs could be added at different times.
- Serum potassium levels must be managed aggressively when using renin-angiotensin-aldosterone inhibitors in combination.
What is the proper workup of a patient with hypertension?
How extensive a workup does a patient with high blood pressure need?
On one hand, we would not want to start therapy on the basis of a single elevated reading, as blood pressure fluctuates considerably during the day, and even experienced physicians often make errors in taking blood pressure that tend to falsely elevate the patient’s readings. Similarly, we would not want to miss the diagnosis of a potentially curable cause of hypertension or of a condition that increases a patient’s risk of cardiovascular disease. But considering that nearly one-third of adults in the United States have hypertension and that another one-fourth have prehypertension (formerly called high-normal blood pressure),1 if we were to launch an intensive workup for every patient with high blood pressure, the cost and effort would be enormous.
Fortunately, for most patients, it is enough to measure blood pressure accurately and repeatedly, perform a focused history and physical examination, and obtain the results of a few basic laboratory tests and an electrocardiogram, with additional tests in special cases.
In this review we address four fundamental questions in the evaluation of patients with a high blood pressure reading, and how to answer them.
ANSWERING FOUR QUESTIONS
The goal of the hypertension evaluation is to answer four questions:
- Does the patient have sustained hypertension? And if so—
- Is the hypertension primary or secondary?
- Does the patient have other cardiovascular risk factors?
- Does he or she have evidence of target organ damage?
DOES THE PATIENT HAVE SUSTAINED HYPERTENSION?
It is important to measure blood pressure accurately, for several reasons. A diagnosis of hypertension has a measurable impact on the patient’s quality of life.2 Furthermore, we want to avoid undertaking a full evaluation of hypertension if the patient doesn’t actually have high blood pressure, ie, systolic blood pressure greater than 140 mm Hg or diastolic pressure greater than 90 mm Hg. However, many people have blood pressures in the prehypertensive range (ie, 120–139 mm Hg systolic; 80–89 mm Hg diastolic). Many people in this latter group can expect to develop hypertension in time, as the prevalence of hypertension increases steadily with age unless effective preventive measures are implemented, such as losing weight, exercising regularly, and avoiding excessive consumption of sodium and alcohol.

The best position to use is sitting, as the Framingham Heart Study and most randomized clinical trials that established the value of treating hypertension used this position for diagnosis and follow-up.6
Proper patient positioning, the correct cuff size, calibrated equipment, and good inflation and deflation technique will yield the best assessment of blood pressure levels. But even if your technique is perfect, blood pressure is a dynamic vital sign, so it is necessary to repeat the measurement, average the values for any particular day, and keep in mind that the pressure is higher (or lower) on some days than on others, so that the running average is more important than individual readings. This leads to two final points about blood pressure measurement:
- Take it right, at least two times on any occasion
- Take it on at least two (preferably three) separate days.
Following up on blood pressure
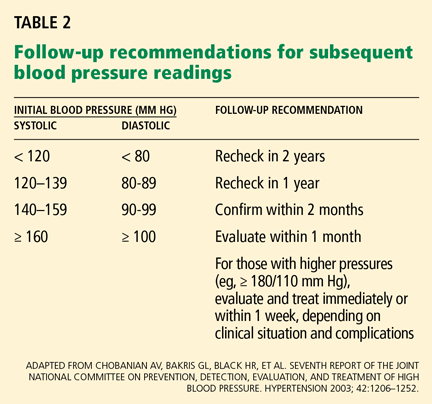
After measuring the blood pressure, it is necessary to plan for follow-up readings, guided by both the blood pressure levels (Table 2) and your clinical judgment.
If the systolic and diastolic blood pressures fall into different categories, you should follow the recommendations for the shorter follow-up time.
IS THE HYPERTENSION PRIMARY OR SECONDARY?
Most patients with hypertension have primary (“essential”) hypertension and are likely to remain hypertensive for life. However, some have secondary hypertension, ie, high blood pressure due to an identifiable cause. Some of these conditions (and the hypertension that they cause) can be cured. For example, pheochromocytoma can be cured if found and removed. Other causes of secondary hypertension, such as parenchymal renal disease, are infrequently cured, and the goal is usually to control the blood pressure with drugs.
The sudden onset of severe hypertension in a patient previously known to have had normal blood pressure raises the suspicion of a secondary form of hypertension, as does the onset of hypertension in a young person (< 25 years) or an older person (> 55 years). However, these ages are arbitrary; with the increasing body mass index in young people, essential hypertension is now more commonly diagnosed in the third decade. And since systolic pressure increases throughout life, we can expect many older patients to develop essential hypertension.7 Indeed, current guidelines are urging us to pay more attention to systolic pressure than in the past.
WHAT IS THE PATIENT’S CARDIOVASCULAR RISK?
The relationship between blood pressure and risk of cardiovascular disease is linear, continuous, and independent of (though additive to) other risk factors.1 For people 40 to 70 years old, each increment of either 20 mm Hg in systolic blood pressure or 10 mm Hg in diastolic blood pressure doubles the risk of cardiovascular disease across the entire range from 115/75 to 185/115 mm Hg.1 If the patient smokes or has elevated cholesterol, other cardiovascular risk factors, or the metabolic syndrome, the risk is even higher.8
The usual goal of antihypertensive treatment is systolic pressure less than 140 mm Hg and diastolic pressure less than 90 mm Hg. However, the target is lower—less than 130/80 mm Hg—for those with diabetes9 or target organ damage such as heart failure or renal disease.1,10 Thus, it is important to try to detect these conditions in the evaluation of the hypertensive patient.
Another reason it is important is that reducing such risk sometimes calls for using (or avoiding) antihypertensive drugs that are likely to alter these factors. For example, the use of beta-blockers in patients with a low level of high-density lipoprotein cholesterol (HDL-C) can lower HDL-C further.11
DOES THE PATIENT HAVE TARGET ORGAN DAMAGE?
Target organ damage is very important to detect because it changes the goal of treatment from primary prevention of adverse target organ outcomes into the more challenging realm of secondary prevention. For example, if a patient has had a stroke, his or her chance of having another stroke in the next 5 years is about 20%. This is much higher than the risk in an average hypertensive patient without such a history. For such patients, the current guidelines1 recommend the combination of a diuretic and an angiotensin-converting enzyme inhibitor, a combination shown to reduce the risk of a second stroke.12 Thus, we need to discover whether the patient had a stroke in the first place.
HISTORY

- The duration (if known) and severity of the hypertension
- The degree of blood pressure fluctuation
- Concomitant medical conditions, especially cardiovascular or renal problems
- Dietary habits
- Alcohol consumption
- Tobacco use
- Level of physical activity
- A family history of hypertension, renal disease, cardiovascular problems, or diabetes mellitus
- Past medications, with particular attention to their side effects and their efficacy in controlling blood pressure
- Current medications, including over-the-counter preparations. One reason: non-steroidal anti-inflammatory drugs other than aspirin can decrease the efficacy of antihypertensive drugs, presumably through mechanisms that inhibit the effects of vasodilatory and natriuretic prostaglandins and potentiate those of angiotensin II.13
PHYSICAL EXAMINATION

The physical examination starts with measurement of height, weight, waist circumference, and blood pressure—in both arms and the leg if coarctation of the aorta is suspected. Measurements with the patient supine, sitting, and standing are usually taken at the first visit, though such an approach is more suited to a hypertension specialty clinic than a primary care setting, in which time constraints usually limit the blood pressure readings to two or three seated values. Most prospective data on the benefits of hypertension treatment are based on a seated blood pressure, so we favor that measurement for follow-up.
Special attention in the physical examination is directed to:
The retina (to assess the vascular impact of the high blood pressure). Look for arteriolar narrowing (grade 1), arteriovenous compression (grade 2), hemorrhages or exudates (grade 3), and papilledema.2 Such findings not only relate to severity (higher grade = more severe blood pressure) but also predict future cardiovascular disease.14
The blood vessels. Bruits in the neck may indicate carotid stenosis, bruits in the abdomen may indicate renovascular disease, and femoral bruits are a sign of general atherosclerosis. Bruits also signal vascular stenosis and irregularity and may be a clue to vascular damage or future loss of target organ function. However, bruits may simply result from vascular tortuosity, particularly with significant flow in the vessel.
Also check the femoral pulses: poor or delayed femoral pulses are a sign of aortic coarctation. The radial artery is about as far away from the heart as the femoral artery; consequently, when palpating both sites simultaneously the pulse should arrive at about the same moment. In aortic coarctation, a palpable delay in the arrival of the femoral pulse may occur, and an interscapular murmur may be heard during auscultation of the back. In these instances, a low leg blood pressure (usually measured by placing a thigh-sized adult cuff on the patient’s thigh and listening over the popliteal area with the patient prone) may confirm the presence of aortic obstruction. When taking a leg blood pressure, the large cuff and the amount of pressure necessary to occlude the artery may be uncomfortable, and one should warn the patient about the discomfort before taking the measurement.
Poor or absent pedal pulses are a sign of peripheral arterial disease.
The heart (to detect gallops, enlargement, or both). Palpation may reveal a displaced apical impulse, which can indicate left ventricular enlargement. A sustained apical impulse may indicate left ventricular hypertrophy. Listen for a fourth heart sound (S4), one of the earliest physical findings of hypertension when physical findings are present. An S4 indicates that the left atrium is working hard to overcome the stiffness of the left ventricle. An S3 indicates an impairment in left ventricular function and is usually a harbinger of underlying heart disease. In some cases, lung rales can also be heard, though the combination of an S3 gallop and rales is an unusual office presentation in the early management of the hypertensive patient.
The lungs. Listen for rales (see above).
The lower extremities should be examined for peripheral arterial pulsations and edema. The loss of pedal pulses is a common finding, particularly in smokers, and is a clue to increased cardiovascular risk.
Strength, gait, and cognition. Perform a brief neurologic examination for evidence of remote stroke. We usually observe our patients’ gait as they enter or leave the examination room, test their bilateral grip strength, and assess their judgment, speech, and memory during the history and physical examination.
A great deal of research has linked high blood pressure to future loss of cognitive function,15 and it is useful to know that impairment is present before beginning treatment, since some patients will complain of memory loss after starting antihypertensive drug treatment.
LABORATORY EVALUATION
Routine tests
The routine evaluation of hypertensive patients should include, at a minimum:
- A hemoglobin or hematocrit measurement
- Urinalysis with microscopic examination
- Serum electrolyte concentrations

- Serum glucose concentration
- A fasting lipid profile
- A 12-lead electrocardiogram (Table 5).

Nonroutine tests
In some cases, other studies may be appropriate, depending on the clinical situation, eg:
- Serum uric acid in those with a history of gout, since some antihypertensive drugs (eg, diuretics) may increase serum uric acid and predispose to further episodes of gout
- Serum calcium in those with a personal or family history of kidney stones, to detect subtle parathyroid excess
- Thyroid-stimulating hormone or other thyroid studies if the history suggests thyroid excess, or if a thyroid nodule is discovered
- Limited echocardiography, which is more sensitive than electrocardiography for detecting left ventricular hypertrophy.
We sometimes use echocardiography if the patient is overweight but seems motivated to lose weight. In these cases we might not start drug therapy right away, choosing rather to wait and see if the patient can lose some weight (which might lower the blood pressure and make drug therapy unnecessary)—but only if the echocardiogram shows that he or she does not have left ventricular hypertrophy.
We also use echocardiography in patients with white-coat hypertension (see below), in whom office pressures are consistently high but whom we have elected to either not treat or not alter treatment. In these cases the echocardiogram serves as a “second opinion” about the merits of not altering therapy and supports this decision when the left ventricular wall thicknesses are normal (and remain so during long-term follow-up). In cases of suspected white-coat hypertension, home or ambulatory blood pressure monitoring is valuable to establish or exclude this diagnosis.1
Urinary albumin excretion. Microalbuminuria is an early manifestation of diabetic nephropathy and hypertension. Although routine urine screening for microalbuminuria is typically done in the management of diabetes, it is still not considered a standard of care, though the growing literature on its role as a cardiovascular risk predictor16–18 and its value as a therapeutic target in diabetes19,20 make it an attractive aid in the overall assessment of patients with hypertension.
Plasma renin activity and serum aldosterone concentrations are useful in screening for aldosterone excess, but are usually reserved as follow-up tests in patients with either hypokalemia or failure to achieve blood pressure control on a three-drug regimen in which at least one drug is a diuretic.1,21
Of note, primary aldosteronism is not as rare as previously thought. In a study of patients referred to hypertension centers, 11% had primary aldosteronism according to prospective diagnostic criteria, almost 5% had curable aldosterone-producing adenomas, and 6% had idiopathic hyperaldosteronism.22
If secondary hypertension is suspected

A search for secondary forms of hypertension is usually considered in patients with moderate or severe hypertension that does not respond to antihypertensive agents. Another situation is in hypertensive patients younger than 25 years, since curable forms of hypertension are more common in this age group. In older patients, the prevalence of secondary hypertension is lower and does not justify the costs and effort of routine elaborate workups unless there is evidence from the history, physical examination, or routine laboratory work for suspecting its presence. An exception to this rule is the need to exclude atherosclerotic renovascular hypertension in an elderly patient. This cause of secondary hypertension is common in the elderly and may be amenable to therapeutic intervention.26
WHEN TO CONSIDER HOME OR AMBULATORY MONITORING
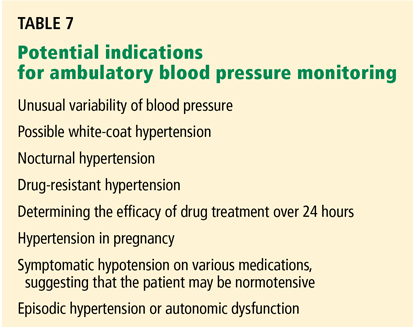
Suspected white-coat hypertension
Blood pressure can be influenced by an environment such as an office or hospital clinic. This has led to the development of ambulatory blood pressure monitors and more use of self-measurement of blood pressure in the home. Blood pressure readings with these techniques are generally lower than those measured in an office or hospital clinic. These methods make it possible to screen for white-coat hypertension. In 10% to 20% of people with hypertensive readings, the blood pressure may be elevated persistently only in the presence of a physician.28 When measured elsewhere, including at work, the blood pressure is not elevated in those with the white-coat effect. Although this response may become less prominent with repeated measurements, it occasionally persists in the office setting, sometimes for years in our experience.
Suspected nocturnal hypertension (’nondipping’ status)
Ambulatory blood pressure is also helpful to screen for nocturnal hypertension. Evidence is accumulating to suggest that hypertensive patients whose pressure remains relatively high at night (“nondippers,” ie, those with less than a 10% reduction at night compared with daytime blood pressure readings) are at greater risk of cardiovascular morbidity than “dippers” (those whose blood pressure is at least 10% lower at night than during the day).29
An early morning surge
Ambulatory monitoring can also detect morning surges in systolic blood pressure,30 a marker of cerebrovascular risk. Generally, these patients have an increase of more than 55 mm Hg in systolic pressure between their sleeping and early-hour waking values, and we may wish to start or alter treatment specifically to address these high morning systolic values.31
‘PIPESTEM’ VESSELS AND PSEUDOHYPERTENSION
Occasionally, one encounters patients with vessels that are stiff and difficult to compress. If the pressure required to compress the brachial artery and stop audible blood flow with a standard blood pressure cuff is greater than the actual blood pressure within the artery as measured invasively, the condition is called pseudohypertension. The stiffness is thought to be due to calcification of the arterial wall.
A way to check for this condition is to inflate the cuff to at least 30 mm Hg above the palpable systolic pressure and then try to “roll” the brachial or radial artery underneath your fingertips, a procedure known as Osler’s maneuver.32 If you feel something that resembles a stiff tube reminiscent of the stem of a tobacco smoker’s pipe (healthy arteries are not palpable when empty), the patient may have pseudohypertension. However, the specificity of Osler’s maneuver has been questioned, particularly in hospitalized elderly patients.33
Pseudohypertension is important because the patients in whom it occurs, usually the elderly or the chronically ill (with diabetes or chronic kidney disease), are prone to orthostatic or postural hypotension, which may be aggravated by increasing their antihypertensive treatment on the basis of a cuff pressure that is actually much higher than the real blood pressure.33
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Wenger NK. Quality of life issues in hypertension: consequences of diagnosis and considerations in management. Am Heart J 1988; 116:628–632.
- McFadden CB, Townsend RR. Blood pressure measurement: common pitfalls and how to avoid them. Consultant 2003; 43:161–165.
- Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit 2006; 11:59–62.
- Mosenkis A, Townsend RR. Sitting on the evidence: what is the proper patient position for the office measurement of blood pressure? J Clin Hypertens (Greenwich) 2005; 7:365–366.
- Vasan RS, Beiser A, Seshadri S, et al. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 2002; 287:1003–1010.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- American Diabetes Association. Treatment of hypertension in adults with diabetes. Diabetes Care 2002; 25:199–201.
- Rosendorff C, Black HR, Cannon CP, et al. Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation 2007; 115:2761–2788.
- Papadakis JA, Mikhailidis DP, Vrentzos GE, Kalikaki A, Kazakou I, Ganotakis ES. Effect of antihypertensive treatment on plasma fibrinogen and serum HDL levels in patients with essential hypertension. Clin Appl Thromb Hemost 2005; 11:139–146.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Fierro-Carrion GA, Ram CV. Nonsteroidal anti-inflammatory drugs (NSAIDs) and blood pressure. Am J Cardiol 1997; 80:775–776.
- Wong TY, McIntosh R. Hypertensive retinopathy signs as risk indicators of cardiovascular morbidity and mortality. Br Med Bull 2005; 73–74:57–70.
- Forette F, Boller F. Hypertension and the risk of dementia in the elderly. Am J Med 1991; 90:14S–19S.
- Schrader J, Luders S, Kulschewski A, et al. Microalbuminuria and tubular proteinuria as risk predictors of cardiovascular morbidity and mortality in essential hypertension: final results of a prospective long-term study (MARPLE Study). J Hypertens 2006; 24:541–548.
- Luque M, de Rivas B, Alvarez B, Garcia G, Fernandez C, Martell N. Influence of target organ lesion detection (assessment of microalbuminuria and echocardiogram) in cardiovascular risk stratification and treatment of untreated hypertensive patients. J Hum Hypertens 2006; 20:187–192.
- Pontremoli R, Leoncini G, Viazzi F, et al. Role of microalbuminuria in the assessment of cardiovascular risk in essential hypertension. J Am Soc Nephrol 2005; 16 suppl 1:S39–S41.
- Erdmann E. Microalbuminuria as a marker of cardiovascular risk in patients with type 2 diabetes. Int J Cardiol 2006; 107:147–153.
- Bakris GL, Sowers JR. Microalbuminuria in diabetes: focus on cardiovascular and renal risk reduction. Curr Diab Rep 2002; 2:258–262.
- Gallay BJ, Ahmad S, Xu L, Toivola B, Davidson RC. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosteronerenin ratio. Am J Kidney Dis 2001; 37:699–705.
- Rossi GP, Bernini G, Caliumi C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006; 48:2293–2300.
- Onusko E. Diagnosing secondary hypertension. Am Fam Physician 2003; 67:67–74.
- Aurell M. Screening for secondary hypertension. Curr Hypertens Rep 1999; 1:461.
- Garovic VD, Kane GC, Schwartz GL. Renovascular hypertension: balancing the controversies in diagnosis and treatment. Cleve Clin J Med 2005; 72:1135–1137.
- Textor SC. Renovascular hypertension in 2007: where are we now? Curr Cardiol Rep 2007; 9:453–461.
- Pickering TG, Shimbo D, Haas D. Ambulatory blood-pressure monitoring. N Engl J Med 2006; 354:2368–2374.
- Angeli F, Verdecchia P, Gattobigio R, Sardone M, Reboldi G. White-coat hypertension in adults. Blood Press Monit 2005; 10:301–305.
- Cicconetti P, Morelli S, De Serra C, et al. Left ventricular mass in dippers and nondippers with newly diagnosed hypertension. Angiology 2003; 54:661–669.
- Kario K, Pickering TG, Umeda Y, et al. Morning surge in blood pressure as a predictor of silent and clinical cerebrovascular disease in elderly hypertensives: a prospective study. Circulation 2003; 107:1401–1406.
- Katakam R, Townsend RR. Morning surges in blood pressure. J Clin Hypertens 2006; 8:450–451.
- Messerli FH. Osler’s maneuver, pseudohypertension, and true hypertension in the elderly. Am J Med 1986; 80:906–910.
- Belmin J, Visintin JM, Salvatore R, Sebban C, Moulias R. Osler’s maneuver: absence of usefulness for the detection of pseudohypertension in an elderly population. Am J Med 1995; 98:42–49.
- Messerli FH, Ventura HO, Amodeo C. Osler’s maneuver and pseudohypertension. N Engl J Med 1985; 312:1548–1551.
How extensive a workup does a patient with high blood pressure need?
On one hand, we would not want to start therapy on the basis of a single elevated reading, as blood pressure fluctuates considerably during the day, and even experienced physicians often make errors in taking blood pressure that tend to falsely elevate the patient’s readings. Similarly, we would not want to miss the diagnosis of a potentially curable cause of hypertension or of a condition that increases a patient’s risk of cardiovascular disease. But considering that nearly one-third of adults in the United States have hypertension and that another one-fourth have prehypertension (formerly called high-normal blood pressure),1 if we were to launch an intensive workup for every patient with high blood pressure, the cost and effort would be enormous.
Fortunately, for most patients, it is enough to measure blood pressure accurately and repeatedly, perform a focused history and physical examination, and obtain the results of a few basic laboratory tests and an electrocardiogram, with additional tests in special cases.
In this review we address four fundamental questions in the evaluation of patients with a high blood pressure reading, and how to answer them.
ANSWERING FOUR QUESTIONS
The goal of the hypertension evaluation is to answer four questions:
- Does the patient have sustained hypertension? And if so—
- Is the hypertension primary or secondary?
- Does the patient have other cardiovascular risk factors?
- Does he or she have evidence of target organ damage?
DOES THE PATIENT HAVE SUSTAINED HYPERTENSION?
It is important to measure blood pressure accurately, for several reasons. A diagnosis of hypertension has a measurable impact on the patient’s quality of life.2 Furthermore, we want to avoid undertaking a full evaluation of hypertension if the patient doesn’t actually have high blood pressure, ie, systolic blood pressure greater than 140 mm Hg or diastolic pressure greater than 90 mm Hg. However, many people have blood pressures in the prehypertensive range (ie, 120–139 mm Hg systolic; 80–89 mm Hg diastolic). Many people in this latter group can expect to develop hypertension in time, as the prevalence of hypertension increases steadily with age unless effective preventive measures are implemented, such as losing weight, exercising regularly, and avoiding excessive consumption of sodium and alcohol.
The best position to use is sitting, as the Framingham Heart Study and most randomized clinical trials that established the value of treating hypertension used this position for diagnosis and follow-up.6
Proper patient positioning, the correct cuff size, calibrated equipment, and good inflation and deflation technique will yield the best assessment of blood pressure levels. But even if your technique is perfect, blood pressure is a dynamic vital sign, so it is necessary to repeat the measurement, average the values for any particular day, and keep in mind that the pressure is higher (or lower) on some days than on others, so that the running average is more important than individual readings. This leads to two final points about blood pressure measurement:
- Take it right, at least two times on any occasion
- Take it on at least two (preferably three) separate days.
Following up on blood pressure
After measuring the blood pressure, it is necessary to plan for follow-up readings, guided by both the blood pressure levels (Table 2) and your clinical judgment.
If the systolic and diastolic blood pressures fall into different categories, you should follow the recommendations for the shorter follow-up time.
IS THE HYPERTENSION PRIMARY OR SECONDARY?
Most patients with hypertension have primary (“essential”) hypertension and are likely to remain hypertensive for life. However, some have secondary hypertension, ie, high blood pressure due to an identifiable cause. Some of these conditions (and the hypertension that they cause) can be cured. For example, pheochromocytoma can be cured if found and removed. Other causes of secondary hypertension, such as parenchymal renal disease, are infrequently cured, and the goal is usually to control the blood pressure with drugs.
The sudden onset of severe hypertension in a patient previously known to have had normal blood pressure raises the suspicion of a secondary form of hypertension, as does the onset of hypertension in a young person (< 25 years) or an older person (> 55 years). However, these ages are arbitrary; with the increasing body mass index in young people, essential hypertension is now more commonly diagnosed in the third decade. And since systolic pressure increases throughout life, we can expect many older patients to develop essential hypertension.7 Indeed, current guidelines are urging us to pay more attention to systolic pressure than in the past.
WHAT IS THE PATIENT’S CARDIOVASCULAR RISK?
The relationship between blood pressure and risk of cardiovascular disease is linear, continuous, and independent of (though additive to) other risk factors.1 For people 40 to 70 years old, each increment of either 20 mm Hg in systolic blood pressure or 10 mm Hg in diastolic blood pressure doubles the risk of cardiovascular disease across the entire range from 115/75 to 185/115 mm Hg.1 If the patient smokes or has elevated cholesterol, other cardiovascular risk factors, or the metabolic syndrome, the risk is even higher.8
The usual goal of antihypertensive treatment is systolic pressure less than 140 mm Hg and diastolic pressure less than 90 mm Hg. However, the target is lower—less than 130/80 mm Hg—for those with diabetes9 or target organ damage such as heart failure or renal disease.1,10 Thus, it is important to try to detect these conditions in the evaluation of the hypertensive patient.
Another reason it is important is that reducing such risk sometimes calls for using (or avoiding) antihypertensive drugs that are likely to alter these factors. For example, the use of beta-blockers in patients with a low level of high-density lipoprotein cholesterol (HDL-C) can lower HDL-C further.11
DOES THE PATIENT HAVE TARGET ORGAN DAMAGE?
Target organ damage is very important to detect because it changes the goal of treatment from primary prevention of adverse target organ outcomes into the more challenging realm of secondary prevention. For example, if a patient has had a stroke, his or her chance of having another stroke in the next 5 years is about 20%. This is much higher than the risk in an average hypertensive patient without such a history. For such patients, the current guidelines1 recommend the combination of a diuretic and an angiotensin-converting enzyme inhibitor, a combination shown to reduce the risk of a second stroke.12 Thus, we need to discover whether the patient had a stroke in the first place.
HISTORY
- The duration (if known) and severity of the hypertension
- The degree of blood pressure fluctuation
- Concomitant medical conditions, especially cardiovascular or renal problems
- Dietary habits
- Alcohol consumption
- Tobacco use
- Level of physical activity
- A family history of hypertension, renal disease, cardiovascular problems, or diabetes mellitus
- Past medications, with particular attention to their side effects and their efficacy in controlling blood pressure
- Current medications, including over-the-counter preparations. One reason: non-steroidal anti-inflammatory drugs other than aspirin can decrease the efficacy of antihypertensive drugs, presumably through mechanisms that inhibit the effects of vasodilatory and natriuretic prostaglandins and potentiate those of angiotensin II.13
PHYSICAL EXAMINATION
The physical examination starts with measurement of height, weight, waist circumference, and blood pressure—in both arms and the leg if coarctation of the aorta is suspected. Measurements with the patient supine, sitting, and standing are usually taken at the first visit, though such an approach is more suited to a hypertension specialty clinic than a primary care setting, in which time constraints usually limit the blood pressure readings to two or three seated values. Most prospective data on the benefits of hypertension treatment are based on a seated blood pressure, so we favor that measurement for follow-up.
Special attention in the physical examination is directed to:
The retina (to assess the vascular impact of the high blood pressure). Look for arteriolar narrowing (grade 1), arteriovenous compression (grade 2), hemorrhages or exudates (grade 3), and papilledema.2 Such findings not only relate to severity (higher grade = more severe blood pressure) but also predict future cardiovascular disease.14
The blood vessels. Bruits in the neck may indicate carotid stenosis, bruits in the abdomen may indicate renovascular disease, and femoral bruits are a sign of general atherosclerosis. Bruits also signal vascular stenosis and irregularity and may be a clue to vascular damage or future loss of target organ function. However, bruits may simply result from vascular tortuosity, particularly with significant flow in the vessel.
Also check the femoral pulses: poor or delayed femoral pulses are a sign of aortic coarctation. The radial artery is about as far away from the heart as the femoral artery; consequently, when palpating both sites simultaneously the pulse should arrive at about the same moment. In aortic coarctation, a palpable delay in the arrival of the femoral pulse may occur, and an interscapular murmur may be heard during auscultation of the back. In these instances, a low leg blood pressure (usually measured by placing a thigh-sized adult cuff on the patient’s thigh and listening over the popliteal area with the patient prone) may confirm the presence of aortic obstruction. When taking a leg blood pressure, the large cuff and the amount of pressure necessary to occlude the artery may be uncomfortable, and one should warn the patient about the discomfort before taking the measurement.
Poor or absent pedal pulses are a sign of peripheral arterial disease.
The heart (to detect gallops, enlargement, or both). Palpation may reveal a displaced apical impulse, which can indicate left ventricular enlargement. A sustained apical impulse may indicate left ventricular hypertrophy. Listen for a fourth heart sound (S4), one of the earliest physical findings of hypertension when physical findings are present. An S4 indicates that the left atrium is working hard to overcome the stiffness of the left ventricle. An S3 indicates an impairment in left ventricular function and is usually a harbinger of underlying heart disease. In some cases, lung rales can also be heard, though the combination of an S3 gallop and rales is an unusual office presentation in the early management of the hypertensive patient.
The lungs. Listen for rales (see above).
The lower extremities should be examined for peripheral arterial pulsations and edema. The loss of pedal pulses is a common finding, particularly in smokers, and is a clue to increased cardiovascular risk.
Strength, gait, and cognition. Perform a brief neurologic examination for evidence of remote stroke. We usually observe our patients’ gait as they enter or leave the examination room, test their bilateral grip strength, and assess their judgment, speech, and memory during the history and physical examination.
A great deal of research has linked high blood pressure to future loss of cognitive function,15 and it is useful to know that impairment is present before beginning treatment, since some patients will complain of memory loss after starting antihypertensive drug treatment.
LABORATORY EVALUATION
Routine tests
The routine evaluation of hypertensive patients should include, at a minimum:
- A hemoglobin or hematocrit measurement
- Urinalysis with microscopic examination
- Serum electrolyte concentrations
- Serum glucose concentration
- A fasting lipid profile
- A 12-lead electrocardiogram (Table 5).
Nonroutine tests
In some cases, other studies may be appropriate, depending on the clinical situation, eg:
- Serum uric acid in those with a history of gout, since some antihypertensive drugs (eg, diuretics) may increase serum uric acid and predispose to further episodes of gout
- Serum calcium in those with a personal or family history of kidney stones, to detect subtle parathyroid excess
- Thyroid-stimulating hormone or other thyroid studies if the history suggests thyroid excess, or if a thyroid nodule is discovered
- Limited echocardiography, which is more sensitive than electrocardiography for detecting left ventricular hypertrophy.
We sometimes use echocardiography if the patient is overweight but seems motivated to lose weight. In these cases we might not start drug therapy right away, choosing rather to wait and see if the patient can lose some weight (which might lower the blood pressure and make drug therapy unnecessary)—but only if the echocardiogram shows that he or she does not have left ventricular hypertrophy.
We also use echocardiography in patients with white-coat hypertension (see below), in whom office pressures are consistently high but whom we have elected to either not treat or not alter treatment. In these cases the echocardiogram serves as a “second opinion” about the merits of not altering therapy and supports this decision when the left ventricular wall thicknesses are normal (and remain so during long-term follow-up). In cases of suspected white-coat hypertension, home or ambulatory blood pressure monitoring is valuable to establish or exclude this diagnosis.1
Urinary albumin excretion. Microalbuminuria is an early manifestation of diabetic nephropathy and hypertension. Although routine urine screening for microalbuminuria is typically done in the management of diabetes, it is still not considered a standard of care, though the growing literature on its role as a cardiovascular risk predictor16–18 and its value as a therapeutic target in diabetes19,20 make it an attractive aid in the overall assessment of patients with hypertension.
Plasma renin activity and serum aldosterone concentrations are useful in screening for aldosterone excess, but are usually reserved as follow-up tests in patients with either hypokalemia or failure to achieve blood pressure control on a three-drug regimen in which at least one drug is a diuretic.1,21
Of note, primary aldosteronism is not as rare as previously thought. In a study of patients referred to hypertension centers, 11% had primary aldosteronism according to prospective diagnostic criteria, almost 5% had curable aldosterone-producing adenomas, and 6% had idiopathic hyperaldosteronism.22
If secondary hypertension is suspected
A search for secondary forms of hypertension is usually considered in patients with moderate or severe hypertension that does not respond to antihypertensive agents. Another situation is in hypertensive patients younger than 25 years, since curable forms of hypertension are more common in this age group. In older patients, the prevalence of secondary hypertension is lower and does not justify the costs and effort of routine elaborate workups unless there is evidence from the history, physical examination, or routine laboratory work for suspecting its presence. An exception to this rule is the need to exclude atherosclerotic renovascular hypertension in an elderly patient. This cause of secondary hypertension is common in the elderly and may be amenable to therapeutic intervention.26
WHEN TO CONSIDER HOME OR AMBULATORY MONITORING
Suspected white-coat hypertension
Blood pressure can be influenced by an environment such as an office or hospital clinic. This has led to the development of ambulatory blood pressure monitors and more use of self-measurement of blood pressure in the home. Blood pressure readings with these techniques are generally lower than those measured in an office or hospital clinic. These methods make it possible to screen for white-coat hypertension. In 10% to 20% of people with hypertensive readings, the blood pressure may be elevated persistently only in the presence of a physician.28 When measured elsewhere, including at work, the blood pressure is not elevated in those with the white-coat effect. Although this response may become less prominent with repeated measurements, it occasionally persists in the office setting, sometimes for years in our experience.
Suspected nocturnal hypertension (’nondipping’ status)
Ambulatory blood pressure is also helpful to screen for nocturnal hypertension. Evidence is accumulating to suggest that hypertensive patients whose pressure remains relatively high at night (“nondippers,” ie, those with less than a 10% reduction at night compared with daytime blood pressure readings) are at greater risk of cardiovascular morbidity than “dippers” (those whose blood pressure is at least 10% lower at night than during the day).29
An early morning surge
Ambulatory monitoring can also detect morning surges in systolic blood pressure,30 a marker of cerebrovascular risk. Generally, these patients have an increase of more than 55 mm Hg in systolic pressure between their sleeping and early-hour waking values, and we may wish to start or alter treatment specifically to address these high morning systolic values.31
‘PIPESTEM’ VESSELS AND PSEUDOHYPERTENSION
Occasionally, one encounters patients with vessels that are stiff and difficult to compress. If the pressure required to compress the brachial artery and stop audible blood flow with a standard blood pressure cuff is greater than the actual blood pressure within the artery as measured invasively, the condition is called pseudohypertension. The stiffness is thought to be due to calcification of the arterial wall.
A way to check for this condition is to inflate the cuff to at least 30 mm Hg above the palpable systolic pressure and then try to “roll” the brachial or radial artery underneath your fingertips, a procedure known as Osler’s maneuver.32 If you feel something that resembles a stiff tube reminiscent of the stem of a tobacco smoker’s pipe (healthy arteries are not palpable when empty), the patient may have pseudohypertension. However, the specificity of Osler’s maneuver has been questioned, particularly in hospitalized elderly patients.33
Pseudohypertension is important because the patients in whom it occurs, usually the elderly or the chronically ill (with diabetes or chronic kidney disease), are prone to orthostatic or postural hypotension, which may be aggravated by increasing their antihypertensive treatment on the basis of a cuff pressure that is actually much higher than the real blood pressure.33
How extensive a workup does a patient with high blood pressure need?
On one hand, we would not want to start therapy on the basis of a single elevated reading, as blood pressure fluctuates considerably during the day, and even experienced physicians often make errors in taking blood pressure that tend to falsely elevate the patient’s readings. Similarly, we would not want to miss the diagnosis of a potentially curable cause of hypertension or of a condition that increases a patient’s risk of cardiovascular disease. But considering that nearly one-third of adults in the United States have hypertension and that another one-fourth have prehypertension (formerly called high-normal blood pressure),1 if we were to launch an intensive workup for every patient with high blood pressure, the cost and effort would be enormous.
Fortunately, for most patients, it is enough to measure blood pressure accurately and repeatedly, perform a focused history and physical examination, and obtain the results of a few basic laboratory tests and an electrocardiogram, with additional tests in special cases.
In this review we address four fundamental questions in the evaluation of patients with a high blood pressure reading, and how to answer them.
ANSWERING FOUR QUESTIONS
The goal of the hypertension evaluation is to answer four questions:
- Does the patient have sustained hypertension? And if so—
- Is the hypertension primary or secondary?
- Does the patient have other cardiovascular risk factors?
- Does he or she have evidence of target organ damage?
DOES THE PATIENT HAVE SUSTAINED HYPERTENSION?
It is important to measure blood pressure accurately, for several reasons. A diagnosis of hypertension has a measurable impact on the patient’s quality of life.2 Furthermore, we want to avoid undertaking a full evaluation of hypertension if the patient doesn’t actually have high blood pressure, ie, systolic blood pressure greater than 140 mm Hg or diastolic pressure greater than 90 mm Hg. However, many people have blood pressures in the prehypertensive range (ie, 120–139 mm Hg systolic; 80–89 mm Hg diastolic). Many people in this latter group can expect to develop hypertension in time, as the prevalence of hypertension increases steadily with age unless effective preventive measures are implemented, such as losing weight, exercising regularly, and avoiding excessive consumption of sodium and alcohol.
The best position to use is sitting, as the Framingham Heart Study and most randomized clinical trials that established the value of treating hypertension used this position for diagnosis and follow-up.6
Proper patient positioning, the correct cuff size, calibrated equipment, and good inflation and deflation technique will yield the best assessment of blood pressure levels. But even if your technique is perfect, blood pressure is a dynamic vital sign, so it is necessary to repeat the measurement, average the values for any particular day, and keep in mind that the pressure is higher (or lower) on some days than on others, so that the running average is more important than individual readings. This leads to two final points about blood pressure measurement:
- Take it right, at least two times on any occasion
- Take it on at least two (preferably three) separate days.
Following up on blood pressure
After measuring the blood pressure, it is necessary to plan for follow-up readings, guided by both the blood pressure levels (Table 2) and your clinical judgment.
If the systolic and diastolic blood pressures fall into different categories, you should follow the recommendations for the shorter follow-up time.
IS THE HYPERTENSION PRIMARY OR SECONDARY?
Most patients with hypertension have primary (“essential”) hypertension and are likely to remain hypertensive for life. However, some have secondary hypertension, ie, high blood pressure due to an identifiable cause. Some of these conditions (and the hypertension that they cause) can be cured. For example, pheochromocytoma can be cured if found and removed. Other causes of secondary hypertension, such as parenchymal renal disease, are infrequently cured, and the goal is usually to control the blood pressure with drugs.
The sudden onset of severe hypertension in a patient previously known to have had normal blood pressure raises the suspicion of a secondary form of hypertension, as does the onset of hypertension in a young person (< 25 years) or an older person (> 55 years). However, these ages are arbitrary; with the increasing body mass index in young people, essential hypertension is now more commonly diagnosed in the third decade. And since systolic pressure increases throughout life, we can expect many older patients to develop essential hypertension.7 Indeed, current guidelines are urging us to pay more attention to systolic pressure than in the past.
WHAT IS THE PATIENT’S CARDIOVASCULAR RISK?
The relationship between blood pressure and risk of cardiovascular disease is linear, continuous, and independent of (though additive to) other risk factors.1 For people 40 to 70 years old, each increment of either 20 mm Hg in systolic blood pressure or 10 mm Hg in diastolic blood pressure doubles the risk of cardiovascular disease across the entire range from 115/75 to 185/115 mm Hg.1 If the patient smokes or has elevated cholesterol, other cardiovascular risk factors, or the metabolic syndrome, the risk is even higher.8
The usual goal of antihypertensive treatment is systolic pressure less than 140 mm Hg and diastolic pressure less than 90 mm Hg. However, the target is lower—less than 130/80 mm Hg—for those with diabetes9 or target organ damage such as heart failure or renal disease.1,10 Thus, it is important to try to detect these conditions in the evaluation of the hypertensive patient.
Another reason it is important is that reducing such risk sometimes calls for using (or avoiding) antihypertensive drugs that are likely to alter these factors. For example, the use of beta-blockers in patients with a low level of high-density lipoprotein cholesterol (HDL-C) can lower HDL-C further.11
DOES THE PATIENT HAVE TARGET ORGAN DAMAGE?
Target organ damage is very important to detect because it changes the goal of treatment from primary prevention of adverse target organ outcomes into the more challenging realm of secondary prevention. For example, if a patient has had a stroke, his or her chance of having another stroke in the next 5 years is about 20%. This is much higher than the risk in an average hypertensive patient without such a history. For such patients, the current guidelines1 recommend the combination of a diuretic and an angiotensin-converting enzyme inhibitor, a combination shown to reduce the risk of a second stroke.12 Thus, we need to discover whether the patient had a stroke in the first place.
HISTORY
- The duration (if known) and severity of the hypertension
- The degree of blood pressure fluctuation
- Concomitant medical conditions, especially cardiovascular or renal problems
- Dietary habits
- Alcohol consumption
- Tobacco use
- Level of physical activity
- A family history of hypertension, renal disease, cardiovascular problems, or diabetes mellitus
- Past medications, with particular attention to their side effects and their efficacy in controlling blood pressure
- Current medications, including over-the-counter preparations. One reason: non-steroidal anti-inflammatory drugs other than aspirin can decrease the efficacy of antihypertensive drugs, presumably through mechanisms that inhibit the effects of vasodilatory and natriuretic prostaglandins and potentiate those of angiotensin II.13
PHYSICAL EXAMINATION
The physical examination starts with measurement of height, weight, waist circumference, and blood pressure—in both arms and the leg if coarctation of the aorta is suspected. Measurements with the patient supine, sitting, and standing are usually taken at the first visit, though such an approach is more suited to a hypertension specialty clinic than a primary care setting, in which time constraints usually limit the blood pressure readings to two or three seated values. Most prospective data on the benefits of hypertension treatment are based on a seated blood pressure, so we favor that measurement for follow-up.
Special attention in the physical examination is directed to:
The retina (to assess the vascular impact of the high blood pressure). Look for arteriolar narrowing (grade 1), arteriovenous compression (grade 2), hemorrhages or exudates (grade 3), and papilledema.2 Such findings not only relate to severity (higher grade = more severe blood pressure) but also predict future cardiovascular disease.14
The blood vessels. Bruits in the neck may indicate carotid stenosis, bruits in the abdomen may indicate renovascular disease, and femoral bruits are a sign of general atherosclerosis. Bruits also signal vascular stenosis and irregularity and may be a clue to vascular damage or future loss of target organ function. However, bruits may simply result from vascular tortuosity, particularly with significant flow in the vessel.
Also check the femoral pulses: poor or delayed femoral pulses are a sign of aortic coarctation. The radial artery is about as far away from the heart as the femoral artery; consequently, when palpating both sites simultaneously the pulse should arrive at about the same moment. In aortic coarctation, a palpable delay in the arrival of the femoral pulse may occur, and an interscapular murmur may be heard during auscultation of the back. In these instances, a low leg blood pressure (usually measured by placing a thigh-sized adult cuff on the patient’s thigh and listening over the popliteal area with the patient prone) may confirm the presence of aortic obstruction. When taking a leg blood pressure, the large cuff and the amount of pressure necessary to occlude the artery may be uncomfortable, and one should warn the patient about the discomfort before taking the measurement.
Poor or absent pedal pulses are a sign of peripheral arterial disease.
The heart (to detect gallops, enlargement, or both). Palpation may reveal a displaced apical impulse, which can indicate left ventricular enlargement. A sustained apical impulse may indicate left ventricular hypertrophy. Listen for a fourth heart sound (S4), one of the earliest physical findings of hypertension when physical findings are present. An S4 indicates that the left atrium is working hard to overcome the stiffness of the left ventricle. An S3 indicates an impairment in left ventricular function and is usually a harbinger of underlying heart disease. In some cases, lung rales can also be heard, though the combination of an S3 gallop and rales is an unusual office presentation in the early management of the hypertensive patient.
The lungs. Listen for rales (see above).
The lower extremities should be examined for peripheral arterial pulsations and edema. The loss of pedal pulses is a common finding, particularly in smokers, and is a clue to increased cardiovascular risk.
Strength, gait, and cognition. Perform a brief neurologic examination for evidence of remote stroke. We usually observe our patients’ gait as they enter or leave the examination room, test their bilateral grip strength, and assess their judgment, speech, and memory during the history and physical examination.
A great deal of research has linked high blood pressure to future loss of cognitive function,15 and it is useful to know that impairment is present before beginning treatment, since some patients will complain of memory loss after starting antihypertensive drug treatment.
LABORATORY EVALUATION
Routine tests
The routine evaluation of hypertensive patients should include, at a minimum:
- A hemoglobin or hematocrit measurement
- Urinalysis with microscopic examination
- Serum electrolyte concentrations
- Serum glucose concentration
- A fasting lipid profile
- A 12-lead electrocardiogram (Table 5).
Nonroutine tests
In some cases, other studies may be appropriate, depending on the clinical situation, eg:
- Serum uric acid in those with a history of gout, since some antihypertensive drugs (eg, diuretics) may increase serum uric acid and predispose to further episodes of gout
- Serum calcium in those with a personal or family history of kidney stones, to detect subtle parathyroid excess
- Thyroid-stimulating hormone or other thyroid studies if the history suggests thyroid excess, or if a thyroid nodule is discovered
- Limited echocardiography, which is more sensitive than electrocardiography for detecting left ventricular hypertrophy.
We sometimes use echocardiography if the patient is overweight but seems motivated to lose weight. In these cases we might not start drug therapy right away, choosing rather to wait and see if the patient can lose some weight (which might lower the blood pressure and make drug therapy unnecessary)—but only if the echocardiogram shows that he or she does not have left ventricular hypertrophy.
We also use echocardiography in patients with white-coat hypertension (see below), in whom office pressures are consistently high but whom we have elected to either not treat or not alter treatment. In these cases the echocardiogram serves as a “second opinion” about the merits of not altering therapy and supports this decision when the left ventricular wall thicknesses are normal (and remain so during long-term follow-up). In cases of suspected white-coat hypertension, home or ambulatory blood pressure monitoring is valuable to establish or exclude this diagnosis.1
Urinary albumin excretion. Microalbuminuria is an early manifestation of diabetic nephropathy and hypertension. Although routine urine screening for microalbuminuria is typically done in the management of diabetes, it is still not considered a standard of care, though the growing literature on its role as a cardiovascular risk predictor16–18 and its value as a therapeutic target in diabetes19,20 make it an attractive aid in the overall assessment of patients with hypertension.
Plasma renin activity and serum aldosterone concentrations are useful in screening for aldosterone excess, but are usually reserved as follow-up tests in patients with either hypokalemia or failure to achieve blood pressure control on a three-drug regimen in which at least one drug is a diuretic.1,21
Of note, primary aldosteronism is not as rare as previously thought. In a study of patients referred to hypertension centers, 11% had primary aldosteronism according to prospective diagnostic criteria, almost 5% had curable aldosterone-producing adenomas, and 6% had idiopathic hyperaldosteronism.22
If secondary hypertension is suspected
A search for secondary forms of hypertension is usually considered in patients with moderate or severe hypertension that does not respond to antihypertensive agents. Another situation is in hypertensive patients younger than 25 years, since curable forms of hypertension are more common in this age group. In older patients, the prevalence of secondary hypertension is lower and does not justify the costs and effort of routine elaborate workups unless there is evidence from the history, physical examination, or routine laboratory work for suspecting its presence. An exception to this rule is the need to exclude atherosclerotic renovascular hypertension in an elderly patient. This cause of secondary hypertension is common in the elderly and may be amenable to therapeutic intervention.26
WHEN TO CONSIDER HOME OR AMBULATORY MONITORING
Suspected white-coat hypertension
Blood pressure can be influenced by an environment such as an office or hospital clinic. This has led to the development of ambulatory blood pressure monitors and more use of self-measurement of blood pressure in the home. Blood pressure readings with these techniques are generally lower than those measured in an office or hospital clinic. These methods make it possible to screen for white-coat hypertension. In 10% to 20% of people with hypertensive readings, the blood pressure may be elevated persistently only in the presence of a physician.28 When measured elsewhere, including at work, the blood pressure is not elevated in those with the white-coat effect. Although this response may become less prominent with repeated measurements, it occasionally persists in the office setting, sometimes for years in our experience.
Suspected nocturnal hypertension (’nondipping’ status)
Ambulatory blood pressure is also helpful to screen for nocturnal hypertension. Evidence is accumulating to suggest that hypertensive patients whose pressure remains relatively high at night (“nondippers,” ie, those with less than a 10% reduction at night compared with daytime blood pressure readings) are at greater risk of cardiovascular morbidity than “dippers” (those whose blood pressure is at least 10% lower at night than during the day).29
An early morning surge
Ambulatory monitoring can also detect morning surges in systolic blood pressure,30 a marker of cerebrovascular risk. Generally, these patients have an increase of more than 55 mm Hg in systolic pressure between their sleeping and early-hour waking values, and we may wish to start or alter treatment specifically to address these high morning systolic values.31
‘PIPESTEM’ VESSELS AND PSEUDOHYPERTENSION
Occasionally, one encounters patients with vessels that are stiff and difficult to compress. If the pressure required to compress the brachial artery and stop audible blood flow with a standard blood pressure cuff is greater than the actual blood pressure within the artery as measured invasively, the condition is called pseudohypertension. The stiffness is thought to be due to calcification of the arterial wall.
A way to check for this condition is to inflate the cuff to at least 30 mm Hg above the palpable systolic pressure and then try to “roll” the brachial or radial artery underneath your fingertips, a procedure known as Osler’s maneuver.32 If you feel something that resembles a stiff tube reminiscent of the stem of a tobacco smoker’s pipe (healthy arteries are not palpable when empty), the patient may have pseudohypertension. However, the specificity of Osler’s maneuver has been questioned, particularly in hospitalized elderly patients.33
Pseudohypertension is important because the patients in whom it occurs, usually the elderly or the chronically ill (with diabetes or chronic kidney disease), are prone to orthostatic or postural hypotension, which may be aggravated by increasing their antihypertensive treatment on the basis of a cuff pressure that is actually much higher than the real blood pressure.33
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Wenger NK. Quality of life issues in hypertension: consequences of diagnosis and considerations in management. Am Heart J 1988; 116:628–632.
- McFadden CB, Townsend RR. Blood pressure measurement: common pitfalls and how to avoid them. Consultant 2003; 43:161–165.
- Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit 2006; 11:59–62.
- Mosenkis A, Townsend RR. Sitting on the evidence: what is the proper patient position for the office measurement of blood pressure? J Clin Hypertens (Greenwich) 2005; 7:365–366.
- Vasan RS, Beiser A, Seshadri S, et al. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 2002; 287:1003–1010.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- American Diabetes Association. Treatment of hypertension in adults with diabetes. Diabetes Care 2002; 25:199–201.
- Rosendorff C, Black HR, Cannon CP, et al. Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation 2007; 115:2761–2788.
- Papadakis JA, Mikhailidis DP, Vrentzos GE, Kalikaki A, Kazakou I, Ganotakis ES. Effect of antihypertensive treatment on plasma fibrinogen and serum HDL levels in patients with essential hypertension. Clin Appl Thromb Hemost 2005; 11:139–146.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Fierro-Carrion GA, Ram CV. Nonsteroidal anti-inflammatory drugs (NSAIDs) and blood pressure. Am J Cardiol 1997; 80:775–776.
- Wong TY, McIntosh R. Hypertensive retinopathy signs as risk indicators of cardiovascular morbidity and mortality. Br Med Bull 2005; 73–74:57–70.
- Forette F, Boller F. Hypertension and the risk of dementia in the elderly. Am J Med 1991; 90:14S–19S.
- Schrader J, Luders S, Kulschewski A, et al. Microalbuminuria and tubular proteinuria as risk predictors of cardiovascular morbidity and mortality in essential hypertension: final results of a prospective long-term study (MARPLE Study). J Hypertens 2006; 24:541–548.
- Luque M, de Rivas B, Alvarez B, Garcia G, Fernandez C, Martell N. Influence of target organ lesion detection (assessment of microalbuminuria and echocardiogram) in cardiovascular risk stratification and treatment of untreated hypertensive patients. J Hum Hypertens 2006; 20:187–192.
- Pontremoli R, Leoncini G, Viazzi F, et al. Role of microalbuminuria in the assessment of cardiovascular risk in essential hypertension. J Am Soc Nephrol 2005; 16 suppl 1:S39–S41.
- Erdmann E. Microalbuminuria as a marker of cardiovascular risk in patients with type 2 diabetes. Int J Cardiol 2006; 107:147–153.
- Bakris GL, Sowers JR. Microalbuminuria in diabetes: focus on cardiovascular and renal risk reduction. Curr Diab Rep 2002; 2:258–262.
- Gallay BJ, Ahmad S, Xu L, Toivola B, Davidson RC. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosteronerenin ratio. Am J Kidney Dis 2001; 37:699–705.
- Rossi GP, Bernini G, Caliumi C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006; 48:2293–2300.
- Onusko E. Diagnosing secondary hypertension. Am Fam Physician 2003; 67:67–74.
- Aurell M. Screening for secondary hypertension. Curr Hypertens Rep 1999; 1:461.
- Garovic VD, Kane GC, Schwartz GL. Renovascular hypertension: balancing the controversies in diagnosis and treatment. Cleve Clin J Med 2005; 72:1135–1137.
- Textor SC. Renovascular hypertension in 2007: where are we now? Curr Cardiol Rep 2007; 9:453–461.
- Pickering TG, Shimbo D, Haas D. Ambulatory blood-pressure monitoring. N Engl J Med 2006; 354:2368–2374.
- Angeli F, Verdecchia P, Gattobigio R, Sardone M, Reboldi G. White-coat hypertension in adults. Blood Press Monit 2005; 10:301–305.
- Cicconetti P, Morelli S, De Serra C, et al. Left ventricular mass in dippers and nondippers with newly diagnosed hypertension. Angiology 2003; 54:661–669.
- Kario K, Pickering TG, Umeda Y, et al. Morning surge in blood pressure as a predictor of silent and clinical cerebrovascular disease in elderly hypertensives: a prospective study. Circulation 2003; 107:1401–1406.
- Katakam R, Townsend RR. Morning surges in blood pressure. J Clin Hypertens 2006; 8:450–451.
- Messerli FH. Osler’s maneuver, pseudohypertension, and true hypertension in the elderly. Am J Med 1986; 80:906–910.
- Belmin J, Visintin JM, Salvatore R, Sebban C, Moulias R. Osler’s maneuver: absence of usefulness for the detection of pseudohypertension in an elderly population. Am J Med 1995; 98:42–49.
- Messerli FH, Ventura HO, Amodeo C. Osler’s maneuver and pseudohypertension. N Engl J Med 1985; 312:1548–1551.
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- Wenger NK. Quality of life issues in hypertension: consequences of diagnosis and considerations in management. Am Heart J 1988; 116:628–632.
- McFadden CB, Townsend RR. Blood pressure measurement: common pitfalls and how to avoid them. Consultant 2003; 43:161–165.
- Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Circulation 2005; 111:697–716.
- Myers MG. Automated blood pressure measurement in routine clinical practice. Blood Press Monit 2006; 11:59–62.
- Mosenkis A, Townsend RR. Sitting on the evidence: what is the proper patient position for the office measurement of blood pressure? J Clin Hypertens (Greenwich) 2005; 7:365–366.
- Vasan RS, Beiser A, Seshadri S, et al. Residual lifetime risk for developing hypertension in middle-aged women and men: The Framingham Heart Study. JAMA 2002; 287:1003–1010.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 2004; 44:720–732.
- American Diabetes Association. Treatment of hypertension in adults with diabetes. Diabetes Care 2002; 25:199–201.
- Rosendorff C, Black HR, Cannon CP, et al. Treatment of hypertension in the prevention and management of ischemic heart disease: a scientific statement from the American Heart Association Council for High Blood Pressure Research and the Councils on Clinical Cardiology and Epidemiology and Prevention. Circulation 2007; 115:2761–2788.
- Papadakis JA, Mikhailidis DP, Vrentzos GE, Kalikaki A, Kazakou I, Ganotakis ES. Effect of antihypertensive treatment on plasma fibrinogen and serum HDL levels in patients with essential hypertension. Clin Appl Thromb Hemost 2005; 11:139–146.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet 2001; 358:1033–1041.
- Fierro-Carrion GA, Ram CV. Nonsteroidal anti-inflammatory drugs (NSAIDs) and blood pressure. Am J Cardiol 1997; 80:775–776.
- Wong TY, McIntosh R. Hypertensive retinopathy signs as risk indicators of cardiovascular morbidity and mortality. Br Med Bull 2005; 73–74:57–70.
- Forette F, Boller F. Hypertension and the risk of dementia in the elderly. Am J Med 1991; 90:14S–19S.
- Schrader J, Luders S, Kulschewski A, et al. Microalbuminuria and tubular proteinuria as risk predictors of cardiovascular morbidity and mortality in essential hypertension: final results of a prospective long-term study (MARPLE Study). J Hypertens 2006; 24:541–548.
- Luque M, de Rivas B, Alvarez B, Garcia G, Fernandez C, Martell N. Influence of target organ lesion detection (assessment of microalbuminuria and echocardiogram) in cardiovascular risk stratification and treatment of untreated hypertensive patients. J Hum Hypertens 2006; 20:187–192.
- Pontremoli R, Leoncini G, Viazzi F, et al. Role of microalbuminuria in the assessment of cardiovascular risk in essential hypertension. J Am Soc Nephrol 2005; 16 suppl 1:S39–S41.
- Erdmann E. Microalbuminuria as a marker of cardiovascular risk in patients with type 2 diabetes. Int J Cardiol 2006; 107:147–153.
- Bakris GL, Sowers JR. Microalbuminuria in diabetes: focus on cardiovascular and renal risk reduction. Curr Diab Rep 2002; 2:258–262.
- Gallay BJ, Ahmad S, Xu L, Toivola B, Davidson RC. Screening for primary aldosteronism without discontinuing hypertensive medications: plasma aldosteronerenin ratio. Am J Kidney Dis 2001; 37:699–705.
- Rossi GP, Bernini G, Caliumi C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006; 48:2293–2300.
- Onusko E. Diagnosing secondary hypertension. Am Fam Physician 2003; 67:67–74.
- Aurell M. Screening for secondary hypertension. Curr Hypertens Rep 1999; 1:461.
- Garovic VD, Kane GC, Schwartz GL. Renovascular hypertension: balancing the controversies in diagnosis and treatment. Cleve Clin J Med 2005; 72:1135–1137.
- Textor SC. Renovascular hypertension in 2007: where are we now? Curr Cardiol Rep 2007; 9:453–461.
- Pickering TG, Shimbo D, Haas D. Ambulatory blood-pressure monitoring. N Engl J Med 2006; 354:2368–2374.
- Angeli F, Verdecchia P, Gattobigio R, Sardone M, Reboldi G. White-coat hypertension in adults. Blood Press Monit 2005; 10:301–305.
- Cicconetti P, Morelli S, De Serra C, et al. Left ventricular mass in dippers and nondippers with newly diagnosed hypertension. Angiology 2003; 54:661–669.
- Kario K, Pickering TG, Umeda Y, et al. Morning surge in blood pressure as a predictor of silent and clinical cerebrovascular disease in elderly hypertensives: a prospective study. Circulation 2003; 107:1401–1406.
- Katakam R, Townsend RR. Morning surges in blood pressure. J Clin Hypertens 2006; 8:450–451.
- Messerli FH. Osler’s maneuver, pseudohypertension, and true hypertension in the elderly. Am J Med 1986; 80:906–910.
- Belmin J, Visintin JM, Salvatore R, Sebban C, Moulias R. Osler’s maneuver: absence of usefulness for the detection of pseudohypertension in an elderly population. Am J Med 1995; 98:42–49.
- Messerli FH, Ventura HO, Amodeo C. Osler’s maneuver and pseudohypertension. N Engl J Med 1985; 312:1548–1551.
KEY POINTS
- To confirm the diagnosis of hypertension, multiple readings should be taken at various times.
- Proper technique is important in measuring blood pressure, including using the correct cuff size, having the patient sit quietly for 5 minutes before taking the pressure, and supporting the arm at the level of the heart.
- If white-coat hypertension is suspected, one can consider ambulatory or home blood pressure measurements to confirm that the hypertension is sustained.
Calciphylaxis Is 'Akin to a Myocardial Infarction'
LAS VEGAS — An evolving understanding of the pathogenesis of calciphylaxis in hospitalized patients may lead to antithrombotic treatment strategies focused on vascular occlusion as well as dialysis- and parathyroid-specific interventions.
“Calciphylaxis is a therapeutic conundrum and also a nightmare,” said Dr. Mark D.P. Davis, professor of dermatology at the Mayo Clinic, Rochester, Minn.
“We urgently need better treatment and preventive strategies,” he stressed at a dermatology seminar sponsored by Skin Disease Education Foundation.
The condition's name, calciphylaxis, reflects an early belief that the introduction of a certain agent (likely during dialysis) induced calcification of vessels, a notion now disputed since the disease can occur in patients without renal insufficiency.
A more accurate name, first proposed by Dr. Patrick Dahl and his associates, is the vascular calcification-cutaneous necrosis syndrome (J. Am. Acad. Dermatol. 1995;33:53–8), which better characterizes calciphylaxis as “akin to a myocardial infarction,” Dr. Davis said. Calcifications in the walls of small arterioles supplying the skin are the first evidence of the disorder. The resultant clots trigger skin infarctions, just as a blockage of a vessel leads to an MI.
Treatment at Mayo focuses on vascular occlusion, along with management of hypercalcemia (with low-calcium dialysate and sodium thiosulfate in dialysis patients), hyperphosphatemia (with phosphate binding agents), hyperparathyroidism (with cinacalcet and bisphosphonates), and pain.
“It's very important to treat vascular occlusions and eliminate these luminal thromboses causing this cutaneous infarct,” he said. “One way to treat an existing clot is to use thrombolytic agents.”
Several Mayo Clinic patients have been treated with infused tissue plasminogen activator (tPA) at doses 1/10 of those used to treat an MI. Because of concern over bleeding, patients are admitted for the 2-week procedure. “We have had some success and are presently reviewing our experience with this approach,” Dr. Davis said.
Anticoagulant medications, including heparin, low-molecular-weight heparin, and warfarin, are also employed so that calciphylaxis patients don't clot more.
Hyperbaric oxygen, which enhances tissue oxygenation and induces vascular neogenesis (and may increase fibrinolytic activity within endothelial cells) also makes sense in the context of calciphylaxis as a disease of vascular calcification/cutaneous infarction.
Dr. M.R. (Pete) Hayden, a calciphylaxis researcher who has published several studies on sodium thiosulfate as a possible treatment, commented later that he is “looking forward excitedly to future papers” on the anticoagulant approach from Dr. Davis and Mayo researchers.
“Indeed, thrombolytic agents may be an important adjunctive intervention along with calcium-chelating agents and phosphate binding agents in appropriate patients because there are so many precipitating variables important to the development of calciphylaxis,” said Dr. Hayden, research professor of internal medicine in the division of endocrinology, diabetes, and metabolism at the University of Missouri, Camdenton.
Other interventions have not fared as well. A comprehensive review of 64 patients treated at the Mayo Clinic failed to find any survival benefit with parathyroidectomy, despite case studies and series that have suggested the surgery is beneficial (J. Am. Acad. Dermatol. 2007;57:365–6).
Debridement was associated with a 1-year survival rate of 62%, versus 27% survival rates in patients who failed to undergo the procedure. Surgical and mechanical debridement are difficult to perform in patients with this “excruciatingly painful” disease, so painless debridement using maggots and ultrasound is being utilized at Mayo, to good effect, Dr. Davis said.
A population-based study conducted in the Rochester area found an incidence of 4.5 cases per million people, per year. About 1% of patients with chronic renal failure and 4% of dialysis patients reportedly have the disease.
Renal insufficiency characterizes the “vast majority” of patients with calciphylaxis, but many patients seen at the Mayo Clinic have underlying liver disease and no history of kidney failure, Dr. Davis noted.
Other findings in patients (with or without renal insufficiency) include hyper- or hypoparathyroidism, calcium-phosphate product greater than 70, obesity (body mass index greater than 30), systemic corticosteroid use, vitamin D deficiency, bone disease, systemic inflammatory state, and malignancy, especially bone metastasis.
SDEF and this news organization are owned by Elsevier.
LAS VEGAS — An evolving understanding of the pathogenesis of calciphylaxis in hospitalized patients may lead to antithrombotic treatment strategies focused on vascular occlusion as well as dialysis- and parathyroid-specific interventions.
“Calciphylaxis is a therapeutic conundrum and also a nightmare,” said Dr. Mark D.P. Davis, professor of dermatology at the Mayo Clinic, Rochester, Minn.
“We urgently need better treatment and preventive strategies,” he stressed at a dermatology seminar sponsored by Skin Disease Education Foundation.
The condition's name, calciphylaxis, reflects an early belief that the introduction of a certain agent (likely during dialysis) induced calcification of vessels, a notion now disputed since the disease can occur in patients without renal insufficiency.
A more accurate name, first proposed by Dr. Patrick Dahl and his associates, is the vascular calcification-cutaneous necrosis syndrome (J. Am. Acad. Dermatol. 1995;33:53–8), which better characterizes calciphylaxis as “akin to a myocardial infarction,” Dr. Davis said. Calcifications in the walls of small arterioles supplying the skin are the first evidence of the disorder. The resultant clots trigger skin infarctions, just as a blockage of a vessel leads to an MI.
Treatment at Mayo focuses on vascular occlusion, along with management of hypercalcemia (with low-calcium dialysate and sodium thiosulfate in dialysis patients), hyperphosphatemia (with phosphate binding agents), hyperparathyroidism (with cinacalcet and bisphosphonates), and pain.
“It's very important to treat vascular occlusions and eliminate these luminal thromboses causing this cutaneous infarct,” he said. “One way to treat an existing clot is to use thrombolytic agents.”
Several Mayo Clinic patients have been treated with infused tissue plasminogen activator (tPA) at doses 1/10 of those used to treat an MI. Because of concern over bleeding, patients are admitted for the 2-week procedure. “We have had some success and are presently reviewing our experience with this approach,” Dr. Davis said.
Anticoagulant medications, including heparin, low-molecular-weight heparin, and warfarin, are also employed so that calciphylaxis patients don't clot more.
Hyperbaric oxygen, which enhances tissue oxygenation and induces vascular neogenesis (and may increase fibrinolytic activity within endothelial cells) also makes sense in the context of calciphylaxis as a disease of vascular calcification/cutaneous infarction.
Dr. M.R. (Pete) Hayden, a calciphylaxis researcher who has published several studies on sodium thiosulfate as a possible treatment, commented later that he is “looking forward excitedly to future papers” on the anticoagulant approach from Dr. Davis and Mayo researchers.
“Indeed, thrombolytic agents may be an important adjunctive intervention along with calcium-chelating agents and phosphate binding agents in appropriate patients because there are so many precipitating variables important to the development of calciphylaxis,” said Dr. Hayden, research professor of internal medicine in the division of endocrinology, diabetes, and metabolism at the University of Missouri, Camdenton.
Other interventions have not fared as well. A comprehensive review of 64 patients treated at the Mayo Clinic failed to find any survival benefit with parathyroidectomy, despite case studies and series that have suggested the surgery is beneficial (J. Am. Acad. Dermatol. 2007;57:365–6).
Debridement was associated with a 1-year survival rate of 62%, versus 27% survival rates in patients who failed to undergo the procedure. Surgical and mechanical debridement are difficult to perform in patients with this “excruciatingly painful” disease, so painless debridement using maggots and ultrasound is being utilized at Mayo, to good effect, Dr. Davis said.
A population-based study conducted in the Rochester area found an incidence of 4.5 cases per million people, per year. About 1% of patients with chronic renal failure and 4% of dialysis patients reportedly have the disease.
Renal insufficiency characterizes the “vast majority” of patients with calciphylaxis, but many patients seen at the Mayo Clinic have underlying liver disease and no history of kidney failure, Dr. Davis noted.
Other findings in patients (with or without renal insufficiency) include hyper- or hypoparathyroidism, calcium-phosphate product greater than 70, obesity (body mass index greater than 30), systemic corticosteroid use, vitamin D deficiency, bone disease, systemic inflammatory state, and malignancy, especially bone metastasis.
SDEF and this news organization are owned by Elsevier.
LAS VEGAS — An evolving understanding of the pathogenesis of calciphylaxis in hospitalized patients may lead to antithrombotic treatment strategies focused on vascular occlusion as well as dialysis- and parathyroid-specific interventions.
“Calciphylaxis is a therapeutic conundrum and also a nightmare,” said Dr. Mark D.P. Davis, professor of dermatology at the Mayo Clinic, Rochester, Minn.
“We urgently need better treatment and preventive strategies,” he stressed at a dermatology seminar sponsored by Skin Disease Education Foundation.
The condition's name, calciphylaxis, reflects an early belief that the introduction of a certain agent (likely during dialysis) induced calcification of vessels, a notion now disputed since the disease can occur in patients without renal insufficiency.
A more accurate name, first proposed by Dr. Patrick Dahl and his associates, is the vascular calcification-cutaneous necrosis syndrome (J. Am. Acad. Dermatol. 1995;33:53–8), which better characterizes calciphylaxis as “akin to a myocardial infarction,” Dr. Davis said. Calcifications in the walls of small arterioles supplying the skin are the first evidence of the disorder. The resultant clots trigger skin infarctions, just as a blockage of a vessel leads to an MI.
Treatment at Mayo focuses on vascular occlusion, along with management of hypercalcemia (with low-calcium dialysate and sodium thiosulfate in dialysis patients), hyperphosphatemia (with phosphate binding agents), hyperparathyroidism (with cinacalcet and bisphosphonates), and pain.
“It's very important to treat vascular occlusions and eliminate these luminal thromboses causing this cutaneous infarct,” he said. “One way to treat an existing clot is to use thrombolytic agents.”
Several Mayo Clinic patients have been treated with infused tissue plasminogen activator (tPA) at doses 1/10 of those used to treat an MI. Because of concern over bleeding, patients are admitted for the 2-week procedure. “We have had some success and are presently reviewing our experience with this approach,” Dr. Davis said.
Anticoagulant medications, including heparin, low-molecular-weight heparin, and warfarin, are also employed so that calciphylaxis patients don't clot more.
Hyperbaric oxygen, which enhances tissue oxygenation and induces vascular neogenesis (and may increase fibrinolytic activity within endothelial cells) also makes sense in the context of calciphylaxis as a disease of vascular calcification/cutaneous infarction.
Dr. M.R. (Pete) Hayden, a calciphylaxis researcher who has published several studies on sodium thiosulfate as a possible treatment, commented later that he is “looking forward excitedly to future papers” on the anticoagulant approach from Dr. Davis and Mayo researchers.
“Indeed, thrombolytic agents may be an important adjunctive intervention along with calcium-chelating agents and phosphate binding agents in appropriate patients because there are so many precipitating variables important to the development of calciphylaxis,” said Dr. Hayden, research professor of internal medicine in the division of endocrinology, diabetes, and metabolism at the University of Missouri, Camdenton.
Other interventions have not fared as well. A comprehensive review of 64 patients treated at the Mayo Clinic failed to find any survival benefit with parathyroidectomy, despite case studies and series that have suggested the surgery is beneficial (J. Am. Acad. Dermatol. 2007;57:365–6).
Debridement was associated with a 1-year survival rate of 62%, versus 27% survival rates in patients who failed to undergo the procedure. Surgical and mechanical debridement are difficult to perform in patients with this “excruciatingly painful” disease, so painless debridement using maggots and ultrasound is being utilized at Mayo, to good effect, Dr. Davis said.
A population-based study conducted in the Rochester area found an incidence of 4.5 cases per million people, per year. About 1% of patients with chronic renal failure and 4% of dialysis patients reportedly have the disease.
Renal insufficiency characterizes the “vast majority” of patients with calciphylaxis, but many patients seen at the Mayo Clinic have underlying liver disease and no history of kidney failure, Dr. Davis noted.
Other findings in patients (with or without renal insufficiency) include hyper- or hypoparathyroidism, calcium-phosphate product greater than 70, obesity (body mass index greater than 30), systemic corticosteroid use, vitamin D deficiency, bone disease, systemic inflammatory state, and malignancy, especially bone metastasis.
SDEF and this news organization are owned by Elsevier.
IgA nephropathy: Challenges and opportunities
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
Much progress has been made in the 40 years since immunoglobulin A (IgA) nephropathy was first described. We now have a reasonably complete understanding of the pathogenesis and mediation of this disease, but its etiology remains obscure and mysterious. New data on its epidemiology continue to emerge that will undoubtedly have clinical significance. We are beginning to perceive—but only dimly—the genetic predisposition to the disease.
Prognostication remains an imperfect science, but we are clearly making progress. The role of pathology in estimating prognosis in individual patients needs to be thoroughly reexamined, based on a uniformly agreed-upon classification scheme. Such work is currently in progress.
Therapy has certainly advanced, and we now have the rudiments of an evidence-based approach to management. However, much more needs to be done to refine these strategies so that they can be better matched to the characteristics of the patients, and there is a great need for novel therapeutic approaches and more information on multidrug regimens in selected patients. Many opportunities exist for improvement in the control of this common cause of chronic kidney disease, but we should not underestimate the challenges that present themselves in the field of IgA nephropathy in 2008 and beyond.
THE SCOPE OF THE PROBLEM
IgA nephropathy, also called Berger disease, is the most common form of primary glomerular disease in the developed world.1,2 Morphologically, it is characterized by diffuse deposition of IgA in the glomerular mesangium and by various degrees of damage of the glomerular capillary network seen on light microscopy.3,4 By some estimates, as many as 5% to 15% (averaging about 10%) of the general population may have IgA deposits in the glomerular mesangium, but only about 1 in 50 people with IgA deposits will actually have some abnormal clinical manifestation (principally recurring bouts of hematuria, with or without accompanying proteinuria) that brings them to the attention of a physician.5
Although not all patients with IgA nephropathy have progressive renal disease, IgA nephropathy is a significant contributor to the incidence of end-stage renal disease (ESRD) in many countries.1–4
DIAGNOSTIC AND PROGNOSTIC CHALLENGES
Since 1968, when IgA nephropathy was first described,6 great strides have been made in clarifying its epidemiology, its pathogenesis, the prognostic factors involved in its progression to ESRD, and its treatment. However, many gaps in our knowledge remain, particularly regarding its etiology, the genetic factors predisposing to it, its therapy, and the problem of recurrent disease in renal transplant recipients.
Can IgA nephropathy be diagnosed without a renal biopsy?
While renal biopsy and immunochemical analysis of renal tissue remain the gold standard for diagnosing IgA nephropathy, new sensitive and reasonably specific noninvasive tests are emerging and may provide another diagnostic approach. One of the most promising new tests is for abnormal circulating levels of abnormally glycosylated IgA subclass 1 (IgA1), which appears to be involved in the pathogenesis of the disease (see below).7 If noninvasive diagnostic techniques can be simplified and their accuracy validated across diverse populations, they offer great promise for use in epidemiologic and genetic studies, in which routine renal biopsy for diagnosis is impractical.
Signs and symptoms of IgA nephropathy are nonspecific
The most common clinical presentation of IgA nephropathy is recurring bouts of macroscopic hematuria, often but not invariably accompanied by proteinuria.2 Persistent asymptomatic hematuria without any detectable proteinuria (so-called isolated hematuria) affects a minority of patients. The red cells in the urine are typically dysmorphic (altered in size and shape compared with normal red cells), as they are in many other glomerulonephritic diseases.
Because low-grade fever and pain in the loins may accompany these bouts of hematuria, the disorder is often initially mistaken for urinary tract infection or urolithiasis. Careful microscopic examination of the urinary sediment for the characteristic dysmorphic erythrocytes that indicate a glomerular disease often provides the crucial clue that a glomerular disorder is the cause of the hematuria.8
However, a somewhat similar presentation may also be seen in thin basement membrane nephropathy, Alport syndrome (hereditary nephritis), and membranoproliferative glomerulonephritis,2 although these disorders can be readily distinguished from IgA nephropathy on examination of renal biopsy material under light, immunofluorescence, and electron microscopy. In addition, serum complement levels are typically reduced in membranoproliferative glomerulonephritis, and a family history of nephritis (without father-to-son transmission), often with deafness, can be obtained in the X-linked form of Alport syndrome. IgA nephropathy can be reliably distinguished from thin basement membrane nephropathy only by renal biopsy and electron microscopy.
Can we better predict which patients with IgA nephropathy will develop renal failure?
Although the rate of progression is very slow, and in only about 50% (or less) of patients does IgA nephropathy progress to ESRD within 25 years of diagnosis, the risk varies considerably among populations.9 Spontaneous clinical remissions are relatively uncommon in adults but much more common among children.
Several factors, if present at the time of discovery or developing within a relatively short time thereafter (usually within 6 months to 1 year), appear to predict a progressive course and, eventually, ESRD.9,10 We need to characterize and validate these risk factors in detail to be able to design and carry out appropriately powered, randomized, controlled clinical trials of treatment.
Unfortunately, cumulatively, the risk factors identified so far explain less than 50% of the variation in observed outcome of IgA nephropathy. Many of the risk factors identified so far are primarily indicators of the extent of disease at a particular time, and it is therefore not surprising that they would have some ability to predict the later behavior of the disease.
Clinical and pathologic risk factors in IgA nephropathy
Although imperfect, the major risk factors auguring a poor prognosis are:
- Proteinuria (> 500 mg/day) that persists for more than 6 months
- Elevated serum creatinine at diagnosis
- Microscopic hematuria that persists for more than 6 months
- Poorly controlled hypertension
- Extensive glomerulosclerosis or interstitial fibrosis or both on renal biopsy.7,10
Extensive crescentic disease also confers a worse short-term prognosis, often accompanied by a rapidly progressive loss of renal function.
Are clinical risk factors more useful than pathologic risk factors in IgA nephropathy?
Of importance, clinical factors, such as persistent proteinuria or declining renal function on follow-up appear to have greater predictive power than pathologic factors for long-term outcome.9–12 Clinical factors, such as decreasing estimated glomerular filtration rate (GFR) after short-term follow-up, persistent moderate to marked proteinuria (500–1,000 mg/day, or more), hyperuricemia, hyperlipidemia, concomitant obesity, poorly controlled hypertension, absence of treatment with angiotensin II inhibitors, and, possibly, persistent micro-hematuria are the most consistent factors independently associated with a poor prognosis in multivariate analysis. Pathologic changes noted in the original diagnostic renal biopsy do not consistently add greatly to the precision of prognosis beyond the analysis of these clinical and laboratory factors.11
A detailed and uniform immunologic and morphologic approach to classifying the pathology of IgA nephropathy may yet uncover some new and very useful prognostic factors, independent of those generated by simple clinical assessment. Efforts are under way, and such a development would greatly improve the accuracy and precision of outcome prediction and reduce the amount of unexplained variation in prognosis observed in groups of patients with IgA nephropathy.
At present, the heterogeneity of participants in clinical trials of therapy, the tendency for the disease to progress slowly, and the variation in prognosis due to unexplained factors pose major challenges in designing and carrying out randomized controlled trials of therapy in IgA nephropathy. If we can find new risk factors that can predict progressive disease earlier, the knowledge will help us in designing future clinical trials, which will be vital if progress is to be made towards controlling IgA nephropathy.
Prognosis in individual patients vs populations with IgA nephropathy
At present, we need a way to determine the prognosis more precisely in individual patients rather than in groups of patients. After all, physicians are called upon to determine the likely outcome in single patients, not in a population. Several prediction formulas have been devised, most of them based on relatively simple clinical factors present at discovery or short-term follow-up.12,13
Conventional pathologic observations have limited utility in such individualized prognostic formulations.12 This is not to say that renal biopsy only offers diagnostic utility and has little if any value as a prognostic tool. However, the challenge is to enhance the prognostic usefulness of renal biopsy by refining the examination of the tissue specimens using modern approaches and to conduct the appropriate correlative studies to confirm the value of new pathologic criteria in prognostication, independent of clinical features alone.
For example, the risk of ESRD is greater if the patient has very extensive (> 50%) crescentic glomerular involvement with a rapidly progressive glomerulonephritic evolution. The risk is less if there are minimal glomerular changes with nephrotic-range proteinuria. Extensive interstitial fibrosis and glomerulosclerosis in the original “diagnostic” renal biopsy merely highlight the existence of prior progressive disease that is likely to continue. The significance of persistent focal necrotizing glomerular lesions (capillaritis) in IgA nephropathy, often associated with persistent microhematuria, is not entirely clear and needs to be specifically explored, especially as it pertains to the need for immunosuppressive therapy added to treatment for hypertension, proteinuria, or both with inhibitors of the renin-angiotensin system (see below).
At present, the most powerful prognostic factor in IgA nephropathy is moderate to severe proteinuria that persists for 6 months or longer.9,10,12 The relationship between the level of proteinuria and the outcome is continuous, ie, the greater the proteinuria, the worse the prognosis. Compared with some other primary glomerular diseases (such as membranous nephropathy or focal and segmental glomerulosclerosis), progressive disease in IgA nephropathy is associated with lower levels of persistent proteinuria (usually 500 mg to 3 g/day).
The estimated GFR at the time IgA nephropathy is discovered is a rather weak independent predictor of outcome (up to a point; see below). Many patients have stable (but reduced) renal function in the long term, especially if they receive angiotensin II inhibitor therapy and can keep their systolic blood pressure between 110 and 120 mm Hg.
How can IgA nephropathy be diagnosed and treated before the ‘point of no return’?
For patients at risk of developing ESRD, the two most critical goals of treatment are to:
- Control blood pressure rigorously, preferably with an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin II receptor antagonist (ARB), or both, and
- Reduce proteinuria to less than 500 mg/day.
If these two goals can be met without undue side effects and if the patient remains compliant in the long term, many patients can avoid ESRD. Patients who cannot achieve these goals despite vigorous attempts become candidates for adjunctive therapy, such as pulse intravenous methylprednisolone (Solu-Medrol) combined with oral prednisone, or in some cases a cytotoxic drug combined with prednisone. Small randomized controlled trials suggest these adjunctive treatments are effective and safe.
Unfortunately, IgA nephropathy can progress silently, and many patients do not receive the diagnosis until late in its course. In such patients, the disease may relentlessly progress even with optimal therapy. The “point of no return” appears to be an estimated GFR of about 30 mL/min/1.73 m2 (stage 4 chronic kidney disease).14
These observations underscore the need for early diagnosis and treatment based on factors that accurately predict an unfavorable outcome. Finding these factors will not be easy, because it will require detailed observation of homogeneous groups of patients over prolonged periods of time. New findings show great promise for identifying patients earlier in the course of disease who are more or less likely to progress to ESRD. The challenge is to translate these findings into rational, safe, and effective therapies applicable across a broad spectrum of disease.
OPPORTUNITIES: GENETICS, PROTEOMICS, NEW TESTS AND TREATMENTS
Genetic studies may lead to novel treatments for IgA nephropathy
Susceptibility to IgA nephropathy has a genetic component to varying degrees, depending on geography and the existence of “founder effects.” Familial forms of IgA nephropathy are more common in northern Italy and in eastern Kentucky. The familial cases may derive from a mutation of a specific gene occurring in a founder many hundreds of years ago. Several genetic loci are strongly associated with IgA nephropathy (usually as an autosomal-dominant trait with highly variable penetrance).15 Familial IgA nephropathy is most likely genetically heterogeneous, and many cases of IgA nephropathy that are believed to be sporadic may actually have a less apparent genetic basis, with skipped generations, lanthanic (covert) disease, and incomplete penetrance.
At present, genetic testing based on genomic or transcriptosomic analysis does not appear to have much diagnostic value except in clearly familial cases, because many loci are involved. Many asymptomatic people have mesangial IgA deposits that could be detected by renal biopsy but not by genetic analysis, and this inability is a major obstacle for genetic susceptibility studies. Indeed, most current genetic studies actually examine susceptibility to the clinical expression of disease rather than susceptibility to the mesangial IgA deposition that underlies the disease.5
The opportunity that lies ahead in genetic testing of IgA nephropathy (including haplotype analysis) appears to be primarily in the elucidation of potential pathogenetic pathways and in the refinement of prognosis and the definition of treatment responsiveness (pharmacogenomics).
If a gene (or group of genes) can be identified that is strongly and consistently associated with IgA nephropathy across diverse populations, its protein product isolated and characterized, and its role in pathogenesis elucidated, then a new era in targeted therapy of IgA nephropathy will be unleashed, much in the same way as the identification of tyrosine phosphatases played a role in the design of targeted therapy in chronic myelogenous leukemia. Early progress is being made in this area, but many obstacles lie in the way.
Proteomics may prove useful in diagnosis and prognosis of IgA nephropathy
Proteomics—the characterization and analysis of the patient’s entire complement of serum and urinary proteins—is a new, exciting, and largely unexplored area in IgA nephropathy. Preliminary studies have shown that this technique may provide a novel noninvasive means of diagnosing IgA nephropathy, and it may have additional value as a prognostic tool.16
Much work needs to be done to standardize how specimens are collected, stored, and shipped and to verify the precision and accuracy of proteomics in diverse populations of patients with IgA nephropathy, patients with other glomerular diseases, and normal subjects to ascertain this technique’s false-negative and false-positive rates.
IgA1 testing may help detect IgA nephropathy early in its course
Abnormally undergalactosylated and oversialyted epitopes at the hinge region of the IgA1 molecule play a critical role in the pathogenesis of sporadic IgA nephropathy.17 This discovery provides a great opportunity for profiling patients suspected of having IgA nephropathy on the basis of sensitive determination of the serum level of these abnormal IgA1 molecules.7
It may be that pathogenic IgA1 molecules (and autoantibodies to them) arise many months or even years before the onset of clinical manifestations of overt IgA nephropathy, similar to the situation known to occur in systemic lupus erythematosus. It is also possible that an abnormality of the disposal of immune complexes created by the interaction of autoantibodies with the abnormally glycosylated IgA1 creates the opportunity for preferential glomerular mesangial deposition of polymeric IgA.
Clearly, the greatest opportunity lies with understanding the fundamental abnormality leading to defective O-linked galactosylation of the serine/threonine residues at the hinge region of IgA1 in IgA nephropathy. In addition, it would be very useful to know if this is a generalized and acquired abnormality or whether it is focal in distribution (eg, in the tonsils, bone marrow, or lymphoid tissue in the gut).
Knowledge of secondary mediators may also lead to new treatments for IgA nephropathy
Detailed knowledge of the participation of specific cell types and the “cytokine milieu” (eg, interleukin 4, interferon) in directing the abnormality toward defective glycosylation would also be very important in designing new approaches to diagnosis and therapy.
A better understanding is slowly emerging of the pathways by which pathogenic immune complexes containing IgA are deposited and cleared, and of the secondary mediator systems evoked by their formation and tissue localization. Interference with these secondary mediator processes, such as alternative or mannose-dependent complement activation, platelet-derived growth factor or transforming growth factor stimulation, also offers a new approach to therapy.
We lack a suitable animal model of IgA nephropathy that mimics all aspects of the human condition, which has impeded progress in this area. A fully humanized mouse model of disease would be a welcome addition to the investigative toolkit.
Prognostic biopsy analysis may be improved in IgA nephropathy
As discussed above, the science of prognostication and stratification of patients with IgA nephropathy into those at high or low risk of ESRD has clearly advanced but is still quite incomplete, especially with respect to individual patients.
Great opportunities lie in refining the value of renal biopsy in prognostication. Although the “snapshot” nature and potential sampling errors intrinsic to diagnostic renal biopsy cannot easily be overcome, at least not without performing multiple and repeated renal biopsies (a very impractical approach to prognostication), refinements in the laboratory seem to offer numerous opportunities for advancement. Much better clinicopathological correlations, especially with respect to outcomes, among well-characterized patients with IgA nephropathy are greatly needed. New nonconventional markers of progression, such as “tubulitis,” deposition of fibroblast-specific proteins, and the proteome of the deposited immunoglobulins and complement show much promise.18
Immunosuppressive therapy could be added to ACE inhibitors or ARBs in IgA nephropathy
The management of IgA nephropathy has clearly advanced over the last several decades, largely as the result of randomized clinical trials.3,19 However, these trials had serious limitations: the numbers of patients were relatively small, follow-up was relatively short, and the findings may not apply to the IgA nephropathy population at large or to specific patients having features that diverge from those in the patients enrolled in the studies.
The value of initial therapy with an ACE inhibitor, an ARB, or both in combination appears well established. However, details of dosage, duration of therapy, and the relative values of monotherapy and combined therapy remain uncertain.
Many opportunities for combining angiotensin II inhibition and immunosuppressive therapy are being explored. By and large, all current therapies are empiric and their long-term effects relatively uncertain, owing to small study size and short duration.
Oral and parenteral glucocorticoids,20 combined regimens of cyclophosphamide (Cytoxan) and azathioprine (Imuran),21 omega-3 fatty acids,22 and anticoagulants and anti-thrombotics3 each have their advocates and their specified target populations.
Tonsillectomy as a treatment has been particularly controversial. While no controlled studies have been performed yet, observational studies (most of them conducted in some prefectures in Japan) have suggested a higher rate of clinical remission with tonsillectomy than with steroid treatment alone.5 However, long-term observations have not shown any consistent effect of tonsillectomy on progression to ESRD.
We hope that a better understanding of the fundamental mechanisms of disease and its mediation will provide an impetus for development of more rational targeted therapy. Evaluating potentially promising targeted therapies will be very difficult. Evaluation of safety and efficacy with long-term use will be a key requirement for a successful novel therapeutic agent.
FOR NOW, AN EMPIRIC APPROACH TO IGA NEPHROPATHY
Start with an angiotensin II inhibitor
The current body of evidence for choosing a particular therapeutic approach for a given patient with IgA nephropathy cannot be regarded as definitive, owing to limitations in the quality and strength of the trials serving as the basis of the evidence. Nonetheless, patients with IgA nephropathy and abnormal protein excretion (> 500 mg/day) should probably always be given angiotensin II inhibitor therapy (an ACE inhibitor, an ARB, or both) if they have no contraindications to it such as a hypersensitivity reaction or pregnancy, as a base for future monitoring and adjuvant therapy.
A response, tentatively defined as a 30% to 50% decline in proteinuria from baseline levels or a decrease to less than 500 mg/day, would be a reason to continue this conservative approach. Lack of a response after several months of observation at maximal tolerated dosage (plus salt restriction or a diuretic) would be a reason for considering adjuvant therapy.
If the patient does not respond to an ACE inhibitor or ARB and his or her estimated GFR is over 70 mL/min/1.73 m2, a trial of oral and parenteral glucocorticoids might be undertaken, as suggested by Pozzi and coworkers.20
On the other hand, if the estimated GFR is in the range of 30 to 70 mL/min/1.73 m2 and declining at a rate that predicts that ESRD will develop in less than 5 to 7 years, this would be a possible indication for low-dose oral cyclo-phosphamide and then azathioprine, as suggested by Ballardie and Roberts.21 Omega-3 fatty acids (Omacor) could also be considered as add-on therapy, particularly for patients with very heavy proteinuria (> 3.0 g/d) and reduced estimated GFR.22
Patients with an estimated GFR of less than 30 mL/min/1.73 m2 and chronic (irreversible) changes on renal biopsy—the point of no return—probably will not respond to any therapy other than an ACE inhibitor, an ARB, or both.
The role of more aggressive immunosuppression
At present, the evidence for using mycophenolate mofetil (CellCept) or calcineurin inhibitors (such as cyclosporin or tacrolimus) is fragmentary or contradictory.3,19,23 Similarly, the benefits of long-term azathioprine therapy are based on observational data only and so it cannot be recommended as evidence-based.24 Opportunities exist for combined therapy (eg, an ACE inhibitor or an ARB or both, combined with omega-3 fatty acids and azathioprine or mycophenolate mofetil), but at present, controlled trials are lacking. Crescentic disease and rapidly progressive glomerulonephritis should probably be treated with combined cyclophosphamide and parental and oral corticosteroids, based on observational data. Patients with IgA nephropathy and minimal change disease with nephrotic syndrome should be treated with oral steroids, but the only data available are observational. Low-protein diets could be tried in the presence of slowly progressive renal disease with estimated GFR less than 30 mL/min/1.73 m2, but there are no controlled trials demonstrating efficacy for this approach in IgA neph-ropathy.
Renal transplantation is very successful
Renal transplantation is a very suitable alternative for patients with IgA nephropathy that progresses to ESRD. Overall success rates are as good or better than those in other primary glomerular diseases. Unfortunately, the disease recurs in the majority of renal grafts and may in some cases lead to loss of the graft.25,26 We need much more information on the factors that predict such recurrences and their undesirable effects on transplantation outcomes.
MUCH WORK TO BE DONE
Much work needs to be done in the field of therapeutics in IgA nephropathy. Much of this effort will hinge on the interests of the pharmaceutical industry in IgA nephropathy as a potential therapeutic market. At present, the prospects for the development of a safe and effective novel therapy for IgA nephropathy (eg, approvable by the US Food and Drug Administration) do not appear great, but this may be overly pessimistic. The nature of the disease mandates long-term observation, agents that are very safe (with low rates of ESRD, death, and transplantation), and dependency on surrogate markers of efficacy. Therefore, designing and executing studies will not be easy.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
- Tomino Y. IgA nephropathy today. Contrib Nephrol 2007; 157:1–255.
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Quart J Med 1987; 245:709–727.
- Lee G, Glassock RJ. Immunoglobulin A nephropathy. In:Ponticelli C, Glassock R, editors. Treatment of Primary Glomerulonephritis. Oxford: Oxford Medical Publication, 1997:187–217.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med 2002; 347:738–748.
- Glassock RJ. Concluding remarks. IgA nephropathy today. Contrib Nephrol 2002; 157:169–173.
- Berger J, Hinglais N. Les dépots intercapillaries d’IgA-IgG. J Urol Nephrol (Paris) 1968; 74:694–700.
- Moldoveanu Z, Wyatt RJ, Lee JY, et al. Patients with IgA nephropa- levels. Kidney Int thy have increased serum galactose deficient IgA1. 2002; 71:1148–1154.
- Kincaid-Smith P, Fairley K. The investigation of hematuria. Semin Nephrol 2005; 25:127–135.
- Coppo R, D’Amico G. Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503–512.
- Donadio JV, Bergstralh EJ, Grande JP, Rademcher DM. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant 2002; 17:1197–1203.
- Cook T. Interpretation of renal biopsies in IgA nephropathy. Contrib Nephrol 2007; 157:44–49.
- Bartosik LP, Lajole G, Sugar L, Cattran D. Predicting progression in IgA nephropathy. Am J Kidney Dis 2001; 58:551–553.
- Rauta V, Finne P, Fagerudd J, et al. Factors associated with progression of IgA nephropathy are related to renal function—a model for estimating risk of progression in mild disease. Clin Nephrol 2002; 58:85–94.
- Komatsu H, Fujimoto S, Sato Y, et al. “Point of no return (PNR)” in progressive IgA nephropathy: significance of blood pressure and proteinuria management up to PNR”. J Nephrol 2005; 18:690–695.
- Schena FP, Cerullo G, Torres DD, et al European IgA Nephropathy Consortium. Searching for IgA nephropathy candidate genes: genetic studies combined with high throughput innovative investigations. Contrib Nephrol 2007; 157:80–89.
- Haubitz M, Wittke S, Weissinger EM, et al. Urine protein patterns can serve as a diagnostic tools in patients with IgA nephropathy. Kidney Int 2005; 67:2313–2320.
- Barratt J, Feehally J, Smith AC. The pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197–217.
- Nishitani Y, Iwano M, Yamaguchi Y, et al. Fibroblast-specific protein 1 is a specific prognostic marker for renal survival in patients with IgAN. Kidney Int 2005; 68:1078–1085.
- Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int 2006; 69:1934–1938.
- Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term follow-up of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–163.
- Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–148.
- Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acid in the treatment of IgA nephropathy. Semin Nephrol 2004; 24:225–243.
- Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int 2004; 65:1842–1849.
- Goumenous DS, Davlouros P, El Nahas AM, et al. Prednis-olone and azathioprine in IgA nephropathy—a ten year follow-up study. Nephron Clin Pract 2003; 93:c58–c68.
- Soler MG, Mir M, Rodriguez E, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc 2005; 37:3705–3709.
- Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol 2004; 24:287–291.
KEY POINTS
- IgA nephropathy tends to progress slowly, and in only about half of patients does it progress to end-stage renal disease within 25 years.
- At present, the factors that predict an accelerated course and progression to end-stage renal disease are persistent proteinuria, elevated serum creatinine at diagnosis, persistent microscopic hematuria, poorly controlled hypertension, and extensive glomerulosclerosis or interstitial fibrosis, or both, on renal biopsy.
- Needed are better diagnostic and prognostic tests and therapies that address the mechanism of the disease.
- The value of treatment with an angiotensin-converting enzyme inhibitor, an angiotensin receptor blocker, or both is well established. If protein excretion does not decrease with this therapy, one can consider adding immunosuppressive therapy in selected patients, but this strategy is still empiric and unproven.
Man, 48, With Excruciating Leg Pain
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
A 48-year-old black man, on hemodialysis since August 2002, presented to his primary care provider (PCP) in July 2006 with excruciating leg pain. According to the patient, the leg pain had worsened during the previous six months and was so severe that he was barely able to walk without pain. He was a full-time night security guard and reported walking three to five miles each night.
The man was undergoing hemodialysis three times per week, necessitated by nephritic range proteinuria. He had a questionable history of diabetes but a known diagnosis of hypertension. Definitive diagnosis through kidney biopsy was not obtained because of the associated risk, the patient's obesity, and his aversion to the procedure.
The patient had recently been hospitalized with shortness of breath and fluid overload. Intensive dialysis allowed a significant drop in his dialysis target weight. He was readmitted a few days later with chills, fever, cough, and shortness of breath. He was diagnosed with bilateral pulmonary emboli. The patient said his hypercoagulation work-up was negative, but he was started on warfarin before discharge.
On current presentation, he had swollen, tender legs and multiple excoriations over the calves, explained by the patient's admitted scratching. His skin was shiny and tight. He was still taking warfarin, with an international normalized ratio of 2.1. The patient denied shortness of breath, pruritus (any more than expected with renal disease), or increased fluid.
In addition to warfarin, he was taking esomeprazole 40 mg/d, extended-release metoprolol 25 mg bid, cinacalcet 90 mg/d, sevelamer 4,000 mg and lanthanum 5,000 mg before every meal, mometasone furoate as needed, hydroxyzine 25 mg every four hours as needed, miconazole powder applied to the feet as needed, and a daily prescription multivitamin complex.
Laboratory tests included normal findings (for a dialysis patient) on the complete blood count; blood urea nitrogen, 101 mg/dL (reference range, 7 to 20 mg/dL); serum creatinine, 16.6 mg/dL (0.8 to 1.4 mg/dL); Kt/V (a measure of adequacy of dialysis), 1.37 (acceptable); calcium, 9.6 mg/dL (8.2 to 10.2 mg/dL); serum phosphorus, 5.6 mg/dL (2.4 to 4.1 mg/dL); intact parathyroid hormone, 359 ng/L (10 to 65 ng/L).
The patient's PCP prescribed oxycodone for the pain and referred him to the vascular clinic for evaluation of his legs. A lower leg duplex scan with ankle/brachial indices performed on July 18 showed significant bilateral peripheral vascular disease. Subsequent magnetic resonance angiography (MRA) showed a questionable adrenal gland mass. Abdominal CT with and without contrast yielded negative results for the adrenal mass but showed a cyst in the right kidney. Although cysts are commonly found in dialysis patients, the vascular surgeon elected to evaluate the cyst with an MRI with gadolinium; the mass was found to be hemorrhagic.
Further vascular work-up continued, including MRI with gadolinium on September 26, 2006, which revealed two-vessel runoff in the right foot and three-vessel runoff in the left foot. According to the vascular consult, there was no area to bypass. The patient was sent back to his PCP. At this point, he was taking oxycodone four times per day and continuing to work full-time as a night security guard.
The patient was then sent to neurology for evaluation. By this time, the severity of his leg pain had increased 90%, with worsening swelling and persistent shininess (see figure). The neurologist was unable to obtain electromyograms due to the severity of the patient's pain and lower extremity swelling. No definitive diagnosis could be made.
About one year later, the man's attending nephrology group received copies of the work-up that the PCP sent to the dialysis center. It was apparent that neither the patient's PCP nor the vascular, radiology, or neurology consultants had seen the FDA warning released in June 20061 regarding the use of gadolinium in patients with renal disease. What had started out as a peripheral neuropathy (either renal or diabetic in etiology) was now a full-blown case of nephrogenic systemic fibrosis (NSF).
Open biopsy performed on October 29, 2007, confirmed the presence of gadolinium in the patient's epidermis. He became the first documented case of NSF in the Washington, DC area.
Discussion
In the late 1990s, several reports of an unknown sclerosing dermopathy in patients with chronic kidney disease began to emerge. In 2000, the new entity was named nephrogenic systemic fibrosis, with a disease course demonstrating systemic involvement that affected multiple organ systems and often resulted in severe joint limitations. A Web-based reporting system for this newly described disease, created by Shawn Cowper, MD, of Yale University,2 made it possible to investigate associated epidemiologic factors.
Neither gender, race, nor age appeared relevant. However, all patients had renal disease—acute, chronic, or transient—and more than 90% of patients were dialysis dependent. Factors since recognized to confirm a diagnosis of NSF are severe renal impairment (ie, glomerular filtration rate [GFR] < 30 mL/min/1.73 m2),3 CD34+ dendritic cells found on deep biopsy,4 and the following clinical manifestations:
• Skin. Burning or itching, reddened or darkened patches; possible skin swelling, hardening, and/or tightening.
• Eyes. Yellow raised spots in the whites of the eyes.
• Bones, joints, muscles. Joint stiffness; limited range of motion in the arms, hands, legs, or feet; pain deep in the hip bone or ribs; and/or muscle weakness.3
Theories abounded on the cause of NSF. While the presence of renal disease is a requirement, dialysis did not seem to be.5 Ten percent of NSF cases are patients who have never been dialyzed, and thousands of dialysis patients never develop NSF. Neither was any temporal correlation to dialysis found: While some patients developed NSF soon after starting dialysis, many had been on dialysis for years before NSF occurred. No association was found between NSF and the type of dialysis (inpatient, outpatient, hemodialysis, or peritoneal dialysis), the filter, manufacturer, dialysate, technique, or dialysis unit.2
Authors of a retrospective study involving two large tissue repositories looked for cases of NSF before 1997, but none were found.6 If dialysis was not causing NSF, and the disease did not appear to have existed before 1997, what renal toxin had been introduced in the 1990s to explain it?
One early suspicion involved erythropoietin (EPO), used to treat anemia in patients with kidney disease. Skin changes had been reported in some patients after initiation of treatment with EPO, and the NSF patients received a significantly higher mean dose of EPO than controls received.7
Ninety percent of patients with NSF had fistula reconstruction or dialysis catheter placement, but these are common in renal disease patients.8 Forty-eight percent of patients had had liver or kidney transplants, and 12% had hypercoagulable states. Most patients with NSF had never received ACE inhibitors. Were the protective antifibrogenic properties of these agents missing?
Mystery Solved
In a triumph for the Internet and its capacity to disseminate information around the world, a breakthrough came in 2006 from a small town in Austria. Grobner9 described nine patients who had received gadodiamide (Omniscan™)–enhanced MRA, five of whom developed NSF. Upon release of this report, researchers reexamined the original cases and detected a clear correlation between gadolinium and NSF. Because the contrast dose given for MRA can be as much as three times that required for routine MRI, the absence of NSF cases before 1997 suddenly made sense.
In May 2006, researchers for the Danish Medicines Agency reported 13 cases of NSF in patients injected with gadodiamide.10 Within months, 28 biopsy-proven cases were reported in St. Louis, six in Texas, and 13 at the University of Wisconsin—all involving patients exposed to gadolinium.11-13 It was apparent that NSF was iatrogenic and could be controlled.
What We Have Learned Since
In subsequent research, it has been found that more than 90% of reported cases of NSF occurred following exposure to gadodiamide—although gadodiamide accounts for only 15% of all gadolinium injections worldwide,14 and this number is decreasing as more cases are reported. The correlation between gadodiamide and NSF is so strong that its manufacturer, GE Healthcare, sent practitioners a letter in June 2006 warning of NSF as an adverse effect of gadolinium exposure.15 Two days later, the FDA issued an advisory on gadolinium-enhanced imaging procedures, recommending prompt hemodialysis after gadolinium exposure and reminding radiologists and nephrologists that gadolinium is not FDA approved for MRA.1
Although the 44% incidence rate of NSF reported by Grobner9 has never been replicated, a retrospective review of all known NSF cases affirmed that more than 90% of patients had been exposed to gadolinium.14 Two 2007 reports published in the Journal of the American Academy of Dermatology demonstrated that gadolinium was detectable in the tissues of patients with NSF.16,17
In Europe, in response to the May 2006 report from the Danish Medicines Agency,10 the European Society of Urogenital Radiology revised its guidelines with a directive that gadodiamide not be administered in any patients who had reduced kidney function or were undergoing dialysis.18 Shortly thereafter, the European Committee for Medicinal Products for Human Use issued a contraindication for gadodiamide use in patients with severe renal impairment and advised that these patients not be given gadolinium unless there was no other choice.19 A contraindication was also issued for gadodiamide use in patients with previous or anticipated liver transplantation.
The American College of Radiology guidelines published in 200720 stated that patients with any level of renal disease should not receive gadodiamide.
In March 2007, GE Healthcare published a paper on NSF, reiterating the safety of gadodiamide while acknowledging that 120 more cases had been reported to them ("usually associated with exposure at high doses").21 The FDA upholds an alert regarding use of all gadolinium-based contrast agents for patients with acute or chronic severe renal insufficiency,3 while stopping short of a ban on gadodiamide in such patients.
How Common Is NSF?
In a 2007 study conducted at the University of Wisconsin, Sadowski et al13 reported 13 cases of gadolinium-induced NSF, 11 involving patients with a GFR below 30 mL/min/1.73 m2 but two with a GFR between 30 and 60 mL/min/1.73 m2 (ie, with renal insufficiency, although the authors noted that renal insufficiency was acute in these two patients). The incidence of NSF was 4.6% among hospitalized patients with a GFR be-low 60 mL/min/1.73 m2 who underwent gadolinium-enhanced MRI at the university hospital's radiology department. A reexamination of the charts of the patients with a GFR between 30 and 60 mL/min/1.73 m2 revealed that these patients had levels below 30 mL/min/1.73 m2 when their gadolinium exposure took place.
In an outpatient population–based calculation performed by Deo et al,22 a 2.4% chance of NSF was determined for each gadolinium exposure. Incidence of NSF was calculated at 4.3 cases per 1,000 patient-years in this population, making NSF as common as contrast-induced nephropathy. Nearly 5% of patients with NSF have an exceedingly rapid and fulminant disease course that may result in death. NSF, of itself, is not a cause of death but may contribute to death by restricting effective ventilation or by restricting mobility to the point of causing an accidental fall that may be further exacerbated by fractures and clotting complications. NSF survivors may experience disabling systemic symptoms. Full recovery occurs only in patients who recover renal function, either naturally or by kidney transplantation.4
Why Is NSF More Common With Gadodiamide?
As of June 2008, five gadolinium-based contrast agents were FDA approved for use with MRI (none with MRA)3: gadobenate (MultiHance®), gadodiamide (Omniscan), gadopentetate (Magnevist®), gadoteridol (ProHance®), and gadoversetamide (Opti-MARK®). More than 90% of NSF cases are associated with gadodiamide. Because this agent is the least stable thermodynamically, it may be more likely than the others to transmetallate.14 All gadolinium chelates are excreted by the kidney, and the decreased renal clearances associated with renal impairment may expose patients to prolonged gadolinium transmetallation, allowing the agent to accumulate in bone and other tissue.
Gadoterate (Dotarem®), a cyclic gadolinium-based agent that is available in Europe but not the US, is considered more stable than other agents. It has been suggested that such agents may be safer choices for patients with decreased renal function.14,19
Strategies to Prevent NSF
In the US and Europe, only a physician who has consulted with a radiologist can write an order for gadolinium use in a patient with a GFR below 30 mL/min/1.73 m2.18,20 European guidelines do not allow use of gadodiamide in such patients.
Although the actual population-based occurrence of NSF is low, the nature of the disease calls for an effort to limit vulnerable patients' exposure to gadolinium (see box). Outside of withholding imaging procedures, the only currently known strategies to reduce the incidence of NSF are to use a more stable, nonchelating gadolinium14 and to remove the gadolinium as soon as possible.3,24
It has been recommended that patients with renal disease who are presently undergoing dialysis be dialyzed within two to three hours of gadolinium exposure, then again within 24 and 48 hours, provided it is clinically safe.20,24 This has been shown to remove 99% of the gadolinium.23
Since peritoneal dialysis clears gadolinium poorly, hemodialysis is recommended for peritoneal dialysis patients after gadolinium exposure, following the regimen outlined above.20
No consensus has been reached regarding the patient with a GFR between 30 and 60 mL/min/1.73 m2, nor for the patient with a lower GFR and no access for dialysis to be administered. Placement of a catheter for two days' dialysis incurs both surgical and renal risks for these patients.8
Patient Outcome
The only known cure for NSF is kidney transplantation, which is associated with a complete cure rate of 40%.4,25 Nevertheless, while this manuscript was in preparation, the patient presented in this case study underwent kidney transplantation. On day 8 postsurgery, he was no longer taking oxycodone, his skin condition was clearing up, and he was feeling considerably better. His health care providers hope for further regression from his disease.
Conclusion
NSF is just one example of iatrogenic conditions that can occur in any hospital, office, or clinic. Health care providers cannot be too vigilant in keeping abreast of warnings from the FDA and other agencies. In this case, several clinicians overlooked a recent, urgent public health advisory, with significant consequences.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
1. US Food and Drug Administration. Public health advisory: gadolinium-containing contrast agents for magnetic resonance imaging (MRI): Omniscan, OptiMARK, Magnevist, ProHance, and MultiHance. www.fda.gov/cder/drug/advisory/gadolinium_agents.htm. Accessed July 24, 2008.
2. Cowper SE, Su L, Bhawan J, et al. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383-393.
3. US Food and Drug Administration. Information for healthcare professionals: gadolinium-based contrast agents for magnetic resonance imaging (marketed as Magnevist, MultiHance, Omniscan, OptiMARK, ProHance). Last updated June 4, 2008. www.fda.gov/cder/drug/InfoSheets/HCP/gcca_200705.htm. Accessed July 24, 2008.
4. International Center for Nephrogenic Fibrosing Dermopathy Research. www.icnfdr.org. Accessed July 24, 2008.
5. DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial. 2006;19(3):191-194.
6. Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614-617.
7. Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med. 2006;145(3):234-235.
8. Miskulin D, Gul A, Rudnick MR, Cowper SE. Nephrogenic systemic fibrosis/nephrogenic fibrosing dermopathy in advanced renal failure. www.uptodate.com/patients/content/topic.do?topicKey=dialysis/48700. Accessed July 24, 2008.
9. Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104-1108.
10. Marckmann P, Skov L, Rossen K, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359-2362.
11. Centers for Disease Control and Prevention. Nephrogenic fibrosing dermopathy associated with exposure to gadolinium-containing contrast agents—St. Louis, Missouri, 2002-2006. MMWR Morb Mortal Wkly Rep. 2007;56(7):137-141.
12. Khurana A, Runge VM, Narayanan M, et al. Nephrogenic systemic fibrosis: a review of 6 cases temporally related to gadodiamide injection (Omniscan). Invest Radiol. 2007;42(2):139-145.
13. Sadowski EA, Bennett LK, Chan MR, et al. Nephrogenic systemic fibrosis: risk factors and incidence estimation. Radiology. 2007;243(1):148-157.
14. Morcos SK. Nephrogenic systemic fibrosis following the administration of extracellular gadolinium based contrast agents: is the stability of the contrast agent molecule an important factor in the pathogenesis of this condition? Br J Radiol. 2007;80(950):73-76.
15. GE Healthcare. Omniscan safety update. http://md.gehealthcare.com/omniscan/safety/index.html. Accessed July 24, 2008.
16. Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27-30.
17. High WA, Ayers RA, Chandler J, et al. Gadolinium is detectable within the tissue of patients with nephrogenic systemic fibrosis. J Am Acad Dermatol. 2007;56(1):21-26.
18. Thomsen H; European Society of Urogenital Radiology. European Society of Urogenital Radiology guidelines on contrast media application. Curr Opin Urol. 2007;17(1):70-76.
19. Bongartz G. Imaging in the time of NFD/NSF: do we have to change our routines concerning renal insufficiency? MAGMA. 2007;20(2):57-62.
20. Kanal E, Barkovich AJ, Bell C, et al; ACR Blue Ribbon Panel on MR Safety. ACR guidance document for safe MR practices: 2007. AJR Am J Roentgenol. 2007;188(6):1447-1474.
21. GE Healthcare Paper on Nephrogenic Systemic Fibrosis (March 2007). http://md.gehealthcare.com/omniscan/GE% 20Healthcare%20Paper%20On%20Nephrogenic%20 Systemic%20Fibrosis.pdf. Accessed July 24, 2008.
22. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development to gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267.
23. Okada S, Katagiri K, Kumazaki T, Yokoyama H. Safety of gadolinium contrast agent in hemodialysis patients. Acta Radiol. 2001;42(3):339-341.
24. Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647-649.
25. Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763-765.
Early Intervention for Diabetes-Related Renal Disease: Don't Let Rising Creatine Block the Way
‘Blood will have blood’

Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.
Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.
Erythropoiesis-stimulating agents (ESAs) changed the landscape, giving us the ability to normalize the hemoglobin level without giving blood products. Patients with renal failure who were making inadequate amounts of endogenous erythropoietin could be given exogenous ESAs. And patients with anemia characterized by resistance to erythropoietin could be given higher doses of an ESA, and the resistance could be overcome. Questions were occasionally raised about the outcomes when boosting the hemoglobin level above 10 g/dL, and seizures and hypertension were reported as complications of therapy, but cost was the major stumbling block to the expanded use of ESAs.
However, as Drs. Sevag Demirjian and Saul Nurko discuss in this issue of the Journal, striving to fully correct the anemia of chronic kidney disease with the use of ESAs may cause unexpected problems—such as more deaths and cardiovascular events.
Why should more patients with chronic kidney disease die if their hemoglobin levels are normalized? It could be another case of “messing with Mother Nature.” Perhaps the decreased erythropoietin production and anemia associated with renal failure are a protective reflex somehow beneficial to patients with decreased renal mass. On the other hand, it seems that the patients who had the most problems with ESAs in randomized trials were actually “resistant” to ESA therapy and were therefore probably given higher ESA doses. More blood may not be the problem, but, rather, too much ESA.
In an editorial in this issue, Dr. Alan Lichtin discusses additional concerns that have arisen with the use of ESAs when treating the anemia associated with malignancy. One issue relates to the expression of erythropoietin receptors by nonerythroid cells: some tumor cells express erythropoietin receptors, and giving high doses of ESAs might stimulate their growth. Dr. Lichtin concludes by saying the ESA story is far from over, and I believe him.